|
NCT02128282
|
Cholangiocarcinoma |
Phase 1
Phase 2 |
Recruiting |
November 2021 |
United States, Arizona
... More >>
Mayo Clinic
Recruiting
Scottsdale, Arizona, United States, 85259-5499
Contact: Mayo Clinic Clinical Trials Office 855-776-0015
Principal Investigator: Mitesh Borad, M.D.
United States, Colorado
University of Colorado- Denver
Recruiting
Aurora, Colorado, United States, 80045
Contact: Amy Szilard 720-848-0702 Amy.Szilard@ucdenver.edu
Principal Investigator: Sarah (Lindsey) Davis, MD
United States, Florida
Mayo Clinic
Recruiting
Jacksonville, Florida, United States, 32224
Contact: Mayo Clinic Clinical Trials Office 855-776-0015
Principal Investigator: Kabir Mody, MD
United States, Minnesota
Mayo Clinic
Recruiting
Rochester, Minnesota, United States, 55905
Contact: Mayo Clinic Clinical Trials Office 855-776-0015
Principal Investigator: Joleen Hubbard, MD
United States, Texas
Texas Oncology - Baylor Charles A. Sammons Cancer Center
Recruiting
Dallas, Texas, United States, 75246
Contact: Tammy Carmical, RN 214-370-1937 tammy.carmical@usoncology.com
Principal Investigator: Carlos Becerra, M.D.
Texas Oncology-Tyler
Recruiting
Tyler, Texas, United States, 75702
Contact: Karen Poe, RN 903-579-9869 karen.poe@usoncology.com
Principal Investigator: Donald A Richards, M.D.
Korea, Republic of
Asan Medical Center
Recruiting
Seoul, Songpa-gu, Korea, Republic of, 138-736
Contact: Heung-Moon Chang, MD 82-3010-3219 ext 3210 changhm@amc.seoul.kr
Contact: Seok kyung Jeong 82-2-3010-5634 jsk0213@amc.seoul.kr
Samsung Medical Center
Recruiting
Seoul, Korea, Republic of
Contact: Eunyou Lee 82-2-3410-0955 ley0709@samsung.com
Principal Investigator: Joon Oh Park, MD
Seoul National University Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Myoungsun Choi 82-2-2072-7612 iamyou3@hanmail.net
Principal Investigator: Do-Youn Oh, MD
Severance Hospital, Yonsei University Health System
Recruiting
Seoul, Korea, Republic of
Contact: So Young Hwang 82-2-2228-8180 syhwang@yuhs.ac
Principal Investigator: Sun Young Rha, MD
Taiwan
China Medical University Hospital
Recruiting
Taichung City, Taiwan
Contact: Pei-Chen Hsu +886-4-2205-2121 peggyshiu0807@gmail.com
Principal Investigator: Li-Yuan Bai, M.D. Less << |
|
NCT00915382
|
Advanced Gastric Cancer |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Department of Oncology, Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT03427359
|
Nasopharyngeal Carcinoma |
Phase 2 |
Completed |
- |
- |
|
NCT02639091
|
Medical Oncology |
Phase 1 |
Active, not recruiting |
June 28, 2019 |
United States, Illinois
... More >>
Chicago, Illinois, United States, 60637
United States, Maryland
Bethesda, Maryland, United States, 20892
United States, Michigan
Detroit, Michigan, United States, 48202
United States, South Carolina
Charleston, South Carolina, United States, 29425
Italy
Milano, Lombardia, Italy, 20089
Milano, Lombardia, Italy, 20133 Less << |
|
NCT00522795
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
United States, Rhode Island
... More >>
Lifespan Hospitals
Providence, Rhode Island, United States, 02903 Less << |
|
NCT02897375
|
Solid Neoplasm
... More >> Stage III Pancreatic Cancer
Stage IIIA Breast Cancer
Stage IIIA Non-Small Cell Lung Cancer
Stage IIIB Breast Cancer
Stage IIIB Non-Small Cell Lung Cancer
Stage IIIC Breast Cancer
Stage IV Breast Cancer
Stage IV Non-Small Cell Lung Cancer
Stage IVA Pancreatic Cancer
Stage IVB Pancreatic Cancer
Sarcoma
Colorectal Cancer
Head and Neck Cancer
Cancer of Unknown Primary
Bladder Cancer
Ovarian Cancer Less << |
Phase 1 |
Recruiting |
December 2021 |
United States, Georgia
... More >>
Emory University/Winship Cancer Institute
Recruiting
Atlanta, Georgia, United States, 30322
Contact: Nakita Brown 404-616-9850 nlbrown@emory.edu
Contact: Monique Williams 404-778-4383 monique.guyinn@emory.edu Less << |
|
NCT01417390
|
Nasopharyngeal Carcinoma |
Phase 2 |
Unknown |
December 2015 |
China, Guangdong
... More >>
Wei-wei Hong
Recruiting
Foshan, Guangdong, China, 528000
Contact: Wei wei Hong, M.D.
Sub-Investigator: Tao Xu, M.D. Less << |
|
NCT01918306
|
- |
|
Terminated(company stopped pro... More >>duction of study drug due to excessive toxicities, lack of efficacy) Less << |
- |
- |
|
NCT02585973
|
Carcinoma, Squamous Cell of He... More >>ad and Neck Less << |
Phase 1 |
Recruiting |
November 2021 |
United States, North Carolina
... More >>
UNC Lineberger Comprehensive Cancer Center
Recruiting
Chapel Hill, North Carolina, United States, 27599
Contact: Dan Goldin 919-966-4432 dan_goldin@med.unc.edu
Contact: Chris Hilliard 919-966-4432 Chris_Hilliard@med.unc.edu Less << |
|
NCT01918306
|
Estrogen Receptor Negative Bre... More >>ast Cancer
Human Epidermal Growth Factor 2 Negative Carcinoma of Breast
Triple Negative Breast Cancer
Recurrent Breast Cancer
Stage IV Breast Cancer
Triple-negative Breast Cancer Less << |
Phase 1
Phase 2 |
Terminated(company stopped pro... More >>duction of study drug due to excessive toxicities, lack of efficacy) Less << |
- |
United States, Alabama
... More >>
University of Alabama
Birmingham, Alabama, United States
United States, California
University of California, San Francisco
San Francisco, California, United States
United States, District of Columbia
Georgetown University
Washington, D.C., District of Columbia, United States
United States, Georgia
Emory University
Atlanta, Georgia, United States
United States, Illinois
University of Chicago
Chicago, Illinois, United States
United States, Indiana
Indiana University
Indianapolis, Indiana, United States
United States, Maryland
John Hopkins University
Baltimore, Maryland, United States
United States, Massachusetts
Dana Farber Cancer Institute
Boston, Massachusetts, United States
United States, Michigan
University of Michigan
Ann Arbor, Michigan, United States
United States, Minnesota
Mayo Clinic
Rochester, Minnesota, United States
United States, New York
Memorial Sloan-Kettering Cancer Center
New York, New York, United States
United States, North Carolina
University of North Carolina
Charlotte, North Carolina, United States
United States, Pennsylvania
University of Pittsburgh
Pittsburgh, Pennsylvania, United States
United States, Tennessee
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States, 37232
United States, Texas
Baylor Breast Center
Houston, Texas, United States
United States, Washington
University of Washington
Seattle, Washington, United States Less << |
|
NCT03168594
|
Neuroendocrine Carcinoma |
Phase 2 |
Recruiting |
May 1, 2020 |
China, Beijing
... More >> Beijing Cancer Hospital
Recruiting
Beijing, Beijing, China, 100142
Contact: Shen Lin, Professor 010-88196561 Linshenpku@163.com
Principal Investigator: Shen Lin, professor Less << |
|
NCT03649048
|
Locally Advanced Head and Neck... More >> Squamous Cell Carcinoma Less << |
Not Applicable |
Not yet recruiting |
February 2025 |
- |
|
NCT01426646
|
Gastric Cancer |
Phase 2 |
Unknown |
September 2016 |
Korea, Republic of
... More >>
Donga university hospital
Recruiting
Busan, Korea, Republic of, 602-715
Contact: Hyuk-Chan Kwon, Professor
Principal Investigator: Hyuk-Chan Kwon, Professor
Ulsan University Hospital
Recruiting
Ulsan, Korea, Republic of, 682-714
Contact: Jin Ho Baek, Professor
Principal Investigator: Jin-Ho Baek, Professor Less << |
|
NCT02407912
|
Pleural Effusion, Malignant |
Not Applicable |
Recruiting |
March 2020 |
China, Guangdong
... More >>
Department of medical oncology
Recruiting
Shantou, Guangdong, China, 515031
Contact: Hongbiao Wang, Master 8613592882093 wanghongbiao123@qq.com Less << |
|
NCT00858663
|
Head and Neck Cancer |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT01345357
|
Solid Tumors or Mantle Cell Ly... More >>mphoma Less << |
Phase 1 |
Completed |
- |
Belgium
... More >> Teva Investigational Site 3
Bruxelles, Belgium
France
Teva Investigational Site 1
Nantes, France
Teva Investigational Site 2
Villejuif, France Less << |
|
NCT01009346
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Terminated(Toxicity) |
- |
United States, Maryland
... More >>
Johns Hopkins University
Baltimore, Maryland, United States, 21231 Less << |
|
NCT00998036
|
Triple Negative Breast Cancer |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Columbia University Medical Center
New York, New York, United States, 10032 Less << |
|
NCT01574092
|
Pediatric High Risk Gliomas |
Phase 2 |
Completed |
- |
Spain
... More >> Hospital Sant Joan De Déu
Esplugues de Llobregat, Barcelona, Spain, 08950 Less << |
|
NCT00522795
|
- |
|
Completed |
- |
- |
|
NCT00148694
|
Breast Cancer |
PHASE2 |
COMPLETED |
2025-05-10 |
Beth Israel Deaconess Medical ... More >>Center, Boston, Massachusetts, 02115, United States|Dana-Farber Cancer Institute, Boston, Massachusetts, 02115, United States|Massachusetts General Hospital, Boston, Massachusetts, 02115, United States Less << |
|
NCT01073839
|
Cholangiocellular Carcinoma |
Phase 1
Phase 2 |
Completed |
- |
Switzerland
... More >>
Zurich, Switzerland Less << |
|
NCT00922896
|
Metastatic Pancreatic Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Dong-A University Hospital
Busan, Korea, Republic of, 602-715
Gyeongsang Unversity Hospital
JinJU, Korea, Republic of
Chung-Ang University Hospital
Seoul, Korea, Republic of, 156-755 Less << |
|
NCT03557112
|
Nasopharyngeal Carcinoma |
Phase 2 |
Recruiting |
December 1, 2021 |
China, Guizhou
... More >> Cancer Hospital of Guizhou Medical University
Recruiting
Guiyang, Guizhou, China, 550000
Contact: Feng Jin, Bachelor 86-13985124806 jinf8865@yahoo.com.cn
Contact: Weili Wu, Master 86-13885124077 wwlmhy@163.com
Sub-Investigator: Weili Wu, master
Sub-Investigator: Yuanyuan Li, master
Sub-Investigator: Jinhua Long, master
Sub-Investigator: Xiuyun Gong, bachelor
Sub-Investigator: Xiaoxiao Chen, bachelor
Sub-Investigator: Yu Chen, master Less << |
|
NCT02632305
|
Unresectable Biliary Tract Can... More >>cer Less << |
Phase 2 |
Recruiting |
June 2020 |
Canada, Alberta
... More >>
Cross Cancer Institute
Recruiting
Edmonton, Alberta, Canada
Contact: Jennifer Spratlin, MD FRCPC Jennifer.Spratlin@albertahealthservices.ca Less << |
|
NCT01579929
|
Lung Cancer |
Phase 1 |
Completed |
- |
United States, New Jersey
... More >>
Memoral Sloan Kettering Cancer Center
Basking Ridge, New Jersey, United States
United States, New York
Memorial Sloan Kettering Cancer Center at Commack
Commack, New York, United States, 11725
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan Kettering Cancer Center at Mercy Medical Center
Rockville Centre, New York, United States, 11570
Memoral Sloan Kettering Cancer Center@Phelps Memorial Hospital
Sleepy Hollow, New York, United States Less << |
|
NCT03371550
|
Locally Advanced Non-small Cel... More >>l Lung Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT03012477
|
Triple-negative Metastatic Bre... More >>ast Cancer Less << |
Phase 2 |
Recruiting |
December 2024 |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Recruiting
Boston, Massachusetts, United States, 02114
Contact: Steven Isakoff, MD, PhD
Contact 617-726-4920 sisakoff@partners.org
Dana-Farber Cancer Institute
Recruiting
Boston, Massachusetts, United States, 02215
Contact: Sara Tolaney, MD, MPH 617-632-3800 stolaney@partners.org
Principal Investigator: Sara Tolaney, MD, MPH Less << |
|
NCT01661114
|
Pancreatic Cancer
... More >> Biliary Cancer Less << |
Phase 2 |
Completed |
- |
United States, Michigan
... More >>
University of Michigan Comprehensive Cancer Center
Ann Arbor, Michigan, United States, 48109 Less << |
|
NCT01661114
|
- |
|
Completed |
- |
- |
|
NCT03660449
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Phase 2 |
Recruiting |
August 2023 |
China, Guangdong
... More >>
Hui Liu
Recruiting
Guangzhou, Guangdong, China, 510000
Contact: Hui Liu, professor +86-020-87343031 liuhui@sysucc.org.cn Less << |
|
NCT02967887
|
Hepatocellular Carcinoma Non-R... More >>esectable Less << |
Phase 2 |
Recruiting |
December 2018 |
Korea, Republic of
... More >>
Korea University Ansan Hospital
Not yet recruiting
Ansan, Korea, Republic of
Contact: Yim, MD., PhD.
Sooncheonhyang University Hospital Bucheon
Not yet recruiting
Bucheon, Korea, Republic of
Contact: Kim, MD., PhD.
The Catholic University of Korea Bucheon ST. Mary's Hospital
Not yet recruiting
Bucheon, Korea, Republic of
Contact: Lee, MD., PhD.
Keimyung University Dongsan Medical Center
Not yet recruiting
Daegu, Korea, Republic of
Contact: Chung, MD., PhD.
The Catholic University of Korea Daejeon ST. Mary's Hospital
Not yet recruiting
Daejeon, Korea, Republic of
Contact: Song, MD., PhD.
Chonnam National University Hwasun Hospital
Not yet recruiting
Hwasun, Korea, Republic of
Contact: Cho, MD., PhD.
Korea University Anam Hospital
Not yet recruiting
Seoul, Korea, Republic of
Contact: Seo, MD., PhD.
Seoul National University Hospital
Not yet recruiting
Seoul, Korea, Republic of
Contact: Yoon, MD., PhD.
Sooncheonhyang University Hospital Seoul
Recruiting
Seoul, Korea, Republic of
Contact: Jang, MD., PhD.
The Catholic University of korea, Seoul ST. Mary's Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Si Hyun Bae, MD., PhD.
Ajou University Hospital
Recruiting
Suwon, Korea, Republic of
Contact: Wang, MD., PhD.
Contact: Cheong, MD., PhD.
The Catholic University of Korea ST. Vincent's Hospital
Not yet recruiting
Suwon, Korea, Republic of
Contact: Song, MD., PhD. Less << |
|
NCT01275183
|
Head and Neck Cancer |
Early Phase 1 |
Completed |
- |
United States, New Mexico
... More >>
University of New Mexico Cancer Center
Albuquerque, New Mexico, United States, 87106 Less << |
|
NCT03296306
|
Bladder Cancer
... More >> Ureter Cancer
Urethral Cancer
Transitional Cell Carcinoma Less << |
Phase 3 |
Recruiting |
February 2022 |
Korea, Republic of
... More >>
Kwonoh Park
Recruiting
Yangsan, Gyeongsangnam-do, Korea, Republic of, 50612
Contact: Kwonoh Park, MD, PhD 1033783529 parkkoh@daum.net
Hallym University Medical Center, Hallym University College of Medicine
Recruiting
Anyang, Korea, Republic of
Contact: Ho Young Kim ksfam@daum.net
Fatima Hospital
Recruiting
Daegu, Korea, Republic of
Contact: Jung Lim Lee junglim3@gmail.com
Keimyeong University Dongsan Medical Center
Recruiting
Daegu, Korea, Republic of
Contact: Jin Young Kim, MD takgu@dsmc.or.kr
Chungnam University Hospital
Recruiting
Daejeon, Korea, Republic of
Contact: Hyo Jin Lee, MD, PhD. cymed@cnu.ac.kr
National Health Insurance Service Ilsan Hospital
Recruiting
Goyang, Korea, Republic of
Contact: Soo Jung Hong suzzy901@nhimc.or.kr
Gil Medical Center
Recruiting
Incheon, Korea, Republic of
Contact: Inkeun Park, MD, PhD ingni79@hanmail.net
Inje University Haeundae Paik Hospital
Recruiting
Pusan, Korea, Republic of
Contact: Il-Hwan Kim onelement@daum.net
Kosin University Hospital
Recruiting
Pusan, Korea, Republic of
Contact: Sung-Hoon Shin ssh1533@daum.net
Pusan National University Hospital, Pusan National University School of Medicine
Recruiting
Pusan, Korea, Republic of
Contact: Hyo Jung Kim leonkim80@naver.com
Asan Medical Center
Recruiting
Seoul, Korea, Republic of
Contact: Jae-Lyun Lee, MD, PhD jaelyun@amc.seoul.kr
Chung Ang University Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Hee Joon Kim
Inje University Sanggye Paik Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Byeong Seok Sohn imbs@paik.ac.kr
Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine
Recruiting
Seoul, Korea, Republic of
Contact: Yun-Gyoo Lee gosciny@gmail.com
Korea University Anam Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Yoon Ji Choi yoonji23@hanmail.net
Seoul National University Hospital, Cancer Research Institute, Seoul National University College of Medicine
Recruiting
Seoul, Korea, Republic of
Contact: Bhum Suk Keam
Contact bhumsuk@snu.ac.kr
VHS medical center
Recruiting
Seoul, Korea, Republic of
Contact: Bong-Seog Kim
Yonsei Cancer Center
Recruiting
Seoul, Korea, Republic of
Contact: Sang Joon Shin ssj338@yuhs.ac
St. Vincent's Hospital, The Catholic University of Korea
Recruiting
Suwon, Korea, Republic of
Contact: Byoung Yong Shim shimby@catholic.ac.kr Less << |
|
NCT01587820
|
Head and Neck Cancer |
Early Phase 1 |
Terminated(lack of enrollment) |
- |
United States, Illinois
... More >>
Southern Illinois University School of Medicine
Springfield, Illinois, United States, 62701
Simmons Cancer Institute at SIU School of Medicine
Springfield, Illinois, United States, 62702 Less << |
|
NCT00580333
|
Breast Cancer |
Phase 2 |
Active, not recruiting |
June 2020 |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02115
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT02291211
|
Stage IV Gastric Cancer |
Phase 2 |
Unknown |
December 2016 |
China, Shaanxi
... More >> IEC of Insititution for National Drug Clinical Trials,Tangdu Hospital,Fourth Military Medical University
Recruiting
Xi'an, Shaanxi, China, 029
Contact: Lina Liu 029-84777631 tangduec@126.com Less << |
|
NCT03068936
|
Nasopharyngeal Carcinoma |
Phase 3 |
Not yet recruiting |
September 2021 |
- |
|
NCT02546934
|
Breast Cancer |
Phase 2 |
Recruiting |
December 2019 |
China
... More >> Fudan University Cancer Hospital
Recruiting
Shanghai, China, 200032
Contact: Xichun Hu, MD,PhD 64175590 ext 5006 huxicun@gmail.com
Contact: Zhonghua Wang, MD 64175590 ext 5000 zhonghuawang95@hotmail.com Less << |
|
NCT01554410
|
Cervical Carcinoma |
Phase 1 |
Unknown |
August 2015 |
United States, California
... More >>
Moores UC San Diego Cancer Center
Recruiting
La Jolla, California, United States, 92093
Contact: Norma Hernandez 858-822-5036 nohernandez@ucsd.edu
Principal Investigator: Loren Mell, MD
Sub-Investigator: Arno Mundt, MD
Sub-Investigator: Catheryn Yashar, MD
Sub-Investigator: Steven Plaxe, MD
Sub-Investigator: Michael McHale, MD
Sub-Investigator: Cheryl Saenz, MD Less << |
|
NCT00580333
|
- |
|
Active, not recruiting |
- |
- |
|
NCT02202772
|
Urothelial Carcinoma of the Ur... More >>inary Bladder Less << |
Phase 1 |
Active, not recruiting |
December 1, 2020 |
United States, New York
... More >>
Columbia University Medical Center- HIP
New York, New York, United States, 10032 Less << |
|
NCT01312311
|
Nasopharyngeal Cancer |
Phase 2 |
Unknown |
- |
Korea, Republic of
... More >>
Samsung Medical Center
Recruiting
Seoul, Korea, Republic of, 135-710
Contact: Myung-Ju Ahn, Pf 822-3410-3459 silkahn@skku.edu Less << |
|
NCT01743365
|
Gastric Cancer
... More >> Gastroesophageal Junction Cancer Less << |
Phase 2 |
Active, not recruiting |
June 2019 |
Greece
... More >> 2nd Dept of Internal Medicine, Agios Savvas Cancer Hospital
Athens, Greece, 11522
Dept of Medical Oncology, 251 Air Force Hospital
Athens, Greece, 11525
2nd Dept of Internal Medicine, General Hospital of Athens "Hippokratio"
Athens, Greece, 11527
Oncology Section, Dept of Clinical Therapeutics, General Hospital of Athens "Alexandra"
Athens, Greece, 11528
Division of Oncology, 2nd Dept of Internal Medicine, University Hospital "Attikon"
Athens, Greece, 12462
2nd Dept of Medical Oncology, Agii Anargiri Cancer Hospital
Athens, Greece, 14564
3rd Dept of Medical Oncology, Agii Anargiri Cancer Hospital
Athens, Greece, 14564
3rd Dept of Medical Oncology, Hygeia Hospital
Athens, Greece, 15123
1st Dept of Medical Oncology, Metropolitan Hospital
Athens, Greece, 18547
2nd Dept of Medical Oncology, Metropolitan Hospital
Athens, Greece, 18547
Dept of Medical Oncology, University Hospital of Heraklion
Heraklion, Greece, 71110
Dept of Medical Oncology, Ioannina University Hospital
Ioannina, Greece, 45110
Division of Oncology, Dept of Internal Medicine, University Hospital of Patras
Patras, Greece, 26504
Dept of Medical Oncology, Papageorgiou General Hospital
Thessaloniki, Greece, 56429
Dept of Medical Oncology, Thermi Clinic S.A
Thessaloniki, Greece, 57001 Less << |
|
NCT02143388
|
Local Advanced High Risk Nasop... More >>haryngeal Carcinoma Less << |
Phase 2
Phase 3 |
Recruiting |
December 2018 |
China, Guangdong
... More >>
Cancer Center, Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China
Contact: Zhao chong, M.D +86-13902206160 zhaochong@sysucc.org.cn Less << |
|
NCT01009346
|
- |
|
Terminated(Toxicity) |
- |
- |
|
NCT03311789
|
Biliary Tract Cancer |
Phase 1
Phase 2 |
Recruiting |
October 2019 |
China
... More >> Department of Biotherapeutic, Chinese PLA General Hospital
Recruiting
Beijing, China
Contact: Weidong Han, Ph.D +86-10-66937463 hanwdrsw@sina.com
Contact: Kaichao Feng, MD +86-10-55499341 timothyfkc@126.com Less << |
|
NCT01372904
|
Cisplatin Ototoxicity |
Phase 4 |
Completed |
- |
Israel
... More >> Clalit Health Services
Haifa, Israel Less << |
|
NCT01182168
|
Bladder Cancer
... More >> Renal Pelvis Cancer
Ureter Cancer Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT00353015
|
Gastrointestinal Cancer
... More >> Carcinoma, Neuroendocrine Less << |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
U.T. M.D. Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT03601975
|
Nasopharyngeal Carcinoma |
Phase 3 |
Recruiting |
July 30, 2021 |
China, Tianjin
... More >> 20th Floor, Block C, Lake Road, Hexi District, Tianjin Medical University Cancer Institute and Hospital .
Recruiting
Tianjin, Tianjin, China, 300600
Contact: Ming Gao, doctor yunspider@126.com Less << |
|
NCT02481518
|
Acute Kidney Injury |
Phase 4 |
Recruiting |
July 2019 |
Thailand
... More >> Faculty of medicine, Ramathibodi Hospital, Mahidol University
Recruiting
Bangkok, Thailand, 10400
Contact: Arkom Nongnuch, MD. +6622011400 oatega@yahoo.com Less << |
|
NCT00257816
|
Cervical Cancer |
Phase 2 |
Completed |
- |
United States, California
... More >>
Chao Family Comprehensive Cancer Center
Orange, California, United States, 92868 Less << |
|
NCT01320254
|
Cholangiocarcinomas |
Phase 2 |
Completed |
- |
Germany
... More >> Esslingen Hospital
Esslingen, Baden-Wuerttemberg, Germany, 73730
University Hospital Freiburg
Freiburg, Baden-Wuerttemberg, Germany, 79106
National Centre for Tumor Diseases (NCT)
Heidelberg, Baden-Wuerttemberg, Germany, 69120
University Hospital Mannheim
Mannheim, Baden-Wuerttemberg, Germany, 68167
Kreiskliniken Reutlingen GmbH
Reutlingen, Baden-Wuerttemberg, Germany, 72764
University Hospital Tuebingen
Tuebingen, Baden-Wuerttemberg, Germany, 72076
Klinikum rechts der Isar der TU München
München, Bavaria, Germany, 81675
University Hospital Regensburg
Regensburg, Bavaria, Germany, 93042
Charité Berlin
Berlin, Berlin-City, Germany, 13353
University Hospital Hamburg-Eppendorf
Hamburg, Free City of Hamburg, Germany, 20246
University Hospital Marburg
Marburg, Hesse, Germany, 35043
Medical School Hannover
Hannover, Lower Saxony, Germany, 30625
University Hospital Essen
Essen, Northrhine-Westfalia, Germany, 45122
University Hospital Köln
Köln, Northrhine-Westfalia, Germany, 50924
University Hospital Mainz
Mainz, Rhineland-Palatinate, Germany, 55131
Magdeburg Hospital
Magdeburg, Saxony-Anhalt, Germany, 39130 Less << |
|
NCT02838745
|
Mesothelioma |
Phase 1 |
Recruiting |
August 2018 |
United States, Texas
... More >>
Baylor St Lukes
Recruiting
Houston, Texas, United States, 77030
Contact: Michelle G Almarez, BBA 713-798-3680 Michelle.Almarez@bcm.edu
Contact: Bryan Burt, MD 713-798-8266 Bryan.Burt@bcm.edu
Principal Investigator: Bryan Burt, MD Less << |
|
NCT02512692
|
Intrahepatic Cholangiocarcinom... More >>a Less << |
Not Applicable |
Recruiting |
August 2021 |
United States, South Carolina
... More >>
Medical University of South Carolina
Recruiting
Charleston, South Carolina, United States, 29425
Contact: Sarah Annand 843-792-9321 hcc-clinical-trials@musc.edu Less << |
|
NCT01261728
|
Urothelial Carcinoma |
Phase 2 |
Recruiting |
December 2019 |
United States, Connecticut
... More >>
Hartford Hospital
Recruiting
Hartford, Connecticut, United States
Contact: Robert Siegel, MD
Principal Investigator: Robert Siegel, MD
United States, Florida
Baptist Alliance MCI
Recruiting
Miami, Florida, United States, 33143
Contact: Antonio Muina, MD 786-596-2000
United States, New Jersey
Memorial Sloan Kettering at Basking Ridge
Recruiting
Basking Ridge, New Jersey, United States, 07920
Contact: Jonathan Coleman, MD 646-422-4432
Memorial Sloan Kettering Bergen
Recruiting
Montvale, New Jersey, United States, 07645
Contact: Jonathan Coleman, MD 646-422-4432
United States, New York
Memorial Sloan Kettering Cancer Center @ Suffolk
Recruiting
Commack, New York, United States, 11725
Contact: Jonathan Coleman, MD 646-422-4432
Memorial Sloan Kettering Westchester
Recruiting
Harrison, New York, United States, 10604
Contact: Jonathan Coleman, MD 646-422-4432
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10065
Contact: Jonathan Coleman, MD 646-422-4432
Contact: Dean Bajornin, MD 646-422-4333
Memorial Sloan Kettering at Mercy Medical Center
Recruiting
Rockville Centre, New York, United States
Contact: Jonathan Coleman, MD 646-422-4432
United States, Pennsylvania
Lehigh Valley Health Network
Recruiting
Allentown, Pennsylvania, United States, 18103
Contact: Jonathan Coleman, MD 646-422-4432 Less << |
|
NCT01650090
|
Pulmonary Relapse of Osteosarc... More >>oma Less << |
Phase 2 |
Active, not recruiting |
December 2018 |
United States, California
... More >>
Children's Hospital Los Angeles
Los Angeles, California, United States, 90027
Stanford University Medical Center
Palo Alto, California, United States, 94304
United States, Florida
H. Lee Moffitt Cancer Center
Tampa, Florida, United States, 33612
United States, Illinois
University of Chicago
Chicago, Illinois, United States, 60637
United States, Louisiana
Ochsner Clinic Foundation
New Orleans, Louisiana, United States, 70121
United States, Massachusetts
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02215
United States, Minnesota
Mayo Clinic
Rochester, Minnesota, United States, 55905
United States, Missouri
Washington University School of Medicine
Saint Louis, Missouri, United States, 63110
United States, New York
The Children's Hospital at Montefiore
Bronx, New York, United States, 10467
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065
United States, Tennessee
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States, 37232
United States, Texas
UT Southwestern Medical Center
Dallas, Texas, United States, 75390
Baylor College of Medicine/Texas Children's Hospital
Houston, Texas, United States, 77030
United States, Utah
Primary Children's Medical Center
Salt Lake City, Utah, United States, 84113
United States, Washington
Seattle Children's Hospital
Seattle, Washington, United States, 98105 Less << |
|
NCT02128906
|
Squamous Cell Carcinoma |
Phase 2 |
Recruiting |
December 2020 |
United States, Pennsylvania
... More >>
University of Pittsburgh
Recruiting
Pittsburgh, Pennsylvania, United States, 15232
Contact: Karen Holeva, RN 412-623-1275 holevakd@upmc.edu Less << |
|
NCT01285674
|
Cisplatin
Oto... More >>toxicity
Intratympanic Steroids Less << |
Not Applicable |
Unknown |
January 2012 |
Israel
... More >> Ziv Medical Center
Recruiting
Safed, Israel, 13100
Contact: Peter Gilbey, MD 972-50-8434014 peter.g@ziv.health.gov.il
Principal Investigator: Peter Gilbey, MD
Sub-Investigator: Khalil Khalil, MD
Sub-Investigator: Jamal Zidan, MD Less << |
|
NCT01444547
|
Esophageal Cancer |
Phase 2 |
Unknown |
December 2015 |
China
... More >> Cancer Institute and Hospital, Chinese Academy of Medical Sciences
Recruiting
Beijing, China, 100021
Contact: Yi Zhou, M.D 8610-87788800 mondaycm@yahoo.com.cn
Contact: Xiao Lv, M.D 8610-87788800 xiaoxiao81473@126.com
Principal Investigator: Jing Huang, M.D.,Ph.D Less << |
|
NCT03079427
|
Cholangiocarcinoma
... More >> Biliary Tract Cancer
Adjuvant Chemotherapy
Capecitabine
Gemcitabine
Cisplatin Less << |
Phase 2 |
Recruiting |
April 2022 |
Korea, Republic of
... More >>
Asan Medical Center, University of Ulsan College of Medicine
Recruiting
Seoul, Korea, Republic of, 05505
Contact: Changhoon Yoo, MD +82-2-3010-1727 yooc@amc.seoul.kr Less << |
|
NCT01801644
|
Bladder Cancer |
Not Applicable |
Completed |
- |
Austria
... More >> Barmherzige Brüder Vienna
Vienna, Austria, 1020 Less << |
|
NCT03193853
|
Triple Negative Breast Cancer |
Phase 2 |
Recruiting |
June 9, 2022 |
United States, Texas
... More >>
Baylor University Medical Center
Recruiting
Dallas, Texas, United States, 75246
Contact: Amy Solis 214-820-8685 amy.solis@bswhealth.org
Contact: Silviya Meletath, MD 214-820-4987 silviya.meletath@bswhealth.org
Principal Investigator: Joyce O'Shaughnessy, MD Less << |
|
NCT01444521
|
Gastric Cancer |
Phase 2 |
Completed |
- |
China
... More >> Cancer Institute and Hospital, Chinese Academy of Medical Sciences
Beijing, China, 100021 Less << |
|
NCT02351765
|
Biliary Tract Cancer
... More >> Gallbladder Cancer
Cholangiocarcinoma
Ampullary Cancer Less << |
Phase 1 |
Active, not recruiting |
February 2019 |
United Kingdom
... More >> Beatson Oncology Centre
Glasgow, United Kingdom
Clatterbridge Cancer Centre
Liverpool, United Kingdom
Imperial College London
London, United Kingdom
University College London
London, United Kingdom
The Christie NHS Foundation Trust
Manchester, United Kingdom Less << |
|
NCT00968435
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
United States, New Jersey
... More >>
Memorial Sloan Kettering Cancer Center at Basking Ridge
Basking Ridge, New Jersey, United States, 07939
United States, New York
Memorial Sloan Kettering Cancer Center at Commack
Commack, New York, United States, 11725
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan Kettering Cancer Center at Mercy Medical Center
Rockville Centre, New York, United States, 11570
Memorial Sloan Kettering Cancer Center at Phelps Memorial Hospital Center
Sleepy Hollow, New York, United States, 10591 Less << |
|
NCT03275857
|
Prostate Cancer |
EARLY_PHASE1 |
COMPLETED |
2023-07-31 |
Wilmot Cancer Institute, Roche... More >>ster, New York, 14642, United States Less << |
|
NCT01096745
|
Metastatic Biliary Tract Cance... More >>r
Locally Advanced Biliary Tract Cancer Less << |
Phase 2 |
Terminated(Planning for random... More >>ized phase III trial for this issue.) Less << |
September 2013 |
Korea, Republic of
... More >>
Gyeongsang University Hospital
Jinju, Gyeongsang Namdo, Korea, Republic of, 660-702 Less << |
|
NCT01872962
|
Nasopharyngeal Carcinoma |
Phase 3 |
Active, not recruiting |
November 2020 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT02254681
|
Intrahepatic Cholangiocarcinom... More >>a Less << |
Phase 2 |
Terminated(lack of funding) |
- |
United States, Minnesota
... More >>
Virginia Piper Cancer Institute
Minneapolis, Minnesota, United States, 55407 Less << |
|
NCT01336842
|
Solid Tumors
... More >>Non-Small Cell Lung Cancer (NSCLC) Less << |
Phase 1 |
Completed |
- |
United States, California
... More >>
University of California Davis Cancer Center
Sacramento, California, United States, 95817
United States, Michigan
Henry Ford Health System
Detroit, Michigan, United States, 48202 Less << |
|
NCT00808145
|
Hepatocellular Carcinoma |
Phase 2 |
Withdrawn(Lack of enrollment, ... More >>many screen failures and non eligible study participants) Less << |
- |
United States, Massachusetts
... More >>
Lahey Clinic
Burlington, Massachusetts, United States, 01805 Less << |
|
NCT02254681
|
- |
|
Terminated(lack of funding) |
- |
- |
|
NCT01982448
|
Triple Negative Breast Cancer |
Phase 2 |
Active, not recruiting |
June 2021 |
United States, Alabama
... More >>
University of Alabama
Birmingham, Alabama, United States, 35294
United States, Indiana
Indiana University- Simon Cancer Center
Indianapolis, Indiana, United States, 46202
United States, Maryland
Johns Hopkins University
Baltimore, Maryland, United States, 21287
United States, Massachusetts
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02215
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02215
South Shore Hospital
Weymouth, Massachusetts, United States, 02190
United States, New Jersey
Memorial Sloan Kettering Cancer Center-Basking Ridge
Basking Ridge, New Jersey, United States, 07920
Memorial Sloan Kettering Cancer Center-Monmouth
Middletown, New Jersey, United States, 07748
United States, New York
Memorial Sloan Kettering Cancer Center-Commack
Commack, New York, United States, 11725
Memorial Sloan Kettering Cancer Center-West Harrison
Harrison, New York, United States, 10604
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan Kettering Cancer Center-Rockville Centre
Rockville Centre, New York, United States, 11570
Memorial Sloan Kettering Cancer Center-Sleepy Hollow
Sleepy Hollow, New York, United States, 10591
United States, North Carolina
University of North Carolina- Lineberger Cancer Center
Chapel Hill, North Carolina, United States, 27599
Duke University
Durham, North Carolina, United States, 27710
United States, Pennsylvania
Universtiy of Pittsburgh- Magee-Womens Hospital
Pittsburgh, Pennsylvania, United States, 15213
United States, Tennessee
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States, 37232
United States, Texas
Baylor College of Medicine
Houston, Texas, United States, 77030
United States, Washington
Seattle Cancer Alliance at EvergreenHealth
Kirkland, Washington, United States, 98034
University of Washignton
Seattle, Washington, United States, 98195 Less << |
|
NCT02207660
|
- |
|
Completed |
- |
Taiwan
... More >> National Taiwan University Hospital
Taipei, Taiwan Less << |
|
NCT01869023
|
Advanced Malignant Pleural Mes... More >>othelioma Less << |
Phase 2 |
Active, not recruiting |
December 2017 |
Mexico
... More >> Instituto Nacional de Cancerología
Mexico City, Mexico Less << |
|
NCT00935961
|
HEAD & NECK Cancer |
Phase 1 |
Completed |
- |
United States, New Jersey
... More >>
Memorial Sloan-Kettering Cancer Center at Basking Ridge
Basking Ridge, New Jersey, United States, 07939
United States, New York
Memorial Sloan-Kettering Cancer Center at Commack
Commack, New York, United States, 11725
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan-Kettering at Mercy Medical Center
Rockville Centre, New York, United States
Memorial Sloan-Kettering Cancer Center at Phelps Memorial Hospital Center
Sleepy Hollow, New York, United States, 10591 Less << |
|
NCT01854203
|
Nasopharyngeal Carcinoma |
Phase 2
Phase 3 |
Unknown |
December 2016 |
China, Guangdong
... More >>
Cancer Center,Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Jian-jun Li, MD +862087343381 lijj@sysucc.org.cn
China
State Key Laboratory of Oncology in South China, Cancer Center, Sun Yat-sen University
Not yet recruiting
Guangzhou, China, 510060
Principal Investigator: Jian-jun Li, M.D.
Sub-Investigator: Li-zhi Liu, M.D. Less << |
|
NCT01387399
|
Ovarian Cancer |
Phase 1 |
Unknown |
July 2013 |
Germany
... More >> University Hospital
Bonn, Germany, 53127 Less << |
|
NCT02392637
|
Biliary Tract Cancer |
Phase 2 |
Active, not recruiting |
April 2019 |
United States, Minnesota
... More >>
Mayo Clinic
Rochester, Minnesota, United States, 55902
United States, Texas
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT02182778
|
Biliary Tract Cancer |
Phase 3 |
Active, not recruiting |
May 2019 |
Japan
... More >> Kyoto University Hospital
Kyoto, Japan, 606-8507 Less << |
|
NCT01670500
|
Breast Cancer |
Phase 2 |
Recruiting |
October 2021 |
United States, Colorado
... More >>
University of Colorado Cancer Center
Recruiting
Aurora, Colorado, United States, 80045
Contact: Virginia Borges, MD 303-724-0186 virginia.borges@ucdenver.edu
United States, Connecticut
Smilow Cancer Hospital Care Center at Derby
Recruiting
Derby, Connecticut, United States, 06418
Contact: Erin Hofstatter, MD erin.hofstatter@yale.edu
Smilow Cancer Hospital Care Center at Guilford
Recruiting
Guilford, Connecticut, United States, 06437
Contact: Erin Hofstatter, MD erin.hofstatter@yale.edu
St. Francis Hospital and Medical Center
Recruiting
Hartford, Connecticut, United States, 06105
Contact: Erin Hofstatter, MD erin.hofstatter@yale.edu
Yale School of Medicine
Recruiting
New Haven, Connecticut, United States, 06520
Contact: Erin Hofstatter, MD 203-737-1600 erin.hofstatter@yale.edu
United States, District of Columbia
Georgetown University Medical Center
Recruiting
Washington, District of Columbia, United States, 20007
Contact: Claudine Isaacs, MD isaacsc@georgetown.edu
Sibley Memorial Hospital
Recruiting
Washington, District of Columbia, United States, 20016-2698
Contact: Antonio Wolff, MD awolff@jhmi.edu
United States, Maryland
Johns Hopkins
Recruiting
Baltimore, Maryland, United States, 21287
Contact: Antonio Wolff, MD awolff@jhmi.edu
United States, Massachusetts
Massachusetts General Hospital
Recruiting
Boston, Massachusetts, United States, 02114
Contact: Steven Isakoff, MD, PhD 617-726-4920 sisakoff@partners.org
Principal Investigator: Steven Isakoff, MD, PhD
Dana-Farber Cancer Institute at Faulkner Hospital
Withdrawn
Boston, Massachusetts, United States, 02130
Beth Israel Deaconess Medical Center
Recruiting
Boston, Massachusetts, United States, 02215
Contact: Nadine Tung, MD 617-667-7081 ntung@bidmc.harvard.edu
Principal Investigator: Nadine Tung, MD
Dana-Farber Cancer Institute
Recruiting
Boston, Massachusetts, United States, 02215
Contact: Judy Garber, MD, MPH 617-632-2282 jegarber@partners.org
Principal Investigator: Judy Garber, MD, MPH
United States, New Hampshire
New Hampshire Oncology-Hematology
Withdrawn
Hooksett, New Hampshire, United States, 03106
United States, New Jersey
Rutgers Cancer Institute of New Jersey
Active, not recruiting
New Brunswick, New Jersey, United States, 08901
United States, New York
Memorial Sloan-Kettering Cancer Center
Withdrawn
New York, New York, United States, 10065
United States, North Carolina
Duke University
Recruiting
Durham, North Carolina, United States, 27708
Contact: P. Kelly Marcom kelly.marcom@dm.duke.edu
United States, Rhode Island
Women and Infants Hospital
Recruiting
Providence, Rhode Island, United States, 02905
Contact: Robert Legare, MD 401-453-7540 rlegare@wihri.org
United States, Texas
MD Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact: Banu Arun, MD 713-792-2817 barun@mdanderson.org
MD Anderson in Katy
Withdrawn
Houston, Texas, United States, 77094
MD Anderson in the Bay Area
Withdrawn
Nassau Bay, Texas, United States, 77058
MD Anderson - Sugar Land
Withdrawn
Sugar Land, Texas, United States, 77478
MD Anderson - The Woodlands
Withdrawn
The Woodlands, Texas, United States, 77384 Less << |
|
NCT01114958
|
Lung Neoplasms
... More >> Neoplasm Metastasis Less << |
Phase 1 |
Unknown |
- |
United States, California
... More >>
University of California, San Diego Moores Cancer Center
Recruiting
La Jolla, California, United States, 92093
Contact: Yuri Matusov 858-246-0357 matusov@ucsd.edu
Principal Investigator: William Read, M.D. Less << |
|
NCT01165385
|
Solid Tumors
... More >>Metastatic Cancer Less << |
Phase 1 |
Completed |
- |
France
... More >> Centre François BACLESSE
Caen, France, 14076
Centre Georges François LECLERC
Dijon, France, 21079
Centre Léon Berard
Lyon, France, 69373 CEDEX 08
Centre René Gauducheau
Nantes Saint Herblain, France, 44805
Institut Curie
Paris, France, 75248 Less << |
|
NCT02723864
|
Neoplasms |
Phase 1 |
Recruiting |
December 31, 2020 |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Recruiting
Bethesda, Maryland, United States, 20892
Contact: For more information at the NIH Clinical Center contact National Cancer Institute Referral Office 888-624-1937
United States, Massachusetts
Dana Farber Cancer Institute
Recruiting
Boston, Massachusetts, United States, 02115
United States, Texas
MD Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030-4096 Less << |
|
NCT00995761
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Gyeongsang National University Hospital
Jinju, Gyeongsang-Nam-Do, Korea, Republic of, 660-702 Less << |
|
NCT01149798
|
Metastatic Breast Cancer |
Phase 2 |
Completed |
- |
China
... More >> Fudan University Cancer Hospital
Shanghai, China, 200032 Less << |
|
NCT03644589
|
Estrogen Receptor Negative
... More >> HER2/Neu Negative
Metastatic Breast Cancer
Progesterone Receptor Negative
Recurrent Breast Carcinoma
Triple Negative Breast Cancer Less << |
Phase 2 |
Not yet recruiting |
October 30, 2022 |
United States, Washington
... More >>
Seattle Cancer Care Alliance, Fred Hutch/University of Washington Cancer Consortium
Not yet recruiting
Seattle, Washington, United States, 98109
Contact: Jennifer Specht, MD Less << |
|
NCT03558087
|
Bladder Cancer |
Phase 2 |
Recruiting |
June 2022 |
United States, New York
... More >>
Icahn School of Medicine: Tisch Cancer Institute at Mount Sinai Medical Center
Recruiting
New York, New York, United States, 10029
Contact: Matthew Galsky, M.D. 212-241-8214 matthew.galsky@mssm.edu Less << |
|
NCT00995761
|
- |
|
Completed |
- |
- |
|
NCT03639467
|
Recurrent Nasopharyngeal Carci... More >>noma
Metastatic Nasopharyngeal Carcinoma Less << |
Phase 1
Phase 2 |
Recruiting |
August 15, 2023 |
China, Beijing
... More >> National Cancer Center/Cancer Hospitial,Chinese Academy of Medical Sciences and Peking Union Medical College
Recruiting
Beijing, Beijing, China, 100021
Contact: Yuankai Shi, MD +86-10-87788293 syuankaipumc@126.com Less << |
|
NCT02030574
|
- |
|
Terminated(The study is being ... More >>closed to accrual secondary to low accrual and an interest in opening up a different trial.) Less << |
- |
- |
|
NCT00916500
|
CERVICAL NEOPLASMS |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Korea Cancer Center Hospital, Korea Institute of Radiological & Medical Sciences
Seoul, Korea, Republic of, 139-706 Less << |
|
NCT00901537
|
Non-small Cell Lung Cancer
... More >> Squamous Cell Carcinoma of the Head and Neck Less << |
Phase 1 |
Terminated(Sponsor terminated ... More >>due to no enrollments within last 4 months.) Less << |
- |
United States, California
... More >>
Loma Linda University Cancer Center
Loma Linda, California, United States, 92354 Less << |
|
NCT02475772
|
Ovarian Cancer |
Phase 1 |
Completed |
- |
Germany
... More >> Ruhr University Bochum, Germany, Marienhospital Herne
Herne, North-Rhine Westphalia, Germany, 44625 Less << |
|
NCT01928680
|
Metastatic Breast Cancer |
Phase 2 |
Unknown |
March 2016 |
China, Beijing
... More >> Cancer Hospital, ChineseAMS
Beijing, Beijing, China, 100021 Less << |
|
NCT02030574
|
Invasive Bladder Cancer
... More >> Bladder Cancer Less << |
Phase 2 |
Terminated(The study is being ... More >>closed to accrual secondary to low accrual and an interest in opening up a different trial.) Less << |
- |
United States, Rhode Island
... More >>
Rhode Island Hospital (including Newport Hospital and East Greenwich)
Providence, Rhode Island, United States, 02903
The Miriam Hospital
Providence, Rhode Island, United States, 02906 Less << |
|
NCT01674842
|
Breast Cancer |
Phase 1 |
Active, not recruiting |
October 2023 |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114
Brigham and Women's Hospital
Boston, Massachusetts, United States, 02115
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115
Dana-Farber at Milford
Milford, Massachusetts, United States, 01757
Dana Farber at South Shore Hospital
Weymouth, Massachusetts, United States, 02190 Less << |
|
NCT03330249
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 2 |
Active, not recruiting |
December 2020 |
France
... More >> Centre Paul Strauss
Strasbourg, France, 67065 Less << |
|
NCT01928680
|
- |
|
Unknown |
- |
- |
|
NCT02250872
|
Cancer
Cispla... More >>tin Adverse Reaction Less << |
Phase 2
Phase 3 |
Unknown |
June 2018 |
Korea, Republic of
... More >>
Seoul National University Bundang Hospital
Recruiting
Seongnam-si, Gyeonggi-do, Korea, Republic of, 463-707
Contact: Hyunjin Cho 82 31 787 7030
Principal Investigator: Ki Young Na, MD PhD Less << |
|
NCT01561586
|
Cervical Cancer |
Phase 3 |
Recruiting |
March 2023 |
China
... More >> Fudan University Shanghai Cancer Center
Recruiting
Shanghai, China
Contact: Gui Hao Ke
Korea, Republic of
Dongnam Inst.of Radiological/Medical Science
Recruiting
Busan, Korea, Republic of
Contact: Sang-IL Park
Soon Chun Hyang University Hospital
Recruiting
Cheonan, Korea, Republic of
Contact: Seob Jeon
Dongsan Medical Center
Recruiting
Daegu, Korea, Republic of
Contact: Chi-Heum Cho
Asan Medical Center
Recruiting
Seoul, Korea, Republic of
Contact: Joo-Hyun Nam
Ewha Womans University Mokdong Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Seung Cheol Kim
Gachon University Gil Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Gwang Beom Lee
Gangnam Severance Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Jae-Hoon Kim
Korea Cancer Center Hospital, Korea Institute of Radiological & Medical Sciences
Recruiting
Seoul, Korea, Republic of
Contact: SANG YOUNG RYU, M.D. 82-2-970-1227 ryu@kcch.re.kr
Principal Investigator: SANG YOUNG RYU, M.D.
Korea University Guro Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Jin Hwa Hong
Kyungpook National University Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Sun-joo Lee
Seoul National University Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Hak Jae Kim
Severance Hospital/Sinchon Severance Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Young Tae Kim
Thailand
Faculty of Medicine, Ramathibodi Hospital, Mahidol University
Recruiting
Bankok, Thailand
Contact: SARIKAPAN WILAILAK, M.D. 66-2-2011451 raswl@mahidol.ac.th
Principal Investigator: SARIKAPAN WILAILAK, M.D.
Vietnam
Ho Chi Minh City Oncology Hospital
Recruiting
Ho Chi Minh, Vietnam
Contact: Thinh Dang huy Quoc Less << |
|
NCT02277184
|
Carcinoma, Squamous Cell of He... More >>ad and Neck Less << |
Phase 1 |
Terminated(the investigator le... More >>ft the institution) Less << |
- |
United States, Pennsylvania
... More >>
UPMC Presbyterian
Pittsburgh, Pennsylvania, United States, 15213
Hillman Cancer Center
Pittsburgh, Pennsylvania, United States, 15232
UPMC Shadyside
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT02994069
|
Carcinoma, Squamous Cell
... More >> Head and Neck Cancer Less << |
Phase 2 |
Recruiting |
October 31, 2028 |
United States, Kentucky
... More >>
University of Kentucky
Recruiting
Lexington, Kentucky, United States, 40536
Contact: Susanne Arnold, MD 859-323-8043 susanne.arnold@uky.edu Less << |
|
NCT01980667
|
Advanced Solid Tumors |
Phase 1 |
Completed |
- |
Switzerland
... More >>
Bellinzona, Switzerland
United Kingdom
London, United Kingdom
Newcastle, United Kingdom Less << |
|
NCT01644994
|
Malignant Pleural Mesothelioma |
Phase 1
Phase 2 |
Recruiting |
August 2020 |
Switzerland
... More >> University Hospital Zurich, Division of Thoracic Surgery
Recruiting
Zurich, ZH, Switzerland, 8091
Contact: Isabelle Opitz, Prof MD +41 44 255 11 11 isabelle.schmitt-opitz@usz.ch Less << |
|
NCT03639168
|
Adenoid Cystic Carcinomas
... More >> Chidamide
Cisplatin Less << |
Phase 2 |
Recruiting |
June 1, 2020 |
China, Shanghai
... More >>
Kai Xue
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Kai Xue, MD 13818659448 xuekaishanghai@126.com Less << |
|
NCT01611727
|
BRCA1 Mutation|Metastatic Brea... More >>st Cancer Less << |
PHASE2 |
COMPLETED |
2025-04-09 |
Pomenarian Medical University,... More >> Szczecin, Poland Less << |
|
NCT00826644
|
Carcinoma, Small Cell |
Phase 3 |
Unknown |
December 2012 |
Korea, Republic of
... More >>
Chonnam National University Hwasun Hospital
Recruiting
Hwasun-gun, Jeonnam, Korea, Republic of
Contact: In-Jae Oh, M.D.,Ph.D. 82-61-379-7617 droij@chonnam.ac.kr Less << |
|
NCT01281696
|
Breast Neoplasms
... More >> Leptomeningeal Metastasis
Brain Metastases Less << |
Phase 2 |
Completed |
- |
Taiwan
... More >> Department of Oncology, National Taiwan University Hospital
Taipei City, Taiwan, 100 Less << |
|
NCT00895245
|
- |
|
Terminated(Study stopped due t... More >>o lack of efficacy in first 6 patients) Less << |
- |
- |
|
NCT02639767
|
Malignant Pleural Mesothelioma |
Phase 1 |
Withdrawn(Lack of accrual) |
- |
United States, New Jersey
... More >>
Memorial Sloan Kettering at Basking Ridge
Basking Ridge, New Jersey, United States, 07920
United States, New York
Memorial Sloan Kettering Cancer Center @ Suffolk
Commack, New York, United States, 11725
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan Kettering at Mercy Medical Center
Rockville Centre, New York, United States
Memoral Sloan Kettering Cancer Center at Phelps
Sleepy Hollow, New York, United States, 10591
Memorial Sloan Kettering West Harrison
West Harrison, New York, United States, 10604 Less << |
|
NCT01893801
|
Stage IV Pancreatic Cancer |
Phase 1
Phase 2 |
Unknown |
August 2014 |
United States, Arizona
... More >>
Scottsdale Health Care
Recruiting
Scottsdale, Arizona, United States, 85260
Contact: Joyce Schaffer, MSN RN AOCNS 480-323-1339 joschaffer@shc.org
Principal Investigator: Gayle S Jameson, MSN ACNP-BC
Sub-Investigator: Ramesh K Ramanathan, MD
Sub-Investigator: Daniel D Von Hoff, MD FACP
Sub-Investigator: Katy B Schroeder, RN BSN OCN
United States, New Jersey
Rutgers - Cancer Institute of New Jersey (CINJ)
Recruiting
New Brunswick, New Jersey, United States, 08901
Contact: Anjali Krishnan, RN 732-235-8996 warrieac@cinj.rutgers.edu
Contact: Tatianna Zelinskaya 732-235-9837 zelinska@cinj.rutgers.edu
Principal Investigator: Elizabeth Popllin, MD
United States, Pennsylvania
Vita Medical Associates, PC
Recruiting
Bethlehem, Pennsylvania, United States, 18015
Contact: Colleen Saitta, NP 610-866-0113 nurses@vitahemonc.org
Contact: Gulyun Zhou, NP 610-866-0113 nurses@vitahemonc.org
Principal Investigator: Anna A Niewiarowska, MD Less << |
|
NCT00895245
|
Nausea and Vomiting
... More >> Stage III Squamous Cell Carcinoma of the Hypopharynx
Stage III Squamous Cell Carcinoma of the Larynx
Stage III Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage III Squamous Cell Carcinoma of the Nasopharynx
Stage III Squamous Cell Carcinoma of the Oropharynx
Stage IV Squamous Cell Carcinoma of the Hypopharynx
Stage IV Squamous Cell Carcinoma of the Larynx
Stage IV Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage IV Squamous Cell Carcinoma of the Nasopharynx
Stage IV Squamous Cell Carcinoma of the Oropharynx Less << |
Phase 2 |
Terminated(Study stopped due t... More >>o lack of efficacy in first 6 patients) Less << |
- |
United States, Washington
... More >>
Fred Hutchinson Cancer Research Center/University of Washington Cancer Consortium
Seattle, Washington, United States, 98109 Less << |
|
NCT00322920
|
Cervix Cancer |
Phase 1 |
Terminated(Low accrual) |
- |
United States, Alabama
... More >>
University of Alabama at Birmingham
Birmingham, Alabama, United States, 35233 Less << |
|
NCT03081039
|
Intrahepatic Cholangiocarcinom... More >>a Less << |
Phase 3 |
Withdrawn(Competing study) |
December 31, 2022 |
United States, Kentucky
... More >>
University of Louisville
Louisville, Kentucky, United States, 40202 Less << |
|
NCT02610556
|
Nasopharyngeal Carcinoma |
Phase 2 |
Recruiting |
January 2022 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Fang-Yun Xie, M.D. +86-020-87342618 xiefy@sysucc.org.cn
Contact: Pu-Yun OuYang, M.D. +86-020-87342618 ouyangpy@sysucc.org.cn Less << |
|
NCT00847015
|
Bladder Cancer
... More >> Urinary Bladder Less << |
Phase 2 |
Completed |
- |
United States, New Jersey
... More >>
Memorial Sloan-Kettering at Basking Ridge
Basking Ridge, New Jersey, United States, 07920
United States, New York
Memorial Sloan-Kettering Cancer Center @ Suffolk
Commack, New York, United States, 11725
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan-Kettering Cancer Center at Mercy Medical Center
Rockville Centre, New York, United States, 11570
Memoral Sloan Kettering Cancer Center@Phelps
Sleepy Hollow, New York, United States Less << |
|
NCT00969761
|
Neoplasms |
Phase 1 |
Completed |
- |
Belgium
... More >> 1230.6.3201 Boehringer Ingelheim Investigational Site
Bruxelles, Belgium
1230.6.3202 Boehringer Ingelheim Investigational Site
Leuven, Belgium Less << |
|
NCT00847015
|
- |
|
Completed |
- |
- |
|
NCT00702299
|
- |
|
Completed |
- |
- |
|
NCT00174837
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 3 |
Completed |
- |
- |
|
NCT02094625
|
Neuroectodermal Tumors, Primit... More >>ive
Liver Neoplasms
Osteosarcoma
Other Childhood Cancers Using Cisplatin-based Regimens Less << |
Phase 1 |
Recruiting |
October 2020 |
United States, California
... More >>
Childrens Hospital Los Angeles
Recruiting
Los Angeles, California, United States, 90027
Principal Investigator: Etan Orgel, MD MS
Sub-Investigator: David R Freyer, DO MS Less << |
|
NCT02734537
|
Head and Neck Squamous Cell Ca... More >>rcinoma
Hypopharyngeal Squamous Cell Carcinoma
Laryngeal Squamous Cell Carcinoma
Laryngeal Squamous Cell Carcinoma, Spindle Cell Variant
Lip and Oral Cavity Squamous Cell Carcinoma
p16INK4a Negative Oropharyngeal Squamous Cell Carcinoma
Stage III Hypopharyngeal Carcinoma AJCC v8
Stage III Laryngeal Cancer AJCC v8
Stage III Lip and Oral Cavity Cancer AJCC v8
Stage III Oral Cavity Verrucous Carcinoma
Stage III Oropharyngeal (p16-Negative) Carcinoma AJCC v8
Stage IVA Hypopharyngeal Carcinoma AJCC v8
Stage IVA Laryngeal Cancer AJCC v8
Stage IVA Lip and Oral Cavity Cancer AJCC v8
Stage IVA Oral Cavity Verrucous Carcinoma
Stage IVA Oropharyngeal (p16-Negative) Carcinoma AJCC v8 Less << |
Phase 2 |
Recruiting |
May 1, 2027 |
- |
|
NCT02422498
|
Locally Recurrent/Metastatic T... More >>riple Negative Breast Cancer Less << |
Phase 2 |
Recruiting |
April 2019 |
United States, New Jersey
... More >>
Memorial Sloan Kettering at Basking Ridge
Recruiting
Basking Ridge, New Jersey, United States, 07920
Contact: Simon Powell, MD, PhD 212-639-3639
Memorial Sloan Kettering Monmouth
Recruiting
Middletown, New Jersey, United States, 07748
Contact: Simon Powell, MD, PhD 212-639-3639
Memorial Sloan Kettering Bergen
Recruiting
Montvale, New Jersey, United States, 07645
Contact: Simon Powell, MD, PhD 212-639-3639
United States, New York
Memorial Sloan Kettering Cancer Center @ Suffolk
Recruiting
Commack, New York, United States, 11725
Contact: Simon Powell, PhD 212-639-3639
Memorial Sloan Kettering Westchester
Recruiting
Harrison, New York, United States, 10604
Contact: Simon Powell, MD, PhD 212-639-3639
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10065
Contact: Simon Powell, MD, PhD 212-639-3639
Contact: George Plitas, MD, PhD 646-888-4587
Memorial Sloan Kettering at Mercy Medical Center
Recruiting
Rockville Centre, New York, United States
Contact: Simon Powell, MD, PhD 212-639-3639 Less << |
|
NCT01287624
|
Breast Cancer |
Phase 3 |
Completed |
- |
China, Shanghai
... More >>
Fudan University Cancer Hospital
Shanghai, Shanghai, China, 200032 Less << |
|
NCT00702299
|
Fallopian Tube Cancer
... More >> Ovarian Cancer
Peritoneal Cavity Cancer Less << |
Phase 1 |
Completed |
- |
United States, Arizona
... More >>
Arizona Oncology - Scottsdale
Scottsdale, Arizona, United States, 85258
Arizona Cancer Center at University of Arizona Health Sciences Center
Tucson, Arizona, United States, 85724-5024 Less << |
|
NCT02308072
|
Head and Neck Cancer |
Phase 1 |
Recruiting |
June 2022 |
United Kingdom
... More >> Guy's and St Thomas' NHS Foundation Trust
Recruiting
London, United Kingdom
Principal Investigator: Maria Teresa Guerrero-Urbano
University College London Hospitals NHS Foundation Trust
Recruiting
London, United Kingdom
Principal Investigator: Martin Forster
Velindre Cancer Centre
Recruiting
Wales, United Kingdom, CF14 2TL
Principal Investigator: Mererid Evans Less << |
|
NCT00770874
|
Cervical Cancer |
Phase 3 |
Completed |
- |
Japan
... More >> Yanagawa Hospital
Chikushimachi, Yanagawa, Fukuoka, Japan, 832-0077
Cancer Institute Hospital
Ariake, Koto-ku, Tokyo, Japan, 135-8550
Korea, Republic of
Konkuk University Medical Center
Hwayang-dong, Gwangjin-gu, Seoul, Korea, Republic of, 143-729
Taiwan
Chang Gung Medical Foundation- Linkou
Fu-Hsing St. Kuei Shan Hsiang, TaoYuan Hsien, Taiwan, 33305 Less << |
|
NCT01355302
|
- |
|
Terminated(Sites not recruitin... More >>g) Less << |
- |
- |
|
NCT01854255
|
Gastric Cancer |
Phase 2 |
Completed |
- |
Germany
... More >> Ruhr University of Bochum
Herne, North Rhine Westphalia, Germany, 44625 Less << |
|
NCT02882308
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 2 |
Recruiting |
May 2019 |
Greece
... More >> Euromedica General Clinic of Thessaloniki
Recruiting
Thessaloníki, Thessaloniki, Greece, 54645
Contact: George Fountzilas, Prof.Emeritus 00302310683136 fountzil@auth.gr
University Hospital "Attikon", 2nd Department of Internal Medicine, Division of Oncology
Recruiting
Athens, Greece, 12462
Contact: Diamanto Psyrri, MD,Ass.Prof 0030 2105831256 dp237@email.med.yale.edu Less << |
|
NCT01689194
|
Locally Advanced Head and Neck... More >> Squamous Cell Carcinoma Less << |
Phase 2 |
Unknown |
December 2017 |
Korea, Republic of
... More >>
Seoul National University Hospital
Seoul, Korea, Republic of, 110-744
Seoul National University Hospital
Seoul, Korea, Republic of Less << |
|
NCT02016274
|
Esophageal Squamous Cell Cance... More >>r Less << |
Phase 2 |
Unknown |
July 2016 |
China, Beijing
... More >> Peking University Cancer Hospital
Recruiting
Beijing, Beijing, China, 100142
Contact: Lin Shen, MD 86-10-88196561 lin100@medmail.com.cn
Contact: Zhihao Lu, MD 86-10-88196561 pppeirain@126.com
Principal Investigator: Lin Shen, MD Less << |
|
NCT02631590
|
Biliary Carcinoma
... More >> Gall Bladder Carcinoma
Cholangiocarcinoma
Gastrointestinal Tumor Less << |
Phase 2 |
Recruiting |
December 2020 |
United States, Florida
... More >>
H. Lee Moffitt Cancer Center and Research Institute
Recruiting
Tampa, Florida, United States, 33612
Contact: Maria Martinez Jimenez 813-745-1946 maria.martinez@moffitt.org
Contact: Richard D. Kim 813-745-1277 richard.kim@moffitt.org
Principal Investigator: Richard D. Kim, M.D.
Sub-Investigator: Rutika Mehta, M.D.
Sub-Investigator: Dae Won Kim, M.D. Less << |
|
NCT02429726
|
Malignant Pleural Effusion |
Phase 2 |
Unknown |
June 2017 |
China, Shanxi
... More >> The First Affiliated Hospital of Xi'an Jiao Tong University
Not yet recruiting
Xian, Shanxi, China, 710061
Contact: wei wang, MD 86-13923853040 wei.wang@163.com
Contact: shuanying yang, MD 86-18149098803 yangshuanying66@163.com Less << |
|
NCT00531687
|
Testicular Cancer |
Phase 2 |
Terminated(slow accrual) |
- |
Denmark
... More >> Department of Oncology 5073, Rigshospitalet
Copenhagen, Denmark, 2100 Less << |
|
NCT00198172
|
Germ Cell Tumor |
Phase 2 |
Terminated(Accrual Goal Met) |
- |
United States, Indiana
... More >>
Indiana University Cancer Center
Indianapolis, Indiana, United States, 46202 Less << |
|
NCT00463788
|
- |
|
Completed |
- |
- |
|
NCT02485548
|
Head and Neck Squamous Cell Ca... More >>ncer Less << |
Phase 3 |
Unknown |
June 2018 |
China, Jiangsu
... More >> Jiangsu Cancer Hospital
Recruiting
Nanjing, Jiangsu, China, 210009
Contact: Xuesong Jiang, M.D. 13851700790@126.com
Sub-Investigator: Xuesong Jiang, M.D. Less << |
|
NCT00463788
|
Breast Neoplasm |
Phase 2 |
Completed |
- |
- |
|
NCT02990468
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Recruiting |
February 2020 |
United States, Nebraska
... More >>
University of Nebraska Medical Center
Not yet recruiting
Omaha, Nebraska, United States, 68198
Contact: Amy Filler-Katz 402-552-2790 afillerkatz@unmc.edu
Principal Investigator: Weining Zhen, MD
United States, North Carolina
Duke University Medical Center
Recruiting
Durham, North Carolina, United States, 27710
Contact: Joan Cahill, RN 919-668-5211 joan.cahill@duke.edu Less << |
|
NCT00960349
|
Gastric Cancer |
Phase 1 |
Completed |
- |
Japan
... More >> Research Site
Nagoya, Aichi, Japan
Research Site
Osakasayama, Osaka, Japan
Research Site
Sunto-gun, Shizuoka, Japan
Research Site
Chuo, Tokyo, Japan Less << |
|
NCT01126749
|
Bladder Cancer |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT00845884
|
Advanced Gastric Cancer |
Phase 1
Phase 2 |
Unknown |
December 2012 |
Israel
... More >> Davidoff Cancer Center, Rabin Medical Center
Not yet recruiting
Petach Tikva, Israel, 49100
Contact: Baruch Brenner, MD 972-3-9378005 brennerb@clalit.org.il
Principal Investigator: Baruch Brenner, MD Less << |
|
NCT01951469
|
Non-small Cell Lung Cancer
... More >> Brain Metastases
EGFR Mutation Less << |
Phase 2 |
Recruiting |
June 2019 |
China, Guangdong
... More >>
Sun Yat-sen University of Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: li-kun Chen, doctor 13798019964 chenlk@sysucc.org.cn
Principal Investigator: li-kun Chen, doctor Less << |
|
NCT00251407
|
Solid Tumor |
Phase 1 |
Completed |
- |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114
Brigham and Women's Hospital
Boston, Massachusetts, United States, 02115
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT02562599
|
Nasopharyngeal Carcinoma |
Phase 2 |
Unknown |
August 2018 |
China, Hubei
... More >> Hubei Cancer hospital
Recruiting
Wu Han, Hubei, China, 430079
Contact: Han Guang, PH.D
Principal Investigator: Hu Desheng, M.D Less << |
|
NCT01282333
|
Advanced Adult Primary Liver C... More >>ancer
Localized Unresectable Adult Primary Liver Cancer
Metastatic Transitional Cell Cancer of the Renal Pelvis and Ureter
Regional Transitional Cell Cancer of the Renal Pelvis and Ureter
Stage III Bladder Cancer
Stage III Pancreatic Cancer
Stage IIIA Non-small Cell Lung Cancer
Stage IIIB Non-small Cell Lung Cancer
Stage IV Bladder Cancer
Stage IV Non-small Cell Lung Cancer
Stage IV Pancreatic Cancer
Transitional Cell Carcinoma of the Bladder
Unresectable Extrahepatic Bile Duct Cancer
Unresectable Gallbladder Cancer Less << |
Phase 1 |
Terminated |
- |
United States, Massachusetts
... More >>
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115
United States, Pennsylvania
Penn State Milton S Hershey Medical Center
Hershey, Pennsylvania, United States, 17033-0850
University of Pittsburgh
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT00169221
|
Head and Neck Cancer |
Phase 2 |
Terminated(Toxicity in an inde... More >>pendent study IMEX. The trial was subsequently terminated (54 pts instead of 140) despite safety analyses showing no excess of toxicity) Less << |
- |
France
... More >> Centre Antoine Lacassagne
Nice, France, 06189 Less << |
|
NCT00571298
|
Malignant Pleural Mesothelioma |
Phase 1 |
Completed |
- |
United States, Massachusetts
... More >>
Brigham and Women's Hospital
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00386815
|
Malignant Pleural Mesothelioma |
Phase 2 |
Completed |
- |
Japan
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Aichi, Japan, 464-8681
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Fukuoka, Japan, 811-1395
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hokkaido, Japan, 060-8648
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hyogo, Japan, 663-8501
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Okayama, Japan, 702-8055
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Osaka, Japan, 589-8511
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Tokyo, Japan, 104-0045 Less << |
|
NCT01614938
|
Nasopharyngeal Carcinoma |
Phase 2 |
Unknown |
August 2014 |
China, Shanghai
... More >>
Fudan University Shanghai Cancer Center
Shanghai, Shanghai, China, 200032 Less << |
|
NCT02537561
|
Solid Tumors
... More >>Carcinoma, Non-Small Cell Lung
Non-Small Cell Lung Cancer
Non-Small-Cell Lung Carcinoma
Nonsmall Cell Lung Cancer Less << |
Phase 1 |
Withdrawn(The pharmaceutical c... More >>ompany did not want to follow through with support for the study.) Less << |
December 2018 |
- |
|
NCT02643407
|
Non-Small Cell Lung Cancer |
Phase 3 |
Unknown |
December 2017 |
China, Guangdong
... More >>
Guangdong Lung Cancer Institute, Guangdong General Hospital, Guangdong Academy of Medical Sciences
Recruiting
Guangzhou, Guangdong, China, 510080
Contact: Haiyan Tu, MD +86 20 83827812 ext 50810 thoraciconcology88@163.com
Contact: Yi-Long Wu, MD +86 20 83827812 ext 51221 syylwu@live.cn
Principal Investigator: Yi-Long Wu, MD
Sub-Investigator: Haiyan Tu, MD Less << |
|
NCT01240590
|
Solid Tumor
A... More >>naplastic Thyroid Cancer Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892 Less << |
|
NCT01126008
|
Head and Neck Cancer |
Phase 2 |
Unknown |
December 2016 |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of, 135-710 Less << |
|
NCT00982436
|
Head and Neck Neoplasms |
Phase 1
Phase 2 |
Unknown |
December 2012 |
United States, Vermont
... More >>
Mountainview Medical Center
Recruiting
Berlin, Vermont, United States, 05602
Contact: John Valentine, MD 802-225-5400 john.valentine@cvmc.org
Principal Investigator: John Valentine, MD
Sub-Investigator: David Ospina, MD
Sub-Investigator: Daniel Fram, MD
Fletcher Allen Health Care
Recruiting
Burlington, Vermont, United States, 05401
Contact: Steven Grunberg, MD 802-847-8400 Steven.Grunberg@vtmednet.org
Contact: Madhuri V Vithala, MD 802-847-8400 Madhuri.Vithala@vtmednet.org
Principal Investigator: Steven M Grunberg, MD
Sub-Investigator: Madhuri V Vithala, MD
Sub-Investigator: Havaleh Gagne, MD
Sub-Investigator: William Brundage, MD
Vermont Center for Cancer Medicine
Recruiting
Colchester, Vermont, United States, 05446
Contact: Christian Thomas, MD 802-655-3400 Christian.Thomas@vtmednet.org
Sub-Investigator: Paul Unger, MD
Sub-Investigator: Dennis Sanders, MD
Sub-Investigator: Johannes Nunnink, MD
Sub-Investigator: Christian Thomas, MD Less << |
|
NCT03579771
|
Resectable Cholangiocarcinoma
... More >>
Stage IB Intrahepatic Cholangiocarcinoma AJCC v8
Stage II Intrahepatic Cholangiocarcinoma AJCC v8
Stage III Intrahepatic Cholangiocarcinoma AJCC v8
Stage IIIA Intrahepatic Cholangiocarcinoma AJCC v8
Stage IIIB Intrahepatic Cholangiocarcinoma AJCC v8 Less << |
Phase 2 |
Recruiting |
September 30, 2024 |
United States, Arizona
... More >>
Mayo Clinic in Arizona
Not yet recruiting
Scottsdale, Arizona, United States, 85259
Contact: Shishir Maithel, MD smaithe@emory.edu
Principal Investigator: Mitesh Borad, MD
United States, Florida
Mayo Clinic in Florida
Not yet recruiting
Jacksonville, Florida, United States, 32224
Contact: Shishir Maithel smaithe@emory.edu
Principal Investigator: Kabir Mody, MD
Principal Investigator: Kristopher Croome, MD
United States, Georgia
Emory University Hospital Midtown
Recruiting
Atlanta, Georgia, United States, 30308
Contact: Shabnam Montazeri 404-686-0242 shabnam.montazeri@emory.edu
Principal Investigator: Shishir Maithel, MD
Emory University Hospital/Winship Cancer Institute
Recruiting
Atlanta, Georgia, United States, 30322
Contact: Tyra Gaines 404-712-3308 tgaine3@emory.edu
Principal Investigator: Shishir Maithel, MD
Emory Saint Joseph's Hospital
Recruiting
Atlanta, Georgia, United States, 30342
Contact: Alicia Escobar 678-843-7029 alicia.m.escobar@emory.edu
Principal Investigator: Shishir Maithel, MD
United States, Minnesota
Mayo Clinic
Not yet recruiting
Rochester, Minnesota, United States, 55905
Contact: Shishir Maithel, MD smaithe@emory.edu
Principal Investigator: Amit Mahipal, MD
Principal Investigator: Rory Smoot, MD
United States, Texas
MD Anderson Cancer Center
Not yet recruiting
Houston, Texas, United States, 77030
Contact: Shishir Maithel, MD smaithe@emory.edu
Principal Investigator: Milind Javle, MD
Principal Investigator: J. Nicolas Vauthey, MD
United States, Washington
Virginia Mason Medical Center
Not yet recruiting
Seattle, Washington, United States, 98101
Contact: Shishir Maithel, MD smaithe@emory.edu
Principal Investigator: Flavio G. Rocha, MD
Principal Investigator: Bruce Lim, MD Less << |
|
NCT02567422
|
Head and Neck Squamous Cell Ca... More >>rcinoma
Human Papillomavirus Negative
Stage III Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma AJCC v6 and v7
Stage III Oropharyngeal Squamous Cell Carcinoma AJCC v7
Stage IV Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma AJCC v7
Stage IV Oropharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVA Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma AJCC v7
Stage IVA Oropharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVB Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma AJCC v7
Stage IVB Oropharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVC Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma AJCC v7
Stage IVC Oropharyngeal Squamous Cell Carcinoma AJCC v7 Less << |
Phase 1 |
Recruiting |
- |
United States, California
... More >>
City of Hope Comprehensive Cancer Center
Recruiting
Duarte, California, United States, 91010
Contact: Site Public Contact 800-826-4673 becomingapatient@coh.org
Principal Investigator: Erminia Massarelli
University of California Davis Comprehensive Cancer Center
Recruiting
Sacramento, California, United States, 95817
Contact: Site Public Contact 916-734-3089
Principal Investigator: Jonathan W. Riess
United States, Connecticut
Smilow Cancer Center/Yale-New Haven Hospital
Recruiting
New Haven, Connecticut, United States, 06510
Contact: Site Public Contact 203-785-5702 canceranswers@yale.edu
Principal Investigator: Barbara A. Burtness
Yale University
Recruiting
New Haven, Connecticut, United States, 06520
Contact: Site Public Contact 203-785-5702 canceranswers@yale.edu
Principal Investigator: Barbara A. Burtness
United States, Georgia
Emory University Hospital Midtown
Recruiting
Atlanta, Georgia, United States, 30308
Contact: Site Public Contact 888-946-7447
Principal Investigator: Conor E. Steuer
Emory University Hospital/Winship Cancer Institute
Recruiting
Atlanta, Georgia, United States, 30322
Contact: Site Public Contact 404-778-1868
Principal Investigator: Conor E. Steuer
United States, Kentucky
University of Kentucky/Markey Cancer Center
Recruiting
Lexington, Kentucky, United States, 40536
Contact: Site Public Contact 859-257-3379
Principal Investigator: Susanne M. Arnold
United States, Maryland
Johns Hopkins University/Sidney Kimmel Cancer Center
Suspended
Baltimore, Maryland, United States, 21287
United States, Ohio
Case Western Reserve University
Suspended
Cleveland, Ohio, United States, 44106
Ohio State University Comprehensive Cancer Center
Recruiting
Columbus, Ohio, United States, 43210
Contact: Site Public Contact 800-293-5066 Jamesline@osumc.edu
Principal Investigator: Darrion L. Mitchell
United States, Virginia
University of Virginia Cancer Center
Recruiting
Charlottesville, Virginia, United States, 22908
Contact: Site Public Contact 434-243-6303 PAS9E@virginia.edu
Principal Investigator: Varinder Kaur
United States, Wisconsin
University of Wisconsin Hospital and Clinics
Recruiting
Madison, Wisconsin, United States, 53792
Contact: Site Public Contact 800-622-8922
Principal Investigator: Justine Yang-Bruce Less << |
|
NCT03387943
|
Poorly Differentiated Thyroid ... More >>Carcinoma Less << |
Phase 2 |
Recruiting |
December 31, 2019 |
China
... More >> Zhejiang Cancer Hospital
Recruiting
Zhejiang, China
Contact: haiyan yang, doctor Less << |
|
NCT01355302
|
Advanced or Metastatic Solid T... More >>umors
Previously Untreated Gastric Cancer Less << |
Phase 1
Phase 2 |
Terminated(Sites not recruitin... More >>g) Less << |
- |
United States, Arizona
... More >>
Arizona Oncology Associates, PC - CASA
Tucson, Arizona, United States, 85715
United States, Florida
Boca Raton Clinical Research Associates, Inc
Plantation, Florida, United States, 33324
United States, Illinois
Robert H. Lurie Comprenhensive Cancer Center of Northwestern University
Chicago, Illinois, United States, 60611
United States, Michigan
University of Michigan Comprehensive Cancer Center
Ann Arbor, Michigan, United States, 48109
Barbara Ann Karmanos Cancer Institute
Detroit, Michigan, United States, 48084
Henry Ford Medical Center
Detroit, Michigan, United States, 48202
United States, North Carolina
University of North Carolina at Chapel Hill
Chapell Hill, North Carolina, United States, 27599
Duke University Medical Center
Durham, North Carolina, United States, 27710
United States, Ohio
Mercy Cancer Centerr at St. Anne
Toledo, Ohio, United States, 43623
Russian Federation
Chelyabinsk Regional Oncology Dispensary
Chelyabinsk, Russian Federation, 454087
GOU VPO St-Petersburg SMA n/a Mechnikov Fed. Agen. of Healthcare and Social Developm.
St Petersburg, Russian Federation, 195067
FSI "SRC of Oncology n. a. N.N.Petrov of Rosmedtekhnologiy"
St Petersburg, Russian Federation, 197758
Ukraine
SI Dnipropetrovsk Medical Academy of MOHU ch of Oncology and Medical Radiology
Dnipropetrovsk, Ukraine, 49102
Municipal Clinical Medical and Prophylactic Institution Donetsk Regional Antitumor Centre
Donetsk, Ukraine, 83092
Kyiv City Clinical Oncological Center
Kyiv, Ukraine, 3115
Lviv State Oncol. Reg. Treatment and Diagnostic Center
Lviv, Ukraine, 79031
United Kingdom
Barts and the London NHS Trust
London, Greater London, United Kingdom, EC1A 7BE
Sarah Cannon Research UK
London, Greater London, United Kingdom, W1G 6AD
The Christie NHS Foundation Trust
Manchester, Greater Manchester, United Kingdom, M20 4BX Less << |
|
NCT01356303
|
Non-small Cell Lung Cancer Met... More >>astatic Less << |
Phase 2 |
Suspended(Difficulty in enroll... More >>ing patients) Less << |
August 2016 |
Saudi Arabia
... More >> King Abdul Aziz Medical City for National Guard Health Affairs
Riyadh, Saudi Arabia, 11426 Less << |
|
NCT02611037
|
Malignant Pleural Mesothelioma... More >>
Mesothelioma Less << |
Phase 2 |
Recruiting |
December 2020 |
United States, Florida
... More >>
H. Lee Moffitt Cancer Center and Research Institute
Recruiting
Tampa, Florida, United States, 22612
Contact: Malesa Pereira 813-745-4090 malesa.pereira@moffitt.org
Contact: Bela Kis 813-745-8425 bela.kis@moffitt.org
Principal Investigator: Bela Kis, M.D., Ph.D.
Sub-Investigator: Scott Antonia, M.D.
Sub-Investigator: Ghassan El-Haddad, M.D.
Sub-Investigator: Jacques-Pierre Fontaine, M.D.
Sub-Investigator: Lary Robinson, M.D.
Sub-Investigator: Tawee Tanvetyanon, M.D.
Sub-Investigator: Benjamin Creelan, M.D.
Sub-Investigator: Junsung Choi, M.D.
Sub-Investigator: Daniel Jeong, M.D.
Sub-Investigator: Brian Morse, M.D. Less << |
|
NCT00984035
|
- |
|
Completed |
- |
United States, Illinois
... More >>
University of Chicago
Chicago, Illinois, United States, 60637 Less << |
|
NCT01855451
|
HPV Positive Oropharyngeal Squ... More >>amous Cell Carcinoma Less << |
Phase 3 |
Active, not recruiting |
June 2022 |
Australia, Australian Capital ... More >>Territory
Canberra Hospital
Canberra, Australian Capital Territory, Australia
Australia, New South Wales
Chris O'Brien Lifehouse
Camperdown, New South Wales, Australia, 2050
Liverpool Hospital
Liverpool, New South Wales, Australia, 2170
St George Hospital
St George, New South Wales, Australia, 2217
Riverina Cancer Care Centre
Wagga Wagga, New South Wales, Australia
Calvary Mater Newcastle
Waratah, New South Wales, Australia
Westmead Hospital
Westmead, New South Wales, Australia, 2145
Australia, Queensland
Royal Brisbane and Womens Hospital
Herston, Queensland, Australia, 4006
Townsville Hospital
Townsville, Queensland, Australia, 4810
Princess Alexandra Hospital
Woolloongabba, Queensland, Australia, 4102
Australia, South Australia
Flinders Medical Centre
Bedford Park, South Australia, Australia, 5042
Australia, Victoria
Peter MacCallum Cancer Centre
East Melbourne, Victoria, Australia, 3002
Austin Hospital
Melbourne N., Victoria, Australia, 3084
Australia, Western Australia
Sir Charles Gairdner
Nedlands, Western Australia, Australia, 6009
New Zealand
Auckland City Hospital
Auckland, New Zealand, 1344
Palmerston North Hospital
Palmerston, New Zealand, 4442 Less << |
|
NCT01240590
|
- |
|
Completed |
- |
- |
|
NCT01091454
|
- |
|
Completed |
- |
- |
|
NCT03345784
|
Endometrioid Adenocarcinoma
... More >> Recurrent Cervical Carcinoma
Stage I Uterine Corpus Cancer AJCC v7
Stage I Vaginal Cancer AJCC v6 and v7
Stage IA Uterine Corpus Cancer AJCC v7
Stage IB Cervical Cancer AJCC v6 and v7
Stage IB Uterine Corpus Cancer AJCC v7
Stage IB2 Cervical Cancer AJCC v6 and v7
Stage II Cervical Cancer AJCC v7
Stage II Uterine Corpus Cancer AJCC v7
Stage II Vaginal Cancer AJCC v6 and v7
Stage IIA Cervical Cancer AJCC v7
Stage IIB Cervical Cancer AJCC v6 and v7
Stage III Cervical Cancer AJCC v6 and v7
Stage III Uterine Corpus Cancer AJCC v7
Stage III Vaginal Cancer AJCC v6 and v7
Stage IIIA Cervical Cancer AJCC v6 and v7
Stage IIIA Uterine Corpus Cancer AJCC v7
Stage IIIB Cervical Cancer AJCC v6 and v7
Stage IIIB Uterine Corpus Cancer AJCC v7
Stage IIIC Uterine Corpus Cancer AJCC v7 Less << |
Phase 1 |
Recruiting |
October 18, 2020 |
United States, California
... More >>
University of California Davis Comprehensive Cancer Center
Recruiting
Sacramento, California, United States, 95817
Contact: Site Public Contact 916-734-3089
Principal Investigator: Vanessa A. Kennedy
United States, Colorado
University of Colorado Hospital
Recruiting
Aurora, Colorado, United States, 80045
Contact: Site Public Contact 720-848-0650
Principal Investigator: Bradley R. Corr
United States, Kentucky
University of Kentucky/Markey Cancer Center
Recruiting
Lexington, Kentucky, United States, 40536
Contact: Site Public Contact 859-257-3379
Principal Investigator: Rachel W. Miller
United States, New Jersey
Rutgers Cancer Institute of New Jersey-Robert Wood Johnson University Hospital
Recruiting
New Brunswick, New Jersey, United States, 08903
Contact: Site Public Contact 732-235-8675
Principal Investigator: Eugenia Girda
Rutgers Cancer Institute of New Jersey
Recruiting
New Brunswick, New Jersey, United States, 08903
Contact: Site Public Contact 732-235-8675
Principal Investigator: Eugenia Girda
United States, North Carolina
Duke University Medical Center
Recruiting
Durham, North Carolina, United States, 27710
Contact: Site Public Contact 888-275-3853
Principal Investigator: James L. Abbruzzese
Canada, Ontario
University Health Network-Princess Margaret Hospital
Recruiting
Toronto, Ontario, Canada, M5G 2M9
Contact: Site Public Contact 416-946-4501 clinical.trials@uhn.on.ca
Principal Investigator: Stephanie Lheureux Less << |
|
NCT02695459
|
Neuroendocrine Carcinomas |
Phase 2 |
Recruiting |
January 2019 |
Netherlands
... More >> NKI-AVL
Recruiting
Amsterdam, Netherlands, 1066CX
Contact: M. Tesselaar, MD
UMCG
Recruiting
Groningen, Netherlands
Contact: A.M.E. Walenkamp, MD
Erasmus Medisch Centrum
Recruiting
Rotterdam, Netherlands
Contact: F. Eskens, MD Less << |
|
NCT01091454
|
Triple-negative Breast Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT01977235
|
Small Cell Lung Carcinoma |
Phase 2 |
Unknown |
September 2017 |
China, Chongqing
... More >>
Chongqing Cancer Hospital
Recruiting
Chongqing, Chongqing, China
Contact: Qiying Li, M.D. 13637808684
Principal Investigator: Qiying Li, M.D.
Principal Investigator: Dairong Li
Xinan Hospital, Third Military Medical University
Recruiting
Chongqing, Chongqing, China
Contact: Wei Xiong, M.D. +86-13512345225
Principal Investigator: Wei Xiong
Xinqiao Hospital, Third Military Medical University
Recruiting
Chongqing, Chongqing, China
Contact: Bo Zhu, M.D. +86-15923366951
Principal Investigator: Bo Zhu, M.D. Less << |
|
NCT02199418
|
Tubular Breast Cancer
... More >> Mucinous Breast Cancer
Invasive Ductal Breast Cancer
Inflammatory Breast Cancer Less << |
Phase 2 |
Completed |
- |
China, Shanghai
... More >>
Fudan University Shanghai Cancer Center
Shanghai, Shanghai, China, 200032
Shanghai Jiaotong University School of Medicine, Renji Hospital
Shanghai, Shanghai, China, 200127 Less << |
|
NCT02906800
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Recruiting |
May 2019 |
France
... More >> Hopital Europeen Georges Pompidou
Recruiting
Paris, Île-de-France, France, 75015
Contact: Stephane Hans, MD stephane.hans@aphp.fr
Principal Investigator: Stephane Hans, MD Less << |
|
NCT03768414
|
Stage III Distal Bile Duct Can... More >>cer AJCC v8
Stage III Gallbladder Cancer AJCC v8
Stage III Intrahepatic Cholangiocarcinoma AJCC v8
Stage IIIA Distal Bile Duct Cancer AJCC v8
Stage IIIA Gallbladder Cancer AJCC v8
Stage IIIA Intrahepatic Cholangiocarcinoma AJCC v8
Stage IIIB Distal Bile Duct Cancer AJCC v8
Stage IIIB Gallbladder Cancer AJCC v8
Stage IIIB Intrahepatic Cholangiocarcinoma AJCC v8
Stage IV Distal Bile Duct Cancer AJCC v8
Stage IV Gallbladder Cancer AJCC v8
Stage IV Intrahepatic Cholangiocarcinoma AJCC v8
Stage IVA Gallbladder Cancer AJCC v8
Stage IVB Gallbladder Cancer AJCC v8
Unresectable Extrahepatic Bile Duct Carcinoma
Unresectable Gallbladder Carcinoma
Unresectable Intrahepatic Cholangiocarcinoma Less << |
Phase 3 |
Recruiting |
January 1, 2024 |
United States, Arizona
... More >>
University of Arizona
Recruiting
Tucson, Arizona, United States, 85724
Contact: Rachna Shroff 520-626-2430 rshroff@email.arizona.edu Less << |
|
NCT02992340
|
Biliary Tract Cancer |
Phase 1
Phase 2 |
Recruiting |
December 2020 |
Korea, Republic of
... More >>
Recruiting
Seoul, Korea, Republic of
Singapore
Recruiting
Singapore, Singapore
Taiwan
Recruiting
Taipei, Taiwan Less << |
|
NCT01093092
|
Adult Solid Neoplasm |
Phase 1 |
Terminated(Study transferring ... More >>to INOVA Health) Less << |
- |
United States, New York
... More >>
Roswell Park Cancer Institute
Buffalo, New York, United States, 14263
United States, Virginia
Emily Couric Clinical Cancer Center
Charlottesville, Virginia, United States, 22903 Less << |
|
NCT01206426
|
- |
|
Completed |
- |
United States, Illinois
... More >>
University of Chicago
Chicago, Illinois, United States, 60637 Less << |
|
NCT01874171
|
Oropharyngeal Squamous Cell Ca... More >>rcinoma Less << |
Phase 3 |
Active, not recruiting |
February 2019 |
Ireland
... More >> St Luke's Hospital
Dublin, Ireland, 6
Beaumont Hospital
Dublin, Ireland
Netherlands
VU University Medical Center
Amsterdam, Netherlands
United Kingdom
Aberdeen Royal Infirmary
Aberdeen, United Kingdom
Royal United Hospital
Bath, United Kingdom, BA1 3NG
Clatterbridge Cancer Centre
Bebington, United Kingdom, CH63 4JY
Bradford Royal Infirmary
Bradford, United Kingdom
Bristol Haematology & Oncology Centre
Bristol, United Kingdom, BS2 8ED
Velindre Hospital
Cardiff, United Kingdom, CF14 2TL
Cheltenham General Hospital
Cheltenham, United Kingdom, GL53 7AN
Colchester General Hospital
Colchester, United Kingdom
Castle Hill Hospital
Cottingham, United Kingdom
University Hospitals Coventry & Warwickshire
Coventry, United Kingdom, CV2 2DX
Royal Derby Hospital
Derby, United Kingdom
Queen Elizabeth Hospital Birmingham
Edgbaston, United Kingdom, B15 2TH
Western General Hospital
Edinburgh, United Kingdom, EH4 2XU
Royal Devon & Exeter Hospital
Exeter, United Kingdom, EX2 5DW
Royal Surrey County Hospital
Guildford, United Kingdom, GU2 7XX
St James's Institute of Oncology
Leeds, United Kingdom, LS9 7TF
Leicester Royal Infirmary
Leicester, United Kingdom, LE1 5WW
University College Hospital
London, United Kingdom, NW1 2PG
Royal Marsden Hospital
London, United Kingdom, SW3 6JJ
James Cook University Hospital
Middlesbrough, United Kingdom, TS4 3BW
New Cross Hospital
New Cross, United Kingdom
Northampton General Hospital
Northampton, United Kingdom
Norfolk & Norwich University Hospital
Norwich, United Kingdom
Nottingham University Hopsital
Nottingham, United Kingdom, NG5 1PB
Glan Clwyd Hospital
Rhyl, United Kingdom
Weston Park Hospital
Sheffield, United Kingdom, S10 2SJ
Royal Shrewsbury Hospital
Shrewsbury, United Kingdom
Royal Marsden Hospital
Sutton, United Kingdom
Singleton Hospital
Swansea, United Kingdom, SA2 8QA
Musgrove Park Hospital
Taunton, United Kingdom Less << |
|
NCT01900158
|
Cholangiocarcinoma |
Phase 1
Phase 2 |
Recruiting |
- |
Germany
... More >> National Center for Tumor Diseases
Active, not recruiting
Heidelberg, Baden-Württemberg, Germany, D-69120
Klinikum rechts der Isar
Recruiting
Munich, Bayern, Germany, 81675
Contact: Hana Alguel, PhD hana.alguel@mri.tum.de
Klinikum der Ludwig-Maximilians-Universität
Recruiting
München, Bayern, Germany, 81377
Contact: Prof. Dr. Jörg Schirra, MD +49 89 4400 73031 Joerg.schirra@med.uni-muenchen.de
Klinikum der Johann Wolfgang Goethe-Universität
Recruiting
Frankfurt am Main, Hessen, Germany, 60590
Contact: Prof. Dr. Jörg Trojan, MD +49 69 6301 87686 trojan@em.uni-frankfurt.de
Universitätsklinikum Essen
Recruiting
Essen, Nordrhein-Westfalen, Germany, 45122
Contact: Dr.med. Stefan Kasper, MD +49 201 723 1631 stefan.kasper@uk-essen.de
Contact: Dr.med. Alexander Dechêne, MD +49 49 201 723 2390 alexander.dechene@uk-essen.de
Klinikum Ludwigshafen
Active, not recruiting
Ludwigshafen, Rheinland-Pfalz, Germany, D-67063
Universitätsklinikum Leipzig
Recruiting
Leipzig, Sachsen, Germany, 04103
Contact: Prof. Dr. Albrecht Hoffmeister, MD + 49341 9712251 Albrecht.Hoffmeister@medizin.uni-leipzig.de
Klinikum Augsburg
Not yet recruiting
Augsburg, Germany, 86156
Contact: Helmut Messmann, MD 0049 821 400-2351 helmut.messmann@klinikum-augsburg.de
Charité, Campus Mitte
Recruiting
Berlin, Germany, D-10117
Contact: Dr. med. Christian Jürgensen, MD 0049 30 450 514 134 Christian.Juergensen@charite.de
Universitätsklinikum Carl Gustav Carus an der Technischen Universität Dresden
Active, not recruiting
Dresden, Germany, 01307
Lithuania
Hospital of Lithuanian University of Health Sciences (LUHS)
Not yet recruiting
Kaunas, Lithuania, LT-50009
Contact: Limas Kupcinskas, MD 0037068640575 l.kupcinskas@gmail.com
National Cancer institute
Not yet recruiting
Vilnius, Lithuania, LT-08660
Contact: Mantas Trakymas, MD 0037061534951 trakymas@gmail.com
Vilnius University hospital Santariskiu Klinikos
Not yet recruiting
Vilnius, Lithuania, LT-08661
Contact: Juozas Stanaitis, MD 0037068661534 juozas.stanaitis@santa.lt
Norway
Oslo Universtiy Hospital
Active, not recruiting
Oslo, Norway, 0310
United Kingdom
University Hospital Aintree
Recruiting
Aintree, Liverpool, United Kingdom, L9 7AL
Contact: Dr Richard Sturgess, MD 0044 (0) 151 529 8157 RICHARD.STURGESS@aintree.nhs.uk
Contact: Dr Dan Palmer, MD 0044 (0)151 706 4177 Daniel.Palmer@liverpool.ac.uk
Hammersmith Hospital
Not yet recruiting
London, United Kingdom, W12OHS
Contact: Christopher Wadsworth
Contact Christopher.Wadsworth@imperial.nhs.uk Less << |
|
NCT00798655
|
Cancer of Head
... More >> Cancer of Head and Neck
Cancer of Neck
Cancer of the Head
Cancer of the Head and Neck
Cancer of the Neck
Head and Neck Cancer
Head Cancer
Head Neoplasms
Head, Neck Neoplasms
Neck Cancer
Neck Neoplasms
Neoplasms, Head
Neoplasms, Head and Neck
Neoplasms, Neck
Neoplasms, Upper Aerodigestive Tract
UADT Neoplasms
Upper Aerodigestive Tract Neoplasms Less << |
Phase 2 |
Completed |
- |
United States, Pennsylvania
... More >>
University of Pittsburgh Cancer Institute
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT02347917
|
Malignant Pleural Mesothelioma... More >>
Non-Small Cell Lung Cancer Less << |
Phase 1
Phase 2 |
Completed |
- |
Japan
... More >> National Cancer Center Hospital East
Chiba, Japan
National Cancer Center Hospital
Tokyo, Japan Less << |
|
NCT00798655
|
- |
|
Completed |
- |
- |
|
NCT01410214
|
Non-small Cell Lung Cancer Sta... More >>ge IIIA Less << |
Phase 2 |
Unknown |
July 2017 |
China, Beijing
... More >> Beijing Cancer Hospital
Recruiting
Beijing, Beijing, China, 100000
Contact: Chao Lv
Principal Investigator: Yue Yang
Chinese PLA General Hospital
Not yet recruiting
Beijing, Beijing, China, 100000
Contact: Tao Wang
Principal Investigator: Xiangyang Chu
China, Guangdong
Sun Yat-Sen University Cancer Center
Not yet recruiting
Guangzhou, Guangdong, China, 510000
Contact: Lanjun Zhang
Principal Investigator: Lanjun Zhang
China, Hebei
Hebei Medical University Fourth Hospital
Recruiting
Shijiazhuang, Hebei, China, 050000
Contact: Xinbo Liu
Principal Investigator: Junfeng Liu
China, Heilongjiang
Harbin Medical University Cancer Hospital
Recruiting
Harbin, Heilongjiang, China, 150000
Contact: Changfa Qu
Principal Investigator: Shidong Xu
China, Jiangsu
The First Affiurted Hospital of Soochow University
Not yet recruiting
Suzhou, Jiangsu, China, 215000
Contact: Haitao Ma
Principal Investigator: Haitao Ma
China, Shandong
Qingdao University Medical College
Not yet recruiting
Qingdao, Shandong, China, 266000
Contact: Yongjie Wang
Principal Investigator: Yi Shen
China, Shanghai
Fudan University Shanghai Cancer Center
Not yet recruiting
Shanghai, Shanghai, China, 200000
Contact: Yihua Sun
Principal Investigator: Haiquan Chen
China, Sichuang
The Second People's Hospital of Sichuan
Recruiting
Chengdu, Sichuang, China, 610000
Contact: Qiang Li
Principal Investigator: Qiang Li
China, Tianji
Tianjin Medical University Cancer Institute and Hospital
Recruiting
Tianji, Tianji, China, 300000
Contact: Xuefeng Kan
Principal Investigator: Changli Wang
China, Zhejiang
Zhejiang Cancer Hospital
Not yet recruiting
Hangzhou, Zhejiang, China, 310000
Contact: Xinming Zhou
Principal Investigator: Weimin Mao Less << |
|
NCT00165503
|
- |
|
Terminated(lack of acurral) |
- |
- |
|
NCT00703638
|
Breast Cancer
... More >> Colorectal Cancer
Head and Neck Cancer
Lung Cancer
Mesothelioma
Pancreatic Cancer
Prostate Cancer
Sarcoma Less << |
Phase 1 |
Completed |
- |
United States, Minnesota
... More >>
Masonic Cancer Center, University of Minnesota
Minneapolis, Minnesota, United States, 55455 Less << |
|
NCT01810367
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
China, Shanghai
... More >>
Cancer hospital Fudan University
Shanghai, Shanghai, China, 200032 Less << |
|
NCT02778191
|
- |
|
Completed |
- |
Netherlands
... More >> VUmc
Amsterdam, Netherlands
University Medical Center Groningen
Groningen, Netherlands, 9713 GZ Less << |
|
NCT00165503
|
Pleural Mesothelioma
... More >> Malignant Pleural Mesothelioma Less << |
Phase 2 |
Terminated(lack of acurral) |
- |
United States, Massachusetts
... More >>
Brigham and Women's Hospital
Boston, Massachusetts, United States, 02115
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00254384
|
Lung Cancer |
Not Applicable |
Active, not recruiting |
October 2019 |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT02216617
|
HNSCC |
Phase 1 |
Completed |
- |
France
... More >> ICM Val d'Aurelle, Montpellier
Montpellier, France, 34298 Less << |
|
NCT01846390
|
Peripheral T-Cell Lymphoma
... More >> Diffuse Large B-Cell Lymphoma Less << |
Phase 1 |
Completed |
- |
Canada, Alberta
... More >>
Tom Baker Cancer Centre
Calgary, Alberta, Canada, T2N 4N2
Canada, British Columbia
BCCA - Vancouver Cancer Centre
Vancouver, British Columbia, Canada, V5Z 4E6
Canada, New Brunswick
Regional Health Authority B, Zone 2
Saint John, New Brunswick, Canada, E2L 4L2
Canada, Nova Scotia
QEII Health Sciences Centre
Halifax, Nova Scotia, Canada, B3H 1V7
Canada, Ontario
Odette Cancer Centre
Toronto, Ontario, Canada, M4N 3M5
Univ. Health Network-Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT01747707
|
Gastric Cancer
... More >> Gastroesophageal Junction Cancer Less << |
Phase 2 |
Unknown |
May 2014 |
China
... More >> Cancer Hospital & Institute, Chinese Academy of Medical Sciences
Recruiting
Beijing, China, 10021
Contact: Yongkun Sun, M.D 8610-87788145 hsunyk@tom.com Less << |
|
NCT02397928
|
Malignant Pleural Mesothelioma |
Phase 2 |
Completed |
- |
Belgium
... More >> Antwerp University Hospital, Thoracic Oncology
Antwerp, Belgium
France
Goustave Roussy - Cancer Campus Grand Paris
Villejuif, France
Germany
Universitätsklinikum Halle (Saale)
Halle, Germany
Italy
A.S.O. "SS Antonio e Biagio e Cesare Arrigo"
Alessandria, Italy
Cliniche Humanitas Gavazzeni
Bergamo, Italy
Ospedale Villa Scassi
Genoa, Italy
Ospedaliera di Perugia
Perugia, Italy
Ospedaliero Universitaria Pisana
Pisa, Italy
Netherlands
Erasmus Mc
Rotterdam, Netherlands
Poland
Medical University Gdansk
Gdansk, Poland
Katedra i Klinika Onkologii Uniwersytetu Medycznegi im. Karola Marcinkowskiego w Poznaniu
Poznań, Poland
Klinika Nowotworów Pluca I Klatki Piersiowej
Warsaw, Poland
Spain
Vall d' Hebron Institute of Oncology (VHIO) Hospital Vall d'Hebron
Barcelona, Spain Less << |
|
NCT02648425
|
Solid Tumor |
Phase 1 |
Completed |
- |
Hong Kong
... More >> Queen Mary Hospital
Hong Kong, Hong Kong
Taiwan
National Taiwan University Hospital
Taipei, Taiwan, 100
Taipei Veterans General Hospital
Taipei, Taiwan, 112 Less << |
|
NCT02452970
|
Cholangiocarcinoma |
Phase 2 |
Active, not recruiting |
December 2018 |
United States, Maryland
... More >>
Johns Hopkins Sidney Kimmel Comprehensive Cancer Center
Baltimore, Maryland, United States, 21287 Less << |
|
NCT02301208
|
Nasopharyngeal Carcinoma |
Phase 3 |
Unknown |
- |
- |
|
NCT03051373
|
Carcinoma, Transitional Cell |
Phase 2 |
Not yet recruiting |
December 31, 2020 |
China
... More >> Chinese PLA General Hospital
Not yet recruiting
Beijing, China, 100022
Contact: Bo Yang shybomb@126.com Less << |
|
NCT00548821
|
Locally Advanced Cervical Canc... More >>er Less << |
PHASE3 |
UNKNOWN |
- |
National University Hospital, ... More >>Singapore, 119074, Singapore Less << |
|
NCT01593306
|
Carcinoma Cervix |
Phase 3 |
Unknown |
February 2013 |
India
... More >> Indira Gandhi Medical College
Recruiting
Shimla, Himachal Pradesh, India, 171001
Contact: Pragyat Thakur, MBBS 919418029244 pragyat28rpgmc@gmail.com
Principal Investigator: Pragyat Thakur, MBBS Less << |
|
NCT02969473
|
Esophageal Neoplasm
... More >> Chemoradiation Less << |
Phase 2 |
Unknown |
May 2017 |
China, Guangdong
... More >>
SYSU Cancer Center
Guangzhou, Guangdong, China, 510000 Less << |
|
NCT01412294
|
Gastric Cancer |
Phase 2 |
Active, not recruiting |
December 2017 |
Japan
... More >> Epidemiological and Clinical Research Information Network
Kyoto, Japan, 606-8392 Less << |
|
NCT02492360
|
- |
|
Withdrawn(Lack of funding) |
December 2019 |
United States, Indiana
... More >>
IU Simon Cancer Center
Indianapolis, Indiana, United States, 46202 Less << |
|
NCT00551512
|
Cancer
Solid ... More >>Tumors Less << |
Phase 1 |
Completed |
- |
United States, Arizona
... More >>
Scottsdale Clinical Research Institute
Scottsdale, Arizona, United States, 85258
United States, Massachusetts
Dana Farber Cancer Institute
Boston, Massachusetts, United States, 02115
United States, Nevada
Nevada Cancer Institute
Las Vegas, Nevada, United States, 89135 Less << |
|
NCT01284413
|
Biliary Tract Cancer |
Phase 1
Phase 2 |
Completed |
- |
Japan
... More >> Kyoto University Hospital
Kyoto, Japan, 606-8507 Less << |
|
NCT00390416
|
Stomach Neoplasms
... More >> Esophageal Neoplasms Less << |
Phase 2 |
Completed |
- |
United States, New Jersey
... More >>
Memoral Sloan Kettering Cancer Center
Basking Ridge, New Jersey, United States
United States, New York
Memorial Sloan-Kettering Cancer Center @ Suffolk
Commack, New York, United States, 11725
Memorial Sloan-Kettering Cancer Center 1275 York Avenue
New York, New York, United States, 10021
Memorial Sloan-Kettering Cancer Center at Mercy Medical Center
Rockville Centre, New York, United States, 11570
Memoral Sloan Kettering Cancer Center@Phelps Memorial Hospital
Sleepy Hollow, New York, United States Less << |
|
NCT00645593
|
Bladder Cancer |
Phase 2 |
Completed |
- |
United States, California
... More >>
City of Hope Cancer Center
Duarte, California, United States, 91010
Kenneth J. Norris Jr. Comprehensive Cancer Center, Keck School of Medicine, University of Southern California
Los Angeles, California, United States, 90033
Stanford University
Stanford, California, United States, 94305
United States, Illinois
Robert H. Lurie Comprehensive Cancer Center, Center of Northwestern University
Chicago, Illinois, United States, 60611
United States, Maryland
Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Baltimore, Maryland, United States, 21231
United States, Massachusetts
Boston Medical Center
Boston, Massachusetts, United States, 02118
Lahey Clinic
Burlington, Massachusetts, United States, 01805
United States, Michigan
University of Michigan
Ann Arbor, Michigan, United States, 48109
Wayne State University/Karmanos Cancer Institute
Detroit, Michigan, United States, 48201
United States, New York
Roswell Park Cancer Institute
Buffalo, New York, United States, 14263-0001
United States, Tennessee
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States, 37232
United States, Texas
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030-3721
United States, Utah
University of Utah Huntsman Cancer Institute
Salt Lake City, Utah, United States, 84112
United States, Washington
University of Washington
Seattle, Washington, United States, 98109 Less << |
|
NCT00390416
|
- |
|
Completed |
- |
- |
|
NCT03093922
|
Urothelial Carcinoma
... More >> Locally Advanced
Unresectable Less << |
Phase 2 |
Suspended(Safety Related) |
March 2019 |
United States, Illinois
... More >>
University of Chicago
Chicago, Illinois, United States, 60637
United States, New Jersey
Memoral Sloan Kettering Basking Ridge
Basking Ridge, New Jersey, United States, 07920
United States, New York
Memorial Sloan Kettering Cancer Center @ Commack
Commack, New York, United States, 11725
Memorial Sloan Kettering Westchester
Harrison, New York, United States, 10604
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
United States, Ohio
Ohio State University
Columbus, Ohio, United States, 43210 Less << |
|
NCT00645593
|
- |
|
Completed |
- |
- |
|
NCT01301248
|
Head and Neck Neoplasms
... More >> AJCC Stage III/IV Less << |
Phase 2 |
Unknown |
June 2011 |
Greece
... More >> Theagenio Cancer Hospital
Thessaloniki, Greece, 54007 Less << |
|
NCT00423865
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT01021150
|
Neoplasms, Malignant |
Phase 1 |
Completed |
- |
Japan
... More >> Sanofi-Aventis Administrative Office
Tokyo, Japan Less << |
|
NCT01097252
|
Cervical Cancers |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Korea Institute of Radiological & Medical Sciences
Seoul, Korea, Republic of, 139-706 Less << |
|
NCT03201861
|
Tubular Breast Cancer
... More >> Mucinous Breast Cancer
Invasive Duct Carcinoma of Breast Less << |
Phase 3 |
Recruiting |
December 31, 2021 |
China, Shanghai
... More >>
Renji Hospital, School of Medicine, Shanghai Jiaotong University
Recruiting
Shanghai, Shanghai, China, 200127
Contact: Yueyao Du, M.D. 86-21-68385569 jessicayy8629@126.com
Contact: Jie Zhang, M.D. 86-21-68385516 doudou090227@163.com Less << |
|
NCT01641575
|
Solid Tumor
N... More >>on-small-cell Lung Cancer
Lung Cancer Less << |
Phase 1 |
Terminated(Registrational stud... More >>y did not meet endpoint so entire program (including CO-101-011) was terminated.) Less << |
- |
United States, Florida
... More >>
Florida Cancer Specialists
Sarasota, Florida, United States, 34232
United States, Tennessee
Tennessee Oncology
Nashville, Tennessee, United States, 37203
United Kingdom
The Beatson West
Glasgow, Scotland, United Kingdom, G12 0YN
University College London Cancer Institute
London, United Kingdom, W1T 4TJ Less << |
|
NCT00324012
|
Adrenocortical Carcinoma |
Phase 2 |
Completed |
- |
Denmark
... More >> Department of Oncology 5073, Rigshospitalet
Copenhagen, Denmark, 2100 Less << |
|
NCT01524991
|
Urothelial Carcinoma |
Phase 2 |
Active, not recruiting |
December 2018 |
United States, California
... More >>
City of Hope: Duarte
Duarte, California, United States, 91010
United States, Indiana
IU Health Goshen Hospital
Goshen, Indiana, United States, 46527
Indiana University Melvin & Bren Simon Cancer Center
Indianapolis, Indiana, United States, 46202
IU Health Central Indiana Cancer Centers
Indianapolis, Indiana, United States, 46219
United States, Nebraska
Nebraska Cancer Specialists
Omaha, Nebraska, United States, 68114
United States, New York
Tisch Cancer Institute at Mount Sinai Medical Center
New York, New York, United States, 10029
United States, Texas
Texas Oncology, PA
Dallas, Texas, United States, 75246
United States, Virginia
Virginia Oncology Associates
Norfolk, Virginia, United States, 23502 Less << |
|
NCT02620839
|
Solid Tumors |
Phase 1 |
Recruiting |
March 2020 |
United States, California
... More >>
UCSF Helen Diller Family Comprehensive Cancer Center
Recruiting
San Francisco, California, United States, 94143-1711
Contact: Pamela N. Munster, MD 877-827-3222 cancertrials@ucsf.edu Less << |
|
NCT00123825
|
Gallbladder Cancer
... More >> Biliary Tract Cancer Less << |
Phase 2 |
Completed |
- |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02115
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00919061
|
- |
|
Completed |
- |
- |
|
NCT00919061
|
Extrahepatic Bile Duct Cancer
... More >>
Gallbladder Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT00165464
|
Esophageal Cancer
... More >> Gastric Cancer
GE Junction Cancer Less << |
Phase 2 |
Completed |
- |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02115
Faulkner Hospital
Boston, Massachusetts, United States, 02130 Less << |
|
NCT00047047
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, Minnesota
... More >>
Mayo Clinic
Rochester, Minnesota, United States, 55905 Less << |
|
NCT02151084
|
Biliary Tract Carcinoma
... More >> Gallbladder Carcinoma Less << |
Phase 2 |
Completed |
- |
Canada, Ontario
... More >>
Princess Margaret Cancer Centre
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT01616875
|
Infiltrating Bladder Urothelia... More >>l Carcinoma Less << |
Phase 2 |
Recruiting |
January 2018 |
United Kingdom
... More >> Bristol Haematology + Oncology Centre, Horfield Road
Recruiting
Bristol, United Kingdom, BS2 8ED
Contact: Sylvia Pearson 0117 3424263 sylvia.pearson@uhbristol.nhs.uk
Contact: Seonaid Wright 0117 342 2069 seonaid.wright@uhbristol.nhs.uk
Principal Investigator: Amit K Bahl
Sub-Investigator: Susan Masson Less << |
|
NCT01445405
|
Carcinoma, Squamous
... More >> Head and Neck Cancer
Oral Cancer
Laryngeal Cancer
Pharyngeal Cancer Less << |
Phase 1 |
Completed |
- |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892 Less << |
|
NCT00165191
|
Adenocarcinoma of Stomach
... More >> Adenocarcinoma of GE Junction
Adenocarcinoma of Esophagus Less << |
Phase 2 |
Completed |
- |
United States, Massachusetts
... More >>
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115
Massachusetts General Hospital
Boston, Massachusetts, United States, 02115 Less << |
|
NCT01483300
|
Breast Cancer |
Phase 2 |
Unknown |
November 2014 |
China, Heilongjiang
... More >>
Cancer Hospital of Harbin Medical University
Recruiting
Harbin, Heilongjiang, China, 150081
Contact: Qingyuan Zhang, MD 86-451-86298276 zhma19650210@163.com
Contact: Xinmei Kang, MD 86-451-86298683 kxm791107@163.com
Principal Investigator: Qingyuan Zhang, MD
Sub-Investigator: Xinmei Kang, MD Less << |
|
NCT00077545
|
Adenocarcinoma of the Esophagu... More >>s
Recurrent Esophageal Cancer
Stage IV Esophageal Cancer Less << |
Phase 2 |
Completed |
- |
United States, Illinois
... More >>
University of Chicago Comprehensive Cancer Center
Chicago, Illinois, United States, 60637-1470 Less << |
|
NCT03502148
|
Oral Squamous Cell Carcinoma |
Phase 1
Phase 2 |
Recruiting |
April 30, 2020 |
United States, Ohio
... More >>
University of Cincinnati Cancer Institute
Recruiting
Cincinnati, Ohio, United States, 45267
Contact: Lisa Schmid, BSN 513-584-0502 wallslm@ucmail.uc.edu
Principal Investigator: Alice Tang, MD
United States, Texas
Baylor College of Medicine
Recruiting
Houston, Texas, United States, 77030
Contact: Vlad Sandulache, MD 713-798-7218 vlad.sandulache@BCM.edu
Ben Taub Hospital
Recruiting
Houston, Texas, United States, 77030
Contact: Vlad Sandulache, MD 713-798-7218 vlad.sandulache@bcm.edu
Harris health System - Smith Clinic
Recruiting
Houston, Texas, United States, 77054
Contact: Vlad Sandulache, MD 713-798-7218 vlad.sandulache@bcm.edu
The University of Texas Health Science Center School of Dentistry
Recruiting
Houston, Texas, United States, 77054
Contact: Simon Young, DDS, MD, PhD 713-486-2568 Simon.Young@uth.tmc.edu Less << |
|
NCT00915850
|
Esophageal Cancer |
Phase 1
Phase 2 |
Unknown |
August 2015 |
Japan
... More >> Wakayama Medical University
Recruiting
Wakayama, Japan, 641-8510
Contact: Makoto Iwahashi, MD 81-73-441-0613 makato@wakayama-med.ac.jp Less << |
|
NCT02460887
|
Nasopharyngeal Carcinoma |
Phase 2 |
Recruiting |
June 2021 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Peiyu Huang, Dr +86-20-87343380 huangpy@sysucc.org.cn
Contact: Donghua Luo, Dr +86-20-87343379 luodh@sysucc.org.cn Less << |
|
NCT01196416
|
- |
|
Completed |
- |
- |
|
NCT01368848
|
NSCLC |
Phase 2 |
Completed |
- |
Austria
... More >> Univ.-Klinik für Innere Medizin V Innsbruck, Abteilung für Hämatologie und Onkologie
Innsbruck, Tirol, Austria, 6020
Landeskrankenhaus Feldkirch
Feldkirch, Vorarlberg, Austria, 6806
Universitaetsklinik der PMU Salzburg, UK f. Innere Medizin III
Salzburg, Austria, 5020 Less << |
|
NCT00104910
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Carcinoma
Cervical Small Cell Carcinoma
Cervical Squamous Cell Carcinoma
Stage IB Cervical Cancer
Stage IIA Cervical Cancer
Stage IIB Cervical Cancer
Stage III Cervical Cancer
Stage IVA Cervical Cancer Less << |
Phase 1 |
Completed |
- |
United States, Georgia
... More >>
Georgia Regents University Medical Center
Augusta, Georgia, United States, 30912
United States, Illinois
University of Chicago Comprehensive Cancer Center
Chicago, Illinois, United States, 60637
United States, Indiana
Indiana University/Melvin and Bren Simon Cancer Center
Indianapolis, Indiana, United States, 46202
United States, Iowa
University of Iowa Hospitals and Clinics
Iowa City, Iowa, United States, 52242
United States, Maryland
Johns Hopkins University/Sidney Kimmel Comprehensive Cancer Center
Baltimore, Maryland, United States, 21287
United States, New Jersey
Cooper Hospital University Medical Center
Camden, New Jersey, United States, 08103
United States, Ohio
MetroHealth Medical Center
Cleveland, Ohio, United States, 44109
Cleveland Clinic Foundation
Cleveland, Ohio, United States, 44195
Riverside Methodist Hospital
Columbus, Ohio, United States, 43214
Lake University Ireland Cancer Center
Mentor, Ohio, United States, 44060
United States, Oklahoma
University of Oklahoma Health Sciences Center
Oklahoma City, Oklahoma, United States, 73104
United States, Rhode Island
Women and Infants Hospital
Providence, Rhode Island, United States, 02905
United States, Texas
Parkland Memorial Hospital
Dallas, Texas, United States, 75235
University of Texas Southwestern Medical Center
Dallas, Texas, United States, 75390
M D Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT01596868
|
Nasopharyngeal Squamous Cell C... More >>arcinoma
Toxicity Due to Radiotherapy Less << |
Phase 2 |
Completed |
- |
China, Shanxi
... More >> Department of Radiation Oncology, Xijing Hospital, Fourth Military
Xi'an, Shanxi, China, 710032 Less << |
|
NCT01196416
|
Recurrent Melanoma
... More >> Stage IV Skin Melanoma Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT00096174
|
- |
|
Unknown |
- |
- |
|
NCT02608073
|
Nasopharyngeal Cancer |
Phase 2 |
Completed |
- |
Hong Kong
... More >>
Hong Kong, Hong Kong, 852
Hong Kong, Hong Kong
Indonesia
Jogjakarta, Indonesia, 55284
Malaysia
Kuala Lumpur, Malaysia, 50603
Taiwan
Kueishan, Taiwan, 333
Taipei, Taiwan, 00112
Taipei, Taiwan, 106
Thailand
Bangkok, Thailand, 10330
Bangkok, Thailand, 10400 Less << |
|
NCT00096174
|
Head and Neck Cancer |
Phase 2 |
Unknown |
July 2016 |
- |
|
NCT01086254
|
Non-small Cell Lung Cancer Sta... More >>ge IV Less << |
Phase 2 |
Completed |
- |
France
... More >> Sanofi-Aventis Investigational Site Number 250002
Caen Cedex, France, 14033
Sanofi-Aventis Investigational Site Number 250003
Marseille Cedex 09, France, 13009
Sanofi-Aventis Investigational Site Number 250004
Toulouse, France, 31059
Sanofi-Aventis Investigational Site Number 250001
Villejuif, France, 94805
Germany
Sanofi-Aventis Investigational Site Number 276003
Essen, Germany, 45122
Sanofi-Aventis Investigational Site Number 276002
Gauting, Germany, 82131
Sanofi-Aventis Investigational Site Number 276001
Großhansdorf, Germany, 22927
Italy
Sanofi-Aventis Investigational Site Number 380003
Livorno, Italy, 57123
Sanofi-Aventis Investigational Site Number 380001
Orbassano, Italy, 10043
Sanofi-Aventis Investigational Site Number 380002
Rozzano, Italy, 20089
Spain
Sanofi-Aventis Investigational Site Number 724001
Badalona, Spain, 08916
Sanofi-Aventis Investigational Site Number 724002
Barcelona, Spain, 08035
United Kingdom
Sanofi-Aventis Investigational Site Number 826001
Newcastle Upon Tyne, United Kingdom, NE7 7DN
Sanofi-Aventis Investigational Site Number 826002
Wolverhampton, United Kingdom, WV10 0QP Less << |
|
NCT02611700
|
Esophageal Squamous Cell Carci... More >>nomas Less << |
Phase 3 |
Recruiting |
June 2019 |
- |
|
NCT01396681
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Seoul National University Bundang Hospital
Seongnam, Korea, Republic of
Seoul National University Hospital
Seoul, Korea, Republic of Less << |
|
NCT00251550
|
Malignant Pleural Mesotherioma |
Phase 1
Phase 2 |
Completed |
- |
Japan
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Aichi, Japan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Ehime, Japan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Fukuoka, Japan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Hokkaido, Japan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Okayama, Japan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Osaka, Japan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Tokyo, Japan Less << |
|
NCT00702481
|
Head and Neck Cancer |
Phase 2 |
Active, not recruiting |
March 2019 |
Singapore
... More >> National Cancer Center Singapore
Singapore, Singapore, 169610 Less << |
|
NCT01109524
|
Lung Neoplasms
... More >> Carcinoma
Cancer of the Lung
Non-Small-Cell Lung Carcinoma Less << |
Phase 2 |
Completed |
- |
United States, Arizona
... More >>
Donald W. Hill M.D., P.C.
Casa Grande, Arizona, United States, 85122
United States, California
Beverly Hills Cancer Center
Beverly Hills, California, United States, 90211
Cancer Care Institute
Los Angeles, California, United States, 90036
Northern California Hematology & Oncology
Oakland, California, United States, 94609
Sharp Clinical Oncology Research
San Diego, California, United States, 92123
United States, Florida
Broward Oncology Associates, P.A.
Ft. Lauderdale, Florida, United States, 33308
Elite Research Institute
Miami, Florida, United States, 33125
United States, Illinois
Edward H. Kaplan, MD & Associates
Skokie, Illinois, United States, 60076
United States, Minnesota
Fairview Southdale Medical Oncology Clinic
Edina, Minnesota, United States, 55435
United States, North Dakota
Mid Dakota Clinic, Pc
Bismarck, North Dakota, United States, 58501
United States, Pennsylvania
Temple University Hospital
Philadelphia, Pennsylvania, United States, 19140
United States, Texas
University Medical Center
Lubbock, Texas, United States, 79415
United States, Washington
Columbia Basin Hematology and Oncology
Kennewick, Washington, United States, 99336
Canada, New Brunswick
The Moncton Hospital
Moncton, New Brunswick, Canada, E1C 6Z8
Canada, Newfoundland and Labrador
Local Institution
Grand Falls-Windsor, Newfoundland and Labrador, Canada, A2A 2E2
Canada, Ontario
The Credit Valley Hospital
Mississauga, Ontario, Canada, L5M 2N1
Sudbury Regional Hospital
Sudbury, Ontario, Canada, P3E 5J1
Thunder Bay Regional Health Sciences Centre (Regional Cancer Care)
Thunder Bay, Ontario, Canada, P7B 6V4
Toronto East General Hospital
Toronto, Ontario, Canada, M4C 3E7
Canada, Quebec
Centre de sante et de services sociaux de Rimouski-Neigette
Rimouski, Quebec, Canada, G5L 5T1
Puerto Rico
Ponce School of Medicine (Caimed Center)
Ponce, Puerto Rico, 00716
Fundacion de Investigacion de Diego
San Juan, Puerto Rico, 00927 Less << |
|
NCT00139269
|
Advanced Squamous Cell Carcino... More >>ma
Squamous Cell Carcinoma of Head and Neck
SSCHN Less << |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT00661778
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Spain
... More >>
Cádiz, Cadiz, Spain, 11009
San Sebastian, Guipuzcoa, Spain, 20080
Palma de Mallorca, Islas Baleares, Spain, 07198
Alcala de Henares, Madrid, Spain, 28805
Sagunto, Valencia, Spain, 46520
Castellon, Spain, 12002
Madrid, Spain, 28036
Madrid, Spain, 28935
Malaga, Spain, 29010
Palencia, Spain, 34005
Valencia, Spain, 46017
Valladolid, Spain, 47005
Valladolid, Spain, 47010
Zamora, Spain, 49021 Less << |
|
NCT02316327
|
Non-Small Cell Lung Cancer (NS... More >>CLC) Less << |
Phase 4 |
Completed |
- |
Spain
... More >> Hospital Clinic
Barcelona, Spain, 08036 Less << |
|
NCT00661778
|
- |
|
Completed |
- |
- |
|
NCT00185835
|
Head and Neck Cancer
... More >> Carcinoma, Squamous Cell Less << |
Phase 1 |
Completed |
- |
United States, California
... More >>
Stanford University School of Medicine
Stanford, California, United States, 94305 Less << |
|
NCT00254904
|
Carcinoma, Non-Small-Cell Lung |
Phase 3 |
Terminated(See Termination Rea... More >>son in Detailed Description) Less << |
- |
- |
|
NCT01029873
|
Metastatic Melanoma |
Phase 1
Phase 2 |
Completed |
- |
United States, California
... More >>
The Angeles Clinic and Research Institute
Los Angeles, California, United States, 90025
United States, Florida
MD Anderson Cancer Center Orlando
Orlando, Florida, United States, 32806
United States, Georgia
Emory University
Atlanta, Georgia, United States, 30322
United States, Illinois
Northwestern University
Chicago, Illinois, United States, 60611
United States, Iowa
University of Iowa Hospitals and Clinics
Iowa City, Iowa, United States, 52242
United States, North Carolina
Carolinas Medical Center-Brumenthal Cancer Center
Charlotte, North Carolina, United States, 28204
United States, Pennsylvania
St. Luke's Hospital and Health Network
Bethlehem, Pennsylvania, United States, 18015
United States, Washington
University of Washington, Seattle Cancer Care Center
Seattle, Washington, United States, 98109 Less << |
|
NCT01109524
|
- |
|
Completed |
- |
- |
|
NCT01830556
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 2 |
Unknown |
November 2015 |
Denmark
... More >> Lena Specht
Recruiting
Copenhagen, Denmark, 2100
Contact: Lena Specht, MD DMSc Professor lena.specht@rh.regionh.dk
Principal Investigator: Lena Specht, MD DMSc Professor
Sweden
Hedda Haugen
Recruiting
Göteborg, Sweden, 413 45
Contact: Hedda Haugen, MD hedda.haugen@vgregion.se
Principal Investigator: Hedda Haugen, MD
Karolinska Universityhospital
Recruiting
Stockholm, Sweden, 17176
Contact: Teresa herlestam-calero-moreno, MD maria.herlestam-calero-moreno@karolinska.se
Principal Investigator: Teresa maria.herlestam-calero-moreno, MD
Karin Söderström
Recruiting
Umeå, Sweden, 90189
Contact: Karin Söderström, D karin.soderstrom@onkologi.umu.se
Principal Investigator: Karin Söderström, MD
Akademiska Universityhospital
Not yet recruiting
Uppsala, Sweden, 751 85
Contact: Silvia Johansson, MD silvia.johansson@onkologi.uu.se
Principal Investigator: Silvia Johansson, MD Less << |
|
NCT00388960
|
Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Belgium
... More >> Algemeen Ziekenhuis Middelheim
Antwerpen, Belgium, 2020
Universitair Ziekenhuis Antwerpen
Edegem, Belgium, 2650
Universiteit Gent
Gent, Belgium, 9000
U.Z. Gasthuisberg
Leuven, Belgium, 3000
Domaine Universitaire du Sart-Tilman
Liege, Belgium, 1BE
Centre Hospitalier Regional de la Citadelle
Liege, Belgium, 4000
Clinique Sainte Elisabeth
Namur, Belgium, 5000
Italy
Instituto Nazionale per la Ricerca sul Cancro
Genova, Italy, 16132
Universita Degli Studi Di Udine
Udine, Italy, 33100
Netherlands
Academisch Medisch Centrum
Amsterdam, Netherlands, 1105 AZ
The Netherlands Cancer Institute Antoni Van Leeuwenhoekziekenhuis
Amsterdam, Netherlands
Medisch Spectrum Twente - Dept of Pulmonary Diseases
Enschede, Netherlands, 7500 KA
Leiden University Medical Centre
Leiden, Netherlands, 2300RC
Academisch Ziekenhuis Maastricht
Maastricht, Netherlands, 6202
Isala Kliniek
Zwolle, Netherlands, 8001
Poland
Medical University of Gdansk - Dept Radiotherapy
Gdansk, Poland, 80211
United Kingdom
Clatterbridge Centre for Oncology NHS Trust
Bebington, Merseyside, United Kingdom, CH684JY
University of Dundee - Ninewells Hospital
Dundee, Scotland, United Kingdom, D01 9SY
Belfast City Hospital
Belfast, United Kingdom, BT9 7AB
Western General Hospital
Edinburgh, United Kingdom, EH4 2XU
Princess Royal Hospital
Hull, United Kingdom, HU8 9HE
Royal Marsden Hospital, London
London, United Kingdom, SM2 5PT
Christie Hospital
Manchester, United Kingdom, M20 4BX
Sir Bobby Robson Cancer Trials Research Centre
Newcastle-Upon-Tyne, United Kingdom, NE4 6BE
Royal Marsden Hospital Lung Unit
Sutton, United Kingdom, (Surrey) SM2 5PT Less << |
|
NCT00623064
|
Bladder Cancer
... More >> Transitional Cell Cancer of the Renal Pelvis and Ureter Less << |
Phase 1 |
Completed |
- |
Denmark
... More >> Rigshospitalet - Copenhagen University Hospital
Copenhagen, Denmark, 2100 Less << |
|
NCT02633176
|
Nasopharyngeal Carcinoma |
Phase 3 |
Recruiting |
- |
China, Fujian
... More >> Fujian Provincial Cancer Hospital
Recruiting
Fuzhou, Fujian, China, 350014
Contact: Cheng Huang, MD cheng671@sina.com
Principal Investigator: Cheng Huang, MD
China, Guangdong
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Zhao Wang, MD +86-20-87343694 wangzhao@sysucc.org.cn
Sub-Investigator: He Huang, MD
The First Affiliated Hospital of Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China, 510080
Contact: Mengping Zhang, MD +86-20-87755766-8576 zhangmp1989@163.com
Principal Investigator: Sheng Ye, MD
China, Guangxi
Guangxi Cancer Hospital
Recruiting
Nanning, Guangxi, China, 530021
Contact: Chaoyong Liang, MD lmlm1995@qq.com
Principal Investigator: Xiaohua Hu, MD
China, Hubei
Union Hospital,Tongji Medical College of Huazhong University of Science & Technology
Recruiting
Wuhan, Hubei, China, 430022
Contact: Gang Wu, MD xhzlwg@163.com
Principal Investigator: Gang Wu, MD
Tongji Hospital,Tongji Medical College of Huazhong University of Science & Technology
Recruiting
Wuhan, Hubei, China, 430030
Contact: Shiying Yu, MD syyu@tjh.tjmu.edu.cn
Principal Investigator: Shiying Yu, MD
China, Jiangsu
Jiangsu Cancer Hospital
Recruiting
Nanjing, Jiangsu, China, 210000
Contact: Jifeng Feng, MD fjif@vip.sina.com
Principal Investigator: Jifeng Feng, MD
China, Shanghai
Fudan University Shanghai Cancer Center
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Xichun Hu, MD xchu2009@hotmail.com
Principal Investigator: Xichun Hu, MD
China, Sichuan
Sichuan Cancer Hospital
Recruiting
Chengdu, Sichuan, China, 610047
Contact: Jinyi Lang, MD langjy610@163.com
Principal Investigator: Jinyi Lang, MD
China, Zhejiang
Zhejiang Cancer Hospital
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: Yuan Zhu, MD zhuyuan63@hotmail.com
Principal Investigator: Yuan Zhu, MD Less << |
|
NCT00639769
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
United States, Georgia
... More >>
Central Georgia Hematology Oncology Associates, P.C.
Macon, Georgia, United States
United States, Tennessee
Erlanger Health System
Chattanooga, Tennessee, United States
Jackson-Madison County Hospital
Jackson, Tennessee, United States
East Tennessee State University
Johnson City, Tennessee, United States
Center for Biomedical Research
Knoxville, Tennessee, United States
Meharry Medical College
Nashville, Tennessee, United States
VA Tennessee Valley Healthcare Center
Nashville, Tennessee, United States
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States Less << |
|
NCT01281852
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Carcinoma
Cervical Squamous Cell Carcinoma, Not Otherwise Specified
Recurrent Cervical Carcinoma
Stage IVB Cervical Cancer Less << |
Phase 1 |
Completed |
- |
- |
|
NCT00639769
|
- |
|
Completed |
- |
- |
|
NCT00821327
|
- |
|
Completed |
- |
- |
|
NCT01342172
|
Urinary Bladder Neoplasms |
Phase 1
Phase 2 |
Terminated(low accrual) |
- |
United States, Maryland
... More >>
National Cancer Institute
Bethesda, Maryland, United States, 20892
United States, New York
Icahn School of Medicine at Mount Sinai
New York, New York, United States, 10029
United States, Utah
Huntsman Cancer Institute/University of Utah
Salt Lake City, Utah, United States, 84112 Less << |
|
NCT00371553
|
Relapsed or Cisplatin-Refracto... More >>ry Germ Cell Cancer Less << |
Phase 2 |
Completed |
- |
Canada, Alberta
... More >>
Tom Baker Cancer Centre
Calgary, Alberta, Canada, T4N 4N2
Canada, British Columbia
BC Cancer Agency - Vancouver Centre
Vancouver, British Columbia, Canada, V5Z 4E6
Canada, Manitoba
Cancer Care Manitoba
Winnipeg, Manitoba, Canada, R3E 0V9
Canada, Nova Scotia
QEII Health Science Center
Halifax, Nova Scotia, Canada, B3H 2Y9
Canada, Ontario
London Regional Cancer Centre
London, Ontario, Canada, N6A 4L6
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G2M9 Less << |
|
NCT01917617
|
Biliary Tract Cancer |
Phase 2 |
Active, not recruiting |
March 2018 |
Japan
... More >> Osaka University, Graduate School of Medicine
Osaka, Japan, 565-0871 Less << |
|
NCT01342172
|
- |
|
Terminated(low accrual) |
- |
- |
|
NCT01611662
|
Distal Urethral Cancer
... More >> Proximal Urethral Cancer
Squamous Cell Carcinoma of the Bladder
Stage II Bladder Cancer
Stage III Bladder Cancer
Urethral Cancer Associated With Invasive Bladder Cancer Less << |
Phase 2 |
Terminated(Extreme Toxicity) |
December 2018 |
United States, Pennsylvania
... More >>
Thomas Jefferson University, Kimmel Cancer Center
Philadelphia, Pennsylvania, United States, 19107
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111-2497 Less << |
|
NCT01611662
|
- |
|
Terminated(Extreme Toxicity) |
- |
- |
|
NCT01821248
|
Biliary Tract Cancer |
Phase 2 |
Recruiting |
February 2019 |
Japan
... More >> Kyoto University Hospital
Recruiting
Kyoto, Japan, 606-8507
Contact: Satoru Seo +81-75-751-4323 rutosa@kuhp.kyoto-u.ac.jp Less << |
|
NCT00349492
|
Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Soonchunhyang University Bucheon Hospital
Bucheon-si, Korea, Republic of, 420-767
Yeungnam University Hospital
Daegu, Korea, Republic of, 705-717
Daegu Catholic University Hospital
Daegu, Korea, Republic of, 705-718
Gyeongsang National University Hospital
Jinju, Korea, Republic of, 660-702
Seoul National University Bundang Hospital
Seongnam-Si, Korea, Republic of, 463-707
Seoul National University Hospital
Seoul, Korea, Republic of, 110-744
Kangbuk Samsung Hospital
Seoul, Korea, Republic of, 110-746
Yonsei Cancer Center
Seoul, Korea, Republic of, 120-752
Seoul Veterans Hospital
Seoul, Korea, Republic of, 134-791
Kangnam St.Mary's Hospital
Seoul, Korea, Republic of, 137-040
Asan Medical Center
Seoul, Korea, Republic of, 138-736
Chung-Ang University Medical Center
Seoul, Korea, Republic of, 151-756
Seoul Municipal Boramae Hospital
Seoul, Korea, Republic of, 156-707
St.Vincent Hospital
Suwon, Korea, Republic of, 422-723 Less << |
|
NCT00537511
|
Carcinoma, Small Cell |
Phase 1
Phase 2 |
Terminated(This study was term... More >>inated for administrative reasons.) Less << |
- |
United States, Ohio
... More >>
Ireland Cancer Center, Case Western Reserve University, Division of Hematology/Oncology
Cleveland, Ohio, United States, 44106
United States, Pennsylvania
Pennsylvania State University
Hershey, Pennsylvania, United States, 17033
United States, Texas
University of Texas Southwestern Medical Center
Dallas, Texas, United States, 75390
Canada, Ontario
Juranvinski Cancer Center - Medical Oncology
Hamilton, Ontario, Canada, L8V 5C2
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9
Canada, Quebec
Mc Gill University - Department of Oncology - Clinical Research Program
Montreal, Quebec, Canada, H2W 1S6 Less << |
|
NCT00982111
|
Non Small Cell Lung Cancer |
Phase 3 |
Suspended |
December 31, 2018 |
- |
|
NCT00537511
|
- |
|
Terminated(This study was term... More >>inated for administrative reasons.) Less << |
- |
- |
|
NCT02061631
|
Squamous Cell Carcinoma of Hea... More >>d and Neck Less << |
Phase 2 |
Completed |
- |
Pakistan
... More >>
Pakistan, Pakistan Less << |
|
NCT01702714
|
Non-Squamous Non-Small Cell Lu... More >>ng Cancer Less << |
Phase 1 |
Withdrawn(Project Team decisio... More >>n) Less << |
February 2016 |
- |
|
NCT00982111
|
- |
|
Suspended |
- |
- |
|
NCT00002913
|
Brenner Tumor
... More >> Ovarian Clear Cell Cystadenocarcinoma
Ovarian Endometrioid Adenocarcinoma
Ovarian Mixed Epithelial Carcinoma
Ovarian Mucinous Cystadenocarcinoma
Ovarian Serous Cystadenocarcinoma
Ovarian Undifferentiated Adenocarcinoma
Stage III Ovarian Epithelial Cancer
Stage IV Ovarian Epithelial Cancer Less << |
Phase 1 |
Completed |
- |
United States, Arizona
... More >>
Gynecologic Oncology Group of Arizona
Phoenix, Arizona, United States, 85012 Less << |
|
NCT00942825
|
Metastatic Non-squamous Non Sm... More >>all Cell Lung Cancer Less << |
Phase 2 |
Completed |
- |
United States, Nevada
... More >>
Nevada Cancer Institute
Las Vegas, Nevada, United States, 89135
United States, Pennsylvania
Penn State Cancer Institute
Hershey, Pennsylvania, United States, 17033
United States, Texas
Mary Crowley Cancer Research Centers
Dallas, Texas, United States, 75246 Less << |
|
NCT01301612
|
Carcinoma
Ade... More >>nocarcinoma
Uterine Cervix Adenosquamous Carcinoma Less << |
Phase 2 |
Withdrawn(Regulatory requireme... More >>nt. A phase III study is being designed.) Less << |
- |
Brazil
... More >> Centro de Pesquisa Clínica da Liga Norte Riograndense contra o Câncer
Natal, Rio Grande do Norte, Brazil, 59075740
Hospital de Caridade de Ijui - ONCOSITE Centro de Pesquisa Clínica em Oncologia
Ijui, Rio Grande do Sul, Brazil, 98700000
Hospital Santa Rita - Núcleo de Novos Tratamentos em Câncer
Porto Alegre, Rio Grande do Sul, Brazil, 90020-160
Caism - Unicamp
Campinas, São Paulo, Brazil, 13083970
Centro de Pesquisas Clínicas da Fundação Amaral Carvalho
Jau, São Paulo, Brazil, 17210000
ICESP
São Paulo, Brazil, 01246000
Hospital Santa Marcelina
São Paulo, Brazil, 08270070 Less << |
|
NCT00139243
|
Squamous Cell Carcinoma
... More >> Carcinoma of Head and/or Neck Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, Massachusetts
... More >>
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115
Massachusetts General Hospital
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00942825
|
- |
|
Completed |
- |
- |
|
NCT03044587
|
Adenocarcinoma Metastatic
... More >> Biliary Tract Cancer
Adenocarcinoma of the Biliary Tract
Adenocarinoma Locally Advanced
Non-Resectable Hepatocellular Carcinoma
Intrahepatic Bile Duct Carcinoma
Extrahepatic Bile Duct Carcinoma Less << |
Phase 2 |
Recruiting |
January 2024 |
Germany
... More >> Universitätsklinikum Ulm
Recruiting
Ulm, Germany, 89081
Contact: Thomas J. Ettrich, Dr. thomas.ettrich@uniklinik-ulm.de Less << |
|
NCT03588533
|
HER-2 Positive Gastric Cancer
... More >>
Metastatic Cancer Less << |
Phase 2 |
Recruiting |
August 31, 2021 |
Korea, Republic of
... More >>
Dong-A University Hospital
Recruiting
Busan, Korea, Republic of, 49201
Contact: Sung Yong Oh, MD +82512402808 drosy@dau.ac.kr
Contact: Enhee Choi, RN +82512405044 dongahicrc8@naver.com Less << |
|
NCT00747799
|
Nasopharyngeal Neoplasms |
Phase 2 |
Completed |
- |
China, Guangdong
... More >>
Cancer Center of Sun-Yat Sen University
Guangzhou, Guangdong, China, 510000 Less << |
|
NCT01407822
|
Non-small Cell Lung Cancer |
Phase 2
Phase 3 |
Active, not recruiting |
December 2022 |
China, Guangdong
... More >>
Guangdong General Hospital
Guangzhou, Guangdong, China, 510080 Less << |
|
NCT01360827
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 1 |
Terminated(Study terminated du... More >>e to potential safety concerns in combination with platinum-based therapies) Less << |
- |
France
... More >> Clinical Research Unit and Pharmacology Lab EA 3035 Institut Claudius Regaud
Toulouse, France Less << |
|
NCT00515411
|
Gastroesophageal Junction Aden... More >>ocarcinoma
Gastric Cancer Less << |
Phase 2 |
Completed |
- |
United States, California
... More >>
City of Hope Cancer Center
Duarte, California, United States, 91010
United States, Florida
Memorial Cancer Institute
Pembroke Pines, Florida, United States, 33025
United States, Georgia
Piedmont Hospital Research Institute
Atlanta, Georgia, United States, 30309
United States, Nebraska
Nebraska Cancer Specialists, Methodist Estabrook Cancer Center
Omaha, Nebraska, United States, 68114
United States, New Jersey
Memoral Sloan Kettering Cancer Center
Basking Ridge, New Jersey, United States
United States, New York
Memorial Sloan-Kettering Cancer Center @ Suffolk
Commack, New York, United States, 11725
Queens Cancer Center of Queens Hospital
Jamaica, New York, United States, 11432
Long Island Jewish Medical Center
New Hyde Park, New York, United States, 11040
Weill Medical College of Cornell University
New York, New York, United States, 10021
Memorial Sloan Kettering Cancer Center 1275 York Avenue
New York, New York, United States, 10065
Memorial Sloan Kettering at Mercy Medical Center
Rockville Centre, New York, United States
Memoral Sloan Kettering Cancer Center@Phelps Memorial Hospital
Sleepy Hollow, New York, United States
United States, Ohio
University Hospital of Cleveland
Cleveland, Ohio, United States, 44106
United States, Pennsylvania
University of Pittsburgh Cancer Institute
Pittsburgh, Pennsylvania, United States, 15232
United States, Wisconsin
Medical College of Wisconsin
Milwaukee, Wisconsin, United States, 53226 Less << |
|
NCT00821327
|
Urothelial Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT01788566
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
United States, Alabama
... More >>
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Birmingham, Alabama, United States, 35294
United States, Florida
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Deerfield Beach, Florida, United States, 33442
United States, Michigan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Farmington Hills, Michigan, United States, 48334
Canada, Alberta
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Calgary, Alberta, Canada, T2N 4N2
Canada, Ontario
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hamilton, Ontario, Canada, L8V 5C2
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
London, Ontario, Canada, N6A 4L6
Canada, Quebec
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Montreal, Quebec, Canada, H3A 1A1
France
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Paris, France, 75970
Mexico
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Durango, Mexico, 34000
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Guadalajara, Mexico, 44200
Netherlands
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Breda, Netherlands, 4818 CK
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Eindhoven, Netherlands, 5623 EJ
Spain
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Madrid, Spain, 28041
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Majadahonda, Spain, 28222
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Sevilla, Spain, 41014
Taiwan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Taichung, Taiwan, 404
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Taipei, Taiwan, 112 Less << |
|
NCT01788566
|
- |
|
Completed |
- |
- |
|
NCT00826891
|
Cervical Cancer |
Phase 2 |
Unknown |
January 2010 |
- |
|
NCT00527761
|
Metastatic Melanoma |
Phase 1 |
Completed |
- |
United States, Texas
... More >>
U.T.M.D. Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT03101566
|
Biliary Tract Neoplasms |
Phase 2 |
Recruiting |
November 1, 2020 |
United States, Georgia
... More >>
Emory University
Recruiting
Atlanta, Georgia, United States, 30322
Contact: Walid Shaib, MD 404-778-1900
Principal Investigator: Walid Shaib, MD
United States, Illinois
Northwestern University
Not yet recruiting
Chicago, Illinois, United States, 60611
Contact: Al B. Benson, III, MD 312-695-0990
Principal Investigator: Al B. Benson, III, MD
United States, Michigan
University of Michigan Comprehensive Cancer Center
Recruiting
Ann Arbor, Michigan, United States, 48109
Contact: Vaibhav Sahai, MBBS, MS 734-936-4991
Principal Investigator: Vaibhav Sahai, MBBS, MS
United States, Pennsylvania
University of Pennsylvania
Not yet recruiting
Philadelphia, Pennsylvania, United States, 19104
Contact: Peter O'Dwyer, MD 215-360-0716
Principal Investigator: Peter O'Dwyer, MD
United States, Texas
University of Texas Southwestern Medical Center
Recruiting
Dallas, Texas, United States, 75390
Contact: Muhammad Beg, MD 214-645-4673
Principal Investigator: Muhammad Beg, MD
United States, Washington
University of Washington
Recruiting
Seattle, Washington, United States, 98109
Contact: David Zhen, MD 206-606-1743
Principal Investigator: David Zehn, MD
United States, Wisconsin
University of Wisconsin
Recruiting
Madison, Wisconsin, United States, 53705
Contact: Dustin Deming, MD 608-265-1700
Principal Investigator: Dustin Deming, MD Less << |
|
NCT00281125
|
Non-Small Cell Lung Cancer and... More >> Pleural Mesothelioma Less << |
Phase 1
Phase 2 |
Terminated(Suspended due to da... More >>ta issues revealed at DSMB meeting. Planned amendment but was never submitted. Study was then closed.) Less << |
- |
United States, Nevada
... More >>
Nevada Cancer Institute
Las Vegas, Nevada, United States, 89135 Less << |
|
NCT00575952
|
Endometrial Adenosquamous Carc... More >>inoma
Endometrial Clear Cell Adenocarcinoma
Endometrial Mixed Adenocarcinoma
Endometrial Serous Adenocarcinoma
Endometrial Squamous Cell Carcinoma
Endometrial Undifferentiated Carcinoma
Recurrent Uterine Corpus Carcinoma
Stage IIIA Uterine Corpus Cancer
Stage IIIC Uterine Corpus Cancer
Stage IVA Uterine Corpus Cancer
Stage IVB Uterine Corpus Cancer Less << |
Phase 1 |
Completed |
- |
United States, Connecticut
... More >>
Hartford Hospital
Hartford, Connecticut, United States, 06102
The Hospital of Central Connecticut
New Britain, Connecticut, United States, 06050
United States, Illinois
University of Chicago Comprehensive Cancer Center
Chicago, Illinois, United States, 60637
United States, Iowa
University of Iowa/Holden Comprehensive Cancer Center
Iowa City, Iowa, United States, 52242
United States, Missouri
Washington University School of Medicine
Saint Louis, Missouri, United States, 63110
United States, New Jersey
Cooper Hospital University Medical Center
Camden, New Jersey, United States, 08103
United States, Ohio
Case Western Reserve University
Cleveland, Ohio, United States, 44106
United States, Oklahoma
University of Oklahoma Health Sciences Center
Oklahoma City, Oklahoma, United States, 73104
United States, Rhode Island
Women and Infants Hospital
Providence, Rhode Island, United States, 02905 Less << |
|
NCT00966914
|
Non-small Cell Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00608205
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
United States, Ohio
... More >>
Case Medical Center, University Hospitals Seidman Cancer Center, Case Comprehensive Cancer Center
Cleveland, Ohio, United States, 44106-5065
Cleveland Clinic Taussig Cancer Institute, Case Comprehensive Cancer Center
Cleveland, Ohio, United States, 44195 Less << |
|
NCT00707473
|
Locally Advanced Nasal Cavity ... More >>and Paranasal Sinus Squamous Cell Carcinoma
Nasal Cavity and Paranasal Sinus Poorly Differentiated Carcinoma
Sinonasal Undifferentiated Carcinoma
Stage II Nasal Cavity and Paranasal Sinus Cancer AJCC v8
Stage III Nasal Cavity and Paranasal Sinus Cancer AJCC v8
Stage IVA Nasal Cavity and Paranasal Sinus Cancer AJCC v8
Stage IVB Nasal Cavity and Paranasal Sinus Cancer AJCC v8 Less << |
Phase 2 |
Recruiting |
June 30, 2019 |
United States, Texas
... More >>
M D Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact: Ehab Y. Hanna 713-745-2672
Principal Investigator: Ehab Y. Hanna Less << |
|
NCT01888302
|
Hepatic Complication |
Phase 1 |
Completed |
- |
United States, Minnesota
... More >>
Mayo Clinic
Rochester, Minnesota, United States, 55905 Less << |
|
NCT00400205
|
Squamous Cell Carcinoma
... More >> Oral Cancer Less << |
Phase 2 |
Terminated(Safety reasons) |
- |
United States, Georgia
... More >>
Emory University Winship Cancer Institute
Atlanta, Georgia, United States, 30322 Less << |
|
NCT00400205
|
- |
|
Terminated(Safety reasons) |
- |
- |
|
NCT00195078
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Terminated |
- |
United States, New York
... More >>
Weill Medical College of Cornell University
New York, New York, United States, 10021 Less << |
|
NCT00002998
|
Breast Cancer |
Phase 2 |
Completed |
- |
United States, Arizona
... More >>
CCOP - Scottsdale Oncology Program
Scottsdale, Arizona, United States, 85259-5404
United States, Illinois
CCOP - Carle Cancer Center
Urbana, Illinois, United States, 61801
United States, Iowa
CCOP - Cedar Rapids Oncology Project
Cedar Rapids, Iowa, United States, 52403-1206
CCOP - Iowa Oncology Research Association
Des Moines, Iowa, United States, 10309-1016
Siouxland Hematology-Oncology
Sioux City, Iowa, United States, 51101-1733
United States, Kansas
CCOP - Wichita
Wichita, Kansas, United States, 67214-3882
United States, Louisiana
CCOP - Ochsner
New Orleans, Louisiana, United States, 70121
United States, Michigan
CCOP - Ann Arbor Regional
Ann Arbor, Michigan, United States, 48106
United States, Minnesota
CCOP - Duluth
Duluth, Minnesota, United States, 55805
Mayo Clinic Cancer Center
Rochester, Minnesota, United States, 55905
CentraCare Clinic
Saint Cloud, Minnesota, United States, 56303
United States, Nebraska
CCOP - Missouri Valley Cancer Consortium
Omaha, Nebraska, United States, 68131
United States, North Dakota
Quain & Ramstad Clinic, P.C.
Bismarck, North Dakota, United States, 58501
CCOP - Merit Care Hospital
Fargo, North Dakota, United States, 58122
Altru Health Systems
Grand Forks, North Dakota, United States, 58201
United States, Ohio
CCOP - Toledo Community Hospital Oncology Program
Toledo, Ohio, United States, 43623-3456
United States, Pennsylvania
CCOP - Geisinger Clinical and Medical Center
Danville, Pennsylvania, United States, 17822-2001
United States, South Dakota
Rapid City Regional Hospital
Rapid City, South Dakota, United States, 57709
CCOP - Sioux Community Cancer Consortium
Sioux Falls, South Dakota, United States, 57105-1080
Canada, Saskatchewan
Saskatchewan Cancer Agency
Regina, Saskatchewan, Canada, S4S 6X3 Less << |
|
NCT00608205
|
- |
|
Completed |
- |
- |
|
NCT01187212
|
Malignant Neoplasm of Stomach
... More >>
Effects of Chemotherapy Less << |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Asan Medical Center
Songpa-gu, Seoul, Korea, Republic of, 138-736 Less << |
|
NCT01595061
|
Stage IIIA Vulvar Cancer AJCC ... More >>v7
Stage IIIB Vulvar Cancer AJCC v7
Stage IIIC Vulvar Cancer AJCC v7
Stage IVA Vulvar Cancer AJCC v7
Vulvar Squamous Cell Carcinoma Less << |
Phase 2 |
Recruiting |
- |
- |
|
NCT02270814
|
Head and Neck Cancer
... More >> Head and Neck Squamous Cell Carcinoma
Cancer of the Head and Neck Less << |
Phase 2 |
Recruiting |
May 31, 2021 |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences
Recruiting
Little Rock, Arkansas, United States, 72205
Contact: Omar Atiq, M.D. 870-535-2800 otatiq@uams.edu
Principal Investigator: Omar Atiq, M.D.
Sub-Investigator: Laura Hutchins, M.D.
Sub-Investigator: Konstantinos Arnaoutakis, M.D.
Sub-Investigator: Issam Makhoul, M.D.
Sub-Investigator: Rangaswamy Govindarajan, M.D.
Sub-Investigator: Fade Mahmoud, M.D.
Sub-Investigator: Peter Emanuel, M.D.
Sub-Investigator: Natasa Milojkovic, M.D.
Sub-Investigator: Pooja Motwani, M.D.
Sub-Investigator: Liudmila Schafer, M.D.
United States, Mississippi
University of Mississippi Medical Center
Recruiting
Jackson, Mississippi, United States, 39216
Contact: Robert Hamilton, M.D. 601-984-5590
Principal Investigator: Robert Hamilton, M.D.
Sub-Investigator: James T Thigpen, M.D.
Sub-Investigator: Barbara Craft, M.D.
Sub-Investigator: Louis Puneky, M.D.
Sub-Investigator: Jennifer Eubanks, M.D.
Sub-Investigator: Natale Sheehan, M.D.
Sub-Investigator: John C Henegan, M.D.
United States, Missouri
Washington University School of Medicine
Recruiting
Saint Louis, Missouri, United States, 63110
Contact: Douglas R Adkins, M.D. 314-362-5654 dadkins@wustl.edu
Principal Investigator: Douglas Adkins, M.D.
Sub-Investigator: Tanya Wildes, M.D.
Sub-Investigator: Peter Oppelt, M.D.
United States, North Dakota
Sanford Roger Maris Cancer Center
Recruiting
Fargo, North Dakota, United States, 58122
Contact: Mark Gitau, M.D. 701-234-6161
Principal Investigator: Mark Gitau, M.D.
Sub-Investigator: Anu Gaba, M.D.
Sub-Investigator: Louis Geeraerts, M.D.
Sub-Investigator: Mahendra Gupta, M.D.
Sub-Investigator: Gerald Gross, M.D.
Sub-Investigator: Susan Hofland, NP
Sub-Investigator: Amit Panwalkar, M.D.
Sub-Investigator: Carrie Pfaff, NP
Sub-Investigator: John Roll, CNS
Sub-Investigator: Howard Russell, M.D.
Sub-Investigator: Denise Snow, M.D.
Sub-Investigator: Preston Steen, M.D.
Sub-Investigator: Shelby Terstriep, M.D.
Sub-Investigator: Matthew Tinguely, M.D.
Sub-Investigator: Radhakrishna Vegunta, M.D.
Sub-Investigator: Lorelei Wolf, NP
United States, South Dakota
Sanford Cancer Center Oncology Clinic and Pharmacy
Recruiting
Sioux Falls, South Dakota, United States, 57104
Contact: Steven Powell, M.D. 605-328-8000
Principal Investigator: Steven Powell, M.D.
Sub-Investigator: Jonathan Bleecker, M.D.
Sub-Investigator: Keely Hack, M.D.
Sub-Investigator: Michael Keppen, M.D.
Sub-Investigator: James Kuzman, M.D.
Sub-Investigator: Miroslaw Mazurczak, M.D.
Sub-Investigator: Amy Sanford, M.D.
Sub-Investigator: Christopher Sumey, M.D.
Sub-Investigator: Heather Casper-McLay, CNP
Sub-Investigator: Avigayil East, CNP
Sub-Investigator: Matthew Graham, CNP
Sub-Investigator: Susan Halbritter, CNP
Sub-Investigator: Kimberly Hamer, CNP
Sub-Investigator: Shauna Jacobs, CNP
Sub-Investigator: Jacqueline Kelley, CNP Less << |
|
NCT01645748
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Chonnam National University Hwasun Hospital
Gwangju, Jeollanamdo, Korea, Republic of, 519-809 Less << |
|
NCT02735928
|
Ovarian Epithelial Cancer Recu... More >>rrent
Platinum-resistant Less << |
Phase 1
Phase 2 |
Recruiting |
October 2018 |
Italy
... More >> Catholic University of Sacred Heart
Recruiting
Rome, Italy, 00168
Contact: Giovanni Scambia, Professor +39 0630156279 clinicaltrials@rm.unicatt.it
Contact: Federica Nardelli, MD +39-06-30156279 nardelli.fede@gmail.com Less << |
|
NCT00832637
|
Hepatocellular Carcinoma
... More >> Cholangiocellular Carcinoma
Cholangiocarcinoma of the Extrahepatic Bile Duct
Bile Duct Cancer
Periampullary Adenocarcinoma
Gallbladder Cancer
Extrahepatic Bile Duct Cancer Less << |
Phase 2 |
Terminated(Low accrual) |
- |
United States, California
... More >>
California Pacific Medical Center
San Francisco, California, United States, 94115
United States, New Mexico
University of New Mexico Cancer Center
Albuquerque, New Mexico, United States, 87131 Less << |
|
NCT00253370
|
Adenocarcinoma of the Gastroes... More >>ophageal Junction
Metastatic Gastric Cancer
Advanced Unresectable Gastric Cancer Less << |
Phase 2 |
Completed |
- |
United States, Massachusetts
... More >>
Eastern Cooperative Oncology Group
Boston, Massachusetts, United States, 02215 Less << |
|
NCT01034189
|
Esophageal Cancer |
Phase 2 |
Unknown |
June 2012 |
Taiwan
... More >> National Taiwan University Hospital
Recruiting
Taipei, Taiwan, 10002
Contact: Chih-Hung Hsu, M.D., Ph.D. 886-2-23123456 ext 67680 chihhunghsu@ntu.edu.tw
Principal Investigator: Chih-Hung Hsu, M.D., Ph.D. Less << |
|
NCT01282151
|
Carcinoma, Non Small Cell Lung |
Phase 3 |
Terminated(Difficulty in recru... More >>itment due to approval of maintenance pemetrexed treatment) Less << |
- |
Korea, Republic of
... More >>
Chonnam National University Hwasun Hospital
Hwasun, Jeonnam, Korea, Republic of, 519-709
Kyungpook National University Medical Center
Daegu, Kyungpook, Korea, Republic of
Hallym University Medical Center
Anyang, Korea, Republic of
Dankook University Hospital
Cheonan, Korea, Republic of, 330-715
Keimyung University Dongsan Center
Daegu, Korea, Republic of, 700-712
Yeungnam Univeristy Hospital
Daegu, Korea, Republic of
Chosun University Hospital
Gwangju, Korea, Republic of
Wonkwang University Hospital
Iksan, Korea, Republic of
Inha University Hospital
Incheon, Korea, Republic of
Pusan National University Hospital
Pusan, Korea, Republic of, 602-739
Kosin University Gospel Hospital
Pusan, Korea, Republic of
Konkuk university medical center
Seoul, Korea, Republic of, 143-729
Korea Cancer Center Hospital
Seoul, Korea, Republic of
Korea University Medical Center
Seoul, Korea, Republic of
Wonju Christian Hospital
Wonju, Korea, Republic of Less << |
|
NCT00253370
|
- |
|
Completed |
- |
- |
|
NCT02560038
|
- |
|
Terminated(slow enrollment; re... More >>source re-allocation) Less << |
- |
- |
|
NCT00265941
|
Head and Neck Cancer |
Phase 3 |
Active, not recruiting |
- |
- |
|
NCT00030576
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Completed |
- |
Canada, Ontario
... More >>
Margaret and Charles Juravinski Cancer Centre
Hamilton, Ontario, Canada, L8V 5C2
Queen's University
Kingston, Ontario, Canada, K7L 3N6
Cancer Care Ontario-London Regional Cancer Centre
London, Ontario, Canada, N6A 4L6
Ottawa Regional Cancer Centre
Ottawa, Ontario, Canada, K1H 1C4
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9
Canada, Quebec
Centre Hospitalier de l'Universite de Montreal
Montreal, Quebec, Canada, H2L-4M1 Less << |
|
NCT02560038
|
Urinary Bladder Neoplasms |
Phase 2 |
Terminated(slow enrollment; re... More >>source re-allocation) Less << |
- |
United States, Texas
... More >>
UTHealth Memorial Hermann Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00265941
|
- |
|
Active, not recruiting |
- |
- |
|
NCT01411098
|
Recurrent Non-small Cell Lung ... More >>Cancer
Stage IIB Non-small Cell Lung Cancer
Stage IIIA Non-small Cell Lung Cancer
Stage IIIB Non-small Cell Lung Cancer Less << |
Phase 1 |
Terminated(Low accrual) |
- |
United States, Washington
... More >>
Fred Hutchinson Cancer Research Center/University of Washington Cancer Consortium
Seattle, Washington, United States, 98109 Less << |
|
NCT01104259
|
Estrogen Receptor-negative Bre... More >>ast Cancer
HER2-negative Breast Cancer
Hereditary Breast/Ovarian Cancer - BRCA1
Hereditary Breast/Ovarian Cancer - BRCA2
Male Breast Cancer
Progesterone Receptor-negative Breast Cancer
Recurrent Breast Cancer
Stage IV Breast Cancer
Triple-negative Breast Cancer Less << |
Phase 1 |
Completed |
- |
United States, Washington
... More >>
Fred Hutchinson Cancer Research Center/University of Washington Cancer Consortium
Seattle, Washington, United States, 98109 Less << |
|
NCT03673072
|
Incidental Gallbladder Carcino... More >>ma
Biliary Tract Cancer Less << |
Phase 3 |
Not yet recruiting |
November 2024 |
- |
|
NCT01437449
|
Head and Neck Cancer |
Phase 2 |
Active, not recruiting |
January 2021 |
United States, California
... More >>
University of California Davis Medical Center
Davis, California, United States, 95616
Stanford University, School of Medicine
Stanford, California, United States, 95305 Less << |
|
NCT00320983
|
Cervical Cancer |
Phase 1 |
Completed |
- |
United States, New Mexico
... More >>
University of New Mexico
Albuquerque, New Mexico, United States, 87131 Less << |
|
NCT00832637
|
- |
|
Terminated(Low accrual) |
- |
- |
|
NCT03095079
|
Melanoma |
Phase 2 |
Recruiting |
October 2018 |
China, Beijing
... More >> Beijing Cancer Hospital
Recruiting
Beijing, Beijing, China, 100000
Contact: Chuanliang Cui, MD +86 13691489319 Less << |
|
NCT03649321
|
Cancer of Pancreas |
Phase 1
Phase 2 |
Not yet recruiting |
May 2022 |
- |
|
NCT00818090
|
Thymoma
Thymi... More >>c Carcinoma Less << |
Phase 2 |
Terminated(marginal statistica... More >>l significance) Less << |
- |
Korea, Republic of
... More >>
Seoul National University Hospital
Seoul, Korea, Republic of Less << |
|
NCT03717155
|
Squamous Non-Small Cell Lung C... More >>ancer Less << |
Phase 2 |
Recruiting |
January 25, 2021 |
Hungary
... More >> Tudogyogyintezet Torokbalint - Onkopulmonologiai es Jarobeteg centrum
Recruiting
Torokbalint, Hungary, 2045
Spain
Hospital Universitario Virgen Macarena - Servicio de Oncologia
Recruiting
Sevilla, Spain, 41009
Contact +49 6151 72 5200 service@merckgroup.com Less << |
|
NCT00234494
|
Bladder Cancer |
Phase 2 |
Completed |
- |
United States, Illinois
... More >>
University of Chicago
Chicago, Illinois, United States, 60637
Medical & Surgical Specialists, LLC
Galesburg, Illinois, United States, 61401
United States, Indiana
Oncology Hematology Associates of SW Indiana
Evansville, Indiana, United States, 47714
Fort Wayne Oncology & Hematology, Inc
Fort Wayne, Indiana, United States, 46815
Indiana University Cancer Center
Indianapolis, Indiana, United States, 46202
Quality Cancer Center (MCGOP)
Indianapolis, Indiana, United States, 46202
Arnett Cancer Care
Lafayette, Indiana, United States, 47904
Northern Indiana Cancer Research Consortium
South Bend, Indiana, United States, 46601
AP&S Clinic
Terre Haute, Indiana, United States, 47804
United States, Missouri
Siteman Cancer Center
St. Louis, Missouri, United States, 63110
United States, Ohio
Oncology Hematology Care, Inc.
Cincinnati, Ohio, United States, 45242 Less << |
|
NCT00234494
|
- |
|
Completed |
- |
- |
|
NCT01326871
|
Transitional Cell Carcinoma of... More >> Bladder
Urethra Cancer
Ureter Cancer
Malignant Tumor of Renal Pelvis Less << |
Phase 1
Phase 2 |
Unknown |
October 2017 |
United States, Arizona
... More >>
The University of Arizona Cancer Center
Tucson, Arizona, United States, 85724
United States, Florida
UF Health Center at Orlando Health
Orlando, Florida, United States, 32806
Martin Health System
Stuart, Florida, United States, 34994
H. Lee Moffitt Cancer Center and Research Institute
Tampa, Florida, United States, 33612
United States, Georgia
Emory University
Atlanta, Georgia, United States, 30322
United States, Illinois
Robert Lurie Comprehensive Cancer Center of Northwestern University
Chicago, Illinois, United States, 60611
United States, Iowa
University of Iowa Hospitals and Clinics
Iowa City, Iowa, United States, 52242
United States, Kansas
University of Kansas Cancer Center
Fairway, Kansas, United States, 66205
United States, Michigan
Karmanos Cancer Center
Detroit, Michigan, United States, 48201
United States, Minnesota
University of Minnesota
Minneapolis, Minnesota, United States, 55455
United States, Missouri
Washington University
St. Louis, Missouri, United States, 63110
United States, North Carolina
Levine Cancer Institute
Charlotte, North Carolina, United States, 28203
United States, Oklahoma
University of Oklahoma Health Science Center
Oklahoma City, Oklahoma, United States, 73104
United States, Pennsylvania
St. Luke's Hospital and Health Network
Easton, Pennsylvania, United States, 18045
Thomas Jefferson University Hospital
Philadelphia, Pennsylvania, United States, 19107
UPMC Cancer Center
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT01971489
|
Adult Solid Neoplasm
... More >> Recurrent Non-Small Cell Lung Carcinoma
Stage IIIA Non-Small Cell Lung Cancer
Stage IIIB Non-Small Cell Lung Cancer
Stage IV Non-Small Cell Lung Cancer Less << |
Phase 1 |
Withdrawn |
- |
United States, New York
... More >>
Roswell Park Cancer Institute
Buffalo, New York, United States, 14263 Less << |
|
NCT00874328
|
Advanced Non-Small Cell Lung C... More >>ancer Less << |
Phase 1
Phase 2 |
Unknown |
December 2012 |
Korea, Republic of
... More >>
Asan Medical Center
Recruiting
Seoul, Korea, Republic of, 138-736
Contact: Dae Ho Lee, M.D. +82-2-3010-3214 leedaeho@amc.seoul.kr Less << |
|
NCT00049166
|
Stage II Squamous Cell Carcino... More >>ma of the Lip and Oral Cavity
Stage II Squamous Cell Carcinoma of the Oropharynx
Stage III Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage III Squamous Cell Carcinoma of the Oropharynx
Stage IV Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage IV Squamous Cell Carcinoma of the Oropharynx Less << |
Phase 1 |
Completed |
- |
United States, Maryland
... More >>
Johns Hopkins University
Baltimore, Maryland, United States, 21287-8936 Less << |
|
NCT01459614
|
Pancreatic Cancer |
Phase 2 |
Active, not recruiting |
November 2019 |
United States, Maryland
... More >>
Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Baltimore, Maryland, United States, 21205 Less << |
|
NCT01095302
|
Neoplasms, Malignant |
Phase 1 |
Completed |
- |
Japan
... More >> Investigational Site Number 392003
Akashi-Shi, Japan
Investigational Site Number 392001
Koto-Ku, Japan
Investigational Site Number 392002
Nagoya-Shi, Japan Less << |
|
NCT00493025
|
- |
|
Terminated(Closed due to early... More >> stopping rule-Low accrual) Less << |
- |
- |
|
NCT01045538
|
Gastric Cancer
... More >> Histone Deacetylase Inhibitor Less << |
Phase 1
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT00794417
|
Advanced Carcinoma
... More >> Non-small Cell Lung Cancer Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, Arizona
... More >>
Arizona Cancer Institute, LLC
Tucson, Arizona, United States, 85715
United States, Arkansas
University of Arkansas for Medical Science
Little Rock, Arkansas, United States, 72205
United States, California
Stanford University Medical Center
Stanford, California, United States, 94305
United States, Florida
Palm Beach Institute of Hematology and Oncology
Boynton Beach, Florida, United States, 33435
United States, Illinois
Edward Hines Jr. VA Medical Center
Hines, Illinois, United States, 60141
United States, Kentucky
Kentucky Cancer Clinic
Hazard, Kentucky, United States, 41701
United States, Maryland
Sidney Kimmel Comprehensive Cancer Center
Baltimore, Maryland, United States, 21231-1000
United States, New Hampshire
Dartmouth-Hitchcock Medical Center
Lebanon, New Hampshire, United States, 03756
United States, New Mexico
UNM Cancer Clinic
Albuquerque, New Mexico, United States, 87131
United States, New York
Montefiore Medical Center
Bronx, New York, United States, 10467
Roswell Park Cancer Institute
Buffalo, New York, United States, 14263
United States, North Carolina
Presbyterian Hospital Center for Cancer Research
Charlotte, North Carolina, United States, 28204
United States, Pennsylvania
Erie Regional Cancer Center
Erie, Pennsylvania, United States, 16505
United States, West Virginia
Schiffler Cancer Center - Medical Oncology Division
Wheeling, West Virginia, United States, 26003
Canada, Ontario
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT00493025
|
Esophageal Cancer |
Phase 2 |
Terminated(Closed due to early... More >> stopping rule-Low accrual) Less << |
- |
United States, Maryland
... More >>
Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Baltimore, Maryland, United States, 21231-2410 Less << |
|
NCT00191763
|
Carcinoma, Non-Small-Cell-Lung... More >> Cancer Less << |
Phase 2 |
Completed |
- |
Turkey
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9AM to 5PM Eastern time (UTC/GMT - 5 hours, EST) or speak with your personal physician.
Istanbul, Turkey Less << |
|
NCT00144989
|
Small-Cell-Lung Cancer |
Phase 3 |
Completed |
- |
Japan
... More >> Aichi Cancer Center Hospital
Nagoya,Chikusa-ku,Kanokoden,1-1, Aichi, Japan, 464-8681
Aichi Cancer Center,Aichi Hospital
Okazaki,Kake-machi,Kuriyado,18, Aichi, Japan, 444-0011
National Cancer Center Hospital East
Kashiwa-shi,Kashiwanoha,6-5-1, Chiba, Japan, 277-8577
National Hospital Organization Shikoku Cancer Center
Matsuyama,Horinouchi,13, Ehime, Japan, 790-0007
Kyushu University Hospital
Higashi-ku,Maidashi,3-1-1, Fukuoka, Japan, 812-8582
Gifu Municipal Hospital
Gifu,Kashima-cho,7-1, Gifu, Japan, 500-8323
Gunma Prefectural Cancer Center
Ota,Takabayashi-nishi-cho,617-1, Gunma, Japan, 373-8550
National Nishigunma Hospital
Shibukawa,Kanai,2854, Gunma, Japan, 377-8511
National Hospital Organization, Dohoku National Hospital
Asahikawa,Hanasaki,7-4048, Hokkaido, Japan, 070-8644
National Hospital Organization Hokkaido Cancer Center
Sapporo,Shiroishi-ku,Kikusui,4-2-3-54, Hokkaido, Japan, 003-0804
Hyogo Medical Center for Adults
Akashi,Kitaouji-cho,13-70, Hyogo, Japan, 673-8558
Kobe City General Hospital
Kobe,Chuo-ku,Minatojimanakamachi,4-6, Hyogo, Japan, 650-0046
Hyogo College of Medicine
Nishinomiya,Mukogawa-cho,1-1, Hyogo, Japan, 663-8501
Ibaraki Kenritsu Chuo Hospital & Cancer Center
Nishi-ibarakigun,Tomobemachi,Koibuchi,6528, Ibaraki, Japan, 309-1793
Kanagawa Cancer Center
Yokohama,Asahi-ku,Nakao,1-1-2, Kanagawa, Japan, 241-0815
Yokohama Mucipical Citizen's Hospital
Yokohama,Hodogaya-ku,Okazawa-cho,56, Kanagawa, Japan, 240-8555
Kumamoto Regional Medical Center Hospital
Kumamoto,Honjo,5-16-10, Kumamoto, Japan, 860-0811
Tohoku University Hospital
Sendai,Aoba-ku,Seiryo-machi,1-1, Miyagi, Japan, 980-0874
Niigata Cancer Center Hospital
Niigata,Kawagishi-cho,2-15-3, Niigata, Japan, 951-8566
Okayama University Hospital
Okayama,Shikata-cho,2-5-1, Okayama, Japan, 700-8558
Osaka Prefectural Medical Center for Respiratory and Allergic Disease
Habikino,Habikino,3-7-1, Osaka, Japan, 583-8588
Rinku General Medical Center
Izumisano,rinku-ohrai-kita,2-23, Osaka, Japan, 598-0048
Graduate School of Medicine, Osaka City University
Osaka,Abeno-ku,Asahi-machi,1-5-7, Osaka, Japan, 545-0051
Osaka Medical Center for Cancer and Cardiovascular Diseases
Osaka,Higashinari-ku,Nakamichi,1-3-3, Osaka, Japan, 537-8511
Osaka City General Hospital
Osaka,Miyakojima-ku,Miyakojimahondori,2-13-22, Osaka, Japan, 534-0021
Osaka General Medical Center
Osaka,Sumiyoshi-ku,Bandai-higashi,3-1-56, Osaka, Japan, 558-8558
Kinki University School of Medicine
Osaka-Sayama,Ohno-higashi,377-2, Osaka, Japan, 589-8511
National Hospital Organization Kinki-Chuo Chest Medical Center
Sakai,Nagasone,1180, Osaka, Japan, 591-8555
National Hospital Organization Toneyama National Hospital
Toyonaka,Toneyama,5-1-1, Osaka, Japan, 560-8552
Saitama Cancer Center
Kita-adachi,Ina,Komuro,818, Saitama, Japan, 362-0806
Sizuoka Cancer Center
Sunto-gun,Nagaizumi-cho,Shimonagakubo,1007, Shizuoka, Japan, 411-8777
Tochigi Cancer Center
Utsunomiya,Yohnan,4-9-13, Tochigi, Japan, 320-0834
National Cancer Center Hospital
Chuo-ku,Tsukiji,5-1-1, Tokyo, Japan, 104-0045
Cancer Institute Hospital
Koto-ku,Ariake,3-10-6, Tokyo, Japan, 135-8550
Toranomon Hospital
Minato-ku,Toranomon,2-2-2, Tokyo, Japan, 105-8470
International Medical Center of Japan
Shinjuku-ku,Toyama,1-21-1, Tokyo, Japan, 162-8655
Yamagata Prefectural Central Hospital
Yamagata,Aoyagi,1800, Yamagata, Japan, 990-2292 Less << |
|
NCT02429466
|
Germ Cell Tumor
... More >> Testis Cancer
Testicular Cancer Less << |
Phase 1 |
Suspended |
December 31, 2019 |
United States, Indiana
... More >>
Indiana University Hospital
Indianapolis, Indiana, United States, 46202
Indiana University Melvin and Bren Simon Cancer Center
Indianapolis, Indiana, United States, 46202 Less << |
|
NCT00477607
|
Ototoxicity
U... More >>nspecified Adult Solid Tumor Less << |
Phase 2
Phase 3 |
Completed |
- |
United States, Oregon
... More >>
VA Medical Center, Portland
Portland, Oregon, United States, 97201
Oregon Health & Science University
Portland, Oregon, United States, 97239 Less << |
|
NCT00165516
|
Pleural Mesothelioma
... More >> Malignant Pleural Mesothelioma Less << |
Phase 2 |
Completed |
- |
United States, Massachusetts
... More >>
Brigham and Women's Hospital
Boston, Massachusetts, United States, 02115
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00477607
|
- |
|
Completed |
- |
- |
|
NCT03225157
|
- |
|
Recruiting |
March 1, 2022 |
United States, Maryland
... More >>
Suburban Hospital
Recruiting
Bethesda, Maryland, United States, 20814
National Institutes of Health Clinical Center
Recruiting
Bethesda, Maryland, United States, 20892
Contact: For more information at the NIH Clinical Center contact Office of Patient Recruitment (OPR) 800-411-1222 ext TTY8664111010 prpl@cc.nih.gov Less << |
|
NCT01460537
|
Neoplasms |
Phase 1 |
Completed |
- |
United States, Florida
... More >>
Tampa, Florida, United States, 33612
United States, Minnesota
Rochester, Minnesota, United States, 55905
United States, North Carolina
Chapel Hill, North Carolina, United States, 27599-7305 Less << |
|
NCT00621361
|
Lung Cancer |
Phase 1 |
Completed |
- |
United States, California
... More >>
Research Site
Sacramento, California, United States
United States, Colorado
Research Site
Denver, Colorado, United States
United States, Kansas
Research Site
Kansas City, Kansas, United States
United States, Texas
Research Site
Houston, Texas, United States Less << |
|
NCT00882583
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Terminated(Low accrual) |
- |
United States, Maryland
... More >>
Johns Hopkins Sidney Kimmel Comprehensive Cancer Center
Baltimore, Maryland, United States, 21287
United States, Ohio
Ohio State University Medical Center
Columbus, Ohio, United States, 43210 Less << |
|
NCT00882583
|
- |
|
Terminated(Low accrual) |
- |
- |
|
NCT03609216
|
Infiltrating Bladder Urothelia... More >>l Carcinoma
Stage II Bladder Urothelial Carcinoma
Stage III Bladder Urothelial Carcinoma Less << |
Phase 2 |
Recruiting |
February 2029 |
- |
|
NCT01326559
|
Nasopharyngeal Carcinoma |
Phase 2 |
Completed |
- |
China
... More >> Department of Clinical Oncology, Queen Mary Hospital(QMH), Hong Kong
Hong Kong, China
Department of Clinical Oncology, Queen Mary Hospital
Hong Kong, China
Department of Clinical Oncology, Tuen Mun Hospital (TMH), Hong Kong
Hong Kong, China
Department of Oncology, Princess Margaret Hospital
Hong Kong, China Less << |
|
NCT03092986
|
Carcinoma, Non-Small-Cell Lung |
Phase 3 |
Not yet recruiting |
April 2018 |
- |
|
NCT02466971
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Carcinoma
Cervical Squamous Cell Carcinoma, Not Otherwise Specified
Stage IB2 Cervical Cancer AJCC v6 and v7
Stage II Cervical Cancer AJCC v7
Stage II Vaginal Cancer AJCC v6 and v7
Stage IIA Cervical Cancer AJCC v7
Stage IIB Cervical Cancer AJCC v6 and v7
Stage III Vaginal Cancer AJCC v6 and v7
Stage IIIB Cervical Cancer AJCC v6 and v7
Stage IVA Cervical Cancer AJCC v6 and v7
Stage IVA Vaginal Cancer AJCC v6 and v7
Vaginal Adenocarcinoma
Vaginal Adenosquamous Carcinoma
Vaginal Squamous Cell Carcinoma, Not Otherwise Specified Less << |
Phase 2 |
Recruiting |
September 30, 2018 |
- |
|
NCT00077883
|
Non-small Cell Lung Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
Univ. of MD, Greenbaum Cancer Center
Baltimore, Maryland, United States, 21201
United States, Tennessee
The Sarah Cannon Cancer Center
Nashville, Tennessee, United States, 37203
United States, Texas
Univ. of TX, MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT01128387
|
- |
|
Terminated(slow accrual) |
- |
- |
|
NCT00084812
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT01128387
|
Cancer |
Phase 1
Phase 2 |
Terminated(slow accrual) |
- |
United States, Wisconsin
... More >>
University of Wisconsin Hospital and Clinics
Madison, Wisconsin, United States, 53792 Less << |
|
NCT00072033
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
Switzerland
... More >> Kantonspital Aarau
Aarau, Switzerland, CH-5001
Universitaetsspital-Basel
Basel, Switzerland, CH-4031
Inselspital Bern
Bern, Switzerland, CH-3010
Spitaeler Chur AG
Chur, Switzerland, CH-7000
Hopital Cantonal Universitaire de Geneve
Geneva, Switzerland, CH-1211
Centre Hospitalier Universitaire Vaudois
Lausanne, Switzerland, CH-1011
Ospedale Civico
Lugano, Switzerland, CH-6900
Kantonsspital - St. Gallen
St. Gallen, Switzerland, CH-9007
City Hospital Triemli
Zurich, Switzerland, 8063 Less << |
|
NCT00909402
|
Stomach Neoplasms
... More >> Esophageal Neoplasms Less << |
Phase 1 |
Completed |
- |
United States, California
... More >>
City Of Hope National Medical Center
Duarte, California, United States, 91010-3012
Usc/Norris Comprehensive Cancer Center
Los Angeles, California, United States, 90033
United States, Texas
The University Of Texas Md Anderson Cancer Center
Houston, Texas, United States, 77030-4009
Canada, Ontario
Local Institution
Toronto, Ontario, Canada, M5G 2M9
France
Local Institution
Villejuif, France, 94805
Netherlands
Local Institution
Amsterdam, Netherlands, 1105 AZ Less << |
|
NCT01480986
|
Progression Free Survival of t... More >>he Treatment Less << |
Phase 2 |
Completed |
- |
China, Beijing
... More >> Beijing Tumor Hospital
Beijing, Beijing, China Less << |
|
NCT02225405
|
Stage IB Non-Small Cell Lung C... More >>arcinoma AJCC v7
Stage II Non-Small Cell Lung Cancer AJCC v7
Stage IIA Non-Small Cell Lung Carcinoma AJCC v7
Stage IIB Non-Small Cell Lung Carcinoma AJCC v7
Stage IIIA Non-Small Cell Lung Cancer AJCC v7 Less << |
Phase 1 |
Active, not recruiting |
April 30, 2020 |
United States, Texas
... More >>
M D Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00216125
|
Non-Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
United States, Illinois
... More >>
Medical & Surgical Specialists, LLC
Galesburg, Illinois, United States, 61401
United States, Indiana
Elkhart Clinic
Elkhart, Indiana, United States, 46515
Oncology Hematology Associates of SW Indiana
Evansville, Indiana, United States, 47714
Fort Wayne Oncology & Hematology, Inc
Fort Wayne, Indiana, United States, 46815
Center for Cancer Care at Goshen Health System
Goshen, Indiana, United States, 46527
Indiana University Cancer Center
Indianapolis, Indiana, United States, 46202
Quality Cancer Center (MCGOP)
Indianapolis, Indiana, United States, 46202
Community Regional Cancer Center
Indianapolis, Indiana, United States, 46256
Medical Consultants, P.C.
Muncie, Indiana, United States, 47303
Center for Cancer Care, Inc., P.C.
New Albany, Indiana, United States, 47150
Northern Indiana Cancer Research Consortium
South Bend, Indiana, United States, 46601
AP&S Clinic
Terre Haute, Indiana, United States, 47804
United States, Missouri
Siteman Cancer Center
St. Louis, Missouri, United States, 63110
United States, Nebraska
Methodist Cancer Center
Omaha, Nebraska, United States, 68114
United States, Texas
US Oncology
Houston, Texas, United States, 77060 Less << |
|
NCT00006105
|
Bladder Cancer
... More >> Drug/Agent Toxicity by Tissue/Organ Less << |
Phase 2 |
Completed |
- |
United States, Illinois
... More >>
University of Chicago Cancer Research Center
Chicago, Illinois, United States, 60637-1470 Less << |
|
NCT00813696
|
Pancreatic Cancer |
Phase 3 |
Unknown |
September 2009 |
Italy
... More >> Ospedal Miulli
Acquaviva delle Fonti, Italy
Ospedale Regionale Torrette
Ancona, Italy
Presidio Osp. Alto Garda e Ledro
ARco, Italy
Centro Riferimento Oncologico
Aviano, Italy
Istituto Oncologico Giovanni Paolo II
Bari, Italy
Azienda Ospedaliera G. Rummo
Benevento, Italy
Ospedale Fatebenefratelli
Benevento, Italy
Ospedale Riuniti
Bergamo, Italy
Ospedale degli Infermi
Biella, Italy
Ospedale Regionale
Bolzano, Italy
Policlinico Universitario
Cagliari, Italy
Ospedale A. Cardarelli
Campobasso, Italy
Ospedale Ramazzini
Carpi, Italy
Centro Catanese di Oncologia
Catania, Italy
Università di Chieti "D'Annunzio"
Chieti, Italy
Ospedale Valduce
Como, Italy
Ospedale Santa Croce
Fano, Italy
Azienda Ospedaliera Universitaria
Ferrara, Italy
Azienda Ospedale Careggi
Firenze, Italy
Azienda Ospedaliera Vito Fazzi
Lecce, Italy
Ospedale Umberto I
Lugo, Italy
Pres. Osp. di Manerbio
Manerbio, Italy
Ospedale L. Sacco
Milano, Italy
Ospedale San Paolo
Milano, Italy
Ospedale G. Moscati
Monteforte Irpino, Italy
Ospedale San Gerardo
Monza, Italy
Istituto Nazionale Tumori
Napoli, Italy
Ospedale Cardarelli
Napoli, Italy
Ospedale Civile
Olbia, Italy
Casa di Cura La Maddalena
Palermo, Italy
Policlinico Giaccone
Palermo, Italy
Ospedale San Salvatore-Muraglia
Pesaro, Italy
Ospedale Guglielmo da Saliceto
Piacenza, Italy
Ospedale San Carlo
Potenza, Italy
Azienda Policlinico S. Andrea
Roma, Italy
Ospedale Fatebenefratelli
Roma, Italy
Ospedale San Raffaele
Roma, Italy
Policlinico Militare Celio
Roma, Italy
Polo Oncologico Istituto Regina Elena
Roma, Italy
Ospedale Civile
Rovereto, Italy
Ospedale Civile
Rovigo, Italy
Centro Oncologico
S. Vito al Tagliamento, Italy
Ospedale Santa Chiara
Trento, Italy
Azienda Ospedaliera Treviglio-Caravaggio
Treviglio, Italy
Ospedale San Bortolo
Vicenza, Italy Less << |
|
NCT00084435
|
Head and Neck Cancer |
Phase 2 |
Withdrawn(Poor accrual and sus... More >>pension of head and neck committee) Less << |
- |
United States, California
... More >>
USC/Norris Comprehensive Cancer Center and Hospital
Los Angeles, California, United States, 90089-9181
United States, Illinois
Decatur Memorial Hospital Cancer Care Institute
Decatur, Illinois, United States, 62526
Regional Cancer Center at Memorial Medical Center
Springfield, Illinois, United States, 62781-0001
United States, Kansas
Kansas Masonic Cancer Research Institute at the University of Kansas Medical Center
Kansas City, Kansas, United States, 66160-7357
United States, Louisiana
Feist-Weiller Cancer Center at Louisiana State University Health Sciences
Shreveport, Louisiana, United States, 71130-3932
United States, Michigan
University of Michigan Comprehensive Cancer Center
Ann Arbor, Michigan, United States, 48109-0942
United States, Mississippi
University of Mississippi Medical Center
Jackson, Mississippi, United States, 39216
United States, Missouri
Saint Louis University Cancer Center
Saint Louis, Missouri, United States, 63110
United States, North Carolina
Rutherford Hospital
Rutherfordton, North Carolina, United States, 28139
United States, Ohio
Adena Regional Medical Center
Chillicothe, Ohio, United States, 45601
Cleveland Clinic Taussig Cancer Center
Cleveland, Ohio, United States, 44195
Riverside Methodist Hospital Cancer Care
Columbus, Ohio, United States, 43214-3998
CCOP - Columbus
Columbus, Ohio, United States, 43215
Grant Riverside Cancer Services
Columbus, Ohio, United States, 43215
Mount Carmel Health - West Hospital
Columbus, Ohio, United States, 43222
Doctors Hospital at Ohio Health
Columbus, Ohio, United States, 43228
Grady Memorial Hospital
Delaware, Ohio, United States, 43015
Community Oncology Group at Cleveland Clinic Cancer Center
Independence, Ohio, United States, 44131
Fairfield Medical Center
Lancaster, Ohio, United States, 43130
Strecker Cancer Center at Marietta Memorial Hospital
Marietta, Ohio, United States, 45750
Licking Memorial Cancer Care Program at Licking Memorial Hospital
Newark, Ohio, United States, 43055
Mercy Medical Center
Springfield, Ohio, United States, 45504
Community Hospital of Springfield and Clark County
Springfield, Ohio, United States, 45505
Mount Carmel Cancer Services at Mount Carmel St. Ann's Hospital
Westerville, Ohio, United States, 43081
Cleveland Clinic - Wooster
Wooster, Ohio, United States, 44691
Genesis - Good Samaritan Hospital
Zanesville, Ohio, United States, 43701
United States, South Carolina
AnMed Health Cancer Center
Anderson, South Carolina, United States, 29621
CCOP - Upstate Carolina
Spartanburg, South Carolina, United States, 29303
Gibbs Regional Cancer Center at Spartanburg Regional Medical Center
Spartanburg, South Carolina, United States, 29303 Less << |
|
NCT00658580
|
Small Cell Lung Carcinoma, Ext... More >>ensive Disease Less << |
Phase 3 |
Completed |
- |
Belgium
... More >> Department of Intensive Care Unit and Thoracic Oncology Institut Jules Bordet
Brussels, Belgium, 1000
France
Department of Pneumology CHRU Lille
Lille, France
Greece
Hellenic Cancer Institute - St Savas Oncology Hospital
Athens, Greece
Spain
Medical Oncology Hospital de Sagunto
Valencia, Spain Less << |
|
NCT00490399
|
Biliary Tract Carcinoma |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Seoul, Korea, Republic of Less << |
|
NCT01998347
|
Esophageal Cancer |
Phase 3 |
Withdrawn(slow enrollment) |
- |
China, Guangdong
... More >>
Department of medical oncology,Cancer hospital of Shantou University Medical colledge
Shantou, Guangdong, China, 515031 Less << |
|
NCT01064648
|
Epithelioid Mesothelioma
... More >> Pleural Malignant Mesothelioma
Recurrent Malignant Mesothelioma
Sarcomatoid Mesothelioma Less << |
Phase 1
Phase 2 |
Active, not recruiting |
- |
- |
|
NCT00678132
|
Neoplasms |
Phase 1 |
Completed |
- |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892 Less << |
|
NCT00930930
|
- |
|
Completed |
- |
- |
|
NCT00930930
|
Breast Cancer |
Phase 2 |
Completed |
- |
United States, Alabama
... More >>
University of Alabama
Birmingham, Alabama, United States, 35249
United States, Mississippi
University of Mississippi Medical Center Research Institute
Jackson, Mississippi, United States, 39213
United States, Pennsylvania
Hershey Medical Center
Hershey, Pennsylvania, United States, 17033
United States, Tennessee
Vanderbilt-Ingram Cancer Center - Cool Springs
Nashville, Tennessee, United States, 37064
MBCCOP - Meharry Medical College - Nashville
Nashville, Tennessee, United States, 37208
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States, 37232-6838
United States, Texas
The Methodist Hospital Research Institute
Houston, Texas, United States, 77030
United States, Virginia
University of Virginia Health Sciences Center
Charlottesville, Virginia, United States, 22098 Less << |
|
NCT00410826
|
Stage III Squamous Cell Carcin... More >>oma of the Hypopharynx
Stage III Squamous Cell Carcinoma of the Larynx
Stage III Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage III Squamous Cell Carcinoma of the Nasopharynx
Stage III Squamous Cell Carcinoma of the Oropharynx
Stage IV Squamous Cell Carcinoma of the Hypopharynx
Stage IV Squamous Cell Carcinoma of the Larynx
Stage IV Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage IV Squamous Cell Carcinoma of the Nasopharynx
Stage IV Squamous Cell Carcinoma of the Oropharynx Less << |
Phase 2 |
Completed |
- |
United States, Alaska
... More >>
Alaska Oncology and Hematology LLC
Anchorage, Alaska, United States, 99508
United States, Florida
University of Miami Miller School of Medicine-Sylvester Cancer Center
Miami, Florida, United States, 33136
United States, New Mexico
University of New Mexico Health Science CCOP
Albuquerque, New Mexico, United States, 87131
United States, North Carolina
University of North Carolina
Chapel Hill, North Carolina, United States, 27599
New Hanover Radiation Oncology Center
Wilmington, North Carolina, United States, 28401
United States, South Carolina
Medical University of South Carolina
Charleston, South Carolina, United States, 29425
United States, Tennessee
University of Tennessee Cancer Institute-Boston Cancer Group PLC
Memphis, Tennessee, United States, 38104
United States, Washington
Fred Hutchinson Cancer Research Center/University of Washington Cancer Consortium
Seattle, Washington, United States, 98109
Multicare Health System
Tacoma, Washington, United States, 98415 Less << |
|
NCT02357147
|
Mesothelioma, Malignant |
Phase 2 |
Active, not recruiting |
November 30, 2018 |
- |
|
NCT00270569
|
Breast Neoplasms |
Phase 2 |
Unknown |
- |
Taiwan
... More >> Far Eastern Memorial Hospital
Recruiting
Taipei, Ban-Ciao, Taiwan, 220
Contact: Kun Huei Yeh, Ph.D. 886-2-89667000 ext 1611 khyeh@mail.femh.org.tw Less << |
|
NCT00270582
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Unknown |
- |
Taiwan
... More >> Chi-Huang Hsiao
Recruiting
Taipei, Ban-Ciao, Taiwan, 220
Contact: CHI HUANG HSIAO, M.D. 886-2-89667000 ext 2102 onchsiao@ms28.hinet.net Less << |
|
NCT00410826
|
- |
|
Completed |
- |
- |
|
NCT01656551
|
Non-small Cell Lung Cancer Met... More >>astatic
Non-small Cell Lung Cancer Stage IIIB Less << |
Phase 3 |
Active, not recruiting |
September 2018 |
- |
|
NCT00680758
|
Breast Cancer |
Phase 1 |
Completed |
- |
United States, Tennessee
... More >>
Vanderbilt-Ingram Cancer Center - Cool Springs
Nashville, Tennessee, United States, 37064
Vanderbilt-Ingram Cancer Center at Franklin
Nashville, Tennessee, United States, 37064
Sarah Cannon Cancer Center at Centennial Medical Center
Nashville, Tennessee, United States, 37203
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States, 37232-6838 Less << |
|
NCT02281006
|
DDP
Head and ... More >>Neck Cancer
Adverse Effect Less << |
Phase 2 |
Terminated(poor accrual) |
- |
Canada
... More >> CHU de Quebec
Quebec, Canada, G1R 2J6 Less << |
|
NCT01386632
|
- |
|
Active, not recruiting |
- |
- |
|
NCT01386632
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 2 |
Active, not recruiting |
January 2019 |
United States, North Dakota
... More >>
Sanford Roger Maris Cancer Center
Fargo, North Dakota, United States, 58122
United States, South Dakota
Sanford Hematology and Oncology
Sioux Falls, South Dakota, United States, 57104 Less << |
|
NCT01771289
|
Non-small Cell Lung Cancer |
Phase 2 |
Unknown |
- |
China, Beijing
... More >> Peking university people's hospital
Recruiting
Beijing, Beijing, China, 100000
Contact: Fan Yang +8688326657 dr.yangf@gmail.com Less << |
|
NCT01083537
|
Ovarian Cancer
... More >> Peritoneal Cancer
Fallopian Tube Cancer
Bowel Obstruction Less << |
Phase 1
Phase 2 |
Terminated(Slow accrual) |
- |
Canada, Ontario
... More >>
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT02341989
|
Testicular Neoplasms
... More >> Seminoma Less << |
Phase 3 |
Recruiting |
December 2020 |
Norway
... More >> Institutt for kreftforskning og molekylær medisin, St Olavs Hospital
Recruiting
Trondheim, Norway
Contact: Torgrim Tandstad, md phd torgrim.tandstad@ntnu.no Less << |
|
NCT01375972
|
Metastatic Biliary Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Asan Medical Center
Seoul, Korea, Republic of, 138736 Less << |
|
NCT00083161
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, Wisconsin
... More >>
Gundersen Lutheran Center for Cancer and Blood
La Crosse, Wisconsin, United States, 54601 Less << |
|
NCT00215995
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 2 |
Completed |
- |
United States, Florida
... More >>
H. Lee Moffitt Cancer Center & Research Institute
Tampa, Florida, United States, 33612
Puerto Rico
Ponce School of Medicine
Ponce, Puerto Rico, 00732
University of Puetro Rico Cancer Center
San Juan, Puerto Rico, 00936-5067 Less << |
|
NCT01194869
|
Breast Cancer |
Phase 2 |
Terminated(Slow accrual, avail... More >>ability of other clinical options) Less << |
- |
United States, Georgia
... More >>
Grady Memorial Hospital
Atlanta, Georgia, United States, 30303
Emory University Hospital Midtown
Atlanta, Georgia, United States, 30308
Emory University Winship Cancer Institute
Atlanta, Georgia, United States, 30322 Less << |
|
NCT00176813
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
United States, Michigan
... More >>
University of Michigan
Ann Arbor, Michigan, United States, 48109 Less << |
|
NCT01194869
|
- |
|
Terminated(Slow accrual, avail... More >>ability of other clinical options) Less << |
- |
- |
|
NCT01088620
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Suspended(the DSMB stopped the... More >> trial due to unacceptable side effects in the experimental arm which has not yet been verified) Less << |
January 2014 |
Germany
... More >> Schwerpunktpraxis für Hämatologie und Internistische Onkologie, Gesundheitszentrum St. Marien GmbH
Amberg, Germany, 92224
Universitätsklinikum Charité - Campus Mitte
Berlin, Germany, 10117
Charité Campus Benjamin Franklin Medizinische Klinik m. S. Hämatologie und Onkologie
Berlin, Germany, 12200
HELIOS Klinikum Emil von Behring - Lungenklinik Heckeshorn
Berlin, Germany, 14165
Augusta-Kranken-Anstalt gGmbH
Bochum, Germany, 44791
Johanniter-Krankenhaus Bonn
Bonn, Germany, 53113
Carl-Thiem-Klinikum Cottbus gGmbH
Cottbus, Germany, 03048
Medizinische Fakultät Carl Gustav Carus der Technischen Universität Dresden Medizinische Klinik 1
Dresden, Germany, 01307
Katholisches Klinikum Duisburg/St. Johannes-Hospital
Duisburg, Germany, 47166
Klinikum Frankfurt (Oder) GmbH
Frankfurt (Oder), Germany, 15236
Krankenhaus Großhansdorf GmbH Onkologischer Schwerpunkt
Großhansdorf, Germany, 22927
Krankenhaus - Martha-Maria Halle-Dölau GmbH
Halle/Saale, Germany, 06120
Universitätsklinikum Halle (Saale), Klinik und Poliklinik für Innere Medizin I
Halle/Saale, Germany, 06120
Universitätsklinikum Jena, Klinik für Innere Medizin I
Jena, Germany, 07740
Onkologische Schwerpunktpraxis Dr. Stauch
Kronach, Germany, 96317
Kliniken der Stadt Köln, Krankenhaus Merheim
Köln, Germany, 51109
UK-SH, Campus Lübeck, Med. Klinik III
Lübeck, Germany, 23538
LMU-Klinikum der Universität München, Medizinische Klinik München-Innenstadt
München, Germany, 80336
Oncologianova GmbH
Recklinghausen, Germany, 45657
Uniklinikum Ulm, Klinik für Innere Medizin II, Pneumologie
Ulm, Germany, 89081 Less << |
|
NCT02964689
|
Advanced Non-small Cell Lung C... More >>ancer
KRAS Gene Mutation
Lung Cancer Less << |
Phase 1 |
Recruiting |
September 2020 |
Switzerland
... More >> Universitaetsspital Basel
Recruiting
Basel, Switzerland, 4031
Contact: Sacha Rothschild, MD +41 (0)61 265 25 25 sacha.rothschild@usb.ch
Principal Investigator: Sacha Rothschild, MD
IOSI Ospedale Regionale di Bellinzona e Valli
Recruiting
Bellinzona, Switzerland, 6500
Contact: Patrizia Froesch, MD +41 (0)91 811 48 40 patrizia.froesch@eoc.ch
Principal Investigator: Patrizia Froesch, MD
Kantonsspital Graubuenden
Recruiting
Chur, Switzerland, CH-7000
Contact: Michael Mark, MD +41 (0)81 256 61 11 michael.mark@ksgr.ch
Principal Investigator: Michael Mark, MD
Kantonsspital St. Gallen
Recruiting
St. Gallen, Switzerland, 9007
Contact: Martin Früh, MD +41 (0)71 494 11 11 martin.frueh@kssg.ch
Principal Investigator: Martin Früh, MD Less << |
|
NCT02624128
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 2 |
Recruiting |
October 2019 |
Italy
... More >> Istituto Nazionale Tumori Fondazione G. Pascale
Recruiting
Napoli, Italy Less << |
|
NCT02325401
|
Head and Neck Cancer |
Phase 1 |
Active, not recruiting |
March 2021 |
United States, Ohio
... More >>
University of Cincinnati Cancer Institute
Cincinnati, Ohio, United States, 45267 Less << |
|
NCT00889733
|
Epithelial Ovarian Cancer |
Phase 1
Phase 2 |
Terminated(Poor recruitment) |
- |
Canada, Ontario
... More >>
University Health Network - Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT00678977
|
Lung Cancer, Non-Small Cell |
Phase 1 |
Completed |
- |
Germany
... More >> GSK Investigational Site
Essen, Nordrhein-Westfalen, Germany, 45122
United Kingdom
GSK Investigational Site
Newcastle Upon Tyne, Northumberland, United Kingdom, NE4 6BE Less << |
|
NCT00492778
|
Endometrial Adenocarcinoma
... More >> Endometrial Adenosquamous Carcinoma
Endometrial Clear Cell Adenocarcinoma
Endometrial Endometrioid Adenocarcinoma, Variant With Squamous Differentiation
Endometrial Serous Adenocarcinoma
Recurrent Uterine Corpus Carcinoma Less << |
Phase 2 |
Recruiting |
- |
- |
|
NCT03382600
|
Gastric Cancer |
Phase 2 |
Recruiting |
August 17, 2020 |
Japan
... More >> National Cancer Center Hospital East ( Site 0001)
Recruiting
Kashiwa, Chiba, Japan, 277-8577
Contact: Study Coordinator +81471331111
National Hospital Organization Shikoku Cancer Center ( Site 0024)
Recruiting
Matsuyama, Ehime, Japan, 791-0280
Contact: Study Coordinator +81899991111
Gunma Prefectural Cancer Center ( Site 0005)
Recruiting
Ohta, Gunma, Japan, 373-8550
Contact: Study Coordinator +81276380771
Hokkaido University Hospital ( Site 0006)
Recruiting
Sapporo, Hokkaido, Japan, 060-8648
Contact: Study Coordinator +81117161161
Hyogo Cancer Center ( Site 0016)
Recruiting
Akashi, Hyogo, Japan, 673-8558
Contact: Study Coordinator +81789291151
Kobe City Medical Center General Hospital ( Site 0015)
Recruiting
Kobe, Hyogo, Japan, 650-0047
Contact: Study Coordinator +81783024321
Ibaraki Prefectural Central Hospital ( Site 0018)
Recruiting
Kasama, Ibaraki, Japan, 309-1793
Contact: Study Coordinator +81296771121
Kitasato University Hospital ( Site 0019)
Recruiting
Sagamihara, Kanagawa, Japan, 252-0375
Contact: Study Coordinator +81427788111
Kanagawa Cancer Center ( Site 0003)
Recruiting
Yokohama, Kanagawa, Japan, 241-8515
Contact: Study Coordinator +81455202222
Tohoku University Hospital ( Site 0023)
Recruiting
Sendai, Miyagi, Japan, 980-8574
Contact: Study Coordinator +81227177000
Kindai University Hospital ( Site 0013)
Recruiting
Osakasayama, Osaka, Japan, 589-8511
Contact: Study Coordinator +81723660221
Osaka University Hospital ( Site 0010)
Recruiting
Suita, Osaka, Japan, 565-0871
Contact: Study Coordinator +81668795111
Saitama Cancer Center ( Site 0004)
Recruiting
Kitaadachi-gun, Saitama, Japan, 362-0806
Contact: Study Coordinator +81487221111
Shizuoka Cancer Center Hospital and Research Institute ( Site 0020)
Recruiting
Sunto-gun, Shizuoka, Japan, 411-8777
Contact: Study Coordinator +81559895222
Jichi Medical University Hospital ( Site 0009)
Recruiting
Shimotsuke, Tochigi, Japan, 329-0498
Contact: Study Coordinator +81285442111
Chiba Cancer Center ( Site 0012)
Recruiting
Chiba, Japan, 260-8717
Contact: Study Coordinator +81432645431
National Hospital Organization Kyushu Cancer Center ( Site 0017)
Recruiting
Fukuoka, Japan, 811-1395
Contact: Study Coordinator +81925413231
Kyushu University Hospital ( Site 0014)
Recruiting
Fukuoka, Japan, 812-8582
Contact: Study Coordinator +81926411151
Gifu University Hospital ( Site 0021)
Recruiting
Gifu, Japan, 501-1194
Contact: Study Coordinator +81582306000
Kochi Health Sciences Center ( Site 0022)
Recruiting
Kochi, Japan, 781-8555
Contact: Study Coordinator +81888373000
Kumamoto University Hospital ( Site 0002)
Recruiting
Kumamoto, Japan, 860-8556
Contact: Study Coordinator +81963442111
Osaka International Cancer Institute ( Site 0011)
Recruiting
Osaka, Japan, 541-8567
Contact: Study Coordinator +81669451181
National Cancer Center Hospital ( Site 0025)
Recruiting
Tokyo, Japan, 104-0045
Contact: Study Coordinator +81335422511
Tokyo Metropolitan Komagome Hospital ( Site 0008)
Recruiting
Tokyo, Japan, 113-8677
Contact: Study Coordinator +81338232101
The Cancer Institute Hospital of JFCR ( Site 0007)
Recruiting
Tokyo, Japan, 135-8550
Contact: Study Coordinator +81335200111 Less << |
|
NCT02439489
|
Solid Tumors |
Phase 1 |
Completed |
- |
Italy
... More >> Ospedale San Raffaele
Milano, MI, Italy, 20100 Less << |
|
NCT02314117
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00082706
|
Bladder Cancer
... More >> Urethral Cancer
Urachal Cancer Less << |
Phase 2 |
Active, not recruiting |
April 2019 |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030-4009 Less << |
|
NCT01295502
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Carcinoma
Cervical Squamous Cell Carcinoma, Not Otherwise Specified
Stage IB Cervical Cancer
Stage IIA Cervical Cancer
Stage IIB Cervical Cancer
Stage IIIA Cervical Cancer
Stage IIIB Cervical Cancer
Stage IVA Cervical Cancer Less << |
Phase 1 |
Active, not recruiting |
- |
United States, California
... More >>
UC Irvine Health/Chao Family Comprehensive Cancer Center
Orange, California, United States, 92868
United States, Connecticut
Hartford Hospital
Hartford, Connecticut, United States, 06102
The Hospital of Central Connecticut
New Britain, Connecticut, United States, 06050
United States, Georgia
Augusta University Medical Center
Augusta, Georgia, United States, 30912
United States, Iowa
University of Iowa/Holden Comprehensive Cancer Center
Iowa City, Iowa, United States, 52242
United States, Ohio
Summa Akron City Hospital/Cooper Cancer Center
Akron, Ohio, United States, 44304
Case Western Reserve University
Cleveland, Ohio, United States, 44106
MetroHealth Medical Center
Cleveland, Ohio, United States, 44109
Cleveland Clinic Foundation
Cleveland, Ohio, United States, 44195
Riverside Methodist Hospital
Columbus, Ohio, United States, 43214
Hillcrest Hospital Cancer Center
Mayfield Heights, Ohio, United States, 44124
United States, Oklahoma
University of Oklahoma Health Sciences Center
Oklahoma City, Oklahoma, United States, 73104
United States, Rhode Island
Women and Infants Hospital
Providence, Rhode Island, United States, 02905
United States, Virginia
Virginia Commonwealth University/Massey Cancer Center
Richmond, Virginia, United States, 23298 Less << |
|
NCT02314117
|
Metastatic Gastric Adenocarcin... More >>oma
Gastroesophageal Junction Adenocarcinoma Less << |
Phase 3 |
Active, not recruiting |
January 31, 2019 |
- |
|
NCT00137852
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT01471470
|
Advanced Gastric Cancer |
Phase 2 |
Active, not recruiting |
December 2019 |
Korea, Republic of
... More >>
Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT03126708
|
Esophageal Cancer, Squamous Ce... More >>ll
Cetuximab Effect
Chemotherapy Effect Less << |
Phase 2 |
Recruiting |
April 2020 |
China, Beijing
... More >> Peking University Cancer Hospital
Recruiting
Beijing, Beijing, China, 100142
Contact: Lin Shen, MD +86-10-88196561 lin100@medmail.com.cn Less << |
|
NCT00957411
|
Cervical Cancer |
Phase 2 |
Completed |
- |
France
... More >> Institut Curie Hopital
Paris, France, 75248 Less << |
|
NCT01873326
|
Germ Cell Tumors |
Phase 2 |
Active, not recruiting |
June 2019 |
United States, California
... More >>
University of Southern California
Los Angeles, California, United States, 90033
Stanford University Medical Center
Stanford, California, United States, 94305-5408
United States, Illinois
University of Chicago
Chicago, Illinois, United States
United States, Minnesota
Mayo Clinic
Rochester, Minnesota, United States, 55905
United States, New Jersey
Memoral Sloan Kettering Cancer Center
Basking Ridge, New Jersey, United States
Memorial Sloan Kettering Monmouth
Middletown, New Jersey, United States, 07748
United States, New York
Memorial Sloan Kettering Cancer Center @ Suffolk
Commack, New York, United States, 11725
Memorial Sloan Kettering Westchester
Harrison, New York, United States, 10604
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan Kettering Cancer Center at Mercy Medical Center
Rockville Centre, New York, United States, 11570
United States, North Carolina
University of North Carolina
Chapel Hill, North Carolina, United States, 27514
United States, Pennsylvania
University of Pittsburgh Medical Center
Pittsburgh, Pennsylvania, United States, 15213 Less << |
|
NCT00660504
|
- |
|
Completed |
- |
- |
|
NCT00497315
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Belgium
... More >> ZNA Middelheim
Antwerpen, Antwerp, Belgium, 2020
University Hospital Antwerp
Edegem, Antwerp, Belgium, 2650
St Augustinus Ziekenhuis
Wilrijk, Antwerp, Belgium, 2610 Less << |
|
NCT00887822
|
Gastric Cancer |
Phase 3 |
Completed |
- |
China
... More >>
Beijing, China, 100021
Beijing, China, 100071
Beijing, China, 100142
Beijing, China, 100853
ChongQing, China, 400042
Guangzhou, China, 510060
Hangzhou, China, 310016
Nanjing, China, 210036
Nanjing, China
Shanghai, China, 200032
Shanghai, China, 200080
Shantou, China, 515041
Shenyang, China, 110001
Wuhan, China, 430030 Less << |
|
NCT01633645
|
Non Small Cell Lung Cancer
... More >> Metastatic Less << |
Phase 2 |
Completed |
- |
Greece
... More >> Air Forces Military Hospital of Athens Athens, Greece
Athens, Greece
METAXA Hospital, B' Pathology Department
Athens, Greece
SOTIRIA Hospital, Medical Oncology Department
Athens, Greece
University Hospital of Crete, Dep of Medical Oncology Heraklion, Greece
Heraklion, Greece
"Theagenion" Anticancer Hospital of Thessaloniki, 2nd Dep of Medical Oncology
Thessaloniki, Greece Less << |
|
NCT00660504
|
Lung Cancer |
Phase 3 |
Completed |
- |
China, Fujian
... More >>
Fuzhou, Fujian, China
China, Gansu
Lanzhou, Gansu, China
China, Guangdong
Guangzhou, Guangdong, China
China, Hunan
Changsha, Hunan, China
China, Jiangsu
Nanjing, Jiangsu, China
China, Jiangxi
Nanchang, Jiangxi, China
China, Jilin
Changchun, Jilin, China
China, Liaoning
Dalian/Shenyang, Liaoning, China
Shenyang, Liaoning, China
China, Shanxi
Xian, Shanxi, China
China, Sichuan
Chengdu, Sichuan, China
China, Zhejiang
Hangzhou, Zhejiang, China
China
Beijing, China
Shanghai, China
Tianjin, China Less << |
|
NCT01105390
|
Advanced Malignant Mesotheliom... More >>a
Epithelial Mesothelioma
Recurrent Malignant Mesothelioma
Sarcomatous Mesothelioma Less << |
Phase 2 |
Withdrawn(withdrawn) |
- |
- |
|
NCT00812266
|
Extensive Disease
... More >> First-Line
Small Cell Lung Cancer Less << |
Phase 3 |
Terminated(Slow accrual) |
- |
Denmark
... More >> Dept. Oncology, Rigshospitalet
Copenhagen, Denmark Less << |
|
NCT00281970
|
Lung Cancer |
Phase 2 |
Unknown |
- |
United States, Maryland
... More >>
Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Baltimore, Maryland, United States, 21231-2410
United States, New York
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021
United States, Tennessee
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States, 37232-6307 Less << |
|
NCT01969877
|
Locally Advanced Head and Neck... More >> Cancer Less << |
Phase 3 |
Active, not recruiting |
November 2024 |
Sweden
... More >> Gävle Hospital
Gävle, Sweden
Sahlgrenska University Hospital
Göteborg, Sweden
County Hospital Ryhov
Jönköping, Sweden
Central Hospital
Karlstad, Sweden
University Hospital Linköping
Linköping, Sweden
Karolinska Universityhospital
Stockholm, Sweden
Norrland University Hospital
Umeå, Sweden
Västmanlands Hospital Västerås
Västerås, Sweden
University Hospital Örebro
Örebro, Sweden Less << |
|
NCT00674167
|
Gastric Cancer |
Phase 2 |
Unknown |
December 2017 |
Singapore
... More >> National University Hospital
Singapore, Singapore Less << |
|
NCT01591135
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Phase 3 |
Completed |
- |
China, Shanghai
... More >>
Fudan University Cancer Center
Shanghai, Shanghai, China Less << |
|
NCT03697239
|
Metastatic Pancreatic Cancer |
Phase 1
Phase 2 |
Not yet recruiting |
October 2022 |
- |
|
NCT00003928
|
Kidney Cancer |
Phase 2 |
Completed |
- |
United States, Ohio
... More >>
Ireland Cancer Center at University Hospitals Case Medical Center, Case Comprehensive Cancer Center
Cleveland, Ohio, United States, 44106-5065 Less << |
|
NCT02409355
|
Non-Small Cell Lung Cancer |
Phase 3 |
Terminated(The study was close... More >>d due to low patient enrollment and the Sponsor's decision to include patients with squamous NSCLC into the GO29431 study, NCT02409342.) Less << |
- |
- |
|
NCT01973101
|
Patients Diagnosed With Advanc... More >>ed Carcinoma of Uterine Cervix t Less << |
Phase 2 |
Unknown |
April 2017 |
Brazil
... More >> ICESP
Recruiting
São Paulo, SP, Brazil
Contact: Maria Del Pilar Estevez Diz, MD 55 11 3893-2000 pesquisa.clinica@icesp.org.br Less << |
|
NCT03435302
|
Melanoma |
Phase 3 |
Recruiting |
February 2019 |
China
... More >> Beijing Cancer Hospital
Recruiting
Beijing, China, 100142
Contact: Bin Lian, MD +86(10)88196951 lianbin0214@126.com
Contact: Lu Si, MD +86(10)88196951 silu.net@hotmail.com
Sub-Investigator: Bin Lian, MD Less << |
|
NCT00744900
|
Lung Cancer |
Phase 2 |
Completed |
- |
France
... More >> Centre Hôspitalier du Pays d'Aix, Service des Maladies Respiratoires
Aix En Provence, France, 13616
CHU d'Angers, Service de Pneumologie
Angers, France, 49033
Médecine 4, C.H.G. de la Fontonne Antibes
Antibes, France, 06606
CHU brest, institut de cancérologie et d'hématologie
Brest, France, 29200
Centre François Baclesse
Caen, France, 14000
Centre Hospitalier René Dubos - Pontoise, Service d'Oncologie - Hématologie Clinique
Cergy Pontoise, France, 95303
Centre Hospitalier, Service de Pneumologie
Charleville, France, 08 000
CHI, Service de Pneumologie
Creteil, France, 94010
Pneumologie, Centre hôspitalier, DRAGUIGNAN
Draguignan, France, 83300
CH GAP
GAP, France, 05000
Centre Hospitalier Départemental, Service de Pneumologie,
La Roche Sur Yon, France, 85000
Hôpital A. Mignot, Service de Pneumologie
Le Chesnay, France, 78157
Hopital de la Croix Rousse
Lyon, France, 69317
Centre Hôspitalier, Service de Pneumo-Neuro
Mantes La Jolie, France, 78200
Hôpital Sainte Margueritte
Marseilles, France, 13274
Serv. de Pneumo-Allergo, CH de Martigues
Martigues, France, 13695
Serv. de Pneumo - Hôpital St Antoine
Paris, France, 75571
Service Pneumologie, Pavillon 1A, CH de Lyon-Sud
Pierre-benite, France, 69495
Hôpital Pontchailloux, Service de Pneumologie.
Rennes, France, 35033
CHG, Service de Pneumologie
Roanne, France, 42300
Luc THIBERVILLE
Rouen, France, 76031
CHU de ROUEN, Hôpital Bois Guillaume, Serv. de Pneumo.
Rouen, France, 76233
Institut de Cancérologie de la Loire
Saint-Priest en Jarez Cedex, France, 42271
CHU, Service du Pr. Carles
Toulouse, France, 31059
Pneumologie, Centre Hospitalier
VILLEFRANCHE sur SAONE, France, 69655 Less << |
|
NCT02409355
|
- |
|
Terminated(The study was close... More >>d due to low patient enrollment and the Sponsor's decision to include patients with squamous NSCLC into the GO29431 study, NCT02409342.) Less << |
- |
- |
|
NCT00374868
|
Urologic Neoplasms |
Phase 1
Phase 2 |
Completed |
- |
Spain
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Barcelona, Spain, 08035
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Madrid, Spain, 28041
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Pamplona, Spain, 31008
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Sabadell, Spain, 08208
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Santander, Spain, 39008 Less << |
|
NCT00374868
|
- |
|
Completed |
- |
- |
|
NCT00415168
|
- |
|
Completed |
- |
- |
|
NCT01528618
|
Nasopharyngeal Neoplasms |
Phase 3 |
Unknown |
June 2017 |
China, Guangdong
... More >>
Department of Medical Oncology,Cancer Center of Sun Yat-Sen University
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT00887822
|
- |
|
Completed |
- |
- |
|
NCT00980603
|
Gastric Cancer |
Phase 2 |
Unknown |
May 2011 |
Korea, Republic of
... More >>
Chungbuk National University Hospital
Recruiting
Chonju, Korea, Republic of, 361-711
Contact: Hye-Suk Han, M.D. sook3529@hanmail.net
Sub-Investigator: Hye-Suk Han, M.D.
Research Institute and Hospital, National Cancer Center Korea
Recruiting
Goyang, Korea, Republic of, 410-769
Contact: Sook Ryun Park, M.D. 82-31-920-1609 ext 1609 sukryun73@hanmail.net
Contact: Young Lan Park, RN 82-31-920-0422 ext 0422 lan0729@hanmail.net
Principal Investigator: Sook Ryun Park, M.D.
Sub-Investigator: Noe Kyeong Kim, M.D., Ph.D.
Sub-Investigator: Youngiee Park, M.D., Ph.D.
Sub-Investigator: Byung-Ho Nam, Ph.D.
Gachon University Gil Hospital
Not yet recruiting
Inchon, Korea, Republic of, 405-760
Contact: Dong Bok Shin, M.D., Ph.D. dbs@gilhospital.com
Principal Investigator: Dong Bok Shin, M.D., Ph.D.
Sub-Investigator: Sun Jin Sym, M.D.
Seoul National University Bundang Hospital
Recruiting
Seongnam, Korea, Republic of, 463-707
Contact: Keun-Wook Lee, M.D., Ph.D. hmodoctor@hanmail.net
Principal Investigator: Keun-Wook Lee, M.D., Ph.D. Less << |
|
NCT02709720
|
Lung Cancer |
Phase 2 |
Active, not recruiting |
April 15, 2021 |
Spain
... More >> Hospital General Universitario de Elche
Elche, Alicante, Spain, 03203
ICO-Badalona
Badalona, Barcelona, Spain, 08916
Hospital Provincial de Castellón
Castelló de la Plana, Castelló, Spain, 12002
H. Son Espases
Palma de Mallorca, Mallorca, Spain, 07014
Hospital Lluís Alcanyís
Xàtiva, Valencia, Spain, 46800
Hospital de Basurto
Bilbao, Vizcaya, Spain, 48013
H.G.U. Alicante
Alicante, Spain, 03010
Hospital de La Santa Creu I Sant Pau
Barcelona, Spain, 08041
Hospital de Jaén
Jaén, Spain, 23007
Hospital Universitario Lucus Augusti
Lugo, Spain, 27003
H. de la Princesa
Madrid, Spain, 28006
H.U. Puerta de Hierro
Madrid, Spain, 28035
Hospital Fundación Jiménez Díaz
Madrid, Spain, 28040
H. Clínico San Carlos
Madrid, Spain
H. Son Llàtzer
Palma de Mallorca, Spain, 07198
H. de Donostia
San Sebastian, Spain, 20014
Hospital Virgen de La Macrena
Sevilla, Spain, 41009
Hospital Clínico Lozano Blesa
Zaragoza, Spain, 50009 Less << |
|
NCT00423930
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT00869752
|
Lung Cancer |
Phase 1
Phase 2 |
Completed |
- |
Canada, Ontario
... More >>
Juravinski Cancer Centre at Hamilton Health Sciences
Hamilton, Ontario, Canada, L8V 5C2
Ottawa Health Research Institute - General Division
Ottawa, Ontario, Canada, K1H 8L6
Univ. Health Network-Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT00772681
|
Head and Neck Neoplasms |
Phase 4 |
Completed |
- |
Turkey
... More >> Sanofi-Aventis Administrative Office
Istanbul, Turkey Less << |
|
NCT00423930
|
- |
|
Completed |
- |
- |
|
NCT02240017
|
Advanced Urothelial Cancer
... More >> Metastatic Urothelial Cancer Less << |
Phase 2
Phase 3 |
Recruiting |
September 2023 |
France
... More >> INSTITUT DE CANCEROLOGIE DE L'OUEST - Site Paul Papin
Recruiting
Angers, France, 49933
Contact: Sophie ABADIE-LACOURTOISIE, MD 33241352734 sophie.abadie-lacourtoisie@ico.unicancer.fr
Principal Investigator: Sophie ABADIE-LACOURTOISIE, MD
Chru Besancon - Hopital Jean Minjoz
Recruiting
Besançon, France, 25030
Contact: Tristan MAURINA, MD 33381668166 tmaurina@chu-besancon.fr
Principal Investigator: Tristan MAURINA, MD
Institut Bergonie
Recruiting
Bordeaux, France, 33076
Contact: Guilhem ROUBAUD, MD 33556333341 g.roubaud@bordeaux.unicancer.fr
Principal Investigator: Guilhem ROUBAUD, MD
Centre Francois Baclesse
Recruiting
Caen, France, 14076
Contact: Florence JOLY, PhD 02 31 45 50 02 f.joly@baclesse.fr
Principal Investigator: Emmanuel SEVIN, PhD
Centre Georges Francois Leclerc
Recruiting
Dijon, France, 21079
Contact: Sylvain LADOIRE, MD 33380737506 sladoire@cgfl.fr
Principal Investigator: Sylvain LADOIRE, MD
CH VERSAILLES - Hôpital André Mignot
Recruiting
Le Chesnay, France, 78157
Contact: Christine ABRAHAM, MD 33139638909 cabraham@ch-versailles.fr
Principal Investigator: Christine ABRAHAM, MD
CHU LIMOGES - Hôpital Dupuytren
Recruiting
Limoges, France, 87042
Contact: Sabrina FALKOWSKI, MD 33555056100 sabrina.falkowski@chu-limoges.fr
Principal Investigator: Sabrina FALKOWSKI, MD
Centre Leon Berard
Recruiting
Lyon, France, 69373
Contact: Aude FLECHON, MD 33478782828 flechon@lyon.unicancer.fr
Principal Investigator: Aude FLECHON, MD
Institut Paoli Calmettes
Recruiting
Marseille, France, 13273
Contact: Gwénaëlle GRAVIS-MESCAM 04 91 22 37 40 gravisg@ipc.unicancer.fr
Ch Mont de Marsan
Recruiting
Mont de Marsan, France, 40024
Contact: Jérôme DAUBA, MD 05 58 05 10 10 jerome.dauba@ch-mt-marsan.fr
Principal Investigator: Jérôme DAUBA, MD
Institut Regional Du Cancer Montpellier
Recruiting
Montpellier, France, 34298
Contact: Diego TOSI, MD 33467612304 diego.tosi@icm.unicancer.fr
Principal Investigator: Diego TOSI, MD
Ap-Hp-Hopital Saint-Louis
Recruiting
Paris, France, 75010
Contact: Hélène GAUTHIER, MD 01 42 49 42 47 helene.gauthier@aphp.frphp.fr
Principal Investigator: Hélène GAUTHIER, MD
INSTITUT DE CANCEROLOGIE DE L'OUEST - Site René Gauducheau
Recruiting
Saint-Herblain, France, 44805
Contact: Frédéric ROLLAND, MD 33240679976 frederic.rolland@ico.unicancer.fr
Principal Investigator: Frédéric ROLLAND, MD
Institut de Cancerologie Lucien Neuwirth
Recruiting
Saint-Priest en Jarez, France, 42271
Contact: Aline GUILLOT, MD 33477917032 aline.guillot@icloire.fr
Principal Investigator: Aline GUILLOT, MD
CHU de STRASBOURG
Recruiting
Strasbourg, France, 67091
Contact: Philippe BARTHELEMY 03 88 11 67 28 philippe.barthelemy@chru-strasbourg.fr
Institut Claudius Regaud
Recruiting
Toulouse, France, 31052
Contact: Loïc MOUREY, PhD 33561424548 mourey.loic@claudiusregaud.fr
Principal Investigator: Loïc MOUREY, MD
Chru Tours
Recruiting
Tours, France, 37044
Contact: Claude LINASSIER, MD 33247479919 claude.linassier@univ-tours.fr
Principal Investigator: Claude LINASSIER, MD
Institut Gustave Roussy
Recruiting
Villejuif, France, 94805
Contact: Yohann LORIOT, MD 33142114211 yohann.loriot@igr.fr
Principal Investigator: Yohann LORIOT, MD Less << |
|
NCT00201760
|
Breast Cancer |
Phase 2 |
Completed |
- |
United States, Ohio
... More >>
Ohio State University
Columbus, Ohio, United States, 43210 Less << |
|
NCT00415168
|
Gastric Cancer |
Phase 2 |
Completed |
- |
Russian Federation
... More >>
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Barnaul, Russian Federation, 656049
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Ivanovo, Russian Federation, 153040
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Kazan, Russian Federation, 420029
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Moscow, Russian Federation, 117997
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Saint Petersburg, Russian Federation, 197758
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Stavropol, Russian Federation, 355001 Less << |
|
NCT00201760
|
- |
|
Completed |
- |
- |
|
NCT00833261
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
United States, Ohio
... More >>
University Hospitals of Cleveland
Cleveland, Ohio, United States, 44106
United States, Texas
Baylor Research Institute
Dallas, Texas, United States, 75204
University of Texas Southwestern Medical Center - Dallas
Dallas, Texas, United States, 75390
United States, Wisconsin
Medical College of Wisconsin
Milwaukee, Wisconsin, United States, 53226 Less << |
|
NCT00885534
|
Melanoma
Skin... More >> Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT02478502
|
Testicular Cancer |
Phase 2 |
Recruiting |
June 2019 |
Denmark
... More >> Rigshospitalet
Recruiting
Copenhagen, Denmark
Contact: Gedske Daugaard, MD, PhD 35453545 ext +45 gedske.daugaard@rh.regionh.dk
Principal Investigator: Gedske Daugaard, MD, PhD
Germany
University Clinic Hamburg Eppendorf
Withdrawn
Hamburg, Germany, 20246
Italy
Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (I.R.S.T)
Active, not recruiting
Meldola, Italy, 47014
Norway
Oslo University Hospital
Recruiting
Oslo, Norway, N-0424
Contact: Helene Negaard, MD, PhD UXHEGA@ous-hf.no
Contact: Marit Husby, MD 23026600 ext +47 UXHUMC@ous-hf.no
Principal Investigator: Helene Negaard, MD, PhD
Sweden
University Hospital of Uppsala, Department of Oncology
Active, not recruiting
Uppsala, Sweden, 75185 Less << |
|
NCT00859339
|
Transitional Cell Carcinoma of... More >> the Bladder Less << |
Phase 2 |
Terminated(Patient Toxicities) |
- |
United States, Florida
... More >>
University of Florida
Gainesville, Florida, United States, 32610
United States, Indiana
Indiana University Simon Cancer Center
Indianapolis, Indiana, United States, 46202
Northern Indiana Cancer Research Consortium
South Bend, Indiana, United States, 46601
United States, Texas
Baylor College of Medicine
Houston, Texas, United States, 77030
United Kingdom
St. Bartholomew's Hospital (Barts)
London, United Kingdom, EC1A 7BE Less << |
|
NCT01269255
|
Gastric Cancer |
Phase 1 |
Completed |
- |
Korea, Republic of
... More >>
Severance Hospital
Seoul, Korea, Republic of, 120-752 Less << |
|
NCT00859339
|
- |
|
Terminated(Patient Toxicities) |
- |
- |
|
NCT01161498
|
Squamous Cell Carcinoma
... More >> Head and Neck Cancer Less << |
Phase 3 |
Terminated(The changing aetiol... More >>ogy of squamous cell carcinoma of the head and neck (SCCHN).) Less << |
- |
United States, Indiana
... More >>
Investigative Clinical Research of Indiana
Indianapolis, Indiana, United States, 46260
United States, Kentucky
James Graham Brown Cancer Center, University of Louisville
Louisville, Kentucky, United States, 40202
United States, Ohio
Gabrail Cancer Center
Canton, Ohio, United States, 44718
United States, South Carolina
Medical Univesity of South Carolina
Charleston, South Carolina, United States, 29425
United States, Virginia
VCU Massey Cancer Center
Richmond, Virginia, United States, 23298
United Kingdom
The Royal Marsden Hospital
London, United Kingdom, SE1 7EH Less << |
|
NCT00885534
|
- |
|
Completed |
- |
- |
|
NCT00510107
|
Stomach Neoplasm
... More >> Stage IV
Recurrent Less << |
Phase 2 |
Unknown |
July 2009 |
Korea, Republic of
... More >>
Gachon University Gil Medical Center
Recruiting
Incheon, Korea, Republic of, 405 760 Less << |
|
NCT01161498
|
- |
|
Terminated(The changing aetiol... More >>ogy of squamous cell carcinoma of the head and neck (SCCHN).) Less << |
- |
- |
|
NCT01041833
|
Non Small Cell Lung Cancer |
Phase 3 |
Unknown |
September 2013 |
Mexico
... More >> Department of Medical Oncology, Instituto Nacional de Cancerología
Not yet recruiting
Mexico, Mexico, 14080
Contact: Oscar Arrieta, M.D. (+52) (55) 5628-0400 ext 832 ogar@servidor.unam.mx
Contact: Oscar Arrieta (+52) (55) 5628-0400 ext 832 ogar@servidor.unam.mx Less << |
|
NCT02016417
|
Nasopharyngeal Carcinoma |
Phase 2 |
Not yet recruiting |
December 2020 |
- |
|
NCT01836341
|
Non-small Cell Lung Cancer |
Phase 1 |
Withdrawn |
April 2015 |
- |
|
NCT03117335
|
Advanced Non-small Cell Lung C... More >>ancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT02370849
|
Stomach Neoplasms |
Phase 2 |
Completed |
- |
China
... More >> Cancer Hospital, Chinese Academy of Medical Sciences
Beijing, China, 100021 Less << |
|
NCT02573493
|
Squamous Cell Carcinoma of the... More >> Head and Neck
Carcinoma, Squamous Cell of the Head and Neck
Cancer of Head and Neck
Cancer of the Head and Neck
Head and Neck Cancer
Neoplasms, Head and Neck Less << |
Phase 2 |
Recruiting |
October 31, 2029 |
United States, Kansas
... More >>
The University of Kansas Cancer Center and Medical Pavilion
Recruiting
Westwood, Kansas, United States, 66205
Contact: Prakash Neupane, M.D. 913-588-1227 pneupane@kumc.edu
Principal Investigator: Prakash Neupane, M.D.
United States, Missouri
Washington University School of Medicine
Recruiting
Saint Louis, Missouri, United States, 63110
Contact: Douglas Adkins, M.D. 314-747-8475 dadkins@wustl.edu
Principal Investigator: Douglas Adkins, M.D.
Sub-Investigator: Mackenzie Daly, M.D.
Sub-Investigator: Hiram Gay, M.D.
Sub-Investigator: Ryan Jackson, M.D.
Sub-Investigator: Jose Zevallos, M.D.
Sub-Investigator: Randall Paniello, M.D.
Sub-Investigator: Jason Rich, M.D.
Sub-Investigator: Wade Thorstad, M.D. Less << |
|
NCT01396668
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Seoul National University Bundang Hospital
Seongnam, Korea, Republic of
Seoul National University Hospital
Seoul, Korea, Republic of Less << |
|
NCT03050437
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Recruiting |
March 2017 |
Korea, Republic of
... More >>
Samsung Medical Center
Recruiting
Seoul, Korea, Republic of, 135-710
Contact: Myung-Ju Ahn, Ph.D. 822-3410-1795 silkahn@skku.edu Less << |
|
NCT02608684
|
Ovarian Cancer |
Phase 2 |
Recruiting |
January 2020 |
United States, California
... More >>
Cedars-Sinai Medical Center
Recruiting
Los Angeles, California, United States, 90048
Contact: Christine Walsh, MD 310-423-3599 Christine.Walsh@cshs.org
Contact: Jaime Richardson 310-423-2133 cancer.trial.info@cshs.org
Principal Investigator: Christine Walsh, MD Less << |
|
NCT00232206
|
Non-small-Cell Lung Carcinoma |
Phase 2 |
Terminated(study terminated du... More >>e to slow accrual) Less << |
- |
United States, Oregon
... More >>
Providence Portland Medical Center
Portland, Oregon, United States, 97213 Less << |
|
NCT00394433
|
- |
|
Completed |
- |
- |
|
NCT00192101
|
Metastatic Breast Cancer |
Phase 2 |
Completed |
- |
Saudi Arabia
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Jeddah, Saudi Arabia Less << |
|
NCT00183807
|
Esophageal Cancer |
Phase 2 |
Terminated(Insufficient Accrua... More >>l) Less << |
- |
United States, California
... More >>
U.S.C. / Norris Comprehensive Cancer Center
Los Angeles, California, United States, 90033 Less << |
|
NCT00865982
|
Gastric Cancer
... More >> Esophageal Cancer Less << |
Phase 2 |
Unknown |
September 2015 |
Germany
... More >> HELIOS-Klinik Bad Saarow
Bad Saarow, Germany
Klinik für Hämatologie, Onkologie und Tumorimmunologie, Charite Campus Buch
Berlin, Germany
Medizinische Klinik mit Schwerpunkt Gastroenterologie, Infektiologie und Rheumatologie, Charite Campus Benjamin-Franklin
Berlin, Germany
Medizinische Klinik mit Schwerpunkt Hämatologie und Onkologie, Charite Campus Virchow Klinikum
Berlin, Germany
Klinik für Innere Medizin Abteilung Hämatologie/Onkologie, Städtisches Klinikum Dessau
Dessau, Germany
Universitätsklinik und Poliklinik für Innere Medizin IV, Martin Luther Universität Halle-Wittenberg
Halle (Saale), Germany
II. Medizinische Klinik und Poliklinik, Universitätsklinikum Schleswig-Holstein Campus Kiel
Kiel, Germany
Internistische Onkologie/ Hämatologie, Städtisches Krankenhaus St. Georg
Leipzig, Germany
3. Medizinische Klinik, Onkologisches Zentrum, Universitätsklinikum Mannheim
Mannheim, Germany Less << |
|
NCT00238407
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
Switzerland
... More >> Hirslanden Klinik Aarau
Aarau, Switzerland, CH-5001
Saint Claraspital AG
Basel, Switzerland, CH-4016
Universitaetsspital-Basel
Basel, Switzerland, CH-4031
Inselspital Bern
Bern, Switzerland, CH-3010
Kantonsspital Bruderholz
Bruderholz, Switzerland, CH-4101
Kantonsspital Graubuenden
Chur, Switzerland, CH-7000
Kantonsspital
Liestal, Switzerland, CH-4410
Kantonsspital - St. Gallen
St. Gallen, Switzerland, CH-9007
Regionalspital
Thun, Switzerland, 3600
City Hospital Triemli
Zurich, Switzerland, CH-8063 Less << |
|
NCT00678535
|
Gastric Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00291850
|
Non-Small Cell Lung Cancer |
Phase 2 |
Terminated(no patient recruitm... More >>ent) Less << |
- |
Austria
... More >> AKH, Universitätsklinik für Innere Medizin 1
Vienna, Austria, 1090
Hungary
Somogy Country Pulmo and Cardio Hospital,
Mosdos, Hungary
Markusovszky Hospital
Szombathely, Hungary
Poland
M. Sklodowska-Curie Memorial Dep. Of Lung and Thoracic Tumours,
Warszawa, Poland, 02-781
Klinika Chirurgii Instytutu Gruzlicy i Chorob Pluc
Warszawa, Poland Less << |
|
NCT00590031
|
- |
|
Completed |
- |
- |
|
NCT01076400
|
Cervical Cancer |
Phase 1
Phase 2 |
Terminated(The sponsor permane... More >>ntly suspended new enrollment into the trial and discontinued the study; which was not related to any concerns over product safety.) Less << |
- |
- |
|
NCT01353482
|
Malignant Pleural Mesothelioma |
Phase 1
Phase 2 |
Withdrawn(UCL CTC were informe... More >>d by Merck Sharp & Dohme on 22.08.11 that support for the trial had been withdrawn in light of results from another trial with trial drug.) Less << |
- |
- |
|
NCT00678535
|
- |
|
Completed |
- |
- |
|
NCT00394433
|
Esophageal Cancer
... More >> Stomach Cancer Less << |
Phase 2 |
Completed |
- |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02115
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00081276
|
Primary Peritoneal Cavity Canc... More >>er
Recurrent Ovarian Epithelial Cancer
Stage III Ovarian Epithelial Cancer
Stage IV Ovarian Epithelial Cancer Less << |
Phase 2 |
Completed |
- |
United States, Pennsylvania
... More >>
Gynecologic Oncology Group
Philadelphia, Pennsylvania, United States, 19103 Less << |
|
NCT00590031
|
Esophageal Carcinoma |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT01076400
|
- |
|
Terminated(The sponsor permane... More >>ntly suspended new enrollment into the trial and discontinued the study; which was not related to any concerns over product safety.) Less << |
- |
- |
|
NCT00851877
|
- |
|
Completed |
- |
- |
|
NCT02375880
|
Carcinoma of Intrahepatic and ... More >>Extra-hepatic Biliary System
Carcinoma of Gallbladder
Bile Duct Cancer
Cholangiocarcinoma Less << |
Phase 1 |
Completed |
- |
United States, California
... More >>
University of Southern California
Los Angeles, California, United States, 90033
United States, Connecticut
Yale University
New Haven, Connecticut, United States, 06520
United States, Massachusetts
Massachusetts General Hospital
Boston, Massachusetts, United States, 02214
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02215
Dana Farber Cancer Institute
Boston, Massachusetts, United States, 02215
United States, New York
Columbia University Medical Center
New York, New York, United States, 10032
United States, Ohio
University Hospitals, Case Medical Center
Cleveland, Ohio, United States, 44106
Cleveland Clinic
Cleveland, Ohio, United States, 44195
United States, Texas
University of Texas Health Science Center at San Antonio
San Antonio, Texas, United States, 78229 Less << |
|
NCT00731380
|
Head and Neck Cancer |
Phase 1 |
Completed |
- |
Canada, Ontario
... More >>
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 3M9 Less << |
|
NCT03583710
|
ENSAT Stage I Adrenal Cortex C... More >>arcinoma
ENSAT Stage II Adrenal Cortex Carcinoma
ENSAT Stage III Adrenal Cortex Carcinoma Less << |
Phase 3 |
Recruiting |
January 22, 2025 |
United States, Texas
... More >>
M D Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact: Mouhammed A. Habra 713-792-2841 mahabra@mdanderson.org
Principal Investigator: Mouhammed A. Habra Less << |
|
NCT00949949
|
Cholangiocarcinoma of the Gall... More >>bladder
Localized Gallbladder Cancer
Unresectable Gallbladder Cancer
Unspecified Adult Solid Tumor, Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, Arizona
... More >>
Mayo Clinic in Arizona
Scottsdale, Arizona, United States, 85259
United States, Florida
Mayo Clinic in Florida
Jacksonville, Florida, United States, 32224-9980
United States, Minnesota
Mayo Clinic
Rochester, Minnesota, United States, 55905 Less << |
|
NCT00895648
|
Mesothelioma
... More >>Pleural Mesothelioma Less << |
Phase 2 |
Terminated(low accrual due to ... More >>competing study for same group of patients) Less << |
- |
Canada, Ontario
... More >>
University Health Network
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT00851877
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, Texas
... More >>
Baylor Research Institute
Dallas, Texas, United States, 75204
University of Texas Southwestern Medical Center
Dallas, Texas, United States, 75239 Less << |
|
NCT01938573
|
Recurrent Bladder Carcinoma
... More >> Stage II Bladder Cancer
Stage III Bladder Cancer
Stage IV Bladder Cancer Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, Washington
... More >>
VA Puget Sound Health Care System
Seattle, Washington, United States, 98101
Fred Hutch/University of Washington Cancer Consortium
Seattle, Washington, United States, 98109 Less << |
|
NCT01388790
|
Gastric Cancer |
Phase 2 |
Completed |
- |
Germany
... More >> Please contact the Merck KGaA Communication Center located in
Darmstadt, Germany Less << |
|
NCT00216125
|
- |
|
Completed |
- |
- |
|
NCT02913066
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Phase 2 |
Recruiting |
March 2019 |
China, Sichaung
... More >>
Xiaobo du
Recruiting
Mianyang, Sichaung, China, 621000
Contact: Xaiobo Du, MD +86 08162230478 duxiaobo2005@126.com Less << |
|
NCT02001519
|
Breast Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT02856568
|
Non-Resectable Cholangiocarcin... More >>oma
Recurrent Cholangiocarcinoma
Stage III Extrahepatic Bile Duct Cancer
Stage III Intrahepatic Cholangiocarcinoma
Stage IIIA Hilar Cholangiocarcinoma
Stage IIIB Hilar Cholangiocarcinoma
Stage IVA Extrahepatic Bile Duct Cancer
Stage IVA Hilar Cholangiocarcinoma
Stage IVA Intrahepatic Cholangiocarcinoma
Stage IVB Extrahepatic Bile Duct Cancer
Stage IVB Hilar Cholangiocarcinoma
Stage IVB Intrahepatic Cholangiocarcinoma
Unresectable Extrahepatic Bile Duct Carcinoma Less << |
Phase 1 |
Withdrawn(Site dropped study) |
October 2021 |
United States, Arizona
... More >>
Mayo Clinic in Arizona
Scottsdale, Arizona, United States, 85259
United States, Florida
Mayo Clinic in Florida
Jacksonville, Florida, United States, 32224-9980
United States, Minnesota
Mayo Clinic
Rochester, Minnesota, United States, 55905 Less << |
|
NCT01938573
|
- |
|
Completed |
- |
- |
|
NCT01384799
|
Head and Neck Cancer |
Phase 1 |
Completed |
- |
United States, California
... More >>
Stanford Cancer Center
Stanford, California, United States, 94305
United States, Colorado
University of Colorado Cancer Center
Aurora, Colorado, United States, 80045
United States, Louisiana
Overton Brooks VA Medical Center
Shreveport, Louisiana, United States, 71101
United States, Massachusetts
Massachusetts General Hospital
Boston, Massachusetts, United States, 02115
United States, Michigan
Henry Ford Health System
Detroit, Michigan, United States, 48202
United States, Pennsylvania
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 01911
United States, Tennessee
Vanderbilt University
Nashville, Tennessee, United States, 37232 Less << |
|
NCT01388790
|
- |
|
Completed |
- |
- |
|
NCT01825603
|
Ampulla of Vater Adenocarcinom... More >>a
Gallbladder Adenocarcinoma
Metastatic Pancreatic Adenocarcinoma
Pancreatic Adenocarcinoma
Stage III Ampulla of Vater Cancer
Stage III Intrahepatic Cholangiocarcinoma
Stage III Pancreatic Cancer
Stage IIIA Gallbladder Cancer
Stage IIIA Hilar Cholangiocarcinoma
Stage IIIB Gallbladder Cancer
Stage IIIB Hilar Cholangiocarcinoma
Stage IV Ampulla of Vater Cancer
Stage IVA Gallbladder Cancer
Stage IVA Hilar Cholangiocarcinoma
Stage IVA Intrahepatic Cholangiocarcinoma
Stage IVA Pancreatic Cancer
Stage IVB Gallbladder Cancer
Stage IVB Hilar Cholangiocarcinoma
Stage IVB Intrahepatic Cholangiocarcinoma
Stage IVB Pancreatic Cancer Less << |
Phase 1 |
Completed |
- |
United States, Nebraska
... More >>
University of Nebraska Medical Center
Omaha, Nebraska, United States, 68198 Less << |
|
NCT03366766
|
Non-Squamous Non-Small Cell Lu... More >>ng Carcinoma
Stage I Non-Small Cell Lung Cancer
Stage IA Non-Small Cell Lung Carcinoma
Stage IB Non-Small Cell Lung Carcinoma
Stage II Non-Small Cell Lung Cancer
Stage IIA Non-Small Cell Lung Carcinoma
Stage IIB Non-Small Cell Lung Carcinoma
Stage IIIA Non-Small Cell Lung Cancer Less << |
Phase 2 |
Recruiting |
July 2022 |
United States, Pennsylvania
... More >>
Abington Hospital - Jefferson Health
Not yet recruiting
Abington, Pennsylvania, United States, 19001
Contact: Mark Sundemeyer, MD 215-481-2400
Principal Investigator: Mark Sundemeyer, MD
Sidney Kimmel Cancer Center at Thomas Jefferson University
Recruiting
Philadelphia, Pennsylvania, United States, 19107
Contact: Ralph Zinner, MD 215-503-5098 ralph.zinner@jefferson.edu
Lankenau Institute for Medical Research
Not yet recruiting
Wynnewood, Pennsylvania, United States, 19096
Contact: Paul Gilman, MD 484-476-4837
Principal Investigator: Paul Gilman, MD Less << |
|
NCT02768558
|
Non-Small Cell Lung Cancer |
Phase 3 |
Active, not recruiting |
October 2024 |
United States, California
... More >>
UC San Diego Moores Cancer Center
La Jolla, California, United States, 92093
Stanford University
Stanford, California, United States, 94305
United States, Florida
University of Florida
Gainesville, Florida, United States, 32610
Mount Sinai Cancer Research Center
Miami Beach, Florida, United States, 33140
United States, Georgia
Nancy N. & J.C. Lewis Cancer & Research Pavilion at St. Joseph's/Candler Hospital
Savannah, Georgia, United States, 31405
United States, Kentucky
University of Kentucky
Lexington, Kentucky, United States, 40536
United States, Maryland
Mercy Medical Cancer Center
Baltimore, Maryland, United States, 21202
United States, Michigan
Henry Ford Health System
Detroit, Michigan, United States, 48202
United States, New York
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
University of Rochester
Rochester, New York, United States, 14642
United States, Ohio
Metro Health Medical Center
Cleveland, Ohio, United States, 44109
United States, Pennsylvania
Reading Hospital/McGlinn Cancer Institute
West Reading, Pennsylvania, United States, 19611
United States, Texas
University of Texas Southwestern Medical Center
Dallas, Texas, United States, 75390
United States, Virginia
Inova Fairfax Hospital
Falls Church, Virginia, United States, 22042
United States, Washington
Virginia Mason
Seattle, Washington, United States, 98101
United States, Wisconsin
University of Wisconsin Hospital and Clinics
Madison, Wisconsin, United States, 53792 Less << |
|
NCT02535312
|
Advanced Malignant Solid Neopl... More >>asm
Advanced Peritoneal Malignant Mesothelioma
Advanced Pleural Malignant Mesothelioma
Recurrent Peritoneal Malignant Mesothelioma
Recurrent Pleural Malignant Mesothelioma
Stage III Non-Small Cell Lung Cancer AJCC v7
Stage III Ovarian Cancer AJCC v6 and v7
Stage III Pleural Malignant Mesothelioma AJCC v7
Stage IIIA Non-Small Cell Lung Cancer AJCC v7
Stage IIIA Ovarian Cancer AJCC v6 and v7
Stage IIIB Lung Non-Small Cell Cancer AJCC v7
Stage IIIB Ovarian Cancer AJCC v6 and v7
Stage IIIC Ovarian Cancer AJCC v6 and v7
Stage IV Non-Small Cell Lung Cancer AJCC v7
Stage IV Ovarian Cancer AJCC v6 and v7
Stage IV Pleural Malignant Mesothelioma AJCC v7
Thymoma
Unresectable Solid Neoplasm Less << |
Phase 1
Phase 2 |
Recruiting |
- |
United States, California
... More >>
City of Hope Comprehensive Cancer Center
Recruiting
Duarte, California, United States, 91010
Contact: Site Public Contact 800-826-4673 becomingapatient@coh.org
Principal Investigator: Marianna Koczywas
Los Angeles County-USC Medical Center
Recruiting
Los Angeles, California, United States, 90033
Contact: Site Public Contact 323-865-0451
Principal Investigator: Anthony El-Khoueiry
USC / Norris Comprehensive Cancer Center
Recruiting
Los Angeles, California, United States, 90033
Contact: Site Public Contact 323-865-0451
Principal Investigator: Anthony El-Khoueiry
Keck Medical Center of USC Pasadena
Recruiting
Pasadena, California, United States, 91105
Contact: Site Public Contact 323-865-0451
Principal Investigator: Anthony El-Khoueiry
University of California Davis Comprehensive Cancer Center
Recruiting
Sacramento, California, United States, 95817
Contact: Site Public Contact 916-734-3089
Principal Investigator: Karen L. Kelly
United States, Colorado
University of Colorado Hospital
Recruiting
Aurora, Colorado, United States, 80045
Contact: Site Public Contact 720-848-0650
Principal Investigator: Stephen Leong
United States, Maryland
University of Maryland/Greenebaum Cancer Center
Recruiting
Baltimore, Maryland, United States, 21201
Contact: Site Public Contact 800-888-8823
Principal Investigator: Edward A. Sausville
United States, Michigan
University of Michigan Comprehensive Cancer Center
Suspended
Ann Arbor, Michigan, United States, 48109
United States, Minnesota
Mayo Clinic
Active, not recruiting
Rochester, Minnesota, United States, 55905
United States, Ohio
Ohio State University Comprehensive Cancer Center
Recruiting
Columbus, Ohio, United States, 43210
Contact: Site Public Contact 800-293-5066 Jamesline@osumc.edu
Principal Investigator: Gregory A. Otterson
United States, Pennsylvania
University of Pittsburgh Cancer Institute (UPCI)
Recruiting
Pittsburgh, Pennsylvania, United States, 15232
Contact: Site Public Contact 412-647-8073
Principal Investigator: Liza C. Villaruz
United States, Tennessee
Vanderbilt University/Ingram Cancer Center
Recruiting
Nashville, Tennessee, United States, 37232
Contact: Site Public Contact 800-811-8480
Principal Investigator: Leora Horn
United States, Wisconsin
University of Wisconsin Hospital and Clinics
Active, not recruiting
Madison, Wisconsin, United States, 53792 Less << |
|
NCT00118131
|
- |
|
Terminated(Slow accrual) |
- |
- |
|
NCT00118131
|
Lung Cancer |
Phase 2 |
Terminated(Slow accrual) |
- |
United States, New Jersey
... More >>
Central Jersey Oncology Center, PA - East Brunswick
East Brunswick, New Jersey, United States, 08816
JFK Medical Center in Edison
Edison, New Jersey, United States, 08818
CentraState Medical Center
Freehold, New Jersey, United States, 07728
Cancer Institute of New Jersey at Hamilton
Hamilton, New Jersey, United States, 08690
Monmouth Medical Center
Long Branch, New Jersey, United States, 07740
Mountainside Hospital Cancer Center
Montclair, New Jersey, United States, 07042
Jersey Shore University Medical Center
Neptune, New Jersey, United States, 07754
Cancer Institute of New Jersey at UMDNJ - Robert Wood Johnson Medical School
New Brunswick, New Jersey, United States, 08903
Saint Peter's University Hospital
New Brunswick, New Jersey, United States, 08903
UMDNJ - University Hospital
Newark, New Jersey, United States, 07103
Raritan Bay Medical Center
Perth Amboy, New Jersey, United States, 08861
Somerset Medical Center
Somerville, New Jersey, United States, 08876
Overlook Hospital
Summit, New Jersey, United States, 07901 Less << |
|
NCT02034968
|
Stage IV Esophageal Squamous C... More >>ell Carcinoma Less << |
Phase 2 |
Withdrawn(No recruitment) |
January 2017 |
China, Zhejiang
... More >>
The first affiliated hospital, Zhejiang University
Hangzhou, Zhejiang, China, 310003 Less << |
|
NCT01005329
|
Endometrial Adenocarcinoma
... More >> Endometrial Adenosquamous Carcinoma
Endometrial Clear Cell Adenocarcinoma
Endometrial Serous Adenocarcinoma
Stage IA Uterine Corpus Cancer AJCC v7
Stage IB Uterine Corpus Cancer AJCC v7
Stage II Uterine Corpus Cancer AJCC v7
Stage IIIA Uterine Corpus Cancer AJCC v7
Stage IIIB Uterine Corpus Cancer AJCC v7
Stage IIIC Uterine Corpus Cancer AJCC v7
Stage IVA Uterine Corpus Cancer AJCC v7
Stage IVB Uterine Corpus Cancer AJCC v7 Less << |
Phase 2 |
Completed |
- |
- |
|
NCT02075112
|
Cancer of Head and Neck |
Phase 1 |
Active, not recruiting |
July 2022 |
United States, Georgia
... More >>
Emory University Hospital Midtown
Atlanta, Georgia, United States, 30308
Emory University Winship Cancer Institute
Atlanta, Georgia, United States, 30322 Less << |
|
NCT00154739
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Taiwan
... More >> Department of Oncology, National Taiwan University Hospital
Taipei, Taiwan, 112 Less << |
|
NCT01262482
|
Advanced or Metastatic Gastric... More >> or Gastroesophageal Junction Adenocarcinoma (Relapsed After a Cisplatin Based Treatment) Less << |
Phase 2 |
Completed |
- |
Spain
... More >> Hospital Clinic de Barcelona
Barcelona, Spain
Hospital Sant Pau
Barcelona, Spain
H. Josep Trueta
Girona, Spain
Centro Oncológico M.D. Anderson Spain
Madrid, Spain
Hospital de Fuenlabrada
Madrid, Spain
Hospital La Paz
Madrid, Spain
Hospital Althaia
Manresa, Spain
Clínica Universitaria de Navarra
Pamplona, Spain
Hospital Parc Taulí
Sabadell, Spain
Hospital General de Valencia
Valencia, Spain Less << |
|
NCT01663285
|
Urothelial Cancer
... More >> Bladder Cancer Less << |
Phase 2 |
Terminated(Low study participa... More >>nt enrollment.) Less << |
- |
United States, Michigan
... More >>
University of Michigan
Ann Arbor, Michigan, United States, 48109
United States, New York
University of Rochester
Rochester, New York, United States, 14642
United States, Oregon
Oregon Health Sciences University
Portland, Oregon, United States, 97239 Less << |
|
NCT00287911
|
Cervical Cancer |
Phase 1 |
Completed |
- |
United States, Minnesota
... More >>
University of Minnesota Cancer Center
Minneapolis, Minnesota, United States, 55455 Less << |
|
NCT00101907
|
Lung Cancer
P... More >>ancreatic Cancer
Esophageal Cancer Less << |
Phase 1 |
Terminated(Study terminated pe... More >>r recommendation of Data Review Team) Less << |
- |
- |
|
NCT00057850
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, Pennsylvania
... More >>
Abramson Cancer Center of the University of Pennsylvania
Philadelphia, Pennsylvania, United States, 19104-4283 Less << |
|
NCT00101907
|
- |
|
Terminated(Study terminated pe... More >>r recommendation of Data Review Team) Less << |
- |
- |
|
NCT01277744
|
Advanced Cancers
... More >> Sarcoma Less << |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00816634
|
First Line Chemotherapy
... More >> Capecitabine Plus Cisplatin Versus Capecitabine Plus Paclitaxel
Advanced or Recurrent Esophageal Squamous Cell Carcinoma Less << |
Phase 2 |
Recruiting |
December 2018 |
Korea, Republic of
... More >>
Samsung Medical Center
Recruiting
Seoul, Korea, Republic of, 135-710
Contact: Young-Hyuck Im, MD, PhD 82-2-3410-3445 imyh00@skku.edu
Principal Investigator: Young-Hyuck Im, MD, PhD
Sub-Investigator: Yeon-Hee Park, MD, PhD Less << |
|
NCT00842244
|
Stomach Neoplasms
... More >> Advanced Gastric Cancer Less << |
Phase 1 |
Completed |
- |
Japan
... More >> Pfizer Investigational Site
Kashiwa, Chiba, Japan
Pfizer Investigational Site
Yufu-city, Oita, Japan
Korea, Republic of
Pfizer Investigational Site
Goyang-si, Gyeonggi-do, Korea, Republic of, 410-769
Pfizer Investigational Site
Seongnam-si, Gyeonggi-do, Korea, Republic of, 463-707
Pfizer Investigational Site
Seoul, Korea, Republic of, 110-744 Less << |
|
NCT02560298
|
Anal Basaloid Carcinoma
... More >> Anal Canal Cloacogenic Carcinoma
Anal Squamous Cell Carcinoma
Metastatic Anal Canal Carcinoma
Recurrent Anal Canal Carcinoma
Stage IIIB Anal Canal Cancer
Stage IV Anal Canal Cancer Less << |
Phase 2 |
Active, not recruiting |
August 2023 |
United States, Pennsylvania
... More >>
ECOG-ACRIN Cancer Research Group
Philadelphia, Pennsylvania, United States, 19103 Less << |
|
NCT00156884
|
Hormone Refractory Prostate Ca... More >>ncer
Bone Metastases Less << |
Phase 2 |
Completed |
- |
Canada, Alberta
... More >>
Tom Baker Cancer Centre
Calgary, Alberta, Canada, T2N 4N2
Canada, British Columbia
BC Cancer Agency
Vancouver, British Columbia, Canada Less << |
|
NCT00842244
|
- |
|
Completed |
- |
- |
|
NCT02919462
|
Carcinoma, Non-Small-Cell Lung... More >>
Secondary
Advanced Stage IIIB
High Thymidylate Synthase Expression Less << |
Phase 2 |
Terminated(low recruitement ra... More >>te) Less << |
- |
Italy
... More >> Ircc Irst
Meldola, FC, Italy, 47014
U.O. Oncologia Medica
Faenza, RA, Italy, 48018
Claudio Dazzi
Ravenna, RA, Italy, 48121
Ospedale di Piacenza, ASL Piacenza
Piacenza, Italy
U.O. Oncologia Ospedale degli Infermi
Rimini, Italy Less << |
|
NCT00079040
|
Extensive Stage Small Cell Lun... More >>g Cancer Less << |
Phase 2 |
Completed |
- |
United States, Massachusetts
... More >>
Eastern Cooperative Oncology Group
Boston, Massachusetts, United States, 02215 Less << |
|
NCT00757172
|
- |
|
Completed |
- |
- |
|
NCT01663285
|
- |
|
Terminated(Low study participa... More >>nt enrollment.) Less << |
- |
- |
|
NCT00924209
|
NSCLC
Stage I... More >>IIA (N2) Less << |
Phase 2 |
Terminated(Study was terminate... More >>d due to poor accrual.) Less << |
- |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892
Croatia
University Hospital for Lung Diseases
Zagreb, Croatia Less << |
|
NCT00924209
|
- |
|
Terminated(Study was terminate... More >>d due to poor accrual.) Less << |
- |
- |
|
NCT00757172
|
Adenocarcinoma of the Gastroes... More >>ophageal Junction
Esophageal Cancer Less << |
Phase 2 |
Completed |
- |
United States, Georgia
... More >>
Curtis and Elizabeth Anderson Cancer Institute at Memorial Health University Medical Center
Savannah, Georgia, United States, 31403-3089
United States, Illinois
Robert H. Lurie Comprehensive Cancer Center at Northwestern University
Chicago, Illinois, United States, 60611-3013
University of Chicago Cancer Research Center
Chicago, Illinois, United States, 60637-1470
Evanston Hospital
Evanston, Illinois, United States, 60201-1781
Simmons Cooper Cancer Institute
Springfield, Illinois, United States, 62794-9677
United States, Kentucky
Central Baptist Hospital
Lexington, Kentucky, United States, 40503-9985
United States, Michigan
William Beaumont Hospital - Royal Oak Campus
Royal Oak, Michigan, United States, 48073
United States, Minnesota
Mayo Clinic Cancer Center
Rochester, Minnesota, United States, 55905
United States, Missouri
Siteman Cancer Center at Barnes-Jewish Hospital - Saint Louis
Saint Louis, Missouri, United States, 63110
United States, New Hampshire
Norris Cotton Cancer Center at Dartmouth-Hitchcock Medical Center
Lebanon, New Hampshire, United States, 03756-0002
United States, North Carolina
Blumenthal Cancer Center at Carolinas Medical Center
Charlotte, North Carolina, United States, 28232-2861
Wake Forest University Comprehensive Cancer Center
Winston-Salem, North Carolina, United States, 27157-1096
United States, Ohio
Good Samaritan Hospital
Dayton, Ohio, United States, 45406
Wayne Hospital
Greenville, Ohio, United States, 45331
Charles F. Kettering Memorial Hospital
Kettering, Ohio, United States, 45429
United States, Oregon
Providence Cancer Center at Providence Portland Medical Center
Portland, Oregon, United States, 97213-2967
Legacy Emanuel Hospital and Health Center and Children's Hospital
Portland, Oregon, United States, 97227
United States, Pennsylvania
Geisinger Cancer Institute at Geisinger Health
Danville, Pennsylvania, United States, 17822-0001
Allegheny Cancer Center at Allegheny General Hospital
Pittsburgh, Pennsylvania, United States, 15212
UPMC Cancer Centers
Pittsburgh, Pennsylvania, United States, 15232
United States, South Carolina
Hollings Cancer Center at Medical University of South Carolina
Charleston, South Carolina, United States, 29425 Less << |
|
NCT00397904
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT02360501
|
Nasopharyngeal Neoplasms |
Phase 2 |
Completed |
- |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT01405586
|
Non-small Cell Lung Cancer Met... More >>astatic
Non-small Cell Lung Cancer Stage IIIB Less << |
Phase 3 |
Active, not recruiting |
September 2018 |
- |
|
NCT03691090
|
Advanced Esophageal Cancer |
Phase 3 |
Not yet recruiting |
October 2021 |
China, Guangdong
... More >>
Cancer Center of Sun-Yat Sen University (CCSYSU)
Not yet recruiting
Guangzhou, Guangdong, China, 510060
Contact: Ruihua Xu Less << |
|
NCT02017600
|
Localized Squamous Cell Carcin... More >>oma of the Esophagus Less << |
Phase 2 |
Terminated |
March 2017 |
Taiwan
... More >> Taipei Veterans General Hospital
Taipei, Taiwan, 112 Less << |
|
NCT00397904
|
- |
|
Completed |
- |
- |
|
NCT00055757
|
Stage IIIB Non-small Cell Lung... More >> Cancer
Stage IV Non-small Cell Lung Cancer Less << |
Phase 2 |
Completed |
- |
United States, Minnesota
... More >>
Mayo Clinic
Rochester, Minnesota, United States, 55905 Less << |
|
NCT03623737
|
Esophageal Squamous Cell Carci... More >>noma
Chemoradiation Less << |
Phase 2
Phase 3 |
Recruiting |
March 1, 2022 |
Taiwan
... More >> National Taiwan University Hospital
Recruiting
Taipei, Taiwan
Contact: Ta-Chen Huang, MD +886-2-2312-3456 e360215@gmail.com Less << |
|
NCT00335998
|
Recurrent Cervical Cancer
... More >> Recurrent Ovarian Epithelial Cancer
Recurrent Vaginal Cancer
Recurrent Vulvar Cancer
Stage III Vaginal Cancer
Stage IIIA Cervical Cancer
Stage IIIA Ovarian Epithelial Cancer
Stage IIIA Vulvar Cancer
Stage IIIB Cervical Cancer
Stage IIIB Ovarian Epithelial Cancer
Stage IIIB Vulvar Cancer
Stage IIIC Ovarian Epithelial Cancer
Stage IIIC Vulvar Cancer
Stage IV Ovarian Epithelial Cancer
Stage IVA Cervical Cancer
Stage IVA Vaginal Cancer
Stage IVB Cervical Cancer
Stage IVB Vaginal Cancer Less << |
Phase 1 |
Completed |
- |
United States, Ohio
... More >>
Case Western Reserve University
Cleveland, Ohio, United States, 44106 Less << |
|
NCT02142010
|
Breast Cancer |
Not Applicable |
Unknown |
- |
China
... More >> Shanghai Jiaotong University School of Medicine, Renji Hospital
Recruiting
Shanghai, China
Contact: Jinsong Lu, MD lujjss@126.com
Principal Investigator: Jinsong Lu, Phd Less << |
|
NCT01116219
|
Lung Cancer |
Phase 2 |
Completed |
- |
Switzerland
... More >> Kantonsspital Aarau
Aarau, Switzerland, CH-5001
Kantonsspital Baden
Baden, Switzerland, 5404
Saint Claraspital AG
Basel, Switzerland, CH-4016
Clinical Cancer Research Center at University Hospital Basel
Basel, Switzerland, CH-4031
Istituto Oncologico della Svizzera Italiana - Ospedale San Giovanni
Bellinzona, Switzerland, CH-6500
Inselspital Bern
Bern, Switzerland, CH-3010
Spitalzentrum Biel
Biel, Switzerland, CH-2501
Kantonsspital Bruderholz
Bruderholz, Switzerland, CH-4101
Kantonsspital Graubuenden
Chur, Switzerland, CH-7000
Kantonsspital Freiburg
Freiburg, Switzerland, 1700
Hopital Cantonal Universitaire de Geneve
Geneva, Switzerland, CH-1211
Centre Pluridisciplinaire d' Oncologie
Lausanne, Switzerland, 1011
Kantonsspital Liestal
Liestal, Switzerland, CH-4410
Kantonsspital Luzern
Luzerne, Switzerland, CH-6000
Kantonsspital Olten
Olten, Switzerland, 4600
Kantonsspital - St. Gallen
St. Gallen, Switzerland, CH-9007
Regionalspital
Thun, Switzerland, 3600
Spital Uster
Uster, Switzerland, 8610
Kantonsspital Winterthur
Winterthur, Switzerland, CH-8401
UniversitaetsSpital Zuerich
Zurich, Switzerland, CH-8044
City Hospital Triemli
Zurich, Switzerland, CH-8063
Onkozentrum Hirslanden
Zürich, Switzerland, 8008 Less << |
|
NCT00069875
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Completed |
- |
United States, Alabama
... More >>
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Birmingham, Alabama, United States
United States, Massachusetts
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Boston, Massachusetts, United States
United States, New York
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Manhasset, New York, United States
United States, North Carolina
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9AM to 5PM Eastern time (UTC/GMT - 5 hours, EST) or speak with your personal physician
Durham, North Carolina, United States
United States, Ohio
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9AM to 5PM Eastern time (UTC/GMT - 5 hours, EST) or speak with your personal physician
Cleveland, Ohio, United States
United States, Texas
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Houston, Texas, United States
Canada, Ontario
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Ottawa, Ontario, Canada
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Toronto, Ontario, Canada
Canada, Quebec
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9AM to 5PM Eastern time (UTC/GMT - 5 hours, EST) or speak with your personal physician
Montreal, Quebec, Canada
Germany
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Gauting, Germany
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Hamburg, Germany
Israel
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Haifa, Israel
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Kfar Saba, Israel
Netherlands
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Amsterdam, Netherlands
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Rotterdam, Netherlands
Spain
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Oviedo, Asturias, Spain
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Badalona, Barcelona, Spain
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Pamplona, Navarra, Spain
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Monday-Friday from 9:00 AM to 5:00 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Madrid, Spain Less << |
|
NCT01517009
|
Gastric Cancer |
Phase 2 |
Completed |
- |
Netherlands
... More >> VieCuri Hospital
Venlo, Limburg, Netherlands, 5912 BL
Elkerliek Hospital
Helmond, Noord Brabant, Netherlands, 5700 AB
Jeroen Bosch Hospital
Den Bosch, Noord-Brabant, Netherlands, 5200 ME
Catharina Hospital
Eindhoven, Noord-Brabant, Netherlands, 5602 ZA
Maxima Medical Centre
Veldhoven, Noord-Brabant, Netherlands, 5500 MB Less << |
|
NCT00079040
|
- |
|
Completed |
- |
- |
|
NCT02348450
|
Small Cell Lung Cancer |
Phase 4 |
Unknown |
December 2018 |
China, Anhui
... More >> The First Affiliated Hospital of AnHui Medical University
Recruiting
Hefei, Anhui, China
Contact: yueyin pan 86-13805695536 yueyinpan@gmail.com
Contact: wei huang 86-13505514938 huangweibsh@163.com
China, Fujian
FuJian Provincial Tumor Hospital
Not yet recruiting
Fuzhou, Fujian, China
Contact: cheng huang 13905010379 cheng671@sina.com
China, Guangdong
Guangdong General Hospital
Active, not recruiting
Guangzhou, Guangdong, China
China, Guangxi
Cancer Hospital Affiliated To GuangXi Medical University
Recruiting
Nanning, Guangxi, China
Contact: xiangqun song 86-13877104657 xiangquns@163.com
Contact: shaozhang zhou 86-18677115144 zhoushaozhang@qq.com
China, Hebei
Fourth hospital of hebei medical university
Recruiting
Shijiazhuang, Hebei, China
Contact: cuimin ding 86-13933083069 wjwdcm@sina.com
Contact: ruijuan li 86-18231196950 lrj0310@sina.com
China, Heilongjiang
The First Affiliated Hospital of HaErBin Medical University
Recruiting
Haerbin, Heilongjiang, China
Contact: gongyan chen
Contact: xuan hong 13946066560 hongxuan_1218@sina.com
China, Henan
HeNan Provincial Tumor Hospital
Recruiting
Zhengzhou, Henan, China
Contact: zhiyong Ma
Contact: xiangtao Yan 18638628118 skyliuyun@126.com
China, Hubei
WuHan Tongji Hospital
Recruiting
WuHan, Hubei, China
Contact: yuan chen
Contact: qian chu qianchu@tjh.tjmu.edu.cn
China, Hunan
HuNan Provincial Tumor Hospital
Recruiting
Changsha, Hunan, China
Contact: jianhua chen
Contact: yongzhong luo 13607443876 65523714@qq.com
China, Jiangsu
JiangSu Provincial Tumor Hospital
Recruiting
Nanjing, Jiangsu, China
Contact: meiqi shi 13809029766 shimeiqi1963@163.com
China, Jiangxi
The First Affiliated Hospital of NanChang University
Active, not recruiting
Nanchang, Jiangxi, China
China, Liaoning
LiaoNing Provincial Tumor Hospital
Active, not recruiting
Shenyang, Liaoning, China
China, Shandong
ShanDong Provincial Tumor Hospital
Recruiting
Jinan, Shandong, China
Contact: qisen guo
Contact: yan guan 15865298279 qhgy219@163.com
Linyi cancer hospital
Recruiting
Linyi, Shandong, China
Contact: jianhua shi 86-15963998868 shijianhualy@126.com
Contact: shuoxin liu 86-15969916696 lyzlyygcp@163.com
China, Shanghai
Changhai Hospital of Shanghai
Recruiting
Shanghai, Shanghai, China
Contact: chong bai
Contact: huijie zhang 18621775835 15236196601@163.com
East Hospital Affiliated To Tongji University
Active, not recruiting
Shanghai, Shanghai, China
Shanghai Chest hospital of Shanghai Jiaotong University
Recruiting
Shanghai, Shanghai, China
Contact: Shun Lu, MD 86 13601813062 shun_lu@hotmail.com
Contact: Zhiwei Chen 86 13916251926 drchenzhiwei@163.com
China, Zhejiang
Hangzhou First People's Hospital
Active, not recruiting
Hangzhou, Zhejiang, China
The second affiliated hospital of zhejiang university school of medicine
Recruiting
Hangzhou, Zhejiang, China
Contact: kai wang 86-13957158572 doctorhuxi@163.com
Contact: liren ding 86-13906535702 lirending@zju.edu.cn
ZheJiang Provincial Tumor Hospital
Active, not recruiting
Hangzhou, Zhejiang, China Less << |
|
NCT00004919
|
Unspecified Childhood Solid Tu... More >>mor, Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, California
... More >>
COG Phase I Consortium
Arcadia, California, United States, 91006-3776 Less << |
|
NCT00846443
|
Local Advanced Non-Small Cell ... More >>Lung Cancer Less << |
Phase 1 |
Unknown |
- |
China, Shanghai
... More >>
Department of Radiation Oncolory, Cancer Hospital, Fudan University
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Liang Xiahou, MB 862164175590 ext 1404 xiahouliang@hotmail.com
Principal Investigator: Xiaolong Fu, MD Less << |
|
NCT00970996
|
Melanoma |
Phase 1 |
Completed |
- |
United States, Texas
... More >>
U.T. M.D. Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00526643
|
Non-Small Cell Lung Cancer |
Phase 3 |
Terminated(scarce enrolment an... More >>d presentation of positive results of similar study in June 2012.) Less << |
- |
Italy
... More >> Azienda Ospedaliera S. Giuseppe Moscati, U.O. di Oncologia Medica
Monteforte Irpino, AV, Italy, 83024
Ospedale Regionale Miulli, Divisione Medicina Interna Sezione Oncologica
Acquaviva delle Fonti, BA, Italy, 70021
Istituto Oncologico di Bari, U.O. di Oncologia Medica e Sperimentale
Bari, BA, Italy, 70126
Istituto Scientifico S. Raffaele
Milano, MI, Italy, 20132
Azienda Ospedaliera C. Poma
Mantova, MN, Italy, 46100
Istituto Oncologico Veneto
Padova, PD, Italy
Ospedale E. Morelli
Sondalo, SO, Italy, 23039
Ospedale Senatore Antonio Perrino
Brindisi, Italy
A.O. Ospedale Mater Domini, Oncoematologia Università Magna Grecia
Catanzaro, Italy
Ospedale F. Veneziale
Isernia, Italy
A.O. Vito Fazzi
Lecce, Italy
Istituto Nazionale dei Tumori , Divisione di Oncologia Medica B
Napoli, Italy, 80131
Ospedale Regional, Unità Operative di Oncologia
Parma, Italy
Ospedale San Camillo - Forlanini
Rome, Italy
Ospedale S. Felice a Cancello
San Felice a Cancello, Italy Less << |
|
NCT01005329
|
- |
|
Completed |
- |
- |
|
NCT00573690
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, North Carolina
... More >>
Lineberger Comprehensive Cancer Center at University of North Carolina - Chapel Hill
Chapel Hill, North Carolina, United States, 27599-7295 Less << |
|
NCT01490437
|
Urothelial Carcinoma |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT00357149
|
Head and Neck Neoplasms |
Phase 2 |
Completed |
- |
Italy
... More >> Sanofi-Aventis
Milan, Italy Less << |
|
NCT02550327
|
Pancreatic Adenocarcinoma |
Early Phase 1 |
Recruiting |
September 2018 |
United States, Texas
... More >>
Baylor Charles A. Sammons Cancer Center
Recruiting
Dallas, Texas, United States, 75246
Contact: Brooke Murphy 214-818-8472 cancer.trials@bswhealth.org
Principal Investigator: Carlos Becerra, MD
Sub-Investigator: David McCollum, MD
Sub-Investigator: Scott Paulson, MD
Sub-Investigator: Scott Celinski, MD Less << |
|
NCT00210041
|
Genital Neoplasms, Male |
Phase 2 |
Completed |
- |
France
... More >> Centre Paul Papin
Angers, France
Institut Bergonie
Bordeaux, France
Centre François Baclesse
Caen, France
CHU Grenoble
Grenoble, France
Centre Léon Bérard
Lyon, France
Institut Paoli Calmette
Marseille, France
Institut Val d'aurelle
Montpellier, France
Institut Curie
Paris, France
Centre Eugène Marquis
Rennes, France
Centre Médico-Chirurgical Foch
Suresnes, France
Institut Claudius Regaud
Toulouse, France Less << |
|
NCT00084734
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Roswell Park Cancer Institute
Buffalo, New York, United States, 14263-0001 Less << |
|
NCT00823186
|
Cervical Cancer |
Phase 1 |
Unknown |
January 2013 |
Taiwan
... More >> Chang Gung Memorial Hospital
Tao Yuan, Lin kou, Taiwan, 333
Chang Gung Memory Hpspital
Chia Yi, Taiwan
Taipei Chang Gung Memory Hospital
Taipei, Taiwan Less << |
|
NCT00083122
|
- |
|
Completed |
- |
- |
|
NCT01939418
|
Metastatic Breast Cancer |
Phase 1
Phase 2 |
Terminated(slow recruitment) |
- |
Korea, Republic of
... More >>
National cancer center
Goyangsi, Gyeonggido, Korea, Republic of, 410-769 Less << |
|
NCT02119650
|
NSCLC (Non-small Cell Lung Car... More >>cinoma) Less << |
Phase 2 |
Terminated(The study was termi... More >>nated as other related studies of ruxolitinib did not provide sufficient efficacy to warrant continuation.) Less << |
- |
- |
|
NCT02119650
|
- |
|
Terminated(The study was termi... More >>nated as other related studies of ruxolitinib did not provide sufficient efficacy to warrant continuation.) Less << |
- |
- |
|
NCT00083122
|
Recurrent Ovarian Epithelial C... More >>ancer
Recurrent Primary Peritoneal Cavity Cancer
Stage IIIA Ovarian Epithelial Cancer
Stage IIIA Primary Peritoneal Cavity Cancer
Stage IIIB Ovarian Epithelial Cancer
Stage IIIB Primary Peritoneal Cavity Cancer
Stage IIIC Ovarian Epithelial Cancer
Stage IIIC Primary Peritoneal Cavity Cancer
Stage IV Ovarian Epithelial Cancer
Stage IV Primary Peritoneal Cavity Cancer Less << |
Phase 2 |
Completed |
- |
United States, Minnesota
... More >>
Mayo Clinic
Rochester, Minnesota, United States, 55905 Less << |
|
NCT00680940
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
India
... More >> Gujarat Cancer & Research Institute, New Civil Hospital Campus
Asarwa, Ahmedabad, India, 380013
Regional Cancer Center, Indira Gandhi Medical College
Shimla, Himachal Pradesh, India, 171001
MNJ Institute of Oncology, Regional Cancer Centre
Red Hills, Hyderabad, India, 500004
Malabar Institute of Medical Science (MIMS)
Calicut, Kerala, India, 673016
B.P. Poddar Hospital and Medical Research Ltd.
New Alipore, Kolkata, India, 700053
Choithram Hospital and Research Centre
Indore, Madhya Pradesh, India, 452014
Regional Cancer Centre, Indira Gandhi Institute of Medical Science
Sheikhpura, Patna, India, 800014
Patel Hospital Pvt. Ltd
Jalandhar, Punjab, India, 144001
Acharya Tulsi Regional Cancer Treatment & Research Institute
Bikaner, Rajasthan, India, 334003
V.N. Cancer Center, GKNM Hospital
Coimbatore, TamilNadu, India, 641037
Netaji Subash Chandra Bose Cancer Research Institute
Kolkata, West Bangal, India, 700016
Bankura Sammilani Medical College
Gobindnagar, West Bengal, India, 722101
Institute of Post Graduate Medical education & Research
Kolkata, West Bengal, India, 700020
Chittaranjan National Cancer Institute
Kolkata, West Bengal, India, 700026 Less << |
|
NCT00143455
|
Small Cell Lung Carcinoma |
Phase 3 |
Completed |
- |
- |
|
NCT00369122
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Carcinoma
Cervical Squamous Cell Carcinoma, Not Otherwise Specified
Stage IB Cervical Cancer AJCC v6 and v7
Stage IIA Cervical Cancer AJCC v7
Stage IIB Cervical Cancer AJCC v6 and v7
Stage III Cervical Cancer AJCC v6 and v7 Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01286766
|
Gastric Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Severance Hospital
Seoul, Korea, Republic of, 120-750 Less << |
|
NCT02241876
|
Head and Neck Neoplasms |
Phase 4 |
Unknown |
- |
Brazil
... More >> State University of Campinas - UNICAMP, Hospital das Clinicas
Not yet recruiting
Campinas, São Paulo, Brazil, 13083-888
Contact: Patricial Moriel 55 (19) 35218884 morielpa@fcm.unicamp.br
Contact: Marília B Visacri 55 (19) 35218884 mariberlofa@gmail.com
Principal Investigator: Marilia B Visacri Less << |
|
NCT02807181
|
Intrahepatic Cholangiocarcinom... More >>a Less << |
Phase 2
Phase 3 |
Recruiting |
June 2021 |
- |
|
NCT01536223
|
Chemoradiation
... More >> Nasopharyngeal Carcinoma Less << |
Phase 3 |
Unknown |
February 2015 |
China, Zhejiang
... More >>
Zhejiang Cancer Hospital
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: Xiaozhong Chen, MD 86-571-88122098 cxzfyun@sina.com
Contact: Weifeng Qin, MD 86-571-88122091 qinweifeng@yahoo.com.cn
Principal Investigator: Xiaozhong Chen, MD Less << |
|
NCT00143455
|
- |
|
Completed |
- |
- |
|
NCT00369122
|
- |
|
Completed |
- |
- |
|
NCT03535727
|
Adenocarcinoma
... More >> Pancreatic Neoplasms
Neoplasm, Glandular
Neoplasms
Neoplasms Pancreatic
Digestive System Neoplasm
Endocrine Gland Neoplasms
Digestive System Disease
Pancreatic Diseases
Endocrine System Diseases Less << |
Phase 1
Phase 2 |
Recruiting |
June 2021 |
United States, Maryland
... More >>
Sidney Kimmel Comprehensive Cancer Center
Recruiting
Baltimore, Maryland, United States, 21231
Contact: Susan Sartorius-Mergenthaler, RN 410-614-3644 Sartosu@jhmi.edu
Contact: Jane Zorzi, RN 410-614-5818 Jzorzi1@jhmi.edu Less << |
|
NCT02409342
|
Non-Squamous Non-Small Cell Lu... More >>ng Cancer, Squamous Non-Small Cell Lung Cancer Less << |
Phase 3 |
Active, not recruiting |
April 23, 2020 |
- |
|
NCT02535325
|
Non-Squamous Non-Small Cell Lu... More >>ng Carcinoma
Stage III Large Cell Lung Carcinoma AJCC v7
Stage III Lung Adenocarcinoma AJCC v7
Stage III Non-Small Cell Lung Cancer AJCC v7
Stage IIIA Large Cell Lung Carcinoma AJCC v7
Stage IIIA Lung Adenocarcinoma AJCC v7
Stage IIIA Non-Small Cell Lung Cancer AJCC v7
Stage IIIB Large Cell Lung Carcinoma AJCC v7
Stage IIIB Lung Adenocarcinoma AJCC v7
Stage IIIB Lung Non-Small Cell Cancer AJCC v7
Stage IV Large Cell Lung Carcinoma AJCC v7
Stage IV Lung Adenocarcinoma AJCC v7
Stage IV Non-Small Cell Lung Cancer AJCC v7 Less << |
Phase 1 |
Recruiting |
- |
United States, Ohio
... More >>
Case Western Reserve University
Recruiting
Cleveland, Ohio, United States, 44106
Contact: Tithi Biswas 800-641-2422
Principal Investigator: Tithi Biswas Less << |
|
NCT03069820
|
Liver Injury, Drug-Induced |
Phase 4 |
Recruiting |
January 1, 2018 |
China, Jiangxi
... More >> The Second Afiliated Hospital of Nanchang University
Recruiting
Nanchang, Jiangxi, China, 330006
Contact: Anwen Liu, MD 13767120022 awliu666@163.com Less << |
|
NCT03410615
|
Oropharyngeal Squamous Cell Ca... More >>rcinoma Less << |
Phase 2 |
Recruiting |
July 2024 |
Canada, Alberta
... More >>
Cross Cancer Institute
Recruiting
Edmonton, Alberta, Canada, T6G 1Z2
Contact: Neil Sun Chua 780 432-8340
Canada, Manitoba
CancerCare Manitoba
Recruiting
Winnipeg, Manitoba, Canada, R3E 0V9
Contact: Andrew Maksymiuk 204 787-2021
Canada, Nova Scotia
QEII Health Sciences Centre
Recruiting
Halifax, Nova Scotia, Canada, B3H 1V7
Contact: Stephanie Snow 902 473-3739
Canada, Ontario
Juravinski Cancer Centre at Hamilton Health Sciences
Recruiting
Hamilton, Ontario, Canada, L8V 5C2
Contact: Brandon M. Meyers 905 387-9495 ext 64604
Kingston Health Sciences Centre
Recruiting
Kingston, Ontario, Canada, K7L 2V7
Contact: Richard W. Gregg 613 549-6666 ext 4509
London Regional Cancer Program
Recruiting
London, Ontario, Canada, N6A 5W9
Contact: Eric W. Winquist 519 685-8261
Ottawa Hospital Research Institute
Recruiting
Ottawa, Ontario, Canada, K1H 8L6
Contact: John Hilton 613 737-7700 ext 70179
Health Sciences North
Recruiting
Sudbury, Ontario, Canada, P3E 5J1
Contact: Lacey Pitre 705 522-6237
University Health Network
Recruiting
Toronto, Ontario, Canada, M5G 2M9
Contact: Anna Spreafico 416 946-4501 ext 4012
Canada, Quebec
The Jewish General Hospital
Recruiting
Montreal, Quebec, Canada, H3T 1E2
Contact: Khalil Sultanem 514 340-8288
CHUQ-Pavillon Hotel-Dieu de Quebec
Not yet recruiting
Quebec City, Quebec, Canada, G1R 2J6
Contact: Andre Fortin 418 691-5264
Canada, Saskatchewan
Allan Blair Cancer Centre
Recruiting
Regina, Saskatchewan, Canada, S4T 7T1
Contact: Osama Souied 306 766-2691 Less << |
|
NCT00530634
|
Lung Cancer |
Phase 2 |
Terminated(Poor accrual) |
- |
- |
|
NCT01565772
|
Non-Small Cell Lung Cancer |
Phase 1 |
Terminated(Slow accrual) |
- |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02214 Less << |
|
NCT00907504
|
Carcinoma, Non-Small-Cell Lung |
Phase 3 |
Withdrawn |
May 2014 |
- |
|
NCT00530634
|
- |
|
Terminated(Poor accrual) |
- |
- |
|
NCT00521521
|
Head and Neck Neoplasms |
Phase 2 |
Completed |
- |
France
... More >> Sanofi-Aventis
Paris, France Less << |
|
NCT02185352
|
Breast Cancer
... More >> Brain Metastases Less << |
Phase 2 |
Recruiting |
July 2019 |
Taiwan
... More >> Kaohsiung Chang Gung Memorial Hospital
Recruiting
Kaohsiung, Taiwan
Contact: Kun-Ming Rau, MD +886-7-7317123 ext 8303 kmrau58@adm.cgmh.org.tw
Principal Investigator: Kun-Ming Rau, MD
Shuang Ho Hospital
Recruiting
New Taipei City, Taiwan
Contact: Tsu-Yi Chao, MD +886-2-22490088 ext 8402 10575@shh.org.tw
Principal Investigator: Tsu-Yi Chao, MD
China Medical University Hospital
Recruiting
Taichung, Taiwan
Contact: Chang-Fang Chiu, MD +886-4-22052121 ext 1921 d5686@mail.cmuh.org.tw
Principal Investigator: Chang-Fang Chiu, MD
National Taiwan University Hospital
Recruiting
Taipei, Taiwan, 100
Contact: Yen-Shen Lu, MD, PhD +886-2-23123456 ext 71673 yslu@ntu.edu.tw
Principal Investigator: Yen-Shen Lu, MD, PhD
Tri-Service General Hospital
Recruiting
Taipei, Taiwan, 114
Contact: Jyh-Cherng Yu, MD +886-2-89723311 ext 16020 doc20106@ndmctsgh.edu.tw
Contact: Ming-Shen Dai, MD, PhD +886-2-89723311 ext 12623 dms1201@gmail.com
Principal Investigator: Jyh-Cherng Yu, MD
Mackay Memorial Hospital
Active, not recruiting
Taipei, Taiwan
Taipei Veterans General Hospital
Recruiting
Taipei, Taiwan
Contact: Ling-Ming Tseng, MD +886-2-28757535 ext 134 lmtseng@vghtpe.gov.tw
Contact: Ta-Chung Chao, MD tcchao@vghtpe.gov.tw
Principal Investigator: Ling-Ming Tseng, MD
Chang Gung Memorial Hospital-LinKou
Recruiting
Taoyuan, Taiwan
Contact: Shin-Cheh Chen, MD +886-2-27135211 ext 3141 chensc@adm.cgmh.org.tw
Principal Investigator: Shin-Cheh Chen, MD Less << |
|
NCT02495493
|
Gastric Adenocarcinoma |
Phase 2 |
Active, not recruiting |
June 2019 |
Korea, Republic of
... More >>
Severance Hospital, Yonsei University Health System, Yonsei Cancer Center
Seoul, Korea, Republic of, 120-752 Less << |
|
NCT00942331
|
Bladder Urothelial Carcinoma
... More >> Distal Urethral Carcinoma
Infiltrating Bladder Urothelial Carcinoma Associated With Urethral Carcinoma
Metastatic Urothelial Carcinoma of the Renal Pelvis and Ureter
Proximal Urethral Carcinoma
Recurrent Bladder Carcinoma
Recurrent Prostate Carcinoma
Recurrent Urethral Carcinoma
Recurrent Urothelial Carcinoma of the Renal Pelvis and Ureter
Regional Urothelial Carcinoma of the Renal Pelvis and Ureter
Stage IV Bladder Cancer AJCC v7
Stage IV Prostate Cancer AJCC v7
Stage IV Urethral Cancer AJCC v7
Ureter Carcinoma Less << |
Phase 3 |
Active, not recruiting |
- |
- |
|
NCT01283217
|
Gastric Adenocarcinoma |
Phase 3 |
Unknown |
March 2016 |
Korea, Republic of
... More >>
Severance Hospital
Recruiting
Seoul, Korea, Republic of, 120-752
Contact: Sun-Young Rha, MD, Ph.D 82-2-2228-8050 rha7655@yuhs.ac Less << |
|
NCT02743923
|
Carcinoma, Non-Small Cell Lung |
Phase 3 |
Recruiting |
April 2020 |
Netherlands
... More >> VUmc Medical Center
Recruiting
Amsterdam, Noord-Holland, Netherlands, 1081HV
Contact: Idris Bahce, MD i.bahce@vumc.nl
Principal Investigator: Idris Bahce, MD
Medical spectrum Twente
Recruiting
Enschede, Overijssel, Netherlands, 7500 KA
Contact: Hugo Schouwink, MD J.Schouwink@mst.nl
Principal Investigator: Hugo Schouwink, MD
Meander Medical Center
Recruiting
Amersfoort, Utrecht, Netherlands, 3818 ES
Contact: Joop van den Brand, MD JJG.vanden.Brand@meandermc.nl
Principal Investigator: Joop van den Brand, MD
ZGT
Recruiting
Almelo, Netherlands, 7609 PP
Contact: Jeske Staal, MD j.staal@zgt.nl
Principal Investigator: Jeske Staal, MD
Antoni van Leeuwenhoek
Recruiting
Amsterdam, Netherlands, 1066CX
Contact: Egbert Smit e.smit@nki.nl
Principal Investigator: Egbert Smit, MD
OLVG
Not yet recruiting
Amsterdam, Netherlands, 1090 HM
Contact: A Moons, MD A.Moons-Pasic@olvg.nl
Principal Investigator: A Moons, MD
Gelre Ziekenhuis
Recruiting
Apeldoorn, Netherlands
Contact: Niels Pronk, MD n.pronk@gelre.nl
Principal Investigator: Niels Pronk, MD
Amphia Hospital
Recruiting
Breda, Netherlands
Contact: Nicolaas van Walree, MD nwalree@amphia.nl
Principal Investigator: Nicolaas van Walree, MD
Jeroen Bosch Hospital
Recruiting
Den Bosch, Netherlands
Contact: Bonne Biesma, MD b.biesma@jbz.nl
Principal Investigator: Bonne Biesma, MD
Haga
Recruiting
Den Haag, Netherlands, 2545 CH
Contact: Pepijn Brocken, MD p.brocken@hagaziekenhuis.nl
Principal Investigator: Pepijn Brocken, MD
Deventer Ziekenhuis
Recruiting
Deventer, Netherlands
Contact: Suzy Samii, MD s.samii@dz.nl
Principal Investigator: Suzy Samii, MD
Albert Schweitzer ziekenhuis
Recruiting
Dordrecht, Netherlands
Contact: Eric van Thiel, MD EricvanThiel@asz.nl
Principal Investigator: Eric van Thiel, MD
Ziekenhuis Gelderse Vallei
Recruiting
Ede, Netherlands
Contact: Wouter de Jong, MD jongw@zgv.nl
Principal Investigator: Wouter de Jong, MD
Maxima Medisch Centrum
Recruiting
Eindhoven, Netherlands, 5631 BM
Contact: Arjen van Henten, MD a.vanhenten@mmc.nl
Principal Investigator: Arjen van Henten, MD
Groene Hart
Recruiting
Gouda, Netherlands
Contact: ten Heuvel, MD alexandra.ten.heuvel@ghz.nl
Principal Investigator: ten Heuvel, MD
UMCG
Recruiting
Groningen, Netherlands, 9713 GZ
Contact: Harry Groen, MD h.j.m.groen@umcg.nl
Principal Investigator: Harry Groen, MD
Martini Ziekenhuis
Recruiting
Groningen, Netherlands
Contact: John van Putten, MD jwg.van.putten@mzh.nl
Principal Investigator: John van Putten, MD
Tergooi ziekenhuizen
Recruiting
Hilversum, Netherlands
Contact: Jan Maarten van Haarst, MD jvanhaarst@tergooi.nl
Principal Investigator: Jan Maarten van Haarst, MD
Spaarne Gasthuis
Recruiting
Hoofddorp, Netherlands, 2130 AT
Contact: Frans Krouwels, MD fkrouwels@spaarnegasthuis.nl
Principal Investigator: Frans Krouwels, MD
Medisch Centrum Leeuwarden
Recruiting
Leeuwarden, Netherlands
Contact: Venmans, MD
Principal Investigator: Venmans, MD
Maastricht University Medical Center
Recruiting
Maastricht, Netherlands
Contact: Anne-Marie Dingemans, MD a.dingemans@mumc.nl
Principal Investigator: Anne-Marie Dingemans, MD
Maasstad ziekenhuis
Recruiting
Rotterdam, Netherlands, 3007 AC
Contact: Susan van 't Westeinde, MD westeindes@maasstadziekenhuis.nl
Contact: , MD
Principal Investigator: Susan van 't Westeinde, MD
St. Fransicus Gasthuis
Recruiting
Rotterdam, Netherlands, 3045 PM
Contact: Wessel Hanselaar, MD w.hanselaar@franciscus.nl
Principal Investigator: Wessel Hanselaar, MD
Medical Center Haaglanden
Recruiting
the Hague, Netherlands
Contact: Klaar Maas, MD k.maas@mchaaglanden.nl
Principal Investigator: Klaar Maas, MD
Diakonessenhuis Utrecht
Recruiting
Utrecht, Netherlands, 3582 KE
Contact: Leontine van Elden, MD lvelden@diakhuis.nl
Principal Investigator: Leontine van Elden, MD
St. Antonius ziekenhuis
Recruiting
Utrecht, Netherlands
Contact: Franz Schramel, MD f.schramel@antoniusziekenhuis.nl
Principal Investigator: Franz Schramel, MD
VieCuri Medisch Centrum voor Noord-Limburg
Recruiting
Venlo, Netherlands
Contact: Vivian van Kampen, MD vvankampen@viecuri.nl
Principal Investigator: Vivian van Kampen, MD
Isala Klinieken
Recruiting
Zwolle, Netherlands, 8000 GK
Contact: Jos Stigt j.a.stigt@isala.nl
Principal Investigator: Jos Stigt, MD Less << |
|
NCT02997332
|
Head and Neck Cancers |
Phase 1 |
Recruiting |
December 2019 |
France
... More >> Gustave Roussy
Recruiting
Villejuif, Val de Marne, France, 94805
Contact: Caroline EVEN, MD 0142116537 ext +33 caroline.even@gustaveroussy.fr
Contact: Matthieu TEXIER 0142116309 ext +33 matthieu.texier@gustaveroussy.fr Less << |
|
NCT03046862
|
Biliary Tract Neoplasms |
Phase 2 |
Recruiting |
May 2020 |
Korea, Republic of
... More >>
Seoul National University Hospital
Recruiting
Seoul, Korea, Republic of, 110-744
Contact: Do-Youn Oh, MD, PhD 82-2-2072-0701 ohdoyoun@snu.ac.kr Less << |
|
NCT00424853
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Completed |
- |
Italy
... More >> Sanofi-Aventis Administrative Office
Milan, Italy Less << |
|
NCT01704690
|
Esophageal Cancer |
Phase 2
Phase 3 |
Terminated(The enrollment of t... More >>he study is much slower than expected.) Less << |
- |
China
... More >> Beijing Cancer Hospital, Peking University Cancer Hospital
Beijing, China, 100142 Less << |
|
NCT00668499
|
Mesothelioma |
Phase 1
Phase 2 |
Withdrawn(Sponsor withdrew sup... More >>port) Less << |
April 2011 |
United States, California
... More >>
USC/Norris Comprehensive Cancer Center
Los Angeles, California, United States, 90033 Less << |
|
NCT00629226
|
Head and Neck Cancer |
Phase 1 |
Completed |
- |
United States, Maryland
... More >>
Warren Grant Magnuson Clinical Center - NCI Clinical Trials Referral Office
Bethesda, Maryland, United States, 20892-1182
United States, Pennsylvania
UPMC Cancer Centers
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT00925743
|
Solid Cancer |
Phase 1 |
Completed |
- |
United States, California
... More >>
Investigational Site Number 840008
Los Angeles, California, United States, 90048
Investigational Site Number 840003
San Diego, California, United States, 92123
United States, Illinois
Investigational Site Number 840010
Decatur, Illinois, United States, 62526
United States, Maryland
Investigational Site Number 840002
Baltimore, Maryland, United States, 21201
United States, Missouri
Investigational Site Number 840006
St Louis, Missouri, United States, 63110
United States, Ohio
Investigational Site Number 840007
Cincinnati, Ohio, United States, 45267-0542
United States, Texas
Investigational Site Number 840005
San Antonio, Texas, United States, 78229 Less << |
|
NCT00832117
|
- |
|
Completed |
- |
- |
|
NCT02607592
|
Carcinoma,Non-Small-Cell Lung |
Phase 3 |
Recruiting |
August 2020 |
China, Guangdong
... More >>
Sun Yat-sen University of cancer center
Recruiting
Guangzhou, Guangdong, China, 510000
Contact: li-kun chen, doctor 13798019964 Less << |
|
NCT01642251
|
Extensive Stage Small Cell Lun... More >>g Carcinoma
Large Cell Lung Carcinoma
Neuroendocrine Carcinoma
Small Cell Carcinoma
Stage IV Non-Small Cell Lung Cancer AJCC v7 Less << |
Phase 1
Phase 2 |
Active, not recruiting |
- |
- |
|
NCT02137343
|
Gastric Cancer |
Phase 3 |
Terminated(All Amgen sponsored... More >> AMG102 clinical studies were terminated following a pre-planned Data Monitoring Committee safety review of study 20070622.) Less << |
- |
Japan
... More >> Research Site
Nagoya-shi, Aichi, Japan, 464-8681
Research Site
Chiba-shi, Chiba, Japan, 260-8717
Research Site
Kashiwa-shi, Chiba, Japan, 277-8577
Research Site
Matsuyama-shi, Ehime, Japan, 791-0280
Research Site
Fukuoka-shi, Fukuoka, Japan, 811-1395
Research Site
Sapporo-shi, Hokkaido, Japan, 060-8648
Research Site
Akashi-shi, Hyogo, Japan, 673-8558
Research Site
Kawasaki-shi, Kanagawa, Japan, 216-8511
Research Site
Osaka-shi, Osaka, Japan, 537-8511
Research Site
Osaka-shi, Osaka, Japan, 540-0006
Research Site
Osakasayama-shi, Osaka, Japan, 589-8511
Research Site
Suita-shi, Osaka, Japan, 565-0871
Research Site
Takatsuki-shi, Osaka, Japan, 569-8686
Research Site
Kitaadachi-gun, Saitama, Japan, 362-0806
Research Site
Suntou-gun, Shizuoka, Japan, 411-8777
Research Site
Utsunomiya-shi, Tochigi, Japan, 320-0834
Research Site
Bunkyo-ku, Tokyo, Japan, 113-8677
Korea, Republic of
Research Site
Goyang-si, Gyeonggi-do, Korea, Republic of, 410-769
Research Site
Hwasun, Korea, Republic of, 519-763
Research Site
Seoul, Korea, Republic of, 110-744
Research Site
Seoul, Korea, Republic of, 120-752
Research Site
Seoul, Korea, Republic of, 135-710
Research Site
Seoul, Korea, Republic of, 136-705
Research Site
Seoul, Korea, Republic of, 137-701
Research Site
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT00832117
|
Non Small Cell Lung Cancer |
Phase 1 |
Completed |
- |
United States, District of Col... More >>umbia
Georgetown University Medical Center
Washington, District of Columbia, United States, 20007
United States, New Jersey
The Cancer Institute Of New Jersey
New Brunswick, New Jersey, United States, 08901
United States, Pennsylvania
Penn State Milton S. Hershey Medical Center
Hershey, Pennsylvania, United States, 17033
Italy
Local Institution
Lucca, Italy, 55100
Local Institution
Meldola (Fc), Italy, 47014
Local Institution
Rimini, Italy, 47900
Local Institution
Viterbo, Italy, 01100 Less << |
|
NCT00428194
|
Cervical Cancer |
Phase 1 |
Withdrawn(Withdrawn due to lac... More >>k of accrual) Less << |
- |
United States, Minnesota
... More >>
University of Minnesota Cancer Center
Minneapolis, Minnesota, United States, 55455 Less << |
|
NCT00436293
|
Nasopharyngeal Neoplasms |
Phase 2 |
Completed |
- |
Hong Kong
... More >> Sanofi-Aventis
Hong Kong, Hong Kong Less << |
|
NCT00941070
|
- |
|
Completed |
- |
- |
|
NCT01281800
|
Mesothelioma |
Phase 2 |
Unknown |
- |
Slovenia
... More >> Institute of Oncology Ljubljana
Recruiting
Ljubljana, Slovenia, 1000
Contact: Viljem Kovac, MD +386 1 5879 504 vkovac@onko-i.si
Sub-Investigator: Matjaz Zwitter, MD, PhD
Principal Investigator: Viljem Kovac, MD Less << |
|
NCT01998529
|
Lung Cancer |
Phase 1 |
Recruiting |
August 2019 |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030 Less << |
|
NCT00118235
|
Extensive Stage Small Cell Lun... More >>g Cancer Less << |
Phase 2 |
Completed |
- |
United States, Illinois
... More >>
Cancer and Leukemia Group B
Chicago, Illinois, United States, 60606
United States, Rhode Island
Rhode Island Hospital
Providence, Rhode Island, United States, 02903 Less << |
|
NCT01911598
|
Head and Neck Cancer |
Phase 1 |
Completed |
- |
United States, Colorado
... More >>
University of Colorado Cancer Center Department of Hematology
Aurora, Colorado, United States, 80045
United States, Illinois
University of Chicago; Hematology/Oncology
Chicago, Illinois, United States, 60637
United States, Massachusetts
Massachusetts General Hospital;Hematology/ Oncology
Boston, Massachusetts, United States, 02114
Belgium
Cliniques Universitaires St-Luc
Bruxelles, Belgium, 1200
UZ Antwerpen
Edegem, Belgium, 2650
UZ Leuven Gasthuisberg
Leuven, Belgium, 3000 Less << |
|
NCT00941070
|
Recurrent Cervical Cancer
... More >> Recurrent Vaginal Cancer
Stage IB Cervical Cancer
Stage II Vaginal Cancer
Stage IIA Cervical Cancer
Stage IIB Cervical Cancer
Stage III Cervical Cancer
Stage III Vaginal Cancer
Stage IVA Cervical Cancer
Stage IVA Vaginal Cancer
Stage IVB Cervical Cancer
Stage IVB Vaginal Cancer
Therapy-related Toxicity Less << |
Phase 2 |
Completed |
- |
United States, Ohio
... More >>
Case Western Reserve University
Cleveland, Ohio, United States, 44106 Less << |
|
NCT02484677
|
Patients With Head and Neck Ca... More >>ncer (ORL) Less << |
Phase 4 |
Unknown |
- |
France
... More >> Assistance Publique Hôpitaux de Marseille
Recruiting
Marseille, France, 13354
Contact: Urielle DESALBRES, Director
Principal Investigator: Sebastien SALAS, Professor Less << |
|
NCT00735904
|
- |
|
Completed |
- |
- |
|
NCT03113188
|
Advanced Solid Tumors |
Phase 1 |
Recruiting |
March 2019 |
United States, Arizona
... More >>
HonorHealth
Recruiting
Scottsdale, Arizona, United States, 85258
Contact: Joyce Schaffer, RN 480-323-1339 clinicaltrials@honorhealth.com
Contact 877-273-3713
United States, Louisiana
Ochsner Clinic Foundation
Recruiting
New Orleans, Louisiana, United States, 70121
Contact: Marc R. Matrana, MD,MSc,FACP 504-703-2654 MaMatrana@ochsner.org
United States, Massachusetts
Dana Faber Cancer Institute
Recruiting
Boston, Massachusetts, United States, 02215
Contact: Solida Pruitt-Thompson, Sr. RC 617-632-6718 SOLIDA_THOMPSON@dfci.harvard.edu Less << |
|
NCT00483223
|
Breast Cancer |
Phase 2 |
Completed |
- |
United States, Alabama
... More >>
University of Alabama-Birmingham
Birmingham, Alabama, United States
United States, California
UCSF
San Francisco, California, United States
United States, District of Columbia
Georgetown - Lombardi Cancer Center
Washington, D.C., District of Columbia, United States
United States, Maryland
Johns Hopkins University Medical Center
Baltimore, Maryland, United States
United States, Massachusetts
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02115
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115
North Shore Medical Center
Peabody, Massachusetts, United States, 01960
United States, New York
Memorial Sloan Kettering Cancer Center
New York, New York, United States
United States, North Carolina
University of North Carolina
Chapel Hill, North Carolina, United States Less << |
|
NCT00173888
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Taiwan
... More >> Department of Oncology, National Taiwan University Hospital
Taipei, Taiwan Less << |
|
NCT00483223
|
- |
|
Completed |
- |
- |
|
NCT02024841
|
Gastric Cancer
... More >> Peritoneal Carcinomatosis Less << |
Phase 1 |
Completed |
- |
China, Hong Kong
... More >>
The University of Hong Kong
Hong Kong, Hong Kong, China Less << |
|
NCT01474642
|
Advanced or Recurrent Esophage... More >>al Squamous Cell Carcinoma Less << |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of Less << |
|
NCT00735904
|
Carcinoma, Non-Small-Cell Lung... More >> (NSCLC) Less << |
Phase 2 |
Completed |
- |
Poland
... More >> Pfizer Investigational Site
Torun, Poland, 87-100
Pfizer Investigational Site
Wodzislaw Sl., Poland, 44-300
Romania
Pfizer Investigational Site
Cluj-Napoca, Cluj, Romania, 400015
Pfizer Investigational Site
Bucuresti, Romania, 022328
Pfizer Investigational Site
Oradea, Romania, 410032
South Africa
Pfizer Investigational Site
Parktown, South Africa, 2193
Ukraine
Pfizer Investigational Site
Dnipropetrovsk, Ukraine, 49102
Pfizer Investigational Site
Donetsk, Ukraine, 83092
Pfizer Investigational Site
Kyiv, Ukraine, 04107
Pfizer Investigational Site
Lviv, Ukraine, 79031 Less << |
|
NCT01491139
|
Carcinoma, Squamous Cell |
Phase 1 |
Withdrawn(Study stopped due to... More >> issues surrounding development and formulation of olaparib) Less << |
- |
- |
|
NCT00148798
|
Non Small Cell Lung Cancer (NS... More >>CLC) Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00148798
|
- |
|
Completed |
- |
- |
|
NCT03410030
|
Pancreatic Cancer
... More >> Pancreas Cancer
Pancreatic Adenocarcinoma Resectable
Pancreatic Ductal Adenocarcinoma
Pancreas Metastases Less << |
Phase 1
Phase 2 |
Recruiting |
December 15, 2019 |
United States, Arizona
... More >>
HonorHealth Research Institute
Recruiting
Scottsdale, Arizona, United States, 85258
Contact: Joyce Schaffer, MSN, AOCNS 480-323-1339 ext option #2 Joyce.Schaffer@HonorHealth.com
Principal Investigator: Gayle S Jameson, RN, MSN, ACNP-BC, AOCN
United States, California
University of California San Diego Moores Cancer Center
Not yet recruiting
San Diego, California, United States, 92093
Contact: Julie Lewis 858-822-4907 jml111@ucsd.edu
Principal Investigator: Hitendra Patel, MD
United States, Georgia
Piedmont Cancer Institute, PC
Not yet recruiting
Atlanta, Georgia, United States, 30318
Contact: Derrick Carter 678-298-3238 dcarter@piedmontcancerinstitute.com
Principal Investigator: Vasileios Assikis, MD
Piedmont Cancer Institute, PC
Not yet recruiting
Fayetteville, Georgia, United States, 30214
Contact: Derrick Carter 678-298-3238 dcarter@piedmontcancerinstitute.com
Principal Investigator: Vasileios Assikis, MD
Piedmont Cancer Institute, PC
Not yet recruiting
Newnan, Georgia, United States, 30265
Contact: Derrick Carter 678-298-3238 dcarter@piedmontcancerinstitute.com
Principal Investigator: Vasileios Assikis, MD Less << |
|
NCT01194453
|
Non-small Cell Lung Cancer
... More >> Efficacy Less << |
Phase 2 |
Completed |
- |
China, Guangdong
... More >>
Cancer Center of Sun Yat-Sen University (CCSU)
Guangzhou, Guangdong, China Less << |
|
NCT01194453
|
- |
|
Completed |
- |
- |
|
NCT02365766
|
Urothelial Carcinoma
... More >> Bladder Cancer Less << |
Phase 1
Phase 2 |
Recruiting |
December 2018 |
United States, Indiana
... More >>
Indiana University Melvin and Bren Simon Cancer Center
Recruiting
Indianapolis, Indiana, United States, 46202
Contact: Costantine Albany, M.D. 317-278-1711 calbany@iu.edu
Contact: Marietta Moore 317-274-7477 marlmoor@iupui.edu
IU Health Central Indiana Cancer Center
Recruiting
Indianapolis, Indiana, United States, 46219
Contact: Deborah McCullough 317-948-1921 dmccullough2@iuhealth.org
Principal Investigator: Andrew Greenspan, MD
Community Regional Cancer Care
Recruiting
Indianapolis, Indiana, United States, 46256
Contact: Kelly Verel 317-497-2836 KVerel@ecommunity.com
Principal Investigator: Radhika Walling, MD
St. Vincent Hospital
Recruiting
Indianapolis, Indiana, United States, 46260
Contact: Deborah Estes 317-338-6594 deborah.estes@ascension.org
Principal Investigator: Niraj Gupta, MD
United States, Missouri
Washington University: Siteman Cancer Center
Recruiting
Saint Louis, Missouri, United States, 63110
Contact: Joel Picus, M.D. 314-362-5740 jpicus@dom.wustl.edu
Contact: Amber Smith, R.N. 314-508-7249 asmith1@wustl.edu
United States, New Mexico
University of New Mexico Cancer Center
Recruiting
Albuquerque, New Mexico, United States, 87106
Contact: Mollie Geske 505-925-0377 mgeske@nmcca.org
Principal Investigator: Richard Lauer, MD
United States, Ohio
University Hospitals Seidman Cancer Center
Recruiting
Cleveland, Ohio, United States, 44106
Contact: Christopher Hoimes, D.O. 216-844-3951 cjh122@case.edu
Contact: Cheryl Eitman 216-844-5393 cheryl.eitman2@uhhospitals.org
United States, Pennsylvania
Thomas Jefferson University: Kimmel Cancer Center
Recruiting
Philadelphia, Pennsylvania, United States, 19107
Contact: Jean Hoffman-Censits, M.D. 215-955-8874 jean.hoffman-censits@jefferson.edu
Contact: Monica Byrnes 215.503.0552 monica.byrnes@jefferson.edu
United States, Virginia
Virginia Oncology Associates
Recruiting
Norfolk, Virginia, United States, 23502
Contact: Mark Fleming, M.D. 757-213-5813 mark.fleming@usoncology.com
Contact: Wendi Gobhart 757.213.5813 wendi.gobhart@usoncology.com Less << |
|
NCT00006942
|
Fallopian Tube Cancer
... More >> Primary Peritoneal Cavity Cancer
Recurrent Ovarian Epithelial Cancer
Stage III Ovarian Epithelial Cancer
Stage IV Ovarian Epithelial Cancer Less << |
Phase 2 |
Completed |
- |
United States, California
... More >>
City of Hope
Duarte, California, United States, 91010 Less << |
|
NCT01000480
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
France
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Le Mans, France, 72000
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Lyon, France, 69373
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Montpellier, France, 34070
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Paris, France, 75015
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Toulouse, France, 31300
Germany
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Berlin, Germany, 14165
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hemer, Germany, 58675
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Köln, Germany, 51109
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Lübeck, Germany, 23538
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Nürnberg, Germany, 90419
Italy
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Avellino, Italy, 50019
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Firenze, Italy, 50139
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Milano, Italy, 20132
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Monza, Italy, 20900
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Orbassano, Italy, 10043
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Perugia, Italy, 06156
Spain
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Barcelona, Spain, 08036
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Madrid, Spain, 28034
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Sabadell, Spain, 08208
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Sevilla, Spain, 41013
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Valencia, Spain, 46010 Less << |
|
NCT00799240
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
United States, Kansas
... More >>
Hutchinson Clinic, PA
Hutchinson, Kansas, United States, 67502
Kansas University Cancer Center
Kansas City, Kansas, United States, 66160
Stormont Vail Healthcare
Topeka, Kansas, United States, 66606
United States, Missouri
VA Medical Center
Kansas City, Missouri, United States, 64128 Less << |
|
NCT00318890
|
Cancer of Head and Neck
... More >> Head Cancer
Head and Neck Cancer
Neck Cancer
Neck Neoplasms Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, Alabama
... More >>
University of Alabama at Birmingham
Birmingham, Alabama, United States, 35294 Less << |
|
NCT00603408
|
- |
|
Terminated(Study was discontin... More >>ued due to lack of accrual.) Less << |
- |
- |
|
NCT00603408
|
Breast Cancer |
Phase 2 |
Terminated(Study was discontin... More >>ued due to lack of accrual.) Less << |
- |
United States, Missouri
... More >>
Washington University School of Medicine
St. Louis, Missouri, United States, 63110 Less << |
|
NCT00814086
|
Chemotherapeutic Agent Toxicit... More >>y
Endometrial Adenocarcinoma
Fallopian Tube Carcinoma
Gastrointestinal Complication
Malignant Ovarian Mixed Epithelial Tumor
Neurotoxicity Syndrome
Ovarian Brenner Tumor
Ovarian Clear Cell Cystadenocarcinoma
Ovarian Mucinous Cystadenocarcinoma
Ovarian Serous Cystadenocarcinoma
Primary Peritoneal Carcinoma
Stage II Ovarian Cancer
Stage III Ovarian Cancer
Stage IV Ovarian Cancer
Undifferentiated Ovarian Carcinoma Less << |
Phase 1 |
Completed |
- |
United States, Illinois
... More >>
University of Chicago Comprehensive Cancer Center
Chicago, Illinois, United States, 60637
United States, Iowa
University of Iowa Hospitals and Clinics
Iowa City, Iowa, United States, 52242
United States, New Jersey
Cooper Hospital University Medical Center
Camden, New Jersey, United States, 08103
United States, Ohio
Riverside Methodist Hospital
Columbus, Ohio, United States, 43214
United States, Oklahoma
Tulsa Cancer Institute
Tulsa, Oklahoma, United States, 74146
United States, Rhode Island
Women and Infants Hospital
Providence, Rhode Island, United States, 02905
United States, Wisconsin
University of Wisconsin Hospital and Clinics
Madison, Wisconsin, United States, 53792 Less << |
|
NCT00301808
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, Michigan
... More >>
Barbara Ann Karmanos Cancer Institute
Detroit, Michigan, United States, 48201-1379
Veterans Affairs Medical Center - Detroit
Detroit, Michigan, United States, 48201 Less << |
|
NCT00565370
|
Advanced Gastric Cancer |
Phase 1
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT00301808
|
- |
|
Completed |
- |
- |
|
NCT02657434
|
Non-Small Cell Lung Cancer |
Phase 3 |
Recruiting |
November 30, 2019 |
- |
|
NCT00496275
|
Non Small Cell Lung Cancer |
Phase 1 |
Completed |
- |
- |
|
NCT02035527
|
Metastatic Squamous Neck Cance... More >>r With Occult Primary Squamous Cell Carcinoma
Recurrent Metastatic Squamous Neck Cancer With Occult Primary
Recurrent Salivary Gland Cancer
Recurrent Squamous Cell Carcinoma of the Hypopharynx
Recurrent Squamous Cell Carcinoma of the Larynx
Recurrent Squamous Cell Carcinoma of the Lip and Oral Cavity
Recurrent Squamous Cell Carcinoma of the Nasopharynx
Recurrent Squamous Cell Carcinoma of the Oropharynx
Recurrent Squamous Cell Carcinoma of the Paranasal Sinus and Nasal Cavity
Recurrent Verrucous Carcinoma of the Larynx
Recurrent Verrucous Carcinoma of the Oral Cavity
Salivary Gland Squamous Cell Carcinoma
Stage IV Squamous Cell Carcinoma of the Hypopharynx
Stage IV Squamous Cell Carcinoma of the Nasopharynx
Stage IVA Salivary Gland Cancer
Stage IVA Squamous Cell Carcinoma of the Larynx
Stage IVA Oral Cavity Squamous Cell Carcinoma
Stage IVA Squamous Cell Carcinoma of the Oropharynx
Stage IVA Squamous Cell Carcinoma of the Paranasal Sinus and Nasal Cavity
Stage IVA Verrucous Carcinoma of the Larynx
Stage IVA Verrucous Carcinoma of the Oral Cavity
Stage IVB Salivary Gland Cancer
Stage IVB Squamous Cell Carcinoma of the Larynx
Stage IVB Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage IVB Squamous Cell Carcinoma of the Oropharynx
Stage IVB Squamous Cell Carcinoma of the Paranasal Sinus and Nasal Cavity
Stage IVB Verrucous Carcinoma of the Larynx
Stage IVB Verrucous Carcinoma of the Oral Cavity
Stage IVC Salivary Gland Cancer
Stage IVC Squamous Cell Carcinoma of the Larynx
Stage IVC Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage IVC Squamous Cell Carcinoma of the Oropharynx
Stage IVC Squamous Cell Carcinoma of the Paranasal Sinus and Nasal Cavity
Stage IVC Verrucous Carcinoma of the Larynx
Stage IVC Verrucous Carcinoma of the Oral Cavity
Tongue Cancer
Untreated Metastatic Squamous Neck Cancer With Occult Primary Less << |
Phase 1 |
Completed |
- |
United States, Ohio
... More >>
Arthur G. James Cancer Hospital and Solove Research Institute at Ohio State University Medical Center
Columbus, Ohio, United States, 43210 Less << |
|
NCT00998166
|
Non-Small Cell Lung Cancer |
Phase 2 |
Terminated(Poor enrollment) |
- |
United States, New York
... More >>
North Shore University Hospital
Lake Success, New York, United States, 11042
Columbia University Medical Center
New York, New York, United States, 10032 Less << |
|
NCT02567409
|
Metastatic Urothelial Carcinom... More >>a of the Renal Pelvis and Ureter
Stage IV Bladder Urothelial Carcinoma AJCC v7 Less << |
Phase 2 |
Recruiting |
- |
- |
|
NCT00565370
|
- |
|
Completed |
- |
- |
|
NCT01450696
|
Gastric Cancer |
Phase 3 |
Terminated(futility following ... More >>planned interim analysis) Less << |
- |
- |
|
NCT01000480
|
- |
|
Completed |
- |
- |
|
NCT01450696
|
- |
|
Terminated(futility following ... More >>planned interim analysis) Less << |
- |
- |
|
NCT02508246
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 1 |
Active, not recruiting |
- |
United States, Washington
... More >>
Fred Hutch/University of Washington Cancer Consortium
Seattle, Washington, United States, 98109 Less << |
|
NCT01369641
|
Cancer |
Not Applicable |
Terminated(Poor accrual) |
- |
United States, Pennsylvania
... More >>
Thomas Jefferson University
Philadelphia, Pennsylvania, United States, 19107 Less << |
|
NCT01369641
|
- |
|
Terminated(Poor accrual) |
- |
- |
|
NCT00012194
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Terminated |
- |
United States, New Hampshire
... More >>
Dartmouth Hitchcock Medical Center
Lebanon, New Hampshire, United States, 03756 Less << |
|
NCT01824459
|
Gastric Cancer |
Phase 3 |
Recruiting |
September 1, 2018 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Ruihua Xu, phD 86-20-87342479 xurh@sysucc.org.cn Less << |
|
NCT00565448
|
Nasopharyngeal Neoplasms
... More >> Carcinoma Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00565448
|
- |
|
Completed |
- |
- |
|
NCT01379807
|
Gastric Adenocarcinoma
... More >> Gastroesophageal Junction Adenocarcinoma Less << |
Phase 2 |
Unknown |
December 2015 |
Spain
... More >> Hospital Arquitecto Mercide
Ferrol, La Coruña, Spain, 15405
Complejo Hospitalario Universitario de Santiago (CHUS)
Santiago de Compostela, La Coruña, Spain, 15706
Complejo Hospitalario Universitario de Vigo (Xeral Cies)
Vigo, Pontevedra, Spain, 36204
Policlínica de Vigo S.A.
Vigo, Pontevedra, Spain, 36211
Complejo Hospitalario Universitario de A Coruña
La Coruña, Spain, 15006
Centro Oncológico de Galícia
La Coruña, Spain, 15009
Hospital Universitario Lucus Augusti
Lugo, Spain, 27003
Complejo Hospitalario Ourense
Ourense, Spain, 32005
Hospital de Pontevedra
Pontevedra, Spain, 36002 Less << |
|
NCT00320515
|
Neoplasm, Gastric |
Phase 1
Phase 2 |
Completed |
- |
Argentina
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Buenos Aires, Argentina, 1264
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Tucumain, Argentina, 4000
Korea, Republic of
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Seoul, Korea, Republic of, 136-705
Mexico
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Ciudad Obregon, Mexico, 85100
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Guadalajara, Mexico, 44280
Taiwan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Tainan, Taiwan, 704
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Taipei, Taiwan, 112
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Tao-Yuan, Taiwan, 333 Less << |
|
NCT01031446
|
- |
|
Completed |
- |
- |
|
NCT03574818
|
Squamous Cell Lung Cancer |
Phase 2 |
Recruiting |
February 26, 2022 |
United States, New York
... More >>
Montefiore Medical Center
Recruiting
Bronx, New York, United States, 10461
Contact: Mariano Bazan, BS 718-862-8840 ext 436 mbazan@montefiore.org
Contact: Anna L Araiza, MPH 718-3796866 aaraiza@montefiore.org Less << |
|
NCT00242749
|
Cancer of Head and Neck |
Phase 2 |
Completed |
- |
Spain
... More >> Research Site
Badalona, Spain
Research Site
Murcia, Spain
Research Site
Málaga, Spain
Research Site
Santander, Spain Less << |
|
NCT01096199
|
Cancer |
Phase 1 |
Terminated |
- |
Singapore
... More >> National University Hospital
Singapore, Singapore, 119074 Less << |
|
NCT00320515
|
- |
|
Completed |
- |
- |
|
NCT02121990
|
Ovarian Cancer
... More >> Primary Peritoneal Cancer
Fallopian Tube Cancer Less << |
Phase 1 |
Active, not recruiting |
April 2019 |
United States, New Jersey
... More >>
Memorial Sloan Kettering Cancer Center at Basking Ridge
Basking Ridge, New Jersey, United States, 07920
United States, New York
Memorial Sloan Kettering Cancer Center @ Suffolk
Commack, New York, United States, 11725
Memorial Sloan Kettering Westchester
Harrison, New York, United States, 10604
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan Kettering Cancer Center at Mercy Medical Center
Rockville Centre, New York, United States, 11570 Less << |
|
NCT01031446
|
Breast Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, Tennessee
... More >>
Erlanger Cancer Center at Erlanger Hospital - Baroness
Chattanooga, Tennessee, United States, 37403
West Tennessee Cancer Center at Jackson-Madison County General Hospital
Jackson, Tennessee, United States, 38301
Baptist Regional Cancer Center at Baptist Riverside
Knoxville, Tennessee, United States, 37901
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States, 37232-6838 Less << |
|
NCT00475657
|
Small Cell Lung Cancer |
Phase 2 |
Terminated(Terminated due to l... More >>ack of efficacy) Less << |
- |
Spain
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Ferrol, Spain, 15405
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
La Coruña, Spain, 15002
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Lugo, Spain, 27004
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Ourense, Spain, 15009
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Santiago de Compostela, Spain, 15706
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Vigo, Spain, 36211 Less << |
|
NCT01073683
|
Larynx Cancer |
Phase 2 |
Unknown |
December 2014 |
Israel
... More >> Rabin MC
Not yet recruiting
Petach Tikva, Israel, 49100
Contact: Aron Popovtzer, MD 039378004 aronp@clalit.org.il
Contact: Salomon Stemmer, MD 039378023 sstemmer@clalit.org.il
Principal Investigator: Aron Popovtzer, MD Less << |
|
NCT00759226
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
Germany
... More >> Charite Universitätsmedizin Berlin
Berlin, Germany, 13353 Less << |
|
NCT01015664
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 1
Phase 2 |
Terminated |
- |
United States, Tennessee
... More >>
Boston Baskin Caner Foundation
Memphis, Tennessee, United States, 38138 Less << |
|
NCT00116896
|
Advanced Solid Tumors |
Phase 1 |
Completed |
- |
United States, Tennessee
... More >>
Vanderbilt Universtiy Medical Center
Nashville, Tennessee, United States, 37232-6307
United States, Texas
Institute for Drug DevelopmentCancer Therapy & Research Center
San Antonio, Texas, United States, 78229 Less << |
|
NCT00072332
|
Esophageal Cancer
... More >> Gastric Cancer
Unspecified Adult Solid Tumor, Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT01374789
|
Urinary Bladder Cancer |
Phase 2 |
Terminated(The study was termi... More >>nated due to insufficient recruitment.) Less << |
- |
Germany
... More >> Bundeswehrkrankenhaus Berlin
Berlin, Germany, 10115
Krankenhaus am Urban
Berlin, Germany, 10967
St.-Josefs-Hospitals Dortmund
Dortmund, Germany, 44263
Universitätsklinikum Dresden
Dresden, Germany, 01307
Universitätsklinikum Düsseldorf
Düsseldorf, Germany, 40225
Universitätsklinikum Erlangen
Erlangen, Germany, 91054
Klinikum Fulda
Fulda, Germany, 36043
Universitätsklinikum Hamburg Eppendorf
Hamburg, Germany, 20246
Medizinische Hochschule Hannover, Urologie
Hannover, Germany, 30625
Universitätskllinikum Heidelberg
Heidelberg, Germany, 69121
Klinikum Kassel
Kassel, Germany, 34215
Heilig-Geist-Krankenhaus
Köln, Germany, 50737
Klinikum Ludwigshafen
Ludwigshafen, Germany, 67063
Universitätsklinikum Mainz
Mainz, Germany, 55131
Uroloische Praxis
Markkleeberg, Germany, 04416
Universitätsklinikum Münster
Münster, Germany, 48149
Johanniter Krankenhaus
Stendal, Germany, 39576
Universitätsklinikum Ulm, Urologische Klinik
Ulm, Germany, 89075
Klinikum Weiden
Weiden, Germany, 92637
Gemeinschaftspraxis für Urologie DGU
Wuppertal, Germany, 742103 Less << |
|
NCT00415324
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, California
... More >>
City of Hope Medical Center
Duarte, California, United States, 91010
City of Hope
Duarte, California, United States, 91010
University of Southern California/Norris Cancer Center
Los Angeles, California, United States, 90033
University of California at Davis Cancer Center
Sacramento, California, United States, 95817 Less << |
|
NCT01487915
|
Advanced Urothelial Carcinoma |
Phase 2 |
Unknown |
- |
Korea, Republic of
... More >>
Keimyeong University Dongsan Medical Center
Recruiting
Daegu, Korea, Republic of, 700-712
Contact: Hong Seok Song, MD, PhD.
Contact: Jin Young Kim, MD
Chungnam University Hospital
Recruiting
Daejeon, Korea, Republic of, 301-721
Contact: Hyo Jin Lee, MD, PhD.
Korea University Anam Hospital
Recruiting
Seoul, Korea, Republic of, 136-705
Contact: Kyeong Hwa Park, MD, PhD
Asan Medical Center
Recruiting
Seoul, Korea, Republic of, 138-736
Contact: Jeong-Mi Ko, MSc crnonc8@amc.seoul.kr
Principal Investigator: Jae-Lyun Lee, MD, PhD.
Sub-Investigator: Jin-Hee Ahn, MD, PhD.
Sub-Investigator: Hanjong Ahn, MD, PhD.
Sub-Investigator: Jun Hyuk Hong, MD, PhD.
Sub-Investigator: Cheryn Song, MD, PhD.
Sub-Investigator: Cheong Soo Kim, MD, PhD.
Chung Ang University Hospital
Not yet recruiting
Seoul, Korea, Republic of, 156-755
Contact: Hee Joon Kim, MD Less << |
|
NCT00595972
|
Gastric Cancer |
Phase 2 |
Completed |
- |
China, Shanghai
... More >>
Fudan University Cancer Hospital
ShangHai, Shanghai, China, 200032 Less << |
|
NCT00449033
|
Carcinoma, Non-Small-Cell Lung |
Phase 3 |
Completed |
- |
- |
|
NCT03707509
|
Nasopharyngeal Carcinoma |
Phase 3 |
Not yet recruiting |
November 1, 2021 |
- |
|
NCT00449033
|
- |
|
Completed |
- |
- |
|
NCT02414685
|
Germ Cell Tumor |
Phase 2 |
Recruiting |
December 2019 |
Slovakia
... More >> National Cancer Institute
Recruiting
Bratislava, Slovakia, 83310
Contact: Michal Mego, Assoc. Prof +421259378 ext 366 michal.mego@nou.sk
Contact: Jozef Mardiak, Prof +421259378 ext 108 jozef.mardiak@nou.sk
Principal Investigator: Jozef Mardiak, Prof
Principal Investigator: Michal Mego, Assoc. Prof Less << |
|
NCT03732677
|
Muscle Invasive Bladder Cancer |
Phase 3 |
Recruiting |
December 18, 2025 |
- |
|
NCT01139775
|
Non Small Cell Lung Cancer |
Phase 1
Phase 2 |
Completed |
- |
Germany
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mannheim, Baden-Wurttemberg, Germany, 68167
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Munster, Nordhein-Westfalen, Germany, 48149
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Coswig, Sachsen, Germany, 01640
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Berlin, Germany, 14165
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Frankfurt, Germany, 60596
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hannover, Germany, 30625
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Heidelberg, Germany, 69126
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Homburg, Germany, 66421
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Immenhausen, Germany, 34376
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Lübeck, Germany, 23538
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mainz, Germany, D-55131
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Rheine, Germany, 48431
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Ulm, Germany, 89081
Spain
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Oviedo, Asturias, Spain, 33006
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mataro, Barcelona, Spain, 08304
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Pozuelo de Alarcon, Madrid, Spain, 28223
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Barcelona, Spain, 08035
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Barcelona, Spain, 08036
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Barcelona, Spain, 08908
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Girona, Spain, 17007
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Madrid, Spain, 28034
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Madrid, Spain, 28046
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Madrid, Spain, 28050
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Sevilla, Spain, 41013 Less << |
|
NCT00458913
|
Malignant Mesothelioma |
Phase 2 |
Completed |
- |
United Kingdom
... More >> Royal Marsden - Surrey
Sutton, England, United Kingdom, SM2 5PT Less << |
|
NCT01139775
|
- |
|
Completed |
- |
- |
|
NCT00192036
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Belgium
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Aalst, Belgium, 9300
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Charleroi, Belgium, 6000
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Gilly, Belgium, 6060
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Haine-St.- Paul, Belgium, 7100
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Herentals, Belgium, 2200
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mechelen, Belgium, 2800
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Ottignies, Belgium, 1340
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Turnhout, Belgium, 2300 Less << |
|
NCT00192036
|
- |
|
Completed |
- |
- |
|
NCT00475657
|
- |
|
Terminated(Terminated due to l... More >>ack of efficacy) Less << |
- |
- |
|
NCT00323830
|
Gastric Cancer
... More >> Surgery Less << |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of Less << |
|
NCT03382561
|
Extensive Stage Lung Small Cel... More >>l Carcinoma
Recurrent Small Cell Lung Carcinoma Less << |
Phase 2 |
Active, not recruiting |
June 2, 2020 |
- |
|
NCT02201355
|
Squamous Cell Carcinoma of the... More >> Head and Neck T1/2N0-2 Less << |
Phase 1 |
Active, not recruiting |
February 2021 |
United States, Texas
... More >>
University of Texas at Southwestern Medical Center
Dallas, Texas, United States, 75390 Less << |
|
NCT00448643
|
Endometrial Cancer |
Phase 1 |
Completed |
- |
United States, Florida
... More >>
University of Miami Sylvester Comprehensive Cancer Center - Miami
Miami, Florida, United States, 33136 Less << |
|
NCT02677727
|
- |
|
Active, not recruiting |
July 26, 2019 |
United States, Indiana
... More >>
Indiana University Health Hospital
Indianapolis, Indiana, United States, 46202
Indiana University Health Melvin and Bren Simon Cancer Center
Indianapolis, Indiana, United States, 46202
United States, Minnesota
Mayo Clinic Department of Medical Oncology
Rochester, Minnesota, United States, 55905 Less << |
|
NCT02639650
|
Gestational Trophoblastic Neop... More >>lasms Less << |
Phase 3 |
Recruiting |
March 2021 |
China, Zhejiang
... More >>
Weiguo Lv
Recruiting
Hangzhou, Zhejiang, China
Contact: Weiguo Lv, Doctor Less << |
|
NCT02395705
|
Esophageal Neoplasm |
Phase 3 |
Recruiting |
July 2022 |
China, Beijing
... More >> Cancer Institute and Hospital, Chinese Academy of Medical Sciences
Recruiting
Beijing, Beijing, China, 100029
Contact: Yousheng Mao, MD. maoysherx@qq.com
Principal Investigator: Yousheng Mao, MD.
Beijing Cancer Hospital
Recruiting
Beijing, Beijing, China, 100142
Contact: Keneng Chen, MD. chenkeneng@bjmu.edu.cn
Principal Investigator: Keneng Chen, MD.
China, Fujian
Fujian Medical University Union Hospital
Recruiting
FuZhou, Fujian, China
Contact: Chun Chen, MD chenchun0209@163.com
Principal Investigator: Chun Chen, MD
China, Guangdong
Sun Yat-sen Uniersity Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Peng Lin, MD & PhD linpeng@sysucc.org.cn
Principal Investigator: Peng Lin, MD & PhD
China, Henan
Anyang cancer hospital
Recruiting
Anyang, Henan, China
Contact: Xiaobing Li, MD
Principal Investigator: Xiaobing Li, MD
Henan Cancer Hospital/The affiliated Cancer Hospital of ZhengZhou university
Recruiting
ZhengZhou, Henan, China, 450008
Contact: Yin Li, M.D.& Ph.D. 8613903838752 liyin825@aliyun.com
Contact: Yan Zheng, M.D.& Ph.D. 8615713660065 sunnyzheng1@126.com
Principal Investigator: Yin Li, M.D.& Ph.D
China, Hunan
Hunan Province Tumor Hospital
Recruiting
Changsha, Hunan, China, 410013
Contact: Gaoming Xiao, MD. xiaogaoming617@163.com
Principal Investigator: Gaoming Xiao
China, Shanghai
Fudan Universitay Shanghai Cancer Center
Recruiting
Shanghai, Shanghai, China
Contact: Jiaqing Xiang, MD j.q.xiang@hotmail.com
Principal Investigator: Jiaqing Xiang, MD
China, Tianjin
Tianjin Medical University Cancer Institute and Hospital
Recruiting
Tianjin, Tianjin, China, 300060
Contact: Zhentao Yu, MD. yuzhtao@hotmail.com
Principal Investigator: Zhentao Yu, MD. Less << |
|
NCT02723838
|
Muscle-invasive Transitional C... More >>ell Carcinoma of the Bladder Less << |
Phase 1 |
Withdrawn(Company has repriori... More >>tized clinical plans to focus on later-stage studies.) Less << |
February 28, 2018 |
- |
|
NCT02985203
|
Lung Neoplasm Malignant |
Phase 2 |
Recruiting |
November 2018 |
China, Guangdong
... More >>
Guangdong General Hospital
Recruiting
Guangzhou, Guangdong, China, 510080
Contact: ZHEN WANG, Dr +8683827812 ext 50811 hunterol@163.com Less << |
|
NCT03143153
|
Various Advanced Cancer |
Phase 3 |
Recruiting |
December 30, 2021 |
- |
|
NCT00772863
|
Ovarian Neoplasms |
Phase 2 |
Completed |
- |
Turkey
... More >> Sanofi aventis administrative office
Istanbul, Turkey Less << |
|
NCT00743964
|
Advanced Gastric Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of, 135 710 Less << |
|
NCT00006389
|
Stage III Gastric Cancer
... More >> Stage IV Gastric Cancer Less << |
Phase 2 |
Completed |
- |
United States, California
... More >>
University of Southern California
Los Angeles, California, United States, 90033-0804 Less << |
|
NCT02544243
|
Metastatic Breast Cancer |
Phase 2 |
Unknown |
- |
- |
|
NCT01089088
|
Bladder Cancer
... More >> Transitional Cell Cancer of the Renal Pelvis and Ureter
Urethral Cancer Less << |
Phase 2 |
Completed |
- |
United Kingdom
... More >> Bristol Haematology and Oncology Centre
Bristol, Avon, United Kingdom, BS2 8ED
Castle Hill Hospital
Cottingham, East Yorkshire, United Kingdom, HU16 5JQ
Royal Bournemouth Hospital
Bournemouth, England, United Kingdom, BH7 7DW
Churchill Hospital
Oxford, Oxfordshire, United Kingdom, OX3 7LJ
Royal Shrewsbury Hospital
Shrewsbury, Shropshire, United Kingdom, SY3 8XQ
The Royal Marsden Hospitals (Surrey)
Sutton, Surrey, United Kingdom, Surrey
St James's University Hospital
Leeds, Yorkshire, United Kingdom, LS9 7TF
Addenbrooke's Hospital
Cambridge, United Kingdom, CB2 0QQ
Velindre Hospital
Cardiff, United Kingdom, CF14 2TL
Beatson West of Scotland Cancer Centre
Glasgow, United Kingdom, G12 0YN
St Bartholomew's Hospital
London, United Kingdom, EC1A 7BE
Hammersmith Hospital
London, United Kingdom, W12 0HS
St Mary's Hospital (Paddington)
London, United Kingdom, W2 1NY
Charing Cross Hospital
London, United Kingdom, W6 8RF
Christie Hospital
Manchester, United Kingdom, M20 4BX
Southampton General Hospital
Southampton, United Kingdom, SO16 6YD Less << |
|
NCT01401192
|
Non-small Cell Lung Cancer |
Phase 2 |
Unknown |
- |
Korea, Republic of
... More >>
Samsung Medical Center
Recruiting
Seoul, Korea, Republic of
Contact: Myung-Ju Ahn, M.D., Ph.D.
Sub-Investigator: Jong-Mu Sun, M.D., Ph.D. Less << |
|
NCT01312324
|
Locally Advanced Stage III or ... More >>IV Thymic Cancer Less << |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of, 135-710 Less << |
|
NCT00510250
|
Cancer of the Cervix |
Phase 1
Phase 2 |
Completed |
- |
Canada, Ontario
... More >>
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT00012012
|
Cervical Cancer
... More >> Radiation Toxicity Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, Florida
... More >>
Integrated Community Oncology Network
Jacksonville Beach, Florida, United States, 32250
Baptist Cancer Institute - Jacksonville
Jacksonville, Florida, United States, 32207
Florida Oncology Associates at Southside Cancer Center
Jacksonville, Florida, United States, 32207
Baptist Medical Center South
Jascksonville, Florida, United States, 32258
Florida Oncology Associates
Orange Park, Florida, United States, 32073
Florida Cancer Center - Palatka
Palatka, Florida, United States, 32177
Flagler Cancer Center
Saint Augustine, Florida, United States, 32086
United States, Michigan
Borgess Medical Center
Kalamazooaa, Michigan, United States, 49001
West Michigan Cancer Center
Kalamazoo, Michigan, United States, 49007-3731
Bronson Methodist Hospital
Kalamazoo, Michigan, United States, 49007
United States, Nevada
University Medical Center of Southern Nevada
Las Vegas, Nevada, United States, 89102
CCOP - Nevada Cancer Research Foundation
Las Vegas, Nevada, United States, 89106
United States, New Jersey
Fox Chase Virtua Health Cancer Program at Virtua Memorial Hospital Marlton
Marlton, New Jersey, United States, 08053
Franklin & Edith Scarpa Regional Cancer Center at South Jersey Healthcare
Vineland, New Jersey, United States, 08360
United States, Ohio
Akron City Hospital
Akron, Ohio, United States, 44309-2090
Cancer Treatment Center
Wooster, Ohio, United States, 44691
United States, Pennsylvania
Mercy Cancer Institute at Mercy Hospital
Pittsburgh, Pennsylvania, United States, 15219 Less << |
|
NCT00002583
|
Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00006389
|
- |
|
Completed |
- |
- |
|
NCT01630174
|
- |
|
Unknown |
- |
China, Shandong
... More >>
Shandong Cancer hospital and institute
Recruiting
Jinan, Shandong, China, 250017
Principal Investigator: Baosheng Li, Ph.D.
Sub-Investigator: Zhongtang Wang, M.D. Less << |
|
NCT00012012
|
- |
|
Completed |
- |
- |
|
NCT00911820
|
Esophageal Cancer
... More >> Gastric Cancer
Stomach Cancer Less << |
Phase 2 |
Active, not recruiting |
December 2019 |
United States, Massachusetts
... More >>
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115
Massachusetts General Hospital
Boston, Massachusetts, United States, 02214
United States, Tennessee
Sarah Cannon Research Institute
Nashville, Tennessee, United States, 37203
United States, Texas
Texas Oncology Research
Dallas, Texas, United States, 75251 Less << |
|
NCT01593475
|
Pancreatic Carcinoma Non-resec... More >>table Less << |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
National Cancer Center, Korea
Goyang-si, Gyeonggi-do, Korea, Republic of, 410-769 Less << |
|
NCT00869310
|
Emesis |
Phase 3 |
Terminated(we terminated the s... More >>tudy before enrolling 303/560 due to a slow accrual) Less << |
- |
Italy
... More >> Fausto Roila
Terni, Italy, 05100 Less << |
|
NCT00084604
|
Adenocarcinoma of the Gastroes... More >>ophageal Junction
Diffuse Adenocarcinoma of the Stomach
Intestinal Adenocarcinoma of the Stomach
Mixed Adenocarcinoma of the Stomach
Recurrent Gastric Cancer
Stage IIIA Gastric Cancer
Stage IIIB Gastric Cancer
Stage IIIC Gastric Cancer
Stage IV Gastric Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT00147160
|
Glioma |
Phase 2 |
Completed |
- |
France
... More >> Institut Gustave-Roussy
Villejuif, France, 94805
United Kingdom
Bristol Royal Hospital for Children
Bristol, United Kingdom, BS2 8BJ Less << |
|
NCT00601510
|
Gastric Cancer |
Phase 1 |
Completed |
- |
Germany
... More >> Klinikum Rechts Der Isar - Technische Universitaet Muenchen
Munich, Germany, D-81675 Less << |
|
NCT00911820
|
- |
|
Active, not recruiting |
- |
- |
|
NCT01017874
|
- |
|
Completed |
- |
- |
|
NCT01285557
|
Metastatic Diffuse Gastric Can... More >>cer Including Carcinoma of the Gastro-esophageal Junction Less << |
Phase 3 |
Unknown |
May 2017 |
- |
|
NCT01017874
|
Non Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
Hong Kong
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Kowloon, Hong Kong
Korea, Republic of
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Incheon, Korea, Republic of, 405-760
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Seoul, Korea, Republic of, 110-744
Singapore
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Singapore, Singapore, 258499
Taiwan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Kuei Shan Hsiang, Taiwan, 33305
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Liouying/Tainan, Taiwan, 736
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Puzih City, Taiwan, 613
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Taichung, Taiwan, 407
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Taipei, Taiwan, 100
Thailand
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Bangkok, Thailand, 10700
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Chiang Mai, Thailand, 50200
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hat Yai, Thailand, 90110 Less << |
|
NCT02641093
|
Head and Neck Cancer |
Phase 2 |
Recruiting |
July 2021 |
United States, Michigan
... More >>
University of Michigan
Recruiting
Ann Arbor, Michigan, United States
Contact: Frank Worden, MD
Principal Investigator: Frank Worden, MD
United States, Ohio
University of Cincinnati Medical Center
Recruiting
Cincinnati, Ohio, United States, 45219
Contact: UC Cancer Institute Clinical Trials Office 513-584-7698 cancer@uchealth.com
Principal Investigator: Trisha Wise-Draper, MD, PhD
Ohio State University
Recruiting
Columbus, Ohio, United States, 43210
Contact: Katrina Banyas, BS 614-685-2127 Katrina.Banyas@osumc.edu
Principal Investigator: Matthew Old, MD
United States, South Carolina
Medical University of South Carolina
Recruiting
Charleston, South Carolina, United States
Contact: Brittanie Weinerman 843-792-6349 weinerma@musc.edu
Principal Investigator: Paul O'Brien, MD Less << |
|
NCT03656536
|
Unresectable Cholangiocarcinom... More >>a
Metastatic Cholangiocarcinoma Less << |
Phase 3 |
Not yet recruiting |
March 2023 |
- |
|
NCT03116971
|
Small Cell Lung Cancer |
Phase 1
Phase 2 |
Terminated(The study has been ... More >>terminated because of low recruitment rate of subjects and also due to change in focus of the study.) Less << |
- |
- |
|
NCT00482014
|
Non-Small-Cell Lung Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, California
... More >>
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Los Angeles, California, United States, 90095
United States, Kansas
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Wichita, Kansas, United States, 67214
United States, Missouri
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
St Louis, Missouri, United States, 63110
United States, Nevada
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Las Vegas, Nevada, United States, 89135
United States, North Carolina
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Burlington, North Carolina, United States, 27216
United States, Tennessee
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Memphis, Tennessee, United States, 38120
United States, Texas
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Corpus Christi, Texas, United States, 78405
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Dallas, Texas, United States, 75390
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Temple, Texas, United States, 76508
India
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Delhi, India, 110085
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Trivandrum, India, 695 011 Less << |
|
NCT00442455
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 3 |
Completed |
- |
Spain
... More >> Hospital de Navarra
Pamplona, Navarra, Spain, 31008
Hospital Clínico San Carlos
Madrid, Spain, 28003
Hospital General Universitario Gregorio Marañón
Madrid, Spain, 28007
Hospital Universitario Clínica Puerta de Hierro
Madrid, Spain, 28035
Hospital Regional Universitario Carlos Haya
Málaga, Spain, 29010 Less << |
|
NCT01275612
|
Solid Tumors
... More >>Acute Kidney Injury Less << |
Phase 1 |
Withdrawn(Patients evaluated s... More >>o far could not be enrolled because they did not meet the primary criterion of acute renal failure provided by the study protocol) Less << |
- |
Italy
... More >> Unit of Oncology - Ospedali Riuniti of Bergamo
Bergamo, BG, Italy, 24128 Less << |
|
NCT01266512
|
Lung Neoplasms |
Phase 2 |
Completed |
- |
Hong Kong
... More >> Sanofi-Aventis Administrative Office
Hong Kong, Hong Kong Less << |
|
NCT00675194
|
Gastric Cancer |
Phase 2 |
Completed |
- |
Germany
... More >> Johannes Gutenberg Universität , I. Medizinische Klinik und Polokilinik
Mainz, Germany, 55101 Less << |
|
NCT01143974
|
Locally Advanced Malignant Neo... More >>plasm Less << |
Phase 4 |
Completed |
- |
China
... More >> Cancer Institute & Hospital. Chinese Academy of Medical Sciences
Beijing, China, 100021 Less << |
|
NCT00482014
|
- |
|
Completed |
- |
- |
|
NCT01953952
|
Stage III Squamous Cell Carcin... More >>oma of the Oropharynx
Stage IVA Squamous Cell Carcinoma of the Oropharynx
Tongue Cancer Less << |
Phase 2 |
Withdrawn(Slow accrual) |
- |
United States, California
... More >>
Stanford University Hospitals and Clinics
Stanford, California, United States, 94305 Less << |
|
NCT00003299
|
Lung Cancer |
Phase 3 |
Completed |
- |
United States, Arizona
... More >>
CCOP - Scottsdale Oncology Program
Scottsdale, Arizona, United States, 85259-5404
United States, Illinois
CCOP - Illinois Oncology Research Association
Peoria, Illinois, United States, 61602
CCOP - Carle Cancer Center
Urbana, Illinois, United States, 61801
United States, Iowa
CCOP - Cedar Rapids Oncology Project
Cedar Rapids, Iowa, United States, 52403-1206
CCOP - Iowa Oncology Research Association
Des Moines, Iowa, United States, 50309-1016
Siouxland Hematology-Oncology
Sioux City, Iowa, United States, 51101-1733
United States, Kansas
CCOP - Wichita
Wichita, Kansas, United States, 67214-3882
United States, Michigan
CCOP - Ann Arbor Regional
Ann Arbor, Michigan, United States, 48106
United States, Minnesota
CCOP - Duluth
Duluth, Minnesota, United States, 55805
Mayo Clinic Cancer Center
Rochester, Minnesota, United States, 55905
CentraCare Clinic
Saint Cloud, Minnesota, United States, 56303
United States, Nebraska
CCOP - Missouri Valley Cancer Consortium
Omaha, Nebraska, United States, 68131
United States, North Dakota
Medcenter One Health System
Bismarck, North Dakota, United States, 58501
CCOP - Merit Care Hospital
Fargo, North Dakota, United States, 58122
Altru Health Systems
Grand Forks, North Dakota, United States, 58201
United States, Ohio
CCOP - Toledo Community Hospital Oncology Program
Toledo, Ohio, United States, 43623-3456
United States, Pennsylvania
CCOP - Geisinger Clinic and Medical Center
Danville, Pennsylvania, United States, 17822-2001
United States, South Dakota
Rapid City Regional Hospital
Rapid City, South Dakota, United States, 57709
CCOP - Sioux Community Cancer Consortium
Sioux Falls, South Dakota, United States, 57105-1080
Canada, Saskatchewan
Allan Blair Cancer Centre
Regina, Saskatchewan, Canada, S4T 7T1 Less << |
|
NCT03268499
|
Hepatocellular Carcinoma |
Phase 2 |
Recruiting |
September 2020 |
Hong Kong
... More >> Department of Imaging and Interventional Radiology, Prince of Wales Hospital, The Chinese University of Hong Kong
Recruiting
Hong Kong, Hong Kong
Contact: Carmen Wong (852)3505 3210 carmenwongsp@cuhk.edu.hk
Contact: Pui Man Chong (852)3505 3211 siuman@cuhk.edu.hk Less << |
|
NCT02421640
|
Nasopharyngeal Carcinoma |
Phase 2 |
Recruiting |
March 2020 |
China, Guangdong
... More >>
Haiqiang Mai
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Qiuyan Chen, MD,PhD 86-20-87343380 chenqy@sysucc.org.cn Less << |
|
NCT00553696
|
- |
|
Completed |
- |
- |
|
NCT00158678
|
Oral Cancer
O... More >>ropharynx Cancer
Hypopharynx Cancer Less << |
Phase 3 |
Active, not recruiting |
December 2018 |
France
... More >> Centre Alexis Vautrin
Vandoeuvre les Nancy, France, 54511
Institut Gustave Roussy
Villejuif, France, 94800 Less << |
|
NCT00237575
|
Bladder Cancer |
Phase 3 |
Terminated |
- |
United States, Kentucky
... More >>
University of Louisville
Louisville, Kentucky, United States, 40202
United States, Nebraska
University of Nebraska
Omaha, Nebraska, United States, 68198-7680
United States, New York
Montefiore Medical Center
Bronx, New York, United States, 10467
New York Medical College
Hawthorne, New York, United States, 10532 Less << |
|
NCT00553696
|
Stomach Neoplasms |
Phase 1 |
Completed |
- |
Japan
... More >> Aichi cancer center central hospital / Medical Oncology
Nagoya, Aichi, Japan, 464-8681
Saku Central Hospital, GI Devision
Saku, Nagano, Japan
Shizuoka Cancer Center
Suntougun, Shizuoka, Japan, 411-8777 Less << |
|
NCT03576417
|
Squamous Cell Carcinoma of Hea... More >>d and Neck Less << |
Phase 3 |
Recruiting |
December 2023 |
France
... More >> Centre Antoine Lacassagne
Recruiting
Nice, France, 06189
Contact: Joël GUIGAY, MD 04 92 03 15 01 ext +33 Joel.Guigay@nice.unicancer.fr Less << |
|
NCT00003377
|
Cervical Cancer |
Phase 1 |
Completed |
- |
United States, Florida
... More >>
University of Miami Sylvester Comprehensive Cancer Center
Miami, Florida, United States, 33136
United States, Illinois
University of Chicago Cancer Research Center
Chicago, Illinois, United States, 60637-1470
United States, Iowa
Holden Comprehensive Cancer Center at University of Iowa
Iowa City, Iowa, United States, 52242
United States, New Jersey
Cancer Institute of New Jersey at the Cooper University Hospital - Voorhees
Camden, New Jersey, United States, 08103
United States, North Carolina
Comprehensive Cancer Center at Wake Forest University
Winston-Salem, North Carolina, United States, 27157
United States, Ohio
MetroHealth's Cancer Care Center at MetroHealth Medical Center
Cleveland, Ohio, United States, 44109
Cleveland Clinic Cancer Center at Fairview Hospital
Cleveland, Ohio, United States, 44111
Cleveland Clinic Taussig Cancer Center
Cleveland, Ohio, United States, 44195
Arthur G. James Cancer Hospital and Solove Research Institute at Ohio State University
Columbus, Ohio, United States, 43210
Riverside Methodist Hospital Cancer Care
Columbus, Ohio, United States, 43214
United States, Oklahoma
Oklahoma University Medical Center
Oklahoma City, Oklahoma, United States, 73104
Cancer Care Associates - Midtown Tulsa
Tulsa, Oklahoma, United States, 74104
United States, Pennsylvania
Abramson Cancer Center of the University of Pennsylvania
Philadelphia, Pennsylvania, United States, 19104 Less << |
|
NCT00003377
|
- |
|
Completed |
- |
- |
|
NCT00548418
|
Cervical Cancer |
Phase 2 |
Completed |
- |
United States, Missouri
... More >>
Washington University School of Medicine
St. Louis, Missouri, United States, 63110
United States, North Carolina
Duke Cancer Institute
Durham, North Carolina, United States
United States, Ohio
The Ohio State University College of Medicine
Columbus, Ohio, United States Less << |
|
NCT00548418
|
- |
|
Completed |
- |
- |
|
NCT01116791
|
Peritoneal Carcinomatosis
... More >> Gastrointestinal Cancer Less << |
Phase 2 |
Terminated(Insufficient enroll... More >>ment in Pancreatic & Biliary arm) Less << |
- |
Belgium
... More >> University Hospitals Leuven
Leuven, Belgium, 3000 Less << |
|
NCT00136955
|
Uterine Cervical Neoplasms |
Phase 2 |
Completed |
- |
Taiwan
... More >> Pfizer Investigational Site
Kaoshiung, Taiwan, 813
Pfizer Investigational Site
Kwei-Shan County, TaoYuan,, Taiwan
Pfizer Investigational Site
Taichung, Taiwan
Pfizer Investigational Site
Taipei, Taiwan, 112
Pfizer Investigational Site
Taipei, Taiwan Less << |
|
NCT02949791
|
Pseudomyxoma Peritonei
... More >> Colorectal Cancer Less << |
Phase 2 |
Completed |
- |
Italy
... More >> Fondazione IRCCS Istituto Nazionale dei Tumori di Milano
Milano, MI, Italy, 20133 Less << |
|
NCT02465736
|
Esophageal Squamous Cell Carci... More >>noma
Esophageal Cancer
Oesophageal Cancer Less << |
Phase 3 |
Recruiting |
July 2023 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Hong Yang, MD, PhD +86+13560405144 yanghong@sysucc.org.cn Less << |
|
NCT01728480
|
Mucositis
Rec... More >>urrent Squamous Cell Carcinoma of the Hypopharynx
Recurrent Squamous Cell Carcinoma of the Larynx
Recurrent Squamous Cell Carcinoma of the Lip and Oral Cavity
Recurrent Squamous Cell Carcinoma of the Nasopharynx
Recurrent Squamous Cell Carcinoma of the Oropharynx
Recurrent Squamous Cell Carcinoma of the Paranasal Sinus and Nasal Cavity
Recurrent Verrucous Carcinoma of the Larynx
Recurrent Verrucous Carcinoma of the Oral Cavity
Stage III Squamous Cell Carcinoma of the Hypopharynx
Stage III Squamous Cell Carcinoma of the Larynx
Stage III Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage III Squamous Cell Carcinoma of the Nasopharynx
Stage III Squamous Cell Carcinoma of the Oropharynx
Stage III Squamous Cell Carcinoma of the Paranasal Sinus and Nasal Cavity
Stage III Verrucous Carcinoma of the Larynx
Stage III Verrucous Carcinoma of the Oral Cavity
Stage IV Squamous Cell Carcinoma of the Hypopharynx
Stage IV Squamous Cell Carcinoma of the Nasopharynx
Stage IVA Squamous Cell Carcinoma of the Larynx
Stage IVA Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage IVA Squamous Cell Carcinoma of the Oropharynx
Stage IVA Squamous Cell Carcinoma of the Paranasal Sinus and Nasal Cavity
Stage IVA Verrucous Carcinoma of the Larynx
Stage IVA Verrucous Carcinoma of the Oral Cavity
Stage IVB Squamous Cell Carcinoma of the Larynx
Stage IVB Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage IVB Squamous Cell Carcinoma of the Oropharynx
Stage IVB Squamous Cell Carcinoma of the Paranasal Sinus and Nasal Cavity
Stage IVB Verrucous Carcinoma of the Larynx
Stage IVB Verrucous Carcinoma of the Oral Cavity
Stage IVC Squamous Cell Carcinoma of the Larynx
Stage IVC Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage IVC Squamous Cell Carcinoma of the Oropharynx
Stage IVC Squamous Cell Carcinoma of the Paranasal Sinus and Nasal Cavity
Stage IVC Verrucous Carcinoma of the Larynx
Stage IVC Verrucous Carcinoma of the Oral Cavity
Tongue Cancer Less << |
Phase 1 |
Withdrawn(Financial Sponsor re... More >>quested termination) Less << |
- |
- |
|
NCT02501954
|
Endometrial Clear Cell Adenoca... More >>rcinoma
Endometrial Serous Adenocarcinoma
Stage IIIA Uterine Corpus Cancer
Stage IIIB Uterine Corpus Cancer
Stage IIIC Uterine Corpus Cancer
Stage IVA Uterine Corpus Cancer Less << |
Phase 3 |
Recruiting |
March 2023 |
United States, New York
... More >>
Women's Cancer Care Associates, LLC
Recruiting
Albany, New York, United States, 12208
Contact: Joyce N Barlin, MD 518-458-1390 jbarlin@womenscancercareassociates.com
Contact: Kimberly A Fredericks, BS, CCRP 518-458-1390 kfredericks@womenscancercareassociates.com
United States, Ohio
The Ohio State University
Recruiting
Columbus, Ohio, United States, 43210
Contact: Amanda Dunson 614-688-8849
Principal Investigator: Ritu Salani, MD
Canada, Quebec
CHUM Hopital Notre-Dame
Recruiting
Montréal, Quebec, Canada, H2L4M1
Contact: Nathalie Grenier 514-890-8000 ext 25165
Principal Investigator: Beatrice Cormier, MD Less << |
|
NCT02563054
|
Gastric Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT01742767
|
NSCLC |
Phase 2 |
Unknown |
April 2015 |
Germany
... More >> University hospital essen
Recruiting
Essen, Northrhine westphalia, Germany, 45122
Contact: Diana Cortes-Incio 02017232011 diana.cortez-incio@uk-essen.de Less << |
|
NCT00354679
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT00354679
|
- |
|
Completed |
- |
- |
|
NCT02363829
|
Uterine Cervix Cancer |
Phase 1 |
Recruiting |
February 2020 |
United States, Pennsylvania
... More >>
Abramson Cancer Center of the University of Pennsylvania
Recruiting
Philadelphia, Pennsylvania, United States, 19104
Principal Investigator: Fiona Simpkins, MD Less << |
|
NCT00349089
|
Non-small Cell Lung Cancer |
Phase 2 |
Unknown |
December 2013 |
Belgium
... More >> Ziekenhuis Oost Limburg
Genk, Belgium, 3600
Department of Pulmonology (Respiratory Tumor Unit), University Hospital Gasthuisberg, Catholic University Leuven, Belgium.
Leuven, Belgium, 3000
Germany
Helios-Klinikum Emil von Behring
Berlin, Germany, 14109
Klinikum Bremen-Ost
Bremen, Germany, 28325
Westdeutsches Tumorzentrum
Essen, Germany, 45122
Department of Hematology and Oncology, University of Goettingen
Goettingen, Germany, 37075
Lungenzentrum Großhansdorf
Großhansdorf, Germany, 22927
Clinic for thoracic diseases at the University of Heidelberg
Heidelberg, Germany, 69120
Lungenklinik Hemer
Hemer, Germany, 58675
Klinik Löwenstein
Loewenstein, Germany, 74245
University of München
Muenchen, Germany
Oldenburg Hospital
Oldenburg, Germany
Dr. Horst Schmidt Klinik
Wiesbaden, Germany, 65199
Luxembourg
Centre Hospitalier du Luxembourg
Luxembourg, Luxembourg, L-1210 Less << |
|
NCT00136955
|
- |
|
Completed |
- |
- |
|
NCT00881114
|
Head and Neck Cancer |
Phase 2 |
Withdrawn(Investigator decided... More >> it was not feasible to conduct this study) Less << |
June 2013 |
- |
|
NCT00842491
|
Advanced Gastric Cancer |
Phase 2 |
Completed |
- |
China, Beijing
... More >> Department of GI Oncology, Peking University, School of Oncology
Beijing, Beijing, China, 100142 Less << |
|
NCT00004083
|
Ovarian Cancer |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Kaplan Cancer Center
New York, New York, United States, 10016 Less << |
|
NCT02223611
|
Non-small Cell Lung Cancer Sta... More >>ge Ⅱ
Non-small Cell Lung Cancer Stage ⅢA Less << |
Phase 2 |
Unknown |
December 2018 |
China, Hebei
... More >> Jun Feng Liu
Not yet recruiting
Shijiazhuang, Hebei, China, 050011
Contact: Xin-Bo Liu, MD +86-311-86095353 lxbbbsj@163.com
Principal Investigator: Jun Feng Liu, MD, PhD Less << |
|
NCT02891447
|
Gastric Adenocarcinoma
... More >> Gastroesophageal Junction Adenocarcinoma
Peritoneal Carcinomatosis Less << |
Phase 2 |
Recruiting |
September 1, 2021 |
United States, Texas
... More >>
M D Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact: Brian D. Badgwell 713-792-6940
Principal Investigator: Brian D. Badgwell Less << |
|
NCT00295503
|
- |
|
Completed |
- |
- |
|
NCT01669707
|
Non-small Cell Lung Cancer |
Phase 2 |
Recruiting |
September 2020 |
China
... More >> The Beijing Chest Hospital
Recruiting
Beijing, China
Contact: Liyan Xu, MD xuliyan2009@yahoo.cn
Principal Investigator: Liyan Xu, MD Less << |
|
NCT01346540
|
Carcinoma, Non-Small-Cell Lung |
Phase 1 |
Completed |
- |
Italy
... More >> 1199.82.39004 Boehringer Ingelheim Investigational Site
Milano, Italy
Netherlands
1199.82.3102 Boehringer Ingelheim Investigational Site
Maastricht, Netherlands
Spain
1199.82.3401 Boehringer Ingelheim Investigational Site
Barcelona, Spain
1199.82.3406 Boehringer Ingelheim Investigational Site
Madrid, Spain
1199.82.3410 Boehringer Ingelheim Investigational Site
Málaga, Spain
United Kingdom
1199.82.4401 Boehringer Ingelheim Investigational Site
London, United Kingdom
1199.82.4402 Boehringer Ingelheim Investigational Site
Manchester, United Kingdom Less << |
|
NCT02703961
|
Cervical Cancer |
Phase 3 |
Recruiting |
February 2021 |
China, Shaanxi
... More >> Xijing hospital, the Fourth Military Medical University
Recruiting
Xi` An, Shaanxi, China, 710032
Contact: Mei Shi, professor 0086-029-84775432 mshi82@fmmu.edu.cn
Contact: ying zhang, doctor 0086-029-84775432 zhangying@fmmu.edu.cn
China, Shanxi
Department of Radiation Oncology, Xijing Hospital, Fourth Military Medical University
Recruiting
Xi'an, Shanxi, China, 710032
Contact: Mei Shi, MM +86-029-84775425 mshi82@fmmu.edu.cn
Principal Investigator: Mei Shi, MM
Department of Radiation Oncology, Xijing Hospital, Fourth Military
Recruiting
Xi'an, Shanxi, China, 710032
Contact: Mei Shi, MD 86-29-84775425 Shimei82@gmail.com Less << |
|
NCT00296322
|
- |
|
Completed |
- |
- |
|
NCT00296322
|
Stomach Cancer |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT00295503
|
Mesothelioma |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
University of Texas Southwestern Medical Center
Dallas, Texas, United States, 75390-8852 Less << |
|
NCT00707161
|
Cancer
Melano... More >>ma Less << |
Phase 2 |
Terminated(discontinued due to... More >> low enrollment) Less << |
- |
United States, Utah
... More >>
Huntsman Cancer Institute
Salt Lake City, Utah, United States, 84112
LDS Hospital
Salt Lake City, Utah, United States, 84143 Less << |
|
NCT00619359
|
Chemotherapy-Induced Nausea an... More >>d Vomiting (CINV) Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00619359
|
- |
|
Completed |
- |
- |
|
NCT00248482
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, Michigan
... More >>
University of Michigan Comprehensive Cancer Center
Ann Arbor, Michigan, United States, 48109-0942
Barbara Ann Karmanos Cancer Institute
Detroit, Michigan, United States, 48201-1379 Less << |
|
NCT00707161
|
- |
|
Terminated(discontinued due to... More >> low enrollment) Less << |
- |
- |
|
NCT02051868
|
Squamous Cell Carcinoma of the... More >> Anus Less << |
Phase 2 |
Unknown |
February 2018 |
United States, Massachusetts
... More >>
Laura Gagnon
Not yet recruiting
Boston, Massachusetts, United States, MA 02215
Contact: Laura Gagnon 617-632-3610 gagnon.laura@jimmy.harvard.edu
Australia, New South Wales
Margot Gorzeman
Not yet recruiting
Sydney, New South Wales, Australia, NSW 1450
Contact: Margot Gorzeman +61 295625359 interAACT@ctc.usyd.edu.au
United Kingdom
Royal Marsden NHS Foundation Trust, London & Sutton
Recruiting
Sutton, United Kingdom, SM2 5PT
Contact: Annette Bryant, BSc (Hons) +44 (0) 0208 661 3637 ext 3637 annette.bryant@rmh.nhs.uk
Principal Investigator: Sheela Rao, MD.FRCP Less << |
|
NCT02595424
|
Gastric Neuroendocrine Carcino... More >>ma
Intestinal Neuroendocrine Carcinoma
Pancreatic Neuroendocrine Carcinoma Less << |
Phase 2 |
Recruiting |
January 1, 2029 |
- |
|
NCT00627835
|
Locally Advanced Squamous Cell... More >> Carcinomas of the Head and Neck (SCCHN) Less << |
Phase 1 |
Withdrawn(Site decided not to ... More >>open this study) Less << |
- |
Canada, British Columbia
... More >>
BC Cancer Agency - Vancouver Centre
Vancouver, British Columbia, Canada, V5Z 4E6 Less << |
|
NCT00981058
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00346801
|
Lung Cancer |
Phase 1 |
Completed |
- |
United States, Texas
... More >>
UT MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00981058
|
Non Small Cell Lung Cancer |
Phase 3 |
Active, not recruiting |
December 31, 2018 |
- |
|
NCT03473574
|
Cholangiocarcinoma
... More >> Gall Bladder Carcinoma
Cholangiocarcinoma Non-resectable
Gallbladder Carcinoma Non-Resectable Less << |
Phase 2 |
Recruiting |
July 2021 |
Germany
... More >> Medizinische Hochschule Hannover, Klinik für Gastroenterologie, Hepatologie & Endokrinologie
Recruiting
Hannover, Germany, 30625
Contact: Arndt Vogel, Prof.Dr. Vogel.Arndt@mh-hannover.de Less << |
|
NCT00262821
|
- |
|
Terminated(Study drug no longe... More >>r available resulting in lack of study drug for participants.) Less << |
- |
- |
|
NCT03067181
|
Adult Germ Cell Tumor
... More >> Childhood Extracranial Germ Cell Tumor
Childhood Germ Cell Tumor
Extragonadal Embryonal Carcinoma
Grade 2 Immature Ovarian Teratoma
Grade 3 Immature Ovarian Teratoma
Malignant Germ Cell Tumor
Stage I Ovarian Choriocarcinoma
Stage I Ovarian Embryonal Carcinoma
Stage I Ovarian Teratoma
Stage I Ovarian Yolk Sac Tumor
Stage I Testicular Choriocarcinoma AJCC v6 and v7
Stage I Testicular Embryonal Carcinoma AJCC v6 and v7
Stage I Testicular Yolk Sac Tumor AJCC v6 and v7
Stage II Ovarian Choriocarcinoma
Stage II Ovarian Embryonal Carcinoma
Stage II Ovarian Yolk Sac Tumor
Stage II Testicular Choriocarcinoma AJCC v6 and v7
Stage II Testicular Embryonal Carcinoma AJCC v6 and v7
Stage II Testicular Yolk Sac Tumor AJCC v6 and v7
Stage III Ovarian Choriocarcinoma
Stage III Ovarian Embryonal Carcinoma
Stage III Ovarian Yolk Sac Tumor
Stage III Testicular Choriocarcinoma AJCC v6 and v7
Stage III Testicular Embryonal Carcinoma AJCC v6 and v7
Stage III Testicular Yolk Sac Tumor AJCC v6 and v7
Stage IV Ovarian Choriocarcinoma
Stage IV Ovarian Embryonal Carcinoma
Stage IV Ovarian Yolk Sac Tumor
Testicular Mixed Choriocarcinoma and Embryonal Carcinoma
Testicular Mixed Choriocarcinoma and Teratoma
Testicular Mixed Choriocarcinoma and Yolk Sac Tumor Less << |
Phase 3 |
Recruiting |
June 30, 2027 |
- |
|
NCT00160875
|
Esophageal Cancer |
Phase 2
Phase 3 |
Completed |
- |
Canada, Ontario
... More >>
University Health Network
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT02537223
|
Squamous Cell Carcinoma of Hea... More >>d and Neck
Locoregionally Advanced Less << |
Phase 1 |
Recruiting |
March 2020 |
Canada, Ontario
... More >>
Princess Margaret Cancer Centre
Recruiting
Toronto, Ontario, Canada, M3G 2M9
Contact: Aaron Hansen, M.D. 416-946-4501 ext 5606
Principal Investigator: Aaron Hansen, M.D. Less << |
|
NCT02170090
|
Cholangiocarcinoma
... More >> Gall Bladder Carcinoma Less << |
Phase 3 |
Recruiting |
April 2022 |
- |
|
NCT01346540
|
- |
|
Completed |
- |
- |
|
NCT01752023
|
- |
|
Terminated(Low Accrual) |
- |
- |
|
NCT02196168
|
- |
|
Terminated(Inadequate accrual ... More >>rate) Less << |
- |
- |
|
NCT00262821
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Cell Carcinoma
Cervical Squamous Cell Carcinoma
Stage IB Cervical Cancer
Stage IIA Cervical Cancer
Stage IIB Cervical Cancer
Stage III Cervical Cancer
Stage IVA Cervical Cancer Less << |
Phase 3 |
Terminated(Study drug no longe... More >>r available resulting in lack of study drug for participants.) Less << |
- |
- |
|
NCT01088347
|
Advanced Cervical Cancer |
Phase 1
Phase 2 |
Completed |
- |
Netherlands
... More >> University Medical Center Groningen
Groningen, Netherlands, 9700 RB Less << |
|
NCT00006036
|
Head and Neck Cancer
... More >> Lung Cancer
Ovarian Cancer
Unspecified Adult Solid Tumor, Protocol Specific Less << |
Phase 1 |
Completed |
- |
Canada, British Columbia
... More >>
British Columbia Cancer Agency
Vancouver, British Columbia, Canada, V5Z 4E6
Canada, Ontario
Cancer Care Ontario-Hamilton Regional Cancer Centre
Hamilton, Ontario, Canada, L8V 5C2
Toronto General Hospital
Toronto, Ontario, Canada, M5G 2C4 Less << |
|
NCT01752023
|
Non-small Cell Lung Cancer Met... More >>astatic Less << |
Phase 2 |
Terminated(Low Accrual) |
- |
United States, Maryland
... More >>
Bayview Medical Center at Johns Hopkins
Baltimore, Maryland, United States, 21224
Johns Hopkins University, SKCCC
Baltimore, Maryland, United States, 21287 Less << |
|
NCT00966706
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
Italy
... More >> San Raffaele Scientific Institute
Milan, Italy, 20132 Less << |
|
NCT02196168
|
Recurrent Hypopharyngeal Squam... More >>ous Cell Carcinoma
Recurrent Laryngeal Squamous Cell Carcinoma
Recurrent Laryngeal Verrucous Carcinoma
Recurrent Lip and Oral Cavity Squamous Cell Carcinoma
Recurrent Metastatic Squamous Cell Carcinoma in the Neck With Occult Primary
Recurrent Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma
Recurrent Oral Cavity Verrucous Carcinoma
Recurrent Oropharyngeal Squamous Cell Carcinoma
Squamous Cell Carcinoma Metastatic in the Neck With Occult Primary
Stage IV Hypopharyngeal Squamous Cell Carcinoma
Stage IVA Laryngeal Squamous Cell Carcinoma
Stage IVA Laryngeal Verrucous Carcinoma
Stage IVA Lip and Oral Cavity Squamous Cell Carcinoma
Stage IVA Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma
Stage IVA Oral Cavity Verrucous Carcinoma
Stage IVA Oropharyngeal Squamous Cell Carcinoma
Stage IVB Laryngeal Squamous Cell Carcinoma
Stage IVB Laryngeal Verrucous Carcinoma
Stage IVB Lip and Oral Cavity Squamous Cell Carcinoma
Stage IVB Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma
Stage IVB Oral Cavity Verrucous Carcinoma
Stage IVB Oropharyngeal Squamous Cell Carcinoma
Stage IVC Laryngeal Squamous Cell Carcinoma
Stage IVC Laryngeal Verrucous Carcinoma
Stage IVC Lip and Oral Cavity Squamous Cell Carcinoma
Stage IVC Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma
Stage IVC Oral Cavity Verrucous Carcinoma
Stage IVC Oropharyngeal Squamous Cell Carcinoma
Tongue Carcinoma Less << |
Phase 2 |
Terminated(Inadequate accrual ... More >>rate) Less << |
- |
Canada, Ontario
... More >>
Juravinski Cancer Centre at Hamilton Health Sciences
Hamilton, Ontario, Canada, L8V 5C2
London Regional Cancer Program
London, Ontario, Canada, N6A 4L6
University Health Network-Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT00087698
|
Pleural Neoplasms |
Phase 2 |
Completed |
- |
United States, California
... More >>
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
San Francisco, California, United States
United States, Illinois
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Chicago, Illinois, United States
United States, Maryland
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Baltimore, Maryland, United States
United States, Massachusetts
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Boston, Massachusetts, United States
United States, Michigan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Detroit, Michigan, United States
United States, New York
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
New York, New York, United States
United States, Texas
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Houston, Texas, United States Less << |
|
NCT00546130
|
Small Cell Lung Cancer |
Phase 2 |
Unknown |
September 2011 |
Japan
... More >> Toho University Sakura Medical Center
Recruiting
Sakura, Chiba, Japan, 285-8741
Contact: Ryoji Kato, MD +81-43-462-8811 ryochan@sakura.med.toho-u.ac.jp
Principal Investigator: Ryoji Kato, MD
Hokkaido University Hospital
Recruiting
Sapporo, Hokkaido, Japan, 060-8648
Contact: Satoshi Oizumi, MD +81-11-716-1161 soizumi@med.hokudai.ac.jp
Principal Investigator: Satoshi Oizumi, MD
Kanazawa University Hospital
Recruiting
Kanazawa, Ishikawa, Japan, 920-8641
Contact: Kazuo Kasahara, MD +81-076-265-2000 kasa1237@med3.m.kanazawa-u.ac.jp
Principal Investigator: Kazuo Kasahara, MD
Kanazawa Medical University Hospital
Recruiting
Uchinada, Ishikawa, Japan, 920-0293
Contact: Hirohisa Toga, MD +81-76-286-3511 toga-h@kanazawa-med.ac.jp
Principal Investigator: Hirohisa Toga, MD
Kinkidaigakuigakubu Nara Hospital
Recruiting
Ikoma, Nara, Japan, 630-0293
Contact: Toshio Shimizu, MD +81-743-77-0880 tshimizu@nara.med.kindai.ac.jp
Principal Investigator: Toshio Shimizu, MD
Kurashiki Central Hospital
Recruiting
Kurashiki, Okayama, Japan, 710-8602
Contact: Hiroshige Yoshioka, MD +81-86-422-0210 hirotin@kchnet.or.jp
Principal Investigator: Hirishige Yoshioka, MD
Osaka Prefectural Medical Center for Respiratory and Allergic Diseases
Recruiting
Habikino, Osaka, Japan, 583-8588
Contact: Tomonori Hirasima, MD +81-957-2121 hirashima@opho.jp
Principal Investigator: Tomonori Hirasima, MD
Kinkidaigakuigakubu Sakai Hospital
Not yet recruiting
Sakai, Osaka, Japan, 590-0132
Contact: Minoru Takada, MD +81-72-299-1120 m-takada@sakai.med.kindai.ac.jp
Principal Investigator: Minoru Takada, MD
NHO Kinki-chuo Chest Medical Center
Recruiting
Sakai, Osaka, Japan, 591-8555
Contact: Akihito Kubo, MD +81-72-252-3021 a-kubo@kch.hosp.go.jp
Principal Investigator: Akihito Kubo, MD
Osaka Medikal College Hospital
Recruiting
Takatsuki, Osaka, Japan, 569-8686
Contact: Takayasu Kurata, MD +81-72-683-1221 ctc002@poh.osaka-med.ac.jp
Principal Investigator: Takayasu Kurata, MD
Hiroshima City Hospital
Not yet recruiting
Hiroshima, Japan, 730-8518
Contact: Yasuo Iwamoto, MD +81-82-221-2291 y-iwamoto@do.enjoy.ne.jp
Principal Investigator: Yasuo Iwamoto, MD
Osaka City General Hospital
Recruiting
Osaka, Japan, 534-0021
Contact: Koji Takeda, MD +81-6-6929-1221 kkk-take@ga2.so-net.ne.jp
Principal Investigator: Koji Takeda, MD
Tokyo Medical University Hospital
Recruiting
Tokyo, Japan, 160-0023
Contact: Masahiro Tsuboi, MD +81-3-3342-6111 mtsuboi@za2.so-net.ne.jp
Principal Investigator: Masahiro Tsuboi, MD
Toyama University Hospital
Recruiting
Toyama, Japan, 930-0194
Contact: Tatsuhiko Kashii, MD, PhD +81-76-434-7808 tkashii@med.u-toyama.ac.jp
Principal Investigator: Tatsuhiko Kashii, MD, PhD
Toyama Red Cross Hospital
Recruiting
Toyama, Japan, 930-0859
Contact: Keiichi Iwase, MD +81-76-433-2222 iwasa@toyama-med.jrc.or.jp
Principal Investigator: Keiichi Iwase, MD Less << |
|
NCT00535509
|
Breast Cancer |
Not Applicable |
Completed |
- |
- |
|
NCT00006482
|
Cervical Cancer |
Phase 2 |
Terminated |
- |
- |
|
NCT00087698
|
- |
|
Completed |
- |
- |
|
NCT00967928
|
Cervical Cancer |
Phase 1 |
Withdrawn(Lack of enrollment) |
December 2010 |
- |
|
NCT00132639
|
Pulmonary Neoplasms |
Phase 2 |
Completed |
- |
Japan
... More >> National Cancer Center
Chuo-ku,Tsukiji,5-1-1, Tokyo, Japan, 104-0045 Less << |
|
NCT00803062
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Carcinoma
Cervical Squamous Cell Carcinoma
Recurrent Cervical Carcinoma
Stage IVB Cervical Cancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT03451331
|
Metastatic Urothelial Cancer |
Phase 2 |
Recruiting |
December 1, 2020 |
United States, New York
... More >>
Icahn School of Medicine: Tisch Cancer Institute at Mount Sinai Medical Center
Recruiting
New York, New York, United States, 10029
Contact: Matthew Galsky, M.D. 212-241-8214 matthew.galsky@mssm.edu
United States, Tennessee
Vanderbilt-Ingram Cancer Center
Recruiting
Nashville, Tennessee, United States, 37232
Contact: Matthew Fister 615-875-9979 matthew.c.fister@vumc.org
Principal Investigator: David Chism, MD Less << |
|
NCT00560573
|
Carcinoma, Non-Small-Cell Lung |
Phase 1 |
Completed |
- |
Belgium
... More >> Pfizer Investigational Site
Charleroi, Belgium, 6000
Ireland
Pfizer Investigational Site
Dublin, Ireland, 8
Spain
Pfizer Investigational Site
Madrid, Spain, 28041
Pfizer Investigational Site
Sevilla, Spain, 41013 Less << |
|
NCT02221999
|
Tubular Breast Cancer
... More >> Mucinous Breast Cancer
Invasive Ductal Breast Cancer
Inflammatory Breast Cancer Less << |
Phase 2
Phase 3 |
Recruiting |
October 2022 |
China, Hebei
... More >> HanDan Central Hospital
Recruiting
Handan, Hebei, China, 056001
Contact: Yinzhou Zhang, MD
China, Hunan
The First Affiliated Hospital of University of South China
Recruiting
Hengyang, Hunan, China, 421001
Contact: Hongguang Liu, MD
China, Jiangsu
The second people's hospital of Kunshan city
Recruiting
Kunshan, Jiangsu, China, 215300
Contact: Bei Gu, MD
China, Shanghai
Zhongshan Hospital
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Hongwei Zhang, MD
Shanghai Jiaotong University School of Medicine, Renji Hospital
Recruiting
Shanghai, Shanghai, China, 200127
Contact: Jinsong Lu, MD lujjss@163.com
Principal Investigator: Jinsong Lu, MD
Yueyang hospital of integrated traditional Chinese and Western medine
Recruiting
Shanghai, Shanghai, China, 200437
Contact: Xiaohong Xue, MD
Armed Police Corps Hospital of Shanghai
Recruiting
Shanghai, Shanghai, China, 201103
Contact: Yumei Ye, MD
China, Zhejiang
Zhoushan hospital
Recruiting
Zhoushan, Zhejiang, China, 316021
Contact: Jing Xu, MD Less << |
|
NCT00560573
|
- |
|
Completed |
- |
- |
|
NCT00387127
|
Neoplasms, Head and Neck |
Phase 2 |
Completed |
- |
- |
|
NCT00122460
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00122460
|
- |
|
Completed |
- |
- |
|
NCT01074970
|
Breast Cancer |
Phase 2 |
Active, not recruiting |
December 2018 |
- |
|
NCT01622660
|
- |
|
Terminated(Safety reasons) |
- |
- |
|
NCT00803062
|
- |
|
Completed |
- |
- |
|
NCT01622660
|
Bladder Cancer |
Phase 2 |
Terminated(Safety reasons) |
- |
United States, New Jersey
... More >>
Memoral Sloan Kettering Cancer Center
Basking Ridge, New Jersey, United States
United States, New York
Memorial Sloan Kettering Cancer Center @ Suffolk
Commack, New York, United States, 11725
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan Kettering Cancer Center at Mercy Medical Center
Rockville Centre, New York, United States, 11570
Memorial Sloan Kettering Cancer Center at Phelps Memorial Hospital Center
Sleepy Hollow, New York, United States, 10591 Less << |
|
NCT02325986
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Phase 2 |
Unknown |
December 2017 |
China, Guangdong
... More >>
SYSU Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510000
Contact: Yujia Zhu, Phd. 18520121368 sysucc@sysucc.org.cn Less << |
|
NCT01441349
|
Small Cell Lung Carcinoma |
Phase 2 |
Recruiting |
December 2020 |
Korea, Republic of
... More >>
National Cancer Center , Korea
Recruiting
Goyang-si, Gyeonggi-do, Korea, Republic of, 410-769
Contact: JONGHEE HAN, RN 82-31-920-0409 hanjonghee@ncc.re.kr
Sub-Investigator: Jin Soo Lee, MD
Sub-Investigator: Heung Tae Kim, MD
Sub-Investigator: Tak Yun, MD
Sub-Investigator: Young Joo Lee, MD
Sub-Investigator: Geon Kook Lee, MD
Sub-Investigator: Soo Hyun Lee, MD Less << |
|
NCT01158248
|
Cervical Cancer |
Phase 2 |
Unknown |
- |
Austria
... More >> Innsbruck Universitaetsklinik
Recruiting
Innsbruck, Austria, A-6020
Contact: Contact Person 43-512-504-24155 Alain.zeimet@i-med.ac.at Less << |
|
NCT02256982
|
- |
|
Terminated(Slow accrual.) |
- |
- |
|
NCT02533765
|
Neoplasms, Germ Cell and Embry... More >>onal Less << |
Phase 2 |
Recruiting |
December 2018 |
Italy
... More >> U.O Oncologia Medica, IRST IRCCS
Recruiting
Meldola, FC, Italy, 47014
Contact: Ugo De Giorgio, MD ugo.degiorgi@irst.emr.it
Istituto Nazionale Tumori IRCCS "Fondazione G.Pascale"
Recruiting
Napoli, Italy, 80131
Contact: Sandro Pignata, MD sandro.pignata@fondazionepascale.it Less << |
|
NCT02256982
|
Unresectable Intrahepatic Chol... More >>angiocarcinoma
Resectable Intrahepatic Cholangiocarcinoma Less << |
Not Applicable |
Terminated(Slow accrual.) |
- |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114 Less << |
|
NCT00387127
|
- |
|
Completed |
- |
- |
|
NCT01373463
|
Nonsquamous Nonsmall Cell Neop... More >>lasm of Lung
Nonsmall Cell Lung Cancer Stage III Less << |
Phase 1 |
Terminated(Investigator left s... More >>ite) Less << |
- |
United States, South Carolina
... More >>
Medical University of South Carolina
Charleston, South Carolina, United States, 29425 Less << |
|
NCT03601871
|
Chemotherapy-induced Nausea an... More >>d Vomiting Less << |
Not Applicable |
Recruiting |
December 30, 2020 |
China, Heilongjiang
... More >>
cancer hospital of Haerbin Medical University
Not yet recruiting
Haerbin, Heilongjiang, China
Contact: Jin Wu, M.D.
China, Jilin
Siping City Cancer Hospital
Not yet recruiting
Siping, Jilin, China
Contact: Zhuohui Qu
Sub-Investigator: Zhuohui Qu
China, Liaoning
Anshan Hospital of First Hospital of China Medical University
Not yet recruiting
Anshan, Liaoning, China
Contact: Mingran Sun, PhD.M.D.
Sub-Investigator: Mingran Sun
Anshan Tumor Hospital
Not yet recruiting
Anshan, Liaoning, China
Contact: Xiuna Zhang, M.D.
Sub-Investigator: Xiuna Zhang
Sub-Investigator: Jinfang Lv
Sub-Investigator: Fugang When
Sub-Investigator: Li Man
Central hospital of Dalian
Not yet recruiting
Dalian, Liaoning, China
Contact: Wei Huo
Sub-Investigator: Wei huo
Sub-Investigator: Min Zhong
Sub-Investigator: Liangwei Yin
Second Affiliated Hospital of Dalian Medical University
Not yet recruiting
Dalian, Liaoning, China
Contact: Zhaoxia Dai, M.D.
Sub-Investigator: Zhaoxia Dai
Sub-Investigator: Jun Chen
The Fifth Hospital of Dalian City
Not yet recruiting
Dalian, Liaoning, China
Contact: Jilai Bian
Sub-Investigator: Jilai Bian
The First Affiliated Hospital of Dalian Medical University
Not yet recruiting
Dalian, Liaoning, China
Contact: Jiwei Liu, PhD;M.D.
Sub-Investigator: Jiwei Liu
Zhuanghe Central Hospital
Not yet recruiting
Dalian, Liaoning, China
Contact: Huali Tang
Sub-Investigator: Huali Tang
Fushun Central Hospital
Recruiting
Fushun, Liaoning, China
Contact: Li Ning
Sub-Investigator: Li Ning
General Hospital of Mining Bureau
Not yet recruiting
Fushun, Liaoning, China
Contact: Yuyang Zhang
Sub-Investigator: Yuyang Zhang
Jinzhou Central Hospital
Not yet recruiting
Jinzhou, Liaoning, China
Contact: Wei Wang
Principal Investigator: Wei Wang
The First Hospital of Liaoning Medical University
Not yet recruiting
Jinzhou, Liaoning, China
Contact: Zhitu Zhu
Sub-Investigator: Zhitu Zhu
Chinese Medicine Hospital of Liaoyang county
Not yet recruiting
Liaoyang, Liaoning, China
Contact: Haifeng Liu, M.D.
Sub-Investigator: Haifeng Liu
Liaoyang Central Hospital
Not yet recruiting
Liaoyang, Liaoning, China
Contact: Jian Zhang, M.D.
Sub-Investigator: Jian Zhang
Petrochemical General Hospital of Liaoyang city
Not yet recruiting
Liaoyang, Liaoning, China
Contact: Hao Chen
Sub-Investigator: Hao Chen
Panjin central Hospital
Not yet recruiting
Panjin, Liaoning, China
Contact: Junwei Zhang
Sub-Investigator: Junwei Zhang
Sub-Investigator: Zhichang Sun
Sub-Investigator: Yu Jiang
Sub-Investigator: Qinghua Gao
Chest Hospital of Shenyang City
Not yet recruiting
Shengyang, Liaoning, China
Contact: Yinyin Li
Sub-Investigator: Yinyin Li
Shengjing Hospital of China Medical University
Not yet recruiting
Shenyang, Liaoning, China, 110004
Contact: Huawei Zou, M.D.
Sub-Investigator: Huawei Zou
Sub-Investigator: Rong Wu
General Hospital of Shenyang Military Region
Not yet recruiting
Shenyang, Liaoning, China
Sub-Investigator: Zhendong Zheng
Liaoning Tumor Hospital & Institute
Not yet recruiting
Shenyang, Liaoning, China
Contact: Yu Tang, M.D.
Sub-Investigator: Yu Tang
The First Hospital of China Medical University
Recruiting
Shenyang, Liaoning, China
Contact: Yunpeng Liu, M.D.;Ph.D.
Principal Investigator: Yunpeng Liu
Sub-Investigator: Xiujuan Qu
Sub-Investigator: Lingyun Zhang
the People'S Hospital
Not yet recruiting
Shenyang, Liaoning, China
Contact: Lijie He, M.D.
Sub-Investigator: Lijie He
Tieling city Central Hospital
Not yet recruiting
Tieling, Liaoning, China
Contact: Huijun Zhang
Sub-Investigator: Huijun Zhang
China
Central Hospital of Anshan City
Not yet recruiting
Anshan, China
Contact: Chuanchun Leng
Sub-Investigator: Chuanchun Leng
Benxi Central Hospital
Not yet recruiting
Benxi, China
Contact: Tiejun Chen
Sub-Investigator: Tiejun Chen
Chaoyang Central Hospital
Not yet recruiting
Chaoyang, China
Contact: Xiujie Cui
Sub-Investigator: Xiujie Cui
Zhongshan Hospital
Not yet recruiting
Dalian, China
Contact: Xuening Ji, M.D.
Sub-Investigator: Xuening Ji
Sub-Investigator: Gang Wang
Sub-Investigator: Tong Zhao
Liaohe Oilfield General Hospital
Not yet recruiting
Panjin, China
Contact: Qiang Chen
Sub-Investigator: Qiang Chen Less << |
|
NCT00066391
|
Penile Cancer |
Phase 2 |
Completed |
- |
Belgium
... More >> U.Z. Gasthuisberg
Leuven, Belgium, B-3000
France
Institut Gustave Roussy
Villejuif, France, F-94805
Hungary
National Institute of Oncology
Budapest, Hungary, 1122
Netherlands
Netherlands Cancer Institute - Antoni van Leeuwenhoek Hospital
Amsterdam, Netherlands, 1066 CX
Poland
Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology
Warsaw, Poland, 02-781
United Kingdom
Bristol Haematology and Oncology Centre
Bristol, England, United Kingdom, BS2 8ED
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Saint Bartholomew's Hospital
London, England, United Kingdom, EC1A 7BE Less << |
|
NCT03723941
|
Adrenocortical Carcinoma |
Phase 3 |
Recruiting |
June 30, 2021 |
Italy
... More >> ASST Spedali Civili di Brescia
Recruiting
Brescia, BS, Italy, 25123
Contact: Alfredo Berruti, MD 0039 030 399 5410 alfredo.berruti@gmail.com Less << |
|
NCT00094081
|
Head and Neck Neoplasms |
Phase 3 |
Completed |
- |
United States, Arizona
... More >>
Foundation for Cancer Research and Education
Phoenix, Arizona, United States, 85013
United States, California
Stanford University Medical Center
Palo Alto, California, United States, 94305
United States, Florida
H-Lee Moffitt Cancer Center and Research
Tampa, Florida, United States, 33612
United States, Illinois
University of Chicago Medical Center
Chicago, Illinois, United States, 60637
United States, Maryland
Johns Hopkins Hospital
Baltimore, Maryland, United States, 21231
United States, Michigan
Harper Hospital
Detroit, Michigan, United States, 48201
United States, New Jersey
VA New Jersey Health Care Medical Center
East Orange, New Jersey, United States, 07018
United States, New York
University of Rochester Medical Center
Rochester, New York, United States, 14620
United States, South Carolina
Medical University of South Carolina
Charleston, South Carolina, United States, 29425
United States, Virginia
Eastern Virginia Medical School
Norfolk, Virginia, United States, 23507 Less << |
|
NCT00126633
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
United States, California
... More >>
UCSF Comprehensive Cancer Center
San Francisco, California, United States, 94115 Less << |
|
NCT00415194
|
- |
|
Completed |
- |
- |
|
NCT00191126
|
Non-Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00933816
|
Hepatocellular Carcinoma |
Phase 1
Phase 2 |
Completed |
- |
Japan
... More >> Kurume University Medical Center
Kurume, Fukuoka, Japan, 839-0863
Ogaki Municipal Hospital
Oogaki, Gifu, Japan, 503-8502
Kinki University School of Medicine
Osaka-Sayama, Osaka, Japan, 589-8511 Less << |
|
NCT00415194
|
Head and Neck Neoplasms |
Phase 3 |
Completed |
- |
- |
|
NCT01999933
|
Cervical Cancer
... More >> Toxicity Due to Radiotherapy Less << |
Phase 2
Phase 3 |
Unknown |
November 2018 |
China, Shaanxi
... More >> Department of Radiation Oncology, Xijing Hospital, Fourth Military Medical University
Recruiting
Xi'an, Shaanxi, China, 710032
Contact: Mei Shi, MD +86-029-84775425 mshifmmu@yahoo.com
Principal Investigator: Mei Shi, MD
Sub-Investigator: Li-Chun Wei, M.D.,Ph.D Less << |
|
NCT03559803
|
Cervical Cancer |
|
UNKNOWN |
2025-12-19 |
Guizhou Province People's Hosp... More >>ital, Guizhou, Guizhou, China|West China Hospital, Sichuan University, Chendu, Sichuan, 610041, China Less << |
|
NCT00003896
|
Fallopian Tube Cancer
... More >> Ovarian Cancer
Peritoneal Cavity Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00003896
|
- |
|
Completed |
- |
- |
|
NCT00771563
|
Non-Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
Belgium
... More >> University Hospital Antwerp
Edegem, Antwerp, Belgium, 2650
Centre Hospitalier Universitaire Sart Tilman
Liège, Belgium, 4000 Less << |
|
NCT01809379
|
Recurrent Ovarian Cancer |
Phase 2 |
Completed |
- |
Germany
... More >> Ruhr University Bochum
Bochum, NRW, Germany, 44623 Less << |
|
NCT00380588
|
Biliary Tract Cancer |
Phase 2 |
Completed |
- |
Japan
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Aichi, Japan, 466-8560
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Chiba, Japan, 277-8577
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Fukuoka, Japan, 811-1395
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hokkaido, Japan, 060-8648
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Kanagawa, Japan, 241-0815
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Shizuoka, Japan, 411-8777
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Tokyo, Japan, 104-0045 Less << |
|
NCT02526316
|
HPV-induced Cancers |
Phase 1 |
Completed |
- |
Germany
... More >> Krankenhaus Nordwest
Frankfurt, Germany, 60488 Less << |
|
NCT02006667
|
- |
|
Completed |
- |
- |
|
NCT00314678
|
Epithelial Ovarian Cancer
... More >> Primary Peritoneal Cancer Less << |
Phase 2 |
Completed |
- |
United States, California
... More >>
Chao Family Comprehensive Cancer Center
Orange, California, United States, 92868 Less << |
|
NCT00220129
|
Oesophageal Carcinoma |
Phase 2 |
Completed |
- |
United Kingdom
... More >> Royal Marsden Hospital
Sutton, Surrey, United Kingdom, SM2 5PT Less << |
|
NCT00380588
|
- |
|
Completed |
- |
- |
|
NCT03020329
|
Nasopharyngeal Carcinoma
... More >> Children Less << |
Phase 2 |
Recruiting |
December 2021 |
China, Guangdong
... More >>
Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: HaiQiang Mai, PhD 8620-38606186 maihq@sysucc.org.cn Less << |
|
NCT00942838
|
Recurrent Ovarian Carcinoma |
Phase 1 |
Withdrawn(PI decision due to u... More >>nder accrual.) Less << |
- |
United States, Utah
... More >>
Huntsman Cancer Institute
Salt Lake City, Utah, United States, 84112 Less << |
|
NCT00002717
|
Ovarian Cancer
... More >> Primary Peritoneal Cavity Cancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT02006667
|
Urinary Tract Cancer |
Phase 2 |
Completed |
- |
Germany
... More >>
Aschersleben, Germany, 06449
Dessau, Germany, 06846
Fulda, Germany, 36043
Leipzig, Germany, 04103
Leipzig, Germany, 04277
Marburg, Germany, 35043
Weiden, Germany, 92637 Less << |
|
NCT00004547
|
Abdominal Neoplasm
... More >> Colonic Neoplasm
Mesothelioma
Peritoneal Neoplasm Less << |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892 Less << |
|
NCT03380468
|
Lung Neoplasms |
Phase 2
Phase 3 |
Recruiting |
December 31, 2025 |
China, Shanghai
... More >>
Fudan University Cancer Center
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Chen Haiquan, MD +86-21 64175590 ext 1707 hqchen1@yahoo.com Less << |
|
NCT02797977
|
Advanced Solid Tumors |
Phase 1
Phase 2 |
Recruiting |
June 2020 |
Spain
... More >> Hospital Universitario Vall d'Hebron
Recruiting
Barcelona, Spain, 08035
Contact: Elena Garralda egarralda@vhio.net
Principal Investigator: Elena Garralda
Hospital Clinic de Barcelona
Recruiting
Barcelona, Spain, 08036
Contact: Javier Garcia Corbacho
Principal Investigator: Javier Garcia Corbacho
START Madrid
Recruiting
Madrid, Spain, 28040
Contact: Victor Moreno victor.moreno@start.stoh.com
Principal Investigator: Victor Moreno
Hospital Universitario 12 de Octobre
Recruiting
Madrid, Spain, 28041
Contact: Daniel Castellano
Principal Investigator: Daniel Castellano
Hospital Universitario Virgen de la Victoria
Recruiting
Málaga, Spain, 29010
Contact: Begona Jimenez Rodrigues
Principal Investigator: Begona Jimenez Rodrigues
Biomedical Research Institute INCLIVA
Recruiting
Valencia, Spain, 46010
Contact: Desamparados Roda Perez
Principal Investigator: Desamparados Roda Perez
United Kingdom
Royal Marsden Hospital
Recruiting
Sutton, London, United Kingdom, SM2 5PT
Contact: Udai Banerji, MD Udai.Banerji@icr.ac.uk
Principal Investigator: Udai Banerji, MD
Belfast City Hospital
Recruiting
Belfast, Northern Ireland, United Kingdom, BT9 7AB
Contact: Melanie Morris 02890638468 melanie.morris@belfasttrust.hscni.net
Principal Investigator: Vicky Coyle
Oxford University Hospitals
Recruiting
Headington, Oxford, United Kingdom, OX3 7LE
Contact: Sarah Blagden
Principal Investigator: Sarah Blagden
Velindre Cancer Centre
Recruiting
Cardiff, Whitchurch, United Kingdom, CF14 2TL
Contact: Rob Jones, MD Robert.Hugh.Jones@wales.nhs.uk
Principal Investigator: Rob Jones, MD
The Clatterbridge Cancer Centre
Recruiting
Bebington, Wirral, United Kingdom, CH63 4JY
Contact: Daniel Palmer
Principal Investigator: Daniel Palmer
The Beatson West of Scotland Cancer Centre
Recruiting
Glasgow, United Kingdom, G12 0YN
Contact: Patricia Roxburgh, MD p.roxburgh@beatson.gla.ac.uk
Principal Investigator: Patricia Roxburgh, MD
Leeds Teaching Hospital of St James University Hospital
Recruiting
Leeds, United Kingdom, LS9 7TF
Contact: David A Anthoney
Principal Investigator: David A Anthoney
University Hospital of Leceister NHS TRUST
Recruiting
Leicester, United Kingdom, LE1 5WW
Contact: Harriet Walter hw191@le.ac.uk
Principal Investigator: Harriet Walter
Guy's and St Thomas'
Recruiting
London, United Kingdom, SE1 9RT
Contact: Debashis Sarker debashis.sarker@kcl.ac.uk
Principal Investigator: Debashis Sarker
Sarah Cannon Research Institute
Recruiting
London, United Kingdom, W1G 6AD
Contact: Hendrik-Tobias Arkenau
Principal Investigator: Hendrik-Tobias Arkenau
University College London
Recruiting
London, United Kingdom, WC1E 6BT
Contact: Rebecca Kristeleit r.kristeleit@ucl.ac.uk
Principal Investigator: Rebecca Kristeleit
The Christie NHS Foundation Trust
Recruiting
Manchester, United Kingdom, M20 4BX
Contact: Louise Carter louise.carter@christie.nhs.uk
Principal Investigator: Louise Carter
Freeman Hospital
Recruiting
Newcastle upon Tyne, United Kingdom, NE7 7DN
Contact: Elizabeth R Plummer Ruth.Plummer@newcastle.ac.uk
Principal Investigator: Elizabeth R Plummer, MD
Sheffield Teaching Hospitals
Recruiting
Sheffield, United Kingdom, S10 2SJ
Contact: Sarah Danson
Principal Investigator: Sarah Danson Less << |
|
NCT02871518
|
Nasopharyngeal Carcinoma |
Phase 2 |
Recruiting |
July 2021 |
China, Guangdong
... More >>
Department of Nasopharyngeal Carcinoma, Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Haiqiang Mai, MD,Ph.D 86-20-8734-3643 maihq@mail.sysu.edu.cn Less << |
|
NCT01124253
|
Non-small Cell Lung Cancer |
Phase 3 |
Completed |
- |
China
... More >> The Lung Cancer Center of shanghai chest Hospital
Shanghai, China, 2000043 Less << |
|
NCT00004547
|
- |
|
Completed |
- |
- |
|
NCT00683514
|
NSCLC |
Phase 3 |
Completed |
- |
Germany
... More >> Pierre Fabre Pharma GmbH
Freiburg, Jechtinger Str. 13, Germany, D-79111 Less << |
|
NCT00085631
|
- |
|
Terminated(Study was closed be... More >>cause of slow accrual) Less << |
- |
- |
|
NCT00085631
|
Cervical Cancer |
Phase 3 |
Terminated(Study was closed be... More >>cause of slow accrual) Less << |
- |
United States, North Carolina
... More >>
Duke Cancer Institute
Durham, North Carolina, United States, 27710 Less << |
|
NCT00673179
|
Osteosarcoma |
Not Applicable |
Terminated(Low accrual.) |
- |
United States, Texas
... More >>
UT MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00268372
|
Head and Neck Cancer |
Phase 3 |
Terminated(SWOG eliminated its... More >> head and neck committee) Less << |
- |
- |
|
NCT01655628
|
Stage IV Nasopharyngeal Carcin... More >>oma Less << |
Phase 2 |
Unknown |
December 2014 |
China, Guangdong
... More >>
Cancer Center, Sun Yat-sen University
Recruiting
Guangzhou city, Guangdong, China, 510080
Contact: Jian-jun Li, M.D. Ph.D 86-2087343381 lijj@sysucc.org.cn
Principal Investigator: Jian-jun Li, M.D. Ph.D. Less << |
|
NCT01588431
|
Carcinoma, Squamous Cell of He... More >>ad and Neck Less << |
Phase 2 |
Active, not recruiting |
March 2022 |
United States, Texas
... More >>
Cancer Therapy and Research Center at UTHSCSA
San Antonio, Texas, United States, 78229 Less << |
|
NCT00673179
|
- |
|
Terminated(Low accrual.) |
- |
- |
|
NCT00220103
|
Adenocarcinoma of Oesophagus |
Phase 2 |
Completed |
- |
United Kingdom
... More >> Royal Marsden NHS Foundation Trust
Sutton, Surrey, United Kingdom Less << |
|
NCT00447421
|
Small Cell Lung Cancer
... More >> Carcinoma, Small Cell
SCLC Less << |
Phase 1
Phase 2 |
Terminated(Interim results of ... More >>another trial showed inferior activity of treatment) Less << |
- |
Belgium
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Leuven, Belgium, 3000
Netherlands
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Amsterdam, Netherlands, 1081 HV
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Den Bosch, Netherlands, 5211 NL
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Rotterdam, Netherlands, 3075 EA Less << |
|
NCT00064077
|
- |
|
Completed |
- |
- |
|
NCT01915134
|
Effects of Chemotherapy
... More >> Stage IVC Nasopharyngeal Carcinoma Less << |
Phase 3 |
Unknown |
December 2016 |
China, Zhejiang
... More >>
Zhejiang Cancer Hospital
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: Bin Li 86-571-88122091 libindoctor@163.com
Contact: Xinglai Feng 86-571-88122092 fengxinglai@hotmail.com
Principal Investigator: Bin Li Less << |
|
NCT00093665
|
Head and Neck Cancer |
Phase 2 |
Unknown |
- |
Japan
... More >> National Hospital Organization - Medical Center of Kure
Hiroshima, Japan, 737-0023
Kanazawa University
Kanazawa, Japan, 920-8641
Kyoto Prefectural University of Medicine
Kyoto, Japan, 602
Aichi Cancer Center
Nagoya, Japan, 464-8681
Nara Medical University Cancer Center
Nara, Japan, 634-8522
Graduate School of Medical Science at the University of Ryukyu
Okinawa, Japan, 903-0215
Mie University School of Medicine
Tsu, Japan, 514-8507 Less << |
|
NCT02432378
|
Cancer of Ovary
... More >> Cancer of the Ovary
Neoplasms, Ovarian
Ovarian Cancer
Ovary Cancer
Ovary Neoplasms Less << |
Phase 1
Phase 2 |
Recruiting |
December 2020 |
United States, Pennsylvania
... More >>
UPMC CancerCenter at Magee-Womens Hospital of UPMC
Recruiting
Pittsburgh, Pennsylvania, United States, 15213
Hillman Cancer Center
Not yet recruiting
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT00109954
|
Liver Cancer |
Phase 3 |
Completed |
- |
United States, Pennsylvania
... More >>
Hillman Cancer Center at University of Pittsburgh Cancer Institute
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT00064077
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Carcinoma
Cervical Squamous Cell Carcinoma
Recurrent Cervical Carcinoma
Stage IVB Cervical Cancer Less << |
Phase 3 |
Completed |
- |
United States, Pennsylvania
... More >>
Gynecologic Oncology Group
Philadelphia, Pennsylvania, United States, 19103 Less << |
|
NCT01733823
|
HNSCC,Larynx, Pharynx and Oral... More >> Cavity Less << |
Phase 1
Phase 2 |
Completed |
- |
Denmark
... More >> Aarhus
Aarhus, Aarhus C, Denmark, 8000
Odense
Odense, Odense C, Denmark, 5000
Aalborg
Aalborg, Denmark, 9100
Herlev
Herlev, Denmark, 2730 Less << |
|
NCT02281708
|
Stage Ib Lung Carcinoma |
Phase 3 |
Recruiting |
September 2020 |
Korea, Republic of
... More >>
Seoul National University Bundang Hospital
Recruiting
Seongnam-si, Gyeonggi-do, Korea, Republic of, 463-707
Contact: Sanghoon Jheon, Ph.D. 82317877140 jheon@snubh.org Less << |
|
NCT00447421
|
- |
|
Terminated(Interim results of ... More >>another trial showed inferior activity of treatment) Less << |
- |
- |
|
NCT01931163
|
Breast Cancer
... More >> Triple Negative Breast Cancer Less << |
Phase 2 |
Active, not recruiting |
August 2019 |
United States, Texas
... More >>
The Methodist Hospital
Houston, Texas, United States, 77030
Houston Methodist Hospital Willowbrook
Houston, Texas, United States, 77070
Houston Methodist Hospital Sugar Land
Sugar Land, Texas, United States, 77479 Less << |
|
NCT02461043
|
Esophageal Cancer |
Phase 3 |
Recruiting |
December 2023 |
China
... More >> Cancer Institute and Hospital, Chinese Academy of Medical Sciences
Recruiting
Beijing, China, 100021
Contact: Xi Wang, M.D
Principal Investigator: Jie He, M.D. Less << |
|
NCT00290966
|
Stomach Neoplasm |
Phase 2
Phase 3 |
Completed |
- |
- |
|
NCT01640860
|
- |
|
Completed |
- |
Korea, Republic of
... More >>
Chonnam National University Hwasun Hospital
Hwasun, Jeolanamdo, Korea, Republic of, 519-809 Less << |
|
NCT01787006
|
Esophageal Cancer |
Phase 2 |
Unknown |
- |
Germany
... More >> Universitätsklinikum Schleswig-Holstein, Campus Lübeck, Klinik für Strahlentherapie
Recruiting
Lübeck, Germany, 23538
Contact: Dirk Rades, Prof. Dr. 0049 451 500-6660 dirk.rades@uksh.de
Principal Investigator: Dirk Rades, Prof. Dr. Less << |
|
NCT03468010
|
Cervical Cancer
... More >> Adjuvant Chemotherapy
Radiation
Lymph Node Metastases Less << |
Phase 3 |
Recruiting |
March 1, 2025 |
China, Guangdong
... More >>
Sun Yat-sen University Affiliated Foshan Hospital
Not yet recruiting
Foshan, Guangdong, China
Contact: Rong Huang, M.D 86-13927736853 nnjbhg@163.com
Hospital of of Guangdong Armed Police Corps
Not yet recruiting
Guangzhou, Guangdong, China
Contact: Zhen-lun Li, M.D 86-13580516205 sfqiu@126.com
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China
Contact: Wei-jun Ye, M.D 86-13538799871 yewj@sysucc.org.cn
Contact: Junyun Li, M.D 86-18824712702 LIJUNY@sysucc.org.cn
The First affiliated Hospital of Guangdong Pharmaceutical University
Not yet recruiting
Guangzhou, Guangdong, China
Contact: Xi-cheng Wang, M.D 86-13902400598 13902400598@126.com
Guangzhou First People's Hospital
Not yet recruiting
Guanzhou, Guangdong, China
Contact: Guo-long Liu, M.D 86-13802527172 liugl@fimmu.com
China, Guangxi
The People's Hospital of Guangxi Zhuang Autonomous Region
Not yet recruiting
Nanning, Guangxi, China
Contact: Jia-xin Chen 86-13978609888 cjx166@163.com
China, Hainan
Hainan General Hospital
Not yet recruiting
Haikou, Hainan, China
Contact: Guang Huang, M.D 86-18089777161 2842749787@qq.com
China, Xinjiang
Xinjiang Medical University Affiliated Tumor Hospital
Not yet recruiting
Ürümqi, Xinjiang, China
Contact: Xiao-wen Li, M.D 86-13899856295 lixiaowen1026@sohu.com Less << |
|
NCT00102505
|
Non-Small-Cell Lung Carcinoma
... More >>
Carcinoma, Bronchogenic Less << |
Phase 1 |
Completed |
- |
United States, Texas
... More >>
The University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00006464
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, California
... More >>
USC/Norris Comprehensive Cancer Center and Hospital
Los Angeles, California, United States, 90033-0804
City of Hope Medical Group
Pasadena, California, United States, 91105
University of California Davis Cancer Center
Sacramento, California, United States, 95817
United States, New Hampshire
Norris Cotton Cancer Center
Lebanon, New Hampshire, United States, 03756-0002 Less << |
|
NCT00293085
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT01590017
|
Cervical Cancer
... More >> Acquired Immunodeficiency Syndrome Less << |
Phase 2 |
Completed |
- |
South Africa
... More >> Clinical HIV Research Unit, a Division of the Wits Health Consortium (Pty) Ltd
Johannesburg, South Africa, 2092
Zimbabwe
University of Zimbabwe Clinical Research Centre / Parirenyatwa Hospital
Harare, Zimbabwe Less << |
|
NCT01372111
|
Head and Neck Cancer |
Phase 2 |
Unknown |
- |
United States, North Carolina
... More >>
Coleman Radiation Oncology Center
Morehead City, North Carolina, United States, 28557
CarolinaEast Cancer Care
New Bern, North Carolina, United States, 28561
South Atlantic Radiation Oncology
Supply, North Carolina, United States, 28462
Coastal Carolina Radiation Oncology
Wilmington, North Carolina, United States, 28401
Zimmer Cancer Center
Wilmington, North Carolina, United States, 28401 Less << |
|
NCT00887159
|
Extensive Stage Small Cell Lun... More >>g Carcinoma
Recurrent Small Cell Lung Carcinoma Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01928160
|
Recurrent Non-small Cell Lung ... More >>Cancer
Stage IV Non-small Cell Lung Cancer Less << |
Phase 2 |
Withdrawn(study not accruing) |
- |
- |
|
NCT01540045
|
- |
|
Completed |
- |
- |
|
NCT00820248
|
- |
|
Completed |
- |
- |
|
NCT00820248
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
Canada, Alberta
... More >>
Cross Cancer Institute
Edmonton, Alberta, Canada, T6G 1Z2
Canada, British Columbia
BCCA - Fraser Valley Cancer Centre
Surrey, British Columbia, Canada, V3V 1Z2
BCCA - Vancouver Cancer Centre
Vancouver, British Columbia, Canada, V5Z 4E6
Canada, Manitoba
CancerCare Manitoba
Winnipeg, Manitoba, Canada, R3E 0V9
Canada, New Brunswick
Atlantic Health Sciences Corporation
Saint John, New Brunswick, Canada, E2L 4L2
Canada, Newfoundland and Labrador
Dr. H. Bliss Murphy Cancer Centre
St. John's, Newfoundland and Labrador, Canada, AIB 3V6
Canada, Ontario
Juravinski Cancer Centre at Hamilton Health Sciences
Hamilton, Ontario, Canada, L8V 5C2
Cancer Centre of Southeastern Ontario at Kingston
Kingston, Ontario, Canada, K7L 5P9
London Regional Cancer Program
London, Ontario, Canada, N6A 4L6
Ottawa Health Research Institute - General Division
Ottawa, Ontario, Canada, K1H 8L6
Northeast Cancer Center Health Sciences
Sudbury, Ontario, Canada, P3E 5J1
Thunder Bay Regional Health Science Centre
Thunder Bay, Ontario, Canada, P7B 6V4
Univ. Health Network-Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9
Canada, Quebec
Hopital Maisonneuve-Rosemont
Montreal, Quebec, Canada, H1T 2M4
McGill University - Dept. Oncology
Montreal, Quebec, Canada, H2W 1S6
CHUQ-Pavillon Hotel-Dieu de Quebec
Quebec City, Quebec, Canada, G1R 2J6
Centre hospitalier universitaire de Sherbrooke
Sherbrooke, Quebec, Canada, J1H 5N4
Canada, Saskatchewan
Allan Blair Cancer Centre
Regina, Saskatchewan, Canada, S4T 7T1
Saskatoon Cancer Centre
Saskatoon, Saskatchewan, Canada, S7N 4H4 Less << |
|
NCT01540045
|
- |
|
Completed |
- |
Mexico
... More >> National Cancer Institute of Mexico
Mexico city, Distrito Federal, Mexico, 14080 Less << |
|
NCT01682031
|
Chemotherapeutic Agent Toxicit... More >>y
Mucositis
Radiation Toxicity
Stage III Squamous Cell Carcinoma of the Hypopharynx
Stage III Squamous Cell Carcinoma of the Larynx
Stage III Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage III Squamous Cell Carcinoma of the Nasopharynx
Stage III Squamous Cell Carcinoma of the Oropharynx
Stage III Squamous Cell Carcinoma of the Paranasal Sinus and Nasal Cavity
Stage IV Squamous Cell Carcinoma of the Hypopharynx
Stage IV Squamous Cell Carcinoma of the Larynx
Stage IV Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage IV Squamous Cell Carcinoma of the Nasopharynx
Stage IV Squamous Cell Carcinoma of the Oropharynx
Stage IV Squamous Cell Carcinoma of the Paranasal Sinus and Nasal Cavity
Xerostomia Less << |
Phase 2 |
Terminated(Due to a lack of fu... More >>nding) Less << |
- |
United States, New York
... More >>
Roswell Park Cancer Institute
Buffalo, New York, United States, 14263
New Zealand
Waikato Hospital
Hamilton, New Zealand, 3204 Less << |
|
NCT01682031
|
- |
|
Terminated(Due to a lack of fu... More >>nding) Less << |
- |
- |
|
NCT00887159
|
- |
|
Completed |
- |
- |
|
NCT00576914
|
Non-small Cell Lung Cancer |
Phase 3 |
Unknown |
June 2012 |
China, Beijing
... More >> The Lung Cancer Center of Cancer Hospital
Recruiting
Beijing, Beijing, China, 100021
Contact: Xinling Liu, Bachelor 86-10-87788495 liuxinling1985@126.com
Contact: Fang Li, M.D. 86-13910145882 nc_lifang@yahoo.com.cn
Sub-Investigator: Xiangru Zhang, M.D. Less << |
|
NCT01669226
|
Bulky Stage IIIC and IV Epithe... More >>lial Ovarian Cancer
Fallopian Tube Cancer
Primary Peritoneal Carcinoma Less << |
Phase 2 |
Completed |
- |
China, Jiangsu
... More >> Zhongda Hospital Southeast University
Nanjing, Jiangsu, China, 210009
Suzhou Municipal Hospital
Suzhou, Jiangsu, China
Wuxi Cancer Hospital
Wuxi, Jiangsu, China
China, Shanghai
Shanghai Zhongshan Hospital
Shanghai, Shanghai, China, 200030
Ren Ji Hospital Affiliated to Shanghai JiaoTong University School of Medicine
Shanghai, Shanghai, China
Shanghai Frist Maternity and Infant Hospital Affiliated to Tongji University
Shanghai, Shanghai, China
China
Fudan University Cancer Hospital
Shanghai, China, 200032 Less << |
|
NCT00010023
|
Cancer |
Phase 1 |
Completed |
- |
United States, New York
... More >>
NYU Cancer Institute at New York University Medical Center
New York, New York, United States, 10016 Less << |
|
NCT01679405
|
Metastatic Disease |
Phase 1 |
Terminated(2016/04/26) |
December 2016 |
Germany
... More >> I. Medizinische Klinik und Poliklinik der Universitätsmedizin
Mainz, Germany, 55131 Less << |
|
NCT00548548
|
- |
|
Completed |
- |
- |
|
NCT00003809
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00590967
|
Cervical Cancer |
Phase 2 |
Terminated(Due to low accrual) |
- |
United States, Missouri
... More >>
Washington University School of Medicine
St. Louis, Missouri, United States, 63110 Less << |
|
NCT00601692
|
Esophageal Cancer |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT00590967
|
- |
|
Terminated(Due to low accrual) |
- |
- |
|
NCT01461759
|
Advanced or Recurrent Endometr... More >>ial Cancer Less << |
Phase 2 |
Recruiting |
December 2017 |
Korea, Republic of
... More >>
Asan Medical Center
Recruiting
Seoul, Korea, Republic of, 138-736
Contact: Joo-Hyun Nam, M.D., Ph.D +82-2-3010-3633 jhnam@amc.seoul.kr Less << |
|
NCT01861496
|
Phase 1: Advanced or Refractor... More >>y Solid Tumours
Phase 2 Part: Metastatic Breast Cancer and Skin Cancer Less << |
Phase 1
Phase 2 |
Recruiting |
May 2019 |
Denmark
... More >> Aalborg Universitetshospital
Withdrawn
Aalborg, Denmark, 9100
The Phase One Unit, The Finsen Centre, Rigshospitalet
Active, not recruiting
Copenhagen, Denmark, DK-2100
Herlev & Gentofte Hospital
Recruiting
Herlev, Denmark, 2730
Contact: Dorte Nielsen, Professor MD +45 38682344 Dorte.Nielsen01@rehionh.dk
Contact: Peter G. Sørensen, MD PhD +4538689044 PeteGrundtvig.Sørensen@regionh.dk
Nordsjællands Hospital Hillerød
Recruiting
Hillerod, Denmark, 3400
Contact: Hella Danoe, MD +45 48297309 Hella.danoe.01@regionh.dk
Vejle Sygehus
Recruiting
Vejle, Denmark, 7100
Contact: Erik Hugger Jakobsen, MD +45 79406015 Erik.hugger.jakobsen@rsyd.dk Less << |
|
NCT00006028
|
Ovarian Cancer
... More >> Primary Peritoneal Cavity Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT03324282
|
Bladder Carcinoma |
Phase 2 |
Recruiting |
December 31, 2022 |
France
... More >> CHU de Besançon
Recruiting
Besançon, France
Contact: Antoine THIERY VUILLEMIN +33 (0)3 81 66 87 05 a.thieryvuillemin@mac.com
CHU de Bordeaux
Recruiting
Bordeaux, France
Contact: Alain RAVAUD
Contact +33 (0)5 56 79 58 08 alain.ravaud@chu-bordeaux.fr
Institut Bergonié
Recruiting
Bordeaux, France
Contact: Guilhem ROUBAUD +33 (0)5 24 07 19 12 G.Roubaud@bordeaux.unicancer.fr
Centre François Baclesse
Recruiting
Caen, France
Contact: Elodie COQUAN +33 (0)2 31 45 50 02 coque@baclesse.unicancer.fr
Centre Léon Bérard
Recruiting
Lyon, France
Contact: Aude FLECHON +33 (0)4 78 78 26 43 aude.flechon@lyon.unicancer.fr
Institut Paoli Calmettes
Not yet recruiting
Marseille, France
Contact: Gwenaëlle GRAVIS +33 (0)4 91 22 37 40 gravisg@ipc.unicancer.fr
Institut de cancérologie de l'Ouest - René Gauducheau
Recruiting
Nantes, France
Contact: Frédéric ROLLAND +33 (0)2 40 67 99 76 frederic.rolland@ico.unicancer.fr
Hôpital Européen Georges-Pompidou, AP-HP
Recruiting
Paris, France
Contact: Stéphane OUDARD +33 (0)1 56 09 34 47 stephane.oudard@aphp.fr
Hôpital Saint-Louis, AP-HP
Recruiting
Paris, France
Contact: Stéphane CULINE +33 (0)1 42 49 42 47 stephane.culine@aphp.fr
CHU de Strasbourg
Recruiting
Strasbourg, France
Contact: Philippe BARTHELEMY +33 (0)3 88 11 67 28 Philippe.barthelemy@chru-strasbourg.fr
Institut Universitaire du Cancer de Toulouse - Oncopole
Recruiting
Toulouse, France
Contact: Christine CHEVREAU +33 (0)5 31 15 51 52 Chevreau.Christine@iuct-oncopole.fr
Institut Gustave Roussy
Not yet recruiting
Villejuif, France
Contact: Yohann LORIOT +33 (0)1 42 11 52 76 yohann.loriot@gustaveroussy.fr Less << |
|
NCT01232452
|
- |
|
Completed |
- |
- |
|
NCT00548548
|
Adenocarcinoma |
Phase 3 |
Completed |
- |
United States, California
... More >>
Tower Cancer Research Fnd
Beverly Hills, California, United States, 90211-1850
Moores UCSD Cancer Center
La Jolla, California, United States, 92093
Kenmar Research Institute LLC
Los Angeles, California, United States, 90057
USC/Norris Cancer Center
Los Angeles, California, United States, 90089
United States, District of Columbia
Georgetown University
Washington, D.C., District of Columbia, United States, 20007
United States, Florida
Florida Cancer Specialists
Fort Myers, Florida, United States, 33916
H. Lee Moffitt Cancer
Tampa, Florida, United States, 33612
United States, Kansas
Cancer Center of Kansas
Wichita, Kansas, United States, 67214-3728
United States, Nebraska
Methodist Cancer Center Onc
Omaha, Nebraska, United States, 68114
United States, New York
Memorial Sloan Kettering
New York, New York, United States, 10021
United States, North Carolina
Duke Univ Medical Center
Durham, North Carolina, United States, 27710
United States, South Carolina
South Carolina Oncology Assoc
Columbia, South Carolina, United States, 10595
United States, Tennessee
The Sarah Cannon Research Inst
Nashville, Tennessee, United States, 37203 Less << |
|
NCT03541252
|
Carcinoma, Basal Cell |
Phase 1
Phase 2 |
Recruiting |
May 2019 |
Denmark
... More >> Department of Dermatology, Bispebjerg Hospital
Recruiting
Copenhagen, Hovedstaden, Denmark, 2400
Contact: Emily C Wenande, MD +4540505590 emilycathrinew@hotmail.com Less << |
|
NCT00063999
|
- |
|
Unknown |
- |
- |
|
NCT01232452
|
Non-Small-Cell Lung Carcinoma |
Phase 2 |
Completed |
- |
- |
|
NCT00248495
|
- |
|
Completed |
- |
- |
|
NCT00248495
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Roswell Park Cancer Institute
Buffalo, New York, United States, 14263-0001 Less << |
|
NCT00736944
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 2 |
Active, not recruiting |
August 31, 2020 |
United States, Missouri
... More >>
Washington University
Saint Louis, Missouri, United States, 63110 Less << |
|
NCT00005965
|
Cervical Cancer |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Albert Einstein Comprehensive Cancer Center
Bronx, New York, United States, 10461
Saint Vincent Catholic Medical Center of New York
New York, New York, United States, 10011
NYU School of Medicine's Kaplan Comprehensive Cancer Center
New York, New York, United States, 10016
New York Presbyterian Hospital - Cornell Campus
New York, New York, United States, 10021
Mount Sinai Medical Center, NY
New York, New York, United States, 10029
New York Medical College
Valhalla, New York, United States, 10595 Less << |
|
NCT00063999
|
Recurrent Uterine Corpus Carci... More >>noma
Stage IIIA Uterine Corpus Cancer
Stage IIIB Uterine Corpus Cancer
Stage IIIC Uterine Corpus Cancer
Stage IVA Uterine Corpus Cancer
Stage IVB Uterine Corpus Cancer Less << |
Phase 3 |
Unknown |
- |
- |
|
NCT00020696
|
Ovarian Cancer
... More >> Primary Peritoneal Cavity Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01069081
|
Breast Cancer
... More >> Metastasis, Neoplasm Less << |
Phase 2 |
Completed |
- |
China, Shanghai
... More >>
Fudan University Cancer Hospital
Shanghai, Shanghai, China, 200032 Less << |
|
NCT03189719
|
Esophageal Neoplasms |
Phase 3 |
Recruiting |
August 22, 2021 |
- |
|
NCT00003345
|
Fallopian Tube Cancer
... More >> Ovarian Cancer
Primary Peritoneal Cavity Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT01379976
|
Non Small Cell Lung Cancer |
Phase 3 |
Terminated(Recruitment was pre... More >>maturely stopped due to difficulty of recruitment encountered by the experimental centers) Less << |
- |
Italy
... More >> Istituto Oncologico del Mediterraneo
Viagrande, CT, Italy, 95029
Azienda Ospedaliera Ospedale S. Anna
Como, Italy, 22020
Azienda Ospedaliera Istituti Ospitalieri
Cremona, Italy, 26100
Azienda Ospedaliera di Desio e Vimercate - Presidio Ospedaliero di Desio
Desio, Italy, 20832
Ospedale Civile
Guastalla, Italy, 42016
Ospedale Alessandro Manzoni
Lecco, Italy, 23900
Azienda Ospedaliera Ospedale Civile di Legnano
Legnano, Italy, 20025
Azienda Ospedaliera Fatebenefratelli e Oftalmico
Milano, Italy, 20121
Istituto Europeo Di Oncologia
Milano, Italy, 20141
Azienda Ospedaliera San Paolo
Milano, Italy, 20142
Azienda Ospedaliera Ospedale San Carlo Borromeo
Milano, Italy, 20153
Azienda Ospedaliero Universitaria di Parma
Parma, Italy, 43126
Fondazione Salvatore Maugeri
Pavia, Italy, 0381 33329
Azienda Ospedaliera Perugia
Perugia, Italy, 06075
Arcispedale S. Maria Nuova
Reggio Emilia, Italy, 42100
IRCCS di Reggio Emilia
Reggio Emilia, Italy, 42123
Azienda Ospedaliera Busto Arsizio - Presidio Ospedaliero di Saronno
Saronno, Italy, 21047
Ospedale SS Annunziata - ASL1
Sassari, Italy, 07100
Azienda Ospedaliera Valtellina e Valchiavenna , Presidio Ospedaliero di Sondrio
Sondrio, Italy, 23100
Azienda Ospedaliera di Pavia, Ospedale Civile di Vigevano
Vigevano, Italy, 27029
Azienda Ospedaliera di Desio e Vimercate - Presidio Ospedaliero di Vimercate
Vimercate, Italy, 20059 Less << |
|
NCT01763788
|
Squamous Non-small Cell Lung C... More >>ancer Less << |
Phase 1
Phase 2 |
Active, not recruiting |
May 2018 |
Japan
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Aichi, Japan, 464-8681
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Chiba, Japan, 277 8577
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Ehime, Japan, 791-0280
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Fukuoka, Japan, 830-0011
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hokkaido, Japan, 070-8644
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hyogo, Japan, 650-0047
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Ishikawa, Japan, 920-8641
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Kanagawa, Japan, 240-0062
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Koto-ku, Japan, 135-8550
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Miyagi, Japan, 980-8574
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Nagasaki, Japan, 852-8501
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Niigata, Japan, 951-8566
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Oita, Japan, 8795593
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Okayama, Japan, 700-8558
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Osaka, Japan, 573-1191
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Saitama, Japan, 362-0806
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Sendai, Japan, 980-0873
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Shizuoka, Japan, 411-8777
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Wakayama, Japan, 641-8510
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Yamaguchi, Japan, 755-0241 Less << |
|
NCT00736944
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00003029
|
Pancreatic Cancer |
Phase 3 |
Completed |
- |
Belgium
... More >> Hopital de Jolimont
Haine Saint Paul, Belgium, 7100
Les Cliniques Saint-Joseph ASBL
Liege, Belgium, B 4000
France
Centre Hospitalier de la Cote Basque
Bayonne, France, 64109
Centre Jean Perrin
Clermont-Ferrand, France, 63011
Centre de Lutte Contre le Cancer, Georges-Francois Leclerc
Dijon, France, 21079
Hopital Perpetuel Secours
Levallois-Perret, France, 92300
Centre Hospital Regional Universitaire de Limoges
Limoges, France, 87042
Hopital Notre-Dame de Bon Secours
Metz, France, 55038
Centre Hospitalier de Montlucon
Montlucon, France, 03109
Clinique Hartmann
Neuilly sur Seine, France, 92200
Hopital Saint-Louis
Paris, France, 75475
Hopital Cochin
Paris, France, 75674
Centre Rene Huguenin
Saint Cloud, France, 92211
Hopital Bellevue
Saint Etienne, France, 42055
Clinique de l'Orangerie
Strasbourg, France, 67010
Hopital Paul Brousse
Villejuif, France, 94804
Israel
Wolfson Medical Center
Holon, Israel, 58100
Italy
Universita G.D'Annunzio Di Chieti
Chieti, Italy, 66100
Portugal
Hospital Fernando Fonseca
Amadora, Portugal, P-2700 Less << |
|
NCT00997906
|
Head and Neck Cancer |
Phase 2
Phase 3 |
Active, not recruiting |
July 2023 |
Singapore
... More >> National Cancer Centre - Singapore
Singapore, Singapore, 169610 Less << |
|
NCT02254278
|
Stage III Oropharyngeal Squamo... More >>us Cell Carcinoma
Stage IVA Oropharyngeal Squamous Cell Carcinoma
Stage IVB Oropharyngeal Squamous Cell Carcinoma
Stage IVC Oropharyngeal Squamous Cell Carcinoma
Tongue Carcinoma Less << |
Phase 2 |
Active, not recruiting |
May 31, 2024 |
- |
|
NCT00068406
|
- |
|
Completed |
- |
- |
|
NCT00068406
|
Stage III Vulvar Cancer
... More >> Stage IVB Vulvar Cancer
Vulvar Squamous Cell Carcinoma Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00001450
|
Carcinoma, Non-Small-Cell Lung... More >>
Lung Neoplasms Less << |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
National Cancer Institute (NCI)
Bethesda, Maryland, United States, 20892 Less << |
|
NCT02967497
|
Epithelial Growth Factor Recep... More >>tor Negative Non-small Cell Lung Cancer Less << |
Phase 2 |
Unknown |
September 2018 |
- |
|
NCT01830270
|
Stomach Neoplasms
... More >> Oesophageal Junction Cancer
Lower Oesophagus Cancer Less << |
Phase 2 |
Terminated |
- |
France
... More >> University hospital of Besançon
Besançon, France, 25000
FNLCC center Georges François Leclerc
Dijon, France, 21000
Hospital of Belfort-Montbeliard
Montbeliard, France, 25200 Less << |
|
NCT01303926
|
Non-squamous Nonsmall Cell Neo... More >>plasm of Lung Less << |
Phase 3 |
Unknown |
June 2012 |
Italy
... More >> "Giovanni Paolo II" Oncology Institute
Bari, BA, Italy, 70124
"San Paolo Hospital" Oncology Service
Bari, BA, Italy
Division of Medical Oncology, "Fatebenefratelli" Hospital
Benevento, BN, Italy
Division of Medical Oncology, "Sen. Perrino" Hospital, Brindisi, Italy
Brindisi, BR, Italy, 72100
7 Division of Medical Oncology, "Casa Sollievo della Sofferenza" Hospital,
San Giovanni Rotondo, FG, Italy
Medical Oncology Division "Vito Fazzi" Hospital
Lecce, Le, Italy
Division of Medical Oncology, "Buccheri-La Ferla" Hospital
Palermo, PA, Italy
Division of Medical Oncology, "La Maddalena" Hospital
Palermo, PA, Italy
Division of Medical Oncology, Castellaneta Hospital
Castellaneta, TA, Italy
Division of Medical Oncology "San Giuseppe Moscati Hospital"
Taranto, TA, Italy
Clinical Trials Office, Department of Medical Sciences, Azienda ULSS 13
Mirano, VE, Italy
National Cancer Institute "G. Pascale" Thoracic Dept.
Napoli, Italy Less << |
|
NCT03340896
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 3 |
Recruiting |
June 30, 2021 |
France
... More >> Hôpital Bretonneau, Service CORad Pôle Henry S Kaplan
Recruiting
Tours, France, 37044
Contact: Gilles CALAIS, Pr + 33 247478265 gilles.calais@univ-tours.fr
Contact: Marie-Hélène GIRARD CALAIS +33 247479121 rc.corad@chu-tours.fr Less << |
|
NCT02709512
|
Mesothelioma |
Phase 2
Phase 3 |
Recruiting |
June 2021 |
- |
|
NCT02036359
|
Non-small Cell Lung Cancer(NSC... More >>LC) Less << |
Phase 2 |
Unknown |
- |
Taiwan
... More >> Department of Oncology, National Taiwan University Hospital
Recruiting
Taipei,, Taiwan
Contact: Chong Jen YU, M.D., Ph.D. jefferycjyu@ntu.edu.tw Less << |
|
NCT00005868
|
Lung Cancer |
Phase 2 |
Completed |
- |
Germany
... More >> Thoraxklinik Rohrbach
Heidelberg, Germany, D-69126
Italy
Istituto Nazionale per la Ricerca sul Cancro
Genoa (Genova), Italy, 16132
Oncologia Medica - Perugia
Perugia, Italy, 06122
Netherlands
Leyenburg Ziekenhuis
's-Gravenhage (Den Haag, The Hague), Netherlands, 2545 CH
Groot Ziekengasthuis 's-Hertogenbosch
's-Hertogenbosch, Netherlands, 5211 NL
Vrije Universiteit Medisch Centrum
Amsterdam, Netherlands, 1001HV
Antoni van Leeuwenhoekhuis
Amsterdam, Netherlands, 1066 CX
Slotervaart Ziekenhuis
Amsterdam, Netherlands, 1066 EC
Onze Lieve Vrouwe Gasthuis
Amsterdam, Netherlands, 1091 HA
Longarts Dr. J.M. Smit Rynstate Hospital
Arnhem, Netherlands, 6800 TA
Ziekenhuis St Jansdal
Harderwijk, Netherlands, 3840 AC
Leiden University Medical Center
Leiden, Netherlands, 2300 CA
Sint Antonius Ziekenhuis
Nieuwegein, Netherlands, 3435 CM
Canisius-Wilhelmina Ziekenhuis
Nijmegen, Netherlands, 6532 SZ
University Medical Center Nijmegen
Nijmegen, Netherlands, NL-6500 HB
University Hospital - Rotterdam Dijkzigt
Rotterdam, Netherlands, 3000 CA
Twee Steden Ziekenhuis Vestiging Tilburg
Tilburg, Netherlands, 5042 AD
Academisch Ziekenhuis Utrecht
Utrecht, Netherlands, 3584 CX
Zaas Medisch Centrum
Zaandam, Netherlands, 1502 DV
Sophia Ziekehuis
Zwolle, Netherlands, 8000 GK
Poland
Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology
Warsaw, Poland, 02-781
United Kingdom
Weston Park Hospital
Sheffield, England, United Kingdom, S1O 2SJ
Royal Marsden Hospital
Sutton, England, United Kingdom, SM2 5PT Less << |
|
NCT03058094
|
NSCLC |
Phase 3 |
Not yet recruiting |
January 2020 |
China, Beijing
... More >> Beijing Cancer Hospital
Not yet recruiting
Beijing, Beijing, China, 100000
Contact: Jian Fang
Chinese General PLA Hospital
Not yet recruiting
Beijing, Beijing, China, 100000
Contact: Yi Hu
Peking Union Medical College Hospital
Not yet recruiting
Beijing, Beijing, China, 100000
Contact: Mengzhao Wang
China, Chongqing
Xinqiao Hospital Army Medical University
Not yet recruiting
Chongqing, Chongqing, China, 400000
Contact: Dong Wang
China, Fujian
Fujian Cancer Hospital
Not yet recruiting
Fuzhou, Fujian, China, 350000
Contact: Cheng Huang
Fuzhou General Hospital of Nanjing Military Command
Not yet recruiting
Fuzhou, Fujian, China, 350000
Contact: Xuenong Ouyang
China, Hebei
Tumor Hospital of Hebei Province
Not yet recruiting
Shijiazhuang, Hebei, China, 050000
Contact: Cuimin Ding
China, Heilongjiang
Harbin Medical University Cancer Hospital
Not yet recruiting
Harbin, Heilongjiang, China, 150000
Contact: Gongyan Chen
China, Henan
Henan Cancer Hospital
Not yet recruiting
Zhengzhou, Henan, China, 450000
Contact: Zhiyong Ma
China, Jiangsu
Jiangsu Province Hospital
Not yet recruiting
Nanjing, Jiangsu, China, 210000
Contact: Yongqian Shu
Nanjing General Hospital
Not yet recruiting
Nanjing, Jiangsu, China, 210000
Contact: Yong Song
China, Liaoning
The First Affiliated Hospital of Dalian Medical University
Not yet recruiting
Dalian, Liaoning, China, 116000
Contact: Jiwei Liu
The second Hospital of Dalian Medical University
Not yet recruiting
Dalian, Liaoning, China, 116000
Contact: Yang Zhang
The First Affiliated Hospital of China Medical University
Not yet recruiting
Shenyang, Liaoning, China, 110000
Contact: Yunpeng Liu
China, Shanghai
Fudan University Shanghai Cancer Center
Not yet recruiting
Shanghai, Shanghai, China, 200000
Contact: Jianhua Chang
China, Sichuan
Sichuan Cancer Hospital & Institute
Not yet recruiting
Chengdu, Sichuan, China, 610000
Contact: Ping Yu
West China Hospital,Sichuan University
Not yet recruiting
Chengdu, Sichuan, China, 610000
Contact: You Lu
China, Tianjin
Tianjin Medical University Cancer Institute & Hospital
Not yet recruiting
Tianjin, Tianjin, China, 300000
Contact: Kai Li
China, Zhejiang
The First Affiliated Hospital,Zhejiang University
Not yet recruiting
Hangzhou, Zhejiang, China, 310000
Contact: Qiong Zhao
Zhejiang Cancer Hospital
Not yet recruiting
Hangzhou, Zhejiang, China, 310000
Contact: Yiping Zhang
China
Cancer Institute and Hospital, Chinese Academy of Medical Sciences
Not yet recruiting
Beijing, China, 100021
Principal Investigator: Yuankai Shi, Professor Less << |
|
NCT01342965
|
Non-Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
China
... More >>
Beijing, China, 100071
Beijing, China, 101149
Changchun, China, 130012
Chongqing, China, 400038
ChongQing, China, 400042
Fuzhou, China, 350014
Guangzhou, China, 510080
Hangzhou, China, 310016
Nanjing, China, 210002
Shanghai, China, 200030
Shanghai, China, 200032
Shanghai, China, 200433
Shantou, China, 515041
Wuhan, China, 430023
Xi'an, China, 710061
Malaysia
Kelantan, Malaysia, 16150
Kuala Lumpur, Malaysia, 50603
Kuala Lumpur, Malaysia, 59100
Nilai, Malaysia, 71800
Pahang, Malaysia, 25100
Petaling Jaya, Selangor, Malaysia, 46050
Petaling Jaya, Malaysia, 46150
Pulau Pinang, Malaysia, 11600
Philippines
Davao, Philippines, 8000
Desmarinas City, Philippines, 4114
Manila, Philippines, 1000
Quezon City, Philippines, 1104
San Juan, Philippines, 1500 Less << |
|
NCT00003132
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
NYU School of Medicine's Kaplan Comprehensive Cancer Center
New York, New York, United States, 10016 Less << |
|
NCT01342965
|
- |
|
Completed |
- |
- |
|
NCT00681967
|
Head and Neck Cancer |
Phase 1 |
Completed |
- |
Switzerland
... More >> Research Site
Basel, Switzerland
Research Site
Bern, Switzerland Less << |
|
NCT00974584
|
Non-Squamous Non-Small Cell Lu... More >>ng Cancer Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Buffalo, New York, United States, 14263
France
Villejuif, France, 94805
Netherlands
Groningen, Netherlands, 9700 RB Less << |
|
NCT00068549
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Carcinoma
Cervical Small Cell Carcinoma
Cervical Squamous Cell Carcinoma
Stage IB Cervical Cancer
Stage IIA Cervical Cancer
Stage IIB Cervical Cancer
Stage III Cervical Cancer
Stage IVA Cervical Cancer Less << |
Phase 1 |
Completed |
- |
United States, Pennsylvania
... More >>
Gynecologic Oncology Group
Philadelphia, Pennsylvania, United States, 19103 Less << |
|
NCT00003264
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
United States, New Jersey
... More >>
Hunterdon Regional Cancer Center
Flemington, New Jersey, United States, 08822
Fox Chase Cancer Center at Virtua Memorial Hospital Burlington County
Mount Holly, New Jersey, United States, 08060
Riverview Medical Center - Booker Cancer Center
Red Bank, New Jersey, United States, 07701
St. Francis Medical Center
Trenton, New Jersey, United States, 08629
United States, Pennsylvania
St. Luke's Network - Bethlehem
Bethlehem, Pennsylvania, United States, 18015
Delaware County Memorial Hospital
Drexel Hill, Pennsylvania, United States, 19026
Harrisburg Polyclinic Medical Center
Harrisburg, Pennsylvania, United States, 17101
Saint Mary Regional Center
Langhorne, Pennsylvania, United States, 19047
North Penn Hospital
Lansdale, Pennsylvania, United States, 19446-1200
Paoli Memorial Hospital
Paoli, Pennsylvania, United States, 19301-1792
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111
Reading Hospital and Medical Center
Reading, Pennsylvania, United States, 19612-6052 Less << |
|
NCT00127049
|
Germ Cell Tumor |
Phase 2 |
Unknown |
- |
France
... More >> Institut Gustave Roussy
Recruiting
Villejuif, France, 94805
Contact: Karim FIZAZI, Dr 33 1 42114559 fizazi@igr.fr Less << |
|
NCT03228537
|
Biphasic Mesothelioma
... More >> Epithelioid Mesothelioma
Stage I Pleural Malignant Mesothelioma AJCC v7
Stage IA Pleural Malignant Mesothelioma AJCC v7
Stage IB Pleural Malignant Mesothelioma AJCC v7
Stage II Pleural Malignant Mesothelioma AJCC v7
Stage III Pleural Malignant Mesothelioma AJCC v7 Less << |
Phase 1 |
Recruiting |
June 1, 2020 |
- |
|
NCT00477711
|
Gastric Cancer |
Phase 2 |
Completed |
- |
China
... More >> Peking University,School of Oncology, Gastrointestinal Department
Beijing, China, 100036 Less << |
|
NCT00431236
|
Nausea and Vomiting, Chemother... More >>apy-Induced Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00191100
|
Cancer of Cervix |
Phase 3 |
Completed |
- |
Argentina
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Buenos Aires, Argentina
Bosnia and Herzegovina
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Sarajevo, Bosnia and Herzegovina
India
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mumbai, India
Mexico
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mexico City, Mexico
Pakistan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Karachi, Pakistan
Peru
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Lima, Peru
Thailand
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Bangkok, Thailand Less << |
|
NCT00651456
|
Mesothelioma |
Phase 2
Phase 3 |
Completed |
- |
- |
|
NCT01217177
|
Locally Advanced Cervical Canc... More >>er Less << |
Phase 1 |
Completed |
- |
Brazil
... More >> Novartis Investigative Site
Rio de Janeiro, RJ, Brazil, 20230-130
Novartis Investigative Site
Rio de Janeiro, Brazil Less << |
|
NCT01025349
|
Metastatic Breast Cancer |
Phase 2 |
Completed |
- |
Taiwan
... More >> Taipei Medical University Hospital
Taipei, Taiwan, 110 Less << |
|
NCT00191100
|
- |
|
Completed |
- |
- |
|
NCT00003182
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Unknown |
- |
United States, Kansas
... More >>
Bethany Medical Center
Kansas City, Kansas, United States, 66102
Heartland Cancer Research Network
Kansas City, Kansas, United States, 66102 Less << |
|
NCT01138137
|
Ovarian Carcinoma, Stage 3 or ... More >>4
Epithelial Ovarian Carcinoma
Primary Peritoneal Carcinoma Less << |
Phase 1 |
Withdrawn(No funding was avail... More >>able for the cost of the IV N-acetylcysteine (NAC).) Less << |
- |
United States, Oregon
... More >>
Oregon Health & Science University
Portland, Oregon, United States, 97239 Less << |
|
NCT00343239
|
Stomach Neoplasms |
Phase 2 |
Completed |
- |
Turkey
... More >> Sanofi-Aventis Administrative Office
Istanbul, Turkey Less << |
|
NCT01042522
|
Ovarian Granulosa Cell Tumor
... More >> Ovarian Gynandroblastoma
Ovarian Sertoli-Leydig Cell Tumor
Ovarian Sex Cord Tumor With Annular Tubules
Ovarian Sex Cord-Stromal Tumor
Ovarian Sex Cord-Stromal Tumor of Mixed or Unclassified Cell Types
Ovarian Steroid Cell Tumor Less << |
Phase 2 |
Recruiting |
- |
- |
|
NCT02029690
|
Pleural Mesothelioma Malignant... More >> Advanced
Peritoneal Mesothelioma Malignant Advanced
Non-squamous Non-small Cell Lung Carcinoma Stage IIIB/IV (NSCLC)
Metastatic Uveal Melanoma
Hepatocellular Carcinoma (HCC)
Glioma
Sarcomatoid Cancers Less << |
Phase 1 |
Active, not recruiting |
February 2019 |
United States, Minnesota
... More >>
Mayo Clinic Rochester
Rochester, Minnesota, United States, 55902
United Kingdom
Centre for Experimental Cancer Medicine (CECM)
London, England, United Kingdom, EC1M 6BQ
Cambridge Hospital
Cambridge, United Kingdom, CB2 0QQ
Guys Hospital
London, United Kingdom, SE1 7EH Less << |
|
NCT00004090
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, Illinois
... More >>
Robert H. Lurie Comprehensive Cancer Center, Northwestern University
Chicago, Illinois, United States, 60611
United States, New Jersey
Cancer Institute of New Jersey
New Brunswick, New Jersey, United States, 08901 Less << |
|
NCT02702700
|
Pleural Effusion, Malignant |
Phase 1 |
Recruiting |
June 2019 |
Switzerland
... More >> Oncology Department, Centre Hospitalier Universitaire Vaudois (CHUV)
Recruiting
Lausanne, Vaud (VD), Switzerland, 1011
Contact: Solange Peters, MD-PhD +41 21 314 44 62 solange.peters@chuv.ch Less << |
|
NCT01192633
|
Sarcoma
Chemo... More >>therapy Less << |
Phase 2 |
Unknown |
January 2011 |
China, Shanghai
... More >>
Fudan University Cancer Hospital
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Zhiguo Luo, PhD
Sub-Investigator: Zhiyu Chen, PhD Less << |
|
NCT00003108
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, District of Col... More >>umbia
Lombardi Cancer Center
Washington, District of Columbia, United States, 20007 Less << |
|
NCT00763646
|
Gastric Junction Adenocarcinom... More >>a
Gastroesophageal Junction Adenocarcinoma Less << |
Phase 2 |
Unknown |
- |
Canada, Quebec
... More >> McGill University
Recruiting
Montreal, Quebec, Canada, H2W 1S6
Contact: Penny Chipman 514398-1444 penny.chipman@mcgill.ca
Contact: Crystal Lameira 514-398-2229 crystal.lameira@mcgill.ca
Principal Investigator: Lorenzo Ferri Less << |
|
NCT01947062
|
Lung Cancer
S... More >>quamous Cell Carcinoma of the Lung
Locally Advanced and Metastatic Squamous Cell Carcinoma of the Lung Less << |
Phase 3 |
Unknown |
October 2014 |
India
... More >> Swami Rama Cancer Hospital & Research Institute
Not yet recruiting
Haldwani, Nainital, Uttarakhand, India, 263139
Contact: Swaroop Revannasiddaiah, MD 91 ext 8971862565 swarooptheone@gmail.com
Principal Investigator: Swaroop Revannasiddaiah, MD
Sub-Investigator: Kailash C Pandey, MD
Sub-Investigator: Sridhar P Susheela, MD Less << |
|
NCT00891761
|
Cancer
Chemot... More >>herapy-Induced Nausea and Vomiting
Nausea and Vomiting
Solid Tumor Cancer Less << |
Phase 3 |
Withdrawn(This study has been ... More >>cancelled prior to enrollment.) Less << |
July 2010 |
United States, Florida
... More >>
GSK Investigational Site
Tampa, Florida, United States, 33614
United States, Minnesota
GSK Investigational Site
Robbinsdale, Minnesota, United States, 55422
Canada, Quebec
GSK Investigational Site
Sherbrooke, Quebec, Canada, J1H 5N4 Less << |
|
NCT00006106
|
Lip and Oral Cavity Cancer
... More >> Head and Neck Cancer
Oropharyngeal Cancer Less << |
Phase 1 |
Withdrawn |
- |
United States, Massachusetts
... More >>
Massachusetts General Hospital Cancer Center
Boston, Massachusetts, United States, 02114
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00716976
|
Brain Tumor
C... More >>entral Nervous System Tumor
Childhood Germ Cell Tumor
Extragonadal Germ Cell Tumor
Liver Cancer
Neuroblastoma
Ototoxicity
Ovarian Cancer
Sarcoma Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00005087
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT02899299
|
Mesothelioma |
Phase 3 |
Active, not recruiting |
September 28, 2021 |
- |
|
NCT01608464
|
Carcinoma of Esophagus |
Phase 2 |
Terminated(poor accrual) |
- |
Saudi Arabia
... More >> King Faisal Specialist Hospital & Research Center
Riyadh, Saudi Arabia, 11211 Less << |
|
NCT00003690
|
Breast Cancer
... More >> Melanoma (Skin)
Prostate Cancer
Unspecified Adult Solid Tumor, Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, Minnesota
... More >>
Mayo Clinic
Rochester, Minnesota, United States, 55905 Less << |
|
NCT00252161
|
Gastric Neoplasm |
Phase 3 |
Completed |
- |
Japan
... More >> Aichi Cancer Center Hospital
Nagoya,Chikusa-ku,Kanokoden,1-1, Aichi, Japan, 464-8681
Fujita Health University
Toyoake,Kutsukake-cho,Dengakugakubo,1-98, Aichi, Japan, 470-1192
National Cancer Center Hospital East
Kashiwa,Kashiwanoha,6-5-1, Chiba, Japan, 277-8577
National Hospital Organization Shikoku Cancer Center
Matsuyama,Horinouchi,13, Ehime, Japan, 790-0007
Gifu Municipal Hospital
Gifu,Kashima-cho,7-1, Gifu, Japan, 500-8323
Hiroshima City Hospital
Hiroshima,Naka-ku,Motomachi,7-33, Hiroshima, Japan, 730-8518
Itami City Hospital
Itami,Koyaike,1-100, Hyogo, Japan, 664-8540
Iwate Medical University
Morioka,Uchimaru,19-1, Iwate, Japan, 020-8505
Kagoshima University,Faculty of Medicine
Kagoshima,Sakuragaoka,8-35-1, Kagoshima, Japan, 890-8520
Kanagawa Cancer Center
Yokohama,Asahi-ku,Nakao,1-1-2, Kanagawa, Japan, 241-0815
Kyoto Second Red Cross Hospital
Kamigyo-ku,Kamanza-Marutamachi,355-5, Kyoto, Japan, 602-8026
Miyagi Cancer Center
Natori,Medeshima-Shiode,Nodayama,47-1, Miyagi, Japan, 981-1293
National Hospital Organization, Sendai Medical Center
Sendai,Miyagino-ku,Miyagino,2-8-8, Miyagi, Japan, 983-8520
Nagaoka Chuo General Hospital
Nagaoka,Kawasaki,2041, Niigata, Japan, 940-8653
Niigata Cancer Center Hospital
Niigata,Kawagishi-cho,2-15-3, Niigata, Japan, 951-8566
Tsubame Rosai Hospital
Tsubame,Sawatari,633, Niigata, Japan, 959-1228
Oita University Fuculty of Medicine
Oita,Hasama-machi,Oogaoka,1-1, Oita, Japan, 879-5593
Osaka National Hospital
Osaka,Chuo-ku,Hoenzaka,2-1-14, Osaka, Japan, 540-0006
Osaka Medical Center for Cancer and Cardiovascular Diseases
Osaka,Higashinari-ku,Nakamichi,1-3-3, Osaka, Japan, 537-8511
Kinki University School of Medicine
Osaka-Sayama,Ohno-higashi,377-2, Osaka, Japan, 589-8511
Sakai Municipal Hospital
Sakai,Minamiyasuicho,1-1-1, Osaka, Japan, 590-0064
Osaka Medical College
Takatsuki,Daigakucho,2-7, Osaka, Japan, 569-0801
Toyonaka Municipal Hospital
Toyonaka,Shibaharacho,4-14-1, Osaka, Japan, 560-0055
Saitama Cancer Center
Kita-adachi,Ina,Komuro,818, Saitama, Japan, 362-0806
National Defense Medical College
Tokorozawa,Namiki,3-2, Saitama, Japan, 359-8513
Shizuoka General Hospital
Shizuoka,Aoi-ku,Kitaando,4-27-1, Sizuoka, Japan, 420-8527
Tokyo Metropolitan Komagome Hospital
Bunkyo-ku,Honkomagome,3-18-22, Tokyo, Japan, 113-8677
Tokyo Medical and Dental University Hospital
Bunkyo-ku,Yushima,1-5-45, Tokyo, Japan, 113-8519
National Cancer Center Hospital
Chuo-ku,Tsukiji,5-1-1, Tokyo, Japan, 104-0045
Cancer Institute Hospital
Koto-ku,Ariake,3-10-6, Tokyo, Japan, 135-8550
International Medical Center of Japan
Shinjuku-ku,Toyama,1-21-1, Tokyo, Japan, 162-8655
Tokyo Metropolitan Bokutoh Hospital
Sumida-ku,Koutoubashi,4-23-15, Tokyo, Japan, 130-8575
Toyama Prefectural Central Hospital
Toyama,nishinagae,2-2-78, Toyama, Japan, 930-8550
Wakayama Medical University, School of Medicine
Wakayama,Kimiidera,811-1, Wakayama, Japan, 641-8510
Yamagata Prefectural Central Hospital
Yamagata,Aoyagi,1800, Yamagata, Japan, 990-2292 Less << |
|
NCT03557164
|
- |
|
Recruiting |
July 1, 2018 |
United Kingdom
... More >> Beatson West of Scotland Cancer Centre
Recruiting
Glasgow, United Kingdom, G12 0YN
Contact: Alan Cameron, MBChB 04143302418 alan.cameron2@glasgow.ac.uk
Contact: Ninian Lang, MBChB PhD 0141 330 1985 ninian.lang@glasgow.ac.uk Less << |
|
NCT00295789
|
Uterine Cervical Neoplasms |
Phase 3 |
Completed |
- |
Japan
... More >> Aichi Cancer Center Hospital
Nagoya,Chikusa-ku,Kanokoden,1-1, Aichi, Japan, 464-8681
Nagoya Medical Center
Nagoya,Naka-ku,Sannomaru,4-1-1, Aichi, Japan, 460-0001
National Hospital Organization Shikoku Cancer Center
Matsuyama,Horinouchi,13, Ehime, Japan, 790-0007
Kyushu University Hospital
Fukuoka,Higashi-ku,Maidashi,3-1-1, Fukuoka, Japan, 812-8582
National Kyushu Cancer Center
Fukuoka,Minami-ku,Notame,3-1-1, Fukuoka, Japan, 811-1395
Kurume University School of Medicine
Kurume, Asahi-machi, 67, Fukuoka, Japan, 830-0011
Gunma Prefectural Cancer Center
Ota,Takabayashi-nishi-cho,617-1, Gunma, Japan, 373-8550
National Hospital Organization Kure Medical Center Chugoku Cancer Center
Kure,Aoyama-cho,3-1, Hiroshima, Japan, 737-0023
Hokkaido University Hospital
North-14 West-5 Kita-ku,Sapporo, Hokkaido, Japan, 060-8648
Sapporo Medical University
S-1,W-16,Chuo-ku,Sapporo, Hokkaido, Japan, 060-8543
Hyogo Medical Center for Adults
Akashi,Kitaouji-cho,13-70, Hyogo, Japan, 673-8558
Institute of Clinical Medicine,Tsukuba University Hospital
Tsukuba,Tennodai,1-1-1, Ibaraki, Japan, 305-8575
Kagoshima City Hospital
Kagoshima,Kajiya-cho,20-17, Kagoshima, Japan, 892-8580
Tohoku University Hospital
Sendai,Aoba-ku,Seiryo-machi,1-1, Miyagi, Japan, 980-8574
Sinshu University
Matsumoto,Asahi,3-1-1, Nagano, Japan, 390-8621
Nagaoka Red Cross Hospital
Nagaoka,Terashima-cho,297-1, Niigata, Japan, 940-2085
Niigata Cancer Center Hospital
Niigata,Kawagishi-cho,2-15-3, Niigata, Japan, 951-8566
Osaka Medical Center for Cancer and Cardiovascular Diseases
Osaka,Higashinari-ku,Nakamichi,1-3-3, Osaka, Japan, 537-8511
Osaka City General Hospital
Osaka,Miyakojima-ku,Miyakojimahondori,2-13-22, Osaka, Japan, 534-0021
Kinki University School of Medicine
Osaka-Sayama,Ohno-higashi,377-2, Osaka, Japan, 589-8511
Faculty of Medicine, Saga University
Saga,Nabeshima,5-1-1, Saga, Japan, 849-8501
Saitama Medical Center, Saitama Medical School
Kawagoe,Komoda,1981, Saitama, Japan, 350-8550
Saitama Cancer Center
Kita-adachi,Ina,Komuro,818, Saitama, Japan, 362-0806
National Defense Medical College
Tokorozawa,Namiki,3-2, Saitama, Japan, 359-8513
Juntendo University School of Medicine
Bunkyo-ku,Hongo,3-1-3, Tokyo, Japan, 113-0033
The University of Tokyo Hospital
Bunkyo-ku,Hongo,7-3-1, Tokyo, Japan, 113-8655
National Cancer Center Hospital
Chuo-ku,Tsukiji,5-1-1, Tokyo, Japan, 104-0045
Cancer Institute Hospital
Koto-ku,Ariake,3-10-6, Tokyo, Japan, 135-8550
Jikei University Hospital
Minato-ku,Nishishinbashi,3-25-8, Tokyo, Japan, 105-8461
Tottori University School of Medicine
Yonago,Nishimachi,36-1, Tottori, Japan, 683-8504 Less << |
|
NCT00005996
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Unknown |
- |
- |
|
NCT01494558
|
Non-Small Cell Lung Cancer |
Phase 2
Phase 3 |
Completed |
- |
- |
|
NCT00733889
|
Esophageal Carcinoma |
Phase 2 |
Completed |
- |
Spain
... More >> Spanish Cooperative Group for Gastrointestinal Tumour Therapy
Madrid, Spain, 28046 Less << |
|
NCT02923258
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 2
Phase 3 |
Recruiting |
December 2022 |
China, Shanghai
... More >>
Shanghai ninth people's hospital
Recruiting
Shanghai, Shanghai, China, 200011
Contact: Guopei Zhu, M.D. Less << |
|
NCT00062439
|
- |
|
Completed |
- |
- |
|
NCT02604784
|
Peritoneal Carcinomatosis |
Phase 1
Phase 2 |
Recruiting |
October 2018 |
Italy
... More >> Department of Surgical Oncology - FPO-IRCCS Institute for Cancer Research and Treatment
Recruiting
Candiolo, Turin, Italy, 10060
Contact: Michele De Simone, MD 011993 ext 3673 michele.desimone@ircc.it
Contact: Marco Vaira, MD 011993 ext 3683 marco.vaira@ircc.it
Principal Investigator: Michele De Simone, MD
Sub-Investigator: Marco Vaira, MD
Sub-Investigator: Manuela Robella, MD Less << |
|
NCT00716976
|
- |
|
Completed |
- |
- |
|
NCT00003580
|
Drug/Agent Toxicity by Tissue/... More >>Organ
Head and Neck Cancer
Oral Complications
Radiation Toxicity Less << |
Phase 2 |
Completed |
- |
United States, Tennessee
... More >>
William F. Bowld Hospital
Memphis, Tennessee, United States, 38103 Less << |
|
NCT00490880
|
Bladder Cancer |
Phase 2 |
Completed |
- |
Poland
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Warsaw, Poland Less << |
|
NCT00620971
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Greece
... More >> University General Hospital of Alexandroupolis, Dep of Medical Oncology
Alexandroupolis, Greece
"Laikon" General Hospital, Medical Oncology Unit, Propedeutic Dep of Internal Medicine
Athens, Greece
"Metaxa's" Anticancer Hospital of Piraeus,1st Dep of Medical Oncology
Athens, Greece
401 Military Hospital, Medical Oncology Unit
Athens, Greece
Air Forces Military Hospital, Dep of Medical Oncology
Athens, Greece
IASO" General Hospital of Athens, 1st Dep of Medical Oncology
Athens, Greece
Sismanogleio General Hospital, 1st, 2nd Dep of Pulmonary Diseases
Athens, Greece
Sotiria" General Hospital, 1st, 3rd, 8th Dep of Pulmonary Diseases
Athens, Greece
"Theagenion" Anticancer Hospital of Thessaloniki
Thessaloniki, Greece Less << |
|
NCT00062439
|
Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00492505
|
Melanoma (Skin) |
Phase 2 |
Unknown |
- |
United States, California
... More >>
San Diego Pacific Oncology and Hematology Associates, Incorporated - Encinitas
Recruiting
Encinitas, California, United States, 92024
Contact: Edward F. McClay, MD 760-452-3340 emcclay@pacificoncology.com Less << |
|
NCT00006000
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, Ohio
... More >>
Arthur G. James Cancer Hospital - Ohio State University
Columbus, Ohio, United States, 43210-1240 Less << |
|
NCT00054444
|
Cervical Adenocarcinoma
... More >> Cervical Squamous Cell Carcinoma
Stage IB Cervical Cancer
Stage IIA Cervical Cancer
Stage IIB Cervical Cancer
Stage III Cervical Cancer
Stage IVA Cervical Cancer Less << |
Phase 1 |
Completed |
- |
United States, Illinois
... More >>
University of Chicago Comprehensive Cancer Center
Chicago, Illinois, United States, 60637
United States, Iowa
University of Iowa Hospitals and Clinics
Iowa City, Iowa, United States, 52242
United States, Missouri
University of Missouri - Ellis Fischel
Columbia, Missouri, United States, 65212
United States, New Jersey
Cooper Hospital University Medical Center
Camden, New Jersey, United States, 08103
United States, Ohio
MetroHealth Medical Center
Cleveland, Ohio, United States, 44109
Arthur G. James Cancer Hospital and Solove Research Institute at Ohio State University Medical Center
Columbus, Ohio, United States, 43210
Riverside Methodist Hospital
Columbus, Ohio, United States, 43214
United States, Oklahoma
University of Oklahoma Health Sciences Center
Oklahoma City, Oklahoma, United States, 73104
Cancer Care Associates-Midtown
Tulsa, Oklahoma, United States, 74104
Tulsa Cancer Institute
Tulsa, Oklahoma, United States, 74146 Less << |
|
NCT00004264
|
Drug/Agent Toxicity by Tissue/... More >>Organ
Lung Cancer Less << |
Phase 1
Phase 2 |
Unknown |
- |
United States, Wisconsin
... More >>
University of Wisconsin Comprehensive Cancer Center
Madison, Wisconsin, United States, 53792 Less << |
|
NCT02976909
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Phase 2 |
Recruiting |
December 2018 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Yu-hong Li, MD, Ph D liyh@sysucc.org.cn
Principal Investigator: Yu-hong Li, MD, Ph D Less << |
|
NCT02633202
|
Nasopharyngeal Carcinoma |
Phase 3 |
Recruiting |
December 2021 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Lei Chen, M.D. +86-20-87343096 chenlei@sysucc.org.cn Less << |
|
NCT01100801
|
Recurrent Gastric Cancer |
Phase 2 |
Unknown |
December 2018 |
Singapore
... More >> National University Hospital
Recruiting
Singapore, Singapore
Contact: Wei Peng Yong, MRCP, MB ChB 65 6772 4670 Wei_Peng_Yong@nuhs.edu.sg Less << |
|
NCT00057928
|
Cervical Cancer |
Phase 3 |
Withdrawn(lack of accrual) |
- |
- |
|
NCT00458887
|
- |
|
Completed |
- |
- |
|
NCT00054626
|
Bladder Cancer |
Phase 3 |
Unknown |
- |
- |
|
NCT03532451
|
Bladder Cancer |
Phase 1 |
Recruiting |
September 2022 |
United States, Texas
... More >>
University of TX Southwestern
Recruiting
Dallas, Texas, United States, 75390
Contact: Allison Beaver Beaver 214-645-8788 allison.beaver@utsouthwestern.edu
Contact: Darren Fleming 214-648-8310 darren.fleming@utsouthwestern.edu
Principal Investigator: Kevin Courtney, MD
United States, Virginia
University of Virginia
Recruiting
Charlottesville, Virginia, United States, 22903
Contact: Jennifer Drake, RN 434-297-7782 jd4cx@virginia.edu
Contact: Stacey Williams 434-243-8588 scw6n@virginia.edu
Principal Investigator: Robert Dreicer, MD Less << |
|
NCT00098995
|
Cervical Cancer |
Phase 1 |
Completed |
- |
Australia, Victoria
... More >>
Peter MacCallum Cancer Centre
East Melbourne, Victoria, Australia, 8006
Canada, Ontario
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT00726986
|
Lung Cancer |
Phase 2 |
Terminated(Extreme toxicity) |
- |
United States, New York
... More >>
Columbia Presbyterian
New York, New York, United States, 10032
United States, Ohio
Lake/University Ireland Cancer Center
Cleveland, Ohio, United States, 44060
Case Medical Center, University Hospitals Seidman Cancer Center, Case Comprehensive Cancer Center
Cleveland, Ohio, United States, 44106-5065
MetroHealth Medical Center
Cleveland, Ohio, United States, 44109
CCF-Fairview Hospital
Cleveland, Ohio, United States, 44111
UHHS Chagrin Highlands Medical Center
Cleveland, Ohio, United States, 44122
Southwest General Health Center
Cleveland, Ohio, United States, 44130
UHHS Westlake Medical Center
Cleveland, Ohio, United States, 44145
Cleveland Clinic Taussig Cancer Institute, Case Comprehensive Cancer Center
Cleveland, Ohio, United States, 44195
UH-Monarch
Mayfield Heights, Ohio, United States, 44124
UH-Firelands
Sandusky, Ohio, United States, 44870 Less << |
|
NCT00004189
|
Lymphoma
Smal... More >>l Intestine Cancer
Unspecified Adult Solid Tumor, Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, Texas
... More >>
University of Texas Health Science Center at San Antonio
San Antonio, Texas, United States, 78229-3900
Cancer Therapy and Research Center
San Antonio, Texas, United States, 78229
St. Luke's Lutheran Hospital
San Antonio, Texas, United States, 78229 Less << |
|
NCT01461746
|
Endometrial Cancer |
Phase 2 |
Recruiting |
December 2017 |
Korea, Republic of
... More >>
Asan Medical Center
Recruiting
Seoul, Korea, Republic of, 138-736
Contact: Joo-Hyun Nam, M.D., Ph.D. +82-2-3010-3633 jhnam@amc.seoul.kr
Contact: Jeong-Yeol Park, M.D., Ph.D. +82-2-3010-3646 obgyjypark@amc.seoul.kr Less << |
|
NCT00726986
|
- |
|
Terminated(Extreme toxicity) |
- |
- |
|
NCT00131586
|
- |
|
Terminated |
- |
Canada, Alberta
... More >>
Cross Cancer Institute
Edmonton, Alberta, Canada, T6G 1Z2 Less << |
|
NCT00229723
|
Neoplasms, Squamous Cell |
Phase 2 |
Completed |
- |
United States, Colorado
... More >>
Research Site
Aurora, Colorado, United States
United States, Texas
Research Site
Dallas, Texas, United States
Belgium
Research Site
Brussels, Belgium
Research Site
Gent, Belgium
Research Site
Leuven, Belgium
Czech Republic
Research Site
Hradec Kralove, Czech Republic
Research Site
Pardubice, Czech Republic
Research Site
Plzen, Czech Republic
Germany
Research Site
Berlin, Germany
Research Site
Essen, Germany
Research Site
Muenster, Germany
Research Site
Saarbrucken, Germany
India
Research Site
Bangalore, India
Research Site
Mumbai, India
Research Site
New Delhi, India
Research Site
Thiruvananthapuram, India
Poland
Research Site
Gliwice, Poland
Research Site
Kraków, Poland
Research Site
Lodz, Poland
Research Site
Lublin, Poland
Research Site
Warszawa, Poland
Serbia
Research Site
Belgrade, Serbia
Research Site
Sremska Kamenica, Serbia
Taiwan
Research Site
Taipei, Taiwan
Research Site
Taoynan, Taiwan Less << |
|
NCT01503385
|
Lung Cancer |
Phase 2 |
Unknown |
December 2016 |
China, Beijing
... More >> Cancer Hospital, Chinese Academy of Medical Sciences
Recruiting
Beijing, Beijing, China, 100021
Contact: Liang Jun, Doctor +861087788503 lj139117@yahoo.com.cn
Contact: Wang luhua, Doctor +861087788799 wlhwq@yahoo.com
Sub-Investigator: Chen Bo, Doctor
Jun Liang
Recruiting
Beijing, Beijing, China, 100021
Contact: Jun Liang, Doctor 8610-87788503 lj139117@yahoo.com.cn Less << |
|
NCT00017459
|
Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00005814
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00090909
|
Lymphoma |
Phase 2 |
Completed |
- |
United States, California
... More >>
Jonsson Comprehensive Cancer Center at UCLA
Los Angeles, California, United States, 90095-1678 Less << |
|
NCT00489944
|
Intraocular Melanoma |
Phase 2 |
Unknown |
- |
United States, California
... More >>
San Diego Pacific Oncology and Hematology Associates, Incorporated - Encinitas
Recruiting
Encinitas, California, United States, 92024
Contact: Edward F. McClay, MD 760-452-3340 emcclay@pacificoncology.com Less << |
|
NCT01077427
|
Resected Pancreatic Adenocarci... More >>noma Less << |
Phase 3 |
Recruiting |
March 2021 |
Germany
... More >> Klinikum Grosshadern, Medical Center, University of Munich
Recruiting
Munich, Bavaria, Germany, 81377
Contact: Rolf D. Issels, MD, PhD +49-89-4400-77776 heat@med.uni-muenchen.de
Principal Investigator: Rolf D. Issels, MD, PhD Less << |
|
NCT01735409
|
Nasopharyngeal Neoplasms |
Phase 1
Phase 2 |
Unknown |
June 2014 |
China, Guangdong
... More >>
Cancer Center, Sun Yat-sen University
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT00246974
|
Bladder Cancer |
Phase 2 |
Completed |
- |
Germany
... More >> Research Site
Aachen, Germany
Research Site
Augsburg, Germany
Research Site
Berlin, Germany
Research Site
Dresden, Germany
Research Site
Freiburg, Germany
Research Site
Halle/ Saale, Germany
Research Site
Hamburg, Germany
Research Site
Hannover, Germany
Research Site
Kassel, Germany
Research Site
Mainz, Germany
Research Site
Mannheim, Germany
Research Site
Münster, Germany
Research Site
Tübingen, Germany
Research Site
Ulm, Germany Less << |
|
NCT00229723
|
- |
|
Completed |
- |
- |
|
NCT01889303
|
Gastric Cancer |
Phase 2 |
Recruiting |
December 2019 |
China, Hubei
... More >> Zhongnan Hospital of Wuhan University
Recruiting
Wuhan, Hubei, China, 430071
Contact: F X Zhou, PHD 86-27-67813155 happyzhoufx@sina.com
Contact: Y F Zhou, PHD 86-27-67823096 yfzhouwhu@163.com Less << |
|
NCT00077311
|
Anemia
Drug/A... More >>gent Toxicity by Tissue/Organ
Lung Cancer
Neutropenia Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01620242
|
Recurrent Head and Neck Cancer... More >>
Metastatic Head and Neck Cancer Less << |
Phase 2 |
Completed |
- |
France
... More >> Centre Oscar Lambret
Lille, France, 59020
Centre Léon Berard
Lyon, France, 69008
Centre Antoine Lacassagne
Nice, France, 06189
Institut Curie
Paris, France, 75231
Institut Gustave Roussy
Villejuif, France, 94805 Less << |
|
NCT03413579
|
Cervical Cancer |
Phase 3 |
Recruiting |
December 2019 |
Algeria
... More >> Centre Pierre et Marie Curie (CPMC)
Recruiting
Algiers, Algeria
Contact: Esma KERBOUA
Principal Investigator: Esma KERBOUA, Prof.
CAC Annaba
Recruiting
Annaba, Algeria
Contact: Ibtissem BOULOUH
Principal Investigator: Ibtissem BOULOUH
CAC Batna
Recruiting
Batna, Algeria
Contact: Wassila BENBRAHIM, Prof.
Principal Investigator: Wassila BENBRAHIM
CHU Frantz Fanon
Recruiting
Blida, Algeria
Contact: Adda BOUNEDJAR, Prof.
Principal Investigator: Adda BOUNEDJAR, Prof
CHU Sidi Belloua
Active, not recruiting
Tizi Ouzou, Algeria Less << |
|
NCT03127774
|
Adrenocortical Carcinoma
... More >> Peritoneal Carcinomatosis Less << |
Phase 2 |
Recruiting |
May 2023 |
United States, New York
... More >>
Columbia University Medical Center
Recruiting
New York, New York, United States, 10032-3729
Contact: Michael Kluger, MD 212-305-6514 mk2462@cumc.columbia.edu
Contact: Vilma Rosario 212-305-6033 vr2222@cumc.columbia.edu
Principal Investigator: Michael Kluger, MD
Sub-Investigator: Antonio T Fojo, MD Less << |
|
NCT01445392
|
Mesothelioma |
Phase 1 |
Terminated |
- |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892 Less << |
|
NCT00651833
|
Advanced Non-Small Cell Lung C... More >>ancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01025089
|
Thymoma
Thymi... More >>c Carcinoma
Clinical Masaoka Stage II to IVA Less << |
Phase 2 |
Active, not recruiting |
December 2019 |
United States, California
... More >>
City of Hope Medical Center
Duarte, California, United States, 91010-3000
United States, New Jersey
Memorial Sloan Kettering at Basking Ridge
Basking Ridge, New Jersey, United States, 07920
United States, New York
Memorial Sloan Kettering Cancer Center @ Suffolk
Commack, New York, United States, 11725
Memorial Sloan Kettering West Harrison
Harrison, New York, United States, 10604
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan Kettering at Mercy Medical Center
Rockville Centre, New York, United States
Memorial Sloan Kettering Cancer Center at Phelps Memorial Hospital Center
Sleepy Hollow, New York, United States, 10591
United States, Texas
Md Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00583674
|
Gastrointestinal Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT03298893
|
Cervical Cancer
... More >> Locally Advanced Cervical Cancer Less << |
Phase 1
Phase 2 |
Recruiting |
April 2020 |
France
... More >> Institut Curie
Recruiting
Paris, France, 75005
Contact: Manuel Rodrigues, MD manuel.rodrigues@curie.fr
Hopital Européen Georges Pompidou
Active, not recruiting
Paris, France, 75015
Institut Curie Hopital René Huguenin
Recruiting
Saint Cloud, France, 9220
Contact: Coraline Dubot, MD coraline.dubot@curie.fr Less << |
|
NCT03067792
|
Inoperable Gastric Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT02657460
|
Malignant Pleural Effusion |
Phase 2 |
Recruiting |
December 2018 |
China, Hubei
... More >> Union Hospital, Tongji Medical College, Huazhong University of Science & Technology
Recruiting
Wuhan, Hubei, China, 430022
Contact: Yang Jin, doctor +86 13554361146 whuhjy@sina.com Less << |
|
NCT00276796
|
Cervical Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT01057277
|
Head and Neck Cancer |
Phase 1 |
Terminated(Pharmaceutical co- ... More >>re aligned their specialties- no longer will fund H&N ca) Less << |
- |
United States, Rhode Island
... More >>
Rhode Island Hospital
Providence, Rhode Island, United States, 02903 Less << |
|
NCT01057277
|
- |
|
Terminated(Pharmaceutical co- ... More >>re aligned their specialties- no longer will fund H&N ca) Less << |
- |
- |
|
NCT00242762
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
Spain
... More >> Research Facility
Granada, Spain
Research Facility
Madrid, Spain
Research Facility
Murcia, Spain
Research Site
Sevilla, Spain Less << |
|
NCT00926640
|
Carcinoma Neuroendocrine
... More >> Small Cell Lung Carcinoma
Malignant Epithelial Neoplasms Less << |
Phase 1 |
Completed |
- |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892 Less << |
|
NCT01177956
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 3 |
Completed |
- |
China
... More >> Cancer Institute & Hospital, Chinese Academy of Medical Sciences
Beijing, China
Jilin Cancer Hospital
Changchun, China
The Xiangya 2nd Hospital of Central South University
Changsha City, China
Fuijan Provincial Tumor Hospital
Fuijian, China
Nanfang Hospital of Nanfang Medical University
Guangzhou, China
Sun Yat-Sen Univesity Cancer Center
Guangzhou, China
Zhejiang Provincial Tumor Hospital
Hangzhou, China
Jiangsu Cancer Hospital
Jiangsu, Nanjing, China
Tumor Hospital of Guangxi Zhuang Autonomous Region / The Tumor Affiliated Hospital of Guangxi Medical University
Nanning City, China
Eye & ENT Hospital of Fundan University
Shanghai, China
Fundan University Shanghai Cancer Center
Shanghai, China
Tongji Hospital of Tongji Medical College of Huazhong University of Science & Technology
Wuhan City, China
Xijing Hospital, the Fourth Military Medical University
Xi'an, China
Korea, Republic of
Clinical Trial Center of Medical Research Institute, Pusan National University Hospital
Busan, Korea, Republic of Less << |
|
NCT00004884
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
Belgium
... More >> Institut Jules Bordet
Brussels, Belgium, 1000
Hopital Universitaire Erasme
Brussels, Belgium, 1070
U.Z. Gasthuisberg
Leuven, Belgium, B-3000
Egypt
National Cancer Institute of Egypt
Cairo, Egypt
France
CHU Ambroise Pare
Boulogne Billancourt, France, F-92104
Centre Alexis Vautrin
Vandoeuvre-les-Nancy, France, 54511
Institut Gustave Roussy
Villejuif, France, F-94805
Germany
Klinikum St. Marien
Amberg, Germany, D-92224
Universitaetsklinik und Strahlenklinik - Essen
Essen, Germany, D-45122
Klinikum der J.W. Goethe Universitaet
Frankfurt, Germany, D-60590
Marien Hospital
Hagen, Germany, 58095
Hermann-Holthusen Institute for Radiotherapy
Hamburg, Germany, D-20099
Medizinische Hochschule Hannover
Hannover, Germany, D-30625
Praxis Innere Medizin
Neustadt, Germany, D-01844
Klinikum der Universitaet Ulm
Ulm, Germany, D-89081
Netherlands
Saint Laurentius Ziekenhuis
Roermond, Netherlands, 6043 CV
Poland
Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology
Warsaw, Poland, 02-781
Turkey
Dokuz Eylul University School of Medicine
Izmir, Turkey, 35340
United Kingdom
Beatson Oncology Centre
Glasgow, Scotland, United Kingdom, G11 6NT Less << |
|
NCT00582205
|
Ovarian Cancer
... More >> Fallopian Tube Cancer
Primary Peritoneal Carcinoma
Uterine Cancer Less << |
Phase 2 |
Terminated(Study completed per... More >> investigator.) Less << |
- |
United States, Oklahoma
... More >>
University of Oklahoma Health Sciences Center
Oklahoma City, Oklahoma, United States, 73104 Less << |
|
NCT00582205
|
- |
|
Terminated(Study completed per... More >> investigator.) Less << |
- |
- |
|
NCT01687413
|
Oropharyngeal Neoplasms |
Phase 3 |
Active, not recruiting |
August 9, 2021 |
United States, Arizona
... More >>
Mayo Clinic Scottsdale
Scottsdale, Arizona, United States, 85259-5499
United States, Missouri
Washington University School of Medicine
Saint Louis, Missouri, United States, 63110 Less << |
|
NCT00304135
|
Extrahepatic Bile Duct Cancer
... More >>
Gallbladder Cancer
Liver Cancer Less << |
Phase 2
Phase 3 |
Completed |
- |
France
... More >> Centre Hospitalier General
Belfort, France, 90000
Centre Hospitalier Pierre Oudot
Bourgoin-Jallieu, France, 38300
Hopital Louis Pasteur
Colmar, France, 68024
Centre Hospitalier de Dax
Dax, France, 40100
Hopital Du Bocage
Dijon, France, 21034
Centre de Lutte Contre le Cancer Georges-Francois Leclerc
Dijon, France, 21079
Centre Hospitalier Departemental
La Roche Sur Yon, France, F-85025
C. H. Du Mans
Le Mans, France, 72037
CHU de la Timone
Marseille, France, 13385
Centre Hospitalier General de Mont de Marsan
Mont-de-Marsan, France, 40000
CHR D'Orleans - Hopital de la Source
Orleans, France, 45100
Hopital Bichat - Claude Bernard
Paris, France, 75018
CHU Pitie-Salpetriere
Paris, France, 75651
Centre Hospitalier Lyon Sud
Pierre Benite, France, 69495
Hopital Sebastopol, C.H.U. de Reims
Reims, France, 51092
Centre Eugene Marquis
Rennes, France, 35062
Hopital Charles Nicolle
Rouen, France, 76031
Centre Hospitalier de Semur en Auxois
Semur en Auxois, France, 21140
Hopital Universitaire Hautepierre
Strasbourg, France, 67098
Centre Hospitalier de Tarbes
Tarbes, France, 65013 Less << |
|
NCT01927744
|
Head and Neck Cancer |
Phase 2 |
Active, not recruiting |
December 2021 |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00632853
|
Lung Cancer |
Phase 3 |
Recruiting |
- |
- |
|
NCT00659113
|
Esophageal Cancer |
Phase 2 |
Unknown |
- |
Korea, Republic of
... More >>
Yonsei Cancer Center at Yonsei University Medical Center
Recruiting
Seoul, Korea, Republic of, 120-752
Contact: Joo-Hang Kim, MD 82-2-2228-8131 kjhang@yuhs.ac Less << |
|
NCT01099358
|
Solid Tumor |
Phase 2 |
Completed |
- |
United States, Arkansas
... More >>
Highlands Oncology Group
Fayetteville, Arkansas, United States, 72703
United States, Kansas
University of Kansas Medical Center
Fairway, Kansas, United States, 66205
United States, Nevada
VA Sierra Nevada Health Care System
Reno, Nevada, United States, 89502
United States, North Carolina
University of North Carolina
Chapel Hill, North Carolina, United States, 27599
United States, Texas
University of Texas Health Science Center - San Antonio
San Antonio, Texas, United States, 78229
Canada
For additional information regarding investigative sites for this trial, contact 1-888-545-5972 Mon - Fri, 9 AM to 4 PM or 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri, 9 AM to 5 PM Eastern Time or speak with your personal physician.
London, Canada, N6A 4L6
For additional information regarding investigative sites for this trial, contact 1-888-545-5972 Mon - Fri, 9 AM to 4 PM or 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri, 9 AM to 5 PM Eastern Time or speak with your personal physician.
Toronto, Canada, M5G 2M9 Less << |
|
NCT01177956
|
- |
|
Completed |
- |
- |
|
NCT00273546
|
Cancer |
Phase 3 |
Completed |
- |
United States, New Jersey
... More >>
Sanofi-Aventis Administrative Office
Bridgewater, New Jersey, United States, 08807
Argentina
Sanofi-Aventis Administrative Office
Buenos Aires, Argentina
Canada
Sanofi-Aventis Administrative Office
Laval, Canada
France
Sanofi-Aventis Administrative Office
Paris, France
Portugal
Sanofi-Aventis Administrative Office
Porto Salvo, Portugal
Russian Federation
Sanofi-Aventis Administrative Office
Moscow, Russian Federation Less << |
|
NCT02263105
|
High-grade Gliomas |
Phase 2 |
Completed |
- |
China, Shanghai
... More >>
Huashan Hospital
Shanghai, Shanghai, China, 200040 Less << |
|
NCT00112710
|
Lung Cancer |
Phase 3 |
Unknown |
- |
United Kingdom
... More >> Institute of Clinical Research - Birmingham
Recruiting
Birmingham, England, United Kingdom, B15 2TT
Contact: Hugh Jarrett 44-121-414-6425
Birmingham Heartlands Hospital
Recruiting
Birmingham, England, United Kingdom, B9 5SS
Contact: Joyce Thompson 44-121-766-6611 Less << |
|
NCT00995293
|
Head and Neck Neoplasms |
Phase 3 |
Completed |
- |
China
... More >> Sanofi Administrative Office
Shanghai, China Less << |
|
NCT00696007
|
Transitional Cell Carcinoma |
Phase 2 |
Withdrawn(Study was unable to ... More >>recruit subjects meeting the study requirements.) Less << |
- |
United States, Massachusetts
... More >>
Lahey Clinic, Inc.
Burlington, Massachusetts, United States, 01805 Less << |
|
NCT00268450
|
- |
|
Terminated |
- |
- |
|
NCT02518802
|
Lung Neoplasms |
Phase 3 |
Recruiting |
January 2020 |
China, Shaanxi
... More >> Tangdu Hospital of the Fourth Millitary Medical University
Recruiting
Xi'an, Shaanxi, China, 710038
Contact: Li Xiaofei, MD +8629,84777436 lxfchest@fmmu.edu.cn
Contact: Yan Xiaolong, MD +862984717558 yanxiaolong@fmmu.edu.cn Less << |
|
NCT00268450
|
Bladder Cancer |
Phase 2 |
Terminated |
- |
United States, South Carolina
... More >>
Hollings Cancer Center at Medical University of South Carolina
Charleston, South Carolina, United States, 29425
McLeod Regional Medical Center
Florence, South Carolina, United States, 29501
Lowcountry Hematology and Oncology, PA
Mount Pleasant, South Carolina, United States, 29464-3233
Gibbs Regional Cancer Center at Spartanburg Regional Medical Center
Spartanburg, South Carolina, United States, 29303 Less << |
|
NCT00003358
|
Fallopian Tube Cancer
... More >> Ovarian Cancer
Primary Peritoneal Cavity Cancer Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00502177
|
- |
|
Completed |
- |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00491127
|
Non-Hodgkin's Lymphoma |
Phase 2 |
Completed |
- |
Spain
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Madrid, Spain Less << |
|
NCT02804646
|
Adenocarcinoma of Lung |
Phase 4 |
Unknown |
December 2018 |
China, Anhui
... More >> Anhui provincial hospital
Recruiting
Hefei, Anhui, China, 230000
Contact: Lejie Cao, prefessor sycaolejie@163.com
Contact: Nana Hu 493305821@qq.com Less << |
|
NCT03415854
|
Pancreatic Cancer
... More >> Pancreatic Ductal Adenocarcinoma
Pancreatic Adenocarcinoma
Pancreas Metastases
Adenocarcinoma Less << |
Phase 2 |
Recruiting |
December 15, 2020 |
United States, Arizona
... More >>
HonorHealth Research Institute
Recruiting
Scottsdale, Arizona, United States, 85258
Contact: Joyce Schaffer, MSN, AOCNS 480-323-1339 ext option #2 Joyce.Schaffer@HonorHealth.com
Principal Investigator: Erkut Borazanci, MD Less << |
|
NCT01099358
|
- |
|
Completed |
- |
- |
|
NCT00087711
|
Non Small Cell Lung Carcinoma |
Phase 3 |
Completed |
- |
- |
|
NCT00079079
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
Canada, Manitoba
... More >>
CancerCare Manitoba
Winnipeg, Manitoba, Canada, R3E 0V9
Canada, Ontario
London Regional Cancer Program at London Health Sciences Centre
London, Ontario, Canada, N6A 4L6
Ottawa Hospital Regional Cancer Centre - General Campus
Ottawa, Ontario, Canada, K1H 8L6
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT01711658
|
Non-HPV Locally Advanced Head ... More >>and Neck Cancer Less << |
Phase 2 |
Active, not recruiting |
March 31, 2023 |
United States, Alabama
... More >>
University of Alabama at Birmingham Comprehensive Cancer Center
Birmingham, Alabama, United States, 35294
United States, California
University of California, San Diego
La Jolla, California, United States, 92093
Sutter General Hospital
Sacramento, California, United States, 95816
University of California San Francisco
San Francisco, California, United States, 94143
United States, Connecticut
Yale University
New Haven, Connecticut, United States, 06520
United States, Georgia
Emory University
Atlanta, Georgia, United States, 30308
United States, Kentucky
James Graham Brown Cancer Center at University of Louisville
Louisville, Kentucky, United States, 40202
United States, Ohio
University Hospitals of Cleveland
Cleveland, Ohio, United States, 44106
Ohio State University Medical Center
Columbus, Ohio, United States, 43210
United States, Oklahoma
University of Oklahoma Health Sciences Center
Oklahoma City, Oklahoma, United States, 73190
United States, Pennsylvania
Fox Chase Cancer Center Buckingham
Furlong, Pennsylvania, United States, 18925
United States, Texas
University of Texas Southwestern Medical School
Dallas, Texas, United States, 75390
University of Texas - MD Anderson Cancer Center
Houston, Texas, United States, 77030-4009
United States, Wisconsin
University of Wisconsin Comprehensive Cancer Center
Madison, Wisconsin, United States, 53792
Medical College of Wisconsin
Milwaukee, Wisconsin, United States, 53226
Canada, Quebec
McGill Cancer Centre at McGill University
Montreal, Quebec, Canada, H2W 1S6 Less << |
|
NCT00072527
|
Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT01839032
|
Lung Cancer |
Phase 2 |
Completed |
- |
France
... More >> CHU CAEN
Caen, France, 14033 Less << |
|
NCT00160030
|
Esophageal Neoplasms |
Phase 2 |
Completed |
- |
France
... More >> Sanofi-Aventis
Paris, France Less << |
|
NCT01616849
|
Stage IV Nasopharyngeal Carcin... More >>oma Less << |
Phase 2 |
Completed |
- |
China, Guangdong
... More >>
Cancer Center, Sun Yat-sen University
Guangzhou, Guangdong, China Less << |
|
NCT00875615
|
Liver Cancer |
Phase 2 |
Completed |
- |
United States, Florida
... More >>
University of Miami Sylvester Comprehensive Cancer Center - Miami
Miami, Florida, United States, 33136 Less << |
|
NCT00875615
|
- |
|
Completed |
- |
- |
|
NCT00215501
|
Solid Tumor |
Phase 1 |
Completed |
- |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114
Brigham and Women's Hospital
Boston, Massachusetts, United States, 02115
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT02940925
|
Nasopharyngeal Carcinoma |
Phase 3 |
Recruiting |
August 2024 |
China, Guangdong
... More >>
SunYat-senU
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Yanqun Xiang, MD +86-18666096623 xiangyq@sysucc.org.cn
Contact: Weixiong Xia, MD +86-18520415699 xiawx@sysucc.org.cn Less << |
|
NCT03769935
|
Progression Free Survival |
Phase 2 |
Recruiting |
July 1, 2020 |
China, Shandong
... More >>
Qingdao Central Hospital, Qingdao Cancer Hospital
Recruiting
Qingdao, Shandong, China, 266042
Contact: youxin ji, M.D. 8653268665078 mdji001@gmail.com
Principal Investigator: ketao lan, M.D.
Sub-Investigator: chunling zhang, M.D.
Sub-Investigator: youxin ji, M.D. Less << |
|
NCT01705184
|
Non-small Cell Lung Cancer Met... More >>astatic
Nonsquamous Nonsmall Cell Neoplasm of Lung Less << |
Phase 2 |
Completed |
- |
France
... More >> Avignon - Institut Sainte-Catherine
Avignon, France, 84918
Caen - Centre François Baclesse
Caen, France, 14000
Caen - CHU Côte de Nacre
Caen, France, 14000
Centre Hospitalier
Chauny, France
CH du Mans
Le Mans, France
Hôpital Nord - Oncologie Multidisciplinaire & Innovations Thérapeutiques
Marseille, France
Mulhouse - CH
Mulhouse, France, 68000
Nantes - Centre René Gauducheau
Nantes, France, 44805
Hopital Tenon - Pneumologie
Paris, France, 75020
HCL - Lyon Sud (Pneumologie)
Pierre Bénite, France, 69495
Rennes - CHU
Rennes, France, 35033
Strasbourg - NHC
Strasbourg, France, 63000 Less << |
|
NCT02107235
|
Head and Neck Neoplasms
... More >> Carcinoma, Squamous Cell Less << |
Phase 1 |
Completed |
- |
United States, Colorado
... More >>
University of Colorado Cancer Center
Aurora, Colorado, United States, 80045
United States, Delaware
Christiana Care Health Services
Newark, Delaware, United States, 19713
United States, New York
Montefiore Medical Center
The Bronx, New York, United States, 10467
United States, Ohio
The Ohio State University Comprehensive Cancer Center
Columbus, Ohio, United States, 43210 Less << |
|
NCT00191308
|
Non-Small Cell Lung Cancer
... More >> Carcinoma Less << |
Phase 2 |
Completed |
- |
Poland
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Bystra Slaska, Poland, 43-360
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Poznan, Poland, 60-569
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Warsaw, Poland, 02-781 Less << |
|
NCT00695032
|
Chemotherapeutic Agent Toxicit... More >>y
Renal Toxicity
Unspecified Adult Solid Tumor, Protocol Specific Less << |
Not Applicable |
Suspended |
- |
France
... More >> Institut Claudius Regaud
Toulouse, France, 31052 Less << |
|
NCT00556621
|
Bladder Cancer |
Phase 1
Phase 2 |
Completed |
- |
France
... More >> Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Montpellier, France, 34298 Less << |
|
NCT01912053
|
Cholangiocarcinoma |
Phase 2 |
Completed |
- |
France
... More >> Hôpital Beaujon - Service de Chirurgie
Clichy, France, 92118
Hôpital Henri Mondor
Créteil, France, 94010
Hôpital saint-Eloi
Montpellier, France, 34295
CHU Nancy - Hôpital Brabois
Nancy, France, 54000
CHU- Hotel Dieu
Nantes, France, 44093
CHU Poitiers
Poitiers, France
Centre Eugene Marquis
Rennes, France, 35042 Less << |
|
NCT00648648
|
Solid Tumors |
Phase 1 |
Completed |
- |
- |
|
NCT01108042
|
Oropharynx Cancer
... More >> Squamous Cell Carcinoma of the Oral Cavity Less << |
Phase 1
Phase 2 |
Completed |
- |
Germany
... More >> Städt. Kliniken Bielefeld gem. GmbH
Bielefeld, Germany, 33604
Friedrich-Schiller-University Jena
Jena, Germany, 07740
Universitätsklinikum Leipzig - Klinik und Poliklinik für HNO-Heilkunde
Leipzig, Germany, 04103
Klinikum Ernst von Bergmann
Potsdam, Germany, 14467 Less << |
|
NCT01296763
|
- |
|
Completed |
- |
- |
|
NCT01296763
|
Pancreatic Cancer |
Phase 1 |
Completed |
- |
United States, Maryland
... More >>
The Johns Hopkins University School of Medicine
Baltimore, Maryland, United States, 21231
United States, New York
Columbia University Medical Center
New York, New York, United States, 10032 Less << |
|
NCT00271323
|
Non-Small-Cell Lung Carcinoma |
Phase 2 |
Terminated |
- |
France
... More >> Sanofi-Aventis
Paris, France Less << |
|
NCT00191308
|
- |
|
Completed |
- |
- |
|
NCT00555672
|
Stomach Neoplasms |
Phase 1 |
Completed |
- |
Spain
... More >> Pfizer Investigational Site
L'hospitalet de Llobregat, Barcelona, Spain, 08907
Pfizer Investigational Site
Barcelona, Spain, 08003
Pfizer Investigational Site
Madrid, Spain, 28041 Less << |
|
NCT00005088
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00625664
|
Urinary Bladder Neoplasms |
Phase 3 |
Completed |
- |
- |
|
NCT01810913
|
Stage III Squamous Cell Carcin... More >>oma of the Hypopharynx
Stage III Squamous Cell Carcinoma of the Larynx
Stage III Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage III Squamous Cell Carcinoma of the Oropharynx
Stage III Verrucous Carcinoma of the Larynx
Stage III Verrucous Carcinoma of the Oral Cavity
Stage IV Squamous Cell Carcinoma of the Hypopharynx
Stage IVA Squamous Cell Carcinoma of the Larynx
Stage IVA Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage IVA Squamous Cell Carcinoma of the Oropharynx
Stage IVA Verrucous Carcinoma of the Larynx
Stage IVA Verrucous Carcinoma of the Oral Cavity
Stage IVB Squamous Cell Carcinoma of the Larynx
Stage IVB Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage IVB Squamous Cell Carcinoma of the Oropharynx
Stage IVB Verrucous Carcinoma of the Larynx
Stage IVB Verrucous Carcinoma of the Oral Cavity
Tongue Cancer Less << |
Phase 2
Phase 3 |
Active, not recruiting |
May 2025 |
- |
|
NCT03503136
|
Nasopharyngeal Carcinoma |
Phase 3 |
Not yet recruiting |
June 2026 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT03527264
|
Cervical Cancer |
Phase 2 |
Not yet recruiting |
December 2023 |
United States, Rhode Island
... More >>
Rhode Island Hospital
Not yet recruiting
Providence, Rhode Island, United States, 02903
Contact: Kayla Rosati 401-863-3000 kayla_rosati@brown.edu
Principal Investigator: Don Dizon, MD
Women and Infants Hospital
Not yet recruiting
Providence, Rhode Island, United States, 02905
Contact: Kayla Rosati 401-863-3000 kayla_rosati@brown.edu Less << |
|
NCT00452881
|
Lung Cancer |
Phase 2 |
Unknown |
December 2014 |
Korea, Republic of
... More >>
National Cancer Center, Korea
Goyang-si, Gyeonggi-do, Korea, Republic of, 411-769 Less << |
|
NCT00614965
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Greece
... More >> Air Forces Military Hospital, Dep of Medical Oncology
Athens, Greece
IASO" General Hospital of Athens, 1st Dep of Medical Oncology
Athens, Greece Less << |
|
NCT00091377
|
Fallopian Tube Cancer
... More >> Ovarian Cancer
Primary Peritoneal Cavity Cancer Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, Connecticut
... More >>
Yale Comprehensive Cancer Center at Yale University School of Medicine
New Haven, Connecticut, United States, 06520-8028
Australia, Victoria
Royal Women's Hospital
Carlton, Victoria, Australia, 3053 Less << |
|
NCT00002659
|
Head and Neck Cancer |
Phase 3 |
Unknown |
- |
- |
|
NCT00655876
|
Esophageal Cancer |
Phase 3 |
Active, not recruiting |
April 2020 |
- |
|
NCT00555672
|
- |
|
Completed |
- |
- |
|
NCT01461772
|
Cervical Cancer |
Phase 3 |
Terminated(Because of very slo... More >>w rate of enrollement) Less << |
- |
Korea, Republic of
... More >>
Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT01910844
|
Metastatic Breast Cancer |
Phase 2 |
Terminated(Because of difficul... More >>ties in recruting) Less << |
- |
France
... More >> Centre Jean Perrin
Clermont-Ferrand, France, 63011 Less << |
|
NCT00768755
|
Carcinoma, Non-Small Cell Lung |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT00939848
|
Biliary Tract Neoplasms |
Phase 2
Phase 3 |
Completed |
- |
United Kingdom
... More >> University College London Hospitals NHS Foundation Trust
London, United Kingdom, NW1 2PQ
The Christie NHS Foundation Trust
Manchester, United Kingdom, M20 4BX Less << |
|
NCT00756444
|
Squamous Cell Carcinoma of Hea... More >>d and Neck Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01762150
|
Renal Cell Carcinoma |
Phase 2 |
Completed |
- |
China, Beijing
... More >> Beijing Cancer Hospital
Beijing, Beijing, China, 100142
China, Guangdong
Sun Yat-sen university cancer center
Guangzhou, Guangdong, China, 510060
China, Liaoning
Shenyang general hospital of Shenyang military command
Shenyang, Liaoning, China, 110016
China, Shanxi
Xijing Hospital
Xi'an, Shanxi, China, 710032
China, Sichuan
West China Hospital
Chengdu, Sichuan, China, 610041 Less << |
|
NCT00360945
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 2 |
Unknown |
- |
France
... More >> Institut Gustave Roussy
Villejuif, France, F-94805
Ireland
Our Lady's Hospital for Sick Children Crumlin
Dublin, Ireland, 12
United Kingdom
Birmingham Children's Hospital
Birmingham, England, United Kingdom, B4 6NH
Institute of Child Health at University of Bristol
Bristol, England, United Kingdom, BS2 8AE
Bristol Royal Hospital for Children
Bristol, England, United Kingdom, BS2 8BJ
Addenbrooke's Hospital at Cambridge University Hospitals NHS Foundation Trust
Cambridge, England, United Kingdom, CB2 2QQ
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Royal Liverpool Children's Hospital, Alder Hey
Liverpool, England, United Kingdom, L12 2AP
Middlesex Hospital
London, England, United Kingdom, W1T 3AA
Great Ormond Street Hospital for Children NHS Trust
London, England, United Kingdom, WC1N 3JH
Central Manchester and Manchester Children's University Hospitals NHS Trust
Manchester, England, United Kingdom, M27 4HA
Sir James Spence Institute of Child Health
Newcastle-Upon-Tyne, England, United Kingdom, NE1 4LP
Queen's Medical Centre
Nottingham, England, United Kingdom, NG7 2UH
Oxford Radcliffe Hospital
Oxford, England, United Kingdom, 0X3 9DU
Children's Hospital - Sheffield
Sheffield, England, United Kingdom, S10 2TH
Southampton University Hospital NHS Trust
Southampton, England, United Kingdom, SO16 6YD
Royal Marsden NHS Foundation Trust - Surrey
Sutton, England, United Kingdom, SM2 5PT
Royal Belfast Hospital for Sick Children
Belfast, Northern Ireland, United Kingdom, BT12 6BE
Royal Aberdeen Children's Hospital
Aberdeen, Scotland, United Kingdom, AB25 2ZG
Royal Hospital for Sick Children
Edinburgh, Scotland, United Kingdom, EH9 1LF
Royal Hospital for Sick Children
Glasgow, Scotland, United Kingdom, G3 8SJ
Childrens Hospital for Wales
Cardiff, Wales, United Kingdom, CF14 4XW Less << |
|
NCT00068744
|
Anal Cancer |
Phase 2
Phase 3 |
Terminated(low accrual) |
- |
Belgium
... More >> Ziekenhuis Netwerk Antwerpen Middelheim
Antwerpen, Belgium, B-2020
Institut Jules Bordet
Brussels, Belgium, 1000
Centre Hospitalier Lyon Sud
Brussels, Belgium, 1200
Cliniques Universitaires Saint-Luc
Brussels, Belgium, 1200
Universitair Ziekenhuis Antwerpen
Edegem, Belgium, B-2650
Cazk Groeninghe - Campus Maria's Voorzienigheid
Kortrijk, Belgium, B-8500
U.Z. Gasthuisberg
Leuven, Belgium, B-3000
Algemeen Ziekenhuis Sint-Augustinus
Wilrijk, Belgium, 2610
Egypt
National Cancer Institute of Egypt
Cairo, Egypt
France
Institut Sainte Catherine
Avignon, France, 84082
Centre Hospitalier Regional de Besancon - Hopital Jean Minjoz
Besancon, France, 25030
Centre de Lutte Contre le Cancer Georges-Francois Leclerc
Dijon, France, 21079
Germany
Urologische Klinik - Universitaetsklinikum Aachen
Aachen, Germany, D-52074
Charite - Campus Charite Mitte
Berlin, Germany, D-10117
Robert Roessle Comprehensive Cancer Center - Charite Campus Buch
Berlin, Germany, D-13122
Universitaetsklinikum Essen
Essen, Germany, D-45122
Universitaetsklinikum Halle
Halle, Germany, D-06097
Onkologische Schwerpunktpraxis - Leer
Leer, Germany, D-26789
Universitaetsklinikum Tuebingen
Tuebingen, Germany, D-72076
Italy
Ospedale Sant Anna
Como, Italy, 22100
Ospedale Busonera - Divisione Oncologia Medica
Padova, Italy, 35128
Netherlands
Arnhems Radiotherapeutisch Instituut
Arnhem, Netherlands, 6815 AD
Dr. Bernard Verbeeten Instituut
Tilburg, Netherlands, 5042 SB
Serbia
Institute of Oncology and Radiology of Serbia
Belgrade, Serbia, 11000 Less << |
|
NCT00655876
|
- |
|
Active, not recruiting |
- |
- |
|
NCT03534804
|
Metastatic Urothelial Carcinom... More >>a
Bladder Cancer Less << |
Phase 2 |
Recruiting |
September 2023 |
United States, Utah
... More >>
Huntsman Cancer Institute
Recruiting
Salt Lake City, Utah, United States, 84112
Contact: Jill Broghammer 801-213-6232 jill.broghammer@hci.utah.edu
Principal Investigator: Neeraj Agarwal, MD Less << |
|
NCT02555644
|
Head and Neck Neoplasms |
Phase 1 |
Recruiting |
February 28, 2019 |
United States, Alabama
... More >>
University of Alabama at Birmingham Medical Center
Recruiting
Birmingham, Alabama, United States, 35249
Contact 205-934-2762
Principal Investigator: Eddy Yang
United States, Texas
University of Texas MD Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact 713-792-6363
Principal Investigator: Mehmet Altan
France
Centre Leon Berard
Recruiting
Lyon, France, 69008
Contact 33478785103
Principal Investigator: Jérôme Fayette
Gustave Roussy
Recruiting
Villejuif Cedex, France, 94805
Contact 33142114931
Principal Investigator: Eric Deutsch Less << |
|
NCT00002876
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 2 |
Completed |
- |
United States, Arizona
... More >>
Mayo Clinic Scottsdale
Scottsdale, Arizona, United States, 85259
United States, Florida
Mayo Clinic Jacksonville
Jacksonville, Florida, United States, 32224
United States, Minnesota
Mayo Clinic Cancer Center
Rochester, Minnesota, United States, 55905 Less << |
|
NCT00768755
|
- |
|
Completed |
- |
- |
|
NCT02236936
|
Squamous Cell Carcinoma of the... More >> Hypopharynx Stage III
Squamous Cell Carcinoma of the Hypopharynx Stage IV
Laryngeal Squamous Cell Carcinoma Stage III
Laryngeal Squamous Cell Carcinoma Stage IV
Oropharyngeal Squamous Cell Carcinoma Stage III
Oropharyngeal Squamous Cell Carcinoma Stage IV
Squamous Cell Carcinoma of the Oral Cavity Stage III
Squamous Cell Carcinoma of the Oral Cavity Stage IV
Locally Advanced Malignant Neoplasm Less << |
Phase 3 |
Recruiting |
September 2021 |
Austria
... More >> Landeskrankenhaus Feldkirch
Recruiting
Feldkirch, Austria, A-6807
Contact: Alexander De Vries, Univ.Doz. MD +43 5522303 ext 3300 alexander.devries@lkhf.at
Principal Investigator: Alexander De Vries, Univ.Doz. MD
Medizinische Universität Graz, HNO Universitätsklinik
Recruiting
Graz, Austria, A-8036
Contact: Dietmar Thurnher, Prof.,MD +43 316 3851 ext 3448 dietmar.thurnher@medunigraz.at
Contact: Sabine Reinisch, MD, Senior +43 316 3851 ext 3448 sabine.reinisch@medunigraz.at
Principal Investigator: Dietmar Thurnher, Prof.,MD
Klinikum Klagenfurt am Wörthersee, HNO-Abteilung
Recruiting
Klagenfurt am Wörthersee, Austria, A-9020
Contact: Thomas J. Primosch, MD +43 463 538 ext 27107 thomasprimosch@gmx.at
Principal Investigator: Thomas J. Primosch, MD
Ordensklinikum Linz - Barmherzigen Schwestern, Abteilung f. HNO, Kopf- und Halschirurgie
Recruiting
Linz, Austria, 4010
Contact: Magdalena Sturm, DI +43 7327677 ext 7521 magdalena.sturm@ordensklinikum.at
Contact: Martin Burian, Prof.Dr. +43 7327677 ext 4828 martin.burian@ordenskliniku.at
Principal Investigator: Martin Burian, Prof.Dr.
Hanuschkrankenhaus
Not yet recruiting
Vienna, Austria, 1140
Contact: Barbara Dixer, MSc +43 1 91021 ext 85435 barbara.dixer@wgkk.at
Contact: Kathrin Holzer, Mag. +43 1 91021 ext 85435 kathrin.holzer@wgkk.at
Principal Investigator: Adelheid Seebacher, Dr.
Landesklinikum Wiener Neustadt
Not yet recruiting
Wiener Neustadt, Austria, 2700
Contact: Laurenz N. Kopf +43 2622 9004 ext 73778 laurenznathanael.kopf@wienerneustadt.lknoe.at
Principal Investigator: Johannes Reiß-Kornfehl, Prof.Dr.
Krankenhaus Hietzing, Sonderabteilung f. Strahlentheapie
Not yet recruiting
Wien, Austria, 1130
Contact: Gerhard Kren, MD +43 1 801 10 ext 2331 gerhard.kren@wienkav.at
Principal Investigator: Tomas-Hendrik Knocke-Abulesz, Prof.Dr. Less << |
|
NCT00933387
|
Oral Cancer |
Phase 2 |
Completed |
- |
Taiwan
... More >> National Health Research of Institutes, Taiwan Cooperative Oncology Group
Tainan, Taiwan Less << |
|
NCT00756444
|
- |
|
Completed |
- |
- |
|
NCT01234324
|
Stomach Neoplasms
... More >> Gastroesophageal Junction Neoplasms Less << |
Phase 2 |
Completed |
- |
Germany
... More >> AIO-Studien gGmbH
Berlin, Germany, 10623 Less << |
|
NCT00259402
|
Esophageal Neoplasms |
Phase 2 |
Completed |
- |
Spain
... More >> Sanofi-Aventis Administrative Office
Barcelona, Spain Less << |
|
NCT00635726
|
Bladder Cancer |
Phase 2 |
Terminated(Due to poor accrual... More >>) Less << |
- |
Greece
... More >> University General Hospital of Alexandroupolis, Dept. of Medical Oncology
Alexandroupolis, Greece
401 Military Hospital, Medical Oncology Unit
Athens, Greece
Air Forces Military Hospital, Dept. of Medical Oncology
Athens, Greece
IASO General Hospital of Athens, 1st Dept. of Medical Oncology
Athens, Greece
Laikon General Hospital, Medical Oncology Unit, Propedeutic Dept. of Internal Medicine
Athens, Greece
Metaxa's Anticancer Hospital of Piraeus, 1st Dept. of Medical Oncology
Piraeus, Greece
Theagenion Anticancer Hospital of Thessaloniki
Thessaloniki, Greece Less << |
|
NCT00005060
|
Gastric Cancer |
Phase 3 |
Completed |
- |
Italy
... More >> European Institute of Oncology
Milan, Italy, 20141
Switzerland
Hopital Cantonal Universitaire de Geneve
Geneva, Switzerland, CH-1211
Zentrum fuer Tumordiagnostikund Praevention
St. Gallen, Switzerland, CH-9006 Less << |
|
NCT00657423
|
Lung Neoplasms |
Phase 3 |
Unknown |
April 2009 |
China, Shaanxi
... More >> Dept. Resp. Diseases, Xijing Hospital
Recruiting
Xi'an, Shaanxi, China, 710032
Contact: Shengqing Li, MD. PHD. 086-029-84771132 shengqingli@gmail.com Less << |
|
NCT00401323
|
Head and Neck Neoplasms
... More >> Neoplasm Recurrence, Local
Neoplasm Metastasis Less << |
Phase 2
Phase 3 |
Completed |
- |
- |
|
NCT00072215
|
Extragonadal Germ Cell Tumor
... More >> Testicular Germ Cell Tumor Less << |
Phase 3 |
Terminated(poor accrual) |
- |
- |
|
NCT00003263
|
Malignant Mesothelioma |
Phase 1 |
Completed |
- |
United States, Louisiana
... More >>
Office of S. Terry Kraus
Marrero, Louisiana, United States, 70072
United States, Pennsylvania
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111
United States, Virginia
Virginia Oncology Associates
Newport News, Virginia, United States, 23606 Less << |
|
NCT00453154
|
Extensive Stage Small Cell Lun... More >>g Carcinoma
Recurrent Small Cell Lung Carcinoma Less << |
Phase 1
Phase 2 |
Active, not recruiting |
- |
- |
|
NCT00103051
|
Lung Cancer |
Phase 2 |
Completed |
- |
Netherlands
... More >> Netherlands Cancer Institute - Antoni van Leeuwenhoek Hospital
Amsterdam, Netherlands Less << |
|
NCT00003055
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00453154
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00144053
|
Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00370890
|
Nasopharyngeal Cancer |
Phase 3 |
Active, not recruiting |
December 2018 |
Hong Kong
... More >> Department of Clinical Oncology, Pamela Youde Nethersole Eastern Hospital
Hong Kong, Hong Kong
Department of Clinical Oncology, Prince of Wales Hospital
Hong Kong, Hong Kong
Department of Clinical Oncology, Queen Elizabeth Hospital
Hong Kong, Hong Kong
Department of Clinical Oncology, Queen Mary Hospital
Hong Kong, Hong Kong
Department of Clinical Oncology, Tuen Mun Hospital
Hong Kong, Hong Kong
Department of Oncology, Princess Margaret Hospital
Hong Kong, Hong Kong Less << |
|
NCT00255476
|
Squamous Cell Cancer
... More >> Cancer of Head and Neck Less << |
Phase 2 |
Completed |
- |
United Kingdom
... More >> Research Site
London, United Kingdom Less << |
|
NCT01624051
|
Lung Cancer |
Phase 2 |
Unknown |
September 2015 |
Canada, Alberta
... More >>
Cross Cancer Institute
Recruiting
Edmonton, Alberta, Canada, T6G 1Z2
Principal Investigator: Michael B Sawyer, MD Less << |
|
NCT00738582
|
Malignant Pleural Mesothelioma |
Phase 2 |
Completed |
- |
- |
|
NCT00154882
|
Breast Cancer |
Phase 2 |
Unknown |
July 2007 |
Taiwan
... More >> Department of Oncology , National Taiwan University Hospital
Recruiting
Taipei, Taiwan
Contact: Yen-Shen Lu, M.D. 886-2-23123456 ext 7009 yslu@ha.mc.ntu.edu.tw
Sub-Investigator: Sung-Hsin Kuo, M.D.
Sub-Investigator: Chia-Chi Lin, M.D. Less << |
|
NCT03668639
|
Chemotherapy-induced Nausea an... More >>d Vomiting
Adverse Event
Cervical Cancer Less << |
Phase 2
Phase 3 |
Recruiting |
February 15, 2020 |
Denmark
... More >> Department of Oncology, Odense University Hospital
Recruiting
Odense, Denmark, 5000
Contact: Christina H. Ruhlmann, MD, PhD 22314446 ext +45 christina.ruhlmann@rsyd.dk
Contact: Annemieke Sibtsen, RN 29427758 ext +45 Annemieke.Sibtsen@rsyd.dk
Principal Investigator: Christina H. Ruhlmann, MD, PhD
Sub-Investigator: Anja Ør Knudsen, MD Less << |
|
NCT02776163
|
Salivary Gland Tumors
... More >> Head and Neck Cancer Less << |
Phase 2 |
Recruiting |
December 2020 |
China, Shanghai
... More >>
Shanghai ninth people's hospital
Recruiting
Shanghai, Shanghai, China, 200011
Contact: Guopei Zhu, M.D. Less << |
|
NCT00004077
|
Testicular Germ Cell Tumor |
Phase 2 |
Completed |
- |
United Kingdom
... More >> Velindre Hospital
Cardiff, Wales, United Kingdom, CF14 2TL Less << |
|
NCT01074697
|
Nausea
Vomiti... More >>ng
Genital Neoplasms, Female Less << |
Phase 3 |
Completed |
- |
Australia
... More >> RAH Cancer Centre, Royal Adelaide Hospital
Adelaide SA, Australia, 5000
Denmark
Department of Oncology
Aarhus, Denmark, 8000
Rigshospitalet, Finsen Centret
Copenhagen, Denmark, 2100
Herlev Hospital
Herlev, Denmark, 2730
Department of Oncology, Odense University Hospital
Odense, Denmark, 5000
Germany
Vivantes Klinikum Neukolln
Berlin, Germany
Universitatsklinikum Schleswig Holstein
Kiel, Germany, 24105
Norway
The Norwegian Radium Hospital
Oslo, Norway, 0310 Less << |
|
NCT02459457
|
Stage III Esophageal Squamous ... More >>Cell Carcinoma Less << |
Phase 3 |
Recruiting |
December 2021 |
China, Shanghai
... More >>
Fudan University Cancer Hospital
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Kuaile Zhao, M.D +86 18017312534 kuaile_z@sina.com Less << |
|
NCT03349710
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 3 |
Active, not recruiting |
November 10, 2022 |
- |
|
NCT00003379
|
Cervical Cancer |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT01614132
|
Medulloblastoma |
Phase 1
Phase 2 |
Active, not recruiting |
March 2021 |
Germany
... More >> Vivantes Netzwerk für Gesundheit GmbH
Berlin, Germany, 10249
Neurologische Universitätsklinik
Bochum, Germany, 44892
Universitätsklinikum Bonn
Bonn, Germany, 53105
Universitätsklinikum Freiburg
Freiburg, Germany, 79106
Medizinische Hochschule Hannover
Hannover, Germany, 30625
Universitätsklinikum Heidelberg
Heidelberg, Germany, 69120
Universitätsklinikum Leipzig
Leipzig, Germany, 04103
Universitätsklinikum Schleswig-Holstein
Lübeck, Germany, 23538
Otto-von-Guericke Universität
Magdeburg, Germany, 39120
Johannes Gutenberg-Universität Mainz
Mainz, Germany, 55131
Universitätsklinikum Münster
Münster, Germany, 48149
Universitätsklinikum Regensburg
Regensburg, Germany, 93053
Katharinenhospital
Stuttgart, Germany, 70174
Universitätsklinikum Tübingen
Tübingen, Germany, 72076
Universitätsklinikum Ulm
Ulm, Germany, 89081 Less << |
|
NCT00082862
|
Pancreatic Cancer |
Phase 2 |
Unknown |
- |
United States, Texas
... More >>
University of Texas Health Science Center at Houston
Recruiting
Houston, Texas, United States, 77030
Contact: Clinical Trials Office - University of Texas Health Science Ce 713-500-9500 Less << |
|
NCT00389688
|
Lung Cancer |
Phase 2 |
Terminated(low accrual) |
- |
Belgium
... More >> Universitair Ziekenhuis Antwerpen
Edegem, Belgium, B-2650 Less << |
|
NCT00077051
|
Head and Neck Cancer |
Phase 1 |
Completed |
- |
United States, Florida
... More >>
University of Miami Sylvester Comprehensive Cancer Center
Miami, Florida, United States, 33136 Less << |
|
NCT00955240
|
Anal Cancer |
Phase 2 |
Unknown |
- |
France
... More >> Institut Bergonie
Bordeaux, France, 33076
CHU Hopital A. Morvan
Brest, France, 29609
Centre Regional Francois Baclesse
Caen, France, 14076
Centre Hospitalier Departemental
La Roche Sur Yon, France, F-85025
Centre Oscar Lambret
Lille, France, 59020
Centre Hospital Regional Universitaire de Limoges
Limoges, France, 87042
Centre Leon Berard
Lyon, France, 69373
Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Montpellier, France, 34298
Centre Regional Rene Gauducheau
Nantes-Saint Herblain, France, 44805
Centre Antoine Lacassagne
Nice, France, 06189
Institut Curie Hopital
Paris, France, 75248
Hopital Saint-Louis
Paris, France, 75475
Hopital Tenon
Paris, France, 75970
Centre Hospitalier Lyon Sud
Pierre Benite, France, 69495
Hopital Charles Nicolle
Rouen, France, 76031
Centre Hospitalier Prive Saint-Gregoire
Saint-Gregoire, France, 35768
Clinique Du Parc
Toulouse, France, 31078
Groupe Oncorad Garonne
Toulouse, France, 31300
Centre Alexis Vautrin
Vandoeuvre-les-Nancy, France, 54511
Institut Gustave Roussy
Villejuif, France, F-94805 Less << |
|
NCT00551122
|
Brain and Central Nervous Syst... More >>em Tumors
Extragonadal Germ Cell Tumor
Ovarian Cancer
Testicular Germ Cell Tumor Less << |
Phase 1
Phase 2 |
Unknown |
- |
United Kingdom
... More >> Queen Elizabeth Hospital at University Hospital of Birmingham NHS Trust
Recruiting
Birmingham, England, United Kingdom, B15 2TH
Contact: Michael H. Cullen, MD 0121-627-2444
Southampton General Hospital
Recruiting
Southampton, England, United Kingdom, SO16 6YD
Contact: G. Mead, MD 44-23-8079-8639
Royal Marsden - Surrey
Recruiting
Sutton, England, United Kingdom, SM2 5PT
Contact: Robert A. Huddart, MD 44-20-8661-3457 robert.huddart@icr.ac.uk Less << |
|
NCT03469531
|
Neoplasms
Cer... More >>vical Adenosquamous Cell Carcinoma
Cervical Squamous Cell Carcinoma in Situ
Stage IB Cervical Cancer
Stage IIA Cervical Cancer
Stage IIB Cervical Cancer
Stage III Cervical Cancer
Stage IVA Cervical Cancer Less << |
Phase 2 |
Recruiting |
December 30, 2021 |
China, Guangdong
... More >>
Zhujiang Hospital
Recruiting
Guangzhou, Guangdong, China, 510282
Contact: junguo bu, MD 13729810406 ljq821028@126.com
Contact: jiqiang li, MD 13631317203 13631317203@163.com Less << |
|
NCT03392571
|
Resectable Pancreatic Cancer |
Phase 2 |
Not yet recruiting |
December 30, 2020 |
- |
|
NCT00112697
|
Pancreatic Cancer |
Phase 2 |
Unknown |
- |
France
... More >> Centre Paul Papin
Angers, France, 49036
Centre Oscar Lambret
Lille, France, 59020
Polyclinique des Quatre Pavillons
Lormont, France, 33310
Marseille Institute of Cancer - Institut J. Paoli and I. Calmettes
Marseille, France, 13273
Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Montpellier, France, 34298
Centre Regional Rene Gauducheau
Nantes-Saint Herblain, France, 44805
Centre Hospitalier Lyon Sud
Pierre Benite, France, 69495
Centre Rene Huguenin
Saint Cloud, France, 92210
Centre Alexis Vautrin
Vandoeuvre-les-Nancy, France, 54511
Institut Gustave Roussy
Villejuif, France, F-94805 Less << |
|
NCT01216644
|
Gastric Cancer |
Phase 2
Phase 3 |
Active, not recruiting |
September 2018 |
Germany
... More >> Krankenhaus Nordwest
Frankfurt, Germany, 60488 Less << |
|
NCT00310011
|
Bladder Cancer
... More >> Transitional Cell Cancer of the Renal Pelvis and Ureter Less << |
Phase 2 |
Completed |
- |
United States, North Carolina
... More >>
Carolinas Hematology-Oncology Associates
Charlotte, North Carolina, United States, 28203-4239
Regional Cancer Center
Greensboro, North Carolina, United States, 27403-1199
Wake Forest University Comprehensive Cancer Center
Winston-Salem, North Carolina, United States, 27157-1096 Less << |
|
NCT01126216
|
Head and Neck Cancer |
Phase 3 |
Active, not recruiting |
June 2019 |
Germany
... More >> Klinikum Coburg, Strahlentherapie, DiaCura
Coburg, Germany, 96450
Universitätsklinikum Düsseldorf, Klinik und Poliklinik für Strahlentherapie und Radiologische Onkologie
Düsseldorf, Germany, 40225
Universitätsklinikum Erlangen, Strahlenklinik
Erlangen, Germany, 91054
Universitätsklinikum Frankfurt, Klinik für Strahlentherapie und Radioonkologie
Frankfurt/M., Germany, 60590
Klinikum am Eichert, Praxis für Strahlentherapie und Klinik für Radioonkologie
Göppingen, Germany, 73035
Universitätsklinikum des Saarlandes, Klinik für Strahlentherapie und Radioonkologie,
Homburg/Saar, Germany, 66421
Universitätsklinikum Schleswig-Holstein, Campus Lübeck, Klinik und Poliklinik für Hals-Nasen- und Ohrenkranke
Lübeck, Germany, 23538
Kliniken Maria Hilf GmbH Mönchengladbach, Klinik für Strahlentherapie
Mönchengladbach, Germany, 41063
Klinikum München Pasing und Perlach, Klinik für HNO
München, Germany, 81241
Brüderkrankenhaus st. Josef Paderborn, Klinik für Strahlentherapie
Paderborn, Germany, 33098
Universitätsklinikum Regensburg, Klinik und Poliklinik für Strahlentherapie
Regensburg, Germany, 93053
Klinikum St. Elisabeth Straubing, Klinik für Hals-Nasen-Ohren-Heilkunde
Straubing, Germany, 94315
MVZ am Klinikum Mutterhaus der Borrmäerinnen, Strahlentherapie
Trier, Germany, 54290 Less << |
|
NCT00045162
|
- |
|
Completed |
- |
- |
|
NCT02567253
|
Ovarian Cancer
... More >> Primary Peritoneal Cancer Less << |
Phase 2 |
Recruiting |
November 2018 |
Belgium
... More >> UZ Ghent
Recruiting
Ghent, East-Flanders, Belgium, 9000
Contact: Wim P Ceelen, MD, PhD, FACS 09 332 62 51 wim.ceelen@ugent.be
Contact: Wouter Willaert, MD, PhD 09 332 8950 wouter.willaert@ugent.be Less << |
|
NCT00259285
|
Non-Small-Cell Lung Cancer |
Phase 2 |
Terminated(Trial was stopped e... More >>arly due to low enrollment.) Less << |
- |
Spain
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Barakaldo, Spain, 48903
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Barcelona, Spain, 08035
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
La Coruña, Spain, 15006
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Madrid, Spain, 28034
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Pamplona, Spain, 31008
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Valencia, Spain, 46014 Less << |
|
NCT01019278
|
Cervical Cancer
... More >> Stage IB Cervical Cancer
Stage IIA Cervical Cancer
Stage IIB Cervical Cancer
Stage III Cervical Cancer
Stage IVA Cervical Cancer Less << |
Phase 2 |
Withdrawn |
- |
- |
|
NCT00084318
|
- |
|
Completed |
- |
- |
|
NCT00045162
|
Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00561054
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Unknown |
- |
Portugal
... More >> fERNANDO bARATA
Recruiting
cOIMBRA, Portugal, 3040
Contact: Fernando Barata, MD 00351 239 800 195 fjssbarata@sapo.pt
Contact: Agostinho Costa, MD 00351 21 7548032 COSTA.AGOSTINHO@CLIX.PT Less << |
|
NCT00084318
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00408694
|
Stage II Nasopharyngeal Kerati... More >>nizing Squamous Cell Carcinoma AJCC v7
Stage III Nasopharyngeal Keratinizing Squamous Cell Carcinoma AJCC v7
Stage III Nasopharyngeal Undifferentiated Carcinoma AJCC v7
Stage IV Nasopharyngeal Keratinizing Squamous Cell Carcinoma AJCC v7
Stage IV Nasopharyngeal Undifferentiated Carcinoma AJCC v7 Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00408694
|
- |
|
Completed |
- |
- |
|
NCT00155259
|
Breast Cancer |
Phase 2 |
Completed |
- |
Taiwan
... More >> Department of Oncology, National Taiwan University Hospital
Taipei, Taiwan Less << |
|
NCT00398385
|
Lung Cancer |
Phase 2 |
Unknown |
- |
Korea, Republic of
... More >>
National Cancer Center - Korea
Recruiting
Goyang, Korea, Republic of, 410-769
Contact: Heungtae T. Kim, MD, PhD 82-31-920-1622 htkim@ncc.re.kr Less << |
|
NCT00178763
|
Pancreatic Neoplasms |
Phase 2 |
Unknown |
June 2013 |
United States, Texas
... More >>
Memorial Hermann Hospital
Recruiting
Houston, Texas, United States, 77030
Contact: Joan M Bull, M.D. 713-500-6815 Joan.M.Bull@uth.tmc.edu
Contact: Esperanza N. Fernandez, N/D 713-500-6774 Esperanza.N.Fernandez@uth.tmc.edu
Principal Investigator: Joan M. Bull, M.D. Less << |
|
NCT00055965
|
Lung Cancer |
Phase 3 |
Unknown |
- |
United Kingdom
... More >> Christie Hospital NHS Trust
Manchester, England, United Kingdom, M20 4BX Less << |
|
NCT02083211
|
Cervical Cancer Recurrent or P... More >>ersistent
Palliative Treatment
Monoclonal Antibody in Cervical Cancer Treatment Less << |
Phase 3 |
Completed |
- |
Mexico
... More >> National Institute of Cancerología
México, Tlalpan, Mexico, 14080 Less << |
|
NCT00251888
|
Cervix Cancer |
Phase 1 |
Completed |
- |
France
... More >> CRLC Val d'Aurelle
Montpellier, France, 34298
Centre Claudius Régaud
Toulouse, France, 31052 Less << |
|
NCT00387699
|
Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00259285
|
- |
|
Terminated(Trial was stopped e... More >>arly due to low enrollment.) Less << |
- |
- |
|
NCT00001569
|
Carcinoma
Per... More >>itoneal Neoplasm Less << |
Phase 1 |
Completed |
- |
United States, Maryland
... More >>
National Cancer Institute (NCI)
Bethesda, Maryland, United States, 20892 Less << |
|
NCT00022191
|
Bladder Cancer
... More >> Transitional Cell Cancer of the Renal Pelvis and Ureter
Urethral Cancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00959387
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
Brazil
... More >> Barretos Cancer Hospital
Barretos, São Paulo, Brazil, 14784-400 Less << |
|
NCT01414608
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Carcinoma
Cervical Squamous Cell Carcinoma, Not Otherwise Specified
Chemotherapeutic Agent Toxicity
Cognitive Side Effects of Cancer Therapy
Psychological Impact of Cancer
Radiation Toxicity
Sexual Dysfunction and Infertility
Stage IB Cervical Cancer
Stage IIA Cervical Cancer
Stage IIB Cervical Cancer
Stage IIIB Cervical Cancer
Stage IVA Cervical Cancer Less << |
Phase 3 |
Active, not recruiting |
- |
- |
|
NCT01977209
|
Non-small Cell Lung Cancer |
Phase 3 |
Unknown |
September 2016 |
China, Chongqing
... More >>
Daping Hospital, Third Military Medical University
Recruiting
Chongqing, Chongqing, China, 400042
Contact: Dong Wang, PH.D. 86-23-68757151 dongwang64@hotmail.com
Principal Investigator: Dong Wang, PH.D.
Chongqing Zhongshan Hospital
Recruiting
Chongqing, Chongqing, China
Contact: Zhixiang Yang, M.D. 13032372491
Principal Investigator: Zhixiang Yang
Fuling Central Hospital
Recruiting
Chongqing, Chongqing, China
Contact: Qi Zhou, M.D. 86-23-7222676
Principal Investigator: Qi Zhou, M.D.
Jiangjin Central Hospital
Recruiting
Chongqing, Chongqing, China
Contact: Debing Xiang, M.D. 86-13320336639
Principal Investigator: Debing Xiang, M.D.
The Second Affiliated Hospital of Medical University Of Chongqing
Recruiting
Chongqing, Chongqing, China
Contact: Xianquan Zhang, M.D. 86-23-63693000
Principal Investigator: Xianquan Zhang, M.D.
Three Gorges Central Hospital
Recruiting
Chongqing, Chongqing, China
Contact: Biyong Ren 13896327099
Principal Investigator: Biyong Ren, M.D. Less << |
|
NCT00006118
|
Bladder Cancer |
Phase 2 |
Unknown |
- |
France
... More >> Hopital Drevon
Dijon, France, 21000
CHR de Grenoble - La Tronche
Grenoble, France, 38043
Hopital Perpetuel Secours
Levallois-Perret, France, 92300
CHU de la Timone
Marseille, France, 13385
Hopital Notre-Dame de Bon Secours
Metz, France, 57038
Hopital Laennec
Paris, France, 75007
Hopital Saint Antoine
Paris, France, 75571
Hopital Tenon
Paris, France, 75970
Polyclinique De Courlancy
Reims, France, F-51100
C.H. Senlis
Senlis, France, 60309
Centre Hospitalier Regional Metz Thionville
Thionville, France, 57126
Monaco
Centre Hospitalier Princesse Grace
Monte Carlo, Monaco, 98000 Less << |
|
NCT00052312
|
Endometrial Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00213642
|
- |
|
Terminated |
- |
- |
|
NCT01048645
|
Non-Small-Cell Lung Carcinoma |
Phase 2 |
Completed |
- |
Mexico
... More >> National Institute of Cancerología
Mexico City, Mexico, 14080 Less << |
|
NCT01187238
|
Nasopharyngeal Carcinoma |
Phase 2 |
Completed |
- |
China, Guangdong
... More >>
Cancer hospital, Sun Yat-sen University
Guangzhou, Guangdong, China, 510060
China, Zhejiang
Zhejiang province cancer hospital
Hangzhou, Zhejiang, China, 310022
China
Cancer hospital, Chinese Academy of Medical Sciences
Beijing, China, 100021 Less << |
|
NCT00003078
|
Cervical Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT03623646
|
Oropharyngeal Squamous Cell Ca... More >>rcinoma Less << |
Phase 2 |
Not yet recruiting |
November 2022 |
- |
|
NCT03748134
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Phase 3 |
Not yet recruiting |
September 30, 2022 |
- |
|
NCT03580395
|
Breast Cancer |
Phase 2
Phase 3 |
Enrolling by invitation |
May 2020 |
China, Hebei
... More >> Fourth Hospital of Hebei Medical University, Tumor Hospital of Hebei Province
Shijiazhuang, Hebei, China, 050011 Less << |
|
NCT00062374
|
Gastric Adenocarcinoma
... More >> Stage II Gastric Cancer
Stage III Gastric Cancer
Stage IV Gastric Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT00062374
|
- |
|
Completed |
- |
- |
|
NCT00773188
|
Non-Squamous Non-Small Cell Lu... More >>ng Cancer Less << |
Phase 1 |
Completed |
- |
Canada, Alberta
... More >>
Calgary, Alberta, Canada, T2N 4N2
Canada, Ontario
Oshawa, Ontario, Canada, L1G 2B9
Ottawa, Ontario, Canada, K1H 8L6
Toronto, Ontario, Canada, M4N 3M5 Less << |
|
NCT03066778
|
Small Cell Lung Cancer
... More >> SCLC Less << |
Phase 3 |
Active, not recruiting |
October 5, 2021 |
- |
|
NCT00458315
|
Unknown Primary Tumors |
Phase 2 |
Withdrawn(never been started) |
May 2012 |
Denmark
... More >> Rigshospitalet, Dept of Oncology
Copenhagen, Denmark, 2100 Less << |
|
NCT00216008
|
Esophageal Adenocarcinomas
... More >> Adenocarcinomas of the Gastroesophageal Junction Less << |
Phase 2 |
Terminated(IRB Closure) |
- |
United States, Florida
... More >>
H. Lee Moffitt Cancer Center & Research Institute
Tampa, Florida, United States, 33612 Less << |
|
NCT00001499
|
Carcinoma, Non-Small-Cell Lung... More >>
Lung Neoplasms Less << |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
National Cancer Institute (NCI)
Bethesda, Maryland, United States, 20892 Less << |
|
NCT00003569
|
- |
|
Completed |
- |
United States, Illinois
... More >>
University of Chicago Cancer Research Center
Chicago, Illinois, United States, 60637-1470
United States, Maryland
Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Baltimore, Maryland, United States, 21231-2410
United States, New York
Roswell Park Cancer Institute
Buffalo, New York, United States, 14263-0001 Less << |
|
NCT01504815
|
Head and Neck Cancer |
Phase 3 |
Recruiting |
September 2021 |
France
... More >> Gustave Roussy Cancer Institute
Recruiting
Villejuif, France, 94800
Contact: Yungan Tao, MD +33 1 42116532 yungan.tao@gustaveroussy.fr
Principal Investigator: Yungan Tao, MD
Netherlands
Netherlands Cancer Institute
Recruiting
Amsterdam, Netherlands, 1066CX
Contact: Olga Hamming-Vrieze, MD +31205122135 o.vrieze@nki.nl
Principal Investigator: Olga Hamming-Vrieze, MD
Universitair Medisch Centrum Groningen
Recruiting
Groningen, Netherlands, 9713 GZ
Contact: Roel Steenbakkers, MD, PhD r.steenbakkers@umcg.nl
Principal Investigator: Roel Steenbakkers, MD, PhD
Maastro Clinic
Recruiting
Maastricht, Netherlands, NL-6229 ET
Contact: Frank Hoebers, MD +31 88 4455521 frank.hoebers@maastro.nl
Principal Investigator: Frank Hoebers, MD
Erasmus Medical Centre
Recruiting
Rotterdam, Netherlands, 3075 EA
Contact: Lisa Tans, MD, PhD l.tans@erasmusmc.nl
Principal Investigator: Lisa Tans, MD, PhD
University Medical Centre Utrecht
Recruiting
Utrecht, Netherlands, NL-3508GA
Contact: Chris Terhaard, MD PhD +31 88 7553137 c.h.j.terhaard@umcutrecht.nl
Principal Investigator: Chris Terhaard, MD PhD
Spain
University Hospital Vall d'Hebron
Recruiting
Barcelona, Spain, 08035
Contact: Jordi Giralt, MD jgiralt@vhebron.net
Contact: Berta Garrido +34 93 2746000 ext 4695 bgarrido@vhebron.net
Principal Investigator: Jordi Giralt, MD
Sweden
Karolinska Institute
Recruiting
Stockholm, Sweden, 17177
Contact: Teresa Herlestam, MD +46 8 51770000 maria.herlestam-calero-moreno@karolinska.se
Principal Investigator: Teresa Herlestam, MD
United Kingdom
Christie Hospital NHS Trust
Recruiting
Manchester, United Kingdom, M20 5BX
Contact: Nick Slevin, MD +44 161 9187939 nick.slevin@christie.nhs.uk
Principal Investigator: Nick Slevin, MD Less << |
|
NCT00417248
|
Non-Small Cell Lung Cancer |
Phase 1
Phase 2 |
Terminated(Negative sorafenib ... More >>results from ESCAPE trial and safety concerns of regimen) Less << |
- |
United States, Illinois
... More >>
Medical & Surgical Specialists, LLC
Galesburg, Illinois, United States, 61401
United States, Indiana
Fort Wayne Oncology & Hematology, Inc
Fort Wayne, Indiana, United States, 46815
Center for Cancer Care at Goshen Health System
Goshen, Indiana, United States, 46527
Indiana University Cancer Center
Indianapolis, Indiana, United States, 46202
Horizon Oncology Center
Lafayette, Indiana, United States, 47905
Medical Consultants, P.C.
Muncie, Indiana, United States, 47303
Northern Indiana Cancer Research Consortium
South Bend, Indiana, United States, 46601
United States, Ohio
Oncology Partners Network
Cincinnati, Ohio, United States, 45247 Less << |
|
NCT00887315
|
Non-small Cell Lung Cancer |
Phase 2 |
Terminated(Slow accrual and lo... More >>ss of sponsor) Less << |
- |
United States, Illinois
... More >>
The University of Chicago
Chicago, Illinois, United States, 60637 Less << |
|
NCT01302834
|
Head and Neck Cancer
... More >> Precancerous Condition Less << |
Phase 3 |
Active, not recruiting |
June 2024 |
- |
|
NCT00887315
|
- |
|
Terminated(Slow accrual and lo... More >>ss of sponsor) Less << |
- |
- |
|
NCT00613626
|
Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
United States, Delaware
... More >>
Helen F. Graham Cancer Center
Newark, Delaware, United States, 19713
United States, Illinois
Northwestern University Feinberg School of Medicine
Chicago, Illinois, United States, 60611
Medical & Surgical Specialists, LLC
Galesburg, Illinois, United States, 61401
United States, Indiana
Cancer Care Center of Southern Indiana
Bloomington, Indiana, United States, 47403
Oncology Hematology Associates of SW Indiana
Evansville, Indiana, United States, 47714
Fort Wayne Oncology & Hematology, Inc
Fort Wayne, Indiana, United States, 46815
IN Onc/Hem Associates
Indianapolis, Indiana, United States, 46202
Indiana University Simon Cancer Center
Indianapolis, Indiana, United States, 46202
St. Vincent Hospital & Health Centers
Indianapolis, Indiana, United States, 46206
IU Health Arnett Cancer Center
Lafayette, Indiana, United States, 47904
Horizon Oncology Researcg
Lafayette, Indiana, United States, 47905
IU Health at Ball Memorial Hospital
Muncie, Indiana, United States, 47303
Monroe Medical Associates
Munster, Indiana, United States, 46321
Northern Indiana Cancer Research Consortium
South Bend, Indiana, United States, 46601
Providence Medical Group
Terre Haute, Indiana, United States, 47802
United States, Nebraska
Methodist Cancer Center
Omaha, Nebraska, United States, 68114
United States, New Jersey
Hematology Oncology Associates S.J., P.A.
Mt. Holly, New Jersey, United States, 08060
United States, Oregon
Providence Portland Medical Center
Portland, Oregon, United States, 97213
United States, Pennsylvania
Pennsylvania Oncology-Hematology Associates
Philadelphia, Pennsylvania, United States, 19106 Less << |
|
NCT00701857
|
Esophageal Cancer |
Phase 1 |
Completed |
- |
United States, Arizona
... More >>
University of Arizona Cancer Center
Tucson, Arizona, United States, 85724 Less << |
|
NCT00584155
|
Hearing Loss |
Phase 1 |
Withdrawn(PI left the universi... More >>ty.) Less << |
- |
United States, Oklahoma
... More >>
University of Oklahoma Health Sciences Center
Oklahoma City, Oklahoma, United States, 73104 Less << |
|
NCT01090466
|
Bladder Cancer
... More >> Transitional Cell Cancer of the Renal Pelvis and Ureter
Urethral Cancer Less << |
Phase 1
Phase 2 |
Completed |
- |
United Kingdom
... More >> Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF Less << |
|
NCT00417248
|
- |
|
Terminated(Negative sorafenib ... More >>results from ESCAPE trial and safety concerns of regimen) Less << |
- |
- |
|
NCT00538681
|
Lung Cancer |
Phase 2 |
Completed |
- |
Belgium
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Leuven, Belgium, 3000
Germany
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Gauting, Germany, 82131
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
GroBhansdorf, Germany, D-22927
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hamburg, Germany, D 21075
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Heidelberg, Germany, 69126
Italy
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Bergamo, Italy, 24128
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Catania, Italy, 95100
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Padova, Italy, 35128
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Trento, Italy, 38100
Poland
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Otwock, Poland, 05-400
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Poznan, Poland, 60-569
Romania
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Bucharest, Romania, 022328 Less << |
|
NCT00046917
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT00625937
|
Head and Neck Cancer |
Phase 2 |
Unknown |
- |
Korea, Republic of
... More >>
Yonsei Cancer Center at Yonsei University Medical Center
Recruiting
Seoul, Korea, Republic of, 120-752
Contact: Joo-Hang Kim, MD 82-2-2228-8131 kjhang@yuhs.ac Less << |
|
NCT00016874
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Albert Einstein Comprehensive Cancer Center
Bronx, New York, United States, 10461
New York Presbyterian Hospital - Cornell Campus
New York, New York, United States, 10021 Less << |
|
NCT00004201
|
Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00226239
|
Cancer |
Phase 2 |
Completed |
- |
United States, Pennsylvania
... More >>
University of Pittsburgh Medical Center
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT01108601
|
Hearing Loss |
Phase 1
Phase 2 |
Unknown |
- |
Canada, Quebec
... More >> Montreal General Hospital
Recruiting
Montreal, Quebec, Canada, H3G 1A1
Contact: Victoria Akinpelu Less << |
|
NCT00613626
|
- |
|
Completed |
- |
- |
|
NCT00226239
|
- |
|
Completed |
- |
- |
|
NCT02446574
|
Esophageal Neoplasms |
Phase 1
Phase 2 |
Completed |
- |
China, Beijing
... More >> Cancer Institute and Hospital, Chinese Academy of Medical Science
Beijing, Beijing, China, 100021 Less << |
|
NCT00326378
|
Carcinoma, Non-Small-Cell Lung |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of, 135-710 Less << |
|
NCT01077713
|
Non-Squamous Non-Small Cell Lu... More >>ng Cancer Less << |
Phase 2 |
Completed |
- |
Italy
... More >>
Rionero in Vulture, Basilicata, Italy, 85028
Catanzaro, Calabria, Italy, 88100
Avellino, Campania, Italy, 83100
Napoli, Campania, Italy, 80131
Bologna, Emilia-Romagna, Italy, 40139
Aviano, Friuli-Venezia Giulia, Italy, 33081
Udine, Friuli-Venezia Giulia, Italy, 33100
Roma, Lazio, Italy, 00128
Roma, Lazio, Italy, 00152
Roma, Lazio, Italy, 00189
Viterbo, Lazio, Italy, 01100
Genova, Liguria, Italy, 16149
Bergamo, Lombardia, Italy, 24128
Milano, Lombardia, Italy, 20142
Monza, Lombardia, Italy, 20052
Rho, Lombardia, Italy, 20017
Sondalo, Lombardia, Italy, 23039
Sondrio, Lombardia, Italy, 23100
Ancona, Marche, Italy, 60121
Pesaro, Marche, Italy, 61122
Novara, Piemonte, Italy, 28100
Catania, Sicilia, Italy, 95100
Lido Di Camaiore, Toscana, Italy, 55043
Perugia, Umbria, Italy, 06156
Mirano, Veneto, Italy, 30035
Padova, Veneto, Italy, 35128 Less << |
|
NCT00789373
|
Non-Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT01077713
|
- |
|
Completed |
- |
- |
|
NCT01192243
|
Toxicity
Non-... More >>small Cell Lung Cancer Less << |
Phase 2 |
Unknown |
- |
China, Shanghai
... More >>
Cancer hospital Fudan University
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Jianhua Chang, MD,PhD 13916619284 changjianhua@hotmail.com Less << |
|
NCT02667743
|
Non-Small Cell Lung Cancer(NSC... More >>LC) Less << |
Phase 3 |
Active, not recruiting |
December 2019 |
China, Guangdong
... More >>
Guangdong General Hospital
Guangzhou, Guangdong, China, 510080
China, Shanghai
Shanghai Chest Hospital
Shanghai, Shanghai, China, 200030
China, Zhejiang
Jiangsu Cancer Hospital
Nanjing, Zhejiang, China, 210009 Less << |
|
NCT00789373
|
- |
|
Completed |
- |
- |
|
NCT02879227
|
Esophagus Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00603915
|
- |
|
Completed |
- |
- |
|
NCT00603915
|
Nasopharyngeal Cancer |
Phase 2 |
Completed |
- |
Canada, Ontario
... More >>
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT00568971
|
Gastric Cancer |
Phase 2 |
Terminated(The study has finis... More >>hed.) Less << |
- |
China, Shanghai
... More >>
Fudan University Cancer Hospital
Shanghai, Shanghai, China, 200032 Less << |
|
NCT02217956
|
Ovarian Adenocarcinoma
... More >> Fallopian Tube Adenocarcinoma
Primary Peritoneal Carcinoma Less << |
Phase 1 |
Completed |
- |
France
... More >> Gustave Roussy
Villejuif, Val de Marne, France, 94805 Less << |
|
NCT00002932
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
United States, California
... More >>
University of California San Diego Cancer Center
La Jolla, California, United States, 92093-0658
UCSF Cancer Center and Cancer Research Institute
San Francisco, California, United States, 94115-0128
United States, Iowa
University of Iowa Hospitals and Clinics
Iowa City, Iowa, United States, 52242
United States, Louisiana
MBCCOP - LSU Medical Center
New Orleans, Louisiana, United States, 70112
United States, Maryland
Marlene & Stewart Greenebaum Cancer Center, University of Maryland
Baltimore, Maryland, United States, 21201
United States, Tennessee
CCOP - Baptist Cancer Institute
Memphis, Tennessee, United States, 38117
University of Tennessee, Memphis Cancer Center
Memphis, Tennessee, United States, 38163
Vanderbilt Cancer Center
Nashville, Tennessee, United States, 37232-6838
United States, Vermont
Fletcher-Allen Health Care
Burlington, Vermont, United States, 05401
Green Mountain Oncology Group
Rutland, Vermont, United States, 05701
United States, Virginia
Naval Medical Center, Portsmouth
Portsmouth, Virginia, United States, 23708-2197
United States, Washington
University Cancer Center
Seattle, Washington, United States, 98195
University of Washington Neutron Facility and Cancer Center
Seattle, Washington, United States, 98195 Less << |
|
NCT00101192
|
- |
|
Completed |
- |
- |
|
NCT00005636
|
Malignant Mesothelioma |
Phase 3 |
Completed |
- |
United States, New Jersey
... More >>
Cancer Institute of New Jersey
New Brunswick, New Jersey, United States, 08901
United States, New York
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021
United States, Ohio
Ireland Cancer Center
Cleveland, Ohio, United States, 44106-5065 Less << |
|
NCT00316862
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
United States, Indiana
... More >>
Elkhart General Hospital
Elkhart, Indiana, United States, 46515
Howard Community Hospital
Kokomo, Indiana, United States, 46904
Center for Cancer Therapy at LaPorte Hospital and Health Services
La Porte, Indiana, United States, 46350
Saint Joseph Regional Medical Center
Mishawaka, Indiana, United States, 46545-1470
CCOP - Northern Indiana CR Consortium
South Bend, Indiana, United States, 46601
Memorial Hospital of South Bend
South Bend, Indiana, United States, 46601
Michiana Hematology-Oncology, PC - South Bend
South Bend, Indiana, United States, 46601
United States, Iowa
Holden Comprehensive Cancer Center at University of Iowa
Iowa City, Iowa, United States, 52242-1002
United States, Maine
CancerCare of Maine at Eastern Maine Medical Center
Bangor, Maine, United States, 04401
Maine Center for Cancer Medicine and Blood Disorders - Scarborough
Scarborough, Maine, United States, 04074
United States, Michigan
Lakeland Regional Cancer Care Center - St. Joseph
Saint Joseph, Michigan, United States, 49085
United States, Nebraska
Methodist Estabrook Cancer Center
Omaha, Nebraska, United States, 68114
United States, New Hampshire
New Hampshire Oncology - Hematology, PA at Payson Center for Cancer Care
Concord, New Hampshire, United States, 03301
New Hampshire Oncology - Hematology, PA - Hooksett
Hooksett, New Hampshire, United States, 03106
Lakes Region General Hospital
Laconia, New Hampshire, United States, 03246
Elliot Regional Cancer Center at Elliot Hospital
Manchester, New Hampshire, United States, 03103
United States, New York
Veterans Affairs Medical Center - Buffalo
Buffalo, New York, United States, 14215
Roswell Park Cancer Institute
Buffalo, New York, United States, 14263-0001
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065
SUNY Upstate Medical University Hospital
Syracuse, New York, United States, 13210
United States, North Carolina
Wayne Memorial Hospital, Incorporated
Goldsboro, North Carolina, United States, 27534
United States, Ohio
Arthur G. James Cancer Hospital and Richard J. Solove Research Institute at Ohio State University Comprehensive Cancer Center
Columbus, Ohio, United States, 43210-1240
United States, Vermont
Mountainview Medical
Berlin, Vermont, United States, 05602
Fletcher Allen Health Care - University Health Center Campus
Burlington, Vermont, United States, 05401 Less << |
|
NCT00004920
|
Malignant Mesothelioma |
Phase 3 |
Completed |
- |
- |
|
NCT03114280
|
Head and Neck Squamous Cell Ca... More >>rcinoma (HNSCC) Less << |
Phase 1
Phase 2 |
Not yet recruiting |
November 2020 |
France
... More >> PEYRADE Frédéric
Not yet recruiting
Nice, France, 06000
Contact: Frédéric PEYRADE +33 4 92031022 frederic.peyrade@nice.unicancer.fr Less << |
|
NCT00316862
|
- |
|
Completed |
- |
- |
|
NCT00154804
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
Taiwan
... More >> Department of Oncology, Nationa Taiwan University Hospital
Taipei, Taiwan Less << |
|
NCT00544362
|
Esophageal Cancer |
Phase 1
Phase 2 |
Completed |
- |
France
... More >> Centre Hospitalier Regional de Besancon - Hopital Jean Minjoz
Besancon, France, 25030
Hopital Saint Andre
Bordeaux, France, 33075
C.H.U. de Brest
Brest, France, 29200
CHR Clermont Ferrand, Hotel Dieu
Clermont-Ferrand, France, 63058
Hopital Du Bocage
Dijon, France, 21034
Federation Francophone de Cancerologie Digestive
Dijon, France, 21079
Centre Oscar Lambret
Lille, France, 59020
Centre Hospital Universitaire Hop Huriez
Lille, France, 59037
CHU de la Timone
Marseille, France, 13385
Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Montpellier, France, 34298
Centre Hospitalier Lyon Sud
Pierre Benite, France, 69495
Centre Hospitalier Regional de Purpan
Toulouse, France, 31059
Centre Alexis Vautrin
Vandoeuvre-les-Nancy, France, 54511 Less << |
|
NCT00489996
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
China
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Shanghai, China Less << |
|
NCT00003723
|
Malignant Mesothelioma |
Phase 2 |
Completed |
- |
- |
|
NCT00101192
|
Cervical Cancer |
Phase 2 |
Completed |
- |
United States, California
... More >>
Providence Saint Joseph Medical Center - Burbank
Burbank, California, United States, 91505
USC/Norris Comprehensive Cancer Center and Hospital
Los Angeles, California, United States, 90089-9181
United States, Georgia
Curtis and Elizabeth Anderson Cancer Institute at Memorial Health University Medical Center
Savannah, Georgia, United States, 31403-3089
United States, Indiana
Indiana University Melvin and Bren Simon Cancer Center
Indianapolis, Indiana, United States, 46202-5289
United States, Kansas
Kansas Masonic Cancer Research Institute at the University of Kansas Medical Center
Kansas City, Kansas, United States, 66160-7357
United States, Louisiana
Ochsner Cancer Institute at Ochsner Clinic Foundation
New Orleans, Louisiana, United States, 70121
Christus Schumpert Cancer Treatment Center
Shreveport, Louisiana, United States, 71101
United States, Mississippi
University of Mississippi Cancer Clinic
Jackson, Mississippi, United States, 39216
United States, New Jersey
Fox Chase Virtua Health Cancer Program at Virtua West Jersey
Voorhees, New Jersey, United States, 08043
United States, North Carolina
Wake Forest University Comprehensive Cancer Center
Winston-Salem, North Carolina, United States, 27157-1096
United States, Ohio
MetroHealth Cancer Care Center at MetroHealth Medical Center
Cleveland, Ohio, United States, 44109
Cleveland Clinic Taussig Cancer Center
Cleveland, Ohio, United States, 44195
United States, Oklahoma
Oklahoma University Cancer Institute
Oklahoma City, Oklahoma, United States, 73104
Cancer Care Associates - Midtown Tulsa
Tulsa, Oklahoma, United States, 74104
United States, Pennsylvania
Fox Chase Cancer Center - Philadelphia
Philadelphia, Pennsylvania, United States, 19111-2497
McGlinn Family Regional Cancer Center at Reading Hospital and Medical Center
Reading, Pennsylvania, United States, 19612-6052
United States, Texas
University of Texas Medical Branch
Galveston, Texas, United States, 77555-0361
M. D. Anderson Cancer Center at University of Texas
Houston, Texas, United States, 77030-4009 Less << |
|
NCT00445861
|
Esophageal Cancer |
Phase 1
Phase 2 |
Completed |
- |
Switzerland
... More >> Kantonsspital - St. Gallen
St. Gallen, Switzerland, CH-9007
Klinik Stephanshorn
St. Gallen, Switzerland, Ch-9016 Less << |
|
NCT02200042
|
Stage III Intrahepatic Cholang... More >>iocarcinoma
Stage IVA Intrahepatic Cholangiocarcinoma Less << |
Phase 3 |
Terminated |
- |
- |
|
NCT01243632
|
Malignant Pleural Mesothelioma |
Phase 2 |
Completed |
- |
Slovenia
... More >> Institute of Oncology Ljubljana
Ljubljana, Slovenia, 1000 Less << |
|
NCT01165021
|
- |
|
Completed |
- |
- |
|
NCT01154920
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 2 |
Active, not recruiting |
July 2020 |
United States, Massachusetts
... More >>
Dana Farber Cancer Institute
Boston, Massachusetts, United States, 20115
United States, Texas
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00466466
|
Small-Cell Lung Cancer |
Phase 1 |
Completed |
- |
United States, Arkansas
... More >>
Highlands Oncology Group
Fayetteville, Arkansas, United States, 72703
United States, Colorado
University of Colorado Health Sciences Center
Aurora, Colorado, United States, 80045
United States, Massachusetts
Dana Faber Cancer Institute
Boston, Massachusetts, United States, 02115
United States, Texas
MD Anderson Cancer Center
Houston, Texas, United States, 77030-4009
Cancer Therapy and Research Center at UTHSCSA
San Antonio, Texas, United States, 78229
France
Novartis Investigative Site
Paris, France Less << |
|
NCT01165021
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Italy
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Milano, Italy, 20133
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Novara, Italy, 28100
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Orbassano, Italy, 10043
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Padova, Italy, 35100
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Pisa, Italy, 56100
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Rome, Italy, 00144
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Sondalo, Italy, 23039 Less << |
|
NCT03133390
|
Urothelial Carcinoma |
Phase 2 |
Recruiting |
January 2019 |
United States, New York
... More >>
New York University School of Medicine
Recruiting
New York, New York, United States, 10016
Contact: Swati Chitkara swati.chitkara@nyumc.org
Principal Investigator: Arjun Balar, MD Less << |
|
NCT01736410
|
HER 2 Positive Advanced Gastri... More >>c Cancer Less << |
Phase 2 |
Completed |
- |
Singapore
... More >> National Cancer Centre Singapore
Singapore, Singapore, 169610 Less << |
|
NCT00898495
|
- |
|
Completed |
- |
- |
|
NCT01848457
|
Osteosarcoma
... More >>Nephrotoxicity
Ototoxicity Less << |
Phase 2 |
Active, not recruiting |
January 2019 |
United States, Pennsylvania
... More >>
Children's Hospital of Philadelphia
Philadelphia, Pennsylvania, United States, 19104 Less << |
|
NCT00083252
|
Melanoma
Skin... More >> Cancer Less << |
Phase 2 |
Completed |
- |
United States, Arkansas
... More >>
University of Arkansas Medical Sciences
Little Rock, Arkansas, United States, 72205
United States, California
Cancer Institute Medical Group, Inc.
Santa Monica, California, United States, 90404
United States, Colorado
University of Colorado Health Sciences Center
Aurora, Colorado, United States, 80045
United States, Florida
Cancer Center of Florida
Ocoee, Florida, United States, 34761
United States, Indiana
Indiana Hematology Oncology Consultants
Indianapolis, Indiana, United States, 46202
United States, New Mexico
New Mexico Cancer Center Alliance
Albuquerque, New Mexico, United States, 87106
United States, New York
NYU School of Medicine
New York, New York, United States, 10016
Fifth Avenue Medical Healthcare
New York, New York, United States, 10128
United States, North Carolina
Carolinas Medical Center
Charlotte, North Carolina, United States, 28203
United States, South Carolina
Cancer Center of the Carolinas
Greenville, South Carolina, United States, 29615
United States, Texas
Mary Crowley Medical Research Center
Dallas, Texas, United States, 75246
Tyler Cancer Center
Tyler, Texas, United States, 75702
United States, Washington
Cancer Care Northwest Research
Spokane, Washington, United States, 99218 Less << |
|
NCT00033449
|
Stage III Squamous Cell Carcin... More >>oma of the Hypopharynx
Stage III Squamous Cell Carcinoma of the Larynx
Stage III Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage III Squamous Cell Carcinoma of the Oropharynx
Stage IV Squamous Cell Carcinoma of the Hypopharynx
Stage IV Squamous Cell Carcinoma of the Larynx
Stage IV Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage IV Squamous Cell Carcinoma of the Oropharynx Less << |
Phase 1 |
Terminated(Administratively co... More >>mplete.) Less << |
- |
United States, Colorado
... More >>
University of Colorado
Denver, Colorado, United States, 80217-3364 Less << |
|
NCT02887248
|
Urothelial Carcinoma
... More >> Bladder Cancer
Transitional Cell Carcinoma Less << |
Phase 2 |
Active, not recruiting |
December 30, 2019 |
United States, Florida
... More >>
Florida Cancer Specialists - South
Fort Myers, Florida, United States, 33916
Florida Cancer Specialists-North
Saint Petersburg, Florida, United States, 33705
Florida Cancer Specialists-East
West Palm Beach, Florida, United States, 33401
United States, Tennessee
Tennessee Oncology
Nashville, Tennessee, United States, 37203
United States, Texas
Center for Cancer and Blood Disorders
Fort Worth, Texas, United States, 76104 Less << |
|
NCT00509366
|
Non Small Cell Lung Cancer |
Phase 2 |
Terminated(Study terminated du... More >>e to reproducibility issues with genomics prediction model.) Less << |
- |
United States, North Carolina
... More >>
Duke University Medical Center
Durham, North Carolina, United States, 27710
Maria Parham Hospital
Henderson, North Carolina, United States, 27536
Scotland HealthCare System (Scotland Memorial Hospital)
Laurinburg, North Carolina, United States, 28352
Southeastern Regional Medical Center, Gibson Cancer Center
Lumberton, North Carolina, United States, 28358
Duke Raleigh Hospital
Raleigh, North Carolina, United States, 27609
United States, South Carolina
Beaufort Memorial Hospital
Beaufort, South Carolina, United States, 29902
Coastal Cancer Center
Myrtle Beach, South Carolina, United States, 29572
United States, Virginia
Community Memorial Health Center
South Hill, Virginia, United States, 23970 Less << |
|
NCT03737123
|
Urothelial Carcinoma |
Phase 2 |
Not yet recruiting |
January 2022 |
- |
|
NCT03383094
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 2 |
Not yet recruiting |
June 2024 |
United States, California
... More >>
UC San Diego Moores Cancer Center
La Jolla, California, United States, 92093 Less << |
|
NCT03071822
|
Peripheral T Cell Lymphoma |
Phase 4 |
Recruiting |
February 28, 2022 |
Hong Kong
... More >> The University of Hong Kong
Recruiting
Hong Kong, Hong Kong
Contact: King Hei Lu, MMedSc 852-22554361 ext 1654 khlu@hku.hk
Contact: Zoe Chan, BNs 852-22551654 zoechan1@hku.hk
Principal Investigator: Yok Lam Kwong, MD(HK)
Sub-Investigator: Thomas Chan, MBBS(HK) Less << |
|
NCT00509366
|
- |
|
Terminated(Study terminated du... More >>e to reproducibility issues with genomics prediction model.) Less << |
- |
- |
|
NCT00095875
|
- |
|
Completed |
- |
- |
|
NCT03029832
|
Urothelial Carcinoma |
Phase 2 |
Completed |
- |
- |
|
NCT03381066
|
Completely Resected NSCLC With... More >> Common EGFR Mutations Less << |
Phase 3 |
Not yet recruiting |
December 2023 |
Korea, Republic of
... More >>
Department of Oncology, Yonsei University College of Medicine
Not yet recruiting
Seoul, Korea, Republic of, 03722
Contact: Byoung Chul Cho, MD 82 2 2228 0880 cbc1971@yuhs.ac Less << |
|
NCT00401492
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT01000415
|
Cervical Cancer |
Phase 3 |
Unknown |
June 2018 |
Thailand
... More >> Gynecologic Oncology Unit, Department of Obstertrics and Gynecology, Faculty of medicine, Prince of Songkla University
Recruiting
Hat-Yai, Songkhla, Thailand, 90110
Contact: Rakchai - Buhachat, MD. 66 74 451201 brakchai@medicine.psu.ac.th
Contact: Chutaporn - Dampan, BSc. 66 74 451201 djutapor@medicine.psu.ac.th
Principal Investigator: Rakchai - Buhachat, MD. Less << |
|
NCT00095875
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
United States, California
... More >>
Rebecca and John Moores UCSD Cancer Center
La Jolla, California, United States, 92093-0658
United States, Colorado
CCOP - Colorado Cancer Research Program
Denver, Colorado, United States, 80224
United States, Florida
Eugene M. and Christine E. Lynn Cancer Institute at Boca Raton Community Hospital - Main Campus
Boca Raton, Florida, United States, 33486
United States, Georgia
Winship Cancer Institute of Emory University
Atlanta, Georgia, United States, 30322
United States, Illinois
Cardinal Bernardin Cancer Center at Loyola University Medical Center
Maywood, Illinois, United States, 60153
United States, Maine
Maine Center for Cancer Medicine and Blood Disorders - Scarborough
Scarborough, Maine, United States, 04074
United States, Maryland
Greenebaum Cancer Center at University of Maryland Medical Center
Baltimore, Maryland, United States, 21201
United States, Massachusetts
Dana-Farber/Harvard Cancer Center at Dana Farber Cancer Institute
Boston, Massachusetts, United States, 02115
United States, Missouri
Siteman Cancer Center at Barnes-Jewish Hospital - Saint Louis
Saint Louis, Missouri, United States, 63110
United States, New Hampshire
Norris Cotton Cancer Center at Dartmouth-Hitchcock Medical Center
Lebanon, New Hampshire, United States, 03756-0002
United States, New Jersey
UMDNJ University Hospital
Newark, New Jersey, United States, 07103
United States, New York
Albert Einstein Cancer Center at Albert Einstein College of Medicine
Bronx, New York, United States, 10461
United States, North Carolina
Blumenthal Cancer Center at Carolinas Medical Center
Charlotte, North Carolina, United States, 28232-2861
United States, Pennsylvania
UPMC Cancer Centers
Pittsburgh, Pennsylvania, United States, 15232
Germany
Klinikum der J.W. Goethe Universitaet
Frankfurt, Germany, D-60590 Less << |
|
NCT01038661
|
Lung Neoplasms |
Phase 3 |
Completed |
- |
China
... More >> Sanofi-Aventis Administrative Office
Shanghai, China Less << |
|
NCT00705874
|
- |
|
Completed |
- |
- |
|
NCT01360086
|
Adenocarcinoma of the Gastroes... More >>ophageal Junction
Gastric Cancer Less << |
Phase 2 |
Completed |
- |
France
... More >> Centre Hospitalier Regional et Universitaire de Lille
Lille, France, 59037
CHU - Robert Debre
Reims, France, 51092 Less << |
|
NCT01211275
|
Malignant Pleural Mesothelioma |
Phase 1
Phase 2 |
Completed |
- |
Netherlands
... More >> Antoni van Leeuwenhoekziekenhuis (NKI-AVL)
Amsterdam, Noord-Holland, Netherlands, 1066 CX Less << |
|
NCT00051480
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
United States, California
... More >>
UCSD Cancer Center
La Jolla, California, United States, 92093-0064
University of California, Irvine
Orange, California, United States, 92868
Palo Alto VA Health Care Systems
Palo Alto, California, United States, 94304
United States, Illinois
The University of Chicago Medical Center
Chicago, Illinois, United States, 60637-1470
United States, Maryland
Johns Hopkins School of Medicine
Baltimore, Maryland, United States, 21231-2410
United States, Missouri
St. Louis University
St. Louis, Missouri, United States, 63110
United States, Ohio
University Hospitals of Cleveland
Cleveland, Ohio, United States, 44106
United States, Texas
US Oncology, Mary Crowley Center
Dallas, Texas, United States, 75246
University of Texas/MD Anderson
Houston, Texas, United States, 77030-4009
Scott & White Center for Cancer Prevention and Care
Temple, Texas, United States, 76508
Tyler Cancer Center
Tyler, Texas, United States, 75702
United States, Virginia
Medical College of Virginia
Richmond, Virginia, United States, 23298-0058 Less << |
|
NCT00705874
|
Cancer |
Phase 1 |
Completed |
- |
United States, Colorado
... More >>
Rocky Mountain Cancer Centre
Denver, Colorado, United States
United States, Florida
Cancer Centres of Florida
Ocoee, Florida, United States, 34761
United States, Indiana
Central Indiana Cancer Centres
Indianapolis, Indiana, United States, 46219
United States, Nevada
Comprehensive Cancer Centres of Nevada
Las Vegas, Nevada, United States, 89169
United States, New York
New York Oncology Hematology PC
Albany, New York, United States, 12206
United States, Ohio
Dayton Oncology and Hematology, PA
Kettering, Ohio, United States, 45409
United States, South Carolina
Cancer Centres of the Carolinas
Greenville, South Carolina, United States, 29605
United States, Texas
Texas Oncology, PA
Dallas, Texas, United States, 75246
Tyler Cancer Centre
Tyler, Texas, United States, 75702
United States, Virginia
Virginia Oncology Associates
Norfolk, Virginia, United States, 23502
United States, Washington
Northwest Cancer Specialists - Vancouver Cancer Centre
Vancouver, Washington, United States, 98684
North Star Lodge Cancer Centre
Yakima, Washington, United States, 98902 Less << |
|
NCT01301716
|
Solid Cancers |
Phase 1 |
Completed |
- |
United States, California
... More >>
Los Angeles, California, United States, 90025
United States, Florida
Tampa, Florida, United States, 33612
United States, Massachusetts
Boston, Massachusetts, United States, 02114
Boston, Massachusetts, United States, 02215
Spain
Madrid, Spain, 28050 Less << |
|
NCT00660270
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
United States, Missouri
... More >>
Washington University School of Medicine
St. Louis, Missouri, United States, 63119 Less << |
|
NCT02906150
|
Non-Small Cell Lung Cancer |
Phase 2 |
Not yet recruiting |
July 2019 |
Italy
... More >> Azienda Sanitaria Locale Frosinone
Not yet recruiting
Frosinone, Italy
Contact: Dott.ssa Teresa Gamucci
Principal Investigator: Dott.ssa Teresa Gamucci
Istituto Nazionale dei Tumori
Not yet recruiting
Milan, Italy
Contact: Dott.ssa Marina Chiara Garassino
Principal Investigator: Dott.ssa Marina Chiara Garassino
Roma_Campus Bio-Medico
Not yet recruiting
Rome, Italy
Contact: Prof. Daniele Santini
Principal Investigator: Prof. Daniele Santini
Roma_Gemelli
Not yet recruiting
Rome, Italy
Contact: Prof. Carlo Antonio Mario Barone
Principal Investigator: Prof. Carlo Antonio Mario Barone
Roma_Regina Apostolorum
Not yet recruiting
Rome, Italy
Contact: Dott. Francesco Angelini
Principal Investigator: Dott. Francesco Angelini
Roma_Tor Vergata
Not yet recruiting
Rome, Italy
Contact: Prof. Mario Roselli
Principal Investigator: Prof. Mario Roselli
Sant'Andrea Hospital
Not yet recruiting
Rome, Italy
Contact: Prof. Paolo Marchetti
Principal Investigator: Prof. Paolo Marchetti
Presidio Sanitario San Camillo
Not yet recruiting
Torino, Italy
Contact: Dott.ssa Maria Rita Migliorino
Principal Investigator: Dott.ssa Maria Rita Migliorino Less << |
|
NCT02171325
|
Small Cell Lung Cancer |
Phase 2 |
Recruiting |
June 2018 |
China, Jilin
... More >> Jilin cancer hospital
Recruiting
Changchun, Jilin, China, 130000
Contact: ying cheng, doctor 86-43185871902 jl.cheng@163.com Less << |
|
NCT01121393
|
Carcinoma, Non-Small-Cell Lung... More >>
Adenocarcinoma Less << |
Phase 3 |
Completed |
- |
China
... More >> Beijing Chao-Yang Hospital
Beijing, China, 100020
307 Hospital of PLA
Beijing, China, 100071
Peking Union Medical College Hospital
Beijing, China, 100730
Beijing Chest Hospital
Beijing, China, 101149
First Hospital of Jilin University
Changchun, China, 130021
Xiangya Hospital, Central South University
Changsha, China, 410008
Hunan Province Tumor Hospital
Changsha, China
West China Hospital
Chengdu, China, 610041
Fujian Provincial Tumor Hospital
Fuzhou, China, 350014
Guangdong General Hospital
Guangzhou, China, 510080
Guangzhou Institute of Respiratory Disease
Guangzhou, China, 510120
NanFang Hosptial
Guangzhou, China, 510515
The Third Affiliated Hospital of Harbin Medical University
Haerbin, China, 150081
Zhejiang Cancer Hospital
Hangzhou, China, 310022
Hubei Cancer Hospital
HongShan, China
Yunnan Provincial Tumor Hospital
Kunming, China, 650118
Lin Yi Tumor Hospital
Linyi, China, 276001
The Affiliated Cancer Hospital, Guangxi Medical University
Nan Ning, China
the 81th Hospital of PLA
Nanjing, China, 210002
Jiangsu Cancer Hospital
Nanjing, China, 210009
The affiliated hospital of medicalcollege qingdao university
Qingdao, China, 266101
Shanghai Changzheng Hospital
Shanghai, China, 200003
Shanghai Chest Hospital
Shanghai, China, 200030
Zhongshan Hospital Fudan University
Shanghai, China, 200032
Shanghai Pulmonary Hospital
Shanghai, China, 200433
Changhai Hospital
Shanghai, China, 200443
The First Hospital of Chinese Medical University
Shenyang, China, 110001
Hebei Provincial Tumor Hospital
Shijiazhuang, China
Tangdu Hospital
Xi'An, China, 710038
Northern Jiangsu People's Hospital
Yangzhou, China, 225001
Korea, Republic of
Kosin University Gospel Hospital
Busan, Korea, Republic of, 602-702
Chungbuk National University Hospital
Cheongju, Korea, Republic of, 361711
Yeungnam University Medical Center
Daegu, Korea, Republic of, 705-717
Konkuk University Medical Center
Seoul, Korea, Republic of, 143-729
Korea University Guro Hospital
Seoul, Korea, Republic of, 152-703
Thailand
Songklanagarind Hospital
Songkla, Thailand, 90110 Less << |
|
NCT03256916
|
Carcinoma Cervix, Stage III |
Phase 3 |
Not yet recruiting |
September 30, 2025 |
India
... More >> Tata Memorial Centre
Not yet recruiting
Mumbai, Maharashtra, India, 400012
Contact: Supriya J Sastri, MD 9930958309 supriyasastri@gmail.com
Contact: Jayant Goda, MD 24177000 ext 7027 godajayantsastri@gmail.com
Principal Investigator: Supriya J Sastri, MD
Principal Investigator: Jayant Goda, MD Less << |
|
NCT02494583
|
Gastric Adenocarcinoma |
Phase 3 |
Active, not recruiting |
June 6, 2020 |
- |
|
NCT01121393
|
- |
|
Completed |
- |
- |
|
NCT00319514
|
Advanced Non-Small Cell Lung C... More >>ancer Less << |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Gachon University Gil Medical Center
Incheon, Korea, Republic of, 405 760 Less << |
|
NCT02096380
|
- |
|
Unknown |
December 2018 |
- |
|
NCT00165490
|
Adenocarcinoma of Esophagus
... More >> Squamous Cell Carcinoma of Esophagus Less << |
Phase 2 |
Completed |
- |
United States, Massachusetts
... More >>
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115
Massachusetts General Hospital
Boston, Massachusetts, United States, 02115 Less << |
|
NCT02436733
|
Malignant Pleural Mesothelioma |
Phase 2 |
Recruiting |
April 2020 |
Belgium
... More >> UZ Antwerpen
Recruiting
Antwerpen, Belgium
Contact: Jan Van Meerbeeck, Prof
UZ Gent
Recruiting
Gent, Belgium
Contact: Veerle Surmont, Prof
France
Assistance Publique - Hopitaux de Marseille - Hopital Nord
Recruiting
Marseille, France
Contact: Pascal-Alexandre Thomas, Prof
Netherlands
The Netherlands Cancer Institute-Antoni Van Leeuwenhoekziekenhuis
Recruiting
Amsterdam, Netherlands
Contact: Paul Baas, Prof
Erasmus MC Hospital
Recruiting
Rotterdam, Netherlands
Contact: Joachim Aerts, Prof Less << |
|
NCT00213486
|
Esophageal Neoplasms |
Phase 2 |
Completed |
- |
France
... More >> CHU de Rouen
Rouen, France, 76031 Less << |
|
NCT02532192
|
Lymphoma |
Phase 1 |
Withdrawn(Withdrawal of study ... More >>support.) Less << |
- |
- |
|
NCT02718742
|
Recurrent Bladder Urothelial C... More >>arcinoma
Stage IV Bladder Urothelial Carcinoma Less << |
Phase 2 |
Withdrawn |
- |
United States, Arizona
... More >>
Mayo Clinic in Arizona
Scottsdale, Arizona, United States, 85259 Less << |
|
NCT00077103
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Terminated(slow accrual) |
- |
United States, Michigan
... More >>
Josephine Ford Cancer Center at Henry Ford Hospital
Detroit, Michigan, United States, 48202
United States, Ohio
Ireland Cancer Center at University Hosptials Case Medical Center, Case Comprehensive Cancer Center
Cleveland, Ohio, United States, 44106-5065
United States, Pennsylvania
Hillman Cancer Center at University of Pittsburgh Cancer Institute
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT00848783
|
- |
|
Terminated(Due to slow accrual... More >>) Less << |
- |
- |
|
NCT00290719
|
Esophageal Cancer |
Phase 1 |
Terminated(low enrollment) |
- |
United States, California
... More >>
UCSF Helen Diller Family Comprehensive Cancer Center
San Francisco, California, United States, 94115 Less << |
|
NCT00430313
|
Liver Cancer
... More >>Liver Metastasis Less << |
Not Applicable |
Active, not recruiting |
January 2021 |
China
... More >> Fudan University Cancer Hospital
Shanghai, China Less << |
|
NCT01221753
|
- |
|
Terminated(Due to slow accrual... More >>) Less << |
- |
- |
|
NCT00721513
|
Head and Neck Cancer |
Phase 2 |
Unknown |
- |
United States, North Carolina
... More >>
Wake Forest University Comprehensive Cancer Center
Winston-Salem, North Carolina, United States, 27157-1096 Less << |
|
NCT00848783
|
Gastric Cancer
... More >> Gastric Adenocarcinoma
Esophageal Cancer Less << |
Phase 2 |
Terminated(Due to slow accrual... More >>) Less << |
- |
United States, California
... More >>
Norris Cancer Center
Los Angeles, California, United States, 90033
United States, New York
Bellevue Hospital
New York, New York, United States, 10016
NYU Cancer Center
New York, New York, United States, 10016 Less << |
|
NCT01578668
|
Lung Adenocarcinoma
... More >> Brain Metastases Less << |
Phase 2 |
Completed |
- |
China, Guangdong
... More >>
The first affiliated hospital of Guangzhou MC
Guangzhou, Guangdong, China, 510120 Less << |
|
NCT03734692
|
Ovarian Cancer Recurrent |
Phase 1
Phase 2 |
Not yet recruiting |
November 20, 2023 |
United States, Pennsylvania
... More >>
Magee-Womens Hospital of UPMC
Not yet recruiting
Pittsburgh, Pennsylvania, United States, 15213
Contact: Robert Edwards, MD 412-641-4212 edwarp@upmc.edu
Contact: Brenda Steele, RN 412-641-3418 steeleb@upmc.edu Less << |
|
NCT01221753
|
Squamous Cell Carcinoma of the... More >> Head and Neck
Human Papilloma Virus Less << |
Phase 2 |
Terminated(Due to slow accrual... More >>) Less << |
- |
United States, Massachusetts
... More >>
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT03668730
|
Nasopharyngeal Carcinoma |
Phase 2 |
Recruiting |
December 30, 2023 |
China, Guangdong
... More >>
Hai Qiang Mai
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Hai Qiang Mai, MD.PHD 020-87343380 maihq@sysucc.org.cn Less << |
|
NCT01200797
|
- |
|
Terminated(Slow accrual was th... More >>e reason for study termination.) Less << |
- |
- |
|
NCT00017004
|
Anemia
Cervic... More >>al Adenocarcinoma
Cervical Adenosquamous Carcinoma
Cervical Squamous Cell Carcinoma
Drug Toxicity
Radiation Toxicity
Stage IIB Cervical Cancer
Stage III Cervical Cancer
Stage IVA Cervical Cancer Less << |
Phase 3 |
Completed |
- |
United States, Arizona
... More >>
Gynecologic Oncology Group of Arizona
Phoenix, Arizona, United States, 85012 Less << |
|
NCT00066742
|
Limited Stage Small Cell Lung ... More >>Cancer Less << |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
Southwest Oncology Group (SWOG) Research Base
San Antonio, Texas, United States, 78245 Less << |
|
NCT00066742
|
- |
|
Completed |
- |
- |
|
NCT02998385
|
HNSCC |
Phase 3 |
Recruiting |
November 2026 |
France
... More >> Hôpital Bégin
Recruiting
Saint-Mandé, France, 94
Contact: François Régis FERRAND, MD francoisregisferrand@gmail.com Less << |
|
NCT00470366
|
Brain and Central Nervous Syst... More >>em Tumors
Extragonadal Germ Cell Tumor
Ovarian Cancer
Teratoma
Testicular Germ Cell Tumor Less << |
Phase 2 |
Completed |
- |
United States, California
... More >>
USC/Norris Comprehensive Cancer Center and Hospital
Los Angeles, California, United States, 90089-9181
United States, New York
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT00470366
|
- |
|
Completed |
- |
- |
|
NCT00842660
|
Cervical Cancer |
Phase 3 |
Unknown |
January 2013 |
Taiwan
... More >> Chong Jong Wang,
Kaohsiung, Taiwan, M.D
Chien-Sheng Tsai
Keelung, Taiwan Less << |
|
NCT00434668
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Belgium
... More >> ZNA Middelheim
Antwerpen, Antwerp, Belgium, 2020
University Hospital Antwerp
Edegem, Antwerp, Belgium, 2650
St Augustinus ziekenhuis
Wilrijk, Antwerp, Belgium, 2610 Less << |
|
NCT01200797
|
Recurrent Fallopian Tube Cance... More >>r
Recurrent Ovarian Epithelial Cancer
Recurrent Primary Peritoneal Cavity Cancer Less << |
Phase 2 |
Terminated(Slow accrual was th... More >>e reason for study termination.) Less << |
- |
United States, Connecticut
... More >>
Hartford Hospital
Hartford, Connecticut, United States, 06102
Oncology Associates PC
Hartford, Connecticut, United States, 06106
United States, Florida
H. Lee Moffitt Cancer Center and Research Institute
Tampa, Florida, United States, 33612
United States, New Jersey
Cancer Institute of New Jersey
New Brunswick, New Jersey, United States, 08903
United States, Tennessee
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States, 37232
United States, Virginia
Virginia Commonwealth University
Richmond, Virginia, United States, 23298 Less << |
|
NCT02885974
|
Bladder Cancer |
Phase 1 |
Recruiting |
June 2020 |
United States, Texas
... More >>
Baylor Clinic
Recruiting
Houston, Texas, United States, 77030
Contact: Edward Yen, MD 713-798-3750
Harris Health System - Smith Clinic
Recruiting
Houston, Texas, United States, 77054
Contact: Aihua Edward Yen, MD 713-798-3750 ay044661@bcm.edu Less << |
|
NCT03193931
|
Cancer of Head and Neck |
Phase 2 |
Recruiting |
September 1, 2023 |
Germany
... More >> Medizinsche Hochschule Hannover
Recruiting
Hannover, Germany, 30625 Less << |
|
NCT00541073
|
Malignant Mesothelioma |
Phase 2 |
Completed |
- |
France
... More >> Centre Oscar Lambret
Lille, France, 59020 Less << |
|
NCT03585998
|
Small Cell Lung Cancer |
Phase 2 |
Recruiting |
December 19, 2021 |
Korea, Republic of
... More >>
Samsung Medical Center
Recruiting
Seoul, MA, Korea, Republic of, 06351
Contact: Jong-Mu Sun, Associate Professor 82234101795 jongmu.sun@samsung.com Less << |
|
NCT00916669
|
Small Cell Lung Cancer |
Phase 2 |
Withdrawn(sponsor withdrew fun... More >>ding prior to patient enrollment) Less << |
- |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02215
North Shore Medical Center
Peabody, Massachusetts, United States, 01970 Less << |
|
NCT01716546
|
Gastric Cancer |
Phase 1
Phase 2 |
Terminated(Not reached the pri... More >>mary endpoint target according to the statistical design) Less << |
- |
Greece
... More >> "IASO" General Hospital of Athens
Athens, Greece
401 Military Hospital of Athens
Athens, Greece
Air Forces Military Hospital of Athens
Athens, Greece
"Ag.Georgios" General Hospital of Chania
Chania, Greece
University Hospital of Crete, Dep of Medical Oncology Heraklion, Greece
Heraklion, Greece
State General Hospital of Larissa
Larissa, Greece
"Metaxa's" Anticancer Hospital of Piraeus, 1st Dep of Medical Oncology
Piraeus, Greece
"Theagenion" Anticancer Hospital of Thessaloniki, 2nd Dep of Medical Oncology
Thessaloniki, Greece
Diabalkaniko General Hospital of Thessaloniki
Thessaloniki, Greece Less << |
|
NCT01679340
|
Gastric Cancer |
Phase 2 |
Unknown |
June 2018 |
China, Beijing
... More >> Beijing Cancer Hospital
Recruiting
Beijing, Beijing, China, 100142
Contact: Shen Lin, Professor 010-88196561 xiaotong10241@sina.com
Principal Investigator: Shen Lin, professor Less << |
|
NCT03255252
|
Cervical Cancer |
Phase 2 |
Not yet recruiting |
January 2020 |
France
... More >> ICM Val d'Aurelle
Not yet recruiting
Montpellier, France, 34298
Contact: BLEUSE Jean-pierre 00467613102 jean-pierre.bleuse@icm.unicancer.fr
Principal Investigator: Olivier RIOU Less << |
|
NCT03319537
|
Mesothelioma |
Phase 1
Phase 2 |
Recruiting |
October 2020 |
United States, New Jersey
... More >>
Memorial Sloan Kettering at Basking Ridge
Recruiting
Basking Ridge, New Jersey, United States, 07920
Contact: Majorie Zauderer, MD 646-888-4656
Memoral Sloan Kettering Monmouth
Recruiting
Middletown, New Jersey, United States, 07748
Contact: Marjorie Zauderer, MD 646-888-4656
Memorial Sloan Kettering Bergen
Recruiting
Montvale, New Jersey, United States, 07645
Contact: Marjorie Zauderer, MD 646-888-4656
United States, New York
Memorial Sloan Kettering Commack
Recruiting
Commack, New York, United States, 11725
Contact: Marjorie Zauderer, MD 646-888-4656
Memorial Sloan Kettering Westchester
Recruiting
Harrison, New York, United States, 10604
Contact: Majorie Zauderer, MD 646-888-4656
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10065
Contact: Marjorie Zauderer, MD 646-888-4656
Contact: Andrea Cercek, MD 646-888-4189
Principal Investigator: Marjorie Zauderer, MD
Memorial Sloan Kettering Rockville Centre
Recruiting
Rockville Centre, New York, United States, 11570
Contact: Marjorie Zauderer, MD 646-888-4656 Less << |
|
NCT00075738
|
Esophageal Cancer |
Phase 2 |
Unknown |
- |
France
... More >> Clinique La Casamance
Abugne, France, 13400
Hopital Saint Andre
Bordeaux, France, 33075
Hopital Drevon
Dijon, France, 21000
Centre Jean Bernard
Le Mans, France, 72000
Centre Hospital Universitaire Hop Huriez
Lille, France, 59037
Clinique Saint Jean
Lyon, France, 69008
Hopital Notre-Dame de Bon Secours
Metz, France, 57038
Hopital Bichat - Claude Bernard
Paris, France, 75018
Hopital Saint Antoine
Paris, France, 75571
Hopital Tenon
Paris, France, 75970
Hopital Haut Leveque
Pessac, France, 33604
Clinique Ste - Marie
Pontoise, France, 95300
Clinique Armoricaine De Radiologie
Saint Brieuc, France, F-22015
Clinique Francois
Saint-Dizier, France, 52100
Centre Medico-Chirurgical Foch
Suresnes, France, 92151 Less << |
|
NCT01336049
|
Esophageal Squamous Cell Cance... More >>r Less << |
Phase 2 |
Unknown |
- |
China, Beijing
... More >> Xiaodong Zhang
Recruiting
Beijing, Beijing, China, 100142
Contact: xiaodong zhang, MD 86-10-88196175 zxd0829@yahoo.com.cn
Principal Investigator: xiaodong zhang, MD Less << |
|
NCT01495676
|
Infiltrating Bladder Urothelia... More >>l Carcinoma Less << |
Phase 2 |
Recruiting |
September 2020 |
France
... More >> CRLC Val d'Aurelle-Paul Lamarque
Recruiting
Montpellier, France, 34000
Contact: Jean-Pierre Bleuse, MD +33 4 67 61 23 44 Jean-Pierre.Bleuse@montpellier.unicancer.fr
Principal Investigator: David Azria, MD, Ph D Less << |
|
NCT01454102
|
Non-small Cell Lung Cancer |
Phase 1 |
Active, not recruiting |
December 29, 2018 |
United States, California
... More >>
Ucla
Santa Monica, California, United States, 90404
United States, Connecticut
Yale University
New Haven, Connecticut, United States, 06520
United States, Florida
H. Lee Moffitt Cancer Center & Research Institute
Tampa, Florida, United States, 33612
United States, Maryland
Johns Hopkins Sidney Kimmel Comprehensive Cancer Center
Baltimore, Maryland, United States, 21287
United States, New York
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
United States, North Carolina
Duke University Medical Center
Durham, North Carolina, United States, 27710
United States, Pennsylvania
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111
United States, Texas
Ut Southwestern Medical Center At Dallas
Dallas, Texas, United States, 75390-8852
United States, Washington
Seattle Cancer Care Alliance
Seattle, Washington, United States, 98109
Canada, Ontario
Local Institution
Hamilton, Ontario, Canada, L8V 5C2
Local Institution
Ottawa, Ontario, Canada, K1H 8L6
Local Institution
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT03117257
|
Locally Advanced Head and Neck... More >> Squamous Cell Carcinoma Less << |
Phase 2 |
Recruiting |
August 20, 2021 |
China, Guizhou
... More >> Cancer Hospital of Guizhou Medical University
Recruiting
Guiyang, Guizhou, China, 550000
Contact: Feng Jin, Bachelor 0851-86512802 jinf8865@yahoo.com.cn
Sub-Investigator: Weili Wu, master
Sub-Investigator: Jinhua Long, master
Sub-Investigator: Yuanyuan Li, master
Sub-Investigator: Xiuyun Gong, Bachelor
Sub-Investigator: Xiaoxiao Chen, Bachelor
Sub-Investigator: Mang Zhang, Bachelor Less << |
|
NCT00511992
|
- |
|
Completed |
- |
- |
|
NCT01665183
|
Cutaneous Melanoma, Uveal Mela... More >>noma, Ovarian Carcinoma or Other Advanced Solid Tumors Less << |
Phase 1 |
Completed |
- |
United States, Texas
... More >>
MD Anderson Cancer Center
Houston, Texas, United States
Taiwan
NCKUH
Tainan, Taiwan, 704 Less << |
|
NCT02396498
|
Stage Ⅲ Gastric Cancer |
Phase 3 |
Unknown |
December 2016 |
China, Shaanxi
... More >> IEC of Insititution for National Drug Clinical Trials,Tangdu Hospital,Fourth Military Medical University
Recruiting
Xi'an, Shaanxi, China, 029
Contact: Lina Liu 029-84777631 tangduec@126.com Less << |
|
NCT00174772
|
Lung Neoplasms |
Phase 2 |
Completed |
- |
Belgium
... More >> Sanofi-Aventis Administrative Office
Diegem, Belgium
Finland
Sanofi-Aventis Administrative Office
Helsinki, Finland
France
Sanofi-Aventis Administrative Office
Paris, France
Italy
Sanofi-Aventis Administrative Office
Milan, Italy
Netherlands
Sanofi-Aventis Administrative Office
Gouda, Netherlands
Spain
Sanofi-Aventis Administrative Office
Barcelona, Spain
Turkey
Sanofi-Aventis Administrative Office
Istanbul, Turkey
United Kingdom
Sanofi-Aventis Administrative Office
Guilford, United Kingdom Less << |
|
NCT02818725
|
Infiltrating Urothelial Carcin... More >>oma
KRAS Gene Mutation Less << |
Phase 3 |
Unknown |
September 2017 |
France
... More >> Hopital Saint Andre
Bordeaux, France, 33075
Institut Bergonie
Bordeaux, France, 33076
Centre Francois Baclesse
Caen, France, 14076
Hopital Henri Mondor
Creteil, France, 94010
Centre Leon Berard
Lyon, France, 69008
Institut Paoli Calmettes
Marseille, France, 13273
Centre Alexis Vautrin
Nancy, France, 54511
Centre Rene Gauducheau
Nantes, France, 44800
Chu de Nimes
Nimes, France, 30029
Institut Curie
Paris, France, 75005
Diaconesses - Croix St Simon
Paris, France, 75012
Pitie Salpetriere
Paris, France, 75013
Centre Hospitalier Lyon Sud
Pierre Benite, France, 69495
Institut Cancerologie de La Loire
St Priest En Jarez, France, 42270
Hopitaux Universitaires
Strasbourg, France, 67091
Institut Claudius Regaud
Toulouse, France, 31052
Institut Gustave Roussy
Villejuif, France, 94805 Less << |
|
NCT00511992
|
Advanced Ovarian Carcinoma
... More >> Primary Peritoneal Carcinoma
Ovarian Carcinosarcoma Less << |
Phase 2 |
Completed |
- |
United States, Oklahoma
... More >>
University of Oklahoma Health Sciences Center
Oklahoma City, Oklahoma, United States, 73104 Less << |
|
NCT02994251
|
Unresectable Intrahepatic Chol... More >>angiocarcinoma Less << |
Phase 2 |
Recruiting |
June 2020 |
United States, Connecticut
... More >>
Smilow Cancer Center
Recruiting
New Haven, Connecticut, United States, 06510
Contact: Todd Schlachter, MD 203-785-4747 todd.schlachter@yale.edu
Sub-Investigator: Hyun Kim, MD
Sub-Investigator: Stacey Stein, MD
Sub-Investigator: Howard S Hochster, MD
Sub-Investigator: Jill Lacy, MD
Sub-Investigator: Jeffrey Pollak, MD Less << |
|
NCT00021333
|
Head and Neck Cancer
... More >> Lung Cancer Less << |
Phase 2 |
Completed |
- |
United States, New Jersey
... More >>
Hunterdon Regional Cancer Center
Flemington, New Jersey, United States, 08822
Kimball Medical Center
Lakewood, New Jersey, United States, 08701
South Jersey Regional Cancer Center
Millville, New Jersey, United States, 08332
Fox Chase Cancer Center at Virtua Memorial Hospital Burlington County
Mount Holly, New Jersey, United States, 08060
Riverview Medical Center - Booker Cancer Center
Red Bank, New Jersey, United States, 07701
Community Medical Center
Toms River, New Jersey, United States, 08755
St. Francis Medical Center
Trenton, New Jersey, United States, 08629
United States, Pennsylvania
Bon Secours-Holy Family Health System
Altoona, Pennsylvania, United States, 16602
Delaware County Memorial Hospital
Drexel Hill, Pennsylvania, United States, 19026
Pinnacle Health Hospitals
Harrisburg, Pennsylvania, United States, 17105-8700
Conemaugh Memorial Hospital
Johnstown, Pennsylvania, United States, 15905
Saint Mary Regional Center
Langhorne, Pennsylvania, United States, 19047
Central Montgomery Medical Center
Lansdale, Pennsylvania, United States, 19446-1200
Paoli Memorial Hospital
Paoli, Pennsylvania, United States, 19301-1792
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111
Pottstown Memorial Regional Cancer Center
Pottstown, Pennsylvania, United States, 19464
Reading Hospital and Medical Center
Reading, Pennsylvania, United States, 19612-6052
Southern Chester County Medical Center
West Grove, Pennsylvania, United States, 19390 Less << |
|
NCT00004063
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1
Phase 2 |
Unknown |
- |
United States, Texas
... More >>
University of Texas Health Science Center at Houston
Recruiting
Houston, Texas, United States, 77225
Contact: Joan M.C. Bull, MD 713-500-6820 joan.m.bull@uth.tmc.edu Less << |
|
NCT00106626
|
Advanced Cancer |
Phase 1 |
Completed |
- |
- |
|
NCT00602082
|
Gastrointestinal Carcinoid Tum... More >>or
Islet Cell Tumor Less << |
Phase 2 |
Completed |
- |
United Kingdom
... More >> Basildon University Hospital
Basildon, England, United Kingdom, SS16 5NL
Addenbrooke's Hospital
Cambridge, England, United Kingdom, CB2 2QQ
Cookridge Hospital
Leeds, England, United Kingdom, LS16 6QB
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Aintree University Hospital
Liverpool, England, United Kingdom, L9 7AL
UCL Cancer Institute
London, England, United Kingdom, NW3 2QG
St. Thomas' Hospital
London, England, United Kingdom, SE1 7EH
Mid Kent Oncology Centre at Maidstone Hospital
Maidstone, England, United Kingdom, ME16 9QQ
Christie Hospital
Manchester, England, United Kingdom, M20 4BX
Clatterbridge Centre for Oncology
Merseyside, England, United Kingdom, CH63 4JY
Northern Centre for Cancer Treatment at Newcastle General Hospital
Newcastle-Upon-Tyne, England, United Kingdom, NE4 6BE
Oxford Radcliffe Hospital
Oxford, England, United Kingdom, 0X3 9DU
Royal Marsden - Surrey
Sutton, England, United Kingdom, SM2 5PT
Southend University Hospital NHS Foundation Trust
Westcliff-On-Sea, England, United Kingdom, SS0 0RY
Edinburgh Cancer Centre at Western General Hospital
Edinburgh, Scotland, United Kingdom, EH4 2XU
Beatson West of Scotland Cancer Centre
Glasgow, Scotland, United Kingdom, G12 0YN
Velindre Cancer Center at Velindre Hospital
Cardiff, Wales, United Kingdom, CF14 2TL Less << |
|
NCT00106626
|
- |
|
Completed |
- |
- |
|
NCT00059761
|
Lung Cancer |
Phase 1 |
Completed |
- |
- |
|
NCT01192165
|
Cancer |
Phase 1 |
Completed |
- |
United States, Arizona
... More >>
GSK Investigational Site
Scottsdale, Arizona, United States, 85259
United States, California
GSK Investigational Site
Duarte, California, United States, 91010
GSK Investigational Site
Sacramento, California, United States, 95817
United States, Colorado
GSK Investigational Site
Denver, Colorado, United States, 80218
United States, Nevada
GSK Investigational Site
Las Vegas, Nevada, United States, 89169
United States, New York
GSK Investigational Site
Albany, New York, United States, 12206
United States, South Carolina
GSK Investigational Site
Greenville, South Carolina, United States, 29605
United States, Tennessee
GSK Investigational Site
Nashville, Tennessee, United States, 37203
United States, Texas
GSK Investigational Site
Dallas, Texas, United States, 75246
GSK Investigational Site
Houston, Texas, United States, 77030
GSK Investigational Site
Tyler, Texas, United States, 75702
United States, Virginia
GSK Investigational Site
Norfolk, Virginia, United States, 23502
United States, Washington
GSK Investigational Site
Vancouver, Washington, United States, 98684
Canada, Ontario
GSK Investigational Site
Toronto, Ontario, Canada, M5G 2M9
France
GSK Investigational Site
Caen Cedex 9, France, 14033
GSK Investigational Site
Marseille cedex 5, France, 13385
GSK Investigational Site
Saint-Herblain cedex, France, 44805
GSK Investigational Site
Toulouse cedex, France, 31052
Korea, Republic of
GSK Investigational Site
Seoul, Korea, Republic of, 120-752
GSK Investigational Site
Seoul, Korea, Republic of, 135-710 Less << |
|
NCT01949870
|
Inoperable Locally Advanced or... More >> Metastatic Biliary Tract Cancer Less << |
Phase 1 |
Terminated(change in the devel... More >>opment strategy) Less << |
- |
Japan
... More >> AZD6244 PhI Japanese Gem/
Kashiwa Shi, Chiba, Japan
AZD6244 PhI Japanese Gem/
Chuo Ku, Tokyo, Japan Less << |
|
NCT01949870
|
- |
|
Terminated(change in the devel... More >>opment strategy) Less << |
- |
- |
|
NCT00169247
|
Larynx Cancer
... More >> Hypopharynx Cancer Less << |
Phase 2 |
Completed |
- |
France
... More >> Centre Oscar Lambret
Lille, France, 59020
Centre René Gauducheau
Nantes, France, 44805
CHU de Tours
Tours, France, 37044 Less << |
|
NCT00716560
|
- |
|
Terminated(Administratively te... More >>rminated.) Less << |
- |
United States, Pennsylvania
... More >>
Thomas Jefferson University Hospital
Philadelphia, Pennsylvania, United States, 19107 Less << |
|
NCT03480971
|
Mucositis
Nep... More >>hrotoxicity
Ototoxicity Less << |
Phase 2 |
Not yet recruiting |
October 2019 |
United States, Maryland
... More >>
University Maryland School of Medicine
Not yet recruiting
Baltimore, Maryland, United States, 21201
Contact: Robert Malyapa, MD 410-328-6080
Principal Investigator: Robert Malyapa, MD Less << |
|
NCT00191789
|
Breast Cancer |
Phase 2 |
Completed |
- |
India
... More >> For additional information regarding investigative sites for this clinical trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9AM to 5PM Eastern Time (UTC/GMT -5 hours, EST), or speak with your personal physician.
Pune, Maharashtra, India, 411001
For additional information regarding investigative sites for this clinical trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9AM to 5PM Eastern Time (UTC/GMT -5 hours, EST), or speak with your personal physician.
Vellore, Tamil Nadu, India, 632004
For additional information regarding investigative sites for this clinical trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9AM to 5PM Eastern Time (UTC/GMT -5 hours, EST), or speak with your personal physician.
Delhi, India, 110029 Less << |
|
NCT00555620
|
Stomach Neoplasms |
Phase 1 |
Completed |
- |
Korea, Republic of
... More >>
Pfizer Investigational Site
Seongnam-si, Gyeonggi-do, Korea, Republic of, 463-707
Pfizer Investigational Site
Goyang, Gyeonggi, Korea, Republic of, 410-769
Pfizer Investigational Site
Seoul, Korea, Republic of, 110-744 Less << |
|
NCT02609503
|
Head and Neck Cancer |
Phase 2 |
Active, not recruiting |
March 19, 2020 |
United States, Maryland
... More >>
John Hopkins Sidney Kimmel Comprehensive Cancer Center
Baltimore, Maryland, United States, 21231
United States, North Carolina
UNC Lineberger Comprehensive Cancer Center
Chapel Hill, North Carolina, United States, 27599
United States, Pennsylvania
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111 Less << |
|
NCT03361865
|
UC (Urothelial Cancer) |
Phase 3 |
Active, not recruiting |
September 2020 |
- |
|
NCT00191789
|
- |
|
Completed |
- |
- |
|
NCT00002559
|
Ovarian Cancer
... More >> Testicular Germ Cell Tumor Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00055835
|
Bladder Cancer
... More >> Urethral Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00555620
|
- |
|
Completed |
- |
- |
|
NCT01222676
|
Bladder Cancer |
Phase 2 |
Unknown |
- |
Italy
... More >> Fondazione Istituto Nazionale dei Tumori
Recruiting
Milan, Italy, 20133
Contact: Contact Person 39-2-2390-2359 roberto.salvioni@istitutotumori.mi.it Less << |
|
NCT01245959
|
Nasopharyngeal Carcinoma |
Phase 3 |
Unknown |
April 2018 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT00952497
|
Gastric Cancer |
Phase 2 |
Completed |
- |
United States, California
... More >>
UCSF Helen Diller Family Comprehensive Cancer Center
San Francisco, California, United States, 94115
United States, Georgia
Central Georgia Cancer Care, P.C.
Macon, Georgia, United States, 31201
United States, Pennsylvania
University of Pennsylvania, Abramson Cancer Center
Philadelphia, Pennsylvania, United States, 19104
United States, Tennessee
The West Clinic
Memphis, Tennessee, United States, 38120
United States, Texas
The University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030
Spain
Hospital Vall d' Hebron
Barcelona, Spain, 08035
Hospital Universitari Germans Trias i Pujol
Barcelona, Spain, 08916
Hospital Universitario Ramon y Cajal
Madrid, Spain, 28034
Hospital Clinico San Carlos
Madrid, Spain, 28040
Hospital Universitario 12 de Octubre
Madrid, Spain, 28041
Hospital Universitario Marques de Valdecilla
Santander, Spain, 39008 Less << |
|
NCT01594099
|
Cervical Cancer |
Phase 2 |
Unknown |
March 2014 |
- |
|
NCT01041404
|
Gastric Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00633568
|
Non-small Cell Lung Carcinoma |
Phase 3 |
Terminated(Slow recruitment) |
- |
Belgium
... More >> Department of Pneumology RHMS Hôpital de la Madeleine
Ath, Belgium, 7800
Department of Pneumology Clinique Saint-Luc
Bouge, Belgium, 5004
Department of Pneumology CHR St Joseph-Warquignies
Boussu, Belgium, 7360
Department of Intensive Care Unit and Thoracic Oncology Institut Jules Bordet
Brussels, Belgium, 1000
Department of Pneumology Hospital Ixelles-Molière
Brussels, Belgium
Department of Pneumology CHU Charleroi
Charleroi, Belgium
Department of Pneumology Hôpital Saint-Joseph
Gilly, Belgium, 6060
Hôpital Ambroise Paré
Mons, Belgium, 7000
Hôpital Vésale - Montigny-le-Tilleul
Montigny-le-Tilleul, Belgium, 6110
Department of Pneumology Centre Hospitalier de Mouscron
Mouscron, Belgium, 7700
CH Peltzer-La Tourelle
Verviers, Belgium, 4800
France
Service de Pneumologie Centre Hospitalier de Douai
Douai, France, 59507
Service de Pneumologie Hôpital de Hayange
Hayange, France, 57700
Pneumology department of CHU Lille
Lille, France
Service de Pneumologie Centre Hospitalier Intercommunal le Raincy-Montfermeil
Montfermeil, France, 93370
Service de Pneumologie CHG Tourcoing
Tourcoing, France, 59208
Cabinet médical Saint-Michel
Valenciennes, France, 59300
Greece
Medical Oncology St Savas Hospital
Athens, Greece, 11522
Spain
Medical Oncology Hospital de Sagunto
Valencia, Spain Less << |
|
NCT01258634
|
Osteosarcoma
... More >>Lung Metastases Less << |
Phase 1 |
Terminated(PI no longer affili... More >>ated with institution; only 2 subjects enrolled) Less << |
- |
United States, Illinois
... More >>
University of Chicago Department of Pediatrics Hematology/Oncology
Chicago, Illinois, United States, 60637 Less << |
|
NCT01882621
|
Non-small Cell Lung Cancer |
Phase 3 |
Unknown |
August 2016 |
China, Guangdong
... More >>
Sun Yat-Sen University Cancer Center
Recruiting
GuangZhou, Guangdong, China, 510030
Contact: Ting Zhou 8602087343786 zhouting@sysucc.org.cn
Sub-Investigator: Yan Huang, Doctor Less << |
|
NCT00952341
|
- |
|
Completed |
- |
- |
|
NCT01466244
|
- |
|
Completed |
- |
- |
|
NCT00022217
|
Head and Neck Cancer |
Phase 2 |
Unknown |
- |
United States, Arizona
... More >>
Veterans Affairs Medical Center - Phoenix (Hayden)
Phoenix, Arizona, United States, 85012
Veterans Affairs Medical Center - Tucson
Tucson, Arizona, United States, 85723
United States, Illinois
Division of Head and Neck Surgery
Evanston, Illinois, United States, 60201
United States, Louisiana
Louisiana State University Health Sciences Center - Shreveport
Shreveport, Louisiana, United States, 71130-3932 Less << |
|
NCT00952341
|
Chemotherapy-induced Nausea an... More >>d Vomiting (CINV) Less << |
Phase 3 |
Completed |
- |
- |
|
NCT01041404
|
- |
|
Completed |
- |
- |
|
NCT00320294
|
Stomach Neoplasm |
Phase 2 |
Unknown |
- |
Korea, Republic of
... More >>
Gachon University Gil Medical Center
Incheon, Korea, Republic of, 405 760 Less << |
|
NCT00130780
|
- |
|
Completed |
- |
- |
|
NCT00130780
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT01131429
|
Neoplasms, Lung
... More >> Carcinoma, Non-Small Cell Lung
Drug Therapy
Genes, EGFR Less << |
Phase 2 |
Unknown |
June 2015 |
China, Beijing
... More >> Chinese PLA General Hospital
Not yet recruiting
Beijing, Beijing, China, 100853
Contact: Liang-An Chen, MD, phD 86-10-66939361 chenla301@263.net
Principal Investigator: Liang-An Chen, MD, phD Less << |
|
NCT00316888
|
Anal Cancer |
Phase 2 |
Active, not recruiting |
May 2022 |
- |
|
NCT02402842
|
Anal Canal Carcinoma |
Phase 2 |
Active, not recruiting |
September 2021 |
France
... More >> University hospital of Besançon
Besançon, France, 25000
FNLCC center Georges François Leclerc
Dijon, France, 21000
Oscar Lambret center
Lille, France, 59000
Jean Mermoz Private Hospital
Lyon, France, 69 008
Hospital of Belfort-Montbeliard
Montbeliard, France, 25200
Regional Institute of Cancer
Montpellier, France, 34 298
Institute of Cancerology of Lorraine
Nancy, France, 54 519
Antoine Lacassagne Center
Nice, France, 06 189
Paris Saint-Joseph Hospital Group
Paris, France, 75 014
Curie Institute
Paris, France, 75 248
Pitié Salpétrière Hospital
Paris, France, 75 651
Mutualist Montsouris Institute
Paris, France, 75 674
European Georges Pompidou Hospital
Paris, France, 75 908
Saint-Antoine Hospital
Paris, France, 75571
University Robert Debré Hospital
Reims, France, 51 092
Paul Strauss Center
Strasbourg, France, 67 065 Less << |
|
NCT00538525
|
Breast Cancer |
Not Applicable |
Completed |
- |
- |
|
NCT00977561
|
Small Cell Lung Carcinoma |
Phase 2 |
Terminated(See termination rea... More >>son in detailed description.) Less << |
- |
- |
|
NCT03121716
|
Nasopharyngeal Carcinoma |
Phase 1 |
Recruiting |
April 2018 |
China
... More >> Jiangsu Hengrui
Recruiting
Shanghai, China
Contact: Qing Yang +8615705155017 yangqing@hrs.com.cn Less << |
|
NCT01714908
|
Non-small Cell Lung Cancer |
Phase 2 |
Active, not recruiting |
October 20, 2018 |
China, Beijing
... More >> Cancer Institute & Hospital, Chinese Academy of Medical Sciences
Beijing, Beijing, China, 100021
Beijing Cancer Hospital
Beijing, Beijing, China, 100142
Chinese PLA General Hospital
Beijing, Beijing, China, 100853
The General Hospital of the People's Liberation Army
Beijing, Beijing, China, 100853
China, Fujian
Fujian Province Cancer Hospital
Fuzhou, Fujian, China, 350008
The First Affiliated Hospital of Xiamen University
Xiamen, Fujian, China, 361003
China, Guizhou
Guizhou Cancer Hospital
Guiyang, Guizhou, China, 550004
China, Hebei
Hebei Medical University Fourth Hospital
Shijiazhuang, Hebei, China, 050011
China, Henan
Henan Cancer Hospital
Zhengzhou, Henan, China, 450008
China, Hubei
Wuhan Union Hospital, Tongji Medical College, Huazhong University of Science and Technology
Wuhan, Hubei, China, 430022
Zhongnan Hospital of Wuhan University
Wuhan, Hubei, China, 430030
Renmin Hospital of Wuhan University
Wuhan, Hubei, China, 430060
China, Liaoning
The First Hospital of China Medical University
Shenyang, Liaoning, China, 110001
China, Shandong
Shandong Cancer Hospital and Institute
Jinan, Shandong, China, 250117
China, Shanghai
Fudan University Shanghai Cancer Center
Shanghai, Shanghai, China, 200032
China, Sichuan
West China Hospital, West China School of Medicine, Sichuan University
Chengdu, Sichuan, China, 610041
China, Tianjin
Tianjin Medical University Cancer Institute and Hospital
Tianjin, Tianjin, China, 300060
China, Zhejiang
The First People's Hospital of Hangzhou
Hangzhou, Zhejiang, China, 310006
Zhejiang Cancer Hospital
Hangzhou, Zhejiang, China, 310022 Less << |
|
NCT00964626
|
Ovarian Cancer |
Phase 2 |
Unknown |
- |
Russian Federation
... More >>
Russian Academy of Medical Sciences Cancer Research Center
Recruiting
Moscow, Russian Federation, 115478
Contact: Sergei A. Tjulandin, MD, PhD 7-495-324-9874 Less << |
|
NCT00977561
|
- |
|
Terminated(See termination rea... More >>son in detailed description.) Less << |
- |
- |
|
NCT00269152
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
France
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Bordeaux, France, 33076
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Paris, France, 75571
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Rennes, France, 35033
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
St Priest En Jarez, France, 42270
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Toulouse, France, 31059
Germany
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Bad Berka, Germany, 99437
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Bielefeld, Germany, D-33604
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Großhansdorf, Germany, D-22927
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Halle, Germany, D-06120
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hannover, Germany, D-30625
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Kassel, Germany, 34125
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Köln, Germany, D-51109
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Loewenstein, Germany, 74245
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mainz, Germany, D-55131
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mannheim, Germany, 68167
Spain
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Barcelona, Spain, 08036
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Madrid, Spain, 28046
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Santiago De Compostela, Spain, 15706 Less << |
|
NCT00400179
|
Gastric Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00316888
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00191841
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Completed |
- |
Russian Federation
... More >>
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Barnaul, Russian Federation
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Moscow, Russian Federation
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
St Petersburg, Russian Federation
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Ufa, Russian Federation Less << |
|
NCT02324543
|
Metastatic Pancreatic Adenocar... More >>cinoma Less << |
Phase 1
Phase 2 |
Active, not recruiting |
January 2020 |
United States, Maryland
... More >>
Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Baltimore, Maryland, United States, 21231 Less << |
|
NCT00072787
|
Gastric Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, California
... More >>
Rosen, Lee
Los Angeles, California, United States, 90025
University of Southern California Norris Comprehensive Cancer Center
Los Angeles, California, United States, 90089
United States, Hawaii
Chong, Clayton
Honolulu, Hawaii, United States, 96817
Straub Clinic and Hospital
Honolulu, Hawaii, United States, 96817
United States, Illinois
Northwestern University Robert H Lurie Comprehensive Cancer Center
Chicago, Illinois, United States, 60611
University of Chicago
Chicago, Illinois, United States, 60637
United States, New Mexico
Lovelace Sandia Health System
Albuquerque, New Mexico, United States, 87108
New Mexico VA Health Care System
Albuquerque, New Mexico, United States, 87108
University of New Mexico - Albuquerque
Albuquerque, New Mexico, United States, 87131
United States, Pennsylvania
Abramson Cancer Center at the University of Pennsylvania
Philadelphia, Pennsylvania, United States, 19104
United States, Texas
MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00269152
|
- |
|
Completed |
- |
- |
|
NCT00536640
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Completed |
- |
Germany
... More >> Klinikum Bayreuth GmbH
Bayreuth, Germany, D-95445
Charite´ Mitte
Berlin, Germany, D-10117
Gemeinschaftskrankenhaus Havelhöhe
Berlin, Germany, D-14089
Helios Klinikum Emil v. Behring
Berlin, Germany, D-14165
Augusta-Krankenanstalten
Bochum, Germany, D-44791
Johanniter-Krankenhaus Bonn
Bonn, Germany, D-53113
Forschungszentrum Borstel
Borstel, Germany, D-23845
St. Johannes Hospital
Duisburg, Germany, D-47166
Krankenhaus Nordwest
Frankfurt am Main, Germany, D-60488
Klinikum Frankfurt (Oder)GmbH
Frankfurt Oder, Germany, D-15232
Städtisches Krankenhaus Frankfurt-Höchst
Frankfurt, Germany, D-65929
Medizinisches Versorgungszentrum Osthessen
Fulda, Germany, D-36043
Universitätsklinikum Greifswald
Greifswald, Germany, D-17487
Krankenhaus Großhansdorf
Großhansdorf, Germany, D-22927
Georg-August-Universität Göttingen
Göttingen, Germany, D-37075
Diakoniekrankenhaus Halle/S.
Halle, Germany, D-06114
Asklepios Klinik Harburg
Hamburg, Germany, D-21075
Thoraxklinik Universitätsklinikum Heidelberg
Heidelberg, Germany, D-69126
Lungenklinik Hemer
Hemer, Germany, 58675
Fachklinik für Lungenerkrankungen Immenhausen
Immenhausen, Germany, D-34376
St. Vincentius-Kliniken Karlsruhe
Karlsruhe, Germany, D-76137
Klinikum Kassel GmbH
Kassel, Germany, 34125
Katholisches Klinikum Haus Marienhof
Koblenz, Germany, 56073
Malteser Krankenhaus St. Hildegardis
Köln, Germany, D-50931
Onkologische Schwerpunktpraxis Dr. Lothar Müller
Leer, Germany, D-26789
Klinikum Lippe-Lemgo
Lemgo, Germany, D-32657
Klinikum Ludwigshafen
Ludwigshafen, Germany, D-67063
Klinik Löwenstein
Löwenstein, Germany, 74245
Universitätsklinikum Schleswig-Holstein
Lübeck, Germany, D-23538
St. Hildegardis Krankenhaus
Mainz, Germany, D-55131
Universitätsklinikum Marburg
Marburg, Germany, D-35033
Johannes Wesling Klinikum Minden
Minden, Germany, 32429
Krankenhaus Barmherzige Brüder
Regensburg, Germany, D-93049
Hanse-Klinikum Stralsund
Stralsund, Germany, D-18410
Onkologische Gemeinschaftspraxis Dr. Nusch
Velbert, Germany, D-42551
Fachkliniken Wangen
Wangen, Germany, D-88239
Helios Klinikum Wuppertal
Wuppertal, Germany, D-42283 Less << |
|
NCT00126269
|
Carcinoma |
Phase 3 |
Unknown |
- |
France
... More >> Institut Gustave Roussy
Recruiting
Villejuif, France, 94805
Contact: Karim FIZAZI, Dr 33 1 42114559 fizazi@igr.fr Less << |
|
NCT01160731
|
Lung Cancer |
Phase 1 |
Withdrawn |
- |
- |
|
NCT00400179
|
- |
|
Completed |
- |
- |
|
NCT01150630
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
Italy
... More >> Istituto Scientifico H. San Raffaele
Milan, Italy, 20132 Less << |
|
NCT00003044
|
Liver Cancer |
Phase 2 |
Unknown |
- |
United States, Maryland
... More >>
Johns Hopkins Oncology Center
Baltimore, Maryland, United States, 21287
United States, Minnesota
Mayo Clinic Cancer Center
Rochester, Minnesota, United States, 55905
Hong Kong
Prince of Wales Hospital
Shatin, New Territories, Hong Kong Less << |
|
NCT02889666
|
Recurrent Non-small Cell Lung ... More >>Cancer Less << |
Phase 1 |
Recruiting |
December 2020 |
China, Jilin
... More >> China-Japan Union Hospital, Jilin University
Recruiting
Changchun, Jilin, China, 130033
Contact: Xian-Ling Cong, M.D 86-0431-89876626 qingzhao_jilin@126.com
China
Shanghai 10th People's Hospital
Recruiting
Shanghai, China, 200072
Contact: Da Fu, Ph.D 86-15921527578 fu800da900@126.com
Shanghai Pulmonary Hospital, Tongji University School of Medicine
Recruiting
Shanghai, China, 200433
Contact: Chun-Yan Wu, M.D 86-65115006-3030 51917533@qq.com Less << |
|
NCT00069966
|
Lymphoma |
Phase 2 |
Unknown |
- |
- |
|
NCT00677118
|
Nasopharyngeal Carcinoma |
Phase 3 |
Unknown |
March 2015 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT00968799
|
Epithelial Ovarian Cancer
... More >> Fallopian Tube Carcinoma Less << |
Not Applicable |
Terminated(poor patient accrua... More >>l) Less << |
- |
Switzerland
... More >> Department of Surgery, Cantonal Hospital St. Gallen
St. Gallen, Switzerland, 9007 Less << |
|
NCT02795988
|
Gastrointestinal Neoplasms
... More >> Adenocarcinoma Less << |
Phase 1
Phase 2 |
Recruiting |
December 2020 |
Georgia
... More >> ARENSIA Exploratory Medicine LLC
Recruiting
Tbilisi, Georgia, 0112
Contact: Lili Ioseliani, MD
Principal Investigator: Marina Maglakelidze, MD
Hong Kong
Department of Medicine, Queen Mary Hospital
Recruiting
Hong Kong, Hong Kong
Contact: Thomas Yau, MD
Moldova, Republic of
ARENSIA Exploratory Medicine IMSP Institutul Oncologic
Recruiting
Chisinau, Moldova, Republic of, 2025
Contact: Natalia Pirtac, MD
Principal Investigator: Lurie Bulat, MD
Taiwan
Chang Gung Memorial Hospital
Recruiting
Taipei, Songshan District, Taiwan, 105
Contact: Wen-Chi Chou, MD
Principal Investigator: Wen-Chi Chou, MD
China Medical University Hospital
Recruiting
Taichung, Taichung City, Taiwan, 40402
Contact: Li-Yuan Bai, MD
Principal Investigator: Li-Yuan Bai, MD
National Cheng-Kung University Hospital
Recruiting
Tainan, Taiwan, 704
Contact: Chia-Jui Yen, MD
Taipei Veterans General Hospital
Recruiting
Taipei, Taiwan, 11217
Contact: Yee Chao, MD
Thailand
Immunology Unit, Research Division, National Cancer Institute
Recruiting
Bangkok, Bankgkok, Thailand, 10400
Contact: Wirote Lausoontornsiri, MD
Principal Investigator: Wirote Lausoontornsiri, MD
Division of Medical Oncology, Department of Medicine, Prince of Songkla University, Songklanagarind Hospital
Recruiting
Hat Yai, Songkhla Province, Thailand, 90110
Contact: Arunee Dechaphunkul, MD
Principal Investigator: Arunee Dechaphunkul, MD
Chulabhorn Hospital
Recruiting
Bangkok, Thailand, 10210
Contact: Teerapat Ungtrakul, MD
Principal Investigator: Teerapat Ungtrakul, MD
Division of Medical Oncology, Department of Medicine, Chulalongkorn University
Recruiting
Bangkok, Thailand, 10330
Contact: Suebpong Tansanvimon, MD
Principal Investigator: Suebpong Tansanvimon, MD
Oncology Unit, Department of Medicine, Rajavithi Hospital
Recruiting
Bangkok, Thailand, 10400
Contact: Jedzada Maneechavakajorn, MD
Principal Investigator: Jedzada Maneechavakajorn, MD
Division of Oncology, Department of Internal Medicine, Faculty of Medicine, Chiang Mai University
Recruiting
Chiang Mai, Thailand, 50200
Contact: Chaiyut Charoentum, MD
Principal Investigator: Chaiyut Charoentum, MD
Srinagarind Hospital
Recruiting
Khon Kaen, Thailand, 40002
Contact: Aumkhae Sookprasert, MD
Principal Investigator: Aumkhae Sookprasert, MD Less << |
|
NCT00968799
|
- |
|
Terminated(poor patient accrua... More >>l) Less << |
- |
- |
|
NCT00588835
|
Tumor |
Phase 4 |
Terminated |
- |
Netherlands
... More >> University Medical Center Groningen
Groningen, Netherlands
Radboud University Nijmegen Medical Centre
Nijmegen, Netherlands Less << |
|
NCT00418535
|
Lymphoma, T-Cell |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of, 135-710 Less << |
|
NCT03317496
|
Non-small Cell Lung Cancer
... More >> Urothelial Cancer Less << |
Phase 2 |
Recruiting |
October 2, 2021 |
- |
|
NCT03102320
|
Neoplasms |
Phase 1 |
Recruiting |
July 28, 2021 |
- |
|
NCT00041015
|
Lung Cancer |
Phase 3 |
Completed |
- |
United States, Ohio
... More >>
Ireland Cancer Center
Cleveland, Ohio, United States, 44106-5065 Less << |
|
NCT00082472
|
NSCLC |
Phase 1
Phase 2 |
Unknown |
- |
United States, Connecticut
... More >>
Yale University Cancer Center
New Haven, Connecticut, United States, 06520
United States, Nevada
Southern Nevada Cancer Research Foundation
Las Vegas, Nevada, United States, 89106
Nevada Cancer Institute
Las Vegas, Nevada, United States, 89135
United States, New York
Montefiore Medical Center
Bronx, New York, United States, 10461
United States, Ohio
Arthur G James Cancer Hospital and Richard Solove Research Institute at Ohio State University
Columbus, Ohio, United States, 43210
United States, Wisconsin
University of Wisconsin Cancer Center
Madison, Wisconsin, United States, 53792 Less << |
|
NCT00001426
|
Ovarian Neoplasm |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892 Less << |
|
NCT02290145
|
Mouth Neoplasms
... More >> Carcinoma, Squamous Cell Less << |
Phase 2 |
Recruiting |
December 2019 |
China, Shanghai
... More >>
Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine
Recruiting
Shanghai, Shanghai, China, 200011
Contact: Lai-ping Zhong, PhD, MD, DDS +86-21-23271699 ext 5160 zhonglaiping@163.com Less << |
|
NCT00006043
|
Ovarian Cancer
... More >> Testicular Germ Cell Tumor Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT01093690
|
Cancer |
Not Applicable |
Completed |
- |
Thailand
... More >> Division of medical oncology, department of medicine Siriraj Hospital
Bangkok, Thailand, 10700 Less << |
|
NCT01604863
|
Cancer |
Phase 1 |
Suspended(Study suspended prio... More >>r to enrollment) Less << |
July 2014 |
United States, Arizona
... More >>
Scottsdale, Arizona, United States, 85258
United States, Massachusetts
Boston, Massachusetts, United States, 02114 Less << |
|
NCT01093690
|
- |
|
Completed |
- |
- |
|
NCT00828386
|
Nasopharyngeal Cancers |
Phase 3 |
Terminated(Low accrual) |
- |
France
... More >> Hôpital de la Pitié-Salpétrière
Paris, France, 75013
Institut Gustave Roussy
Villejuif, France, 94805
Morocco
University of Casablanca
Casablanca, Morocco
Romania
University of Cluj
Cluj, Romania
Tunisia
Hôpital Habib Bourguiba
Sfax, Tunisia Less << |
|
NCT01833832
|
Adrenocortical Carcinoma
... More >> Peritoneal Carcinomatosis Less << |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892 Less << |
|
NCT00005840
|
Endometrial Clear Cell Adenoca... More >>rcinoma
Endometrial Serous Adenocarcinoma
Stage III Uterine Corpus Cancer
Stage IV Uterine Corpus Cancer Less << |
Phase 1 |
Completed |
- |
United States, Pennsylvania
... More >>
Gynecologic Oncology Group
Philadelphia, Pennsylvania, United States, 19103 Less << |
|
NCT01424709
|
Non-small Cell Lung Cancer |
Phase 2 |
Unknown |
December 2014 |
China
... More >> Medical Department, Shanghai Pulmonary Hospital
Shanghai, China, 200433 Less << |
|
NCT01936376
|
- |
|
Unknown |
June 2015 |
United States, Massachusetts
... More >>
Harvard Medical School - Brigham and Women's Hospital
Not yet recruiting
Boston, Massachusetts, United States, 02115
Contact: Research Coordinator1
Contact: Research Coordinator2
Principal Investigator: Sushrut Waikar, MD
Dana-Farber Cancer Institute
Recruiting
Boston, Massachusetts, United States, 02215
Contact: Research Coordinator1
Contact: Research Coordinator2
Principal Investigator: Robert Haddad, MD
United States, Texas
University of Texas MD Anderson
Recruiting
Houston, Texas, United States, 77030
Contact: Research Coordinator1
Contact: Research Coordinator2
Principal Investigator: Bonnie Glisson, MD
Sub-Investigator: Ala Abudayyeh, MD Less << |
|
NCT00040625
|
- |
|
- |
- |
United States, New Jersey
... More >>
For additional information regarding investigative sites for this trial, contact the Clinical Trials Support Center at 1-877-CTLILLY(1-877-285-4559) or speak with your personal physician.
Basking Ridge, New Jersey, United States
Brazil
For additional information regarding investigative sites for this trial, contact the Clinical Trials support Center at 1-877-CTLILLY (1-877-285-4559) or speak with your personal physician
Porto Alegre, RS, Brazil
For additional information regarding investigative sites for this trial, contact the Clinical Trials support Center at 1-877-CTLILLY (1-877-285-4559) or speak with your personal physician
Sao Paulo, Brazil
Egypt
"For additional information regarding investigative sites for this trial, contact the Clinical Trials Support Center at 1-877-CTLILLY (1-877-285-4559) or speak with your personal physician."
Cairo, Egypt
For additional information regarding investigative sites for this trial, contact the Clinical Trials support Center at 1-877-CTLILLY (1-877-285-4559) or speak with your personal physician
Cairo, Egypt
Saudi Arabia
For additional information regarding investigative sites for this trial, contact the Clinical Trials support Center at 1-877-CTLILLY (1-877-285-4559) or speak with your personal physician
Jeddah, Saudi Arabia Less << |
|
NCT00811447
|
Stomach Neoplasms |
Phase 3 |
Completed |
- |
China
... More >> Sanofi-Aventis Administrative Office
Shanghai, China Less << |
|
NCT01029678
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
France
... More >> CHU Brest
Brest, France
Centre Hospitalier GAP
GAP, France
Département de Pathologie Respiratoire du CHU de Limoges
Limoges, France
CH de Meaux
Meaux, France
CHU Reims
Reims, France Less << |
|
NCT03258554
|
Head and Neck Squamous Cell Ca... More >>rcinoma
Squamous Cell Carcinoma of Unknown Primary Origin
Stage III Hypopharyngeal Squamous Cell Carcinoma AJCC v7
Stage III Laryngeal Squamous Cell Carcinoma AJCC v6 and v7
Stage III Oral Cavity Squamous Cell Carcinoma AJCC v6 and v7
Stage III Oropharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVA Hypopharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVA Laryngeal Squamous Cell Carcinoma AJCC v7
Stage IVA Oral Cavity Squamous Cell Carcinoma AJCC v6 and v7
Stage IVA Oropharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVB Hypopharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVB Laryngeal Squamous Cell Carcinoma AJCC v7
Stage IVB Oral Cavity Squamous Cell Carcinoma AJCC v6 and v7
Stage IVB Oropharyngeal Squamous Cell Carcinoma AJCC v7 Less << |
Phase 2
Phase 3 |
Suspended(Scheduled Interim Mo... More >>nitoring) Less << |
December 31, 2025 |
- |
|
NCT02812420
|
Hydronephrosis
... More >> Infiltrating Bladder Urothelial Carcinoma Sarcomatoid Variant
Infiltrating Bladder Urothelial Carcinoma, Micropapillary Variant
Infiltrating Bladder Urothelial Carcinoma, Plasmacytoid Variant
Infiltrating Renal Pelvis Urothelial Carcinoma, Sarcomatoid Variant
Renal Pelvis Urothelial Carcinoma
Stage II Bladder Urothelial Carcinoma AJCC v6 and v7
Stage II Renal Pelvis Cancer AJCC v7
Stage II Ureter Cancer AJCC v7
Stage II Urethral Cancer AJCC v7
Stage III Bladder Urothelial Carcinoma AJCC v6 and v7
Stage III Renal Pelvis Cancer AJCC v7
Stage III Ureter Cancer AJCC v7
Stage III Urethral Cancer AJCC v7
Stage IV Renal Pelvis Cancer AJCC v7
Stage IV Ureter Cancer AJCC v7
Stage IV Urethral Cancer AJCC v7
Ureter Urothelial Carcinoma
Urethral Urothelial Carcinoma Less << |
Phase 1 |
Recruiting |
March 31, 2020 |
United States, Texas
... More >>
M D Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact: Jianjun Gao 713-792-2830
Principal Investigator: Jianjun Gao Less << |
|
NCT00003157
|
Gastric Cancer
... More >> Pancreatic Cancer Less << |
Phase 1 |
Completed |
- |
United States, Arizona
... More >>
CCOP - Scottsdale Oncology Program
Scottsdale, Arizona, United States, 85259-5404
United States, Illinois
CCOP - Illinois Oncology Research Association
Peoria, Illinois, United States, 61602
CCOP - Carle Cancer Center
Urbana, Illinois, United States, 61801
United States, Iowa
CCOP - Cedar Rapids Oncology Project
Cedar Rapids, Iowa, United States, 52403-1206
CCOP - Iowa Oncology Research Association
Des Moines, Iowa, United States, 50309-1016
Siouxland Hematology-Oncology
Sioux City, Iowa, United States, 51101-1733
United States, Kansas
CCOP - Wichita
Wichita, Kansas, United States, 67214-3882
United States, Minnesota
CCOP - Duluth
Duluth, Minnesota, United States, 55805
Mayo Clinic Cancer Center
Rochester, Minnesota, United States, 55905
CentraCare Clinic
Saint Cloud, Minnesota, United States, 56303
CCOP - Metro-Minnesota
Saint Louis Park, Minnesota, United States, 55416
United States, Nebraska
CCOP - Missouri Valley Cancer Consortium
Omaha, Nebraska, United States, 68131
United States, North Dakota
Medcenter One Health System
Bismarck, North Dakota, United States, 58501
Altru Health Systems
Grand Forks, North Dakota, United States, 58201
United States, Ohio
CCOP - Toledo Community Hospital Oncology Program
Toledo, Ohio, United States, 43623-3456
United States, Pennsylvania
CCOP - Geisinger Clinic and Medical Center
Danville, Pennsylvania, United States, 17822-2001
United States, South Dakota
Rapid City Regional Hospital
Rapid City, South Dakota, United States, 57709
CCOP - Sioux Community Cancer Consortium
Sioux Falls, South Dakota, United States, 57105-1080 Less << |
|
NCT02445872
|
Chemotherapy-Induced Nausea an... More >>d Vomiting
Lung Cancer Less << |
Phase 2 |
Unknown |
May 2016 |
China, Zhejiang
... More >>
The first affiliated hospital, Zhejiang University
Hangzhou, Zhejiang, China, 310003 Less << |
|
NCT00006096
|
Vulvar Cancer |
Phase 3 |
Terminated |
- |
- |
|
NCT00109850
|
Esophageal Cancer |
Phase 2 |
Terminated(Closed due to poor ... More >>accrual) Less << |
- |
- |
|
NCT02001896
|
Non-Small Cell Lung Cancer |
Phase 3 |
Unknown |
June 2015 |
China, Xinjiang
... More >>
Xinjiang medical university
Not yet recruiting
Urumqi, Xinjiang, China, XJTHLC001
Contact: Li Shan, Master 13609989394 shanlinew319@163.com
Sub-Investigator: Qiang Wang, Master Less << |
|
NCT00026403
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00550901
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
- |
|
NCT00686114
|
Esophageal Cancer |
Phase 3 |
Unknown |
December 2014 |
China, Zhejiang
... More >>
1st affliated hospital of Wen Zhou Medical college
Recruiting
Wen Zhou, Zhejiang, China, 325000
Contact: Shixiu Wu, M.D. +86057788069372 wushixiu@medmail.com.cn
Contact: Xuebang Zhang, M.D. baxuza@126.com
Principal Investigator: Shixiu Wu, MD Less << |
|
NCT00109850
|
- |
|
Terminated(Closed due to poor ... More >>accrual) Less << |
- |
- |
|
NCT01215136
|
Transitional Cell Carcinoma
... More >> Bladder Carcinoma
Urothelial Carcinoma Less << |
Phase 2 |
Active, not recruiting |
June 2018 |
United States, Alabama
... More >>
University of Alabama Hematology Oncology Clinic at Medical West
Birmingham, Alabama, United States, 35294
United States, Illinois
Northwestern University, Robert H. Lurie Comprehensive Cancer Center
Chicago, Illinois, United States, 60611
United States, Indiana
Cancer Care Center of Southern Indiana
Bloomington, Indiana, United States, 47403
Indiana University Melvin and Bren Simon Cancer Center
Indianapolis, Indiana, United States, 46202
IU Health Central Indiana Cancer Centers
Indianapolis, Indiana, United States, 46219
United States, Michigan
Metro Health Cancer Care
Wyoming, Michigan, United States, 49519
United States, Nebraska
Nebraska Cancer Specialists
Omaha, Nebraska, United States, 68114
United States, New York
Icahn School of Medicine: Tisch Cancer Institute at Mount Sinai Medical Center
New York, New York, United States, 10029
United States, South Carolina
MUSC Hollings Cancer Center
Charleston, South Carolina, United States, 29425
United States, Texas
University of Texas Medical Branch
Galveston, Texas, United States, 77555
United States, Virginia
Virginia Oncology Associates
Norfolk, Virginia, United States, 23502 Less << |
|
NCT00857246
|
- |
|
Completed |
- |
- |
|
NCT02335411
|
Gastric Adenocarcinoma
... More >> Gastroesophageal Junction Adenocarcinoma Less << |
Phase 2 |
Active, not recruiting |
May 27, 2019 |
- |
|
NCT00191139
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
United States, Colorado
... More >>
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
Denver, Colorado, United States, 80218
United States, Indiana
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
South Bend, Indiana, United States, 46601
United States, Maryland
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
Baltimore, Maryland, United States, 21237
United States, Michigan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
Kalamazoo, Michigan, United States, 49048
United States, Missouri
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
Kansas City, Missouri, United States, 64128
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
St Louis, Missouri, United States, 63141
United States, Ohio
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
Cleveland, Ohio, United States, 44106
United States, Oregon
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
Portland, Oregon, United States, 97213
United States, Pennsylvania
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
Philadelphia, Pennsylvania, United States, 19111
United States, Tennessee
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
Memphis, Tennessee, United States, 38120
Argentina
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
Rosario, Argentina, 2000
China
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
Beijing, China, 100021
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
Chengdu, China, 610041
Korea, Republic of
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
Seoul, Korea, Republic of, 138-736
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal ph
Suwon-City, Korea, Republic of, 442-723 Less << |
|
NCT01755845
|
Cervical Cancer |
Phase 3 |
Unknown |
December 2016 |
China, Zhejiang
... More >>
The First Affiliated Hospital of Wenzhou Medical College
Recruiting
Wenzhou, Zhejiang, China, 325000
Contact: congying xie +86-577-88069316 wzxiecongying@163.com
Principal Investigator: congying xie, MD Less << |
|
NCT00857246
|
Gastric Cancer
... More >> Stomach Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
NYU Cancer Center
New York, New York, United States, 10016 Less << |
|
NCT00599755
|
- |
|
Completed |
- |
- |
|
NCT00599755
|
Carcinoma
Non... More >>-small Cell Lung Cancer Less << |
Phase 1 |
Completed |
- |
- |
|
NCT01501149
|
Nasal and Nasal-type NK/T-cell... More >> Lymphoma Less << |
Phase 4 |
Recruiting |
May 2019 |
China, Henan
... More >> Oncology Department of The First Affiliated Hospital of Zhengzhou University
Recruiting
Zhengzhou, Henan, China, 450052
Contact: Mingzhi Zhang, Pro,Dr mingzhi_zhang@126.com
Principal Investigator: Mingzhi Zhang, Pro,Dr Less << |
|
NCT00867009
|
Non-Small-Cell Lung Cancer |
Phase 2 |
Completed |
- |
Austria
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Graz, Austria, 8036
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Salzburg, Austria, 5020
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Wels, Austria, 4600
Germany
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Berlin, Germany, 13125
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Halle, Germany, D-06120
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Homburg, Germany, 66421
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mannheim, Germany, 68167
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Tuebingen, Germany, 72076
Greece
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Athens, Greece, 11527
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Patras, Greece, 26500
Italy
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Bari, Italy, 70126
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Bergamo, Italy, 24128
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Palermo, Italy, 90146
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Potenza, Italy, 85028
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Torino, Italy, 10100
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Treviso, Italy, 31100
Netherlands
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Amsterdam, Netherlands, 1081 HV
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Groningen, Netherlands, 9713 GZ
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Maastricht, Netherlands, 6229 HX
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
S-Hertogenbosch, Netherlands, 5211 NL
Spain
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Barcelona, Spain, 08003
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Lleida, Spain, 25198
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Madrid, Spain, 28034
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Reus, Spain, 43201 Less << |
|
NCT00757549
|
Head and Neck Cancer |
Early Phase 1 |
Completed |
- |
United States, Minnesota
... More >>
Mayo Clinic
Rochester, Minnesota, United States, 55905 Less << |
|
NCT00402051
|
Non-Small-Cell Lung Cancer |
Phase 2 |
Completed |
- |
Germany
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Coswig, Germany, D-01640
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Frankfurt, Germany, 65929
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Großhansdorf, Germany, D-22927
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Halle, Germany, D-06120
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hamburg, Germany, D-21075
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hofheim, Germany, D-65719
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Homburg/Saar, Germany, 66421
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Immenhausen, Germany, D-34376
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Leipzig, Germany, 04207
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Luebeck, Germany, 23538
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mainz, Germany, 55131
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Muenchen, Germany, 81664 Less << |
|
NCT00191139
|
- |
|
Completed |
- |
- |
|
NCT00817895
|
Hepatocellular Carcinoma |
Not Applicable |
Completed |
- |
China, Shanghai
... More >>
Eastern Hepatobiliary Surgery Hospital
Shanghai, Shanghai, China, 200438 Less << |
|
NCT00402051
|
- |
|
Completed |
- |
- |
|
NCT00985998
|
Non-small Cell Lung Cancer |
Phase 1 |
Withdrawn(no paticipants enrol... More >>led) Less << |
- |
China, Beijing
... More >> Cancer Institute & Hospital, Chinese Academy of Medical Sciences
Beijing, Beijing, China, 100000 Less << |
|
NCT00169182
|
Larynx Cancer
... More >> Hypopharynx Cancer Less << |
Phase 3 |
Completed |
- |
France
... More >> CHU Bretonneau
Tours, France, 37044 Less << |
|
NCT00868491
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
Slovenia
... More >> Institute of Oncology Ljubljana
Ljubljana, Slovenia, SI-1000 Less << |
|
NCT00867009
|
- |
|
Completed |
- |
- |
|
NCT01194635
|
- |
|
Completed |
- |
- |
|
NCT00290953
|
Lung Cancer
P... More >>ulmonary Neoplasms
Small Cell Lung Cancer Less << |
Phase 2
Phase 3 |
Completed |
- |
Argentina
... More >> Sanofi-Aventis Administrative Office
Buenos Aires, Argentina
Australia
Sanofi-Aventis Administrative Office
Macquarie Park, Australia
Belgium
Sanofi-Aventis Administrative Office
Diegem, Belgium
Brazil
Sanofi-Aventis Administrative Office
Sao Paulo, Brazil
France
Sanofi-Aventis Administrative Office
Paris, France
Germany
Sanofi-Aventis Administrative Office
Berlin, Germany
Hungary
Sanofi-Aventis Administrative Office
Budapest, Hungary
Italy
Sanofi-Aventis Administrative Office
Milano, Italy
Mexico
Sanofi-Aventis Administrative Office
Mexico, Mexico
Netherlands
Sanofi-aventis adminsitrative office
Gouda, Netherlands
Poland
Sanofi-Aventis Administrative Office
Warszawa, Poland
Russian Federation
Sanofi-Aventis Administrative Office
Moscow, Russian Federation
Spain
Sanofi-Aventis Administrative Office
Barcelona, Spain
United Kingdom
Sanofi-Aventis Administrative Office
Guildford Surrey, United Kingdom Less << |
|
NCT03240016
|
Urothelial Carcinoma |
Phase 2 |
Recruiting |
October 1, 2021 |
United States, Michigan
... More >>
University of Michigan Comprehensive Cancer Center
Recruiting
Ann Arbor, Michigan, United States, 48109
Contact: Ajjai Alva, M.D. 734-936-0091 ajjai@umich.edu
Principal Investigator: Ajjai Alva, MD Less << |
|
NCT00940069
|
Non-small-cell Lung Cancer
... More >> Adenocarcinoma Less << |
Phase 2 |
Completed |
- |
China, Hunan
... More >> HuNan province tumor hospital
ChangSha, Hunan, China, 410013 Less << |
|
NCT00616785
|
Lung Cancer |
Phase 2 |
Unknown |
- |
Korea, Republic of
... More >>
Yonsei Cancer Center at Yonsei University Medical Center
Recruiting
Seoul, Korea, Republic of, 120-752
Contact: Joo-Hang Kim, MD +82-10-4507-6063 kjhang@yuhs.ac Less << |
|
NCT00402545
|
Head and Neck Neoplasms |
Phase 1 |
Completed |
- |
United States, Massachusetts
... More >>
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00480597
|
Metastatic Breast Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT01639521
|
Anterior Urethral Cancer
... More >> Localized Transitional Cell Cancer of the Renal Pelvis and Ureter
Posterior Urethral Cancer
Recurrent Bladder Cancer
Recurrent Urethral Cancer
Regional Transitional Cell Cancer of the Renal Pelvis and Ureter
Stage III Bladder Cancer
Transitional Cell Carcinoma of the Bladder
Ureter Cancer
Urethral Cancer Associated With Invasive Bladder Cancer Less << |
Phase 2 |
Withdrawn(Lack of funding) |
- |
- |
|
NCT00409006
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
China
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Fujian, China, 350014
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Qingdao, China, 266003
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Shanghai, China, 201900
Korea, Republic of
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Seoul, Korea, Republic of, 137-701
Taiwan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Changhua, Taiwan, 500
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Taipei, Taiwan, 100
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Tao-Yuan, Taiwan, 333 Less << |
|
NCT03613168
|
Cholangiocarcinoma
... More >> Biliary Tract Cancer
HER-2 Protein Overexpression
HER-2 Gene Amplification Less << |
Phase 2 |
Not yet recruiting |
December 1, 2020 |
- |
|
NCT00052494
|
Lung Cancer |
Phase 1 |
Completed |
- |
Canada, Ontario
... More >>
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT03544814
|
Lung Adenocarcinoma
... More >> EGFR Activating Mutation Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00409006
|
- |
|
Completed |
- |
- |
|
NCT00268671
|
Head and Neck Neoplasms |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT01859741
|
Stage IV Small Cell Lung Cance... More >>r Less << |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT02610010
|
Nasopharyngeal Carcinoma |
Phase 3 |
Recruiting |
November 2025 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Fang-Yun Xie, M.D. +86-020-87342618 xiefy@sysucc.org.cn
Contact: Pu-Yun OuYang, M.D. +86-020-87342618 ouyangpy@sysucc.org.cn Less << |
|
NCT00156286
|
Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
United States, Michigan
... More >>
Univeristy of Michigan Cancer Center
Ann Arbor, Michigan, United States, 48109 Less << |
|
NCT00003930
|
Bladder Cancer |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT03267940
|
Cholangiocarcinoma Non-resecta... More >>ble
Cholangiocarcinoma, Intrahepatic
Cholangiocarcinoma, Extrahepatic
Gallbladder Adenocarcinoma Less << |
Phase 1 |
Recruiting |
February 29, 2020 |
- |
|
NCT02041819
|
Squamous Cell Carcinoma of Eso... More >>phagus Less << |
Phase 2 |
Withdrawn(No recruitment) |
January 2017 |
China, Zhejiang
... More >>
The first affiliated hospital, Zhejiang University
Zhejiang, Zhejiang, China, 310003 Less << |
|
NCT02120807
|
Stage IV Lung Adenocarcinoma |
Phase 1 |
Active, not recruiting |
April 2019 |
United States, New Jersey
... More >>
Memorial Sloan Kettering at Basking Ridge
Basking Ridge, New Jersey, United States, 07920
United States, New York
Memorial Sloan Kettering Cancer Center @ Suffolk
Commack, New York, United States, 11725
Memorial Sloan Kettering West Harrison
Harrison, New York, United States, 10604
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan Kettering at Mercy Medical Center
Rockville Centre, New York, United States
Memorial Sloan Kettering Cancer Center at Phelps Memorial Hospital Center
Sleepy Hollow, New York, United States, 10591 Less << |
|
NCT00999700
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 3 |
Active, not recruiting |
December 2021 |
Italy
... More >> Ospedale Civile Ss. Antonio E Biagio
Alessandria, Italy
Ausl Della Valle D' Aosta
Aosta, Italy
Policlinico S. Orsola-Malpighi
Bologna, Italy
Asl 8 - Ospedale Businco
Cagliari, Italy
A.S.O. S. Croce E Carle
Cuneo, Italy, 12100
Irccs - Aou S. Martino - Oncology
Genoa, Italy
Irccs - Aou San Martino - Radiotherapy
Genoa, Italy
Asl 3 Genovese
Genova, Italy
E.O. Ospedali Galliera
Genova, Italy
Azienda Ospedaliera Villa Scassi - Asl3
Italy, Italy
Istituto Nazionale Dei Tumori
Milano, Italy
Azienda Ospedaliero Universitaria Di Modena
Modena, Italy
Ospedale S. Giacomo
Novi Ligure (al), Italy
Azienda Ospedaliero-Universitaria Di Parma
Parma, Italy
Azienda Ospedaliera Ospedali Riuniti Di Fano
Pesaro, Italy
Arcispedale S. Maria Nuova
Reggio Emilia, Italy
Irccs Centro Di Riferimento Oncologico Di Basilicata (Crob)
Rionero in Vulture (pz), Italy
Ospedale S. Filippo Neri
Roma, Italy
Ospedale S. Paolo
Savona, Italy
A.O. Universitaria S. Giovanni Battista-Molinette Di Torino Torino (to)
Turin, Italy
A.O. Universitaria S. Giovanni Battista-Molinette Di Torino
Turin, Italy Less << |
|
NCT01530997
|
Carcinoma, Squamous Cell
... More >> Head and Neck Neoplasms
Oropharyngeal Neoplasms Less << |
Phase 2 |
Active, not recruiting |
January 2022 |
United States, Colorado
... More >>
Penrose Cancer Center
Colorado Springs, Colorado, United States, 80907
United States, Florida
University of Florida, Department of Radiation Oncology
Gainesville, Florida, United States, 32610-0385
United States, North Carolina
University of North Carolina at Chapel Hill, Department of Radiation Oncology
Chapel Hill, North Carolina, United States, 27599
Rex Healthcare
Raleigh, North Carolina, United States, 27607
Rex Cancer Center of Wakefield
Raleigh, North Carolina, United States, 27614 Less << |
|
NCT00269997
|
Anemia
Neopla... More >>sms Less << |
Phase 2 |
Completed |
- |
- |
|
NCT03100955
|
Progression Free Survival |
Phase 3 |
Recruiting |
August 1, 2019 |
China, Shandong
... More >>
Affiliated Hospital of Qingdao University
Recruiting
Qingdao, Shandong, China, 266001
Contact: Zhuang Yu, MD (86)18661805688 yuzhuang2002@163.com
Principal Investigator: Zhuang Yu, MD, Ph.D Less << |
|
NCT03502681
|
Metastatic Urothelial Cell Can... More >>cer Less << |
Phase 1 |
Recruiting |
July 2019 |
United States, Pennsylvania
... More >>
Penn State Cancer Intsitute
Recruiting
Hershey, Pennsylvania, United States, 17033
Contact: Heather Rutherford 717-531-0003
Principal Investigator: Monika Joshi Less << |
|
NCT01192750
|
- |
|
Completed |
- |
- |
|
NCT00066222
|
Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT01530997
|
- |
|
Active, not recruiting |
- |
- |
|
NCT02821533
|
Hepatocellular Carcinoma |
Phase 2 |
Terminated(Poor case accrual) |
- |
Hong Kong
... More >> Department of Imaging and Interventional Radiology, Prince of Wales Hospital, The Chinese University of Hong Kong
Hong Kong, Hong Kong Less << |
|
NCT03480672
|
HNSCC |
Phase 2 |
Recruiting |
August 2022 |
Germany
... More >> Department of Head Medicine and Oral Health, University of Leipzig
Recruiting
Leipzig, Germany, 04103
Contact: Andreas Dietz, Prof. Dr. Less << |
|
NCT00774319
|
Head and Neck Cancer |
Phase 2 |
Unknown |
April 2012 |
Netherlands
... More >> Netherlands Cancer Institute
Recruiting
Amsterdam, Netherlands
Contact: J.P. de Boer 31 20 512 9111 j.d.boer@nki.nl
Principal Investigator: J.P. de Boer
University Medical Center Nijmegen st Radboud
Recruiting
Nijmegen, Netherlands, 6525 GH
Principal Investigator: C.M.L van Herpen, Md, Phd Less << |
|
NCT00296335
|
Stomach Cancer |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT00296335
|
- |
|
Completed |
- |
- |
|
NCT00427102
|
Squamous Cell Carcinoma
... More >> Head and Neck Cancer
Resected Head and Neck Cancer
Mucositis Less << |
Phase 1 |
Terminated(No subjects enrolle... More >>d and PI went to another facility) Less << |
- |
- |
|
NCT00066222
|
- |
|
Completed |
- |
- |
|
NCT00477867
|
Ovarian Cancer |
Not Applicable |
Withdrawn(Trial was never acti... More >>vated) Less << |
- |
- |
|
NCT02705612
|
Cervical Cancer |
Phase 2 |
Unknown |
September 2018 |
China, Shaanxi
... More >> Department of Radiation Oncology, Xijing Hospital, Fourth Military Medical University
Recruiting
Xi'an, Shaanxi, China, 710032
Contact: Mei Shi, MD +86-029-84775425 mshifmmu@yahoo.com
Principal Investigator: Mei Shi, MD
Sub-Investigator: Li-Chun Wei, M.D.,Ph.D Less << |
|
NCT01697072
|
Gastric Cancer |
Phase 3 |
Terminated(All Amgen sponsored... More >> AMG102 clinical studies were terminated following a pre-planned Data Monitoring Committee safety review of study 20070622.) Less << |
- |
- |
|
NCT01611506
|
Gastric Cancer |
Phase 1
Phase 2 |
Terminated(Outcome of EXPAND s... More >>tudy (no benefit from adding cetuximab to the first-line chemotherapy in advanced gastric cancer in the overall patient population)) Less << |
- |
Switzerland
... More >> Centre Hospitalier Universitaire Vaudois
Lausanne, Switzerland Less << |
|
NCT00034268
|
Carcinoma, Non-Small-Cell Lung |
Phase 3 |
Completed |
- |
- |
|
NCT00002654
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
Switzerland
... More >> Universitaetsspital
Zurich, Switzerland, CH-8091 Less << |
|
NCT00005819
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, Ohio
... More >>
Ireland Cancer Center at University Hospitals Case Medical Center, Case Comprehensive Cancer Center
Cleveland, Ohio, United States, 44106-5065 Less << |
|
NCT02558959
|
Biliary Tract Neoplasms |
Phase 2 |
Completed |
- |
China, Zhejiang
... More >>
First affiliated hospital, Zhejiang University
Hangzhou, Zhejiang, China, 310006 Less << |
|
NCT00354549
|
Lung Cancer |
Phase 2 |
Completed |
- |
Switzerland
... More >> Universitaetsspital-Basel
Basel, Switzerland, CH-4031
Oncology Institute of Southern Switzerland
Bellinzona, Switzerland, CH-6500 Less << |
|
NCT02579564
|
Malignant Hydrothorax
... More >> Non Small Cell Lung Cancer Less << |
Phase 3 |
Unknown |
December 2018 |
- |
|
NCT00433498
|
Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00506155
|
- |
|
Completed |
- |
- |
|
NCT02586207
|
Head and Neck Cancer
... More >> Squamous Cell Carcinoma
Oral Cavity Cancer
Oropharynx Cancer
Hypopharynx Cancer
Larynx Cancer
Laryngeal Cancer Less << |
Phase 1 |
Active, not recruiting |
September 2021 |
United States, California
... More >>
UCSD Moores Cancer Center
La Jolla, California, United States, 92093-0698
United States, North Dakota
Sanford-Bismarck Medical Center
Bismarck, North Dakota, United States, 58501
Sanford-Roger Maris Cancer Center
Fargo, North Dakota, United States, 58122
United States, South Dakota
Sanford Health Cancer Center
Sioux Falls, South Dakota, United States, 57104 Less << |
|
NCT02778308
|
Gallbladder Cancer |
Not Applicable |
Unknown |
December 2017 |
India
... More >> GIPMER
Recruiting
New Delhi, India, 110002
Contact: Sundeep S Saluja, MCh (718599259 sundeepsaluja@yahoo.co.in Less << |
|
NCT02012062
|
Nasopharyngeal Carcinoma |
Phase 3 |
Unknown |
June 2018 |
China
... More >> Fudan University Shanghai Cancer Center
Recruiting
Shanghai, China, 200032
Contact: Lin Kong, MD 86 18017312533 konglinj@gmail.com
Principal Investigator: Jiade J Lu, MD
Principal Investigator: Jianji Pan, MD
Sub-Investigator: Shaojun Lin, MD
Principal Investigator: Qin Lin, MD
Sub-Investigator: Chaosu Hu, MD Less << |
|
NCT00540280
|
Carcinoma, Non-Small-Cell Lung |
Phase 3 |
Suspended(Follow up only) |
October 2013 |
- |
|
NCT01297101
|
- |
|
- |
- |
- |
|
NCT00506155
|
Bladder Cancer |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
UT MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00190801
|
Gastrointestinal Neoplasms |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9AM to 5PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Goyang-Si, Korea, Republic of
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9AM to 5PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
In Cheon, Korea, Republic of
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9AM to 5PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Seoul, Korea, Republic of Less << |
|
NCT00404326
|
Cervical Cancer |
Phase 2 |
Completed |
- |
Mexico
... More >> National Institute of Cancerologia
Mexico City, Tlalpan, Mexico, 14080 Less << |
|
NCT00716417
|
Neoplasms |
Phase 1 |
Completed |
- |
Belgium
... More >> 1200.37.3202 Boehringer Ingelheim Investigational Site
Bruxelles, Belgium
1200.37.3201 Boehringer Ingelheim Investigational Site
Edegem, Belgium
1200.37.3203 Boehringer Ingelheim Investigational Site
Gent, Belgium Less << |
|
NCT00716417
|
- |
|
Completed |
- |
- |
|
NCT00191815
|
Breast Cancer |
Phase 2 |
Completed |
- |
Germany
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon- Fri from 9AM to 5PM Eastern time (UTC/ GMT - 5hours, EST), or speak with your personal physician
Munich, Germany, 81377
Russian Federation
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon- Fri from 9AM to 5PM Eastern time (UTC/ GMT - 5hours, EST), or speak with your personal physician
Moscow, Russian Federation, 115478 Less << |
|
NCT00191815
|
- |
|
Completed |
- |
- |
|
NCT00942357
|
Endometrial Clear Cell Adenoca... More >>rcinoma
Endometrial Serous Adenocarcinoma
Stage IA Uterine Corpus Cancer
Stage IB Uterine Corpus Cancer
Stage II Uterine Corpus Cancer
Stage IIIA Uterine Corpus Cancer
Stage IIIB Uterine Corpus Cancer
Stage IIIC Uterine Corpus Cancer
Stage IVA Uterine Corpus Cancer Less << |
Phase 3 |
Active, not recruiting |
- |
- |
|
NCT01265147
|
Nasopharyngeal Carcinoma |
Phase 2 |
Unknown |
December 2017 |
China
... More >> Daping Hospital
Not yet recruiting
Chong Qing, China
Contact: Zhenzhou Yang, Dr.
Principal Investigator: Zhenzhou Yang, Dr. Less << |
|
NCT03086993
|
Bile Duct Cancer
... More >> Intrahepatic Cholangiocarcinoma Less << |
Phase 2
Phase 3 |
Recruiting |
May 2023 |
United States, North Carolina
... More >>
Duke Health
Recruiting
Durham, North Carolina, United States, 27710
Contact: Kim Turnage, MSN 919-684-3381
Principal Investigator: Sabino Zani, MD
United States, Ohio
Ohio State University/Teaching Hospital
Active, not recruiting
Columbus, Ohio, United States, 43210
United States, Tennessee
West Cancer Center
Recruiting
Memphis, Tennessee, United States, 38120
Contact: Catherine Morningstar, BS 901-683-0055 cmorningstar@WESTCLINIC.com
Principal Investigator: Evan Glazer, MD, PhD Less << |
|
NCT01874678
|
Non-small-cell Lung Cancer (NS... More >>CLC) Less << |
Not Applicable |
Completed |
- |
Taiwan
... More >> Dalin Tzu Chi General Hospital
Chiayi, Taiwan
E-Da Hospital
Kaohsiung, Taiwan
China Medical University Hospital
Taichung, Taiwan
Taichung Veterans General Hospital
Taichung, Taiwan
Taipei Veterans General Hospital
Taipei, Taiwan Less << |
|
NCT00452634
|
Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
National Cancer Center, Korea
Goyang-si, Gyenggi, Korea, Republic of, 411-769 Less << |
|
NCT02944357
|
Infiltrating Bladder Urothelia... More >>l Carcinoma
Stage II Bladder Urothelial Carcinoma
Stage III Bladder Urothelial Carcinoma
Stage IV Bladder Urothelial Carcinoma Less << |
Not Applicable |
Withdrawn |
- |
United States, Minnesota
... More >>
Mayo Clinic
Rochester, Minnesota, United States, 55905 Less << |
|
NCT02344095
|
Epithelial Ovarian Carcinoma
... More >> Fallopian Tube Carcinoma
Primary Peritoneal Carcinoma Less << |
Phase 1 |
Unknown |
October 2015 |
Korea, Republic of
... More >>
Seoul National University Bundang Hospital
Recruiting
Seongnam-si, Gyeonggi-do, Korea, Republic of, 463707
Contact: Kidong Kim kidong.kim.md@gmail.com
Sub-Investigator: Kidong Kim Less << |
|
NCT01854749
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Phase 2 |
Completed |
- |
China, Shanghai
... More >>
Cancer hospital Fudan University
Shanghai, Shanghai, China, 200032 Less << |
|
NCT02326285
|
Non-squamous Non-small Cell Lu... More >>ng Cancer Stage II
Non-squamous Non-small Cell Lung Cancer Stage IIIA
Non-squamous Non-small Cell Lung Cancer Stage IIIB
Activating EGFR Mutation
NSCLC Less << |
Phase 2
Phase 3 |
Terminated(Due to low patient ... More >>enrollment was stopped. Only one patient could be enrolled. 37 patients were pre-screened, but not into the inclusion criteria (wt-EGFR)) Less << |
- |
Germany
... More >> Pius-Hospital
Oldenburg, Germany, 26121 Less << |
|
NCT00041106
|
Metastatic Transitional Cell C... More >>ancer of the Renal Pelvis and Ureter
Recurrent Transitional Cell Cancer of the Renal Pelvis and Ureter Less << |
Phase 2 |
Completed |
- |
United States, Illinois
... More >>
Cancer and Leukemia Group B
Chicago, Illinois, United States, 60606 Less << |
|
NCT03772028
|
Ovarian Cancer |
Phase 3 |
Not yet recruiting |
February 1, 2025 |
- |
|
NCT01154140
|
Non Squamous Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT02996214
|
Lung Squamous Cell Carcinoma |
Phase 4 |
Not yet recruiting |
June 2020 |
- |
|
NCT01154140
|
- |
|
Completed |
- |
- |
|
NCT00304278
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
United States, Illinois
... More >>
Simmons Cooper Cancer Institute/SIU School of Medicine
Springfield, Illinois, United States, 62702 Less << |
|
NCT02276248
|
Stage I/II Extranodal NK/T-cel... More >>l Lymphoma Less << |
Phase 2 |
Completed |
- |
China, Beijing
... More >> Cancer Hospital and Institute, Chinese Academy of Medical Sciences (CAMS) and Peking Union Medical College (PUMC)
Beijing, Beijing, China, 100021 Less << |
|
NCT00304278
|
- |
|
Completed |
- |
- |
|
NCT00385606
|
Advanced Non-Small Cell Lung C... More >>ancer Less << |
Phase 2
Phase 3 |
Completed |
- |
Italy
... More >> Azienda Ospedaliera S. Giuseppe Moscati, U.O. di Oncologia Medica
Monteforte Irpino, AV, Italy, 83024
Ospedale Regionale Miulli, Divisione Medicina Interna Sezione Oncologica
Acquaviva delle Fonti, BA, Italy, 70021
IRCCS Oncologico Bari, Oncologia Medica
Bari, BA, Italy, 70126
Istituto Oncologico di Bari, U.O. di Oncologia Medica e Sperimentale
Bari, BA, Italy, 70126
Policlinico Universitario, Oncologia Medica II
Cagliari, CA, Italy, 09042
Ospedale A. Cardarelli
Campobasso, CB, Italy, 86100
Ospedale Mariano Santo, U.O. di Oncologia Medica
Cosenza, CS, Italy, 87100
Ospedale Umberto di Frosinone
Frosinone, FR, Italy, 03031
Ospedale Civile di Legnano
Legnano, MI, Italy
Ospedale S. Paolo
Milano, MI, Italy, 20142
Policlinico Universitario P. Giaccone
Palermo, PA, Italy, 90100
Casa di Cura La Maddalena S.p.A., Dipartimento Oncologico
Palermo, PA, Italy, 90146
Istituto Oncologico Veneto
Padova, PD, Italy
Ospedale Civile Umberto I, Day Hospital Oncoematologico
Nocera Inferiore, SA, Italy, 84014
Divisione di Oncologia Medica, U.S.L.L. 13
.Noale, VE, Italy, 30033
Ospedale S. Bortolo ULSS 6, U.O. di Oncologia Medica
Vicenza, VI, Italy, 36100
Ospedale L. Sacco
Milano, Italy
Istituto Nazionale dei Tumori , Divisione di Oncologia Medica B
Napoli, Italy, 80131
Istituto Nazionale dei Tumori, Divisione di Oncologia Medica C
Napoli, Italy, 80131
Second University of Naples
Napoli, Italy
Ospedale Santa Corona
Pietre Ligure, Italy
Istituto Regina Elena, Divisione di Oncologia Medica
Roma, Italy, 00144
Ospedale S. Giovanni Calibita Gatebenefratelli
Roma, Italy, 00186
Ospedale San Camillo - Forlanini
Rome, Italy
Azienda Ospedaliera Di Busto Arsizio
Saronno, Italy Less << |
|
NCT01289522
|
Squamous Cell Head and Neck Ca... More >>rcinoma
Recurrent or Metastatic Disease Less << |
Phase 2 |
Completed |
- |
Belgium
... More >> Cliniques Universitaires
Bruxelles, Belgium
Clinique Sainte Elisabeth
Namur, Belgium
Clinique universitaire de Mont Godinne UCL
Yvoir, Belgium
France
Hôpital Saint André
Bordeaux, France
Centre Jean Perrin,
Clermont-Ferrand, France
Centre G-F Leclerc
Dijon, France
Centre Hospitalier de la Dracénie
Draguignan, France
Centre Hospitalier de Bretagne Sud
Lorient, France
Centre Léon Bérard
Lyon, France
Hôpital de la Timone
Marseille, France
Centre Henri Becquerel
Rouen, France
Hôpital Foch
Suresnes, France
CHU Bretonneau
Tours, France
Institut Gustave Roussy
Villejuif, France Less << |
|
NCT02322593
|
Gastric Cancer |
Phase 3 |
Active, not recruiting |
May 2019 |
Japan
... More >> Taiho Pharmaceutical Co., Ltd selected site
Ibaraki, Japan, 305-8576
Taiho Pharmaceutical Co., Ltd selected site
Kumamoto, Japan, 860-8556
Taiho Pharmaceutical Co., Ltd selected site
Tokyo, Japan, 104-0045
Korea, Republic of
Taiho Pharmaceutical Co., Ltd selected site
Seoul, Korea, Republic of, 110-744
Taiho Pharmaceutical Co., Ltd selected site
Seoul, Korea, Republic of, 120-752
Taiho Pharmaceutical Co., Ltd selected site
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT02337712
|
Small Cell Lung Cancer |
Phase 2 |
Recruiting |
December 2020 |
China, Guangdong
... More >>
Sun yat-sen university cancer center
Recruiting
Guangzhou, Guangdong, China, 510000
Contact: Bo Qiu, Attending +86-020-87343031 qiubo@sysucc.org.cn Less << |
|
NCT00188266
|
Stomach Neoplasms |
Phase 1
Phase 2 |
Completed |
- |
Canada, Ontario
... More >>
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT01289522
|
- |
|
Completed |
- |
- |
|
NCT00334815
|
Adenosquamous Lung Carcinoma
... More >> Large Cell Lung Carcinoma
Lung Adenocarcinoma
Minimally Invasive Lung Adenocarcinoma
Squamous Cell Lung Carcinoma
Stage IIIA Non-Small Cell Lung Cancer AJCC v7
Stage IIIB Lung Non-Small Cell Cancer AJCC v7 Less << |
Phase 2 |
Active, not recruiting |
- |
- |
|
NCT00334815
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00526396
|
Small Cell Lung Cancer |
Phase 3 |
Unknown |
July 2016 |
Italy
... More >> Azienda Sanitaria S. Giuseppe Moscati
Monteforte Irpino, AV, Italy
Azienda Ospedaliera G. Rummo
Benevento, BN, Italy
Casa di Cura La Maddalena S.p.A., Dipartimento Oncologico
Palermo, PA, Italy, 90146
Istituto Oncologico Veneto
Padova, PD, Italy
Ospedale E. Morelli
Sondalo, SO, Italy, 23039
Ospedale San Lazzaro
Alba, Italy
Ospedale Mater Domini
Catanzara, Italy
Ospedale L. Sacco Polo Universitario
Milano, Italy
Istituto Nazionale dei Tumori
Napoli, Italy
Ospedale Cotugno
Napoli, Italy
Ospedale Guglielmo da Saliceto
Piacenza, Italy
Ospedale Maggiore
Trieste, Italy Less << |
|
NCT01542931
|
Stage III Oral Cavity Squamous... More >> Cell Carcinoma
Stage IVA Oral Cavity Squamous Cell Carcinoma Less << |
Phase 2
Phase 3 |
Completed |
- |
China, Shanghai
... More >>
Ninth People's Hospital, School of Medicine, Shanghai Jiao Tong University
Shanghai, Shanghai, China, 200011 Less << |
|
NCT00720083
|
Head and Neck Cancer |
Phase 2 |
Terminated(Withdrawal of drug ... More >>supply.) Less << |
- |
- |
|
NCT00423852
|
Brain and Central Nervous Syst... More >>em Tumors
Extragonadal Germ Cell Tumor
Ovarian Cancer
Teratoma
Testicular Germ Cell Tumor Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT03472274
|
Bladder Cancer |
Phase 2 |
Not yet recruiting |
December 2022 |
Spain
... More >> ICO Badalona
Not yet recruiting
Badalona, Spain
Contact: Albert Font
Hospital Clinic
Not yet recruiting
Barcelona, Spain
Contact: Begoña Mellado
Hospital de la Santa Creu i Sant Pau
Not yet recruiting
Barcelona, Spain
Contact: Pablo Maroto
ICO L'Hospitalet
Not yet recruiting
L'Hospitalet De Llobregat, Spain
Contact: Francisco J García del Muro
Hospital 12 de Octubre
Not yet recruiting
Madrid, Spain
Contact: Daniel Castellano
Hospital Clinico San Carlos
Not yet recruiting
Madrid, Spain
Contact: Javier Puente
Hospital Ramon y Cajal
Not yet recruiting
Madrid, Spain
Contact: Teresa Alonso-Gordoa
MD Anderson Cancer Center
Not yet recruiting
Madrid, Spain
Contact: Enrique Grande
Hospital Marqués de Valdecilla
Not yet recruiting
Santander, Spain
Contact: Ignacio Durán
Instituto Valenciano de Oncología
Not yet recruiting
Valencia, Spain
Contact: Miguel A Climent Less << |
|
NCT00030862
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00066482
|
Childhood Germ Cell Tumor
... More >> Extragonadal Germ Cell Tumor Less << |
Not Applicable |
Completed |
- |
- |
|
NCT00578864
|
Neuroblastoma |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
Texas Children's Hospital
Houston, Texas, United States, 77030 Less << |
|
NCT00423852
|
- |
|
Completed |
- |
- |
|
NCT00578864
|
- |
|
Completed |
- |
- |
|
NCT02711553
|
Biliary Tract Cancer
... More >> Metastatic Cancer
Advanced Cancer Less << |
Phase 2 |
Active, not recruiting |
May 31, 2020 |
- |
|
NCT00505492
|
Uterine Neoplasms |
Phase 2 |
Terminated(Slow accrual.) |
- |
United States, Texas
... More >>
UT MD Anderson Cancer Center
Houston, Texas, United States, 77030
United States, Washington
University of Washington School of Medicine
Seattle, Washington, United States, 98104 Less << |
|
NCT00720083
|
- |
|
Terminated(Withdrawal of drug ... More >>supply.) Less << |
- |
- |
|
NCT00122746
|
Cancer of the Cervix |
Phase 3 |
Unknown |
- |
India
... More >> Dept. of Atomic Energy, Tata Memorial Centre
Recruiting
Mumbai, India
Contact: Reena Engineer, MD reena_engineer@rediffmail.com
Principal Investigator: Reena Engineer, MD
South Africa
Johannesburg Hospital
Active, not recruiting
Johannesburg, South Africa
Tanzania
Ocean Road Cancer Institute
Recruiting
Dar Es Salaam, Tanzania
Contact: Twalib Ngoma, MD ngoma@uccmail.co.tz
Principal Investigator: Twalib Ngoma, MD
Uganda
Radiotherapy Centre
Recruiting
Kampala, Uganda
Contact: Joseph Kigula, MD jbkigula@yahoo.com
Principal Investigator: Joseph Kigula, MD
Zimbabwe
Radiotherapy Centre
Active, not recruiting
Harare, Zimbabwe Less << |
|
NCT02901301
|
Gastric Cancer |
Phase 1
Phase 2 |
Recruiting |
March 2019 |
Korea, Republic of
... More >>
Severance Hospital, Yonsei University Health System
Recruiting
Seoul, Korea, Republic of, 03722
Contact: Hyo Song Kim Less << |
|
NCT01907100
|
Mesothelioma |
Phase 3 |
Completed |
- |
- |
|
NCT02161419
|
Small Cell Lung Carcinoma |
Phase 2 |
Terminated |
- |
- |
|
NCT00191854
|
Breast Cancer |
Phase 2 |
Completed |
- |
Brazil
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Santo André, Brazil, 09060-020
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Sao Paulo, Brazil, 05403-900
China
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Beijing, China, 100071
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hangzhou, China, 310016
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Nan Jing, China, 210009
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Wu Han, China, 430030
India
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Delhi, India, 110085
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Kolkata, India, 700053
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Ludhiana, India, 141001
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mumbai, India, 400016
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Pune, India, 411001
Korea, Republic of
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Seoul, Korea, Republic of, 138-736
Mexico
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Cuernavaca, Mexico, 62200
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mexico City, Mexico, 01120
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Villahermosa, Mexico, 86090
Turkey
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Antalya, Turkey
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Gaziantep, Turkey
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Kayseri, Turkey, 38039 Less << |
|
NCT00253591
|
Lung Cancer |
Phase 2
Phase 3 |
Completed |
- |
United Kingdom
... More >> Wansbeck General Hospital
Ashington, England, United Kingdom, NE63 9JJ
Bristol Haematology and Oncology Centre
Bristol, England, United Kingdom, BS2 8ED
Cheltenham General Hospital
Cheltenham, England, United Kingdom, GL53 7AN
Derbyshire Royal Infirmary
Derby, England, United Kingdom, DE1 2QY
University Hospital of North Durham
Durham, England, United Kingdom, DH1 5TW
Queen Elizabeth Hospital
Gateshead, England, United Kingdom, NE9 6SX
Northern Centre for Cancer Treatment at Newcastle General Hospital
Newcastle-Upon-Tyne, England, United Kingdom, NE4 6BE
Mount Vernon Cancer Centre at Mount Vernon Hospital
Northwood, England, United Kingdom, HA6 2RN
Kings Mill Hospital
Nottinghamshire, England, United Kingdom, NG17 4JL
Nottingham City Hospital NHS Trust
Nottingham, England, United Kingdom, NG5 1PB
Churchill Hospital
Oxford, England, United Kingdom, OX3 7LJ
Cancer Research Centre at Weston Park Hospital
Sheffield, England, United Kingdom, S1O 2SJ
Great Western Hospital
Swindon, England, United Kingdom, SN3 6BB
Worcester Royal Hospital
Worcester, England, United Kingdom, WR5 1DD
Yeovil District Hospital
Yeovil, England, United Kingdom, BA21 4AT
Beatson West of Scotland Cancer Centre
Glasgow, Scotland, United Kingdom, G12 0YN
Crosshouse Hospital
Kilmarnock, Scotland, United Kingdom, KA2 OBE
Velindre Cancer Center at Velindre Hospital
Cardiff, Wales, United Kingdom, CF14 2TL
Llandough Hospital
Llandough, Wales, United Kingdom, CF64 2XX
Royal Gwent Hospital
Newport Gwent, Wales, United Kingdom, NP9 2UB Less << |
|
NCT00384878
|
Stomach Neoplasms |
Phase 2 |
Unknown |
- |
Taiwan
... More >> Kun Huei Yeh, Ph.D
Recruiting
Taipei, Ban-Ciao, Taiwan, 220
Contact: Kun Huei Yeh, Ph.D. 886-2-89667000 ext 1611 khyeh@mail.femh.org.tw Less << |
|
NCT01884259
|
Squamous Cell Carcinoma of the... More >> Hypopharynx Stage III
Squamous Cell Carcinoma of the Hypopharynx Stage IV
Squamous Cell Carcinoma of the Larynx Stage III
Squamous Cell Carcinoma of the Larynx Stage IV
Squamous Cell Carcinoma of the Oropharynx Stage III
Squamous Cell Carcinoma of the Oropharynx Stage IV
Squamous Cell Carcinoma of the Oral Cavity Stage III
Squamous Cell Carcinoma of the Oral Cavity Stage IV Less << |
Phase 2 |
Active, not recruiting |
June 2019 |
Austria
... More >> Landeskrankenhaus Feldkirch
Feldkirch, Austria, A-6807
Landesklinikum Krems
Krems, Austria, A-3500
Krankenhaus d. Barmherzigen Schwestern Linz
Linz, Austria, A-4010
Kepler Universitätsklinikum, Med Campus III. Klinik für Interne 3 - Schwerpunkt Hämatologie u. Onkologie
Linz, Austria, A-4021
PMU Salzburg
Salzburg, Austria, 5020
Hanusch Krankenhaus Wien
Vienna, Austria, 1140
Universität f. Strahlentherape, AKH Wien
Vienna, Austria, A-1090
Klinikum Kreuzschwestern Wels GmbH
Wels, Austria, A-4600 Less << |
|
NCT00006116
|
Lung Cancer |
Phase 2 |
Unknown |
- |
France
... More >> Clinique Sainte Catherine
Avignon, France, 84082
C.H.G. Beauvais
Beauvais, France, 60021
Centre Hospitalier Departmental Felix Guyon
Bellepierre, France, 97400
Centre Hospitalier General
Brive, France
Centre Hospitalier Emile Roux
Eaubonne, France, 95602
Centre Jean Bernard
Le Mans, France, 72000
C.H. Mans
Le Mans, France, 72037
Hopital Perpetuel Secours
Levallois-Perret, France, 92300
Clinique St. Faron
Mareuil Les Meaux, France, 77100
Clinique de Docteur Terrioux
Meaux, France, 77100
Clinique Hartmann
Neuilly sur Seine, France, 92200
American Hospital of Paris
Neuilly Sur Seine, France, F-92202
Clinique Du Mont Louis
Paris, France, 75011
Hopital Saint Antoine
Paris, France, 75571
Hopital Tenon
Paris, France, 75970
Hopital Claude Gallien
Quincy Sous Senart, France, 91480
Groupe Medical Saint Remy
Reims, France, 51096
Clinique les Bleuets
Reims, France, 51100
Polyclinique De Courlancy
Reims, France, F-51100
Centre Rene Huguenin
Saint Cloud, France, 92210
Centre du Rouget
Sarcelles, France, 95250 Less << |
|
NCT02999893
|
Oesophageal Carcinoma |
Phase 1
Phase 2 |
Recruiting |
April 30, 2023 |
Australia, Victoria
... More >>
Monash Medical Centre
Recruiting
Clayton, Victoria, Australia, 3000
Contact: Benjamin Markman
Principal Investigator: Benjamin Markman, MBBS, FRACP
Austin Health
Not yet recruiting
Heidelberg, Victoria, Australia, 3000
Contact: Niall Tebbutt
Principal Investigator: Niall Tebbutt, MBBS, FRACP
Peter MacCallum Cancer Centre
Recruiting
Melbourne, Victoria, Australia, 3002
Contact: Lara Lipton, MSc lara.lipton@petermac.org
Principal Investigator: Lara Lipton, MBBS, FRACP
Sub-Investigator: Michael Michael, MBBS, FRACP
Alfred Hospital
Not yet recruiting
Prahran, Victoria, Australia, 3000
Contact: Andrew Haydon
Principal Investigator: Andrew Haydon, MBBS, FRACP
Sunshine Hospital Western Health
Not yet recruiting
Sunshine, Victoria, Australia, 3000
Contact: Lara Lipton lara.lipton@petermac.org
Principal Investigator: Lara Lipton, MBBS, FRACP Less << |
|
NCT02309658
|
Cancer of Cervix |
Phase 2 |
Completed |
- |
Brazil
... More >> Instituto de Medicina Integral Fernando Figueira
Recife, Pernambuco, Brazil, 50070550 Less << |
|
NCT00191854
|
- |
|
Completed |
- |
- |
|
NCT00002813
|
Cervical Cancer |
Phase 2 |
Completed |
- |
United States, California
... More >>
Chao Family Comprehensive Cancer Center
Orange, California, United States, 92868
United States, Hawaii
MBCCOP - Hawaii
Honolulu, Hawaii, United States, 96813
United States, Maryland
Johns Hopkins Oncology Center
Baltimore, Maryland, United States, 21231
United States, Michigan
CCOP - Ann Arbor Regional
Ann Arbor, Michigan, United States, 48106
United States, New York
Cancer Center of Albany Medical Center
Albany, New York, United States, 12208
United States, North Carolina
Comprehensive Cancer Center of Wake Forest University Baptist Medical Center
Winston-Salem, North Carolina, United States, 27157-1082
United States, South Carolina
CCOP - Upstate Carolina
Spartanburg, South Carolina, United States, 29303
United States, Tennessee
Brookview Research, Inc.
Nashville, Tennessee, United States, 37203
United States, Texas
University of Texas - MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT01646853
|
Esophageal Neoplasms |
Not Applicable |
Unknown |
- |
China, Jiangsu
... More >> Jiangsu Province Hospital
Nanjing, Jiangsu, China, 210029 Less << |
|
NCT00057837
|
Extensive Stage Small Cell Lun... More >>g Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00057837
|
- |
|
Completed |
- |
- |
|
NCT01016561
|
Cervical Cancer |
Not Applicable |
Unknown |
- |
United States, Pennsylvania
... More >>
Abramson Cancer Center of the Unviersity of Pennsylvania
Philadelphia, Pennsylvania, United States, 19104 Less << |
|
NCT01047007
|
Solid Tumors |
Phase 1 |
Terminated(The study has been ... More >>terminated due to business reasons.) Less << |
- |
- |
|
NCT00334594
|
Malignant Mesothelioma |
Phase 2 |
Completed |
- |
Germany
... More >> Universitaetsklinikum Freiburg
Freiburg, Germany, D-79106
Switzerland
Kantonsspital Aarau
Aarau, Switzerland, CH-5001
Kantonsspital Baden
Baden, Switzerland, CH-5404
Universitaetsspital-Basel
Basel, Switzerland, CH-4031
Spital Tiefenau
Bern 4, Switzerland, 3004
Inselspital Bern
Bern, Switzerland, CH-3010
Kantonsspital Bruderholz
Bruderholz, Switzerland, CH-4101
Kantonsspital Graubuenden
Chur, Switzerland, CH-7000
Centre Hospitalier Universitaire Vaudois
Lausanne, Switzerland, CH-1011
Kantonsspital Olten
Olten, Switzerland, CH-4600
Kantonsspital - St. Gallen
St. Gallen, Switzerland, CH-9007
SpitalSTS AG Simmental-Thun-Saanenland
Thun, Switzerland, 3600
UniversitaetsSpital Zuerich
Zurich, Switzerland, CH-8091 Less << |
|
NCT00209690
|
Esophageal Cancer |
Phase 1
Phase 2 |
Completed |
- |
Japan
... More >> ・ Hokkaido University Hospital (Hokkaido University Graduate School of Medicine / School of Medicine)
Sapporo, Hokkaido, Japan, 060-8638 Less << |
|
NCT01372202
|
Esophageal Cancer |
Phase 2 |
Active, not recruiting |
June 2019 |
United States, Maryland
... More >>
Ronan Kelly, M.D.
Baltimore, Maryland, United States, 21287 Less << |
|
NCT03617913
|
Bladder Carcinoma Infiltrating... More >> the Muscle of the Bladder Wall
Stage II Bladder Cancer AJCC v8
Stage II Renal Pelvis Cancer AJCC v8
Stage II Ureter Cancer AJCC v8
Stage II Urethral Cancer AJCC v8
Stage III Bladder Cancer AJCC v8
Stage III Renal Pelvis Cancer AJCC v8
Stage III Ureter Cancer AJCC v8
Stage III Urethral Cancer AJCC v8
Stage IIIA Bladder Cancer AJCC v8
Stage IIIB Bladder Cancer AJCC v8
Urethral Urothelial Carcinoma Less << |
Phase 2 |
Recruiting |
July 31, 2025 |
United States, Arizona
... More >>
Mayo Clinic in Arizona
Recruiting
Scottsdale, Arizona, United States, 85054
Contact: Clinical Trials Referral Office 855-776-0015
Principal Investigator: Parminder Singh, M.D
United States, Florida
Mayo Clinic in Florida
Recruiting
Jacksonville, Florida, United States, 32224
Contact: Clinical Trials Referral Office 855-776-0015
Principal Investigator: Winston W. Tan, M.D.
United States, Minnesota
Mayo Clinic
Recruiting
Rochester, Minnesota, United States, 55905
Contact: Clinical Trials Referral Office 855-776-0015
Principal Investigator: Brian A. Costello, M.D. Less << |
|
NCT01087970
|
Head and Neck Neoplasms |
Phase 2 |
Completed |
- |
- |
|
NCT03330028
|
Diseases of Oesophagus Stomach... More >> and Duodenum Less << |
Phase 1 |
Recruiting |
April 2022 |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact bbadgwell@mdanderson.org Less << |
|
NCT01047007
|
- |
|
Terminated(The study has been ... More >>terminated due to business reasons.) Less << |
- |
- |
|
NCT02474368
|
Recurrent Head and Neck Carcin... More >>oma
Head and Neck Cancer Metastatic
Head or Neck Cancer Less << |
Phase 1 |
Recruiting |
June 2021 |
United States, Massachusetts
... More >>
Dana-Farber Cancer Institute
Recruiting
Boston, Massachusetts, United States, 02115
Contact: Jonathan Schoenfeld, MD, MPH 617-632-5734 Jdschoenfeld@partners.org
Principal Investigator: Jonathan Schoenfeld, MD, MPH Less << |
|
NCT01087970
|
- |
|
Completed |
- |
- |
|
NCT01372202
|
- |
|
Active, not recruiting |
- |
- |
|
NCT02025036
|
Stage III Esophageal Squamous ... More >>Cell Carcinoma
Stage II Esophageal Squamous Cell Carcinoma Less << |
Phase 3 |
Recruiting |
December 2020 |
China, Henan
... More >> The First Affiliated Hospital of Henan University of Science and Technology
Recruiting
Luoyang, Henan, China, 471003
Contact: Shegan Gao, M.D Ph.D +86(379)64811906 gsg112258@163.com
Contact: Tanyou Shan, master +86(379)64815350 shantanyou@163.com
Sub-Investigator: Dan Zhou, M.D.M.S Less << |
|
NCT00042510
|
Stomach Neoplasms
... More >> Esophageal Neoplasms Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01160744
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Completed |
- |
- |
|
NCT01920061
|
Neoplasm |
Phase 1 |
Recruiting |
January 2019 |
- |
|
NCT01675765
|
Malignant Pleural Mesothelioma |
Phase 1 |
Active, not recruiting |
December 2018 |
United States, California
... More >>
University of California at San Francisco
San Francisco, California, United States, 94115
United States, Florida
H. Lee Moffitt Cancer Center
Tampa, Florida, United States, 33612
United States, Illinois
University of Chicago Medical Center
Chicago, Illinois, United States, 60637
United States, Maryland
National Cancer Institute
Bethesda, Maryland, United States, 20892
United States, Pennsylvania
University of Pennsylvania Abramson Cancer Center
Philadelphia, Pennsylvania, United States, 19104 Less << |
|
NCT00072124
|
Melanoma (Skin) |
Phase 3 |
Completed |
- |
United States, Maryland
... More >>
Warren Grant Magnuson Clinical Center - NCI Clinical Studies Support
Bethesda, Maryland, United States, 20892-1182 Less << |
|
NCT00623558
|
Head and Neck Neoplasm |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Seoul National University Hospital
Seoul, Korea, Republic of, 110-744 Less << |
|
NCT00209716
|
Esophageal Cancer |
Phase 1
Phase 2 |
Completed |
- |
Japan
... More >> ・ Hokkaido University Hospital (Hokkaido University Graduate School of Medicine)
Sapporo, Hokkaido, Japan, 060-8638 Less << |
|
NCT00949650
|
Carcinoma, Non-Small-Cell Lung... More >>
Adenocarcinoma Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00275951
|
Gastric Cancer |
Phase 2 |
Completed |
- |
Taiwan
... More >> Department of Oncology, National Taiwan University Hospital
Taipei, Taiwan, 100 Less << |
|
NCT01160744
|
- |
|
Completed |
- |
- |
|
NCT00949650
|
- |
|
Completed |
- |
- |
|
NCT01639001
|
NSCLC (Non-small Cell Lung Can... More >>cer) Less << |
Phase 3 |
Active, not recruiting |
January 2019 |
- |
|
NCT00985855
|
Non-Small-Cell Lung Carcinoma |
Phase 2 |
Completed |
- |
France
... More >> Institut de Cancerologie de la Loire
St Priest en Jarez, France, 42271 Less << |
|
NCT01557959
|
- |
|
Completed |
- |
- |
|
NCT01639001
|
- |
|
Active, not recruiting |
- |
- |
|
NCT01557959
|
Adenocarcinoma of the Lung
... More >> Adenosquamous Cell Lung Cancer
Bronchoalveolar Cell Lung Cancer
Large Cell Lung Cancer
Non-small Cell Lung Cancer
Recurrent Non-small Cell Lung Cancer
Squamous Cell Lung Cancer
Stage IIIB Non-small Cell Lung Cancer
Stage IV Non-small Cell Lung Cancer Less << |
Phase 2 |
Completed |
- |
United States, North Carolina
... More >>
Wake Forest University Health Sciences
Winston-Salem, North Carolina, United States, 27157 Less << |
|
NCT03047265
|
Nasopharyngeal Carcinoma |
Phase 2 |
Active, not recruiting |
June 1, 2027 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT00313690
|
Lung Cancer |
Phase 1
Phase 2 |
Withdrawn(Recruitment difficul... More >>ties) Less << |
- |
- |
|
NCT00154700
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
Taiwan
... More >> Department of Oncology, National Taiwan University Hospital
Taipei, Taiwan, 100 Less << |
|
NCT00024323
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, Arizona
... More >>
Arizona Clinical Research Center
Tucson, Arizona, United States, 85712
United States, Connecticut
Yale Comprehensive Cancer Center
New Haven, Connecticut, United States, 06520-8028 Less << |
|
NCT03288350
|
Gastric Adenocarcinoma
... More >> Esophageal Adenocarcinoma Less << |
Phase 2 |
Recruiting |
December 2023 |
Canada, Quebec
... More >> McGill University Health Centre
Recruiting
Montréal, Quebec, Canada, H4A 3J1
Contact: Penny Chipman 514-934-1934 ext 64802 penny.chipman@muhc.mcgill.ca
Principal Investigator: Thierry Alcindor, MD, MSc Less << |
|
NCT00547157
|
Cancer
Head a... More >>nd Neck Cancer
Oncology
Squamous Cell Carcinoma Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01824004
|
Gastric Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Chonnam National University Hwasun Hospital
Hwasun-Eup, Jeollanam-do, Korea, Republic of, 519-809 Less << |
|
NCT02786641
|
Nasopharyngeal Carcinoma |
Phase 3 |
Not yet recruiting |
August 2021 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT02177695
|
Bladder Cancer |
Phase 2 |
Recruiting |
October 2022 |
- |
|
NCT02289807
|
Nasopharyngeal Carcinoma |
Not Applicable |
Not yet recruiting |
March 2020 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT00536614
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
Italy
... More >> A.O. Treviglio-Caravaggio, P.le Ospedale n1
Treviglio, Bergamo, Italy, 24047
Ospedale S.Gerardo, Via Donizetti, 106
Monza, Milano, Italy, 20052
A.O. Ospedale Umberto I - Università - Località Torretta
Ancona, Italy, 60020
Ospedali Riuniti, Largo Barozzi, 1
Bergamo, Italy, 24128
Casa di Cura di Poliambulanza, Via Bissolati 57
Brescia, Italy, 25100
A.O. Careggi-Università, Viale Pieraccini, 17
Firenze, Italy, 50139
Azienda USL 6 - Viale Alfieri, 36
Livorno, Italy, 57121
A.O. Carlo Poma - Via Albertoni, 1
Mantova, Italy, 46100
A.O. Cà Granda, Piazza Ospedale Maggiore, 3
Milano, Italy, 20162
Università Campus Biomedico, Via Emilio Longoni, 83
Roma, Italy, 00155 Less << |
|
NCT01633203
|
- |
|
Active, not recruiting |
September 2018 |
Chile
... More >> Instituto Nacional del Cáncer
Santiago, RM, Chile, 8380455 Less << |
|
NCT00662311
|
Stage IIIA Non-small Cell Lung... More >> Cancer
Stage IIIB Non-small Cell Lung Cancer Less << |
Phase 1
Phase 2 |
Terminated |
- |
United States, Washington
... More >>
Fred Hutchinson Cancer Research Center/University of Washington Cancer Consortium
Seattle, Washington, United States, 98109 Less << |
|
NCT02099786
|
Cisplatin Ototoxicity
... More >> Hearing Loss Less << |
Not Applicable |
Completed |
- |
United States, Oregon
... More >>
VA Portland Health Care System, Portland, OR
Portland, Oregon, United States, 97239 Less << |
|
NCT00547157
|
- |
|
Completed |
- |
- |
|
NCT02980991
|
Lung Adenocarcinoma |
Phase 2 |
Unknown |
April 2018 |
China
... More >> Fudan University Shanghai Cancer Center
Recruiting
Shanghai, China, 200032
Contact: Haiquan Chen, MD +86-21 64175590 ext 1707 hqchen1@yahoo.com
Contact: Xinghua Cheng, MD,PhD +8618017317567 chengxinghua_001@163.com
Principal Investigator: Haiquan Chen, MD Less << |
|
NCT00033657
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01488838
|
Malignant Salivary Gland Tumor... More >>s Less << |
Phase 2 |
Completed |
- |
Saudi Arabia
... More >> King Faisal Specialist Hospital& Research Center
Riyadh, Saudi Arabia, 11211 Less << |
|
NCT00033657
|
- |
|
Completed |
- |
- |
|
NCT03071848
|
- |
|
Completed |
- |
Argentina
... More >> Hospital General de Agudos J. A. Penna ; Breast Pathology
Buenos Aires, Argentina, 1437
Instituto Ángel H. Roffo - Universidad de Buenos Aires
Buenos Aires, Argentina, B1650
Hospital Julio C. Perrando
Chaco, Argentina
Hospital General de Agudos Juan Antonio Fernandez
Ciudad Autonoma de Buenos Aires, Argentina, 1425
Hospital Pablo Soria
Jujuy, Argentina
Centro Oncologico Riojano Integral (CORI)
La Rioja, Argentina, F5300COE
Hospital Interzonal General De Agudos "Luisa C. de Gandulfo"
Lomas de Zamora, Argentina
Hospital Privado de Comunidad; Oncology
Mar Del Plata, Argentina, 7600
Hosp Provincial D. Centenarios; Oncology Dept
Rosario, Argentina, S2002KDS
CENICLAR
Rosario, Argentina
Policlínico regional de San Luis
San Luis, Argentina
Centro Medico San Roque
San Miguel de Tucuman, Argentina, T4000IAK Less << |
|
NCT00191334
|
Ovarian Cancer |
Phase 2 |
Completed |
- |
Russian Federation
... More >>
For additional information regarding investigative sites for thsi trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Irkutsk, Russian Federation
For additional information regarding investigative sites for thsi trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Ivanovo, Russian Federation
For additional information regarding investigative sites for thsi trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Moscow, Russian Federation
For additional information regarding investigative sites for thsi trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
St. Petersburg, Russian Federation
For additional information regarding investigative sites for thsi trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Stavropol, Russian Federation Less << |
|
NCT03567642
|
Lung Cancer |
Phase 1 |
Recruiting |
June 2020 |
United States, New Jersey
... More >>
Memoral Sloan Kettering Cancer Center
Recruiting
Basking Ridge, New Jersey, United States, 07920
Contact: Helena Yu, MD 646-888-4274
Memorial Sloan Kettering Monmouth
Recruiting
Middletown, New Jersey, United States, 07748
Contact: Helena Yu, MD 646-888-4274
United States, New York
Memorial Sloan Kettering Commack
Recruiting
Commack, New York, United States, 11725
Contact: Helena Yu, MD 646-888-4274
Memorial Sloan Kettering Westchester
Recruiting
Harrison, New York, United States, 10604
Contact: Helena Yu, MD 646-888-4274
Principal Investigator: Helena Yu, MD
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10065
Contact: Helena Yu, MD 646-888-4274
Contact: Mark Kris, MD 646-888-4205
Principal Investigator: Helena Yu, MD
Memorial Sloan Kettering Rockville Centre
Recruiting
Rockville Centre, New York, United States, 11570
Contact: Helena Yu, MD 646-888-4274 Less << |
|
NCT00662311
|
- |
|
Terminated |
- |
- |
|
NCT00191334
|
- |
|
Completed |
- |
- |
|
NCT00705016
|
Squamous Cell Cancer |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT00001427
|
Ovarian Neoplasms |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
National Cancer Institute (NCI)
Bethesda, Maryland, United States, 20892 Less << |
|
NCT00001272
|
Ovarian Neoplasms |
Phase 1 |
Completed |
- |
United States, Maryland
... More >>
National Cancer Institute (NCI)
Bethesda, Maryland, United States, 20892 Less << |
|
NCT00994097
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Italy
... More >> Istituto Nazionale per la ricerca sul cancro
Genoa, Italy, 16132
Fondazione San Raffaele del Monte Tabor
Milan, Italy, 20132
Istituto Nazionale dei Tumori
Milan, Italy, 20133
Istituto Europeo Oncologico
Milan, Italy Less << |
|
NCT00705016
|
- |
|
Completed |
- |
- |
|
NCT00002774
|
Squamous Neck Carcinoma of the... More >> Head and Neck Cancer (SCCHN) Less << |
Phase 2 |
Completed |
- |
United States, California
... More >>
Veterans Affairs Medical Center - Palo Alto
Palo Alto, California, United States, 94304
Stanford University Medical Center
Stanford, California, United States, 94305-5408 Less << |
|
NCT01573338
|
Small Cell Lung Carcinoma |
Phase 1
Phase 2 |
Terminated |
- |
United States, Missouri
... More >>
Saint Louis, Missouri, United States, 63110
United States, New York
Buffalo, New York, United States, 14263-0001
United States, Ohio
Cleveland, Ohio, United States, 44195
France
Caen Cedex, France, 14033
Lyon Cedex, France, 69008
Marseille, France, 13005
Villejuif Cedex, France, 94805
Korea, Republic of
Seoul, Korea, Republic of, 03080
Seoul, Korea, Republic of, 120-752 Less << |
|
NCT00190476
|
Non-small-cell Lung Cancer |
Phase 3 |
Terminated |
- |
Japan
... More >> Aichi Cancer Center Hospital
Nagoya,Chikusa-ku,Kanokoden,1-1, Aichi, Japan, 464-8681
Aichi Cancer Center,Aichi Hospital
Okazaki,Kake-machi,Kuriyado,18, Aichi, Japan, 444-0011
National Cancer Center Hospital East
Kashiwa,Kashiwanoha,6-5-1, Chiba, Japan, 277-8577
National Hospital Organization Shikoku Cancer Center
Matsuyama,Horinouchi,13, Ehime, Japan, 790-0007
Kyushu University Hospital
Higashi-ku,Maidashi,3-1-1, Fukuoka, Japan, 812-8582
Gifu Municipal Hospital
Gifu,Kashima-cho,7-1, Gifu, Japan, 500-8323
Gunma Prefectural Cancer Center
Ota,Takabayashi-nishi-cho,617-1, Gunma, Japan, 373-8550
National Nishigunma Hospital
Shibukawa,Kanai,2854, Gunma, Japan, 377-8511
National Hospital Organization, Dohoku National Hospital
Asahikawa,Hanasaki,7-4048, Hokkaido, Japan, 070-8644
National Hospital Organization Hokkaido Cancer Center
Sapporo,Shiroishi-ku,Kikusui,4-2-3-54, Hokkaido, Japan, 003-0804
Hyogo Medical Center for Adults
Akashi,Kitaouji-cho,13-70, Hyogo, Japan, 673-8558
Hyogo College of Medicine
Nishinomiya,Mukogawa-cho,1-1, Hyogo, Japan, 663-8501
Ibaraki Kenritsu Chuo Hospital & Cancer Center
Nishi-ibarakigun,Tomobemachi,Koibuchi,6528, Ibaraki, Japan, 309-1793
Kanagawa Cancer Center
Yokohama,Asahi-ku,Nakao,1-1-2, Kanagawa, Japan, 241-0815
Yokohama Mucipical Citizen's Hospital
Yokohama,Hodogaya-ku,Okazawa-cho,56, Kanagawa, Japan, 240-8555
Kumamoto Regional Medical Center Hospital
Kumamoto,Honjo,5-16-10, Kumamoto, Japan, 860-0811
Niigata Cancer Center Hospital
Niigata,Kawagishi-cho, 2-15-3, Niigata, Japan, 951-8566
Osaka Prefectural Medical Center for Respiratory and Allergic Disease
Habikino,Habikino,3-7-1, Osaka, Japan, 583-8588
Osaka Medical Center for Cancer and Cardiovascular Diseases
Osaka,Higashinari-ku,Nakamichi,1-3-3, Osaka, Japan, 537-8511
Osaka City General Hospital
Osaka,Miyakojima-ku,Miyakojimahondori,2-13-22, Osaka, Japan, 534-0021
Kinki University School of Medicine
Osaka-Sayama,Ohno-higashi,377-2, Osaka, Japan, 589-8511
National Hospital Organization Kinki-Chuo Chest Medical Center
Sakai,Nagasone,1180, Osaka, Japan, 591-8555
National Hospital Organization Toneyama National Hospital
Toyonaka,Toneyama,5-1-1, Osaka, Japan, 560-8552
Saitama Cancer Center
Kita-adachi,Ina,Komuro,818, Saitama, Japan, 362-0806
Sizuoka Cancer Center
Sunto-gun,Nagaizumi-cho,Shimonagakubo,1007, Shizuoka, Japan, 411-8777
Tochigi Cancer Center
Utsunomiya,Yohnan,4-9-13, Tochigi, Japan, 320-0834
National Cancer Center Hospital
Chuo-ku,Tsukiji,5-1-1, Tokyo, Japan, 104-0045
International Medical Center of Japan
Shinjuku-ku,Toyama,1-21-1, Tokyo, Japan, 162-8655
Yamagata Prefectural Central Hospital
Yamagata,Aoyagi,1800, Yamagata, Japan, 990-2292 Less << |
|
NCT01262859
|
Head and Neck Neoplasms |
Phase 2 |
Withdrawn(PI relocated to anot... More >>her institution. No subjects went on treatment.) Less << |
- |
United States, Pennsylvania
... More >>
Hillman Cancer Center
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT00389155
|
Bladder Cancer
... More >> Transitional Cell Carcinoma
Metastasis Less << |
Phase 2
Phase 3 |
Completed |
- |
- |
|
NCT03061630
|
Urinary Bladder |
Phase 2 |
Active, not recruiting |
February 2020 |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of, 06351 Less << |
|
NCT03234153
|
Muscle-invasive Bladder Cancer |
Phase 2 |
Recruiting |
March 2025 |
Switzerland
... More >> Departement of Medical Oncology, University Hospital Berne
Recruiting
Berne, Switzerland, 3010
Contact: Julian Schardt, MD +41 31 632 8195 Julian.Schardt@insel.ch Less << |
|
NCT03326947
|
Neoplasm Neck |
Phase 2 |
Not yet recruiting |
January 1, 2023 |
China, Shanghai
... More >>
Ninth People's Hospital, School of Medicine, Shanghai Jiao Tong University
Shanghai, Shanghai, China, 200011 Less << |
|
NCT00490659
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Poland
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Lublin, Poland Less << |
|
NCT02162537
|
Non-small Cell Lung Cancer Met... More >>astatic
Non-small Cell Lung Cancer
Adenocarcinoma of Lung Metastatic to Brain
Cerebral Metastases Less << |
Phase 3 |
Recruiting |
January 2019 |
France
... More >> Centre Hospitalier du Pays d'Aix
Recruiting
Aix En Provence, France, 13613
Contact: Jacques LE TREUT jletreut@ch-aix.fr
Principal Investigator: Jacques LE TREUT
Centre Hospitalier Victor Dupouy
Recruiting
Argenteuil, France, 95100
Contact: Christine RAYNAUD christine.raynaud@ch-argenteuil.fr
Principal Investigator: Christine RAYNAUD
Centre d'Oncologie et de Radiothérapie du Pays Basque
Recruiting
Bayonne, France, 64100
Contact: Aurélien BLOUET aurelien.blouet@oncologie-pays-basque.org
Principal Investigator: Aurélien BLOUET
Centre Hospitalier
Recruiting
Beauvais, France, 60021
Contact: Jacky CREQUIT Jacky.crequit@orange.fr
Principal Investigator: Jacky CREQUIT
Hôpital Avicenne
Not yet recruiting
Bobigny, France, 93000
Contact: Antoine CARPENTIER antoine.carpentier@avc.aphp.fr
Principal Investigator: Antoine CARPENTIER
HIA de Clermont-Tonnerre
Not yet recruiting
Brest, France, 22240
Contact: Nicolas PALEIRON nicolas.paleiron@free.fr
Principal Investigator: Nicolas PALEIRON
CHU
Recruiting
Brest, France, 29200
Contact: Gilles ROBINET gilles.robinet@chu-brest.fr
Principal Investigator: Gilles ROBINET
Centre François Baclesse
Recruiting
Caen, France, 14000
Contact: Radj GERVAIS r.gervais@baclesse.fr
Principal Investigator: Radj GERVAIS
Centre Hospitalier
Recruiting
Charleville Meziere, France, 08000
Contact: Stéphane CHOUABE Schouabe@ch-charleville-mezieres.fr
Principal Investigator: Stéphane CHOUABE
Centre Hospitalier Laennec
Recruiting
Creil, France, 60109
Contact: Sandrine LOUTSKI-VETTESE sandrine.loutski@ch-creil.fr.fr
Principal Investigator: Sandrine LOUTSKI-VETTESE
Centre Hospitalier Intercommunal
Recruiting
Creteil, France, 94010
Contact: Isabelle MONNET isabelle.monnet@chicreteil.fr
Principal Investigator: Isabelle MONNET
Centre Hospitalier
Recruiting
Draguignan, France, 83300
Contact: Hervé LE CAER herve.lecaer@ch-draguignan.fr
Principal Investigator: Hervé LE CAER
Centre Hospitalier
Recruiting
GAP, France, 05000
Contact: Pascal THOMAS pascal.thomas@chicas-gap.fr
Principal Investigator: Pascal THOMAS
Centre Hospitalier Robert Boulin
Recruiting
Libourne, France, 33500
Contact: Samir ABDICHE samir.abdiche@ch-libourne.fr
Principal Investigator: Samir ABDICHE
Hôpital Le Cluzeau
Recruiting
Limoges, France, 87042
Contact: Alain VERGNENEGRE avergne@unilim.fr
Principal Investigator: Alain VERGNENEGRE
Centre Hospitalier Régional
Recruiting
Longjumeau, France, 91161
Contact: Gérard OLIVIERO gerard.oliviero@ch-longjumeau.fr
Principal Investigator: Gérard OLIVIERO
Centre Hospitalier de Bretagne Sud
Recruiting
Lorient, France, 56322
Contact: Régine LAMY r.lamy@ch-bretagne-sud.fr
Principal Investigator: Régine LAMY
Centre Léon Bérard
Recruiting
Lyon, France, 69373
Contact: Maurice PEROL maurice.perol@lyon.unicancer.fr
Principal Investigator: Maurice PEROL
Centre Hospitalier Les Chanaux
Recruiting
Macon, France, 71000
Contact: Dominique ARPIN doarpin@ch-macon.fr
Principal Investigator: Dominique ARPIN
Centre Hospitalier F. QUESNAY
Recruiting
Mantes La Jolie, France, 78200
Contact: Jean-Bernard AULIAC j-b.auliac@ch-mantes-la-jolie.rss.fr
Principal Investigator: Jean-Bernard AULIAC
Institut Paoli Calmette
Recruiting
Marseille, France, 13000
Contact: Anne MADROSZYC madroszyca@marseille.fnclcc.fr
Principal Investigator: Anne MADROSZYC
Hôpital Nord APHM
Recruiting
Marseille, France, 13915
Contact: Laurent GREILLIER laurent.greillier@mail.ap-hm.fr
Principal Investigator: Laurent GREILLIER
Centre Hospitalier
Recruiting
Meaux, France, 77108
Contact: Chrystèle LOCHER ch-locher@ch-meaux.fr
Principal Investigator: Chrystèle LOCHER
Centre Hospitalier Intercommunal
Recruiting
Meulan, France, 78250
Contact: Etienne LEROY-TERQUEM etiennelt@hotmail.fr
Principal Investigator: Etienne LEROY-TERQUEM
Clinique du Pont de Chaume
Recruiting
Montauban, France, 82000
Contact: Franck THOUVENY fthouveny@clinique-pontdechaume.fr
Principal Investigator: Franck THOUVENY
Centre Hospitalier
Recruiting
Perigueux, France, 24019
Contact: Jean-Yves DELHOUME jy.delhoume@ch-perigeux.fr
Principal Investigator: Jean-Yves DELHOUME
Centre Catalan d'Oncologie
Recruiting
Perpignan, France, 66000
Contact: Sabine VIEILLOT sabinevieillot@gmail.com
Principal Investigator: Sabine VIEILLOT
Centre Hospitalier René Dubos
Recruiting
Pontoise, France, 95303
Contact: Gislaine FRABOULET gislaine.fraboulet@ch-pontoise.fr
Principal Investigator: Gislaine FRABOULET
Centre Hospitalier de la Région d'Annecy (CHRA)
Recruiting
Pringy, France, 74374
Contact: Chantal DECROISETTE cdecroisette@ch-annecy.fr
Principal Investigator: Chantal DECROISETTE
Centre Hospitalier Intercommunal de Cornouaille
Recruiting
Quimper, France, 29107
Contact: Anne-Marie CHIAPPA am.chiappa@ch-cornouaille.fr
Principal Investigator: CHIAPPA Anne-Marie
Hôpital Pontchailloux
Recruiting
Rennes, France, 35033
Contact: Hervé LENA herve.lena@chu-rennes.fr
Principal Investigator: Hervé LENA
Hôpital Charles Nicolle
Recruiting
Rouen, France, 79031
Contact: Suzanna BOTA Suzanna.bota@chu-rouen.fr
Principal Investigator: Suzanna BOTA
Clinique Mutualiste de l'Estuaire
Recruiting
Saint Nazaire, France, 44600
Contact: Philippe DEGUIRAL philippe.deguiral@mla.fr
Principal Investigator: Philippe DEGUIRAL
Institut de Cancérologie de la Loire (I.C.L)
Recruiting
Saint Priest En Jarez, France, 42271
Contact: Pierre FOURNEL pierre.fournel@icloire.fr
Principal Investigator: Pierre FOURNEL
Centre Hospitalier de Salon de Provence
Recruiting
Salon de Provence, France, 13658
Contact: Jacques LE TREUT jletreut@ch-aix.fr
Principal Investigator: Jacques LE TREUT
Centre Hospitalier
Recruiting
Sens, France, 89108
Contact: GONZALEZ Gilles ggonzalez@ch-sens.fr
Principal Investigator: GONZALEZ Gilles
Centre Paul Strauss
Recruiting
Strasbourg, France, 67000
Contact: Roland SCHOTT rschott@strasbourg.unicancer.fr
Principal Investigator: Roland SCHOTT
Hopital d'Instruction des Armées Sainte Anne
Recruiting
Toulon, France, 83800
Contact: Henri BERARD hberard@libertysurf.fr
Principal Investigator: Henri BERARD
Hôpital Larrey
Recruiting
Toulouse, France, 31059
Contact: Laurence BIGAY-GAME bigaygame.l@chu-toulouse.fr
Principal Investigator: Laurence BIGAY-GAME
Clinique Pasteur
Recruiting
Toulouse, France, 31076
Contact: Olivier GALLOCHER o.gallocher@clinique-pasteur.com
Principal Investigator: Olivier GALLOCHER
Centre Hospitalier
Recruiting
Villefranche Sur Saone, France, 69655
Contact: Lionel FALCHERO lfalchero@ch-villefranche.fr
Principal Investigator: Lionel FALCHERO Less << |
|
NCT01405079
|
Non-small Cell Lung Cancer |
Phase 3 |
Active, not recruiting |
December 2020 |
China
... More >> Guangdong General Hospital
Guangzhou, China Less << |
|
NCT01425736
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 2 |
Completed |
- |
China, Guangdong
... More >>
First Affiliated Hospital of Sun Yat-sen University
Guangzhou, Guangdong, China, 510000
China, Jiangsu
Institute of Stomatology of Nanjing Medical University
Nanjing, Jiangsu, China, 210000
Wuxi People's Hospital; Nanjing Medical University
Wuxi, Jiangsu, China, 214000
Xuzhou Central Hospital of Xuzhou city,Dongnan University
Xuzhou, Jiangsu, China, 221009
China, Shanghai
9th People's Hospital, School of Stomatology,Shanghai Jiaotong University
Shanghai, Shanghai, China, 200011 Less << |
|
NCT00861094
|
Esophageal Cancer |
Phase 2
Phase 3 |
Completed |
- |
France
... More >> CHR de Besancon - Hopital Saint-Jacques
Besancon, France, 25030
Hopital Saint Andre
Bordeaux, France, 33075
Institut Bergonie
Bordeaux, France, 33076
Hopital Ambroise Pare
Boulogne-Billancourt, France, F-92104
Polyclinique Du Parc
Caen, France, 14052
Centre Regional Francois Baclesse
Caen, France, 14076
Hopital Du Bocage
Dijon, France, 21034
Centre de Lutte Contre le Cancer Georges-Francois Leclerc
Dijon, France, 21079
Centre Oscar Lambret
Lille, France, 59020
Centre Hospital Regional Universitaire de Limoges
Limoges, France, 87042
Centre Leon Berard
Lyon, France, 69373
CHU de la Timone
Marseille, France, 13385
Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Montpellier, France, 34298
Centre Antoine Lacassagne
Nice, France, 06189
Hopital Europeen Georges Pompidou
Paris, France, 75015
Hopital Saint-Louis
Paris, France, 75475
Hopital Tenon
Paris, France, 75970
CHU Poitiers
Poitiers, France, 86021
Institut Jean Godinot
Reims, France, 51056
CHU - Robert Debre
Reims, France, 51092
Centre Eugene Marquis
Rennes, France, 35042
Hopital Charles Nicolle
Rouen, France, 76031
Clinique Armoricaine De Radiologie
Saint Brieuc, France, F-22015
Centre Paul Strauss
Strasbourg, France, 67065
Institut Claudius Regaud
Toulouse, France, 31052
Clinique Du Parc
Toulouse, France, 31078
Centre Alexis Vautrin
Vandoeuvre-les-Nancy, France, 54511
Institut Gustave Roussy
Villejuif, France, F-94805 Less << |
|
NCT00068575
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
M. D. Anderson Cancer Center at University of Texas
Houston, Texas, United States, 77030-4009 Less << |
|
NCT00068575
|
- |
|
Completed |
- |
- |
|
NCT02383966
|
Carcinoma, Squamous Cell of He... More >>ad and Neck Less << |
Phase 3 |
Active, not recruiting |
November 30, 2018 |
Germany
... More >> Please contact the Merck KGaA Communication Center
Darmstadt, Germany Less << |
|
NCT00295672
|
Lung Neoplasm |
Phase 2 |
Completed |
- |
France
... More >> CHU of Brest
Brest, France, 29250
CH GAP
GAP, France, 05000
CHU de LIMOGES
Limoges, France, 87042
Hopital de la Croix Rousse
Lyon, France, 69317
Hôpital Sainte Margueritte
Marseilles, France, 13274
CHU Hôpital Nord
Saint Etienne, France, 42055 Less << |
|
NCT00262769
|
Extrahepatic Bile Duct Cancer
... More >>
Gallbladder Cancer Less << |
Phase 3 |
Completed |
- |
United Kingdom
... More >> Basingstoke and North Hampshire NHS Foundation Trust
Basingstoke, England, United Kingdom, RG24 9NA
Queen Elizabeth Hospital at University Hospital of Birmingham NHS Trust
Birmingham, England, United Kingdom, B15 2TH
Addenbrooke's Hospital
Cambridge, England, United Kingdom, CB2 2QQ
Cumberland Infirmary
Carlisle, England, United Kingdom, CA2 7HY
Gloucestershire Oncology Centre at Cheltenham General Hospital
Cheltenham, England, United Kingdom, GL53 7AN
Derbyshire Royal Infirmary
Derby, England, United Kingdom, DE1 2QY
Princess Alexandra Hospital
Essex, England, United Kingdom, CM20 1QX
Gloucestershire Royal Hospital
Gloucester, England, United Kingdom, GL1 3NN
Princess Royal Hospital at Hull and East Yorkshire NHS Trust
Hull, England, United Kingdom, HU8 9HE
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Helen Rollason Cancer Care Centre at North Middlesex Hospital
London, England, United Kingdom, N18 1QX
Royal Marsden - London
London, England, United Kingdom, SW3 6JJ
Hammersmith Hospital
London, England, United Kingdom, W12 OHS
UCL Cancer Institute
London, England, United Kingdom, WC1E 6DD
University College of London Hospitals
London, England, United Kingdom, WIT 3AA
Maidstone Hospital
Maidstone, England, United Kingdom, ME16 9QQ
Clatterbridge Centre for Oncology
Merseyside, England, United Kingdom, CH63 4JY
Mount Vernon Cancer Centre at Mount Vernon Hospital
Northwood, England, United Kingdom, HA6 2RN
Nottingham City Hospital
Nottingham, England, United Kingdom, NG5 1PB
Portsmouth Oncology Centre at Saint Mary's Hospital
Portsmouth Hants, England, United Kingdom, PO3 6AD
Cancer Research Centre at Weston Park Hospital
Sheffield, England, United Kingdom, S10 2SJ
Belfast City Hospital Trust Incorporating Belvoir Park Hospital
Belfast, Northern Ireland, United Kingdom, BT9 7AB
Aberdeen Royal Infirmary
Aberdeen, Scotland, United Kingdom, AB25 2ZN
Velindre Cancer Center at Velindre Hospital
Cardiff, Wales, United Kingdom, CF14 2TL
Glan Clwyd Hospital
Rhyl, Denbighshire, Wales, United Kingdom, LL 18 5UJ Less << |
|
NCT00183885
|
Hepatocellular Carcinoma
... More >> Liver Cancer Less << |
Phase 2 |
Active, not recruiting |
May 28, 2019 |
United States, California
... More >>
U.S.C. / Norris Comprehensive Cancer Center
Los Angeles, California, United States, 90033 Less << |
|
NCT01222312
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Germany
... More >> Krankenhaus Nordwest
Frankfurt/Main, Germany, 60488 Less << |
|
NCT00261703
|
Head and Neck Neoplasms |
Phase 2
Phase 3 |
Completed |
- |
Portugal
... More >> Sanofi-Aventis
Porto Salvo, Portugal
Spain
Sanofi-Aventis
Barcelona, Spain Less << |
|
NCT00886028
|
Malignant Pleural Mesothelioma |
Phase 2 |
Unknown |
June 2009 |
Mexico
... More >> National Institute of Cancerología
Mexico city, Distrito Federal, Mexico, 14080 Less << |
|
NCT03028766
|
Hypopharynx Squamous Cell Carc... More >>inoma
Oral Cavity Squamous Cell Carcinoma
Larynx Cancer Less << |
Phase 1 |
Recruiting |
December 2019 |
United Kingdom
... More >> University Hospital Birmingham Nhs Foundation Trust
Recruiting
Birmingham, West Midlands, United Kingdom, B15 2TH
Principal Investigator: James Good
Beatson West of Scotland Cancer Centre
Recruiting
Glasgow, United Kingdom, G12 0YN
Principal Investigator: Stefano Schipani
University College London Hospitals
Recruiting
London, United Kingdom, W1T 7HA
Principal Investigator: Martin Forster
Clatterbridge Cancer Centre
Not yet recruiting
Wirral, United Kingdom, CH63 4JY
Principal Investigator: Joseph Sacco Less << |
|
NCT00016367
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
University of Texas - MD Anderson Cancer Center
Houston, Texas, United States, 77030-4009 Less << |
|
NCT00191620
|
Non-Small-Cell Lung Carcinoma |
Phase 2 |
Completed |
- |
Argentina
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/ GMT - 5 hours, EST), or speak with your personal physician
Buenos Aires, Argentina
Brazil
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/ GMT - 5 hours, EST), or speak with your personal physician
Sao Paulo, Brazil
Chile
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/ GMT - 5 hours, EST), or speak with your personal physician
Santiago, Chile
Mexico
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/ GMT - 5 hours, EST), or speak with your personal physician
Mexico City, Mexico Less << |
|
NCT01139281
|
Ototoxicity
H... More >>earing Loss Less << |
Phase 2 |
Completed |
- |
Brazil
... More >> Hospital de Base do Distrito Federal (HBDF)
Brasília, DF, Brazil, 70000-000 Less << |
|
NCT01585805
|
BRCA1 Gene Mutation
... More >> BRCA2 Gene Mutation
Metastatic Pancreatic Adenocarcinoma
PALB2 Gene Mutation
Pancreatic Adenocarcinoma
Recurrent Pancreatic Carcinoma
Stage III Pancreatic Cancer AJCC v6 and v7
Stage IV Pancreatic Cancer AJCC v6 and v7 Less << |
Phase 2 |
Recruiting |
- |
United States, Illinois
... More >>
University of Chicago Comprehensive Cancer Center
Recruiting
Chicago, Illinois, United States, 60637
Contact: Hedy L. Kindler 773-702-0360 hkindler@medicine.bsd.uchicago.edu
Principal Investigator: Hedy L. Kindler
Ingalls Memorial Hospital
Recruiting
Harvey, Illinois, United States, 60426
Contact: Mark F. Kozloff 708-339-4800 mkozloff@bsd.uchicago.edu
Principal Investigator: Mark F. Kozloff
United States, Michigan
University of Michigan Comprehensive Cancer Center
Recruiting
Ann Arbor, Michigan, United States, 48109
Contact: Mark M. Zalupski 734-615-3969 zalupski@umich.edu
Principal Investigator: Mark M. Zalupski
United States, Missouri
Mercy Hospital Saint Louis
Completed
Saint Louis, Missouri, United States, 63141
United States, New Jersey
Memorial Sloan Kettering Basking Ridge
Recruiting
Basking Ridge, New Jersey, United States, 07920
Contact: Han Xiao 212-639-7202
Sub-Investigator: Han Xiao
Sub-Investigator: Sree B. Chalasani
Sub-Investigator: Afsheen Iqbal
United States, New York
Memorial Sloan Kettering Commack
Recruiting
Commack, New York, United States, 11725
Contact: Jahan Aghalar 212-639-7202
Sub-Investigator: Jahan Aghalar
Sub-Investigator: Avni M. Desai
Sub-Investigator: Juliana W. Eng
Sub-Investigator: John J. Fiore
Sub-Investigator: Stuart M. Lichtman
Memorial Sloan Kettering Westchester
Recruiting
Harrison, New York, United States, 10604
Contact: Parisa Momtaz kashanie@mskcc.org
Sub-Investigator: Parisa Momtaz
Sub-Investigator: Philip C. Caron
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10065
Contact: Eileen M. O'Reilly 646-888-4182 oreillye@mskcc.org
Principal Investigator: Eileen M. O'Reilly
Memorial Sloan Kettering Rockville Centre
Recruiting
Rockville Centre, New York, United States, 11570
Contact: Arlyn J. Apollo 212-639-7202
Sub-Investigator: Arlyn J. Apollo
Sub-Investigator: Pamela R. Drullinsky
Sub-Investigator: Zoe Goldberg
Sub-Investigator: Kenneth K. Ng
Sub-Investigator: Tiffany Troso-Sandoval
Canada, Ontario
University Health Network-Princess Margaret Hospital
Recruiting
Toronto, Ontario, Canada, M5G 2M9
Contact: Neesha Dhani 416-946-4501 neesha.dhani@uhn.ca
Principal Investigator: Neesha Dhani
Israel
Shaare Zedek Medical Center
Recruiting
Jerusalem, Israel, 91031
Contact: Amiel Segal 972-2-666-6331 segal@szmc.org.il
Principal Investigator: Amiel Segal
Chaim Sheba Medical Center
Recruiting
Tel Hashomer, Israel, 52621
Contact: Talia Golan 972-3-530-3030 tailia.golan@sheba.health.gov.il
Principal Investigator: Talia Golan Less << |
|
NCT00754858
|
Lung Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Yonsei Cancer Center at Yonsei University Medical Center
Seoul, Korea, Republic of, 120-752 Less << |
|
NCT00320359
|
Lung Cancer, Small Cell |
Phase 3 |
Completed |
- |
- |
|
NCT00005830
|
Endometrial Adenocarcinoma
... More >> Endometrial Clear Cell Adenocarcinoma
Endometrial Serous Adenocarcinoma
Stage III Uterine Corpus Cancer
Stage IV Uterine Corpus Cancer Less << |
Phase 1 |
Completed |
- |
United States, Pennsylvania
... More >>
Gynecologic Oncology Group
Philadelphia, Pennsylvania, United States, 19103 Less << |
|
NCT00717951
|
Advanced Breast Cancer |
Phase 2 |
Unknown |
May 2010 |
- |
|
NCT02133612
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
China, Beijing
... More >> Xiao Lv
Beijing, Beijing, China, 100021 Less << |
|
NCT01671449
|
Gastric Cancer |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Chungbuk National University Hospital
Cheongju-si, Chungcheongbuk-do, Korea, Republic of, 361-711
Seoul National University Bundang Hospital
Bundang-gu, Seongnam-si, Gyeonggi-do, Korea, Republic of, 463-707
National Cancer Center
Ilsan, Gyeonggi-do, Korea, Republic of
Chonnam National University Hwasun Hospital
Hwasun-eup, Hwasun-gun, Jeollanam-do, Korea, Republic of, 519-763
Dongnam Institute of Radiological and Medical Sciences
Busan, Korea, Republic of, 619-953
Yeungnam University Medical Center
Daegu, Korea, Republic of, 705-717
Gangneung Asan Hospital
Gangneung-si, Korea, Republic of, 210-711
Seoul St. Mary's hospital of the Catholic University of Korea
Seoul, Korea, Republic of, 137-701
Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT02278250
|
Solid Tumor
A... More >>dvanced Solid Tumor Less << |
Phase 1 |
Recruiting |
September 23, 2019 |
United States, Massachusetts
... More >>
Research site
Recruiting
Boston, Massachusetts, United States, 02114
United States, Tennessee
Research site 1
Recruiting
Nashville, Tennessee, United States, 37203
Research site 2
Recruiting
Nashville, Tennessee, United States, 37232
Netherlands
Research site
Recruiting
Rotterdam, Netherlands, 3075 EA
United Kingdom
Research site
Recruiting
London, United Kingdom, W1G 6AD
Research site
Recruiting
Sutton, United Kingdom Less << |
|
NCT01100944
|
Thymoma
Thymi... More >>c Carcinoma Less << |
Phase 1
Phase 2 |
Terminated(Principal investiga... More >>tor left the National Institutes of Health (NIH).) Less << |
- |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892 Less << |
|
NCT00976768
|
Advanced Gastric Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Asan medical center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT00003627
|
Head and Neck Cancer |
Phase 3 |
Unknown |
- |
France
... More >> Centre Oscar Lambret
Lille, France, 59020
Institut J. Paoli and I. Calmettes
Marseille, France, 13273
CHU de la Timone
Marseille, France, 13385
Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Montpellier, France, 34298
CRLCC Nantes - Atlantique
Nantes-Saint Herblain, France, 44805
Centre Antoine Lacassagne
Nice, France, 06189
Institut Jean Godinot
Reims, France, 51056
Centre Henri Becquerel
Rouen, France, 76038
Centre Rene Huguenin
Saint Cloud, France, 92211
Centre Hospitalier Universitaire Bretonneau de Tours
Tours, France, 37044
Centre Alexis Vautrin
Vandoeuvre-les-Nancy, France, 54511 Less << |
|
NCT02595905
|
Breast Carcinoma Metastatic in... More >> the Brain
Deleterious BRCA1 Gene Mutation
Deleterious BRCA2 Gene Mutation
Estrogen Receptor Negative
HER2/Neu Negative
Progesterone Receptor Negative
Recurrent Breast Carcinoma
Stage IV Breast Cancer AJCC v6 and v7
Triple-Negative Breast Carcinoma Less << |
Phase 2 |
Recruiting |
- |
- |
|
NCT00088816
|
Gastric Cancer |
Phase 2 |
Unknown |
- |
Japan
... More >> Shimane Prefectural Central Hospital
Izumo-shi, Shimane, Japan, 693-0068
Kyoto University Hospital
Kyoto, Japan, 606-8507
National Hospital Organization - Kyoto Medical Center
Kyoto, Japan, 612-0861
Kyoto-Katsura Hospital
Kyoto, Japan, 615-8256
Kitano Hospital
Osaka, Japan, 530-8480
Kansai Denryoku Hospital
Osaka, Japan, 553-0003
Yamato Municipal Hospital
Yamato City Kanagawa, Japan, 242-8602 Less << |
|
NCT01501136
|
Nasal and Nasal-type NK/T-cell... More >> Lymphoma Less << |
Phase 4 |
Recruiting |
May 2019 |
China, Henan
... More >> Oncology Department of The First Affiliated Hospital of Zhengzhou University
Recruiting
Zhengzhou, Henan, China, 450052
Contact: Mingzhi Zhang, Pro,Dr 13838565629 mingzhi_zhang@126.com
Principal Investigator: Mingzhi Zhang, Pro,Dr Less << |
|
NCT01627379
|
Esophageal Squamous Cell Cance... More >>r Less << |
Phase 3 |
Terminated(Sponsor decision du... More >>e to recommendation of the IDMC.) Less << |
- |
Germany
... More >> Johannes-Gutenberg-Universität Mainz, I. Medizinische Klinik und Poliklinik
Mainz, Rheinland-Pfalz, Germany, 55101 Less << |
|
NCT00003587
|
Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00047008
|
- |
|
Active, not recruiting |
- |
- |
|
NCT02513355
|
Non-small Cell Lung Cancer |
Phase 4 |
Unknown |
- |
China, Jiangsu
... More >> Jiangsu Taizhou People's Hospital
Recruiting
Nanjing City, Jiangsu, China, 225499
Contact: Qingfeng Yin, Clinical Manager 0086013912903257 y_qingfeng@163.com
Contact: Xiaolei Zhou, Manager 0086013776639377 zhouxiaolei@simcere.com Less << |
|
NCT00324298
|
Drug/Agent Toxicity by Tissue/... More >>Organ
Testicular Germ Cell Tumor Less << |
Phase 3 |
Completed |
- |
United Kingdom
... More >> Basildon University Hospital
Basildon, England, United Kingdom, SS16 5NL
Addenbrooke's Hospital
Cambridge, England, United Kingdom, CB2 2QQ
Essex County Hospital
Colchester, England, United Kingdom, C03 3NB
Ipswich Hospital
Ipswich, England, United Kingdom, IP4 5PD
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Saint Bartholomew's Hospital
London, England, United Kingdom, EC1A 7BE
University College of London Hospitals
London, England, United Kingdom, WIT 3AA
Norfolk and Norwich University Hospital
Norwich, England, United Kingdom, NR4 7UY
Royal Marsden - Surrey
Sutton, England, United Kingdom, SM2 5PT
Southend University Hospital NHS Foundation Trust
Westcliff-On-Sea, England, United Kingdom, SS0 0RY Less << |
|
NCT01100944
|
- |
|
Terminated(Principal investiga... More >>tor left the National Institutes of Health (NIH).) Less << |
- |
- |
|
NCT00047008
|
Head and Neck Cancer |
Phase 3 |
Active, not recruiting |
- |
- |
|
NCT00512096
|
- |
|
Completed |
- |
- |
|
NCT00512096
|
Penile Cancer |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
U.T.M.D. Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT02952586
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 3 |
Recruiting |
May 18, 2022 |
- |
|
NCT00182611
|
Gastric Cancer |
Phase 3 |
Completed |
- |
Japan
... More >> Yamato Municipal Hospital
Yamatotakada, Nara, Japan, 635-8501
Shimane Prefectural Central Hospital
Izumo-shi, Shimane, Japan, 693-0068
Fukui Red Cross Hospital
Fukui, Japan, 918-8501
Kyoto University Hospital
Kyoto, Japan, 606-8507
National Hospital Organization - Kyoto Medical Center
Kyoto, Japan, 612-0861
Kitano Hospital
Osaka, Japan, 530-8480 Less << |
|
NCT03533582
|
Childhood Hepatocellular Carci... More >>noma
Childhood Malignant Liver Neoplasm
Elevated Alpha-Fetoprotein
Hepatoblastoma
SMARCB1 Gene Mutation Less << |
Phase 2
Phase 3 |
Recruiting |
March 31, 2025 |
- |
|
NCT03267498
|
Nasopharyngeal Carcinoma |
Phase 2 |
Recruiting |
October 15, 2020 |
United States, California
... More >>
Helen Diller Family Comprehensive Center, UCSF
Recruiting
San Francisco, California, United States, 94115
Contact: Clinical Trials Office 877-827-3222 cancertrials@ucsf.edu
Principal Investigator: Sue Yom, M.D. Less << |
|
NCT02879513
|
Invasive Ductal Breast Cancer
... More >>
Tubular Breast Cancer
Mucinous Breast Cancer
Inflammatory Breast Cancer Less << |
Phase 3 |
Recruiting |
December 2022 |
China, Shanghai
... More >>
Shanghai Jiaotong University School of Medicine, Renji Hospital
Recruiting
Shanghai, Shanghai, China, 200127
Contact: Jinsong Lu, MD lujjss@163.com Less << |
|
NCT02104986
|
Extra Cranial Non Seminomateou... More >>s Malignant Germ Cell Tumour Less << |
Phase 2 |
Recruiting |
May 2024 |
France
... More >> CHU
Not yet recruiting
Amiens, France, 80054
Principal Investigator: Catherine DEVOLDERE
CHU
Recruiting
Angers, France, 49033
Principal Investigator: Isabelle PELLIER
CHRU
Not yet recruiting
Besançon, France, 25030
Principal Investigator: Véronique LAITHIER
CHU
Recruiting
Bordeaux, France, 33076
Principal Investigator: Cécile VERITE
Chu Morvan
Not yet recruiting
Brest, France, 26609
Principal Investigator: Philippe LEMOINE
CHU
Not yet recruiting
Caen, France, 14033
Principal Investigator: Odile MINCKES
Chu Estaing
Not yet recruiting
Clermont-ferrand, France, 63003
Principal Investigator: Eric DORE
CHU
Not yet recruiting
Dijon, France, 21079
Principal Investigator: GERARD COUILLAULT
Centre Oscar Lambret
Recruiting
Lille, France, 59000
Principal Investigator: HELENE SUDOUR
CHU
Not yet recruiting
Limoges, France, 87042
Principal Investigator: CHRISTOPHE PIGUET
Hopital de La Timone
Recruiting
Marseille, France, 13385
Principal Investigator: ANGELIQUE ROME
CHU
Not yet recruiting
Montpellier, France, 34295
Principal Investigator: NICOLAS KALFA
CHU
Recruiting
Nancy, France, 54511
Principal Investigator: PASCAL CHASTAGNER
CHU
Recruiting
Nantes, France, 44093
Principal Investigator: ESTELLE THEBAUD
CHU
Recruiting
Nice, France, 06202
Principal Investigator: MARILYNE POIREE
Institut Curie
Not yet recruiting
Paris, France, 75005
Principal Investigator: DANIEL ORBACH
Hopital Trousseau
Not yet recruiting
Paris, France, 75012
Principal Investigator: SYLVIE FASOLA
CHU
Not yet recruiting
Poitiers, France, 86021
Principal Investigator: FREDERIC MILLOT
CHU
Not yet recruiting
Reims, France, 51092
Principal Investigator: MARTINE MUNZER
CHU
Not yet recruiting
Rennes, France, 35203
Principal Investigator: VIRGINIE GANDEMER
CHU
Recruiting
Rouen, France, 76031
Principal Investigator: CECILE DUMESNIL DE MARICOURT
Hopital Felix Guyon
Not yet recruiting
Saint Denis, France, 97405
Principal Investigator: MATHILDE JEHANNE
CHRU
Recruiting
Saint Etienne, France, 42055
Principal Investigator: CLAIRE BERGER
CHRU
Not yet recruiting
Strasbourg, France, 67098
Principal Investigator: PATRICK LUTZ
CHU
Not yet recruiting
Toulouse, France, 31059
Principal Investigator: MARIE PIERRE CASTEIX
CHRU
Not yet recruiting
Tours, France, 37044
Principal Investigator: ODILE LEJARS
Institut Gustave Roussy
Not yet recruiting
Villejuif, France, 94805
Principal Investigator: VERONIQUE MINARD Less << |
|
NCT00003996
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 2 |
Completed |
- |
United States, Arizona
... More >>
CCOP - Scottsdale Oncology Program
Scottsdale, Arizona, United States, 85259-5404
United States, Illinois
CCOP - Illinois Oncology Research Association
Peoria, Illinois, United States, 61602
CCOP - Carle Cancer Center
Urbana, Illinois, United States, 61801
United States, Iowa
CCOP - Cedar Rapids Oncology Project
Cedar Rapids, Iowa, United States, 52403-1206
CCOP - Iowa Oncology Research Association
Des Moines, Iowa, United States, 50309-1016
Siouxland Hematology-Oncology
Sioux City, Iowa, United States, 51101-1733
United States, Kansas
CCOP - Wichita
Wichita, Kansas, United States, 67214-3882
United States, Louisiana
CCOP - Ochsner
New Orleans, Louisiana, United States, 70121
United States, Minnesota
CCOP - Duluth
Duluth, Minnesota, United States, 55805
Mayo Clinic Cancer Center
Rochester, Minnesota, United States, 55905
CentraCare Clinic
Saint Cloud, Minnesota, United States, 56303
CCOP - Metro-Minnesota
Saint Louis Park, Minnesota, United States, 55416
United States, Nebraska
CCOP - Missouri Valley Cancer Consortium
Omaha, Nebraska, United States, 68131
United States, North Dakota
Quain & Ramstad Clinic, P.C.
Bismarck, North Dakota, United States, 58501
Altru Health Systems
Grand Forks, North Dakota, United States, 58201
United States, Ohio
CCOP - Toledo Community Hospital Oncology Program
Toledo, Ohio, United States, 43623-3456
United States, Pennsylvania
CCOP - Geisinger Clinical and Medical Center
Danville, Pennsylvania, United States, 17822-2001
United States, South Dakota
Rapid City Regional Hospital
Rapid City, South Dakota, United States, 57709
CCOP - Sioux Community Cancer Consortium
Sioux Falls, South Dakota, United States, 57105-1080 Less << |
|
NCT00045617
|
Lung Cancer |
Phase 2 |
Terminated(Closed due to lack ... More >>of drug availability.) Less << |
- |
- |
|
NCT01167192
|
Breast Neoplasms |
Phase 2 |
Terminated |
- |
United States, Missouri
... More >>
Washington University School of Medicine
St. Louis, Missouri, United States, 63110 Less << |
|
NCT01167192
|
- |
|
Terminated |
- |
- |
|
NCT02513342
|
Advanced Lung Squamous Carcino... More >>ma Less << |
Phase 4 |
Unknown |
April 2018 |
China, Jiangsu
... More >> Jinling Hospital
Recruiting
Nanjing, Jiangsu, China, 210002
Contact: Yin Qingfeng, Manager 0086013912903257 y_qingfeng@163.com
Contact: Zhou Xiaolei, Manager 0086013776639377 zhouxiaolei@simcere.com
Principal Investigator: Song Yong, director
The first affiliated hospital of soochow university
Recruiting
Soochow, Jiangsu, China, 215006
Contact: Yin Qingfeng, mananger 08613912903257 y_qingfeng@163.com
Contact: Zhou Xiaolei, mananger 08613776639377 zhouxiaolei@simcere.com
Principal Investigator: Huang Jianan, director Less << |
|
NCT00963807
|
- |
|
Completed |
- |
- |
|
NCT00963807
|
Recurrent Non-Small Cell Lung ... More >>Carcinoma
Stage IB Non-Small Cell Lung Carcinoma
Stage IIA Non-Small Cell Lung Carcinoma
Stage IIB Non-Small Cell Lung Carcinoma
Stage IIIA Non-Small Cell Lung Cancer
Stage IV Non-Small Cell Lung Cancer Less << |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
Johns Hopkins University/Sidney Kimmel Comprehensive Cancer Center
Baltimore, Maryland, United States, 21287
United States, Ohio
Ohio State University Comprehensive Cancer Center
Columbus, Ohio, United States, 43210 Less << |
|
NCT02157116
|
Non-small Cell Lung Cancer |
Phase 2 |
Terminated(Insufficient accrua... More >>l to meet analysis goals) Less << |
- |
United States, North Carolina
... More >>
Duke University Medical Center
Durham, North Carolina, United States, 27710 Less << |
|
NCT02157116
|
- |
|
Terminated(Insufficient accrua... More >>l to meet analysis goals) Less << |
- |
- |
|
NCT02936388
|
Uveal Melanoma |
Phase 2 |
Unknown |
December 2018 |
Germany
... More >> Charité - University Medicine Berlin, Dept. of Haematology, Medical Oncology and Tumor Immunology, Campus Benjamin Franklin
Recruiting
Berlin, Germany, 12200
Contact: Caroline Anna Peuker +49 30 450 513470 caroline-anna.peuker@charite.de
Contact: Sebastian Ochsenreither sebastian.ochsenreither@charite.de Less << |
|
NCT00003242
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00292955
|
Cancer of the Cervix |
Phase 2 |
Unknown |
- |
United States, Missouri
... More >>
Washinton University School of Medicine
Suspended
St. Louis, Missouri, United States, 63110
United States, Virginia
University of Virginia Cancer Center
Recruiting
Charlottesville, Virginia, United States, 22908 Less << |
|
NCT02512315
|
Nasopharyngeal Carcinoma |
Phase 3 |
Recruiting |
December 2024 |
China, Guangdong
... More >>
Cancer Center of Guangzhou Medical University
Recruiting
Guangzhou, Guangdong, China, 510030
Contact: Tong-chong Zhou, M.D +86-13808841276 noveaie@163.com
Contact: Hai-hua Peng, M.D +86-15011978349 phhnihao@126.com
Principal Investigator: Tong-chong Zhou, M.D
Sub-Investigator: Hai-hua Peng, M.D
First Affiliated Hospital of Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Bi-Xiu Wen, M.D +86-18903056780 wenbix@mail.sysu.edu.cn
Contact: Cheng-tao Wang, M.D +86-13711132767 ct_wang@163.com
Principal Investigator: Bi-Xiu Wen, M.D
Sub-Investigator: Cheng-tao Wang, M.D
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Yuan-hong Gao, M.D +86-13560182168 gaoyh@sysucc.org.cn
Contact: Hui Chang, M.D +86-13480295989 changhui@sysucc.org
Principal Investigator: Yuan-hong Gao, M.D
Sub-Investigator: Li-xia Lu, M.D
Sub-Investigator: Qing Liu, Ph.D
Sub-Investigator: Hui Chang, M.D
Sub-Investigator: Xin-hua Jiang, M.D
Sub-Investigator: Wei-wei Xiao, M.D
Sub-Investigator: Qi-wen Li, M.D
Sub-Investigator: Lu-ning Zhang, M.D
Sub-Investigator: Xin Yu, M.D
Shenzhen People's Hospital
Recruiting
Shenzhen, Guangdong, China, 518020
Contact: Ya-jie Liu, M.D +86-13823394075 liuyj-dr@sohu.com
Contact: Shi-hai Wu, M.D +86-13632935732 jiangxiwu84@126.com
Principal Investigator: Ya-jie Liu, M.D
Sub-Investigator: Shi-hai Wu, M.D Less << |
|
NCT01860040
|
Non-squamous Cell Non-Metastat... More >>ic Non-Small Cell Lung Cancer
Squamous Cell Non-Metastatic Non-Small Cell Lung Cancer Less << |
Phase 2 |
Terminated(Poor accrual, no da... More >>ta to analyze) Less << |
- |
United States, Arizona
... More >>
Western Regional Medical Center
Goodyear, Arizona, United States, 85338 Less << |
|
NCT00014196
|
Lung Cancer |
Phase 2 |
Terminated(This study was clos... More >>ed early due to poor accrual.) Less << |
- |
- |
|
NCT01004250
|
- |
|
Completed |
- |
- |
|
NCT00324415
|
Anal Cancer |
Phase 2 |
Completed |
- |
United States, California
... More >>
Rebecca and John Moores UCSD Cancer Center
La Jolla, California, United States, 92093-0658
UCLA Clinical AIDS Research and Education (CARE) Center
Los Angeles, California, United States, 90095-1793
United States, Massachusetts
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02215
United States, Missouri
Siteman Cancer Center at Barnes-Jewish Hospital - Saint Louis
Saint Louis, Missouri, United States, 63110
United States, New York
Albert Einstein Cancer Center at Albert Einstein College of Medicine
Bronx, New York, United States, 10461
United States, Pennsylvania
Joan Karnell Cancer Center at Pennsylvania Hospital
Philadelphia, Pennsylvania, United States, 19106
United States, Washington
Benaroya Research Institute at Virginia Mason Medical Center
Seattle, Washington, United States, 98101 Less << |
|
NCT01064921
|
Stage III Squamous Cell Carcin... More >>oma of the Oropharynx
Stage IV Squamous Cell Carcinoma of the Oropharynx Less << |
Phase 1 |
Completed |
- |
United States, Ohio
... More >>
The Ohio State University Comprehensive Cancer Center
Columbus, Ohio, United States, 43210 Less << |
|
NCT00329472
|
Carcinoma, Non-Small-Cell Lung |
Phase 3 |
Withdrawn(poor enrollment) |
July 2010 |
- |
|
NCT01004250
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Denmark
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Copenhagen, Denmark, 2200
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Herlev, Denmark, 2730
Germany
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Coswig, Germany, 01640
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Frankfurt, Germany, 60431
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hamburg, Germany, 22087
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hamm, Germany, 59071
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hannover, Germany, 30625
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Magdeburg, Germany, D-39120
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mannheim, Germany, 68167
Italy
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Lecce, Italy, 73100
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Padova, Italy, 35128
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Rome, Italy, 00144
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Rozzano, Italy, 20089
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Trento, Italy, 38100
Spain
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Jaen, Spain, 23007
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Madrid, Spain, 28046
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Pozuelo De Alarcon, Spain, 28223
Sweden
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Malmo, Sweden, 205 02
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Solna, Sweden, 17176 Less << |
|
NCT00746655
|
Hepatocellular Carcinoma |
Not Applicable |
Withdrawn(Unable to recruit) |
- |
United States, Pennsylvania
... More >>
UPMC Presbyterian/Montifore
Pittsburgh, Pennsylvania, United States, 15213
Hillman Cancer Center
Pittsburgh, Pennsylvania, United States, 15232
UPMC Cancer Pavilion
Pittsburgh, Pennsylvania, United States, 15232
UPMC Shadyside Radiation Oncology
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT00324415
|
- |
|
Completed |
- |
- |
|
NCT01057589
|
- |
|
Completed |
- |
- |
|
NCT01566435
|
Head and Neck Neoplasms |
Phase 2 |
Active, not recruiting |
October 31, 2023 |
United States, Missouri
... More >>
Washington University School of Medicine
Saint Louis, Missouri, United States, 63110 Less << |
|
NCT01860040
|
- |
|
Terminated(Poor accrual, no da... More >>ta to analyze) Less << |
- |
- |
|
NCT02328716
|
Peritoneal Carcinomatosis From... More >> Ovarian Cancer
Fallopian Tube Carcinoma
Primary Peritoneal Carcinoma Less << |
Phase 3 |
Recruiting |
December 2018 |
Spain
... More >> Hospital Clínico Universitario Virgen de la Arrixaca
Recruiting
El Palmar., Murcia, Spain, 30120
Contact: Pedro Cascales Campos 968369500
Principal Investigator: Pedro Cascales Campos, M.D Less << |
|
NCT02486718
|
Non-Small Cell Lung Cancer |
Phase 3 |
Active, not recruiting |
September 25, 2026 |
- |
|
NCT01057589
|
Head and Neck Neoplasms |
Phase 2 |
Completed |
- |
Belgium
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Edegem, Belgium, 2650
France
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Saint Herblain, France, 44805
Germany
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Dresden, Germany, 01307
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Essen, Germany, 45122
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hannover, Germany, 30625
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Leipzig, Germany, 04103
Italy
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Milano, Italy, 20133
Spain
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Barcelona, Spain, 08041
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Madrid, Spain, 28041
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Pamplona, Spain, 31008
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Valencia, Spain, 46014
United Kingdom
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Cardiff, South Glamorgan, United Kingdom, CF14 2TL
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
London, United Kingdom, SW3 6JJ Less << |
|
NCT01664975
|
Peripheral T-cell Lymphoma |
Phase 4 |
Completed |
- |
China, Henan
... More >> Oncology Department of The First Affiliated Hospital of Zhengzhou University
Zhengzhou, Henan, China, 450052 Less << |
|
NCT02707666
|
Pleural Mesothelioma |
Phase 1 |
Recruiting |
September 2018 |
United States, Illinois
... More >>
University of Chicago
Recruiting
Chicago, Illinois, United States, 60637
Contact: Mehwish Ahmad 773-834-1472 mahmad3@medicine.bsd.uchicago.edu
Contact: Buerkley Rose, RN 773 834 4002 brose@medicine.bsd.uchicago.edu
Principal Investigator: Hedy Kindler, M.D. Less << |
|
NCT02716961
|
Bladder Cancer
... More >> Gemcitabine and Cisplatin Chemotherapy
Epirubicin Instillation
Recurrence
Prognosis Less << |
Not Applicable |
Recruiting |
December 2020 |
China, Jiangsu
... More >> The first affiliated hospital of Nanjing Medical University
Recruiting
Nanjing, Jiangsu, China, 210000
Contact: Qiang Lu, PhD 13505196501 dxhlvqiang@163.com
Contact: Pengchao Li, PhD 13584025756 superkulian@aliyun.com Less << |
|
NCT01918124
|
Prosthesis Survival |
Phase 2 |
Completed |
- |
China, Shanghai
... More >>
Shanghai Cancer Center
Shanghai, Shanghai, China, 200000 Less << |
|
NCT01566435
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00599131
|
Cancer of Larynx |
Phase 2 |
Terminated(The study was disco... More >>ntinued prematurely due to early stopping rules.) Less << |
- |
United States, Michigan
... More >>
University of Michigan Comprehensive Cancer Center
Ann Arbor, Michigan, United States, 48109-0848 Less << |
|
NCT01664975
|
- |
|
Completed |
- |
- |
|
NCT00147147
|
Gastric Neoplasm |
Phase 3 |
Completed |
- |
Japan
... More >> Gastric Surgery Division, National Cancer Center Hospital
Chuo-ku, Tokyo, Japan, 104-0045 Less << |
|
NCT00008112
|
Cervical Cancer |
Phase 2 |
Unknown |
- |
Netherlands
... More >> Academisch Medisch Centrum
Amsterdam, Netherlands, 1105 AZ
University Hospital - Rotterdam Dijkzigt
Rotterdam, Netherlands, 3000 CA
Rotterdam Cancer Institute
Rotterdam, Netherlands, 3075 EA
Academisch Ziekenhuis Utrecht
Utrecht, Netherlands, 3584 CX Less << |
|
NCT01312350
|
Hypopharyngeal Cancer |
Phase 2 |
Unknown |
December 2016 |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of, 135-710 Less << |
|
NCT03641547
|
Oesophageal Adenocarcinoma
... More >> Squamous Cell Carcinoma
Solid Tumor Less << |
Phase 1 |
Not yet recruiting |
May 2023 |
United Kingdom
... More >> The Churchill Hospital, Oxford University Hospitals Trust
Not yet recruiting
Oxford, United Kingdom, OX3 7LE
Contact: Maria A Hawkins, MD FRCR MRCP Less << |
|
NCT01210131
|
Small Cell Lung Cancer (SCLC) |
Not Applicable |
Withdrawn |
August 2014 |
- |
|
NCT01569919
|
Malignant
Ple... More >>ural
Mesothelioma Less << |
Phase 2 |
Unknown |
December 2014 |
United Kingdom
... More >> Velindre Cancer Centre
Recruiting
Cardiff, South Wales, United Kingdom, CF14 2TL
Sub-Investigator: Mick Button Less << |
|
NCT00599131
|
- |
|
Terminated(The study was disco... More >>ntinued prematurely due to early stopping rules.) Less << |
- |
- |
|
NCT00509561
|
Esophageal Cancer |
Phase 2
Phase 3 |
Unknown |
- |
- |
|
NCT01487226
|
Cervical Cancer |
Phase 2 |
Unknown |
January 2015 |
Korea, Republic of
... More >>
KGOG
Recruiting
Seoul, Korea, Republic of
Contact: Eunkyung Park 8225125420 koreagynonco@gmail.com Less << |
|
NCT02575547
|
- |
|
Active, not recruiting |
December 2020 |
China, Guangdong
... More >>
Cancer Center, Sun Yat-sen University
Guangzhou, Guangdong, China Less << |
|
NCT00537875
|
Nausea
Vomiti... More >>ng Less << |
Not Applicable |
Completed |
- |
Iran, Islamic Republic of
... More >>
National Research Institute of Tuberculosis and Lung Disease (NRITLD)
Tehran, Iran, Islamic Republic of, P.O: 19575/154 Less << |
|
NCT01659554
|
Recurrent Ovarian Cancer
... More >> Fallopian Tube Cancer Less << |
Phase 2 |
Terminated(Principal Investiga... More >>tor left institution) Less << |
- |
United States, New York
... More >>
Columbia University Medical Center
New York, New York, United States, 10032 Less << |
|
NCT01880359
|
Locally Advanced Head and Neck... More >> HPV Negative Squamous Cell Cancers Less << |
Phase 3 |
Active, not recruiting |
December 2020 |
Australia
... More >> Royal Brisbane And Women's Hospital
Brisbane, Australia, QLD 4029
Princess Alexandra Hospital - University Of Queensland
Brisbane, Australia, QLD 4102
Royal North Shore Hospital
St Leonards, Australia, NSW 2065
Belgium
Hopitaux Universitaires Bordet-Erasme - Institut Jules Bordet
Brussels, Belgium, 1000
Cliniques Universitaires Saint-Luc
Brussels, Belgium, 1200
U.Z. Leuven - Campus Gasthuisberg
Leuven, Belgium, 3000
France
Centre Georges-Francois-Leclerc
Dijon, France, 21079
CHU de Tours - Hopital Bretonneau
Tours, France, 37044
Institut Gustave Roussy
Villejuif, France, 94805
Germany
Charite - Universitaetsmedizin Berlin - Campus Virchow-Klinikum
Berlin, Germany
Ludwig-Maximilians-Universitaet Muenchen - Klinikum der Universitaet Muenchen - Campus Grosshadern
Muenchen, Germany, 81377
Netherlands
Vrije Universiteit Medisch Centrum
Amsterdam, Netherlands, 7007MB
Radboud University Medical Center Nijmegen
Nijmegen, Netherlands, 6500 H
Poland
Medical University Of Gdansk
Gdansk, Poland, 80 211
The Great Poland Cancer Centre
Poznan, Poland
Maria Sklodowska-Curie Memorial Cancer Centre
Warsaw, Poland
Lower Silesian Oncology Centre
Wrocław, Poland
Switzerland
Hôpitaux universitaires de Genève - HUG - site de Cluse-Roseraie
Geneva, Switzerland, 1211
UniversitaetsSpital Zurich
Zurich, Switzerland, 8091 Less << |
|
NCT03298763
|
Adenocarcinoma of Lung |
Phase 1
Phase 2 |
Not yet recruiting |
March 1, 2024 |
United Kingdom
... More >> University College London Hospital
Not yet recruiting
London, United Kingdom Less << |
|
NCT00004033
|
Malignant Mesothelioma |
Phase 2 |
Completed |
- |
United States, New York
... More >>
NYU School of Medicine's Kaplan Comprehensive Cancer Center
New York, New York, United States, 10016
United States, Texas
University of Texas - MD Anderson Cancer Center
Houston, Texas, United States, 77030-4009 Less << |
|
NCT01659554
|
- |
|
Terminated(Principal Investiga... More >>tor left institution) Less << |
- |
- |
|
NCT00520091
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00109928
|
- |
|
Completed |
- |
- |
|
NCT00352105
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, Ohio
... More >>
Cleveland Clinic Taussig Cancer Institute, Case Comprehensive Cancer Center
Cleveland, Ohio, United States, 44195 Less << |
|
NCT00352105
|
- |
|
Completed |
- |
- |
|
NCT03701451
|
Nasopharyngeal Carcinoma |
Phase 1
Phase 2 |
Recruiting |
December 1, 2020 |
China, Guangxi
... More >> Guilin Hospital of Traditional Chinese Medicine
Recruiting
Guilin, Guangxi, China, 541002
Contact: Ruihua Xiong, Ph.D.
Guilin Medical University
Recruiting
Guilin, Guangxi, China
Contact: Rongjun Zhang, Mster +8618977330177 41974954@qq.com
Contact: Wei Jiang, Ph.D +8613788561863 Less << |
|
NCT01005680
|
Non Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
China
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Beijing, China, 100730
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Changchun, China, 130012
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Dalian, China, 116023
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Guang Zhou, China, 510080
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Nanjing, China, 210002
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Nanning, China, 530000
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Shanghai, China, 200433
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Sichuan, China, 610041 Less << |
|
NCT00379665
|
Carcinoma, Non-Small-Cell Lung |
PHASE2 |
COMPLETED |
2025-09-12 |
Cancer Treatment Centers of Am... More >>erica ? Southwestern Regional Medical Center, Tulsa, Oklahoma, 74133, United States Less << |
|
NCT00109928
|
Lymphoma |
Phase 2 |
Completed |
- |
- |
|
NCT03123445
|
Non Small Cell Lung Cancer |
Phase 2 |
Not yet recruiting |
September 2019 |
- |
|
NCT01005680
|
- |
|
Completed |
- |
- |
|
NCT01216111
|
- |
|
- |
- |
- |
|
NCT02986503
|
- |
|
Completed |
- |
Germany
... More >> Cooperative Osteosarcoma Study Group
Stuttgart, Germany, D-70176
Italy
Istituti Ortopedici Rizzoli - Unit of Chemotherapy of Muscoloskeletal Tumors
Bologna, Italy, 40136
Sweden
Scandinavian Sarcoma Group
Lund, Sweden, SE-221 85 Less << |
|
NCT00191490
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Completed |
- |
Italy
... More >> For additional information regarding investigative sites for this trial, please call 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559), Monday-Friday, 9:00 am to 5:00 pm Eastern time (UTC/GMT - 5 hours, EST) or speak with your physician
Sesto Fiorentino, Florence, Italy, 50019 Less << |
|
NCT02429687
|
Ovarian Germ Cell Cancer
... More >> Ovarian Neoplasms
Ovarian Cancer Less << |
Phase 3 |
Recruiting |
May 2024 |
China, Shandong
... More >>
Qilu Hospital of Shandong University
Recruiting
Jinan, Shandong, China, 250012
Contact: Beihua Kong, MD. PhD. +8618560081888 kongbeihua@sdu.edu.cn Less << |
|
NCT00051506
|
Lung Neoplasms |
Phase 2 |
Completed |
- |
United States, Arkansas
... More >>
For additional information regarding investigative sites for this trial, contact the Clinical Trials Support Center at 1-877-CTLILLY(1-877-285-4559) or speak with your personal physician.
Springdale, Arkansas, United States
United States, California
For additional information regarding investigative sites for this trial, contact the Clinical Trials Support Center at 1-877-CTLILLY(1-877-285-4559) or speak with your personal physician.
Los Angeles, California, United States
United States, Florida
For additional information regarding investigative sites for this trial, contact the Clinical Trials Support Center at 1-877-CTLILLY(1-877-285-4559) or speak with your personal physician.
Fort Myers, Florida, United States
For additional information regarding investigative sites for this trial, contact the Clinical Trials Support Center at 1-877-CTLILLY(1-877-285-4559) or speak with your personal physician.
Miami, Florida, United States
United States, New York
For additional information regarding investigative sites for this trial, contact the Clinical Trials Support Center at 1-877-CTLILLY(1-877-285-4559) or speak with your personal physician.
Latham, New York, United States
United States, North Carolina
For additional information regarding investigative sites for this trial, contact the Clinical Trials Support Center at 1-877-CTLILLY(1-877-285-4559) or speak with your personal physician.
Burlington, North Carolina, United States
For additional information regarding investigative sites for this trial, contact the Clinical Trials Support Center at 1-877-CTLILLY(1-877-285-4559) or speak with your personal physician.
Chapel Hill, North Carolina, United States
United States, South Carolina
For additional information regarding investigative sites for this trial, contact the Clinical Trials Support Center at 1-877-CTLILLY(1-877-285-4559) or speak with your personal physician.
Charleston, South Carolina, United States Less << |
|
NCT00454519
|
Stomach Neoplasms
... More >> Colorectal Neoplasms
Neoplasm Metastasis
Mesothelioma Less << |
Phase 2 |
Unknown |
- |
China, Hubei
... More >> Cancer Center of Wuhan University & Department of Oncology, Zhongnan Hospital of Wuhan University
Recruiting
Wuhan, Hubei, China, 430071
Contact: Yan Li, M.D., Ph.D +86-27-67813152 ext 3152 liyansd2@163.com
Contact: Yonemura Yutaka, M.D., Ph.D +81-072-433-2131 y.yonemura@coda.ocn.ne.jp
Sub-Investigator: Guo-Liang Yang, M.D
Sub-Investigator: Fu-Lin Cheng, M.D.
Principal Investigator: Yan Li, M.D., Ph.D
Sub-Investigator: Mao-Hui Feng, M.D., Ph.D
Sub-Investigator: Shibo Masaya, M.D. Less << |
|
NCT02429700
|
Ovarian Sex Cord Stromal Tumor... More >>
Ovarian Neoplasms
Ovarian Cancer Less << |
Phase 3 |
Recruiting |
May 2024 |
China, Shandong
... More >>
Qilu Hospital of Shandong University
Recruiting
Jinan, Shandong, China
Contact: Beihua Kong, MD. PhD. Less << |
|
NCT01903018
|
Radiation Induced Mucositis in... More >> Head and Neck Cancer Less << |
Phase 2 |
Completed |
- |
India
... More >> Bharat Cancer Hospital & Research Institute
Surat, Gujarat, India, 395010
Sri Venkateshwara Hospitals
Bangalore, Karnataka, India, 560068
Mazumdar Shaw Cancer Center
Bangalore, Karnataka, India, 560099
Tata Memorial Hospital
Mumbai, Maharashtra, India, 400012
Curie Manavta Cancer Centre
Nashik, Maharashtra, India, 422004
Ruby Hall Clinic
Pune, Maharashtra, India, 411001
Meenakshi Mission Hosp. & Res. Centre
Madurai, Tamil Nadu, India, 625107 Less << |
|
NCT01088581
|
Hepatocellular Carcinoma |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Severance Hospital
Seoul, Korea, Republic of, 120-752 Less << |
|
NCT03304756
|
Triple Negative Breast Cancer ... More >>Patients Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00101842
|
Carcinoma, Transitional Cell |
Phase 1
Phase 2 |
Completed |
- |
United States, Texas
... More >>
For additional information regarding investigative sites for this trial, contact the Clinical Trials Support Center at 1-877-CTLILLY (1-877-285-4559) or speak with your personal physician.
Dallas, Texas, United States Less << |
|
NCT02949700
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 1
Phase 2 |
Recruiting |
December 2021 |
United States, Texas
... More >>
Baylor College of Medicine
Recruiting
Houston, Texas, United States, 77030
Contact: Vlad Sandulache, MD, PhD 713-798-7218 vlad.sandulache@bcm.edu
Michael E. DeBakey Veterans Affairs Medical Center
Recruiting
Houston, Texas, United States, 77030
Contact: Vlad Sandulache, MD, PhD 713-798-7218 vlad.sandulache@bcm.edu
Harris Health System - Smith Clinic
Recruiting
Houston, Texas, United States, 77054
Contact: Vlad Sandulache, MD, PhD 713-798-7218 vlad.sandulache@bcm.edu Less << |
|
NCT00003621
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 2 |
Completed |
- |
United States, Arizona
... More >>
CCOP - Scottsdale Oncology Program
Scottsdale, Arizona, United States, 85259-5404
United States, Illinois
CCOP - Illinois Oncology Research Association
Peoria, Illinois, United States, 61602
United States, Iowa
CCOP - Cedar Rapids Oncology Project
Cedar Rapids, Iowa, United States, 52403-1206
CCOP - Iowa Oncology Research Association
Des Moines, Iowa, United States, 50309-1016
Siouxland Hematology-Oncology
Sioux City, Iowa, United States, 51101-1733
United States, Kansas
CCOP - Wichita
Wichita, Kansas, United States, 67214-3882
United States, Louisiana
CCOP - Ochsner
New Orleans, Louisiana, United States, 70121
United States, Minnesota
CCOP - Duluth
Duluth, Minnesota, United States, 55805
Mayo Clinic Cancer Center
Rochester, Minnesota, United States, 55905
CentraCare Clinic
Saint Cloud, Minnesota, United States, 56303
CCOP - Metro-Minnesota
Saint Louis Park, Minnesota, United States, 55416
United States, Nebraska
CCOP - Missouri Valley Cancer Consortium
Omaha, Nebraska, United States, 68131
United States, North Dakota
Quain & Ramstad Clinic, P.C.
Bismarck, North Dakota, United States, 58501
Altru Health Systems
Grand Forks, North Dakota, United States, 58201
United States, Ohio
CCOP - Toledo Community Hospital Oncology Program
Toledo, Ohio, United States, 43623-3456
United States, Pennsylvania
CCOP - Geisinger Clinic and Medical Center
Danville, Pennsylvania, United States, 17822-2001
United States, South Dakota
Rapid City Regional Hospital
Rapid City, South Dakota, United States, 57709
CCOP - Sioux Community Cancer Consortium
Sioux Falls, South Dakota, United States, 57105-1080 Less << |
|
NCT00777491
|
Bladder Cancer |
Phase 2 |
Active, not recruiting |
January 2023 |
United States, Georgia
... More >>
Georgia Cancer Center for Excellence at Grady Memorial Hospital
Atlanta, Georgia, United States, 30303
Winship Cancer Institute of Emory University
Atlanta, Georgia, United States, 30322
United States, Idaho
Saint Alphonsus Cancer Care Center at Saint Alphonsus Regional Medical Center
Boise, Idaho, United States, 83706
United States, Illinois
Cancer Institute at St. John's Hospital
Springfield, Illinois, United States, 62702
United States, Indiana
Parkview Regional Cancer Center at Parkview Health
Fort Wayne, Indiana, United States, 46805
United States, Maryland
St. Agnes Hospital Cancer Center
Baltimore, Maryland, United States, 21229
United States, Massachusetts
Hudner Oncology Center at Saint Anne's Hospital - Fall River
Fall River, Massachusetts, United States, 02721
United States, Michigan
Saint Joseph Mercy Cancer Center
Ann Arbor, Michigan, United States, 48106-0995
University of Michigan Comprehensive Cancer Center
Ann Arbor, Michigan, United States, 48109-0942
West Michigan Cancer Center
Kalamazoo, Michigan, United States, 49007-3731
Canada, Quebec
McGill Cancer Centre at McGill University
Montreal, Quebec, Canada, H2W 1S6 Less << |
|
NCT00425191
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Italy
... More >> Sanofi-Aventis
Milan, Italy Less << |
|
NCT01525771
|
Stage IV Gastric Cancer With M... More >>etastasis Less << |
Phase 1
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Asan Medical Center
Seoul, Korea, Republic of Less << |
|
NCT00239304
|
Head and Neck Neoplasms |
Phase 1
Phase 2 |
Completed |
- |
Finland
... More >> Research Centre
Helsinki, Finland Less << |
|
NCT03217747
|
Malignant Neoplasm of Breast
... More >> Malignant Neoplasms of Bone and Articular Cartilage
Malignant Neoplasms of Digestive Organs
Malignant Neoplasms of Eye Brain and Other Parts of Central Nervous System
Malignant Neoplasms of Female Genital Organs
Malignant Neoplasms of Ill-defined Secondary and Unspecified Sites
Malignant Neoplasms of Independent (Primary) Multiple Sites
Malignant Neoplasms of Lip Oral Cavity and Pharynx
Malignant Neoplasms of Male Genital Organs
Malignant Neoplasms of Mesothelial and Soft Tissue
Malignant Neoplasms of Respiratory and Intrathoracic Organs
Malignant Neoplasms of Thyroid and Other Endocrine Glands
Malignant Neoplasms of Urinary Tract
Neoplasms of Uncertain or Unknown Behavior Less << |
Phase 1
Phase 2 |
Recruiting |
August 2023 |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact anaing@mdanderson.org Less << |
|
NCT00006487
|
Lung Cancer |
Phase 1 |
Completed |
- |
- |
|
NCT01326468
|
Head and Neck Cancer |
Not Applicable |
Withdrawn(funding) |
- |
- |
|
NCT02845323
|
Urothelial Carcinoma
... More >> Bladder Cancer Less << |
Phase 2 |
Recruiting |
January 2020 |
United States, Maryland
... More >>
Johns Hopkins Hospital
Recruiting
Baltimore, Maryland, United States, 21205
Contact: Noah Hahn, MD 443-287-2886 nhahn4@jhmi.edu
Contact: Rana Sullivan, RN 410-614-6337 tomalra@jhmi.edu Less << |
|
NCT01023347
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Chungnam National University Hospital
Daejeon, Jung-gu, Korea, Republic of, 301-721 Less << |
|
NCT01857726
|
Hepatocellular Carcinoma |
Not Applicable |
Withdrawn(No participants were... More >> enrolled) Less << |
December 2016 |
Korea, Republic of
... More >>
Seoul National University Hospital
Seoul, Korea, Republic of Less << |
|
NCT03015727
|
Locally Advanced Nasopharyngea... More >>l Carcinoma Less << |
Phase 3 |
Recruiting |
December 2024 |
China, Zhejiang
... More >>
Xiaozhong Chen
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: Xiaozhong Chen, MD +86-571-88128202 cxzfyun@sina.com Less << |
|
NCT00699517
|
Sarcoma |
Phase 3 |
Completed |
- |
- |
|
NCT00505635
|
- |
|
Terminated(Slow accrual) |
- |
- |
|
NCT03272217
|
Urothelial Carcinoma |
Phase 2 |
Active, not recruiting |
April 2021 |
United States, Minnesota
... More >>
HealthPartners Institute
Minneapolis, Minnesota, United States, 55440
United States, New York
New York University Clinical Cancer Center
New York, New York, United States, 10016
United States, Tennessee
West Cancer Center University of Tennessee
Memphis, Tennessee, United States, 38120 Less << |
|
NCT01902875
|
- |
|
Unknown |
- |
China, Jiangsu
... More >> Nanjing General Hospital of Nanjing Military Command(Jinling Hospital)
Recruiting
Nanjing, Jiangsu, China, 210002
Contact: Longbang Chen, M.D. dr.chenlb@nju.edu.cn
Principal Investigator: Longbang Chen, M.D. Less << |
|
NCT00217698
|
Lung Cancer |
Phase 2 |
Completed |
- |
Switzerland
... More >> Saint Claraspital AG
Basel, Switzerland, CH-4016
Universitaetsspital-Basel
Basel, Switzerland, CH-4031
Inselspital Bern
Bern, Switzerland, CH-3010
Kantonsspital Bruderholz
Bruderholz, Switzerland, CH-4101
Spitaeler Chur AG
Chur, Switzerland, CH-7000
Hopital Cantonal Universitaire de Geneve
Geneva, Switzerland, CH-1211
Centre Hospitalier Universitaire Vaudois
Lausanne, Switzerland, CH-1011
Kantonsspital
Liestal, Switzerland, CH-4410
Kantonsspital - St. Gallen
St. Gallen, Switzerland, CH-9007
Regionalspital
Thun, Switzerland, 3600
Kantonsspital Winterthur
Winterthur, Switzerland, CH-8400
City Hospital Triemli
Zurich, Switzerland, 8063
Klinik Hirslanden
Zurich, Switzerland, CH-8008
UniversitaetsSpital Zuerich
Zurich, Switzerland, CH-8091 Less << |
|
NCT02764593
|
Head and Neck Squamous Cell Ca... More >>rcinoma (HNSCC) Less << |
Phase 1 |
Active, not recruiting |
March 2021 |
United States, California
... More >>
Stanford Cancer Institute
Palo Alto, California, United States, 94304
United States, Florida
University of Florida Cancer Center at Orlando Health
Orlando, Florida, United States, 32806
United States, Georgia
Emory University/Winship Cancer Institute
Atlanta, Georgia, United States, 30322
United States, Kentucky
University of Louisville
Louisville, Kentucky, United States, 40202
United States, Ohio
University Hospitals Cleveland Medical Center
Cleveland, Ohio, United States, 44106
Ohio State University
Columbus, Ohio, United States, 43210
United States, Oregon
Providence Portland Medical Center
Portland, Oregon, United States, 97213
United States, Pennsylvania
UPMC - Shadyside Hospital
Pittsburgh, Pennsylvania, United States, 15232
United States, Texas
MD Anderson Cancer Center
Houston, Texas, United States, 77030
United States, Virginia
Inova Fairfax Hospital
Falls Church, Virginia, United States, 22042
United States, Wisconsin
University of Wisconsin Hospital and Clinics
Madison, Wisconsin, United States, 53792 Less << |
|
NCT01003964
|
Non Small Cell Lung Cancer |
Phase 2 |
Unknown |
December 2014 |
Korea, Republic of
... More >>
National Cancer Center, Korea
Goyang-si, Gyeonggi-do, Korea, Republic of Less << |
|
NCT01081041
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00331760
|
Cervical Cancer
... More >> Endometrial Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT02969083
|
Upper Tract Urothelial Carcino... More >>ma Less << |
Phase 2 |
Recruiting |
October 2023 |
Netherlands
... More >> Leiden University Medical Centre
Recruiting
Leiden, South Holland, Netherlands Less << |
|
NCT00309972
|
Lung Cancer |
Phase 3 |
Completed |
- |
United Kingdom
... More >> Clatterbridge Centre for Oncology
Merseyside, England, United Kingdom, CH63 4JY Less << |
|
NCT00843934
|
Carcinoma, Hepatocellular |
Phase 2
Phase 3 |
Unknown |
February 2015 |
Japan
... More >> Department of Digestive Surgery, Nihon University School of Medicine
Recruiting
Itabashi, Tokyo, Japan, 173-8610
Contact: Masashi Fujii, MD +81332931711 ext 207 masashi.fujii@gioncology.jp
Contact: Tadatoshi Takayama, MD +81339728111 ext 2471 Takayama.Tadatoshi@nihon-u.ac.jp Less << |
|
NCT00331760
|
- |
|
Completed |
- |
- |
|
NCT00215644
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 2 |
Completed |
- |
Germany
... More >> Research Site
Essen, Germany
Research Site
Hamburg, Germany
Research Site
Oldenburg, Germany
Research Site
Recklinghausen, Germany
Spain
Research Site
A Coruna, Spain
Research Site
Barcelona, Spain
Research Site
Cadiz, Spain
Research Site
Valencia, Spain
Switzerland
Research Site
Bern, Switzerland
Research Site
Geneva, Switzerland
Research Site
Lausanne, Switzerland
Research Site
St. Gallen, Switzerland
United Kingdom
Research Site
Northwood, Middlesex, United Kingdom
Research Site
Bournemouth, United Kingdom
Research Site
Cambridge, United Kingdom
Research Site
Chelmsford, United Kingdom
Research Site
Guildford, United Kingdom
Research Site
Leicester, United Kingdom
Research Site
London, United Kingdom
Research Site
Newcastle, United Kingdom
Research Site
Northwood, United Kingdom
Research Site
Portsmouth, United Kingdom Less << |
|
NCT00215644
|
- |
|
Completed |
- |
- |
|
NCT01327235
|
Malignant Pleural Effusion
... More >> Malignant Ascites Less << |
Phase 2 |
Completed |
- |
China
... More >> The 81st Hospital of Chinese PLA
Nanjing, China
The Affiliated Hospital of Medical College, QingDao University
Qingdao, China
Fudan University Shanghai Cancer Center
Shanghai, China Less << |
|
NCT00249977
|
Solid Tumors
... More >>Cancer Less << |
Phase 1 |
Completed |
- |
United States, New Mexico
... More >>
University of New Mexico
Albuquerque, New Mexico, United States, 87131 Less << |
|
NCT03773302
|
Advanced Cholangiocarcinoma |
Phase 3 |
Not yet recruiting |
October 31, 2022 |
- |
|
NCT01081041
|
- |
|
Completed |
- |
- |
|
NCT02298595
|
Carcinoma, Squamous
... More >> Squamous Cell Carcinoma
Oropharyngeal Neoplasms
Oropharyngeal Cancer Less << |
Phase 1
Phase 2 |
Withdrawn |
August 2024 |
- |
|
NCT00588237
|
- |
|
Completed |
- |
- |
|
NCT00505635
|
Melanoma |
Phase 2 |
Terminated(Slow accrual) |
- |
United States, Texas
... More >>
U.T.M.D. Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT01204307
|
Advanced Non-Squamous Non-Smal... More >>l Cell Lung Cancer Less << |
Phase 2 |
Completed |
- |
Taiwan
... More >> Chang Gung Memorial Hospital, Kaohsiung Branch
Kaohsiung, Taiwan
Chang Gung Memorial Hospital
Taipei, Taiwan, 10507
McKay Memorial Hospital
Taipei, Taiwan
Shin Kong Wu Ho-Su Memorial Hospital
Taipei, Taiwan Less << |
|
NCT01453660
|
- |
|
Active, not recruiting |
October 2019 |
United States, New York
... More >>
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT00588237
|
Ovarian Cancer
... More >> Primary PERITONEUM
Fallopian Tube Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT02973386
|
Local Advanced High Risk Nasop... More >>haryngeal Carcinoma Less << |
Phase 2
Phase 3 |
Recruiting |
December 2024 |
China, Guangdong
... More >>
Cancer Center, Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China
Contact: Zhao chong, M.D +86-13902206160 zhaochong@sysucc.org.cn Less << |
|
NCT02725918
|
Non-small Cell Lung Cancer |
Phase 2 |
Recruiting |
June 2020 |
China, Guangdong
... More >>
Guangdong Lung cancer institute
Recruiting
Guangzhou, Guangdong, China, 510080
Contact: Qing Zhou, MD 86 13544561166 gzzhouqing@126.com Less << |
|
NCT01187615
|
Small Cell Lung Carcinoma |
Phase 1 |
Terminated |
- |
United States, Connecticut
... More >>
New Haven, Connecticut, United States, 06519
United States, Nevada
Las Vegas, Nevada, United States, 89135-3011
United States, New York
New York, New York, United States, 10065
United States, North Carolina
Chapel Hill, North Carolina, United States, 27599 Less << |
|
NCT00064259
|
Adenocarcinoma of the Esophagu... More >>s
Adenocarcinoma of the Gastroesophageal Junction
Diffuse Adenocarcinoma of the Stomach
Intestinal Adenocarcinoma of the Stomach
Mixed Adenocarcinoma of the Stomach
Recurrent Esophageal Cancer
Recurrent Gastric Cancer
Squamous Cell Carcinoma of the Esophagus
Stage III Esophageal Cancer
Stage IIIA Gastric Cancer
Stage IIIB Gastric Cancer
Stage IIIC Gastric Cancer
Stage IV Esophageal Cancer
Stage IV Gastric Cancer Less << |
Phase 1
Phase 2 |
Terminated(Discontinued develo... More >>pment of G3139 (oblimersen)) Less << |
- |
United States, New York
... More >>
Montefiore Medical Center
Bronx, New York, United States, 10467-2490 Less << |
|
NCT00716391
|
Head and Neck Cancer |
Phase 3 |
Unknown |
December 2013 |
Spain
... More >> Hospital Puerta del Mar
Recruiting
Almeria, Almería, Spain
Principal Investigator: Esperanza Arriola, MD
Hospital Germans Trias i Pujol
Recruiting
Badalona, Barcelona, Spain, 08916
Principal Investigator: Beatriz Cirauqui, MD
Hospital Durán i Reynals
Recruiting
Hospitalet de Ll., Barcelona, Spain
Principal Investigator: Ricard Mesía, MD
Hospital de Manresa
Recruiting
Manresa, Barcelona, Spain
Principal Investigator: Esther Casado
Hospital Mútua de Terrassa
Recruiting
Terrassa, Barcelona, Spain
Principal Investigator: Romà Bastús, MD
Hospital Son Dureta
Recruiting
Palma de Mallorca, Mallorca, Spain, 07014
Principal Investigator: José Fuster, MD
Hospital de Sagunto
Recruiting
Sagunto, Valencia, Spain
Principal Investigator: José M Vicent, MD
Hospital general Universitario
Recruiting
Alicante, Spain
Principal Investigator: Juan L. Martí Ciriquián, MD
Hospital Clínic i Provincial de Barcelona
Recruiting
Barcelona, Spain, 08036
Principal Investigator: Juan J Grau, MD
Hospital de Basurto
Recruiting
Bilbao, Spain
Principal Investigator: Elena Galve, MD
Hospital General Yagüe
Recruiting
Burgos, Spain
Principal Investigator: Carlos García Girón, MD
Hospital San Pedro de Alcántara
Terminated
Cáceres, Spain
Hospital Dr. Trueta (ICO Girona)
Recruiting
Girona, Spain, 17007
Principal Investigator: José Rubio, MD
Oncogranada
Terminated
Granada, Spain, 18014
H. Virgen de las Nieves
Recruiting
Granada, Spain
Principal Investigator: Antonio L Irigoyen
Hospital General de Jaén
Not yet recruiting
Jaén, Spain
Principal Investigator: Miguel A Moreno, MD
Hospital Xeral Calde
Recruiting
Lugo, Spain
Principal Investigator: J. Ramón Mel, MD
Hospital Universitari Arnau de Vilanova
Recruiting
Lérida, Spain, 25198
Principal Investigator: Santiaago Miguel Sanz, MD
Hospital Clínico San Carlos
Recruiting
Madrid, Spain, 28040
Principal Investigator: José A García, MD
Hospital Universitario 12 de Octubre
Recruiting
Madrid, Spain, 28041
Principal Investigator: Ricardo Hitt, MD
Clínica Quirón
Recruiting
Madrid, Spain
Principal Investigator: Ramón Pérez Carrión, MD
Fundación Jiménez Díaz
Recruiting
Madrid, Spain
Principal Investigator: Victoria Casado, MD
Hospital Gregorio Marañon
Recruiting
Madrid, Spain
Principal Investigator: Yolanda Escobar, MD
Hospital La Paz
Recruiting
Madrid, Spain
Principal Investigator: Beatriz Castelo, MD
Hospital Son Llàtzer
Recruiting
Mallorca, Spain
Principal Investigator: Belén González, MD
Hospital Universitario Central de Asturias
Recruiting
Oviedo, Spain, 33006
Principal Investigator: Enrique estrada, MD
Hospital Universitario de Salamanca
Recruiting
Salamanca, Spain, 37007
Sub-Investigator: Juan J Cruz, MD
Principal Investigator: Elvira del Barco Morillo, MD
Sub-Investigator: Juan Carlos Adansa Klain, MD
Hospital Universitario Marques de Valdecilla
Recruiting
Santander, Spain, 39008
Principal Investigator: Almudena García Castaño, MD
Hospital Clínico de Santiago
Not yet recruiting
Santiago de Compostela, Spain
Principal Investigator: Jorge J García, MD
Hospital General de Segovia
Recruiting
Segovia, Spain
Principal Investigator: Beatriz Esteban, MD
Hospital Arnau de Vilanova
Recruiting
Valencia, Spain, 46015
Principal Investigator: Oscar Juan Vidal
Hospital General Universitario
Recruiting
Valencia, Spain
Principal Investigator: Alfonso Berrocal, MD
Hospital La Fe
Recruiting
Valencia, Spain
Contact: Miguel Pastor Borgoñon, MD
Principal Investigator: Miguel Pastor Borgoñon, MD
Hospital Xeral Cies
Recruiting
Vigo, Spain, 36024
Principal Investigator: Javier Castellanos, MD
Hospital de Meixoeiro
Recruiting
Vigo, Spain, 36200
Principal Investigator: Carlos Grande, MD
Hospital Provincial de Zamora
Recruiting
Zamora, Spain, 49021
Principal Investigator: Yolanda López, MD
Hospital Clínico Lozano Blesa
Recruiting
Zaragoza, Spain
Principal Investigator: Julio Lambea, MD
Hospital Miguel Servet
Recruiting
Zaragoza, Spain
Principal Investigator: Javier Martínez Trufero, MD
Hospital Nuestra Señora de Sonsoles
Recruiting
Ávila, Spain
Contact: Elena Filipovich, MD
Principal Investigator: Elena Filipovich, MD Less << |
|
NCT03002064
|
Esophagus Cancer
... More >> Chemotherapy Effects Less << |
Phase 3 |
Recruiting |
December 2020 |
China, Guangdong
... More >>
Cancer center of Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Rui-Hua Xu, MD, PhD 86-020-87343333 xurh@sysucc.org.cn
Contact: Miao-Zhen Qiu, MD, PhD 86-020-87342490 qiumzh@sysucc.org.cn
Sub-Investigator: Miao-zhen Qiu, MD
Principal Investigator: Rui-hua Xu, MD, PhD Less << |
|
NCT01088906
|
Carcinoma, Non Small Cell Lung |
Phase 2 |
Terminated(No safety reasons. ... More >>Low recruitment.) Less << |
- |
Spain
... More >> H. Clínica Benidorm
Benidorm, Alicante, Spain, 03501
H. General de Elche
Elche, Alicante, Spain, 03202
H. Germans Trias i Pujol
Badalona, Barcelona, Spain, 088916
H. Insular de Gran Canarias
Las Palmas de Gran Canarias, Gran Canarias, Spain, 35016
H. Universitario de Canarias
La Laguna, Tenerife, Spain, 38320
MD Anderson
Madrid, Spain, 28033
Hospital 12 de Octubre
Madrid, Spain, 28041
H. Morales Messeguer
Murcia, Spain, 30008 Less << |
|
NCT00064259
|
- |
|
Terminated(Discontinued develo... More >>pment of G3139 (oblimersen)) Less << |
- |
- |
|
NCT00337532
|
Head and Neck Cancer |
Phase 2
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Local Insitution
Seoul, Korea, Republic of
Local Institution
Seoul, Korea, Republic of Less << |
|
NCT01641172
|
Testicular Cancer |
Phase 4 |
Completed |
- |
Netherlands
... More >> University Medical Center Groningen
Groningen, Netherlands, 9713 GZ Less << |
|
NCT00003596
|
Anal Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT03366415
|
Nasopharyngeal Carcinoma |
Phase 3 |
Recruiting |
December 2023 |
China, Shanghai
... More >>
Fudan University Shanghai Cancer Center
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Chaosu Hu, MD,PhD +86 18017312302 Less << |
|
NCT00046969
|
Anemia
Cervic... More >>al Cancer Less << |
Phase 4 |
Completed |
- |
Germany
... More >> Martin Luther Universitaet
Halle, Germany, D-06097 Less << |
|
NCT03535207
|
Esophageal Cancer |
Not Applicable |
Recruiting |
April 1, 2021 |
China, Shanghai
... More >>
Shanghai Genernal Hospital
Recruiting
Shanghai, Shanghai, China, 210000
Contact: Ningning Chen, MD 37798364 ext 8119 ningcnn@163.com
Contact: yong Liu, MD 37798364 drliuyrt@163.com Less << |
|
NCT01123473
|
Adenocarcinoma of the Gastroes... More >>ophageal Junction
Gastric Cancer Less << |
Phase 2 |
Terminated(company withdrew in... More >>terest) Less << |
- |
Belgium
... More >> Institut Jules Bordet
Brussels, Belgium
U.Z. Gasthuisberg
Leuven, Belgium
Germany
Johannes Gutenberg Universitaetskliniken
Mainz, Germany
Portugal
I.P.O. Francisco Gentil - Centro De Lisboa
Lisboa, Portugal Less << |
|
NCT01784549
|
Non Small Cell Lung Cancer |
Phase 2 |
Unknown |
December 2015 |
Italy
... More >> IRCCS Azienda Ospedaliera Universitaria San Martino - IST Istituto Nazionale per la Ricerca sul Cancro
Recruiting
Genoa, Italy, 16132
Contact: Francesco Grossi, MD +393355255484 fg1965@libero.it Less << |
|
NCT00817258
|
Nasopharyngeal Carcinoma |
Phase 2 |
Unknown |
December 2009 |
China
... More >> Department of Radiation Oncology, Cancer Hospital, Fudan University
Recruiting
Shanghai, China, 200032
Contact: Jing Yuan 8621-64175590 ext 6511 skelly_sh@hotmail.com Less << |
|
NCT03699956
|
Carcinoma, Small Cell
... More >> Carcinoma, Small Cell Lung Less << |
Phase 3 |
Not yet recruiting |
May 1, 2021 |
- |
|
NCT00887549
|
- |
|
Completed |
- |
- |
|
NCT00887549
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Ireland
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Dublin, Ireland
United Kingdom
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Derby, Derbyshire, United Kingdom, DE22 3NE
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Exeter, Devon, United Kingdom, EX2 5DW
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Maidstone, Kent, United Kingdom, ME16 9QQ
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Preston, Lancashire, United Kingdom, PR2 9HT
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Liverpool, Merseyside, United Kingdom, L14 3PE
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Northwood, Middlesex, United Kingdom, HA6 2RN
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Scunthorpe, North Lincolnshire, United Kingdom, DN15 7BH
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Belfast, Northern Ireland, United Kingdom, BT9 7BL
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Nottingham, Nottinghamshire, United Kingdom, NG5 1PD
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Aberdeen, Scotland, United Kingdom, AB25 2ZN
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Wolverhampton, West Midlands, United Kingdom, WV10 0QP
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Grimsby, United Kingdom
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Manchester, United Kingdom, M23 9LT Less << |
|
NCT03389438
|
Urinary Bladder Neoplasm |
Phase 2 |
Recruiting |
January 2021 |
China
... More >> Huanxing ward, Cancer Hospital Chinese Academy of Medical Sciences
Recruiting
Beijing, China
Contact: Dong Wang, Master +86-13701017215 wangdong1199@126.com Less << |
|
NCT00003332
|
Pancreatic Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, New York
... More >>
NYU School of Medicine's Kaplan Comprehensive Cancer Center
New York, New York, United States, 10016 Less << |
|
NCT02281955
|
Carcinoma, Squamous Cell
... More >> Head and Neck Neoplasms
Oropharyngeal Neoplasms Less << |
Phase 2 |
Active, not recruiting |
November 2024 |
United States, Florida
... More >>
University of Florida
Gainesville, Florida, United States, 32610
United States, North Carolina
University of North Carolina at Chapel Hill, Department of Radiation Oncology
Chapel Hill, North Carolina, United States, 27599
Pardee Memorial Hospital
Hendersonville, North Carolina, United States, 28791
High Point Regional Health
High Point, North Carolina, United States, 27262
Rex Healthcare
Raleigh, North Carolina, United States, 27607 Less << |
|
NCT00227539
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, Washington
... More >>
Seattle Cancer Care Alliance
Seattle, Washington, United States, 98109-1023
University of Washington School of Medicine
Seattle, Washington, United States, 98195 Less << |
|
NCT00002472
|
Brain and Central Nervous Syst... More >>em Tumors
Pediatric Germ Cell Tumor
Extragonadal Germ Cell Tumor Less << |
Phase 2 |
Completed |
- |
United States, Arizona
... More >>
Mayo Clinic Scottsdale
Scottsdale, Arizona, United States, 85259
United States, Florida
Mayo Clinic - Jacksonville
Jacksonville, Florida, United States, 32224
United States, Minnesota
Mayo Clinic Cancer Center
Rochester, Minnesota, United States, 55905 Less << |
|
NCT02268695
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 2 |
Active, not recruiting |
December 2018 |
France
... More >> Institut Sainte Catherine
Avignon, France, 84082
Centre Hospitalier de la Dracénie
Draguignan, France
Centre Médical de Forcilles
Férolles-Attilly, France, 77150
Clinique des Ormeaux
Le Havre, France, 76600
Centre Hospitalier de Bretagne Sud (CHBS)
Lorient, France, 56322
Centre Léon Bérard
Lyon, France, 69008
Hôpital de la Timone
Marseille, France, 13385
ICM Val d'Aurelle, Montpellier
Montpellier, France, 34298
Centre Antoine-Lacassagne
Nice, France, 06189
Val de Grace
Paris, France, 75005
Centre Eugene Marquis
Rennes, France, 35042
Centre Henri Becquerel
Rouen, France, 76038
Institut de Cancérologie de l'Ouest (ICO) René Gauducheau
Saint Herblain, France, 44805
L'Institut de Cancérologie de Lorraine (ICL) Alexis Vautrin
Vandoeuvre les Nancy, France, 54511
Gustave Roussy
Villejuif, France, 94805
Germany
Charité Campus Benjamin Franklin
Berlin, Germany, 12203
Spain
Instituto Catalá de Oncologia (ICO)
Barcelona, Spain, 08907 Less << |
|
NCT00592501
|
Nasopharyngeal Carcinoma |
Phase 2 |
Active, not recruiting |
December 2019 |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00062322
|
Lung Cancer |
Phase 1 |
Completed |
- |
United States, Pennsylvania
... More >>
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111-2497 Less << |
|
NCT02092298
|
Gastrointestinal Cancer |
Phase 2 |
Active, not recruiting |
May 2019 |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT03534713
|
Cervical Cancer TNM Staging Re... More >>gional Lymph Nodes (N) Less << |
Phase 3 |
Not yet recruiting |
December 2024 |
- |
|
NCT02285413
|
Melanoma |
Phase 2 |
Completed |
- |
Netherlands
... More >> Radboud University Nijmegen Medical Centre
Nijmegen, Gelderland, Netherlands, 6500 HB Less << |
|
NCT00379717
|
Non-Small-Cell Lung Carcinoma |
Phase 1
Phase 2 |
Unknown |
- |
Belgium
... More >> UZ Brussel
Recruiting
Jette, Belgium, 1090
Contact: Samuel Bral, MD 0032 2 477 64 15 samuel.bral@uzbrussel.be
Contact: Nicolas Fontaine, Mr 0032 2 477 54 61 nicolas.fontaine@uzbrussel.be Less << |
|
NCT02983097
|
Diffuse Large B-cell Lymphoma ... More >>(DLBCL)
Follicular Lymphoma Grade III (FL III°)
Mantle Cell Lymphoma (MCL), Blastoid Variant
Burkitt Lymphoma (BL)
Aggressive Marginal Zone Lymphoma (MZL) Less << |
Phase 1
Phase 2 |
Terminated(Phase II no scienti... More >>fic interests are given anymore) Less << |
- |
Germany
... More >> Diakonie Krankenhaus Bremen
Bremen, Germany, 28239
Klinikum Chemnitz
Chemnitz, Germany, 09113
Universitätsklinikum Essen
Essen, Germany, 45122
Klinikum Frankfurt/Oder
Frankfurt/Oder, Germany, 15236
Universitätsklinikum Göttingen
Göttingen, Germany, 37075
Asklepios Klinik St. Georg
Hamburg, Germany, 20099
Asklepios Klinik Altona
Hamburg, Germany, 22763
Universitätsklinikum Heidelberg
Heidelberg, Germany, 69120
Universitätsklinikum des Saarlandes
Homburg, Germany, 66421
Westpfalz Klinikum
Kaiserslautern, Germany, 67665
Städtisches Klinikum Karlsruhe
Karlsruhe, Germany, 76133
LMU Klinikum München-Großhadern
München, Germany, 81377 Less << |
|
NCT01084083
|
Head and Neck Cancer
... More >> Precancerous Condition Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01084083
|
- |
|
Completed |
- |
- |
|
NCT00190554
|
Esophageal Neoplasms
... More >> Carcinoma, Squamous Cell Less << |
Phase 3 |
Terminated |
- |
Japan
... More >> Aichi Cancer Center Hospital
Nagoya,Chikusa-ku,Kanokoden,1-1, Aichi, Japan, 464-8681
Chiba University, Graduate School of Medicine
Chiba,Chuo-ku,Inohana,1-8-1, Chiba, Japan, 260-8670
Tokyo Dental College Ichikawa General Hospital
Ichikawashi,Sugano,5-11-13, Chiba, Japan, 2728513
National Cancer Center Hospital East
Kashiwa,Kashiwanoha,6-5-1, Chiba, Japan, 277-8577
National Hospital Organization Shikoku Cancer Center
Matsuyama,Horinouchi,13, Ehime, Japan, 790-0007
Kurume University School of Medicine
Kurume,Asahi-machi,67, Fukuoka, Japan, 830-0011
Hiroshima City Asa Hospital
Hiroshima,Asakitaku,KabeminamiHiroshima,2-1-1, Hiroshima, Japan, 731-0293
Iwate Medical University
Morioka,Uchimaru,19-1, Iwate, Japan, 020-8505
Tokai University School of Medicine
Isehara,Shimokasuya,143, Kanagawa, Japan, 259-1193
Kanagawa Cancer Center
Yokohama,Asahi-ku,Nakao,1-1-2, Kanagawa, Japan, 241-0815
Kyoto University Hospital
Kyoto,Sakyo-ku,Syogoinkawara,54, Kyoto, Japan, 606-8507
Niigata University Medical and Dental Hospital
Niigata,Asahimachi-dori,1-754, Niigata, Japan, 951-8520
Niigata Cancer Center Hospital
Niigata,Kawagishi-cho,2-15-3, Niigata, Japan, 951-8566
Osaka National Hospital
Osaka,Chuo-ku,Hoenzaka,2-1-14, Osaka, Japan, 540-0006
Osaka Medical Center for Cancer and Cardiovascular Diseases
Osaka,Higashinari-ku,Nakamichi,1-3-3, Osaka, Japan, 537-8511
Sizuoka Cancer Center
Sunto-gun,Nagaizumi-cho,Shimonagakubo,1007, Shizuoka, Japan, 411-8777
Tochigi Cancer Center
Utsunomiya,Yohnan,4-9-13, Tochigi, Japan, 320-0834
Juntendo University School of Medicine
Bunkyo-ku,Hongo,3-1-3, Tokyo, Japan, 113-0033
Tokyo Medical and Dental University Hospital
Bunkyo-ku,Yushima,1-5-45, Tokyo, Japan, 113-8519
National Cancer Center Hospital
Chuo-ku,Tsukiji,5-1-1, Tokyo, Japan, 104-0045
National Hospital Organization Tokyo Medical Center
Meguro-ku,Higashigaoka,2-5-1, Tokyo, Japan, 152-8902
Toranomon Hospital
Minato-ku,Toranomon,2-2-2, Tokyo, Japan, 105-8470
Tokyo Women's Medical University
Shinjuku-ku,Kawada-cho,8-1, Tokyo, Japan, 162-8666
Keio University Hospital
Shinjuku-ku,Shinanomachi,35, Tokyo, Japan, 160-8582 Less << |
|
NCT02795884
|
NSCLC |
Phase 3 |
Withdrawn(Because of reconside... More >>ration of using erlotinib(EGFR Tyrosine kinase inhibitor) as adjuvant aim) Less << |
November 2020 |
- |
|
NCT03177174
|
Drug-Related Side Effects and ... More >>Adverse Reactions Less << |
Not Applicable |
Recruiting |
June 1, 2020 |
China
... More >> Chinese PLA general hospital
Recruiting
Beijing, China
Contact: LUO yanrong, doctor +8601066936933 lyr961lyr@163.com Less << |
|
NCT00227539
|
- |
|
Completed |
- |
- |
|
NCT02985658
|
- |
|
- |
- |
United States, Washington
... More >>
Seattle Cancer Care Allliance
Seattle, Washington, United States, 98109
Contact: Jennifer Specht, MD 206-606-6329 Less << |
|
NCT00173862
|
Transitional Cell Carcinoma |
Phase 2 |
Completed |
- |
Taiwan
... More >> Department of Oncology , National Taiwan University Hospital
Taipei, Taiwan, 100 Less << |
|
NCT00166881
|
Gastric Cancer |
Phase 2 |
Completed |
- |
Taiwan
... More >> Department of Oncology, National Taiwan University Hospital
Taipei, Taiwan, 100 Less << |
|
NCT00002684
|
Bladder Cancer
... More >> Urethral Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT02088515
|
Squamous Cell Carcinoma |
Phase 4 |
Completed |
- |
China, Hunan
... More >> Hunan Xiangya Hospital
Changsha, Hunan, China
China, Jiangsu
Nanjing Military General Hospital
Nanjing, Jiangsu, China
China, Shanghai
Shanghai Chest hospital
Shanghai, Shanghai, China, 200000
China, Shanxi
Xijing Hospital
Xian, Shanxi, China Less << |
|
NCT00208936
|
Esophageal Diseases |
Phase 2 |
Terminated |
- |
United States, Georgia
... More >>
Emory University Winship Cancer Institute
Atlanta, Georgia, United States, 30322 Less << |
|
NCT00227630
|
Malignant Mesothelioma |
Phase 2 |
Completed |
- |
Belgium
... More >> Universitair Ziekenhuis Antwerpen
Edegem, Belgium, B-2650
Italy
Istituto Nazionale per la Ricerca sul Cancro
Genoa, Italy, 16132
Azienda Ospedaliera Di Parma
Parma, Italy, 43100
Universita Degli Studi di Udine
Udine, Italy, 33100
Netherlands
Sint Antonius Ziekenhuis
Nieuwegein, Netherlands, 3435 CM
Daniel Den Hoed Cancer Center at Erasmus Medical Center
Rotterdam, Netherlands, 3008 AE
United Kingdom
Princess Royal Hospital at Hull and East Yorkshire NHS Trust
Hull, England, United Kingdom, HU8 9HE
Edinburgh Cancer Centre at Western General Hospital
Edinburgh, Scotland, United Kingdom, EH4 2XU Less << |
|
NCT00801736
|
Lung Cancer |
Phase 3 |
Terminated(The antibody used d... More >>id not appear prognostic/predictive based on interim results.) Less << |
- |
United Kingdom
... More >> University College London Hospitals
London, United Kingdom Less << |
|
NCT01479504
|
Nasopharyngeal Carcinoma |
Phase 3 |
Unknown |
December 2017 |
China, Guangxi
... More >> Department of Radiotherapy,the First Affiliated Hospital of Guangxi Medical University
Recruiting
Nanning, Guangxi, China, 530021
Contact: Wang R Sheng, M.D. 8613807806008 wrsgx@yahoo.com.cn
Contact: Wu Fang, M.D. 8613978880156 96160f@163.com
Principal Investigator: Wang R Sheng, M.D.
Principal Investigator: Wu Fang, M.D. Less << |
|
NCT00335543
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
Austria
... More >> Innsbruck Universitaetsklinik
Innsbruck, Austria, A-6020
Allgemeines Krankenhaus - Universitatskliniken
Vienna, Austria, A-1090
Germany
Robert Roessle Comprehensive Cancer Center - Charite Campus Buch
Berlin, Germany, D-13125
Knappschaft Krankenhaus
Bochum, Germany, D-44892
DIAKO Ev. Diakonie Krankenhaus gGmbH
Bremen, Germany, D-28239
Krankenhaus Dresden - Friedrichstadt
Dresden, Germany, D-01008
Universitaet Erlangen
Erlangen, Germany, D-91054
Arbeitsgruppe Lebermetastasen und Tumoren in der Chirurgischen Arbeitsgemeinschaft Onkologie
Frankfurt, Germany, 60590
Klinik am Eichert
Goeppingen, Germany, D-73035
Chirurgische Universitaetsklinik
Heidelberg, Germany, D-69120
Universitaet Leipzig
Leipzig, Germany, D-04103
Staedtisches Klinikum Magdeburg
Magdeburg, Germany, D-39130
Klinikum der Universitaet Muenchen - Grosshadern Campus
Munich, Germany, D-81377
Klinikum Nuernberg - Klinikum Nord
Nuremberg, Germany, D-90419
Klinikum der Universitaet Regensburg
Regensburg, Germany, D-93053
Universitaetsklinikum Tuebingen
Tuebingen, Germany, D-72076
Switzerland
Kantonsspital - St. Gallen
St. Gallen, Switzerland, CH-9007 Less << |
|
NCT03663205
|
Non-Small Cell Lung Cancer |
Phase 3 |
Recruiting |
September 23, 2020 |
China
... More >> Shanghai Chest Hospital
Recruiting
Shanghai, China Less << |
|
NCT00993655
|
Fallopian Tube Cancer
... More >> Metastatic Cancer
Ovarian Cancer
Peritoneal Cavity Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01648023
|
Unresectable Intrahepatic Chol... More >>angiocarcinoma Less << |
Phase 2 |
Recruiting |
July 2020 |
United States, Kentucky
... More >>
University of Louisville
Recruiting
Louisville, Kentucky, United States, 40202
Contact: Mary Healey, CCRC 502-629-3327 mary.healey@louisville.edu
Contact: Robert Martin, MD, PhD 502-629-3355 robert.martin@louisville.edu
Principal Investigator: Robert Martin, MD, PhD Less << |
|
NCT00002993
|
Sarcoma |
Phase 2 |
Terminated |
- |
- |
|
NCT00004865
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
United States, New Jersey
... More >>
Cooper Cancer Institute
Camden, New Jersey, United States, 08103
Kimball Medical Center
Lakewood, New Jersey, United States, 08701
Monmouth Medical Center
Long Branch, New Jersey, United States, 07740 Less << |
|
NCT02227940
|
Advanced Malignant Solid Neopl... More >>asm
ALK Positive
Metastatic Pancreatic Adenocarcinoma
Stage III Pancreatic Cancer
Stage IV Pancreatic Cancer Less << |
Phase 1 |
Recruiting |
January 11, 2019 |
United States, New York
... More >>
Roswell Park Cancer Institute
Recruiting
Buffalo, New York, United States, 14263
Contact: Roswell Park 877-275-7724 ASKRPCI@RoswellPark.org
Principal Investigator: Renuka V. Iyer Less << |
|
NCT03684811
|
Cohort 1a and 1b: Glioma
... More >> Cohort 1a and 1b: Glioblastoma Multiforme
Cohort 2a and 2b: Hepatobiliary Tumors (Hepatocellular Carcinoma, Bile Duct Carcinoma, Intrahepatic Cholangiocarcinoma, Other Hepatobiliary Carcinomas)
Cohort 3a and 3b: Chondrosarcoma
Cohort 4a and 4b: Intrahepatic Cholangiocarcinoma
Cohort 5a: Other Solid Tumors With IDH1 Mutations Less << |
Phase 1
Phase 2 |
Recruiting |
April 1, 2022 |
United States, Florida
... More >>
University of Miami, Sylvester Comprehensive Cancer Center
Recruiting
Miami, Florida, United States, 33136
Contact: Tamara Leon 305-243-0865 txl351@med.miami.edu Less << |
|
NCT00002488
|
Lymphoma |
Phase 2 |
Unknown |
- |
Canada, Ontario
... More >>
Ottawa Regional Cancer Center - General Division
Ottawa, Ontario, Canada, K1H 8L6
Ottawa Regional Cancer Centre - Civic Campus
Ottawa, Ontario, Canada, K1Y 4K7 Less << |
|
NCT00113386
|
Lung Cancer |
Phase 3 |
Terminated(This study terminat... More >>ed early due to low accrual.) Less << |
- |
- |
|
NCT00993655
|
- |
|
Completed |
- |
- |
|
NCT00792701
|
Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00113386
|
- |
|
Terminated(This study terminat... More >>ed early due to low accrual.) Less << |
- |
- |
|
NCT00792701
|
- |
|
Completed |
- |
- |
|
NCT00003624
|
Cervical Cancer
... More >> Endometrial Cancer
Fallopian Tube Cancer
Neurotoxicity
Ovarian Cancer
Primary Peritoneal Cavity Cancer
Sarcoma Less << |
Phase 2 |
Terminated |
- |
United States, California
... More >>
Chao Family Comprehensive Cancer Center
Orange, California, United States, 92868
United States, District of Columbia
Vincent T. Lombardi Cancer Research Center, Georgetown University
Washington, District of Columbia, United States, 20007
United States, Georgia
Emory University Hospital - Atlanta
Atlanta, Georgia, United States, 30322
United States, Illinois
University of Chicago Cancer Research Center
Chicago, Illinois, United States, 60637
United States, Indiana
Indiana University Cancer Center
Indianapolis, Indiana, United States, 46202-5265
United States, Missouri
Washington University School of Medicine
Saint Louis, Missouri, United States, 63110
United States, North Carolina
Lineberger Comprehensive Cancer Center, UNC
Chapel Hill, North Carolina, United States, 27599-7295
United States, Ohio
Cleveland Clinic Cancer Center
Cleveland, Ohio, United States, 44195 Less << |
|
NCT00996567
|
Cancer |
Phase 2 |
Completed |
- |
Belgium
... More >> UZ Antwerpen
Antwerpen, Belgium
University Hospital Ghent
Ghent, Belgium, 9000
AZ St. Maarten
Mechelen, Belgium, 2800
Netherlands
AMC Heerlen
Heerlen, Netherlands Less << |
|
NCT02177838
|
Stage III Squamous Cell Carcin... More >>oma of the Hypopharynx
Stage III Squamous Cell Carcinoma of the Larynx
Stage III Squamous Cell Carcinoma of the Oropharynx
Stage III Verrucous Carcinoma of the Larynx
Stage IV Squamous Cell Carcinoma of the Hypopharynx
Stage IVA Squamous Cell Carcinoma of the Larynx
Stage IVA Squamous Cell Carcinoma of the Oropharynx
Stage IVA Verrucous Carcinoma of the Larynx
Stage IVB Squamous Cell Carcinoma of the Larynx
Stage IVB Squamous Cell Carcinoma of the Oropharynx
Stage IVB Verrucous Carcinoma of the Larynx
Tongue Cancer Less << |
Not Applicable |
Suspended(MedOnc turnover at t... More >>he institution) Less << |
June 2020 |
United States, New Jersey
... More >>
Rutgers Cancer Institute of New Jersey
New Brunswick, New Jersey, United States, 08903
New Jersey Medical School
Newark, New Jersey, United States, 07103 Less << |
|
NCT01820754
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
United States, North Carolina
... More >>
Duke University Medical Center
Durham, North Carolina, United States, 27710 Less << |
|
NCT01820754
|
- |
|
Completed |
- |
- |
|
NCT01797900
|
Nasopharyngeal Neoplasms |
Phase 2 |
Completed |
- |
China, Beijing
... More >> Department of Radiation Oncology, Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College
Beijing, Beijing, China, 100021 Less << |
|
NCT00776698
|
Non-Squamous Non-Small Cell Lu... More >>ng Cancer Less << |
Phase 1 |
Completed |
- |
Netherlands
... More >>
Amsterdam, Netherlands, 1007 MB
Groningen, Netherlands, 9713 GZ
Maastricht, Netherlands, 6229 HX
United Kingdom
Aberdeen, United Kingdom, AB9 2ZB
Manchester, United Kingdom, M2O 4BX Less << |
|
NCT00522886
|
Non-small Cell Lung Cancer |
Phase 1 |
Completed |
- |
Netherlands
... More >> Maastricht Radiation Oncology, MAASTRO clinic
Maastricht, Limburg, Netherlands Less << |
|
NCT00816816
|
Nasopharyngeal Carcinoma |
Phase 2 |
Unknown |
June 2010 |
China
... More >> Department of Radiation Oncology, Cancer Hospital, Fudan University
Recruiting
Shanghai, China, 200032
Contact: Jing Yuan 8621-64175590 ext 6511 skelly_sh@hotmail.com Less << |
|
NCT00002888
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT02878889
|
Nasopharyngeal Carcinoma |
Phase 2 |
Recruiting |
- |
China, Zhejiang
... More >>
Xiaozhong Chen
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: Xiaozhong Chen, MD +86-571-88122098 cxzfyun@sina.com
Zhejiang Cancer Hospital
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: Xiaozhong Chen, MD 86-571-88122098 cxzfyun@sina.com
Principal Investigator: Xiaozhong Chen, MD Less << |
|
NCT00011960
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00470340
|
Liver Neoplasms
... More >> Liver Cirrhosis
Carcinoma, Hepatocellular
Recurrence
Neoplasm Recurrence, Local Less << |
Phase 3 |
Terminated(during the study) |
- |
France
... More >> Urc La Pitie Salpetriere,
Poissy, France, 75013 Less << |
|
NCT01276730
|
Cervical Cancer |
Phase 2 |
Completed |
- |
India
... More >> Chittaranjan National Cancer Institute
Kolkata, India, 700026 Less << |
|
NCT00816855
|
Nasopharyngeal Carcinoma |
Phase 2 |
Unknown |
June 2010 |
China
... More >> Department of Radiation Oncology, Cancer Hospital, Fudan University
Recruiting
Shanghai, China, 200032
Contact: Jing Yuan 8621-64175590 ext 6511 skelly_sh@hotmail.com Less << |
|
NCT02651441
|
Lung Cancer
N... More >>on-small Cell Lung Cancer Less << |
Phase 1
Phase 2 |
Recruiting |
August 2019 |
China, Guangdong
... More >>
Affiliated Tumor Hospital of Guangzhou Medical University
Recruiting
Guangzhou, Guangdong, China, 510010
Contact: Yuan Z Wang, Professor 020-66673666 WangZY@QQ.com Less << |
|
NCT00330499
|
Transitional Cell Carcinoma of... More >> Urinary Bladder Less << |
Phase 3 |
Completed |
- |
Australia, New South Wales
... More >>
Liverpool Hospital
Liverpool, New South Wales, Australia, 1871
Calvary Mater Newcastle
Newcastle, New South Wales, Australia, 2298
Nepean Cancer Care Centre
Penrith, New South Wales, Australia, 2751
Prince of Wales Hospital
Randwick, New South Wales, Australia, 2031
Westmead Hospital
Wentworthville, New South Wales, Australia, 2145
Australia, Queensland
Mater Centre - South Brisbane
Brisbane, Queensland, Australia, 4120
Townsville Hospital
Douglas, Queensland, Australia, 4814
Royal Brisbane Hospital
Herston, Queensland, Australia, 4029
East Coast Cancer Centre
Tugun, Queensland, Australia, 4224
Princess Alexandra Hospital
Woolloongabba, Queensland, Australia, 4102
Australia, South Australia
Royal Adelaide Hospital
Adelaide, South Australia, Australia, 5000
Australia, Tasmania
Launceston General Hospital
Launceston, Tasmania, Australia, 7250
Australia, Victoria
Peter MacCallum Cancer Centre
East Melbourne, Victoria, Australia, 3002
Andrew Love Cancer Care Centre, Geelong Hospital
Geelong, Victoria, Australia, 3220
Alfred Hospital
Prahran, Victoria, Australia, 3181
Australia, Western Australia
Sir Charles Gairdner Hospital
Nedlands, Western Australia, Australia, 6009
Royal Perth Hospital
Perth, Western Australia, Australia, 6000
New Zealand
Auckland Hospital
Auckland, New Zealand, 1001
Christchurch Hospital
Christchurch, New Zealand, 4710
Dunedin Hospital
Dunedin, New Zealand
Palmerston North Hospital
Palmerston North, New Zealand
Wellington Hospital
Wellington, New Zealand, 7902 Less << |
|
NCT00215514
|
Stomach Cancer
... More >> Gastric Cancer
Gastro-esophageal Junction Cancer Less << |
Early Phase 1 |
Completed |
- |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114
Brigham and Women's Hospital
Boston, Massachusetts, United States, 02115
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00980954
|
Cervical Cancer |
Phase 3 |
Recruiting |
August 2026 |
- |
|
NCT03765918
|
Head and Neck Neoplasms |
Phase 3 |
Not yet recruiting |
January 24, 2026 |
- |
|
NCT03067610
|
Head and Neck Cancer
... More >> Carcinoma, Squamous Cell
Neoplasms, Oral
Neoplasms, Pharyngeal Less << |
Phase 2 |
Recruiting |
December 31, 2026 |
United States, Texas
... More >>
University of Texas Southwestern Medical Center - Dallas
Recruiting
Dallas, Texas, United States, 75390 Less << |
|
NCT00971932
|
- |
|
Completed |
- |
- |
|
NCT00971932
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 2 |
Completed |
- |
Japan
... More >> Research Site
Aichi, Japan
National Cancer Center East Hospital
Chiba, Japan
Research Site
Ehime, Japan
Research Site
Hokkaido, Japan
Research Site
Kanagawa, Japan
Tokai University
Kanagawa, Japan
Research Site
Osaka, Japan
Research Site
Shizuoka, Japan
Research Site
Tochigi, Japan
Research Site
Tokyo, Japan Less << |
|
NCT00634595
|
Head and Neck Squamous Carcino... More >>ma
Nasopharyngeal Carcinoma Less << |
Phase 2 |
Unknown |
December 2010 |
China, Guangdong
... More >>
Cancer center, Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Xubin Lin, MD, PhD linxubin@mail.sysu.edu.cn Less << |
|
NCT00686959
|
Non Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00686959
|
- |
|
Completed |
- |
- |
|
NCT00941135
|
Laryngeal Neoplasms |
Phase 2 |
Terminated |
May 2013 |
Spain
... More >> Hospital Carlos Haya
Malaga, Andalucía, Spain, 29010
Clinica Universitaria de Navarra
Pamplona, Navarra, Spain, 31008
Hospital de Navarra
Pamplona, Navarra, Spain, 31008
Fundación Jiménez Díaz
Madrid, Spain, 28040 Less << |
|
NCT00002568
|
Ovarian Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00004199
|
Lung Cancer |
Phase 3 |
Completed |
- |
United States, California
... More >>
Agouron Pharmaceuticals, Inc.
La Jolla, California, United States, 92037 Less << |
|
NCT00178698
|
Neuroendocrine Cancer
... More >> Small Cell Lung Cancer
Non-Small Cell Lung Cancer
Gastric Cancer Less << |
Phase 2 |
Unknown |
June 2014 |
United States, Texas
... More >>
Memorial Hermann Hospital
Recruiting
Houston, Texas, United States, 77030
Contact: Joan M Bull, M.D. 713-600-6820 Joan.M.Bull@uth.tmc.edu
Contact: Esperanza N Fernandez 713-500-6774 Esperanza.N.Fernandez@uth.tmc.edu
Principal Investigator: Joan M Bull, M.D. Less << |
|
NCT01214343
|
Advanced Hepatocellular Carcin... More >>oma
Carcinoma
Carcinoma, Hepatocellular
Liver Neoplasms
Neoplasms Less << |
Phase 3 |
Unknown |
September 2013 |
Japan
... More >> National Cancer Center Hospital East
Recruiting
Kashiwa, Chiba, Japan, 277-8577
Contact: Masafumi Ikeda, Dr.
Principal Investigator: Masafumi Ikeda, Dr.
Kurume University Medical Center
Recruiting
Kurume, Fukuoka, Japan, 839-0863
Contact: Masatoshi Tanaka, Professor
Principal Investigator: Masatoshi Tanaka, Professor
Ogaki Municipal Hospital
Recruiting
Ogaki, Gifu, Japan, 503-8502
Contact: Takashi Kumada, Dr
Principal Investigator: Takashi Kumada, Dr.
Sapporo Medical University
Recruiting
Sapporo, Hokkaido, Japan, 060-8556
Contact: Junji Kato, Dr.
Principal Investigator: Junji Kato, Dr.
Sapporo-Kosei General Hospital
Recruiting
Sapporo, Hokkaido, Japan, 060-8556
Contact: Takumi Ohmura, Dr.
Principal Investigator: Takumi Ohmura, Dr.
Japanese Red Cross Takamatsu Hospital
Recruiting
Takamatsu, Kagawa, Japan, 760-0017
Contact: Chikara Ogawa, Dr.
Principal Investigator: Chikara Ogawa, Dr.
Mie University Hospital
Recruiting
Tsu, Mie, Japan, 514-8507
Contact: Katsuya Shiraki, Dr.
Principal Investigator: Katsuya Shiraki, Dr.
National Hospital Organization Nagasaki Medical Center
Recruiting
Ohmura, Nagasaki, Japan, 856-8562
Contact: Hiromi Ishibashi, Dr.
Principal Investigator: Hiromi Ishibashi, Dr.
Kawasaki Medical School Hospital
Recruiting
Kurashiki, Okayama, Japan, 701-0192
Contact: Keisuke Hino, Dr.
Principal Investigator: Keisuke Hino, Dr.
Ikeda Municipal Hospital
Recruiting
Ikeda, Osaka, Japan, 563-8510
Contact: Yasuharu Imai, Dr.
Principal Investigator: Yasuharu Imai, Dr.
Kinki University Hospital
Recruiting
Osaka-Sayama, Osaka, Japan, 589-8511
Contact: Masatoshi Kudo, Professor +81-72-366-0221 ext 3149 m-kudo@med.kindai.ac.jp
Contact: Kazuomi Ueshima, Dr. +81-72-366-0221 ext 3525 kaz-ues@med.kindai.ac.jp
Principal Investigator: Masatoshi Kudo, Professor
Sub-Investigator: Kazuomi Ueshima, Dr.
Principal Investigator: Kazuto Nishio, Professor
Osaka University Hospital
Recruiting
Suita, Osaka, Japan, 565-0871
Contact: Hiroaki Nagano, Professor
Principal Investigator: Hiroaki Nagano, Professor
Kyorin University Hospital
Recruiting
Mitaka, Tokyo, Japan, 181-8611
Contact: Junji Furuse, Professor
Principal Investigator: Junji Furuse, Professor
Musashino Red Cross Hospital
Recruiting
Musashino, Tokyo, Japan, 180-8610
Contact: Namiki Izumi, Dr.
Principal Investigator: Namiki Izumi, Dr.
Juntendo University Nerima Hospital
Recruiting
Nerima, Tokyo, Japan, 177-0033
Contact: Shigehiro Kokubu, Dr.
Principal Investigator: Shigehiro Kokubu, Dr.
Yamaguchi University Hospital
Recruiting
Ube, Yamaguchi, Japan, 755-8505
Contact: Isao Sakaida, Prof
Principal Investigator: Isao Sakaida, Professor
Chiba University Hospital
Recruiting
Chiba, Japan, 260-8677
Contact: Fumihiko Kanai, Dr.
Principal Investigator: Fumihiko Kanai, Dr.
Gifu Municipal Hospital
Recruiting
Gifu, Japan, 500-8513
Contact: Eiichi Tomita, Dr.
Principal Investigator: Eiichi Tomita, Dr.
Hiroshima City Hospital
Recruiting
Hiroshima, Japan, 730-8518
Contact: Yoshiyuki Kobayashi, Dr.
Principal Investigator: Yoshiyuki Kobayashi, Dr.
Hiroshima University Hospital
Recruiting
Hiroshima, Japan, 734-8551
Contact: Hiroshi Aikata, Dr.
Principal Investigator: Hiroshi Aikata, Dr.
Kumamoto University Hospital
Recruiting
Kumamoto, Japan, 860-8556
Contact: Yutaka Sasaki, Professor
Principal Investigator: Yutaka Sasaki, Professor
Kyoto University Hospital
Recruiting
Kyoto, Japan, 606-8507
Contact: Etsuro Hatano, Professor
Principal Investigator: Etsuro Hatano, Professor
Center for Gastroenterological and Hepatological Diseases
Recruiting
Miyazaki, Japan, 880-0003
Contact: Hidemori Sakamoto, Dr.
Principal Investigator: Hidemori Sakamoto, Dr.
Saiseikai Niigata Dai-ni Hospital
Recruiting
Niigata, Japan, 950-1104
Contact: Toru Ishikawa, Dr.
Principal Investigator: Toru Ishikawa, Dr.
Niigata University Medical and Dental Hospital
Recruiting
Niigata, Japan, 951-8520
Contact: Kouhei Akazawa
Principal Investigator: Kouhei Akazawa
Okayama University Hospital
Recruiting
Okayama, Japan, 700-8558
Contact: Kazuhide Yamamoto, Professor
Principal Investigator: Kazuhide Yamamoto, Professor
Osaka Red Cross Hospital
Recruiting
Osaka, Japan, 543-8555
Contact: Ikuo Osaki, Dr.
Principal Investigator: Ikuo Osaki, Dr.
The University of Tokushima Faculty of Medicine
Recruiting
Tokushima, Japan, 770-8503
Contact: Tetsuji Takayama, Dr.
Principal Investigator: Tetsuji Takayama, Dr.
Kyoundo Hospital
Recruiting
Tokyo, Japan, 101-0062
Contact: Shuntaro Obi, Dr.
Principal Investigator: Shuntaro Obi, Dr.
National Cancer Center Hospital
Recruiting
Tokyo, Japan, 104-0045
Contact: Takushi Okusaka, Dr.
Principal Investigator: Takushi Okusaka, Dr. Less << |
|
NCT00055601
|
Bladder Cancer |
Phase 2 |
Completed |
- |
United States, Utah
... More >>
LDS Hospital
Salt Lake City, Utah, United States, 84103
Utah Cancer Specialists at UCS Cancer Center
Salt Lake City, Utah, United States, 84106 Less << |
|
NCT01683864
|
Gastric Cancer
... More >> Peritoneal Carcinomatosis Less << |
Phase 2
Phase 3 |
Terminated(Recruitment problem... More >>s) Less << |
- |
Germany
... More >> University of Tuebingen
Tuebingen, BW, Germany, 72076 Less << |
|
NCT00055601
|
- |
|
Completed |
- |
- |
|
NCT00003037
|
Lung Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00794547
|
Non Small Cell Lung Cancer |
Phase 1
Phase 2 |
Terminated |
- |
United States, Michigan
... More >>
VA Ann Arbor Healthcare System
Ann Arbor, Michigan, United States, 48105
St. Joseph Mercy Hospital
Ann Arbor, Michigan, United States, 48106
University of Michigan Comprehensive Cancer Center
Ann Arbor, Michigan, United States, 48109
United States, New York
Roswell Park Cancer Institute
Buffalo, New York, United States, 14263 Less << |
|
NCT00794547
|
- |
|
Terminated |
- |
- |
|
NCT00992199
|
Advanced Gastric Cancer |
Phase 2 |
Unknown |
December 2011 |
China, Shanghai
... More >>
Fudan University Cancer Hospital
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Jin Li, MD 86(021)64175590 ext 5108 fudanlijin@163.com
Contact: Xiaodong Zhu, MD 86(021)64175590 ext 5008 xddr001@163.com
Principal Investigator: Jin Li, MD
Sub-Investigator: Haiyi Guo, MD
Sub-Investigator: xiaodnog Zhu, MD Less << |
|
NCT01933568
|
Inoperable Locally Advanced No... More >>n Small Cell Lung Cancer Less << |
Phase 1 |
Completed |
- |
Netherlands
... More >> The Netherlands Cancer Institute
Amsterdam, Netherlands, 1066 CX Less << |
|
NCT01345279
|
- |
|
Completed |
- |
- |
|
NCT01683864
|
- |
|
Terminated(Recruitment problem... More >>s) Less << |
- |
- |
|
NCT03515538
|
Oral Mucositis |
Phase 2 |
Recruiting |
June 2020 |
United States, California
... More >>
John Wayne Cancer Institute @ Providence St. John's Health Center
Recruiting
Santa Monica, California, United States, 90401
Contact: Mini Gill, RN BSN 310-582-7437 Jaya.Gill@providence.org
Principal Investigator: Santosh Kesari, MD, PhD
United States, Colorado
Centura Health Research Center
Recruiting
Denver, Colorado, United States, 80210
Contact: Sheryl Giambartolomei, RN 303-765-3536 sherylgiambartolomei@centura.org
Principal Investigator: Seth Reiner, MD
United States, District of Columbia
George Washington University
Recruiting
Washington, District of Columbia, United States, 20037
Contact: Richard Lush, PhD 202-994-3647 rmlush3@email.gwu.edu
Principal Investigator: Sharad Goyal, MD
United States, New York
Montefiore Medical Center
Recruiting
Bronx, New York, United States, 10467
Contact: Alyssa Asaro 718-920-5636 aasaro@montefiore.org
Principal Investigator: Rafi Kabarriti, MD
United States, North Carolina
East Carolina University
Recruiting
Greenville, North Carolina, United States, 27834
Contact: Debra Peaden, CCRP peadend@ecu.edu
Principal Investigator: Brian C Muzyka, DMD, MD, MBA
United States, Ohio
University of Cincinnati Medical Center
Recruiting
Cincinnati, Ohio, United States, 45267
Contact: Vinita Takiar, MD 513-584-7661 Vinita.Takiar@UCHealth.com
Principal Investigator: Vinita Takiar, MD
Ohio State University
Recruiting
Columbus, Ohio, United States, 43210
Contact: Nancy Curtis 614-685-8029 Nancy.Curtis@osumc.edu
Principal Investigator: Marcelo Bonomi, MD
United States, Pennsylvania
Thomas Jefferson University
Recruiting
Philadelphia, Pennsylvania, United States, 19107
Contact: Dawn Poller 215-955-1964 Dawn.Poller@jefferson.edu
Principal Investigator: Voichita Bar-Ad, MD
United States, Tennessee
Ballad Health
Recruiting
Johnson City, Tennessee, United States, 37604
Contact: Anna Yakubenko 423-431-5647 Anna.Yakubenko@balladhealth.org
Principal Investigator: Kyle T. Colvett, MD Kyle T Colvett, MD Less << |
|
NCT00003237
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00045604
|
Lung Cancer |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00003298
|
- |
|
Completed |
- |
- |
|
NCT00009932
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Unknown |
- |
United States, Ohio
... More >>
Arthur G. James Cancer Hospital - Ohio State University
Columbus, Ohio, United States, 43210-1240 Less << |
|
NCT03389477
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 2 |
Recruiting |
June 30, 2026 |
United States, Missouri
... More >>
Washington University School of Medicine
Recruiting
Saint Louis, Missouri, United States, 63110
Contact: Douglas R Adkins, M.D. 314-362-4471 dadkins@wustl.edu
Principal Investigator: Douglas R Adkins, M.D.
Sub-Investigator: Olivia Aranha, M.D.
Sub-Investigator: Mackenzie Daly, M.D.
Sub-Investigator: Hiram Gay, M.D.
Sub-Investigator: Ryan Jackson, M.D.
Sub-Investigator: Peter Oppelt, M.D.
Sub-Investigator: Randal Paniello, M.D.
Sub-Investigator: Patrik Pipkorn, M.D.
Sub-Investigator: Jason Rich, M.D.
Sub-Investigator: Wade Thorstad, M.D.
Sub-Investigator: Jose Zevallos, M.D., MPH Less << |
|
NCT01064479
|
Metastatic Squamous Cell Carci... More >>noma of the Hypopharynx
Metastatic Squamous Cell Carcinoma of the Larynx
Metastatic Squamous Cell Carcinoma of the Oral Cavity
Metastatic Squamous Cell Carcinoma of the Oropharynx
Recurrent Hypopharyngeal Squamous Cell Carcinoma
Recurrent Laryngeal Squamous Cell Carcinoma
Recurrent Oral Cavity Squamous Cell Carcinoma
Recurrent Oropharyngeal Squamous Cell Carcinoma
Stage IV Hypopharyngeal Squamous Cell Carcinoma AJCC v7
Stage IV Laryngeal Squamous Cell Carcinoma AJCC v7
Stage IV Oral Cavity Squamous Cell Carcinoma AJCC v6 and v7
Stage IV Oropharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVA Hypopharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVA Laryngeal Squamous Cell Carcinoma AJCC v7
Stage IVA Oral Cavity Squamous Cell Carcinoma AJCC v6 and v7
Stage IVA Oropharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVB Hypopharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVB Laryngeal Squamous Cell Carcinoma AJCC v7
Stage IVB Oral Cavity Squamous Cell Carcinoma AJCC v6 and v7
Stage IVB Oropharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVC Hypopharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVC Laryngeal Squamous Cell Carcinoma AJCC v7
Stage IVC Oral Cavity Squamous Cell Carcinoma AJCC v6 and v7
Stage IVC Oropharyngeal Squamous Cell Carcinoma AJCC v7 Less << |
Phase 2 |
Active, not recruiting |
February 28, 2019 |
United States, Texas
... More >>
M D Anderson Cancer Center
Houston, Texas, United States, 77030
MD Anderson Regional Care Center-Katy
Houston, Texas, United States, 77094
MD Anderson Regional Care Center-Bay Area
Nassau Bay, Texas, United States, 77058
MD Anderson Regional Care Center-Sugar Land
Sugar Land, Texas, United States, 77478
MD Anderson Regional Care Center-The Woodlands
The Woodlands, Texas, United States, 77384 Less << |
|
NCT03040999
|
Head and Neck Neoplasms |
Phase 3 |
Recruiting |
June 29, 2023 |
- |
|
NCT01711515
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Carcinoma
Cervical Squamous Cell Carcinoma, Not Otherwise Specified
Positive Para-Aortic Lymph Node
Positive Pelvic Lymph Node
Stage IB2 Cervical Cancer AJCC v6 and v7
Stage II Cervical Cancer AJCC v7
Stage IIA Cervical Cancer AJCC v7
Stage IIB Cervical Cancer AJCC v6 and v7
Stage IIIB Cervical Cancer AJCC v6 and v7
Stage IVA Cervical Cancer AJCC v6 and v7 Less << |
Phase 1 |
Active, not recruiting |
- |
United States, California
... More >>
Los Angeles County-USC Medical Center
Los Angeles, California, United States, 90033
USC / Norris Comprehensive Cancer Center
Los Angeles, California, United States, 90033
UC Irvine Health/Chao Family Comprehensive Cancer Center
Orange, California, United States, 92868
University of California Davis Comprehensive Cancer Center
Sacramento, California, United States, 95817
United States, Connecticut
Hartford Hospital
Hartford, Connecticut, United States, 06102
The Hospital of Central Connecticut
New Britain, Connecticut, United States, 06050
United States, Georgia
Augusta University Medical Center
Augusta, Georgia, United States, 30912
United States, New Mexico
University of New Mexico Cancer Center
Albuquerque, New Mexico, United States, 87102
United States, New York
Montefiore Medical Center-Einstein Campus
Bronx, New York, United States, 10461
United States, Ohio
Case Western Reserve University
Cleveland, Ohio, United States, 44106
Cleveland Clinic Foundation
Cleveland, Ohio, United States, 44195
United States, Oklahoma
University of Oklahoma Health Sciences Center
Oklahoma City, Oklahoma, United States, 73104
United States, Pennsylvania
Thomas Jefferson University Hospital
Philadelphia, Pennsylvania, United States, 19107
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111
United States, Rhode Island
Women and Infants Hospital
Providence, Rhode Island, United States, 02905
United States, Virginia
Virginia Commonwealth University/Massey Cancer Center
Richmond, Virginia, United States, 23298 Less << |
|
NCT00304070
|
Stage I Adrenocortical Carcino... More >>ma
Stage II Adrenocortical Carcinoma
Stage III Adrenocortical Carcinoma
Stage IV Adrenocortical Carcinoma Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00003298
|
Gastric Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT03138720
|
Resectable Pancreatic Cancer
... More >> Unresectable Pancreatic Cancer
Pancreatic Adenocarcinoma Less << |
Phase 2 |
Recruiting |
March 1, 2020 |
United States, Arizona
... More >>
HonorHealth Research Institute
Recruiting
Scottsdale, Arizona, United States, 85258
Contact: Joyce Schaffer, MSN, AOCNS 480-323-1339 ext option #2 Joyce.Schaffer@HonorHealth.com
Principal Investigator: Erkut Borazanci, MD Less << |
|
NCT02074189
|
Bladder Cancer |
Phase 3 |
Enrolling by invitation |
April 2019 |
China, Chongqing
... More >>
Southwest hospital,China
Chongqing, Chongqing, China, 400030 Less << |
|
NCT00006011
|
Endometrial Adenocarcinoma
... More >> Endometrial Adenosquamous Carcinoma
Endometrial Clear Cell Adenocarcinoma
Endometrial Endometrioid Adenocarcinoma, Variant With Squamous Differentiation
Endometrial Serous Adenocarcinoma
Stage III Uterine Corpus Cancer Less << |
Phase 3 |
Completed |
- |
United States, Pennsylvania
... More >>
Gynecologic Oncology Group
Philadelphia, Pennsylvania, United States, 19103 Less << |
|
NCT00006011
|
- |
|
Completed |
- |
- |
|
NCT00304070
|
- |
|
Completed |
- |
- |
|
NCT00020787
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 3 |
Completed |
- |
United States, California
... More >>
Jonsson Comprehensive Cancer Center, UCLA
Los Angeles, California, United States, 90095-1781 Less << |
|
NCT00961415
|
- |
|
Completed |
- |
- |
|
NCT02435186
|
Ovarian Epithelial Cancer
... More >> Fallopian Tube Cancer
Primary Peritoneal Cancer Less << |
Phase 2 |
Unknown |
August 2018 |
China, Liaoning
... More >>
xijing hospital in China Medical University
Not yet recruiting
Shenyang, Liaoning, China, 110022
Contact: xiuqing Li, MD +86 18940251286 2403621353@qq.com Less << |
|
NCT01185847
|
Non-Squamous Non-Small Cell Lu... More >>ng Cancer Less << |
Phase 2 |
Completed |
- |
Belgium
... More >>
Charleroi, Belgium, B6000
Leuven, Belgium, 3000
Liège, Belgium, 4000
France
Caen, France, 14033
Marseille, France, 13915
Saint Herblain, France, 44805
Toulouse, France, 31400
Germany
Bad Berka, Germany, 99437
Gauting, Germany, 82131
Grosshansdorf, Germany, 22927
Heidelberg, Germany, 69126
Köln, Germany, 51109
Mainz, Germany, 55131
Mannheim, Germany, 68167
Italy
Milano, Lombardia, Italy, 20162
Milan, Lombardia, Italy, 20132
Ancona, Marche, Italy
Lido Di Camaiore, Toscana, Italy, 55043
Livorno, Toscana, Italy, 57100
Perugia, Umbria, Italy, 06156
Poland
Lodz, Poland, 93-509
Otwock, Poland, 05-400
Warszawa, Poland, 02-781
Spain
Barcelona, Spain, 08003
Barcelona, Spain, 08035
Madrid, Spain, 28040
Madrid, Spain, 28041
Madrid, Spain, 28046
Madrid, Spain, 28050
Malaga, Spain, 29010
Sevilla, Spain, 41013
United Kingdom
London, United Kingdom, SE1 9RT Less << |
|
NCT00961415
|
Non-Squamous Non-Small Cell Lu... More >>ng Cancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00450138
|
Head and Neck Cancer |
Phase 1 |
Completed |
- |
United States, Colorado
... More >>
Research Site
Denver, Colorado, United States
United States, Illinois
Research Site
Chicago, Illinois, United States
United States, Texas
Research Site
Dallas, Texas, United States
Research Site
Houston, Texas, United States Less << |
|
NCT03700476
|
Nasopharyngeal Neoplasms |
Phase 3 |
Not yet recruiting |
January 2025 |
China, Guangdong
... More >>
Panyu central hospital
Not yet recruiting
Guangzhou, Guangdong, China, 510060
Contact: Guo-Rong Zhou, MD
SUN YAT-SEN UNIVERSITY cANCER CENTER
Guangzhou, Guangdong, China, 510060
China, Guangxi
Cancer Hospital of Guangxi Medical University
Not yet recruiting
Nanning, Guangxi, China
Contact: Xiao-Dong Zhu, MD
China, Guizhou
Cancer Hospital of Guizhou Medical University
Not yet recruiting
Guiyang, Guizhou, China
Contact: Feng Jin, MD
China, Hubei
Union Hospital Affiliated with Tongji Medical College of Huazhong University of Science and Technology
Not yet recruiting
Wuhan, Hubei, China
Contact: Kun-Yu Yang, MD
China, Hunan
Xiangya Hospital Central South University
Not yet recruiting
Changsha, Hunan, China
Contact: Liang-Fang Shen, MD
China, Shanxi
Xijing Hospital, Fourth Military Medical University
Not yet recruiting
Xi'an, Shanxi, China
Contact: Mei SHI, MD Less << |
|
NCT01670890
|
Malignant Gliomas |
Phase 2 |
Unknown |
- |
China, Beijing
... More >> Beijing Tiantan Hospital Affiliated to Capital Medial University
Recruiting
Beijing, Beijing, China, 100050
Contact: Jiang tao, MD 86-010-67021832 jiangtao369@sohu.com
Peking Union Medical College Hospital
Recruiting
Beijing, Beijing, China, 100730
Contact: wang ren zhi, MD 86-010-69156071 wangrz@126.com
Principal Investigator: Ma wen bin, MD
Sub-Investigator: wang yu, MD
China, Tianjin
Tianjin medical university general university
Recruiting
Tianjin, Tianjin, China, 300052
Contact: Yang xue jun, MD ydenny@yahoo.com Less << |
|
NCT02954536
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 2 |
Recruiting |
November 2019 |
United States, New Jersey
... More >>
Memoral Sloan Kettering Cancer Center
Recruiting
Basking Ridge, New Jersey, United States
Contact: Yelena Janjigian, MD 646-888-4186
Principal Investigator: Yelena Janjigian, MD
Memorial Sloan Kettering Monmouth
Recruiting
Middletown, New Jersey, United States, 07748
Contact: Yelena Janjigian, MD 646-888-4186
United States, New York
Memorial Sloan Kettering Cancer Center @ Suffolk
Recruiting
Commack, New York, United States, 11725
Contact: Yelena Janjigian, MD 646-888-4186
Principal Investigator: Yelena Janjigian, MD
Memorial Sloan Kettering Westchester
Recruiting
Harrison, New York, United States, 10604
Contact: Yelena Janjigian, MD 646-888-4186
Memorial Sloan Kettering Cancer Center 1275 York Avenue
Recruiting
New York, New York, United States, 10065
Contact: Yelena Janjigian, MD 646-888-4186
Contact: David Ilson, MD, PhD 646-888-4183
Principal Investigator: Yelena Janjigian, MD
Memorial Sloan Kettering at Mercy Medical Center
Recruiting
Rockville Centre, New York, United States
Contact: Yelena Janjigian, MD 646-888-4186
Principal Investigator: Yelena Janjigian, MD
United States, Texas
Md Anderson Cancer Center
Not yet recruiting
Houston, Texas, United States, 77030
Contact: Mariela Blum Murphy, MD 713-792-2828
Principal Investigator: Mariela Blum Murphy, MD Less << |
|
NCT03413358
|
Non-small-Cell Lung Cancer |
Not Applicable |
Recruiting |
October 31, 2019 |
China, Beijing
... More >> CHINA ACADENY OF CHINESE MEDICAL SCIENCES Guang'anmen Hospital
Recruiting
Beijing, Beijing, China, 100053
Contact: Hongsheng Lin, Professor 010-88001192 drlinhongsheng@163.com
Contact: , Professor Less << |
|
NCT03600831
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Phase 2
Phase 3 |
Recruiting |
December 31, 2023 |
China, Jiangsu
... More >> Chinese Peiple's Liberation Army No.82 Hospital
Recruiting
Huai'an, Jiangsu, China, 223300
Contact: Qian-wen Li, MD
Huai'an second peiple's Hospital
Recruiting
Huai'an, Jiangsu, China, 223300
Contact: Li-qing Zhou, Phd
Lianshui County Peoples Hospital
Recruiting
Huai'an, Jiangsu, China, 223300
Contact: juan pu, MD
xuyi peiple's Hospital
Recruiting
Xu Yi, Jiangsu, China, 211700
Contact: Lian Wang, MD Less << |
|
NCT00682383
|
Unresectable Locally Advanced ... More >>NSCLC Less << |
Phase 2 |
Completed |
- |
United States, Florida
... More >>
Mount Sinai Medical Center
Miami Beach, Florida, United States, 33140 Less << |
|
NCT00313872
|
Gastric Cancer
... More >> Metastases Less << |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of Less << |
|
NCT02022098
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Not Applicable |
Active, not recruiting |
April 2019 |
France
... More >> Centre Hospitalier de Bretagne Sud - HÔPITAL DU SCORFF
Lorient, Bp 2233, France, 56322
C.H.U. Sud Amiens
Amiens, France, 80054
ICO Paul Papin
Angers Cedex 02, France, CS 10059
Institut Sainte-Catherine
Avignon Cedex 9, France, 84918
CHU Côte de Nacre
CAEN Cedex 09, France, CS30001
Centre Jean Perrin
Clermont-Ferrand Cedex 01, France, BP 392
Centre Georges François Leclerc
Dijon, France, 21079
CHU Grenoble
Grenoble, France, BP 217
CHD Vendée
La Roche Sur Yon cedex 9, France, F- 85925
Centre Guillaume le Conquérant
Le Havre, France, 76600
Centre Jean Bernard
Le Mans, France
Centre Léon Berard
Lyon, France
Hôpital Nord Franche-Comté
Montbéliard, France, 25200
ICM - Val D'Aurelle
Montpellier, France, 34298
Institut Curie
Paris, France, 75248
Centre Henri Becquerel
Rouen, France, 76038
Institut de Cancérologie de l'Ouest (ICO) René Gauducheau
Saint-Herblain, France, BP 217
Institut de Cancérologie Lucien Neuwirth (ICLN)
Saint-Priest en Jarez, France, BP 60008
Institut Claudius Regaud
Toulouse, France, 31052
L'Institut de Cancérologie de Lorraine (ICL) Alexis Vautrin
Vandoeuvre-lès-Nancy, France, CS 30519
Institut Gustave Roussy
Villejuif, France, 94000
Switzerland
Inselspital Bern, Universitätsklinik für Radio-Onkologie, Freiburgstrasse 4
Bern, Switzerland, 3010
Centre Hospitalier Universitaire Vaudois (CHUV)
Lausanne, Switzerland, 1011 Less << |
|
NCT01454089
|
Urologic Neoplasms
... More >> Metastatic Bladder Cancer
Urinary Tract Neoplasms Less << |
Phase 2 |
Completed |
- |
- |
|
NCT02901483
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 1
Phase 2 |
Recruiting |
December 2020 |
Taiwan
... More >> Keelung Chang Gung Memorial Hospital (Lovers Lake Branch)
Recruiting
Keelung, Taiwan Less << |
|
NCT02000531
|
Carcinoma, Non-Small-Cell Lung |
Phase 4 |
Completed |
- |
China
... More >>
Beijing, China, 101149
Changchun, China, 130012
Chongqing, China, 400038
ChongQing, China, 400042
Fuzhou, China, 350014
Guangzhou, China
Nanjing, China
Shanghai, China, 200030
Shanghai, China, 200433
Shantou, China, 515041
Wuhan, China, 430023 Less << |
|
NCT00028743
|
Fallopian Tube Cancer
... More >> Ovarian Cancer
Peritoneal Cavity Cancer Less << |
Phase 3 |
Completed |
- |
Canada, Alberta
... More >>
Tom Baker Cancer Centre
Calgary, Alberta, Canada, T2N 4N2
Cross Cancer Institute
Edmonton, Alberta, Canada, T6G 1Z2
Canada, British Columbia
BCCA - Cancer Centre for the Southern Interior
Kelowna, British Columbia, Canada, V1Y 5L3
Lions Gate Hospital
North Vancouver, British Columbia, Canada, V7L 2L7
BCCA - Fraser Valley Cancer Centre
Surrey, British Columbia, Canada, V3V 1Z2
BCCA - Vancouver Cancer Centre
Vancouver, British Columbia, Canada, V5Z 4E6
Canada, Manitoba
CancerCare Manitoba
Winnipeg, Manitoba, Canada, R3E 0V9
Canada, New Brunswick
The Moncton Hospital
Moncton, New Brunswick, Canada, E1C 6Z8
Atlantic Health Sciences Corporation
Saint John, New Brunswick, Canada, E2L 4L2
Canada, Newfoundland and Labrador
Dr. H. Bliss Murphy Cancer Centre
St. John's, Newfoundland and Labrador, Canada, AIB 3V6
Canada, Nova Scotia
QEII Health Sciences Center
Halifax, Nova Scotia, Canada, B3H 1V7
Canada, Ontario
Juravinski Cancer Centre at Hamilton Health Sciences
Hamilton, Ontario, Canada, L8V 5C2
Cancer Centre of Southeastern Ontario at Kingston
Kingston, Ontario, Canada, K7L 5P9
Grand River Regional Cancer Centre
Kitchener, Ontario, Canada, N2G 1G3
London Regional Cancer Program
London, Ontario, Canada, N6A 4L6
Ottawa Health Research Institute - General Division
Ottawa, Ontario, Canada, K1H 8L6
Niagara Health System
St. Catharines, Ontario, Canada, L2R 7C6
Northeast Cancer Center Health Sciences
Sudbury, Ontario, Canada, P3E 5J1
Thunder Bay Regional Health Science Centre
Thunder Bay, Ontario, Canada, P7B 6V4
Univ. Health Network-Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9
Windsor Regional Cancer Centre
Windsor, Ontario, Canada, N8W 2X3
Canada, Prince Edward Island
PEI Cancer Treatment Centre,Queen Elizabeth Hospital
Charlottetown, Prince Edward Island, Canada, C1A 8T5
Canada, Quebec
CHUM - Hopital Notre-Dame
Montreal, Quebec, Canada, H2L 4M1
Hopital du Sacre-Coeur de Montreal
Montreal, Quebec, Canada, H4J 1C5
CHUQ-Pavillon Hotel-Dieu de Quebec
Quebec City, Quebec, Canada, G1R 2J6
Centre hospitalier universitaire de Sherbrooke
Sherbrooke, Quebec, Canada, J1H 5N4
Canada, Saskatchewan
Allan Blair Cancer Centre
Regina, Saskatchewan, Canada, S4T 7T1
Saskatoon Cancer Centre
Saskatoon, Saskatchewan, Canada, S7N 4H4 Less << |
|
NCT02000531
|
- |
|
Completed |
- |
- |
|
NCT00122772
|
Cervix Cancer |
Phase 3 |
Completed |
- |
Austria
... More >> University of Vienna; Department of Radiotherapy and Radiobiology
Vienna, Austria
Brazil
rmandade de Santa Casa de Misericordia de Porto Alegre; Hospital Santa Rita
Porto Alegre, Brazil
Canada, Ontario
Peel Regional Cancer Centre
Mississauga, Ontario, Canada
India
Department of Atomic Energy (DAE); Tata Memorial Centre (TMC); Tata
Mumbai, India
Korea, Republic of
National Cancer Center
Seoul, Korea, Republic of
Macedonia, The Former Yugoslav Republic of
Radiotherapy and Oncology University Clinic
Skopje, Macedonia, The Former Yugoslav Republic of
Morocco
Institut National d'Oncologie
Rabat, Morocco
Pakistan
Bahawalpur Institute of Nuclear Medicine and Oncology (BINO)
Bahawalpur, Pakistan
Peru
Instituto Nacional de Enfermedades Neoplásicas
Lima, Peru
South Africa
Department of Radiation Oncology, Groote Schuur Hospital
Cape Town, South Africa
United Kingdom
Christie Hospital; NHS Trust
Manchester, United Kingdom Less << |
|
NCT01487499
|
Small Cell Lung Cancer |
PHASE3 |
TERMINATED |
2025-03-15 |
University of Florida, Gainesv... More >>ille, Florida, 32610, United States Less << |
|
NCT03449693
|
Febrile Neutropenia |
Phase 2 |
Recruiting |
December 2019 |
Mexico
... More >> Hospital Infantil de Mexico Dr. Federico Gomez
Recruiting
Cuauhtémoc, Ciudad De México, Mexico, 06720
Contact: Victoria E Barrios, QFB +52 1 2291290208 victoria.barrios@cinvestav.mx
Contact: Miguel A Palomo, MD Less << |
|
NCT00123318
|
Gastric Cancer |
Phase 3 |
Completed |
- |
Australia, New South Wales
... More >>
Liverpool Hospital
Liverpool, New South Wales, Australia, 1871
Calvary Mater Newcastle
Newcastle, New South Wales, Australia, 2298
Nepean Cancer Care Centre
Penrith, New South Wales, Australia, 2751
Prince of Wales Hospital
Randwick, New South Wales, Australia, 2031
Royal Prince Alfred Hospital
Sydney, New South Wales, Australia, 2050
Royal North Shore Hospital
Sydney, New South Wales, Australia, 2069
Westmead Hospital
Sydney, New South Wales, Australia, 2145
Australia, Queensland
Mater QRI
Brisbane, Queensland, Australia
Royal Brisbane Hospital
Herston, Queensland, Australia, 4029
East Coast Cancer Centre
Tugun, Queensland, Australia, 4224
Princess Alexandra Hospital
Woolloongabba, Queensland, Australia, 4102
Australia, Tasmania
Launceston General Hospital
Launceston, Tasmania, Australia, 7250
Australia, Victoria
Box Hill Hospital
Box Hill, Victoria, Australia
Andrew Love Cancer Care Centre, Geelong Hospital
Geelong, Victoria, Australia, 3220
Austin Health
Melbourne, Victoria, Australia, 3081
Peter MacCallum Cancer Centre
Melbourne, Victoria, Australia, 8006
Alfred Hospital
Prahran, Victoria, Australia, 3181
Australia, Western Australia
Sir Charles Gairdner Hospital
Perth, Western Australia, Australia, 6009
New Zealand
Christchurch Hospital
Christchurch, New Zealand, 4710 Less << |
|
NCT00916097
|
Head and Neck Neoplasms |
Phase 2 |
Completed |
- |
Algeria
... More >> Sanofi-Aventis Administrative Office
Alger, Algeria
Morocco
Sanofi-Aventis Administrative Office
Casablanca, Morocco
Tunisia
Sanofi-Aventis Administrative Office
Megrine, Tunisia Less << |
|
NCT00006248
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT03201146
|
Lung Cancer
N... More >>on Small Cell Lung Cancer
Combination Chemotherapy
Apatinib Less << |
Phase 1
Phase 2 |
Recruiting |
August 1, 2020 |
China, Sichuan
... More >> West China Hospital, Sichuan University
Recruiting
Chengdu, Sichuan, China, 610041
Contact: You Lu, MD 86-028-85422114 radyoulu@hotmail.com
Contact: Meijuan Huang, MD 86-028-85422114 hmj107@163.com Less << |
|
NCT01076231
|
Stage IIIA Non-small Cell Lung... More >> Cancer Less << |
Phase 1
Phase 2 |
Unknown |
- |
United States, Pennsylvania
... More >>
Abramson Cancer Center of The University of Pennsylvania
Philadelphia, Pennsylvania, United States, 19104 Less << |
|
NCT00539669
|
Ovarian Neoplasms |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Sanofi-Aventis
Seoul, Korea, Republic of Less << |
|
NCT01487499
|
Small Cell Lung Cancer |
PHASE3 |
TERMINATED |
2025-03-15 |
University of Florida, Gainesv... More >>ille, Florida, 32610, United States Less << |
|
NCT01783951
|
Gastric Cancer |
Phase 1
Phase 2 |
Completed |
- |
China, Beijing
... More >> Capital Medical University Cancer Center
Beijing, Beijing, China, 100038 Less << |
|
NCT03147404
|
Neuroendocrine Carcinoma, Grad... More >>e 3 Less << |
Phase 2 |
Recruiting |
February 2020 |
Korea, Republic of
... More >>
Samsung Medical Center
Recruiting
Seoul, Korea, Republic of, 06351
Contact: seungtae kim, MD,PhD Less << |
|
NCT03387774
|
Oral Mucositis (Ulcerative) Du... More >>e to Radiation Less << |
Phase 3 |
Recruiting |
November 30, 2018 |
China, Guangdong
... More >>
Cancer Center, Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China
Contact: zhao chong, M.D +86-13902206160 zhaochong@sysucc.org.cn Less << |
|
NCT02595879
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Carcinoma
Cervical Squamous Cell Carcinoma, Not Otherwise Specified
Stage IB Vulvar Cancer AJCC v7
Stage IB2 Cervical Cancer AJCC v6 and v7
Stage II Vulvar Cancer AJCC v7
Stage IIA1 Cervical Cancer AJCC v7
Stage IIA2 Cervical Cancer AJCC v7
Stage IIB Cervical Cancer AJCC v6 and v7
Stage IIIA Cervical Cancer AJCC v6 and v7
Stage IIIA Vulvar Cancer AJCC v7
Stage IIIB Cervical Cancer AJCC v6 and v7
Stage IIIB Vulvar Cancer AJCC v7
Stage IIIC Vulvar Cancer AJCC v7
Stage IVA Cervical Cancer AJCC v6 and v7
Stage IVA Vulvar Cancer AJCC v7
Vulvar Adenocarcinoma
Vulvar Squamous Cell Carcinoma Less << |
Phase 1 |
Suspended(Drug Supply Issues) |
- |
United States, Pennsylvania
... More >>
University of Pittsburgh Cancer Institute (UPCI)
Pittsburgh, Pennsylvania, United States, 15232
United States, Virginia
University of Virginia Cancer Center
Charlottesville, Virginia, United States, 22908 Less << |
|
NCT02243436
|
CLASSICAL HODGKIN LYMPHOMA |
Phase 1
Phase 2 |
Active, not recruiting |
January 2019 |
Spain
... More >> Hospital Universitario Central de Asturias
Oviedo, Asturias, Spain, 33006
Institut Català d'Oncologia, Hospital Germans Trias i Pujol
Badalona, Barcelona, Spain, 08916
Institut Català d'Oncologia, Hospital Duran i Reynals
L'Hospitalet de Llobregat, Barcelona, Spain, 08908
Hospital Son Espases
Palma, Islas Baleares, Spain
Hospital del Mar
Barcelona, Spain, 08003
Hospital Clinic i Provincial de Barcelona
Barcelona, Spain, 08036
Hospital General Universitario Gregorio Marañon
Madrid, Spain, 28007
Hospital Universitario Ramón y Cajal
Madrid, Spain, 28034
Hospital Universitario 12 de Octubre
Madrid, Spain, 28041
Hospital Universitario La Paz
Madrid, Spain, 28046
Hospital Universitario de Salamanca
Salamanca, Spain, 37007
Hospital Universitario de Canarias
Santa Cruz de Tenerife, Spain, 38320
Hospital Universitario Virgen del Rocío
Sevilla, Spain, 41013
Hospital Clínico de Valencia
Valencia, Spain, 46010 Less << |
|
NCT03306901
|
Esophageal Cancer |
Not Applicable |
Recruiting |
December 31, 2025 |
Korea, Republic of
... More >>
National Cancer Center
Not yet recruiting
Goyang-si, Korea, Republic of
Contact: Jong Mog Lee +82-10-7236-1078 jongmog@ncc.re.kr
Pusan National University Hospital
Not yet recruiting
Pusan, Korea, Republic of, 06351
Contact: Jihyung Kang +82-2-2008-4259 sammy_jkang@naver.com
Seoul National University Bundang Hospital
Not yet recruiting
Seongnam, Korea, Republic of
Contact: Jihyung Kang +82-2-2008-4259 sammy_jkang@naver.com
Asan Medical Center
Not yet recruiting
Seoul, Korea, Republic of
Contact: Jihyung Kang +82-2-2008-4259 sammy_jkang@naver.com
Gangnam Severance Hospital
Not yet recruiting
Seoul, Korea, Republic of
Contact: Jihyung Kang +82-2-2008-4259 sammy_jkang@naver.com
Samsung Medical Center
Recruiting
Seoul, Korea, Republic of
Contact: Jihyung Kang +82-2-2008-4259 sammy_jkang@naver.com
Seoul St. Mary's Hospital
Not yet recruiting
Seoul, Korea, Republic of
Contact: Jihyung Kang +82-2-2008-4259 sammy_jkang@naver.com Less << |
|
NCT01443078
|
- |
|
Completed |
- |
- |
|
NCT00027703
|
Advanced Malignant Mesotheliom... More >>a
Epithelial Mesothelioma
Localized Malignant Mesothelioma
Recurrent Malignant Mesothelioma
Sarcomatous Mesothelioma Less << |
Phase 2 |
Completed |
- |
United States, Illinois
... More >>
University of Chicago
Chicago, Illinois, United States, 60637 Less << |
|
NCT01046461
|
Solid Tumour
... More >>Postoperative Nausea and Vomiting Less << |
Phase 2 |
Unknown |
June 2012 |
Korea, Republic of
... More >>
Hallym University Sacred Heart Hospital
Anyang-si, Gyeonggi-do, Korea, Republic of, 431-070 Less << |
|
NCT01982955
|
Non-small Cell Lung Cancer |
Phase 1
Phase 2 |
Active, not recruiting |
October 16, 2018 |
Germany
... More >> Please contact the Merck KGaA Communication Center located in
Darmstadt, Germany Less << |
|
NCT02031250
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 2 |
Recruiting |
December 2020 |
United States, Michigan
... More >>
Veterans Affairs (VA) Ann Arbor Healthcare System
Recruiting
Ann Arbor, Michigan, United States, 48105
Contact: Nicky Chalpe 734-845-3713 Niharika.Chalpe@va.gov
University of Michigan Hospital
Recruiting
Ann Arbor, Michigan, United States, 48109
Contact: Michelle Mierzwa, M.D. mmierzwa@med.umich.edu
Principal Investigator: Michelle Mierzwa, M.D. Less << |
|
NCT01443078
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, New Jersey
... More >>
Memoral Sloan Kettering Cancer Center
Basking Ridge, New Jersey, United States
United States, New York
Memorial Sloan-Kettering Cancer Center @ Suffolk
Commack, New York, United States, 11725
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan-Kettering at Mercy Medical Center
Rockville Centre, New York, United States
Memorial Sloan-Kettering Cancer Center at Phelps Memorial Hospital Center
Sleepy Hollow, New York, United States, 10591 Less << |
|
NCT00423449
|
Non-Small Cell Lung Cancer |
Phase 1 |
Completed |
- |
- |
|
NCT02902432
|
Nasopharyngeal Neoplasms |
Phase 2 |
Active, not recruiting |
December 2018 |
China, Hunan
... More >> the Second Xiangya Hospital
Changsha, Hunan, China, 410011 Less << |
|
NCT00717938
|
Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
Sweden
... More >> Gävle hospital
Gävle, Sweden, 801 87
Sahlgrenska University Hospital
Göteborg, Sweden, 413 45
Helsingborg Hospital
Helsingborg, Sweden
Ryhov Hospital, Jönköping
Jönköping, Sweden
Blekinge Hospital
Karlskrona, Sweden, 371 85
Central Hospital
Karlstad, Sweden, 651 85
Central Hospital
Kristianstad, Sweden, 291 85
University Hospital Linköping
Linköping, Sweden, 581 85
University Hospital Department of Respiratory Medicine
Lund, Sweden, 221 85
University Hospital MAS
Malmö, Sweden, 205 02
Karolinska University Hospital
Stockholm, Sweden, 171 76
Norrlands University Hospital
Umeå, Sweden
Akademiska hospital Uppsala
Uppsala, Sweden, 751 85
Central Hospital
Växjö, Sweden, 351 85
Ystad hospital
Ystad, Sweden, 271 82
University Hospital, Örebro
Örebro, Sweden Less << |
|
NCT02187315
|
Nasopharyngeal Carcinoma |
Phase 2 |
Recruiting |
December 2019 |
China, Guizhou
... More >> The Affiliated Hospital of Guiyang Medical College/The Affiliated Cancer Hospital of Guiyang Medical College/Guizhou Cancer Hospital
Recruiting
Guiyang, Guizhou, China, 550001
Contact: Feng Jin, Professor +86 13985124806 jinf8865@gmail.com Less << |
|
NCT02060656
|
Diffuse Large B-Cell Lymphoma |
Phase 2 |
Unknown |
- |
United Kingdom
... More >> Royal Marsden NHS Foundation Trust - London and Surrey
Recruiting
London, United Kingdom, SM2 5PT
Contact: Bijal Patel, Msc +44 (0)208 661 3808 bijal.patel@rmh.nhs.uk
Principal Investigator: David Cunningham, MD FRCP Less << |
|
NCT01512407
|
Hepatocellular Carcinoma |
Phase 3 |
Unknown |
January 2018 |
China
... More >> Prince of Wales Hospital
Recruiting
Hong Kong, China
Contact: Yue Sun Cheung, MBChB (852) 26321411 yuesun@surgery.cuhk.edu.hk
Principal Investigator: Yue Sun Cheung, MBChB Less << |
|
NCT00006027
|
Endometrial Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00719550
|
Esophagogastric Junction Adeno... More >>carcinoma
Gastric Cancer
Esophageal Cancer Less << |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT00907543
|
Esophageal Neoplasms |
Phase 2
Phase 3 |
Recruiting |
November 2018 |
Canada, Ontario
... More >>
London Health Sciences Centre
Recruiting
London, Ontario, Canada, N6A 5W9
Contact: Richard Malthaner, MD 519-667-6835 richard.malthaner@lhsc.on.ca
Contact: Deb Lewis, BSc 519-685-8500 ext 75685 deb.lewis@lhsc.on.ca
Principal Investigator: Richard Malthaner, MD Less << |
|
NCT00423449
|
- |
|
Completed |
- |
- |
|
NCT00003742
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT01544179
|
Non-Small Cell Lung Cancer |
Phase 3 |
Active, not recruiting |
December 31, 2018 |
- |
|
NCT01088802
|
Squamous Cell Carcinoma of Oro... More >>pharynx Less << |
Phase 2 |
Active, not recruiting |
January 2022 |
United States, Maryland
... More >>
The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Baltimore, Maryland, United States, 21231 Less << |
|
NCT01755897
|
Uterine Cervical Neoplasms
... More >> Cervical Cancer
Uterine Cervical Cancer Less << |
Phase 3 |
Active, not recruiting |
December 2018 |
China, Hubei
... More >> Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology
Wuhan, Hubei, China, 430030
China, Shandong
Qilu Hospital, Shandong University
Jinan, Shandong, China, 250012
China, Zhejiang
Women's Hospital, School of Medicine, Zhejiang University
Hangzhou, Zhejiang, China, 310006 Less << |
|
NCT00002521
|
Lymphoma |
Phase 2 |
Completed |
- |
United States, Pennsylvania
... More >>
Fox Chase - Temple Cancer Center
Philadelphia, Pennsylvania, United States, 19111-2442 Less << |
|
NCT01544179
|
- |
|
Active, not recruiting |
- |
- |
|
NCT01268878
|
- |
|
Completed |
- |
France
... More >> Institut Sainte Catherine
Avignon, France, 84082
Centre Hospitalier Universitaire
Besancon, France, 25000
Polyclinique de Bordeaux Nord
Bordeaux, France, 33077
Centre Hospitalier Universitaire
Caen, France, 14033
Centre Jean Perrin
Clermont Ferrand, France, 63011
Centre Hospitalier
Draguignan, France, 83300
Centre Hospitalier de Vendée
La Roche sur Yon, France, 85925
Centre Guillaume le Conquerant
Le Havre, France, 76600
Centre Oscar Lambret
Lille, France, 59020
Centre Hospitalier
Lorient, France, 56000
Centre Leon Berard
Lyon, France, 69008
Centre Antoine Lacassagne
Nice, France, 06189
Groupe Hospitalier Pitié Salpétrière
Paris, France, 75651
Institut Jean Godinot
Reims, France, 51056
Centre Eugene Marquis
Rennes, France, 35042
Clinique Armoricaine
Saint Brieuc, France, 22015
Centre René Gauducheau
Saint Herblain, France, 44885
Centre Hospitalier Bretonneau
Tours, France, 37044 Less << |
|
NCT00002722
|
Gastric Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT03556839
|
Carcinoma of the Cervix, Stage... More >> IVB Less << |
Phase 3 |
Recruiting |
December 2023 |
Spain
... More >> Intitut Català d' Oncolgia L' Hospitalet
Not yet recruiting
Hospitalet de LLobregat, Barcelona, Spain, 08907
Contact: Beatriz Pardo, MD
Principal Investigator: Beatriz Pardo, MD
Parc Taulí
Not yet recruiting
Sabadell, Barcelona, Spain, 08208
Contact: Yolanda García, MD
Principal Investigator: Yolanda García, MD
Hospital Universitario Donostia- Donostia Unibertsitate Ospitalea
Not yet recruiting
Donostia, Gipuzkoa, Spain, 20014
Contact: Nerea Anzizar, MD
Principal Investigator: Nerea Anzizar, MD
Hospital Universitario Son Espases
Not yet recruiting
Palma De Mallorca, Islas Baleares, Spain, 07120
Contact: Jesús Alarcón, MD
Principal Investigator: Jesús Alarcón, MD
Hospital de la Vall d'Hebron
Recruiting
Barcelona, Spain, 08035
Contact: Ana Oaknin, MD PhD +34 932746085 aoaknin@vhio.net
Principal Investigator: Ana Oaknin, MD PhD
H. Clínic Barcelona
Not yet recruiting
Barcelona, Spain, 08036
Contact: Lydia Gaba, MD
Principal Investigator: Lydia Gaba, MD
Hospital Reina Sofía Cordoba
Not yet recruiting
Cordoba, Spain, 14004
Contact: Maria Jesus Rubio, MD
Principal Investigator: Maria Jesus Rubio, MD
ICO Girona
Not yet recruiting
Girona, Spain, 17007
Contact: Pilar Barretina, MD
Principal Investigator: Pilar Barretina, MD
Hospital Ramon y Cajal
Not yet recruiting
Madrid, Spain, 28034
Contact: Eva Guerra, MD
Principal Investigator: Eva Guerra, MD
Hospital Universitario 12 de Octubre
Not yet recruiting
Madrid, Spain, 28041
Contact: Luis Manso Sánchez, MD
Principal Investigator: Luis Manso Sánchez, MD
Hospital Universitario La Paz
Recruiting
Madrid, Spain, 28046
Contact: Andrés Redondo, MD
Principal Investigator: Andrés Redondo, MD
Hospital Clinico Universitario Virgen Arrixaca
Not yet recruiting
Murcia, Spain, 30120
Contact: Jerónimo Martínez, MD
Principal Investigator: Jerónimo Martínez, MD
Complejo Hospitalario Regional de Málaga
Not yet recruiting
Málaga, Spain, 29010
Contact: Enrique Sáez, MD
Principal Investigator: Enrique Sáez, MD
Hosptial Clinico Universitario de Santiago de Compostela
Not yet recruiting
Santiago de Compostela, Spain, 15706
Contact: Juan Cueva, MD
Principal Investigator: Juan Cueva, MD
Hospital Virgen de la Salud
Not yet recruiting
Toledo, Spain
Contact: Carmen Esteban, MD
Principal Investigator: Carmen Esteban, MD
Instituto Valenciano de Oncología
Not yet recruiting
Valencia, Spain, 46009
Contact: Ignacio Romero, MD
Principal Investigator: Ignacio Romero, MD
Hospital Clínico Universitario de Valencia
Not yet recruiting
Valencia, Spain, 46010
Contact: José A Pérez Fidalgo, MD
Principal Investigator: José A Pérez Fidalgo, MD
Hospital Miguel Servet
Not yet recruiting
Zaragoza, Spain, 50009
Contact: Ana Herrero, MD
Principal Investigator: Ana Herrero, MD Less << |
|
NCT00006048
|
Lung Cancer |
Phase 3 |
Unknown |
- |
United States, Delaware
... More >>
AstraZeneca Pharmaceuticals LP
Wilmington, Delaware, United States, 19850-5437 Less << |
|
NCT00193882
|
Esophagus Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00191230
|
Carcinoma, Non Small Cell Lung |
Phase 2 |
Completed |
- |
United States, North Carolina
... More >>
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Chapel Hill, North Carolina, United States Less << |
|
NCT03750539
|
Cervix Cancer |
Not Applicable |
Active, not recruiting |
November 10, 2021 |
Mexico
... More >> David Cantu de Leon
Mexico City, Tlalpan, Mexico, 14080 Less << |
|
NCT03071289
|
Cervical Cancer
... More >> Lymphocyst
Radiation Therapy Less << |
Phase 3 |
Recruiting |
March 31, 2025 |
China, Guangdong
... More >>
Sun Yat-sen University Affiliated Foshan Hospital
Not yet recruiting
Foshan, Guangdong, China, 528000
Contact: Rong Huang, M.D 86-13927736853 nnjbhg@163.com
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Wei-jun Ye, M.D 86-13538799871 yewj@sysucc.org.cn
Contact: Hui Chang, M.D 86-13480295989 changhui@sysucc.org.cn
The First affiliated Hospital of Guangdong Pharmaceutical University
Not yet recruiting
Guangzhou, Guangdong, China, 510080
Contact: Xi-cheng Wang, M.D 86-13902400598 13902400598@126.com
Guangzhou First People's Hospital
Not yet recruiting
Guangzhou, Guangdong, China, 510180
Contact: Guo-long Liu, M.D 86-13802527172 liugl@fimmu.com
Hospital of of Guangdong Armed Police Corps
Not yet recruiting
Guangzhou, Guangdong, China, 510507
Contact: Zhen-lun Li, M.D 86-13580516205 sfqiu@126.com
China, Guangxi
The People's Hospital of Guangxi Zhuang Autonomous Region
Not yet recruiting
Nanning, Guangxi, China, 530021
Contact: Jia-xin Chen, M.D 86-13978609888 cjx166@163.com
China, Hainan
Hainan General Hospital
Not yet recruiting
Haikou, Hainan, China, 570311
Contact: Guang Huang, M.D 86-18089777161 2842749787@qq.com
China, Xinjiang
Xinjiang Medical University Affiliated Tumor Hospital
Not yet recruiting
Wulumuqi, Xinjiang, China, 830000
Contact: Xiao-wen Li, M.D 86-13899856295 lixiaowen1026@sohu.com Less << |
|
NCT00770393
|
Squamous Cell Carcinoma |
Phase 2
Phase 3 |
Completed |
- |
France
... More >> CALLOC'H
Aix Les Bains, France
LITAS
Le Puy, France
PIGNAT
Lyon, France, 69000
MAYAUD
Montbrison, France
Crampette
Montpellier, France
Lallemant
Nîmes, France
LACHEB
Roanne, France Less << |
|
NCT00005049
|
Colorectal Cancer
... More >> Gastric Cancer
Gastrointestinal Carcinoid Tumor
Gastrointestinal Stromal Tumor
Ovarian Cancer
Peritoneal Cavity Cancer
Small Intestine Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
NYU School of Medicine's Kaplan Comprehensive Cancer Center
New York, New York, United States, 10016 Less << |
|
NCT00005086
|
Bladder Cancer |
Phase 2 |
Completed |
- |
United States, Illinois
... More >>
University of Chicago Cancer Research Center
Chicago, Illinois, United States, 60637-1470
Louis A. Weiss Memorial Hospital
Chicago, Illinois, United States, 60640
Oncology/Hematology Associates of Central Illinois, P.C.
Peoria, Illinois, United States, 61602
United States, Indiana
Fort Wayne Medical Oncology and Hematology, Inc.
Fort Wayne, Indiana, United States, 46885-5099 Less << |
|
NCT00003322
|
Ovarian Cancer
... More >> Primary Peritoneal Cavity Cancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00002493
|
Endometrial Cancer
... More >> Psychosocial Effects of Cancer and Its Treatment Less << |
Phase 3 |
Completed |
- |
- |
|
NCT03682068
|
Unresectable Locally Advanced ... More >>Urothelial Cancer
Metastatic Urothelial Cancer Less << |
Phase 3 |
Recruiting |
April 27, 2022 |
- |
|
NCT01181401
|
Squamous Cell Carcinoma of the... More >> Head
Squamous Cell Carcinoma of the Neck Less << |
Phase 2 |
Unknown |
December 2012 |
Germany
... More >> Charité Universitaetsmedizin Berlin, CVK, CBF
Recruiting
Berlin, Germany, 13353
Contact: Carmen Stromberger, MD 0049 (0) 30 450 ext 657105 carmen.stromberger@charite.de
Sub-Investigator: Carmen Stromberger, MD
Sub-Investigator: Jan-Dirk Raguse, MD
Sub-Investigator: Eva-Tessina Becker, MD
Sub-Investigator: Ulrich Keilholz, MD, PhD
Sub-Investigator: Lutz Moser, MD
Sub-Investigator: Heidi Olze, MD, PD
University Medical Center Hamburg - Eppendorf
Not yet recruiting
Hamburg, Germany, 20246
Contact: Rainald Knecht, MD, PhD 0049 (0) 40 / 74105 ext 2360 r.knecht@uke.de
Sub-Investigator: Philippe Schafhausen, MD
Sub-Investigator: Cordula Petersen, MD, PD
Medizinische Hochschule Hannover
Not yet recruiting
Hannover, Germany, 30625
Contact: Viktor Grünwald Grünwald, MD, PD 0049 (0) 511 5324077 / 9196 Gruenwald.Viktor@mh-hannover.de
Sub-Investigator: Viktor Grünwald, MD, PD
Universitätsklinikum Gießen und Marburg
Not yet recruiting
Marburg, Germany, 35043
Contact: Rita Engenhart-Cabillic, MD, PD 0049 (0) 6421 58-66434 engenhar@med.uni-marburg.de
Sub-Investigator: Rita Engenhart-Cabillic, MD, PD
Universitätsklinikum Regensburg
Not yet recruiting
Regensburg, Germany, 93053
Contact: Oliver Koelbel, MD, PD 0049 (0941) 944 9410 oliver.koelbl@klinik.uni-regensburg.de
Sub-Investigator: Oliver Koelbl, MD, PD
Sub-Investigator: Hermann Hilber, MD Less << |
|
NCT03612219
|
Nasopharyngeal Carcinoma |
Phase 2 |
Recruiting |
July 28, 2021 |
China, Guangxi
... More >> People's Hospital of Baise
Enrolling by invitation
Baise, Guangxi, China
Guangxi Naxishan Hospital
Enrolling by invitation
Guilin, Guangxi, China
Guilin Medical University
Recruiting
Guilin, Guangxi, China
Contact: Wei Jiang, Ph.D +8613788561863 weijiang@glmc.edu.cn
People's Hospital of Laibin
Enrolling by invitation
Laibin, Guangxi, China
People's Hospital of Lingshan
Enrolling by invitation
Linshan, Guangxi, China
Wuzhou Red Cross Hospital
Enrolling by invitation
Wuzhou, Guangxi, China Less << |
|
NCT00539630
|
Carcinoma, Squamous Cell |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Sanofi-Aventis
Seoul, Korea, Republic of Less << |
|
NCT02908477
|
Oropharynx Cancer |
Phase 3 |
Recruiting |
September 15, 2024 |
United States, Arizona
... More >>
Mayo Clinic in Arizona
Recruiting
Scottsdale, Arizona, United States, 85259
Contact: Clinical Trials Referral Office 855-776-0015
United States, Minnesota
Mayo Clinic in Rochester
Recruiting
Rochester, Minnesota, United States, 55905
Contact: Clinical Trials Referral Office 855-776-0015 Less << |
|
NCT03260712
|
Biliary Tract Cancer
... More >> Metastatic Cancer
Advanced Cancer
Gallbladder Cancer Less << |
Phase 2 |
Not yet recruiting |
June 1, 2021 |
- |
|
NCT02353260
|
Head and Neck Cancer |
Phase 2 |
Unknown |
September 2017 |
China, Jiangsu
... More >> Xuzhou Central Hospital
Recruiting
Xuzhou, Jiangsu, China, 221000
Contact: Jian Meng, MD
Principal Investigator: Jian Meng, MD Less << |
|
NCT00140556
|
Head and Neck Cancer
... More >> Pharynx Cancer Less << |
Early Phase 1 |
Completed |
- |
United States, North Carolina
... More >>
Department of Radiation Oncology; Duke University Medical Center
Durham, North Carolina, United States, 27710 Less << |
|
NCT00140556
|
- |
|
Completed |
- |
- |
|
NCT00005791
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT03577704
|
Advanced Solid Tumors |
Phase 1
Phase 2 |
Recruiting |
October 30, 2021 |
China, Shanghai
... More >>
Shanghai East Hospital
Recruiting
Shanghai, Shanghai, China, 200123
Contact: Jin E Li, Phd (021)38804518 ext 22228 lijin@csco.org.cn
Contact: Jin E Li lijin@csco.org.cn
Principal Investigator: Jin Li, PhD
Sub-Investigator: Ye Guo, PhD Less << |
|
NCT00003144
|
Drug/Agent Toxicity by Tissue/... More >>Organ
Unspecified Adult Solid Tumor, Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
NYU School of Medicine's Kaplan Comprehensive Cancer Center
New York, New York, United States, 10016 Less << |
|
NCT00004100
|
Lung Cancer |
Phase 3 |
Unknown |
- |
Italy
... More >> Ospedale San Lazzaro
Alba, Italy, 12051
Ospedale Civile Avellino
Avellino, Italy
Ospedale G. Di Maria - Avola (SR)
Avola (SR), Italy
Azienda Ospedaliena G. Rummo
Benevento, Italy, 82100
Ospedale Cardarelli - Campobasso
Campobasso, Italy
Ospedale Civile Cosenza
Cosenza, Italy, 87100
Ospedale San Martino/Cliniche Universitarie Convenzionate
Genoa, Italy, 16132
Ospedale Gen. Provinciale Santa Maria Goretti
Latina, Italy, 04100
Ospedale di Legnano
Legnano, Italy, 20025
Ospedale Maggiore Lodi
Lodi, Italy, I-20075
Istituto Di Science Biomediche San Paolo
Milano, Italy, 20142
Ospedale Di Gabargnate Milanese
Milan, Italy, 20024
Ospedale San Carlo Borromeo
Milan, Italy, 20153
Federico II University Medical School
Naples, Italy, 80131
Istituto Nazionale per lo Studio e la Cura dei Tumori
Naples, Italy, 80131
Ospedale S. Gennora USL 42
Naples, Italy, 80136
Ospedale Vincenzo Monaldi
Napoli, Italy, 80131
ASL 2 - Napoli
Napoli, Italy
Universita di Palermo
Palermo, Italy, 90141
Ospedale La Maddalena - Palermo
Palermo, Italy
Ospedale S. Francesco - Paola
Paola (CS), Italy
Ospedale Agnelli
Pinerolo, Italy, 10064
Ospedale San Carlo
Potenza, Italy, 85100
Ospedali Riuniti
Reggio Calabria, Italy, 89100
U.S.S.L. 33
Rho, Italy
Ospedale Oncologieo G. Fortunato
Rionero, Italy
Istituti Fisioterapici Ospitalieri - Roma
Rome, Italy, 00161
Ospedale Civile - Rovereto
Rovereto, Italy
Ospedale San Bortolo
Vicenza, Italy, 36100 Less << |
|
NCT00198367
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
France
... More >> Centre Hospitalier Lyon-Sud - Radiotherapie/Oncologie
Pierre-Benite, France Less << |
|
NCT01285037
|
Cancer |
Phase 1 |
Completed |
- |
United States, Arizona
... More >>
Arizona Cancer Center
Tucson, Arizona, United States, 85724
United States, District of Columbia
Georgetown University Medical Center
Washington, District of Columbia, United States, 20007
United States, New York
Mount Sinai Medical Center
New York, New York, United States, 10029
United States, Pennsylvania
University of Pennsylvania Hospital
Philadelphia, Pennsylvania, United States, 19104
Thomas Jefferson University
Philadelphia, Pennsylvania, United States, 19107
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111
United States, Rhode Island
Rhode Island Hospital
Providence, Rhode Island, United States, 02903 Less << |
|
NCT03175939
|
Nasopharyngeal Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT03738228
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Carcinoma
Cervical Squamous Cell Carcinoma, Not Otherwise Specified
Positive Lymph Node
Stage IB2 Cervical Cancer AJCC v8
Stage II Cervical Cancer AJCC v8
Stage IIA Cervical Cancer AJCC v8
Stage IIA1 Cervical Cancer AJCC v8
Stage IIA2 Cervical Cancer AJCC v8
Stage IIB Cervical Cancer AJCC v8
Stage IIIB Cervical Cancer AJCC v8
Stage IVA Cervical Cancer AJCC v8 Less << |
Phase 1 |
Recruiting |
October 31, 2019 |
United States, Pennsylvania
... More >>
NRG Oncology
Recruiting
Philadelphia, Pennsylvania, United States, 19103
Contact: Jyoti S. Mayadev 858-822-6040 jmayadev@ucsd.edu
Principal Investigator: Jyoti S. Mayadev Less << |
|
NCT00004003
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
United States, Michigan
... More >>
Barbara Ann Karmanos Cancer Institute
Detroit, Michigan, United States, 48201-1379 Less << |
|
NCT03351842
|
Chemotherapy, Adjuvant
... More >> Lung Adenocarcinoma, Stage I
Treatment
Histological Type of Neoplasm Less << |
Phase 2 |
Recruiting |
September 1, 2024 |
China
... More >> Thoracic Surgery Department of Shanghai Pulmonary Hospital
Recruiting
Shanghai, China, 200000
Contact: Yingran Shen 86-18117166317 elaineshen91@gmail.com Less << |
|
NCT01967875
|
Stomach Neoplasms |
Phase 2 |
Unknown |
May 2016 |
China, Liaoning
... More >>
The Fourth Hospital of Anshan
Not yet recruiting
Anshan, Liaoning, China
The First Hospital of Dalian Medical University
Not yet recruiting
Dalian, Liaoning, China
The Second Hospital of Dalian Medical University
Not yet recruiting
Dalian, Liaoning, China
The First Hospital of Liaoning Medical University
Not yet recruiting
Jinzhou, Liaoning, China
The First Hospital of China Medical University
Recruiting
Shenyang, Liaoning, China, 110001
Contact: Yunpeng Liu, MD., PhD. 86-24-83282312 cmuliuyunpeng@hotmail.com
Liaoning Tumor Hospital
Not yet recruiting
Shenyang, Liaoning, China
Shengjing Hospital of China Medical University
Not yet recruiting
Shenyang, Liaoning, China Less << |
|
NCT02350712
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 1 |
Completed |
- |
United Kingdom
... More >> The Royal Marsden Hospital
Sutton, Surrey, United Kingdom, SM2 5PT
University College London Hospital
London, United Kingdom, NW1 2BU
The Royal Marsden Hospital
London, United Kingdom, SW3 6JJ Less << |
|
NCT00003202
|
Lung Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, North Carolina
... More >>
Comprehensive Cancer Center at Wake Forest University
Winston-Salem, North Carolina, United States, 27157-1082 Less << |
|
NCT03373019
|
Chidamide
Lym... More >>phoma, B-Cell
Lymphoma, Large B-Cell, Diffuse
Neoplasm by Histology
Neoplasms
Lymphoproliferative Disorders
Lymphatic Diseases
Immunoproliferative Disorders
Immune System Diseases
Lymphoma, Non-Hodgkin
Cyclophosphamide
Rituximab
Gemcitabine
Cisplatin
Dexamethasone
HDAC Inhibitor Less << |
Phase 2 |
Recruiting |
March 1, 2021 |
China, Shanghai
... More >>
Kai Xue
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Kai Xue, MD 13818659448 xuekaishanghai@126.com
China
Kai Xue
Recruiting
Shanghai, China
Contact: Kai Xue, MD 13818659448 xuekaishanghai@126.com
Contact: Junning Cao, MD 13818659448 Less << |
|
NCT01409174
|
Melanoma |
Phase 1 |
Terminated(Slow accrual, close... More >>d in Phase I.) Less << |
- |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00601705
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 2 |
Unknown |
- |
United States, Ohio
... More >>
Cleveland Clinic Taussig Cancer Institute, Case Comprehensive Cancer Center
Cleveland, Ohio, United States, 44106-5065 Less << |
|
NCT01147809
|
Thrombocytopaenia |
Phase 2 |
Completed |
- |
- |
|
NCT00080795
|
Bladder Cancer
... More >> Transitional Cell Cancer of the Renal Pelvis and Ureter
Urethral Cancer Less << |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
M.D. Anderson Cancer Center at University of Texas
Houston, Texas, United States, 77030-4009 Less << |
|
NCT00006374
|
Lung Cancer |
Phase 2 |
Withdrawn(Slow accrual) |
- |
- |
|
NCT01147809
|
- |
|
Completed |
- |
- |
|
NCT00815308
|
- |
|
Completed |
- |
- |
|
NCT00815308
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
China, Shandong
... More >>
Department of Radiation Oncology, Shandong Cancer Hospital and Institute
Jinan, Shandong, China, 250117 Less << |
|
NCT00005850
|
Anxiety Disorder
... More >> Depression
Fatigue
Lung Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00980759
|
Cervical Cancer |
Phase 2 |
Active, not recruiting |
November 2019 |
Korea, Republic of
... More >>
National Cancer Center, Korea
Goyang-si, Gyeonggi-do, Korea, Republic of, 410-769 Less << |
|
NCT02324296
|
- |
|
Completed |
- |
- |
|
NCT00989651
|
Fallopian Tube Carcinosarcoma
... More >>
Fallopian Tube Clear Cell Adenocarcinoma
Fallopian Tube Endometrioid Adenocarcinoma
Fallopian Tube Mucinous Adenocarcinoma
Fallopian Tube Serous Neoplasm
Fallopian Tube Transitional Cell Carcinoma
Fallopian Tube Undifferentiated Carcinoma
Ovarian Brenner Tumor
Ovarian Carcinosarcoma
Ovarian Clear Cell Adenocarcinoma
Ovarian Endometrioid Adenocarcinoma
Ovarian Mucinous Adenocarcinoma
Ovarian Seromucinous Tumor
Ovarian Serous Adenocarcinoma
Ovarian Transitional Cell Carcinoma
Ovarian Undifferentiated Carcinoma
Primary Peritoneal Serous Adenocarcinoma
Stage IIA Fallopian Tube Cancer AJCC v6 and v7
Stage IIA Ovarian Cancer AJCC V6 and v7
Stage IIB Fallopian Tube Cancer AJCC v6 and v7
Stage IIB Ovarian Cancer AJCC v6 and v7
Stage IIC Fallopian Tube Cancer AJCC v6 and v7
Stage IIC Ovarian Cancer AJCC v6 and v7
Stage IIIA Fallopian Tube Cancer AJCC v7
Stage IIIA Ovarian Cancer AJCC v6 and v7
Stage IIIA Primary Peritoneal Cancer AJCC v7
Stage IIIB Fallopian Tube Cancer AJCC v7
Stage IIIB Ovarian Cancer AJCC v6 and v7
Stage IIIB Primary Peritoneal Cancer AJCC v7
Stage IIIC Fallopian Tube Cancer AJCC v7
Stage IIIC Ovarian Cancer AJCC v6 and v7
Stage IIIC Primary Peritoneal Cancer AJCC v7
Stage IV Fallopian Tube Cancer AJCC v6 and v7
Stage IV Ovarian Cancer AJCC v6 and v7
Stage IV Primary Peritoneal Cancer AJCC v7 Less << |
Phase 1 |
Active, not recruiting |
- |
- |
|
NCT01472653
|
Head and Neck Cancer |
Phase 2 |
Unknown |
December 2016 |
Slovenia
... More >> Institute of Oncology Ljubljana
Recruiting
Ljubljana, Slovenia, SI-1000
Contact: Primož Strojan, Prof. pstrojan@onko-i.si
Sub-Investigator: Marta Dremelj, MD, MSc
Sub-Investigator: Igor Fajdiga, MD, PhD
Sub-Investigator: Cvetka Kuhar Grašič, MD, PhD
Sub-Investigator: Jančar Boris, MD, MSc
Sub-Investigator: Simona Jereb, MD
Sub-Investigator: Katarina Karner, MD, MSc
Sub-Investigator: Barbara Žumer, MD Less << |
|
NCT00005638
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 1 |
Completed |
- |
United States, California
... More >>
USC/Norris Comprehensive Cancer Center and Hospital
Los Angeles, California, United States, 90033-0804
United States, New York
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT02934503
|
Small Cell Lung Cancer (SCLC) |
Phase 2 |
Recruiting |
October 2020 |
United States, New York
... More >>
Laura and Isaac Perlmutter Cancer Center at NYU Langone
Recruiting
New York, New York, United States, 10016
Contact: Leena Gandhi, MD, Ph.D. 212-731-6363 Leena.Gandhi@nyumc.org
Principal Investigator: Leena Gandhi, MD, Ph.D. Less << |
|
NCT02445391
|
Estrogen Receptor Negative
... More >> HER2/Neu Negative
Invasive Breast Carcinoma
Progesterone Receptor Negative
Stage II Breast Cancer
Stage IIA Breast Cancer
Stage IIB Breast Cancer
Stage III Breast Cancer
Stage IIIA Breast Cancer
Stage IIIB Breast Cancer
Stage IIIC Breast Cancer
Triple-Negative Breast Carcinoma Less << |
Phase 3 |
Recruiting |
May 31, 2024 |
- |
|
NCT00003111
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, Connecticut
... More >>
Yale Comprehensive Cancer Center
New Haven, Connecticut, United States, 06520-8028 Less << |
|
NCT00003803
|
Lung Cancer |
Phase 3 |
Terminated(low accrual) |
- |
Belgium
... More >> Algemeen Ziekenhuis Middelheim
Antwerp, Belgium, 2020
Hopital de Jolimont
Haine Saint Paul, Belgium, 7100
France
CHR de Grenoble - La Tronche
Grenoble, France, 38043
Germany
Mutterhaus der Borromaerinnen
Trier, Germany, D-54219
Netherlands
Medisch Centrum Haaglanden
's-Gravenhage (Den Haag, The Hague), Netherlands, 2501 CK
Antoni van Leeuwenhoekhuis
Amsterdam, Netherlands, 1066 CX
Academisch Medisch Centrum
Amsterdam, Netherlands, 1105 AZ
Gelre Ziekenhuizen - Lokatie Lukas
Apeldoorn, Netherlands, 7334 DZ
Amphia Ziekenhuis
Breda, Netherlands, 4810 EV
Amphia Ziekenhuis - locatie Molengracht
Breda, Netherlands, 4818 CK
Reinier de Graaf Group
Delft, Netherlands, NL 2600 GA
Radiotherapeutisch Instituut-(Riso)
Deventer, Netherlands, 7400 AC
Catharina Ziekenhuis
Eindhoven, Netherlands, 5602 ZA
Twee Steden Ziekenhuis Vestiging Tilburg
Tilburg, Netherlands, 5042 AD
Dr. Bernard Verbeeten Instituut
Tilburg, Netherlands, 5042 SB
Sophia Ziekehuis
Zwolle, Netherlands, 8000 GK
United Kingdom
Western General Hospital
Edinburgh, Scotland, United Kingdom, EH4 2XU Less << |
|
NCT00622349
|
Carcinoma, Non-Small-Cell Lung |
Phase 3 |
Completed |
- |
Belgium
... More >> Department of Intensive Care Unit and Thoracic Oncology Institut Jules Bordet
Brussels, Belgium, 1000
France
Pneumology department of CHU Lille
Lille, France
Greece
Hellenic Cancer Institute - St Savas Oncology Hospital
Athens, Greece
Spain
Medical Oncology Hospital de Sagunto
Valencia, Spain Less << |
|
NCT02047201
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 2 |
Completed |
- |
Lithuania
... More >> Lithuanian University of Health Sciences
Kaunas, Lithuania, LT-44307 Less << |
|
NCT03579758
|
Stage I Gallbladder Cancer AJC... More >>C v8
Stage II Gallbladder Cancer AJCC v8
Stage IIA Gallbladder Cancer AJCC v8
Stage IIB Gallbladder Cancer AJCC v8
Stage III Gallbladder Cancer AJCC v8
Stage IIIA Gallbladder Cancer AJCC v8
Stage IIIB Gallbladder Cancer AJCC v8 Less << |
Phase 3 |
Not yet recruiting |
January 31, 2026 |
United States, Georgia
... More >>
Emory University Hospital Midtown
Not yet recruiting
Atlanta, Georgia, United States, 30308
Contact: Shabnam Montazeri 404-686-0242 shabnam.montazeri@emory.edu
Principal Investigator: Shishir Maithel, MD
Emory University Hospital/Winship Cancer Institute
Not yet recruiting
Atlanta, Georgia, United States, 30322
Contact: Sheena Abraham 404-778-2670 sheena.abraham@emory.edu
Principal Investigator: Shishir Maithel, MD
Emory Saint Joseph's Hospital
Not yet recruiting
Atlanta, Georgia, United States, 30342
Contact: Alicia Escobar 678-843-7029 alicia.m.escobar@emory.edu
Principal Investigator: Shishir Maithel, MD Less << |
|
NCT03623087
|
Non-Hodgkin's Lymphoma, Relaps... More >>ed
Non-Hodgkin T-cell Lymphoma
Natural Killer/T-Cell Lymphoma, Nasal and Nasal-Type Less << |
Phase 3 |
Recruiting |
June 30, 2020 |
Hong Kong
... More >> The University of Hong Kong
Recruiting
Hong Kong, Hong Kong
Contact: King Hei Lu, MMedSc 852-22554361 ext 1654 khlu@hku.hk
Contact: Zoe Chan, BNs 852-22551654 zoechan1@hku.hk
Principal Investigator: Yok Lam Kwong, MD(HK)
Sub-Investigator: Thomas Chan, MBBS(HK) Less << |
|
NCT03664024
|
Non-Small Cell Lung Cancer |
Phase 2 |
Recruiting |
March 4, 2022 |
United States, Maryland
... More >>
Harry & Jeanette Weinberg Cancer Institute ( Site 0901)
Recruiting
Baltimore, Maryland, United States, 21237
Contact: Study Coordinator 443-777-7364
Canada, Quebec
CISSS de la Monteregie-Centre ( Site 0801)
Recruiting
Greenfield Park, Quebec, Canada, J4V 2H1
Contact: Study Coordinator 45046650003226
Hungary
Orszagos Koranyi TBC es Pulmonologiai Intezet ( Site 0400)
Recruiting
Budapest, Hungary, 1121
Contact: Study Coordinator +36302005233
Petz Aladar Megyei Oktato Korhaz ( Site 0402)
Recruiting
Gyor, Hungary, 9024
Contact: Study Coordinator +3696418244
Jasz Nagykun Szolnok Megyei Hetenyi Geza Korhaz Rendelointezet ( Site 0401)
Recruiting
Szolnok, Hungary, 5900
Contact: Study Coordinator +36209323256
Israel
Meir Medical Center ( Site 0501)
Recruiting
Kfar-Saba, Israel, 4428132
Contact: Study Coordinator +97297472414
Chaim Sheba Medical Center ( Site 0500)
Recruiting
Ramat Gan, Israel, 5262000
Contact: Study Coordinator +97235307096
Spain
Hospital Universitario Insular de Gran Canaria ( Site 0700)
Recruiting
Las Palmas de Gran Canaria, Gran Canaria, Spain, 35001
Contact: Study Coordinator +34928441738
H.U. Vall de Hebron ( Site 0701)
Recruiting
Barcelona, Spain, 08035
Contact: Study Coordinator +34932746085
Hospital General Universitario 12 de Octubre ( Site 0703)
Recruiting
Madrid, Spain, 28041
Contact: Study Coordinator +34914692313
Hospital Universitario Central de Asturias ( Site 0704)
Recruiting
Oviedo, Spain, 33011
Contact: Study Coordinator +34609804227 Less << |
|
NCT01220583
|
Head and Neck Cancer |
Phase 2 |
Recruiting |
October 2028 |
- |
|
NCT01059188
|
Lung Cancer |
Phase 2 |
Active, not recruiting |
June 2021 |
Switzerland
... More >> Saint Claraspital AG
Basel, Switzerland, CH-4016
Universitaetsspital-Basel
Basel, Switzerland, CH-4031
Istituto Oncologico della Svizzera Italiana - Ospedale San Giovanni
Bellinzona, Switzerland, CH-6500
Inselspital Bern
Bern, Switzerland, CH-3010
Spitalzentrum Biel
Biel, Switzerland, CH-2501
Kantonsspital Bruderholz
Bruderholz, Switzerland, CH-4101
Kantonsspital Graubuenden
Chur, Switzerland, CH-7000
Hopital Fribourgeois
Fribourg, Switzerland, 1708
Hopital Cantonal Universitaire de Geneve
Geneva, Switzerland, CH-1211
Centre Hospitalier Universitaire Vaudois
Lausanne, Switzerland, CH-1011
Kantonsspital Liestal
Liestal, Switzerland, CH-4410
Kantonsspital - St. Gallen
St. Gallen, Switzerland, CH-9007
Regionalspital
Thun, Switzerland, 3600
Kantonsspital Winterthur
Winterthur, Switzerland, CH-8400
UniversitaetsSpital Zuerich
Zurich, Switzerland, CH-8091 Less << |
|
NCT00022308
|
Lung Cancer |
Phase 1 |
Completed |
- |
United States, New Jersey
... More >>
South Jersey Regional Cancer Center
Millville, New Jersey, United States, 08332
Fox Chase Cancer Center at Virtua Memorial Hospital Burlington County
Mount Holly, New Jersey, United States, 08060
Riverview Medical Center - Booker Cancer Center
Red Bank, New Jersey, United States, 07701
Community Medical Center
Toms River, New Jersey, United States, 08755
St. Francis Medical Center
Trenton, New Jersey, United States, 08629
United States, Pennsylvania
Delaware County Memorial Hospital
Drexel Hill, Pennsylvania, United States, 19026
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111
Reading Hospital and Medical Center
Reading, Pennsylvania, United States, 19612-6052 Less << |
|
NCT02116231
|
Effects of Chemotherapy |
Phase 2 |
Unknown |
May 2018 |
China, Guangxi
... More >> Cancer Hospital of Guangxi Medical University
Not yet recruiting
Nanning, Guangxi, China, 530021
Contact: Xiaodong Zhu, Doctor zhuxiaodong83@163.com
Principal Investigator: Song Qu, Doctor
Principal Investigator: Ling Li, Master Less << |
|
NCT00004103
|
Gastric Cancer |
Phase 2 |
Completed |
- |
United States, New York
... More >>
NYU Cancer Institute at New York University Medical Center
New York, New York, United States, 10016 Less << |
|
NCT00137358
|
Cervical Cancer |
Phase 1 |
Withdrawn(unable to obtain fun... More >>ding to conduct study) Less << |
- |
United States, Alabama
... More >>
University of Alabama at Birmingham
Birmingham, Alabama, United States, 35294 Less << |
|
NCT00002984
|
Esophageal Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, North Carolina
... More >>
Lineberger Comprehensive Cancer Center, UNC
Chapel Hill, North Carolina, United States, 27599-7295 Less << |
|
NCT01756170
|
Cervical Cancer |
Phase 3 |
Unknown |
December 2016 |
China, Zhejiang
... More >>
The First Affiliated Hospital of Wenzhou Medical College
Recruiting
Wenzhou, Zhejiang, China, 325000
Contact: congying xie, MD +86-577-88069316 wzxiecongying@163.com
Principal Investigator: congying xie, MD Less << |
|
NCT03709147
|
Advanced LKB1-inactive Lung Ad... More >>enocarcinoma Less << |
Phase 2 |
Not yet recruiting |
September 10, 2023 |
Italy
... More >> Marina Chiara Garassino
Not yet recruiting
Milan, Italy, 20133
Contact: MARINA CHIARA GARASSINO, MD 0223903813 ext +39 marina.garassino@istitutotumori.mi.it Less << |
|
NCT00150670
|
Gastric Cancer |
Phase 3 |
Completed |
- |
Japan
... More >> East Hospital, Kitasato University
2-1-1, Asamizodai, Sagamihara, Kanagawa, Japan Less << |
|
NCT01356368
|
Non-Small Cell Lung Cancer |
Phase 2 |
Terminated(slow accrual rate) |
- |
Saudi Arabia
... More >> King Abdul Aziz Medical City for National Guard Health Affairs
Riyadh, Saudi Arabia Less << |
|
NCT00003811
|
Childhood Germ Cell Tumor
... More >> Drug/Agent Toxicity by Tissue/Organ
Extragonadal Germ Cell Tumor
Ovarian Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00172380
|
NSCLC |
Phase 2 |
Completed |
- |
Taiwan
... More >> Department of Internal Medicine, National Taiwan University Hospital
Taipei, Taiwan, 100 Less << |
|
NCT02667925
|
Ovarian Cancer |
Not Applicable |
Withdrawn(Because of the obsol... More >>escence of the study) Less << |
- |
France
... More >> Centre Jean Perrin
Clermont-Ferrand, France, 63000 Less << |
|
NCT03619824
|
Nasopharyngeal Neoplasms |
Phase 2 |
Not yet recruiting |
March 2024 |
China, Guangdong
... More >>
SUN YAT-SEN UNIVERSITY cANCER CENTER
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT02429037
|
Advanced Head and Neck Cancer |
Phase 2 |
Unknown |
December 2018 |
China, Jiangsu
... More >> Jiangsu cancer hospital
Not yet recruiting
Nanjing, Jiangsu, China, 210000
Contact: Xia He, MD, PhD 86-13601458518 13601458515@qq.com
Contact: Jianfeng Wu, MD, PhD 86-13923753040 595864485@qq.com Less << |
|
NCT02891083
|
Esophageal Neoplasms |
Phase 3 |
Recruiting |
January 2021 |
China
... More >> Shanghai Chest Hospital
Recruiting
Shanghai, China, 200030
Contact: Wentao Fang, MD +86-21-62821990 ext 2901 vwtfang12@shchest.org
Contact: Xufeng Guo, MD +86-21-62821900 ext 2608 shandagxf@126.com Less << |
|
NCT00367497
|
Lymphoma, Non-Hodgkin |
Phase 2 |
Terminated(The stopping rule w... More >>as applied because of low response rates.) Less << |
- |
Japan
... More >> Keio University School of Medicine
Tokyo, Japan, 160-8582 Less << |
|
NCT00407186
|
Gastric Cancer |
Phase 3 |
Active, not recruiting |
December 2025 |
Netherlands
... More >> Nederlands Kanker Instituut/Antoni van Leeuwenhoek Ziekenhuis
Amsterdam, Netherlands, 1066 CX Less << |
|
NCT01502202
|
Non Small Cell Lung Cancer
... More >> Adenocarcinoma Less << |
Phase 2 |
Unknown |
February 2015 |
Korea, Republic of
... More >>
National Cancer Center
Recruiting
Goyang-si, Gyenggido, Korea, Republic of, 410-769
Contact: Sung Jin Yoon, RN +82-31-920-0405 jinijiniya@ncc.re.kr
Contact: Jin Soo Lee, MD. Ph.D. +82-31-920-1501 jslee@ncc.re.kr Less << |
|
NCT00006260
|
Breast Cancer |
Phase 2 |
Completed |
- |
United States, Ohio
... More >>
Ireland Cancer Center at University Hospitals Case Medical Center, Case Comprehensive Cancer Center
Cleveland, Ohio, United States, 44106-5055 Less << |
|
NCT00667342
|
Osteosarcoma
... More >>Malignant Fibrous Histiocytoma (MFH) of Bone Less << |
Phase 2 |
Active, not recruiting |
May 2019 |
United States, California
... More >>
Rady Children's Hospital and Health Center
San Diego, California, United States, 92123
United States, Maryland
Johns Hopkins - Sidney Kimmel Comprehensive Cancer Center
Baltimore, Maryland, United States, 21231
NCI/NIH - Pediatric Oncology Branch
Bethesda, Maryland, United States, 20892
United States, Tennessee
St Jude Children's Research Hospital
Memphis, Tennessee, United States, 38105
United States, Texas
MD Anderson Cancer Center
Houston, Texas, United States, 77030-4009 Less << |
|
NCT01670409
|
Esophageal Neoplasms |
Phase 2 |
Completed |
- |
China, Guangdong
... More >>
Cancer Hospital, Shantou University Medical College
Shantou, Guangdong, China, 515031 Less << |
|
NCT00823719
|
Lymphoma, Large-Cell, Diffuse |
Phase 2 |
Completed |
- |
- |
|
NCT00719524
|
Neoplasms |
Phase 1 |
Completed |
- |
France
... More >> Investigational Site Number 250001
Villejuif, France, 94805
Italy
Investigational Site Number 380001
Milano, Italy, 20133
Switzerland
Investigational Site Number 756001
Bellinzona, Switzerland, 6500 Less << |
|
NCT02825940
|
Neoplasms |
Phase 1 |
Recruiting |
January 10, 2020 |
China
... More >> Beijing Cancer Hospital
Active, not recruiting
Beijing, China, 100142
Jilin Cancer Hospital
Recruiting
Changchun, China, 130012
Cancer Center, Sun Yat-sen University of Medical Sciences; Department of Medical Oncology
Recruiting
Guangzhou, China, 510060
Sir Run Run Shaw Hospital
Recruiting
Hangzhou, China, 310016
Harbin Medical University Cancer Hospital
Recruiting
Harbin, China, 150081
Harbin Medical University Cancer Hospital
Not yet recruiting
Harbin, China, 150081
Fudan University Shanghai Cancer Center
Recruiting
Shanghai, China, 200032
Zhongshan Hospital Fudan University
Recruiting
Shanghai, China, 200032 Less << |
|
NCT00823719
|
- |
|
Completed |
- |
- |
|
NCT00667342
|
- |
|
Active, not recruiting |
- |
- |
|
NCT02047487
|
- |
|
Unknown |
August 2018 |
China
... More >> PLAGH
Recruiting
Beijing, China, 100853
PLA307
Recruiting
Beijing, China Less << |
|
NCT00498979
|
Stage IV Melanoma |
Phase 1 |
Completed |
- |
United States, Ohio
... More >>
Cleveland Clinic Taussig Institute, Case Comprehensive Cancer Center
Cleveland, Ohio, United States, 44195 Less << |
|
NCT00490360
|
Cancer of the Pancreatic Head |
Phase 2 |
Completed |
- |
Switzerland
... More >> University Hospital of Zurich, Department of Surgery
Zurich, Switzerland Less << |
|
NCT00369954
|
Fallopian Tube Cancer
... More >> Ovarian Cancer
Peritoneal Cavity Cancer Less << |
Phase 2 |
Withdrawn(Trial was never acti... More >>vated) Less << |
- |
- |
|
NCT02741856
|
Oesophageal Cancer |
Phase 2
Phase 3 |
Recruiting |
April 2023 |
United Kingdom
... More >> Aberdeen Royal Infirmary
Recruiting
Aberdeen, United Kingdom
Contact: Lucy Wells
Bristol Haematology & Oncology
Recruiting
Bristol, United Kingdom
Contact: Stephen Falk
Addenbrooke's Hospital
Recruiting
Cambridge, United Kingdom
Contact: Susan Harden
Kent and Canterbury
Not yet recruiting
Canterbury, United Kingdom
Contact: Mathilda Cominos
Velindre Cancer Care Centre
Recruiting
Cardiff, United Kingdom, CF14 2TL
Contact: Carys Morgan 02920 615 888 carys.morgan6@wales.nhs.uk
Cheltenham General Hospital
Recruiting
Cheltenham, United Kingdom
Contact: Charles Candish
University Hospital Coventry
Recruiting
Coventry, United Kingdom
Contact: Sharmila Sothi
Derby Teaching Hospitals NHS Trust
Recruiting
Derby, United Kingdom
Contact: Prantik Das
Glan Clwyd Hospital
Recruiting
Glan Clwyd, United Kingdom
Contact: Angel Garcia
Beatson West of Scotland Cancer Centre
Recruiting
Glasgow, United Kingdom
Contact: David McIntosh
Gloucestershire Royal Hospital
Recruiting
Gloucester, United Kingdom
Contact: Charles Candish
Castle Hill Hospital
Recruiting
Hull, United Kingdom
Contact: Raj Roy
The Clatterbridge Cancer Centre nhs Foundation Trust
Recruiting
Liverpool, United Kingdom
Contact: Raj Sripadam
Guy's and St Thomas'
Recruiting
London, United Kingdom
Contact: Asad Qureshi
Imperial College Healthcare NHS Trust
Recruiting
London, United Kingdom
Contact: Danielle Power
North Middlesex Hospital
Recruiting
London, United Kingdom
Contact: Lucinda Melcher
The Royal Marsden Hospitals (Fulham)
Recruiting
London, United Kingdom
Contact: Katharine Aitken
The James Cook University Hospital
Recruiting
Middlesbrough, United Kingdom
Contact: David Wilson
Churchill Hospital
Recruiting
Oxford, United Kingdom
Contact: Somnath Mukherjee
Peterborough and Stamford Hospitals NHS Foundation Trust
Recruiting
Peterborough, United Kingdom
Contact: Catherine Jephcott
Sheffield Teaching Hospitals - Weston Park Hospital
Recruiting
Sheffield, United Kingdom
Contact: Jon Wadsley
University Hospital Southampton NHS Foundation Trust
Recruiting
Southampton, United Kingdom
Contact: Andrew Bateman
The Royal Marsden Hospitals (Sutton, Surrey)
Recruiting
Sutton, United Kingdom
Contact: Katharine Aitken
Singleton Hospital
Recruiting
Swansea, United Kingdom
Contact: Sarah Gwynne
Worcestershire Royal Hospital
Recruiting
Worcester, United Kingdom
Contact: Cheng Boon
Wrexham Maelor
Recruiting
Wrexham, United Kingdom
Contact: Simon Gollins Less << |
|
NCT00017173
|
Head and Neck Cancer |
Phase 2 |
Terminated(Terminated for poor... More >> accrual.) Less << |
- |
United States, Kansas
... More >>
Kansas Masonic Cancer Research Institute at the University of Kansas Medical Center
Kansas City, Kansas, United States, 66160-7357
United States, Kentucky
Markey Cancer Center at University of Kentucky Chandler Medical Center
Lexington, Kentucky, United States, 40536-0293
United States, Michigan
Barbara Ann Karmanos Cancer Institute
Detroit, Michigan, United States, 48201-1379 Less << |
|
NCT00054054
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00004913
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, Illinois
... More >>
Robert H. Lurie Comprehensive Cancer Center, Northwestern University
Chicago, Illinois, United States, 60611-3013
United States, Indiana
Indiana University Cancer Center
Indianapolis, Indiana, United States, 46202-5289
United States, Pennsylvania
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111 Less << |
|
NCT02627807
|
Nasopharyngeal Neoplasms |
Not Applicable |
Active, not recruiting |
December 2022 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT00003888
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00002546
|
Sarcoma |
Phase 3 |
Completed |
- |
- |
|
NCT02422563
|
Cervical Cancer |
Phase 3 |
Not yet recruiting |
October 2025 |
- |
|
NCT02573584
|
- |
|
Recruiting |
May 2021 |
Netherlands
... More >> Nederlands Kanker Instituut - Antoni van Leeuwenhoek ziekenhuis (NKI-AVL)
Recruiting
Amsterdam, Netherlands
Contact: J. Kerst, MD PhD
University Medical Center Groningen (UMCG)
Recruiting
Groningen, Netherlands
Contact: J.A. Gietema, MD PhD j.a.gietema@umcg.nl
Contact: S. Lubberts, MD s.lubberts@umcg.nl
Maastricht University Medical Center (MUMC)
Recruiting
Maastricht, Netherlands
Contact: M. Aarts, MD PhD
Portugal
Instituto Portuges de Oncologia Francisco Gentil (IPOLFG)
Recruiting
Lisbon, Portugal
Contact: M. Brito, MD PhD
Switzerland
UniversitatsSpital Zurich
Recruiting
Zurich, Switzerland
Contact: J. Beyer, MD PhD
Contact: C. Fankhauser, MD PhD Less << |
|
NCT00436644
|
Ovarian Cancer
... More >> Peritoneal Cavity Cancer Less << |
Phase 2 |
Completed |
- |
United States, Arizona
... More >>
Mayo Clinic Arizona
Scottsdale, Arizona, United States, 85254
United States, Florida
Mayo Clinic in Jacksonville
Jacksonville, Florida, United States, 32224
United States, Minnesota
Mayo Clinic Cancer Center
Rochester, Minnesota, United States, 55905 Less << |
|
NCT02404571
|
Lymphoma, T-Cell, Peripheral |
Phase 2 |
Unknown |
December 2016 |
China, Beijing
... More >> Cancer Hospital and Institute, Chinese Academy of Medical Sciences (CAMS) and Peking Union Medical College (PUMC)
Recruiting
Beijing, Beijing, China, 100021
Contact: Mei Dong (86)13811929322 dongmei030224@163.com Less << |
|
NCT03027284
|
Advanced Cancer
... More >> Metastatic Cancer
Biliary Tract Carcinoma
Cholangiocarcinoma
Gall Bladder Carcinoma
Solid Tumor
Non-Hodgkin's Lymphoma Less << |
Phase 1 |
Active, not recruiting |
February 2, 2019 |
Japan
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Chiba, Japan, 277 8577
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician
Tokyo, Japan, 104-0045 Less << |
|
NCT00436644
|
- |
|
Completed |
- |
- |
|
NCT01225523
|
Squamous Carcinoma of Esophagu... More >>s
Esophagus Disorders Less << |
Phase 1 |
Completed |
- |
China, Shaanxi
... More >> First Affiliated Hospital of College of Medicine of Xi'an Jiao Tong University
Xi'an, Shaanxi, China, 710061 Less << |
|
NCT00508664
|
Squamous Cell Carcinoma of the... More >> Hypopharynx
Larynx Carcinoma Less << |
Phase 2 |
Completed |
- |
Austria
... More >> Universitätsklinik für HNO
Graz, Austria, 8036
Landeskrankenhaus Klagenfurt
Klagenfurt, Austria, A-9020
Allgemeines Krankenhaus der Stadt Wien
Vienna, Austria, 1090
Germany
Helios Klinikum Erfurt GmbH Klinik für HNO-Heilkunde, Plastische Operationen
Erfurt, Thüringen, Germany, 99089
Universitätsklinik Aachen
Aachen, Germany, 52074
Charité, Campus Benjamin Franklin
Berlin, Germany, 12200
Klinikum Neukölln, Vivantes GmbH
Berlin, Germany, 12351
Klinikum Bielefeld-Mitte
Bielefeld, Germany, 33604
Malteser Krankenhaus St. Anna gGmbH, HNO-Klinik
Duisburg, Germany, 47259
Klinikum Fulda gAG, Klinik für Hals-Nasen-Ohrenkrankheiten
Fulda, Germany, 36043
Medizinische Hochschule Hannover
Hannover, Germany, 30625
Klinikum Hannover Nordstadt
Hannover, Germany
Universitätsklinikum Heidelberg
Heidelberg, Germany, 69120
Universitätsklinikum Jena
Jena, Germany, 07743
St. Vincentius Kliniken
Karlsruhe, Germany
Klinikum Kassel GmbH
Kassel, Germany, 34125
Katholisches Klinikum Koblenz Marienhof
Koblenz, Germany, 56073
Universitätsklinik Köln
Köln, Germany, 50924
Universitätsklinikum Leipzig
Leipzig, Germany, 04103
Universtitätsklinikum Schleswig-Holstein
Lübeck, Germany, 23538
Klinikum Großhadern
München, Germany, 81377
Medizinische Fakultät der Westfälischen Wilhelms-Universität Münster
Münster, Germany, 48149
Südharz-Krankenhaus Nordhausen gGmbH
Nordhausen, Germany
Klinikum Ernst von Bergmann gGmbH
Potsdam, Germany, 14467
Universtitätsklinikum Regensburg
Regensburg, Germany
Klinikum Stuttgart Katharinenhospital, Klinik für HNO-Krankheiten
Stuttgart, Germany, 70174
Bayerischen Julius Maximillians-Universtät Würzburg
Würzburg, Germany, 97080 Less << |
|
NCT01230476
|
Nasopharyngeal Carcinoma |
Phase 2 |
Terminated |
- |
Malaysia
... More >> University Malaya Medical Centre
Kuala Lumpur, Wilayah Persekutuan, Malaysia, 59100 Less << |
|
NCT03747419
|
Bladder Cancer
... More >> Muscle Invasive Bladder Cancer Less << |
Phase 2 |
Not yet recruiting |
January 31, 2031 |
United States, Massachusetts
... More >>
Brigham and Women's Hospital
Not yet recruiting
Boston, Massachusetts, United States, 02215
Contact: Kent Mouw, MD, PhD 617-525-7382 kent_mouw@dfci.harvard.edu
Principal Investigator: Kent Mouw, MD, PhD Less << |
|
NCT01551589
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Not Applicable |
Completed |
- |
China, Guangxi
... More >> Guangxi Tumor Hospital
Nanning, Guangxi, China, 530021
China, Guizhou
GuiZhou Cancer Hospital
Guiyang, Guizhou, China, 550000
China, Sichuan
The Second People's Hospital of Sichuan
Chengdu, Sichuan, China, 610000
China, Yunnan
Yunnan Tumor Hospital, The Third Affiliated Hospital of KUNMING Medical University
Kunming, Yunnan, China, 652100 Less << |
|
NCT03675737
|
Stomach Neoplasms |
Phase 3 |
Recruiting |
February 2, 2024 |
United States, Maryland
... More >>
Greater Baltimore Medical Center ( Site 0102)
Recruiting
Baltimore, Maryland, United States, 21204
Contact: Study Coordinator 443-849-3122
United States, Minnesota
Minnesota Oncology Hematology, PA ( Site 8000)
Recruiting
Minneapolis, Minnesota, United States, 55404
Contact: Study Coordinator 281-863-4643
United States, Virginia
Oncology & Hematology Assoc. SW Virginia, Inc., DBA Blue Ridge Cancer Care ( Site 8001)
Recruiting
Roanoke, Virginia, United States, 24014
Contact: Study Coordinator 281-863-4643
United States, Washington
Wenatchee Valley Clinic [Wenatchee, WA] ( Site 0116)
Recruiting
Wenatchee, Washington, United States, 98801
Contact: Study Coordinator 509-665-5800
Australia, New South Wales
Southern Medical Day Care Centre ( Site 2303)
Recruiting
Wollongong, New South Wales, Australia, 4020
Contact: Study Coordinator
Canada, Quebec
McGill University Health Centre ( Site 0208)
Recruiting
Montreal, Quebec, Canada, H4A 3J1
Contact: Study Coordinator 5149341934
France
C.H.R.U. de Brest - Hopital Morvan ( Site 1007)
Recruiting
Brest, France, 29200
Contact: Study Coordinator +33298223428
Israel
Rabin Medical Center ( Site 1302)
Recruiting
Petah Tikva, Israel, 4941492
Contact: Study Coordinator +97239378077
Chaim Sheba Medical Center ( Site 1304)
Recruiting
Ramat Gan, Israel, 5262000
Contact: Study Coordinator +97235307776
Sourasky Medical Center ( Site 1306)
Recruiting
Tel Aviv, Israel, 6423906
Contact: Study Coordinator +97236973082
Korea, Republic of
Seoul National University Hospital ( Site 2803)
Recruiting
Seoul, Korea, Republic of, 03080
Contact: Study Coordinator +82220720701
Severance Hospital Yonsei University Health System ( Site 2800)
Recruiting
Seoul, Korea, Republic of, 03722
Contact: Study Coordinator +82222288132
Asan Medical Center ( Site 2802)
Recruiting
Seoul, Korea, Republic of, 05505
Contact: Study Coordinator +82230103230
Samsung Medical Center ( Site 2801)
Recruiting
Seoul, Korea, Republic of, 06351
Contact: Study Coordinator +82234101779
Switzerland
Luzern Kantonsspital ( Site 1904)
Recruiting
Luzern, Switzerland, 6000
Contact: Study Coordinator +41412055875
Taiwan
National Cheng Kung University Hospital ( Site 2901)
Recruiting
Tainan, Taiwan, 704
Contact: Study Coordinator +886623535354559 Less << |
|
NCT00003087
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT01926236
|
Biliary Tract Cancer
... More >> Gallbladder Cancer
Cholangiocarcinoma
Ampullary Cancer Less << |
Phase 3 |
Active, not recruiting |
February 2019 |
United Kingdom
... More >> Queen Elizabeth Hospital
Birmingham, United Kingdom
Bristol Haematology & Oncology Centre
Bristol, United Kingdom
North Cumbria University Hospitals
Carlisle, United Kingdom
Beatson West of Scotland Cancer Centre
Glasgow, United Kingdom, G12 0YN
Castle Hill Hospital
Hull, United Kingdom
St James' Hospital
Leeds, United Kingdom
Clatterbridge Cancer Centre
Liverpool, United Kingdom
Guy's and St Thomas' Hospital
London, United Kingdom
Hammersmith Hospital
London, United Kingdom
Royal Free Hospital
London, United Kingdom
University College London
London, United Kingdom
Maidstone Hospital
Maidstone, United Kingdom
The Christie NHS Foundation Trust
Manchester, United Kingdom
Nottingham City Hospital
Nottingham, United Kingdom
Churchill Hospital
Oxford, United Kingdom
Weston Park Hospital
Sheffield, United Kingdom
Southampton General Hospital
Southampton, United Kingdom Less << |
|
NCT02412670
|
Localized Urothelial Carcinoma... More >> of the Renal Pelvis and Ureter
Recurrent Urothelial Carcinoma of the Renal Pelvis and Ureter
Regional Urothelial Carcinoma of the Renal Pelvis and Ureter Less << |
Phase 2 |
Active, not recruiting |
- |
- |
|
NCT02951767
|
- |
|
Active, not recruiting |
- |
- |
|
NCT02951767
|
Bladder Cancer |
Phase 2 |
Active, not recruiting |
May 18, 2019 |
- |
|
NCT03690921
|
Anal Canal Squamous Cell Carci... More >>noma Less << |
Phase 2 |
Recruiting |
May 23, 2022 |
United States, Texas
... More >>
M D Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact: Emma B. Holliday 713-563-2300
Principal Investigator: Emma B. Holliday Less << |
|
NCT03472560
|
Non-Small Cell Lung Cancer
... More >> Urothelial Cancer Less << |
Phase 2 |
Recruiting |
September 18, 2020 |
United States, Arizona
... More >>
Arizona Oncology Associates- Saguaro Cancer Center
Recruiting
Glendale, Arizona, United States, 85308
Arizona Oncology Associates- Biltmore Cancer Center
Recruiting
Phoenix, Arizona, United States, 85016
Arizona Oncology Associates- Deer Valley Cancer Center
Recruiting
Phoenix, Arizona, United States, 85027
Arizona Oncology Associates- East Valley Cancer Center
Recruiting
Tempe, Arizona, United States, 85284
United States, Michigan
Karmanos Cancer Institute
Not yet recruiting
Detroit, Michigan, United States, 48201
Karmanos Cancer Institute
Not yet recruiting
Farmington Hills, Michigan, United States, 48334
United States, Missouri
Oncology Hematology Associates
Recruiting
Springfield, Missouri, United States, 65807
United States, Nebraska
Oncology Hematology West, PC dba Nebraska Cancer Specialists
Recruiting
Omaha, Nebraska, United States, 68114
Oncology Hematology West, PC dba Nebraska Cancer Specialists
Recruiting
Omaha, Nebraska, United States, 68130
United States, Ohio
Toledo Clinic Cancer Center- Toledo
Not yet recruiting
Toledo, Ohio, United States, 43623
United States, South Carolina
Saint Francis Hospital
Recruiting
Greenville, South Carolina, United States, 29601
Saint Francis Hospital Cancer Center
Recruiting
Greenville, South Carolina, United States, 29607
Hungary
Orszagos Onkologiai Intezet
Not yet recruiting
Budapest, Hungary, H-1122
Pecsi Tudomanyegyetem Klinikai Kozpont
Not yet recruiting
Pecs, Hungary, H-7624
Tudogyogyintezet Torokbalint
Recruiting
Torokbalint, Hungary, H-2045
Korea, Republic of
National Cancer Center
Recruiting
Goyang-si, Gyeonggi-do, Korea, Republic of, 10408
Samsung Medical Center
Recruiting
Gangnam-gu, Seoul, Korea, Republic of, 06351
Asan Medical Center
Not yet recruiting
Songpa-gu, Seoul, Korea, Republic of, 05505
Severance Hospital, Yonsei University Health System
Recruiting
Seoul, Korea, Republic of, 03722
Taiwan
Chi Mei Hospital, Liouying
Recruiting
Tainan City, Liouying District, Taiwan, 73657
National Cheng Kung University Hospital
Not yet recruiting
Tainan, Taiwan, 704 Less << |
|
NCT03105596
|
B-cell Non-Hodgkin's Lymphoma |
Phase 2 |
Not yet recruiting |
September 30, 2019 |
- |
|
NCT00090207
|
Nausea
Vomiti... More >>ng Less << |
Phase 4 |
Completed |
- |
- |
|
NCT01914900
|
Locally Advanced Resectable Or... More >>al Cavity Squamous Cell Cancer Less << |
Phase 2 |
Unknown |
March 2015 |
Italy
... More >> Istituto Nazionale Tumori
Recruiting
Milano, Italy, 20133
Contact: Paolo Bossi, MD +390223902765 paolo.bossi@istitutotumori.mi.it
Principal Investigator: Lisa Licitra, MD Less << |
|
NCT00700440
|
Nasopharyngeal Carcinoma |
Phase 2 |
Completed |
- |
China, Guangdong
... More >>
Cancer Center, Sun Yat-sen University
Guangzhou, Guangdong, China, 510080 Less << |
|
NCT02279576
|
Penile Squamous Cell Carcinoma... More >> Stage IV Less << |
Phase 2 |
Terminated(The low recruitment... More >> of patients will not allow to complete the study with the required number of patients within reasonable time.) Less << |
- |
Spain
... More >> Institut Català D'Oncologia L'Hospitalet
Hospitalet de Llobregat, Barcelona, Spain, 08908
Complejo Hospitalario de Navarra
Pamplona, Navarra, Spain, 31008
Hospital de La Santa Creu I Sant Pau
Barcelona, Spain, 08025
Complejo Hospitalario Regional Reina Sofía
Córdoba, Spain, 14004
Hospital Universitario Lucus Augusti
Lugo, Spain, 27003
Hospital Clínico San Carlos
Madrid, Spain, 28040
Hospital General Universitario J.M. Morales Meseguer
Murcia, Spain, 30008
Instituto Valenciano de Oncología
Valencia, Spain, 46009 Less << |
|
NCT01714037
|
Non-small Cell Lung Cancer |
Phase 1 |
Terminated |
- |
France
... More >> Centre GF Leclerc
Dijon, France
Centre Léon Bérard
Lyon, France
Institut de Cancérologie de l'Ouest- Institut René Gauduchau
Nantes, France
Institut Claudius Regaud
Toulouse, France
Spain
Hospital Universitari Vall d'Hebron
Barcelona, Spain
Hospital Puerta de Hierro Majadahonda
Madrid, Spain
Hospital Universitario Virgen del Rocío
Seville, Spain
United Kingdom
Freeman Hospital
Newcastle, United Kingdom Less << |
|
NCT03036098
|
Urothelial Cancer |
Phase 3 |
Recruiting |
December 23, 2022 |
- |
|
NCT00700440
|
- |
|
Completed |
- |
- |
|
NCT02516813
|
Advanced Solid Tumors |
Phase 1 |
Recruiting |
January 8, 2021 |
United States, California
... More >>
Research site
Recruiting
Fresno, California, United States, 93720
United States, Florida
Research site
Recruiting
Fort Lauderdale, Florida, United States, 33308
Research site
Recruiting
Miami, Florida, United States, 33136-1002
United States, Montana
Research site
Recruiting
Billings, Montana, United States, 59101
United States, New York
Research site
Recruiting
Bronx, New York, United States, 10461
United States, Pennsylvania
Research site
Recruiting
Philadelphia, Pennsylvania, United States, 19107
United States, Texas
Research site
Not yet recruiting
Houston, Texas, United States, 77030
United States, Washington
Research site
Completed
Tacoma, Washington, United States, 98405
Belgium
Research site
Active, not recruiting
Leuven, Belgium
Denmark
Research site
Recruiting
Copenhagen, Denmark, 2100
Research site
Recruiting
Herlev, Denmark
Germany
Research site
Active, not recruiting
Freiburg, Baden Wuerttemberg, Germany, 79106
Research site
Recruiting
Heidelberg, Baden Wuerttemberg, Germany
Research site
Recruiting
Mannheim, Baden Wuerttemberg, Germany
Research site
Recruiting
Tuebingen, Baden Wuerttemberg, Germany, 72076
Research site
Recruiting
Mainz, Rhineland-Palatinate, Germany, 55131
Research site
Recruiting
Dresden, Sachsen, Germany, 1307
Research site
Recruiting
Kiel, Schleswig-Holstein, Germany, 24105
Research site
Recruiting
Berlin, Germany, 12200
Netherlands
Research site
Recruiting
Amsterdam, Netherlands, 1066 CX
Research site 1
Active, not recruiting
Amsterdam, Netherlands
Norway
Research site
Active, not recruiting
Oslo, Norway
Sweden
Research site
Recruiting
Solna, Sweden, 17176 Less << |
|
NCT00993616
|
- |
|
Completed |
- |
- |
|
NCT02607631
|
Thymic Epithelial Tumors |
Phase 2 |
Unknown |
August 2018 |
- |
|
NCT03390595
|
Metastatic Urothelial Cancer |
Phase 2 |
Recruiting |
April 2020 |
Spain
... More >> Hospital General Universitario de Elche
Recruiting
Elche, Alicante, Spain, 03203
Contact: Federico José Vázquez, MD, PhD +34 966 616 250 fvazquezmd@gmail.com
Principal Investigator: Federico José Vázquez, MD, PhD
Instituto Catalán de Oncología (ICO L'Hospitalet) - H. Durán i Reynals
Recruiting
L'Hospitalet De Llobregat, Barcelona, Spain, 08907
Contact: Xavier García del Muro, MD, PhD +34 932 607 332 garciadelmuro@iconcologia.net
Principal Investigator: Xavier García del Muro, MD, PhD
Fundación Althaia - Xarxa Assistencial Univ. de Manresa
Recruiting
Manresa, Barcelona, Spain, 08243
Contact: Montserrat Domènech, MD, PhD +34 938 75 93 00 mdomenech@althaia.cat
Principal Investigator: Montserrat Domènech, MD, PhD
Hospital Universitario Parc Tauli
Recruiting
Sabadell, Barcelona, Spain, 08208
Contact: Teresa Bonfill, MD, PhD +34 935 537 134 tbonfill@tauli.cat
Principal Investigator: Teresa Bonfill, MD, PhD
Sub-Investigator: Enrique Gallardo, MD, PhD
Clínica Universidad de Navarra
Recruiting
Pamplona, Navarra, Spain, 31008
Contact: José Luís Pérez, MD, PhD +34 948 25 54 00 jlgracia@unav.es
Principal Investigator: José Luís Pérez, MD, PhD
Hospital del Mar
Recruiting
Barcelona, Spain, 08003
Contact: Alejo Rodríguez-Vida, MD, PhD +34 932 483 139 arodriguezvida@hospitaldelmar.cat
Principal Investigator: Alejo Rodríguez-Vida, MD, PhD
Hospital de la Santa Creu i Sant Pau
Recruiting
Barcelona, Spain, 08025
Contact: José Pablo Maroto, MD, PhD +34 935 537 134 JMaroto@santpau.cat
Principal Investigator: José Pablo Maroto, MD, PhD
Hospital Universitario Vall d'Hebron
Recruiting
Barcelona, Spain, 08035
Contact: Rafael Morales, MD, PhD +34 934 89 30 00 rmorales@vhio.net
Sub-Investigator: Joan Carles Galceran, MD, PhD
Hospital Clìnic de Barcelona
Recruiting
Barcelona, Spain, 08036
Contact: Begoña Mellado, MD, PhD +34 93 227 54 02 BMELLADO@clinic.cat
Principal Investigator: Begoña Mellado, MD, PhD
Hospital Universitario 12 de Octubre
Recruiting
Madrid, Spain, 28026
Contact: Daniel Castellano, MD,PhD +34 913 90 80 00 cdanicas@hotmail.com
Principal Investigator: Daniel Castellano, MD, PhD
Hospital Clínico San Carlos
Recruiting
Madrid, Spain, 28040
Contact: Javier Puente, MD, PhD +34 913 30 30 01 jpuente.hcsc@salud.madrid.org
Principal Investigator: Javier Puente, MD, PhD
Hospital Universitario La Paz
Recruiting
Madrid, Spain, 28046
Contact: Álvaro Pinto, MD, PhD +34 91 727 75 16 alvaropintomarin@gmail.com
Principal Investigator: Álvaro Pinto, MD, PhD
Hospital General Universitario Morales Meseguer
Recruiting
Murcia, Spain, 30008
Contact: Enrique González, MD, PhD +34 968 360 900 ext 3807 enrique.gonzalezbilla@gmail.com
Principal Investigator: Enrique González, MD, PhD
Hospital Universitario Virgen del Rocío
Recruiting
Sevilla, Spain, 41013
Contact: Begoña Pérez, MD, PhD +34 955 01 30 68 bperezv@gmail.com
Principal Investigator: Begoña Pérez, MD, PhD
Fundación Instituto Valenciano de Oncología
Recruiting
Valencia, Spain, 46009
Contact: Miguel Ángel Climent, MD, PhD +34 961 11 40 00 macliment@fivo.org
Principal Investigator: Miguel Ángel Climent, MD, PhD Less << |
|
NCT01584284
|
Cancer of Head and Neck |
Phase 1 |
Completed |
- |
United States, California
... More >>
Moores UC San Diego Cancer Center
La Jolla, California, United States, 92037 Less << |
|
NCT00040534
|
Neoplasms |
Phase 1 |
Terminated |
- |
- |
|
NCT00993616
|
Brenner Tumor
... More >> Fallopian Tube Cancer
Ovarian Clear Cell Cystadenocarcinoma
Ovarian Endometrioid Adenocarcinoma
Ovarian Mixed Epithelial Carcinoma
Ovarian Mucinous Cystadenocarcinoma
Ovarian Serous Cystadenocarcinoma
Ovarian Undifferentiated Adenocarcinoma
Primary Peritoneal Cavity Cancer
Recurrent Ovarian Epithelial Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00606021
|
Non Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Egypt
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Assiut, Egypt, 0000
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Cairo, Egypt, 11372
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Mounofia, Egypt, 32514
Lebanon
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Beirut, Lebanon
Saudi Arabia
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Riyadh, Saudi Arabia, 11211 Less << |
|
NCT03322267
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Phase 2 |
Recruiting |
January 2025 |
Taiwan
... More >> National Taiwan University Hospital
Recruiting
Taipei City, Taiwan, 10002
Contact: Chih-Hung Hsu, MD, PhD +886-2-23123456 ext 67680 chihhunghsu@ntu.edu.tw Less << |
|
NCT02215265
|
Human Papillomavirus (HPV)-Pos... More >>itive Oropharyngeal Cancer Less << |
Phase 2
Phase 3 |
Recruiting |
February 2024 |
United Kingdom
... More >> Poole Hospital NHS Foundation Trust
Recruiting
Poole, Dorset, United Kingdom, BH15 2JB
Contact: Emma King, Dr emmabarker@doctors.org.uk
Royal United Hospitals Bath NHS Foundation Trust
Recruiting
Bath, United Kingdom, BA1 3NH
Contact: Emma De Winton, Dr emma.dewinton@nhs.net
Queen Elizabeth Hospital
Not yet recruiting
Birmingham, United Kingdom, B15 2TH
Contact: Andrew Hartley, Dr
University Hospitals Bristol NHS Foundation Trust
Recruiting
Bristol, United Kingdom, BS2 8ED
Contact: Matthew Beasley, Dr Matthew.Beasley@UHBristol.nhs.uk
Cambridge University Hospitals NHS Foundation Trust
Recruiting
Cambridge, United Kingdom, CB2 0QQ
Contact: Irune Ekpemi, Dr ekpemi.irune@addenbrookes.nhs.uk
Velindre Cancer Centre
Not yet recruiting
Cardiff, United Kingdom, CF14 2TL
Contact: Waheeda Owadally, Dr Waheeda.Owadally@wales.nhs.uk
HPV Research Group Section of Pathology Cardiff University ,School of Medicine
Not yet recruiting
Cardiff, United Kingdom, CF14 4XN
Contact: Ned Powell, Dr PowellNG@cardiff.ac.uk
Cardiff and Vale University Local Health Board
Recruiting
Cardiff, United Kingdom, CF14 4XW
Contact: David Owens, Dr david.owens2@wales.nhs.uk
Wales Cancer Trials Unit
Not yet recruiting
Cardiff, United Kingdom, CF14 4YS
Contact: Chris Hurt 02920687471 HurtCN@cf.ac.uk
Velindre NHS Trust
Not yet recruiting
Cardiff, United Kingdom, CF142TL
Contact: Nachi Palaniappan, Dr Nachi.Palaniappan@wales.nhs.uk
Castle Hill Hospital
Recruiting
Cottingham, United Kingdom, HU16 5JQ
Contact: Lorcan O'Toole, Dr lorcan.otoole@hey.nhs.uk
Derby Teaching Hospitals NHS Foundation Trust
Recruiting
Derby, United Kingdom, DE22 3DT
Contact: Sean Mortimore, Dr sean.mortimore@nhs.net
Aintree University Hospitals NHS Foundation Trust
Recruiting
Liverpool, United Kingdom, L3 9TA
Contact: Terry Jones, Professor T.M.Jones@liverpool.ac.uk
Cwm Taf University Health Board
Recruiting
Llantrisant, United Kingdom, CF72 8XR
Contact: Sandeep Berry, Dr sandeep.berry2@wales.nhs.uk
Barts Health NHS Trust
Not yet recruiting
London, United Kingdom, E1 1RD
Contact: Paul Stimpson, Dr paul.stimpson2@bartshealth.nhs.uk
Central Manchester University Hospital NHS Foundation Trust
Not yet recruiting
Manchester, United Kingdom, M13 9WL
Contact: Jarrod Homer, Professor jarrod.j.homer@manchester.ac.uk
The Christie NHS Foundation Trust
Not yet recruiting
Manchester, United Kingdom, M20 4BX
Contact: David Thomson, Dr david.thomson@christie.nhs.uk
The James Cook University Hospital
Recruiting
Middlesbrough, United Kingdom, TS4 3BW
Contact: Shane Lester, Mr shanelester@nhs.net
Newcastle upon Tyne Hospitals NHS Foundation Trust
Recruiting
Newcastle upon Tyne, United Kingdom, NE7 7DN
Contact: Rebecca Goranova, Dr Rebecca.goronova@nuth.nhs.uk
Institute of Health and Society
Not yet recruiting
Newcastle, United Kingdom, NE2 4AX
Contact: Jo Patterson, Dr joanne.patterson@newcastle.ac.uk
Centre for Oral Health Research Newcastle University
Not yet recruiting
Newcastle, United Kingdom, NE24BW
Contact: Conrad Robinson, Dr max.robinson@newcastle.ac.uk
Oxford University Hospitals NHS Foundation Trust
Recruiting
Oxford, United Kingdom, OX3 7LD
Contact: Stuart Winter, Dr stuart.winter@ouh.nhs.uk
Queen Alexandra Hospital
Recruiting
Portsmouth, United Kingdom, PO6 3LY
Contact: Costa Repanos, Mr costa.repanos@porthosp.nhs.uk
Royal Berkshire Hospital
Not yet recruiting
Reading, United Kingdom, RG1 5AN
Contact: Alice Freebairn, Dr alice.freebairn@royalberkshire.nhs.uk
Sunderland Royal Hospital
Recruiting
Sunderland, United Kingdom, SR4 7TP
Contact: Helen Cocks, Dr helen.cocks@chsft.nhs.uk
Abertawe Bro Morgannwg University Health Board
Recruiting
Swansea, United Kingdom, SA2 8QA
Contact: Conor Marnane, Dr conor.marnane@wales.nhs.uk Less << |
|
NCT00070096
|
Brain and Central Nervous Syst... More >>em Tumors
Extragonadal Germ Cell Tumor
Ovarian Cancer
Testicular Germ Cell Tumor Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00859495
|
Pleural Mesothelioma |
Phase 2 |
Unknown |
December 2014 |
United States, New York
... More >>
Columbia University Medical Center
Recruiting
New York, New York, United States, 10032 Less << |
|
NCT02773485
|
Cholangiocarcinoma |
Phase 3 |
Recruiting |
June 2022 |
India
... More >> Advanced Centre of Treatment Research and Education In Cancer,Tata Memorial Centre
Recruiting
Navi Mumbai, Maharashtra, India, 410210
Contact: Dr Supriya Chopra, MD 91-22-27405000 ext 5491 schopra@actrec.gov.in
Principal Investigator: Supriya Chopra, MD Less << |
|
NCT03692780
|
Chemotherapy-Induced Myelosupp... More >>ression Less << |
Phase 1
Phase 2 |
Not yet recruiting |
June 2021 |
- |
|
NCT00630240
|
- |
|
Completed |
- |
Taiwan
... More >> Division of Hepatobiliary Medicine, Department of Internal Medicine, Kaohsiung Medical University Hospital, No. 100 Tzyou 1st Road
Kaohsiung, Taiwan, 807 Less << |
|
NCT01565109
|
Gastric Adenocarcinoma |
Phase 2 |
Unknown |
March 2018 |
France
... More >> Institut Sainte Catherine
Recruiting
Avignon, France, 84000
Contact: LAURENT MINEUR, DOCTOR 04.90.27.62.68 l.mineur@isc84.org Less << |
|
NCT03737968
|
Stage II-IVB Operable HNSCC Or... More >>al Cavity
Hypopharynx
Oropharynx
Larynx Less << |
Phase 2 |
Not yet recruiting |
December 2022 |
- |
|
NCT00606021
|
- |
|
Completed |
- |
- |
|
NCT01899989
|
Non-small Cell Lung Cancer |
Phase 1 |
Recruiting |
July 2019 |
United States, New Jersey
... More >>
Memorial Sloan Kettering Cancer Center at Basking Ridge
Recruiting
Basking Ridge, New Jersey, United States, 07920
Contact: Andreas Rimner, MD 212-639-6025
Memorial Sloan Kettering Monmouth
Recruiting
Middletown, New Jersey, United States, 07748
Contact: Andreas Rimner, MD 212-639-6025
Memorial Sloan Kettering Bergen
Recruiting
Montvale, New Jersey, United States, 07645
Contact: Andreas Rimner, MD 212-639-6025
United States, New York
Memorial Sloan Kettering Cancer Center @ Suffolk
Recruiting
Commack, New York, United States, 11725
Contact: Andreas Rimner, MD 212-639-6025
Memorial Sloan Kettering Westchester
Recruiting
Harrison, New York, United States, 10604
Contact: Andreas Rimner, MD 212-639-6025
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10065
Contact: Andreas Rimner, MD 212-639-6025
Contact: Jamie Chaft, MD 646-888-4545
Principal Investigator: Andreas Rimner, MD
Memorial Sloan Kettering at Mercy Medical Center
Recruiting
Rockville Centre, New York, United States
Contact: Andreas Rimner, MD 212-639-6025
Contact: Jamie Chaft, MD 646-888-4545
Principal Investigator: Andreas Rimner, MD Less << |
|
NCT01203735
|
Locally Advanced Inoperable No... More >>n-small-lung Cancer Less << |
Phase 1
Phase 2 |
Unknown |
February 2015 |
Israel
... More >> Soroka University Medical Center
Recruiting
Beer Sheva, Israel, 84101
Contact: Konstantin Lavrenkov, MD, PhD +97286400537 constant@bgu.ac.il
Contact: Julia Dudnik, MD +97286400537 juliad@clalit.org.il
Sub-Investigator: Vladimir Gavrilov, MD, PhD
Sub-Investigator: Julia Dudnik, MD
Sub-Investigator: Kerenr Rouvinov, MD Less << |
|
NCT02890030
|
- |
|
Active, not recruiting |
December 2018 |
United States, Indiana
... More >>
Indiana University Melvin and Bren Simon Cancer Center
Indianapolis, Indiana, United States, 46202 Less << |
|
NCT02662504
|
Malignant Pleural Mesothelioma |
Phase 2 |
Completed |
- |
France
... More >> CHRU de Lille Hôpital Calmette
Lille, France Less << |
|
NCT00004873
|
Gastric Cancer |
Phase 2 |
Completed |
- |
Switzerland
... More >> Hopital Cantonal Universitaire de Geneva
Geneva, Switzerland, CH-1211 Less << |
|
NCT02739204
|
Oral Squamous Cell Carcinoma |
Phase 2 |
Unknown |
December 2017 |
Taiwan
... More >> Department of Otolaryngology
Recruiting
Taichung, Taiwan, 40447
Contact: Ming-Hsul Tsai, M.D. Ph.D. 886-4-22052121 ext 4436 minghsui@mail.cmuh.org.tw
Principal Investigator: Chun-Hung Hua, M.D. Less << |
|
NCT02344979
|
- |
|
Unknown |
June 2015 |
China, Shanghai
... More >>
Shanghai Chest Hospital
Shanghai, Shanghai, China, 200030 Less << |
|
NCT00002882
|
Melanoma
Skin... More >> Cancer Less << |
Phase 3 |
Completed |
- |
United States, Texas
... More >>
University of Texas - MD Anderson Cancer Center
Houston, Texas, United States, 77030-4009 Less << |
|
NCT03734809
|
NPC |
Phase 2 |
Not yet recruiting |
December 31, 2023 |
Hong Kong
... More >> Department of Clinical Oncology, Prince of Wales Hospital
Not yet recruiting
Hong Kong, Hong Kong
Contact: Anthony TC Chan, MD 3505 2119 anthonytcchan@cuhk.edu.hk
Contact: Candy Yuen, RN 3505 1040 sf_yuen@clo.cuhk.edu.hk
Singapore
National Cancer Centre of Singapore
Not yet recruiting
Singapore, Singapore
Contact: Darren WT Lim, MD darren.lim.w.t@singhealth.com.sg
Contact: Yoke Lim YL Soong, MD Soong.yoke.lim@Singhealth.com.sg Less << |
|
NCT02124148
|
Neoplasm Metastasis
... More >> Colorectal Neoplasms
Breast Cancer Less << |
Phase 1 |
Recruiting |
February 27, 2020 |
United States, Florida
... More >>
Florida Cancer Specialists
Recruiting
Sarasota, Florida, United States, 34232
Contact 9413779993
Principal Investigator: Manish Rajni Patel
United States, Oklahoma
University of Oklahoma Health Sciences Center
Recruiting
Oklahoma City, Oklahoma, United States, 73104
Contact 405-271-4022
Principal Investigator: Kathleen Moore
United States, Tennessee
Sarah Cannon Research Institute SCRI
Recruiting
Nashville, Tennessee, United States, 37203
Contact 615-329-7615
Principal Investigator: SMO Sarah Cannon Research Inst.
Tennessee Oncology PLLC
Recruiting
Nashville, Tennessee, United States, 37203
Contact 6153297274
Principal Investigator: Johanna Chock Bendell
United States, Texas
University of Texas MD Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact 713-563-5844
Principal Investigator: David S Hong Less << |
|
NCT03524508
|
Metastatic Biliary Tract Cance... More >>r Less << |
Phase 2 |
Recruiting |
March 2021 |
Korea, Republic of
... More >>
Asan Medical Center
Recruiting
Seoul, Korea, Republic of, 05505
Contact: Changhoon Yoo, MD
Principal Investigator: Changhoon Yoo, MD Less << |
|
NCT01303978
|
Chemotherapy-induced Nausea an... More >>d Vomiting Less << |
Phase 2 |
Completed |
- |
Denmark
... More >> Herlev Hospital
Copenhagen, Denmark
Rigshospitalet
Copenhagen, Denmark
Odense University Hospital
Odense, Denmark
United Kingdom
University Hospital of South Manchester NHS Trust
Manchester, United Kingdom Less << |
|
NCT02633800
|
Head and Neck Neoplasms |
Phase 2 |
Completed |
- |
Belgium
... More >> Institut Jules Bordet
Brussels, Belgium, 1000
Univeristair Ziekenhuis Antwerpen
Edegem, Belgium, 2650
UZ Leuven
Leuven, Belgium, 3000
France
Institut Curie
Paris, Cedex, France, 75248
Institut de Cancerologie de l'Ouest
Angers cedex 02, France, 49055
Centre Hospitalier de Bordeaux - Hôpital Saint André
Bordeaux, France, 33075
Hopital Croix-Rousse
Lyon, France, 69004
Centre Leon Berard
Lyon, France, 69373
CHU Hopital de la Timone
Marseille, France, 13005
Hopital Saint Joseph
Marseille, France, 13008
Centre de Cancerologie du Grand Montpellier
Montpellier, France, 34070
Institut de Cancerologie de l'Ouest
Saint-Herblain Cedex, France, 44805
Gustave Roussy
Villejuif, France, 94805
Germany
Charite Universitatsmedizin Berlin
Berlin, Germany, 12200/12203
Medizinische Hochschule Hannover
Hannover, Germany, 30625
Klinikum der Universitat Munchen
München, Germany, 81377
Hungary
Orszagos Onkologiai Intezet
Budapest, Hungary, 1122
Debreceni Egyetem Orvos-es Egeszsegtudomanyi Centrum
Debrecen, Hungary, 4032
Bacs-Kiskun Megyei Korhaz
Kecskemet, Hungary, 6000
Borsod Abauj Zemplen Megyei Korhaz es Egyetemi Oktato Korhaz
Miskolc, Hungary, 3526
Josa Andras Oktatokorhaz
Nyíregyháza, Hungary, 4400
Poland
Centrum Onkologii im. Prof. Franciszka Lukaszczyka w Bydgoszczy
Bydgoszcz, Poland, 85-796
Przychodnia Lekarska "KOMED"
Konin, Poland, 62-500
Regionalny Osrodek Onkologiczny Szpital im. M. Kopernika w Lodzi
Lodz, Poland, 93-513
Romania
Medisprof SRL
Cluj-Napoca, Romania, 400058
Centrul de Oncologie Sfantul Nectarie
Craiova, Romania, 200347
Institutul Regional de Oncologie Iasi
Iasi, Romania, 700483
United Kingdom
University College London Hospitals NHS Foundation Trust - University College Hospital
London, United Kingdom, NW1 2PG
Guy's and St Thomas' NHS Foundation Trust - St Thomas' Hospital
London, United Kingdom, SE1 7EH
The Royal Marsden NHS Foundation Trust
London, United Kingdom, SW3 6JJ
Weston Park Hospital
Sheffield, United Kingdom, S10 2SJ
The Shrewsbury and Telford Hospital NHS Trust
Shrewsbury, United Kingdom, SY3 8XQ
Southampton General Hospital
Southampton, United Kingdom, SO16 6YD
The Royal Marsden NHS Foundation Trust
Sutton, United Kingdom, SM2 5PT Less << |
|
NCT02933099
|
Chemotherapy-induced Nausea an... More >>d Vomiting Less << |
Phase 3 |
Unknown |
December 2018 |
- |
|
NCT00052962
|
Gastrointestinal Neoplasm |
Phase 3 |
Completed |
- |
United States, Maryland
... More >>
National Cancer Institute (NCI)
Bethesda, Maryland, United States, 20892 Less << |
|
NCT00052962
|
- |
|
Completed |
- |
- |
|
NCT02728492
|
Non-small Cell Lung Cancer
... More >> Epithelial Ovarian Cancer Less << |
Phase 1 |
Completed |
- |
Russian Federation
... More >>
Russian Oncological Research Center n.a. N. N. Blokhin RAMS
Moscow, Russian Federation, 115478
State Budgetary Healthcare Institution of Stavropol Territory "Pyatigorsk oncology dispensary"
Pyatigorsk, Russian Federation, 357502
Saint-Peterburg State Budgetary healthcare Institution "City Clinical Oncology Dispensary"
Saint-Petersburg, Russian Federation, 197022
BioEq LLC
Saint-Petersburg, Russian Federation, 197342
State Budget Institution of healthcare "Saint-Petersburg clinical research and practical centre of specialized medical aid (oncology)"
Saint-Petersburg, Russian Federation, 197758
State Healthcare Institution of Yaroslavl region "Regional Clinical oncology hospital"
Yaroslavl, Russian Federation, 150054 Less << |
|
NCT00020709
|
Adenocarcinoma of the Lung
... More >> Bronchoalveolar Cell Lung Cancer
Large Cell Lung Cancer
Squamous Cell Lung Cancer
Stage IIIA Non-small Cell Lung Cancer
Stage IIIB Non-small Cell Lung Cancer Less << |
Phase 3 |
Completed |
- |
United States, Texas
... More >>
Southwest Oncology Group (SWOG) Research Base
San Antonio, Texas, United States, 78245 Less << |
|
NCT03321188
|
Ovarian Cancer
... More >> Epithelial Ovarian Cancer
Fallopian Tube Cancer Less << |
Phase 2 |
Recruiting |
December 15, 2020 |
United States, Kansas
... More >>
The University of Kansas Cancer Center
Recruiting
Kansas City, Kansas, United States, 66160-7357
Contact: Kerry Hepler 913-945-7552 ctnursenav@kumc.edu Less << |
|
NCT00002536
|
Cervical Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00945451
|
Lung Cancer |
Not Applicable |
Recruiting |
December 2021 |
France
... More >> Centre Antoine Lacassagne
Recruiting
Nice, France, 06189
Contact: Pierre-Yves Bondiau, MD, PhD 33-4-9203-1261 Less << |
|
NCT00301301
|
- |
|
Completed |
- |
United Kingdom
... More >> Royal Marsden NHS trust
Sutton, Surrey, United Kingdom, SM2 5PT Less << |
|
NCT00411138
|
Endometrial Cancer |
Phase 3 |
Active, not recruiting |
December 2020 |
Netherlands
... More >> Leiden University Medical Center
Leiden, Netherlands, 2300 RC Less << |
|
NCT00113581
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 1 |
Completed |
- |
United Kingdom
... More >> The Royal Marsden Hospital
London, United Kingdom Less << |
|
NCT03464968
|
Biliary Cancer Metastatic |
Phase 2 |
Recruiting |
December 25, 2020 |
Korea, Republic of
... More >>
Seoul National University Bundang Hospital
Recruiting
Seongnam, Gyeonggi, Korea, Republic of, 13620
Contact: Jin Won Kim, MD, PhD 82-31-787-8209 jwkim@snubh.org Less << |
|
NCT03062800
|
Advanced Nsclc |
Phase 2 |
Recruiting |
October 2018 |
China, Shandong
... More >>
Qilu hospital of Shandong University
Recruiting
Jinan Shi, Shandong, China, 250012
Contact: Shuguang Li, MD. +86 18560082862 lishuguang96@163.com
Principal Investigator: Xiuwen Wang, MD. Less << |
|
NCT01558947
|
Gastric Cancer |
Phase 2 |
Unknown |
June 2013 |
China, Zhejiang
... More >>
Zhejiang Cancer Hospital
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: xiangdong Cheng, MD +86 571 88122516 abdsurg@hotmail.com
Principal Investigator: xiangdong Cheng, MD Less << |
|
NCT00002601
|
Sarcoma |
Phase 2 |
Completed |
- |
United States, California
... More >>
Cancer Center and Beckman Research Institute, City of Hope
Duarte, California, United States, 91010-3000 Less << |
|
NCT00006111
|
Bladder Cancer
... More >> Transitional Cell Cancer of the Renal Pelvis and Ureter
Urethral Cancer Less << |
Phase 2 |
Unknown |
- |
France
... More >> Institut Sainte Catherine
Avignon, France, 84082
Centre Oscar Lambret
Lille, France, 59020
Centre Leon Berard
Lyon, France, 69373
Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Montpellier, France, 34298
Centre Antoine Lacassagne
Nice, France, 06189
C.H.U. - Hopital Gaston Doumergue
Nimes, France, 30006
Centre Eugene Marquis
Rennes, France, 35064
Centre Rene Huguenin
Saint Cloud, France, 92211
Centre Hospitalier General de Saint Nazaire
Saint Nazaire, France, 44600
Institut Claudius Regaud
Toulouse, France, 31052
Centre Alexis Vautrin
Vandoeuvre-les-Nancy, France, 54511 Less << |
|
NCT00002601
|
- |
|
Completed |
- |
- |
|
NCT01160185
|
- |
|
Completed |
- |
- |
|
NCT00014209
|
Lymphoma |
Phase 2 |
Completed |
- |
Canada, Alberta
... More >>
Tom Baker Cancer Center - Calgary
Calgary, Alberta, Canada, T2N 4N2
Cross Cancer Institute
Edmonton, Alberta, Canada, T6G 1Z2
Canada, New Brunswick
Moncton Hospital
Moncton, New Brunswick, Canada, E1C 6ZB
Canada, Newfoundland and Labrador
Newfoundland Cancer Treatment and Research Foundation
St. Johns, Newfoundland and Labrador, Canada, A1B 3V6
Canada, Ontario
Cancer Care Ontario-Hamilton Regional Cancer Centre
Hamilton, Ontario, Canada, L8V 5C2
Kingston Regional Cancer Centre
Kingston, Ontario, Canada, K7L 5P9
Cancer Care Ontario-London Regional Cancer Centre
London, Ontario, Canada, N6A 4L6
Credit Valley Hospital
Mississauga, Ontario, Canada, L5M 2N1
Northwestern Ontario Regional Cancer Centre, Thunder Bay
Thunder Bay, Ontario, Canada, P7A 7T1
Toronto Sunnybrook Regional Cancer Centre
Toronto, Ontario, Canada, M4N 3M5
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9
Humber River Regional Hospital
Weston, Ontario, Canada, M9N 1N8
Cancer Care Ontario - Windsor Regional Cancer Centre
Windsor, Ontario, Canada, N8W 2X3 Less << |
|
NCT01688700
|
Esophageal Squamous Cell Carci... More >>noma Resectable Less << |
Phase 2 |
Unknown |
September 2017 |
China, Zhejiang
... More >>
Zhejiang Cancer Hospital
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: Yun Fan, doctor 0086-571-88122192 fanyun@csco.org.cn
Principal Investigator: Weimin Mao, doctor Less << |
|
NCT01730222
|
Pancreatic Cancer |
Phase 1
Phase 2 |
Completed |
- |
Italy
... More >> IRCCS S Raffaele
Milan, Italy, 20132 Less << |
|
NCT00003118
|
Esophageal Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT01379339
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Icahn School of Medicine at Mount Sinai
New York, New York, United States, 10029 Less << |
|
NCT02158988
|
Malignant Neoplasm of Stomach
... More >>
Secondary Malignant Neoplasm of Peritoneum
Secondary Malignant Neoplasm of Other and Unspecified Sites Less << |
Phase 3 |
Recruiting |
September 2020 |
Germany
... More >> Klinik für Allgemein-, Visceral-, Gefäß- und ThoraxchirurgieCharité Campus Mitte
Recruiting
Berlin, Germany, 10117
Contact: Beate Rau +49 (0)30 450 622 ext 214 beate.rau@charite.de Less << |
|
NCT00524498
|
Hepatocellular Carcinoma |
Phase 2 |
Completed |
- |
Japan
... More >>
Chubu region, Japan
Chugoku region, Japan
Hokkaido region, Japan
Kanto region, Japan
Kinki region, Japan
Kyushu region, Japan
Tohoku region, Japan Less << |
|
NCT01125020
|
Liver Transplantation
... More >> Hepatocellular Carcinoma
Tumor Recurrence and Metastasis
Survival
Adjuvant Chemotherapy Less << |
Not Applicable |
Unknown |
December 2010 |
China, Shanghai
... More >>
Shanghai First People's Hospital
Recruiting
Shanghai, Shanghai, China, 200080
Contact: Zhi-Hai Peng, MD 0086-021-63240090 ext 3132 pengpzh@hotmail.com
Principal Investigator: Jun-Wei Fan, MD Less << |
|
NCT01631357
|
Lung Cancer |
Phase 2
Phase 3 |
Unknown |
December 2016 |
China, Tianjin
... More >> Tianjin Medical University Cancer Institute and Hospital
Active, not recruiting
Tianjin, Tianjin, China, 300060 Less << |
|
NCT00525200
|
Esophageal Cancer |
Phase 3 |
Completed |
- |
Austria
... More >> Landesklinikum St. Pölten
St. Polten, Lower Austria, Austria, 3100
Landesklinikum Wiener Neustadt
Wiener Neustadt, Lower Austria, Austria, 2700
Medical University Innsbruck
Innsbruck, Tirol, Austria
Landesklinikum Feldkirch
Feldkirch, Vorarlberg, Austria
Landeskrankenhaus Leoben
Leoben, Austria, 8790
Krankenhaus der Elisabethinen
Linz, Austria, 4020
Krankenhaus der Barmherzigen Brüder
Stankt Veit, Austria, 9300
Rudolfstiftung
Vienna, Austria, 1030
Medical University of Vienna
Vienna, Austria, 1090
Kaiser Franz Josef Spital
Vienna, Austria, 1100
Hanusch Krankenhaus
Vienna, Austria, 1140
SMZ OST
Vienna, Austria
Wilhelminenspital
Vienna, Austria Less << |
|
NCT02957266
|
Cervical Cancer |
Phase 3 |
Recruiting |
December 2020 |
Azerbaijan
... More >> National Center of Oncology
Recruiting
Baku, Azerbaijan, AZ1011
Contact: Aziz Aliyev, Professor +994504807021 internationalnoc@gmail.com Less << |
|
NCT00474669
|
Ovarian Carcinoma |
Phase 1 |
Completed |
- |
United States, Kentucky
... More >>
James Graham Brown Cancer Center
Louisville, Kentucky, United States, 40202 Less << |
|
NCT01450761
|
- |
|
Completed |
- |
- |
|
NCT02250404
|
- |
|
Withdrawn(PI left institution) |
August 22, 2019 |
United States, California
... More >>
Los Angeles County-USC Medical Center
Los Angeles, California, United States, 90033
USC Norris Comprehensive Cancer Center
Los Angeles, California, United States, 90033 Less << |
|
NCT01450761
|
Small Cell Lung Carcinoma |
Phase 3 |
Completed |
- |
- |
|
NCT00310128
|
AIDS-related Lymphoma
... More >> Adult Non-Hodgkin's Lymphoma
Anaplastic Large Cell Lymphoma Less << |
Phase 2 |
Withdrawn(Drug supply unavaila... More >>ble) Less << |
- |
- |
|
NCT00193921
|
Non Small Cell Lung Carcinoma |
Phase 2 |
Completed |
- |
Australia, New South Wales
... More >>
Calvary Mater Newcastle
Newcastle, New South Wales, Australia, 2298
Australia, Queensland
Mater Misericordiae Hospital
Brisbane, Queensland, Australia, 4101
Princess Alexandra Hospital
Brisbane, Queensland, Australia, 4102
North Queensland Oncology Service
Townsville, Queensland, Australia, 4810
The John Flynn Hospital
Tugun, Queensland, Australia, 4224
Australia, Victoria
Frankston Hospital
Frankston, Victoria, Australia
Peter MacCallum Cancer Centre
Melbourne, Victoria, Australia, 8006
Border Medical Oncology
Wondonga, Victoria, Australia Less << |
|
NCT00550784
|
Fallopian Tube Cancer
... More >> Ovarian Cancer
Peritoneal Cavity Cancer Less << |
Phase 1 |
Completed |
- |
- |
|
NCT00301028
|
- |
|
Completed |
- |
- |
|
NCT00778908
|
Nasopharyngeal Carcinoma |
Phase 2
Phase 3 |
Completed |
- |
China, Guangxi
... More >> People's Hospital of Guangxi Zhuang Autonomous Region
Nanning, Guangxi, China, 530021 Less << |
|
NCT02603003
|
Non-small Cell Lung Cancer(NSC... More >>LC) Less << |
Phase 1 |
Active, not recruiting |
December 2019 |
- |
|
NCT02760667
|
Head and Neck Cancer |
Phase 2 |
Active, not recruiting |
June 2020 |
United States, District of Col... More >>umbia
George Washington University-Medical Faculty Associates
Washington, District of Columbia, United States, 20037 Less << |
|
NCT00817583
|
Nasopharyngeal Carcinoma |
Phase 2 |
Unknown |
July 2011 |
China, Shanghai
... More >>
Department of Radiation Oncology, Cancer Hospital, Fudan University
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Xiaoshen Wang, M.D. 86-21-64175590 ext 6516 wangxiaoshen@gmail.com
Contact: TingTing Xu, M.D. 86-21-64175590 ext 6511 littlepuppyxtt@gmail.com Less << |
|
NCT00301028
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
M.D. Anderson Cancer Center at University of Texas
Houston, Texas, United States, 77030-4009 Less << |
|
NCT00930891
|
Small Cell Lung Cancer |
Phase 2
Phase 3 |
Completed |
- |
- |
|
NCT02784171
|
Mesothelioma |
Phase 2 |
Recruiting |
December 2019 |
Canada, Alberta
... More >>
Tom Baker Cancer Centre
Recruiting
Calgary, Alberta, Canada, T2N 4N2
Contact: Desiree Hao 403 521-3706
Cross Cancer Institute
Recruiting
Edmonton, Alberta, Canada, T6G 1Z2
Contact: Quincy Chu 780 432-8248
Canada, British Columbia
BCCA - Cancer Centre for the Southern Interior
Recruiting
Kelowna, British Columbia, Canada, V1Y 5L3
Contact: Delia Sauciuc 250 712-3900 ext 683930
BCCA - Fraser Valley Cancer Centre
Recruiting
Surrey, British Columbia, Canada, V3V 1Z2
Contact: Christopher Lee 604 930-4017
Canada, Manitoba
CancerCare Manitoba
Recruiting
Winnipeg, Manitoba, Canada, R3E 0V9
Contact: David Dawe 204 787-8644
Canada, Ontario
Juravinski Cancer Centre at Hamilton Health Sciences
Recruiting
Hamilton, Ontario, Canada, L8V 5C2
Contact: John Goffin 905 387-9495
London Regional Cancer Program
Recruiting
London, Ontario, Canada, N6A 5W9
Contact: Mark D. Vincent 519 685-8640
Ottawa Hospital Research Institute
Recruiting
Ottawa, Ontario, Canada, K1H 8L6
Contact: Paul Wheatley-Price 613 737-7700 ext 79859
University Health Network
Recruiting
Toronto, Ontario, Canada, M5G 2M9
Contact: Penelope A. Bradbury 416 946-4501 ext 3544
Canada, Quebec
CHUM-Centre Hospitalier de l'Universite de Montreal
Recruiting
Montreal, Quebec, Canada, H2X 3E4
Contact: Marie Florescu 514 890-8444
The Research Institute of the McGill University
Recruiting
Montreal, Quebec, Canada, H4A 3J1
Contact: Scott Owen 514 398-8307
University Institute of Cardiology and
Recruiting
Quebec City, Quebec, Canada, G1V 4G5
Contact: Catherine Labbe 418 656-8711 ext 5504
Canada, Saskatchewan
Allan Blair Cancer Centre
Recruiting
Regina, Saskatchewan, Canada, S4T 7T1
Contact: Mussawar Iqbal 306 766-2691
Italy
Oncologia SS Antonio e Biagio Alessandria
Recruiting
Alessandria, AL, Italy, 15121
Contact: Federica Grosso
Azienda Ospedaliera San Giuseppe Moscati
Recruiting
Avellino, AV, Italy, 83100
Contact: Gridelli Cesare
Oncologia Medica Humanitas Gavazzeni Bergamo
Not yet recruiting
Bergamo, BG, Italy, 24125
Contact: Giovanni Luca Ceresoli
Azienda Ospedaliera Garibaldi Nesima
Recruiting
Catania, CT, Italy, 95123
Contact: Roberto Bordonaro
Oncologia Medica IRCCS Arcispedale Maria
Recruiting
Reggio Emilia, RE, Italy, 42123
Contact: Maria Pagano
Istituti Fisioterapici Ospitalieri IFO Istituto
Recruiting
Rome, RM, Italy, 00144
Contact: Fabiana Letizia Cecere
PO A Perrino ASL Brindisi - UOC Oncologia Medica
Recruiting
Brindisi, Italy, 72100
Contact: Saverio Cinieri
AOU Policlinico Vittorio Emanuele UOC di Oncologia
Recruiting
Catania, Italy, 95125
Contact: Hector Jose Soto Parra 2 8224-4459
U.O. di Oncologia Ospedale Villa Scassi
Recruiting
Genova, Italy, 16149
Contact: Manlio Mencoboni 010 410-2639
Intstituto Scientifico Romangnolo
Recruiting
Meldola, Italy, 47014
Contact: Marco Angelo Burgio 054 373-9100
Instituto Nazionale Tumori
Recruiting
Milan, Italy
Contact: Maria Chiara Garassino
U.O.C. di Oncologia U.L.S.S. 13
Recruiting
Mirano, Italy, 30035
Contact: Francesco Rosetti 3904 1579-4002
Azienda Ospedaliera di Rilievo Nazionale
Not yet recruiting
Napoli, Italy, 80131
Contact: Giacomo Carteni
Dott. Fortunato Ciardiello,Cattedra Oncologia Medica
Recruiting
Napoli, Italy, 80131
Contact: Fortunato Ciardiello 081 566-6745
Unita Sperimentazioni Cliniche Istituto per lo
Recruiting
Napoli, Italy, 80131
Contact: Alessandro Morabito 081 590-3571
Azienda USL di Piacenza, Ospedale Gugliemimo Salieto
Recruiting
Piacenza, Italy, 29100
Contact: Luigi Cavanna 052 330-2697 Less << |
|
NCT00274911
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 2 |
Unknown |
- |
Ireland
... More >> Our Lady's Hospital for Sick Children Crumlin
Dublin, Ireland, 12
United Kingdom
Birmingham Children's Hospital
Birmingham, England, United Kingdom, B4 6NH
Institute of Child Health at University of Bristol
Bristol, England, United Kingdom, BS2 8AE
Addenbrooke's Hospital
Cambridge, England, United Kingdom, CB2 2QQ
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Royal Liverpool Children's Hospital, Alder Hey
Liverpool, England, United Kingdom, L12 2AP
Royal London Hospital
London, England, United Kingdom, E1 1BB
Great Ormond Street Hospital for Children
London, England, United Kingdom, WC1N 3JH
Royal Manchester Children's Hospital
Manchester, England, United Kingdom, M27 4HA
Sir James Spence Institute of Child Health at Royal Victoria Infirmary
Newcastle-Upon-Tyne, England, United Kingdom, NE1 4LP
Queen's Medical Centre
Nottingham, England, United Kingdom, NG7 2UH
Oxford Radcliffe Hospital
Oxford, England, United Kingdom, 0X3 9DU
Children's Hospital - Sheffield
Sheffield, England, United Kingdom, S10 2TH
Southampton General Hospital
Southampton, England, United Kingdom, SO16 6YD
Royal Marsden - Surrey
Sutton, England, United Kingdom, SM2 5PT
Royal Belfast Hospital for Sick Children
Belfast, Northern Ireland, United Kingdom, BT12 6BE
Royal Aberdeen Children's Hospital
Aberdeen, Scotland, United Kingdom, AB25 2ZG
Royal Hospital for Sick Children
Edinburgh, Scotland, United Kingdom, EH9 1LF
Royal Hospital for Sick Children
Glasgow, Scotland, United Kingdom, G3 8SJ
Childrens Hospital for Wales
Cardiff, Wales, United Kingdom, CF14 4XW Less << |
|
NCT00003691
|
Endometrial Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT02726399
|
Gastric Cancer
... More >> Gastroesophageal Junction Cancer Less << |
Phase 2 |
Active, not recruiting |
March 2019 |
United States, New Jersey
... More >>
Memoral Sloan Kettering Cancer Center
Basking Ridge, New Jersey, United States
Memorial Sloan Kettering Monmouth
Middletown, New Jersey, United States, 07748
United States, New York
Memorial Sloan Kettering Cancer Center @ Suffolk
Commack, New York, United States, 11725
Memorial Sloan Kettering Westchester
Harrison, New York, United States, 10604
Memorial Sloan Kettering Cancer Center 1275 York Avenue
New York, New York, United States, 10065
Memorial Sloan Kettering at Mercy Medical Center
Rockville Centre, New York, United States Less << |
|
NCT00421096
|
Uterine Cervical Cancer |
Phase 2 |
Terminated(Problems of recruit... More >>ment) Less << |
- |
France
... More >> Centre Leonard de Vinci
Dechy, France, 59187
Centre Oscar Lambret
Lille, France, 59020 Less << |
|
NCT00276666
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 2 |
Unknown |
- |
Ireland
... More >> Our Lady's Hospital for Sick Children Crumlin
Dublin, Ireland, 12
United Kingdom
Birmingham Children's Hospital
Birmingham, England, United Kingdom, B4 6NH
Institute of Child Health at University of Bristol
Bristol, England, United Kingdom, BS2 8AE
Addenbrooke's Hospital
Cambridge, England, United Kingdom, CB2 2QQ
Cookridge Hospital
Leeds, England, United Kingdom, LS16 6QB
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Royal Liverpool Children's Hospital, Alder Hey
Liverpool, England, United Kingdom, L12 2AP
Royal London Hospital
London, England, United Kingdom, E1 1BB
Great Ormond Street Hospital for Children
London, England, United Kingdom, WC1N 3JH
Royal Manchester Children's Hospital
Manchester, England, United Kingdom, M27 4HA
Sir James Spence Institute of Child Health at Royal Victoria Infirmary
Newcastle-Upon-Tyne, England, United Kingdom, NE1 4LP
Queen's Medical Centre
Nottingham, England, United Kingdom, NG7 2UH
Oxford Radcliffe Hospital
Oxford, England, United Kingdom, 0X3 9DU
Children's Hospital - Sheffield
Sheffield, England, United Kingdom, S10 2TH
Southampton General Hospital
Southampton, England, United Kingdom, SO16 6YD
Royal Marsden - Surrey
Sutton, England, United Kingdom, SM2 5PT
Royal Belfast Hospital for Sick Children
Belfast, Northern Ireland, United Kingdom, BT12 6BE
Royal Aberdeen Children's Hospital
Aberdeen, Scotland, United Kingdom, AB25 2ZG
Royal Hospital for Sick Children
Edinburgh, Scotland, United Kingdom, EH9 1LF
Royal Hospital for Sick Children
Glasgow, Scotland, United Kingdom, G3 8SJ
Childrens Hospital for Wales
Cardiff, Wales, United Kingdom, CF14 4XW Less << |
|
NCT02409186
|
Prosthesis Survival |
Phase 3 |
Recruiting |
December 2021 |
China, Beijing
... More >> Cancer Hospital Chinese Academy of Medical Sciences
Recruiting
Beijing, Beijing, China, 100021
Contact: Lvhua Wang 13601283715 wlhwq@yahoo.com
Contact: Zongmei Zhou 13801389769 zhouzongmei2013@163.com
Principal Investigator: Lvhua Wang
Sub-Investigator: Zongmei Zhou
Beijing Cancer Hospital
Recruiting
Beijing, Beijing, China, 100142
Contact: Guangying Zhu 13717999977 zgypu@yahoo.com.cn
Contact: Anhui Shi 13901136511 anhuidoctor@163.com
Principal Investigator: Guangying Zhu
Sub-Investigator: Anhui Shi
China, Guangdong
Cancer Hospital of Shantou University Medical College
Not yet recruiting
Shantou, Guangdong, China, 515041
Contact: Zhixiong Lin 13829638278 zxlin5@qq.com
Contact: Zhining Yang 13750443575 36407342@qq.com
Principal Investigator: Zhixiong Lin
Sub-Investigator: Zhining Yang
China, Guangxi
The Guangxi Zhuang Autonomous Region Cancer Hospita
Not yet recruiting
Nanning, Guangxi, China, 530021
Contact: Xiaodong Zhu 13978873616 zhuxiaodong83@163.com
Contact: Long Chen 13977129168 clong6@126.com
Principal Investigator: Xiaodong Zhu
Sub-Investigator: Long Chen
China, Guizhou
Affiliated Hospital of Zunyi Medical College
Not yet recruiting
Zunyi, Guizhou, China, 563099
Contact: Hu Ma 18385034657 mahuab@163.com
Contact: Xiaoli Gou 15121201843 526392247@qq.com
Principal Investigator: Hu Ma
Sub-Investigator: Xiaoli Gou
China, Hebei
Fourth Hospital of Hebei Medical University Tumor
Recruiting
Shijiazhuang, Hebei, China, 50011
Contact: Chun Han 13831105846 hanchun@yahoo.com.cn
Contact: Jun Wang 13931182128 wangjunzr@163.com
Principal Investigator: Chun Han
Sub-Investigator: Jun Wang
China, Henan
Henan Cancer Hospital
Recruiting
Zhengzhou, Henan, China, 450008
Contact: Jianhua Wang 13938278827 huajianye@sina.cn
Contact: Yongshun Chen 15286831671 yongshun2007@163.com
Principal Investigator: Jianhua Wang
Sub-Investigator: Yongshun Chen
China, Jiangsu
Jiangsu Cancer Hospital
Recruiting
Nanjing, Jiangsu, China, 210000
Contact: Jun Zhu 15380882705 zhujdr@126.com
Contact: Hong Ji 18625155033 dr_jihong@163.com
Principal Investigator: Jun Zhu
Sub-Investigator: Hong Ji
Affiliated Hospital of Jiangsu University
Recruiting
Zhenjiang, Jiangsu, China, 212013
Contact: Dai Chunhua, PhD 13952850012
Principal Investigator: Dai Chunhua, PhD
China, Liaoning
The First Hospital of China Medical University
Recruiting
Shenyang, Liaoning, China, 310022
Contact: Guang Li 13804058616 13804058616@163.com
Contact: Jun Dang 13898150850 dangjunsy@163.com
Principal Investigator: Guang Li
Sub-Investigator: Jun Dang
China, Shandong
Shandong Cancer Hospital and Institute
Recruiting
Jinan, Shandong, China, 250117
Contact: Xue Meng, Ph.D, M.D 86-531-67626142 mengxue5409@126.com
Sub-Investigator: Xue Meng, Ph.D, M.D
China, Shanghai
Renji Hospital Shanghai Jiao Tong University School of Medicine
Not yet recruiting
Shanghai, Shanghai, China, 200127
Contact: Ming Ye 13901814744 renjiyeming@gmail.com
Contact: Xin Xu 13917978366 157198711@qq.com
Principal Investigator: Ming Ye
Sub-Investigator: Xin Xu
China, Zhejiang
The First Hospital of Zhejiang Province
Not yet recruiting
Hangzhou, Zhejiang, China, 310003
Contact: Senxiang Yan 13957162839 yansenxiang@zju.edu.cn
Contact: Haogang Yu 13516721749 jusn3@163.com
Principal Investigator: Senxiang Yan
Sub-Investigator: Haogang Yu
Zhejiang Cancer Hospital
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: Ming Chen 18758875572 chenming@sysucc.org.cn
Contact: Yujin Xu 13858037993 xuyj@zjcc.org.cn
Principal Investigator: Ming Chen
Sub-Investigator: Yujin Xu Less << |
|
NCT03406299
|
Biliary Tract Neoplasms |
Phase 2 |
Recruiting |
December 31, 2020 |
Taiwan
... More >> Taiwan Cooperative Oncology Group, National Health Research Institutes
Recruiting
Taipei, Taiwan
Contact: Tsang-Wu Liu, MD +886-2-26534401 ext 25150 walter@nhri.org.tw
Principal Investigator: Li-Tzong Chen, MD PHD
Principal Investigator: Nai-Jung Chiang, MD Less << |
|
NCT03332069
|
Cervical Cancer |
Phase 3 |
Recruiting |
July 31, 2020 |
South Africa
... More >> Charlotte Maxeke Johannesburg Academic Hospital
Recruiting
Johannesburg, Gauteng, South Africa, 2163
Contact: Jeffrey A Kotzen, MBBCH +27825747385
Contact: Carrie A Minnaar, Masters +27721234292 Less << |
|
NCT00083915
|
Multiple Myeloma |
Phase 3 |
Completed |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences/MIRT
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT00083915
|
- |
|
Completed |
- |
- |
|
NCT03389295
|
Nasopharyngeal Carcinoma |
Phase 2 |
Recruiting |
October 2020 |
China, Shanghai
... More >>
Fudan University Shanghai Cancer Center
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Xiayun He, MD +86-18017312167 hexiayun1962@163.com
Contact: Fen Xue, MD +86-18505123563 xuefen1992@126.com Less << |
|
NCT00352118
|
Head and Neck Cancer |
Phase 2 |
Terminated(Low dose radiation ... More >>treatment was not appropriate for these patients.) Less << |
- |
United States, Minnesota
... More >>
Masonic Cancer Center at University of Minnesota
Minneapolis, Minnesota, United States, 55455 Less << |
|
NCT00352118
|
- |
|
Terminated(Low dose radiation ... More >>treatment was not appropriate for these patients.) Less << |
- |
- |
|
NCT03519295
|
Anal Cancer |
Phase 2 |
Recruiting |
June 2022 |
France
... More >> CHRU Jean Minjoz
Recruiting
Besançon, France, 25000
Contact: Stefano KIM, MD
CHU Bordeaux - Hôpital Haut Lévêque
Recruiting
Bordeaux, France
Contact: Denis SMITH
Centre François Baclesse
Recruiting
Caen, France
Contact: Aurélie PARZY, MD
Centre Georges François Leclerc
Recruiting
Dijon, France
Contact: françois GHIRINGHELLI, MD
Centre Oscar Lambert
Recruiting
Lille, France
Contact: Farid EL HAJBI, MD
Centre Léon Bérard
Recruiting
Lyon, France
Contact: Christelle de la Fouchardière, MD
Hôpital Jean Mermoz
Recruiting
Lyon, France
Contact: Jérôme DESRAME, MD
Hopital Montbéliard
Recruiting
Montbéliard, France
Contact: Christophe BORG, MD
ICM Val d'Aurelle
Recruiting
Montpellier, France
Contact: Emmanuelle SAMALIN, MD
Centre Antoine Lacassagne
Recruiting
Nice, France
Contact: Eric FRANCOIS, MD
Hôpital Saint Antoine
Recruiting
Paris, France, 7514
Contact: Daniel LOPEZ TRABADA ATAZ
Groupe Hospitalier Saint Joseph
Recruiting
Paris, France
Contact: Nabil BABA HAMED, MD
Hôpital Européen Georges Pompidou
Recruiting
Paris, France
Contact: Simon PERNOT
Hôpital Henri Mondor
Not yet recruiting
Paris, France
Contact: Isabelle BAUMGAERTNER, MD
Hôpital saint Louis
Recruiting
Paris, France
Contact: Nelson LOURENCO, MD
Insitut Curie
Recruiting
Paris, France
Contact: Bruno BUECHER
CHU Robert Debré Reims
Recruiting
Reims, France
Contact: Olivier BOUCHE, MD
Centre Paul Strauss
Recruiting
Strasbourg, France
Contact: Meher BENABDELGHANI, MD Less << |
|
NCT00400114
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
Canada, Ontario
... More >>
The Ottawa Hospital Regional Cancer Centre
Ottawa, Ontario, Canada, K1H 8L6
University Health Network (Princess Margaret & Toronto General Hospitals)
Toronto, Ontario, Canada, M5G 2C4 Less << |
|
NCT00842712
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Completed |
- |
- |
|
NCT00002711
|
Esophageal Cancer |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT02759575
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Recruiting |
January 2020 |
United States, Ohio
... More >>
University of Cincinnati Medical Center
Recruiting
Cincinnati, Ohio, United States, 45219
Contact: UC Cancer Institute Clinical Trials Office 513-584-7698 cancer@uchealth.com
Principal Investigator: Vinita Takiar, MD Less << |
|
NCT00842712
|
- |
|
Completed |
- |
- |
|
NCT02403531
|
Neoplasms |
Phase 2 |
Active, not recruiting |
December 2018 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT00578760
|
Hearing Loss
... More >>Ototoxicity Less << |
Not Applicable |
Unknown |
February 2010 |
Canada, Ontario
... More >>
Princess Margaret Hospital
Not yet recruiting
Toronto, Ontario, Canada
Sub-Investigator: Emma Barker, FRCS, PhD Less << |
|
NCT01167738
|
Pancreatic Cancer |
Phase 2 |
Terminated(concern of detrimen... More >>tal effect) Less << |
- |
Italy
... More >> Istituto Scientifico H. San Raffaele
Milan, Italy, 20132 Less << |
|
NCT00005601
|
Lymphoma |
Phase 2 |
Completed |
- |
United States, Arizona
... More >>
CCOP - Scottsdale Oncology Program
Scottsdale, Arizona, United States, 85259-5404
United States, Florida
Mayo Clinic
Jacksonville, Florida, United States, 32224
United States, Illinois
CCOP - Illinois Oncology Research Association
Peoria, Illinois, United States, 61602
United States, Iowa
CCOP - Cedar Rapids Oncology Project
Cedar Rapids, Iowa, United States, 52403-1206
CCOP - Iowa Oncology Research Association
Des Moines, Iowa, United States, 50309-1016
Siouxland Hematology-Oncology
Sioux City, Iowa, United States, 51101-1733
United States, Kansas
CCOP - Wichita
Wichita, Kansas, United States, 67214-3882
United States, Louisiana
CCOP - Ochsner
New Orleans, Louisiana, United States, 70121
United States, Michigan
CCOP - Ann Arbor Regional
Ann Arbor, Michigan, United States, 48106
United States, Minnesota
CCOP - Duluth
Duluth, Minnesota, United States, 55805
Mayo Clinic Cancer Center
Rochester, Minnesota, United States, 55905
CentraCare Health Plaza
Saint Cloud, Minnesota, United States, 56303
CCOP - Metro-Minnesota
Saint Louis Park, Minnesota, United States, 55416
United States, Nebraska
CCOP - Missouri Valley Cancer Consortium
Omaha, Nebraska, United States, 68106
United States, North Dakota
Medcenter One Health System
Bismarck, North Dakota, United States, 58501
CCOP - Merit Care Hospital
Fargo, North Dakota, United States, 58122
United States, Ohio
CCOP - Toledo Community Hospital
Toledo, Ohio, United States, 43623-3456
United States, Pennsylvania
CCOP - Geisinger Clinic and Medical Center
Danville, Pennsylvania, United States, 17822-2001
United States, South Dakota
Rapid City Regional Hospital
Rapid City, South Dakota, United States, 57709
CCOP - Sioux Community Cancer Consortium
Sioux Falls, South Dakota, United States, 57104
Canada, Saskatchewan
Allan Blair Cancer Centre
Regina, Saskatchewan, Canada, S4T 7T1 Less << |
|
NCT03025477
|
Ovarian Cancer, Epithelial |
Phase 2 |
Recruiting |
October 2029 |
France
... More >> Centre Georges François Leclerc
Recruiting
Dijon, France
Contact: Leïla BENGRINE, MD
Principal Investigator: Leïla BENGRINE, MD
Institut Hospitalier Franco-Britannique
Recruiting
Levallois-Perret, France
Contact: Aimery De GRAMONT, MD
Principal Investigator: Aimery De GRAMONT, MD
Groupe Hospitalier Diaconesses Croix Saint Simon
Recruiting
Paris, France
Contact: Richard VILLET, MD
Principal Investigator: Richard VILLET, MD
Hôpital Saint Antoine
Recruiting
Paris, France
Contact: Honorine GERVAIS, MD
Principal Investigator: Honorine GERVAIS, MD
Sub-Investigator: Delphine COCHEREAU, MD
Hôpital Poissy Saint Germain
Recruiting
Poissy, France
Contact: Cyrille HUCHON, MD
Principal Investigator: Cyrille HUCHON, MD
CHU Poitiers
Recruiting
Poitiers, France
Contact: Nadia RABAN
Principal Investigator: Nadia RABAN, MD
Centre Hospitalier Senlis
Recruiting
Senlis, France
Contact: Elisabeth CAROLA, MD
Principal Investigator: Elisabeth CAROLA, MD Less << |
|
NCT01627288
|
Cancer of the Cervix
... More >> Cervical Neoplasms Less << |
Not Applicable |
Active, not recruiting |
June 2020 |
United States, North Carolina
... More >>
Radiation Oncology, DUMC
Durham, North Carolina, United States, 27710 Less << |
|
NCT00705627
|
Nasopharyngeal Carcinoma |
Phase 3 |
Unknown |
June 2017 |
China, Guangdong
... More >>
Department of Nasopharyngeal Carcinoma, Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Sumei Cao, Ph. D 86-20-8734-5685 caosumei@mail.sysu.edu.cn
Principal Investigator: Minghuang Hong, MD Less << |
|
NCT00002783
|
Gastric Cancer |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT02630264
|
Head and Neck Neoplasms |
Phase 3 |
Unknown |
December 2016 |
China, Guangdong
... More >>
Guangzhou DB
Recruiting
Gaungzhou, Guangdong, China, 510663
Contact: Chao Zhang georgezhang2015@sina.com Less << |
|
NCT03612791
|
Locally Advanced Cervical Canc... More >>er Less << |
Phase 2 |
Recruiting |
July 2022 |
France
... More >> Gustave Roussy
Recruiting
Villejuif, Val De Marne, France, 94805
Contact: Cyrus CHARGARI, MD, PhD 0142114211 ext +33 cyrus.chargari@gustaveroussy.fr Less << |
|
NCT02425397
|
- |
|
Unknown |
March 2016 |
France
... More >> Institut Curie
Paris, France, 75005 Less << |
|
NCT00146276
|
Bladder Cancer
... More >> Carcinoma, Transitional Cell Less << |
Phase 3 |
Unknown |
- |
Germany
... More >> Saarland University
Recruiting
Homburg/Saar, Saarland, Germany, 66421
Contact: Jan Lehmann, MD +49-(0)6841-1624700 jan.lehmann@uniklinikum-saarland.de
Contact: Michael Stöckle, MD +49-(0)6841-1624702 michael.stoeckle@uniklinikum-saarland.de
Principal Investigator: Michael Stöckle, MD
Sub-Investigator: Jan Lehmann, MD Less << |
|
NCT03515837
|
Non-small Cell Lung Cancer |
Phase 3 |
Recruiting |
June 15, 2023 |
- |
|
NCT02381847
|
Malignant Neoplasm of Stomach |
Phase 3 |
Recruiting |
March 2020 |
China, Jiangsu
... More >> Meng Wang
Recruiting
Nanjing, Jiangsu, China, 210000
Contact: Meng Wang 86-13815890469 wangmeng001@263.net
Contact: Wenxian Guan 86-25-83106666 guan_wx@163.com
Principal Investigator: Wenxian Guan, MD
Sub-Investigator: Baorui Liu, MD
Sub-Investigator: Meng Wang, MD Less << |
|
NCT01791374
|
Part 1- Advanced Solid Tumors
... More >>
Part 2- Advanced or Metastatic Gastric Cancer
Part 2- Advanced or Metastatic GEJ Less << |
Phase 1 |
Completed |
- |
Japan
... More >> Research Site
Nagoya-shi, Aichi, Japan, 464-8681
Research Site
Kashiwa-shi, Chiba, Japan, 277-8577
Research Site
Matsuyama-shi, Ehime, Japan, 791-0280
Research Site
Sapporo-shi, Hokkaido, Japan, 060-8648
Research Site
Kawasaki-shi, Kanagawa, Japan, 216-8511
Research Site
Kitaadachi-gun, Saitama, Japan, 362-0806
Research Site
Suntou-gun, Shizuoka, Japan, 411-8777 Less << |
|
NCT00039910
|
Non-Hodgkin Lymphoma
... More >> Hodgkin Disease
Thrombocytopenia Less << |
Phase 3 |
Completed |
- |
- |
|
NCT03635567
|
Cervical Cancer |
Phase 3 |
Recruiting |
November 23, 2022 |
- |
|
NCT01406249
|
Gastric Cancer |
Phase 2 |
Active, not recruiting |
December 2017 |
Japan
... More >> Epidemiological and Clinical Research Information Network
Kyoto, Japan, 606-8392 Less << |
|
NCT02262325
|
Recurrent Non-small Cell Lung ... More >>Cancer
Stage IIA Non-small Cell Lung Cancer
Stage IIB Non-small Cell Lung Cancer
Stage IIIA Non-small Cell Lung Cancer
Stage IIIB Non-small Cell Lung Cancer Less << |
Phase 2 |
Recruiting |
December 31, 2018 |
United States, Ohio
... More >>
Arthur G. James Cancer Hospital and Solove Research Institute at Ohio State University Medical Center
Recruiting
Columbus, Ohio, United States, 43210
Contact: Terence M. Williams 614-293-5557 terence.williams@osumc.edu
Principal Investigator: Terence M. Williams Less << |
|
NCT02578680
|
Non-Small-Cell Lung Carcinoma |
Phase 3 |
Active, not recruiting |
April 15, 2019 |
- |
|
NCT03246750
|
Extranodal NK/T-cell Lymphoma |
Phase 1
Phase 2 |
Not yet recruiting |
February 2022 |
- |
|
NCT00544414
|
Head and Neck Cancer |
Phase 2 |
Active, not recruiting |
January 2019 |
- |
|
NCT00984997
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT02578680
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00981656
|
Bladder Cancer |
Phase 2 |
Active, not recruiting |
October 2023 |
United States, Georgia
... More >>
Emory Crawford Long Hospital
Atlanta, Georgia, United States, 30308
Winship Cancer Institute of Emory University
Atlanta, Georgia, United States, 30322
United States, Maryland
St. Agnes Hospital Cancer Center
Baltimore, Maryland, United States, 21229
United States, Massachusetts
Hudner Oncology Center at Saint Anne's Hospital - Fall River
Fall River, Massachusetts, United States, 02721
United States, New Hampshire
Norris Cotton Cancer Center at Dartmouth-Hitchcock Medical Center
Lebanon, New Hampshire, United States, 03756-0002
United States, New York
Beth Israel Medical Center - Petrie Division
New York, New York, United States, 10003-3803
United States, Ohio
Summa Center for Cancer Care at Akron City Hospital
Akron, Ohio, United States, 44309-2090
Barberton Citizens Hospital
Barberton, Ohio, United States, 44203
Cancer Care Center, Incorporated
Salem, Ohio, United States, 44460
Cancer Treatment Center
Wooster, Ohio, United States, 44691
United States, Pennsylvania
Fox Chase Cancer Center - Philadelphia
Philadelphia, Pennsylvania, United States, 19111-2497
United States, Texas
University of Texas Medical Branch
Galveston, Texas, United States, 77555-0361
United States, Vermont
Norris Cotton Cancer Center - North
Saint Johnsbury, Vermont, United States, 05819 Less << |
|
NCT00003376
|
Bladder Cancer
... More >> Transitional Cell Cancer of the Renal Pelvis and Ureter
Urethral Cancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT02662062
|
Bladder Cancer |
Phase 2 |
Recruiting |
January 2024 |
Australia, New South Wales
... More >>
Chris O'Brien Lifehouse
Recruiting
Camperdown, New South Wales, Australia, 2050
Principal Investigator: Peter Grimison, MBBS, FRACP
Prince of Wales Hospital
Recruiting
Randwick, New South Wales, Australia, 2013
Contact: Christie Norris, RN, MPH +61 2 9382 2588 Christie.Norris@health.nsw.gov.au
Principal Investigator: Elizabeth Hovey, MBBS, FRACP
Royal North Shore Hospital
Not yet recruiting
St Leonards, New South Wales, Australia, 2065
Contact: Clare Banks Clare.Banks@health.nsw.gov.au
Contact: George Hruby george.hruby@health.nsw.gov.au
Principal Investigator: George Hruby
Australia, Victoria
Austin Health
Recruiting
Heidelberg, Victoria, Australia, 3084
Contact: Andrew Weickhardt, MBBS, FRACP +61 39496 5763 andrew.weickhardt@onjcri.org.au
Contact: Jenni Flynn, BNur +61 3 9496 3352 jenni.flynn@austin.org.au
Principal Investigator: Andrew Weickhardt, MBBS, FRACP
Peter MacCallum Cancer Centre
Terminated
Melbourne, Victoria, Australia, 3002
Australia, Western Australia
Sir Charles Gairdner Hospital
Recruiting
Nedlands, Perth, Western Australia, Australia, 6009
Contact: Judy Innes-Rowe +61 0408 818 656 judy.innes-rowe@health.wa.gov.au
Contact: Jennifer McConnell, BNur +61 8 6383 3187 jennifer.mcconnell@health.wa.gov.au
Principal Investigator: Siobhan Ng, MBBS, FRACP Less << |
|
NCT03775265
|
Bladder Carcinoma Infiltrating... More >> the Muscle of the Bladder Wall
Bladder Urothelial Carcinoma
Stage II Bladder Cancer AJCC v8
Stage III Bladder Cancer AJCC v8
Stage IIIA Bladder Cancer AJCC v8 Less << |
Phase 3 |
Not yet recruiting |
June 1, 2025 |
- |
|
NCT01230996
|
Locally Advanced Cervical Canc... More >>er Less << |
Phase 1
Phase 2 |
Active, not recruiting |
April 2018 |
United Kingdom
... More >> Saint Bartholomew's Hospital
London, England, United Kingdom, EC1A 7BE
Hammersmith Hospital
London, England, United Kingdom
Royal Marsden - London
London, England, United Kingdom
Sheffield Teaching Hospitals NHS Foundation Trust
Sheffield, United Kingdom Less << |
|
NCT01461057
|
Gastric Cancer |
Phase 2 |
Completed |
- |
Belgium
... More >> Cliniques Universitaires St-Luc
Bruxelles, Belgium, 1200
UZ Leuven Gasthuisberg
Leuven, Belgium, 3000
Czechia
Masaryk Memorial Cancer Institute; Oncological Clinic
Brno, Czechia, 656 53
Fakultni Nemocnice Hradec Kralove; Dept of Radiotherapy & Oncology
Hradec Kralove, Czechia, 500 05
Fakultni nemocnice Olomouc; Onkologicka klinika
Olomouc, Czechia, 779 00
Fakultní Nemocnice V Motole; Radioterapeuticko-Onkologicke Oddeleni
Praha 5, Czechia, 150 06
University Hospital Na Bulovce; Institut of Radiation Oncology
Praha 8, Czechia, 180 81
France
Hopital Morvan
Brest, France, 29200
CRLCC Val dAurelle Paul Lam
Montpellier cedex 5, France, 34298
Hopital Robert Debre; DERMATOLOGIE
Reims, France, 51092
Germany
Charité-Universitätsm. Berlin; Med. Klinik mit Schwerpunkt Hämatologie, Onkologie und Tumorimmunolo.
Berlin, Germany, 10117
Klinikum Braunschweig; Medizinische Klinik III; Klinik für Hämatologie und Onkologie
Braunschweig, Germany, 38114
Krankenhaus Nordwest; Klinik f. Onkologie und Hämatologie
Frankfurt, Germany, 60488
Klinikum Mannheim III. Medizinische Klinik
Mannheim, Germany, 68167
Italy
Campus Universitario S.Venuta; Centro Oncologico T.Campanella
Catanzaro, Calabria, Italy, 88100
Irccs Istituto Nazionale Dei Tumori (Int);S.C. Medicina Oncologica 2
Milano, Lombardia, Italy, 20133
A.O. Universitaria Pisana; Oncologia
Pisa, Toscana, Italy, 56100
Korea, Republic of
Seoul National Uni Hospital; Dept. of Internal Medicine/Hematology/Oncology
Seoul, Korea, Republic of, 03080
Yonsei Medical Center; Dept. of Medicine , Division of Hemato-Oncology
Seoul, Korea, Republic of, 03722
Asan Medical Center; Medical Oncology
Seoul, Korea, Republic of, 05505
Netherlands
Academish Ziekenhuis Maastricht (Azm); Inwendige Geneeskunde
Maastricht, Netherlands, 6229 HX
Spain
Hospital del Mar; Servicio de Oncologia
Barcelona, Spain, 08003
Hospital Universitario La Paz; Servicio de Oncologia
Madrid, Spain, 28046
Hospital Clinico Universitario de Valencia; Servicio de Onco-hematologia
Valencia, Spain, 46010 Less << |
|
NCT00960284
|
Pancreatic Cancer |
Phase 2
Phase 3 |
Completed |
- |
Italy
... More >> Istituto Scientifico H. San Raffaele
Milan, Italy, 20132 Less << |
|
NCT01336543
|
Stage III Non-small Cell Lung ... More >>Cancer Less << |
Phase 2 |
Withdrawn(Low accrual, small p... More >>atient population at center.) Less << |
- |
United States, Louisiana
... More >>
LSU Health Sciences Center, 1501 Kings Highway
Shreveport, Louisiana, United States, 71103 Less << |
|
NCT02977169
|
Non-small Cell Lung Cancer Sta... More >>ge IIIA
Radiotherapy Less << |
Phase 2 |
Recruiting |
December 2019 |
China, Shanghai
... More >>
Shanghai Chest Hospital
Recruiting
Shanghai, Shanghai, China
Contact: Xiaolong Fu, PhD +862122200000 ext 3602 xlfu1964@126.com Less << |
|
NCT00883779
|
Non-Squamous Non-Small Cell Lu... More >>ng Cancer Less << |
Phase 3 |
Completed |
- |
China
... More >>
Beijing, China, 100021
Beijing, China, 100142
Beijing, China, 101149
Guangzhou, China, 510060
Guangzhou, China
Hangzhou, China
Nanjing, China, 210029
Shanghai, China, 200030
Shanghai, China, 200433
Hong Kong
Hong Kong, Hong Kong, 852
Hong Kong, Hong Kong
Shatin, Hong Kong
Indonesia
Jakarta, Indonesia, 13230
Jogjakarta, Indonesia, 55284
Surabaya, Indonesia, 60286
Korea, Republic of
Gyeonggi-do, Korea, Republic of, 410-769
Philippines
Manila, Philippines, 1000
Pasig City, Philippines, 1605
Quezon City, Philippines, 1104
Taiwan
Taichung, Taiwan, 407
Taipei, Taiwan, 100
Taipei, Taiwan, 116
Taipei, Taiwan
Thailand
Bangkok, Thailand, 10400
Bangkok, Thailand, 10700
Chiang Mai, Thailand, 50200 Less << |
|
NCT00957086
|
Carcinoma, Squamous Cell of He... More >>ad and Neck Less << |
Phase 3 |
Recruiting |
June 2021 |
- |
|
NCT00003027
|
Melanoma (Skin) |
Phase 3 |
Completed |
- |
- |
|
NCT00039338
|
Cervical Cancer |
Phase 3 |
Active, not recruiting |
- |
Austria
... More >> Karl-Franzens-University Graz
Graz, Austria
Kaiser Franz Josef Hospital
Vienna, Austria
Belgium
Universitair Ziekenhuis Antwerpen
Edegem, Belgium
U.Z. Gasthuisberg
Leuven, Belgium
Centre Hospitalier Regional de la Citadelle
Liege, Belgium, 4000
France
Centre Regional Francois Baclesse
Caen, France
Italy
Istituto Europeo Di Oncologia
Milano, Italy
Ospedale San Gerardo
Monza, Italy
Istituto Nazionale Per Lo Studio E La Cura Dei Tumori
Naples, Italy
Ospedale Mauriziano Umberto I
Torino, Italy, 10128
Clinica Universitaria
Turin, Italy, 10126
Ospedale di Circolo e Fondazione Macchi
Varese, Italy
Netherlands
Academisch Medisch Centrum at University of Amsterdam
Amsterdam, Netherlands, 1105 AZ
Vrije Universiteit Medisch Centrum
Amsterdam, Netherlands
Medisch Spectrum Twente
Enschede, Netherlands
Leiden University Medical Center
Leiden, Netherlands
Universitair Medisch Centrum St. Radboud - Nijmegen
Nijmegen, Netherlands
Erasmus MC - Daniel den Hoed Cancer Center
Rotterdam, Netherlands, 3008 AE
Universitair Medisch Centrum - Academisch Ziekenhuis
Utrecht, Netherlands
Portugal
Hospitais da Universidade de Coimbra (HUC)
Coimbra, Portugal
Spain
Hospital Universitario San Carlos
Madrid, Spain
United Kingdom
Queen Elizabeth The Queen Mother Hospital
Margate, England, United Kingdom
NHS Greater Glasgow and Clyde - Beatson West of Scotland Cancer Centre
Glasgow, Scotland, United Kingdom
Mid Kent Oncology Centre
Maidstone, Kent, United Kingdom Less << |
|
NCT00460265
|
Recurrent and/or Metastatic He... More >>ad and Neck Cancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT01102231
|
Stage III Non-small Cell Lung ... More >>Cancer Less << |
Phase 2 |
Completed |
- |
France
... More >> Clinique de l'Europe
Amiens, France
Centre Hospitalier
Annemasse, France
CHU Besancon - Pneumologie
Besancon, France, 25000
Bordeaux - Polyclinique Nord
Bordeaux, France, 33300
Caen - Centre François Baclesse
Caen, France, 14000
Caen - CHU Côte de Nacre
Caen, France, 14000
CH
Chartres, France
CH
Cholet, France
CHU
Clermont-Ferrand, France
CH
Colmar, France
Clinique des Cèdres
Cornebarrieu, France
Dijon - CAC
Dijon, France, 21000
CHU Grenoble
Grenoble, France, 38000
Institut d'Oncologie Hartmann
Levallois, France
CHU (Hôpital Calmette) - Pneumologie
Lille, France, 59000
CH
Longjumeau, France
Clinique des 4 Pavillons
Lormont, France
Hôpital Louis Pradel
Lyon, France
Hôpital Nord
Marseille, France
Centre Hospitalier
Montélimar, France
CHU
Nancy, France
CH
Nevers, France
Centre Hospitalier
Niort, France
APHP - Hopital Tenon - Pneumologie
Paris, France, 75020
Hôpital du Val de Grâce
Paris, France
Hôpital Saint-Joseph
Paris, France
Perpignan - Centre Catalan d'Oncologie
Perpignan, France, 66000
HCL - Lyon Sud
Pierre Bénite, France, 69495
CHU
Poitiers, France
Centre Hospitalier
Rambouillet, France
Reims - CHU
Reims, France, 51092
Institut Jean Godinot
Reims, France
Centre Frederic Joliot
Rouen, France
Centre Etienne Dolet
Saint-Nazaire, France
Hôpitaux Universitaires - Nouvel Hôpital Civil
Strasbourg, France, 63000
Suresnes - Hopital Foch
Suresnes, France, 92151
Tours - CHU
Tours, France, 37000
Institut Gustave Roussy
Villejuif, France, 94800 Less << |
|
NCT00883779
|
- |
|
Completed |
- |
- |
|
NCT00003582
|
Carcinoma of Unknown Primary
... More >> Head and Neck Cancer Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, Florida
... More >>
Sylvester Cancer Center, University of Miami
Miami, Florida, United States, 33136 Less << |
|
NCT00460265
|
- |
|
Completed |
- |
- |
|
NCT00433563
|
Lung Cancer |
Not Applicable |
Active, not recruiting |
February 2019 |
United Kingdom
... More >> The Christie NHS Foundation Trust
Manchester, United Kingdom, M20 4BX Less << |
|
NCT00394966
|
Nausea
Vomiti... More >>ng Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00360971
|
Head and Neck Cancer
... More >> Mucositis
Pain
Radiation Toxicity Less << |
Phase 3 |
Terminated(Due to positive pre... More >>liminary results from other palifermin studies.) Less << |
- |
- |
|
NCT01063478
|
Non-Small Cell Lung Cancer |
Phase 1 |
Terminated(Poor accrual) |
- |
United States, Illinois
... More >>
University of Chicago Medical Center
Chicago, Illinois, United States, 60637 Less << |
|
NCT01720563
|
Cancer-related Fatigue |
Phase 2 |
Terminated(Change study drug d... More >>osage form) Less << |
- |
Taiwan
... More >> Chang Gung Memorial Hospital
Taoyuan, Taiwan, 333 Less << |
|
NCT01424839
|
Intracranial Germ Cell Tumors |
Phase 4 |
Unknown |
October 2018 |
Germany
... More >> University Hospital Muenster
Recruiting
Muenster, Germany, 48129
Contact: Gabriele Calaminus, MD +492518358060 gabriele.calaminus@ukmuenster.de
Contact: Carmen Teske +492518358055 carmen.teske@ukmuenster.de
Principal Investigator: Gabriele Calaminus, MD Less << |
|
NCT00310739
|
- |
|
Unknown |
- |
Japan
... More >> Hamamatsu University School of Medicine
Recruiting
Hamamatsu, Shizuoka, Japan, 431-3192
Contact: Takayuki Tsuji, MD +81-53-435-2261 tsu@hama-med.ac.jp Less << |
|
NCT01734798
|
Locally Advanced Bladder Cance... More >>r Less << |
Phase 3 |
Completed |
- |
Egypt
... More >> National cancer Institute-Cairo University
Cairo, Egypt Less << |
|
NCT01040312
|
- |
|
Unknown |
March 2014 |
Japan
... More >> Department of Obstetrics and Gynecology, National Defense Medical College Hospital
Tokorozawa, Saitama, Japan, 359-8513 Less << |
|
NCT02672865
|
Locally Advanced Malignant Neo... More >>plasm
Gastric Cancer Less << |
Phase 1 |
Recruiting |
December 2018 |
United States, California
... More >>
Loma Linda University Cancer Center
Recruiting
Loma Linda, California, United States, 92354
Contact: Maheswari Senthil, MD 909-558-4000 ext 15460 msenthil@llu.edu Less << |
|
NCT01809210
|
Locally Advanced or Metastatic... More >> NSCL Cancer Stage IIIB IV Less << |
Phase 1 |
Completed |
- |
United Kingdom
... More >> Research Site
Glasgow, United Kingdom, G12 0YN
Research Site
London, United Kingdom, W1G 6AD
Research Site
Manchester, United Kingdom, M20 4BX
Research Site
Newcastle upon Tyne, United Kingdom, NE7 7DN Less << |
|
NCT00003004
|
Unspecified Adult Solid Tumor,... More >> Protocol Specific Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT03579004
|
Esophageal Neoplasms
... More >> Squamous Cell Carcinoma
Concurrent Chemoradiotherapy Less << |
Phase 2 |
Recruiting |
July 10, 2020 |
Russian Federation
... More >>
Alexey Tryakin
Recruiting
Moscow, Russian Federation, 115478
Contact: Alexey Tryakin, MD, PhD 4993249259 ext +7 atryakin@mail.ru
Russian Cancer Research Center named after N.N.Blokhin RAMS
Recruiting
Moscow, Russian Federation, 115478
Contact: Alexey Tryakin, PhD +74993249259 atryakin@mail.ru
Contact: Ilya Pokataev +74993241219 pokia@mail.ru
Principal Investigator: Alexey Tryakin, PhD Less << |
|
NCT01809210
|
- |
|
Completed |
- |
- |
|
NCT01993979
|
Transitional Cell Carcinoma of... More >> Ureter Less << |
Phase 3 |
Active, not recruiting |
May 2022 |
- |
|
NCT00003642
|
Bladder Cancer |
Phase 2 |
Terminated(low accrual) |
- |
France
... More >> Institut Bergonie
Bordeaux, France, 33076
CHR de Grenoble - La Tronche
Grenoble, France, 38043
Netherlands
Dr. Bernard Verbeeten Instituut
Tilburg, Netherlands, 5042 SB Less << |
|
NCT00003053
|
Lung Cancer |
Phase 3 |
Unknown |
- |
- |
|
NCT02115464
|
Lung Cancer |
Phase 2 |
Recruiting |
December 2020 |
Canada, Alberta
... More >>
Cross Cancer Institute
Recruiting
Edmonton, Alberta, Canada
Principal Investigator: Wilson Roa
Canada, Manitoba
CancerCare Manitoba
Recruiting
Winnipeg, Manitoba, Canada, R3E 0V9
Principal Investigator: Naseer Ahmed
Canada, Ontario
Juravinski Cancer Centre
Recruiting
Hamilton, Ontario, Canada
Principal Investigator: Theos Tsakiridis
Cancer Centre of Southeastern Ontario at Kingston General Hospital
Recruiting
Kingston, Ontario, Canada
Principal Investigator: Andrew Robinson
Grand River Regional Cancer Centre
Active, not recruiting
Kitchener, Ontario, Canada
Walker Family Cancer Centre - Niagara Health System
Recruiting
St. Catharines, Ontario, Canada
Principal Investigator: Theos Tsakiridis
Canada, Quebec
Montreal General Hospital - McGill
Recruiting
Montreal, Quebec, Canada
Principal Investigator: Bassam Abdulkarim Less << |
|
NCT02444949
|
Nasopharyngeal Neoplasms |
Phase 3 |
Unknown |
June 2017 |
- |
|
NCT01133678
|
Head and Neck Cancer |
Phase 2 |
Active, not recruiting |
October 2018 |
United States, Illinois
... More >>
The University of Chicago Medical Center
Chicago, Illinois, United States, 60637
NorthShore University Health System
Evanston, Illinois, United States, 60201 Less << |
|
NCT01004978
|
Hepatocellular Carcinoma
... More >> Unresectable Hepatocellular Carcinoma Less << |
Phase 3 |
Active, not recruiting |
- |
- |
|
NCT03423849
|
Breast Cancer
... More >> Chemotherapy Effect
Disease-free Survival Less << |
Phase 2
Phase 3 |
Not yet recruiting |
February 8, 2020 |
- |
|
NCT02749526
|
Esophageal |
Phase 2 |
Unknown |
December 2016 |
China, Jiangsu
... More >> The First Affiliated Hospital with Nanjing Medical University
Recruiting
Nanjing, Jiangsu, China, 210000
Contact: Yin Qingfeng, Manager 13912903257 363994906@qq.com Less << |
|
NCT00450203
|
Oesophagogastric Cancer |
Phase 2
Phase 3 |
Unknown |
December 2017 |
United Kingdom
... More >> Royal Bournemouth Hospital
Recruiting
Bournemouth, England, United Kingdom, BH7 7DW
Contact: Tom Geldart 44-1202-726-088
Bradford Royal Infirmary
Active, not recruiting
Bradford, England, United Kingdom, BD9 6RJ
Bristol Haematology and Oncology Centre
Recruiting
Bristol, England, United Kingdom, BS2 8ED
Contact: Stephen J. Falk, MD 44-117-928-3074 stephen.falk@ubht.nhs.uk
Addenbrooke's Hospital
Active, not recruiting
Cambridge, England, United Kingdom, CB2 2QQ
Cumberland Infirmary
Active, not recruiting
Carlisle, England, United Kingdom, CA2 7HY
Doncaster Royal Infirmary
Recruiting
Doncaster, England, United Kingdom, DN2 5LT
Contact: Jonathan Wadsley 44-1302-366-666
St. Luke's Cancer Centre at Royal Surrey County Hospital
Recruiting
Guildford, England, United Kingdom, GU2 7XX
Contact: Gary W. Middleton 44-1483-570-122 gmiddleton@royalsurrey.nhs.uk
Huddersfield Royal Infirmary
Recruiting
Huddersfield, West Yorks, England, United Kingdom, HD3 3EA
Contact: Jo Dent 44-1484-342-000
Leeds Cancer Centre at St. James's University Hospital
Recruiting
Leeds, England, United Kingdom, LS9 7TF
Contact: Matthew T. Seymour, MA, MD, FRCP 44-113-206-6400
Lincoln County Hospital
Active, not recruiting
Lincoln, England, United Kingdom, LN2 5QY
Aintree University Hospital
Recruiting
Liverpool, England, United Kingdom, L9 7AL
Contact: David Smith, MD 44-151-525-5980
Saint Bartholomew's Hospital
Recruiting
London, England, United Kingdom, EC1A 7BE
Contact: Sarah Slater, MD 44-20-7601-8391
St. George's Hospital
Active, not recruiting
London, England, United Kingdom, SW17 0QT
St. Mary's Hospital
Active, not recruiting
London, England, United Kingdom, W2 1NY
Mid Kent Oncology Centre at Maidstone Hospital
Recruiting
Maidstone, England, United Kingdom, ME16 9QQ
Contact: Justin Waters, MD 44-1622-729-000
Christie Hospital
Recruiting
Manchester, England, United Kingdom, M20 4BX
Contact: Was Mansoor, MD 44-845-226-3000
Clatterbridge Centre for Oncology
Recruiting
Merseyside, England, United Kingdom, CH63 4JY
Contact: David Smith, MD 44-151-334-1155 david.smith@ccotrust.nhs.uk
Northern Centre for Cancer Treatment at Newcastle General Hospital
Recruiting
Newcastle-Upon-Tyne, England, United Kingdom, NE4 6BE
Contact: Fareeda Coxon, MD 44-191-256-3551 fareeda.coxon@nuth.nhs.uk
Derriford Hospital
Active, not recruiting
Plymouth, England, United Kingdom, PL6 8DH
Dorset Cancer Centre
Active, not recruiting
Poole Dorset, England, United Kingdom, BH15 2JB
Berkshire Cancer Centre at Royal Berkshire Hospital
Recruiting
Reading, England, United Kingdom, RG1 5AN
Contact: Joss Adams, MD 44-118-322-7878
Rochdale Infirmary
Active, not recruiting
Rochdale, England, United Kingdom, 0L12 0NB
Salisbury District Hospital
Recruiting
Salisbury, England, United Kingdom, SP2 8BJ
Contact: Tim J. Iveson, MD 44-1722-336-262 ext. 4688
Wexham Park Hospital
Recruiting
Slough, Berkshire, England, United Kingdom, SL2 4HL
Contact: Marcia Hall, MD 44-1753-634-364 marcia.hall@nhs.net.uk
Southampton General Hospital
Recruiting
Southampton, England, United Kingdom, SO16 6YD
Contact: Tim J. Iveson, MD 44-23-8079-6802 t.iveson@soton.ac.uk
Royal Marsden - Surrey
Recruiting
Sutton, England, United Kingdom, SM2 5PT
Contact: David Cunningham, MD 44-20-8661-3279 david.cunningham@rmh.nhs.uk
Aberdeen Royal Infirmary
Recruiting
Aberdeen, Scotland, United Kingdom, AB25 2ZN
Contact: Russell Petty, MD 44-84-5456-6000
Velindre Cancer Center at Velindre Hospital
Recruiting
Cardiff, Wales, United Kingdom, CF14 2TL
Contact: Tom Crosby, MD 44-29-2031-6292
Basingstoke and North Hampshire Hospital
Recruiting
Basingstoke, United Kingdom
Principal Investigator: Charlotte Rees
Birmingham Heartlands Hospital
Recruiting
Birmingham, United Kingdom
Principal Investigator: Jo Dent
Castle Hill Hospital
Recruiting
Cottingham, United Kingdom
Principal Investigator: Mohan Hingorani
University Hospitals Coventry and Warwickshire
Recruiting
Coventry, United Kingdom
Principal Investigator: Sharmila Sothi
Beatson West of Scotland Cancer Centre
Recruiting
Glasgow, United Kingdom
Principal Investigator: Janet Graham
St James Hospital
Recruiting
Leeds, United Kingdom
Principal Investigator: Matt Seymour
Leicester Royal Infirmary
Recruiting
Leicester, United Kingdom
Principal Investigator: Anne Thomas
Norfolk and Norwich University Hospital
Recruiting
Norwich, United Kingdom
Principal Investigator: Tom Roques
Churchill Hospital
Recruiting
Oxford, United Kingdom
Principal Investigator: Kinnari Patel
Queens Hospital
Recruiting
Romford, United Kingdom
Principal Investigator: Sherif Raouf
Weston Park
Recruiting
Sheffield, United Kingdom
Principal Investigator: Suzanne Darby
Great Western Hospital
Recruiting
Swindon, United Kingdom
Principal Investigator: Claire Blesing
Musgrove Park Hospital
Recruiting
Taunton, United Kingdom
Principal Investigator: Emma Cattell Less << |
|
NCT01055301
|
Multiple Myeloma
... More >> Plasma Cell Myeloma Less << |
Phase 2 |
Withdrawn(lack of accrual) |
- |
United States, Louisiana
... More >>
Tulane Cancer Center Office of Clinical Research
Alexandria, Louisiana, United States, 71315-3198
Hematology-Oncology Clinic
Baton Rouge, Louisiana, United States, 70809
United States, Michigan
Barbara Ann Karmanos Cancer Institute
Detroit, Michigan, United States, 48201-1379
United States, Mississippi
University of Mississippi Cancer Clinic
Jackson, Mississippi, United States, 39216
United States, Washington
Island Hospital Cancer Care Center at Island Hospital
Anacortes, Washington, United States, 98221
St. Joseph Cancer Center
Bellingham, Washington, United States, 98225
Olympic Hematology and Oncology
Bremerton, Washington, United States, 98310
Highline Medical Center Cancer Center
Burien, Washington, United States, 98166
Columbia Basin Hematology
Kennewick, Washington, United States, 99336
Skagit Valley Hospital Cancer Care Center
Mount Vernon, Washington, United States, 98274
Harrison Poulsbo Hematology and Onocology
Poulsbo, Washington, United States, 98370
Harborview Medical Center
Seattle, Washington, United States, 98104
Minor and James Medical, PLLC
Seattle, Washington, United States, 98104
Fred Hutchinson Cancer Research Center
Seattle, Washington, United States, 98109
Group Health Central Hospital
Seattle, Washington, United States, 98112
Swedish Cancer Institute at Swedish Medical Center - First Hill Campus
Seattle, Washington, United States, 98122-4307
University Cancer Center at University of Washington Medical Center
Seattle, Washington, United States, 98195
North Puget Oncology at United General Hospital
Sedro-Woolley, Washington, United States, 98284
Cancer Care Northwest - Spokane South
Spokane, Washington, United States, 99202
Evergreen Hematology and Oncology, PS
Spokane, Washington, United States, 99218
Wenatchee Valley Medical Center
Wenatchee, Washington, United States, 98801-2028 Less << |
|
NCT00496652
|
Cancer of the Head and Neck |
Phase 3 |
Completed |
- |
Denmark
... More >> Department of Experimental Clinical Oncology, Aarhus University Hospital
Aarhus, Denmark, 8000 N Less << |
|
NCT00030810
|
Lung Cancer |
Phase 2 |
Completed |
- |
Switzerland
... More >> Centre Hospitalier Universitaire Vaudois
Lausanne, Switzerland, CH-1011 Less << |
|
NCT00559351
|
Esophageal Neoplasms
... More >> Squamous Cell Cancer Less << |
Phase 3 |
Terminated(difficulty in recru... More >>itment) Less << |
- |
Poland
... More >> 2nd Department of General, Gastrointestinal Surgery & Surgical Oncology of the Digestive Tract, Medical University of Lublin, Poland
Lublin, Lubelskie, Poland, 20-081 Less << |
|
NCT00875849
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
France
... More >> Centre Antoine Lacassagne
Nice, France, 06189 Less << |
|
NCT01034332
|
Esophageal Cancer |
Phase 2 |
Unknown |
December 2009 |
Taiwan
... More >> National Taiwan University Hospital
Recruiting
Taipei, Taiwan, 100
Contact: Chin-Hung Hsu, MD 02-23123456 ext 7680 chihhunghsu@ntu.edu.tw Less << |
|
NCT03042429
|
Neuroblastoma |
Phase 3 |
Completed |
- |
Germany
... More >> University of Cologne
Koln, Germany, 50924 Less << |
|
NCT02949895
|
Small Cell Lung Cancer |
Phase 1 |
Completed |
- |
Japan
... More >> Local Institution
Takatsuki-shi, Osaka, Japan, 5698686
Local Institution
Chuo-ku, Tokyo, Japan, 1040045 Less << |
|
NCT03287375
|
Peritoneum; Carcinomatosis
... More >> Peritoneal Neoplasms
Peritoneal Metastases
Chemotherapy Effect
Chemotherapeutic Toxicity
Quality of Life
Histologic Progression Less << |
Phase 2 |
Recruiting |
December 1, 2020 |
Denmark
... More >> Department of Surgery, Odense University Hospital
Recruiting
Odense, Denmark, 5000
Principal Investigator: Michael B Mortensen, MD, Ph.D. Less << |
|
NCT02283489
|
Head and Neck Neoplasms |
Phase 2 |
Unknown |
October 2016 |
China, Sichuan
... More >> West China Hospital, Sichuan University
Recruiting
Chengdu, Sichuan, China, 610041
Contact: Liqun Zou, M.D.
Principal Investigator: Liqun Zou, M.D.
Sichuan Provincial People's Hospital
Recruiting
Chengdu, Sichuan, China, 610072
Contact: Ke Xie, M.D.
Principal Investigator: Ke Xie, M.D.
China
Chongqing Cancer Hospital
Recruiting
Chongqing, China, 400030
Contact: Xiaohong Zhou, M.D.
Principal Investigator: Xiaohong Zhou, M.D.
Shanghai Ninth People's Hospital Affiliated Shanghai JiaoTong University School of Medicine
Recruiting
Shanghai, China, 200011
Contact: Wei Guo, M.D.
Principal Investigator: Wei Guo, M.D. Less << |
|
NCT01103544
|
- |
|
Completed |
- |
Germany
... More >> Pierre Fabre Pharma GmbH
Freiburg, Germany Less << |
|
NCT01828099
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00052429
|
- |
|
Completed |
- |
- |
|
NCT00052429
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan - Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT02120118
|
Solid Tumors |
Phase 2 |
Withdrawn(Taiwan FDA asked for... More >> seperating this protocol into one disease site per protocol.) Less << |
February 2017 |
Taiwan
... More >> Shin Kong Wu Ho-Su Memorial Hospital
Taipei, Taiwan, 11101 Less << |
|
NCT02072148
|
Human Papilloma Virus
... More >> Oropharyngeal Squamous Cell Carcinoma Less << |
Not Applicable |
Recruiting |
March 2019 |
United States, New York
... More >>
Mount Sinai Beth Israel
Recruiting
New York, New York, United States, 10003
Principal Investigator: Raymond Chai, MD
Icahn School of Medicine at Mount Sinai
Recruiting
New York, New York, United States, 10029
Principal Investigator: Brett A Miles, DDS, MD Less << |
|
NCT02320448
|
Peritoneal Carcinomatosis |
Phase 2 |
Completed |
- |
Denmark
... More >> Odense University Hospital
Odense, Denmark, 5000 Less << |
|
NCT00588770
|
Recurrent Hypopharyngeal Squam... More >>ous Cell Carcinoma
Recurrent Laryngeal Squamous Cell Carcinoma
Recurrent Laryngeal Verrucous Carcinoma
Recurrent Lip and Oral Cavity Squamous Cell Carcinoma
Recurrent Metastatic Squamous Cell Carcinoma in the Neck With Occult Primary
Recurrent Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma
Recurrent Oral Cavity Verrucous Carcinoma
Recurrent Oropharyngeal Squamous Cell Carcinoma
Recurrent Salivary Gland Carcinoma
Salivary Gland Squamous Cell Carcinoma
Squamous Cell Carcinoma Metastatic in the Neck With Occult Primary
Stage IV Hypopharyngeal Squamous Cell Carcinoma AJCC v7
Stage IV Major Salivary Gland Cancer AJCC v7
Stage IVA Laryngeal Squamous Cell Carcinoma AJCC v7
Stage IVA Laryngeal Verrucous Carcinoma AJCC v7
Stage IVA Lip and Oral Cavity Squamous Cell Carcinoma AJCC v6 and v7
Stage IVA Major Salivary Gland Cancer AJCC v7
Stage IVA Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma AJCC v7
Stage IVA Oral Cavity Cancer AJCC v6 and v7
Stage IVA Oropharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVB Laryngeal Squamous Cell Carcinoma AJCC v7
Stage IVB Laryngeal Verrucous Carcinoma AJCC v7
Stage IVB Lip and Oral Cavity Squamous Cell Carcinoma AJCC v6 and v7
Stage IVB Major Salivary Gland Cancer AJCC v7
Stage IVB Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma AJCC v7
Stage IVB Oral Cavity Cancer AJCC v6 and v7
Stage IVB Oropharyngeal Squamous Cell Carcinoma AJCC v7
Stage IVC Laryngeal Squamous Cell Carcinoma AJCC v7
Stage IVC Laryngeal Verrucous Carcinoma AJCC v7
Stage IVC Lip and Oral Cavity Squamous Cell Carcinoma AJCC v6 and v7
Stage IVC Major Salivary Gland Cancer AJCC v7
Stage IVC Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma AJCC v7
Stage IVC Oral Cavity Cancer AJCC v6 and v7
Stage IVC Oropharyngeal Squamous Cell Carcinoma AJCC v7
Tongue Carcinoma
Untreated Metastatic Squamous Cell Carcinoma to Neck With Occult Primary Less << |
Phase 3 |
Active, not recruiting |
- |
- |
|
NCT02194556
|
EGFR Positive Non-small Cell L... More >>ung Cancer
Adenocarcinoma Less << |
Phase 4 |
Unknown |
July 2017 |
China, Guangxi
... More >> First Affiliated Hospital of Guangxi Medical University
NanNing, Guangxi, China, 530021 Less << |
|
NCT01743482
|
Germ Cell Tumors
... More >> Measurable Disease
Relapse or Progression After 2 or 3 Chemotherapy Regimens.
Relapse or Progression After High-dose Chemotherapy. Less << |
Phase 2 |
Unknown |
April 2015 |
Italy
... More >> Fondazione IRCCS Istituto Nazionale dei Tumori
Not yet recruiting
Milano, Mi, Italy, 20133
Contact: Andrea Necchi, MD +39 02 2390 2402 andrea.necchi@istitutotumori.mi.it
Sub-Investigator: Patrizia Giannatempo, MD
Principal Investigator: Andrea Necchi, MD Less << |
|
NCT02987699
|
HepatoCellular Carcinoma |
Phase 2 |
Unknown |
May 2017 |
China, Guangdong
... More >>
Cancer Center, Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Ming Shi, MD. shiming@sysu.edu.cn
Principal Investigator: shi Ming, MD. Less << |
|
NCT01828099
|
Non-Small Cell Lung Cancer |
Phase 3 |
Active, not recruiting |
April 11, 2022 |
- |
|
NCT01249352
|
Esophageal Cancer
... More >> Adenocarcinoma Less << |
Phase 2
Phase 3 |
Completed |
- |
Brazil
... More >> Hospital Universitário de Brasília
Brasília, DF, Brazil
Hospital Evangélico do Cachoeiro do Itapemirim
Cachoeiro do Itapemirim, ES, Brazil
Santa Casa de Misericórdia de BH
Belo Horizonte, MG, Brazil
Hospital Erasto Gaetner
Curitiba, PR, Brazil
Hospital Geral de Bonsucesso
Rio de Janeiro, RJ, Brazil
Instituto Nacional do Câncer (INCA)
Rio de Janeiro, RJ, Brazil
Hospital da cidade de Passo Fundo
Passo Fundo, RS, Brazil
Hospital de Clínicas de Porto Alegre
Porto Alegre, RS, Brazil
Hospital Nossa Senhora da Conceição
Porto Alegre, RS, Brazil
Centro de Novos Tratamentos de Itajaí
Itajaí, SC, Brazil
Hospital Municipal São José
Joinville, SC, Brazil
Hospital Amaral Carvalho
Jau, SP, Brazil
Centro Oncológico Mogi das Cruzes
Mogi das Cruzes, SP, Brazil
Faculdade de Medicina do ABC / CEPHO
Santo André, SP, Brazil
Centro de Estudos de Investigações Clíncas (CEIC)
São Caetano do Sul, SP, Brazil
Hospital de Base
São José do Rio Preto, SP, Brazil
Hospital Santa Marcelina
São Paulo, SP, Brazil
Hospital São Paulo (UNIFESP)
São Paulo, SP, Brazil
Instituto do Câncer do Estado de São Paulo
São Paulo, SP, Brazil Less << |
|
NCT03706287
|
Lung Cancer Metastatic |
Phase 1
Phase 2 |
Not yet recruiting |
March 10, 2021 |
China, Sichuan
... More >> Sichuan Cancer Hospital
Not yet recruiting
Chengdu, Sichuan, China, 610041
Contact: juan li, doctor +8613880276636 dr.lijuan@gmail.com
Contact: rui shi, doctor +8613880898008 shirui729@hotmail.com
Principal Investigator: ping yu, doctor
Principal Investigator: juan li, doctor
China
Chengdu fifth people's hospital
Not yet recruiting
Chengdu, China
Contact: mei mei
Principal Investigator: mei mei
People's hospital of deyang city
Deyang, China
The affiliated hospital of southwest medical university
Not yet recruiting
Luzhou, China
Contact: Sheng Lin
Principal Investigator: Sheng Lin
Nanchong central hospital
Not yet recruiting
Nanchong, China
Contact: xin hu
Principal Investigator: xin hu
Suining central hospital
Not yet recruiting
Suining, China
Contact: qiang zhou
Principal Investigator: qiang zhou Less << |
|
NCT01516996
|
Oropharyngeal Cancer
... More >> Hypopharyngeal Cancer Less << |
Phase 2 |
Unknown |
March 2018 |
China, Gansu
... More >> Gansu Province Medical Science Institute
Active, not recruiting
Lanzhou, Gansu, China, 730050
China, Guangxi
Guangxi Tumor Hospital
Active, not recruiting
Nanning, Guangxi, China, 530021
China, Guizhou
GuiZhou Cancer Hospital
Active, not recruiting
Guiyang, Guizhou, China, 550004
China, Neimenggu
Neimenggu Tumor Hospital
Active, not recruiting
Baotou, Neimenggu, China, 014030
China, Ningxia
The Tumor Affiliated Hospital of Ningxia Medical University General Hospita
Active, not recruiting
Yinchuan, Ningxia, China, 750004
China, Qinghai
Qinghai Five Hospital
Active, not recruiting
Xining, Qinghai, China
China, Shanxi
Xijing Hospital
Active, not recruiting
Xi-an, Shanxi, China, 710032
ShanXi Cancer Hospital
Active, not recruiting
Xian, Shanxi, China, 710061
China, Sichuan
The Second People's Hospital of Sichuan
Recruiting
Chengdu, Sichuan, China, 610041
Contact: Peng Xu, M.D. 15828312322 xupeng4618@163.com
West China Hospital, Sichuan University
Active, not recruiting
Chengdu, Sichuan, China, 610041
Daping Hospital and the Research Institute of Surgery of the Third Military Medical University
Active, not recruiting
Chongqing, Sichuan, China, 400042
China, Xinjiang
Xinjiang tumor hospital, The Third Affiliated Hospital of Xinjiang Medical University
Active, not recruiting
Wulumuqi, Xinjiang, China, 830000
China, Yunnan
Yunnan Tumor Hospital, The Third Affiliated Hospital of KUNMING Medical University
Active, not recruiting
Kunming, Yunnan, China, 652100 Less << |
|
NCT01840579
|
Solid Tumor
N... More >>on-small Cell Lung Cancer
Small Cell Lung Cancer Less << |
Phase 1 |
Active, not recruiting |
April 17, 2020 |
- |
|
NCT02178241
|
Metastatic Ureter Carcinoma
... More >> Metastatic Urethral Carcinoma
Stage III Bladder Urothelial Carcinoma AJCC v6 and v7
Stage III Ureter Cancer AJCC v7
Stage III Urethral Cancer AJCC v7
Stage IV Bladder Urothelial Carcinoma AJCC v7
Stage IV Ureter Cancer AJCC v7
Stage IV Urethral Cancer AJCC v7
Ureter Urothelial Carcinoma
Urethral Urothelial Carcinoma Less << |
Phase 2 |
Active, not recruiting |
December 31, 2018 |
United States, California
... More >>
City of Hope Comprehensive Cancer Center
Duarte, California, United States, 91010
Los Angeles County-USC Medical Center
Los Angeles, California, United States, 90033
USC / Norris Comprehensive Cancer Center
Los Angeles, California, United States, 90033
Keck Medical Center of USC Pasadena
Pasadena, California, United States, 91105
University of California Davis Comprehensive Cancer Center
Sacramento, California, United States, 95817
United States, Colorado
University of Colorado Hospital
Aurora, Colorado, United States, 80045
United States, District of Columbia
MedStar Georgetown University Hospital
Washington, District of Columbia, United States, 20007
United States, Florida
Moffitt Cancer Center P2C
Tampa, Florida, United States, 33612
United States, Illinois
University of Chicago Comprehensive Cancer Center
Chicago, Illinois, United States, 60637
UC Comprehensive Cancer Center at Silver Cross
New Lenox, Illinois, United States, 60451
United States, Minnesota
Mayo Clinic Cancer Center P2C
Rochester, Minnesota, United States, 55905
United States, New York
Roswell Park Cancer Institute
Buffalo, New York, United States, 14263
United States, Ohio
Case Western Reserve University
Cleveland, Ohio, United States, 44106
Cleveland Clinic Foundation
Cleveland, Ohio, United States, 44195
Ohio State University Comprehensive Cancer Center
Columbus, Ohio, United States, 43210
United States, Pennsylvania
University of Pittsburgh Cancer Institute (UPCI)
Pittsburgh, Pennsylvania, United States, 15232
United States, Texas
University of Texas M D Anderson Cancer Center P2C
Houston, Texas, United States, 77030
Canada, Ontario
University Health Network Princess Margaret Cancer Center P2C
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT02501278
|
Uterine Cervical Neoplasms |
Phase 2 |
Withdrawn(company (Inovio) is ... More >>no longer able to support the study) Less << |
May 2021 |
Switzerland
... More >> Centre Hospitalier Universitaire Vaudois
Lausanne, Switzerland Less << |
|
NCT01459484
|
Osteosarcoma |
Phase 2
Phase 3 |
Recruiting |
February 2020 |
Italy
... More >> I.R.C.C. - Unit of Medical Oncology
Not yet recruiting
Candiolo, Torino, Italy, 10060
Contact: Giovanni Grignani, MD +39.011.9933 ext 278 giovanni.grignani@ircc.it
Contact: Luisa Gioeni, Pharmacist +39.011.9933 ext 278 luisa.gioeni@ircc.it
Sub-Investigator: Giovanni Grignani, MD
Principal Investigator: Massimo Aglietta, MD
Presidio Sanitario Gradenigo
Not yet recruiting
Torino, TO, Italy, 10153
Contact: Alessandro Comandone, MD +390118151 ext 211 alessandro.comandone@gradenigo.it
Principal Investigator: Alessandro Comandone, MD
Istituto Ortopedico Rizzoli
Recruiting
Bologna, Italy, 40136
Contact: Stefano Ferrari, MD +390516366 ext 411 stefano.ferrari@ior.it
Contact: Emanuela Marchesi, PhD +390516366 ext 400 emanuela.marchesi@ior.it
Principal Investigator: Stefano Ferrari, MD
A.O. Universitaria Meyer
Recruiting
Firenze, Italy, 50139
Contact: Angela Tamburini, MD +3905556 ext 621 a.tamburini@meyer.it
Principal Investigator: Angela Tamburini, MD
Istituto Giannina Gaslini
Not yet recruiting
Genova, Italy
Contact: Carla Manzitti, MD
Principal Investigator: Carla Manzitti, MD
FONDAZIONE IRCCS Istituto Nazionale dei Tumori
Recruiting
Milano, Italy
Contact: Rossella Bertulli, MD
Principal Investigator: Rossella Bertulli, MD
FONDAZIONE IRCCS Istituto Nazionale dei Tumori
Recruiting
Milano, Italy
Contact: Roberto Luksch, MD
Principal Investigator: Roberto Luksch, MD
Istituto Nazionale Tumori "Fondazione G. Pascale"
Not yet recruiting
Napoli, Italy, 80131
Contact: Gaetano Apice, MD
Principal Investigator: Gaetano Apice, MD
Azienda Ospedaliera di Padova
Recruiting
Padova, Italy
Contact: Gianni Bisogno, MD
Principal Investigator: Gianni Bisogno, MD
Istituti Fisioterapici Ospitalieri di Roma
Recruiting
Roma, Italy
Contact: Virginia Ferraresi, MD
Principal Investigator: Virginia Ferraresi, MD
Ospedale Pediatrico Bambin Gesu'
Recruiting
Roma, Italy
Contact: Raffaele Cozza, MD
Principal Investigator: Raffaele Cozza, MD
Ospedale Infantile Regina Margherita - Unit of Paediatric Oncoematology
Recruiting
Torino, Italy, 10126
Contact: Franca Fagioli, MD +39.011.3135 ext 230 franca.fagioli@unito.it
Principal Investigator: Franca Fagioli, MD Less << |
|
NCT00002461
|
Lymphoma |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Albert Einstein Comprehensive Cancer Center
Bronx, New York, United States, 10461 Less << |
|
NCT02083679
|
Carcinoma, Non-Small-Cell Lung |
Phase 1 |
Terminated(Sponsor the return ... More >>rights of the compound to the collaboration partner for further clinical development) Less << |
- |
United States, Massachusetts
... More >>
Please Contact U.S. Medical Information Located in
Rockland, Massachusetts, United States
Germany
Please contact the Merck KGaA Communication Center located in
Darmstadt, Germany Less << |
|
NCT02788461
|
Non-Small Cell Lung Cancer |
Not Applicable |
Recruiting |
April 2025 |
Canada, Ontario
... More >>
London Regional Cancer Program
Recruiting
London, Ontario, Canada, N6A 4L6
Contact: David Palma, MD 519-685-8500 david.palma@lhsc.on.ca
Stronach Regional Cancer Centre at Southlake Regional Health Centre
Recruiting
Newmarket,, Ontario, Canada, L3Y 2P9
Contact: Mojgan Taremi, MD, FRCPC 905-895-4521 ext 6595 mtaremi@southlakeregional.org
Princess Margaret Cancer Centre
Recruiting
Toronto, Ontario, Canada, M5G 2M9
Contact: Alex Sun, MD, FRCPC (416) 946-2000 alex.sun@rmp.uhn.on.ca
Canada, Ont
Kingston General Hospital
Recruiting
Kingston, Ont, Canada, K7L 2V7
Contact: Allison Ashworth, MD, FRCPC 613-544-2631 ext 8106 allison.ashworth@krcc.on.ca
Canada, Quebec
McGill University Health Centre, Glen site Cedars Cancer Center
Recruiting
Montreal,, Quebec, Canada, H4A 3J1
Contact: Sergio L. Faria, MD, PhD (514) 934-1934 ext 36157
Contact: Marianna Perna (514) 934-1934 ext 43191 marianna.perna@muhc.mcgill.ca
Canada
CHU de Quebec - L'Hôtel-Dieu de Québec
Recruiting
Quebec, Canada, G2L 2Z3
Contact: Anne Dagnault, MD PhD 1 418-525-4444, ext 15264 anne.dagnault@mail.chuq.qc.ca
Contact: Josee Allard; (418) 525-4444 ext 16730 Josee.Allard@chudequebec.ca Less << |
|
NCT01031420
|
Muscle Invasive Bladder Cancer... More >>
High Risk Urothelial Carcinoma of the Upper Urinary Tracts Less << |
Phase 2 |
Completed |
- |
United States, Pennsylvania
... More >>
Thomas Jefferson University Hospital
Philadelphia, Pennsylvania, United States, 19107
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111 Less << |
|
NCT02083679
|
- |
|
Terminated(Sponsor the return ... More >>rights of the compound to the collaboration partner for further clinical development) Less << |
- |
- |
|
NCT03425643
|
Non-small Cell Lung Cancer |
Phase 3 |
Recruiting |
June 29, 2026 |
- |
|
NCT01687439
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
China
... More >> Fudan University Shanghai Cancer Center
Shanghai, China Less << |
|
NCT02365805
|
Breast Cancer |
Phase 2 |
Completed |
- |
Spain
... More >> Hospital Universitario Puerta del Mar
Cádiz, Spain, 11009
Hospital Universitario Reina Sofía
Córdoba, Spain, 14004
Hospital Universitario Juan Ramón Jimenez
Huelva, Spain, 21005
Complejo Hospitalario de Jaén
Jaén, Spain, 23007
Hospital Universitario Virgen Macarena
Seville, Spain, 41007
Hospital Universitario Virgen del Rocío
Seville, Spain, 41013
Hospital Universitario Nuestra Señora de Valme
Seville, Spain, 41014 Less << |
|
NCT00201383
|
Oral Cavity
S... More >>quamous Cell Carcinoma Less << |
Phase 3 |
Completed |
- |
Taiwan
... More >> National Taiwan University Hospital
Taipei, Taiwan, 100 Less << |
|
NCT00626639
|
Mucositis
Hea... More >>d and Neck Cancer Less << |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT00532818
|
Metastatic Cervical Cancer |
Phase 3 |
Unknown |
December 2010 |
Mexico
... More >> Instituto Nacional de Cancerologia
Recruiting
Mexico City, Distrito Federal, Mexico, 14080
Principal Investigator: Lucely Cetina, MD Less << |
|
NCT00863512
|
Lung Cancer |
Phase 3 |
Terminated |
- |
- |
|
NCT02867865
|
Gall Bladder Cancers |
Phase 2
Phase 3 |
Recruiting |
August 2022 |
India
... More >> Tata Memorial Centre
Recruiting
Mumbai, Maharashtra, India, 400012
Contact: Reena Engineer, MD +912224177165 reena_engineer@rediffmail.com
Contact: ShyamKishore Shrivastava, MD +912224177163 shrivastavask@tmc.gov.in
Principal Investigator: Reena Engineer, MD Less << |
|
NCT00626639
|
- |
|
Completed |
- |
- |
|
NCT00416507
|
Pancreatic Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00003401
|
Multiple Myeloma and Plasma Ce... More >>ll Neoplasm Less << |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
Marlene & Stewart Greenebaum Cancer Center, University of Maryland
Baltimore, Maryland, United States, 21201 Less << |
|
NCT00003211
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 2 |
Completed |
- |
United States, Tennessee
... More >>
St. Jude Children's Research Hospital
Memphis, Tennessee, United States, 38105
United States, Texas
Texas Children's Cancer Center
Houston, Texas, United States, 77030-2399
Australia, New South Wales
Children's Hospital at Westmead
Westmead, New South Wales, Australia, 2145
Australia, Victoria
Royal Children's Hospital
Parkville, Victoria, Australia, 3052 Less << |
|
NCT00863512
|
- |
|
Terminated |
- |
- |
|
NCT01107639
|
Adenocarcinoma of the Gastroes... More >>ophageal Junction
Esophageal Cancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00504751
|
Non-Hodgkin's Lymphoma |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Weill Cornell Medical College
New York, New York, United States, 10021 Less << |
|
NCT00002481
|
Lymphoma |
Phase 2 |
Unknown |
- |
United States, Wisconsin
... More >>
St. Luke's Medical Center
Milwaukee, Wisconsin, United States, 53215 Less << |
|
NCT00002854
|
Cancer |
Phase 1 |
Completed |
- |
United States, California
... More >>
City of Hope Comprehensive Cancer Center
Duarte, California, United States, 91010-3000 Less << |
|
NCT00003402
|
Leukemia
Lymp... More >>homa Less << |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
Marlene & Stewart Greenebaum Cancer Center, University of Maryland
Baltimore, Maryland, United States, 21201 Less << |
|
NCT03215719
|
Oropharyngeal Carcinoma
... More >> HPV Positive Oropharyngeal Squamous Cell Carcinoma Less << |
Phase 2 |
Recruiting |
July 2021 |
United States, New York
... More >>
New York University School of Medicine
Recruiting
New York, New York, United States, 10016
Contact: Sunita Latchman 646-754-4590 Sunita.Latchman@nyumc.org
Principal Investigator: Kenneth Hu, MD Less << |
|
NCT02899728
|
Extensive Stage Lung Small Cel... More >>l Carcinoma Less << |
Phase 2 |
Suspended(Other - Pending prot... More >>ocol amendment) Less << |
April 7, 2020 |
United States, California
... More >>
Los Angeles County-USC Medical Center
Los Angeles, California, United States, 90033
USC / Norris Comprehensive Cancer Center
Los Angeles, California, United States, 90033
Keck Medical Center of USC Pasadena
Pasadena, California, United States, 91105
United States, Florida
Mayo Clinic in Florida
Jacksonville, Florida, United States, 32224-9980
Moffitt Cancer Center
Tampa, Florida, United States, 33612
United States, Georgia
Emory University Hospital Midtown
Atlanta, Georgia, United States, 30308
Emory University Hospital/Winship Cancer Institute
Atlanta, Georgia, United States, 30322
United States, Kentucky
University of Kentucky/Markey Cancer Center
Lexington, Kentucky, United States, 40536
United States, Massachusetts
Massachusetts General Hospital Cancer Center
Boston, Massachusetts, United States, 02114
Brigham and Women's Hospital
Boston, Massachusetts, United States, 02115
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02215
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02215
United States, Minnesota
Mayo Clinic
Rochester, Minnesota, United States, 55905
United States, Missouri
Siteman Cancer Center at West County Hospital
Creve Coeur, Missouri, United States, 63141
Washington University School of Medicine
Saint Louis, Missouri, United States, 63110
Siteman Cancer Center at Christian Hospital
Saint Louis, Missouri, United States, 63136
Siteman Cancer Center at Saint Peters Hospital
Saint Peters, Missouri, United States, 63376
United States, New York
Laura and Isaac Perlmutter Cancer Center at NYU Langone
New York, New York, United States, 10016
United States, Ohio
Case Western Reserve University
Cleveland, Ohio, United States, 44106
Ohio State University Comprehensive Cancer Center
Columbus, Ohio, United States, 43210
United States, Pennsylvania
Thomas Jefferson University Hospital
Philadelphia, Pennsylvania, United States, 19107
University of Pittsburgh Cancer Institute (UPCI)
Pittsburgh, Pennsylvania, United States, 15232
United States, Virginia
University of Virginia Cancer Center
Charlottesville, Virginia, United States, 22908
Virginia Commonwealth University/Massey Cancer Center
Richmond, Virginia, United States, 23298 Less << |
|
NCT00002642
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, Tennessee
... More >>
Vanderbilt Cancer Center
Nashville, Tennessee, United States, 37232-6838 Less << |
|
NCT02285543
|
Mouth Neoplasms
... More >> Carcinoma, Squamous Cell Less << |
Phase 2 |
Active, not recruiting |
December 2021 |
China, Shanghai
... More >>
Ninth People's Hospital, School of Medicine, Shanghai Jiao Tong University
Shanghai, Shanghai, China, 200011 Less << |
|
NCT00049348
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00003399
|
Multiple Myeloma and Plasma Ce... More >>ll Neoplasm Less << |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
Marlene & Stewart Greenebaum Cancer Center, University of Maryland
Baltimore, Maryland, United States, 21201 Less << |
|
NCT00504751
|
- |
|
Completed |
- |
- |
|
NCT02715531
|
Solid Tumor |
Phase 1 |
Recruiting |
January 28, 2021 |
- |
|
NCT01993810
|
Stage II Non-Small Cell Lung C... More >>ancer AJCC v7
Stage IIA Non-Small Cell Lung Carcinoma AJCC v7
Stage IIB Non-Small Cell Lung Carcinoma AJCC v7
Stage III Non-Small Cell Lung Cancer AJCC v7
Stage IIIA Non-Small Cell Lung Cancer AJCC v7
Stage IIIB Non-Small Cell Lung Cancer AJCC v7 Less << |
Phase 3 |
Recruiting |
December 31, 2025 |
United States, Florida
... More >>
University of Florida Health Science Center - Jacksonville
Recruiting
Jacksonville, Florida, United States, 32209
Contact: Site Public Contact 412-339-5294 Roster@nrgoncology.org
Principal Investigator: Bradford S. Hoppe
United States, Illinois
Northwestern Medicine Cancer Center Warrenville
Suspended
Warrenville, Illinois, United States, 60555
United States, Maryland
Maryland Proton Treatment Center
Recruiting
Baltimore, Maryland, United States, 21201
Contact: Site Public Contact 410-369-5226 info@mdproton.com
Principal Investigator: Charles B. Simone
University of Maryland/Greenebaum Cancer Center
Recruiting
Baltimore, Maryland, United States, 21201
Contact: Site Public Contact 800-888-8823
Principal Investigator: Charles B. Simone
Upper Chesapeake Medical Center
Recruiting
Bel Air, Maryland, United States, 21014
Contact: Site Public Contact 443-643-3010
Principal Investigator: Jack J. Hong
Central Maryland Radiation Oncology in Howard County
Recruiting
Columbia, Maryland, United States, 21044
Contact: Site Public Contact 443-546-1300
Principal Investigator: Charles B. Simone
Tate Cancer Center
Recruiting
Glen Burnie, Maryland, United States, 21061
Contact: Site Public Contact 410-553-8100
Principal Investigator: Charles B. Simone
United States, Massachusetts
Massachusetts General Hospital Cancer Center
Suspended
Boston, Massachusetts, United States, 02114
Mass General/North Shore Cancer Center
Suspended
Danvers, Massachusetts, United States, 01923
United States, Missouri
Siteman Cancer Center at West County Hospital
Recruiting
Creve Coeur, Missouri, United States, 63141
Contact: Site Public Contact 800-600-3606 info@siteman.wustl.edu
Principal Investigator: Jeffrey D. Bradley
Washington University School of Medicine
Recruiting
Saint Louis, Missouri, United States, 63110
Contact: Site Public Contact 800-600-3606 info@siteman.wustl.edu
Principal Investigator: Jeffrey D. Bradley
United States, New Jersey
Memorial Sloan Kettering Basking Ridge
Recruiting
Basking Ridge, New Jersey, United States, 07920
Contact: Site Public Contact 212-639-7592
Principal Investigator: Abraham J. Wu
ProCure Proton Therapy Center-Somerset
Suspended
Somerset, New Jersey, United States, 08873
United States, New York
Memorial Sloan Kettering Commack
Recruiting
Commack, New York, United States, 11725
Contact: Site Public Contact 212-639-7592
Principal Investigator: Abraham J. Wu
Memorial Sloan Kettering Westchester
Recruiting
Harrison, New York, United States, 10604
Contact: Site Public Contact 212-639-7592
Principal Investigator: Abraham J. Wu
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10065
Contact: Site Public Contact 212-639-7592
Principal Investigator: Abraham J. Wu
Memorial Sloan Kettering Rockville Centre
Recruiting
Rockville Centre, New York, United States, 11570
Contact: Site Public Contact 212-639-7592
Principal Investigator: Abraham J. Wu
Memorial Sloan Kettering Sleepy Hollow
Suspended
Sleepy Hollow, New York, United States, 10591
United States, Pennsylvania
University of Pennsylvania/Abramson Cancer Center
Recruiting
Philadelphia, Pennsylvania, United States, 19104
Contact: Site Public Contact 800-474-9892
Principal Investigator: Samuel Swisher-McClure
Thomas Jefferson University Hospital
Active, not recruiting
Philadelphia, Pennsylvania, United States, 19107
United States, Tennessee
Tennessee Cancer Specialists-Dowell Springs
Recruiting
Knoxville, Tennessee, United States, 37909
Contact: Site Public Contact 865-244-3209
Principal Investigator: J. B. Wilkinson
United States, Texas
M D Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact: Site Public Contact 877-312-3961
Principal Investigator: Quynh-Nhu Nguyen
MD Anderson Regional Care Center-Katy
Suspended
Houston, Texas, United States, 77094
MD Anderson Regional Care Center-Bay Area
Suspended
Nassau Bay, Texas, United States, 77058
MD Anderson Regional Care Center-Sugar Land
Suspended
Sugar Land, Texas, United States, 77478
MD Anderson Regional Care Center-The Woodlands
Suspended
The Woodlands, Texas, United States, 77384
United States, Washington
ProCure Proton Therapy Center-Seattle
Recruiting
Seattle, Washington, United States, 98133
Contact: Site Public Contact 412-339-5294 Roster@nrgoncology.org
Principal Investigator: Ramesh Rengan
University of Washington Medical Center
Recruiting
Seattle, Washington, United States, 98195
Contact: Site Public Contact 800-804-8824
Principal Investigator: Ramesh Rengan Less << |
|
NCT02359058
|
Stomach Neoplasms |
Phase 1 |
Completed |
- |
Japan
... More >> For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/GMT-5 hours, EST), or speak with your personal physician.
Aichi, Japan, 464-8681
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/GMT-5 hours, EST), or speak with your personal physician.
Chiba, Japan, 277-8577
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/GMT-5 hours, EST), or speak with your personal physician.
Ehime, Japan, 791-0280
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon-Fri from 9 AM to 5 PM Eastern Time (UTC/GMT-5 hours, EST), or speak with your personal physician
Osaka, Japan, 565-0871 Less << |
|
NCT02359058
|
- |
|
Completed |
- |
- |
|
NCT02886195
|
EGFR Gene Mutation |
Phase 3 |
Enrolling by invitation |
July 2019 |
- |
|
NCT03251313
|
Triple Negative Breast Cancer |
Phase 1 |
Not yet recruiting |
December 2020 |
- |
|
NCT00715611
|
Mesothelioma |
Phase 2 |
Recruiting |
July 2019 |
United States, Florida
... More >>
Moffitt Cancer Center
Recruiting
Tampa, Florida, United States, 33612
Contact: Bradford Perez, MD 888-663-3488
Principal Investigator: Bradford Perez, MD
United States, Massachusetts
Brigham and Women's Hospital
Recruiting
Boston, Massachusetts, United States, 02115
Contact: Raymond Mak, MD 617-632-3591
United States, Minnesota
Mayo Clinic Cancer Center
Recruiting
Rochester, Minnesota, United States, 55905
Contact: Nina Garces, MD 507-538-3270
United States, New Jersey
Memorial Sloan Kettering at Basking Ridge
Recruiting
Basking Ridge, New Jersey, United States, 07920
Contact: Andreas Rimner, MD 212-639-6025
Memorial Sloan Kettering Monmouth
Recruiting
Middletown, New Jersey, United States, 07748
Contact: Andreas Rimner, MD 212-639-6025
Memorial Sloan Kettering Bergen
Recruiting
Montvale, New Jersey, United States, 07645
Contact: Andreas Rimner, MD 212-639-6025
United States, New York
Memorial Sloan Kettering Cancer Center @ Suffolk
Recruiting
Commack, New York, United States, 11725
Contact: Andreas Rimner, MD 212-639-6025
Memorial Sloan Kettering West Harrison
Recruiting
Harrison, New York, United States, 10604
Contact: Andreas Rimner, MD 212-639-6025
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10065
Contact: Andreas Rimner, MD 212-639-6025
Contact: Valerie Rusch, MD 212-639-5873
Principal Investigator: Andreas Rimner, MD
Memorial Sloan Kettering at Mercy Medical Center
Recruiting
Rockville Centre, New York, United States
Contact: Andreas Rimner, MD 212-639-6025
Memorial Sloan Kettering Cancer Center at Phelps Memorial Hospital Center
Recruiting
Sleepy Hollow, New York, United States, 10591
Contact: Andreas Rimner, MD 212-639-6025
United States, Texas
Md Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact: Daniel Gomez, MD
Principal Investigator: Daniel Gomez, MD Less << |
|
NCT00003105
|
Bladder Cancer
... More >> Transitional Cell Cancer of the Renal Pelvis and Ureter Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00201396
|
Nasopharyngeal Carcinoma |
Phase 3 |
Completed |
- |
Taiwan
... More >> Kaohsiung Medical University Hospital
Kaohsiung, Taiwan, 80708
China Medical University Hospital
Taichung, Taiwan, 40447
National Taiwan University Hospital
Taipei, Taiwan, 115
Chang-Gung Memorial Hospital(Lin-Kou),
Taoyuan, Taiwan, 333 Less << |
|
NCT02542111
|
Diffuse Large B-cell Lymphoma |
Phase 2 |
Recruiting |
May 2018 |
China, Shanghai
... More >>
Ruijin Hospital
Recruiting
Shanghai, Shanghai, China
Contact: Pengpeng XU, MD 64370045 ext 610707 Less << |
|
NCT00003240
|
Lung Cancer |
Phase 3 |
Unknown |
- |
United Kingdom
... More >> Middlesex Hospital- Meyerstein Institute
London, England, United Kingdom, WIT 3AA Less << |
|
NCT02959385
|
Stage II Esophageal Cancer
... More >> Stage III Esophageal Cancer Less << |
Phase 2 |
Recruiting |
September 2021 |
China, Tianjin
... More >> Department of Radiation Oncology, Tianjin Medical University Cancer Hospital
Recruiting
Tianjin, Tianjin, China, 300060
Contact: QING SONG PANG, M.D +86-22-23340123-1314 pangqingsong@yahoo.com.cn Less << |
|
NCT00014573
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 2 |
Completed |
- |
United States, Michigan
... More >>
Barbara Ann Karmanos Cancer Institute
Detroit, Michigan, United States, 48201-1379 Less << |
|
NCT02567383
|
Recurrent Head and Neck Cancer |
Phase 2 |
Recruiting |
March 2020 |
Taiwan
... More >> Shin Kong Wu Ho-Su Memorial Hospital
Recruiting
Taipei, Taiwan
Contact: Kwan-Hwa Chi, M.D. 886-2-28332211 ext 2274 M006565@ms.skh.org.tw
Contact: Kai-Lin Yang, M.D. 886-2-28332211 ext 2275 M011360@ms.skh.org.tw Less << |
|
NCT00022178
|
Carcinoma of Unknown Primary |
Phase 3 |
Unknown |
- |
United Kingdom
... More >> Christie Hospital N.H.S. Trust
Manchester, England, United Kingdom, M20 4BX Less << |
|
NCT01218048
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 2 |
Completed |
- |
United States, Pennsylvania
... More >>
UPCI - Hillman Cancer Center
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT01218048
|
- |
|
Completed |
- |
- |
|
NCT00004887
|
Lung Cancer |
Phase 3 |
Unknown |
- |
United Kingdom
... More >> Christie Hospital N.H.S. Trust
Manchester, England, United Kingdom, M20 4BX Less << |
|
NCT00069953
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00069953
|
- |
|
Completed |
- |
- |
|
NCT03473756
|
Urothelial Carcinoma |
Phase 1
Phase 2 |
Recruiting |
July 25, 2022 |
- |
|
NCT02520219
|
Peripheral T-cell Lymphomas |
Phase 2 |
Unknown |
December 2015 |
China, Jiangsu
... More >> Jiangsu province tumor hospital
Recruiting
Nanjing City, Jiangsu, China, 210002
Contact: Qingfeng Yin, Clinical Manager 0086013912903257 y_qingfeng@163.com
Contact: Xiaolei Zhou, Manager 0086013776639377 zhouxiaolei@simcere.com Less << |
|
NCT00016341
|
Endometrial Cancer |
Phase 3 |
Terminated |
- |
- |
|
NCT02641847
|
Triple Negative Breast Cancer
... More >>
Breast Cancer Less << |
Phase 2
Phase 3 |
Recruiting |
- |
China, Shanghai
... More >>
Fudan University Shanghai Cancer Center
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Zhi-Min Shao, MD.PhD. 86-18017312288 zhimingshao@yahoo.com Less << |
|
NCT01793701
|
Locally Advanced Cervical Canc... More >>er Less << |
Phase 1
Phase 2 |
Active, not recruiting |
April 2018 |
United Kingdom
... More >> Barts Health NHS Trust
London, United Kingdom
Hammersmith Hospital
London, United Kingdom
Royal Marsden
London, United Kingdom
Sheffield Teaching Hospitals NHS Foundation Trust
Sheffield, United Kingdom Less << |
|
NCT03331575
|
Unresectable Stage III Non-sma... More >>ll-cell Lung Cancer Less << |
Phase 3 |
Recruiting |
November 2020 |
China, Shanghai
... More >>
Shanghai Chest Hospital
Recruiting
Shanghai, Shanghai, China
Contact: Xiaolong Fu, PhD +862122200000 ext 3602 xlfu1964@126.com
Contact: Qin Zhang, PhD +8618017321606 ext 3602 zhangq0616@163.com Less << |
|
NCT03540017
|
Ovarian Cancer|Fallopian Tube ... More >>Cancer|Primary Peritoneal Carcinoma Less << |
PHASE1 |
RECRUITING |
2025-07-05 |
Long Island Jewish Medical Cen... More >>ter, New Hyde Park, New York, 11040, United States Less << |
|
NCT00128037
|
Pulmonary Neoplasm |
Phase 2 |
Completed |
- |
Japan
... More >> National Cancer Center
Chuo-ku, Tokyo, Japan, 104-0045 Less << |
|
NCT03023436
|
Stomach Neoplasm |
Phase 3 |
Recruiting |
June 2022 |
China, Beijing
... More >> Peking University Cancer Hospital
Recruiting
Beijing, Beijing, China, 100-142
Contact: Xiangqian Su, M.D., Ph.D. +86-138-0126-2916
Principal Investigator: Xiangqian Su, M.D., Ph.D
China, Fujian
Fujian Provincial Hospital
Recruiting
Fuzhou, Fujian, China, 350-014
Contact: Liangxiang Huang, M.D., Ph.D. 0086-138-5902-0211
Principal Investigator: Liangxiang Huang, M.D., Ph.D.
China, Guangdong
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510-060
Contact: Zhiwei Zhou, M.D., Ph.D. +86-139-0222-2859 zhouzhw@sysucc.org.cn
Principal Investigator: Zhiwei Zhou, M.D., Ph.D.
Guangdong General Hospital
Recruiting
Guangzhou, Guangdong, China, 510-080
Contact: Yong Li, M.D., Ph.D. +86-138-2217-7479 yuan821007@126.com
Principal Investigator: Yong Li, M.D., Ph.D.
Cancer Center of Guangzhou Medical University
Recruiting
Guangzhou, Guangdong, China, 510-095
Contact: Shuzhong Cui, M.D., Ph.D. +86-138-0251-3800 cuishuzhong@126.com
Principal Investigator: cuishuzhong@126.com Cui, M.D., Ph.D.
Zhujiang Hospital of Southern Medical University
Recruiting
Guangzhou, Guangdong, China, 510-280
Contact: Jinlong Yu, M.D., Ph.D. +86-131-8909-7861 yujinlong640506@163.com
Principal Investigator: Jinlong Yu, M.D., Ph.D.
Nanfang Hospital, Southern Medical University
Recruiting
Guangzhou, Guangdong, China, 510-515
Contact: Guoxin Li, M.D., Ph.D. +86-138-0277-1450 gzliguoxin@163.com
Contact: Hao Liu, M.D., Ph.D. +86-138-2215-8578 liuhaofbi@163.com
Principal Investigator: Guoxin Li, M.D., Ph.D.
The Third Affiliated Hospital of Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China, 510-630
Contact: Hong-Bo Wei, M.D., Ph.D. 0086-18922102969 drweihb@126.com
Contact: Bo Wei, M.D. 0086-13527794069 sanpi2013@163.com
Principal Investigator: Hong-Bo Wei, M.D., Ph.D.
The Sixth Affiliated Hospital of Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China, 510-655
Contact: Junsheng Peng, M.D., Ph.D. +86-138-0296-3578 pengsir1010@126.com
Principal Investigator: Junsheng Peng, M.D., Ph.D.
Meizhou People's Hospital
Recruiting
Meizhou, Guangdong, China, 514-031
Contact: Zuguang Wu, M.D. +86-135-0252-3063 wuzg1913@163.com
Principal Investigator: Zuguang Wu, M.D.
Zhongshan City People Hospital
Recruiting
Zhongshan, Guangdong, China, 528-403
Contact: Hong Chen, M.D., Ph.D. +86-135-9084-3781 cenhong228@sohu.com
Principal Investigator: Hong Chen, M.D., Ph.D.
China, Shanghai
Zhongshan Hospital, Fudan University
Recruiting
Shanghai, Shanghai, China, 200-032
Contact: Yihong Sun, M.D., Ph.D. +86-137-0173-5406 yihongsun@medmail.com.cn
Principal Investigator: Yihong Sun, M.D., Ph.D.
China, Sichuan
West China Hospital, Sichuan University
Recruiting
Chengdu, Sichuan, China, 610-041
Contact: Jiankun Hu, M.D., Ph.D. +86-189-8060-1504 hujkwch@126.com
Principal Investigator: Jiankun Hu, M.D.,Ph.D. Less << |
|
NCT02741570
|
Head and Neck Cancer |
Phase 3 |
Recruiting |
August 15, 2025 |
- |
|
NCT00360971
|
- |
|
Terminated(Due to positive pre... More >>liminary results from other palifermin studies.) Less << |
- |
- |
|
NCT01633541
|
Laryngeal Cancer |
Phase 2 |
Active, not recruiting |
December 2018 |
United States, Michigan
... More >>
University of Michigan Comprehensive Cancer Center
Ann Arbor, Michigan, United States, 48109 Less << |
|
NCT00808899
|
Neuroblastoma |
Phase 2 |
Terminated(Voluntarily closed ... More >>and terminated by the PI due to lack of feasibility) Less << |
- |
United States, Tennessee
... More >>
St. Jude Children's Research Hospital
Memphis, Tennessee, United States, 38105 Less << |
|
NCT03708042
|
Stage II Esophageal Cancer
... More >> Stage III Esophageal Cancer Less << |
Phase 3 |
Not yet recruiting |
December 1, 2023 |
- |
|
NCT00199758
|
Non Small Cell Lung Carcinoma |
Phase 2 |
Completed |
- |
France
... More >> Service de Pneumologie
Beauvais, France
Service de Pneumologie
Bordeaux, France
Service de Pneumologie
Charleville Mezière, France
Service de Pneumologie
Créteil, France
Service de Pneumologie
Draguignan, France
Service de Pneumologie
Elbeuf, France
Service de Pneumologie
Gap, France
Service de Pathologie Respiratoire
Limoges, France
Service de Pneumologie, Hôpital de la Croix Rousse
Lyon, France
Service de Pneumologie
Mantes La Jolie, France
Département des Maladies Respiratoires
Marseille, France
Service de Pneumologie-Allergologie
Martigues, France
Service de Pneumologie
Meaux, France
Service de Pneumologie
Mulhouse, France
Service de Pneumologie - Hôpital St Antoine
Paris, France
Service de Pneumologie, Hôpital Pontchailloux
Rennes, France
Hôpital Charles Nicolle, Service de Pneumologie
Rouen, France
Service de Pneumologie, Hôpital Bois Guillaume
Rouen, France
Service de Pneumologie, Hôpital Nord
saint Etienne, France
Service de Pathologie Respiratoire
Toulon naval, France
Service de Pneumologie
Villefranche, France Less << |
|
NCT00002522
|
Lymphoma |
Phase 2 |
Completed |
- |
United States, Pennsylvania
... More >>
Fox Chase - Temple Cancer Center
Philadelphia, Pennsylvania, United States, 19111-2442 Less << |
|
NCT02876861
|
High-Grade Upper Tract Urothel... More >>ial Carcinoma Less << |
Phase 2 |
Recruiting |
August 2020 |
China, Hunan
... More >> Xiangya Hospital of Central South Univeristy
Recruiting
Changsha, Hunan, China, 410008
Contact: Long Wang, M.D. Ph.D. 008613873151645 doctorwanglong@163.com Less << |
|
NCT00113399
|
- |
|
Terminated(Terminated early du... More >>e to low accrual.) Less << |
- |
- |
|
NCT00006038
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00113399
|
Head and Neck Cancer |
Phase 3 |
Terminated(Terminated early du... More >>e to low accrual.) Less << |
- |
- |
|
NCT00468169
|
Head and Neck Cancer |
Phase 2 |
Unknown |
November 2012 |
United States, Illinois
... More >>
University of Chicago
Chicago, Illinois, United States, 60637 Less << |
|
NCT03306121
|
Nasopharyngeal Carcinoma |
Phase 3 |
Recruiting |
November 9, 2023 |
China, Guangdong
... More >>
Sun Yat-sen Universitty Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Hai-Qiang Mai, MD,PhD +862087343643 maihq@sysucc.org.cn
Principal Investigator: Hai-Qiang Mai, MD,PhD Less << |
|
NCT02987998
|
Stage IIIA Non-Small Cell Lung... More >> Cancer Less << |
Phase 1 |
Recruiting |
January 2020 |
United States, Ohio
... More >>
University Hospitals Seidman Cancer Center, Case Comprehensive Cancer Center
Withdrawn
Cleveland, Ohio, United States, 44106
Cleveland Clinic Taussig Cancer Institute, Case Comprehensive Cancer Center
Recruiting
Cleveland, Ohio, United States, 44195
Contact: Nathan Pennell, MD, PhD 216-445-9282 penneln@ccf.org Less << |
|
NCT00808899
|
- |
|
Terminated(Voluntarily closed ... More >>and terminated by the PI due to lack of feasibility) Less << |
- |
- |
|
NCT02595554
|
Cervical Carcinoma Stage IIB |
Phase 3 |
Recruiting |
December 2020 |
China, Guangdong
... More >>
Department of Gynecologic Oncology, Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Jihong Liu, Ph.D. 86-20-87343102 Liujih@mail.sysu.edu.cn
Contact: He Huang, Ph.D. 86-20-87343104 huangh@sysucc.org.cn Less << |
|
NCT03249519
|
Cervical Cancer |
Not Applicable |
Recruiting |
December 31, 2099 |
Germany
... More >> Universitätsklinikum Erlangen, Strahlenklinik
Recruiting
Erlangen, Germany, 91054
Contact: Oliver Ott, MD ++49 9131 85 ext 33968 st-studiensekretariat@uk-erlangen.de Less << |
|
NCT02076477
|
Oligo-metastatic Stage IV Non-... More >>small Cell Lung Cancer Less << |
Phase 3 |
Unknown |
- |
China, Sichuan
... More >> Sichuan Cancer Hospital & Institute
Recruiting
Chengdu, Sichuan, China, 610041
Contact: TAO LI, MD, PhD 86-18908178818 litaoxmf@163.com
Principal Investigator: TAO LI, MD, PhD
Sub-Investigator: XIAOHU WANG, MD, PhD
Sub-Investigator: XIAOBO DU, MD, PhD
Sub-Investigator: BING LU, MD, PhD Less << |
|
NCT02082522
|
Hilar Cholangiocarcinoma |
Phase 3 |
Terminated(Accrual rate remain... More >>ing too low) Less << |
- |
- |
|
NCT00138658
|
Non-small Cell Lung Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, California
... More >>
LAC-USC Medical Center
Los Angeles, California, United States, 90033
University of Southern California Norris
Los Angeles, California, United States, 90033
United States, New York
New York Oncology Hematology
Albany, New York, United States, 12208
United States, Oregon
Oregon Health and Science University
Portland, Oregon, United States, 97239
United States, South Carolina
Cancer Centers of the Carolinas
Greenville, South Carolina, United States, 29605
United States, Texas
Mary Crowley Medical Research Center
Dallas, Texas, United States, 75246
Canada, Alberta
Tom Baker Cancer Centre
Calgary, Alberta, Canada
Canada, British Columbia
BC Cancer Agency, Fraser Valley Centre
Surrey, British Columbia, Canada
BC Cancer Agency, Vancouver Center
Vancouver, British Columbia, Canada
Canada, Newfoundland and Labrador
Dr. H. Bliss Murphy Cancer Center
St. Johns, Newfoundland and Labrador, Canada
Canada, Ontario
Royal Victoria Hospital of Barrie
Barrie, Ontario, Canada
London Regional Cancer Centre
London, Ontario, Canada
Ottawa Hospital
Ottawa, Ontario, Canada
Toronto Sunnybrook Regional Cancer Center
Toronto, Ontario, Canada
Canada, Quebec
Hopital Laval
Ste-Foy, Quebec, Canada Less << |
|
NCT01631136
|
Advanced Non Small Cell Lung C... More >>ancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT01534949
|
Diffuse Large B-Cell Lymphoma |
Phase 1 |
Terminated |
- |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of Less << |
|
NCT01802021
|
Carcinoma, Non-Small-Cell Lung |
Phase 2
Phase 3 |
Unknown |
February 2014 |
Taiwan
... More >> Center for traditional chinese medicine, Chang Gung Memorial Hospital
Recruiting
Gueishan Township, Taoyuan County, Taiwan, 33378
Contact: Tse-Hung Huang, M.D. +886-3196200 ext 2613 tsehunghuang_1089@yahoo.com.tw
Principal Investigator: Tse-Hung Huang, M.D. Less << |
|
NCT01134861
|
Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT01536392
|
Cancer of the Cervix |
Phase 3 |
Active, not recruiting |
March 2020 |
United States, Texas
... More >>
Lyndon B. Johnson General Hospital
Houston, Texas, United States, 77026
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00627432
|
Bladder Cancer |
Phase 2 |
Suspended |
- |
France
... More >> Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Montpellier, France, 34298 Less << |
|
NCT03274414
|
Paranasal Sinus Cancer
... More >> Nasal Cavity Tumor
Nasal Cavity Adenocarcinoma Less << |
Phase 2 |
Recruiting |
September 1, 2020 |
United States, New Jersey
... More >>
Memoral Sloan Kettering Basking Ridge
Recruiting
Basking Ridge, New Jersey, United States, 07920
Contact: Marc Cohen, MD 212-639-3769
Memoral Sloan Kettering Monmouth
Recruiting
Middletown, New Jersey, United States, 07748
Contact: Marc Cohen, MD 212-639-3769
Memorial Sloan Kettering Bergen
Recruiting
Montvale, New Jersey, United States, 07645
Contact: Marc Cohen, MD 212-639-3769
United States, New York
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10021
Contact: Marc Cohen, MD 212-639-3769 Less << |
|
NCT03212651
|
Bladder Cancer |
Phase 2 |
Recruiting |
May 2019 |
France
... More >> Gustave Roussy
Recruiting
Villejuif, Val de Marne, France, 94805
Contact: Yohann LORIOT, MD 0142115276 ext +33 yohann.loriot@gustaveroussy.fr
Principal Investigator: Yohann LORIOT, MD Less << |
|
NCT01222572
|
Non-small Cell Lung Cancer
... More >> Lung Cancer
NSCLC Less << |
Phase 1
Phase 2 |
Terminated(Slow accrual due to... More >> restrictive eligibility criteria) Less << |
- |
United States, Massachusetts
... More >>
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00138658
|
- |
|
Completed |
- |
- |
|
NCT01222572
|
- |
|
Terminated(Slow accrual due to... More >> restrictive eligibility criteria) Less << |
- |
- |
|
NCT00921635
|
Cervical Cancer
... More >> Anemia Less << |
Phase 3 |
Suspended |
- |
- |
|
NCT00675597
|
Lung Cancer |
Not Applicable |
Completed |
- |
United States, New Jersey
... More >>
Memorial Sloan-Kettering at Basking Ridge
Basking Ridge, New Jersey, United States, 07920
United States, New York
Memorial Sloan-Kettering Cancer Center @ Suffolk
Commack, New York, United States, 11725
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan-Kettering at Mercy Medical Center
Rockville Centre, New York, United States
Memorial Sloan-Kettering Cancer Center at Phelps Memorial Hospital Center
Sleepy Hollow, New York, United States, 10591 Less << |
|
NCT00675597
|
- |
|
Completed |
- |
- |
|
NCT00003852
|
Childhood Germ Cell Tumor
... More >> Extragonadal Germ Cell Tumor
Ovarian Cancer
Testicular Germ Cell Tumor Less << |
Phase 2 |
Terminated(lack of patient inc... More >>lusion) Less << |
- |
France
... More >> Centre Paul Papin
Angers, France, 49036
CHR de Besancon - Hopital Jean Minjoz
Besancon, France, 25030
Institut Bergonie
Bordeaux, France, 33076
Centre Regional Francois Baclesse
Caen, France, 14076
Centre Jean Perrin
Clermont-Ferrand, France, 63011
Centre de Lute Contre le Cancer,Georges-Francois Leclerc
Dijon, France, 21079
CHR de Grenoble - La Tronche
Grenoble, France, 38043
Clinique Saint Michel
La Rochelle, France, 17000
Centre Leon Berard
Lyon, France, 69373
Institut J. Paoli and I. Calmettes
Marseille, France, 13273
Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Montpellier, France, 34298
Centre Antoine Lacassagne
Nice, France, 06189
Hopital d'Instruction des Armees du Val de Grace
Paris, France
Institut Jean Godinot
Reims, France, 51056
Centre Henri Becquerel
Rouen, France, 76038
Centre Rene Huguenin
Saint Cloud, France, 92211
Hopitaux Universitaire de Strasbourg
Strasbourg, France, 67091
Institut Gustave Roussy
Villejuif, France, F-94805 Less << |
|
NCT00616109
|
Extensive-Stage Small Cell Lun... More >>g Cancer Less << |
Phase 2 |
Completed |
- |
United States, Michigan
... More >>
University of Michigan Comprehensive Cancer Center
Ann Arbor, Michigan, United States, 48109 Less << |
|
NCT02212574
|
Medulloblastoma |
Not Applicable |
Recruiting |
May 2022 |
United States, Arizona
... More >>
Phoenix Childrens Hospital Hematology/Oncology
Recruiting
Phoenix, Arizona, United States, 85016-7710
Contact: Michael Etzl, Dr 602-546-0920 metzl@phoenixchildrens.com
United States, Colorado
Children's Hospital Colorado Center for Cancer & Blood Disorders
Recruiting
Aurora, Colorado, United States, 80045
Contact: Kathleen Dorris, M.D., M.S. 720-777-6772 kathleen.dorris@childrenscolorado.org
United States, Florida
M D Anderson Cancer Center-Orlando Pediatric Hematology/Oncology
Not yet recruiting
Orlando, Florida, United States, 32806
Contact: Amy Smith, Dr 321-841-8588 amy.smith@orlandohealth.com
All Children's Hospital Pediatric Hematology/Oncology
Recruiting
Saint Petersburg, Florida, United States, 33701
Contact: Stacie Stapleton, Dr 727-767-4176 Stacie.Stapleton@allkids.org
United States, Georgia
Children's Healthcare of Atlanta- Egleston Pediatric Neuro-Oncology
Recruiting
Atlanta, Georgia, United States, 30322
Contact: Tobey MacDonald, Dr 404-727-1447 tobey.macdonald@emory.edu
United States, Illinois
Ann and Robert H Lurie Children's Hospital of Chicago Hematology/Oncology
Recruiting
Chicago, Illinois, United States, 60611
Contact: Stewart Goldman, Dr 312-227-4844 sgoldman@northwestern.edu
United States, Maryland
Johns Hopkins University
Recruiting
Baltimore, Maryland, United States, 21287
Contact: Kenneth Cohen, MD, MBA 410-614-5055 kcohen@jhmi.edu
Contact: Tammy Scott, RN 4140-614-5990 scottta@jhmi.edu
United States, Massachusetts
Dana-Farber Cancer Institute
Recruiting
Boston, Massachusetts, United States, 02215
Contact: Pratiti Bandopadhayay, MBBS, PhD
United States, Missouri
Washington University School of Medicine Pediatric Hematology/Oncology
Recruiting
Saint Louis, Missouri, United States, 63110
Contact: Joshua Rubin, Dr 314-454-6018 rubin_j@kids.wustl.edu
United States, New Jersey
Hackensack University Medical Center
Recruiting
Hackensack, New Jersey, United States, 07601
Contact: Derek Hanson, MD 551-996-5437 dhanson@hackensackumc.org
United States, New York
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10065
Contact: Kevin De Braganca, MD 212-639-3449 debragak@mskcc.org
United States, North Carolina
Duke University Medical Center
Recruiting
Durham, North Carolina, United States, 27705
Contact: David Ashley, MD, PhD david.ashley@duke.edu
Principal Investigator: David Ashley, MD, PhD
United States, Ohio
Nationwide Children's Hospital
Recruiting
Columbus, Ohio, United States, 43205
Contact: Mohamed S AbdelBaki, MBBCh 614-722-3250 mohamed.abdelbaki@nationwidechildrens.org
United States, Oregon
Oregon Health and Science University Pediatric Hematology/Oncology
Not yet recruiting
Portland, Oregon, United States, 97239-3098
Contact: Kellie Nazemi, Dr 503-494-0714 nazemik@ohsu.edu
United States, Washington
Seattle Children's Hospital Hematology/Oncology
Recruiting
Seattle, Washington, United States, 98105
Contact: Sarah Leary, Dr 206-987-2106 sarah.leary@seattlechildrens.org
United States, Wisconsin
Childrens Hospital of Wisconsin (Medical College of Wisconsin)
Recruiting
Milwaukee, Wisconsin, United States, 53226
Contact: Jeffrey Knipstein, MD 414-955-4108 jknipstein@jhmi.edu Less << |
|
NCT00616109
|
- |
|
Completed |
- |
- |
|
NCT00429754
|
Nausea
Vomiti... More >>ng Less << |
Phase 4 |
Withdrawn(change of study popu... More >>lation and chemotherapeutic regimen) Less << |
- |
- |
|
NCT02750670
|
Lymphoma, Non-Hodgkin's |
Phase 2 |
Active, not recruiting |
May 2020 |
Canada, Ontario
... More >>
Princess Margaret Cancer Centre
Toronto, Ontario, Canada Less << |
|
NCT03432598
|
Locally Advanced or Metastatic... More >> Lung Cancer Less << |
Phase 2 |
Recruiting |
July 2019 |
China, Beijing
... More >> Chinese Academy of Medical Sciences Tumor Hospital
Recruiting
Beijing, Beijing, China, 100021
Contact: Lijuan Zhao 861087788495 cancergcp@163.com
Principal Investigator: Jie Wang, MD
Beijing Cancer Hospital
Recruiting
Beijing, Beijing, China, 100142
Contact: Jun Huang 861088196023 hj19840804@163.com
Principal Investigator: Jun Zhao, MD
Beijing Chest Hospital
Recruiting
Beijing, Beijing, China, 133500
Contact: Jing Liu 861089509157 liujingworkbj@126.com
Principal Investigator: Zhe Liu, MD
China, Henan
Henan Cancer Hospital
Recruiting
Zhengzhou, Henan, China, 450008
Contact: Hongxia Li 8637165588007 51lihongxia@sina.com
Principal Investigator: Zhiyong Ma, MD
China, Jiangsu
Jaingsu People's Hospital
Recruiting
Nanjing, Jiangsu, China, 210029
Contact: Mingmin Sun 862568136221 jsphkj@163.com
Principal Investigator: Yongqian Shu, MD
China, Jilin
Jilin University First Hospital
Recruiting
Changchun, Jilin, China, 130021
Contact: Fei Wang 8643188786014 wangfei5780@126.com
Principal Investigator: Ying Cheng, MD
Jilin Cancer Hospital
Recruiting
Changchun, Jilin, China, 132000
Contact: Yan Xi 8643185879120 417064629@qq.com
Principal Investigator: Ying Cheng, MD Less << |
|
NCT02397486
|
Head and Neck Neoplasms |
Phase 2 |
Completed |
- |
Egypt
... More >> Ain Shams University Hospitals
Cairo, Egypt Less << |
|
NCT00965172
|
Sarcoma
Oral ... More >>Mucositis Less << |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
UT MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT03243825
|
Cervical Cancer |
Not Applicable |
Completed |
- |
China
... More >> Cancer Hospital, Chinese Acadamy of Medical Sceinces
Beijing, China, 100021 Less << |
|
NCT00502463
|
Head and Neck Cancer |
Not Applicable |
Unknown |
December 2013 |
Austria
... More >> LKH Feldkirch, Dept. of Radiooncology
Feldkirch, Austria, 6807
Medical University of Graz, Dept. of Radiooncology
Graz, Austria, 8036
LKH Leoben Dept. of Hemato-Oncology
Leoben, Austria, 8700
Medical University of Vienna, Dept. of Medicine I
Vienna, Austria, 1090 Less << |
|
NCT02591030
|
Bile Duct Cancer |
Phase 2
Phase 3 |
Recruiting |
September 2023 |
France
... More >> Chu Picardie
Recruiting
Amiens, France
Principal Investigator: Vincent Heutefeuille
Ico Paul Papin
Recruiting
Angers, France
Principal Investigator: Olivier Capitain
CH Victor Dupouy
Recruiting
Argenteuil, France
Principal Investigator: Arnaud BOUTAN-LAROZE
CH de la Côte Basque
Recruiting
Bayonne, France, 64109
Principal Investigator: Franck AUDEMAR
CHRU Besançon
Recruiting
Besançon, France
Principal Investigator: Marine Jarry
Hôpital Avicenne
Not yet recruiting
Bobigny, France
Principal Investigator: Thomas Aparicio
Hôpital Saint-André
Recruiting
Bordeaux, France, 33000
Principal Investigator: Jean-Frédéric Blanc
Polyclinique Nord
Recruiting
Bordeaux, France, 33000
Principal Investigator: Cedric Lecaille
Hôpital Duchenne
Recruiting
Boulogne sur Mer, France
Principal Investigator: Vincent Bourgeois
Centre François Baclesse
Recruiting
Caen, France
Principal Investigator: Aurélie PARZY
Chu D'Estaing
Recruiting
Clermont-Ferrand, France
Principal Investigator: Caroline Petorin
Hôpitaux Civils
Recruiting
Colmar, France
Principal Investigator: Laurianne Plastaras
Ch Sud Francilien
Recruiting
Corbeil-Essonnes, France, 91100
Principal Investigator: Samy LOUAFI
Centre de radiothérapie et oncologie du Parc
Recruiting
Dijon, France, 21000
Principal Investigator: Ariane DARUT-JOUVE
Centre Georges François Leclerc
Recruiting
Dijon, France
Principal Investigator: François GHIRINGHELLI
Hôpital Michallon
Recruiting
Grenoble, France
Principal Investigator: Thomas DECAENS
CHD Vendée
Recruiting
La Roche sur Yon, France
Principal Investigator: Roger Faroux
Clinique du cap d'Or
Recruiting
La Seyne sur Mer, France
Principal Investigator: Mounira EL DEMERY
CH Le Kremlin Bicetre
Recruiting
Le Kremlin-Bicêtre, France
Principal Investigator: Anne THIROT-BIDAULT
CHRU Lille
Recruiting
Lille, France
Principal Investigator: Stéphane CATTAN
Centre Hospitalier de Longjumeau
Recruiting
Longjumeau, France, 91160
Principal Investigator: Samy LOUAFI
Clinique de la Sauvegarde
Not yet recruiting
Lyon, France, 69000
Principal Investigator: Isabelle Moullet
Centre Léon Bérard
Recruiting
Lyon, France
Principal Investigator: Françoise DESSEIGNE
Hôpital de la Croix Rousse
Recruiting
Lyon, France
Principal Investigator: Marielle Guillet
Hôpital Lyon Sud
Recruiting
Lyon, France
Principal Investigator: Marion Chauvenet
Hôpital Saint-Joseph
Recruiting
Marseille, France
Principal Investigator: Hervé Perrier
Centre Catherine de Sienne
Recruiting
Nantes, France
Principal Investigator: Hélène Castanie
CHR Orléans
Recruiting
Orléans, France
Principal Investigator: Jérome Meunier
HEGP
Recruiting
Paris, France
Principal Investigator: Julien TAIEB
Hôpital Cochin
Recruiting
Paris, France
Principal Investigator: Romain Coriat
Hôpital Saint-Jean
Recruiting
Perpignan, France
Principal Investigator: Faiza KHEMISSA-AKOUZ
CHU La Miletrie
Recruiting
Poitiers, France
Principal Investigator: David Tougeron
CHU Reims
Recruiting
Reims, France
Principal Investigator: Alexandra Heurgue
Centre Eugène Marquis
Recruiting
Rennes, France
Principal Investigator: Julien Edeline
Hôpital Drôme Nord
Recruiting
Romans sur Isère, France
Principal Investigator: Marie-Claude Goutttebel
Chu Rouen
Recruiting
Rouen, France
Principal Investigator: Pierre Michel
CHU Saint-Etienne
Recruiting
Saint-Etienne, France, 42055
Principal Investigator: Jean-Marc Phelip, PhD
CH Saint-Jean de Luz
Recruiting
Saint-Jean de Luz, France, 64500
Principal Investigator: Etienne Mauriac
CH Saint-Quentin
Recruiting
Saint-Quentin, France
Principal Investigator: Innocenti DADAMESSI
Ch Robert Morlevat
Recruiting
Semur en Auxois, France
Principal Investigator: Arnaud Patenotte
CAC Paul Strauss
Recruiting
Strasbourg, France, 67065
Principal Investigator: Meher BEN ABDELGHANI
CHU Tours
Recruiting
Tours, France
Principal Investigator: Thierry Lecomte Less << |
|
NCT00439426
|
Nasopharyngeal Cancer |
Phase 2 |
Completed |
- |
Morocco
... More >>
Agadir, Morocco, 80000
Casablanca, Morocco, 20502
Marrakech, Morocco, 40000
Rabat, Morocco, 10000
Rabat, Morocco, 6213 Less << |
|
NCT00312819
|
Non-small Cell Lung Cancer |
Phase 2
Phase 3 |
Completed |
- |
Israel
... More >> Oncology Institute Meir Medical Center
Kfar Saba, Israel Less << |
|
NCT01857232
|
CINV |
Phase 2 |
Completed |
- |
Denmark
... More >> Odense University Hospital
Odense, Denmark Less << |
|
NCT00452985
|
- |
|
Completed |
- |
Bangladesh
... More >> Sanofi-Aventis
Dhaka, Bangladesh Less << |
|
NCT01665417
|
EGFR Positive Non-small Cell L... More >>ung Cancer
Adenocarcinoma Less << |
Phase 4 |
Recruiting |
December 2017 |
China, Beijing
... More >> Chinese People's Liberation Army (PLA) General Hospital
Recruiting
Beijing, Beijing, China, 100853
Contact: Jiao Shunchang, MD 0086-13801380677
Principal Investigator: Jiao Shunchang, MD Less << |
|
NCT00162721
|
Uterine Sarcoma |
Phase 3 |
Unknown |
- |
France
... More >> Institut Gustave Roussy
Recruiting
Villejuif, France, 94800
Contact: Patricia Pautier, Dr 33 1 42 11 4340 pautier@igr.fr Less << |
|
NCT02665039
|
Urothelial Carcinoma
... More >> Bladder Cancer
Renal Pelvis Cancer
Ureter Cancer
Urethra Cancer Less << |
Phase 2 |
Active, not recruiting |
January 2019 |
Denmark
... More >> Department of Oncology, Rigshospitalet
Copenhagen, Denmark, DK-2100
Sweden
Department of Oncology, Karolinska University Hospital
Stockholm, Sweden Less << |
|
NCT00139230
|
Squamous Cell Carcinoma
... More >> Carcinoma of Head/Neck Less << |
Phase 2 |
Completed |
- |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Boston, Massachusetts, United States, 02114
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00198432
|
NSCLC Stage IIIB
... More >> Concomitant Radiochemotherapy Less << |
Phase 2 |
Completed |
- |
France
... More >> CHU Saint-Etienne Pneumologie
Saint-Etienne, France, 42000 Less << |
|
NCT01104714
|
- |
|
Completed |
- |
France
... More >> CH d'Alès
Alès, France, 30103
CHU de Bordeaux - Groupe Hospitalier Saint-André
Bordeaux, France, 33075
CHU de Bordeaux - Groupe Hospitalier Pellegrin
Bordeaux, France, 33076
CHU de Grenoble
Grenoble cedex 09, France, 38043
Centre de Lutte Contre le Cancer - Centre Oscar Lambret
Lille Cedex, France, 59020
CHU de Montpellier - Hôpital Gui de Chauliac
Montpellier cedex 5, France, 34295
Centre Regional de Lutte Contre le Cancer - Val d'Aurelle - Paul Lamarque
Montpellier cedex 5, France, 34298
Centre Antoine Lacassagne
Nice cedex 2, France, 06189
CHU de Nîmes - Hôpital Universitaire Carémeau
Nîmes, France, 30029
CHRU de Toulouse - Hôpital Purpan
Toulouse Cedex 9, France, 31059
Centre de Lutte Contre le Cancer - Institut Claudius Regaud
Toulouse Cedex, France, 31052
Pharmacologie Clinique, expérim. des anticancéreux, CLCC Claudius Regaud
Toulouse, France, 31052
CHRU de Toulouse - Hôpital de Rangueil
Toulouse, France, 31059
CHRU de Toulouse - Hôpital Larrey
Toulouse, France, 31059 Less << |
|
NCT02074241
|
- |
|
Enrolling by invitation |
April 2019 |
China, Chongqing
... More >>
Southwest hospital,Chian
Chongqing, Chongqing, China, 400030 Less << |
|
NCT02240238
|
Solid Tumors |
Phase 1
Phase 2 |
Recruiting |
November 30, 2019 |
United States, California
... More >>
California Cancer Associates for Research and Excellence
Recruiting
Encinitas, California, United States, 92024
UC San Diego Moores Cancer Center
Recruiting
La Jolla, California, United States, 92037
Pacific Hematology Oncology Associates
Recruiting
San Francisco, California, United States, 94115
United States, Illinois
Northwestern University Feinberg School of Medicine
Recruiting
Chicago, Illinois, United States, 60611
United States, Massachusetts
Tufts Medical Center
Recruiting
Boston, Massachusetts, United States, 02111
United States, North Carolina
University of North Carolina at Chapel Hill
Recruiting
Chapel Hill, North Carolina, United States, 27599
United States, Ohio
University Hospitals Case Medical Center
Recruiting
Cleveland, Ohio, United States, 44121
United States, Oklahoma
University of Oklahoma Health Sciences Center
Recruiting
Oklahoma City, Oklahoma, United States, 73104
United States, Texas
University of Texas Southwestern Medical Center
Recruiting
Dallas, Texas, United States, 75390
MD Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Bulgaria
Multiprofile Hospital for Active Treatment Serdika EOOD
Recruiting
Sofia, Sofia-Grad, Bulgaria, 1632
Complex Oncology Center - Shumen EOOD
Recruiting
Shumen, Bulgaria, 9700
Italy
Istituto Scientifico Romagnolo Per Lo Studio E La Cura Dei Tumori IRST
Recruiting
Meldola, Italy, 47014
ASST Grande Ospedale Metropolitano Niguarda - Presidio Ospedaliero Ospedale Niguarda Ca' Granda
Recruiting
Milano, Italy, 20162
Poland
Wojewodzki Szpital Specjalistyczny im. Ludwika Rydygiera w Krakowie
Recruiting
Krakow, Poland, 31826
Med-Polonia Sp. z o.o.
Recruiting
Poznan, Poland, 60693
Romania
Fundeni Clinical Institute
Recruiting
Bucharest, Romania, 22328
Coltea Clinical Hospital
Recruiting
Bucharest, Romania, 30171
Prof Dr I Chiricuta Institute of Oncology
Recruiting
Cluj-Napoca, Romania, 400015
Oncology Center Sfantul Nectarie
Recruiting
Craiova, Romania, 200347
Euroclinic Oncology Center SRL
Recruiting
Iasi, Romania, 700106
Institutul Regional de Oncologie Iasi
Recruiting
Iasi, Romania, 700483 Less << |
|
NCT01118039
|
Bladder Cancer
... More >> Transitional Cell Cancer of the Renal Pelvis and Ureter
Urethral Cancer Less << |
Phase 2 |
Unknown |
- |
Spain
... More >> Hospital Del Mar
Recruiting
Barcelona, Spain, 08003
Contact: Contact Person 34-39-3248-3861 XVillanueva@imas.imim.es
Hospital de la Santa Cruz i Sant Pau
Recruiting
Barcelona, Spain, 08025
Contact: Contact Person 34-93-553-7118 jmaroto@santpau.cat
Hospital Universitario San Carlos
Recruiting
Madrid, Spain, 28040
Contact: Contact Person 34-91-330-3000, Ext. 7935 jgonzalezl.hcsc@salud.madrid.org
Hospital Universitario 12 de Octubre
Recruiting
Madrid, Spain, 28041
Contact: Contact Person 34-91-469-2313 cdanicas@hotmail.com Less << |
|
NCT00883480
|
Non-Small Cell Lung Cancer |
Not Applicable |
Completed |
- |
Spain
... More >> Ico-Hospital Universitarios Germans Trias I Pujol
Badalona, Barcelona, Spain, 08916
Hospital Carlos Haya
Málaga, Spain Less << |
|
NCT03404791
|
Bladder Cancer TNM Staging Pri... More >>mary Tumor (T) T2A
Bladder Cancer TNM Staging Primary Tumor (T) T2B
Bladder Cancer TNM Staging Primary Tumor (T) T3A
Bladder Cancer TNM Staging Primary Tumor (T) T3B Less << |
Phase 1 |
Recruiting |
May 2019 |
United States, Arizona
... More >>
Mayo Clinic
Recruiting
Phoenix, Arizona, United States, 85054
Contact: Clinical Trails Office - All Mayo Clinic Locations 855-776-0015
United States, Georgia
North Georgia Urology
Recruiting
Dalton, Georgia, United States, 30720
Contact: Tennille Emery 423-315-7240 tennille.emery@elligodirect.com
United States, Maryland
Chesapeake Urology
Enrolling by invitation
Hanover, Maryland, United States, 21076
United States, Michigan
Michigan Institute of Urology
Recruiting
Troy, Michigan, United States, 48084
Contact: Danielle Osterhout, MLS(ASCP)CM 248-786-0467 OsterhoutD@michiganurology.com
United States, New York
University of Rochester Medical Center
Recruiting
Rochester, New York, United States, 14642
Contact: Mare Perevich, BSN,CCRC 585-275-0989 mare_perevich@urmc.rochester.edu
United States, Tennessee
Urology Associates of Nashville
Recruiting
Nashville, Tennessee, United States, 37209
Contact: Rick Trotter, CRC 615-250-9268 crtrotter@ua-pc.com
Vanderbilt University Medical Center
Recruiting
Nashville, Tennessee, United States, 37232
Contact: Pam Steele, RN,BSN,CCRC 615-434-2120 pamela.steele@vanderbilt.edu
United States, Texas
North Austin Urology
Recruiting
Austin, Texas, United States, 78750
Contact: Atilano Mendoza 737-931-0927 atilano.mendoza@elligodirect.com
United States, Virginia
Urology of Virginia
Recruiting
Virginia Beach, Virginia, United States, 23462
Contact: Laurie Jackson 757-452-3461 LaurieJackson@urologyofva.net Less << |
|
NCT02167711
|
Cholangio Carcinoma |
Phase 2 |
Recruiting |
October 2018 |
Hong Kong
... More >> Department of Clinical Oncology, Prince of Wales Hospital
Recruiting
Hong Kong, Hong Kong
Contact: Stephen L Chan, FRCP 35052166 l_chan@clo.cuhk.edu.hk
Contact: Jane Koh, RN 35051142 jane@clo.cuhk.edu.hk Less << |
|
NCT02508389
|
Radiation Induced Oral Mucosit... More >>is Less << |
Phase 2 |
Active, not recruiting |
August 2019 |
- |
|
NCT02016209
|
Non Small Cell Lung Cancer |
Phase 2 |
Unknown |
March 2016 |
China, Zhejiang
... More >>
The first affiliated hospital, Zhejiang University
Not yet recruiting
Hangzhou, Zhejiang, China, 310003
Contact: Qiong Zhao, PhD 0571-87236802 doczq.2008@gmail.com
Principal Investigator: Qiong Zhao, PhD Less << |
|
NCT03629925
|
Squamous NSCLC |
Phase 3 |
Not yet recruiting |
August 2020 |
- |
|
NCT00637624
|
Carcinoma, Non-Small-Cell Lung... More >>
Carcinoma, Small Cell Lung
Mesothelioma Less << |
Not Applicable |
Unknown |
February 2012 |
Netherlands
... More >> Rijnstate Hospital
Recruiting
Arnhem, Gelderland, Netherlands, 6800TA
Contact: Idris Bahce, MD +31263788888 ext 3652 I.Bahce@VUmc.nl
Principal Investigator: Idris Bahce, MD Less << |
|
NCT02985255
|
Tongue Neoplasms
... More >> Squamous Cell Carcinoma
Anterior Tongue Squamous Cell Carcinoma Less << |
Phase 2 |
Unknown |
June 2017 |
India
... More >> HealthCare Global Enterprises Ltd
Recruiting
Bangalore, Karnataka, India, 560027
Contact: Vishal US Rao, MS 00919739774949 drvishalrao@yahoo.com
Contact: Sataksi Chatterjee, MS 00918971966903 sataksis2007@gmail.com
Sub-Investigator: Shekhar Patil, DM
Sub-Investigator: Radheshyam Naik, DM
Sub-Investigator: Vijay Agarwal, Phd
Sub-Investigator: Ajai BS Kumar, MS
Sub-Investigator: Kumara Swamy, MD
Sub-Investigator: Vikram Maiya, MD
Sub-Investigator: Ravi Nayar, MS
Sub-Investigator: Sataksi Chaterjee, MS
Sub-Investigator: Raghavendra M Rao, PhD
Sub-Investigator: Amritanshu Ram, PhD Less << |
|
NCT03390946
|
Osteosarcoma
... More >>Feasibility
Treatment Response
Biomarker Less << |
Phase 2 |
Not yet recruiting |
December 31, 2020 |
Korea, Republic of
... More >>
National Cancer Center
Not yet recruiting
Goyang-si, Gyeonggi, Korea, Republic of, 10408
Contact: Byung-Kiu Park, M.D., Ph.D. 82-31-920-1240 bkpark@ncc.re.kr
Contact: Mi Mi Kwon, RN 82-31-920-1240 rnjsalal@ncc.re.kr Less << |
|
NCT03662841
|
Carcinoma, Hepatocellular |
Not Applicable |
Recruiting |
March 2021 |
Hong Kong
... More >> Department of Imaging and Interventional Radiology, Prince of Wales Hospital, The Chinese University of Hong Kong
Recruiting
Hong Kong, Hong Kong
Contact: Carmen Wong (852)3505 3210 carmenwongsp@cuhk.edu.hk
Contact: Pui Man Chong (852)3505 3211 siuman@cuhk.edu.hk Less << |
|
NCT00226473
|
Metastatic Melanoma |
Phase 4 |
Unknown |
- |
Germany
... More >> Skin Cancer Unit, German Cancer Ressearch Center and Dept. of Dermatology, University Hospital of Mannheim
Recruiting
Mannheim, Baden-Württemberg, Germany, D-68167
Contact: Dirk Schadendorf, MD +49-621-3832127 d.schadendorf@dkfz.de
Principal Investigator: Dirk Schadendorf, MD
Dept. of Dermatology, University Hospital Tuebingen
Recruiting
Tuebingen, Baden-Württemberg, Germany, D-72076
Contact: Claus Garbe, MD +49-7071-2980872 studienzentrum-derm-onko@med.uni-tuebingen.de
Principal Investigator: Claus Garbe, MD
Dept. of Dermatology, University of Frankfurt
Recruiting
Frankfurt, Hessen, Germany, D-60590
Contact: Konstanze Spieth, MD +49-69-6301-5311 konstanze.spieth@kgu.de
Principal Investigator: Konstanze Spieth, MD
Dept. of Dermatology, Medical Center Buxtehude
Recruiting
Buxtehude, Niedersachsen, Germany, D-21641
Contact: Peter Mohr, MD +49-4161-7036297 mohrpe@cs.com
Principal Investigator: Peter Mohr, MD
Dept. of Dermatology, Hildesheim
Recruiting
Hildesheim, Niedersachsen, Germany, D-31134
Contact: Michael Tronnier, MD +49-05121-894821
Principal Investigator: Michael Tronnier, MD
Dept. of. Dermatology, University of Saarland, Homburg
Recruiting
Homburg, Saaland, Germany, D-66421
Contact: Wolfgang Tilgen, MD +49-6841-3800 hawtil@med-rz-sb.de
Sub-Investigator: Dorothea Tadler, MD
Dept. of Dermatology, University Otto von Guericke
Recruiting
Magdeburg, Saxony-Anhalt, Germany, D-39120
Contact: Jens Ulrich, MD +49-391-6715428 jens.ulrich@medizin.uni-magdeburg.de
Contact: Vassiliki Bekou, MD +49-391-6715264 vassiliki.bekou@medizin.uni-magdeburg.de
Sub-Investigator: Vassiliki Bekou, MD
Dept. of Dermatology, University of Schleswig-Holstein
Recruiting
Kiel, Schleswig-Holstein, Germany, D-24105
Contact: Axel Hauschild, MD +49-431-596-1613 ahauschild@dermatology.uni.kiel.de
Principal Investigator: Axel Hauschild, MD
Dept. of Dermatology, University of Schleswig-Holstein, Campus Lübeck
Recruiting
Lübeck, Schleswig-Holstein, Germany, D-23538
Contact: Sven Krengel, MD +49-451-5002517 svenkrengel@gm.net
Principal Investigator: Sven Krengel, MD
Dept. of Dermatology, Helios Clinic Erfurt
Recruiting
Erfurt, Thuringia, Germany, 99012
Contact: Ruthild Linse, MD +49-361-7814300
Principal Investigator: Ruthild Linse, MD
Sub-Investigator: Yvonne Kellner, MD
Dept. of Dermatology, University of Jena
Recruiting
Jena, Thuringia, Germany, D-07740
Contact: Martin Kaatz, MD +49-3641-937302 kaatz@derma.uni-jena.de
Principal Investigator: Martin Kaatz, MD
Dept. of Dermatology, Charité Berlin
Recruiting
Berlin, Germany, D-10098
Contact: Uwe Trefzer, MD +49-30-450518063 uwe.trefzer@charite.de
Contact: Maja Hofmann, MD +49-30-450518063 maja.hofmann@charite.de
Principal Investigator: Uwe Trefzer, MD
Dept. of Dermatology, Vivantes Clinics
Recruiting
Berlin, Germany, D-12351
Contact: Peter Kohl, MD +49-30-60043601 p.kohl@vivantes.de
Principal Investigator: Peter Kohl, MD Less << |
|
NCT02258659
|
Human Papilloma Virus Infectio... More >>n
Stage III Squamous Cell Carcinoma of the Oropharynx
Stage IVA Squamous Cell Carcinoma of the Oropharynx
Stage IVB Squamous Cell Carcinoma of the Oropharynx Less << |
Phase 2 |
Active, not recruiting |
December 2021 |
United States, Illinois
... More >>
University of Chicago
Chicago, Illinois, United States, 60637 Less << |
|
NCT03520491
|
Bladder Cancer |
Phase 2 |
Recruiting |
January 2021 |
United States, New Jersey
... More >>
Memoral Sloan Kettering Basking Ridge
Recruiting
Basking Ridge, New Jersey, United States, 07920
Contact: Min Yuen Teo, MB BCh 914-367-7393
United States, New York
Memorial Sloan Kettering Westchester
Recruiting
Harrison, New York, United States, 10604
Contact: Min Yuen Teo, MB BCh 914-367-7393
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10065
Contact: Min Yuen Teo, MB BCh 914-367-7393
Contact: Jonathan Rosenberg, MD 646-888-4741 Less << |
|
NCT00112996
|
Neurotoxicity
... More >> Unspecified Adult Solid Tumor, Protocol Specific
Unspecified Childhood Solid Tumor, Protocol Specific Less << |
Phase 3 |
Completed |
- |
United States, Arkansas
... More >>
Hembree Mercy Cancer Center at St. Edward Mercy Medical Center
Fort Smith, Arkansas, United States, 72913
United States, Indiana
Horizon Oncology Center
Lafayette, Indiana, United States, 47905
United States, Kansas
CCOP - Wichita
Wichita, Kansas, United States, 67214-3882
United States, Louisiana
Cabrini Center for Cancer Care at Christus St. Frances Cabrini Hospital
Alexandria, Louisiana, United States, 71301
United States, Michigan
CCOP - Kalamazoo
Kalamazoo, Michigan, United States, 49007-3731
United States, Minnesota
CCOP - Metro-Minnesota
St. Louis Park, Minnesota, United States, 55416
United States, Missouri
Cancer Research for the Ozarks
Springfield, Missouri, United States, 65804
United States, Oregon
CCOP - Columbia River Oncology Program
Portland, Oregon, United States, 97225
United States, Pennsylvania
CCOP - Main Line Health
Wynnewood, Pennsylvania, United States, 19096
United States, South Carolina
CCOP - Greenville
Greenville, South Carolina, United States, 29615
United States, Texas
University of Texas M.D. Anderson CCOP Research Base
Houston, Texas, United States, 77030-4009
CCOP - Scott and White Hospital
Temple, Texas, United States, 76508
United States, Wisconsin
Marshfield Clinic - Marshfield Center
Marshfield, Wisconsin, United States, 54449 Less << |
|
NCT01191892
|
Bladder Cancer
... More >> Transitional Cell Cancer of the Renal Pelvis and Ureter
Ureter Cancer
Urethral Cancer Less << |
Phase 2 |
Unknown |
December 2014 |
United Kingdom
... More >> Beatson West of Scotland Cancer Centre
Recruiting
Glasgow, Scotland, United Kingdom, G12 0YN
Contact: Contact Person 44-141-301-7095 Rob.Jones@ggc.scot.nhs.uk
Wales Cancer Trials Unit
Recruiting
Cardiff, Wales, United Kingdom, CF11 9LJ
Contact: Contact Person 44-29-2019-6800
Ayr Hospital
Recruiting
Ayr, United Kingdom, KA66DX
Contact: Margaret McKernan 01563 825749 margaret.mckernan@aaaht.scot.nhs.uk
Principal Investigator: Jawaher Ansari
Royal Bournemouth General Hospital
Recruiting
Bournemouth, United Kingdom, BH7 7DW
Contact: Susannah Brock susannah.brock@poole.nhs.uk
Principal Investigator: Susannah Brock
Queens Hospital
Recruiting
Burton upon Trent, United Kingdom, DE13 0RB
Contact: Pugazhenthi Pattu 01332 340131 pugazhenthi.pattu@nhs.net
Principal Investigator: Pugazhenthi Pattu
Velindre Hospital
Recruiting
City and County of Cardiff, United Kingdom, CF142TL
Contact: Dr Lester, Jason 44-2920 196159 jason.lester2@wales.nhs.uk
Principal Investigator: Jason Lester
Western General Hospital
Recruiting
Edinburgh, United Kingdom, EH4 2XU
Contact: Duncan McLaren duncan.mclaren@luht.scot.nhs.uk
Principal Investigator: Duncan McLaren
Calderdale Royal Infirmary
Recruiting
Halifax, United Kingdom, HX30PW
Contact: Lisa Gledhill 01484 342925 lisa.gledhill@cht.nhs.uk
Principal Investigator: Ursula Hofmann
Huddersfield Royal Infirmary
Recruiting
Huddersfield, United Kingdom, HD3 3EA
Contact: Uschi Hofmann 0484 847299 uschi.hofmann@cht.nhs.uk
Principal Investigator: Uschi Hofmann
The Royal Lancaster Infirmary
Recruiting
Lancaster, United Kingdom, LA1 4RP
Contact: Alison Birtle 01524 583219 alison.birtle@lhtr.nhs.uk
Principal Investigator: Alison Birtle
St. James's University Hospital
Recruiting
Leeds, United Kingdom, LS9 7TF
Contact: Satinder Jagdev 0113 2067645 satinder.jagdev@leedsth.nhs.uk
Principal Investigator: Satinder Jagdev
The Royal Free Hospital
Not yet recruiting
London, United Kingdom, NW3 2QG
Principal Investigator: Maria Vilarino-Varela
St Marys Hospital
Not yet recruiting
London, United Kingdom, W21NY
Contact: Gillian Hornzee Gillian.Hornzee@imperial.nhs.uk
Principal Investigator: Simon Stewart
Charing Cross Hospital
Not yet recruiting
London, United Kingdom, W68RF
Contact: Simon Stewart simon.stewart@imperial.nhs.uk
Principal Investigator: Simon Stewart
Christie Hospital
Recruiting
Manchester, United Kingdom, M20 4BX
Contact: Tony Eliott 01619187214 tony.eliott@christie.nhs.uk
Principal Investigator: Tony Eliott
Mount Vernon Hospital
Recruiting
Northwood Middlesex, United Kingdom, HA6 2RN
Contact: Peter Hoskin 01923 844533 peter.hoskin@nhs.net
Principal Investigator: Peter Hoskin
Churchill Hospital
Recruiting
Oxford, United Kingdom, OX37LJ
Contact: Georgina Rogers patpandrewprotheroe@oncology.ox.ac.uk
Principal Investigator: Andrew Protheroe
Weston Park Hospital
Recruiting
Sheffield, United Kingdom, S102SJ
Contact: Linda Evans 0114 226500 linda.evans@sth.nhs.uk
Principal Investigator: Linda Evans
Southampton General Hospital
Recruiting
Southampton, United Kingdom, S016 6YD
Contact: Simon Crabb s.j.crabb@southampton.ac.uk
Principal Investigator: Simon Crabb
Royal Surrey County Hospital
Recruiting
Surrey, United Kingdom, GU27XX
Contact: Katie Wood 01483 571122 katiewood@nhs.net
Principal Investigator: Katie Wood
The Royal Marsden Hospital
Recruiting
Surrey, United Kingdom, KT2 7QB
Contact: Robert Huddart 020 8661 3457 robert.huddart@icr.ac.uk
Principal Investigator: Robert Huddart Less << |
|
NCT03171493
|
Urothelial Carcinoma |
Phase 1 |
Recruiting |
December 2019 |
United States, Minnesota
... More >>
Mayo Clinic
Recruiting
Rochester, Minnesota, United States, 55905
Contact: Tessa Kroeninger 507-538-6107 kroeninger.tessa@mayo.edu
Contact: Tanner Miest, MD, PhD miest.tanner@mayo.edu
Principal Investigator: Bradley Leibovich, MD
Sub-Investigator: Tanner Miest, MD, PhD Less << |
|
NCT01192230
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Unknown |
- |
China, Shanghai
... More >>
Cancer hospital Fudan University
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Jianhua Chang, MD,PhD 13916619284 changjianhua@hotmail.com Less << |
|
NCT01969578
|
Salivary Gland Cancer |
Phase 2 |
Recruiting |
June 2021 |
Austria
... More >> Medical University Vienna - General Hospital AKH
Recruiting
Vienna, Austria, 1090
Principal Investigator: Thorsten Fuereder
Belgium
ZNA Middelheim
Recruiting
Antwerp, Belgium, 2020
Principal Investigator: Dirk Schrijvers
Hopitaux Universitaires Bordet-Erasme - Institut Jules Bordet
Recruiting
Brussels, Belgium, 1000
Principal Investigator: Yassine Lalami
Cliniques Universitaires Saint-Luc
Recruiting
Brussels, Belgium, 1200
Principal Investigator: Jean-Pascal Machiels
Universitair Ziekenhuis Antwerpen
Recruiting
Edegem, Belgium, 2650
Principal Investigator: Pol Specenier
U.Z. Leuven - Campus Gasthuisberg
Recruiting
Leuven, Belgium
Principal Investigator: Paul Clement
France
CHU de Bordeaux - Groupe Hospitalier Saint-André - Hopital Saint-Andre
Recruiting
Bordeaux, France, 33075
Principal Investigator: Laurence Digue
Institut régional du Cancer Montpellier
Recruiting
Montpellier, France
Principal Investigator: Didier Cupissol
Institut de Cancerologie de l'Ouest (ICO) - Centre Rene Gauducheau
Recruiting
Nantes, France, 44805
Principal Investigator: Frederic Rolland
CHU de Nantes - Hotel Dieu
Recruiting
Nantes, France
Principal Investigator: Olivier Malard
Centre Antoine Lacassagne
Recruiting
Nice, France, 06189
Principal Investigator: Joel Guigay
Assistance Publique - Hopitaux de Paris - Hopital Tenon
Recruiting
Paris, France, 75020
Principal Investigator: Bertrand Baujat
Institut Universitaire du Cancer de Toulouse (IUCT) Oncopole - Institut Claudius Regaud
Recruiting
Toulouse, France, 31059
Principal Investigator: Jean Pierre Delord
Institut de Cancérologie de Lorraine
Recruiting
Vandoeuvre-Les-Nancy, France, 54519
Principal Investigator: Marie-Christine Kaminsky-Forrett
Gustave Roussy
Recruiting
Villejuif, France
Principal Investigator: Caroline Even
Germany
Charite - Universitaetsmedizin Berlin - Campus Benjamin Franklin
Recruiting
Berlin, Germany, 12200
Principal Investigator: Sebastian Ochsenreither
Universitaetsklinikum Jena-Radiation Therapy and Radiooncology Clinic
Recruiting
Jena, Germany, 07747
Principal Investigator: Orlando Guntinas-Lichius
Universitaetsklinikum Leipzig-Ambulanzen/Sprechstunden
Recruiting
Leipzig, Germany
Principal Investigator: Andreas Dietz
Greece
Athens University - Attikon University General Hospital
Recruiting
Athens, Greece, 12462
Principal Investigator: Amanda Psyrri
Hungary
National Institute Of Oncology
Recruiting
Budapest, Hungary, 1122
Principal Investigator: Erika Hitre
Italy
Azienda Ospedaliera Papa Giovanni XXIII
Recruiting
Bergamo, Italy, 24127
Principal Investigator: Cecilia Moro
Fondazione IRCCS Istituto Nazionale dei Tumori
Recruiting
Milan, Italy
Principal Investigator: Laura Locati
Azienda Provinciale per i Servizi Sanitari - Ospedale Santa Chiara
Recruiting
Trento, Italy, 38100
Principal Investigator: Alessia Caldara
Netherlands
Spaarne Gasthuis - Vrije Universiteit Medisch Centrum
Recruiting
Amsterdam, Netherlands, 1007MB
Principal Investigator: Jan Buter
University Medical Center Groningen (UMCG)
Recruiting
Groningen, Netherlands, 9713 GZ
Principal Investigator: Sjoukje Oosting-Lenstra
Radboud University Medical Center Nijmegen
Recruiting
Nijmegen, Netherlands
Principal Investigator: Carla van Herpen Less << |
|
NCT03467360
|
Small Cell Lung Cancer |
Phase 1 |
Not yet recruiting |
December 30, 2022 |
- |
|
NCT03159143
|
Metastatic Transitional Cell C... More >>ancer of the Urothelial Tract Less << |
Phase 2 |
Completed |
- |
United States, Pennsylvania
... More >>
University of Pittsburgh Cancer Institute
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT00392327
|
Anaplastic Medulloblastoma
... More >> Medulloblastoma
Untreated Childhood Medulloblastoma Less << |
Phase 3 |
Suspended(Temporarily stopped ... More >>for assessment. Contact ctsucontact@westat.com for specifics) Less << |
- |
- |
|
NCT00828841
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT03159143
|
- |
|
Completed |
- |
- |
|
NCT02580994
|
Small Cell Lung Cancer (SCLC) |
Phase 2 |
Recruiting |
August 2020 |
Egypt
... More >> Ain Shams University Faculty of Medicine - Ain Shams University Hospital
Not yet recruiting
Cairo, Egypt, 11331
Contact: Omar Abdel-Rahman
Principal Investigator: Omar Abdel-Rahman, MD
National Cancer Institute
Not yet recruiting
Cairo, Egypt, 11796
Contact: Rabab Gaafar
Principal Investigator: Rabab Gaafar, MD, PhD
France
Centre Hospitalier d'Avignon - Hopital Duffaut
Recruiting
Avignon, France, 84902
Contact: Nicolas Cloarec cloarec.nicolas@ch-avignon.fr
Principal Investigator: Nicolas Cloarec, MD
CHU de Brest
Recruiting
Brest, France, 29200
Contact: Gilles Robinet gilles.robinet@chu-brest.fr
Principal Investigator: Gilles Robinet, MD
Centre Regional Francois Baclesse
Recruiting
Caen, France, 14076
Contact: Radji Gervais r.gervais@baclesse.unicancer.fr
Principal Investigator: Radji Gervais, MD
Centre Hopitalier Intercommunal De Creteil
Recruiting
Créteil, France, 94010
Contact: Isabelle Monnet, MD Isabelle.Monnet@chicreteil.fr
Principal Investigator: Isabelle Monnet, MD
Centre Leon Berard
Recruiting
Lyon, France, 69008
Contact: Jerome Fayette jerome.fayette@lyon.unicancer.fr
Principal Investigator: Jerome Fayette, MD
Assistance Publique - Hopitaux de Marseille - Hopital Nord
Recruiting
Marseille, France, 13015
Contact: Laurent Greillier laurent.greiller@ap-hm.fr
Principal Investigator: Laurent Greillier, MD, PhD
Centre Hospitalier D'Annecy
Recruiting
Metz-Tessy, France, 74370
Contact: Chantal Decroisette cdecroisette@ch-annecygenevois.fr
Principal Investigator: Chantal Decroisette, MD
Assistance Publique - Hopitaux de Paris - Hopital Avicenne
Recruiting
Paris, France, 93009
Contact: Boris Duchemann boris.duchemann@aphp.fr
Principal Investigator: Boris Duchemann, MD
Centre Paul Strauss
Recruiting
Strasbourg, France, 67065
Contact: Roland Schott rschott@strasbourg.unicancer.fr
Principal Investigator: Roland Schott, MD
Gustave Roussy
Recruiting
Villejuif, France, 94805
Contact: Benjamin Besse Benjamin.besse@gustaveroussy.fr
Principal Investigator: Benjamin Besse, MD, PhD
Italy
Ospedale Cannizzaro
Recruiting
Catania, Italy, 95126
Contact: Giuseppe Banna
Principal Investigator: Giuseppe Banna, MD
Santa Croce e Carle General Hospital
Not yet recruiting
Cuneo, Italy, 12100
Contact: Ida Colantonio colantonio.i@ospedale.cuneo.it
Principal Investigator: Ida Colantonio, MD
Azienda Ospedaliero-Universitaria Careggi
Not yet recruiting
Firenze, Italy, 50134
Contact: Lorenzo Livi
Principal Investigator: Lorenzo Livi, MD, PhD
Istituto Europeo di Oncologia
Recruiting
Milano, Italy, 20141
Contact: Filippo De Marinis filippo.demarinis@ieo.it
Principal Investigator: Filippo De Marinis, MD, PhD
Ospedale San Paolo
Recruiting
Milano, Italy, 20142
Contact: Andrea Luciani andrea.luciani@asst-santipaolocarlo.it
Principal Investigator: Andrea Luciani, MD
Ospedale S. Luigi Gonzaga - Universita Di Torino
Recruiting
Orbassano, Italy, 10043
Contact: Silvia Novello silvia.novello@unito.it
Principal Investigator: Silvia Novello, MD, PhD
Azienda Ospedaliero-Universitaria "Santa Maria della Misericordia" di Udine
Not yet recruiting
Udine, Italy, 33100
Contact: Alessandro Follador alessandro.follador@asuiud.sanita.fvg.it
Principal Investigator: Alessandro Follador, MD
Azienda Ospedaliera Universitaria Integrata Verona - Ospedale Borgo Roma
Not yet recruiting
Verona, Italy, 37134
Contact: Emilio Bria emilio.bria@univr.it
Principal Investigator: Emilio Bria, MD, PhD
United Kingdom
NHS Greater Glasgow and Clyde - Beatson West of Scotland Cancer Centre - Gartnavel General Hospital
Not yet recruiting
Glasgow, United Kingdom, G12 0YN
Contact: Nicola Steele Nicola.Steele@ggc.scot.nhs.uk
Principal Investigator: Nicola Steele, MD
The Christie NHS Foundation Trust
Not yet recruiting
Manchester, United Kingdom, M20 4BX
Contact: Raffaele Califano raffaele.califano@christie.nhs.uk
Principal Investigator: Raffaele Califano, MD
Newcastle Hospitals NHS Trust - Freeman Hospital, Northern Centre For Cancer Care
Not yet recruiting
Newcastle Upon Tyne, United Kingdom, NE7 7DN
Contact: Alastair Greystoke alastair.greystoke@newcastle.ac.uk
Principal Investigator: Alastair Greystoke, MD
Sheffield Teaching Hospitals NHS Foundation Trust - Weston Park Hospital
Not yet recruiting
Sheffield, United Kingdom, S10 2SJ
Contact: Robin Young r.j.young@sheffield.ac.uk
Principal Investigator: Robin Young, MD
University Hospital Southampton NHS Foundation Trust - Southampton General Hospital
Not yet recruiting
Southampton, United Kingdom, SO16 6YD
Contact: Judith Cave Judith.Cave@uhs.nhs.uk
Principal Investigator: Judith Cave, MD
Royal Marsden Hospital - Sutton, Surrey
Not yet recruiting
Sutton, United Kingdom, SM2 5PT
Contact: Jaishree Bhosle Jaishree.Bhosle@rmh.nhs.uk
Principal Investigator: Jaishree Bhosle, MD Less << |
|
NCT03082586
|
Esophageal Cancer |
Not Applicable |
Completed |
- |
China, Shanghai
... More >>
the Ethic Committee of Shanghai General Hospital
Shanghai, Shanghai, China, 210000 Less << |
|
NCT00454636
|
Gastric Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00828841
|
- |
|
Completed |
- |
- |
|
NCT02579616
|
Biliary Tract Cancer |
Phase 2 |
Active, not recruiting |
February 1, 2019 |
Japan
... More >>
Nagoya, Aichi, Japan
Kashiwa, Chiba, Japan
Yokohama, Kanagawa, Japan
Ina-machi, Saitama, Japan
Chuo-ku, Tokyo, Japan
Koto-ku, Tokyo, Japan
Mitaka, Tokyo, Japan Less << |
|
NCT00454636
|
- |
|
Completed |
- |
- |
|
NCT00405405
|
Head and Neck Cancer
... More >> Parotid Gland Cancer
Thyroid Gland Cancer
Melanoma
Stage IV Head and Neck Cancer Less << |
Phase 1 |
Completed |
- |
United States, Pennsylvania
... More >>
Thomas Jefferson University
Philadelphia, Pennsylvania, United States, 19107 Less << |
|
NCT02467426
|
- |
|
Completed |
- |
- |
|
NCT02812641
|
Stage III Esophageal Squamous ... More >>Cell Carcinoma Less << |
Phase 2 |
Recruiting |
December 2021 |
Taiwan
... More >> National Taiwan University Hospital
Recruiting
Taipei, Taiwan, 100
Contact: Jason Chia-Hsien Cheng +886-2-23123456 ext 66696 jasoncheng@ntu.edu.tw Less << |
|
NCT00042835
|
Adenocarcinoma of the Lung
... More >> Bronchoalveolar Cell Lung Cancer
Large Cell Lung Cancer
Squamous Cell Lung Cancer
Stage IIIA Non-small Cell Lung Cancer
Stage IIIB Non-small Cell Lung Cancer Less << |
Phase 1 |
Terminated(Administratively co... More >>mplete.) Less << |
- |
United States, Illinois
... More >>
University of Chicago Comprehensive Cancer Center
Chicago, Illinois, United States, 60637-1470 Less << |
|
NCT00201344
|
Nasopharyngeal Carcinoma |
Phase 3 |
Terminated |
- |
Taiwan
... More >> Veterans General Hospital-Taipei
Taipei, Taiwan, 112 Less << |
|
NCT01579630
|
NSCLC |
Phase 2
Phase 3 |
Unknown |
June 2014 |
Taiwan
... More >> Veterans General Hospital -Taipei
Taipei, Taiwan, 112 Less << |
|
NCT02572843
|
NSCLC Non-small Cell Lung Canc... More >>er Less << |
Phase 2 |
Recruiting |
December 2021 |
Germany
... More >> Uniklinik Köln
Recruiting
Köln, Germany, 50937
Contact: Jürgen Wolf, Prof +49 221 478-89050 juergen.wolf@uk-koeln.de
Principal Investigator: Jürgen Wolf, Prof
Universitätsklinikum Gießen und Marburg
Recruiting
Marburg, Germany, 35043
Contact: Jorge Riera Knorrenschild, MD +49 (06421) 58-62784 rierakno@med.uni-marburg.de
Principal Investigator: Jorge Riera Knorrenschild, MD
Switzerland
Stadtspital Triemli
Recruiting
Zürich, Zurich, Switzerland, 8063
Contact: Donat Dürr, MD +41 44 416 34 80 donat.duerr@triemli.zuerich.ch
Principal Investigator: Donat Dürr, MD
Kantonsspital Aarau
Recruiting
Aarau, Switzerland, CH-5001
Contact: Wolf-Dieter Janthur, MD +41 62 838 55 80 wolf-dieter.janthur@ksa.ch
Principal Investigator: Wolf-Dieter Janthur, MD
Kantonsspital Baden
Recruiting
Baden, Switzerland, 5404
Contact: Christine Waibel, MD +41 (0)56 486 27 62 christine.waibel@ksb.ch
Principal Investigator: Christine Waibel, MD
St. Claraspital Basel
Recruiting
Basel, Switzerland, 4016
Contact: Martin Buess, MD +41 (0)61 685 83 68 martin.buess@claraspital.ch
Principal Investigator: Martin Buess, MD
Universitaetsspital Basel
Recruiting
Basel, Switzerland, 4031
Contact: Sacha Rothschild, MD +41 61 265 25 25 sacha.rothschild@usb.ch
Principal Investigator: Sacha Rothschild, MD
IOSI Ospedale Regionale di Bellinzona e Valli
Recruiting
Bellinzona, Switzerland, 6500
Contact: Patrizia Froesch, MD +41 (0)91 811 48 40 patrizia.froesch@eoc.ch
Principal Investigator: Patrizia Froesch, MD
Inselspital Bern
Recruiting
Bern, Switzerland, CH-3010
Contact: Adrian Ochsenbein, Prof +41 31 632 81 69 adrian.ochsenbein@sec.insel.ch
Principal Investigator: Adrian Ochsenbein, Prof
Hôpital du Valais
Not yet recruiting
Brig, Switzerland, 3900
Contact: Reinhard Zenhäusern, MD +41 (0)27 604 36 60 reinhard.zenhaeusern@hopitalvs.ch
Principal Investigator: Reinhard Zenhäusern, MD
Kantonsspital Bruderholz
Not yet recruiting
Bruderholz, Switzerland, CH-4101
Contact: Lorenz M. Jost, MD 41-61-436-3636 lorenz.jost@ksbh.ch
Principal Investigator: Lorenz M. Jost, MD
Kantonsspital Graubuenden
Recruiting
Chur, Switzerland, CH-7000
Contact: Michael Mark, MD +41 81 256 61 11 michael.mark@ksgr.ch
Principal Investigator: Michael Mark, MD
Spital Thurgau AG
Terminated
Frauenfeld, Switzerland, CH-8500
Hopital Fribourgeois HFR
Recruiting
Fribourg, Switzerland, 1708
Contact: Daniel Betticher, Prof +41(0)26 426 72 40 daniel.betticher@h-fr.ch
Principal Investigator: Daniel Betticher, Prof
Hopital Cantonal Universitaire de Geneve
Recruiting
Geneva, Switzerland, CH-1211
Contact: Nicolas Mach +41 (0)22 372 98 81 nicolas.mach@hcuge.ch
Principal Investigator: Nicolas Mach, MD
CCAC Lausanne
Recruiting
Lausanne, Switzerland, 1004
Contact: Pierre Bohanes, MD +41 (21) 646 79 94 pbohanes@bluewin.ch
Principal Investigator: Pierre Bohanes, MD
Centre hospitalier universitaire vaudois CHUV
Recruiting
Lausanne, Switzerland, CH-1011
Contact: Solange Peters, Prof +41 79 556 01 92 solange.peters@chuv.ch
Principal Investigator: Solange Peters, Prof
Luzerner Kantonsspital
Recruiting
Luzern, Switzerland, CH-6000
Contact: Oliver Gautschi, PD MD +41 41 205 58 60 oliver.gautschi@luks.ch
Principal Investigator: Oliver Gautschi, MD
Kantonsspital St. Gallen
Recruiting
St. Gallen, Switzerland, 9007
Contact: Martin Früh, MD +41 (0)71 494 63 25 martin.frueh@kssg.ch
Principal Investigator: Martin Früh, MD
Regionalspital
Recruiting
Thun, Switzerland, 3600
Contact: Daniel Rauch, MD +41 33 226 26 45 daniel.rauch@spitalstsag.ch
Principal Investigator: Daniel Rauch, MD
Kantonsspital Winterthur
Recruiting
Winterthur, Switzerland, CH-8401
Contact: Miklos Pless, Prof 41-52-266-2552 miklos.pless@ksw.ch
Principal Investigator: Miklos Pless, Prof
Klinik Hirslanden Onkozentrum Zürich
Recruiting
Zurich, Switzerland, CH-8032
Contact: Thomas von Briel, MD +41 (0)44 387 21 11 vonbriel@onkozentrum.ch
Principal Investigator: Thomas von Briel, MD
UniversitaetsSpital Zuerich
Recruiting
Zurich, Switzerland, CH-8091
Contact: Christian Britschgi, MD +41 (44) 255 22 14 christian.britschgi@usz.ch
Principal Investigator: Christian Britschgi, MD Less << |
|
NCT00785421
|
Pancreatic Cancer |
Phase 2
Phase 3 |
Completed |
- |
Germany
... More >> Universitätsmedizin - Berlin - Charite
Berlin, Germany, 13353 Less << |
|
NCT03572829
|
Vomiting
Naus... More >>ea Post Chemotherapy
Head Neck Cancer Less << |
Phase 2 |
Recruiting |
December 20, 2019 |
China
... More >> National Cancer Center/ Cancer Hospital, Chinese Academy of Medcal science and Peking Union Medical College
Recruiting
Beijing, China
Contact: Ye Zhang, M.D. 8610-87787625 drzye1983@163.com Less << |
|
NCT00911092
|
Esophageal Cancer |
Phase 4 |
Completed |
- |
France
... More >> Centre Paul Papin
Angers, France, 49933
Centre Hospitalier Universitaire
Brest, France, 29200
Centre François BACLESSE
Caen, France, 14046
Centre Oscar Lambret
Lille, France, 59020
Centre Eugène Marquis
Rennes, France, 35042
CHU - Hopital Charles Nicolle
Rouen, France, 76031 Less << |
|
NCT01693718
|
Head and Neck |
Phase 2 |
Completed |
- |
United States, Illinois
... More >>
The University of Chicago
Chicago, Illinois, United States, 60653 Less << |
|
NCT03568097
|
Small Cell Lung Carcinoma |
Phase 2 |
Recruiting |
August 2021 |
Greece
... More >> Agii Anargiri Cancer Hospital
Not yet recruiting
Néa Kifisiá, Athens, Greece, 14564
Contact: Gerasimos Aravantinos, MD 0030 210 3501283 garavantinos@yahoo.gr
Agii Anargiri Cancer Hospital
Recruiting
Néa Kifisiá, Athens, Greece, 14564
Contact: Eleni Res, MD 0030 210 3501273 nellieres@yahoo.gr
Metropolitan Hospital
Recruiting
Néo Fáliro, Athens, Greece, 18547
Contact: Helena Linardou, MD 0030 210 4809852 elinardou@otenet.gr Less << |
|
NCT01612351
|
Head and Neck Cancer
... More >> Squamous Cell Carcinoma of the Head and Neck Less << |
Phase 2 |
Active, not recruiting |
November 2031 |
United States, North Carolina
... More >>
The University of North Carolina at Chapel Hill
Chapel Hill, North Carolina, United States, 27599 Less << |
|
NCT00398307
|
Renal Impairment |
Phase 1 |
Completed |
- |
United States, Arizona
... More >>
Premiere Oncology of Arizona
Scottsdale, Arizona, United States, 85260
United States, California
LAC/USC Medical Center
Los Angeles, California, United States, 90033
Premiere Oncology
Santa Monica, California, United States, 90404
United States, Connecticut
Yale Cancer Center
New Haven, Connecticut, United States, 06520
United States, Kentucky
University of Kentucky/Division of Hematology/Oncology and Blood Marrow Transplantation
Lexington, Kentucky, United States, 40536
United States, Maryland
University of Maryland/Greenebaum Cancer Center
Baltimore, Maryland, United States, 21201
United States, Missouri
Washington University School of Medicine
St Louis, Missouri, United States, 63110
United States, New Mexico
Cancer Research & Treatment Center/University of New Mexico
Albuquerque, New Mexico, United States, 87131
United States, Texas
The Institute for Drug Development
San Antonio, Texas, United States, 78245 Less << |
|
NCT03664115
|
Lung Cancer |
Phase 2 |
Recruiting |
December 2, 2020 |
Egypt
... More >> oncology department Ain shams university
Recruiting
Cairo, Egypt, 11591
Contact: Amr sh Tawfik, MD
Contact: Asmaa WH Mohamed, Master 01003538597 drasmaa_wahid@hotmail.com Less << |
|
NCT00398424
|
Impaired Hepatic Function |
Phase 1 |
Completed |
- |
United States, Arizona
... More >>
Premiere Oncology of Arizona
Scottsdale, Arizona, United States, 85260
United States, Connecticut
Yale Cancer Center
New Haven, Connecticut, United States, 06520
United States, Kentucky
University of Kentucky/Division of Hematology/Oncology and Blood Marrow Transplantation
Lexington, Kentucky, United States, 40536
United States, Maryland
University of Maryland/Greenebaum Cancer Center
Baltimore, Maryland, United States, 21201
United States, Missouri
Washington University School of Medicine
St Louis, Missouri, United States, 63110
United States, Texas
The Institute for Drug Development
San Antonio, Texas, United States, 78245 Less << |
|
NCT02861872
|
- |
|
Unknown |
December 2017 |
Netherlands
... More >> Radboudumc
Recruiting
Nijmegen, Netherlands, 6500HB
Contact: Mark Rietveld, M.D. MSc. +31243610353 mark.rietveld@radboudumc.nl
Contact: Peter van Essen, MSc. +31243610353 peter.vanessen@radboudumc.nl
Principal Investigator: Nelleke Ottevanger, M.D. PhD. Less << |
|
NCT03639090
|
- |
|
Recruiting |
May 1, 2019 |
United States, Oklahoma
... More >>
University of Oklahoma HSC, Department of Urology
Recruiting
Oklahoma City, Oklahoma, United States, 73104
Contact: Jonathan Heinlen, M.D. 405-271-6900 jonathan-heinlen@ouhsc.edu
Contact: Felicia Kiplinger, B.A. 405-271-6900 felicia-kiplinger@ouhsc.edu Less << |
|
NCT03367871
|
Cervical Cancer |
Phase 2 |
Recruiting |
October 2025 |
United States, Florida
... More >>
University of Miami
Recruiting
Miami, Florida, United States, 33136
Contact: Marilyn Huang, MD 305-243-2233 m.huang@med.miami.edu Less << |
|
NCT00697905
|
Nasopharyngeal Carcinoma |
Phase 2 |
Completed |
- |
Malaysia
... More >> Ipoh Specialist Centre
Ipoh, Malaysia
Johor Specialist Centre
Johor Bahru, Malaysia
Sabah Medical Centre
Kota Kinabalu, Malaysia
Tung Shin Hospital
Kuala Lumpur, Malaysia
University Malaya Medical Centre
Kuala Lumpur, Malaysia
Hospital Universiti Sains Malaysia
Kubang Kerian, Malaysia
Normah Medical Specialist Centre
Kuching, Malaysia
Likas Hospital
Likas, Malaysia
NCI Cancer Hospital
Nilai, Malaysia
Loh Guan Lye Specialist Centre
Penang, Malaysia
Penang General Hospital
Penang, Malaysia Less << |
|
NCT01612351
|
- |
|
Active, not recruiting |
- |
- |
|
NCT02337530
|
Non-Small Cell Lung Cancer |
Phase 2 |
Active, not recruiting |
December 31, 2018 |
Canada, Alberta
... More >>
Tom Baker Cancer Centre
Calgary, Alberta, Canada, T2N 4N2
Cross Cancer Institute
Edmonton, Alberta, Canada, T6G 1Z2
Canada, British Columbia
BCCA - Abbotsford Centre
Abbotsford, British Columbia, Canada, V2S 0C2
BCCA - Vancouver Cancer Centre
Vancouver, British Columbia, Canada, V5Z 4E6
Canada, New Brunswick
Regional Health Authority B, Zone 2
Saint John, New Brunswick, Canada, E2L 4L2
Canada, Ontario
Juravinski Cancer Centre at Hamilton Health Sciences
Hamilton, Ontario, Canada, L8V 5C2
Cancer Centre of Southeastern Ontario at Kingston
Kingston, Ontario, Canada, K7L 5P9
Lakeridge Health Oshawa
Oshawa, Ontario, Canada, L1G 2B9
Ottawa Hospital Research Institute
Ottawa, Ontario, Canada, K1H 8L6
Mount Sinai Hospital
Toronto, Ontario, Canada, M5G 1X5
University Health Network
Toronto, Ontario, Canada, M5G 2M9
Canada, Quebec
CHUM - Hopital Notre-Dame
Montreal, Quebec, Canada, H2L 4M1 Less << |
|
NCT01427478
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 3 |
Active, not recruiting |
September 2021 |
France
... More >> Centre Paul Papin
Angers, France, 49933
Institut Sainte-Catherine
Avignon, France, 84000
CHU Bordeaux - Hôpital Saint-André
Bordeaux, France, 33075
Polyclinique de Bordeaux Nord
Bordeaux, France, 33300
CHRU Brest - Hôpital Morvan
Brest, France, 29609
Centre François Baclesse
Caen, France, 14076
CHIC Créteil
Créteil, France, 94010
Centre Guillaume le Conquérant
Le Havre, France, 76600
Centre Hospitalier Bretagne Sud
Lorient, France, 56100
Centre Léon Bérard
Lyon, France, 69373
AP-HM La Timone Adultes
Marseille, France, 13386 Cedex
Centre Antoine Lacassagne
Nice, France, 06189
CHU Poitiers
Poitiers, France, 86021
Centre Eugène Marquis
Rennes, France, 44229
Centre Henri Becquerel
Rouen, France, 76038
Institut de Cancérologie de la Loire
Saint Priest en Jarez, France, 42270
Institut de Cancérologie de l'Ouest
Saint-Herblain, France, 44805
Pôle Hospitalier Mutualiste- Centre Etienne Dolet
Saint-Nazaire, France, 44600
Strasbourg Oncologie Libérale
Strasbourg, France, 67000
Hopitaux du Léman
Thonon-les-bains, France, 74203
Clinique Pasteur Bâtiment l'Atrium
Toulouse, France, 31076
Institut Claudius Regaud
Toulouse, France, 71000
CHU TOURS (Hôpital Bretonneau)
Tours, France, 37044
Centre de Radiothérapie Marie Curie
Valence, France, 26000
Institut de Cancérologie de lorraine (ICL)
Vandoeuvre-les-Nancy, France, 54519 cedex
Institut Gustave Roussy
Villejuif, France, 94805 Less << |
|
NCT00448240
|
Head and Neck Cancer
... More >> Carcinoma, Squamous Cell Less << |
Phase 2
Phase 3 |
Terminated(Low Accrual, Fundin... More >>g) Less << |
January 2015 |
- |
|
NCT02504229
|
Gastric Cancer |
Phase 2 |
Unknown |
- |
China, Beijing
... More >> Chinese Academy of Medical Sciences Tumor Hospital
Recruiting
Beijing, Beijing, China
Contact: jinwan Wang lyang69@sina.com
Contact: lin Yang lyang69@sina.com
Principal Investigator: jinwan Wang
Principal Investigator: lin Yang Less << |
|
NCT00169572
|
Nausea and Vomiting
... More >> Chemotherapy-Induced Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01096368
|
Cellular Ependymoma
... More >> Childhood Anaplastic Ependymoma
Childhood Infratentorial Ependymoma
Childhood Supratentorial Ependymoma
Clear Cell Ependymoma
Ependymoma
Newly Diagnosed Childhood Ependymoma
Papillary Ependymoma Less << |
Phase 3 |
Recruiting |
- |
- |
|
NCT01059552
|
Locally Advanced Non-small Cel... More >>l Lung Cancer Less << |
Phase 1 |
Active, not recruiting |
December 1, 2018 |
United States, Florida
... More >>
Moffitt Cancer Center
Tampa, Florida, United States, 33612
United States, Pennsylvania
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111
United States, South Carolina
Medical University of South Carolina
Charleston, South Carolina, United States, 29425 Less << |
|
NCT02937519
|
Esophageal Neoplasms |
Phase 2 |
Recruiting |
June 2018 |
China, Guizhou
... More >> Cancer Hospital of Guizhou Medical University
Recruiting
Guiyang, Guizhou, China, 550000
Contact: Feng Jin, Bachelor 0851-86512802 jinf8865@yahoo.com.cn
Principal Investigator: Feng Jin, Bachelor
Sub-Investigator: Ling Guo, Doctor
Sub-Investigator: Ling Wu, Bachelor
Sub-Investigator: Weili Wu, master
Sub-Investigator: Jinhua Long, master
Sub-Investigator: Yuanyuan Li, master
Sub-Investigator: Xiuyun Gong, Bachelor
Sub-Investigator: Xiaoxiao Chen, Bachelor
Sub-Investigator: Zhuoling Li, master
Sub-Investigator: Hang Jiang, Bachelor Less << |
|
NCT02195453
|
Non-Small Cell Lung Cancer |
Phase 4 |
Unknown |
December 2016 |
- |
|
NCT00775385
|
Carcinoma, Non-Small-Cell Lung |
Phase 2
Phase 3 |
Completed |
- |
France
... More >> Centre Hospitalier
Aix en Provence, France
Clinique de L'Europe
Amiens, France
Angers - CHU
Angers, France, 49000
Centre Hospitalier
Béziers, France
Centre F. Baclesse
Caen, France, 14000
CHU - Pneumologie
Caen, France, 14000
Centre Hospitalier
Chauny, France
Chevilly Larue - CH
Chevilly Larue, France
Hôpital Percy-Armées - Pneumologie
Clamart, France, 92140
Clermont Ferrand - CHU
Clermont Ferrand, France, 63000
Colmar - CH
Colmar, France, 68000
Dax - CH
Dax, France, 40107
Ermont - Clinique Claude Bernard
Ermont, France
CHU Grenoble - pneumologie
Grenoble, France, 38000
Le Mans - CHG
Le Mans, France
Centre Léon Bérard
Lyon, France, 69000
Lyon - Hôpital Louis Pradel (Pneumologie)
Lyon, France
Mantes La Jolie - CH
Mantes La Jolie, France, 78200
APHM - Hôpital Sainte Marguerite
Marseille, France, 13000
Institut Paoli Calmette
Marseille, France
Metz - Belle Isle
Metz, France
Mont de Marsan - CH
Mont de Marsan, France, 40000
Montpellier - Clinique Clémentville
Montpellier, France, 34070
Mulhouse - CH
Mulhouse, France, 68000
Nancy - Polyclinique Gentilly
Nancy, France
Nevers - CH
Nevers, France, 58033
Paris - Saint Louis
Paris, France, 75000
APHP - Hopital Tenon - Pneumologie
Paris, France, 75020
Pau - CH
Pau, France, 64046
HCL - Lyon Sud (Pneumologie)
Pierre Bénite, France, 69495
Reims - CHU
Reims, France, 51092
Rennes - CHU
Rennes, France
Centre Hospitalier
Saint-Quentin, France
Nouvel Hopital Civil
Strasbourg, France, 63000
Hôpital Foch
Suresnes, France
Centre Hospitalier Intercommunal
Toulon, France
CHU Toulouse - Pneumologie
Toulouse, France
Clinique Pasteur
Toulouse, France
Tours - CHU
Tours, France, 37000
Nancy - CHU
Vandoeuvre lès Nancy, France, 54500
Institut Gustave Roussy
Villejuif, France, 94805
Centre Hospitalier Intercommunal
Villeneuve-Saint-Georges, France Less << |
|
NCT00568607
|
Lymphoma |
Phase 2 |
Unknown |
December 2010 |
China, Shanghai
... More >>
Fudan University Cancer Hospital
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Biyun Wang, MD 8613701748410 wangbiyun@msn.com
Sub-Investigator: Biyun Wang, M.D. Less << |
|
NCT03598517
|
Non-small Cell Lung Cancer |
Not Applicable |
Recruiting |
July 1, 2020 |
China, Shanghai
... More >>
Shanghai Genernal Hospital
Recruiting
Shanghai, Shanghai, China, 210000
Contact: Ningning Cheng 37798364 ext 8119 ningcnn@163.com
Contact: yong Liu 37798364 ext 8110 drliuyrt@163.com Less << |
|
NCT03652142
|
- |
|
Recruiting |
May 1, 2020 |
Greece
... More >> Attikon Hospital
Recruiting
Athens, Chaidari, Greece
Contact: AMANDA PSYRRI, MD +2105831664 psyrri237@yahool.com Less << |
|
NCT03406650
|
Urothelial Cancer |
Phase 2 |
Recruiting |
April 2026 |
Switzerland
... More >> Kantonspital Aarau
Recruiting
Aarau, Switzerland, CH-5001
Contact: Tobias Wehrhahn, MD +41 62 838 59 27 tobias.wehrhahn@ksa.ch
Principal Investigator: Tobias Wehrhahn, MD
Kantonsspital Baden
Recruiting
Baden, Switzerland, CH-5404
Contact: Andreas Erdmann, MD +41 56 486 27 62 andreas.erdmann@ksb.ch
Principal Investigator: Andreas Erdmann, MD
Universitaetsspital Basel
Recruiting
Basel, Switzerland, 4031
Contact: Sacha Rothschild, MD +41 61 265 25 25 sacha.rothschild@usb.ch
Principal Investigator: Sacha Rothschild, MD
Inselspital
Recruiting
Bern, Switzerland, 3010
Contact: Roland Seiler, MD +41 31 632 20 45 roland.seiler@insel.ch
Principal Investigator: Roland Seiler, MD
Kantonsspital Graubünden
Recruiting
Chur, Switzerland, 7000
Contact: Richard Cathomas, MD +41 81 256 61 11 Richard.Cathomas@ksgr.ch
Principal Investigator: Richard Cathomas, MD
Spital Thurgau AG
Not yet recruiting
Frauenfeld, Switzerland, CH-8500
Contact: Regina Woelkly, MD +41 52 723 76 91 regina.woelkly@stgag.ch
Principal Investigator: Regina Woelkly, MD
Centre Hospitalier Universitaire Vaudois CHUV
Recruiting
Lausanne, Switzerland, CH-1011
Contact: Dominik Berthold, MD +41 21 314 80 83 Dominik.Berthold@chuv.ch
Principal Investigator: Dominik Berthold, MD
Luzerner Kantonsspital
Recruiting
Luzern, Switzerland, 6000
Contact: Christian Riklin, MD +41 41 205 58 73 christian.riklin@luks.ch
Principal Investigator: Christian Riklin, MD
Kantonsspital St. Gallen
Recruiting
St. Gallen, Switzerland, CH-9007
Contact: Sabine Schmid, MD +41 71 494 11 11 sabine.schmid@kssg.ch
Principal Investigator: Sabine Schmid, MD
Kantonsspital Winterthur
Not yet recruiting
Winterthur, Switzerland, 8401
Contact: Miklos Pless, Prof +41 52 266 36 40 miklos.pless@ksw.ch
Principal Investigator: Miklos Pless, Prof
City Hospital Triemli
Not yet recruiting
Zurich, Switzerland, 8063
Contact: Mathias Schmid, MD +41 44 466 11 11 mathias.schmid@triemli.zuerich.ch
Principal Investigator: Mathias Schmid, MD
UniversitaetsSpital Zuerich
Not yet recruiting
Zurich, Switzerland, 8091
Contact: Axel Mischo, MD +41 44 255 22 14 axel.mischo@usz.ch
Principal Investigator: Axel Mischo, MD
Klinik Hirslanden - Onkozentrum Hirslanden
Recruiting
Zürich, Switzerland, 8032
Contact: Anita Hirschi, MD +41 44 387 37 80 ahirschi@onkozentrum.ch
Principal Investigator: Anita Hirschi, MD
OnkoZentrum Zürich
Recruiting
Zürich, Switzerland, 8038
Contact: Ulf Petrausch, MD +41 43 344 33 33 ulf.petrausch@ozh.ch
Principal Investigator: Ulf Petrausch, MD Less << |
|
NCT00611962
|
Neoplasms, Germ Cell and Embry... More >>onal Less << |
Phase 2 |
Completed |
- |
France
... More >> Sanofi-Aventis
Lyon, France Less << |
|
NCT00478699
|
Non-small-cell Lung Cancer |
Phase 3 |
Completed |
- |
Spain
... More >> Hospital de Elche
Elche, Alicante, Spain, 03202
Hospital Central de Asturias
Oviedo, Asturias, Spain
Ico - H. Germans Trias I Pujol
Badalona, Barcelona, Spain, 08916
Hospital D'Althaia
Manresa, Barcelona, Spain, 08243
Hospital de Mataró
Mataró, Barcelona, Spain, 08034
Hospital Insular de Gran Canaria
Las Palmas de Gran Canaria, Gran Canaria, Spain
Hospital Clin. Univ. Santiago de Compostela
Santiago de Compostela, La Coruna, Spain, 15076
Hospital San Pedro
Logrono, La Rioja, Spain, 26004
F.H.Alcorcon
Alcorcon, Madrid, Spain, 28922
Hospital Severo Ochoa
Leganés, Madrid, Spain, 28911
Hospital de La Ribera
Alzira, Valencia, Spain, 46600
Hospital de Cruces
Baracaldo, Vizcaya, Spain, 48903
Hospital de Basurto
Bilbao, Vizcaya, Spain
H.G.U. Alicante
Alicante, Spain, 03010
Hospital de La Santa Creu I Sant Pau
Barcelona, Spain, 08025
Hospital Clínic de Barcelona
Barcelona, Spain, 08036
Hospital Univ. Sagrat Cor
Barcelona, Spain, 08036
Instituto Universitario Dexeus
Barcelona, Spain, 28036
Hospital Provincial de Castellón
Castellón, Spain
Hospital Reina Sofía
Córdoba, Spain, 14004
Ico-Girona (Hospital Josep Trueta)
Girona, Spain, 17007
Hospital Virgen de Las Nieves
Granada, Spain, 18015
Hospital de Jaén
Jaén, Spain, 23007
Hospital de León
Leon, Spain
Hospital Arnau de Vilanova
Lleida, Spain, 46015
Hospital 12 de Octubre
Madrid, Spain, 28006
Hospital Clínico San Carlos
Madrid, Spain, 28006
Hospital de La Princesa
Madrid, Spain, 28006
Md Anderson Internacional
Madrid, Spain, 28033
Fundación Jiménez Díaz
Madrid, Spain, 28040
Hospital La Paz
Madrid, Spain, 28046
Hospital Puerta de Hierro
Madrid, Spain, 28222
Hospital Carlos Haya
Malaga, Spain, 29010
Hospital Morales Messeguer
Murcia, Spain, 30008
Clínica Rotger
Palma de Mallorca, Spain, 07012
Hospital Son Dureta
Palma de Mallorca, Spain, 07014
Hospital Son Llátzer
Palma de Mallorca, Spain, 07198
Hospital Clinico de Salamanca
Salamanca, Spain, 37007
Hospital de Donostia
San Sebastián, Spain, 20014
Instituto Oncológico de San Sebastián
San Sebastián, Spain
Hospital Universitario de Canarias
Santa Cruz de Tenerife, Spain
H. Arnau de Vilanova
Valencia, Spain, 46015
Hospital General de Valencia
Valencia, Spain
Instituto Valenciano de Oncología
Valencia, Spain
Hospital Provincial de Zamora
Zamora, Spain, 49012
Hospital Lozano Blesa
Zaragoza, Spain
Hospital Miguel Servet
Zaragoza, Spain Less << |
|
NCT01828736
|
Recurrent Bladder Cancer
... More >> Stage IV Bladder Cancer
Transitional Cell Carcinoma of the Bladder Less << |
Phase 2 |
Completed |
- |
Belgium
... More >> Cliniques saint Luc - Université Catholique de Louvain
Bruxelles, Belgium, 1200
France
CHU de Besançon
Besançon, France, 25000
CHU Hôpital Saint André
Bordeaux, France, 33000
Hôpital Jean Perrin
Clermont Ferrand, France, 63000
Centre Hospitalier Départemental de la Vendée
La Roche-sur-yon, France, 85000
Clinique Victor Hugo
Le Mans, France, 72000
CHU Hôpital La Timone
Marseille, France, 13005
Institut Paoli Calmettes
Marseille, France, 13009
Clinique Hartmann
Neuilly-Sur-Seine, France, 92200
Curie Institute
Paris, France, 75005
Hôpital Saint Louis
Paris, France, 75010
Groupe Hospitalier Saint Joseph Paris
Paris, France, 75014
Hôpital Cochin
Paris, France, 75014
Hôpital Européen Georges Pompidou
Paris, France, 75015
Hôpital Foch
Suresnes, France, 92151 Less << |
|
NCT01936974
|
Ovarian Carcinoma
... More >> Fallopian Tube Carcinoma
Peritoneal Carcinoma Less << |
Phase 2 |
Terminated(PI Decision) |
- |
United States, Arizona
... More >>
Western Regional Medical Center
Goodyear, Arizona, United States, 85338 Less << |
|
NCT00933673
|
Lymphoma |
Phase 2 |
Unknown |
June 2012 |
China, Shanghai
... More >>
Fudan University Cancer Hospital
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Ye Guo, MD 862164175590 pattrick_guo@msn.com
Principal Investigator: Ye Guo, MD Less << |
|
NCT02875314
|
Medulloblastoma
... More >> Central Nervous System Embryonal Tumors Less << |
Phase 4 |
Recruiting |
September 2020 |
- |
|
NCT03621696
|
HPV Related Oropharyngeal Squa... More >>mous Cell Carcinoma Less << |
Phase 2 |
Recruiting |
July 31, 2024 |
United States, Missouri
... More >>
Washington University School of Medicine
Recruiting
Saint Louis, Missouri, United States, 63110
Contact: Douglas Adkins, M.D. 314-747-8475 dadkins@wustl.edu
Principal Investigator: Douglas Adkins, M.D.
Sub-Investigator: Wade Thorstad, M.D.
Sub-Investigator: Jose Zevallos, M.D.
Sub-Investigator: Mackenzie Daly, M.D.
Sub-Investigator: Ryan Jackson, M.D.
Sub-Investigator: Jason Rich, M.D.
Sub-Investigator: Peter Oppelt, M.D.
Sub-Investigator: Randall Paniello, M.D.
Sub-Investigator: Patrick Pipkorn, M.D.
Sub-Investigator: Hiram Gay, M.D.
Sub-Investigator: Esther Lu, Ph.D.
Sub-Investigator: Rebecca Chernock, M.D.
Sub-Investigator: Samir El-Mofty, DMD, M.S., Ph.D. Less << |
|
NCT00101582
|
Mucositis
Sol... More >>id Tumors
Stomatitis
Head and Neck Cancer
Squamous Cell Carcinoma Less << |
Phase 3 |
Completed |
- |
- |
|
NCT01566240
|
Cervical Cancer |
Phase 3 |
Recruiting |
September 2021 |
Mexico
... More >> Instituto Nacional de Cancerologia (INCAN)
Recruiting
Mexico City, Mexico
Principal Investigator: Dolores Dr Gallardo
United Kingdom
North Devon District Hospital
Recruiting
Barnstaple, Devon, United Kingdom, EX31 4JB
Principal Investigator: Jennifer Forrest
University College London Hospital
Recruiting
London, Greater London, United Kingdom, NW1 2BU
Principal Investigator: Mary McCormack
Weston Park Hospital
Recruiting
Sheffield, South Yorkshire, United Kingdom, S10 2SJ
Principal Investigator: Simon Pledge
Belfast City Hospital
Recruiting
Belfast, United Kingdom
Principal Investigator: Ursula Dr McGiven
Pilgrim Hospital
Recruiting
Boston, United Kingdom
Principal Investigator: Miguel Dr Panades
Royal Sussex County Hospital
Recruiting
Brighton, United Kingdom
Principal Investigator: Kate Dr Lankester
Velindre Cancer Centre
Recruiting
Cardiff, United Kingdom
Principal Investigator: Louise Dr Hanna
Cheltenham General Hospital
Recruiting
Cheltenham, United Kingdom
Principal Investigator: Audrey Dr Cook
Royal Derby Hospital
Recruiting
Derby, United Kingdom
Principal Investigator: Mojca Dr Persic
Royal Devon and Exeter NHS Foundation Trust
Recruiting
Exeter, United Kingdom, EX2 5DY
Principal Investigator: Jennifer Forrest
Beatson WOSCC
Recruiting
Glasgow, United Kingdom
Principal Investigator: Nick Dr Reed
Gloucester Royal Hospital
Recruiting
Gloucester, United Kingdom
Principal Investigator: Audrey Dr Cook
Grantham and District Hospital
Recruiting
Grantham, United Kingdom
Principal Investigator: Miguel Dr Panades
Castle Hill Hospital
Recruiting
Hull, United Kingdom
Principal Investigator: Faheem Dr Bashir
Leicester Royal Infirmary
Recruiting
Leicester, United Kingdom
Principal Investigator: David Dr Peel
Lincoln County Hospital
Recruiting
Lincoln, United Kingdom
Principal Investigator: Miguel Dr Panades
Guy's and St Thomas' NHS Foundation Trust
Recruiting
London, United Kingdom
Principal Investigator: Vinod Dr Mullassery
Imperial College Healthcare NHS Trust
Recruiting
London, United Kingdom
Contact: Sophie Kathirgamanathan
Principal Investigator: Gemma EMinowicz
St Bart's Hospital
Recruiting
London, United Kingdom
Principal Investigator: Melanie Dr Powell
The Christie NHS Foundation Trust
Recruiting
Manchester, United Kingdom, M20 4BX
Principal Investigator: Susan Davidson
James Cook University Hospital
Recruiting
Middlesbrough, United Kingdom
Principal Investigator: Madhavi Dr Adusumalli
Norfolk and Norwich University Hospital
Recruiting
Norfolk, United Kingdom
Principal Investigator: Robert Dr Wade
Northampton General Hospital
Recruiting
Northampton, United Kingdom
Principal Investigator: Anupama Dr Gore
Nottingham University Hospitals NHS Trust
Recruiting
Nottingham, United Kingdom, NG5 1PB
Principal Investigator: Anjana Anand
Derriford Hospital
Recruiting
Plymouth, United Kingdom
Contact: Irene Harvey
Principal Investigator: Sidharth Dubey
Southampton General Hospital
Recruiting
Southampton, United Kingdom
Principal Investigator: Victoria Dr Lankaster
Royal Stoke University Hospital
Recruiting
Stoke-On-Trent, United Kingdom
Principal Investigator: Rajanee Dr Bhana
The Clatterbridge Cancer Centre
Recruiting
Wirral, United Kingdom
Principal Investigator: Karen Dr Whitmarsh
New Cross Hospital
Recruiting
Wolverhampton, United Kingdom
Principal Investigator: Rozenne Dr Allerton Less << |
|
NCT02582697
|
Germ Cell Tumor |
Phase 3 |
Recruiting |
July 2023 |
Australia, New South Wales
... More >>
Calvary Mater Newcastle
Recruiting
Newcastle, New South Wales, Australia, 2298
Contact: Louise Plowman Louise.Plowman@calvarymater.org.au
Contact: Girish Mallesara girish.mallesara@calvarymater.org.au
Principal Investigator: Girish Mallesara
Royal North Shore Hospital
Recruiting
St Leonards, New South Wales, Australia, 2065
Contact: Susan Kirby-Lewis Susan.KirbyLewis@health.nsw.gov.au
Contact: Alexander Guminski aguminski@nsccahs.health.nsw.gov.au
Principal Investigator: Alexander Guminski
Prince of Wales Hospital
Recruiting
Sydney, New South Wales, Australia, 2031
Contact: Daisy Buchanan Daisy.buchanan@sesiahs.health.nsw.gov.au
Contact: Julie Howard Julie.howard@sesiahs.health.nsw.gov.au
Principal Investigator: Elizabeth Hovey
Chris O'Brien Lifehouse
Recruiting
Sydney, New South Wales, Australia, 2050
Contact: Melissa Quaggiott melissa.mcmahon@lh.org.au
Contact: Peter Grimison peter.grimison@lh.org.au
Principal Investigator: Peter Grimison
Macquarie Cancer Clinical Trials
Recruiting
Sydney, New South Wales, Australia, 2109
Contact: Louise Francisco louise.francisco@mq.edu.au
Contact: Radhika Butala radhika.butala@mq.edu.au
Principal Investigator: Howard Gurney
Concord Repatriation General Hospital
Recruiting
Sydney, New South Wales, Australia, 2139
Contact: Kathy Hall Kathy.Hall@sswahs.nsw.gov.au
Contact: Martin Stockler martin.stockler@sydney.edu.au
Principal Investigator: Martin Stockler
Westmead Hospital
Recruiting
Sydney, New South Wales, Australia, 2145
Contact: Vicky Wegener vicky.wegener@sydney.edu.au
Contact: Howard Gurney howard_gurney@wmi.usyd.edu.au
Principal Investigator: Howard Gurney
Nepean Hospital
Recruiting
Sydney, New South Wales, Australia, 2751
Contact: Jeremy Jones jeremy.jones@swahs.health.nsw.gov.au
Contact: Amanda Stevanovic amanda.stevanovic@swahs.health.nsw.gov.au
Principal Investigator: Amanda Stevanovic
Tweed Hospital
Recruiting
Tweed Heads, New South Wales, Australia, 2485
Contact: Charmayne Chorlton Charmayne.Chorlton@ncahs.health.nsw.gov.au
Contact: Ehtesham Abdi eaabdi@mac.com
Principal Investigator: Ehtesham Abdi
SAN Clinical Trials Unit
Recruiting
Wahroonga, New South Wales, Australia, 2076
Contact: James McQuilan James.McQuillan@sah.org.au
Contact: Gavin Marx gmarx@nhog.com.au
Principal Investigator: Gavin Marx
Australia, Queensland
Royal Brisbane & Women's Hospital
Recruiting
Brisbane, Queensland, Australia, 4029
Contact: Natasha Roberts natasha.roberts@health.qld.gov.au
Contact: David Wyld david.wyld@health.qld.gov.au
Principal Investigator: David Wyld
Princess Alexandra
Recruiting
Woolloongabba, Queensland, Australia, 4102
Contact: Paul Baxter Paul.Baxter@health.qld.gov.au
Contact: Euan Walpole Euan.Walpole@health.qld.gov.au
Principal Investigator: Euan Walpole
Australia, South Australia
Royal Adelaide Hospital
Recruiting
Adelaide, South Australia, Australia, 5000
Contact: Hazel Bourke hazel.bourke@health.sa.gov.au
Contact: Thean Hsiang Tan hsiang.tan@health.sa.gov.au
Principal Investigator: Thean Tan
Flinders Medical Centre
Recruiting
Bedford Park, South Australia, Australia, 5042
Contact: Alex Scott-Hoy Alex.Scott-Hoy@health.sa.gov.au
Contact: Ganessan Kichenadasse Ganessan.Kichenadasse@health.sa.gov.au
Principal Investigator: Ganessan Kichenadasse
Australia, Tasmania
Royal Hobart Hospital
Recruiting
Hobart, Tasmania, Australia, 7000
Contact: Lesley Oliver lesley.oliver@dhhs.tas.gov.au
Contact: David Boadle david.boadle@dhhs.tas.gov.au
Principal Investigator: David Boadle
Australia, Victoria
Box Hill Hospital
Recruiting
Box Hill, Victoria, Australia, 3128
Contact: Lauren Mitchell lauren.mitchell@monash.edu
Contact: Philip Parente Phillip.parente@monash.edu
Principal Investigator: Philip Parente
Peter MacCallum Cancer Centre
Recruiting
East Melbourne, Victoria, Australia, 3002
Contact: Jennifer Petersen jennifer.petersen@petermac.org
Contact: Guy Toner guy.toner@petermac.org
Principal Investigator: Guy Toner
Austin Health
Recruiting
Heidelberg, Victoria, Australia, 3084
Contact: Jaren Caine Jaren.Caine@austin.org.au
Contact: Andrew Weickhardt Andrew.Weickhardt@ludwig.edu.au
Principal Investigator: Andrew Weickhardt
Sunshine Hospital
Recruiting
St Albans, Victoria, Australia, 3021
Contact: Jessica Tanner Jessica.Tanner@wh.org.au
Contact: Shirley Wong shirleys.wong@mh.org.au
Principal Investigator: Shirley Wong
Border Medical Oncology
Recruiting
Wodonga, Victoria, Australia, 3690
Contact: Lauren Callow lcallow@bordermedonc.com.au
Contact: Craig Underhill cunderhill@bordermedonc.com.au
Principal Investigator: Craig Underhill
Australia, Western Australia
Fiona Stanley Hospital
Recruiting
Murdoch, Western Australia, Australia, 6847
Contact: Jaye Harding jaye.harding@health.wa.gov.au
Contact: Simon Troon simon.troon@health.wa.gov.au
Principal Investigator: Simon Troon
New Zealand
Auckland Hospital
Recruiting
Grafton, Auckland, New Zealand, 1142
Contact: Andrew Conley andrewcon@adhb.govt.nz
Contact: Fritha Hanning FrithaH@adhb.govt.nz
Principal Investigator: Fritha Hanning
Palmerston North Hospital
Recruiting
Roslyn, Palmerston North, New Zealand, 4442
Contact: Sarah Holwell Sarah.Holwell@midcentraldhb.govt.nz
Contact: Gary Forgeson Garry.Forgeson@midcentraldhb.govt.nz
Principal Investigator: Gary Forgeson
Christchurch Hospital
Recruiting
Christchurch, New Zealand, 8011
Contact: Elizabeth Thompson liz.thompson@cdhb.health.nz
Contact: Mark Jeffrey Mark.jeffery@cdhb.health.nz
Principal Investigator: Mark Jeffrey
Dunedin Hospital
Recruiting
Dunedin, New Zealand, 9054
Contact: Rachel McLay-Barnes ONCRESEARCH@southerndhb.govt.nz
Contact: David Perez david.perez@southerndhb.govt.nz
Principal Investigator: David Perez Less << |
|
NCT01936974
|
- |
|
Terminated(PI Decision) |
- |
- |
|
NCT00267046
|
- |
|
Completed |
- |
- |
|
NCT00262951
|
Pancreatic Cancer |
Phase 2 |
Terminated(Closed at a planned... More >> interim analysis by meeting a predefined toxicity endpoint) Less << |
- |
United States, Minnesota
... More >>
Masonic Cancer Center at University of Minnesota
Minneapolis, Minnesota, United States, 55455 Less << |
|
NCT00267046
|
Sarcoma
Oral ... More >>Mucositis Less << |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
U.T.M.D. Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00021320
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
United States, Pennsylvania
... More >>
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111-2497 Less << |
|
NCT00101582
|
- |
|
Completed |
- |
- |
|
NCT00620269
|
Lung Cancer
N... More >>SCLC Less << |
Phase 2 |
Unknown |
March 2015 |
Korea, Republic of
... More >>
National Cancer Center, Korea
Recruiting
Goyang-si, Gyenggi-do, Korea, Republic of, 411-769
Contact: Jin Soo Lee, M.D. +82-31-920-1601
Principal Investigator: Jin Soo Lee, M.D.
Sub-Investigator: Heung Tae Kim, M.D.
Sub-Investigator: Ji-Youn Han, M.D.
Sub-Investigator: Geon Kook Lee, M.D.
Sub-Investigator: Tak Yun, M.D.
Sub-Investigator: Kwan Ho Cho, M.D.
Sub-Investigator: Hong Ryull Pyo, M.D.
Sub-Investigator: Bin Hwangbo, M.D.
Sub-Investigator: Hee Seok Lee, M.D.
Sub-Investigator: Kun young Lim, M.D.
Sub-Investigator: Kyong-Ah Yoon, PH.D.
Sub-Investigator: Byung-Ho Nam, Ph.D.
Sub-Investigator: Young Ho Yun, M.D.
Sub-Investigator: Jae-ill Zo, M.D. Less << |
|
NCT01970722
|
Recurrent Uterine Corpus Cance... More >>r
Recurrent Fallopian Tube Cancer
Recurrent Ovarian Cancer
Recurrent Primary Peritoneal Cancer
Stage IIIA Uterine Corpus Cancer
Stage IIIA Fallopian Tube Cancer
Stage IIIA Ovarian Cancer
Stage IIIA Primary Peritoneal Cavity Cancer
Stage IIIB Uterine Corpus Cancer
Stage IIIB Fallopian Tube Cancer
Stage IIIB Ovarian Cancer
Stage IIIB Primary Peritoneal Cavity Cancer
Stage IIIC Uterine Corpus Cancer
Stage IIIC Fallopian Tube Cancer
Stage IIIC Ovarian Cancer
Stage IIIC Primary Peritoneal Cavity Cancer
Stage IV Fallopian Tube Cancer
Stage IV Ovarian Cancer
Stage IV Primary Peritoneal Cavity Cancer
Stage IVA Uterine Corpus Cancer
Stage IVB Uterine Corpus Cancer Less << |
Phase 1 |
Recruiting |
May 2020 |
United States, California
... More >>
City of Hope Corona
Recruiting
Corona, California, United States, 92879
Contact: Cheryl Corpus 626-256-4673 ext 81529
Contact: Misagh Karimi, MD 626-256-4673
Principal Investigator: Misagh Karimi, MD
City of Hope Medical Center
Recruiting
Duarte, California, United States, 91010
Contact: Thanh Dellinger 800-826-4673
Principal Investigator: Thanh Dellinger
City of Hope Rancho Cucamonga
Recruiting
Rancho Cucamonga, California, United States, 91730
Contact: Valerie Estala 626-256-4673 ext 81699
Principal Investigator: Behnam Ebrahimi, MD Less << |
|
NCT01760811
|
Squamous Cell Carcinoma |
Phase 1
Phase 2 |
Unknown |
December 2017 |
Israel
... More >> Rabin Medical Center
Not yet recruiting
Petah Tiqva, Israel, 49100
Contact: Aron Popovtzer, MD 9729378044 aronp@clalit.org.il
Sub-Investigator: Dror Limon, MD
Sub-Investigator: Salomon Stemmer, MD
Sub-Investigator: Rafael Feinmesser, MD
Sub-Investigator: Thomas Spitzer, MD
Sub-Investigator: Gideon Bachar, MD
Principal Investigator: Aron Popovtzer, MD Less << |
|
NCT00262951
|
- |
|
Terminated(Closed at a planned... More >> interim analysis by meeting a predefined toxicity endpoint) Less << |
- |
- |
|
NCT03314935
|
Biliary Tract Cancer (BTC)
... More >> Colorectal Cancer (CRC)
Endometrial Cancer
Gastroesophageal Cancer (GC)
Ovarian Cancer
Solid Tumors Less << |
Phase 1
Phase 2 |
Recruiting |
May 27, 2020 |
United States, Alabama
... More >>
University of Alabama
Not yet recruiting
Birmingham, Alabama, United States, 35294-3300
USA Mitchell Cancer Center
Recruiting
Mobile, Alabama, United States, 36604
United States, California
UC Davis - Comprehensive Cancer Centre
Not yet recruiting
Sacramento, California, United States, 95817
United States, Georgia
Northwest Georgia Oncology Centers
Recruiting
Marietta, Georgia, United States, 30060
United States, Texas
The University of Texas MD Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
START San Antonio
Recruiting
San Antonio, Texas, United States, 78229
Belgium
Grand Hopital de Charleroi - Department of Medical Oncology
Not yet recruiting
Brussels, Belgium, 6000
Institut Jules Bordet - Clinical Trials Conduct Unit
Recruiting
Brussels, Belgium, B-1000
United Kingdom
University of Liverpool
Not yet recruiting
Liverpool, United Kingdom, L69 3BX
UCL Cancer Institute
Recruiting
London, United Kingdom, WC1E 6BT
The Christie NHS Foundation Trust
Recruiting
Manchester, United Kingdom, M20 4BX Less << |
|
NCT00003907
|
- |
|
Completed |
- |
- |
|
NCT03456063
|
Non-Small-Cell Lung |
Phase 3 |
Recruiting |
November 20, 2024 |
- |
|
NCT00577057
|
Nasopharyngeal Neoplasms |
Not Applicable |
Unknown |
August 2013 |
China
... More >> Pamela Youde Nethersole Eastern Hospital
Not yet recruiting
Hong Kong, China
Prince of Wales Hospital
Not yet recruiting
Hong Kong, China
Princess Margaret Hospital
Not yet recruiting
Hong Kong, China
Sub-Investigator: Ashley Cheng, Dr
Queen Elizabeth Hospital
Not yet recruiting
Hong Kong, China
Sub-Investigator: Roger Ngan, Dr
Queen Mary Hospital
Not yet recruiting
Hong Kong, China
Sub-Investigator: Dora Kwong, Dr
Tuen Mun Hospital
Not yet recruiting
Hong Kong, China
Sub-Investigator: Stewart Y Tung, Dr Less << |
|
NCT00085735
|
- |
|
Completed |
- |
- |
|
NCT03386838
|
Head and Neck Cancer |
Phase 3 |
Terminated(Business objectives... More >> have changed.) Less << |
- |
United States, Pennsylvania
... More >>
Allegheny General Hospital
Pittsburgh, Pennsylvania, United States, 15212-0000 Less << |
|
NCT01711541
|
Head and Neck Squamous Cell Ca... More >>rcinoma
Stage IVA Oropharyngeal Carcinoma AJCC v7
Stage IVB Oropharyngeal Carcinoma AJCC v7 Less << |
Phase 1
Phase 2 |
Active, not recruiting |
- |
United States, Illinois
... More >>
Northwestern University
Chicago, Illinois, United States, 60611
University of Chicago Comprehensive Cancer Center
Chicago, Illinois, United States, 60637
NorthShore University HealthSystem-Evanston Hospital
Evanston, Illinois, United States, 60201
United States, Missouri
Washington University School of Medicine
Saint Louis, Missouri, United States, 63110
United States, North Carolina
UNC Lineberger Comprehensive Cancer Center
Chapel Hill, North Carolina, United States, 27599 Less << |
|
NCT00301782
|
Extragonadal Germ Cell Tumor
... More >> Teratoma
Testicular Germ Cell Tumor Less << |
Phase 2 |
Completed |
- |
United Kingdom
... More >> Queen Elizabeth Hospital at University Hospital of Birmingham NHS Trust
Birmingham, England, United Kingdom, B15 2TH
Bristol Haematology and Oncology Centre
Bristol, England, United Kingdom, BS2 8ED
Addenbrooke's Hospital
Cambridge, England, United Kingdom, CB2 2QQ
Gloucestershire Oncology Centre at Cheltenham General Hospital
Cheltenham, England, United Kingdom, GL53 7AN
Walsgrave Hospital
Coventry, England, United Kingdom, CV2 2DX
Royal Devon and Exeter Hospital
Exeter, England, United Kingdom, EX2 5DW
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Saint Bartholomew's Hospital
London, England, United Kingdom, EC1A 7BE
University College of London Hospitals
London, England, United Kingdom, WIT 3AA
Christie Hospital
Manchester, England, United Kingdom, M20 4BX
Clatterbridge Centre for Oncology
Merseyside, England, United Kingdom, CH63 4JY
Nottingham City Hospital
Nottingham, England, United Kingdom, NG5 1PB
Rosemere Cancer Centre at Royal Preston Hospital
Preston, England, United Kingdom, PR2 9HT
Berkshire Cancer Centre at Royal Berkshire Hospital
Reading, England, United Kingdom, RG1 5AN
Southampton General Hospital
Southampton, England, United Kingdom, SO16 6YD
Royal Marsden - Surrey
Sutton, England, United Kingdom, SM2 5PT
Beatson West of Scotland Cancer Centre
Glasgow, Scotland, United Kingdom, G11 6NT
Raigmore Hospital
Inverness, Scotland, United Kingdom, 1V2 3UJ
Velindre Cancer Center at Velindre Hospital
Cardiff, Wales, United Kingdom, CF14 2TL Less << |
|
NCT00871013
|
Myeloma |
Phase 2 |
Recruiting |
December 2020 |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences
Recruiting
Little Rock, Arkansas, United States, 72205
Contact: Nathan Petty 501-526-6990 ext 2435 pettynathanm@uams.edu
Sub-Investigator: Frits van Rhee, M.D.,Ph.D.
Sub-Investigator: Maurizio Zangari, MD
Sub-Investigator: Atul Kothari, MD
Sub-Investigator: Carolina Schinke, MD
Sub-Investigator: Faith Davies, MD
Sub-Investigator: Gareth Morgan, MD
Sub-Investigator: Juan Crescencio, MD
Sub-Investigator: Mary Burgess, MD
Sub-Investigator: Meera Mohan, MD
Sub-Investigator: Muthukumar Radhakrishnan, MD
Sub-Investigator: Pankaj Mathur, MD
Sub-Investigator: Sharmilan Thanendrajan, MD Less << |
|
NCT02114229
|
Malignant Rhabdoid Tumor
... More >> Atypical Teratoid Rhabdoid Tumor Less << |
Phase 2 |
Recruiting |
May 2027 |
United States, California
... More >>
Children's Hospital of Los Angeles
Withdrawn
Los Angeles, California, United States, 90027
Lucille Packard Children's Hospital at Stanford University Medical Center
Recruiting
Palo Alto, California, United States, 94304
Contact: Sonia Partap, MD 650-723-0993 spartap@stanford.edu
Principal Investigator: Sonia Partap, MD
Rady Children's Hospital
Recruiting
San Diego, California, United States, 92123
Contact: John Crawford, MD, MS 858-576-1700 jcrawford@rchsd.org
Principal Investigator: John Crawford, MD, MS
United States, Colorado
Children's Hospital Colorado
Recruiting
Aurora, Colorado, United States, 80045
Contact: Kathleen Dorris, MD 720-777-6672 kathleen.dorris@childrenscolorado.org
Principal Investigator: Kathleen Dorris, MD
United States, District of Columbia
Children's National Medical Center
Recruiting
Washington, District of Columbia, United States, 20010
Contact: Lindsay B. Kilburn, MD 202-476-2800 lkilburn@childrensnational.org
Principal Investigator: Lindsay B. Kilburn, MD
United States, Florida
UF Cancer Center at Orlando Health
Recruiting
Orlando, Florida, United States, 32806
Contact: Amy A. Smith, MD 321-841-7246 amy.smith@orlandohealth.com
Principal Investigator: Amy A. Smith, MD
United States, Georgia
Children's Healthcare of Atlanta
Recruiting
Atlanta, Georgia, United States, 30322
Contact: Sindy Moon 404-785-1441 cindy.moon@choa.org
Contact: Jaclyn Smith 404-785-0692 jaclyn.smith@choa.org
Principal Investigator: Dolly Aguilera, MD
United States, Minnesota
Children's Hospital and Clinics of Minnesota
Recruiting
Minneapolis, Minnesota, United States, 55102
Contact: Anne Bendel, MD 651-220-6732 anne.bendel@childrensmn.org
Principal Investigator: Anne Bendel, MD
United States, Tennessee
St. Jude Children's Research Hospital
Recruiting
Memphis, Tennessee, United States, 38105
Contact: Tabatha E. Doyle, RN 901-595-2544 tabatha.doyle@stjude.org
Principal Investigator: Santhosh Upadhyaya, MD
United States, Texas
Texas Children's Hospital
Recruiting
Houston, Texas, United States, 77030
Contact: Patricia Baxter, MD 832-824-1000 pabaxter@txch.org
Principal Investigator: Patricia Baxter, MD
United States, Washington
Seattle Children's Hospital
Recruiting
Seattle, Washington, United States, 98105
Contact: Sarah Leary, MD 866-987-2000 sarah.leary@seattlechildrens.org
Principal Investigator: Sarah Leary, MD Less << |
|
NCT02157792
|
Advanced Solid Tumor |
Phase 1 |
Recruiting |
September 18, 2019 |
- |
|
NCT01496742
|
Non-Squamous Non-Small Cell Lu... More >>ng Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00580983
|
Head and Neck Cancer |
Not Applicable |
Completed |
- |
United States, Michigan
... More >>
University of Michigan
Ann Arbor, Michigan, United States, 48109-5010 Less << |
|
NCT02607423
|
Stage IIIA Non-Small Cell Lung... More >> Cancer Less << |
Phase 2 |
Withdrawn(The study failed to ... More >>meet its accrual targets.) Less << |
- |
United States, Pennsylvania
... More >>
ECOG-ACRIN Cancer Research Group
Philadelphia, Pennsylvania, United States, 19103 Less << |
|
NCT00580983
|
- |
|
Completed |
- |
- |
|
NCT00869232
|
Multiple Myeloma |
Phase 2 |
Active, not recruiting |
October 2019 |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT03588494
|
Quality of Life |
Phase 2
Phase 3 |
Not yet recruiting |
September 1, 2021 |
China, Sichuan
... More >> Affiliated Hospital of North Sichuan Medical College
Not yet recruiting
Nanchong, Sichuan, China, 600000
Contact: Daiyuan Ma, M.D 868172246171 angenpn@gmail.com
Principal Investigator: xin hu, M.D
Principal Investigator: xiangdong fang, M.D Less << |
|
NCT02576574
|
First Line Non-Small Cell Lung... More >> Cancer Less << |
Phase 3 |
Active, not recruiting |
September 5, 2024 |
- |
|
NCT00003907
|
Liver Cancer
... More >>Metastatic Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT02280993
|
Hodgkin Lymphoma
... More >> Refractory
Relapse Less << |
Phase 1 |
Active, not recruiting |
May 2020 |
Denmark
... More >> Rigshospitalet
Copenhagen, Denmark
France
Centre Hospitalier Universitaire
Lille, France
Hospices Civils de Lyon
Lyon, France
Centre Hospitalier et Universitaire
Nantes, France
Hôpital Saint Louis
Paris, France
Institut Gustave Roussy
Paris, France
Netherlands
Academic Medical Center
Amsterdam, Netherlands, 1105 AZ
Free University Medical Center
Amsterdam, Netherlands
University Medical Center Groningen
Groningen, Netherlands
Erasmus Medical Center
Rotterdam, Netherlands
United Kingdom
Cambridge University Hospitals NHS Foundation Trust | Addenbrooke's Hospital
Cambridge, United Kingdom
King's College Hospital NHS Foundation Trust
London, United Kingdom
Christie Hospital, Manchester
Manchester, United Kingdom Less << |
|
NCT01108666
|
Non Small Cell Lung Cancer |
Phase 1 |
Unknown |
- |
United States, Pennsylvania
... More >>
Abramson Cancer Center of the University of Pennsylvania
Philadelphia, Pennsylvania, United States, 19104 Less << |
|
NCT02995850
|
Gastric Cancer With Peritoneal... More >> Metastasis (PCI<12) Less << |
Phase 1
Phase 2 |
Recruiting |
May 2023 |
Korea, Republic of
... More >>
Department of Surgery, Yonsei University College of Medicine
Recruiting
Seoul, Korea, Republic of, 03722
Contact: Woo Jin Hyung, MD 82-2-2228-2129 WJHYUNG@yuhs.ac Less << |
|
NCT01843049
|
Esophageal Cancer |
Phase 1
Phase 2 |
Unknown |
- |
China
... More >> Fudan University Shanghai Cancer Center
Recruiting
Shanghai, China Less << |
|
NCT01278602
|
Diffuse Large B-cell Lymphoma |
Phase 2 |
Unknown |
February 2014 |
China
... More >> Fudan University Shanghai Cancer Center
Recruiting
Shanghai, China, 200032
Contact: Ye Guo, MD pattrick_guo@msn.com
Principal Investigator: Ye Guo, MD Less << |
|
NCT02116530
|
- |
|
Completed |
- |
- |
|
NCT02205047
|
Malignant Neoplasm of Stomach
... More >>
Malignant Neoplasm of Cardio-esophageal Junction of Stomach
Epidermal Growth Factor Receptor (EGFR) Protein Overexpression Less << |
Phase 2 |
Recruiting |
September 2024 |
- |
|
NCT00670631
|
Multiple Myeloma |
Phase 2 |
Completed |
- |
- |
|
NCT00083681
|
Multiple Myeloma |
Phase 2 |
Completed |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences/MIRT
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT02784795
|
Solid Tumor
B... More >>reast Cancer
Colon Cancer
Cholangiocarcinoma
Soft Tissue Sarcoma Less << |
Phase 1 |
Active, not recruiting |
January 31, 2019 |
United States, Florida
... More >>
Sylvester Comprehensive Cancer Center
Miami, Florida, United States, 33136
United States, Michigan
Karmanos Cancer Institute
Detroit, Michigan, United States, 48201
United States, New York
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
United States, Texas
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030
Denmark
Rigshospitalet
Copenhagen, København Ø, Denmark, 2100
France
Institut Bergonie
Bordeaux, France, 33076
Centre Leon Berard
Lyon Cedex 08, France, 69373
Gustave Roussy
Villejuif Cedex, France, 94805
Spain
Hospital Universitari Vall d'Hebron
Barcelona, Spain, 08035
Fundacion Jimenez Diaz
Madrid, Spain, 28040
Hospital Madrid Norte Sanchinarro
Madrid, Spain, 28050 Less << |
|
NCT02116530
|
Hematopoietic/Lymphoid Cancer
... More >>
Nausea and Vomiting
Unspecified Adult Solid Tumor, Protocol Specific Less << |
Phase 3 |
Completed |
- |
- |
|
NCT03288545
|
Carcinoma, Transitional Cell
... More >> Urinary Bladder Neoplasms
Urologic Neoplasms
Renal Pelvis Neoplasms
Urothelial Cancer
Ureteral Neoplasms
Urethral Neoplasms Less << |
Phase 1 |
Recruiting |
September 2024 |
United States, California
... More >>
University of California at San Francisco
Recruiting
San Francisco, California, United States, 94134
Stanford Cancer Center / Blood & Marrow Transplant Program
Recruiting
Stanford, California, United States, 94305
United States, Colorado
University of Colorado Hospital / University of Colorado
Recruiting
Aurora, Colorado, United States, 80045-0510
United States, Connecticut
Yale Cancer Center
Recruiting
New Haven, Connecticut, United States, 06520
United States, Florida
University of Miami
Recruiting
Miami, Florida, United States, 33136
United States, Georgia
Winship Cancer Institute / Emory University School of Medicine
Recruiting
Atlanta, Georgia, United States, 30322
United States, Illinois
Cardinal Bernardin Cancer Center / Loyola University Medical Center
Recruiting
Maywood, Illinois, United States, 60153
United States, Kansas
University of Kansas Cancer Center
Recruiting
Westwood, Kansas, United States, 66205
United States, Michigan
University of Michigan Comprehensive Cancer Center
Recruiting
Ann Arbor, Michigan, United States, 48109
United States, Minnesota
University of Minnesota
Recruiting
Minneapolis, Minnesota, United States, 55455
United States, New Jersey
Hackensack University Medical Center
Recruiting
Hackensack, New Jersey, United States, 07601
United States, New York
Roswell Park Cancer Institute
Recruiting
Buffalo, New York, United States, 14263
Weill Cornell Medical College
Recruiting
New York, New York, United States, 10065
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10087-9049
United States, North Carolina
UNC Lineberger Comprehensive Cancer Center / University of North Carolina
Recruiting
Chapel Hill, North Carolina, United States, 27599
Levine Cancer Institute
Recruiting
Charlotte, North Carolina, United States, 28204
United States, Ohio
Case Western Reserve University / University Hospitals Case Medical Center
Recruiting
Cleveland, Ohio, United States, 44106
United States, South Carolina
Medical University of South Carolina/Hollings Cancer Center
Recruiting
Charleston, South Carolina, United States, 29425 Less << |
|
NCT00670631
|
- |
|
Completed |
- |
- |
|
NCT01151761
|
Cholangiocarcinoma
... More >> Hepatobiliary Neoplasm
Liver Cancer
Bile Duct Cancer
Cancer of Gallbladder Less << |
Phase 2 |
Terminated(Poor accrual) |
- |
United States, California
... More >>
Stanford University School of Medicine
Stanford, California, United States, 94305 Less << |
|
NCT01151761
|
- |
|
Terminated(Poor accrual) |
- |
- |
|
NCT00003941
|
Mediastinal Cancer
... More >> Metastatic Cancer
Testicular Germ Cell Tumor Less << |
Phase 3 |
Completed |
- |
- |
|
NCT01440270
|
Locally Advanced Malignant Neo... More >>plasm
Oral Cancer
Oropharyngeal Carcinoma Less << |
Phase 2 |
Active, not recruiting |
October 2019 |
China, Shanghai
... More >>
Eye & Ear Hospital of Fudan University
Shanghai, Shanghai, China
Fudan University Shanghai Cancer Center
Shanghai, Shanghai, China
Ninth People's Hospital, School of Medicine, Shanghai Jiao Tong University
Shanghai, Shanghai, China
Shanghai Changzheng Hospital
Shanghai, Shanghai, China
Shanghai First People's Hospital
Shanghai, Shanghai, China
Songjiang Hospital Affiliated to Shanghai Jiao Tong University School of Medicine
Shanghai, Shanghai, China
Tongji University Dongfang Hospital
Shanghai, Shanghai, China Less << |
|
NCT02358863
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Not Applicable |
Terminated(Issues with recruit... More >>ment.) Less << |
- |
United States, District of Col... More >>umbia
Georgetown University
Washington, District of Columbia, United States, 20057 Less << |
|
NCT01033578
|
Hepatocellular Carcinoma |
Not Applicable |
Unknown |
December 2010 |
China
... More >> Liver Cancer Insitute, Zhongshan Hospital
Recruiting
Shanghai, China, 200032
Contact: Jia Fan, Doctor 86-21-64041990 ext 2375 jiafan99@yahoo.com
Contact: Xiaoying Wang, Doctor 86-21-64041990 ext 2918 xiaoyingwang@fudan.edu.cn
Principal Investigator: Jia Fan, Doctor Less << |
|
NCT02738723
|
Small Cell Lung Cancer |
Phase 2 |
Recruiting |
January 2020 |
China, Chongqing
... More >>
Daping Hospital, Third Military Medical University
Recruiting
Chongqing, Chongqing, China, 400042
Contact: Xueqin Yang, PH.D. 86-23-68757151 yangxueqin@hotmail.com
Principal Investigator: Xueqin Yang, PH.D. Less << |
|
NCT02830139
|
Malignant Neoplasm of Colorect... More >>um Less << |
Phase 2 |
Not yet recruiting |
July 2021 |
- |
|
NCT01545687
|
Head and Neck Cancer
... More >> Oral Complications of Radiation Therapy
Pain
Weight Changes Less << |
Phase 3 |
Withdrawn |
- |
- |
|
NCT00085735
|
Untreated Childhood Medullobla... More >>stoma Less << |
Phase 3 |
Completed |
- |
- |
|
NCT02720614
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT03727061
|
Recurrent Head and Neck Carcin... More >>oma
Locally Advanced Head and Neck Carcinoma Less << |
Phase 2 |
Not yet recruiting |
November 1, 2022 |
- |
|
NCT00642824
|
Non-Squamous Non-Small Cell Lu... More >>ng Cancer Less << |
Phase 2 |
Terminated(Terminated due to l... More >>ow enrollment. No statistical analyses were performed.) Less << |
- |
Spain
... More >>
Alcorcon, Spain, 28922
Alcoy, Spain, 03804
Alicante, Spain, 03010
Badalona, Spain, 08915
Barakaldo, Spain, 48903
Burgos, Spain, 09005
Elche, Spain, 03203
Girona, Spain, 17007
Huesca, Spain, 22004
La Laguna, Spain, 38320
Las Palmas de Gran Canaria, Spain, 35016
Lugo, Spain, 27004
Madrid, Spain, 28006
Madrid, Spain, 28040
Madrid, Spain, 28046
Murcia, Spain, 30008
Palma de Mallorca, Spain, 07014
Pamplona, Spain, 31008
Salamanca, Spain, 37007
Valencia, Spain, 41014
Valencia, Spain, 46009
Zaragoza, Spain, 50009 Less << |
|
NCT01689558
|
1、Enough Cases
... More >> 2、Elekta Precise 1343 Digital Control Electron Linear Accelerator
Can Undertake Nasopharyngeal Carcinoma Specimens in the Materia,
Image Department of Nose Pharynx Ministry MRI Dynamic Testing, Less << |
Phase 2
Phase 3 |
Unknown |
January 2017 |
China, Guizhou
... More >> Cancer Hospital of Guizhou province
Recruiting
Guiyang, Guizhou, China, 550002
Contact: Feng Jin, Professor +86 13985124806 jinf8865@gmail.com
Principal Investigator: Feng Jin, Professor Less << |
|
NCT02358863
|
- |
|
Terminated(Issues with recruit... More >>ment.) Less << |
- |
- |
|
NCT02095119
|
Uterine Cervical Cancer |
Phase 1
Phase 2 |
Completed |
- |
Mexico
... More >> Instituto Nacional de Cancerología
Mexico City, Federal District, Mexico, 14080 Less << |
|
NCT03502785
|
Urothelial Carcinoma |
Phase 1
Phase 2 |
Recruiting |
September 2020 |
United States, Alaska
... More >>
Alaska Clinical Research Center, LLC
Recruiting
Anchorage, Alaska, United States, 99503
Contact: Study Coordinator Talia Wyckoff 907-276-1455 TWyckoff@akmed.com
United States, Florida
H. Lee Moffitt Cancer Center & Research Institute, Inc.
Recruiting
Tampa, Florida, United States, 33612
Contact: Study Coordinator Yazmin Rodriguez 813-745-3353 Yazmin.Rodriguez@moffitt.org
United States, Indiana
Indiana University Simon Cancer Center
Recruiting
Indianapolis, Indiana, United States, 46202
Contact: Clinical Research Nurse Christin Snow, RN, BSN 317-274-5830 chsnow@iu.edu
United States, Michigan
Karmanos Cancer Institute
Recruiting
Detroit, Michigan, United States, 48201
Contact: Study Coordinator Veena Ramakrishna 313-576-9703 ramakriv@karmanos.org
United States, Missouri
Washington University School of Medicine in St. Louis
Recruiting
Saint Louis, Missouri, United States, 63110
Contact: Study Coordinator Andrew Atkocius 314-747-1343 a.atkocius@wustl.edu
United States, New York
Columbia University, Herbert Irving Comprehensive Cancer Center
Recruiting
New York, New York, United States, 10032
Contact: Study Coordinator Bridget James 212-304-5543 bb2771@cumc.columbia.edu
United States, Pennsylvania
University of Pittsburgh Medical Center
Recruiting
Pittsburgh, Pennsylvania, United States, 15232
Contact: Study Coordinator Daniel Mocan, RN 412-235-1319 mocand@upmc.edu
United States, South Carolina
Greenville Memorial Hospital
Recruiting
Greenville, South Carolina, United States, 29615
Contact: Study Coordinator Gina Norris 864-241-6251 gnorris@ghs.org
Spain
Hospital Universitario 12 de Octubre
Recruiting
Madrid, Spain, 28041
Contact: Principal Investigator Daniel Castellano dcastellano.ec@gmail.com Less << |
|
NCT00738452
|
Lung Cancer |
Phase 1 |
Completed |
- |
United States, California
... More >>
City of Hope Comprehensive Cancer Center
Duarte, California, United States, 91010-3000 Less << |
|
NCT00257829
|
Cervical Cancer |
Phase 2 |
Withdrawn(This study was close... More >>d due to lack of funding.) Less << |
- |
- |
|
NCT01917695
|
Carcinoma Cervix |
Phase 3 |
Unknown |
August 2018 |
India
... More >> Government Royapettah Hospital
Recruiting
Chennai, Tamil Nadu, India, 600014IndiaNaduI6
Contact: Rajaraman Ramamurthy, MS MCh +919444046168 rajaanu1@rediffmail.com
Contact: Subbiah Shanmugam, MS MCh +919360206030 subbiah_doctor@yahoo.co.uk
Principal Investigator: Rajaraman Ramamurthy, MS MCh
Sub-Investigator: Subbiah Shanmugam, MS MCh Less << |
|
NCT02801487
|
Nasopharyngeal Carcinoma |
Phase 1
Phase 2 |
Recruiting |
March 2021 |
China, Shanghai
... More >>
Shanghai Proton and Heavy Ion Center
Recruiting
Shanghai, Shanghai, China, 201315
Contact: Lin Kong, MD lin.kong@sphic.org.cn
Contact: Jiyi Hu, MD jiyi.hu@sphic.org.cn
Principal Investigator: Jiade J Lu, MD Less << |
|
NCT02296684
|
Cancer of Head and Neck
... More >> Head and Neck Cancer
Neoplasms, Head and Neck
Carcinoma, Squamous Cell of Head and Neck
Squamous Cell Carcinoma of the Head and Neck
Squamous Cell Carcinoma, Head and Neck Less << |
Phase 2 |
Recruiting |
March 31, 2023 |
United States, Massachusetts
... More >>
Dana Farber Cancer Institute
Recruiting
Boston, Massachusetts, United States, 02215
Contact: Ravindra Uppaluri, M.D., Ph.D. 617-632-3091
Principal Investigator: Ravindra Uppaluri, M.D., Ph.D.
Sub-Investigator: Robert Haddad, M.D.
Sub-Investigator: Nicole Chau, M.D.
Sub-Investigator: Jochen Lorch, M.D.
Sub-Investigator: Guilherme Rabinowitz, M.D.
Sub-Investigator: Glenn Hanna, M.D.
Sub-Investigator: Jason Glass, NP
Sub-Investigator: Patricia McHugh, RN
Sub-Investigator: Michelle Flynn, RN
Sub-Investigator: Jennifer McColgan, PA
Sub-Investigator: Jonathan Schoenfeld, M.D., MPH
Sub-Investigator: Danielle Margalit, M.D., MPH
Sub-Investigator: Roy Tishler, M.D., Ph.D.
Sub-Investigator: Donald Annino, M.D.
Sub-Investigator: Laura Goguen, M.D.
Sub-Investigator: Sonia Patel, NP, MPH
Sub-Investigator: Elizabeth Castronovo, NP
United States, Missouri
Washington University School of Medicine
Recruiting
Saint Louis, Missouri, United States, 63110
Contact: Douglas R Adkins, M.D. 314-362-4471 dadkins@wustl.edu
Sub-Investigator: Max Artyomov, Ph.D.
Sub-Investigator: Rebecca Chernock, M.D.
Sub-Investigator: Hiram Gay, M.D.
Sub-Investigator: Nsangou Ghogomu, M.D.
Sub-Investigator: Dorina Kallogjeri, M.D., M.P.H.
Sub-Investigator: Brian Nussenbaum, M.D.
Sub-Investigator: Randall Paniello, M.D.
Sub-Investigator: Jay Piccirillo, M.D.
Sub-Investigator: Jason Rich, M.D.
Sub-Investigator: Robert Schreiber, Ph.D.
Sub-Investigator: Wade Thorstad, M.D.
Sub-Investigator: Tanya Wildes, M.D.
Sub-Investigator: Gavin P Dunn, M.D., Ph.D.
United States, New York
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10065
Contact: Luc Morris, M.D. 212-639-3049
Principal Investigator: Luc Morris, M.D.
Sub-Investigator: Lara Dunn, M.D.
Sub-Investigator: Sean McBride, M.D.
Sub-Investigator: Jatin Shah, M.D.
Sub-Investigator: Bhuvanesh Singh, M.D., Ph.D.
Sub-Investigator: Snehal Patel, M.D.
Sub-Investigator: Jennifer Cracchiolo, M.D.
Sub-Investigator: Benjamin Roman, M.D.
Sub-Investigator: Marc Cohen, M.D.
Sub-Investigator: Vaios Hatzoglou, M.D.
Sub-Investigator: Nora Katabi, M.D.
Sub-Investigator: Nancy Lee, M.D.
Sub-Investigator: Daniel Higginson, M.D. Less << |
|
NCT00030771
|
Lung Cancer |
Phase 3 |
Active, not recruiting |
December 2037 |
Germany
... More >> Klinik Loewenstein gGmbH
Löwenstein, Germany, 74245
Klinikum der Stadt Mannheim
Mannheim, Germany, D-68135
Serbia
Institut za plucne bolesti
Sremska Kamenica, Serbia, 21204
Institute of Oncology
Sremska Kamenica, Serbia, 21204
Switzerland
Kantonsspital Aarau
Aarau, Switzerland, CH-5001
Kantonsspital Baden
Baden, Switzerland, CH-5404
Kantonsspital
Baden, Switzerland, CH-5404
Saint Claraspital AG
Basel, Switzerland, CH-4016
Universitaetsspital-Basel
Basel, Switzerland, CH-4031
Istituto Oncologico della Svizzera Italiana - Ospedale Regionale Bellinzona e Valli
Bellinzona, Switzerland, 6500
Inselspital Bern
Bern, Switzerland, CH-3010
Kantonsspital Bruderholz
Bruderholz, Switzerland, CH-4101
Kantonsspital Graubuenden
Chur, Switzerland, CH-7000
Kantonsspital Freiburg
Freiburg, Switzerland, 1708
Hopital Cantonal Universitaire de Geneve
Geneva, Switzerland, CH-1211
Centre Pluridisciplinaire d' Oncologie
Lausanne, Switzerland, 1011
Kantonsspital Liestal
Liestal, Switzerland, CH-4410
Kantonsspital Olten
Olten, Switzerland, CH-4600
FMH Onkologie/Haematologie
Rheinfelden, Switzerland, 4310
Kantonsspital - St. Gallen
St. Gallen, Switzerland, CH-9007
Regionalspital
Thun, Switzerland, 3600
Kantonsspital Winterthur
Winterthur, Switzerland, CH-8400
Onkozentrum
Zurich, Switzerland, 8038
City Hospital Triemli
Zurich, Switzerland, CH-8063
UniversitaetsSpital Zuerich
Zurich, Switzerland, CH-8091 Less << |
|
NCT01732640
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 1
Phase 2 |
Terminated(Slow accrual and hi... More >>gh levels of toxicity lead to early termination.) Less << |
- |
United States, Maryland
... More >>
Johns Hopkins Sidney Kimmel Comprehensive Cancer Center
Baltimore, Maryland, United States, 21287
United States, Tennessee
Vanderbilt Ingram Cancer Center
Nashville, Tennessee, United States, 37232 Less << |
|
NCT01732640
|
- |
|
Terminated(Slow accrual and hi... More >>gh levels of toxicity lead to early termination.) Less << |
- |
- |
|
NCT03092518
|
Gastric Adenocarcinoma
... More >> Esophagogastric Junction
Gastric Cancer Less << |
Phase 2 |
Recruiting |
October 1, 2020 |
United States, Maryland
... More >>
National Institutes of Health Clinical Center
Recruiting
Bethesda, Maryland, United States, 20892 Less << |
|
NCT03523234
|
Small-cell Lung Cancer |
Not Applicable |
Recruiting |
July 2023 |
China, Shanghai
... More >>
Shanghai Pulmonary Hospital
Recruiting
Shanghai, Shanghai, China, 200000
Contact: Peng Zhang, doctor Less << |
|
NCT00379262
|
Nasopharyngeal Carcinoma |
Phase 3 |
Active, not recruiting |
December 2018 |
China
... More >> Cancer Center, Sun Yat Sen University
Guangzhou, China
Department of Clinical Oncology, Pamela Youde Nethersole Eastern Hospital
Hong Kong, China
Department of Clinical Oncology, Prince of Wales Hospital
Hong Kong, China
Department of Clinical Oncology, Princess Margaret Hospital
Hong Kong, China
Department of Clinical Oncology, Queen Elizabeth Hospital
Hong Kong, China
Department of Clinical Oncology, Queen Mary Hospital
Hong Kong, China
Department of Clinical Oncology, Tuen Mun Hospital
Hong Kong, China Less << |
|
NCT01709487
|
Ovarian Carcinoma
... More >> Fallopian Tube Carcinoma
Primary Peritoneal Carcinoma Less << |
Phase 1
Phase 2 |
Completed |
- |
Belgium
... More >> Institut Jules Bordet
Brussels, Belgium, 1000
Hopital de la Citadelle
Liège, Belgium, 4000 Less << |
|
NCT00002839
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
Belgium
... More >> Algemeen Ziekenhuis Middelheim
Antwerp, Belgium, 2020
Universitair Ziekenhuis Antwerpen
Edegem, Belgium, B-2650
France
CHR de Besancon - Hopital Jean Minjoz
Besancon, France, 25030
Centre Regional Francois Baclesse
Caen, France, 14076
Centre Hospitalier Universitaire de Dijon
Dijon, France, 21033
Centre de Lutte Contre le Cancer, Georges-Francois Leclerc
Dijon, France, 21079
Centre Oscar Lambret
Lille, France, 59020
Centre Hospitalier Regional et Universitaire de Lille
Lille, France, 59037
CRLCC Nantes - Atlantique
Nantes-Saint Herblain, France, 44805
Centre Antoine Lacassagne
Nice, France, 06189
Hopital Charles Nicolle
Rouen, France, 76031
Centre Alexis Vautrin
Vandoeuvre-les-Nancy, France, 54511
Israel
Rambam Medical Center
Haifa, Israel, 31096
Italy
Istituto Nazionale per lo Studio e la Cura dei Tumori
Milano (Milan), Italy, 20133
Ospedale Civile Monselice
Monselice, Padova, Italy, 35043
Azienda Ospedaliera "Santa Maria Degli Angeli"
Pordenone, Italy, 33170
Netherlands
Vrije Universiteit Medisch Centrum
Amsterdam, Netherlands, 1007 MB
Leiden University Medical Center
Leiden, Netherlands, 2300 CA
Academisch Ziekenhuis Maastricht
Maastricht, Netherlands, 6202 AZ
Switzerland
Centre Hospitalier Universitaire Vaudois
Lausanne, Switzerland, CH-1011 Less << |
|
NCT02976051
|
Locally Advanced Head and Neck... More >> Cancer
Radiotherapy Less << |
Phase 2 |
Recruiting |
December 2021 |
Denmark
... More >> Department of Experimental Clinical Oncology, Aarhus University Hospital
Recruiting
Aarhus, Denmark, 8000 C
Contact: Jens Overgaard, Prof., MD +45 7846 2629 jens@oncology.au.dk
Contact: Mette Saxø, MD +89492619 mette.saksoe@oncology.au.dk Less << |
|
NCT00258323
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
United States, Ohio
... More >>
Cleveland Clinic Taussig Cancer Institute, Case Comprehensive Cancer Center
Cleveland, Ohio, United States, 44195 Less << |
|
NCT03718741
|
Bladder Cancer |
Phase 2 |
Recruiting |
November 1, 2023 |
Italy
... More >> Istituto Clinico Humanitas
Recruiting
Rozzano, Milano, Italy, 20089
Contact: Giuseppe D'Agostino, MD Less << |
|
NCT00401609
|
Small Cell Lung Cancer |
Phase 1
Phase 2 |
Completed |
- |
Italy
... More >> Azienda Ospedaliera S. Giuseppe Moscati, U.O. di Oncologia Medica
Monteforte Irpino, AV, Italy, 83024
IRCCS Oncologico Bari, Oncologia Medica
Bari, BA, Italy, 70126
Ospedale A. Cardarelli
Campobasso, CB, Italy, 86100
Ospedale Mariano Santo, U.O. di Oncologia Medica
Cosenza, CS, Italy, 87100
Ospedale Umberto di Frosinone
Frosinone, FR, Italy, 03031
Umberto I SS. Trinita' Ospedale
Frosinone, FR, Italy
Ospedale San Martino
Genova, GE, Italy
Ospedale Serbelloni
Gorgonzola, MI, Italy
Policlinico Universitario P. Giaccone
Palermo, PA, Italy, 90100
Ospedale La ferla
Palermo, PA, Italy
Policlinico Giaccone
Palermo, PA, Italy
Istituto Oncologico Veneto
Padova, PD, Italy
Ospedale Civile
Polla, SA, Italy
Divisione di Oncologia Medica, U.S.L.L. 13
Noale, VE, Italy, 30033
Ospedale L. Sacco
Milano, Italy
Istituto Nazionale dei Tumori , Divisione di Oncologia Medica B
Napoli, Italy, 80131
Ospedale Monaldi
Napoli, Italy
Azienda Sanitaria Locale 2
Pozzuoli, Italy
Istituto Regina Elena, Divisione di Oncologia Medica
Roma, Italy, 00144
Ospedale S. Giovanni Calibita Fatebenefratelli
Roma, Italy, 00186
Ospedale San Camillo - Forlanini
Rome, Italy Less << |
|
NCT02820857
|
Neuroendocrine Carcinomas |
Phase 2 |
Recruiting |
March 4, 2022 |
France
... More >> Service d'Hépato-Gastroenterologie et Oncologie Digestive, Hôpital Sud, CHU d'Amiens
Recruiting
Amiens, France, 80054
Contact: Vincent HAUTEFEUILLE, MD
Principal Investigator: Vincent HAUTEFEUILLE, MD
Service d'Hépatogastroentérologie, CHU d'Angers
Recruiting
Angers, France, 49933
Contact: Guillaume ROQUIN, MD
Principal Investigator: Guillaume ROQUIN, MD
Service de Gastroentérologie et Pancréatologie, Hôpital Beaujon, APHP
Recruiting
Clichy, France, 92118
Contact: Olivia HENTIC, MD
Principal Investigator: Olivia HENTIC, MD
Service d'Hépato-Gastroentérologie et Oncologie Digestive, CHU de Dijon
Recruiting
Dijon, France, 21000
Contact: Côme LEPAGE, MD
Principal Investigator: Côme LEPAGE, MD
Service d'Hépatogastroentéologie, Hôpital Michallon, CHU de Grenoble
Recruiting
Grenoble, France, 38043
Contact: Victoire GRANGER, MD
Principal Investigator: Victoire GRANGER, MD
Département de Cancérologie Urologique et Digestive, Centre Oscar Lambret
Recruiting
Lille, France, 59020
Contact: Farid EL HAJBI, MD
Principal Investigator: Farid EL HAJBI, MD
Service d'Oncologie Médicale - Hôpital Edouard Herriot - Hospices Civils de Lyon
Recruiting
Lyon, France, 69437
Contact: Thomas WALTER, MD 4 72 11 73 98 ext +33 thomas.walter@chu-lyon.fr
Principal Investigator: Thomas WALTER, MD
Service d'Hépato-Gastroentérologie et d'Oncologie Digestive, Hôpital de la Timone, APHM
Recruiting
Marseille, France, 13365
Contact: Laetitia DAHAN, MD
Principal Investigator: Laetitia DAHAN, MD
Service d'Oncologie Médicale, Hôpital Saint Eloi, CHU de Montpellier
Recruiting
Montpellier, France, 34298
Contact: Eric ASSENAT, MD
Principal Investigator: Eric ASSENAT, MD
Service d'Hépato-Gastroentérologie et d'Oncologie Digestive, Hôpital de la Salpêtrière, APHP
Recruiting
Paris, France, 75013
Contact: Olivier DUBREUIL, MD
Principal Investigator: Olivier DUBREUIL, MD
Service de Gastroentérologie, Hôpital Cochin, APHP
Recruiting
Paris, France, 75014
Contact: Romain CORIAT, MD
Principal Investigator: Romain CORIAT, MD
Service d'Hépato-Gastroenterologie et Oncologie Digestive, Hôpital Européen Georges Pompidou, APHP
Recruiting
Paris, France, 75015
Contact: Céline LEPERE, MD
Principal Investigator: Céline LEPERE, MD
Service d'Hépato-Gastroentérologie et d'Oncologie Digestive, Hôpital Haut Lévêque, CHU Bordeaux
Recruiting
Pessac, France, 33604
Contact: Denis SMITH, MD
Principal Investigator: Denis SMITH, MD
Pôle Régional de Cancérologie, CHU de Poitiers
Recruiting
Poitiers, France, 86021
Contact: Aurélie FERRU, MD
Principal Investigator: Aurélie FERRU, MD
Service d'Hépato-Gastroentérologie et Cancérologie, Hôpital Robert Debré, CHU de Reims
Recruiting
Reims, France, 51100
Contact: Guillaume CADIOT, MD
Principal Investigator: Guillaume CADIOT, MD
Service d'Hépatogastroentérologie, Hôpital Pontchaillou, CHU de Rennes
Recruiting
Rennes, France, 35000
Contact: Astrid LIEVRE, MD
Principal Investigator: Astrid LIEVRE, MD
Service de Gastroentérologie, CHU de Rouen
Recruiting
Rouen, France, 76031
Contact: Alice GANGLOFF, MD
Principal Investigator: Alice GANGLOFF, MD
Service de Gastro-Entérologie, Hôpital Nord, CHU de ST-Etienne
Recruiting
Saint-Priest-en-Jarez, France, 42270
Contact: Nadia BOUARIOUA, MD
Principal Investigator: Nadia BOUARIOUA, MD
Service d'Oncologie Médicale, Hôpital Civil, CHU de Strasbourg
Recruiting
Strasbourg, France, 67200
Contact: Jean-Emmanuel KURTZ, MD
Principal Investigator: Jean-Emmanuel KURTZ, MD
Service d'Oncologie Endocrinienne, Institut Gustave Roussy
Recruiting
Villejuif, France, 94805
Contact: Eric BAUDIN, MD
Principal Investigator: Eric BAUDIN, MD Less << |
|
NCT00002875
|
Brain Tumors
... More >>Central Nervous System Tumors Less << |
Phase 3 |
Completed |
- |
- |
|
NCT01416961
|
Lung Cancer |
Phase 1 |
Withdrawn(Neutron therapy has ... More >>become unavailable) Less << |
- |
- |
|
NCT03514849
|
Small-cell Lung Cancer |
Not Applicable |
Recruiting |
March 2026 |
China, Shanghai
... More >>
Shanghai Pulmonary Hospital
Recruiting
Shanghai, Shanghai, China, 200000
Contact: Peng Zhang, doctor Less << |
|
NCT03632798
|
Recurrent Ovarian Cancer |
Phase 3 |
Recruiting |
January 31, 2022 |
United States, Mississippi
... More >>
Univeristy of Mississippi Medical Center
Recruiting
Jackson, Mississippi, United States, 39216
Contact: Kelly J Wilkinson, MD 888-815-2005 kjeanes@umc.edu
United States, West Virginia
Charleston Area Medical Center
Recruiting
Charleston, West Virginia, United States, 25326
Contact: Stephen Bush, MD 304-925-4200
Contact: Tina Garten, RN 304-388-9995 Tina.Garten@camc.org
Edwards Comprehensive Cancer Center - Cabell Huntington Hospital
Recruiting
Huntington, West Virginia, United States, 25705
Contact: Nadim Bou Zgheib, MD 304-399-6600 zgheib@marshall.edu
Contact: Barbara Payne, RN 304-399-6617 Barbara.Payne@chhi.org Less << |
|
NCT02687958
|
Neuroendocrine Carcinoma |
Phase 2 |
Recruiting |
May 2017 |
Italy
... More >> S.C. Oncologia - Policlinico
Active, not recruiting
Modena, Emilia - Romagna, Italy, 41124
U.O. Oncologia Medica 1 - AOU Pisana
Recruiting
Pisa, Tuscany, Italy, 56126
Contact: Cinzia Orlandini, SC
Principal Investigator: Sergio Ricci, MD
Azienda Ospedaliera "Spedali Civili di Brescia"
Active, not recruiting
Brescia, Italy, 25123
Azienda Ospedaliera Universitaria Careggi
Recruiting
Florence, Italy, 50134
Contact: Luisa Di Cerbo, SC +390557947298 luisa.dicerbo@libero.it
Contact: Alessandro Di Costanzo, SC +390557947298 adicostanzo.oncmed@hotmail.com
Principal Investigator: Francesco Di Costanzo, MD
Principal Investigator: Lorenzo Antonuzzo, MD
Istituto Europeo di Oncologia
Active, not recruiting
Milano, Italy, 20141 Less << |
|
NCT00413283
|
Lung Cancer
C... More >>hemotherapy-Induced Thrombocytopenia
Non-Small Cell Lung Cancer
Cancer
Lung Neoplasms
Oncology
Solid Tumors
Thrombocytopenia Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00413283
|
- |
|
Completed |
- |
- |
|
NCT00193804
|
- |
|
Active, not recruiting |
May 2017 |
India
... More >> Tata Memorial Hospital
Mumbai, Maharastra, India, 400 012 Less << |
|
NCT02264990
|
Non-squamous Non-small Cell Lu... More >>ng Cancer Less << |
Phase 3 |
Active, not recruiting |
July 26, 2019 |
- |
|
NCT01719926
|
Colorectal Neoplasms
... More >> Esophageal Neoplasms
Ovarian Neoplasms Less << |
Phase 1 |
Completed |
- |
Netherlands
... More >> Meander Medisch Centrum
Amersfoort, Utrecht, Netherlands, 3813TZ
the Netherlands Cancer Institute
Amsterdam, Netherlands, 1066 CX
UMC Utrecht
Utrecht, Netherlands, 3584CX
Switzerland
Oncology Institute of Southern Switzerland
Bellinzona, Switzerland, CH-6500 Less << |
|
NCT01362127
|
Carcinoma, Squamous Cell
... More >> Adenocarcinoma of the Esophagus and Gastric Cardia Less << |
Phase 2 |
Active, not recruiting |
June 2018 |
Sweden
... More >> Gastrocentrum. Department of surgery, Karolinska University Hospital
Stockholm, Sweden, 14186 Less << |
|
NCT03519971
|
Non-Small Cell Lung Cancer |
Phase 3 |
Recruiting |
August 1, 2022 |
- |
|
NCT00193739
|
Cancer of Cervix |
Phase 3 |
Active, not recruiting |
July 31, 2019 |
India
... More >> Tata Memorial Hospital
Mumbai, Maharashtra, India, 400012 Less << |
|
NCT00381706
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT02052960
|
Carcinoma, Squamous Cell of He... More >>ad and Neck Less << |
Phase 2 |
Active, not recruiting |
December 15, 2017 |
Belgium
... More >> Investigator
Antwerp, Belgium, 2650
Investigator
Brussels, Belgium, 1000
Investigator
Brussels, Belgium, 1200
Investigator
Gent, Belgium, 9000
France
Investigator
Avignon, France, 84918
Investigator
Lille, France, 59020
Investigator
Lyon, France, 69008
Investigator
Nice, France, 06189
Investigator
St Herblain, France, 44805
Germany
Investigator
Aachen, Germany, 52074
Investigator
Berlin, Germany, 12203
Investigator
Dresden, Germany, 01307
Investigator
Essen, Germany, 45122
Investigator
Hamburg, Germany, 20246
Investigator
Hannover, Germany, 30625
Investigator
Leipzig, Germany, 04103
Italy
Investigator
Milan, Italy, 20142
Investigator
Pavia, Italy, 27100
Poland
Investigator
Bydgoszcz, Poland, 85-796
Investigator
Krakow, Poland
Investigator
Lodz, Poland, 93-513
Investigator
Lublin, Poland, 20-090
Investigator
Warsaw, Poland, 2781
Romania
Investigator
Brasov, Romania, 500091
Investigator
Clui-Napoca, Romania, 400015
Investigator
Cluj-Napoca, Romania, 400015
Investigator
Craiova, Romania, 200385
Investigator
Oradea, Romania, 410469
Investigator
Ploiesti, Romania, 100011
Investigator
Timisoara, Romania, 300167
Investigator
Timisoara, Romania, 300239
Spain
Investigator
Barcelona, Spain, 08036
Investigator
Madrid, Spain, 28050
Investigator
Madrid, Spain, 28911
Investigator
Valencia, Spain, 46009 Less << |
|
NCT03077243
|
Carcinoma, Squamous Cell
... More >> Head and Neck Neoplasms
Oropharyngeal Neoplasms Less << |
Phase 2 |
Recruiting |
February 2025 |
United States, Florida
... More >>
University of Florida
Recruiting
Gainesville, Florida, United States, 32610
Contact: Robert Amdur, MD 352-265-0287 amdurr@shands.ufl.edu
University of Florida Proton Therapy Institute
Recruiting
Jacksonville, Florida, United States, 32206
Contact: Roi Dagan, MD 904-588-1445 rdagan@floridaproton.org
United States, North Carolina
University of North Carolina at Chapel Hill, Department of Radiation Oncology
Recruiting
Chapel Hill, North Carolina, United States, 27599
Contact: Bhishamjit Chera, MD 984-974-0400 bchera@med.unc.edu
Contact: Rebecca Green, MSW (984) 974-8440 rlgreen@med.unc.edu
Rex Healthcare
Recruiting
Raleigh, North Carolina, United States, 27607
Contact: Nathan Sheets, MD 919-784-1251 nathan.sheets@unchealth.unc.edu Less << |
|
NCT01294280
|
- |
|
Recruiting |
- |
United States, New York
... More >>
SUNY Upstate Medical University
Recruiting
Syracuse, New York, United States, 13210
Contact: Stephen Graziano, MD 315-464-4353 Less << |
|
NCT02055144
|
- |
|
Recruiting |
March 2018 |
Italy
... More >> IRCCS Azienda Ospedaliera Universitaria San Martino - IST Istituto Nazionale per la Ricerca sul Cancro
Recruiting
Genoa, Italy, 16132
Contact: Francesco Grossi, MD +393355255484 fg1965@libero.it Less << |
|
NCT02858804
|
Mantle Cell Lymphoma |
Phase 4 |
Recruiting |
June 2020 |
China
... More >> Shuhua Yi
Recruiting
Tianjin, China, 300020
Contact: Shuhua Yi, Doc 86-22-23909106 yishuhua@ihcams.ac.cn
Contact: Lugui Qiu, Doc 86-22-23909172 qiulg@ihcams.ac.cn Less << |
|
NCT00381706
|
- |
|
Completed |
- |
- |
|
NCT00455572
|
Lung Cancer, Non-Small Cell |
Phase 1 |
Terminated(Study early termina... More >>tion was due to slow recruitment and difficulties at achieving the required enrolment for the study.) Less << |
- |
Belgium
... More >> GSK Investigational Site
Genk, Belgium, 3600
GSK Investigational Site
Leuven, Belgium, 3000
GSK Investigational Site
Liège, Belgium, 4000
Canada, Alberta
GSK Investigational Site
Edmonton, Alberta, Canada, T6G 1Z2
Canada, Quebec
GSK Investigational Site
Greenfield Park, Quebec, Canada, J4V 2H1
GSK Investigational Site
Montreal, Quebec, Canada, H4J 1C5
France
GSK Investigational Site
Montpellier, France, 34295
GSK Investigational Site
Pierre Benite, France, 69495
GSK Investigational Site
Saint Herblain, France, 44805
GSK Investigational Site
Strasbourg, France, 67091
Germany
GSK Investigational Site
Hemer, Nordrhein-Westfalen, Germany, 58675
GSK Investigational Site
Mainz, Rheinland-Pfalz, Germany, 55131
GSK Investigational Site
Halle, Sachsen-Anhalt, Germany, 06120
GSK Investigational Site
Grosshansdorf, Schleswig-Holstein, Germany, 22927
GSK Investigational Site
Bad Berka, Thueringen, Germany, 99437
GSK Investigational Site
Hamburg, Germany, 20246
Italy
GSK Investigational Site
Udine, Friuli-Venezia-Giulia, Italy, 33100
GSK Investigational Site
Roma, Lazio, Italy, 00152
GSK Investigational Site
Genova, Liguria, Italy, 16132
GSK Investigational Site
Milano, Lombardia, Italy, 20132
GSK Investigational Site
Milano, Lombardia, Italy, 20141
United Kingdom
GSK Investigational Site
Wythenshawe, Greater Manchester, United Kingdom, M23 9LT
GSK Investigational Site
Bebington, Wirral, United Kingdom, CH63 4JY
GSK Investigational Site
London, United Kingdom, SE1 9RT
GSK Investigational Site
Nottingham, United Kingdom, NG5 1PB
GSK Investigational Site
Southampton, United Kingdom, SO16 6YD Less << |
|
NCT00180791
|
Medulloblastoma
... More >> Brain Tumors
Neuroectodermal Tumors, Primitive Less << |
Phase 2 |
Unknown |
- |
France
... More >> Institut Gustave Roussy
Recruiting
Villejuif, France, 94800
Contact: Chantal Kalifa, MD 33 1 42 11 4166 kalifa@igr.fr Less << |
|
NCT02974426
|
Non-small Cell Lung Cancer Sta... More >>ge IIIA
Radiotherapy Less << |
Phase 3 |
Recruiting |
December 2020 |
China, Shanghai
... More >>
Shanghai Chest Hospital
Recruiting
Shanghai, Shanghai, China
Contact: Xiaolong Fu, PhD +862122200000 ext 3602 xlfu1964@126.com Less << |
|
NCT00003912
|
Liver Cancer |
Phase 3 |
Completed |
- |
Italy
... More >> Azienda Ospedaliera di Padova
Padova, Italy, 35128 Less << |
|
NCT00002555
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
Belgium
... More >> Institut Jules Bordet
Brussels, Belgium, 1000
France
Centre de Lute Contre le Cancer,Georges-Francois Leclerc
Dijon, France, 21079
CHR de Grenoble - La Tronche
Grenoble, France, 38043
Centre Oscar Lambret
Lille, France, 59020
CRLCC Nantes - Atlantique
Nantes-Saint Herblain, France, 44805
Hopital Jean Bernard
Poitiers, France, 86021
Institut Gustave Roussy
Villejuif, France, F-94805
Italy
Istituto Nazionale per lo Studio e la Cura dei Tumori
Milan, Italy, 20133
Istituti Fisioterapici Ospitalieri - Roma
Rome, Italy, 00161
Netherlands
University Medical Center Nijmegen
Nijmegen, Netherlands, NL-6252 HB
Poland
Medical University of Gdansk
Gdansk, Poland, 80-211
Slovenia
Institute of Oncology, Ljubljana
Ljubljana, Slovenia, Sl-1000
Spain
Ciudad Sanitaria Vall D'Hebron
Barcelona, Spain, 08035
Switzerland
Ospedale San Giovanni
Bellinzona, Switzerland, CH-6500
Inselspital, Bern
Bern, Switzerland, CH-3010
Centre Hospitalier Universitaire Vaudois
Lausanne, Switzerland, CH-1011
Universitaetsspital
Zurich, Switzerland, CH-8091
Turkey
Dokuz Eylul University School of Medicine
Izmir, Turkey, 35340
United Kingdom
Nottingham City Hospital NHS Trust
Nottingham, England, United Kingdom, NG5 1PB Less << |
|
NCT00002550
|
Lung Cancer |
Phase 3 |
Completed |
- |
United States, Illinois
... More >>
CCOP - Carle Cancer Center
Urbana, Illinois, United States, 61801
United States, Indiana
Indiana University Cancer Center
Indianapolis, Indiana, United States, 46202-5289
Veterans Affairs Medical Center - Indianapolis (Roudebush)
Indianapolis, Indiana, United States, 46202
United States, Iowa
CCOP - Cedar Rapids Oncology Project
Cedar Rapids, Iowa, United States, 52403-1206
United States, Michigan
CCOP - Ann Arbor Regional
Ann Arbor, Michigan, United States, 48106
United States, Nebraska
CCOP - Missouri Valley Cancer Consortium
Omaha, Nebraska, United States, 68106
United States, New York
University of Rochester Cancer Center
Rochester, New York, United States, 14642
United States, Ohio
Ireland Cancer Center
Cleveland, Ohio, United States, 44106-5065
CCOP - Toledo Community Hospital Oncology Program
Toledo, Ohio, United States, 43623-3456
United States, Pennsylvania
Hahnemann University Hospital
Philadelphia, Pennsylvania, United States, 19102-1192
University of Pittsburgh Cancer Institute
Pittsburgh, Pennsylvania, United States, 15213-3489
United States, Tennessee
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States, 37232-6838
United States, Wisconsin
CCOP - Green Bay
Green Bay, Wisconsin, United States, 54301
Medical College of Wisconsin
Milwaukee, Wisconsin, United States, 53226
Veterans Affairs Medical Center - Milwaukee (Zablocki)
Milwaukee, Wisconsin, United States, 53295
South Africa
Pretoria Academic Hospitals
Pretoria, South Africa, 0001 Less << |
|
NCT01667302
|
Extranodal NK/T-cell Lymphoma,... More >> Nasal Type Less << |
Phase 2 |
Unknown |
December 2016 |
China, Shanghai
... More >>
Fudan University Cancer Hospital
Recruiting
SHanghai, Shanghai, China, 200032
Contact: Ye Guo, MD 862164175590 pattrick_guo@msn.com Less << |
|
NCT02853305
|
Urothelial Carcinoma Associate... More >>d 1 RNA, Human Less << |
Phase 3 |
Active, not recruiting |
May 31, 2020 |
- |
|
NCT02621333
|
Lung Adenocarcinoma |
Phase 2 |
Unknown |
September 2018 |
China, Henan
... More >> Henan Cancer Hospital/The affiliated Cancer Hospital of ZhengZhou university
Recruiting
ZhengZhou, Henan, China, 450008
Contact: Quanli Gao, M.D. +8615038171966 guaoquanli2015@126.com
Contact: Lingdi Zhao, M.D. +8637165587483 lingdi1010@sohu.com
Principal Investigator: Quanli Gao, M.D. Less << |
|
NCT00210171
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Terminated |
- |
France
... More >> Centre de radiothérapie d'Agen
Agen, France, 47000
Clinique Tivoli
Bordeaux, France, 33000
Polyclinique Bordeaux Nord
Bordeaux, France, 33000
Centre Hospitalier Universitaire de Bordeaux
Bordeaux, France, 33076
Institut Bergonié - Centre Régional de Luttre Contre le Cancer de Bordeaux et du Sud Ouest
Bordeaux, France, 33076
Hôpital Robert Boulin
Libourne, France, 33500
Clinique Francheville
Perigueux, France, 24000 Less << |
|
NCT00174629
|
Lung Neoplasms |
Phase 3 |
Completed |
- |
Germany
... More >> Sanofi-Aventis
Berlin, Germany
Spain
Sanofi-Aventis
Barcelona, Spain
Switzerland
Sanofi-Aventis
Genève, Switzerland Less << |
|
NCT00186680
|
Breast Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, California
... More >>
Stanford University School of Medicine
Stanford, California, United States, 94305 Less << |
|
NCT03360760
|
Osteosarcoma of Pelvis |
Not Applicable |
Recruiting |
February 2025 |
China, Beijing
... More >> Peking University People's Hospital
Recruiting
Beijing, Beijing, China, 100034
Contact: Jie Xu, M.D. 86 15901040835 xujie_pkuph@sina.com Less << |
|
NCT00002558
|
- |
|
Completed |
- |
- |
|
NCT00050973
|
Non-small Cell Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00453232
|
Extragonadal Germ Cell Tumor
... More >> Teratoma
Testicular Germ Cell Tumor Less << |
Phase 2 |
Completed |
- |
United Kingdom
... More >> Addenbrooke's Hospital
Cambridge, England, United Kingdom, CB2 2QQ
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Saint Bartholomew's Hospital
London, England, United Kingdom, EC1A 7BE
Northern Centre for Cancer Treatment at Newcastle General Hospital
Newcastle-Upon-Tyne, England, United Kingdom, NE4 6BE
Churchill Hospital
Oxford, England, United Kingdom, OX3 7LJ
Edinburgh Cancer Centre at Western General Hospital
Edinburgh, Scotland, United Kingdom, EH4 2XU
Beatson West of Scotland Cancer Centre
Glasgow, Scotland, United Kingdom, G11 6NT Less << |
|
NCT00003994
|
Childhood Hepatoblastoma
... More >> Recurrent Childhood Liver Cancer
Stage I Childhood Liver Cancer Less << |
Phase 3 |
Completed |
- |
United States, California
... More >>
Children's Oncology Group
Arcadia, California, United States, 91006-3776 Less << |
|
NCT01212354
|
Locally Advanced Head and Neck... More >> Cancer Less << |
Phase 3 |
Recruiting |
September 2025 |
Germany
... More >> Klinik für RadioOnkologie Strahlentherapie
Recruiting
Munich, Bavaria, Germany, 81675
Contact: Steffi U. Pigorsch, Dr. med. +49 (0)89-4140 ext -5611 steffi.pigorsch@lrz.tum.de
Contact: Sabine Barta, Dr. +49 (0)89-4140 ext 7787 sabine.barta@mri.tum.de
Principal Investigator: Steffi U. Pigorsch, Dr. med.
Sub-Investigator: Stephanie Combs, Prof. Dr. Less << |
|
NCT00002558
|
Extragonadal Germ Cell Tumor
... More >> Ovarian Cancer
Testicular Germ Cell Tumor Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT00003231
|
Lung Cancer |
Phase 2 |
Completed |
- |
Switzerland
... More >> Kantonspital Aarau
Aarau, Switzerland, 5001
Office of Walter Weber-Stadelman
Basel, Switzerland, CH 4051
University Hospital
Basel, Switzerland, CH-4031
Inselspital, Bern
Bern, Switzerland, CH-3010
Hopital Cantonal Universitaire de Geneva
Geneva, Switzerland, CH-1211
Istituto Oncologico della Svizzera Italiana
Lugano, Switzerland, CH-6900
Burgerspital, Solothurn
Solothurn, Switzerland, 4500
City Hospital Triemli
Zurich, Switzerland, 8063
Klinik Hirslanden
Zurich, Switzerland, CH-8008 Less << |
|
NCT00578526
|
Urothelial Cancer
... More >> Bladder Cancer
Adult Less << |
Phase 2 |
Completed |
- |
Canada, Alberta
... More >>
Tom Baker Cancer Centre
Calgary, Alberta, Canada, T2N 4N2
Cross Cancer Institute
Edmonton, Alberta, Canada, T6G 1Z2
Canada, British Columbia
British Columbia Cancer Agency - Vancouver Centre
Vancouver, British Columbia, Canada, V5Z 4E6
Canada, Ontario
Juravinski Cancer Centre
Hamilton, Ontario, Canada, L8V 5C2
London Health Sciences Centre
London, Ontario, Canada, T6A 4L6 Less << |
|
NCT02937116
|
Cancer, Solid Tumor |
Phase 1 |
Unknown |
December 2018 |
- |
|
NCT00303758
|
Pancreatic Cancer |
Phase 3 |
Completed |
- |
France
... More >> Centre Hospitalier d'Abbeville
Abbeville, France, 80101
Hopital Duffaut
Avignon, France, 84902
Centre Hospitalier de Blois
Blois, France, 41016
Centre Hospitalier Docteur Duchenne
Boulogne Sur Mer, France, 62200
Centre Hospitalier Universitaire Ambroise Pare - Boulogne
Boulogne, France, F-92104
C.H. Bourg En Bresse
Bourg En Bresse, France, 01012
Centre Hospitalier Pierre Oudot
Bourgoin-Jallieu, France, 38300
Centre Hospitalier Universitaire d'Amiens
Caen, France, 14076
Centre Hospitalier de Chalons-en-Champagne
Chalons-en-Champagne, France, 51000
CHR Clermont Ferrand, Hotel dieu
Clermont-Ferrand, France, 63003
Hopital Beaujon
Clichy, France, 92118
Hopital Louis Pasteur
Colmar, France, 68024
Hopital Du Bocage
Dijon, France, 21034
Centre de Lutte Contre le Cancer Georges-Francois Leclerc
Dijon, France, 21079
Centre Hospitalier Draguignan
Draguignan, France, 83300
Centre Hospitalier De Dunkerque - CHD
Dunkerque, France, 59240
Centre Hospitalier Intercommunal St. Aubin les Elbeuf
Elbeuf, France, 76503
CHU de Grenoble - Hopital de la Tronche
Grenoble, France, 38043
Clinique Pasteur
Guilherand Granges, France, 07500
Hopital Robert Boulin
Libourne, France, 33500
Centre Hospital Universitaire Hop Huriez
Lille, France, 59037
Centre Hospitalier Regional et Universitaire de Lille
Lille, France, 59037
Hopital Edouard Herriot
Lyon, France, 69437
Hopital Saint Joseph
Marseille, France, 13008
CHU de la Timone
Marseille, France, 13385
CHU Nord
Marseille, France, 13915
Centre Hospitalier de Martigues
Martigues, France, 13698
Centre Hospitalier General de Mont de Marsan
Mont-de-Marsan, France, 40000
Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Montpellier, France, 34298
CHR Hotel Dieu
Nantes, France, 44093
CHR D'Orleans - Hopital de la Source
Orleans, France, 45100
Hopital Europeen Georges Pompidou
Paris, France, 75015
Hopital Bichat - Claude Bernard
Paris, France, 75018
Hopital Haut Leveque
Pessac, France, 33604
Clinique Ste - Marie
Pontoise, France, 95301
CHU - Robert Debre
Reims, France, 51092
Centre Eugene Marquis
Rennes, France, 35064
Hopital Charles Nicolle
Rouen, France, 76031
Clinique Armoricaine De Radiologie
Saint Brieuc, France, F-22015
Centre Joliot Curie Des Docteurs Jean-Christophe Chardon Jacques Hernandez Et Laurent Gasnault
Saint Martin Boulogne, France, 62280
Centre Hospitalier de Saint-Quentin
Saint-Quentin, France, 02321
Centre Hospitalier de Semur en Auxois
Semur en Auxois, France, 21140
Centre Hospitalier de Soissons
Soissons cedex, France, 02209
Hopital Universitaire Hautepierre
Strasbourg, France, 67098
Centre Hospitalier de Tarbes
Tarbes, France, 65013
Nouvelle Clinique Generale
Valence, France, 26000 Less << |
|
NCT00014274
|
Bladder Cancer
... More >> Transitional Cell Cancer of the Renal Pelvis and Ureter
Urethral Cancer Less << |
Phase 2
Phase 3 |
Completed |
- |
- |
|
NCT00463515
|
Non-Small-Cell-Lung Cancer |
Phase 2 |
Completed |
- |
Belgium
... More >> ZNA Middelheim
Antwerpen, Antwerp, Belgium
University Hospital Antwerp
Edegem, Antwerp, Belgium
Sint Augustinus Ziekenhuis
Wilrijk, Antwerp, Belgium Less << |
|
NCT00070629
|
Carcinoma, Non-Small Cell Lung |
Phase 2 |
Completed |
- |
- |
|
NCT03321539
|
Nasopharyngeal Carcinoma |
Phase 3 |
Recruiting |
December 31, 2025 |
China, Guangdong
... More >>
Guangzhou Medical University
Recruiting
Guangzhou, Guangdong, China, 510000
Contact: Dong-Ping Chen, MD +862066673626 chen_dpgz@163.com
The First Affiliated Hospital of Guangdong Pharmaceutical University
Recruiting
Guangzhou, Guangdong, China, 510000
Contact: Xi-Cheng Wang, MD +862061321397 13902400598@126.com
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Hai-Qiang Mai, MD,PhD +862087343643 maihq@sysucc.org.cn Less << |
|
NCT00062270
|
Lung Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, Alabama
... More >>
University of Alabama at Birmingham Comprehensive Cancer Center
Birmingham, Alabama, United States, 35294-3300 Less << |
|
NCT02549261
|
Non-small Cell Lung Cancer |
Not Applicable |
Unknown |
December 2015 |
China, Beijing
... More >> Cancer Institute & Hospital, Chinese Academy of Medical Sciences
Beijing, Beijing, China Less << |
|
NCT02283476
|
Lung Cancer |
Phase 3 |
Unknown |
November 2017 |
China
... More >> Tianjin Cancer Hospital
Not yet recruiting
TianJin, China, 300060
Contact: Jinhuai Xue, Master 8613502065304 xuejh1210@sina.com
Principal Investigator: Kai Li, Doctor Less << |
|
NCT01599013
|
Bladder Transitional Cell Carc... More >>inoma Stage IV Less << |
Phase 2 |
Completed |
- |
France
... More >> Pierre Fabre Research Institute
Boulogne, France Less << |
|
NCT02621970
|
Nasopharyngeal Carcinoma |
Phase 3 |
Not yet recruiting |
January 2024 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT03604965
|
Locally Advanced Nasopharyngea... More >>l Carcinoma Less << |
Phase 3 |
Recruiting |
July 21, 2020 |
China, 贵州省
... More >> Cancer Hospital of Guizhou Medical University
Recruiting
Guiyang, 贵州省, China, 550000
Contact: Yuanyuan Li, master 085186512802 lilyuanyuan@qq.com
Sub-Investigator: Feng Jin, Bachelor
Sub-Investigator: Weili Wu, Master
Sub-Investigator: Jinhua Long, Master
Sub-Investigator: Xiuling Luo, Bachelor
Sub-Investigator: Xiuyun Gong, Bachelor
Sub-Investigator: Yanfang Cen, Bachelor Less << |
|
NCT00002735
|
Head and Neck Cancer |
Phase 2 |
Terminated(Terminated due to p... More >>oor accrual.) Less << |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences
Little Rock, Arkansas, United States, 72205
United States, California
USC/Norris Comprehensive Cancer Center
Los Angeles, California, United States, 90033-0800
United States, Colorado
University of Colorado Cancer Center
Denver, Colorado, United States, 80262
United States, Kansas
University of Kansas Medical Center
Kansas City, Kansas, United States, 66160-7357
United States, Louisiana
MBCCOP - LSU Medical Center
New Orleans, Louisiana, United States, 70112
United States, Michigan
Veterans Affairs Medical Center - Ann Arbor
Ann Arbor, Michigan, United States, 48105
University of Michigan Comprehensive Cancer Center
Ann Arbor, Michigan, United States, 48109-0752
Veterans Affairs Medical Center - Detroit
Detroit, Michigan, United States, 48201-1932
Barbara Ann Karmanos Cancer Institute
Detroit, Michigan, United States, 48201
Henry Ford Hospital
Detroit, Michigan, United States, 48202
United States, Missouri
Veterans Affairs Medical Center - Kansas City
Kansas City, Missouri, United States, 64128
United States, New York
Veterans Affairs Medical Center - Brooklyn
Brooklyn, New York, United States, 11209
United States, Ohio
Cleveland Clinic Cancer Center
Cleveland, Ohio, United States, 44195
United States, Oklahoma
Oklahoma Medical Research Foundation
Oklahoma City, Oklahoma, United States, 73104
Veterans Affairs Medical Center - Oklahoma City
Oklahoma City, Oklahoma, United States, 73104
United States, Texas
University of Texas Medical Branch
Galveston, Texas, United States, 77555-1329
Veterans Affairs Medical Center - Temple
Temple, Texas, United States, 76504
CCOP - Scott and White Hospital
Temple, Texas, United States, 76508 Less << |
|
NCT00003133
|
Bladder Cancer
... More >> Transitional Cell Cancer of the Renal Pelvis and Ureter Less << |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT01867086
|
Stage III Ovarian Cancer
... More >> Stage IV Ovarian Cancer Less << |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
Mary Crowley Cancer Research Centers
Dallas, Texas, United States, 75230 Less << |
|
NCT01548573
|
Multiple Myeloma |
Phase 2 |
Terminated(Met study stopping ... More >>rules) Less << |
- |
United States, Iowa
... More >>
University of Iowa Holden Comprehensive Cancer Center
Iowa City, Iowa, United States, 52242 Less << |
|
NCT02724579
|
Medulloblastoma
... More >> Untreated Childhood Medulloblastoma Less << |
Phase 2 |
Recruiting |
May 31, 2025 |
- |
|
NCT02940990
|
NSCLC
SBRT
... More >> GM-CSF Less << |
Phase 2 |
Not yet recruiting |
- |
China, Shanghai
... More >>
Shanghai Chest Hospital
Not yet recruiting
Shanghai, Shanghai, China
Contact: Xiaolong Fu, PhD +862122200000 ext 3602 xlfu1964@126.com Less << |
|
NCT00276705
|
Liver Cancer |
Phase 2 |
Unknown |
- |
Ireland
... More >> Our Lady's Hospital for Sick Children Crumlin
Dublin, Ireland, 12
United Kingdom
Birmingham Children's Hospital
Birmingham, England, United Kingdom, B4 6NH
Institute of Child Health at University of Bristol
Bristol, England, United Kingdom, BS2 8AE
Addenbrooke's Hospital
Cambridge, England, United Kingdom, CB2 2QQ
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Royal Liverpool Children's Hospital, Alder Hey
Liverpool, England, United Kingdom, L12 2AP
Royal London Hospital
London, England, United Kingdom, E1 1BB
Great Ormond Street Hospital for Children
London, England, United Kingdom, WC1N 3JH
Royal Manchester Children's Hospital
Manchester, England, United Kingdom, M27 4HA
Sir James Spence Institute of Child Health at Royal Victoria Infirmary
Newcastle-Upon-Tyne, England, United Kingdom, NE1 4LP
Queen's Medical Centre
Nottingham, England, United Kingdom, NG7 2UH
Oxford Radcliffe Hospital
Oxford, England, United Kingdom, 0X3 9DU
Children's Hospital - Sheffield
Sheffield, England, United Kingdom, S10 2TH
Southampton General Hospital
Southampton, England, United Kingdom, SO16 6YD
Royal Marsden - Surrey
Sutton, England, United Kingdom, SM2 5PT
Royal Belfast Hospital for Sick Children
Belfast, Northern Ireland, United Kingdom, BT12 6BE
Royal Aberdeen Children's Hospital
Aberdeen, Scotland, United Kingdom, AB25 2ZG
Royal Hospital for Sick Children
Edinburgh, Scotland, United Kingdom, EH9 1LF
Royal Hospital for Sick Children
Glasgow, Scotland, United Kingdom, G3 8SJ
Childrens Hospital for Wales
Cardiff, Wales, United Kingdom, CF14 4XW Less << |
|
NCT01867086
|
- |
|
Completed |
- |
- |
|
NCT02098954
|
Carcinoma, Non-Small Cell Lung... More >>
EGFR Gene Mutation Less << |
Phase 2 |
Unknown |
December 2016 |
China, Hunan
... More >> Hunan Province Tumor Hospital
Recruiting
Changsha, Hunan, China
Contact: Nong Yang, MD +86 731 89762323 yangnong0217@163.com
Contact: Ming Zhou, MD +86 731 89762321 zhouming243@gmail.com
Principal Investigator: Nong Yang, MD Less << |
|
NCT00498953
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Completed |
- |
Belgium
... More >> Institut Jules Bordet
Brussels, Belgium, 1000 Less << |
|
NCT01878617
|
Medulloblastoma |
Phase 2 |
Recruiting |
December 2026 |
- |
|
NCT01175356
|
- |
|
Completed |
- |
- |
|
NCT01175356
|
Disseminated Neuroblastoma
... More >> Ganglioneuroblastoma
Localized Resectable Neuroblastoma
Localized Unresectable Neuroblastoma
Regional Neuroblastoma
Stage 4S Neuroblastoma Less << |
Not Applicable |
Completed |
- |
- |
|
NCT01142414
|
Head and Neck Cancer |
Phase 3 |
Withdrawn |
- |
- |
|
NCT00003141
|
Brain Tumors
... More >>Central Nervous System Tumors
Neuroblastoma
Sarcoma Less << |
Phase 1 |
Completed |
- |
- |
|
NCT00416676
|
Neuroblastoma |
Phase 3 |
Unknown |
- |
- |
|
NCT00031590
|
Brain Tumors
... More >>Central Nervous System Tumors
Medulloblastoma
Primitive Neuroectodermal Tumor Less << |
Phase 2 |
Terminated(The study was termi... More >>nated prematurely due to slow accrual.) Less << |
- |
United States, California
... More >>
Lucile Packard Children's Hospital at Stanford University Medical Center
Palo Alto, California, United States, 94304
United States, Georgia
Winship Cancer Institute of Emory University
Atlanta, Georgia, United States, 30322
United States, Pennsylvania
Children's Hospital of Philadelphia
Philadelphia, Pennsylvania, United States, 19104-4318 Less << |
|
NCT01783197
|
Non Small Cell Lung Cancer |
Phase 1 |
Active, not recruiting |
December 2018 |
Canada, British Columbia
... More >>
BCCA - Vancouver Cancer Centre
Vancouver, British Columbia, Canada, V5Z 4E6
Canada, Ontario
Juravinski Cancer Centre at Hamilton Health Sciences
Hamilton, Ontario, Canada, L8V 5C2
Cancer Centre of Southeastern Ontario at Kingston
Kingston, Ontario, Canada, K7L 5P9
Ottawa Hospital Research Institute
Ottawa, Ontario, Canada, K1H 8L6 Less << |
|
NCT02240771
|
Hepatocellular Carcinoma |
Phase 2
Phase 3 |
Unknown |
March 2015 |
India
... More >> AII India Institute of Medical Sciences
Recruiting
New Delhi, Delhi, India, 110029
Contact: Subrat K Acharya, DM +91-11-26594934 subratacharya2004@yahoo.com
Contact: Shalimar . ., DM +91-9968405815 drshalimar@yahoo.com
Principal Investigator: Subrat K Acharya, DM
Sub-Investigator: Shashi B. Paul, Ph.D
Sub-Investigator: Shivanand R Gamanagatti, M.D
Sub-Investigator: Sreenivas Vishnubhatla, Ph.D Less << |
|
NCT00238160
|
Localized Resectable Adult Pri... More >>mary Liver Cancer
Stage III Childhood Liver Cancer Less << |
Phase 3 |
Withdrawn |
- |
Japan
... More >> Kyoto Prefectural University of Medicine
Kyoto, Japan, 602-8566
Kyoto City Hospital
Kyoto, Japan, 604-8845
Kyoto University Hospital
Kyoto, Japan, 606-8501
Kyoto-Katsura Hospital
Kyoto, Japan, 615-8256 Less << |
|
NCT00186849
|
Neuroblastoma |
Phase 2 |
Completed |
- |
United States, Tennessee
... More >>
St. Jude Children's Research Hospital
Memphis, Tennessee, United States, 38105 Less << |
|
NCT01909856
|
Cancer |
Phase 2 |
Completed |
- |
China, Hubei
... More >> Tongji Hospital,Tongji Medical College, Huazhong University of Science and Technology
Wuhan, Hubei, China, 430030 Less << |
|
NCT00530179
|
Diffuse Large B Cell Lymphoma |
Not Applicable |
Unknown |
- |
Canada, Alberta
... More >>
Tom Baker Cancer Centre
Calgary, Alberta, Canada, T4C 2H5
Canada, Ontario
Ottawa General Hospital
Ottawa, Ontario, Canada, K1H 8L6
Canada, Saskatchewan
Saskatchewan Cancer Agency
Regina, Saskatchewan, Canada, S7N 4H4 Less << |
|
NCT00554463
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, Florida
... More >>
University of Florida Shands Cancer Center
Gainesville, Florida, United States, 32610-0232
CCOP - Mount Sinai Medical Center
Miami Beach, Florida, United States, 33140
United States, Kentucky
Lucille P. Markey Cancer Center at University of Kentucky
Lexington, Kentucky, United States, 40536-0093
United States, Maryland
Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Baltimore, Maryland, United States, 21231-2410
United States, Montana
Northern Rockies Radiation Oncology Center
Billings, Montana, United States, 59101
United States, Nebraska
Methodist Estabrook Cancer Center
Omaha, Nebraska, United States, 68114
United States, Ohio
McDowell Cancer Center at Akron General Medical Center
Akron, Ohio, United States, 44307
Summa Center for Cancer Care at Akron City Hospital
Akron, Ohio, United States, 44309-2090
Charles M. Barrett Cancer Center at University Hospital
Cincinnati, Ohio, United States, 45267
Cleveland Clinic Taussig Cancer Center
Cleveland, Ohio, United States, 44195
Cancer Research UK Medical Oncology Unit at Churchill Hospital & Weatherall Institute of Molecular Medicine - Oxford
Salem, Ohio, United States, 44460
Cancer Treatment Center
Wooster, Ohio, United States, 44691
United States, Pennsylvania
McGlinn Family Regional Cancer Center at Reading Hospital and Medical Center
Reading, Pennsylvania, United States, 19612-6052
United States, Wisconsin
Veterans Affairs Medical Center - Milwaukee
Milwaukee, Wisconsin, United States, 53295 Less << |
|
NCT00554463
|
- |
|
Completed |
- |
- |
|
NCT00006461
|
Untreated Childhood Medullobla... More >>stoma
Untreated Childhood Supratentorial Primitive Neuroectodermal Tumor Less << |
Phase 3 |
Completed |
- |
United States, California
... More >>
Children's Oncology Group
Arcadia, California, United States, 91006-3776 Less << |
|
NCT03169764
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 1
Phase 2 |
Not yet recruiting |
March 2019 |
- |
|
NCT00732498
|
Lymphoma |
Phase 2 |
Active, not recruiting |
December 31, 2019 |
United States, Arizona
... More >>
The University of Arizona Cancer Center
Tucson, Arizona, United States, 85724 Less << |
|
NCT00293358
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 3 |
Completed |
- |
Ireland
... More >> Our Lady's Hospital for Sick Children
Dublin, Ireland, 12
United Kingdom
Birmingham Children's Hospital
Birmingham, England, United Kingdom, B4 6NH
Institute of Child Health at University of Bristol
Bristol, England, United Kingdom, BS2 8AE
Addenbrooke's Hospital at Cambridge University Hospitals NHS Foundation Trust
Cambridge, England, United Kingdom, CB2 2QQ
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Royal Liverpool Children's Hospital, Alder Hey
Liverpool, England, United Kingdom, L12 2AP
Royal London Hospital
London, England, United Kingdom, E1 1BB
Great Ormond Street Hospital for Children NHS Trust
London, England, United Kingdom, WC1N 3JH
Central Manchester and Manchester Children's University Hospitals NHS Trust
Manchester, England, United Kingdom, M27 4HA
Sir James Spence Institute of Child Health
Newcastle-Upon-Tyne, England, United Kingdom, NE1 4LP
Queen's Medical Centre
Nottingham, England, United Kingdom, NG7 2UH
Oxford Radcliffe Hospital
Oxford, England, United Kingdom, 0X3 9DU
Children's Hospital - Sheffield
Sheffield, England, United Kingdom, S10 2TH
Southampton General Hospital
Southampton, England, United Kingdom, SO16 6YD
Royal Marsden NHS Foundation Trust - Surrey
Sutton, England, United Kingdom, SM2 5PT
Royal Belfast Hospital for Sick Children
Belfast, Northern Ireland, United Kingdom, BT12 6BE
Royal Aberdeen Children's Hospital
Aberdeen, Scotland, United Kingdom, AB25 2ZG
Royal Hospital for Sick Children
Edinburgh, Scotland, United Kingdom, EH9 1LF
Royal Hospital for Sick Children
Glasgow, Scotland, United Kingdom, G3 8SJ
Childrens Hospital for Wales
Cardiff, Wales, United Kingdom, CF14 4XW Less << |
|
NCT01032967
|
Esophageal Cancers |
Phase 3 |
Completed |
- |
- |
|
NCT02445209
|
HER2 Negative Gastric Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00645632
|
Sarcoma |
Not Applicable |
Completed |
- |
- |
|
NCT02099188
|
Unresectable Sinonasal Tumors |
Phase 2 |
Active, not recruiting |
January 2024 |
Italy
... More >> Presidio Ospedaliero Spedali Civili di Brescia
Brescia, BS, Italy, 25125
Fondazione IRCCS Istituto Nazionale Tumori
Milano, MI, Italy, 20133
IRCCS Policlinico San Matteo
Pavia, PV, Italy, 27100
A.O. Ospedale di Circolo e Fondazione Macchi
Varese, VA, Italy, 21100
Azienda Ospedaliera "Maggiore della Carità"
Novara, Italy, 28100 Less << |
|
NCT00911222
|
- |
|
Completed |
- |
Germany
... More >> iOMEDICO AG
Freiburg, Baden-Württemberg, Germany, 79108 Less << |
|
NCT02099175
|
Sinonasal Tumors |
Phase 2 |
Active, not recruiting |
January 2024 |
Italy
... More >> Presidio Ospedaliero Spedali Civili di Brescia
Brescia, BS, Italy, 25125
Fondazione IRCCS Istituto Nazionale Tumori
Milano, MI, Italy, 20133
IRCCS Policlinico San Matteo
Pavia, PV, Italy, 27100
A.O. Ospedale di Circolo e Fondazione Macchi
Varese, VA, Italy, 21100
Azienda Ospedaliera "Maggiore della Carità"
Novara, Italy, 28100 Less << |
|
NCT00004188
|
Neuroblastoma |
Phase 3 |
Completed |
- |
- |
|
NCT00278278
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 3 |
Unknown |
- |
United States, Texas
... More >>
M. D. Anderson Cancer Center at University of Texas
Houston, Texas, United States, 77030-4009 Less << |
|
NCT03169738
|
Non-small Cell Lung Cancer |
Phase 1
Phase 2 |
Not yet recruiting |
March 2019 |
- |
|
NCT01111942
|
Head and Neck Cancer |
Early Phase 1 |
Terminated(lack of enrollment) |
- |
United States, Illinois
... More >>
Southern Illinois University School of Medicine
Springfield, Illinois, United States, 62701 Less << |
|
NCT00931645
|
Chronic Lymphocytic Leukemia |
Phase 3 |
Completed |
- |
France
... More >> DBIM Hopital Saint Louis
Paris, France, 75000 Less << |
|
NCT03197571
|
Urothelial Carcinoma |
Phase 1
Phase 2 |
Not yet recruiting |
March 2019 |
- |
|
NCT02710734
|
Urothelial Carcinoma of the Bl... More >>adder Less << |
Phase 2 |
Recruiting |
April 2023 |
United States, Pennsylvania
... More >>
Fox Chase Cancer Center
Recruiting
Philadelphia, Pennsylvania, United States, 19111
Contact: Daniel Geynisman, MD 215-728-4300 daniel.geynisman@fccc.edu
Principal Investigator: Daniel Geynisman, MD Less << |
|
NCT00003937
|
Cardiac Toxicity
... More >> Sarcoma Less << |
Phase 3 |
Completed |
- |
- |
|
NCT01719536
|
NSCLC |
Phase 3 |
Unknown |
December 2016 |
- |
|
NCT01548573
|
- |
|
Terminated(Met study stopping ... More >>rules) Less << |
- |
- |
|
NCT00085098
|
- |
|
Completed |
- |
- |
|
NCT03175666
|
Triple Negative Breast Cancer |
Phase 1
Phase 2 |
Not yet recruiting |
March 2019 |
- |
|
NCT02777788
|
Cancer |
Phase 2
Phase 3 |
Active, not recruiting |
December 2019 |
- |
|
NCT03342352
|
Head and Neck Cancer |
Phase 3 |
Withdrawn(Study was cancelled ... More >>prior to enrolling any patients.) Less << |
- |
- |
|
NCT00085098
|
Brain Tumor
C... More >>entral Nervous System Tumor Less << |
Phase 3 |
Completed |
- |
- |
|
NCT02142738
|
Non-Small Cell Lung Carcinoma |
Phase 3 |
Active, not recruiting |
May 31, 2019 |
- |
|
NCT01849783
|
Extramedullary Plasmacytoma
... More >> Isolated Plasmacytoma of Bone
Light Chain Deposition Disease
Primary Systemic Amyloidosis
Stage I Multiple Myeloma
Stage II Multiple Myeloma
Stage III Multiple Myeloma Less << |
Phase 2 |
Active, not recruiting |
December 2020 |
United States, Iowa
... More >>
University of Iowa Hospitals and Clinics
Iowa City, Iowa, United States, 52242 Less << |
|
NCT01211002
|
Non-small Cell Lung Cancer |
Phase 4 |
Unknown |
December 2012 |
China
... More >> Daping Hospital
Not yet recruiting
Chongqin, China
Contact: zhenzhou yang, M.D. yangzhenzhou@sohu.com
Principal Investigator: zhenzhou yang, M.D. Less << |
|
NCT00514293
|
Lung Cancer |
Phase 2 |
Unknown |
- |
United States, California
... More >>
R. Nandan M.D. Incorporated
Recruiting
Lakewood, California, United States, 90712
Contact: Raghu Nandan, MD 562-272-7630 traghu9@hotmail.com Less << |
|
NCT00705549
|
Non-Small-Cell Lung Cancer |
Phase 2 |
Terminated(Due to poor accrual... More >>) Less << |
- |
Greece
... More >> University General Hospital of Alexandroupolis, Dept. of Medical Oncology
Alexandroupolis, Greece
"Laikon" General Hospital, Medical Oncology Unit, Propedeutic Dep of Internal Medicine
Athens, Greece
401 Military Hospital, Medical Oncology Unit
Athens, Greece
Air Forces Military Hospital, Dep of Medical Oncology
Athens, Greece
IASO" General Hospital of Athens, 1st Dep of Medical Oncology
Athens, Greece
Sismanogleio General Hospital, 1st, 2nd Dep of Pulmonary Diseases
Athens, Greece
Sotiria" General Hospital, 1st, 3rd, 8th Dep of Pulmonary Diseases
Athens, Greece
"Metaxa's" Anticancer Hospital of Piraeus,1st Dep of Medical Oncology
Piraeus, Greece
"Theagenion" Anticancer Hospital of Thessaloniki
Thessaloniki, Greece Less << |
|
NCT02142738
|
- |
|
Active, not recruiting |
- |
- |
|
NCT03086681
|
Cervical Carcinoma |
Phase 3 |
Not yet recruiting |
February 2020 |
China, Guangxi
... More >> First Affiliated Hospital of Guangxi Medical University
Not yet recruiting
Nanning, Guangxi, China, 530021
Contact: Yong Zhang, M.D. 0086-13607884001 zhangyonggx@163.com
Contact: Fang Wu, M.D.
Sub-Investigator: Jian Li, M.D.
Sub-Investigator: Hemin Lu, M.D.
Sub-Investigator: Haixin Huang, Master
Sub-Investigator: Zhanxiong Luo, Master
Sub-Investigator: Meilian Liu, Master
Sub-Investigator: Gaojuan Lin, Master
Sub-Investigator: Sihui Liao, bachelor
Sub-Investigator: Hongqian Wang, bachelor Less << |
|
NCT00416858
|
Esophageal Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT03532737
|
Locally Advanced Head and Neck... More >> Cancer Less << |
Phase 2 |
Recruiting |
October 31, 2022 |
Kuwait
... More >> Kuwait Cancer Control Center
Recruiting
Kuwait, Kuwait
Contact: Mustafa El-Sherify Less << |
|
NCT03498521
|
Cancer of Unknown Primary Site |
Phase 2 |
Recruiting |
April 16, 2022 |
- |
|
NCT03174275
|
Carcinoma, Squamous Cell
... More >> Oral Cancer
Oropharynx Cancer
Larynx Cancer
Lip Cancer
Esophageal Cancer Less << |
Phase 2 |
Recruiting |
September 1, 2026 |
United States, North Carolina
... More >>
Lineberger Comprehensive Cancer Center at University of North Carolina Chapel Hill
Recruiting
Chapel Hill, North Carolina, United States, 27599
Contact: Jared Weiss, MD 919-843-7718 jared_weiss@med.unc.edu
Principal Investigator: Jared Weiss, MD Less << |
|
NCT02124421
|
Stage III Ovarian Cancer
... More >> Stage IV Ovarian Cancer
Epithelial Ovarian Cancer
Fallopian Tube Cancer
Primary Peritoneal Carcinoma
Ovarian Carcinoma
Fallopian Tube Carcinoma Less << |
Phase 2 |
Recruiting |
April 2020 |
United States, Maryland
... More >>
Mercy Medical Center
Recruiting
Baltimore, Maryland, United States, 21202
Sub-Investigator: Teresa Diaz-Montes, M.D., M.P.H
Principal Investigator: Armando Sardi, M.D. Less << |
|
NCT01553942
|
Lung Cancer |
Phase 2 |
Recruiting |
December 2020 |
United States, Massachusetts
... More >>
Dana-Farber Cancer Institute
Recruiting
Boston, Massachusetts, United States, 02215
Contact: Geoffrey Oxnard, MD 617-632-6049 goxnard@partners.org
Principal Investigator: Geoffrey Oxnard, MD
Massachusetts General Hospital
Recruiting
Boston, Massachusetts, United States, 02215
Contact: Lecia V Sequist, MD MPH 617-724-4000 lvsequist@partners.org
Principal Investigator: Lecia V Sequist, MD MPH Less << |
|
NCT03192644
|
Hepatocellular Carcinoma |
Phase 3 |
Recruiting |
December 31, 2024 |
China
... More >> Cancer Center of Sun Yat-Sen University
Recruiting
Guangzhou, China
Contact: Shaohua Li, MD lishaoh@sysucc.org.cn Less << |
|
NCT03656965
|
Drug Use for Unapproved Schedu... More >>le Less << |
Phase 3 |
Active, not recruiting |
May 2021 |
Kuwait
... More >> Kuwait Cancer Control Center
Kuwait, Kuwait Less << |
|
NCT01066234
|
Non-small Cell Lung Cancer |
Phase 2 |
Unknown |
March 2017 |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of Less << |
|
NCT00002492
|
Sarcoma |
Phase 2 |
Completed |
- |
United States, Florida
... More >>
Sylvester Cancer Center, University of Miami
Miami, Florida, United States, 33136 Less << |
|
NCT03323463
|
HPV-Associated Oropharyngeal S... More >>quamous Cell Carcinoma
Squamous Cell Carcinoma of the Neck Less << |
Phase 2 |
Recruiting |
October 2020 |
United States, New Jersey
... More >>
Memoral Sloan Kettering Basking Ridge
Recruiting
Basking Ridge, New Jersey, United States, 07920
Contact: Nancy Lee, MD 212-639-3341
Memoral Sloan Kettering Monmouth
Recruiting
Middletown, New Jersey, United States, 07748
Contact: Nancy Lee, MD 212-639-3341
Memorial Sloan Kettering Bergen
Recruiting
Montvale, New Jersey, United States, 07645
Contact: Nancy Lee, MD 212-639-3341
United States, New York
Memorial Sloan Kettering Commack
Recruiting
Commack, New York, United States, 11725
Contact: Nancy Lee, MD 212-639-3341
Memoral Sloan Kettering Westchester
Recruiting
Harrison, New York, United States, 10604
Contact: Nancy Lee, MD 212-639-3341
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10021
Contact: Nancy Lee, MD 212-639-3341
Memorial Sloan Kettering Rockville Centre
Recruiting
Rockville Centre, New York, United States, 11570
Contact: Nancy Lee, MD 212-639-3341 Less << |
|
NCT00969306
|
Small Cell Lung Cancer |
Phase 1 |
Completed |
- |
Netherlands
... More >> NKI/AvL
Amsterdam, Netherlands
VU Medical Center
Amsterdam, Netherlands
Maastricht Radiation Oncology
Maastricht, Netherlands
Maastricht University Medical Center
Maastricht, Netherlands Less << |
|
NCT01042041
|
Hepatocellular Carcinoma
... More >> Liver Cancer
Localized Unresectable Liver Cancer Less << |
Phase 1 |
Completed |
- |
United States, Pennsylvania
... More >>
Abramson Cancer Center of The University of Pennsylvania
Philadelphia, Pennsylvania, United States, 19104 Less << |
|
NCT02580253
|
Hepatocellular Carcinoma |
Phase 2 |
Not yet recruiting |
December 2018 |
China, Zhejiang
... More >>
First affiliated hospital, Zhejiang University
Hangzhou, Zhejiang, China, 310006 Less << |
|
NCT01733589
|
Stage III Non-small-Cell Lung ... More >>Cancer Less << |
Phase 1
Phase 2 |
Completed |
- |
China, Zhejiang
... More >>
Zhejiang Cancer Hospital
Hangzhou, Zhejiang, China, 310022 Less << |
|
NCT03150628
|
Gastric Cancer
... More >> Peritoneal Carcinomatosis
HIPEC Less << |
Phase 2 |
Suspended(Conflicting recruitm... More >>ent with PERISCOPE II study (NCT03348150)) Less << |
June 1, 2025 |
Netherlands
... More >> University Medical Center Utrecht
Utrecht, Netherlands, 3584 CX Less << |
|
NCT03107182
|
HPV-Related Squamous Cell Carc... More >>inoma
HNSCC Less << |
Phase 2 |
Recruiting |
June 2020 |
United States, Illinois
... More >>
University of Chicago Medical Center
Recruiting
Chicago, Illinois, United States, 60637
Contact: Sara Kochanny 773-702-2336 skochanny@medicine.bsd.uchicago.edu
Principal Investigator: Tanguy Seiwert, MD Less << |
|
NCT00576056
|
Carcinoma, Hepatocellular |
Phase 2 |
Terminated(Bayer Healthcare is... More >> no supplying the study drug) Less << |
- |
United States, Pennsylvania
... More >>
University of Pittsburgh Cancer Institute
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT01549730
|
Cervix Cancer |
Not Applicable |
Active, not recruiting |
May 2019 |
Canada, Ontario
... More >>
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT03004287
|
Multiple Myeloma |
Phase 2 |
Recruiting |
January 2021 |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences
Recruiting
Little Rock, Arkansas, United States, 72205
Contact: Nathan Petty 501-526-6990 ext 2435 pettynathanm@uams.edu Less << |
|
NCT03070366
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 2 |
Recruiting |
October 2021 |
France
... More >> Hôpital Nord Franche Comté
Recruiting
Montbéliard, France, 25209
Contact: Xu Shan SUN, MD (0)3 81 98 88 68 ext +33 xssun@chbm.fr Less << |
|
NCT01434394
|
Locally Advanced Malignant Neo... More >>plasm
Oral Cancer
Oropharyngeal Carcinoma
Effects of Chemotherapy Less << |
Phase 2
Phase 3 |
Completed |
- |
China, Shanghai
... More >>
Ninth People's Hospital, School of Medicine, Shanghai Jiao Tong University
Shanghai, Shanghai, China, 200011 Less << |
|
NCT03700905
|
Head and Neck Cancer |
Phase 3 |
Recruiting |
May 2024 |
Germany
... More >> Universitätsklinikum Ulm
Recruiting
Ulm, Baden-Württemberg, Germany, 89075
Contact: Dr. med. Simon Laban +49(0)731 500-59501 simon.laban@uniklinik-ulm.de
Contact: PD Dr. med. Jens Greve +49(0)731 500-59501 jens.greve@uniklinik-ulm.de
Technische Universität München, Klinikum rechts der Isar
Not yet recruiting
München, Bayern, Germany, 81675
Contact: Dr. med. Folker Scheller +49(0) 089-4140-5647 folker.schneller@tum.de
Contact: PD Dr. med. Andreas Knopf +49(0) 089-41405383 Andreas.Knopf@mri.tum.de
Universitätsklinikum Gießen
Not yet recruiting
Gießen, Hessen, Germany, 35392
Contact: Prof. Dr. med. Claus Wittekind +49 (0) 641-985-43725 claus.wittekindt@hno.med.uni-giessen.de
Contact: Dr. med. Christine Langer +49 (0) 641/985-43732 christine.langer@hno.med.uni-giessen.de
Klinikum Bielefeld
Not yet recruiting
Bielefeld, Nordrhein-Westfalen, Germany, 330604
Contact: PD Dr. med. Martin Görner +49 (0) 521-581-3601 Martin.goerner@klinikumbielefeld.de
Contact: Dr. med. Kai Wegehenkel +49 (0) 521-581-3613 Kai.wegehenkel@klinikumbielefeld.de
HELIOS Klinikum Erfurt GmbH
Not yet recruiting
Erfurt, Thüringen, Germany, 99089
Contact: Dr. med. Kristin Möller +49(0) 361 781-72536 kristin.moeller@helios-kliniken.de
Contact: Dr. med. Stefanie Pratsch +49(0) 361 781-72538 Stefanie.Pratsch@helios-kliniken.de
Universitätsklinikum Hamburg Eppendorf
Recruiting
Hamburg, Germany, 20246
Contact: PD Dr. med. Chia- Jung Busch +49(0)40 7410 50038 cjbusch@uke.de
Contact: Dr. med. Philippe Schafhausen +49(0)40 7410 57122 schafhausen@uke.de
Katholisches Marienkrankenhaus Hamburg
Recruiting
Hamburg, Germany, 22087
Contact: Dr.med. Peter Ebeling +49(0)40 25 46-25 02 ebeling.innere@marienkrankenhaus.org
Contact: Dr. med. Gunnar Hapke +49(0)40 25 46 - 25 31 hapke.innere@marienkrankenhaus.org Less << |
|
NCT00052910
|
- |
|
Completed |
- |
- |
|
NCT00576056
|
- |
|
Terminated(Bayer Healthcare is... More >> no supplying the study drug) Less << |
- |
- |
|
NCT02672254
|
- |
|
Completed |
- |
- |
|
NCT00104676
|
Extragonadal Germ Cell Tumor
... More >> Teratoma
Testicular Germ Cell Tumor Less << |
Phase 3 |
Unknown |
- |
United States, Texas
... More >>
M. D. Anderson Cancer Center at University of Texas
Recruiting
Houston, Texas, United States, 77030-4009
Contact: Clinical Trials Office - M. D. Anderson Cancer Center at the U 713-792-3245
France
Centre Paul Papin
Recruiting
Angers, France, 49100
Contact: Remy Delva 33-49-800-918-507
Institut Bergonie
Recruiting
Bordeaux, France, 33076
Contact: Nguyen Binh Bui, MD 33-5-5633-3813
C.H.U. de Brest
Recruiting
Brest, France, 29609
Contact: Jean-Pierre Malhaire, MD 33-298-223-333 ext. 233-95
Centre Regional Francois Baclesse
Recruiting
Caen, France, 14076
Contact: Emmanuel Sevin, MD 33-2-3145-5000 e.sevin@baclesse.fr
CHU de Grenoble - Hopital de la Tronche
Recruiting
Grenoble, France, 38043
Contact: Francois Ringeisen 33-4-7676-5537
Centre Oscar Lambret
Recruiting
Lille, France, 59020
Contact: Armelle Caty, MD 33-32-029-5959 acaty@o-lambret.fr
Centre Leon Berard
Recruiting
Lyon, France, 69008
Contact: Aude Flechon 33-4-78-78-26-45
Marseille Institute of Cancer - Institut J. Paoli and I. Calmettes
Recruiting
Marseille, France, 13273
Contact: Gwenaelle Gravis, MD 33-4-9122-3740 gravisg@marseille.fnclcc.fr
Hopital Notre-Dame de Bon Secours
Recruiting
Metz, France, 57038
Contact: Christian Platini, MD 33-3-8755-3554 cplatini@chr-metz-thionville.fr
Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Recruiting
Montpellier, France, 34298
Contact: Stephane Culine, MD 33-4-6761-3755 stculine@valdorel.fnclcc.fr
CRLCC Nantes - Atlantique
Recruiting
Nantes-Saint Herblain, France, 44805
Contact: Frederic Rolland, MD 33-2-40-67-99-76 F-rolland@nantes.fnclcc.fr
Centre Antoine Lacassagne
Recruiting
Nice, France, 06189
Contact: Antoine Thyss, MD 33-04-9203-1538 antoine.thyss@cal.nice.fnclcc.fr
Hopital Europeen Georges Pompidou
Recruiting
Paris, France, 75015
Contact: Stephane Oudard, MD, PhD 33-1-56-093-476 stephane.oudard@hop.egp.ap-hop-paris.fr
Hopital Tenon
Recruiting
Paris, France, 75970
Contact: Jean-Pierre Lotz, MD 33-1-5601-6058 jean-pierre.lotz@tnn.ap-hop-paris.fr
Institut Jean Godinot
Recruiting
Reims, France, 51056
Contact: Jean-Christophe Eymard, MD 33-03-2650-4444 jc.eymard@reims.fnclcc.fr
Centre Eugene Marquis
Recruiting
Rennes, France, 35042
Contact: Pierre Kerbrat, MD, PhD 33-299-253-280 kerbrat@rennes.fnclcc.fr
Centre Hospitalier de Rodez
Recruiting
Rodez, France, 12027
Contact: Laurent Mosser 33-05-6575-1212
Centre Henri Becquerel
Recruiting
Rouen, France, 76038
Contact: Paule Chinet-Charrot 33-2-3208-2222
Institut Claudius Regaud
Recruiting
Toulouse, France, 31052
Contact: Christine Chevreau-Dalbianco, MD 33-56-142-4114 chevreau.christine@claudiusregaud.fr
Centre Hospitalier Universitaire Bretonneau de Tours
Recruiting
Tours, France, 37044
Contact: Claude Linassier, MD, PhD 33-2-4747-3827 linassier@med.univ-tours.fr
Centre Alexis Vautrin
Recruiting
Vandoeuvre-les-Nancy, France, 54511
Contact: Lionnel Geoffrois, MD 33-3-8359-8400
Institut Gustave Roussy
Recruiting
Villejuif, France, F-94805
Contact: Karim Fizazi, MD, PhD 33-1-4211-4559 fizazi@igr.fr
Slovakia
National Cancer Institute - Bratislava
Recruiting
Bratislava, Slovakia, 833 10
Contact: Maria Reckova, MD 421-2-5937-8366 maria.reckova@nou.sk Less << |
|
NCT00052910
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT03745222
|
Carcinoma, Non-Small-Cell Lung |
Phase 3 |
Recruiting |
October 26, 2023 |
United States, Maryland
... More >>
Center For Cancer and Blood Disorders
Not yet recruiting
Bethesda, Maryland, United States, 20817
United States, Washington
Medical Oncology Associates, P.S.
Recruiting
Spokane, Washington, United States, 99208 Less << |
|
NCT02568033
|
Non Small Cell Lung Cancer |
Early Phase 1 |
Unknown |
October 2017 |
United States, New Jersey
... More >>
MD Anderson Cancer Center at Cooper
Recruiting
Camden, New Jersey, United States, 08103
Contact: Kim Krieger 856-735-6237 Less << |
|
NCT01672229
|
Graft Versus Host Disease |
Phase 1 |
Active, not recruiting |
December 2019 |
United States, California
... More >>
University of California Comprehensive Cancer Center
Sacramento, California, United States, 95817 Less << |
|
NCT01857934
|
Neuroblastoma |
Phase 2 |
Recruiting |
July 2021 |
United States, Tennessee
... More >>
St. Jude Children's Research Hospital
Recruiting
Memphis, Tennessee, United States, 38105
Contact: Wayne L. Furman, MD 866-278-5833 referralinfo@stjude.org
Principal Investigator: Wayne L. Furman, MD Less << |
|
NCT03387111
|
Squamous Cell Carcinoma |
Phase 1
Phase 2 |
Recruiting |
December 2019 |
United States, California
... More >>
Chan Soon-Shiong Institute for Medicine
Recruiting
El Segundo, California, United States, 90245
Contact: NantKWest Clinical Review Team 800-988-6083 clinical.trials@NantKwest.com Less << |
|
NCT00003573
|
Brain Tumors
... More >>Central Nervous System Tumors Less << |
Phase 2 |
Completed |
- |
- |
|
NCT03387085
|
Triple Negative Breast Cancer |
Phase 1
Phase 2 |
Recruiting |
January 2020 |
United States, California
... More >>
Chan Soon-Shiong Institute for Medicine
Recruiting
El Segundo, California, United States, 90245 Less << |
|
NCT02041533
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00867178
|
Medulloblastoma
... More >> Pineoblastoma
Supratentorial Embryonal Tumor, Not Otherwise Specified
Untreated Childhood Medulloblastoma
Untreated Childhood Pineoblastoma
Untreated Childhood Supratentorial Primitive Neuroectodermal Tumor Less << |
Not Applicable |
Active, not recruiting |
- |
United States, California
... More >>
Children's Hospital Los Angeles
Los Angeles, California, United States, 90027
Lucile Packard Children's Hospital Stanford University
Palo Alto, California, United States, 94304
United States, District of Columbia
Children's National Medical Center
Washington, District of Columbia, United States, 20010
United States, Illinois
Lurie Children's Hospital-Chicago
Chicago, Illinois, United States, 60611
United States, Maryland
National Cancer Institute Pediatric Oncology Branch
Bethesda, Maryland, United States, 20892
United States, New York
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
United States, North Carolina
Duke University Medical Center
Durham, North Carolina, United States, 27710
United States, Ohio
Cincinnati Children's Hospital Medical Center
Cincinnati, Ohio, United States, 45229
United States, Pennsylvania
Children's Hospital of Philadelphia
Philadelphia, Pennsylvania, United States, 19104
Children's Hospital of Pittsburgh of UPMC
Pittsburgh, Pennsylvania, United States, 15224
United States, Tennessee
Pediatric Brain Tumor Consortium
Memphis, Tennessee, United States, 38105
St. Jude Children's Research Hospital
Memphis, Tennessee, United States, 38105
United States, Texas
M D Anderson Cancer Center
Houston, Texas, United States, 77030
Texas Children's Hospital
Houston, Texas, United States, 77030
United States, Washington
Seattle Children's Hospital
Seattle, Washington, United States, 98105 Less << |
|
NCT03275194
|
HPEC
Ovarian ... More >>Cancer
Women's Health: Neoplasm of Ovary
Chemotherapy Effect Less << |
Phase 2 |
Recruiting |
December 1, 2025 |
Mexico
... More >> National Cancer Institute of Mexico
Recruiting
Mexico City, Mexico, 14080
Contact: Rosa A Salcedo-Hernández, M.Sc 525534265921 rosasalher@gmail.com
Contact: Leonardo S Lino-Silva 525534265921 saul.lino.sil@gmail.com Less << |
|
NCT02054819
|
Non-small Cell Lung Cancer |
Phase 2 |
Terminated(Didn't meet accrual... More >> goal, change in standard of care.) Less << |
- |
United States, North Carolina
... More >>
Leo W. Jenkins Cancer Center
Greenville, North Carolina, United States, 27834 Less << |
|
NCT01858922
|
Hodgkin Disease |
Phase 2 |
Active, not recruiting |
March 2021 |
United States, Texas
... More >>
Texas Children's Hospital
Houston, Texas, United States, 77030 Less << |
|
NCT02393248
|
Lung Cancer
S... More >>olid Tumor
Gastric Cancer
UC (Urothelial Cancer)
Endometrial Cancer
Multiple Myeloma
MPN (Myeloproliferative Neoplasms)
Breast Cancer
Cholangiocarcinoma Less << |
Phase 1
Phase 2 |
Recruiting |
January 2020 |
United States, Alabama
... More >>
University of Alabama
Recruiting
Tuscaloosa, Alabama, United States, 35205
United States, California
Cedars Sinai
Terminated
Los Angeles, California, United States, 90048
United States, Connecticut
Yale Cancer Center
Terminated
New Haven, Connecticut, United States, 06520
United States, District of Columbia
Georgetown University - Lombardi Comprehensive Cancer Center
Terminated
Washington, District of Columbia, United States, 20007
United States, Florida
Hematology - Oncology Associates of Treasure Coast
Recruiting
Port Saint Lucie, Florida, United States, 34952
United States, Michigan
University of Michigan Health System
Recruiting
Ann Arbor, Michigan, United States, 48109
United States, Missouri
Washington University
Recruiting
Saint Louis, Missouri, United States, 63110
United States, New Jersey
Hackensack University Medical Center
Recruiting
Hackensack, New Jersey, United States, 07601
United States, North Carolina
Duke Cancer Institute
Recruiting
Durham, North Carolina, United States, 27710
United States, Ohio
Ohio State University Comprehensive Cancer Center
Recruiting
Columbus, Ohio, United States, 43210
Signal Point Clinical Research Center
Terminated
Middletown, Ohio, United States, 45042
United States, South Carolina
Greenville Health System
Recruiting
Greenville, South Carolina, United States, 29605
United States, Texas
Mary Crowley Cancer Research Center
Recruiting
Dallas, Texas, United States, 75230
MD Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
South Texas Accelerated Research Therapeutics, LLC
Recruiting
San Antonio, Texas, United States, 78229
Denmark
Onkologisk Klinik Rigshospitalet
Recruiting
Copenhagen, Denmark, 2100 Less << |
|
NCT02041533
|
Stage IV or Recurrent Non-Smal... More >>l Cell Lung Cancer Less << |
Phase 3 |
Active, not recruiting |
October 1, 2018 |
- |
|
NCT02448797
|
Non-small Cell Lung Cancer |
Phase 3 |
Recruiting |
December 2022 |
China, Beijing
... More >> 307 Hospital of PLA
Recruiting
Beijing, Beijing, China, 100071
Principal Investigator: Xiao-qing Liu, M.D.
China, Fujian
Fujian Provincal Cancer Hospital
Recruiting
Fuzhou, Fujian, China, 350014
Contact: Huang Cheng, M.D. 0086-13905010397
Principal Investigator: Huang Cheng, M.D.
China, Guangdong
The First Affiliated Hospital Of Guangzhou Medical Collage
Recruiting
Guangzhou, Guangdong, China, 510120
Contact: He Jianxing 0086-13802777270
Principal Investigator: He Jianxing, M.D.
Shenzhen People's Hospital
Recruiting
Shenzhen, Guangdong, China, 518020
Contact: Zheng Wang, MD 13502886638
Principal Investigator: Zheng Wang, MD
China, Liaoning
The First Hospital of China Medical University
Recruiting
Shenyang, Liaoning, China, 110001
Contact: Liu Yunpeng, M.D. 0086-13898865122
Principal Investigator: Liu Yunpeng, M.D.
First Affiliated Hospital of China Medical University
Active, not recruiting
Shenyang, Liaoning, China, 150081
China, Shanghai
Shanghai Pulmonary Hospital
Recruiting
Shanghai, Shanghai, China, 200433
Principal Investigator: Cai-Cun Zhou
China, Zhejiang
The First Affiliated Hospital of Medical School of Zhejiang University
Recruiting
Hangzhou, Zhejiang, China, 310000
Contact: Zhou Jianying, M.D. 0086-13750881678 drzjy@163.com
Principal Investigator: Zhou Jianying, M.D. Less << |
|
NCT00003203
|
Brain Tumors
... More >>Central Nervous System Tumors
Neuroblastoma Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00025298
|
Drug/Agent Toxicity by Tissue/... More >>Organ
Head and Neck Cancer
Oral Complications
Radiation Toxicity Less << |
Phase 2 |
Terminated(low accrual) |
- |
Belgium
... More >> Institut Jules Bordet
Brussels, Belgium, 1000
Universitair Ziekenhuis Antwerpen
Edegem, Belgium, B-2650
U.Z. Gasthuisberg
Leuven, Belgium, B-3000
France
CRLCC Nantes - Atlantique
Nantes-Saint Herblain, France, 44805
Centre Antoine Lacassagne
Nice, France, 06189
Italy
Ospedale Santa Croce
Cuneo, Italy, 12100
Istituto Nazionale per la Ricerca sul Cancro
Genoa (Genova), Italy, 16132
Istituto Nazionale per lo Studio e la Cura dei Tumori
Milano (Milan), Italy, 20133
Istituto Nazionale per lo Studio e la Cura dei Tumori
Naples, Italy, 80131
Spain
Hospital General de Jerez
Jerez, Spain, 11407
Hospital Universitario 12 de Octubre
Madrid, Spain, 28041
Turkey
Istanbul University-Institute of Oncology
Istanbul, Turkey, 34390
United Kingdom
Beatson Oncology Centre
Glasgow, Scotland, United Kingdom, G11 6NT Less << |
|
NCT02279134
|
Esophageal Neoplasms
... More >> Esophageal Cancer Less << |
Phase 2
Phase 3 |
Recruiting |
October 2022 |
China, Beijing
... More >> Cancer Institute and Hospital, Chinese Academy of Medical Science
Recruiting
Beijing, Beijing, China, 100021
Contact: Zefen Xiao, MD 8610-87787463 xiaozefen@sina.com
Contact: Shufei Yu, MD 86-15911070901 sophie314@yeah.net Less << |
|
NCT00209222
|
Lymphoma, Mantle-Cell |
Phase 3 |
Unknown |
December 2014 |
France
... More >> Groupe D´Etudes des Lymphomes De l´Adulte (GELA)
Recruiting
Paris, France, F-75743
Contact: Guylène Chartier +33-1-42499811 Guylene.chartier@chu-stlouis.fr
Contact: Olivier Hermine, PhD +33-1-44 49 52 83 hermine@necker.fr
Principal Investigator: Olivier Hermine, PhD
Germany
German Low Grade Study Group (Glsg)
Recruiting
Munich, Germany, D-81377
Contact: Michael Unterhalt, Dr. +49-89-7095 ext 4915 Michael.Unterhalt@med.uni-muenchen.de
Contact: Martin Dreyling, PhD +49-89-7095 ext 2202 Martin.Dreyling@med.uni-muenchen.de
Principal Investigator: Wolfgang Hiddemann, PhD
Poland
The Maria Sklodowska Memorial, Cancer Center - Inst. of Oncology
Recruiting
Warszawa, Poland, PL-02-781
Contact: Jan Walewski, MD +48-22-546-2223 walewski@coi.waw.pl
Contact: Marek P Nowacki, MD +48-22-546-2223
Principal Investigator: Jan Walewski, MD Less << |
|
NCT00354393
|
Malignant Mesothelioma |
Phase 2 |
Unknown |
- |
United States, Ohio
... More >>
Case Comprehensive Cancer Center
Cleveland, Ohio, United States, 44106-5065 Less << |
|
NCT00053352
|
Childhood Embryonal Tumor
... More >> Childhood Extracranial Germ Cell Tumor
Childhood Extragonadal Germ Cell Tumor
Childhood Malignant Ovarian Germ Cell Tumor
Childhood Malignant Testicular Germ Cell Tumor
Childhood Teratoma
Ovarian Embryonal Carcinoma
Ovarian Yolk Sac Tumor
Stage II Malignant Testicular Germ Cell Tumor
Stage IIA Ovarian Germ Cell Tumor
Stage IIB Ovarian Germ Cell Tumor
Stage IIC Ovarian Germ Cell Tumor
Stage III Malignant Testicular Germ Cell Tumor
Stage IIIA Ovarian Germ Cell Tumor
Stage IIIB Ovarian Germ Cell Tumor
Stage IIIC Ovarian Germ Cell Tumor
Testicular Choriocarcinoma and Yolk Sac Tumor
Testicular Embryonal Carcinoma Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00053352
|
- |
|
Completed |
- |
- |
|
NCT01261299
|
Carcinoma, Non-Small-Cell Lung... More >>
Urinary Bladder Neoplasms Less << |
Early Phase 1 |
Terminated(Contract ended) |
- |
United States, California
... More >>
University of California Los Angeles
Los Angeles, California, United States, 90095
University of California, Davis
Sacramento, California, United States, 95817 Less << |
|
NCT01822496
|
Stage III Non-Small Cell Lung ... More >>Cancer AJCC v7
Stage IIIA Non-Small Cell Lung Cancer AJCC v7
Stage IIIB Non-Small Cell Lung Cancer AJCC v7 Less << |
Phase 2 |
Terminated |
- |
- |
|
NCT03088540
|
Carcinoma,Non-Small-Cell Lung
... More >>
Lung Carcinomas, Non-Small-Cell
Non-small-cell Lung Carcinoma
Nonsmall Cell Lung Cancer Less << |
Phase 3 |
Recruiting |
September 20, 2022 |
- |
|
NCT00663741
|
Gastric Cancer |
Phase 1 |
Completed |
- |
Japan
... More >>
Kashiwa, Chiba, Japan, 277-8577
Kobe, Hyogo, Japan, 650-0017
Chuo-ku, Tokyo, Japan, 104-0045 Less << |
|
NCT03043872
|
Small Cell Lung Carcinoma Exte... More >>nsive Disease Less << |
Phase 3 |
Recruiting |
September 30, 2020 |
- |
|
NCT00057785
|
Head and Neck Cancer
... More >> Oral Complications of Radiation Therapy
Radiation Toxicity Less << |
Phase 2 |
Completed |
- |
United States, Alabama
... More >>
Comprehensive Cancer Center at University of Alabama at Birmingham
Birmingham, Alabama, United States, 35294
United States, California
University of California Davis Cancer Center
Davis, California, United States, 95616
Radiological Associates of Sacramento Medical Group, Incorporated
Sacramento, California, United States, 95815
UCSF Comprehensive Cancer Center
San Francisco, California, United States, 94115
United States, Georgia
Northeast Georgia Medical Center
Gainesville, Georgia, United States, 30501
United States, Minnesota
Mayo Clinic Cancer Center
Rochester, Minnesota, United States, 55905
United States, Missouri
Siteman Cancer Center at Barnes-Jewish Hospital
St Louis, Missouri, United States, 63110
United States, New Jersey
Monmouth Medical Center
Long Branch, New Jersey, United States, 07740
United States, New Mexico
Albuquerque Regional Medical Center at Lovelace Sandia Health System
Albuquerque, New Mexico, United States, 87102
United States, Ohio
Akron City Hospital
Akron, Ohio, United States, 44304
United States, Pennsylvania
Kimmel Cancer Center at Thomas Jefferson University - Philadelphia
Philadelphia, Pennsylvania, United States, 19107
Fox Chase-Temple Cancer Center
Philadelphia, Pennsylvania, United States, 19111-2497
CCOP - MainLine Health
Wynnewood, Pennsylvania, United States, 19096
United States, Texas
M.D. Anderson Cancer Center at University of Texas
Houston, Texas, United States, 77030
Wilford Hall Medical Center
Lackland AFB, Texas, United States, 78236
United States, Utah
McKay-Dee Hospital Center
Ogden, Utah, United States, 84403
United States, Wisconsin
Medical College of Wisconsin Cancer Center
Milwaukee, Wisconsin, United States, 53226 Less << |
|
NCT03622827
|
Uterine Cervical Neoplasms |
Phase 2 |
Recruiting |
July 30, 2025 |
China, Jiangsu
... More >> The Affiliated Suzhou Hospital of Nanjing Medical University
Recruiting
Suzhou, Jiangsu, China, 215001
Contact: Ke Gu, M.D., Ph.D 86-13405070898 dr.guke@hotmail.com
Contact: Zhiliang Ding, M.D. 86-18913535515 Less << |
|
NCT00193791
|
Cancer of Cervix |
Phase 3 |
Active, not recruiting |
May 2018 |
India
... More >> Tata Memorial Hospital
Mumbai, Maharastra, India, 400 012 Less << |
|
NCT00057785
|
- |
|
Completed |
- |
- |
|
NCT00152828
|
Cervix Neoplasms |
Phase 1
Phase 2 |
Completed |
- |
Canada, Ontario
... More >>
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9 Less << |
|
NCT00094497
|
Carcinoma, Adrenal Cortical |
Phase 3 |
Completed |
- |
United States, Maryland
... More >>
National Cancer Institute - Center for Cancer Research
Bethesda, Maryland, United States
United States, Michigan
University of Michigan, Department of Internal Medicine
Ann Arbor, Michigan, United States, 48109
Australia
Royal Adelaide Hospital
Adelaide, Australia, SA 5000
Austria
University of Graz
Graz, Austria, 8036
France
Clinique Marc Linquette
Lille, France
Centre Leon Berard
Lyon, France
Hospital de Marseille la timone
Marseille, France, 13385
Cochin Hospital
Paris, France, 75679
Hospital Bordeaux haut leveque
Pessac, France, 33600
Institut Gustave Roussy
Villejuif, France, 94805
Germany
Charité-University, Dept. of Endocrinology; Campus Benjamin Franklin
Berlin, Germany
Charité-Universitätsmedizin Berlin - Campus Mitte
Berlin, Germany
Dept. of Medicine III
Dresden, Germany
University of Duesseldorf, Dept. of Endocrrinology
Duesseldorf, Germany, 40001
Zentrum für Innere Medizin - Endokrinologie des Universitätsklinikum Essen
Essen, Germany
Endokrinologie Medizinische Hochschule Hannover
Hannover, Germany
Otto-von-Guericke University; Dept. of Endocrinology
Magdeburg, Germany, 39120
Dept of Medicine I
Mainz, Germany
University of Munich, Dept. of Internal Medicine (Innenstadt)
Munich, Germany, 80336
University of Wuerzburg - Dept. of Medicine
Wuerzburg, Germany, 97080
Italy
University of Turin, Dept of Internal Medicine
Orbassano, Italy, 10043
Clinica Endocrinologica, Università di Padova, Azienda Ospedaliera di Padova
Padova, Italy
Netherlands
Vrije Universiteit Medisch Centrum
Amsterdam, Netherlands, 1007
Academisch Medisch Centrum; Dept. of Endocrinology
Amsterdam, Netherlands, 1105 AZ
Maxima Medisch Centrum; Dept. of Internal Medicine
Eindhoven, Netherlands, 5631 BM
University Hospital Groningen; Dept. of Internal Medine
Groningen, Netherlands, 9700
Leiden University Medical Center
Leiden, Netherlands
Sweden
Department of Oncology, Sahlgrenska University Hospital
Gothenburg, Sweden
Department of Oncology, Linköping University Hospital
Linköping, Sweden
Department of Medicine, The Jubileum Institute, Lund University
Lund, Sweden
Dept of Surgery, Karolinska Hospital, Stockholm
Stockholm, Sweden
Uppsala University Hospital - Dept of Medical Sciences
Uppsala, Sweden, 751 85 Less << |
|
NCT01052844
|
Vomiting
Cisp... More >>latin Adverse Reaction Less << |
Phase 3 |
Completed |
- |
Brazil
... More >> Faculdade de Medicina do ABC
Santo André, São Paulo, Brazil, 09060-650 Less << |
|
NCT00094497
|
- |
|
Completed |
- |
- |
|
NCT03723343
|
- |
|
Not yet recruiting |
June 30, 2021 |
China, Guizhou
... More >> Cancer Hospital of Guizhou Medical University
Recruiting
Guiyang, Guizhou, China, 550000
Contact: Jinhua Long, Master 085186512802 longjinhua100@sina.cn
Principal Investigator: Feng Jin
Principal Investigator: Weili Wu
Principal Investigator: Yuanyuan Li
Principal Investigator: hong Tang
Principal Investigator: Xiuling Luo
Principal Investigator: Xiuyun Gong
Principal Investigator: Xiaoxiao Chen Less << |
|
NCT01052844
|
- |
|
Completed |
- |
- |
|
NCT00951496
|
Malignant Ovarian Mixed Epithe... More >>lial Tumor
Ovarian Brenner Tumor
Ovarian Clear Cell Cystadenocarcinoma
Ovarian Endometrioid Adenocarcinoma
Ovarian Mucinous Cystadenocarcinoma
Ovarian Serous Cystadenocarcinoma
Stage IIA Fallopian Tube Cancer
Stage IIA Ovarian Cancer
Stage IIB Fallopian Tube Cancer
Stage IIB Ovarian Cancer
Stage IIC Fallopian Tube Cancer
Stage IIC Ovarian Cancer
Stage IIIA Fallopian Tube Cancer
Stage IIIA Ovarian Cancer
Stage IIIA Primary Peritoneal Cancer
Stage IIIB Fallopian Tube Cancer
Stage IIIB Ovarian Cancer
Stage IIIB Primary Peritoneal Cancer
Stage IIIC Fallopian Tube Cancer
Stage IIIC Ovarian Cancer
Stage IIIC Primary Peritoneal Cancer
Undifferentiated Ovarian Carcinoma Less << |
Phase 3 |
Active, not recruiting |
March 15, 2022 |
- |
|
NCT00084838
|
Central Nervous System Tumor, ... More >>Pediatric Less << |
Phase 2 |
Completed |
- |
United States, California
... More >>
Stanford Cancer Center
Stanford, California, United States, 94305-5826
United States, Connecticut
Yale Cancer Center
New Haven, Connecticut, United States, 06520-8028
United States, Georgia
AFLAC Cancer Center and Blood Disorders Service of Children's Healthcare of Atlanta - Scottish Rite Campus
Atlanta, Georgia, United States, 30342
United States, Illinois
Children's Memorial Hospital - Chicago
Chicago, Illinois, United States, 60614
United States, Maryland
Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Baltimore, Maryland, United States, 21231-2410
United States, Massachusetts
Children's Hospital Boston
Boston, Massachusetts, United States, 02115
Dana-Farber/Harvard Cancer Center at Dana Farber Cancer Institute
Boston, Massachusetts, United States, 02115
United States, Minnesota
Children's Hospitals and Clinics of Minnesota - Minneapolis
Minneapolis, Minnesota, United States, 55404
United States, Nevada
Sunrise Hospital and Medical Center
Las Vegas, Nevada, United States, 89109
United States, Ohio
Cleveland Clinic Taussig Cancer Center
Cleveland, Ohio, United States, 44195
United States, Pennsylvania
Children's Hospital of Philadelphia
Philadelphia, Pennsylvania, United States, 19104
United States, Texas
Simmons Comprehensive Cancer Center at University of Texas Southwestern Medical Center - Dallas
Dallas, Texas, United States, 75390 Less << |
|
NCT00167401
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Completed |
- |
United States, New York
... More >>
University of Rochester, Dept. Radiation Oncology
Rochester, New York, United States, 14642 Less << |
|
NCT03206684
|
Cervical Cancer |
Phase 4 |
Not yet recruiting |
September 2018 |
China, Shaanxi
... More >> Department of Radiation Oncology, Xijing Hospital, Fourth Military Medical University
Not yet recruiting
Xi'an, Shaanxi, China, 710032
Contact: Mei Shi, MD +86-029-84775425 Shimei82@gmail.com Less << |
|
NCT03628716
|
Bladder Cancer |
Phase 2 |
Recruiting |
November 2023 |
United States, Massachusetts
... More >>
Massachusetts General Hospital
Not yet recruiting
Boston, Massachusetts, United States, 02114
Contact: Michelle Wright MWright10@mgh.harvard.edu
Sub-Investigator: Richard Lee, MD
Principal Investigator: Guru Sonpavde, MD
Dana Farber Cancer Institute (DFCI)
Not yet recruiting
Boston, Massachusetts, United States, 02215
Contact: Alyssa Klein Alyssa_Klein@DFCI.Harvard.edu
Principal Investigator: Guru Sonpavde, MD
United States, Utah
University of Utah - Huntsman Cancer Institute
Recruiting
Salt Lake City, Utah, United States, 84112
Contact: Sally Fairbairn sally.fairbairn@hci.utah.edu
Principal Investigator: Benjamin Maughan, MD Less << |
|
NCT00951496
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00401401
|
Head and Neck Cancer
... More >> Squamous Cell Cancer Less << |
Phase 1
Phase 2 |
Terminated(MTD was established... More >> and patients completed 16 months safety f-up and response assessments. It is considered of limited value to follow patients for 3 years.) Less << |
- |
United States, Oregon
... More >>
Oregon Health Sciences Center
Portland, Oregon, United States, 97239
Belgium
St-Luc University Hospital
Brussels, Belgium
University Hospital Gasthuisberg
Leuven, Belgium
France
Centre Georges-Francois Leclerc Hospital
Dijon, France
Hopital Bretonneau Clinique d'Oncologie et Radiothérapie
Tours, France
Netherlands
Nijmegen University Hospital
Nijmegen, Netherlands
Sweden
Lund University Hospital
Lund, Sweden Less << |
|
NCT00401401
|
- |
|
Terminated(MTD was established... More >> and patients completed 16 months safety f-up and response assessments. It is considered of limited value to follow patients for 3 years.) Less << |
- |
- |
|
NCT02972216
|
- |
|
Completed |
- |
- |
|
NCT01752517
|
- |
|
Unknown |
December 2015 |
China, Beijing
... More >> Department of Respiratory Medicine, Peking Union Medical College Hospital
Recruiting
Beijing, Beijing, China, 100730
Contact: Mengzhao Wang, MD 010-69155039 ext +86 mengzhaowang@sina.com
Contact: Jing Zhao, MD 010-69158206 ext +86 pumchzj@sina.com
Sub-Investigator: Wei Zhong, MD
Sub-Investigator: Jinmei Luo, MD Less << |
|
NCT00003637
|
Head and Neck Cancer |
Phase 3 |
Unknown |
- |
Singapore
... More >> National Cancer Centre - Singapore
Singapore, Singapore, 169610 Less << |
|
NCT00004141
|
Melanoma (Skin) |
Phase 2 |
Completed |
- |
United States, Illinois
... More >>
University of Chicago Cancer Research Center
Chicago, Illinois, United States, 60637-1470 Less << |
|
NCT01798004
|
Disseminated Neuroblastoma
... More >> Localized Resectable Neuroblastoma
Localized Unresectable Neuroblastoma
Regional Neuroblastoma
Stage 4S Neuroblastoma Less << |
Not Applicable |
Suspended(CRM 31684 - CTEP PIO... More >> requested) Less << |
October 2018 |
- |
|
NCT00030745
|
Malignant Mesothelioma |
Phase 2 |
Completed |
- |
Switzerland
... More >> UniversitaetsSpital
Zurich, Switzerland, CH-8091 Less << |
|
NCT01798004
|
- |
|
Suspended(CRM 31684 - CTEP PIO... More >> requested) Less << |
- |
- |
|
NCT00005022
|
Lung Cancer |
Phase 1 |
Completed |
- |
- |
|
NCT02662634
|
Non-small Cell Lung Cancer (NS... More >>CLC) Less << |
Phase 2 |
Unknown |
March 2018 |
United States, Nebraska
... More >>
Cancer Research Network of Nebraska / Oncology Associates
Recruiting
Omaha, Nebraska, United States, 68118
Contact: Tony Romero, MS, CCRC 402-697-2229 tromero@gucancer.com
Principal Investigator: Stephen Lemon, MD Less << |
|
NCT00006051
|
Head and Neck Cancer |
Phase 2 |
Unknown |
- |
France
... More >> Centre Jean Perrin
Clermont-Ferrand, France, 63011
Hopital Jean Monnet
Epinal, France, 88021
CRLCC Nantes - Atlantique
Nantes-Saint Herblain, France, 44805
Centre Hospitalier Universitaire Bretonneau de Tours
Tours, France, 37044
Centre Alexis Vautrin
Vandoeuvre-les-Nancy, France, 54511
Institut Gustave Roussy
Villejuif, France, F-94805 Less << |
|
NCT03358004
|
Triple Negative Breast Cancer |
Phase 2 |
Terminated(Low accrual rate) |
- |
Italy
... More >> AOU Ospedali riuniti di Ancona
Torrette, Ancona, Italy, 60126
ASST Monza
Monza, MB, Italy, 20052
Azienda Ospedaliero-Universitaria Pisana
Pisa, PI, Italy, 56126
Azienda Ospedaliero Universitaria di Parma
Parma, PR, Italy, 43126
Ospedale Civile di Guastalla
Reggio Emilia, RE, Italy, 42016
Ospedale Martini ASL Torino 1
Torino, TO, Italy, 10141
Istituto Tumori Giovanni Paolo II
Bari, Italy, 70124
ASST Papa Giovanni XXIII
Bergamo, Italy, 24127
A. Ospedaliero universitaria di Bologna
Bologna, Italy, 40138
Azienda Sanitaria Locale Brindisi
Brindisi, Italy, 72100
ASST - Cremona
Cremona, Italy, 26100
A.O. San Croce e Carle
Cuneo, Italy, 12100
Ospedale Vito Fazzi
Lecce, Italy, 73100
Istituto Europeo di Oncologia
Milano, Italy, 20141
Policlinico di Modena
Modena, Italy, 41124
Policlinico Paolo Giaccone
Palermo, Italy, 90127
Casa di Cura La Maddalena
Palermo, Italy, 90146
Ospedale Felice Lotti
Pontedera, Italy, 56025
Istituto Nazionale Regina Elena
Roma, Italy, 0144
Azienda Ospedaliero Universitaria di Sassari
Sassari, Italy, 07100
Portugal
Instituto Portugues Oncologia de Coimbra
Coimbra, Portugal, 3000-075
CHLN Hospital Santa Maria
Lisboa, Portugal, 1349-035
Hospital de S. Francisco Xavier
Lisboa, Portugal, 1449-005
Hospital Beatriz Angelo
Loures, Portugal, 2674-514
Centro Hospitalar Do Porto
Porto, Portugal, 4099-001
Centro Hospitalar De Sao Joao EPE
Porto, Portugal, 4200
Instituto Portugues Oncologia de Porto
Porto, Portugal, 4200
Spain
Hospital Vall d'Hebron
Barcelona, Spain, 08035
Hospital Virgen de la Salud
Toledo, Spain, 45071
Hospital Clinico Universitario Lozano Blesa
Zaragoza, Spain, 50009 Less << |
|
NCT00324805
|
Stage IB Non-Small Cell Lung C... More >>arcinoma
Stage IIA Non-Small Cell Lung Carcinoma
Stage IIB Non-Small Cell Lung Carcinoma
Stage IIIA Non-small Cell Lung Cancer Less << |
Phase 3 |
Active, not recruiting |
October 16, 2023 |
- |
|
NCT02204306
|
Metastatic Gastric Cancer |
Phase 2 |
Unknown |
December 2016 |
China, Hunan
... More >> Institute of Clinical Pharmacology
Recruiting
Changsha, Hunan, China, 410005
Contact: Yijing He, MD, PhD +86-15874812612 yijinghe@gmail.com
Principal Investigator: Zihua Chen, MD
Principal Investigator: Meizuo Zhong, MD
Principal Investigator: Jing Huang, MD, PhD
Principal Investigator: Xianli Ying, MD
Principal Investigator: Fang Xiang, MD, PhD
Principal Investigator: Keli Yan, MD
Principal Investigator: Weisheng Ye, MD
Principal Investigator: Xianrang Song, MD Less << |
|
NCT00003589
|
Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT03188497
|
Acute Toxic Effects
... More >> Tumor Responses Less << |
Phase 1 |
Completed |
- |
China, Guangdong
... More >>
the Fifth Hospital Affiliated to Sun Yat-Sen University
Zhuhai, Guangdong, China, 519000 Less << |
|
NCT00563927
|
Nasopharyngeal Neoplasms
... More >> Carcinoma, Squamous Cell Less << |
Not Applicable |
Unknown |
October 2009 |
China
... More >> Pamela Youde Nethersole Eastern Hospital
Hong Kong, China
Queen Elizabeth Hospital
Hong Kong, China
Queen Mary Hospital
Hong Kong, China
Tuen Mun Hospital
Hong Kong, China Less << |
|
NCT00324805
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00002897
|
Esophageal Cancer |
Phase 3 |
Completed |
- |
Italy
... More >> University of Padua
Padua, Italy, 35128 Less << |
|
NCT01887340
|
Seminoma |
Phase 2 |
Recruiting |
June 2026 |
France
... More >> Gustave Roussy
Recruiting
Villejuif, Val de Marne, France, 94805
Contact: Yohann LORIOT, MD 0142115276 ext +33 yohann.loriot@gustaveroussy.fr Less << |
|
NCT00002999
|
Head and Neck Cancer |
Phase 2 |
Withdrawn |
- |
- |
|
NCT00004209
|
Lung Cancer |
Phase 3 |
Unknown |
- |
United Kingdom
... More >> Royal Marsden NHS Trust
London, England, United Kingdom, SW3 6JJ
Aberdeen Royal Infirmary
Aberdeen, Scotland, United Kingdom, AB25 2ZN Less << |
|
NCT01782339
|
Germ Cell Tumour |
Phase 2 |
Recruiting |
January 2021 |
United Kingdom
... More >> St Bartholomew's Hospital
Recruiting
London, United Kingdom, EC1A 7BE
Contact: GAMMA coordinator
Principal Investigator: Jonathan Shamash, MD FRCP Less << |
|
NCT00256295
|
Cancer of the Head and Neck |
Phase 2 |
Terminated(Low accrual) |
- |
United States, California
... More >>
Chao Family Comprehensive Cancer Center
Orange, California, United States, 92868 Less << |
|
NCT01726452
|
Adenocarcinoma of the Oesophag... More >>us
Adenocarcinoma of the Oesophago-gastric Junction
Oesophageal Tumours
Junctional Tumours
Oesophageal Cancer Less << |
Phase 3 |
Recruiting |
- |
Denmark
... More >> Rigshospitalet
Recruiting
Blegdamsvej 9, Denmark, 2100 København Ø
Contact: Contact Person +45 35 45 35 45
Principal Investigator: Dr Lene Baeksgaard
France
Centre Hospitalier Régional, Universitaire de Lille 2 Avenue Oscar Lambret, 59000
Not yet recruiting
Lille, France
Contact: Contact Person +33 3 20 44 41 45
Principal Investigator: Prof Guillaume Piessen
Ireland
Belfast Health and Social Care Trust, Northern Ireland Cancer Centre, Belfast CityHospital
Recruiting
Belfast, Ireland, BT9 7AB
Contact: contact + 44 028 9097 2756
Principal Investigator: Dr Richard Turkingon
Cork University Hospital
Recruiting
Cork, Ireland
Contact: Contact Person +353 21-4271971
Principal Investigator: Dr Derek Power
Beaumont Hospital
Recruiting
Dublin, Ireland
Contact: Contact Person +353 1 809 3000
Principal Investigator: Prof Liam Grogan
SLRON- St Luke's Radiation Oncology Network
Recruiting
Dublin, Ireland
Contact: Contact Person +353 1 4065000
Principal Investigator: Dr Moya Cunningham
St. James's Hospital
Recruiting
Dublin, Ireland
Contact: Contact Person +353 1 410 3000
Principal Investigator: Prof John Reynolds
University Hospital Galway
Recruiting
Galway, Ireland
Contact: Contact Person +353 91 524222
Principal Investigator: Dr Greg Leonard
United Kingdom
The Royal Bournemouth Hospital
Recruiting
The Royal Bournemouth And Christchurch Hospitals NHS Foundation, Bournemouth, United Kingdom, BH7 7DW
Contact: Contact Person +44 1202 726127
Principal Investigator: Joanne Parkinson
Cambridge University Hospitals NHS Foundation Trust
Recruiting
Addenbrooke's Hospital, Box 279(s4), Cambridge Biomedical Camp, Cambridge, United Kingdom, CB2 0QQ
Contact: Contact Person +44 1223 256620
Principal Investigator: Dr Hugo Ford
Velindre Cancer Centre
Recruiting
Velindre NHS Trust, Velindre Road, Whitchurch, Cardiff, United Kingdom, CF14 2TL
Contact: Contact Person +44 29 20 615 888
Principal Investigator: Carys Morgan
University Hospitals Coventry & Warwickshire
Recruiting
Clifford Bridge Road, Walsgrave, Coventry, United Kingdom, CV2 2DX
Contact: Contact Person +44 2476 966 202
Principal Investigator: Sharmila Sothi
Hull and East Yorkshire Hospitals NHS Trust, Castle Hill Hospital,
Recruiting
Cottingham, East Riding Of Yorkshire, United Kingdom, HU16 5JQ
Contact: Contact Person +44 23 8120 8448
Principal Investigator: Dr Rajarshi Roy
Portsmouth Hospitals NHS Trust
Recruiting
Southwick Hill Road, Cosham, Hampshire, United Kingdom, PO6 3LY
Contact: Contact Person +44 2392 286000
Principal Investigator: Freddie Bartlett
Mount Vernon Cancer Centre
Recruiting
E & N Hertfordshire NHS Trust, Rickmansworth Road, Northwood, Middlesex, United Kingdom, HA6 2RN
Contact: Contact Person +44 203 826 2063
Principal Investigator: Mark Harrison
Nottingham City Hospital
Recruiting
Nottingham University Hospitals NHS Trust, Hucknall Road, Nottingham, United Kingdom, HG5 1PB
Contact: Contact Person +44 115 969 1169
Principal Investigator: Simon Parsons
Oxford University Hospital NHS Trust Churchill Hospital
Recruiting
Headington, Oxfordshire, United Kingdom, OX3 7LE
Contact: Contact Person +44 1865 741841
Principal Investigator: Somnath Mukherjee
University Hospital Southampton NHS Foundation Trust
Recruiting
Southampton General Hospital, Division A Cancer Care, Mp307, T, Southampton, United Kingdom, SO16 6YD
Contact: Contact Person +44 23 8120 8448
Principal Investigator: Dr Andrew Bateman
Royal Surrey County Hospital
Recruiting
Guildford, Surrey, United Kingdom, GU2 7XX
Contact: Contact Person +44 1483 571122
Principal Investigator: Shaun Preston
Worcestershire Royal Hospital
Recruiting
Worcestershire Oncology Centre, Charles Hastings Way, Worcester, United Kingdom, WR5 1DD
Contact: Contact Person +44 1905 733194
Principal Investigator: Mr Martin Wadley
University Hospitals Bristol NHS Foundation Trust
Recruiting
Bristol, United Kingdom, BS2 8ED
Contact: Contact Person +44 117 342 0231
Principal Investigator: Dr Stephen Falk
NHS Lothian, Edinburgh Cancer Centre,
Recruiting
Edinburgh, United Kingdom, EH4 2XU
Contact: Contact Person +44 131 242 3327
Principal Investigator: Dr Sorcha Campbell
Imperial College Healthcare NHS Trust St Mary's Hospital
Recruiting
London, United Kingdom, W2 1NY
Contact: Contact Person +44 20 3312 6666
Principal Investigator: Prof George Hanna
The Newcastle upon Tyne Hospital NHS Foundation TrustFreeman Hospital, Freeman Road, High Heaton
Recruiting
Newcastle upon Tyne, United Kingdom, NE7 7DN
Contact: Contact Person +44 191 233 6161
Principal Investigator: Prof Michael Griffin Less << |
|
NCT03721757
|
Squamous Cell Carcinoma of the... More >> Oral Cavity Less << |
Phase 2 |
Not yet recruiting |
November 2021 |
- |
|
NCT02164942
|
- |
|
Terminated(Futility) |
- |
United States, North Carolina
... More >>
Carolinas Healthcare System
Charlotte, North Carolina, United States, 28203 Less << |
|
NCT00600210
|
Cervical Cancer |
Phase 2 |
Withdrawn(low patient accrual) |
- |
United States, North Carolina
... More >>
East Carolina University School of Medicine
Greenville, North Carolina, United States, 27834 Less << |
|
NCT03101995
|
Cervical Cancer |
Phase 2 |
Recruiting |
July 1, 2019 |
Mexico
... More >> National Institute of Cancer
Recruiting
Mexico City, DF, Mexico, 14080
Contact: Lucely Cetina, MD, MSc +521555 4851237 micuentalucely@yahoo.com Less << |
|
NCT03006432
|
OESOPHAGO-GASTRIC CARCINOMA |
Phase 3 |
Recruiting |
August 2022 |
- |
|
NCT00661011
|
Non Small Cell Lung Carcinoma |
Phase 2 |
Terminated(The study was termi... More >>nated due to too slow recruitment) Less << |
- |
Belgium
... More >> Department of Pneumology RHMS Hôpital de la Madeleine
Ath, Belgium, 7800
Department of Pneumology CHR St Joseph-Warquignies
Boussu, Belgium, 7360
Department of Intensive Care Unit and Thoracic Oncology Institut Jules Bordet
Brussels, Belgium, 1000
Department of Pneumology Hospital Ixelles-Molière
Brussels, Belgium
Department of Pneumology CHU Charleroi
Charleroi, Belgium, 6000
Department of Pneumology Hôpital Saint-Joseph
Gilly, Belgium, 6060
Hôpital Ambroise Paré
Mons, Belgium, 7000
Hôpital Vésale - Montigny-le-Tilleul
Montigny-le-Tilleul, Belgium, 6110
Department of Pneumology Centre Hospitalier de Mouscron
Mouscron, Belgium, 7700
CH Peltzer-La Tourelle
Verviers, Belgium, 4800
Greece
Medical Oncology St Savas Hospital
Athens, Greece, 11522
Spain
Medical Oncology Hospital de Sagunto
Valencia, Spain Less << |
|
NCT02842307
|
Chemotherapy-induced Nausea an... More >>d Vomiting Less << |
Not Applicable |
Completed |
- |
China, Beijing
... More >> Dongzhimen Hospital, Beijing University of Chinese medicine
Beijing, Beijing, China, 100007
Guanganmen Hospital,China Academy of Chinese Medical Sciences
Beijing, Beijing, China, 100053
China
Xiyuan Hospital, China Academy of Chinese Medical Sciences
Beijing, China, 100091 Less << |
|
NCT01534585
|
Nasopharyngeal Carcinoma |
Phase 1
Phase 2 |
Completed |
- |
China, Zhejiang
... More >>
Taizhou Hospital, Wenzhou Medical College
Taizhou, Zhejiang, China, 317000 Less << |
|
NCT01639625
|
Squamous Cell Carcinoma of the... More >> Cervix
Adenocarcinoma of the Cervix Less << |
Phase 2 |
Completed |
- |
Argentina
... More >> Instituto de Oncologia Angel H. Roffo
Ciudad Autonoma de Buenos Aires, Buenos Aires, Argentina, C1417DTB Less << |
|
NCT00256295
|
- |
|
Terminated(Low accrual) |
- |
- |
|
NCT00347256
|
Paranasal Sinus Neoplasms |
Not Applicable |
Withdrawn |
- |
- |
|
NCT00186277
|
Bladder Cancer |
Phase 2 |
Completed |
- |
United States, California
... More >>
Stanford University School of Medicine
Stanford, California, United States, 94305 Less << |
|
NCT02880007
|
Cervical Adenocarcinoma
... More >> Stage IB Cervical Cancer
Stage II Cervical Cancer
Stage III Cervical Cancer Less << |
Phase 2 |
Recruiting |
February 2020 |
France
... More >> CHU de Besançon
Recruiting
Besancon, France, 25030
Contact: BARON Marie-Hélène, MD
Institut Bergonié
Recruiting
Bordeaux, France, 33076
Contact: THOMAS Laurence, MD
Contact: PETIT Adeline, MD
Centre Georges-François Leclerc
Recruiting
Dijon, France, 21079
Contact: PEIGNAUX Karine, MD
Contact: MARTIN Etienne, MD
Centre Léon Bérard
Recruiting
Lyon, France, 69373
Contact: POMMIER Pascal, MD
Contact: CARRIE Christian, MD
Centre Val d'Aurelle
Recruiting
Montpellier, France, 34298
Contact: KERR Christine, MD
Centre Paul Strauss
Recruiting
Strasbourg, France, 67065
Contact: POP Marius, MD
Centre Claudius Regaud
Recruiting
Toulouse, France, 31052
Contact: DELANNES Martine, MD
Contact: DUCASSOU Anne, MD
CHRU Tours - Hôpital Bretonneau
Recruiting
Tours, France, 37044
Contact: BARILLOT Isabelle, MD
Institut de Cancérologie de Lorraine
Recruiting
Vandoeuvre-les-Nancy, France, 54519
Contact: PEIFFERT Didier, MD
Contact: BRUNAUD Claire, MD Less << |
|
NCT01326767
|
Carcinoma, Non-Small-Cell Lung |
Phase 3 |
Completed |
- |
Germany
... More >> CESAR Study Center
Bochum, Germany
CESAR study center
Bonn, Germany
CESAR Study Center
Essen, Germany
CESAR study center
Gerlingen, Germany
CESAR study center
Großhansdorf, Germany
CESAR Study Center
Halle an der Saale, Germany
CESAR Study Center
Leer, Germany
CESAR Study Center
Löwenstein, Germany
CESAR Study Center
Munich, Germany
CESAR Study Center
Tübingen, Germany
Switzerland
Kantonsspital St. Gallen
St. Gallen, Switzerland, 9007 Less << |
|
NCT02635360
|
Cervical Cancer |
Phase 2 |
Recruiting |
May 2021 |
United States, Alabama
... More >>
University of South Alabama Mitchell Cancer Institute
Recruiting
Mobile, Alabama, United States, 36604
Contact: Joanie Broemmelsiek 251-445-9866 jbroemmelsiek@health.southalabama.edu
Contact: Tammy Mitchem-Welch 251-445-9649 twelch@health.southalabama.edu
Principal Investigator: Jennifer Scalici, MD
United States, Maryland
Johns Hopkins
Recruiting
Baltimore, Maryland, United States, 21287
Contact: Marquis Simms 410-955-8849 msimms10@jhmi.edu
United States, Missouri
Washington University, School of Medicine
Recruiting
Saint Louis, Missouri, United States, 63108
Contact: Jehan Ganachaud, M.Sc. 314-747-9202 ganachaud.j@wustl.edu
United States, North Carolina
Levine Cancer Institute
Recruiting
Charlotte, North Carolina, United States, 28204
Contact: Heather Neagle, RN, BSN 980-442-2303 Heather.Neagle@Carolinashealthcare.org
United States, Oklahoma
University of Oklahoma
Recruiting
Oklahoma City, Oklahoma, United States, 73104
Contact: Braden Curry 405-271-8777 braden-curry@ouhsc.edu
United States, Virginia
University of Virginia
Recruiting
Charlottesville, Virginia, United States, 22908
Contact: Sheena Clift 434-924-2745 SHC2WN@hscmail.mcc.virginia.edu
Principal Investigator: Linda Duska, MD
INOVA Fairfax Hospital
Recruiting
Falls Church, Virginia, United States, 22042
Contact: Kelly Jeffords 703-208-6606 Kelly.Jeffords@inova.org
Virginia Commonwealth University
Recruiting
Richmond, Virginia, United States, 23298
Contact: Melanie R Hamilton mrhamilton2@vcu.edu Less << |
|
NCT01462474
|
Locally Advanced Nasopharyngea... More >>l Carcinoma Less << |
Phase 1 |
Completed |
- |
China, Guangdong
... More >>
Department of Medical Oncology, Cancer Center, Sun Yet-sen University
Guangzhou, Guangdong, China Less << |
|
NCT00450762
|
Breast Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT03061058
|
Stomach Neoplasms
... More >> Chemotherapy Effect
Chemotherapeutic Toxicity Less << |
Phase 3 |
Recruiting |
December 2019 |
China, Anhui
... More >> Ma'anshan People's Hospital
Recruiting
Ma'anshan, Anhui, China
Contact: Fenglin Zhang, MD
Contact: Fangbo Cui, MD
China, Jiangsu
Jiangyin People's Hospital
Recruiting
Jiangyin, Jiangsu, China
Contact: Weisheng Shen, MD
Contact: Lei Xi, MD
Nanjing Gaochun People's Hospital
Recruiting
Nanjing, Jiangsu, China, 210000
Contact: Rongfu Wei, MD
Contact: Yajun Xin, MD
Nanjing Lishui People's Hospital
Recruiting
Nanjing, Jiangsu, China, 210000
Contact: Chunlan Nie, MD
Contact: Hui Xu, MD
The Comprehensive Cancer Center of Nanjing Drum Tower Hospital
Recruiting
Nanjing, Jiangsu, China, 210008
Contact: Yang Yang, MD,PhD,MSCR 0086-18602568379 wing_young7@hotmail.com
Contact: Baorui Liu, MD, PhD 0086-13770621908 baoruiliu07@163.com
Suqian People's Hospital
Recruiting
Suqian, Jiangsu, China
Contact: Chuanwen You, MD
Contact: Qing Zhu, MD
Xuzhou Central Hospital
Recruiting
Xuzhou, Jiangsu, China
Contact: Sanyuan Sun, MD
Contact: Yuan Yuan, MD
Affiliated Hospital of Jiangsu University
Recruiting
Zhenjiang, Jiangsu, China
Contact: Xiaoqing Li, MD
Contact: Meilian Cheng, MD Less << |
|
NCT03418038
|
High Grade B-Cell Lymphoma Wit... More >>h MYC and BCL2 or BCL6 Rearrangements
Recurrent Diffuse Large B-Cell Lymphoma
Recurrent Hodgkin Lymphoma
Recurrent Lymphoma
Refractory Diffuse Large B-Cell Lymphoma
Refractory Lymphoma Less << |
Phase 2 |
Recruiting |
March 15, 2024 |
United States, Arizona
... More >>
Mayo Clinic in Arizona
Not yet recruiting
Scottsdale, Arizona, United States, 85259
Contact: Clinical Trials Referral Office 855-776-0015
Principal Investigator: Craig B. Reeder
United States, Florida
Mayo Clinic in Florida
Not yet recruiting
Jacksonville, Florida, United States, 32224-9980
Contact: Clinical Trials Referral Office 855-776-0015
Principal Investigator: Taimur Sher
United States, Iowa
University of Iowa/Holden Comprehensive Cancer Center
Not yet recruiting
Iowa City, Iowa, United States, 52242
Contact: Umar Farooq umar-farooq@uiowa.edu
Principal Investigator: Umar Farooq
United States, Minnesota
Mayo Clinic
Recruiting
Rochester, Minnesota, United States, 55905
Contact: Clinical Trials Referral Office 855-776-0015
Principal Investigator: Thomas E. Witzig Less << |
|
NCT00453115
|
Lung Cancer |
Phase 2 |
Unknown |
May 2011 |
Korea, Republic of
... More >>
National Cancer Center, Korea
Goyang-si, Gyeonggi-dog, Korea, Republic of, 411-769 Less << |
|
NCT02188615
|
Squamous Cell Esophageal Carci... More >>noma Less << |
Phase 2 |
Recruiting |
July 2019 |
China, Zhejiang
... More >>
Thaizhou Hospital
Recruiting
Linhai, Zhejiang, China, 317000
Contact: Cheng chu Zhu, professor +86-576-85199876 zhucc669266@163.com
Principal Investigator: Bao fu Chen, professor Less << |
|
NCT03219762
|
Small Cell Lung Cancer |
Phase 2 |
Recruiting |
January 2019 |
Italy
... More >> UO di Oncologia Ematologia, Azienda Ospedaliero Universitaria di Ferrara
Recruiting
Cona, Ferrara, Italy, 44124
Principal Investigator: Antonio Frassoldati, MD
Sezione Pneumo-Oncologica - Medicina Interna I; IRCCS "Casa Sollievo della Sofferenza"
Not yet recruiting
San Giovanni Rotondo, Foggia, Italy, 71013
Principal Investigator: Vito D'Alessandro, MD
Oncologia Medica, IRST. Istituto Scientifico Romagnolo per lo studio e la cura dei Tumori, IRCCS di Meldola
Recruiting
Meldola, Forlì-Cesena, Italy, 47014
Principal Investigator: Angelo Del Monte, MD
Oncologia Medica - Ospedale Versilia
Not yet recruiting
Lido di Camaiore, Lucca, Italy, 55043
Principal Investigator: Andrea Camerini, MD
SC di Oncologia Medica, A.O. San Gerardo di Monza
Not yet recruiting
Monza, MB, Italy, 20900
Principal Investigator: Diego Cortinovis, MD
UO Medicina Oncologica Ospedale di Carpi (MO)
Recruiting
Carpi, Modena, Italy, 41012
Principal Investigator: Lucia Longo, MD
UOC di Oncologia Medica - Ospedale di Saronno
Not yet recruiting
Saronno, Varese, Italy, 21047
Principal Investigator: Claudio Verusio, MD
UOC Oncologia Medica, Azienda ULSS21 di Legnago
Not yet recruiting
Legnago, Verona, Italy, 37045
Principal Investigator: Andrea Bonetti, MD
Oncologia Medica, Ospedale Sacro Cuore - Don Calabria - Negrar (VR)
Not yet recruiting
Negrar, Verona, Italy, 37024
Principal Investigator: Stefania Gori, MD
SC Oncologia - ASO "SS Antonio e Biagio e Cesare Arrigo,Alessandria
Not yet recruiting
Alessandria, Italy, 15121
Principal Investigator: Perluigi Piovano
Cliniche Humanitas Gavazzeni
Not yet recruiting
Bergamo, Italy, 24125
Principal Investigator: Giovanni Luca Ceresoli
Oncologia Medica, Ospedale Papa Giovanni XXIII
Recruiting
Bergamo, Italy, 24127
Principal Investigator: Anna Bettini, MD
UO di Oncologia Medica, Azienda Ospedaliero-Universitaria S. Orsola Malpighi di Bologna,
Recruiting
Bologna, Italy, 40138
Principal Investigator: Andrea Ardizzoni, MD
Divisione di Oncologia Medica - Ospedale di Bolzano,
Recruiting
Bolzano, Italy, 39100
Principal Investigator: Emanuela Vattemi, MD
UOC Oncologia Medica PO A.Perino ASL di Brindisi
Recruiting
Brindisi, Italy, 72100
Principal Investigator: Saverio Cinieri, MD
SC di Oncologia, Istituti Ospitalieri di Cremona
Not yet recruiting
Cremona, Italy, 26100
Principal Investigator: Matteo Brighenti, MD
Dipartimento di Oncologia Medica A.O. Santa Croce e Carle Ospedale Carle
Recruiting
Cuneo, Italy, 12100
Principal Investigator: Ida Colantonio, MD
Azienda Ospedaliera Careggi, UO di Oncologia Medica
Not yet recruiting
Firenze, Italy, 50139
Principal Investigator: Francesco Di Costanzo, MD
UOC Oncologia, Azienda USL di Imola, Ospedale Santa Maria della Scaletta
Not yet recruiting
Imola, Italy, 40028
Principal Investigator: Antonio Maestri, MD
Dipartimento Oncologico, Azienda USL 2 di Lucca, Ospedale San Luca
Not yet recruiting
Lucca, Italy, 55100
Principal Investigator: Carmelo Tibaldi, MD
Azienda Ospedaliero-Universitaria di Modena-Policlinico, U.O. di Oncologia Medica ed Ematologia
Recruiting
Modena, Italy, 41100
Principal Investigator: Fausto Barbieri, MD
S. C. di Oncologia Medica AORN "Antonio Cardarelli"
Not yet recruiting
Napoli, Italy, 80131
Principal Investigator: Ferdinando Riccardi, MD
UOC di Oncologia Medica di Parma, Azienda Ospedaliero Universitaria di Parma
Not yet recruiting
Parma, Italy, 43126
Principal Investigator: Marcello Tiseo, MD
Dipartimento di Oncologia e Ematologia, UO di Oncologia Medica Azienda USL di Piacenza
Recruiting
Piacenza, Italy, 29121
Principal Investigator: Luigi Cavanna, MD
Dipartimento di Oncologia Ematologia. UO di Oncologia Medica, AUSL della Romagna, Ospedale Santa Maria delle Croci di Ravenna
Recruiting
Ravenna, Italy, 48121
Principal Investigator: Claudio Dazzi, MD
Azienda Sanitaria Ospedaliera Molinette, U.O. di Oncologia Medica
Not yet recruiting
Torino, Italy, 10126
Principal Investigator: Libero Ciuffreda, MD Less << |
|
NCT01026467
|
- |
|
Completed |
- |
United States, New York
... More >>
Syracuse VA Medical Center
Syracuse, New York, United States, 13210
United States, North Carolina
Wake Forest University Health Sciences
Winston-Salem, North Carolina, United States, 27157 Less << |
|
NCT01898494
|
Human Papilloma Virus Infectio... More >>n
Stage III Squamous Cell Carcinoma of the Oropharynx
Stage IVA Squamous Cell Carcinoma of the Oropharynx
Stage IVB Squamous Cell Carcinoma of the Oropharynx Less << |
Phase 2 |
Active, not recruiting |
February 2023 |
- |
|
NCT00336024
|
Untreated Childhood Medullobla... More >>stoma
Untreated Childhood Supratentorial Primitive Neuroectodermal Tumor Less << |
Phase 3 |
Completed |
- |
- |
|
NCT02200354
|
Advanced Non-small Cell Lung C... More >>ancer Less << |
Phase 2 |
Recruiting |
March 2019 |
Japan
... More >> Kobe City Medical Center General Hospital
Recruiting
Kobe, Hyogo, Japan, 650-0046
Contact: Daichi Fujimoto, MD +81-78-302-4321 ext 773 daichianzen@yahoo.co.jp Less << |
|
NCT02862535
|
Gastric Adenocarcinoma
... More >> Gastroesophageal Junction Adenocarcinoma Less << |
Phase 1 |
Active, not recruiting |
July 2019 |
Japan
... More >>
Nagoya, Japan
Osaka, Japan
Tokyo, Japan Less << |
|
NCT01684904
|
Esophagus Cancer |
Not Applicable |
Recruiting |
August 2024 |
United States, California
... More >>
Loma Linda University Medical Center
Recruiting
Loma Linda, California, United States, 92354
Contact: Gary Yang, MD 909-558-4280 gyang@llu.edu
Principal Investigator: Gary Yang, MD Less << |
|
NCT00336024
|
- |
|
Completed |
- |
- |
|
NCT03221426
|
Gastric Cancer
... More >> Gastroesophageal Junction Cancer Less << |
Phase 3 |
Recruiting |
July 26, 2023 |
- |
|
NCT02497053
|
Malignant Pleural Mesothelioma |
Phase 2 |
Unknown |
- |
Egypt
... More >> Ain Shams University Hospitals
Recruiting
Cairo, Egypt
Contact: Omar Abdel-Rahman, MD 26858397 omar.abdelrhman@med.asu.edu.eg
Contact: Ahmed Nagi, MD ahmedalynagy@yahoo.com
Principal Investigator: Omar Abdel-Rahman, MD
Sub-Investigator: Ahmed Nagi, MD Less << |
|
NCT03163979
|
Cervical Cancer |
Phase 2
Phase 3 |
Recruiting |
July 31, 2019 |
China, Jiangsu
... More >> Nanjing Medical University Affiliated Suzhou Hospital
Recruiting
Suzhou, Jiangsu, China, 215000
Contact: Ke Gu, doctor +86-512-62364013 dr.guke@hotmail.com Less << |
|
NCT00142038
|
Stomach Neoplasm
... More >> Neoplasm Metastasis Less << |
Phase 2 |
Completed |
- |
Germany
... More >> Charité, Universitätsmedizin Berlin, Campus Virchow-Klinikum, Dept. of Hematology and Oncology
Berlin, Germany, 13353 Less << |
|
NCT02633514
|
Thymoma and Thymic Carcinoma |
Phase 3 |
Unknown |
- |
China, Shanghai
... More >>
Kailiang Wu
Recruiting
Shanghai, Shanghai, China, 20032
Contact: Kailiang Wu, M.D. Ph. D. +86 64175590 ext 86722 wukailiang@aliyun.com Less << |
|
NCT03193788
|
Bladder Cancer
... More >> Ureter Cancer
Transitional Cell Carcinoma Less << |
Phase 3 |
Recruiting |
June 2020 |
Korea, Republic of
... More >>
Hallym University Medical Center, Hallym University College of Medicine
Recruiting
Anyang, Korea, Republic of
Contact: Ho Young Kim ksfam@daum.net
Keimyeong University Dongsan Medical Center
Recruiting
Daegu, Korea, Republic of, 700-712
Contact: Jin Young Kim, MD takgu@dsmc.or.kr
Fatima Hospital
Recruiting
Daegu, Korea, Republic of
Contact: Jung Lim Lee junglim3@gmail.com
Chungnam University Hospital
Recruiting
Daejeon, Korea, Republic of, 301-721
Contact: Hyo Jin Lee, MD, PhD. cymed@cnu.ac.kr
National Health Insurance Service Ilsan Hospital
Recruiting
Goyang, Korea, Republic of
Contact: Soo Jung Hong suzzy901@nhimc.or.kr
Hallym University Dongtan Sacred Heart Hospital
Recruiting
Hwaseong-si, Korea, Republic of
Contact: Hyun Ae Jung hyunaejung@hallym.or.kr
Gil Medical Center
Recruiting
Incheon, Korea, Republic of, 21565
Contact: Inkeun Park, MD ingni79@hanmail.net
Dong-A University Medical Center
Recruiting
Pusan, Korea, Republic of
Contact: Suee Lee
Inje University Haeundae Paik Hospital
Recruiting
Pusan, Korea, Republic of
Contact: Il-Hwan Kim onelement@daum.net
Pusan National University Hospital, Pusan National University School of Medicine
Recruiting
Pusan, Korea, Republic of
Contact: Hyo Jung Kim leonkim80@naver.com
Asan Medical Center
Recruiting
Seoul, Korea, Republic of, 138-736
Contact: Jae-Lyun Lee, MD, PhD 82 2 3010 5977 jaelyun@amc.seoul.kr
Chung Ang University Hospital
Recruiting
Seoul, Korea, Republic of, 156-755
Contact: Hee Joon Kim, MD heejun.dino11@gmail.com
Inje University Sanggye Paik Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Byeong Seok Sohn imbs@paik.ac.kr
Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine
Recruiting
Seoul, Korea, Republic of
Contact: Yun-Gyoo Lee gosciny@gmail.com
Korea University Anam Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Yoon Ji Choi yoonji23@hanmail.net
Seoul National University Hospital, Cancer Research Institute, Seoul National University College of Medicine
Recruiting
Seoul, Korea, Republic of
Contact: Bhum Suk Keam
Contact bhumsuk@snu.ac.kr
Seoul St. Mary's Hospital, College of Medicine, The Catholic University of Korea
Recruiting
Seoul, Korea, Republic of
Contact: In-Ho Kim ihkmd@naver.com
Yonsei Cancer Center
Recruiting
Seoul, Korea, Republic of
Contact: Sang Joon Shin ssj338@yuhs.ac
St. Vincent's Hospital, The Catholic University of Korea
Recruiting
Suwon, Korea, Republic of
Contact: Byoung Yong Shim shimby@catholic.ac.kr
Uijeongbu St Mary's hospital, Catholic university of Korea
Recruiting
Uijeongbu, Korea, Republic of
Contact: Yoon Ho Ko koyoonho@catholic.ac.kr
Pusan National University Yangsan Hospital
Recruiting
Yangsan, Korea, Republic of
Contact: Kwonoh Park parkkoh@hanmail.net Less << |
|
NCT00084838
|
- |
|
Completed |
- |
- |
|
NCT00960518
|
Hepatitis B Virus
... More >> Hepatocellular Carcinoma Less << |
Phase 2 |
Unknown |
August 2015 |
China, Heilongjiang
... More >>
The Fourth Affiliated Hospital of Haerbin Medical University
Recruiting
Ha'er'bin, Heilongjiang, China, 150001
Contact: Baozhong Shen, MD +86-451-82576888 drbaozhong.shen@gmail.com
Principal Investigator: Baozhong Shen, MD
China, Shanghai
Shanghai 10th Hospital of Tongji University
Recruiting
Shanghai, Shanghai, China, 200025
Contact: Maoquan Li, MD, PhD +86-21-6630058 drmaoquan.li@gmail.com
Principal Investigator: Maoquan Li, MD, PhD Less << |
|
NCT02489903
|
Small Cell Carcinoma
... More >> Carcinoma, Non-Small-Cell Lung
Neuroendocrine Tumors
Ovarian Epithelial Cancer Less << |
Phase 2 |
Recruiting |
July 2020 |
United States, California
... More >>
Stanford University
Recruiting
Palo Alto, California, United States, 94304
Contact: Melanie San Pedro-Salcedo 650-724-1388 msanpedro@stanford.edu
Principal Investigator: Sukhmani Padda, MD
United States, Connecticut
VA Connecticut Cancer Center
Recruiting
West Haven, Connecticut, United States, 06516
Contact: Jessica Jordan 203-932-5711 ext 3287 jessica.jordan@va.gov
Principal Investigator: Michal Rose, MD
United States, Indiana
Memorial Hospital of South Bend
Recruiting
South Bend, Indiana, United States, 46601
Contact: Amie Lake 574-647-6784 ALake@beaconhealthsystem.org
Contact: Mary Jane Danner 574-647-7309 MDanner@beaconhealthsystem.org
Principal Investigator: Thomas J Reid, MD, PhD
United States, Kentucky
Baptist Health
Recruiting
Lexington, Kentucky, United States, 40503
Contact: Stephanie Cason 859-260-3197 stephanie.cason@bhsi.com
Contact: Micheal F Stephens 859-260-6456 micheal.stephens@BHSI.com
Principal Investigator: Arvinda Padmanabhan, MD
United States, Maryland
Walter Reed National Military Medical Center
Recruiting
Bethesda, Maryland, United States, 20889
Contact: Sonja Skeete 301-319-4599 sonja.t.skeete.ctr@mail.mil
Principal Investigator: Karen Zeman, MD
United States, Michigan
Henry Ford Allegiance Health
Recruiting
Jackson, Michigan, United States, 49201
Contact: Kris Strzalkowski, RN 517-205-1522 KSTRZAL1@hfhs.org
Contact: Natalie Peterson, RN OCN (517) 205-1549 npeters8@hfhs.org
Principal Investigator: Malcolm Trimble, MD
United States, Missouri
Washington University
Recruiting
Saint Louis, Missouri, United States, 63110
Contact: Dan Blume 314-362-0872 dan.blume@wustl.edu
Principal Investigator: Daniel Morgensztern, MD
United States, Ohio
University of Cincinnati Cancer Institute
Recruiting
Cincinnati, Ohio, United States, 45267
Contact: Lauren Hendricks 513-584-5725 hendrilu@ucmail.uc.edu
Principal Investigator: John C Morris, MD
United States, Tennessee
University of Tennessee Medical Center
Withdrawn
Knoxville, Tennessee, United States, 37920
United States, Virginia
Virginia Cancer Specialists
Recruiting
Fairfax, Virginia, United States, 22031
Contact: Karin Choquette, MSN, RN 703-208-9268 Karin.Choquette@USONCOLOGY.COM
Principal Investigator: Alexander Spira, MD
United States, West Virginia
West Virginia University
Recruiting
Morgantown, West Virginia, United States, 26506
Contact: Carla Ross, RN 304-581-1158 cjross@hsc.wvu.edu
Principal Investigator: Patrick Ma, MD Less << |
|
NCT00577993
|
Lymphoma |
Phase 3 |
Completed |
- |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT02896582
|
Mantle Cell Lymphoma |
Phase 2 |
Recruiting |
March 2025 |
France
... More >> CHU d'Amiens
Recruiting
Amiens, France, 80480
Principal Investigator: Jean-Pierre Marolleau, Dr
CHU d'Angers
Recruiting
Angers, France, 49000
Principal Investigator: Marie-Pierre Moles-Moreau, Dr
CH d'Avignon
Recruiting
Avignon, France, 84902
Principal Investigator: Hacene Zerazhi, Dr
CHU de Caen
Recruiting
Caen, France, 14033
Principal Investigator: Christophe Fruchart, Dr
CHU de Clermont Ferrand
Recruiting
Clermont Ferrand, France, 63000
Principal Investigator: Victoria Cacheux, Dr
Hopital Henri Mondor
Recruiting
Créteil, France, 94010
Principal Investigator: Corinne Haioun
CHU de Dijon - Hôpital le Bocage
Recruiting
Dijon, France, 21034
Principal Investigator: Olivier Casasnovas, Dr
CHU de Grenoble
Recruiting
Grenoble, France, 38700
Principal Investigator: Rémy Gressin, Dr
CHD Vendée
Recruiting
La Roche sur Yon, France, 85925
Principal Investigator: Nadine Morineau, Dr
Clinique Victor Hugo
Recruiting
Le Mans, France, 72000
Principal Investigator: Katell Le Du, Dr
CHRU Lille - Hôpital Claude Huriez
Recruiting
Lille, France, 59037
Principal Investigator: Franck MORSCHHAUSER, Pr
CHU Limoges
Recruiting
Limoges, France, 87042
Principal Investigator: Julie Abraham, Dr
CHU Montpellier
Recruiting
Montpellier, France, 34295
Principal Investigator: Guillaume Cartron, Dr
CHU Nantes
Recruiting
Nantes, France, 44093
Principal Investigator: Steven Le Gouill, Pr
Hôpital Saint Louis
Recruiting
Paris, France, 75475
Principal Investigator: Catherine THIEBLEMONT
APHP - Hopital Necker
Recruiting
Paris, France, 75743
Principal Investigator: Richard Delarue, Dr
CH Perpignan
Recruiting
Perpignan, France, 66046
Principal Investigator: Laurence Sanhes, Dr
CHU de Haut Leveque
Recruiting
Pessac, France, 33604
Principal Investigator: Kamal Bouabdallah, Dr
CHU Lyon Sud
Recruiting
Pierre Bénite, France, 69130
Principal Investigator: Gilles Salles
CHU de Poitiers
Recruiting
Poitiers, France, 86021
Principal Investigator: Vincent Delwail, Dr
Centre Hospitalier Annecy-Genevois
Recruiting
Pringy, France, 74374
Principal Investigator: Nicolas Daguindau, Dr
CHU Robert Debré
Recruiting
Reims, France, 51092
Principal Investigator: Alain Delmer, Pr
CHU Pontchaillou
Recruiting
Rennes, France, 35033
Principal Investigator: Thierry Lamy de la Chapelle, Pr
Centre Henri Becquerel
Recruiting
Rouen, France, 76038
Principal Investigator: Hervé Tilly, Dr
Institut de Cancérologie de Loire
Recruiting
Saint priest en Jarez, France, 42271
Principal Investigator: Jérome Cornillon, Dr
CHU de Strasbourg
Recruiting
Strasbourg, France, 67091
Principal Investigator: Luc-Matthieu Fornecker, Dr
I.U.C.T Oncopole
Recruiting
Toulouse, France, 31100
Principal Investigator: Lucie Oberic, Dr
CHRU Bretonneau
Recruiting
Tours, France, 37044
Principal Investigator: Caroline Dartigeas, Dr
CHU de Brabois
Recruiting
Vandoeuvre les Nancy, France
Principal Investigator: Pierre Feugier, Dr
Gustave Roussy Cancer Campus
Recruiting
Villejuif, France, 94805
Principal Investigator: Vincent Ribrag, Dr Less << |
|
NCT01248299
|
Squamous Cell Carcinoma of Eso... More >>phagus Less << |
Phase 2 |
Unknown |
September 2016 |
France
... More >> CHU Brest
Brest, France, 29200
Centre François BACLESSE
Caen, France, 14076
Centre Georges François Leclerc
Dijon, France, 21079
CHU Dijon
Dijon, France, 21079
Centre Oscar Lambret
Lille, France, 59020
CHU Lille
Lille, France, 59035
CHU La Timone
Marseille, France, 13385
Centre Val d'Aurelle
Montpellier, France, 34298
Centre René Gauducheau
Nantes Saint-herblain, France, 44805
Centre Antoine Lacassagne
Nice, France, 06189
Centre Eugène Marquis
Rennes, France, 35042
Clinique de la Theuillerie
Ris Orangis, France, 91130
CHU Rouen
Rouen, France, 76031
Clinique de l'Armoricaine
St-Brieuc, France, 22000
Centre Paul Strauss
Strasbourg, France, 67000
Centre Alexis Vautrin
Vandoeuvre-les-nancy, France, 54511
Centre Hospitalier Intercommunal
Villeneuve St Georges, France, 94190 Less << |
|
NCT03520231
|
Urothelial Carcinoma
... More >> Kidney Cancer
Ureter Cancer
Bladder Cancer Less << |
Phase 2 |
Recruiting |
June 1, 2020 |
Canada, Ontario
... More >>
Princess Margaret Cancer Centre
Recruiting
Toronto, Ontario, Canada, M5G 2M9
Contact: Srikala Sridhar, M.D. 416-946-4501 ext 2662 srikala.sridhar@uhn.ca
Principal Investigator: Srikala Sridhar, M.D. Less << |
|
NCT02771743
|
Newly Diagnosed High Risk Neur... More >>oblastoma Less << |
Phase 2 |
Recruiting |
March 2023 |
Korea, Republic of
... More >>
Samsung Medical Center
Recruiting
Seoul, Korea, Republic of
Contact: Ki Woong Sung 82-2-3410-3529 kwsped@skku.edu
Principal Investigator: Ki Woong Sung Less << |
|
NCT02340936
|
Diffuse Large B-cell Lymphoma |
Phase 1
Phase 2 |
Active, not recruiting |
September 2019 |
Spain
... More >> ICO- Hospital Universitari Germans Trias i Pujol
Badalona, Barcelona, Spain, 08916
ICO- Hospital Duran i Reynals
L´Hospitalet de Llobregat, Barcelona, Spain, 08908
Hospital del Mar
Barcelona, Spain, 08003
Hospital Clinic i Provincial de Barcelona
Barcelona, Spain, 08036
Hospital Universitario 12 de Octubre
Madrid, Spain, 28041
Hospital Regional Universitario de Málaga
Málaga, Spain, 29010
Alejandro Martín
Salamanca, Spain, 37007
Hospital Universitario Virgen del Rocío
Sevilla, Spain, 41013
Hospital Universitario La Fe
Valencia, Spain, 46026 Less << |
|
NCT00554788
|
Extraocular Retinoblastoma |
Phase 3 |
Active, not recruiting |
- |
- |
|
NCT03581786
|
Recurrent or Metastatic NPC |
Phase 3 |
Recruiting |
August 21, 2020 |
- |
|
NCT02829008
|
Metastatic Breast Cancer |
Phase 2 |
Active, not recruiting |
April 2020 |
China, Beijing
... More >> Cancer Hospital, ChineseAMS
Beijing, Beijing, China, 100021 Less << |
|
NCT02436707
|
Lymphoma |
Phase 2 |
Recruiting |
December 2019 |
Canada, Alberta
... More >>
Tom Baker Cancer Centre
Recruiting
Calgary, Alberta, Canada, T2N 4N2
Contact: Douglas A. Stewart 403 521-3347
Cross Cancer Institute
Recruiting
Edmonton, Alberta, Canada, T6G 1Z2
Contact: Neil Sun Chua 780 432-8340
Canada, British Columbia
BCCA - Vancouver Cancer Centre
Suspended
Vancouver, British Columbia, Canada, V5Z 4E6
Canada, Manitoba
CancerCare Manitoba
Suspended
Winnipeg, Manitoba, Canada, R3E 0V9
Canada, New Brunswick
The Moncton Hospital
Recruiting
Moncton, New Brunswick, Canada, E1C 6Z8
Contact: Nizar Abdel-Samad 506 870-2404
The Vitalite Health Network - Dr. Leon Richard
Suspended
Moncton, New Brunswick, Canada, E1C 8X3
Canada, Nova Scotia
QEII Health Sciences Centre
Recruiting
Halifax, Nova Scotia, Canada, B3H 1V7
Contact: Stephen Couban 902 473-8562
Canada, Ontario
Juravinski Cancer Centre at Hamilton Health Sciences
Recruiting
Hamilton, Ontario, Canada, L8V 5C2
Contact: Graeme Fraser 905 575-7820
Kingston Health Sciences Centre
Recruiting
Kingston, Ontario, Canada, K7L 2V7
Contact: Annette Hay 613 533-6430 ext 77094
Ottawa Hospital Research Institute
Suspended
Ottawa, Ontario, Canada, K1H 8L6
Odette Cancer Centre
Suspended
Toronto, Ontario, Canada, M4N 3M5
University Health Network
Recruiting
Toronto, Ontario, Canada, M5G 2M9
Contact: John Kuruvilla 416 946-2827
Canada, Quebec
CIUSSS de l'Est-de-I'lle-de-Montreal
Suspended
Montreal, Quebec, Canada, H1T 2M4
CHUM-Centre Hospitalier de l'Universite de Montreal
Suspended
Montreal, Quebec, Canada, H2X 3E4
CHU de Quebec-Hopital l'Enfant-Jesus (HEJ)
Recruiting
Quebec City, Quebec, Canada, G1J 1Z4
Contact: Jean-Francois Larouche 418 649-5726
Canada, Saskatchewan
Allan Blair Cancer Centre
Suspended
Regina, Saskatchewan, Canada, S4T 7T1 Less << |
|
NCT02031601
|
Non-Small-Cell Lung Cancer |
Phase 4 |
Unknown |
December 2017 |
China, Shandong
... More >>
Yu Li
Recruiting
Jinan, Shandong, China, 250012
Contact: Yu Li Less << |
|
NCT00818350
|
Urinary Bladder Neoplasms |
Phase 1
Phase 2 |
Unknown |
January 2011 |
- |
|
NCT01683175
|
Non-small Cell Lung Cancer Sta... More >>ge III Less << |
Phase 2 |
Active, not recruiting |
October 2020 |
China, Tianjin
... More >> Tianjin Medical University Cancer Institute and Hospital
Tianjin, Tianjin, China, 300060
China
Beijing Cancer Hospital
Beijing, China
Chinese PLA General Hospital
Beijing, China
The second people's hospital of Sichuan
Chengdu, China
Fujian Medical University Union Hospital
Fuzhou, China
Sun Yat-Sen University Cancer Center
Guangzhou, China
Zhejiang Cancer Hospital
Hangzhou, China
The third affiliated hospital of Harbin Medical Univer
Harbin, China
The affiliated hospital of medical college Qingdao University
Qingdao, China
Fudan University Shanghai Cancer Center
Shanghai, China
Zhongshan Hospital Fudan University
Shanghai, China
Liaoning Cancer Hospital & Institute
Shenyang, China
Hebei Provincial Tumor Hospital
Shijiazhuang, China
The first affiliated hospital of Soochow University
Suzhou, China
The fourth military medical university,Tangdu Hospital
Xi'an, China Less << |
|
NCT03219333
|
Carcinoma, Transitional Cell
... More >> Urinary Bladder Neoplasms
Urologic Neoplasms
Renal Pelvis Neoplasms
Urothelial Cancer
Ureteral Neoplasms
Urethral Neoplasms Less << |
Phase 2 |
Recruiting |
May 2025 |
- |
|
NCT01283204
|
Recurrent or Metastatic Gastri... More >>c Cancer Less << |
Phase 2 |
Unknown |
August 2016 |
Korea, Republic of
... More >>
Severance Hospital
Seoul, Korea, Republic of, 120-752 Less << |
|
NCT02784015
|
Unresectable Localized Soft Ti... More >>ssue Sarcoma Less << |
Phase 2 |
Recruiting |
May 2023 |
Korea, Republic of
... More >>
Samsung Medical Center
Recruiting
Seoul, Korea, Republic of
Contact: Ki Woong Sung 82-2-3410-3529 kwsped@skku.edu
Principal Investigator: Ki Woong Sung Less << |
|
NCT00256269
|
Esophageal Cancer
... More >> Gastroesophageal Cancer Less << |
Phase 2 |
Terminated(Study was terminate... More >>d by funding entity) Less << |
- |
United States, California
... More >>
Chao Family Comprehensive Cancer Center
Orange, California, United States, 92868 Less << |
|
NCT02862457
|
Neoplasms
Car... More >>cinoma, Non-Small-Cell Lung Less << |
Phase 1 |
Recruiting |
November 30, 2020 |
Japan
... More >> MSD K.K.
Recruiting
Chiyoda-Ku, Tokyo, Japan, 102-8667
Contact: Japan Call Center 81-3-6272-1957 Less << |
|
NCT03631784
|
Non-small Cell Lung Cancer |
Phase 2 |
Recruiting |
January 12, 2022 |
United States, California
... More >>
St Joseph Heritage Healthcare ( Site 1403)
Recruiting
Santa Rosa, California, United States, 95403
Contact: Study Coordinator 707-521-3830
United States, Indiana
Parkview Cancer Institute ( Site 1415)
Recruiting
Fort Wayne, Indiana, United States, 46845
Contact: Study Coordinator 833-724-8356
United States, Massachusetts
UMass Memorial Medical Center ( Site 1417)
Recruiting
Worcester, Massachusetts, United States, 01655
Contact: Study Coordinator 508-334-5539
United States, Michigan
Henry Ford Hospital ( Site 1418)
Recruiting
Detroit, Michigan, United States, 48202
Contact: Study Coordinator 313-916-2635
United States, Nebraska
St. Francis Cancer Treatment Center ( Site 1421)
Recruiting
Grand Island, Nebraska, United States, 68803
Contact: Study Coordinator 308-398-5450
United States, New Jersey
Rutgers Cancer Institute of New Jersey ( Site 1422)
Recruiting
New Brunswick, New Jersey, United States, 08903
Contact: Study Coordinator 732-235-6048
United States, Oklahoma
CTCA Southwestern ( Site 1428)
Recruiting
Tulsa, Oklahoma, United States, 74133
Contact: Study Coordinator 918-286-5448
Australia, New South Wales
Blacktown Hospital Western Sydney Local Health District ( Site 0204)
Recruiting
Blacktown, New South Wales, Australia, 2148
Contact: Study Coordinator +61403170371
MNCCI Port Macquarie Base Hospital ( Site 0200)
Recruiting
Port Macquarie, New South Wales, Australia, 2444
Contact: Study Coordinator +61265801818
Australia, Victoria
Ballarat Health Services ( Site 0206)
Recruiting
Ballarat, Victoria, Australia, 3350
Contact: Study Coordinator +61353204735
Korea, Republic of
Chungbuk National University Hospital ( Site 1003)
Recruiting
Cheongju si, Chungcheongbuk Do, Korea, Republic of, 28644
Contact: Study Coordinator +82432696015
Samsung Medical Center ( Site 1001)
Recruiting
Seoul, Korea, Republic of, 06351
Contact: Study Coordinator +82234103459
Ulsan University Hospital ( Site 1000)
Recruiting
Ulsan, Korea, Republic of, 44033
Contact: Study Coordinator +82522508832
Poland
Centrum Onkologii im. Prof. Franciszka Lukaszczyka ( Site 0811)
Recruiting
Bydgoszcz, Poland, 85-796
Contact: Study Coordinator +48523743029
Osrodek Badan Klinicznych przy Szpitalu Specjalistycznym ( Site 0802)
Recruiting
Krakow, Poland, 31-826
Contact: Study Coordinator +48721295010
Centrum Onkologii Instytut im. Marii Sklodowskiej Curie ( Site 0800)
Recruiting
Warszawa, Poland, 02-781
Contact: Study Coordinator +48225463066
Russian Federation
Republican Clinical Oncology Dispensary of Tatarstan MoH ( Site 0910)
Recruiting
Kazan, Russian Federation, 420029
Contact: Study Coordinator +79172662851
N.N. Blokhin National Medical Oncology Research Centre ( Site 0902)
Recruiting
Moscow, Russian Federation, 115478
Contact: Study Coordinator +79031990755
National Medical Research Center of Oncology n.a. N. N. Petrov ( Site 0904)
Recruiting
St. Petersburg, Russian Federation, 197758
Contact: Study Coordinator +79117500005
Republican Clinical Oncology Dispensary of Republic of Bashkortostan ( Site 0903)
Recruiting
Ufa, Russian Federation, 450054
Contact: Study Coordinator +79050058625
Spain
Hospital Universitari Vall d Hebron ( Site 1101)
Recruiting
Barcelona, Spain, 08035
Contact: Study Coordinator +34934894158
Hospital Clinic de Barcelona ( Site 1100)
Recruiting
Barcelona, Spain, 08036
Contact: Study Coordinator +34932275402
Clinica Universitaria de Navarra ( Site 1102)
Recruiting
Madrid, Spain, 28027
Contact: Study Coordinator +349135319027502 Less << |
|
NCT03053856
|
Stage IIIA Non-small Cell Lung... More >> Cancer Less << |
Phase 2 |
Not yet recruiting |
August 13, 2021 |
Korea, Republic of
... More >>
Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine
Seoul, Korea, Republic of, 135-710 Less << |
|
NCT03469557
|
Esophageal, Gastric, or Gastro... More >>esophageal Junction Carcinoma Less << |
Phase 2 |
Recruiting |
March 23, 2019 |
China, Beijing
... More >> The 307th Hospital of Military Chinese People's Liberation Army
Recruiting
Beijing, Beijing, China, 100071
Contact: Jinchao Hu, BS 86-10-66947797 jgb947797@163.com
Principal Investigator: Jianming Xu, MD
China, Heilongjiang
The Tumor Hospital Affiliated to Harbin Medical University
Recruiting
Ha'erbin, Heilongjiang, China, 150006
Contact: Weilu Liu, BS 86-451-86298104 sylcyj609@163.com
Principal Investigator: Yuxian Bai, MD
China, Hubei
Tongji Medical College Huanzhong University of Science&Technology
Recruiting
Wuhan, Hubei, China, 430030
Contact: Chang Shu, BS 86-27-83663625 lsjhe12345@163.com
Principal Investigator: Xianglin Yuan, MD
China, Jiangsu
Clinical Medical School, Yangzhou University Affliated Hospital to Yangzhou University
Recruiting
Yangzhou, Jiangsu, China, 225001
Contact: Hua Zhu, BS 86-1805106131 yzsbhgcp@163.com
Principal Investigator: Buhai Wang, MD
China, Shanxi
The First Affiliated Hospital of Xi'an Jiaotong University
Recruiting
Xi'an, Shanxi, China, 710000
Contact: Mingying Lu, BS 86-29-85324094 Lumy3215@163.com
Principal Investigator: Xiaoen Li, MD
China, Zhejiang
The First Affiliated Hospital, Zhejiang University
Recruiting
Hangzhou, Zhejiang, China, 310003
Contact: Lihua Wu, BS 86-571-87236560 zyct79@163.com
Principal Investigator: Nong Xu, MD Less << |
|
NCT02066220
|
Brain Tumors |
Phase 2
Phase 3 |
Recruiting |
April 2024 |
- |
|
NCT01732939
|
- |
|
Unknown |
May 2014 |
China, Beijing
... More >> Hospital affiliated academy military medical science
Recruiting
Beijing, Beijing, China, 100071
Contact: Tao Wang, MD 8610-66947430
Contact: Shaohua ZHANG, MD 8610-66947173
Principal Investigator: Zefei Jiang, MD Less << |
|
NCT03421353
|
Carcinoma, Non-small-cell Lung... More >>
Neoplasms
Carcinoma, Bronchogenic
Bronchial Neoplasms
Thoracic Neoplasms
Lung Neoplasms
Respiratory Tract Neoplasms Less << |
Phase 1
Phase 2 |
Recruiting |
April 1, 2020 |
United States, Florida
... More >>
Research Site
Withdrawn
Tampa, Florida, United States, 33612
United States, Indiana
Research Site
Recruiting
Lafayette, Indiana, United States, 47905
United States, Oklahoma
Research Site
Recruiting
Oklahoma City, Oklahoma, United States, 73104
United States, Tennessee
Research Site
Recruiting
Nashville, Tennessee, United States, 37203
United States, Texas
Research Site
Recruiting
Dallas, Texas, United States, 75230
Research Site
Recruiting
Houston, Texas, United States, 77030
United States, Virginia
Research Site
Not yet recruiting
Fairfax, Virginia, United States, 22031
United Kingdom
Research Site
Not yet recruiting
London, United Kingdom, W1G 6AD Less << |
|
NCT00936364
|
Fasting in Malignant Solid Tum... More >>ors Less << |
Not Applicable |
Recruiting |
July 9, 2020 |
United States, California
... More >>
USC/Norris Comprehenseive Cancer Center
Recruiting
Los Angeles, California, United States, 90033
Contact: America Casillas-Lopez 323-865-0804 America.Casillas-Lopez@med.usc.edu
Principal Investigator: David Quinn, MD Less << |
|
NCT02741388
|
B-cell Lymphoma |
Phase 1 |
Recruiting |
June 2020 |
Belgium
... More >> Institut Jules Bordet
Recruiting
Bruxelles, Belgium
Contact: Marie MAEREVOET, MD +32 25 41 32 12
Principal Investigator: Marie MAEREVOET, MD
France
CHU Dijon
Recruiting
Dijon, France
Contact: Olivier CASASNOVAS, MD +33380295041
Principal Investigator: Olivier CASASNOVAS, MD
CHRU de Lille - Hôpital Claude Huriez
Recruiting
Lille, France
Contact: Franck MORSCHHAUSER, MD +33320445713
Principal Investigator: Franck MORSCHHAUSER, MD
CHU Montpellier - Hôpital Saint Eloi
Recruiting
Montpellier, France
Contact: Guillaume CARTRON, MD +33467338079
Principal Investigator: Guillaume CARTRON, MD
CHU Nancy - Hôpital de Brabois
Recruiting
Nancy, France
Contact: Pierre FEUGIER, MD +33383153282
Principal Investigator: Pierre FEUGIER, MD
Hôpital Saint-Louis
Recruiting
Paris, France
Contact: Catherine THIEBLEMONT, MD +33142499236
Principal Investigator: Catherine THIEBLEMONT, MD
CHU Bordeaux - Centre François Magendie
Recruiting
Pessac, France
Contact: Kamal BOUABDALLAH +33557656511
Principal Investigator: Kamal BOUABDALLAH, MD
Centre Henri Becquerel
Recruiting
Rouen, France
Contact: Hervé TILLY, MD +33232082202
Principal Investigator: Hervé TILLY, MD Less << |
|
NCT00005838
|
Adenocarcinoma of the Lung
... More >> Adenosquamous Cell Lung Cancer
Large Cell Lung Cancer
Squamous Cell Lung Cancer
Stage IIIA Non-small Cell Lung Cancer
Stage IIIB Non-small Cell Lung Cancer Less << |
Phase 3 |
Completed |
- |
United States, Texas
... More >>
M D Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT02822404
|
Kidney Diseases |
Not Applicable |
Active, not recruiting |
September 2020 |
France
... More >> CHU Toulouse
Toulouse, France, 31059 Less << |
|
NCT02103257
|
EGFR Positive Non-small Cell L... More >>ung Cancer
Adenocarcinoma Less << |
Phase 4 |
Unknown |
September 2017 |
China, Gansu
... More >> First Hospital of Lanzhou University
Active, not recruiting
Lanzhou, Gansu, China, 730000
Lanzhou military region general hospital
Active, not recruiting
Lanzhou, Gansu, China, 730050
China, Guangdong
General Hospital of Guangzhou Military Command
Active, not recruiting
Guangzhou, Guangdong, China, 510010
Cancer Hospital of Sun Yat-sen
Active, not recruiting
Guangzhou, Guangdong, China, 510060
First Affiliated Hospital of Guangzhou Medical College
Active, not recruiting
Guangzhou, Guangdong, China, 510120
Jiangmen central hospital
Active, not recruiting
Jiangmen, Guangdong, China, 529030
The university of Hong Kong-Shenzhen Hospital
Active, not recruiting
Shenzhen, Guangdong, China, 518000
Shenzhen People's Hospital
Active, not recruiting
Shenzhen, Guangdong, China, 518020
Medical Oncology,Shenzhen Second People's Hospital
Active, not recruiting
Shenzhen, Guangdong, China, 518035
Thoracic Surgery,Shenzhen Second People's Hospital
Active, not recruiting
Shenzhen, Guangdong, China, 518035
Peking University Shenzhen Hospital
Active, not recruiting
Shenzhen, Guangdong, China, 518036
Guangdong Agribusiness Center Hospital
Active, not recruiting
Zhanjiang, Guangdong, China, 524009
China, Guangxi
First Affiliated Hospital of Guangxi Medical University
Active, not recruiting
Nanning, Guangxi, China, 530021
China, Hainan
Hainan Provincal Nong Ken Hospital
Active, not recruiting
Haikou, Hainan, China, 570311
Hainan Provincial People's Hospital
Active, not recruiting
Haikou, Hainan, China, 570311
China, Henan
First Affiliated Hospital of Zhengzhou University
Active, not recruiting
Zhengzhou, Henan, China, 450052
China, Ningxia
Medical Oncology,General Hospital of Ningxia Medical University
Active, not recruiting
Yinchuan, Ningxia, China, 750004
Radiation Oncology,General Hospital of Ningxia Medical University
Active, not recruiting
Yinchuan, Ningxia, China, 750004
Respiratory medicine,General Hospital of Ningxia Medical University
Active, not recruiting
Yinchuan, Ningxia, China, 750004
China, Shanxi
Baoji Central Hospital
Active, not recruiting
Baoji, Shanxi, China, 721008
3201 Hospital, Hanzhong, Shanxi
Active, not recruiting
Hanzhong, Shanxi, China, 723000
Shaanxi province people's hospital
Active, not recruiting
Xi'an, Shanxi, China, 710000
Second Affiliated Hospital of Xi'an Jiaotong University
Active, not recruiting
Xi'an, Shanxi, China, 710004
Xi'an Chang'an Hospital
Active, not recruiting
Xi'an, Shanxi, China, 710018
Tangdu Hospital,Fourth Military Medical University
Recruiting
Xi'an, Shanxi, China, 710038
Contact: Helong Zhang, MD 13709202616
Principal Investigator: Helong Zhang, MD
First Affiliated Hospital of Xi'an Jiaotong University
Active, not recruiting
Xi'an, Shanxi, China, 710061
Shanxi Cancer Hospital
Active, not recruiting
Xi'an, Shanxi, China, 710061
China, Xinjiang
Urumqi General Hospital of Lanzhou Military Region General Hospital
Active, not recruiting
Urumqi, Xinjiang, China, 830000
Xinjiang medical university affiliated tumor hospital
Active, not recruiting
Urumqi, Xinjiang, China, 830000
First Affiliated Hospital of Xinjiang Medical University
Active, not recruiting
Urumqi, Xinjiang, China, 830054
Autonome Region Xinjiang Uygur Chinese medicine hospital
Active, not recruiting
Urumqi, Xinjiang, China, 830099 Less << |
|
NCT00718913
|
Gastric Cancer |
Phase 2 |
Completed |
- |
Greece
... More >> University Hospital of Crete Dept of Medical Oncology
Heraklion, Crete, Greece
University General Hospital of Alexandroupolis, Dep of Medical Oncology
Alexandroupolis, Greece
"IASO" General Hospital of Athens, 1st Dep of Medical Oncology
Athens, Greece
"Laikon" General Hospital, Medical Oncology Unit, Propedeutic Dep of Internal Medicine
Athens, Greece
401 Military Hospital of Athens
Athens, Greece
Air Forces Military Hospital of Athens
Athens, Greece
General Hospital of Larissa Dept of Medical Oncology
Larissa, Greece
"Metaxa's" Anticancer Hospital of Piraeus, 1st Dep of Medical Oncology
Piraeus, Greece Less << |
|
NCT02352792
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Not Applicable |
Recruiting |
December 2022 |
Germany
... More >> Tuebingen university, radiation oncology
Recruiting
Tuebingen, Baden-Württemberg, Germany, 72076
Contact: Stefan Welz, Dr. med. +49 7071 29 86144 stefan.welz@med.uni-tuebigen.de Less << |
|
NCT03167177
|
Melanoma |
Phase 1
Phase 2 |
Not yet recruiting |
March 2019 |
- |
|
NCT02972372
|
Esophagus Cancer |
Not Applicable |
Recruiting |
June 2021 |
China, Henan
... More >> Anyang Tumor Hospital
Recruiting
Anyang, Henan, China
Contact: Fuyou Zhou, MD,PhD +86 13939998799 jiaruinuo@163.com
The First Affiliated Hospital, and College of Clinical Medicine of Henan University of Science and Technology
Recruiting
Luoyang, Henan, China, 471003
Contact: Shegan Gao, MD, PhD +86 18638859977 gsg112258@163.com
Contact: Ruinuo Jia, MD +86 18537950766 jiaruinuo@163.com
Principal Investigator: Ruinuo Jia, MD
Sub-Investigator: Xinshuai Wang, MD, PhD
Sub-Investigator: Guoqiang Kong, MD
Sub-Investigator: Baoping Chang, MD, PhD
Sub-Investigator: Jiachun Sun, MD, PhD
Sub-Investigator: Xiaozhi Yuan, MD
Sub-Investigator: Dan Zhou, MD
Sub-Investigator: Ruina Yang, MD
Sub-Investigator: Jing Ren, MD
Sub-Investigator: Shuoguo Li, MD
Sub-Investigator: Wei Wang, MD
Sub-Investigator: Shiyuan Song, MD
Sub-Investigator: Weijiao Yin, MD Less << |
|
NCT00117572
|
Cancer of the Pharynx
... More >> Cancer of the Larynx
Cancer of the Nasal Cavity
Paranasal Sinus Neoplasms
Cancer of the Oral Cavity Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00117572
|
- |
|
Completed |
- |
- |
|
NCT02339753
|
Pediatric Solid Tumor |
Phase 2 |
Unknown |
December 2016 |
Korea, Republic of
... More >>
Seoul National University Hospital
Recruiting
Seoul, Korea, Republic of
Principal Investigator: HeeYeong shin Less << |
|
NCT02815592
|
Small Cell Lung Cancer |
Phase 1
Phase 2 |
Active, not recruiting |
October 2, 2019 |
Canada, Alberta
... More >>
Local Institution
Edmonton, Alberta, Canada, T6G 1Z2
Canada, Ontario
Local Institution
Ottawa, Ontario, Canada, K1H 8L6
Spain
Local Institution
Barcelona, Spain, 08035
Local Institution
Madrid, Spain, 28041
Local Institution
Majadahonda - Madrid, Spain, 28222
Local Institution
Malaga, Spain, 29010 Less << |
|
NCT00791154
|
Lung Cancer
S... More >>mall Cell Lung Cancer
Solid Tumors
Extensive-stage Small Cell Lung Cancer Less << |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT03167164
|
Merkel Cell Carcinoma |
Phase 1
Phase 2 |
Not yet recruiting |
March 2019 |
- |
|
NCT01053390
|
Gallbladder Neoplasms |
Phase 3 |
Completed |
- |
China
... More >> Xinhua hospital affiliated shanghai jiaotong
Shanghai, China Less << |
|
NCT03574324
|
Locally Advanced Nasopharyngea... More >>l Carcinoma Less << |
Phase 3 |
Recruiting |
May 24, 2023 |
China, 贵州省
... More >> Cancer Hospital of Guizhou Medical University
Recruiting
Guiyang, 贵州省, China, 550000
Contact: Yuanyuan Li, master 085186512802 lilyuanyuan@qq.com
Sub-Investigator: Feng Jin, Bachelor
Sub-Investigator: Weili Wu, Master
Sub-Investigator: Jinhua Long, Master
Sub-Investigator: Xiuling Luo, Bachelor
Sub-Investigator: Xiuyun Gong, Bachelor
Sub-Investigator: Hang Jiang, Master
Sub-Investigator: Yanfang Cen, Bachelor Less << |
|
NCT03673735
|
Squamous Cell Head and Neck Ca... More >>rcinoma Less << |
Phase 3 |
Not yet recruiting |
December 2027 |
- |
|
NCT02629718
|
PFS
OS
... More >> Quality of Life Less << |
Phase 3 |
Recruiting |
December 2022 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center, Department of Gynecologic Oncology
Recruiting
GuangZhou, Guangdong, China, 510060
Contact: Ting Wan, Ph.D Less << |
|
NCT00135135
|
Neuroblastoma |
Phase 2 |
Completed |
- |
United States, Tennessee
... More >>
St. Jude Children's Research Hospital
Memphis, Tennessee, United States, 38105 Less << |
|
NCT03358472
|
Head and Neck Cancer |
Phase 3 |
Active, not recruiting |
June 2020 |
- |
|
NCT00766480
|
Esophageal Cancer |
Not Applicable |
Unknown |
- |
Japan
... More >> Aichi Cancer Center
Recruiting
Nagoya, Aichi, Japan, 464-8681
Contact: Shunzo Hatooka 81-52-762-6111
National Cancer Center Hospital East
Recruiting
Kashiwa, Chiba, Japan, 277-8577
Contact: Keiko Minashi 81-4-7133-1111
Kurume University School of Medicine
Recruiting
Kurume, Fukuoka, Japan, 830-0011
Contact: Toshiaki Tanaka 81-942-35-3311
Gunma University Graduate School of Medicine
Recruiting
Maebashi-shi, Gunma, Japan, 371-8511
Contact: Hiroyuki Kuwano, MD, PhD 81-27-220-8224 hkuwano@med.gunma-u.ac.jp
Hyogo College of Medicine
Recruiting
Nishinomiya, Hyogo, Japan, 663-8501
Contact: Koushi Oh 81-798-45-6582
Iwate Medical University Hospital
Recruiting
Morioka, Iwate, Japan, 020-8505
Contact: Kenichiro Ikeda 81-19-651-5111
Kawasaki Medical School
Recruiting
Kurashiki, Okayama, Japan, 701-01
Contact: Toshihiro Hirai 81-86-462-1199
Graduate School of Medical Science at the University of Ryukyu
Recruiting
Nishiharacho, Okinawa, Japan, 903-0215
Contact: Tadashi Nishimaki 81-98-895-3331
Kinki University School of Medicine
Recruiting
Osakasayama, Osaka, Japan, 589-8511
Contact: Takushi Yasuda 81-72-366-0221
Osaka University Graduate School of Medicine
Recruiting
Suita, Osaka, Japan, 565-0871
Contact: Masaichi Ohira 81-6-6645-3837
National Kyushu Cancer Center
Recruiting
Fukuoka, Japan, 811-1395
Contact: Yasushi Toh, MD, PhD 81-92-541-3231
Hiroshima City Asa Hospital
Recruiting
Hiroshima, Japan, 731-0293
Contact: Hidenori Mukaida 81-82-815-5211
Kagoshima University
Recruiting
Kagoshima, Japan, 890-8520
Contact: Shoji Natsugoe 81-99-275-5361
Kyoto University Hospital
Recruiting
Kyoto, Japan, 606-8507
Contact: Michihide Mitsumori 81-75-751-3417
Niigata University Medical and Dental Hospital
Recruiting
Niigata, Japan, 951-8510
Contact: Tatsuo Kanda, MD 81-25-227-2228
Niigata Cancer Center Hospital
Recruiting
Niigata, Japan, 951-8566
Contact: Hiroshi Yabusaki 81-25-266-5111
Osaka Medical Center for Cancer and Cardiovascular Diseases
Recruiting
Osaka, Japan, 537-8511
Contact: Ryu Ishihara 81-6-6972-1181
Mita Hospital at the International University of Health and Welfare
Recruiting
Tokyo, Japan, 108-8329
Contact: Ken-ichi Mafune, MD 81-3-3451-8121 mafune@iuhw.ac.jp
Tokyo Women's Medical University
Recruiting
Tokyo, Japan, 162-8666
Contact: Tsutomu Nakamura 81-3-3353-8111
Toyama University Hospital
Recruiting
Toyama, Japan, 930-8555
Contact: Yutaka Shimada, MD, PhD 81-76-434-7331 Less << |
|
NCT00003269
|
Breast Cancer
... More >> Drug/Agent Toxicity by Tissue/Organ
Lung Cancer
Lymphoma
Ovarian Cancer
Unspecified Adult Solid Tumor, Protocol Specific Less << |
Phase 2 |
Completed |
- |
United States, California
... More >>
Scripps Clinic
La Jolla, California, United States, 92037 Less << |
|
NCT00004164
|
Head and Neck Cancer |
Phase 2 |
Unknown |
- |
United States, Massachusetts
... More >>
Massachusetts General Hospital Cancer Center
Boston, Massachusetts, United States, 02114
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00586846
|
- |
|
Completed |
- |
- |
|
NCT02402920
|
Extensive Stage Small Cell Lun... More >>g Carcinoma
Limited Stage Small Cell Lung Carcinoma
Neuroendocrine Neoplasm Less << |
Phase 1 |
Recruiting |
July 31, 2023 |
United States, Texas
... More >>
M D Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact: James Welsh 713-563-2300
Principal Investigator: James Welsh Less << |
|
NCT00617656
|
LUNG CANCER |
Phase 3 |
Terminated(No safety reasons. ... More >>Interim analysis shows that the hypothesis superiority of the experimental arm over the control arm- would not be confirmed.) Less << |
- |
Spain
... More >> H. Germans Trias i Pujol
Badalona, Barcelona, Spain, 08916 Less << |
|
NCT00586846
|
Osteosarcoma |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT01437488
|
Urothelial Carcinoma |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
National Cancer Institute
Bethesda, Maryland, United States, 20892
United States, Pennsylvania
University of Pennsylvania
Philadelphia, Pennsylvania, United States, 19104
Thomas Jefferson University
Philadelphia, Pennsylvania, United States, 19107 Less << |
|
NCT01136031
|
Advanced Gastric Cancer |
Phase 1
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Seoul National University Hospital
Seoul, Korea, Republic of, 110-744 Less << |
|
NCT01247246
|
Oral Mucositis |
Phase 2 |
Unknown |
June 2014 |
- |
|
NCT03737994
|
ALK Gene Rearrangement
... More >> ALK Positive
Non-Squamous Non-Small Cell Lung Carcinoma
Stage IV Lung Cancer AJCC v8
Stage IVA Lung Cancer AJCC v8
Stage IVB Lung Cancer AJCC v8 Less << |
Phase 2 |
Not yet recruiting |
December 13, 2025 |
- |
|
NCT01524575
|
Metastatic Pancreatic Cancer
... More >> ERCC1 Less << |
Phase 2 |
Unknown |
January 2014 |
United States, Hawaii
... More >>
University of Hawaii
Recruiting
Honolulu, Hawaii, United States, 96813
Contact: Jared D Acoba, MD 808-531-8521 jacoba@hawaii.edu Less << |
|
NCT01437488
|
- |
|
Completed |
- |
- |
|
NCT00365183
|
Non-Small Cell Lung Carcinoma |
Phase 2 |
Terminated |
- |
United States, California
... More >>
Huntington Beach, California, United States
Long Beach, California, United States
United States, Illinois
Chicago, Illinois, United States
United States, Maryland
Baltimore, Maryland, United States
United States, Minnesota
Minneapolis, Minnesota, United States
United States, Missouri
St. Louis, Missouri, United States
United States, New Mexico
Albuquerque, New Mexico, United States
Santa Fe, New Mexico, United States
United States, New York
Armonk, New York, United States
United States, Ohio
Columbus, Ohio, United States
United States, Washington
Seattle, Washington, United States Less << |
|
NCT02819752
|
Squamous Cell Carcinoma of the... More >> Head and Neck
Squamous Cell Carcinoma Less << |
Phase 1 |
Unknown |
December 2018 |
- |
|
NCT00606502
|
- |
|
Completed |
- |
- |
|
NCT03250520
|
Brain Tumor, Pediatric, Brains... More >>tem Glioma
Brain Tumor, Pediatric, Recurrent Less << |
Early Phase 1 |
Active, not recruiting |
February 28, 2021 |
Mexico
... More >> Hospital Infantil de Mexico Federico Gomez
DF, Mexico Less << |
|
NCT03200340
|
Stomatitis |
Phase 2 |
Recruiting |
June 20, 2019 |
United States, Arkansas
... More >>
NEA Baptist Cancer Center
Recruiting
Jonesboro, Arkansas, United States, 72401
Contact: Emily Carvel
United States, Georgia
Winship Cancer Institute of Emory University
Recruiting
Atlanta, Georgia, United States, 30322
Contact: Swathi Chinarangari
United States, Illinois
NorthShore University HealthSystem
Recruiting
Evanston, Illinois, United States, 60201
Contact: Michelle Britto
United States, Indiana
Goshen Center for Cancer Care
Recruiting
Goshen, Indiana, United States, 46526
Contact: Jill Borsa
United States, Kansas
Cancer Center of Kansas
Recruiting
Wichita, Kansas, United States, 67214
Contact: Pat Stone
United States, Kentucky
University of Kentucky, Chandler Medical Center, CCTS
Recruiting
Lexington, Kentucky, United States, 40536
Contact: Linda Rice
United States, Maryland
Greater Baltimore Medical Center
Recruiting
Baltimore, Maryland, United States, 21204
Contact: Melissa Loomis
United States, Missouri
Washington University School of Medicine
Recruiting
Saint Louis, Missouri, United States, 63110-1010
Contact: Josh Smith
United States, Oregon
Oregon Health and Science University
Recruiting
Portland, Oregon, United States, 97239
Contact: John Minger
United States, Pennsylvania
St. Luke's Cancer Center - Anderson Campus
Recruiting
Easton, Pennsylvania, United States, 18045
Contact: Jillian Timer
United States, Texas
Hope Cancer Center of East Texas
Recruiting
Tyler, Texas, United States, 75701
Contact: Grace Loredo
Baylor Scott & White Medical Center - Hillcrest
Recruiting
Waco, Texas, United States, 76712
Contact: Dedra Preece Less << |
|
NCT03644342
|
Metastatic Carcinoma of the Ce... More >>rvix Less << |
Phase 1
Phase 2 |
Not yet recruiting |
March 2, 2026 |
United States, Texas
... More >>
Baylor College of Medicine
Not yet recruiting
Houston, Texas, United States, 77030
Contact: Michelle S Ludwig, MD, MPH, PhD 713-566-3757 Michelle.Ludwig@bcm.edu
Harris Health System - Smith Clinic
Houston, Texas, United States, 77054 Less << |
|
NCT00771810
|
Thrombocytopenia
... More >> Neutropenia
Lymphopenia
Anemia Less << |
Phase 2 |
Completed |
- |
United States, Alabama
... More >>
University of Southern Alabama Mitchell Cancer Institute
Mobile, Alabama, United States, 36604
United States, California
USC - LAC Medical Center
Los Angeles, California, United States, 90033
University of California - Irvine, Chao Family Comprehensive Cancer Center
Orange, California, United States, 92868
United States, Illinois
Rush University Medical Center
Chicago, Illinois, United States, 60612
United States, Kansas
Associates in Women's Health
Wichita, Kansas, United States, 67208
United States, New York
Schwartz Gynecologic Oncology, PLLC
Brightwaters, New York, United States, 11718 Less << |
|
NCT00771810
|
- |
|
Completed |
- |
- |
|
NCT00723658
|
Lymphoma |
Phase 2 |
Withdrawn(lack of accrual) |
- |
United States, Connecticut
... More >>
Saint Francis/Mount Sinai Regional Cancer Center at Saint Francis Hospital and Medical Center
Hartford, Connecticut, United States, 06105
United States, Indiana
St. Francis Hospital and Health Centers - Beech Grove Campus
Beech Grove, Indiana, United States, 46107
Reid Hospital & Health Care Services
Richmond, Indiana, United States, 47374
United States, Kansas
Lawrence Memorial Hospital
Lawrence, Kansas, United States, 66044
Wesley Medical Center
Wichita, Kansas, United States, 67214
United States, Michigan
Saint Joseph Mercy Cancer Center
Ann Arbor, Michigan, United States, 48106-0995
CCOP - Michigan Cancer Research Consortium
Ann Arbor, Michigan, United States, 48106
Oakwood Cancer Center at Oakwood Hospital and Medical Center
Dearborn, Michigan, United States, 48123-2500
Barbara Ann Karmanos Cancer Institute
Detroit, Michigan, United States, 48201-1379
Genesys Hurley Cancer Institute
Flint, Michigan, United States, 48503
Hurley Medical Center
Flint, Michigan, United States, 48503
Van Elslander Cancer Center at St. John Hospital and Medical Center
Grosse Pointe Woods, Michigan, United States, 48236
Foote Memorial Hospital
Jackson, Michigan, United States, 49201
Sparrow Regional Cancer Center
Lansing, Michigan, United States, 48912-1811
St. Mary Mercy Hospital
Livonia, Michigan, United States, 48154
St. Joseph Mercy Oakland
Pontiac, Michigan, United States, 48341-2985
Mercy Regional Cancer Center at Mercy Hospital
Port Huron, Michigan, United States, 48060
Seton Cancer Institute at Saint Mary's - Saginaw
Saginaw, Michigan, United States, 48601
St. John Macomb Hospital
Warren, Michigan, United States, 48093
United States, Ohio
Grandview Hospital
Dayton, Ohio, United States, 45405
Good Samaritan Hospital
Dayton, Ohio, United States, 45406
David L. Rike Cancer Center at Miami Valley Hospital
Dayton, Ohio, United States, 45409
CCOP - Dayton
Dayton, Ohio, United States, 45420
Blanchard Valley Medical Associates
Findlay, Ohio, United States, 45840
Middletown Regional Hospital
Franklin, Ohio, United States, 45005-1066
Charles F. Kettering Memorial Hospital
Kettering, Ohio, United States, 45429
UVMC Cancer Care Center at Upper Valley Medical Center
Troy, Ohio, United States, 45373-1300
Clinton Memorial Hospital
Wilmington, Ohio, United States, 45177
Ruth G. McMillan Cancer Center at Greene Memorial Hospital
Xenia, Ohio, United States, 45385 Less << |
|
NCT03135977
|
Extensive-stage Small Cell Lun... More >>g Cancer
Lung Cancer Less << |
Phase 2 |
Recruiting |
December 2018 |
China, Sichuan
... More >> Sicchuan cancer hospital
Recruiting
Chendu, Sichuan, China, 610000
Contact: xiaohong Cai, Doctor 8618036672884 xionghong_cai@hotmail.com Less << |
|
NCT01718327
|
Unresectable and Advanced Chol... More >>angiocarcinoma Less << |
Phase 2 |
Completed |
- |
France
... More >> Hôpital Beaujon
Clichy, France, 92118
Hôpital privé Jean Mermoz
Lyon, France, 69008
CHU La Timone
Marseille, France, 13005
Hôpital Saint Antoine
Paris, France, 75012
Institut Mutualiste Montsouris
Paris, France, 75014
Institut Gustave Roussy
Villejuif, France, 94805 Less << |
|
NCT02305654
|
Squamous Cell Carcinoma of the... More >> Penis, Usual Type Less << |
Phase 3 |
Recruiting |
July 2022 |
United Kingdom
... More >> The Royal Marsden NHS Foundation Trust
Recruiting
London, United Kingdom, SM2 5PT
Contact: Dr Alison Tree
St George's Hospital NHS Foundation Trust
Recruiting
London, United Kingdom, SW17 0QT
Contact: Mr Nick Watkin Less << |
|
NCT03627728
|
Gastric Cancer |
Phase 2 |
Recruiting |
December 2021 |
Italy
... More >> Struttura Complessa di OncologiaIRCCS- Istituto in Tecnologie Avanzate e Modelli Assistenziali in Oncologia Arcispedale Santa Maria Nuova
Recruiting
Reggio Emilia, Italy, 42123
Contact: gnoni roberta, biologist +390522295181 gnoni.roberta@ausl.re.it Less << |
|
NCT00200681
|
Multiple Myeloma |
Phase 3 |
Completed |
- |
France
... More >> CHU de Nantes, Service d'Hématologie
Nantes, France, 44093 Less << |
|
NCT02289547
|
Stomach Neoplasms |
Phase 3 |
Recruiting |
January 2019 |
Korea, Republic of
... More >>
St. Vincent's Hospital
Recruiting
Suwon, Gyeonggi-do, Korea, Republic of, 442-723
Contact: Byoungyong Shim, Ph.D., M.D 82-31-249-8457
Principal Investigator: Byoungyong Shim, M.D., Ph.D
Buchon St. Mary's Hospital
Recruiting
Buchon, Korea, Republic of
Contact: Cuk Jin Lee
Daejeon St. Mary's Hospital
Recruiting
Daejeon, Korea, Republic of
Contact: Ji Chan Park
Incheon St. Mary's Hospital
Recruiting
Incheon, Korea, Republic of
Contact: Jeo Ho Byen
Seoul St. Mary's Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Young Seon Hong
St. Mary's Hospital
Recruiting
Seoul, Korea, Republic of
Contact: In Sook Woo
Bundang Seoul National hospital
Recruiting
Sungnam, Korea, Republic of
Contact: Keun Wook Lee
Ujeongbu St. Mary's Hospital
Recruiting
Ujeongbu, Korea, Republic of
Contact: Yoon Ho Ko Less << |
|
NCT01993966
|
- |
|
Unknown |
December 2016 |
Taiwan
... More >> Department of Urology, National Taiwan University Hospital
Not yet recruiting
Taipei, No. 7, Chung Shans. Rd.,, Taiwan, 100
Contact: Kuo-How Huang, M.D.,Ph.D. 886-2-23123456
Contact: Chin-Ling Huang 886-2-23123456 ext 65233 d95447004@ntu.edu.tw
Department of Urology, National Taiwan University Hospital
Not yet recruiting
Taipei, No. 7, Chung Shans. Rd., Taiwan, 100
Contact: Kuo-How Huang, M.D.,Ph.D. 886-2-23123456 ext 65952 khhuang123@ntu.edu.tw Less << |
|
NCT00042952
|
Ovarian Dysgerminoma
... More >> Recurrent Malignant Testicular Germ Cell Tumor
Recurrent Ovarian Germ Cell Tumor
Stage II Malignant Testicular Germ Cell Tumor
Stage II Ovarian Germ Cell Tumor
Stage III Malignant Testicular Germ Cell Tumor
Stage III Ovarian Germ Cell Tumor
Testicular Seminoma Less << |
Phase 2 |
Terminated(Administratively co... More >>mplete.) Less << |
- |
United States, Illinois
... More >>
Cancer and Leukemia Group B
Chicago, Illinois, United States, 60606 Less << |
|
NCT02945852
|
Small Cell Lung Cancer |
Phase 2 |
Unknown |
December 2018 |
China, Zhejiang
... More >>
Zhejiang Cancer Hospital
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: Yun Fan, MD 0086-571-88122192 fanyun@csco.org.cn
Principal Investigator: Yun Fan, MD Less << |
|
NCT01201265
|
- |
|
Completed |
- |
- |
|
NCT01201265
|
Breast Cancer |
Phase 2 |
Completed |
- |
India
... More >>
Ahmedabad, India, 380009
Bangalore, India, 560029
Bangalore, India, 560054
Delhi, India, 110029
Gandhinagar, India, 382428
Mumbai, India, 400012
Mumbai, India, 400016
Mumbai, India, 400020
New Delhi, India, 110011
New Delhi, India, 110085
Pune, India, 411004
Vellore, India, 632004 Less << |
|
NCT03180177
|
Epithelial Ovarian Cancer
... More >> Fallopian Tube Cancer
Primary Peritoneal Carcinoma Less << |
Phase 3 |
Not yet recruiting |
July 1, 2022 |
China, Guangdong
... More >>
Department of Abdominal Surgery (Section 2), Affiliated Tumor Hospital of Guangzhou Medical University
Not yet recruiting
Guangzhou, Guangdong, China, 510095
Contact: shuzhong cui, M.D 0086-138-0251-3800 cuishuzhong@126.com
Contact: Xianzi Yang, M.D 0086-188-9853-4167 7097359@qq.com
Principal Investigator: shuzhong cui, M.D Less << |
|
NCT02643056
|
Advanced Head and Neck Squamou... More >>s Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01543412
|
Pancreatic Adenocarcinoma |
Phase 2 |
Completed |
- |
Italy
... More >> A.O. Treviglio-Caravaggio, P.le Ospedale n1
Treviglio, Bergamo, Italy, 24047
Azienda Ospedaliera "Di Liegro"
Gaeta, Latina, Italy, 04024
A.O. Ospedale S.Paolo
Milano, MI, Italy, 20100
A.O. S.Salvatore
Pesaro, PS, Italy, 61100
Ospedale Morelli
Sondalo, SO, Italy, 23100
Ospedali Riuniti Umberto I - GM Lancisi-G Salesi
Ancona, Italy, 60126
Ospedali Riuniti, Largo Barozzi, 1
Bergamo, Italy, 24128
Fondazione Poliambulanza, Via Bissolati 57
Brescia, Italy, 25100
A.O. Carlo Poma - Via Albertoni, 1
Mantova, Italy, 46100 Less << |
|
NCT01486992
|
Advanced Esophageal Carcinoma |
Phase 2 |
Unknown |
November 2012 |
China, Beijing
... More >> Zhang Xiaodong
Recruiting
Beijing, Beijing, China, 100142
Contact: XIAODONG ZHANG, MD 861088196175 zxd0829@yahoo.com.cn Less << |
|
NCT00002565
|
Lymphoma |
Phase 3 |
Completed |
- |
United States, Texas
... More >>
University of Texas - MD Anderson Cancer Center
Houston, Texas, United States, 77030
Brazil
Faculdade de Medicina do ABC
Sao Paulo, Brazil, 01224--010
Chile
Clinica Alemana
Santiago, Chile, 5951 Less << |
|
NCT00930605
|
Peripheral T-cell Lymphoma |
Phase 2 |
Completed |
- |
Thailand
... More >> King Chulalongkorn Memorial Hospital
Bangkok, Thailand Less << |
|
NCT00392886
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 3 |
Unknown |
- |
- |
|
NCT01743157
|
Metastatic Melanoma |
Phase 1
Phase 2 |
Completed |
- |
United States, California
... More >>
San Francisco Oncology Associates
San Francisco, California, United States, 94115 Less << |
|
NCT03719924
|
Squamous Cell Carcinoma |
Phase 2 |
Not yet recruiting |
April 15, 2022 |
- |
|
NCT03663166
|
Carcinoma, Non-Small-Cell Lung |
Phase 1
Phase 2 |
Recruiting |
March 1, 2027 |
United States, Florida
... More >>
H. Lee Moffitt Cancer Center and Research Institute
Recruiting
Tampa, Florida, United States, 33612
Contact: Austin Lannon 813-745-2169 Austin.Lannon@moffitt.org
Principal Investigator: Bradford Perez, MD
Sub-Investigator: Scott Antonia, MD, PhD
Sub-Investigator: Alberto Chiappori, MD
Sub-Investigator: Benjamin Creelan, MD
Sub-Investigator: Thomas Dilling, MD
Sub-Investigator: Jhanelle Gray, MD
Sub-Investigator: Eric Haura, MD
Sub-Investigator: Stephen Rosenberg, MD, MS
Sub-Investigator: Michael Shafique, MD
Sub-Investigator: Tawee Tanvetyanon, MD, MPH Less << |
|
NCT03373058
|
Epithelial Ovarian Cancer
... More >> Fallopian Tube Cancer
Primary Peritoneal Carcinoma Less << |
Phase 3 |
Not yet recruiting |
July 1, 2021 |
China, Guangdong
... More >>
Affiliated Tumor Hospital of Guangzhou Medical University
Not yet recruiting
Guangzhou, Guangdong, China, 510095
Contact: shuzhong cui, M.D 0086-138-0251-3800 cuishuzhong@126.com
Contact: Zhen Tang, M.M 0086-159-2081-9128 57932181@qq.com
Principal Investigator: shuzhong cui, M.D Less << |
|
NCT01044420
|
Advanced Esophageal Carcinoma |
Phase 2 |
Unknown |
June 2012 |
China, Beijing
... More >> Zhang Xiaodong
Recruiting
Beijing, Beijing, China, 100142
Contact: zhang xiaodong, MD 86-10-88196175 zxd0829@yahoo.com.cn Less << |
|
NCT00161174
|
- |
|
Completed |
- |
Netherlands
... More >> University Medical Centre Groningen
Groningen, Netherlands, 9713 GZ Less << |
|
NCT00514540
|
Prostate Cancer |
Phase 2 |
Completed |
- |
United States, California
... More >>
University of California-San Francisco
San Francisco, California, United States, 94143
United States, Texas
UT MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT02195973
|
Recurrent Ovarian Cancer |
Phase 1 |
Completed |
- |
United States, Alabama
... More >>
University of Alabama at Birmingham
Birmingham, Alabama, United States, 35294 Less << |
|
NCT02817113
|
Head and Neck Neoplasms |
Phase 1 |
Unknown |
December 2017 |
Taiwan
... More >> National Taiwan University Hospital
Not yet recruiting
Taipei, Taiwan, 100
Contact: Ruey-Long Hong, MD PhD
Taipei Veterans General Hospital
Recruiting
Taipei, Taiwan, 112
Contact: Muh-Hwa Yang, MD PhD
Chang Gung Memorial Hospital, Linkou Branch
Recruiting
Taoyuan, Taiwan, 333
Contact: Hung-Ming Wang, MD PhD Less << |
|
NCT00319839
|
Head and Neck Cancer |
Phase 2 |
Terminated(slow accrual) |
- |
United States, California
... More >>
Chao Family Comprehensive Cancer Center
Orange, California, United States, 92868 Less << |
|
NCT00319839
|
- |
|
Terminated(slow accrual) |
- |
- |
|
NCT03498196
|
Bladder Cancer
... More >> Metastatic Bladder Cancer
Invasive Bladder Cancer Less << |
Phase 1
Phase 2 |
Recruiting |
January 2025 |
United States, Texas
... More >>
Baylor Clinic
Not yet recruiting
Houston, Texas, United States, 77030
Contact: Jennifer M. Taylor, MD 713-798-4001 jennifer.taylor@bcm.edu
Baylor College of Medicine
Not yet recruiting
Houston, Texas, United States, 77030
Contact: Jennifer M. Taylor, M.D. 713-798-4001 jennifer.taylor@bcm.edu
Sub-Investigator: Martha P. Mims, M.D., Ph.D.
Sub-Investigator: Anita L. Sabichi, M.D.
Sub-Investigator: Aihua E. Yen, M.D.
Sub-Investigator: Seth Lerner, M.D.
Sub-Investigator: Guilherme Godoy, M.D.
Ben Taub General Hospital
Recruiting
Houston, Texas, United States, 77030
Contact: Jennifer M. Taylor, MD 713-798-4001 jennifer.taylor@bcm.edu
Sub-Investigator: Martha Mims, MD, PhD
Sub-Investigator: Aihua E. Yen, MD
Sub-Investigator: Seth Lerner, MD
Sub-Investigator: Guilherme Godoy, MD
Sub-Investigator: Anita L. Sabichi, MD
Michael E. DeBakey Veteran Affairs Medical Center
Not yet recruiting
Houston, Texas, United States, 77030
Contact: Jennifer M. Taylor, MD 713-791-1414 ext 26429 jennifer.taylor@bcm.edu
Sub-Investigator: Anita L. Sabichi, MD
Sub-Investigator: Aihua E. Yen, MD
Sub-Investigator: Guilherme Godoy, MD Less << |
|
NCT02603159
|
Stage III Esophageal Squamous ... More >>Cell Carcinoma
Stage II Esophageal Squamous Cell Carcinoma Less << |
Phase 3 |
Recruiting |
December 2019 |
China, Henan
... More >> The First Affiliated Hospital of Henan University of Science and Technology
Recruiting
Luoyang, Henan, China, 471003
Contact: shegan gao, doctor 0379 64811906 gsg112258@163.com
Contact: tanyou shan, master 0379 64815350 shantanyou@163.com Less << |
|
NCT00638937
|
- |
|
Completed |
- |
- |
|
NCT01963052
|
Metastatic Urothelial Cancer |
Phase 1 |
Active, not recruiting |
March 2019 |
United States, Alabama
... More >>
Site US00006
Birmingham, Alabama, United States, 35294
United States, Connecticut
Site US00002
New Haven, Connecticut, United States, 06510
United States, Michigan
Site US00001
Detroit, Michigan, United States, 48201
United States, Missouri
Site US00003
Saint Louis, Missouri, United States, 63110
United States, New York
Site US00009
Buffalo, New York, United States, 14263
United States, Pennsylvania
Site US00011
Pittsburgh, Pennsylvania, United States, 15232
United States, Tennessee
Site US00010
Nashville, Tennessee, United States, 37212
United States, Washington
Site US00008
Seattle, Washington, United States, 98109
Canada, British Columbia
Site CA00004
Vancouver, British Columbia, Canada, V5Z 1H5
Canada, Ontario
Site CA00007
Hamilton, Ontario, Canada, L8V 5C2
Site CA00005
Toronto, Ontario, Canada, M4N 3M5 Less << |
|
NCT00256230
|
Stage IV Melanoma |
Phase 1
Phase 2 |
Completed |
- |
United States, California
... More >>
Chao Family Comprehensive Cancer Center
Orange, California, United States, 92868 Less << |
|
NCT00134030
|
Localized Osteosarcoma
... More >> Metastatic Osteosarcoma Less << |
Phase 3 |
Active, not recruiting |
- |
- |
|
NCT02335424
|
Urothelial Cancer |
Phase 2 |
Active, not recruiting |
September 28, 2020 |
- |
|
NCT00638937
|
Recurrent Non-small Cell Lung ... More >>Cancer
Stage IIIB Non-small Cell Lung Cancer
Stage IV Non-small Cell Lung Cancer Less << |
Phase 2 |
Completed |
- |
Canada, Ontario
... More >>
Juravinski Cancer Centre at Hamilton Health Sciences
Hamilton, Ontario, Canada, L8V 5C2
The Ottawa Hospital Cancer Centre (Ottawa Health Research Institute) Civic Campus
Ottawa, Ontario, Canada, K1Y 4E9
University Health Network-Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9
Canada, Quebec
McGill University Department of Oncology
Montreal, Quebec, Canada, H2W 1S6 Less << |
|
NCT00336583
|
- |
|
Completed |
- |
- |
|
NCT03631199
|
Non-small Cell Lung Cancer |
Phase 3 |
Not yet recruiting |
October 21, 2022 |
- |
|
NCT00704639
|
Head and Neck Cancer |
Phase 1
Phase 2 |
Completed |
- |
Australia, Victoria
... More >>
Peter MacCallum Cancer Centre
Melbourne, Victoria, Australia, 3002 Less << |
|
NCT01467921
|
Curatively-resected, Node-posi... More >>tive Esophageal Squamous Cell Carcinoma Less << |
Phase 2 |
Unknown |
- |
Korea, Republic of
... More >>
Samsung Medical Center
Recruiting
Seoul, Korea, Republic of
Contact: Mi yeon kwon, RN +82-2-3410-1248 miyeon.kwon@samsung.com Less << |
|
NCT00336583
|
Non-Hodgkin's Lymphoma |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT02229656
|
Laryngeal Cancer Stage II
... More >> Laryngeal Cancer Stage III
Carcinoma, Squamous Cell
Head and Neck Neoplasms Less << |
Phase 1 |
Recruiting |
August 2020 |
Netherlands
... More >> The Netherlands Cancer Institute
Recruiting
Amsterdam, Netherlands, 1066 CX
Contact: Marcel Verheij, MD, PhD +31 20 512 2153 m.verheij@nki.nl
Contact: Rosemarie de Haan, MD +31 20 512 9085 ro.d.haan@nki.nl
Principal Investigator: Marcel Verheij, MD, PhD
Sub-Investigator: Margot Tesselaar, MD, PhD
Sub-Investigator: Abrahim Al-Mamgani, MD, PhD Less << |
|
NCT01836575
|
Non Small Cell Lung Carcinoma |
Phase 3 |
Completed |
- |
United States, Florida
... More >>
The Mount Sinai Comprehensive Cancer Center
Miami Beach, Florida, United States, 33140
Brazil
Instituto do Câncer do Ceará - ICC
Fortaleza, Ceará, Brazil
Hospital Lifecenter
Belo Horizonte, Minas Gerais, Brazil
Hospital Caridade de Ijuí - CACON
Ijuí, RS, Brazil
Hospital São Lucas
Porto Alegre, RS, Brazil
Centro de Pesquisas Oncológicas - CEPON
Florianópolis, Santa Catarina, Brazil
Hospital Amaral Carvalho
Jaú, São Paulo, Brazil
INCA
Rio de Janeiro, Brazil
Instituto do Câncer Arnaldo Vieira de Carvalho
São Paulo, Brazil Less << |
|
NCT03399643
|
- |
|
Active, not recruiting |
December 20, 2021 |
Germany
... More >> Praxis Dr.med. Wolfgang Hölzer
Berlin, Germany, 13055
Urologie Schlosscarree
Braunschweig, Germany, 38100
Coub Centrum F. Operative Urologie Bremen; Prof. Dr. Gerald Mickisch & Dr. Med. Roland Mattes
Bremen, Germany, 28277
Urologische Praxis Dr. Krieger
Chemnitz, Germany, 09127
Zeisigwaldkliniken Bethanien
Chemnitz, Germany, 09130
St. Joseph-Stift Dresden; Praxis für Urologie
Dresden, Germany, 01307
St. Georg Klinikum Eisenach GmbH
Eisenach, Germany, 99817
St. Franziskus-Hospital; Malteser Krankenhaus; Medizinische Klinik I
Flensburg, Germany, 24939
Klinikum Frankfurt Höchst GmbH; Klinik Innere Medizin 3 - Hämatologie, Onkologie, Palliativmedizin
Frankfurt am Main, Germany, 65929
Dres.Jochen Wilke und Harald Wagner
Fürth, Germany, 90766
Krankenhaus Martha-Maria Halle-Dölau, Klinik für Urologie
Halle (Saale), Germany, 06120
Urologische Praxis Michael Steinacker
Halle, Germany, 06120
Asklepios Klinik Altona, Abteilung für Urologie
Hamburg, Germany, 22763
Onkologische Praxis in Heidenheim
Heidenheim, Germany, 89522
St. Bernward-Krankenhaus
Hildesheim, Germany, 31134
Onkologische Praxis Landshut
Landshut, Germany, 84036
Gemeinschaftspraxis; Onkologisches Zentrum Lebach; Caritas Krankenhaus Lebach
Lebach, Germany, 66822
ÜBAG MVZ Mitte / MVZ Delitzsch GmbH, Standort Leipzig
Leipzig, Germany, 04103
Sankt Elisabeth Krankenhaus; Urlogische Abt.
Leipzig, Germany, 04277
Drk Krankenhaus Luckenwalde
Luckenwalde, Germany, 14943
Universitätsklinikum Schleswig-Holstein / Campus Lübeck; Klinik für Urologie
Lübeck, Germany, 23538
Urologikum Lübeck
Lübeck, Germany, 23566
Urologischen Praxis in Lüneburg
Lüneburg, Germany, 21335
Medizinische Fakultät Mannheim, Universitätsklinikum Mannheim, Klinik für Urologie
Mannheim, Germany, 68167
Praxis Markkleeberg
Markkleeberg, Germany, 04416
Mühlenkreiskliniken; Johannes Wesling Klinikum Minden; Klinik für Hämatologie, Onkologie und Pallia.
Minden, Germany, 32429
Städt.Kliniken München GmbH Klinikum Bogenhausen
München, Germany, 81925
Praxis Dr. Uhlig, Naunhof
Naunhof, Germany, 04683
Brüderkrankenhaus St. Josef Paderborn, Klinik für Hämatologie/Onkologie
Paderborn, Germany, 33098
Praxis für Hämatologie & Onkologie
Saarbruecken, Germany, 66113
MVZ für Hämatologie, Onkologie, Strahlentherapie und Palliativmedizin -; Klinik Dr. Hancken
Stade, Germany, 21680
Krankenhaus der Barmherzigen Brüder Trier; Innere Medizin I, Hämatologie / Internistische Onkologie
Trier, Germany, 54292
Urologische Praxis
Wedel, Germany, 22880
Gemeinschaftspraxis für Hämatologie und Onkologie Westerstede Aurich Rhauder
Westerstede, Germany, 26655 Less << |
|
NCT01487720
|
Prostate Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT01379482
|
Peritoneal Carcinomatosis
... More >> Gastric Cancer Less << |
Phase 2 |
Completed |
- |
Sweden
... More >> Uppsala University
Uppsala, Sweden Less << |
|
NCT00231842
|
Uterine Cancer |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Montefiore Medical Center
Bronx, New York, United States, 10461 Less << |
|
NCT00953420
|
Nasopharyngeal Carcinoma |
Phase 2 |
Completed |
- |
United States, Texas
... More >>
Houston Methodist Hospital
Houston, Texas, United States, 77030
MD Anderson Cancer Center
Houston, Texas, United States, 77030
Texas Children's Hospital
Houston, Texas, United States, 77030 Less << |
|
NCT01383148
|
Non-Small-Cell Lung Carcinoma |
Phase 2
Phase 3 |
Terminated |
- |
- |
|
NCT00130936
|
Gastric Cancer
... More >> Esophageal Cancer
Tumors Less << |
Phase 1
Phase 2 |
Terminated |
- |
Canada, Alberta
... More >>
Cross Cancer Institute
Edmonton, Alberta, Canada, T6G 1Z2 Less << |
|
NCT00533533
|
Gastric Cancer |
Phase 1
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of, 135-710 Less << |
|
NCT01921426
|
Squamous Cell Carcinoma of the... More >> Oral Cavity
Squamous Cell Carcinoma of the Oropharynx Less << |
Phase 1 |
Completed |
- |
United States, Florida
... More >>
Lakeland Regional Cancer Center
Lakeland, Florida, United States, 33805
United States, Iowa
University of Iowa
Iowa City, Iowa, United States, 52242
United States, Missouri
Washington University School of Medicine
St. Louis, Missouri, United States, 63110
United States, Nebraska
University of Nebraska Medical Center
Omaha, Nebraska, United States, 68198
United States, New York
University of Rochester Medical Center
Rochester, New York, United States, 14642
United States, North Carolina
Wake Forest Baptist Health
Winston-Salem, North Carolina, United States, 27157
United States, Ohio
Summa Health System- Cooper Cancer Center
Akron, Ohio, United States, 44304
United States, Pennsylvania
St. Luke's Cancer Center
Easton, Pennsylvania, United States, 18045
Allegheny General Hospital
Pittsburgh, Pennsylvania, United States, 15212
United States, Washington
Cancer Care Northwest
Spokane, Washington, United States, 99216 Less << |
|
NCT02921854
|
Non Small Cell Lung Cancer |
Not Applicable |
Recruiting |
January 2019 |
Belgium
... More >> Unuversity Hospitals Leuven
Not yet recruiting
Leuven, Belgium, 3000
Contact: Maarten Lambrecht +32 16 34 76 00 maarten.lambrecht@uzleuven.be
Principal Investigator: Maarten Lambrecht
Netherlands
MAASTRO clinic
Recruiting
Maastricht, Netherlands, 6229 ET
Contact: Chantal Overhof-Wedick +31 88 44 55 686 chantal.overhof@maastro.nl
Principal Investigator: Dirk De Ruysscher, MD, PhD Less << |
|
NCT00176735
|
Pancreatic Cancer |
Not Applicable |
Terminated |
- |
- |
|
NCT03762161
|
Bladder Cancer |
Phase 2 |
Not yet recruiting |
December 2023 |
United States, Kansas
... More >>
University of Kansas Cancer Center - Clinical Research Center
Not yet recruiting
Fairway, Kansas, United States, 66208
Contact: kerry hepler 913-945-7552 ctnursenav@kumc.edu
Principal Investigator: Rahul Parikh, MD
University of Kansas Cancer Center, Westwood Campus
Not yet recruiting
Kansas City, Kansas, United States, 66205
Contact: Clinical Trials Nurse Navigator 913-945-7552 ctnursenav@kumc.edu
Principal Investigator: Rahul Parikh, MD Less << |
|
NCT01613677
|
Treatment for Metastatic or Lo... More >>cally Advanced Cervical Cancer Less << |
Phase 2 |
Withdrawn |
September 2017 |
Brazil
... More >> Novartis Investigative Site
Rio de Janiero, RJ, Brazil, 20231-050 Less << |
|
NCT01966913
|
Germ Cell Tumor |
Phase 1
Phase 2 |
Recruiting |
September 2018 |
France
... More >> Hopital Tenon
Recruiting
Paris, France, 75012 Less << |
|
NCT00130520
|
- |
|
Completed |
- |
- |
|
NCT03006614
|
Breast Cancer Model
... More >> Effects of Chemotherapy
Breast Cancer Less << |
Phase 3 |
Recruiting |
March 2023 |
China, Guangdong
... More >>
Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Yanxia Shi, doctor 13609058827 shiyx@sysucc.org.cn Less << |
|
NCT00606502
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT02499367
|
Breast Cancer |
Phase 2 |
Recruiting |
August 2022 |
Netherlands
... More >> Antoni van Leeuwenhoek
Recruiting
Amsterdam, Netherlands, 1066 CX
Contact: Marleen Kok, MD +3120512 ext 7901 m.kok@nki.nl
Contact: Ingrid AM Mandjes, MSc +3120512 ext 2667 i.mandjes@nki.nl
Principal Investigator: Marleen kok, MD Less << |
|
NCT00130520
|
Ovarian Neoplasms |
Phase 2 |
Completed |
- |
United States, Arizona
... More >>
University of Arizona Cancer Center
Tucson, Arizona, United States, 85724 Less << |
|
NCT01364493
|
Gastric Cancer |
Phase 2 |
Completed |
- |
China, Beijing
... More >> Lin Shen
Beijing, Beijing, China, 100142 Less << |
|
NCT00080028
|
Head and Neck Cancer
... More >> Oropharynx Cancer
Larynx Cancer
Hypopharynx Cancer Less << |
Phase 1 |
Terminated |
- |
United States, North Carolina
... More >>
Duke University Medical Center
Durham, North Carolina, United States
United States, Texas
University of Texas, San Antonio Health Science Center
San Antonio, Texas, United States Less << |
|
NCT01259479
|
Solid Tumors
... More >>Brain Tumors
Brain Metastases Less << |
Phase 1 |
Completed |
- |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892 Less << |
|
NCT01330186
|
- |
|
Unknown |
May 2012 |
Denmark
... More >> Department of Oncology Herlev Hospital
Recruiting
Herlev, Denmark, 2730
Contact: Eva Serup-Hansen, MD +4538689084 evseha01@heh.regionh.dk
Contact: Hanne Havsteen, MD +4538682287 hahav@heh.regionh.dk
Principal Investigator: Eva Serup-Hansen, MD Less << |
|
NCT01503372
|
Advanced Gastric Cancer |
Phase 2 |
Unknown |
May 2015 |
Germany
... More >> Charite University medicine
Recruiting
Berlin, Germany, 13353
Contact: Mario Lorenz + 49 30-450 553 889 magenkarzinom@charite.de
Contact: Daniela Bohnen + 49 30-450 553 889 magenkarzinom@charite.de
Principal Investigator: Peter Thuss-Patience, Dr.
Sub-Investigator: Kirstin Breithaupt, Dr. Less << |
|
NCT00787488
|
Ovarian Cancer |
Not Applicable |
Withdrawn(Study not funded) |
- |
- |
|
NCT01998919
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Australia
... More >>
Campbelltown, Australia, 2560
Camperdown, Australia, 2050
Liverpool, Australia, 2170
China
Guangzhou, China, 510060
Guangzhou, China, 510080
Shanghai, China, 200030
Shanghai, China, 200433
Hong Kong
Hong Kong, Hong Kong
Indonesia
Jakarta, Indonesia, 10410
Jakarta, Indonesia, 10430
Jogjakarta, Indonesia, 55284
Semarang, Indonesia, 50136
Korea, Republic of
Kyunggi-do, Korea, Republic of, 411-769
Philippines
Manila, Philippines, 1000
Metro Manila, Philippines, 1502
Taiwan
Taipei, Taiwan, 100
Taipei, Taiwan
Thailand
Bangkok, Thailand, 10400
Bangkok, Thailand, 10700 Less << |
|
NCT00965250
|
Thymoma
Thymi... More >>c Carcinoma
Thymic Carcinoid
Thymic Neuroendocrine Tumors Less << |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892
United States, New York
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00325351
|
Ovarian Neoplasms |
Phase 2 |
Completed |
- |
United States, Arizona
... More >>
Tucson, Arizona, United States, 85724
United States, Arkansas
Little Rock, Arkansas, United States, 72205
United States, California
Bakersfield, California, United States, 93309
La Jolla, California, United States, 92093
San Diego, California, United States, 92121
United States, Georgia
Savannah, Georgia, United States, 31404
United States, Indiana
South Bend, Indiana, United States, 46617
United States, Maryland
Baltimore, Maryland, United States, 21204
United States, New Mexico
Albuquerque, New Mexico, United States, 87131
United States, North Carolina
Winston-Salem, North Carolina, United States, 27157
United States, Ohio
Cleveland, Ohio, United States, 44109
United States, Oklahoma
Oklahoma City, Oklahoma, United States, 73104
United States, Tennessee
Knoxville, Tennessee, United States, 37920
United States, Virginia
Roanoke, Virginia, United States, 24014
Canada, Alberta
Calgary, Alberta, Canada, T2N 4N2
Canada, Ontario
Toronto, Ontario, Canada, M4N 3M5 Less << |
|
NCT00759824
|
Small Cell Lung Carcinoma |
Phase 2 |
Completed |
- |
Belgium
... More >> Department of Intensive Care Unit and Thoracic Oncology Institut Jules Bordet
Brussels, Belgium, 1000
Department of Pneumology CHU Charleroi
Charleroi, Belgium, 6000
Department of Pneumology Hôpital Saint-Joseph
Gilly, Belgium, 6060
Hôpital Ambroise Paré
Mons, Belgium, 7000
Department of Pneumology Centre Hospitalier de Mouscron
Mouscron, Belgium, 7700 Less << |
|
NCT02128243
|
Unresectable, Locally Advanced... More >> or Metastatic, Adenocarcinoma of the Stomach, or of the Gastro Esophageal Junction
Gastric Neoplasm
Gastroesophageal Junction Adenocarcinoma
Esophageal Adenocarcinoma
Gastric Adenocarcinoma
Esophageal Neoplasms Less << |
Phase 2 |
Recruiting |
September 2018 |
Germany
... More >> NCT-Med. Onkologie
Recruiting
Heidelberg, Baden-Württemberg, Germany, 69120
Contact: Georg Martin Haag, Dr. +49-6221 ext 5637630 GeorgMartin.Haag@med.uni-heidelberg.de Less << |
|
NCT03178552
|
Non-Small Cell Lung Cancer |
Phase 2
Phase 3 |
Recruiting |
January 2, 2022 |
- |
|
NCT01998919
|
- |
|
Completed |
- |
- |
|
NCT00995488
|
- |
|
Terminated(Terminated prematur... More >>ely due to low accrual.) Less << |
- |
- |
|
NCT00589290
|
Thymoma
Thymi... More >>c Carcinoma Less << |
Phase 2 |
Completed |
- |
United States, Indiana
... More >>
Indiana University Cancer Center
Indianapolis, Indiana, United States, 46202-5262
United States, Maryland
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892 Less << |
|
NCT02855944
|
Ovarian Cancer
... More >> Epithelial Ovarian Cancer
Fallopian Tube Cancer
Peritoneal Cancer Less << |
Phase 3 |
Recruiting |
June 2024 |
- |
|
NCT00589290
|
- |
|
Completed |
- |
- |
|
NCT00995488
|
Urothelial Cancer
... More >> Bladder Cancer Less << |
Phase 2 |
Terminated(Terminated prematur... More >>ely due to low accrual.) Less << |
- |
United States, Michigan
... More >>
University of Michigan Comprehensive Cancer Center
Ann Arbor, Michigan, United States, 48109 Less << |
|
NCT03575013
|
Urothelial Carcinoma |
Phase 1 |
Recruiting |
May 1, 2020 |
United States, Iowa
... More >>
University of Iowa Hospitals and Clinics
Recruiting
Iowa City, Iowa, United States, 52242
Contact: Rohan Garje, MD 319-356-1770 rohan-garje@uiowa.edu Less << |
|
NCT00742924
|
Sarcoma |
Phase 1 |
Completed |
- |
- |
|
NCT02285530
|
Mouth Neoplasms
... More >> Carcinoma, Squamous Cell Less << |
Phase 2 |
Recruiting |
December 2021 |
China, Shanghai
... More >>
Ninth People's Hospital, School of Medicine, Shanghai Jiao Tong University
Recruiting
Shanghai, Shanghai, China, 200011
Contact: Lai-ping Zhong, MD, PhD +86-21-23271699 ext 5160 zhonglaiping@163.com
Principal Investigator: Lai-ping Zhong, MD, PhD Less << |
|
NCT00965250
|
- |
|
Completed |
- |
- |
|
NCT02986568
|
Cervical Cancer
... More >> Uterine Cancer Less << |
Not Applicable |
Recruiting |
- |
Korea, Republic of
... More >>
Seoul National University Hospital
Recruiting
Seoul, Korea, Republic of
Contact: Seungmee Lee, MD 82-2-2072-2821 yumenoarika@naver.com
Contact: Hee Seung Kim, MD 82-2-2072-4863 bboddi0311@gmail.com
Principal Investigator: Hee Seung Kim, MD Less << |
|
NCT01326923
|
Head and Neck Squamous Cell Ca... More >>ncer Less << |
Phase 2 |
Terminated(Principle Investiga... More >>tor left the institution.) Less << |
- |
United States, Louisiana
... More >>
LSU Health Sciences Center
Shreveport, Louisiana, United States, 71103 Less << |
|
NCT00742924
|
- |
|
Completed |
- |
- |
|
NCT02064491
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Finland
... More >> Helsinki University Hospital
Espoo, Finland
Helsinki University Hospital
Helsinki, Finland
Oulu University Hospital
Oulu, Finland
Pori Central Hospital
Pori, Finland
Tampere University Hospital
Tampere, Finland
Turku University Hopital
Turku, Finland
Vaasa Central Hospital
Vaasa, Finland Less << |
|
NCT00321334
|
Non-small Cell Lung Cancer |
Phase 3 |
Completed |
- |
China, Guangdong
... More >>
Chinese Society of Lung Cancer
Guangzhou, Guangdong, China, 510080 Less << |
|
NCT00004917
|
Anemia
Head a... More >>nd Neck Cancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00006356
|
Ovarian Cancer |
Phase 3 |
Terminated(low accrual) |
- |
Italy
... More >> Spedali Civili
Brescia, Italy, 25123 Less << |
|
NCT02128230
|
Multiple Myeloma |
Phase 2 |
Recruiting |
May 2025 |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences
Recruiting
Little Rock, Arkansas, United States, 72205
Contact: Nathan M Petty 501-526-6990 ext 2435 pettynathanm@uams.edu
Contact: Doug Steward 501-526-6990 ext 2452 dmsteward@uams.edu
Sub-Investigator: Michele Fox, MD
Sub-Investigator: Maurizio Zangari, MD
Sub-Investigator: Atul Kothari, MD
Sub-Investigator: Carolina Schinke, MD
Sub-Investigator: Faith Davies, MD
Sub-Investigator: Gareth Morgan, MD
Sub-Investigator: Juan Crescencio, MD
Sub-Investigator: Mary Burgess, MD
Sub-Investigator: Meera Mohan, MD
Sub-Investigator: Muthukumar Radhakrishnan, MD
Sub-Investigator: Pankaj Mathur, MD
Sub-Investigator: Sandra Susanibar-Adaniya, MD
Sub-Investigator: Shadiqul Hoque, MD
Sub-Investigator: Sharmilan Thanendrarajan, MD Less << |
|
NCT00002894
|
Ovarian Cancer
... More >> Primary Peritoneal Cavity Cancer Less << |
Phase 3 |
Completed |
- |
United Kingdom
... More >> Middlesex Hospital- Meyerstein Institute
London, England, United Kingdom, WIT 3AA Less << |
|
NCT01377298
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 2 |
Unknown |
May 2014 |
Taiwan
... More >> National Taiwan University Hospital
Recruiting
Taipei, Taiwan, 100
Contact: Ruey-Long Hong, MD +886 2 23123456 ext 67510 rlhong@ntu.edu.tw
Sub-Investigator: Jo-Pai Chen, MD
Principal Investigator: Ruey-Long Hong, MD Less << |
|
NCT02309242
|
Sarcoma, Soft Tissue
... More >> Osteosarcoma Tumor Less << |
Not Applicable |
Unknown |
October 2018 |
Belgium
... More >> UZ Leuven , pediatric oncology
Recruiting
Leuven, Vl Brabant, Belgium, 3000
Contact: Anne Uyttebroeck, MD, PhD +32 16 343972 anne.uyttebroeck@uzleuven.be
Contact: Charlotte Sleurs, Msc +32 16 340650 charlotte.sleurs@uzleuven.be
Principal Investigator: Anne Uyttebroeck, MD, PhD Less << |
|
NCT02492503
|
Metastatic Carcinoma to the Ut... More >>erine Cervix
Recurrent Carcinoma Cervix
Cervix Carcinoma Recurrent Less << |
Phase 1
Phase 2 |
Unknown |
August 2015 |
India
... More >> AIIMS
New Delhi, India, 110029 Less << |
|
NCT00568815
|
Lymphoma |
Phase 2 |
Completed |
- |
China, Shanghai
... More >>
Fudan University Cancer Hospital
Shanghai, Shanghai, China, 200032 Less << |
|
NCT01118676
|
Locally Advanced Non Small Cel... More >>l Lung Cancer (NSCLC)* Less << |
Phase 1 |
Completed |
- |
France
... More >> CHU Toulouse Larrey
Toulouse, France, 31000
Institut Claudius Regaud
Toulouse, France, 31000 Less << |
|
NCT00155766
|
Cervical Cancer |
Phase 1 |
Unknown |
- |
Taiwan
... More >> National Taiwan University Hospital
Recruiting
Taipei, Taiwan
Contact: Chi-An Chen, MD 886-2-2312-3456 ext 5157 cachen@ha.mc.ntu.edu.tw Less << |
|
NCT03582475
|
Bladder Small Cell Neuroendocr... More >>ine Carcinoma
Castration-Resistant Prostate Carcinoma
Metastatic Bladder Urothelial Carcinoma
Metastatic Urethral Urothelial Carcinoma
Prostate Carcinoma Metastatic in the Bone
Prostate Neuroendocrine Neoplasm
Prostate Small Cell Carcinoma
Stage III Bladder Cancer AJCC v8
Stage III Prostate Cancer AJCC v8
Stage III Urethral Cancer AJCC v8
Stage IV Bladder Cancer AJCC v8
Stage IV Prostate Cancer AJCC v8
Stage IV Urethral Cancer AJCC v8
Stage IVA Bladder Cancer AJCC v8
Stage IVB Bladder Cancer AJCC v8
Ureter Small Cell Carcinoma
Urothelial Carcinoma Less << |
Phase 1 |
Not yet recruiting |
September 1, 2021 |
United States, California
... More >>
UCLA / Jonsson Comprehensive Cancer Center
Not yet recruiting
Los Angeles, California, United States, 90095
Contact: Arnold Chin 310-206-6453
Principal Investigator: Arnold Chin Less << |
|
NCT01304784
|
HER-2 Gene Amplification |
Phase 1 |
Completed |
- |
United States, Colorado
... More >>
Rocky Mountain Cancer Centers
Denver, Colorado, United States, 80218
United States, Georgia
Georgia Cancer Specialists
Atlanta, Georgia, United States, 30342
United States, Indiana
Central Indiana Cancer Centers
Indianapolis, Indiana, United States, 46151
Horizon Oncology Research, Inc
Lafayette, Indiana, United States, 47908
United States, Nevada
Comprehensive Cancer Centers of Nevada
Las Vegas, Nevada, United States, 89135
United States, New York
New York Oncology/Hematology
Albany, New York, United States, 12206
United States, Ohio
Innovation Center - Kettering Medical Center Health Network
Kettering, Ohio, United States, 45429
United States, Pennsylvania
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111
United States, South Carolina
GHS Institute of Transitional Oncology Research
Greenville, South Carolina, United States, 29605
United States, Texas
Texas Oncology Cancer Center
Austin, Texas, United States, 78731
Texas Oncology PA North/Sammans Cancer Center
Dallas, Texas, United States, 75246
Texas Oncology - Tyler
Tyler, Texas, United States, 75702
United States, Virginia
Virginia Oncology Associates
Norfolk, Virginia, United States, 23502
United States, Washington
Evergreen Hematology and Oncology
Spokane, Washington, United States, 99218
Northwest Cancer Specialists-Vancouver Cancer Center
Vancouver, Washington, United States, 98684 Less << |
|
NCT02938546
|
Non-small-cell Lung Cancer |
Phase 3 |
Not yet recruiting |
January 2023 |
- |
|
NCT01715233
|
Metastatic Esophageal Cancer
... More >> Gastroesophageal Cancer
Gastric Cancer Less << |
Phase 2 |
Recruiting |
November 2019 |
United States, Maryland
... More >>
SKCCC at Johns Hopkins
Recruiting
Baltimore, Maryland, United States, 21287
Contact: Charles P Raines, CRNP, MSN 410-502-3696 craines1@jhmi.edu Less << |
|
NCT02394652
|
Uterine Cervical Neoplasms
... More >> Squamous Cell Carcinoma
Adenocarcinoma
Carcinoma, Adenosquamous Less << |
Phase 2 |
Recruiting |
December 2019 |
Canada, Alberta
... More >>
Tom Baker Cancer Centre
Not yet recruiting
Calgary, Alberta, Canada, T2N 4N2
Contact: Corinne Doll, M.D. 403-521-3685
Cross Cancer Institute
Not yet recruiting
Edmonton, Alberta, Canada, T6G 1Z2
Contact: Ericka Wiebe, M.D. 480-432-8755
Canada, Ontario
Princess Margaret Cancer Centre
Recruiting
Toronto, Ontario, Canada, M5G 2M9
Contact: Kathy Han, M.D., M.Sc. 416-946-2919
Canada, Quebec
Hôpital Maisonneuve-Rosemont
Not yet recruiting
Montréal, Quebec, Canada, H1T 2M4
Contact: Eve-Lyne Marchand, M.D., Ph.D. 514-252-3425
Centre Hospitalier De L'Université de Montréal
Not yet recruiting
Montréal, Quebec, Canada, H2L 4M1
Contact: Maroie Barkati, M.D. 514-890-8000 ext 28820 Less << |
|
NCT00005834
|
Multiple Myeloma |
Phase 3 |
Terminated(poor accrual) |
- |
- |
|
NCT03144466
|
Cervix Cancer |
Phase 1 |
Suspended(Slow recruitment.) |
June 2019 |
United Kingdom
... More >> The Royal Marsden NHS Foundation Trust
London, United Kingdom, SW3 6JJ Less << |
|
NCT00500760
|
Head and Neck Cancer
... More >> Squamous Cell Carcinoma Less << |
Phase 2 |
Completed |
- |
- |
|
NCT02807636
|
Urothelial Carcinoma |
Phase 3 |
Active, not recruiting |
November 17, 2020 |
- |
|
NCT03604653
|
- |
|
Recruiting |
May 2021 |
United States, New Jersey
... More >>
Holy Name Medical Center
Recruiting
Teaneck, New Jersey, United States, 07666
Contact: Sung Kwon, MD 201-541-5989 steve-kwonmd@mail.holyname.org
Contact: Julie Schwegman, PAC 201-541-5989 schwegman@holyname.org
Principal Investigator: Sung Kwon, MD
Principal Investigator: Sharyn Lewin, MD
Principal Investigator: Maria Schiavone, MD Less << |
|
NCT01926483
|
Non-small Cell Lung Cancer Sta... More >>ge IIIA Less << |
Phase 2 |
Unknown |
June 2016 |
China, Zhejiang
... More >>
Zhejiang Cancer Hospital
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: Xu Yaping +0086-571-88122082 xuyaping1207@gmail.com Less << |
|
NCT00500760
|
- |
|
Completed |
- |
- |
|
NCT00003606
|
Lung Cancer |
Phase 3 |
Unknown |
- |
France
... More >> Hopital Arnaud de Villeneuve
Montpellier, France, 34295 Less << |
|
NCT01028729
|
Non-small Cell Lung Cancer (NS... More >>CLC) Less << |
Phase 4 |
Unknown |
August 2011 |
China, Zhejiang
... More >>
Zhejiang Cancer Hospital
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: Yiping Zhang, Dr. 86-0571-88122188 Less << |
|
NCT03100409
|
Uterine Cervical Neoplasm
... More >> Dietary Modification
Radiation Toxicity
Gastrointestinal Complication Less << |
Not Applicable |
Recruiting |
August 2020 |
Mexico
... More >> National Cancer Institute of Mexico
Recruiting
Mexico City, Tlalpan, Mexico, 14080
Contact: LUCELY CETINA, MD,MSc +5215531952791 lucelycetina.incan@gmail.com
Contact: Angel D Castro, RDN, PhD +5215541940650 angeldenisse@gmail.com Less << |
|
NCT02358031
|
Recurrent Head and Neck Cancer... More >>
Metastatic Head and Neck Cancer Less << |
Phase 3 |
Active, not recruiting |
January 17, 2020 |
- |
|
NCT01612286
|
Nasopharyngeal Carcinoma |
Phase 2 |
Unknown |
May 2014 |
China, Zhejiang
... More >>
Zhejiang cancer hospital
Hangzhou, Zhejiang, China, 310022 Less << |
|
NCT01209520
|
Lung Cancer
N... More >>on Small Cell Lung Carcinoma
Hypermethylation Less << |
Not Applicable |
Completed |
- |
United States, Florida
... More >>
University of Miami Sylvester Comprehensive Cancer Center
Miami, Florida, United States, 33136 Less << |
|
NCT01209520
|
- |
|
Completed |
- |
- |
|
NCT01091636
|
Epithelial Ovarian Cancer |
Phase 2
Phase 3 |
Unknown |
December 2017 |
Korea, Republic of
... More >>
National Cancer Center
Goyang, Gyeonggi, Korea, Republic of, 410-769 Less << |
|
NCT01171170
|
Non Small Cell Lung Cancer |
Phase 2 |
Unknown |
August 2014 |
Netherlands
... More >> VU medisch centrum
Recruiting
Amsterdam, Netherlands
Contact: Smit ef.smit@vumc.nl
Principal Investigator: Egbert F. Smit, MD PhD
Amphia Ziekenhuis
Recruiting
Breda, Netherlands
Contact: Aerts jaerts@amphia.nl
Principal Investigator: Joachim G. Aerts, MD PhD
Jeroen Bosch Ziekenhuis
Recruiting
Den Bosch, Netherlands
Contact: Biesma b.biesma@jbz.nl
Principal Investigator: Bonne Biesma, MD PhD
Catharina-Ziekenhuis
Recruiting
Eindhoven, Netherlands
Contact: van den Borne ben.vd.borne@cze.nl
Principal Investigator: Ben E. van den Borne, MD PhD
Martini Ziekenhuis
Recruiting
Groningen, Netherlands
Contact: van Putten jwg.van.putten@mzh.nl
Principal Investigator: John W. van Putten, MD PhD
Maastricht UMC
Recruiting
Maastricht, Netherlands, 6202 AZ
Contact: Anne-Marie C. Dingemans, MD PhD a.dingemans@mumc.nl
Principal Investigator: Anne-Marie C. Dingemans, MD PhD
HagaZiekenhuis
Recruiting
The Hague, Netherlands
Contact: Codrington h.codrington@hagaziekenhuis.nl
Principal Investigator: Henk E. Codrington, MD
Isala Klinieken
Recruiting
Zwolle, Netherlands
Contact: Stigt j.a.stigt@isala.nl
Principal Investigator: Jos A. Stigt, MD Less << |
|
NCT00691236
|
Osteosarcoma |
Phase 2
Phase 3 |
Unknown |
August 2013 |
India
... More >> Tata Memorial Hospital
Recruiting
Mumbai, Maharashtra, India, 400012
Principal Investigator: Manish G Agarwal, M.S(Orth) Less << |
|
NCT01209507
|
- |
|
Completed |
- |
United States, Colorado
... More >>
University of Colorado Denver Anschutz Medical Campus
Aurora, Colorado, United States, 80045 Less << |
|
NCT00580372
|
Multiple Myeloma |
Phase 2 |
Completed |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences/MIRT
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT00580372
|
- |
|
Completed |
- |
- |
|
NCT01946867
|
Head and Neck Cancer |
Phase 1 |
Unknown |
August 2017 |
France
... More >> Centre Oscar Lambret
Recruiting
Lille, France, 59000
Contact: Xavier Mirabel, MD
Institut Curie
Recruiting
Paris, France, 75005
Contact: Christophe LE TOURNEAU, MD-PhD
Contact: José RODRIGUEZ, MD
Principal Investigator: Christophe LE TOURNEAU, MD-PhD
Sub-Investigator: José RODRIGUEZ, MD
Sub-Investigator: Valentin CALUGARU, MD-PhD
CHU Pontchaillou
Recruiting
Rennes, France, 35033
Contact: Franck JEGOUX, Pr, MD
Spain
START MADRID, Hospital Fundacion Jimenez Diaz
Recruiting
Madrid, Spain
Contact: Victor MORENO, MD-PhD
START MADRID, Hospital Universitario Madrid Norte Sanchinarro
Recruiting
Madrid, Spain
Contact: Emiliano CALVO, MD-PhD Less << |
|
NCT03612232
|
Adrenocortical Carcinoma |
Phase 2 |
Not yet recruiting |
June 2022 |
Germany
... More >> University Hospital Würzburg
Not yet recruiting
Würzburg, Germany, 97080
Contact: Matthias Kroiss, MD, PhD +4993120139740 Kroiss_M@ukw.de
Contact: Martin Fassnacht, MD +4993120139200 Fassnacht_M@ukw.de Less << |
|
NCT01167530
|
Non-small Cell Lung Cancer
... More >> Locally Advanced Disease Less << |
Phase 1 |
Unknown |
July 2014 |
France
... More >> Institut Gustave Roussy
Recruiting
Villejuif, France, 94800
Contact: Eric DEUTSCH, MD 33 1 42114413 eric.deutsch@igr.fr
Contact: Jean-Pierr PIGNON, MD 33 1 42114565 jean-pierre.pignon@igr.fr Less << |
|
NCT01253057
|
- |
|
Unknown |
December 2011 |
Taiwan
... More >> Taipei Medical University WanFang Hospital
Taipei, Taiwan Less << |
|
NCT00228488
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
Hong Kong
... More >>
Hong Kong, Hong Kong
Singapore
Singapore, Singapore Less << |
|
NCT02468661
|
Non-Small Cell Lung Cancer |
Phase 1
Phase 2 |
Recruiting |
March 31, 2020 |
- |
|
NCT00198393
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Completed |
- |
France
... More >> CHU Avicenne - Oncologie
Bobigny, France, 93000
CHU Grenoble - pneumologie
Grenoble, France, 38000 Less << |
|
NCT03603184
|
Endometrial Cancer |
Phase 3 |
Recruiting |
July 2022 |
Italy
... More >> AO SS Antonio e Biagio e Cesare Arrigo
Not yet recruiting
Alessandria, Italy
Contact: Vittorio Fusco
Policlinico S. Orsola Malpighi
Not yet recruiting
Bologna, Italy
Contact: Claudio Zamagni
Azienda Sanitaria dell'Alto Adige
Not yet recruiting
Bolzano, Italy
Contact: Maria R Lusso
ASST degli Spedali Civili di Brescia
Not yet recruiting
Brescia, Italy
Contact: Germana Tognon
Fondazione Poliambulanza
Not yet recruiting
Brescia, Italy
Contact: Chiara Abeni
AOU Cagliari, Policlinico Universitario
Not yet recruiting
Cagliari, Italy
Contact: Elena Massa
Ente ospedaliero Ospedali Galliera
Not yet recruiting
Genova, Italy
Contact: Andrea De Censi
ASST di Lecco
Not yet recruiting
Lecco, Italy
Contact: Antonio Ardizzoia
Ospedale San Luca
Not yet recruiting
Lucca, Italy
Contact: Editta Baldini
Istituto Europeo di Oncologia
Recruiting
Milan, Italy
Contact: Nicoletta Colombo
Ospedale San Gerardo
Not yet recruiting
Monza, Italy
Contact: Andrea A Lissoni
Istituto Oncologico Veneto (IOV)
Not yet recruiting
Padova, Italy
Contact: Giulia Tasca
AOU di Parma
Not yet recruiting
Parma, Italy
Contact: Antonino Musolino
AOU Pisana
Not yet recruiting
Pisa, Italy
Contact: Angiolo Gadducci
AO Arcispedale Santa Maria Nuova
Not yet recruiting
Reggio Emilia, Italy
Contact: Alessandra Bologna
Policlinico Umberto I, Università di Roma "La Sapienza"
Not yet recruiting
Roma, Italy
Contact: Pierluigi Benedetti Panici
Ospedale SS Trinità
Not yet recruiting
Sora, Italy
Contact: Filomena Narducci
AO Ordine Mauriziano
Not yet recruiting
Torino, Italy
Contact: Annamaria Ferrero
AOU Città della Salute e della Scienza di Torino - Ospedale Sant'Anna
Not yet recruiting
Torino, Italy
Contact: Paolo Zola
ASST Valtellina e Alto Lario
Not yet recruiting
Varese, Italy
Contact: Nicoletta Donadello Less << |
|
NCT03063450
|
Mesothelioma |
Phase 3 |
Recruiting |
July 2021 |
United Kingdom
... More >> Aberdeen Royal Infirmary
Recruiting
Aberdeen, United Kingdom
Contact: Gillian Price
Belfast City Hospital
Not yet recruiting
Belfast, United Kingdom
Contact: Gerry Hanna
Royal Bournemouth Hospital
Recruiting
Bournemouth, United Kingdom
Contact: Tom Geldart
Addenbrooke's Hospital
Not yet recruiting
Cambridge, United Kingdom
Contact: David Gilligan
Velindre Cancer Centre
Recruiting
Cardiff, United Kingdom
Contact: Jason Lester
Ninewells Hospital
Not yet recruiting
Dundee, United Kingdom
Contact: Angela Scott
Beatson West of Scotland Cancer Centre
Recruiting
Glasgow, United Kingdom
Contact: Nicola Steele
Harrogate District Hospital
Recruiting
Harrogate, United Kingdom
Contact: Samuel Chan
University Hospitals Morecambe Bay
Recruiting
Lancaster, United Kingdom
Contact: Amy Ford
Leicester Royal Infirmary
Recruiting
Leicester, United Kingdom
Contact: Dean Fennell
Barts Cancer Institute
Not yet recruiting
London, United Kingdom
Contact: Peter Szlosarek
University College London Hospitals NHS Foundation Trust
Recruiting
London, United Kingdom
Contact: Dionysis Papadatos-Pastos
Wythenshawe Hospital
Recruiting
Manchester, United Kingdom
Contact: Raffaele Califano
Mount Vernon Cancer Centre
Recruiting
Northwood, United Kingdom
Contact: Andreas Polychronis
Churchill Hospital
Not yet recruiting
Oxford, United Kingdom
Contact: Denis Talbot
Southampton General Hospital
Recruiting
Southampton, United Kingdom
Contact: Christian Ottensmeier
Southend Hospital
Recruiting
Southend-on-Sea, United Kingdom
Contact: Gairin Dancey
Lister Hospital
Recruiting
Stevenage, United Kingdom
Contact: Andreas Polychronis
Musgrove Park Hospital
Recruiting
Taunton, United Kingdom
Contact: Petra Jankowska Less << |
|
NCT02272816
|
Metastatic Seminoma |
Phase 2 |
Active, not recruiting |
December 2017 |
United Kingdom
... More >> St Bartholomew's Hospital
London, United Kingdom, EC1A 7BE
Mount Vernon Hosoital
London, United Kingdom, HA6 2RN Less << |
|
NCT01271322
|
Adenocarcinomas of the Esophag... More >>ogastric Junction Less << |
Phase 2 |
Terminated |
- |
Germany
... More >> Nationales Centrum für Tumorerkrankungen
Heidelberg, Germany, 69120 Less << |
|
NCT02533700
|
Peripheral T-Cell Lymphoma
... More >> Angioimmunoblastic T Cell Lymphoma
ALK-negative Anaplastic Large Cell Lymphoma
Enteropathy Associated T Cell Lymphoma
Subcutaneous Panniculitis Like T Cell Lymphoma
Acute Adult T-Cell Leukemia/Lymphoma Less << |
Phase 4 |
Unknown |
December 2017 |
China, Shandong
... More >>
Shandong Provincial Hospital
Recruiting
Jinan, Shandong, China, 250021
Contact: Xin Wang, MD, PHD 86-531-13156012606 xinw@sdu.edu.cn
Contact: Yujie Jiang, MD 86-531-13370506886 yujiejiang05@126.com Less << |
|
NCT03134846
|
Head and Neck Squamous Cell Ca... More >>rcinoma
Margin Assessment Less << |
Phase 1
Phase 2 |
Recruiting |
January 2021 |
Netherlands
... More >> University Medical Center Groningen
Recruiting
Groningen, Netherlands, 9713 GZ
Contact: Max J.H. Witjes, MD, PhD m.j.h.witjes@umcg.nl
Contact: Floris J. Voskuil, MD +31-50-3610030 f.j.voskuil@umcg.nl Less << |
|
NCT01222936
|
Small Cell Lung Carcinoma |
Phase 2 |
Completed |
- |
Germany
... More >> Klinik für Onkologie und Haematologie
Frankfurt am Main, Germany, 60488
Klinikum Kassel Innere Medizin
Kassel, Germany, 34125
Italy
Azienda Ospedaliera "S. G. Moscati"
Avellino, AV, Italy, 83100
Istituto Nazionale Ricerca sul Cancro
Genova, GE, Italy, 16132
U.O. di Oncologia Medica
Palermo, PA, Italy, 90141
Ospedale Maggiore di Parma
Parma, PR, Italy, 43100
Azienda Ospedaliera San Camillo Forlanini
Rome, RM, Italy, 00151
Az. San. Ospedaliera Molinette S. Giovanni Battista di Torino
Torino, TO, Italy, 10126 Less << |
|
NCT03089892
|
- |
|
Recruiting |
December 31, 2018 |
Taiwan
... More >> Tri-Service General Hospital
Recruiting
Taipei, Taiwan
Contact: Cheng-Chang Chang, M.D., Ph.D.
Principal Investigator: Cheng-Chang Chang, M.D., Ph.D.
Sub-Investigator: Mu-Hsien Yu, M.D., Ph.D.
Sub-Investigator: Yu-Chi Wang, M.D., Ph.D.
Sub-Investigator: Salvatore Giovanni Vitale, M.D. Less << |
|
NCT00107614
|
Waldenstrom Macroglobulinemia |
Phase 2 |
Terminated(Terminated due to p... More >>oor accrual.) Less << |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences/MIRT
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT01709006
|
Head and Neck Cancer |
Not Applicable |
Active, not recruiting |
December 31, 2020 |
Canada, Quebec
... More >> Notre-Hame Hospital of the CHUM
Montreal, Quebec, Canada, H2L 4M1 Less << |
|
NCT00864266
|
Non Small Cell Lung Carcinoma |
Not Applicable |
Active, not recruiting |
March 2021 |
Belgium
... More >> Department of Pneumology Clinique Saint-Luc
Bouge, Belgium, 5004
Department of Intensive Care Unit and Thoracic Oncology Institut Jules Bordet
Brussels, Belgium, 1000
Service de Pneumologie Hôpital Erasme
Brussels, Belgium, 1070
Department of Pneumology Hôpital Saint-Joseph
Gilly, Belgium, 6060
Hôpital Ambroise Paré
Mons, Belgium, 7000
Department of Pneumology Centre Hospitalier de Mouscron
Mouscron, Belgium, 7700 Less << |
|
NCT00469898
|
- |
|
Completed |
- |
- |
|
NCT00469898
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, Kentucky
... More >>
Owensboro Medical Health System
Owensboro, Kentucky, United States, 42303
United States, Tennessee
Memorial Health Care System
Chattanooga, Tennessee, United States, 37404
West Tennessee Cancer Center at Jackson-Madison County General Hospital
Jackson, Tennessee, United States, 38301
Tennessee Cancer Specialists
Knoxville, Tennessee, United States, 37901
St. Thomas Health Services
Nashville, Tennessee, United States, 37205
MBCCOP - Meharry Medical College - Nashville
Nashville, Tennessee, United States, 37208
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States, 37232-6838
Canada
British Columbia Cancer Agency - Vancouver Cancer Centre
Vancouver, Canada, V52 4 Less << |
|
NCT00107614
|
- |
|
Terminated(Terminated due to p... More >>oor accrual.) Less << |
- |
- |
|
NCT03049410
|
Bladder Cancer |
Not Applicable |
Recruiting |
February 15, 2020 |
United Kingdom
... More >> North Bristol NHS Trust
Recruiting
Bristol, United Kingdom
Principal Investigator: Edward Rowe
University College London Hospitals NHS Foundation Trust
Recruiting
London, United Kingdom, NW1 2BU
Contact: Pramit Khetrapal p.khetrapal@ucl.ac.uk
Principal Investigator: Prof John Kelly
Guy's Hospital
Recruiting
London, United Kingdom
Principal Investigator: Shamim Khan
Royal Berkshire Hospital
Recruiting
Reading, United Kingdom
Principal Investigator: Philip Charlesworth
Sheffield Teaching Hospitals NHS Foundation Trust
Recruiting
Sheffield, United Kingdom, S10 2JF
Contact: Louise Goodwin louise.goodwin@sth.nhs.uk
Principal Investigator: Prof James Catto
Lister Hospital
Recruiting
Stevenage, United Kingdom
Principal Investigator: Nikhil Vasdev Less << |
|
NCT02424045
|
T-cell Lymphoma |
Phase 2 |
Completed |
- |
- |
|
NCT03344328
|
- |
|
Not yet recruiting |
September 30, 2018 |
France
... More >> CHU Clermont-Ferrand
Not yet recruiting
Clermont-Ferrand, France, 63003
Contact: Patrick LACARIN 04 73 75 11 95 placarin@chu-clermmontferrand.fr
Principal Investigator: David BALAYSSAC Less << |
|
NCT01609114
|
- |
|
Unknown |
- |
Taiwan
... More >> Far Eastern Memorial Hospital
New Taipei city, Taiwan, 220 Less << |
|
NCT00905983
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Unknown |
September 2011 |
Spain
... More >> Hospital Virgen de los Lirios
Alcoy, Alicante, Spain, 3804
Hospital Clínica de Benidorm
Benidorm, Alicante, Spain, 03501
Hospital General de Elda
Elda, Alicante, Spain, 03600
Hospital Althaia, Xarxa Asistencial de Manresa
Manresa, Barcelona, Spain, 08243
Hospital Provincial de Castellón
Castellón de la Plana, Castellón, Spain, 12002
Hospital de Sagunto
Sagunto, Valencia, Spain, 46520
Hospital San Juan de Alicante
Alicante, Spain, 03550
Hospital Universitario La Fe
Valencia, Spain, 46009
Instituto Valenciano de Oncología
Valencia, Spain, 46009
Hospital Arnau de Vilanova
Valencia, Spain, 46015
Hospital Universitario Dr. Peset
Valencia, Spain, 46017 Less << |
|
NCT03470350
|
Colorectal Cancer Metastatic |
Phase 1
Phase 2 |
Recruiting |
June 1, 2020 |
Netherlands
... More >> Antoni van Leeuwenhoek (NKI-AVL)
Recruiting
Amsterdam, Noord-Holland, Netherlands, 1066 CX
Contact: S Huijberts, MD +31 (0)20-5129111 s.huijberts@nki.nl
Contact: N Steeghs, MD +31 (0)20-5129111 n.steeghs@nki.nl
Principal Investigator: N Steeghs, Dr.
Sub-Investigator: S Huijberts, MD Less << |
|
NCT01472419
|
- |
|
Completed |
- |
- |
|
NCT03529422
|
Larynx
Lip
... More >> Oral Cancer
Digestive Organs--Diseases Less << |
Phase 1 |
Recruiting |
February 2026 |
United States, North Carolina
... More >>
UNC Lineberger Comprehensive Cancer Center
Recruiting
Chapel Hill, North Carolina, United States, 27599
Contact: Dan Goldin 919-445-6241 dan_goldin@med.unc.edu
Principal Investigator: Jared Weiss, MD Less << |
|
NCT00058071
|
Gestational Trophoblastic Tumo... More >>r
Neurotoxicity
Peripheral Neuropathy
Unspecified Adult Solid Tumor, Protocol Specific Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00664105
|
Lung Cancer |
Phase 2 |
Terminated(Treatment became st... More >>andard.) Less << |
- |
United States, Florida
... More >>
M.D. Anderson Cancer Center, Orlando
Orlando, Florida, United States, 32806
United States, Maryland
Chesapeake Oncology Hematology Associates
Baltimore, Maryland, United States, 21225
United States, Ohio
University Hospital of Cleveland
Cleveland, Ohio, United States, 44106
United States, Pennsylvania
Lehigh Valley Hospital - John & Dorothy Morgan Cancer Center
Allentown, Pennsylvania, United States, 18103
United States, Tennessee
Erlanger Health System
Chattanooga, Tennessee, United States, 37403
Clarksville Regional Hematology Oncology Group
Clarksville, Tennessee, United States, 37043
Jackson Madison County Hospital
Jackson, Tennessee, United States, 38301
Tennessee Cancer Specialists
Knoxville, Tennessee, United States, 37920
University of Tennessee Medical Center
Knoxville, Tennessee, United States, 37920
The West Clinic, PC
Memphis, Tennessee, United States, 38120
St. Thomas Health Services
Nashville, Tennessee, United States, 37205
Meharry Medical College
Nashville, Tennessee, United States, 37208
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States, 37232
United States, Texas
UT Southwestern Medical Center
Dallas, Texas, United States, 75390
United States, Washington
Swedish Cancer Institute
Seattle, Washington, United States, 98104 Less << |
|
NCT00664105
|
- |
|
Terminated(Treatment became st... More >>andard.) Less << |
- |
- |
|
NCT02690012
|
Hypomagnesemia |
Not Applicable |
Unknown |
December 2017 |
Canada, Ontario
... More >>
The Ottawa Hospital Cancer Centre
Recruiting
Ottawa, Ontario, Canada, K1H8L6 Less << |
|
NCT02353936
|
Squamous Cell Carcinoma of Eso... More >>phagus Less << |
Phase 2 |
Recruiting |
January 2019 |
Korea, Republic of
... More >>
Severance hospital
Recruiting
Seoul, Korea, Republic of, 120-752
Contact: Hye Ryun Kim, MD 82-2-2228-8125 nobelg@yuhs.ac Less << |
|
NCT00770588
|
Non-small Cell Lung Cancer (NS... More >>CLC) Less << |
Phase 4 |
Completed |
- |
China, Beijing
... More >> Research Site
Beijing, Beijing, China
China, Fujian
Research Site
Fuzhou, Fujian, China
China, Guangdong
Research Site
Guangzhou, Guangdong, China
China, Guangxi
Research Site
Nanning, Guangxi, China
China, Henan
Research Site
Zhengzhou, Henan, China
China, Hubei
Research Site
Wuhan, Hubei, China
China, Jiangsu
Research Site
Nanjing, Jiangsu, China
China, Jilin
Research Site
Changchun, Jilin, China
China, Liaoning
Research Site
Shengyang, Liaoning, China
China, Shanghai
Research Site
Shanghai, Shanghai, China
China, Shanxi
Research Site
Xi'an, Shanxi, China
China, Sichuan
Research Site
Chengdu, Sichuan, China
China, Tianjin
Research Site
Tianjin, Tianjin, China
China, Zhejiang
Research Site
Hangzhou, Zhejiang, China Less << |
|
NCT00770588
|
- |
|
Completed |
- |
- |
|
NCT00049322
|
Liver Cancer |
Phase 2 |
Completed |
- |
United States, California
... More >>
Jonsson Comprehensive Cancer Center at UCLA
Los Angeles, California, United States, 90095-1781 Less << |
|
NCT00049322
|
- |
|
Completed |
- |
- |
|
NCT02105350
|
Biliary Tract Carcinoma
... More >> Gallbladder Carcinoma Less << |
Phase 1 |
Withdrawn(Study was never init... More >>iated) Less << |
- |
- |
|
NCT02127255
|
Chemotherapy-induced Nausea an... More >>d Vomiting Less << |
Not Applicable |
Completed |
- |
China
... More >> Xiyuan Hospital, China Academy of Chinese Medical Sciences
Beijing, China, 100091 Less << |
|
NCT00462735
|
- |
|
Completed |
- |
- |
|
NCT00068653
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, Michigan
... More >>
Barbara Ann Karmanos Cancer Institute
Detroit, Michigan, United States, 48201-1379 Less << |
|
NCT02016287
|
Esophageal Squamous Cell Cance... More >>r
Elderly Patients Less << |
Phase 2 |
Unknown |
July 2016 |
China, Beijing
... More >> Peking University Cancer Hospital
Recruiting
Beijing, Beijing, China, 100142
Contact: Lin Shen, MD 86-10-88196561 lin100@medmail.com.cn
Contact: Zhihao Lu, MD 86-10-88196561 pppeirain@126.com
Principal Investigator: Lin Shen, MD Less << |
|
NCT01099085
|
Gastric Cancer |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Samsung Cancer Center
Seoul, Korea, Republic of Less << |
|
NCT00462735
|
Head and Neck Cancer
... More >> Cancer of the Pharynx
Cancer of the Larynx
Nose Neoplasms
Paranasal Sinus Neoplasms
Cancer of the Oral Cavity Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Mount Sinai School of Medicine
New York, New York, United States, 10029 Less << |
|
NCT01243775
|
- |
|
Completed |
- |
- |
|
NCT02430298
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
Thailand
... More >> Ubon Ratchathani Cancer Hospital
Ubon Ratchathani, Thailand, 34000 Less << |
|
NCT01243775
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Chonnam National University Hwasun Hospital
Hwasun, Jeonnam, Korea, Republic of, 519-809 Less << |
|
NCT00479674
|
Breast Cancer |
Phase 2 |
Completed |
- |
United States, North Carolina
... More >>
Presbyterian Health Care
Charlotte, North Carolina, United States, 28204
Northeast Oncology Associates
Concord, North Carolina, United States, 28205
Duke University Medical Center
Durham, North Carolina, United States, 27710
Forsyth Regional Cancer Center
Winston-Salem, North Carolina, United States, 27103-3019 Less << |
|
NCT00479674
|
- |
|
Completed |
- |
- |
|
NCT03577132
|
Urothelial Carcinoma |
Not Applicable |
Not yet recruiting |
May 31, 2022 |
- |
|
NCT02108652
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00080444
|
Vomiting |
Phase 3 |
Completed |
- |
- |
|
NCT02108652
|
Bladder Cancer |
Phase 2 |
Active, not recruiting |
May 18, 2019 |
- |
|
NCT00002487
|
Ovarian Cancer |
Phase 2 |
Unknown |
- |
Canada, Ontario
... More >>
Ottawa Regional Cancer Center - General Division
Ottawa, Ontario, Canada, K1H 8L6
Ottawa Regional Cancer Centre - Civic Campus
Ottawa, Ontario, Canada, K1Y 4K7 Less << |
|
NCT02735057
|
Oesophagus Cancer
... More >> Elderly Patients Less << |
Phase 1
Phase 2 |
Recruiting |
April 2024 |
France
... More >> Centre Hospitalier Universitaire de Besançon
Recruiting
Besancon, France, 25000
Contact: Sophie DEPIERRE sdepierre@chu-besancon.fr
Principal Investigator: Gilles CREHANGE, PU-PH Less << |
|
NCT02433639
|
Biliary Tract Cancer |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Seoul National University Hospital
Seoul, Korea, Republic of, 110-744 Less << |
|
NCT03598218
|
Nasopharyngeal Carcinoma |
Not Applicable |
Recruiting |
July 13, 2021 |
China
... More >> Guangxi Naxishan Hospital
Recruiting
Guilin, China
Contact: Yu-fei Pan, M.D.
Linshan people's hospital
Recruiting
Linshan, China
Contact: Yi-xin Su
Wuzhou Red Cross Hospital
Recruiting
Wuzhou, China
Contact: Bin Zhang, M.D. Less << |
|
NCT01152853
|
Advanced Gastric Cancer
... More >> HER2 Less << |
Phase 2 |
Completed |
- |
Korea, Republic of
... More >>
Seoul National University Hospital
Seoul, Korea, Republic of, 110-744 Less << |
|
NCT00659269
|
- |
|
Completed |
- |
- |
|
NCT00659269
|
Cancer |
Phase 3 |
Completed |
- |
United States, New Mexico
... More >>
University of New Mexico Cancer Center @ Lovelace Medical Center
Albuquerque, New Mexico, United States, 87102
Hematology Oncology Associates
Albuquerque, New Mexico, United States, 87106
Cancer Center at Presbyterian Hospital
Albuquerque, New Mexico, United States, 87110
University of New Mexico Cancer Center
Albuquerque, New Mexico, United States, 87131
New Mexico Cancer Care Associates
Santa Fe, New Mexico, United States, 87505 Less << |
|
NCT02229916
|
- |
|
Recruiting |
September 2029 |
Switzerland
... More >> Kantonsspital St.Gallen; Onkologie/Haematologie
Recruiting
St.Gallen, Switzerland, 9007
Contact: Christian Rothermundt, Dr. med. +41 71 494 11 11 christian.rothermundt@kssg.ch
Principal Investigator: Christian Rothermundt, Dr. med. Less << |
|
NCT03079440
|
Neuroendocrine Tumors
... More >> Neuroendocrine Carcinoma
Temozolomide
Capecitabine Less << |
Phase 2 |
Recruiting |
October 2020 |
Korea, Republic of
... More >>
Asan Medical Center, University of Ulsan College of Medicine
Recruiting
Seoul, Korea, Republic of, 05505
Contact: Changhoon Yoo, MD +82-2-3010-1727 yooc@amc.seoul.kr Less << |
|
NCT01922232
|
- |
|
Withdrawn |
- |
United States, New York
... More >>
NYU Langone Medical Center
New York, New York, United States, 10016 Less << |
|
NCT00068289
|
Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT03158129
|
Non-Small Cell Lung Cancer |
Phase 2 |
Recruiting |
June 2022 |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact tcascone@mdanderson.org Less << |
|
NCT00041626
|
Carcinoma, Squamous Cell |
Phase 3 |
Unknown |
- |
United States, Arkansas
... More >>
University of Arkansas
Recruiting
Little Rock, Arkansas, United States, 72205
Contact: Karen Duvall 501-526-7129 kaduvall@uams.edu
Principal Investigator: James Suen, MD
United States, Colorado
Unversity of Colorado Cancer Center
Recruiting
Aurora, Colorado, United States, 80045
Contact: Brittney Hines 720-848-0678 Brittany.Hines@uchsc.edu
Principal Investigator: Madeleine Kane, MD
United States, Florida
University of Miami Hospital and Sylvester Comprehensive Cancer Center
Recruiting
Miami, Florida, United States, 33136
Contact: Andrea Gachupin-Garcia 305-243-3379 agachupin@med.miami.edu
Principal Investigator: Jarrad Goodwin, MD
United States, Kentucky
Norton Healthcare Pavilion
Recruiting
Louisville, Kentucky, United States, 40202
Contact: Daniela Neamtu 502-629-4679 daniela.neamtu@nortonhealthcare.org
Principal Investigator: John Hamm, MD
United States, Maryland
Center Center of GBMC
Recruiting
Baltimore, Maryland, United States, 21204
Contact: Lauren Titus 443-849-3285 ltitus@gbmc.org
Principal Investigator: Marshall Levine, MD
United States, South Carolina
WJB Dorn VA Medical Center
Recruiting
Columbia, South Carolina, United States, 29209
Contact: Justin Reynolds 803-776-4000 ext 6074 Justin.Reynolds@va.gov
Principal Investigator: William Hrushesky, MD
United States, Texas
Mary Crowley Medical Research Center
Recruiting
Dallas, Texas, United States, 75201
Contact: Arlen Waclawczyk 214-658-1985 awaclawczyk@mcmrc.com
Principal Investigator: John Nemunaitis, MD
University of Texas, MD Andersen Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact: Aida Berryman 713-745-1428 atberryman@mdanderson.org
Principal Investigator: Gary Clayman, MD Less << |
|
NCT00183820
|
Testicular Cancer
... More >> Germ Cell Neoplasm Less << |
Phase 2 |
Completed |
- |
United States, California
... More >>
Norris Comprehensive Cancer Center
Los Angeles, California, United States, 90033 Less << |
|
NCT03170570
|
Cervical Cancer
... More >> Radiation Toxicity
Recurrent Cervical Carcinoma Less << |
Not Applicable |
Recruiting |
January 31, 2020 |
China, Guangdong
... More >>
Sun Yat-Sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: xinping cao, MD (86)13602736388 caoxp@sysucc.org.cn Less << |
|
NCT02002195
|
- |
|
Unknown |
May 2016 |
Italy
... More >> Azienda Ospedaliera Ospedali Riuniti Marche Nord, Presidio Ospedaliero San Salvatore
Recruiting
Pesaro, PU, Italy, 61122
Principal Investigator: Vincenzo Catalano, MD
Azienda Ospedaliera Ospedali Riuniti Marche Nord, Presidio San Salvatore,
Recruiting
Pesaro, Italy, 61122
Contact: Vincenzo Catalano, MD 0039072136 ext 4001 catalano_v@yahoo.it
Principal Investigator: Vincenzo Catalano, MD Less << |
|
NCT02236195
|
Urothelial Carcinoma |
Phase 2 |
Completed |
- |
United States, Alabama
... More >>
University of Alabama
Birmingham, Alabama, United States, 35294
United States, California
City of Hope National Medical Center
Duarte, California, United States, 91010
United States, Florida
Florida Cancer Specialists
Fort Myers, Florida, United States, 33905
United States, Illinois
University of Chicago
Chicago, Illinois, United States, 60637
United States, Maryland
Johns Hopkins University
Baltimore, Maryland, United States, 21287
United States, Massachusetts
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02215
United States, Michigan
University of Michigan
Ann Arbor, Michigan, United States, 48109
United States, Missouri
Washington University
Saint Louis, Missouri, United States, 63110
United States, New York
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
University of Rochester
Rochester, New York, United States, 14627
United States, North Carolina
University of North Carolina
Chapel Hill, North Carolina, United States, 27599
United States, Ohio
Cleveland Clinic Foundation
Cleveland, Ohio, United States, 44195
Ohio State University
Columbus, Ohio, United States, 43210
United States, Utah
Huntsman Cancer Institute
Salt Lake City, Utah, United States, 84112 Less << |
|
NCT00604461
|
Mesothelioma |
Phase 1
Phase 2 |
Terminated(Slow Accrual) |
- |
United States, Florida
... More >>
H. Lee Moffitt Cancer Center & Research Institute
Tampa, Florida, United States, 33612 Less << |
|
NCT01937208
|
Esophageal Carcinoma |
Phase 3 |
Unknown |
July 2017 |
China, Zhejiang
... More >>
Zhejiang Cancer Hospital
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: xiao zheng, MD 0086-571-88122078 zhengxiao@medmail.com
Principal Investigator: ming chen, PHD Less << |
|
NCT00867347
|
Bladder Cancer |
Phase 3 |
Unknown |
- |
United Kingdom
... More >> Institute of Cancer Research - Sutton
Sutton, England, United Kingdom, SM2 5PT Less << |
|
NCT03059667
|
Small Cell Lung Cancer
... More >> Small Cell Lung Cancer Limited Stage
Small Cell Lung Cancer Extensive Stage Less << |
Phase 2 |
Active, not recruiting |
December 2018 |
France
... More >> Annemasse - CH
Ambilly, France, 74100
Angers - CHU
Angers, France, 49000
CH
Colmar, France
CHRU Grenoble
Grenoble, France
Centre Hospitalier - Pneumologie
Le Mans, France, 72000
Lorient - CHBS
Lorient, France
Montpellier - CHRU
Montpellier, France, 34295
Mulhouse - CH
Mulhouse, France, 68000
Centre Antoine Lacassagne
Nice, France
AP-HP Hopital Tenon - Pneumologie
Paris, France, 75020
APHP - Paris Bichat
Paris, France, 75877
GH Paris Saint-Joseph
Paris, France
CHG de Pau
Pau, France
HCL - Lyon Sud (Pneumologie)
Pierre Bénite, France, 69495
Rouen - CHU
Rouen, France, 76000
Centre Hospitalier
Saint-Quentin, France
Toulouse - CHU Larrey
Toulouse, France
CHU Tours - Pneumologie
Tours, France Less << |
|
NCT02456714
|
Cholangiocarcinoma |
Phase 2 |
Active, not recruiting |
December 1, 2018 |
Netherlands
... More >> Academic medical center Amsterdam
Amsterdam, Netherlands, 1105AZ Less << |
|
NCT03377400
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Phase 2 |
Recruiting |
December 30, 2019 |
Korea, Republic of
... More >>
Samsung Medical Center
Recruiting
Seoul, MA, Korea, Republic of, 06351
Contact: Jong-Mu Sun, Associate Professor 82234101795 jongmu.sun@samsung.com Less << |
|
NCT01845779
|
Metastatic Anal Canal Cancer
... More >> Human Papillomavirus Less << |
Not Applicable |
Recruiting |
January 2020 |
France
... More >> Medical Oncology - University Hospital of Besançon
Recruiting
Besançon, France, 25000
Contact: Christophe BORG, Prof +3381615615 christophe.borg@efs.sante.fr
Contact: Marion JACQUIN m1jacquin@chu-besancon.fr
Principal Investigator: Christophe BORG, Prof
Sub-Investigator: Jean-François BOSSET, Prof
Sub-Investigator: Thierry Nguyen, Doctor Less << |
|
NCT01207102
|
Metastatic Breast Cancer |
Phase 2 |
Terminated(Low enrollment and ... More >>there is insufficient data to publish.) Less << |
- |
United States, North Carolina
... More >>
Duke University Medical Center
Durham, North Carolina, United States, 27710
China
Peking University School of Oncology/Beijing Cancer Hospital
Beijing, China, 100142 Less << |
|
NCT01207102
|
- |
|
Terminated(Low enrollment and ... More >>there is insufficient data to publish.) Less << |
- |
- |
|
NCT02423811
|
Cancer Stem Cell |
Phase 2 |
Unknown |
- |
Taiwan
... More >> National Cheng Kung University Hospital
Recruiting
Tainan, Taiwan, 704
Contact: Forn-Chia Lin, M.D., Ph.D. 886-6-2353535 ext 2463 fornchia@mail.ncku.edu.tw Less << |
|
NCT00794339
|
Cervical Cancer |
Phase 2 |
Active, not recruiting |
December 2017 |
United States, California
... More >>
USC/Norris Comprehensive Cancer Center and Hospital
Los Angeles, California, United States, 90089-9181
United States, Missouri
Siteman Cancer Center at Barnes-Jewish Hospital - Saint Louis
Saint Louis, Missouri, United States, 63110 Less << |
|
NCT02131480
|
Uterus Leiomyosarcoma
... More >> Soft Tissue Leiomyosarcoma Less << |
Phase 2 |
Completed |
- |
France
... More >> Gustave Roussy Cancer Campus Grand Paris
Villejuif, Val de Marne, France, 94805 Less << |
|
NCT02783885
|
- |
|
Recruiting |
September 2020 |
Saudi Arabia
... More >> King Abdul Aziz Medical City for National Guard Health Affairs
Recruiting
Riyadh, Saudi Arabia, 11426
Contact: Abdularahman Jazieh, MD,MPH 0096612520088 ext 14688 jazieha@ngha.med.sa
Contact: Oncology Research 0096612520088 ext 14601 oncologyresearch@ngha.med.sa
King Abdul Aziz Medical City for National Guard
Recruiting
Riyadh, Saudi Arabia
Contact: Nagham RZ Sheblaq 009668011111 ext 53352 sheblaqn@ngha.med.sa
Contact 009668011111 ext 53352 sheblaqn@ngha.med.sa
Principal Investigator: Nagham RZ Sheblaq, BS.c Less << |
|
NCT00066768
|
Recurrent Non-small Cell Lung ... More >>Cancer
Stage IIIB Non-small Cell Lung Cancer
Stage IV Non-small Cell Lung Cancer Less << |
Phase 1 |
Completed |
- |
United States, Ohio
... More >>
Ohio State University Medical Center
Columbus, Ohio, United States, 43210 Less << |
|
NCT01453504
|
Hodgkin Lymphoma |
Phase 1
Phase 2 |
Recruiting |
December 2018 |
Germany
... More >> 1st Dept. of Medicine, Cologne University Hospital
Recruiting
Cologne, Germany
Contact: Michael Fuchs ghsg@uk-koeln.de Less << |
|
NCT00511446
|
Stomach Neoplasms |
Phase 2 |
Completed |
- |
Germany
... More >> Charite - Universitatsmedizin Berlin
Berlin, Germany, 13353
Medizinische Universitätsklinik - Knappschaftskrankenhaus
Bochum, Germany, 44892
Städtische Kliniken Esslingen
Esslingen, Germany, 73730
MVZ Osthessen
Fulda, Germany, 36043
Martin-Luther-University Halle-Wittenberg
Halle (Saale), Germany, 06120
Städt. Klinikum St. Georg
Leipzig, Germany, 04129
OSP Lörrach-Rheinfelden
Lörrach, Germany, 79539
Universitätsklinikum Mainz
Mainz, Germany, 55101
Universitätsklinikum Mannheim
Mannheim, Germany, 68167
Universitätsklinik Ulm
Ulm, Germany, 89081 Less << |
|
NCT00190840
|
Carcinoma, Non-Small Cell Lung |
Phase 2 |
Completed |
- |
Australia, New South Wales
... More >>
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Wollongong, New South Wales, Australia, 2500
Australia, Queensland
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Chermside, Queensland, Australia, 4032
Australia, South Australia
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Ashford, South Australia, Australia, 5035
Australia, Victoria
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Fitzroy, Victoria, Australia, 3065
China
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hong Kong, China
Egypt
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Alexandria, Egypt
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Cairo, Egypt, 11796
India
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Hyderabad, Andhra Pradesh, India, 500004
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Ludhiana, Punjab, India, 141001
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Chandigarh, India, 160012
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Chennai, India, 600035
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Delhi, India, 110085
Korea, Republic of
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Goyang-Si, Gyeonggi-Do, Korea, Republic of, 411764
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Seoul, Korea, Republic of
Romania
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Oradea, Bihor, Romania, 3700
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Craiova, Dolj, Romania, 1100
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Bucuresti, Romania, 72435
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Iasi, Romania, 6600
Taiwan
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Taipei, Taiwan
Turkey
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Eskisehir, Turkey Less << |
|
NCT01523574
|
Peripheral Neuropathy |
Phase 2 |
Completed |
- |
Brazil
... More >> Faculdade de Medicina do ABC
Santo Andre, Sao Paulo, Brazil, 09060-870
Faculdade de Medicina do ABC
Santo André, São Paulo, Brazil, 09060-870 Less << |
|
NCT02665702
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Phase 2 |
Recruiting |
September 2019 |
China, Shanghai
... More >>
Cancer hospital Fudan University
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Chang jian hua, PD 13916619284 changjianhua@163.com Less << |
|
NCT00604461
|
- |
|
Terminated(Slow Accrual) |
- |
- |
|
NCT00980460
|
Alpha Fetoprotein Increased
... More >> PRETEXT Stage 1 Hepatoblastoma
PRETEXT Stage 2 Hepatoblastoma
PRETEXT Stage 3 Hepatoblastoma
PRETEXT Stage 4 Hepatoblastoma Less << |
Phase 3 |
Active, not recruiting |
December 31, 2018 |
- |
|
NCT02114359
|
Gastric Cancer |
Phase 3 |
Recruiting |
February 2020 |
Korea, Republic of
... More >>
Hallym University Sacred Heart Hospital
Recruiting
Anyang, Korea, Republic of, 431-796
Contact: ZANG, M.D. fhdzang@hallym.ac.kr
Principal Investigator: ZANG, M.D.
Inje University Haeundae Paik Hospital
Recruiting
Busan, Korea, Republic of
Contact: LEE, M.D.
Principal Investigator: LEE, M.D.
Chungbuk National University Hospital
Not yet recruiting
Cheongju, Korea, Republic of, 361-711
Contact: HAN, M.D.
Principal Investigator: HAN, M.D.
Kyungpook National University Medical Center
Recruiting
Daegu, Korea, Republic of, 702-210
Contact: KIM, M.D.
Principal Investigator: KIM, M.D.
Yeongnam University Medical Center
Recruiting
Daegu, Korea, Republic of
Contact: KOH, M.D.
Principal Investigator: KOH, M.D.
National Cancer Center
Recruiting
Goyang, Korea, Republic of, 410-769
Contact: PARK, M.D. youngiee@dreamwiz.com
Principal Investigator: PARK, M.D.
Gachon University Gil Medical Center
Not yet recruiting
Incheon, Korea, Republic of, 400-713
Contact: SIM, M.D.
Principal Investigator: SIM, M.D.
Inje University Pusan Paik Hospital
Recruiting
Pusan, Korea, Republic of, 614-735
Contact: KIM, M.D.
Principal Investigator: KIM, M.D.
Seoul National University Bundang Hospital
Recruiting
Seongnam, Korea, Republic of, 463-707
Contact: LEE, M.D. hmodoctor@hanmail.net
Principal Investigator: LEE, M.D.
Asan Medical Center
Recruiting
Seoul, Korea, Republic of, 138-736
Contact: RYU, M.D.
Principal Investigator: RYU, M.D.
SMG-SNU Boramae Medical Center
Recruiting
Seoul, Korea, Republic of, 156-707
Contact: CHOI, M.D.
Principal Investigator: CHOI, M.D.
Inje University Sanggye Paik Hospital
Recruiting
Seoul, Korea, Republic of
Contact: SOHN, M.D.
Principal Investigator: SOHN, M.D.
Kangbuk Samsung Hospital
Recruiting
Seoul, Korea, Republic of
Contact: KOO, M.D.
Principal Investigator: KOO, M.D.
The Catholic University of Korea Uijeongbu St. Mary's Hospital
Recruiting
Uijeongbu, Korea, Republic of
Contact: KO, M.D.
Principal Investigator: KO, M.D. Less << |
|
NCT01749072
|
Non-small Cell Lung Cancer
... More >> Effects of Chemotherapy Less << |
Phase 2 |
Unknown |
December 2017 |
China, Beijing
... More >> Department of Respiratory Medicine, Peking Union Medical College Hospital
Recruiting
Beijing, Beijing, China, 100730
Contact: Mengzhao Wang, MD 010-69155039 ext +86 mengzhaowang@sina.com
Contact: Jing Zhao, MD 010-69158206 ext +86 pumchzj@sina.com
Sub-Investigator: Wei Zhong, MD
Sub-Investigator: Jinmei Luo, MD Less << |
|
NCT01070290
|
Gastric Cancer |
Phase 2 |
Withdrawn |
- |
- |
|
NCT01870609
|
Malignant Pleural Mesothelioma |
Phase 2 |
Terminated(Interim analysis-DS... More >>MB stated good safety profile but lack of efficacy) Less << |
- |
- |
|
NCT03719989
|
Diffuse Large B Cell Lymphoma
... More >>
Relapsed Non Hodgkin Lymphoma
Refractory Non-Hodgkin Lymphoma Less << |
Phase 2 |
Not yet recruiting |
November 30, 2023 |
Korea, Republic of
... More >>
Seoul National University Hospital
Not yet recruiting
Seoul, Korea, Republic of, 03080
Contact: Junshik Hong, MD 82220723383 alertjun@hanmail.net Less << |
|
NCT00446290
|
Stomach Cancer |
Phase 1 |
Completed |
- |
Korea, Republic of
... More >>
Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT01453205
|
Diffuse Large B-Cell Lymphoma |
Phase 2 |
Completed |
- |
- |
|
NCT01453205
|
- |
|
Completed |
- |
- |
|
NCT00446290
|
- |
|
Completed |
- |
- |
|
NCT00686985
|
Small Cell Lung Cancer |
Phase 2 |
Unknown |
July 2010 |
Netherlands
... More >> B. Biesma
Recruiting
's-Hertogenbosch, Brabant, Netherlands, 5211NL
Contact: B. Biesma, Dr. 31-073-699-2615 b.biesma@jbz.nl
Contact: F. Schramel, Dr. 31-0609-9111 f.schramel@antonius.net
Sub-Investigator: H van der Heijden, Dr.
Sub-Investigator: J.N.H. Timmer - Bonte, Dr.
Sub-Investigator: H Smit, Dr.
Sub-Investigator: H.J.M. Groen, Prof. Dr. Less << |
|
NCT01655823
|
Pain
Peripher... More >>al Neuropathy
Neuropathic Pain Less << |
Phase 2 |
Terminated(Interim analysis co... More >>mpleted and decided to terminate and proceed to Phase 3 trial.) Less << |
- |
- |
|
NCT02557529
|
Head and Neck Neoplasms
... More >> Weight Loss Less << |
Phase 2 |
Unknown |
May 2018 |
Denmark
... More >> Department of Oncology, Copenhagen University hospital, Herlev
Recruiting
Herlev, Denmark, 2730
Contact: Julie Gehl 004538683868 julie.gehl@regionh.dk
Contact: Camilla K Lonkvist 004538689571
Principal Investigator: Julie Gehl Less << |
|
NCT01655823
|
- |
|
Terminated(Interim analysis co... More >>mpleted and decided to terminate and proceed to Phase 3 trial.) Less << |
- |
- |
|
NCT00039039
|
Lung Cancer |
Phase 3 |
Unknown |
- |
Italy
... More >> Ospedale Civile di Asti
Asti, Italy, 14100
Centro di Riferimento Oncologico - Aviano
Aviano, Italy, 33081
Ospedali Riuniti di Bergamo
Bergamo, Italy, 24100
Spedali Civili di Brescia
Brescia, Italy, 25133
Civic Hospital of Carrara
Carrara, Italy, I-54033
Ospedale Regionale A. Pugliese
Catanzaro, Italy, 88100
Ospedale Mariano Santo
Cosenza, Italy, 87100
Azienda Istituti Ospitalieri
Cremona, Italy, 26100
Ospedale Santa Croce
Cuneo, Italy, 12100
Ospedale Galliera Oncologia
Genoa, Italy, 16100
Istituto Nazionale per la Ricerca sul Cancro
Genoa, Italy, 16132
Ospedale San Martino
Genoa, Italy, 16132
Ospedale Civile di Ivrea
Ivrea, Italy, 10015
Ospedale Santa Maria Goretti
Latina, Italy, 04100
Ospedale Umberto I
Lugo DI Romagna, Italy, 48022
Istituto Policlinico San Donato
Milan, Italy, 20097
Istituto Nazionale per lo Studio e la Cura dei Tumori
Milan, Italy, 20133
University of Modena Hospital and Reggio Emilia School of Medicine
Modena, Italy, 41100
Nuovo Ospedale San Gerardo at University of Milano-Bicocca
Monza, Italy, 20052
Azienda Ospedale S. Luigi at University of Torino
Orbassano, Italy, 10043
Azienda Ospedaliera di Padova
Padova, Italy, 35128
Azienda Ospedaliera Di Parma
Parma, Italy, 43100
Policlinico Monteluce
Perugia, Italy, 06122
Ospedale Santa Chiara Pisa
Pisa, Italy, 56100
Ospedale Sta. Maria Delle Croci
Ravenna, Italy, 48100
Ospedale Carlo Forlanini
Rome, Italy, 00149
Ospedale S. Camillo-Forlanini
Rome, Italy, 00152
Azienda Policlinico Umberto Primo
Rome, Italy, 00161
Istituti Fisioterapici Ospitalieri - Roma
Rome, Italy, 00161
Istituto Regina Elena
Rome, Italy, 00161
Ospedale Casa Sollievo della Sofferenza
San Giovanni - Rotondo, Italy, 71013
Azienda Ospedaliera S. Maria
Terni, Italy, 051100
Azienda Sanitaria Ospedale San Giovanni Battista Molinette di Torino
Turin, Italy, 10123
Ospedale Evangelico Valdese
Turin, Italy, 10125
Ospedale Ostetrico Ginecologica Sant Anna
Turin, Italy, 10125
Azienda Ospedaliera Santa Maria della Misericordia
Udine, Italy, 33100
Ospedale San Bortolo
Vicenza, Italy, 36100 Less << |
|
NCT00085202
|
- |
|
Active, not recruiting |
- |
- |
|
NCT01404156
|
Esophageal Cancer
... More >> Adenocarcinoma, Esophageal
Adenocarcinoma, Gastroesophageal Junction Less << |
Phase 2
Phase 3 |
Recruiting |
April 2021 |
Canada, Manitoba
... More >>
Health Sciences Centre / CancerCare Manitoba
Recruiting
Winnipeg, Manitoba, Canada, R3A 1R9 Less << |
|
NCT00274937
|
- |
|
Completed |
- |
- |
|
NCT02434614
|
Nasopharyngeal Carcinoma |
Phase 3 |
Active, not recruiting |
December 2021 |
China, Guangxi
... More >> Affiliated Hospital of Youjiang Medical University for Nationalities
Baise, Guangxi, China
Guigang People's Hospital
Guigang, Guangxi, China
Guangxi Naxishan Hospital
Guilin, Guangxi, China
Liuzhou People's Hospital
Liuzhou, Guangxi, China
Nanning Monority Hospital
Nanning, Guangxi, China
Wuzhou Red Cross Hospital
Wuzhou, Guangxi, China Less << |
|
NCT00274937
|
Stage I Lymphoepithelioma of t... More >>he Nasopharynx
Stage I Squamous Cell Carcinoma of the Nasopharynx
Stage II Lymphoepithelioma of the Nasopharynx
Stage II Squamous Cell Carcinoma of the Nasopharynx
Stage III Lymphoepithelioma of the Nasopharynx
Stage III Squamous Cell Carcinoma of the Nasopharynx
Stage IV Lymphoepithelioma of the Nasopharynx
Stage IV Squamous Cell Carcinoma of the Nasopharynx Less << |
Phase 3 |
Completed |
- |
- |
|
NCT02349958
|
Ovarian Cancer
... More >> Peritoneal Cancer
Fallopian Tube Cancer
Uterine Cancer
Mesotheliomas
Gastrointestinal Cancers
Cervical Cancer Less << |
Phase 2 |
Recruiting |
January 2018 |
United States, California
... More >>
James Lilja
Recruiting
Los Gatos, California, United States, 95032
Contact: Heather McAvoy, LVN 408-827-4274 info@bayareago.com
Principal Investigator: James Lilja, MD
Sub-Investigator: Augusto Bastidas, MD Less << |
|
NCT03017326
|
Hepatoblastoma
... More >> Carcinoma, Hepatocellular Less << |
Phase 3 |
Recruiting |
December 2022 |
United Kingdom
... More >> Royal Aberdeen Children's Hospital
Recruiting
Aberdeen, United Kingdom, AB25 2ZG
Contact: Hugh Bishop, MBChB
Addenbrooke's Hospital
Recruiting
Cambridge, United Kingdom, CB2 0QQ
Contact: Amos Burke, MB ChB amos.burke@addenbrookes.nhs.uk Less << |
|
NCT01283178
|
Salivary Gland Squamous Cell C... More >>arcinoma
Stage II Salivary Gland Cancer
Stage II Squamous Cell Carcinoma of the Hypopharynx
Stage II Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage II Squamous Cell Carcinoma of the Oropharynx
Stage II Verrucous Carcinoma of the Oral Cavity
Stage III Salivary Gland Cancer
Stage III Squamous Cell Carcinoma of the Hypopharynx
Stage III Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage III Squamous Cell Carcinoma of the Oropharynx
Stage III Verrucous Carcinoma of the Oral Cavity
Stage IV Salivary Gland Cancer
Stage IV Squamous Cell Carcinoma of the Hypopharynx
Stage IV Squamous Cell Carcinoma of the Lip and Oral Cavity
Stage IV Squamous Cell Carcinoma of the Oropharynx
Stage IV Verrucous Carcinoma of the Oral Cavity Less << |
Phase 1 |
Terminated(The study was termi... More >>nated. The data analysis was futile with only 3 accruals. PI opted to terminate with IRB and withdraw FDA IND.) Less << |
- |
United States, Virginia
... More >>
Virginia Commonwealth University
Richmond, Virginia, United States, 23298 Less << |
|
NCT01461057
|
- |
|
Completed |
- |
- |
|
NCT00059826
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
United States, Florida
... More >>
University of Florida Shands Cancer Center
Gainesville, Florida, United States, 32610-0232
United States, Illinois
Rush University Medical Center
Chicago, Illinois, United States, 60612
United States, Kentucky
James Graham Brown Cancer Center at University of Louisville
Louisville, Kentucky, United States, 40202
United States, Massachusetts
Massachusetts General Hospital Cancer Center
Boston, Massachusetts, United States, 02114
Brigham and Women's Hospital
Boston, Massachusetts, United States, 02115
Dana-Farber/Harvard Cancer Center at Dana Farber Cancer Institute
Boston, Massachusetts, United States, 02115
United States, Minnesota
Fairview University Medical Center - University Campus
Minneapolis, Minnesota, United States, 55455
United States, New York
Roswell Park Cancer Institute
Buffalo, New York, United States, 14263-0001
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021
James P. Wilmot Cancer Center at University of Rochester Medical Center
Rochester, New York, United States, 14642
United States, Tennessee
Vanderbilt-Ingram Cancer Center
Nashville, Tennessee, United States, 37232
United States, Texas
Presbyterian Hospital of Dallas
Dallas, Texas, United States, 75231
Baylor University Medical Center - Houston
Houston, Texas, United States, 77030
United States, Wisconsin
Medical College of Wisconsin Cancer Center
Milwaukee, Wisconsin, United States, 53226 Less << |
|
NCT00870870
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
United States, Alabama
... More >>
ImClone Investigational Site
Anniston, Alabama, United States, 36207
United States, California
ImClone Investigational Site
La Jolla, California, United States, 92093
ImClone Investigational Site
Orange, California, United States, 92868
United States, Florida
ImClone Investigational Site
Orlando, Florida, United States, 32806
United States, Georgia
ImClone Investigational Site
Atlanta, Georgia, United States, 30341
United States, Illinois
ImClone Investigational Site
Chicago, Illinois, United States, 60612
ImClone Investigational Site
Chicago, Illinois, United States, 60637
United States, New Mexico
ImClone Investigational Site
Albuquerque, New Mexico, United States, 87131
United States, New York
ImClone Investigational Site
New York, New York, United States, 10011
ImClone Investigational Site
New York, New York, United States, 10021
ImClone Investigational Site
New York, New York, United States, 10032
United States, Ohio
ImClone Investigational Site
Cincinnati, Ohio, United States, 45247 Less << |
|
NCT00870870
|
- |
|
Completed |
- |
- |
|
NCT00779714
|
Melanoma |
Phase 3 |
Unknown |
April 2013 |
Germany
... More >> Dept of Dermatology, University of Aachen
Recruiting
Aachen, Germany, 52074
Principal Investigator: Hans F Merk, Prof. (MD)
Dept of Dermatology, University of Berlin Charite
Recruiting
Berlin, Germany, 10117
Principal Investigator: Uwe Trefzer, PD (MD)
Dept of Dermatology, University of Bochum
Recruiting
Bochum, Germany, 44791
Principal Investigator: Norbert Brockmeyer, Prof. (MD)
Medizinisches Zentrum Bonn Friedensplatz
Recruiting
Bonn, Germany, 53111
Principal Investigator: Uwe Reinhold, Prof. (MD)
Dept of Dermatology, University of Essen
Recruiting
Essen, Germany, 45147
Principal Investigator: Dirk Schadendorf, Prof. (MD)
Dept of Dermatology, University of Frankfurt
Recruiting
Frankfurt / Main, Germany, 60590
Principal Investigator: Roland Kaufmann, Prof. (MD)
Dermatology, Klinikum Frankfurt/Oder
Recruiting
Frankfurt/Oder, Germany, 15236
Principal Investigator: Anett Milling, Dr. (MD)
Dept of Dermatology, University of Hannover
Recruiting
Hannover, Germany, 30449
Principal Investigator: Ralf Gutzmer, PD (MD)
Dept of Dermatology, Saarland University
Recruiting
Homburg/Saar, Germany, 66421
Principal Investigator: Knuth Rass, Dr. (MD)
Dept of Dermatology, University of Jena
Recruiting
Jena, Germany, 07740
Principal Investigator: Martin Kaatz, Dr. (MD)
Dept of Dermatology, University of Schleswig-Holstein Campus Kiel
Recruiting
Kiel, Germany, 24105
Principal Investigator: Axel Hauschild, Prof. (MD)
Dermatology, Klinikum Ludwishafen
Recruiting
Ludwigshafen, Germany, 67063
Principal Investigator: Edgar Dippel, Prof. (MD)
Dept of Dermatology, University of Schleswig-Holstein Campus Luebeck
Recruiting
Luebeck, Germany, 23538
Principal Investigator: Patrick Terheyden, Dr. (MD)
Dept of Dermatology, Univeristy of Magdeburg
Recruiting
Magdeburg, Germany, 39120
Principal Investigator: Martin Leverkus, Prof. (MD)
Dept of Dermatology, University of Mainz
Recruiting
Mainz, Germany, 55131
Principal Investigator: Carmen Loquai, Dr. (MD)
Dept of Dermatology, University of Mannheim
Recruiting
Mannheim, Germany, 68167
Principal Investigator: Jessica Hassel, Dr. (MD)
Dept of Dermatology, University of Marburg
Recruiting
Marburg, Germany, 35033
Principal Investigator: Michael Hertl, Prof. (MD)
Dept of Dermatology, University of Muenchen
Recruiting
Muenchen, Germany, 80337
Principal Investigator: Carola Berking, Prof. (MD)
Dept of Dermatology, University of Muenster
Recruiting
Muenster, Germany, 48149
Principal Investigator: Cord Sunderkoetter, Prof. (MD)
Dept of Medical Oncology, Fachklinik Hornheide
Recruiting
Muenster, Germany, 48157
Principal Investigator: Michael Fluck, Dr. (MD)
Dermatology, Klinikum Dorothea Christiane Erxleben
Recruiting
Quedlinburg, Germany, 06484
Principal Investigator: Jens Ulrich, PD (MD)
Dept of Dermatology, University of Tuebingen
Recruiting
Tuebingen, Germany, 72086
Principal Investigator: Claus Garbe, Prof. (MD)
Dept of Dermatology, University of Wuerzburg
Recruiting
Wuerzburg, Germany, 97080
Principal Investigator: Selma Ugurel, Prof. (MD) Less << |
|
NCT00002836
|
Breast Cancer |
Phase 3 |
Completed |
- |
United States, Texas
... More >>
University of Texas - MD Anderson Cancer Center
Houston, Texas, United States, 77030-4009 Less << |
|
NCT01391572
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Phase 2 |
Unknown |
- |
China, Shanghai
... More >>
Fudan University Cancer Center
Recruiting
Shanghai, Shanghai, China, 200032
Contact: Xu-Wei Cai, M.D., Ph.D. 8621-64175590 ext 1504 birdhome2000@hotmail.com
Principal Investigator: Xiao-Long Fu, M.D., Ph.D. Less << |
|
NCT00053105
|
Lymphoma |
Phase 1 |
Unknown |
- |
United States, Arizona
... More >>
Arizona Clinical Research Center
Tucson, Arizona, United States, 85712
United States, Arkansas
Highlands Oncology Group
Springdale, Arkansas, United States, 72764
United States, California
USC/Norris Comprehensive Cancer Center and Hospital
Los Angeles, California, United States, 90033-0800
United States, Maryland
Marlene and Stewart Greenebaum Cancer Center, University of Maryland
Baltimore, Maryland, United States, 21201
United States, Ohio
Ireland Cancer Center
Cleveland, Ohio, United States, 44106-5055
United States, Tennessee
Boston Baskin Cancer Group, University Tennessee
Memphis, Tennessee, United States, 38104
United States, Texas
University of Texas - MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00002615
|
Gastric Cancer |
Phase 3 |
Completed |
- |
United Kingdom
... More >> Epsom General Hospital
Epsom, Surrey, United Kingdom, KT18 7EG Less << |
|
NCT00003576
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
Singapore
... More >> National Cancer Centre - Singapore
Singapore, Singapore, 169610 Less << |
|
NCT00005825
|
Lung Cancer |
Phase 2 |
Unknown |
- |
United States, California
... More >>
Rajendra Prasad M.D., Inc.
Lakewood, California, United States, 90712 Less << |
|
NCT00021112
|
Lung Cancer |
Phase 2 |
Terminated(low accrual) |
- |
Belgium
... More >> Algemeen Ziekenhuis Middelheim
Antwerp, Belgium, 2020
Universitair Ziekenhuis Antwerpen
Edegem, Belgium, B-2650
Germany
Thoraxklinik Rohrbach
Heidelberg, Germany, D-69126
Netherlands
Gelre Ziekenhuizen - Lokatie Lukas
Apeldoorn, Netherlands, 7334 DZ
Sint Antonius Ziekenhuis
Nieuwegein, Netherlands, 3435 CM
Rotterdam Cancer Institute
Rotterdam, Netherlands, 3000 CA
University Hospital - Rotterdam Dijkzigt
Rotterdam, Netherlands, 3000 CA
Academisch Ziekenhuis Utrecht
Utrecht, Netherlands, 3584 CX
Poland
Medical University of Gdansk
Gdansk, Poland, 80-211
National Institute of Tuberculosis and Lung Diseases
Warsaw, Poland Less << |
|
NCT01836029
|
Carcinoma, Squamous Cell of He... More >>ad and Neck Less << |
Phase 2 |
Unknown |
September 2016 |
- |
|
NCT00004099
|
Gastric Cancer |
Phase 3 |
Terminated(low accrual) |
- |
Belgium
... More >> Institut Jules Bordet
Brussels, Belgium, 1000
Egypt
National Cancer Institute of Egypt
Cairo, Egypt
Germany
Robert Roessle Klinik
Berlin, Germany, D-13122
Medizinische Klinik I
Dresden, Germany, D-01307
Universitaetsklinik Duesseldorf
Duesseldorf, Germany, D-40225
Department of Medicine III
Erlangen, Germany, D-91054
Kliniken Essen - Mitte
Essen, Germany, D-45136
Evangelisches Bethesda Krankenhaus GmbH
Essen, Germany, D-45355
Krankenhaus Nordwest
Frankfurt, Germany, D-60488
Klinikum der J.W. Goethe Universitaet
Frankfurt, Germany, D-60590
Universitatsklinik - Saarland
Homburg, Germany, D-66421
Klinik & Poliklinik fur Strahlentherapie der Universitat zu Koln
Koln, Germany, D-50924
Kreiskrankenhaus Meissen
Meissen, Germany, D-01657
Westfaelische Wilhelms-Universitaet
Muenster, Germany, DOH-48149
Klinikum Rechts Der Isar/Technische Universitaet Muenchen
Munich, Germany, D-81675
Staedtisches Krankenhaus
Solingen, Germany, D-42653
Netherlands
Academisch Ziekenhuis Maastricht
Maastricht, Netherlands, 6202 AZ
Portugal
Instituto Portugues de Oncologia Centro do Porto, SA
Porto, Portugal, 4200 Less << |
|
NCT00030459
|
Malignant Mesothelioma |
Phase 2 |
Unknown |
- |
United Kingdom
... More >> Princess Royal Hospital
Hull, England, United Kingdom, HU8 9HE
Leeds Teaching Hospital Trust
Leeds, England, United Kingdom, LS1 3EX
Saint Bartholomew's Hospital
London, England, United Kingdom, EC1A 7BE
Medical Research Council Clinical Trials Unit
London, England, United Kingdom, NW1 2DA
Royal Marsden Hospital
Sutton, England, United Kingdom, SM2 5PT
Aberdeen Royal Infirmary
Aberdeen, Scotland, United Kingdom, AB25 2ZN
Stobhill General Hospital
Glasgow, Scotland, United Kingdom, G21 3UW
Dorothy House Foundation
Bradford-Onavon, United Kingdom, BA15 2LE
St. Peters Hospital
Chertsey Surrey, United Kingdom, KT 16 OPZ Less << |
|
NCT00025090
|
Anal Cancer |
Phase 3 |
Completed |
- |
United Kingdom
... More >> Northwick Park Hospital
Harrow, England, United Kingdom, HA1 3UJ
Ipswich Hospital
Ipswich, England, United Kingdom, IP4 5PD
Cookridge Hospital
Leeds, England, United Kingdom, LS16 6QB
Saint Bartholomew's Hospital
London, England, United Kingdom, EC1A 7BE
Cancer Research UK and University College London Cancer Trials Centre
London, England, United Kingdom, NW1 2ND
James Cook University Hospital
Middlesbrough, England, United Kingdom, TS4 3BW
Mount Vernon Cancer Centre at Mount Vernon Hospital
Northwood, England, United Kingdom, HA6 2RN
Nottingham City Hospital
Nottingham, England, United Kingdom, NG5 1PB
Royal Marsden - Surrey
Sutton, England, United Kingdom, SM2 5PT
Southend University Hospital NHS Foundation Trust
Westcliff-On-Sea, England, United Kingdom, SS0 0RY
Aberdeen Royal Infirmary
Aberdeen, Scotland, United Kingdom, AB25 2ZN
Velindre Cancer Center at Velindre Hospital
Cardiff, Wales, United Kingdom, CF14 2TL
Cancer Research Centre at Weston Park Hospital
Sheffield, United Kingdom, S1O 2SJ Less << |
|
NCT00006212
|
Lung Cancer |
Phase 1 |
Unknown |
- |
United States, New York
... More >>
Center for Molecular Medicine
Garden City, New York, United States, 11530 Less << |
|
NCT00003652
|
Anal Cancer |
Phase 3 |
Unknown |
- |
France
... More >> Centre Paul Papin
Angers, France, 49036
Institut Sainte Catherine
Avignon, France, 84082
Institut Bergonie
Bordeaux, France, 33076
CHU Ambroise Pare
Boulogne Billancourt, France, F-92104
C.H. Bourg En Bresse
Bourg En Bresse, France, 01012
Centre Regional Francois Baclesse
Caen, France, 14076
Centre Hospitalier
Chalon Sur Saone, France, F-71321
Hopital Louis Pasteur
Colmar, France, 68024
Centre Hospitalier Universitaire Henri Mondor
Creteil, France, 94010
Centre de Lutte Contre le Cancer Georges-Francois Leclerc
Dijon, France, 21079
Centre Hospitalier Departemental
La Roche Sur Yon, France, F-85025
Centre Oscar Lambret
Lille, France, 59020
Centre Hospital Regional Universitaire de Limoges
Limoges, France, 87042
Centre Leon Berard
Lyon, France, 69373
Institut J. Paoli and I. Calmettes
Marseille, France, 13273
CHU de la Timone
Marseille, France, 13385
Hopital Clinique Claude Bernard
Metz, France, 57072
Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Montpellier, France, 34298
Centre Hospitalier de Mulhouse
Mulhouse, France, 68051
Centre D'Oncologie De Gentilly
Nancy, France, 54100
Centre Regional Rene Gauducheau
Nantes-Saint Herblain, France, 44805
Centre Catherine de Sienne
Nante, France, 02
Centre Antoine Lacassagne
Nice, France, 06189
CHR D'Orleans - Hopital de la Source
Orleans, France, 45067
Hopital Europeen Georges Pompidou
Paris, France, 75015
Hopital Bichat - Claude Bernard
Paris, France, 75018
Hopital Robert Debre
Paris, France, 75019
Institut Curie - Section Medicale
Paris, France, 75248
Hopital Saint-Louis
Paris, France, 75475
CHU Pitie-Salpetriere
Paris, France, 75651
Hopital Tenon
Paris, France, 75970
Hopital Jean Bernard
Poitiers, France, 86021
Clinique Ste - Marie
Pontoise, France, 95301
Institut Jean Godinot
Reims, France, 51056
Centre Eugene Marquis
Rennes, France, 35042
Hopital Charles Nicolle
Rouen, France, 76031
Centre Henri Becquerel
Rouen, France, 76038
Clinique Armoricaine De Radiologie
Saint Brieuc, France, F-22015
Centre Rene Huguenin
Saint Cloud, France, 92211
Centre Hospitalier General de Saint Nazaire
Saint Nazaire, France, 44600
Clinique de l'Orangerie
Strasbourg, France, 67010
Centre Paul Strauss
Strasbourg, France, 67065
Centre Hospitalier Regional Metz Thionville
Thionville, France, 57126
Institut Claudius Regaud
Toulouse, France, 31052
Clinique Fleming
Tours, France, 37000
Nouvelle Clinique Generale
Valence, France, 26000
Centre Alexis Vautrin
Vandoeuvre-les-Nancy, France, 54511
Institut Gustave Roussy
Villejuif, France, F-94805 Less << |
|
NCT02753140
|
Carcinoma, Squamous Cell of He... More >>ad and Neck Less << |
Phase 2 |
Unknown |
September 2018 |
- |
|
NCT03269162
|
NSCLC |
Phase 3 |
Active, not recruiting |
September 30, 2019 |
- |
|
NCT00085202
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 3 |
Active, not recruiting |
January 2023 |
United States, North Carolina
... More >>
Duke Comprehensive Cancer Center
Durham, North Carolina, United States, 27710
United States, Pennsylvania
Children's Hospital of Philadelphia
Philadelphia, Pennsylvania, United States, 19104
United States, Tennessee
St. Jude Children's Research Hospital
Memphis, Tennessee, United States, 38105
United States, Texas
Texas Children's Cancer Center and Hematology Service at Texas Children's Hospital
Houston, Texas, United States, 77030-2399
Australia, New South Wales
Sydney Children's Hospital
Randwick, New South Wales, Australia, 2031
Children's Hospital at Westmead
Westmead, New South Wales, Australia, 2145
Australia, Queensland
Lady Cilento Children's Hospital, Brisbane
Brisbane, Queensland, Australia, 4029
Australia, Victoria
Royal Children's Hospital
Parkville, Victoria, Australia, 3052
Canada, Ontario
Hospital for Sick Children
Toronto, Ontario, Canada, M5G 1X8 Less << |
|
NCT00113932
|
Multiple Myeloma |
Phase 2 |
Completed |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT00559156
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT01063946
|
Neoplasms, Malignant |
Phase 1 |
Completed |
- |
Belgium
... More >> Sanofi-Aventis Administrative Office
Brussels, Belgium Less << |
|
NCT02636556
|
Thymoma and Thymic Carcinoma |
Phase 2 |
Recruiting |
December 2017 |
China, Shanghai
... More >>
Kailiang Wu
Recruiting
Shanghai, Shanghai, China, 20032
Contact: Kailiang Wu, M.D. Ph. D. +86 64175590 ext 86722 wukailiang@aliyun.com Less << |
|
NCT02976142
|
Gastric Cancer, Metastatic |
Phase 2 |
Not yet recruiting |
December 2020 |
- |
|
NCT01263886
|
Non-small Cell Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00004160
|
Lung Cancer |
Phase 1
Phase 2 |
Completed |
- |
United States, Alabama
... More >>
University of Alabama Comprehensive Cancer Center
Birmingham, Alabama, United States, 35294 Less << |
|
NCT01262183
|
Irresectable Squamous Cell or ... More >>Adenocarcinoma of the Oesophagus Less << |
Phase 2 |
Terminated(low recruitment num... More >>bers) Less << |
October 2012 |
Netherlands
... More >> University Medical Centre Nijmegen
Nijmegen, Gelderland, Netherlands, 6500 HB Less << |
|
NCT03760575
|
Mesotheliomas Pleural |
Phase 1 |
Not yet recruiting |
November 2021 |
United States, Pennsylvania
... More >>
Abramson Cancer Center of the University of Pennsylvania
Not yet recruiting
Philadelphia, Pennsylvania, United States, 19104
Contact: Evan W Alley 588-216-0098 penncancertrials@emergingmed.com Less << |
|
NCT02132143
|
Lung Adenocarcinoma Patients W... More >>ith Postoperative in pN2 Less << |
Not Applicable |
Unknown |
January 2017 |
China
... More >> The People's Hospital of Guangxi Zhuang Autonomous Region
Recruiting
Nanning Shi, China, 530021
Contact: Hui Lin 8613878133266 linhui33266@sina.com Less << |
|
NCT01704716
|
Neuroblastoma |
Phase 3 |
Recruiting |
September 2024 |
- |
|
NCT01525927
|
Oropharyngeal Neoplasms |
Phase 2 |
Terminated(Principal Investiga... More >>tor left institution. IRB approval lapsed.) Less << |
- |
United States, New York
... More >>
Long Island Jewish Medical Center
New Hyde Park, New York, United States, 11040 Less << |
|
NCT01525927
|
- |
|
Terminated(Principal Investiga... More >>tor left institution. IRB approval lapsed.) Less << |
- |
- |
|
NCT00112346
|
Non-Small-Cell Lung Carcinoma |
Phase 2 |
Completed |
- |
- |
|
NCT03345810
|
Carcinoma, Non-Small-Cell Lung... More >>
Metastatic Lung Cancer
Non Small Cell Lung Cancer
Lung Adenocarcinoma Metastatic
Large Cell Lung Carcinoma Metastatic Less << |
Phase 2 |
Recruiting |
December 2022 |
Germany
... More >> Gesundheitszentrum St. Marien GmbH
Recruiting
Amberg, Germany, 92224
Contact: Ludwig Fischer von Weikersthal, Dr. +49 9621 381637 weikersthal.ludwig@klinikum-amberg.de
DRK-Kliniken Berlin Mitte
Recruiting
Berlin-Mitte, Germany, 13359
Contact: Vincent van Laak, Dr. +49 30 30356303 v.laak@drk-kliniken-berlin.de
Ev. Lungenklinik Berlin
Recruiting
Berlin, Germany, 13125
Contact: Christian Grohé, Prof. Dr. +49 30 94802112 christian.grohe@pgdiakonie.de
Klinikum Darmstadt
Recruiting
Darmstadt, Germany, 64283
Contact: Helga Bernhard, Prof. Dr. +49 61511076651 helga.bernhard@mail.klinikum-darmstadt.de
Universitätsklinikum Carl-Gustav-Carus
Recruiting
Dresden, Germany, 01307
Contact: Martin Wermke, Dr. +49 351 4587566 martin.wermke@uniklinikum-dresden.de
Klinikum Esslingen
Recruiting
Esslingen, Germany, 73730
Contact: Martin Faehling, Dr. +49 711 31032402 m.faehling@klinikum-es.de
Universitätsklinikum Frankfurt
Recruiting
Frankfurt am Main, Germany, 60590
Contact: Wolfgang Gleiber, Dr. +49 69 63016337 wolfgang.gleiber@kgu.de
Klinik Schillerhöhe
Recruiting
Gerlingen, Germany, 70839
Contact: Martin Kimmich, Dr. +49 7156 2037719 martin.kimmich@klinik-schillerhoehe.de
Universitätsmedizin Greifswald
Recruiting
Greifswald, Germany, 17475
Contact: Michael Boesche, Dr. +49 3834 8680683 boeschem@uni-greifswald.de
Onkodoc GmbH
Recruiting
Gütersloh, Germany, 33332
Contact: Philipp Schütt, PD Dr. +49 5241 8328150 schuett@onkologie-guetersloh.de
Krankenhaus Martha-Maria Halle Dölau
Recruiting
Halle (Saale), Germany, 06120
Contact: Wolfgang Schütte, Prof. Dr. +49 345 5591440 wolfgang.schuette@Martha-Maria.de
Department of Thoracic Oncology, Thoraxklinik at Heidelberg University Hospital
Recruiting
Heidelberg, Germany
Contact: Jonas Kuon, Dr +49 6221 396 8008 Jonas.Kuon@med.uni-heidelberg.de
Lungenklinik Hemer
Recruiting
Hemer, Germany, 58675
Contact: Monika Serke, Dr +49 2372 9082201 monika.serke@lkhemer.de
"Vincentius-Diakonissen-Kliniken gAG
Recruiting
Karlsruhe, Germany, 76137
Contact: Christian Meyer zum Büschenfelde, Prof. Dr. +49 721 81083014 christian.mzb@vincentius-ka.de
Kliniken der Stadt Köln gGmbH
Recruiting
Köln, Germany, 51109
Contact: Walburga Engel-Riedel, Dr. +49 221 890713962 engelriedelw@kliniken-koeln.de
Ortenau-Klinikum Lahr
Active, not recruiting
Lahr, Germany, 77933
Ev. Diakonissenkrankenhaus Leipzig
Recruiting
Leipzig, Germany, 04177
Contact: Sylvia Gütz, Dr. +49 341 4443621 Sylvia.Guetz@ediacon.de
Klinikum Ludwigsburg
Recruiting
Ludwigsburg, Germany, 71640
Contact: Matthias Ulmer, Dr. +49 7141 9994467 matthias.ulmer@kliniken-lb.de
Klinik Löwenstein gGmbH
Recruiting
Löwenstein, Germany, 74245
Contact: J.R. Fischer, PD Dr. +49 7130 154207 jr.fischer@klinik-loewenstein.de
Klinikum der Universität München
Recruiting
München, Germany, 81377
Contact: Dominik Modest, PD Dr. +40 89 44000 dominik.modest@med.uni-muenchen.de
Pius Hospital Oldenburg
Recruiting
Oldenburg, Germany, 26121
Contact: Frank Griesinger, Prof. Dr. +49 441 2291691 frank.griesinger@pius-hospital.de
Krankenhaus Barmherzige Brüder
Recruiting
Regensburg, Germany, 93049
Contact: Nicolas Moosmann, Dr. +49 941 3692151 nicolas.moosmann@barmherzige-regensburg.de
"Klinikum Rheine
Not yet recruiting
Rheine, Germany, 48341
Contact: Nicolas Dickgreber, Dr. +49 5971-422701 n.dickgreber@mathias-spital.de
Marienhospital
Recruiting
Stuttgart, Germany, 70199
Contact: Claudio Denzlinger, Prof. Dr. claudio.denzlinger@vinzenz.de
Krankenhaus der Barmherzigen Brüder
Recruiting
Trier, Germany, 54292
Contact: Heinz Kirchen, Dr. +49 651 2082662 h.kirchen@bk-trier.de
Universitätsklinikum Ulm
Recruiting
Ulm, Germany, 89081
Contact: Cornelia Kropf-Sanchen, Dr. cornelia.kropf-sanchen@uniklinik-ulm.de
Schwarzwald-Baar Klinikum
Not yet recruiting
Villingen-Schwenningen, Germany, 78052
Contact: Anja Rückert, Dr. +49 7721-934013 anja.rueckert@sbk-vs.de
SHG-Kliniken-Völklingen
Not yet recruiting
Völklingen, Germany, 66333
Contact: Harald Schäfer, Prof. Dr. +49 6898 122351 h.schaefer@vk.shg-kliniken.de
Hämatologisch-Onkologische Praxis Würselen
Not yet recruiting
Würselen, Germany, 52146
Contact: Christoph Maintz, Dr. +49 2405-48920 c.maintz@aachen-onkologie.de
Klinikum Würzburg Mitte gGmbH
Not yet recruiting
Würzburg, Germany, 97074
Contact: Jens Kern, Dr. +49 931 7912814 jens.kern@missioklinik.de Less << |
|
NCT01769391
|
Squamous Non Small Cell Lung C... More >>ancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01769391
|
- |
|
Completed |
- |
- |
|
NCT02145390
|
Bladder Cancer
... More >> Bladder Carcinoma
Transition Cell Cancer
Muscle Invasive Bladder Carcinoma Less << |
Not Applicable |
Terminated(Investigator Decisi... More >>on - Insufficient Accrual) Less << |
- |
United States, Florida
... More >>
University of Miami Sylvester Comprehensive Cancer Center
Miami, Florida, United States, 33136 Less << |
|
NCT01125995
|
Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
Keunchil Park
Seoul, Korea, Republic of, 135-710
Asan Medican Center
Seoul, Korea, Republic of Less << |
|
NCT02996617
|
Lymphoma,Non-Hodgkin |
Phase 4 |
Unknown |
December 2018 |
China, Shandong
... More >>
Department of Hematology, Provincial Hospital Affiliated to Shandong University
Recruiting
Jin'an, Shandong, China, 250012
Contact: Xin Wang, MD, PHD 86-531-68778331 xinw007@126.com
Contact: changqing zhen, MD 86-531-68778331 zcq1521@163.com Less << |
|
NCT00486460
|
Pancreatic Cancer |
Phase 3 |
Unknown |
- |
Israel
... More >> Sourasky Medical Center
Recruiting
Tel Aviv, Israel, 64239
Contact: Nadir Arber, M.D, MSc, MHA 03-6953179 nadir@tasmc.health.gov.il Less << |
|
NCT01717924
|
Signet Ring Cell Gastric Adeno... More >>carcinoma Less << |
Not Applicable |
Recruiting |
April 2024 |
France
... More >> General and digestive surgical department, Claude Huriez Hospital, University Hospital
Recruiting
Lille cedex, France, 59037
Contact: christophe mariette, MD,PhD +33320444407 christophe.mariette@chru-lille.fr
Principal Investigator: christophe mariette Less << |
|
NCT02055924
|
B-cell Lymphoma |
Phase 1 |
Terminated(ANSM decision due t... More >>o veino occlusive disease (security alert)) Less << |
- |
Belgium
... More >> Universite Catholique de Louvain Saint Luc
Bruxelles, Belgium
CHU de Liège
Liège, Belgium, 04000
CHU UCL Namur asbl
Yvoir, Belgium, 5530
France
Centre François Baclesse
Caen, France, 14076
Hôpital Henri Mondor
Créteil, France, 94010
CHU de Dijon - Hôpital le Bocage
Dijon, France, 21034
CHRU de Lille - Hôpital Claude Huriez
Lille, France, 59037
Centre Léon Bérard
Lyon, France, 69373
CHU Montpellier
Montpellier, France, 34295
CHU de Nantes
Nantes, France, 44093
Hôpital Saint Louis
Paris, France, 75475
Centre François Magendie - Hôpital du Haut Lévêque
Pessac, France, 33604
Centre Hospitalier Lyon Sud
Pierre Bénite, France, 69495
CHU Pontchaillou
Rennes, France, 35003
Centre Henri Becquerel
Rouen, France, 76038 Less << |
|
NCT02765919
|
Squamous Cell Carcinoma of Cer... More >>vix Less << |
Phase 3 |
Completed |
- |
Bangladesh
... More >> BSM Medical University
Dhaka, Bangladesh, 1200 Less << |
|
NCT01170923
|
Non-small Cell Lung Cancer |
Phase 2 |
Unknown |
- |
- |
|
NCT00666783
|
Chemotherapy-Induced Nausea an... More >>d Vomiting Less << |
Phase 2 |
Completed |
- |
China, Shaanxi
... More >> Xijng Hospital
Xi,an, Shaanxi, China, 710032 Less << |
|
NCT02557035
|
Chemotherapy-Induced Nausea an... More >>d Vomiting Less << |
Phase 3 |
Completed |
- |
- |
|
NCT02145390
|
- |
|
Terminated(Investigator Decisi... More >>on - Insufficient Accrual) Less << |
- |
- |
|
NCT02557035
|
- |
|
Completed |
- |
- |
|
NCT00970385
|
Peripheral T Cell Lymphoma |
Phase 3 |
Completed |
- |
France
... More >> Dr REMY GRESSIN
Grenoble, France, 38043 Less << |
|
NCT01074021
|
Nasopharyngeal Cancer |
Phase 3 |
Unknown |
August 2016 |
China, Fujian
... More >> Fujian Medical University Union Hospital
Fuzhou, Fujian, China, 350001
Fujian Provincial Cancer Hospital
Fuzhou, Fujian, China
Xiamen First Hospital
Xiamen, Fujian, China, 361003
China, Guangdong
The Affiliated Cancer Hospital of Guangzhou Medical Hospital
Guangzhou, Guangdong, China, 510095
China, Guangxi
The First Affiliated Hospital of Guangxi Medical University
Nanning, Guangxi, China
Affiliated Tumor Hospital of Guangxi Medical University
Xining, Guangxi, China, 530021
China, Hebei
The Fourth Hebei Province Hospital
Shijiazhuang, Hebei, China, 050011
China, Heilongjiang
The Affiliated Cancer Hospital of Haerbin Medical University
Haerbin, Heilongjiang, China, 150081
China, Henan
Henan Cancer Hospital
Zhengzhou, Henan, China, 450008
China, Hubei
Hubei Province Caner Hospital
Wuhan, Hubei, China, 430079
Wuhan Union Hospital
Wuhan, Hubei, China
China, Liaoning
Shengjing Hospital of China Medical University
Shenyang, Liaoning, China, 110003
Liaoning Province Cancer Hospital
Shenyang, Liaoning, China, 110042
China, Shandong
Shandong Caner Hospital
Jinan, Shandong, China, 250117
China, Sichuan
Sichuan Province Cancer Hospital
Chengdu, Sichuan, China
West China School of Medicine/West China Hospital of Sichuan University (WCSM/WCH)
Chengdu, Sichuan, China
China, Zhejiang
Zhejiang Cancer Hospital
Hangzhou, Zhejiang, China, 310022
China
Cancer Institute & Hospital.Chinese Academy of Medical Sciences
Beijing, China, 100021
Peking Union Medical College Hospital
Beijing, China, 100032
Beijing Cancer Hospital
Beijing, China, 100142
The General Hospital of the People's Liberation Army
Beijing, China, 100853
The Affiliated Renji Hosptial of ShanghaiJiao Tong University
Shanghai, China, 200001
Fudan University Shanghai Cancer Center
ShangHai, China
Tianjin Cancer Hospital
Tianjin, China, 300060 Less << |
|
NCT03579082
|
Diffuse Large B Cell Lymphoma |
Phase 4 |
Recruiting |
December 1, 2019 |
China, Henan
... More >> Oncology Department of The First Affilliated Hospital of Zhengzhou University
Recruiting
Zhengzhou, Henan, China, 450052
Contact: Mingzhi Zhang, Pro,Dr 13838565629 mingzhi_zhang@126.com
Contact: Mingzhi Zhang, Pro,Drc 13838565629 mingzhi_zhang@126.com Less << |
|
NCT00570531
|
Loco-regional Esophageal Cance... More >>r Less << |
Phase 2 |
Terminated(The study was unabl... More >>e the accrue the required number of patients within a reasonable time.) Less << |
- |
- |
|
NCT02516241
|
Urothelial Cancer |
Phase 3 |
Recruiting |
September 23, 2019 |
- |
|
NCT00570531
|
- |
|
Terminated(The study was unabl... More >>e the accrue the required number of patients within a reasonable time.) Less << |
- |
- |
|
NCT01770418
|
Lung Neoplasms |
Phase 1
Phase 2 |
Recruiting |
- |
United States, Florida
... More >>
University of Florida Proton Therapy Institute
Recruiting
Jacksonville, Florida, United States, 32206
Contact: Samantha Sago 904-588-1529 SSago@floridaproton.org
Principal Investigator: Brad Hoppe, MD
United States, Illinois
Northwestern Medicine Chicago Proton Center
Recruiting
Warrenville, Illinois, United States, 60555
Contact: Megan Boroczk 630-315-1797 Megan.Boroczk@nm.org
Principal Investigator: John Chang, MD
United States, Maryland
Maryland Proton Treatment Center
Recruiting
Baltimore, Maryland, United States, 21201
Contact: Carl Brown 410-364-5353 CarlBrown@umm.edu
Principal Investigator: Pranshu Mohindra, MD
United States, New Jersey
Princeton ProCure Management LLC
Recruiting
Somerset, New Jersey, United States, 08873
Contact: Heba Darwish 732-357-2676 heba.darwish@nj.procure.com
Principal Investigator: Brian Chon, MD
United States, Oklahoma
ProCure Proton Therapy Center
Recruiting
Oklahoma City, Oklahoma, United States, 73142
Contact: Kayla Tarrant 405-773-6775 research@okc.procure.com
Principal Investigator: Gary Larson, MD
United States, Virginia
Hampton University Proton Therapy Institute
Recruiting
Hampton, Virginia, United States, 23666
Contact: Donna Sternberg, RN, BSN, OCN 757-251-6839 Donna.Sternberg@hamptonproton.org
Principal Investigator: Christopher Sinesi, MD Less << |
|
NCT02471027
|
- |
|
Not yet recruiting |
June 2019 |
China, Beijing
... More >> Beijing Obstetrics and Gynecology Hospital, Capital Medical University
Beijing, Beijing, China, 100000 Less << |
|
NCT01945879
|
Pancreatic Cancer |
Phase 1
Phase 2 |
Completed |
- |
Germany
... More >> Charité - Universitätsmedizin Berlin
Berlin, Germany, 13353 Less << |
|
NCT03274284
|
Bladder Cancer |
Phase 4 |
Not yet recruiting |
December 2019 |
- |
|
NCT00954512
|
- |
|
Terminated(This study was term... More >>inated for business reasons.) Less << |
- |
- |
|
NCT01187290
|
Non-small Cell Lung Cancer |
Phase 2 |
Unknown |
June 2013 |
- |
|
NCT01553071
|
Solid Tumor |
Phase 1 |
Recruiting |
- |
United States, Texas
... More >>
University of Texas Southwestern
Not yet recruiting
Dallas, Texas, United States, 75390
Contact: Dong Ying 214-648-5107 ying.dong@utsouthwestern.edu
Contact: Marcella Aguilar 214-648-1479 marcella.aguilar@utsouthwestern.edu
Principal Investigator: David Gerber, MD
Joe Arrington Cancer Center/Covenant Children's Hospital
Recruiting
Lubbock, Texas, United States, 79410
Contact: Christy Ratheal 806-725-8131 cratheal@covhs.org
Principal Investigator: Isaac Tafur, MD
University Medical Center
Recruiting
Lubbock, Texas, United States, 79415
Contact: Melanie Bisbee 806-775-8590 melanie.bisbee@umchealthsystem.com
Principal Investigator: Sanjay Awasthi, MD Less << |
|
NCT00128817
|
Larynx Neoplasms |
Phase 3 |
Terminated(Slow Accrual) |
May 2015 |
India
... More >> Tata Memorial Hospital
Mumbai, Maharashtra, India, 400012 Less << |
|
NCT00954512
|
Neoplasms |
Phase 1
Phase 2 |
Terminated(This study was term... More >>inated for business reasons.) Less << |
- |
- |
|
NCT02350361
|
Lung Cancer |
Phase 2
Phase 3 |
Unknown |
March 2016 |
China
... More >> Peking University Third Hospital
Recruiting
Beijing, China
Contact: Li Liang, MD 13241870816 Less << |
|
NCT02014831
|
Squamous Cell Carcinoma of the... More >> Penis Less << |
Phase 2 |
Withdrawn(Industry decline to ... More >>supply study drug) Less << |
- |
France
... More >> CHRU Strasbourg
Strasbourg, Bas Rhin, France, 67098
Institut Paoli Calmettes
Marseille, Bouches du Rhône, France, 13273
Institut Bergonié
Bordeaux, Gironde, France, 33076
Institut Claudius Regaud
Toulouse, Haute Garonne, France, 31052
Institut Curie
Paris, Ile de France, France, 75005
APHP Hôpital Saint Louis
Paris, Ile de France, France, 75475
Centre Léon Bérard
Lyon, Rhône, France, 69373 Less << |
|
NCT01950689
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 3 |
Recruiting |
June 2021 |
United Kingdom
... More >> Cheltenham General Hospital
Recruiting
Cheltenham, Gloucestershire, United Kingdom, GL53 7AN
Principal Investigator: Warren Grant
Clatterbridge Centre for Oncology
Recruiting
Bebington, Merseyside, United Kingdom, CH63 4JY
Principal Investigator: David Husband
St James' Hospital
Recruiting
Leeds, West Yorkshire, United Kingdom, LS9 7TF
Principal Investigator: Mehmet Sen
Belfast City Hospital
Recruiting
Belfast, United Kingdom, BT9 7AB
Contact: Fiona McCloskey
Queen Elizabeth Hospital
Suspended
Birmingham, United Kingdom, B15 2TH
Bradford Teaching Hospitals NHS Foundation Trust
Recruiting
Bradford, United Kingdom, BD9 6RJ
Principal Investigator: Michael Ho
Bristol Haematology and Oncology centre
Recruiting
Bristol, United Kingdom, BS2 8ED
Principal Investigator: Hoda Booz
Addenbrookes Hospital
Recruiting
Cambridge, United Kingdom, CB2 0QQ
Principal Investigator: Gill Barnett
Velindre Cancer Centre
Recruiting
Cardiff, United Kingdom, CF14 2TL
Principal Investigator: Mererid Evans
University Hospitals Coventry and Warwickshire
Recruiting
Coventry, United Kingdom, CV2 2DX
Principal Investigator: Andrew Chan
The Beatson West of Scotland Cancer Centre
Recruiting
Glasgow, United Kingdom, G12 0YN
Principal Investigator: Stefano Schipani
The Royal Surrey County Hospital
Recruiting
Guildford, United Kingdom, GU2 7XX
Principal Investigator: Katie Wood
Leicester Royal Infirmary
Recruiting
Leicester, United Kingdom, LE1 5WW
Principal Investigator: Thiagarajan Sridhar
University College London Hospital
Recruiting
London, United Kingdom, NW1 2PG
Principal Investigator: Ruheena Mendes
The Royal Marsden
Recruiting
London, United Kingdom, SW3 6JJ
Principal Investigator: Chris Nutting
The Christie NHS Foundation Trust
Recruiting
Manchester, United Kingdom, M20 4BX
Principal Investigator: Nick Slevin
The James Cook University Hospital
Suspended
Middlesborough, United Kingdom, TS4 3BW
Mount Vernon
Recruiting
Middlesex, United Kingdom, HA6 2RN
Principal Investigator: Catherine Lemon
Freeman Hospital
Withdrawn
Newcastle, United Kingdom, NE7 7DN
Nottingham University Hospitals
Recruiting
Nottingham, United Kingdom, NG5 1PB
Principal Investigator: Judith Christian
Weston Park Hospital
Recruiting
Sheffield, United Kingdom, S10 2SJ
Principal Investigator: Bernadette Foran
Singleton Hospital
Recruiting
Swansea, United Kingdom, SA2 8QA
Principal Investigator: Russell Banner
York Hospital
Recruiting
York, United Kingdom, Y031 8HE
Principal Investigator: Robin Prestwich Less << |
|
NCT01692496
|
Advanced and / or Metastatic L... More >>iposarcoma Less << |
Phase 2 |
Completed |
- |
Germany
... More >> HELIOS Klinikum Berlin-Buch (Sarkomzentrum Berlin-Brandenburg)
Berlin, Germany, D-10707
Universitätsklinikum EssenInnere Klinik (Tumorforschung)
Essen, Germany, D-45147
Medizinische Hochschule Hannover (Zentrum Innere Medizin)
Hannover, Germany, D-30625
Universitätsmedizin Mannheim (Sarkomzentrum)
Manheim, Germany, D-68169
Klinikum Großhadern der LMU Universität München (Med. Klinik und Poliklinik II)
Munich, Germany, D-80336
Spain
Hospital Central de Asturias
Oviedo, Asturias, Spain, 33011
Hospital Universitario de Canarias
La Laguna, Islas Canarias, Spain, 38320
Hospital de Navarra
Pamplona, Navarra, Spain, 31008
Complejo Hospitalario Universitario de Vigo
Vigo, Pontevedra, Spain, 36204
Hospital Vall d'Hebrón
Barcelona, Spain, 08035
Hospital 12 de Octubre
Madrid, Spain, 28041
Hospital Universitari Son Espases
Palma de Mallorca, Spain, 07010
Hospital Universitario Virgen del Rocío
Sevilla, Spain, 41013
Instituto Valenciano de Oncología
Valencia, Spain, 46009
Hospital Miguel Servet
Zaragoza, Spain, 50009 Less << |
|
NCT00450502
|
Neoplasms |
Phase 1 |
Completed |
- |
United States, Maryland
... More >>
National Institutes of Health Clinical Center, 9000 Rockville Pike
Bethesda, Maryland, United States, 20892 Less << |
|
NCT02509806
|
Gastric Cancer |
Phase 2
Phase 3 |
Recruiting |
December 2017 |
China, Hebei
... More >> Fourth Affiliated Hospital of Hebei Medical University
Recruiting
Shijiazhuang, Hebei, China, 050011
Principal Investigator: Zhao Qun, MD Less << |
|
NCT00003143
|
Lymphoma |
Phase 2 |
Completed |
- |
United States, California
... More >>
Jonsson Comprehensive Cancer Center, UCLA
Los Angeles, California, United States, 90095-1781 Less << |
|
NCT03679338
|
Nonresectable Bile Duct Cancer |
Phase 2 |
Recruiting |
November 2021 |
France
... More >> Institut Paoli-Calmettes
Recruiting
Marseille, France, 13009
Contact: GENRE Dominique, MD + 33 4 91 22 37 78 drci.up@ipc.unicancer.fr
Contact: COURNIER Sandra + 33 4 91 22 37 78 drci.up@ipc.unicancer.fr
Principal Investigator: BORIES Erwan, MD Less << |
|
NCT00539877
|
Stomach Neoplasms |
Phase 1 |
Completed |
- |
Korea, Republic of
... More >>
Samsung Medical Center
Seoul, Korea, Republic of, 135-710 Less << |
|
NCT00127920
|
Ovarian Neoplasms |
Phase 2 |
Completed |
- |
United States, California
... More >>
Gynecologic Oncology Associates
Newport Beach, California, United States, 92663
United States, Florida
Florida Hospital College of Health Sciences
Orlando, Florida, United States, 32803 Less << |
|
NCT02679963
|
Non Small Cell Lung Cancer |
Phase 2 |
Recruiting |
January 2021 |
France
... More >> Gustave Roussy Cancer Campus
Recruiting
Villejuif, Val de Marne, France, 94805
Contact: Valérie Mairot 0142113730 ext +33 Valerie.mairot@gustaveroussy.fr Less << |
|
NCT01163552
|
Pleural Metastases
... More >> Breast Cancer
Colon Cancer
Ovarian Cancer
Uterine Cancer
Renal Cell Cancer
Thymic Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
St. Luke's-Roosevelt Hospital Center
New York, New York, United States, 10019 Less << |
|
NCT00815945
|
Mesenchymal Tumor
... More >> Carcinosarcoma
Leiomyosarcoma Less << |
Phase 2 |
Completed |
- |
Germany
... More >> Charité, Campus Virchow Klinikum, Frauenklinik
Berlin, Germany, 13353
Malteser Krankenhaus, Gynäkologie und Geburtshilfe
Bonn, Germany, 53123
Klinikum Bremen-Mitte gGmbH, Frauenklinik
Bremen, Germany, 28177
Universitätsklinikum Carl Gustav Carus, Klinik und Poliklinik für Frauenheilkunde und Geburtshilfe
Dresden, Germany, 01307
Evangelisches Krankenhaus Düsseldorf, Frauenklinik
Duesseldorf, Germany, 40217
Universitätsklinikum Essen, Frauenklinik
Essen, Germany, 45122
Klinikum der J. W. Goethe-Universität, Klinik für Gynäkologie und Geburtshilfe
Frankfurt, Germany, 60590
University Hospital Hamburg-Eppendorf
Hamburg, Germany, 20251
Gynäkologisch-onkologische Praxis
Hannover, Germany, 30177
St. Vincentius Kliniken AG, Frauenklinik
Karlsruhe, Germany, 76135
Universitätsklinikum Giessen und Marburg GmbH, Klinik für Gynäkologie, Gynäkologische Endokrinologie und Onkologie
Marburg, Germany, 35043
Klinikum Großhadern, Frauenklinik
München, Germany, 81377
Universitätsklinikum Tübingen, Frauenklinik
Tübingen, Germany, 72076
Universitätsklinikum Ulm, Universitätsfrauenklinik
Ulm, Germany, 89075
Dr. Horst Schmidt Kliniken GmbH, Klinik für Gynäkologie und gynäkologische Onkologie
Wiesbaden, Germany, 65199 Less << |
|
NCT03509012
|
Carcinoma, Squamous Cell of He... More >>ad and Neck
Carcinoma, Non-Small-Cell Lung
Small Cell Lung Carcinoma Less << |
Phase 1 |
Recruiting |
April 4, 2022 |
United States, Arizona
... More >>
Research Site
Recruiting
Tucson, Arizona, United States, 85719
United States, Colorado
Research Site
Recruiting
Aurora, Colorado, United States, 80045
United States, Missouri
Research Site
Not yet recruiting
Saint Louis, Missouri, United States, 63110
United States, New Jersey
Research Site
Withdrawn
Middletown, New Jersey, United States, 07748
Japan
Research Site
Recruiting
Koto-ku, Japan, 135-8550
Research Site
Recruiting
Sunto-gun, Japan, 411-8777
Korea, Republic of
Research Site
Recruiting
Seoul, Korea, Republic of, 03080
Research Site
Recruiting
Seoul, Korea, Republic of, 03722
Research Site
Recruiting
Seoul, Korea, Republic of, 05505
Research Site
Recruiting
Seoul, Korea, Republic of, 135-710
Taiwan
Research Site
Not yet recruiting
Kaohsiung, Taiwan, 807
Research Site
Not yet recruiting
Taipei, Taiwan, 10002 Less << |
|
NCT00937612
|
Head and Neck Neoplasms
... More >> Carcinoma, Squamous Cell Less << |
Not Applicable |
Terminated(Study group showed ... More >>better results. Poor Accrual.) Less << |
- |
Taiwan
... More >> Chang Gung Memorial Hospital, Keelung branch
Keelung, Taiwan, 222
Chang Gung Memorial Hospital, Linkou Branch
Taoyuan, Taiwan, 333
Department of Radiation Therapy, Chang Gung Memorial Hospital, Linkou branch
Taoyuan, Taiwan, 333 Less << |
|
NCT02118415
|
NSCLC Stage IIIA/B |
Phase 2 |
Unknown |
February 2018 |
Germany
... More >> Klinikum rechts der Isar, Strahlentherapie und Radiologische Onkologie
Recruiting
Munich, Germany, 81675
Contact: Hanno Specht, Dr. +49 (0)89 4140 ext 5910 Hanno.Specht@mri.tum.de Less << |
|
NCT02284308
|
NSCLC |
Not Applicable |
Recruiting |
July 2023 |
Belgium
... More >> University Hospital Gent
Not yet recruiting
Gent, Belgium, 9000
Contact: Veerle Surmont
Netherlands
Radiotherepiegroep
Recruiting
Arnhem, Gelderland, Netherlands, 6815 AD
Principal Investigator: Marcel Stam
Caniusius-Wilhelmina Hospital
Recruiting
Nijmegen, Gelderland, Netherlands, 6532 SZ
Principal Investigator: Yvonne Berk
Ziekenhuisgroep Twente
Recruiting
Almelo, Overijssel, Netherlands, 7609 PP
Principal Investigator: Jeske Staal
Deventer Hospital
Recruiting
Deventer, Overijssel, Netherlands, 7416 SE
Principal Investigator: Suzy Samii
Radiotherapiegroep
Recruiting
Deventer, Overijssel, Netherlands, 7416 SE
Principal Investigator: Ernest Vonk
Gelre ziekenhuis
Recruiting
Apeldoorn, Netherlands
Contact: N Pronk, Dr.
Principal Investigator: N Pronk
Rijnstate Ziekenhuis
Recruiting
Arnhem, Netherlands, 6815 AD
Contact: Niels Claessens
Amphia Ziekenhuis
Not yet recruiting
Breda, Netherlands, 4818 CK
Contact: Joachim Aerts
Medical Centre Haaglanden
Recruiting
Den Haag, Netherlands, 2512 VA
Contact: Klaar Maas
Catharina Hospital Eindhoven
Recruiting
Eindhoven, Netherlands, 5623 EJ
Contact: Ben van den Borne
Zuyderland
Recruiting
Heerlen, Netherlands, 6419 PC
Principal Investigator: Gerben Bootsma
Leiden University Medical Centre
Not yet recruiting
Leiden, Netherlands, 2333 ZC
Contact: Rajen Ramai
MAASTRO clinic
Recruiting
Maastricht, Netherlands, 6229 ET
Contact: Judith van Loon, MD, PhD +31 88 44 55 666 judith.vanloon@maastro.nl
Contact: Nadja Drummen +31 88 44 55 817 nadja.drummen@maastro.nl
Principal Investigator: Judith van Loon
Maastricht University Medical Center
Recruiting
Maastricht, Netherlands, 6229 HX
Contact: Anne-Marie Dingemans, MD, PhD +31 43 387 65 43 a.dingemans@maastro.nl
Principal Investigator: Anne-Marie Dingemans
University Medical Centre Nijmegen
Recruiting
Nijmegen, Netherlands, 6525 GA
Principal Investigator: Olga Schuurbiers
Laurentius Hospital
Recruiting
Roermond, Netherlands, 6043 VC
Contact: Cordula Pitz
VieCuri Medical Centre
Recruiting
Venlo, Netherlands, 5912 BL
Contact: Marcel Westenend Less << |
|
NCT03223740
|
Locally Advanced Gastric Carci... More >>noma Less << |
Phase 3 |
Recruiting |
October 2021 |
China, Shanxi
... More >> Xijing Hospital, GI institute
Active, not recruiting
Xi'an, Shanxi, China, 710032
China, Sichuan
friendship Hospital
Recruiting
Chengdu, Sichuan, China, 610000
Contact: lili deng, MD +8613880926676
ziyang People's Hospital
Recruiting
Siyang, Sichuan, China
Contact: min wei, MD +8613980386078 Less << |
|
NCT00159471
|
Pancreatic Cancer |
Not Applicable |
Terminated(Insufficient Accrua... More >>l) Less << |
- |
United States, California
... More >>
U.S.C / Norris Comprehensive Cancer Center
Los Angeles, California, United States, 90033 Less << |
|
NCT01653067
|
Diffuse Large B-Cell Lymphoma |
Phase 2 |
Unknown |
July 2018 |
Germany
... More >> University Hospital Freiburg
Freiburg, Baden-Württemberg, Germany, 79106
University of Heidelberg Hospital
Heidelberg, Baden-Württemberg, Germany, 69120
University Hospital Ulm
Ulm, Baden-Württemberg, Germany, 89081
University Hospital Erlangen
Erlangen, Bayern, Germany, 91054
Ludwig-Maximilians-University of Munich
Munich, Bayern, Germany, 81377
Technische Universität München
Munich, Bayern, Germany, 81675
Johann Wolfgang Goethe University Hospitals, Frankfurt
Frankfurt, Hessen, Germany, 60590
Johannes Guttenberg University Mainz
Mainz, Rheinland-Pfalz, Germany, 55101
Charité University Berlin
Berlin, Germany, 12200 Less << |
|
NCT02644252
|
Lung Cancer |
Phase 3 |
Recruiting |
December 2019 |
Denmark
... More >> Department of Oncology, Vejle Hospital
Recruiting
Vejle, Denmark
Contact: Christa H Nyhus, MD christa.haugaard.nyhus@rsyd.dk
Principal Investigator: Lisbeth Bertelsen, MD Less << |
|
NCT03547973
|
Urothelial Carcinoma |
Phase 2 |
Recruiting |
May 2020 |
United States, Connecticut
... More >>
Smilow Cancer Hospital at Yale-New Haven
Recruiting
New Haven, Connecticut, United States, 06510
Contact: Ebony Williams 203-785-5720 ebony.williams@yale.edu
Contact: Shelby DeCarlo 203-737-6299 shelby.decarlo@yale.edu
Principal Investigator: Daniel Petrylak, MD
United States, Nebraska
Urology Cancer Center
Recruiting
Omaha, Nebraska, United States, 68130
Contact: Stacy Moore 402-697-2229 smoore@gucancer.com
Principal Investigator: Luke Nordquist, MD
United States, New York
Weill Cornell Medicine
Recruiting
New York, New York, United States, 10021
Contact: Pryianka Patel prp2013@med.cornell.edu
Contact: Monique Tarrant mot2013@med.cornell.edu
Principal Investigator: Scott T. Tagawa, MD, MS Less << |
|
NCT00301548
|
Breast Neoplasms
... More >> Carcinoma Less << |
Phase 2 |
Completed |
- |
- |
|
NCT02937272
|
Solid Tumor |
Phase 1 |
Recruiting |
May 3, 2021 |
United States, California
... More >>
University of California, San Francisco
Recruiting
San Francisco, California, United States, 94115
Contact 415-514-6568
Principal Investigator: Pamela Munster
United States, Kentucky
University of Louisville
Recruiting
Louisville, Kentucky, United States, 40202
Contact 502-562-4158
Principal Investigator: Rebecca Redman
United States, New York
NYU Clinical Cancer Center
Not yet recruiting
New York, New York, United States, 10016
Contact 212-263-2441
Principal Investigator: Dimitris Placantonakis
Weill Cornell Medical College
Recruiting
New York, New York, United States, 10021
Contact 646-962-6137
Principal Investigator: Howard Fine
United States, Texas
University of Texas MD Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact 713-563-5844
Principal Investigator: Timothy Yap
Australia, New South Wales
Calvary Mater Newcastle
Recruiting
Newcastle, New South Wales, Australia, 2298
Contact 61243401581
Principal Investigator: Andre van der Westhuizen
St Vincent's Hospital
Recruiting
Sydney, New South Wales, Australia, 2010
Contact 6193555655
Principal Investigator: Anthony Joshua
Australia, Queensland
Greenslopes Private Hospital
Recruiting
Greenslopes, Queensland, Australia, 4120
Contact 61733947111
Principal Investigator: Victoria Atkinson
Canada, Ontario
Princess Margaret Hospital
Recruiting
Toronto, Ontario, Canada, M5G 2M9
Contact 4169462277
Principal Investigator: Warren P Mason
France
Hopital Saint-Louis
Recruiting
Paris, Cedex 10, France, 75475
Contact 33171207466
Principal Investigator: Antoine Carpentier
CHRU de Lille- Hôpital Roger Salengro
Recruiting
Lille, France, 59037
Contact 33662688717
Principal Investigator: Emilie Le Rhun
Hôpital Saint Joseph
Recruiting
Paris, France, 75014
Contact 330144127456
Principal Investigator: ERIC RAYMOND
Gustave Roussy
Recruiting
Villejuif Cedex, France, 94805
Contact 33142114338
Principal Investigator: Christophe Massard
Germany
Universitätsklinikum Würzburg
Recruiting
Würzburg, Bayern, Germany, 97080
Contact 4993120140160
Principal Investigator: Mario Löhr
Universitätsklinikum Essen
Recruiting
Essen, Nordrhein-Westfalen, Germany, 45122
Contact 492017232024
Principal Investigator: Jörg Hense
Charité Universitätsmedizin Berlin
Recruiting
Berlin, Germany, 12203
Contact 493084452648
Principal Investigator: Sebastian Ochsenreither
Italy
Istituto Clinico Humanitas
Recruiting
Rozzano, Milano, Italy, 20089
Contact 390282244080
Principal Investigator: Armando Santoro
Ospedale Policlinico Giambattista Rossi, Borgo Roma
Recruiting
Verona, Italy, 37134
Contact 390458128148
Principal Investigator: Davide Melisi
Japan
National Cancer Center Hospital
Not yet recruiting
Chuo-Ku, Tokyo, Japan, 104-0045
Contact 81120360605
Principal Investigator: Shunsuke Kondo
Spain
Hospital Universitari Vall d'Hebron
Recruiting
Barcelona, Spain, 08035
Contact 34934894374
Principal Investigator: Elena Garralda Cabanas
Hospital Universitario 12 de Octubre
Recruiting
Madrid, Spain, 28041
Contact 34913908000
Principal Investigator: Juan Manuel Sepúlveda Sánchez
Hospital Clínico Universitario de Valencia
Recruiting
Valencia, Spain, 46010
Contact 34961973528
Principal Investigator: Andres Cervantes Ruiperez
Switzerland
Ospedale Regionale di Bellinzona e Valli - Bellinzona
Recruiting
Bellinzona, Ticino, Switzerland, 6500
Contact 41918118931
Principal Investigator: Anastasios Stathis
Universitätsspital Zürich
Recruiting
Zürich, Switzerland, 8091
Contact 41442552214
Principal Investigator: Christian Britschgi Less << |
|
NCT02178397
|
NSCLC Patients With EGFR Activ... More >>ating Mutation Less << |
Phase 3 |
Terminated(lack of recruitment... More >>) Less << |
- |
France
... More >> CH
Aix En Provence, France
CH
Amiens, France
CHRU
Angers, France, 49933
CH
Annecy, France, 74374
CHU
Brest, France, 29609
Centre François BACLESSE
Caen, France, 14000
CHIC
Creteil, France, 94000
CH
Draguignan, France, 83007
CH
Elbeuf, France
CHIC
GAP, France, 05007
Ch La Roche/Yon
La Roche/yon, France
Centre Hospitalier
Le MANS, France, 72037
Centre Oscar Lambret
Lille, France, 59020
CHU
Limoges, France, 87042
CH
Longjumeau, France, 91160
CH
Lorient, France, 35632
CH
Macon, France, 71018
CH
Mantes La Jolie, France, 78201
Institut Paoli Calmettes
Marseille, France, 13273
AP-HM - Hôpital Nord
Marseille, France
CH
Meaux, France, 77104
Centre Hospitalier Intercommunal
Meulan, France, 78250
CH
PAU, France
Centre Catalan
Perpignan, France
CHU
Rennes, France, 35033
CH
Roanne, France
CHU
Rouen, France
CH
Salon de Provence, France
CH
Sens, France, 89100
CH
St BRIEUC, France
Institut de Cancérologie de la Loire
St ETIENNE, France, 42271
Centre Paul Strauss
Strasbourg, France, 67065
CH
Tarbes, France
Hôpital d'instruction des Armées Sainte-Anne
Toulon, France, 83041
CH
Villefranche, France, 69655 Less << |
|
NCT02817958
|
Penile Cancer
... More >> Squamous Carcinoma Less << |
Phase 2 |
Recruiting |
September 2022 |
France
... More >> ICO-Paul Papin
Recruiting
Angers, France, 49055
Contact: Emmanuelle BOMPAS, Doctor +33(0)2 41 35 27 00 emmanuelle.bompas@ico.unicancer.fr
Chr Besancon
Recruiting
Besançon, France, 25000
Contact: Tristant MAURINA, Doctor +33(0)3 81 66 93 67 t1maurina@chu-besancon.fr
Hôpital SAINT ANDRE
Recruiting
Bordeaux, France, 33075
Contact: Amaury DASTE, Doctor amaury.daste@chu-bordeaux.fr
Centre FRANCOIS BACLESSE
Recruiting
Caen, France, 14076
Contact: Florence JOLY, Prof +33 2 31 45 50 02 f.joly@baclesse.unicancer.fr
Chru Gabriel Montpied
Recruiting
Clermont Ferrand, France, 63000
Contact: Laurent GUY, Professor +33(0)4 73 75 14 97 lguy@chu-clermontferrand.fr
Ch de Limoges
Recruiting
Limoges, France, 87042
Contact: Aurélien DESCAZEAUD, Professor +33(0)5 55 05 80 15 aurelien.descazeaud@chu-limoges.fr
Centre Leon Berard
Recruiting
Lyon, France, 69008
Contact: Aude FLECHON, Doctor +33(0)4 78 78 26 43 aude.flechon@lyon.unicancer.fr
Chu Lyon Sud
Recruiting
Lyon, France, 69310
Contact: Jean-Etienne TERRIER, Dr +33 4 72 67 88 01 jean-etienne.terrier01@chu-lyon.fr
Institut Paoli-Calmettes
Recruiting
Marseille, France, 13273
Contact: Gwenaelle GRAVIS, Doctor +33(0)4 91 22 37 40 gravisg@ipc.unicancer.fr
Institut de Cancerologie de Lorraine
Recruiting
Nancy, France, 54519
Contact: Vincent MASSARD, Dr +33 3 23 59 84 61 v.massard@nancy.unicancer.fr
Clinique Urologique- Chu Hotel Dieu
Recruiting
Nantes, France, 44093
Contact: Jérôme RIGAUD, Professor +33(0)2 40 08 39 10 jrigaud@chu-nantes.fr
Institut de Cancerologie Du Gard - Centre Oncogard
Recruiting
Nîmes, France, 300029
Contact: Angélique CHAPELLE, Doctor +33(0)4 30 06 10 14 a.chapelle@oncogard.com
Hopital Saint Louis
Recruiting
Paris, France, 75010
Contact: Stéphane CULINE, Professor +33(0)1 42 49 42 47 stephane.culine@aphp.fr
Chu de Rouen
Recruiting
Rouen, France, 76031
Contact: Christian PFISTER, Professor +33(0)2 32 88 81 63 christian.pfister@chu-rouen.fr
ICO-René Gauducheau
Recruiting
saint Herblain, France, 44805
Contact: Emmanuelle BOMPAS, Doctor +33(0)2 40 67 97 00 emmanuelle.bompas@ico.unicancer.fr
Hopitaux Universitaires de Strasbourg
Recruiting
Strasbourg, France, 67091
Contact: Philippe BARTHELEMY, Doctor philippe.barthelemy@chru-strasbourg.fr
Institut Claudius Regaud
Recruiting
Toulouse, France, 31059
Contact: Loic MOUREY, Doctor +33(0)5 31 15 51 67 mourey.loic@iuct-oncopole.fr Less << |
|
NCT02215447
|
Gastrointestinal Neuroendocrin... More >>e Carcinomas Less << |
Phase 2 |
Active, not recruiting |
October 2020 |
Australia, New South Wales
... More >>
Royal North Shore Hospital
St Leonards, New South Wales, Australia, 2065
Australia, Victoria
Barwon Health
Geelong, Victoria, Australia, 3220
Royal Melbourne Hospital
Parkville, Victoria, Australia, 3050 Less << |
|
NCT01048970
|
Lung Cancer |
Phase 2
Phase 3 |
Unknown |
December 2010 |
Mexico
... More >> National Institute of Cancerologia
Recruiting
Mexico City, Mexico, 14080
Contact: Oscar Arrieta, MD (+52) (55) 5628-0400 ext 832 ogar@servidor.unam.mx
Contact: Karla Sanchez, MsC (+52) (55) 5414-7200 ext 4216 ksanchez@medicasur.org.mx
National Institute of Cancerología
Recruiting
Mexico City, Mexico, 14080
Contact: Oscar Arrieta Less << |
|
NCT00920920
|
- |
|
Active, not recruiting |
December 2020 |
Austria
... More >> MUV
Vienna, Austria, 1090 Less << |
|
NCT01376752
|
Ovarian Epithelial Cancer Recu... More >>rrent Less << |
Phase 3 |
Recruiting |
December 2020 |
Belgium
... More >> Institut Jules Bordet
Recruiting
Brussels, Belgium
Principal Investigator: Gabriel LIBERALE
France
CHU d'AMIENS
Recruiting
Amiens, France
Principal Investigator: Sophie SANGUIN
Centre Paul Papin
Recruiting
Angers Cedex 9, France, 49933
Contact: Gérard LORIMIER, Doctor g.lorimier@angers.fnclcc.fr
Principal Investigator: Romuald WERNERT
Centre Hospitaleir Universitaire Jean Minjoz
Not yet recruiting
Besancon, France, 25000
Contact: Elsa KALBACHER, Dr ekalbacher@gmail.com
Principal Investigator: Elsa KALBACHER
Institut Bergonie
Recruiting
Bordeaux Cedex, France, 33076
Contact: Frédéric GUYON, Doctor guyon@bergonie.org
Principal Investigator: Frédéric GUYON
Centre Francois Baclesse
Not yet recruiting
Caen Cedex 3, France, 14076
Contact: Jean-Marc GUILLOIT, Doctor jm.guilloit@baclesse.fr
Principal Investigator: Jean-Marc GUILLOIT
Centre Jean Perrin
Recruiting
Clermont-ferrand Cedex 1, France, 63011
Contact: Christian POMEL, Pr Christophe.POMEL@cjp.fr
Principal Investigator: Christian POMEL
Centre hospitalier de Dijon
Recruiting
Dijon, France
Principal Investigator: Jean-Marc ROUSSELET
CHU Grenoble
Not yet recruiting
Grenoble, France, 38043
Contact: Fabien PETITPERRIN FPetitperrin@chu-grenoble.fr
Principal Investigator: Fabien PETITPERRIN
CHU de Grenoble
Recruiting
Grenoble, France
Principal Investigator: Cristina COSTAN
Centre Oscar Lambret
Recruiting
Lille, France, 59020
Contact: Eric LEBLANC, Dr e-leblanc@o-lambret.fr
Principal Investigator: Eric LEBLANC
Centre Hospitalier Universitaire Dupuytren
Recruiting
Limoges, France, 87042
Contact: Sylvaine DURAND-FONTANIER, Doctor sylvainedurand-fontanier@voila.fr
Principal Investigator: Sylvaine DURAND-FONTANIER
Centre Leon Berard
Recruiting
Lyon Cedex 08, France, 69373
Contact: Pierre MEEUS, Doctor MEEUS@lyon.fnclcc.fr
Principal Investigator: Pierre MEEUS
Institut Paoli Calmettes
Recruiting
Marseille Cedex 9, France, 13273
Contact: Gilles HOUVENAEGHEL, Pr houvenaeghelg@marseille.fnclcc.fr
Principal Investigator: Gilles HOUVENAEGHEL
AP-HM - Hôpital de la Timone
Recruiting
Marseille, France
Principal Investigator: Aubert AGOSTINI
CRLC Val d'Aurelle
Recruiting
Montpellier, France, 34298
Contact: François QUENET francois.quenet@montpellier.unicancer.fr
Principal Investigator: François QUENET
Centre Hospitalier Universitaire Nice - Hopital L'Archet 2
Not yet recruiting
Nice Cedex 3, France, 06202
Contact: Jean-Marc BEREDER, Dr ber224@chu-nice.fr
Principal Investigator: Jean-Marc BEREDER
Hopital Lariboisiere
Not yet recruiting
Paris, France, 75010
Contact: Marc POCARD marc.pocard@lrb.aphp.fr
Principal Investigator: Marc POCARD
Hopital Tenon
Not yet recruiting
Paris, France, 75020
Contact: Valeria LOI, Dr valeria.loi@tnn.aphp.fr
Principal Investigator: Valeria LOI
Hôpital Européen Georges Pompidou
Recruiting
Paris, France
Principal Investigator: Fabrice LECURU
Institut Curie
Recruiting
Paris, France
Principal Investigator: Pascale MARIANI
Institute Curie
Recruiting
Paris, France
Contact: Bernard BARANGER bernard.baranger@curie.net
Principal Investigator: Bernard BARANGER
Centre Hopsitalier Lyon Sud
Recruiting
Pierre-benite Cedex, France, 69495
Contact: Olivier GLEHEN, Professor olivier.glehen@chu-lyon.fr
Principal Investigator: Olivier GLEHEN
CHU - Hôpital de la Milétrie
Recruiting
Poitiers, France
Principal Investigator: Cédric NADEAU
Centre Jean Godinot
Not yet recruiting
Reims Cedex, France, 51056
Contact: David KERE, Dr david.kere@reims.fnclcc.fr
Principal Investigator: David KERE
Centre Hospitalier Universitaire Saint Etienne- Hopital Nord
Not yet recruiting
Saint-etienne, France, 42055
Contact: Karine ABBOUD, Dr abboud_go@yahoo.fr
Principal Investigator: Karine ABBOUD
Ico-Centre Rene Gauducheau
Recruiting
Saint-herblain Cedex, France, 44805
Contact: Jean-Marc CLASSE, Professor jm-classe@nantes.fnclcc.fr
Principal Investigator: Jean-Marc CLASSE
CHU Hautepierre
Not yet recruiting
Strasbourg, France, 67098
Contact: Cécile BRIGAND cecile.brigand@chru-strasbourg.fr
Principal Investigator: Cécile BRIGAND
Institut Claudius Regaud
Recruiting
Toulouse Cedex, France, 31052
Contact: Gwenaël FERRON, Doctor ferron.gwenael@claudiusregaud.fr
Principal Investigator: Gwenaël FERRON
Centre Alexis Vautrin
Recruiting
Vandoeuvre-les-nancy Cedex, France, 54511
Contact: Frédéric MARCHAL, Pr f.marchal@nancy.fnclcc.fr
Principal Investigator: Frédéric MARCHAL
Institut Gustave Roussy
Recruiting
Villejuif, France, 94805
Contact: Sébastien GOUY, Doctor sebastien.gouy@igr.fr
Principal Investigator: Sébastien GOUY
Spain
Hospital Universitari Germans Trias I Pujol
Recruiting
Badalona, Spain
Principal Investigator: Juan José TORRENT Less << |
|
NCT00540735
|
Cholangiocarcinoma |
Phase 3 |
Terminated(Big difficulties to... More >> enroll patients) Less << |
- |
France
... More >> UH Amiens
Amiens, France, 80 000
UH Angers
Angers, France, 49933
H Beaujon
Clichy, France, 92 118
UH Henri Mondor
Créteil, France, 94 010
UH Kremlin Bicêtre
Le Kremlin Bicêtre, France, 94 275
UH Claude Huriez
Lille, France, 59 037
Clinique Sainte Anne
Lyon, France, 69 003
UH La Timone
Marseille, France, 13 385
UH Marseille Nord
Marseille, France, 13 915
H Metz-Thionville
Metz, France, 57 038
UH Nantes
Nantes, France, 44 000
Clinique du Trocadéro
Paris, France, 75 116
UH Cochin
Paris, France, 75 679
UH La Milétrie
Poitiers, France, 86 021
UH Charles Nicolle
Rouen, France, 76 031
UH Hôpital Civil
Strasbourg, France, 67 091
UH Purpan
Toulouse, France, 31 059
UH Rangueil
Toulouse, France, 31 059 Less << |
|
NCT00772694
|
Testicular Cancer |
Phase 2 |
Unknown |
December 2011 |
Poland
... More >> Chemotherapy Unit, Dept of Urology, Instituite of Oncology
Recruiting
Warsaw, Poland, 02781
Principal Investigator: Iwona A Skoneczna, MD
Sub-Investigator: Agnieszka Chaladaj-Kujawska, MD Less << |
|
NCT02721576
|
Esophageal Neoplasms |
Phase 2 |
Recruiting |
December 2018 |
China, Shandong
... More >>
Qianfoshan Hospital of Shandong
Recruiting
Jinan, Shandong, China, 250117
Contact: Jiandong Zhang 13583123486
Shandong Cancer Hospital and Institute
Recruiting
Jinan, Shandong, China, 250117
Contact: Jinming Yu 8653167626891
Jining NO.1 People's Hospital
Recruiting
Jining, Shandong, China, 250117
Contact: Peipei Wu 15853770581
Liaocheng People Hospital
Recruiting
Liaocheng, Shandong, China
Contact: mingjun Li
Qingdao Center Medical Group
Recruiting
Qingdao, Shandong, China, 250117
Contact: Xuezhen Ma 18660221959
Rizhao City People 's Hospital
Recruiting
Rizhao, Shandong, China
Contact: Yufeng Li
Fei Cheng People's Hospital
Recruiting
Tai'an, Shandong, China, 250117
Contact 13518688689
An Qiu People's Hospital
Recruiting
Weifang, Shandong, China, 250117
Contact: Chuandong Wang 15605368369
The Fourth People's Hospitalof Zibo
Recruiting
Zibo, Shandong, China, 250117
Contact: Qiang Zhang 13345221111 Less << |
|
NCT00264056
|
Advanced Metastastic Malignant... More >> Melanoma
Refractory to First-Line Chemotherapy Irresectable Progressive Soft Tissue Metastases Less << |
Not Applicable |
Unknown |
- |
Germany
... More >> Fachklinik Hornheide at the University of Münster, Germany
Recruiting
Münster, Germany, D-48157
Contact: Jens Atzpodien, MD PhD 49-251-3287-431 jens.atzpodien@fachklinik-hornheide.de
Sub-Investigator: Martina Reitz, Dr Less << |
|
NCT02721550
|
Esophageal Neoplasms |
Phase 2 |
Recruiting |
December 2018 |
China, Shandong
... More >>
Qianfoshan Hospital of Shandong
Recruiting
Jinan, Shandong, China, 250117
Contact: Jiandong Zhang 13583123486
Shandong Cancer Hospital and Institute
Recruiting
Jinan, Shandong, China, 250117
Contact: Jinming Yu
Jining NO.1 People's Hospital
Recruiting
Jining, Shandong, China, 250117
Contact: Peipei Wu 15853770581
Liaocheng People Hospital
Recruiting
Liaocheng, Shandong, China
Contact: Mingjun Li
Qingdao Center Medical Group
Recruiting
Qingdao, Shandong, China, 250117
Contact: Xuezhen Ma
Rizhao City People 's Hospital
Recruiting
Rizhao, Shandong, China
Contact: Yufeng Li
Fei Cheng People's Hospital
Recruiting
Tai'an, Shandong, China, 250117
Contact: Wenyuan Zhao 13518688689
An Qiu People's Hospital
Recruiting
Weifang, Shandong, China, 250117
Contact: Chuandong Wang 15605368369
The Fourth People's Hospitalof Zibo
Recruiting
Zibo, Shandong, China, 250117
Contact: Qiang Zhang 13345221111 Less << |
|
NCT00760591
|
- |
|
Unknown |
- |
Korea, Republic of
... More >>
Soon Chun Hyang University Hospital
Recruiting
Seoul, Korea, Republic of, 140-743
Contact: Soon Hyo Kwon, MD +8-02-709-9491 ksoonhyo@hosp.sch.ac.kr
Principal Investigator: Dong Cheol Han, MD Less << |
|
NCT03325738
|
- |
|
Recruiting |
June 30, 2018 |
France
... More >> PEYRADE Frédéric
Recruiting
Nice, France, 06189
Contact: Frédéric PEYRADE +33492031022 frederic.peyrade@nice.unicancer.fr Less << |
|
NCT03582800
|
Systemic Sclerosis
... More >> Dermatomyositis
iPPSD2 Less << |
Phase 2 |
Not yet recruiting |
January 2022 |
France
... More >> CHU de BORDEAUX
Not yet recruiting
Bordeaux, France, 33000
Contact: Marie-Elise TRUCHETET, MD marie-elise.truchetet@chu-bordeaux.fr
Principal Investigator: Marie-Elise TRUCHETET, MD
ApHp - Hôpital Bicêtre
Not yet recruiting
Le Kremlin-Bicêtre, France, 94270
Contact: Agnès LINGLART, MD agnes.linglart@aphp.fr
Principal Investigator: Agnès LINGLART, MD
CHU de Limoges
Not yet recruiting
Limoges, France, 87042
Contact: Vincent GUIGONIS, MD vincent.guigonis@unilim.fr
Contact: Claire BAHANS claire.bahans@chu-limoges.fr
Principal Investigator: Vincent GUIGONIS, MD
Sub-Investigator: Anne-Laure FAUCHAIS, MD
Sub-Investigator: Pascalle VERGNE-SALLE, MD
Sub-Investigator: Didier MORIAU, MD
CHU de MONTPELLIER
Not yet recruiting
Montpellier, France, 34000
Contact: Alain LE QUELLEC, MD a-lequellec@chu-montpellier.fr
Principal Investigator: Alain LE QUELLEC, MD
ApHp - hôpital Lariboisière
Not yet recruiting
Paris, France, 75000
Contact: Korng EA, MD korngea@yahoo.fr
Principal Investigator: Korng EA, MD Less << |
|
NCT03456076
|
Carcinoma, Non-Small-Cell Lung |
Phase 3 |
Recruiting |
September 30, 2026 |
- |
|
NCT02727868
|
Patient With Breast Cancer |
Not Applicable |
Unknown |
March 2018 |
France
... More >> Assistance Publique Hôpitaux de Marseille
Recruiting
Marseille, France, 13354
Contact: Urielle DESALBRES, Director 0491382747 drci@ap-hm.fr Less << |
|
NCT03691441
|
Oropharyngeal Cancer |
Phase 4 |
Recruiting |
June 5, 2023 |
Germany
... More >> Universitätsklinikum Hamburg Eppendorf
Recruiting
Hamburg, Germany, 20246
Contact: Chia- Jung Busch, PD Dr. 004940741050038 cjbusch@uke.de
Contact: Andreas Krüll, PD Dr. 004940741055425 kruell@uke.de Less << |
|
NCT01216124
|
- |
|
- |
- |
- |
|
NCT02721563
|
Esophageal Neoplasms |
Phase 2 |
Recruiting |
December 2018 |
China, Shandong
... More >>
Qianfoshan Hospital of Shandong
Recruiting
Jinan, Shandong, China, 250117
Contact: Jiandong Zhang 13583123486
Qilu Hospital of Shandong University
Not yet recruiting
Jinan, Shandong, China, 250117
Contact: Yufeng Cheng 13869199969
Shandong Cancer Hospital and Institute
Recruiting
Jinan, Shandong, China, 250117
Contact: Jinming Yu 8653167626891
Jining NO.1 People's Hospital
Recruiting
Jining, Shandong, China, 250117
Contact: Peipei Wu 15853770581
Liaocheng People 's Hospital
Recruiting
Liaocheng, Shandong, China
Contact: Mingjun Li, MD
Qingdao Center Medical Group
Recruiting
Qingdao, Shandong, China, 250117
Contact: Xuezhen Ma 18660221959
Rizhao City People 's Hospital
Recruiting
Rizhao, Shandong, China
Contact: yufeng li
Fei Cheng People's Hospital
Recruiting
Tai'an, Shandong, China, 250117
Contact: Wenyuan Zhao 13518688689
An Qiu People's Hospital
Recruiting
Weifang, Shandong, China, 250117
Contact: Chuandong Wang 15605368369
The Fourth People's Hospitalof Zibo
Recruiting
Zibo, Shandong, China, 250117
Contact: Qiang Zhang 13345221111 Less << |
|
NCT03500133
|
Pediatric Hodgkin's Disease |
Phase 4 |
Recruiting |
October 31, 2032 |
Argentina
... More >> Hospital JP Garrahan
Recruiting
Buenos Aires, CF, Argentina, C1245AAL
Contact: Carolina Cernadas, MD 541141226335 coordinacioninvestigacion@garrahan.gov.ar
Contact: Guillermo L Chantada, MD, PhD 541141226000 gchantada@yahoo.com
Sub-Investigator: Elizabeth Alfaro, MD
Sub-Investigator: Myriam Guitter, MD
Sub-Investigator: Cristian Sanchez La Rosa, MD
Sub-Investigator: María Sara Felice, MD Less << |
|
NCT02455843
|
Non Small Cell Lung Cancer |
Phase 3 |
Recruiting |
May 2018 |
China, Shandong
... More >>
Binzhou Medical University Hospital
Recruiting
Binzhou, Shandong, China, +86 256600
Contact: Shao shui Chen, M.D +86 15169959936 byfychenss@126.com
Dezhou People's Hospital
Recruiting
Dezhou, Shandong, China, +86 253000
Contact: Yu ting Dong +86 13969286066 dyt1963@163.com
The Second People's Hospital of Dezhou
Recruiting
Dezhou, Shandong, China, +86 253000
Contact: Chun tang Wang, M.D +86 13181378288 dzeyxwct@sina.com
Jinan Military General Hospital
Recruiting
Jinan, Shandong, China, +83 250001
Contact: Jing wang Bi, M.D +86 15963119538 jingwangbi@outlook.com
Affliated Hospital of Shandong Academy of Medical Sciences
Recruiting
Jinan, Shandong, China, +86 250001
Contact: Ya hong Sun, M.D 13606415915 sunyahong0915@163.com
Shandong Provincial Hospital
Recruiting
Jinan, Shandong, China, +86 250001
Contact: Xin Ye, M.D +86 0531-68773171 yexintaian2014@163.com
Affliated Hospital of Jining Medical University
Recruiting
Jining, Shandong, China, +86 272000
Contact: Jun ye Wang, M.D +86 13563771996 jiningwangjunye@163.com
Liaocheng Cancer Hospital
Recruiting
Liaocheng, Shandong, China, +86 252000
Contact: Qing liang Feng, M.D +86 15339949567 fql-123@sohu.com
The People's Hospital of Pingyi Country
Recruiting
Linyi, Shandong, China, +86 276000
Contact: Xing lu Xu, M.D +86 18265398816 xlk2082@126.com
Affliated Hospital of Taishan Medical University
Recruiting
Taian, Shandong, China, +86 271000
Contact: Ben hua Zhang, M.D +86 15169887577 zhangbenhua1964@163.com
The People's Liberation Army 88 Hospital
Recruiting
Taian, Shandong, China, +86 271000
Contact: Li cheng Zhang, M.D +86 13605383651 zhanglc88@aliyun.com
Weifang People's Hospital
Recruiting
Weifang, Shandong, China, +86 262000
Contact: Guo hua Yu, M.D +86 13685368817 ghyry@126.com
Yantai Yuhuangding Hospital
Recruiting
Yantai, Shandong, China, +86 264000
Contact: Liang ming Zhang, M.D +86 18660079893 zhanglmdr@163.com
Tengzhou center of people's hospital
Recruiting
Zaozhuang, Shandong, China, +86 277000
Contact: Kai xian Zhang, M.D +86 18663069829 kaixianzhang@aliyun.com Less << |
|
NCT00856830
|
Small Cell Lung Cancer
... More >> Extensive Stage Lung Cancer
Chemonaive Less << |
Phase 1
Phase 2 |
Completed |
- |
United States, Alabama
... More >>
University of Alabama at Birmingham
Birmingham, Alabama, United States, 35294 - 0104
United States, Georgia
Georgia Cancer Specialists
Marietta, Georgia, United States, 30060 Less << |
|
NCT03443921
|
Pancreatic Cancer
... More >> Locally Advanced Pancreatic Cancer
Neoadjuvant Therapy
Borderline Resectable Pancreatic Cancer Less << |
Not Applicable |
Not yet recruiting |
March 2021 |
China, Jiangsu
... More >> The First Affiliated Hospital of Nanjing Medical University
Not yet recruiting
Nanjing, Jiangsu, China, 210029
Contact: Yi Miao, MD, PhD +86-25-68136508 miaoyi@njmu.edu.cn Less << |
|
NCT02185768
|
Liver Cancer |
Phase 2 |
Completed |
- |
France
... More >> CHU Amiens
Amiens, France
CHU d'ANGERS
Angers, France, 49933
CHU - Hôpital François Mitterand
Dijon, France, 21079
Hôpital La Croix Rousse
Lyon, France, 69317
Hôpital Edouard Herriot
Lyon, France, 69437
CHU St Eloi
Montpellier, France, 34295
Hôpital de l'Archet II
Nice, France, 06202 Less << |
|
NCT01590160
|
Lung Cancer - Malignant Pleura... More >>l Mesothelioma Less << |
Phase 1
Phase 2 |
Active, not recruiting |
November 2018 |
United Kingdom
... More >> UCL Cancer Trials Centre
London, United Kingdom, W1T 4TJ Less << |
|
NCT02036164
|
Uterine Cervical Cancer |
Phase 3 |
Recruiting |
January 2019 |
Thailand
... More >> Ratchaburi Hospital
Active, not recruiting
Muang, Ratchaburi, Thailand, 70000
Bhumibol Adulyadej Hospital
Recruiting
Bangkok, Thailand, 10220
Contact: Piyawan Pariyawateekul, MD 6689-0353777 nootpiya@hotmail.com
Contact: Apiradee Kridakara, MD 66-86-3671487 akrid7@hotmail.com
Principal Investigator: Piyawan Pariyawateekul, MD
Principal Investigator: Apiradee Kridakara, MD
Department of Obstetrics and Gynecology, Department of Radiology, Epidemiology Unit, Navamindradhiraj University
Recruiting
Bangkok, Thailand, 10300
Contact: Siriwan Tangjitgamol, MD 66-86-3841431 siriwanonco@yahoo.com
Contact: Kanisa Rongsriyam, MD 66-8181-68789 kanisa_r@hotmail.com
Principal Investigator: Siriwan Tangjitgamol, MD
Principal Investigator: Kanisa Rongsriyam, MD
Sub-Investigator: Jakkapan Khunnarong, MD
Sub-Investigator: Sunamchok Srijaipracharoen, MD
Sub-Investigator: Busaba Supawattanabodee, MSc
Sub-Investigator: Suphakarn Techapongsatorn, MD
Sub-Investigator: Kanyarat Katanyoo, MD
Health Intervention and Technology Assessment Program
Not yet recruiting
Bangkok, Thailand, 11000
Contact: Yot Teerawattananon, MD 66-84-6760080 yot.t@hitap.net
Principal Investigator: Yot Teerawattananon, MD
Department of Radiology, Department of Obstetrics and Gynecology, Chiang Mai University
Recruiting
Chiang Mai, Thailand, 50200
Contact: Ekkasit Tharavijitkul, MD 966-89-4338709 paan_31@hotmail.com
Principal Investigator: Ekkasit Tharavijitkul, MD
Sub-Investigator: Vichan Lordvithaya, MD
Sub-Investigator: Jatupol Srisomboon, MD
Chonburi Cancer Hospital
Recruiting
Chonburi, Thailand, 20000
Contact: Chokaew Tovanabutra, MD 66-81-4726633 chokaewt@hotmail.com
Principal Investigator: Chokaew Tovanabutra, MD
Sub-Investigator: Kittisak Chomprasert, MD
Lampang Cancer Hospital
Recruiting
Lampang, Thailand, 52000
Contact: Tussawan Asakit, MD 66-81-6715013 tussawan_a1@hotmail.com
Principal Investigator: Tussawan Asakit, MD
Sub-Investigator: Kannika Paengchit, MD
Lopburi Cancer Hospital
Recruiting
Lopburi, Thailand, 15000
Contact: Jirasak Sukhaboon, MD 66-89-7970600 fodjiji@gmail.com
Principal Investigator: Jirasak Sukhaboon, MD
Sub-Investigator: Panit Chiewaratanapong, MD
Department of Obstetrics and Gynecology, Department of Radiology, Prince of Songkla University
Recruiting
Songkla, Thailand, 90110
Contact: Thiti Atjimakul, MD 6681-2773793 thitiatjimakul@yahoo.com
Contact: Jitti Hanprasertpong, MD 66-89-6540214 hjitti@medicine.psu.ac.th
Principal Investigator: Thiti Atjimakul, MD
Principal Investigator: Duangjai Sangthawan, MD
Sub-Investigator: Jitti hanprasertpong, MD
Ubonratchathani Cancer Hospital
Recruiting
Ubonratchathani, Thailand, 34000
Contact: Taywin Chottetanaprasith, MD 66-81-9359756 taywinc@hotmail.com
Principal Investigator: Taywin Chottetanaprasith, MD
Sub-Investigator: Metee Wongsena, MD
Udonthani Cancer Hopital
Recruiting
Udonthani, Thailand, 41330
Contact: Sirentra Wanglikitkoon, MD 6684-6570028 sirentra@hotmail.com
Principal Investigator: Sirentra Wanglikitkoon, MD
Sub-Investigator: Somkit Penpattanagul, MD Less << |
|
NCT02208583
|
Hormone Refractory Prostate Ca... More >>ncer Less << |
Not Applicable |
Unknown |
December 2016 |
China, Tianjin
... More >> Department of Interventional Oncology, Tianjin Medical University Cancer Institute and Hospital
Recruiting
Tianjin, Tianjin, China, 300060
Contact: Wang HaiTao, Ph.D +86-18630955984 peterrock2000@126.com
Principal Investigator: Wang HaiTao, Ph.D Less << |
|
NCT00856830
|
- |
|
Completed |
- |
- |
|
NCT00532025
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Terminated |
- |
Germany
... More >> SIRN investigational trial site
Essen, Germany
SIRN investigational trial site
Halle (Saale), Germany
SIRN investigational trial site
Loewenstein, Germany
SIRN investigational trial site Johannes Gutenberg Universität
Mainz, Germany
SIRN investigational trial site Katholisches Klinikum
Mainz, Germany
SIRN investigational trial site
Rotenburg (Wuemme), Germany Less << |
|
NCT01726374
|
Stage I Testicular Non-Seminom... More >>atous Germ Cell Tumor Less << |
Phase 3 |
Active, not recruiting |
August 2019 |
United Kingdom
... More >> Guy's Hospital
London, England, United Kingdom, SE1 9RT
Northampton General Hospital NHS Trust
Northampton, England, United Kingdom, NN6 8BJ
Velindre Cancer Center at Velindre Hospital
Cardiff, Wales, United Kingdom, CF14 2TL
Aberdeen Royal Infirmary
Aberdeen, United Kingdom
Ysbyty Gwynedd
Bangor, United Kingdom
Queen Elizabeth Hospital
Birmingham, United Kingdom
Royal Sussex County Hospital
Brighton, United Kingdom
Bristol Haematology and Oncology Centre
Bristol, United Kingdom
Queen's Hospital
Burton-on-Trent, United Kingdom
Addenbrooke's Hospital
Cambridge, United Kingdom
Cheltenham General Hospital
Cheltenham, United Kingdom
Gloucestershire Royal Hospital
Cheltenham, United Kingdom
University Hospitals Coventry and Warwickshire NHS Trust
Coventry, United Kingdom
Royal Derby Hospital
Derby, United Kingdom
Western General Hospital
Edinburgh, United Kingdom
Royal Devon and Exeter Hospital
Exeter, United Kingdom
Beatson West of Scotland Cancer Centre
Glasgow, United Kingdom
Royal Surrey County Hospital
Guildford, United Kingdom
Castle Hill Hospital
Hull, United Kingdom
Ipswich Hospital
Ipswich, United Kingdom
St James's University Hospital
Leeds, United Kingdom
Leicester Royal Infirmary
Leicester, United Kingdom
Lincoln County Hospital
Lincoln, United Kingdom
Clatterbridge Centre for Oncology
Liverpool, United Kingdom
Royal Liverpool University Hospital
Liverpool, United Kingdom
St Bartholomew's Hospital
London, United Kingdom
University College Hospital
London, United Kingdom
Maidstone Hospital
Maidstone, United Kingdom
James Cook University Hospital
Middlesbrough, United Kingdom
Norfolk and Norwich University Hospital
Norwich, United Kingdom
Nottingham City Hospital
Nottingham, United Kingdom
Churchill Hospital
Oxford, United Kingdom
Weston Park Hospital
Sheffield, United Kingdom
Southampton General Hospital
Southampton, United Kingdom
Royal Marsden Hospital
Sutton, United Kingdom Less << |
|
NCT01218516
|
Adenocarcinoma of the Lung |
Phase 2 |
Completed |
- |
- |
|
NCT02272699
|
Esophageal Squamous Cell Carci... More >>noma Less << |
Phase 2
Phase 3 |
Suspended(Because there are qu... More >>estions about adjuvant radiotherapy in this study, the clinical trial is under re-design.) Less << |
December 2018 |
China, Beijing
... More >> Beijing Cancer Hospital & Peking University Cancer Hospital
Beijing, Beijing, China, 100142 Less << |
|
NCT03701373
|
Gastric Cancer Stage IV
... More >> Cancer of Stomach Less << |
Phase 2 |
Recruiting |
December 2018 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Rui-hua Xu +86-20-87343295 xurh@sysucc.org.cn
Contact: Ming-ming He, MD +86-13632350737 hemm@sysucc.org.cn Less << |
|
NCT01313663
|
Lung Cancer, Small Cell |
Phase 2 |
Terminated |
- |
- |
|
NCT01233843
|
Squamous Cell Head and Neck Ca... More >>rcinoma Less << |
Phase 3 |
Active, not recruiting |
May 2019 |
France
... More >> GIRARD CALAIS Marie-Helene
Tours, France, 37044 Less << |
|
NCT02577341
|
Non-small-cell Lung Cancer |
Phase 2 |
Recruiting |
July 2020 |
China, Guangdong
... More >>
Sun yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510000
Contact: Bo Qiu, Attending 020-87343031 qiubo@sysucc.org.cn Less << |
|
NCT01924819
|
Gastric Cancer |
Phase 2
Phase 3 |
Recruiting |
December 2020 |
Australia, New South Wales
... More >>
Calvary Mater Newcastle
Recruiting
Newcastle, New South Wales, Australia
Liverpool Hospital
Recruiting
Sydney, New South Wales, Australia
Nepean Hospital
Recruiting
Sydney, New South Wales, Australia
Prince of Wales Hospital
Recruiting
Sydney, New South Wales, Australia
Royal North Shore Hospital
Recruiting
Sydney, New South Wales, Australia
Royal Prince Alfred Hospital
Recruiting
Sydney, New South Wales, Australia
St George Hospital
Recruiting
Sydney, New South Wales, Australia
Westmead Hospital
Recruiting
Sydney, New South Wales, Australia
The Tweed Hospital
Recruiting
Tweed Heads, New South Wales, Australia
Wollongong Hospital
Recruiting
Wollongong, New South Wales, Australia
Australia, Queensland
Princess Alexandra Hospital
Recruiting
Brisbane, Queensland, Australia
Australia, South Australia
Flinders Medical Centre
Recruiting
Adelaide, South Australia, Australia
Australia, Tasmania
Royal Hobart Hospital
Recruiting
Hobart, Tasmania, Australia
Launceston General Hospital
Recruiting
Launceston, Tasmania, Australia
Australia, Victoria
Geelong Hospital
Recruiting
Geelong, Victoria, Australia
Austin Hospital
Recruiting
Melbourne, Victoria, Australia
Monash Medical Centre
Recruiting
Melbourne, Victoria, Australia
Peter MacCallum Cancer Centre
Recruiting
Melbourne, Victoria, Australia
St Vincent's Hospital
Recruiting
Melbourne, Victoria, Australia
Australia, Western Australia
Sir Charles Gairdner Hospital
Recruiting
Nedlands, Western Australia, Australia
Royal Perth Hospital
Recruiting
Perth, Western Australia, Australia
New Zealand
Auckland Hospital
Recruiting
Auckland, New Zealand
Christchurch Hospital
Recruiting
Christchurch, New Zealand
Dunedin Hospital
Recruiting
Dunedin, New Zealand
Waikato Hospital
Recruiting
Waikato, New Zealand Less << |
|
NCT00896181
|
Stage II Lymphoepithelioma of ... More >>the Nasopharynx
Stage II Squamous Cell Carcinoma of the Nasopharynx
Stage III Lymphoepithelioma of the Nasopharynx
Stage III Squamous Cell Carcinoma of the Nasopharynx
Stage IV Lymphoepithelioma of the Nasopharynx
Stage IV Squamous Cell Carcinoma of the Nasopharynx Less << |
Phase 2 |
Active, not recruiting |
July 2020 |
United States, California
... More >>
Stanford University Hospitals and Clinics
Stanford, California, United States, 94305 Less << |
|
NCT01284335
|
Advanced Solid Tumors |
Phase 1 |
Terminated(Study was terminate... More >>d due to the termination of tasisulam development.) Less << |
- |
United States, Alabama
... More >>
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Birmingham, Alabama, United States, 35233
United States, Arkansas
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Fayetteville, Arkansas, United States, 72703
United States, Colorado
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Denver, Colorado, United States, 80218
United States, Florida
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Orlando, Florida, United States, 32806
United States, Illinois
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Park Ridge, Illinois, United States, 60068
United States, Indiana
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Indianapolis, Indiana, United States, 46219
United States, Nevada
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Las Vegas, Nevada, United States, 89169
United States, New York
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Albany, New York, United States, 12206
United States, Ohio
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Kettering, Ohio, United States, 45429
United States, Oregon
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Portland, Oregon, United States, 97213
United States, South Carolina
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Greenville, South Carolina, United States, 29605
United States, Texas
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Austin, Texas, United States, 78731
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Dallas, Texas, United States, 75246
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
The Woodlands, Texas, United States, 77380
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Tyler, Texas, United States, 75702
United States, Virginia
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Norfolk, Virginia, United States, 23502
United States, Washington
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Spokane, Washington, United States, 99218
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Vancouver, Washington, United States, 98684
For additional information regarding investigative sites for this trial, contact 1-877-CTLILLY (1-877-285-4559, 1-317-615-4559) Mon - Fri from 9 AM to 5 PM Eastern Time (UTC/GMT - 5 hours, EST), or speak with your personal physician.
Yakima, Washington, United States, 98902 Less << |
|
NCT01313663
|
- |
|
Terminated |
- |
- |
|
NCT01284335
|
- |
|
Terminated(Study was terminate... More >>d due to the termination of tasisulam development.) Less << |
- |
- |
|
NCT00572169
|
Multiple Myeloma |
Phase 3 |
Active, not recruiting |
August 2019 |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences/Myeloma Institute
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT03085914
|
Solid Tumors
... More >>Colorectal Cancer (CRC)
Adenocarcinoma (PDAC)
Lung Cancer
UC (Urothelial Cancer)
Head and Neck Cancer Less << |
Phase 1
Phase 2 |
Active, not recruiting |
January 2020 |
United States, Arizona
... More >>
Mayo Clinic Arizona
Phoenix, Arizona, United States, 85054
United States, California
University of California San Diego Medical Center, Moores Cancer Center
La Jolla, California, United States, 92093
The Angeles Clinic and Research Institute
Los Angeles, California, United States, 90025
United States, Florida
Mayo Clinic
Jacksonville, Florida, United States, 32224
United States, Illinois
University of Chicago
Chicago, Illinois, United States, 60637
United States, Minnesota
Mayo Clinic
Rochester, Minnesota, United States, 55905
United States, Missouri
Washington University School of Medicine
Saint Louis, Missouri, United States, 63110
United States, New York
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
United States, North Carolina
Carolina Bio-Oncology Institute, PLLC
Huntersville, North Carolina, United States, 28078
United States, Oregon
Oregon Health and Science University
Portland, Oregon, United States, 97239
United States, Pennsylvania
University of Pennsylvania Health System
Philadelphia, Pennsylvania, United States, 19104
University of Pittsburgh Cancer Institute
Pittsburgh, Pennsylvania, United States, 15237
United States, Tennessee
Tennessee Oncology - Nashville; The Sarah Cannon Research Institute
Nashville, Tennessee, United States, 37203
Vanderbilt University; Henry Joyce Cancer Clinic
Nashville, Tennessee, United States, 37232
United States, Texas
MD Anderson Cancer Center
Houston, Texas, United States, 77030
United States, Utah
Huntsman Cancer Institute at University of Utah
Salt Lake City, Utah, United States, 84112 Less << |
|
NCT02788201
|
Urothelial Carcinoma
... More >> Bladder Cancer
Urinary Bladder Neoplasms Less << |
Phase 2 |
Recruiting |
July 1, 2020 |
United States, Maryland
... More >>
National Institutes of Health Clinical Center
Recruiting
Bethesda, Maryland, United States, 20892
Contact: For more information at the NIH Clinical Center contact National Cancer Institute Referral Office 888-624-1937 Less << |
|
NCT00002835
|
Lymphoma |
Phase 3 |
Completed |
- |
United States, Texas
... More >>
University of Texas - MD Anderson Cancer Center
Houston, Texas, United States, 77030-4009 Less << |
|
NCT03348904
|
Lung Cancer |
Phase 3 |
Terminated(Study halted premat... More >>urely and will not resume; participants are no longer being examined or receiving intervention.) Less << |
- |
United States, California
... More >>
Pacific Cancer Medical Center, Inc
Anaheim, California, United States, 92801
United States, Kansas
Cancer Center of Kansas
Wichita, Kansas, United States, 67214 Less << |
|
NCT01838538
|
Ovarian Cancer With Malignant ... More >>Ascites Less << |
Phase 2 |
Unknown |
- |
China, Beijing
... More >> First Hospital Affiliated to the PLA General Hospital, Beijing,China
Recruiting
Beijing, Beijing, China, 100048
Contact: Nan Du, PhD 861068989123 dunan05@yahoo.com.cn
Principal Investigator: Hui Zhao, PhD Less << |
|
NCT01336192
|
Non Small Cell Lung Cancer |
Phase 4 |
Unknown |
April 2013 |
China, Shanghai
... More >>
Shanghai Pulmonary Hospital Medical Oncology Department
Recruiting
Shanghai, Shanghai, China, 200433
Contact: Ying Xu, MD 86-21-65115006 ext 1053 xuying2186@hotmail.com
Principal Investigator: Di Zheng, MD Less << |
|
NCT00956930
|
Liver Cancer |
Phase 2 |
Completed |
- |
United States, Illinois
... More >>
Northwestern University, Northwestern Memorial Hospital
Chicago, Illinois, United States, 60611-3013 Less << |
|
NCT00025259
|
- |
|
Completed |
- |
- |
|
NCT01093066
|
Urothelial Carcinoma |
Phase 2 |
Active, not recruiting |
December 2022 |
France
... More >> CH du Pays d'Aix-en-Provence
Aix-en-Provence, France
Clinique AXIUM - AIX EN PROVENCE
Aix-en-Provence, France
CHU Bordeaux
Bordeaux, France, 33000
Clinique Saint-Augustin
Bordeaux, France
Institut Bergonie
Bordeaux, France
CHU Caen
Caen, France, 14000
Crlcc Francois Baclesse
Caen, France, 54500
CHU Créteil
Créteil, France
Polyclinique de Lisieux
Lisieux, France
APHM - Marseille - Hôpital de la Conception
Marseille, France, 13 385
APHM - Marseille - Hôpital la Timone
Marseille, France
CRLC Marseille
Marseille, France
Hôpital Européen - Marseille
Marseille, France
Hôpitaux privés de Metz
Metz, France
Chu Nancy
Nancy, France
Crlc Nancy
Nancy, France
Chu Nantes
Nantes, France
APHP - Saint-Louis
Paris, France
APHP- Hôpital Tenon
Paris, France
CHU Poitiers
Poitiers, France, 86000
Chu Reims
Reims, France
Institut Jean Godinot - Reims
Reims, France
Clinique Mutualiste Chirurgicale
Saint-etienne, France, 42055
ICO - SITE Gauducheau - ICL Nantes
Saint-Herblain, France, 44 805
ICLN
Saint-priest En Jarez, France, 42270
CHU Saint-Etienne
Saint-Étienne, France
Hôpitaux du Léman - Thonon-les-Bains
Thonon-les-Bains, France
CHI Toulon
Toulon, France
CHU Toulouse
Toulouse, France, 31059
INSTITUT CLAUDIUS REGAUD - CRLC Toulouse
Toulouse, France, 31059
Polyclinique Du Cotentin
Équeurdreville-Hainneville, France Less << |
|
NCT02575859
|
Colorectal Neoplasm
... More >> Metastasis Less << |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT02542293
|
Non Small Cell Lung Carcinoma ... More >>NSCLC Less << |
Phase 3 |
Active, not recruiting |
March 29, 2019 |
- |
|
NCT00576199
|
Liver Cancer |
Phase 2 |
Completed |
- |
Hong Kong
... More >>
Hong Kong, Hong Kong, 0
Hong Kong, Hong Kong, 852
Hong Kong, Hong Kong Less << |
|
NCT00202969
|
Gastric Cancer |
Phase 3 |
Completed |
- |
China, Beijing
... More >> Beijing Cancer Hospital
No.52 Fu-Cheng Road, Hai-dian District, Beijing, China Less << |
|
NCT00054067
|
Cervical Cancer |
Phase 3 |
Terminated |
- |
- |
|
NCT00003400
|
Prostate Cancer |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
Marlene & Stewart Greenebaum Cancer Center, University of Maryland
Baltimore, Maryland, United States, 21201 Less << |
|
NCT00025259
|
Childhood Lymphocyte-Depleted ... More >>Classical Hodgkin Lymphoma
Childhood Mixed Cellularity Classical Hodgkin Lymphoma
Childhood Nodular Lymphocyte Predominant Hodgkin Lymphoma
Childhood Nodular Sclerosis Classical Hodgkin Lymphoma
Stage I Childhood Hodgkin Lymphoma
Stage II Childhood Hodgkin Lymphoma
Stage III Childhood Hodgkin Lymphoma
Stage IV Childhood Hodgkin Lymphoma Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00077389
|
Liver Cancer |
Phase 2 |
Unknown |
- |
France
... More >> Institut Gustave Roussy
Villejuif, France, F-94805
Ireland
Our Lady's Hospital for Sick Children Crumlin
Dublin, Ireland, 12
Netherlands
Emma Kinderziekenhuis
Amsterdam, Netherlands, NL-1100 DE
United Kingdom
Birmingham Children's Hospital
Birmingham, England, United Kingdom, B4 6NH
Institute of Child Health at University of Bristol
Bristol, England, United Kingdom, BS2 8AE
Addenbrooke's Hospital
Cambridge, England, United Kingdom, CB2 2QQ
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Children's Cancer and Leukaemia Group
Leicester, England, United Kingdom, LE1 6TH
Royal Liverpool Children's Hospital, Alder Hey
Liverpool, England, United Kingdom, L12 2AP
Middlesex Hospital
London, England, United Kingdom, W1T 3AA
Great Ormond Street Hospital for Children
London, England, United Kingdom, WC1N 3JH
Royal Manchester Children's Hospital
Manchester, England, United Kingdom, M27 4HA
Sir James Spence Institute of Child Health at Royal Victoria Infirmary
Newcastle-Upon-Tyne, England, United Kingdom, NE1 4LP
Queen's Medical Centre
Nottingham, England, United Kingdom, NG7 2UH
Oxford Radcliffe Hospital
Oxford, England, United Kingdom, 0X3 9DU
Children's Hospital - Sheffield
Sheffield, England, United Kingdom, S10 2TH
Southampton General Hospital
Southampton, England, United Kingdom, SO16 6YD
Royal Marsden - Surrey
Sutton, England, United Kingdom, SM2 5PT
Royal Belfast Hospital for Sick Children
Belfast, Northern Ireland, United Kingdom, BT12 6BE
Royal Aberdeen Children's Hospital
Aberdeen, Scotland, United Kingdom, AB25 2ZG
Royal Hospital for Sick Children
Edinburgh, Scotland, United Kingdom, EH9 1LF
Royal Hospital for Sick Children
Glasgow, Scotland, United Kingdom, G3 8SJ
Childrens Hospital for Wales
Cardiff, Wales, United Kingdom, CF14 4XW Less << |
|
NCT00003701
|
Bladder Cancer
... More >> Urethral Cancer Less << |
Phase 3 |
Completed |
- |
United States, Georgia
... More >>
Veterans Affairs Medical Center - Atlanta (Decatur)
Decatur, Georgia, United States, 30033
United States, Indiana
Indiana University Hospitals
Indianapolis, Indiana, United States, 46202
United States, New Jersey
Hunterdon Regional Cancer Center
Flemington, New Jersey, United States, 08822
Hackensack University Medical Center
Hackensack, New Jersey, United States, 07601
South Jersey Hospital - Millville
Millville, New Jersey, United States, 08332
Fox Chase Cancer Center at Virtua-Memorial Hospital Burlington County
Mount Holly, New Jersey, United States, 08060
Riverview Medical Center
Red Bank, New Jersey, United States, 07701
Overlook Hospital
Summit, New Jersey, United States, 07902-0220
United States, New York
Veterans Affairs Medical Center - New York
New York, New York, United States, 10010
NYU School of Medicine's Kaplan Comprehensive Cancer Center
New York, New York, United States, 10016
United States, Ohio
Cleveland Clinic Taussig Cancer Center
Cleveland, Ohio, United States, 44195
United States, Pennsylvania
Hahnemann University Hospital
Philadelphia, Pennsylvania, United States, 19102-1192
United States, Tennessee
Vanderbilt Cancer Center
Nashville, Tennessee, United States, 37232-6838 Less << |
|
NCT00002919
|
Bladder Cancer |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00281905
|
Brain and Central Nervous Syst... More >>em Tumors
Neuroblastoma Less << |
Phase 2 |
Unknown |
- |
Ireland
... More >> Our Lady's Hospital for Sick Children
Dublin, Ireland, 12
United Kingdom
Birmingham Children's Hospital
Birmingham, England, United Kingdom, B4 6NH
Institute of Child Health at University of Bristol
Bristol, England, United Kingdom, BS2 8AE
Addenbrooke's Hospital at Cambridge University Hospitals NHS Foundation Trust
Cambridge, England, United Kingdom, CB2 2QQ
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Royal Liverpool Children's Hospital, Alder Hey
Liverpool, England, United Kingdom, L12 2AP
Royal London Hospital
London, England, United Kingdom, E1 1BB
Great Ormond Street Hospital for Children NHS Trust
London, England, United Kingdom, WC1N 3JH
Central Manchester and Manchester Children's University Hospitals NHS Trust
Manchester, England, United Kingdom, M27 4HA
Sir James Spence Institute of Child Health
Newcastle-Upon-Tyne, England, United Kingdom, NE1 4LP
Queen's Medical Centre
Nottingham, England, United Kingdom, NG7 2UH
Oxford Radcliffe Hospital
Oxford, England, United Kingdom, 0X3 9DU
Children's Hospital - Sheffield
Sheffield, England, United Kingdom, S10 2TH
Southampton General Hospital
Southampton, England, United Kingdom, SO16 6YD
Royal Marsden NHS Foundation Trust - Surrey
Sutton, England, United Kingdom, SM2 5PT
Royal Belfast Hospital for Sick Children
Belfast, Northern Ireland, United Kingdom, BT12 6BE
Royal Aberdeen Children's Hospital
Aberdeen, Scotland, United Kingdom, AB25 2ZG
Royal Hospital for Sick Children
Edinburgh, Scotland, United Kingdom, EH9 1LF
Royal Hospital for Sick Children
Glasgow, Scotland, United Kingdom, G3 8SJ
Childrens Hospital for Wales
Cardiff, Wales, United Kingdom, CF14 4XW Less << |
|
NCT00576199
|
- |
|
Completed |
- |
- |
|
NCT00003214
|
Ovarian Cancer |
Phase 3 |
Completed |
- |
Switzerland
... More >> Kantonspital Aarau
Aarau, Switzerland, 5001
Frauenspital, Basel
Basel, Switzerland, 4031
Office of Walter Weber-Stadelman
Basel, Switzerland, CH 4051
University Hospital
Basel, Switzerland, CH-4031
Inselspital, Bern
Bern, Switzerland, CH-3010
Hopital Cantonal Universitaire de Geneva
Geneva, Switzerland, CH-1211
Istituto Oncologico della Svizzera Italiana
Lugano, Switzerland, CH-6900
Burgerspital, Solothurn
Solothurn, Switzerland, 4500
City Hospital Triemli
Zurich, Switzerland, 8063
Klinik Hirslanden
Zurich, Switzerland, CH-8008 Less << |
|
NCT00343408
|
Colorectal Cancer
... More >> Lung Cancer Less << |
Phase 1 |
Completed |
- |
Canada, Ontario
... More >>
Ottawa Hospital Regional Cancer Centre - General Campus
Ottawa, Ontario, Canada, K1H 8L6
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9
Canada, Quebec
Jewish General Hospital - Montreal
Montreal, Quebec, Canada, H3T 1E2 Less << |
|
NCT00003364
|
Lung Cancer |
Phase 3 |
Completed |
- |
United Kingdom
... More >> Heatherwood Hospital
Ascot, England, United Kingdom, SL5 8AA
Royal Sussex County Hospital
Brighton, England, United Kingdom, BN2 5BE
Addenbrooke's NHS Trust
Cambridge, England, United Kingdom, CB2 2QQ
Broomfield Hospital
Chelmsford, Essex, England, United Kingdom, CM1 5ET
Essex County Hospital
Colchester, England, United Kingdom
Royal Free Hospital
Hampstead, London, England, United Kingdom, NW3 2QG
Cookridge Hospital
Leeds, England, United Kingdom, LS16 6QB
Saint Bartholomew's Hospital
London, England, United Kingdom, EC1A 7BE
Guy's and St. Thomas' Hospitals Trust
London, England, United Kingdom, SE1 9RT
Charing Cross Hospital
London, England, United Kingdom, W6 8RF
Middlesex Hospital- Meyerstein Institute
London, England, United Kingdom, WIT 3AA
Mount Vernon Hospital
Northwood, England, United Kingdom, HA6 2RN
Peterborough Hospitals Trust
Peterborough, England, United Kingdom, PE3 6DA
Southend NHS Trust Hospital
Westcliff-On-Sea, England, United Kingdom Less << |
|
NCT00312728
|
- |
|
Completed |
- |
- |
|
NCT00231582
|
Testicular Neoplasms |
Phase 2 |
Completed |
- |
France
... More >> Hôpital TENON, Service d'Oncologie Médicale
Paris, France, 75020 Less << |
|
NCT00005583
|
Endometrial Cancer |
Phase 3 |
Completed |
- |
Belgium
... More >> Institut Jules Bordet
Brussels, Belgium, 1000
Universitair Ziekenhuis Antwerpen
Edegem, Belgium, B-2650
Cazk Groeninghe - Campus Maria's Voorzienigheid
Kortrijk, Belgium, B-8500
U.Z. Gasthuisberg
Leuven, Belgium, B-3000
France
Centre Henri Becquerel
Rouen, France, 76038
Ireland
Coombe Women's Hospital
Dublin, Ireland, 8
St. James's Hospital
Dublin, Ireland, 8
Italy
Istituto Nazionale per lo Studio e la Cura dei Tumori
Naples, Italy, 80131
Azienda Ospedaliera Di Parma
Parma, Italy, 43100
Fondazione I.R.C.C.S. Policlinico San Matteo
Pavia, Italy, 27100
Ospedale di Circolo e Fondazione Macchi
Varese, Italy, 21100
Netherlands
Onze Lieve Vrouwe Gasthuis
Amsterdam, Netherlands, 1091 HA
Medisch Spectrum Twente
Enschede, Netherlands, 7500 KA
Universitair Medisch Centrum St. Radboud - Nijmegen
Nijmegen, Netherlands, NL-6500 HB
Norway
Norwegian Radium Hospital
Oslo, Norway, N-0310
Poland
Medical University of Gdansk
Gdansk, Poland, 80-211
Portugal
Hospitais da Universidade de Coimbra (HUC)
Coimbra, Portugal, 3049
South Africa
Groote Schuur Hospital
Cape Town, South Africa, 7925
Spain
Hospital Universitario San Carlos
Madrid, Spain, 28040
Hospital Universitario Central de Asturias
Oviedo, Spain, 33006
United Kingdom
Nottingham City Hospital NHS Trust
Nottingham, England, United Kingdom, NG5 1PB
Centre for Cancer Research and Cell Biology at Belfast City Hospital
Belfast, Northern Ireland, United Kingdom, BT9 7AB
Western Infirmary
Glasgow, Scotland, United Kingdom, G11 6NT Less << |
|
NCT00312728
|
Non-Small Cell Lung Cancer
... More >> Brain Neoplasms Less << |
Phase 2 |
Completed |
- |
- |
|
NCT01466257
|
- |
|
Completed |
- |
- |
|
NCT00734877
|
Multiple Myeloma |
Phase 3 |
Recruiting |
September 2020 |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences, Myeloma Institute for Research and Therapy
Recruiting
Little Rock, Arkansas, United States, 72205
Contact: Nathan Petty 501-526-6990 ext 2435 pettynathanm@uams.edu
Sub-Investigator: Frits van Rhee, MD, PhD
Sub-Investigator: Michele Fox, MD
Sub-Investigator: Maurizio Zangari, MD
Sub-Investigator: Atul Kothari, MD
Sub-Investigator: Carolina Schinke, MD
Sub-Investigator: Faith Davies, MD
Sub-Investigator: Gareth Morgan, MD
Sub-Investigator: Juan Crescencio, MD
Sub-Investigator: Mary Burgess, MD
Sub-Investigator: Meera Mohan, MD
Sub-Investigator: Muthukumar Radhakrishnan, MD
Sub-Investigator: Pankaj Mathur, MD
Sub-Investigator: Sharmilan Thanendrajan, MD
Sub-Investigator: Shadiqul Hogue, MD
Sub-Investigator: Sandra Susanibar-Adaniya, MD Less << |
|
NCT01703910
|
Adenocarcinoma of Colon
... More >> Adenocarcinoma of Rectum
Metastatic Disease Less << |
Phase 2 |
Completed |
- |
Spain
... More >> Hospital Universitario de Fuenlabrada
Fuenlabrada, Madrid, Spain, 28950
Hospital Madrid Norte Sanchinarro
Madrid, Spain, 28050 Less << |
|
NCT00058370
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Not Applicable |
Active, not recruiting |
February 2019 |
United States, New York
... More >>
Herbert Irving Comprehensive Cancer Center at Columbia University Medical Center
New York, New York, United States, 10032
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT02759250
|
Cancer |
Phase 1 |
Completed |
- |
Belgium
... More >> UZG - Universitair Ziekenhuis Gent
Gent, Belgium Less << |
|
NCT00002823
|
Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00938145
|
Bladder Cancer |
Not Applicable |
Recruiting |
December 31, 2018 |
United States, Ohio
... More >>
Ohio State University Medical Center
Recruiting
Columbus, Ohio, United States, 43210
Contact: Michael Knopp, MD, PhD 614-293-9998 knopp.16@osu.edu Less << |
|
NCT00820755
|
- |
|
Completed |
- |
- |
|
NCT03032614
|
Breast Cancer Stage IV
... More >> Ovarian Cancer
BRCA1 Mutation
BRCA2 Mutation Less << |
Phase 2 |
Withdrawn(Lack of funding) |
April 30, 2020 |
- |
|
NCT00820755
|
Non-Small Cell Lung Cancer (NS... More >>CLC) Less << |
Phase 3 |
Completed |
- |
Germany
... More >> Central Contact
Darmstadt, Germany Less << |
|
NCT01305772
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 2 |
Terminated(This study was term... More >>inated for low accrual.) Less << |
- |
United States, North Carolina
... More >>
University of North Carolina at Chapel Hill
Chapel Hill, North Carolina, United States, 27514
Duke University Medical Center
Durham, North Carolina, United States, 27710 Less << |
|
NCT02644408
|
Stage III Esophageal Squamous ... More >>Cell Carcinoma
Stage II Esophageal Squamous Cell Carcinoma Less << |
Phase 3 |
Recruiting |
December 2020 |
China, Henan
... More >> The First Affiliated Hospital of Henan University of Science and Technology
Recruiting
Luoyang, Henan, China, 471003
Contact: shegan gao, doctor 0379 64811906 gsg112258@163.com
Contact: tanyou shan, master 0379 64815350 shantanyou@163.com Less << |
|
NCT00002524
|
Lymphoma |
Phase 2 |
Completed |
- |
United States, Florida
... More >>
MD Anderson Cancer Center Orlando
Orlando, Florida, United States, 32806
United States, Texas
University of Texas - MD Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT03549715
|
Infiltrating Bladder Urothelia... More >>l Carcinoma Less << |
Phase 1
Phase 2 |
Not yet recruiting |
February 2022 |
France
... More >> Hôpital Européen Georges Pompidou
Not yet recruiting
Paris, Ile De France, France, 75015
Contact: Constance THIBAULT, MD 00 33 1 56 09 59 92 constance.thibault@aphp.fr
Principal Investigator: Constance THIBAULT, MD
Hôpital Saint André, CHU de Bordeaux
Not yet recruiting
Bordeaux, France, 33075
Contact: Marine GROSS-GOUPIL, MD marine.gross-goupil@chu-bordeaux.fr
Sasu Roc37
Not yet recruiting
Chambray Les Tours, France, 37170
Contact: Pierre COMBE, MD
CHU Henri-Mondor
Not yet recruiting
Créteil, France, 94000
Contact carolina.saldana@aphp.fr
Principal Investigator: Carolina SALDANA, MD
Centre Leon Berard
Not yet recruiting
Lyon, France, 69008
Contact: Aude FLECHON, MD
Institut Paoli Calmettes
Not yet recruiting
Marseille, France, 13009
Contact: Gwaenaelle Gravis, MD GRAVISG@marseille.fnclcc.fr
Centre Antoine Lacassagne
Not yet recruiting
Nice, France, 06100
Contact: Delphine BORCHIELLINI, MD Delphine.BORCHIELLINI@nice.unicancer.fr
Hôpital Cochin
Not yet recruiting
Paris, France, 75679
Contact: Olivier HUILLARD, MD olivier.huillard@aphp.fr
Centre Eugene Marquis
Not yet recruiting
Rennes, France, 35042
Contact: Brigitte LAGUERRE, MD b.laguerre@rennes.unicancer.fr
Hia Begin
Not yet recruiting
Saint-Mandé, France, 94160
Contact: Francois-Regis FERRAND, MD
Hôpitaux universitaires de Strasbourg
Not yet recruiting
Strasbourg, France, 67000
Contact: Philippe BARTHELEMY, MD philippe.barthelemy@chru-strasbourg.fr
Institut Claudius Regaud
Not yet recruiting
Toulouse, France, 31059
Contact: Damien POUESSEL, MD
Institut Gustave Roussy
Not yet recruiting
Villejuif, France, 94805
Contact: Yohann LORIOT Less << |
|
NCT01975766
|
Head and Neck Neoplasms |
Not Applicable |
Terminated(Poor accrual) |
- |
United States, Iowa
... More >>
Holden Comprehensive Cancer Center
Iowa City, Iowa, United States, 52242 Less << |
|
NCT01305772
|
- |
|
Terminated(This study was term... More >>inated for low accrual.) Less << |
- |
- |
|
NCT00721799
|
Mouth Neoplasms
... More >> Oropharyngeal Neoplasms
Laryngeal Neoplasms
Head and Neck Neoplasms Less << |
Phase 2 |
Active, not recruiting |
June 30, 2020 |
United States, Iowa
... More >>
University of Iowa Hospitals and Clinics
Iowa City, Iowa, United States, 52242 Less << |
|
NCT02805530
|
Carcinoma, Non-Small-Cell Lung... More >>
Synchronous Neoplasms Less << |
Not Applicable |
Recruiting |
December 2017 |
Mexico
... More >> Instituto Nacional de Cancerologia
Recruiting
Mexico City, Mexico, 14080
Contact: Oscar Arrieta 015556280400 ogarrieta@gmail.com Less << |
|
NCT01755585
|
- |
|
Unknown |
- |
- |
|
NCT03674424
|
Non-metastatic Muscle Invasive... More >> Bladder Cancer Less << |
Phase 2 |
Recruiting |
April 1, 2023 |
Belgium
... More >> Institut Jules Bordet
Recruiting
Brussels, Belgium, 1000
Contact: Thierry Gil, MD 0032 2 541 31 88 thierry.gil@bordet.be
CHU de Liège Sart Tilman
Not yet recruiting
Liège, Belgium, 4000
Contact: Brieuc Sautois, MD 0032 4 366 82 03 Brieuc.Sautois@ulg.ac.be Less << |
|
NCT02701231
|
Lung Neoplasms |
Not Applicable |
Recruiting |
December 2020 |
China, Beijing
... More >> Chinese PLA General Hospital
Recruiting
Beijing, Beijing, China, 100853
Contact: Liang-An Chen, MD, phD 8610-55499027 chenla301@263.net
Principal Investigator: Liang-An Chen, MD, phD
China, Shandong
Affiliated hospital of Shandong University of traditional Chinese medicine
Not yet recruiting
Jinan, Shandong, China, 250011
Shandong Provincial Hospital
Not yet recruiting
Jinan, Shandong, China, 250021
China, Shanxi
Xijing Hospital affiliated to the Fourth Military Medical University
Not yet recruiting
Xi'an, Shanxi, China, 710032 Less << |
|
NCT03503864
|
Neuroblastoma |
Phase 2 |
Recruiting |
December 30, 2029 |
China, Guangdong
... More >>
Sun Yat-sen Memorial Hospital, Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China, 510120
Contact: Yang Li, Professor +8602081332456 drliyang@126.com Less << |
|
NCT03529279
|
Nasopharyngeal Carcinoma |
Phase 3 |
Not yet recruiting |
June 12, 2026 |
- |
|
NCT02431403
|
Lymphoma, Non-Hodgkin |
Phase 1
Phase 2 |
Recruiting |
- |
Korea, Republic of
... More >>
Dong-A University Hospital
Recruiting
Busan, Korea, Republic of
Contact: Ji Sook Park dongahicrc5@naver.com
Inje University Busan Paik Hospital
Recruiting
Busan, Korea, Republic of
Contact: Jin Hwa Kong +82-51-890-6987 velika-jh@nate.com
Kosin University Gospel Hospital
Recruiting
Busan, Korea, Republic of
Contact: Saet Byeol Park siou7777@naver.com
Pusan National University Hospital
Recruiting
Busan, Korea, Republic of
Contact: Younghee Kim +82-51-240-7899 shinyspring@naver.com
Ulsan University Hospital
Recruiting
Ulsan, Korea, Republic of
Contact: Mi Sun Jeon uuh3103@naver.com Less << |
|
NCT00460551
|
- |
|
Terminated(Due to consideratio... More >>ns regarding the appropriate therapeutic regimen for these patients.) Less << |
- |
- |
|
NCT00721799
|
- |
|
Active, not recruiting |
- |
- |
|
NCT02469493
|
Lung Neoplasms |
Not Applicable |
Unknown |
- |
China, Shaanxi
... More >> Xijing Hospital
Recruiting
Xi'an, Shaanxi, China, 710032
Contact: Yang 86(029)-84775343 Less << |
|
NCT02063412
|
- |
|
Withdrawn(Slow recruitment pac... More >>e) Less << |
- |
- |
|
NCT00432315
|
Carcinoma, Squamous Cell |
Phase 2 |
Completed |
- |
Austria
... More >> Sanofi-Aventis Administrative Office
Vienna, Austria Less << |
|
NCT01989858
|
Gastric Adenocarcinoma |
Phase 3 |
Terminated(lack of clinical ca... More >>ses) Less << |
- |
Italy
... More >> Ospedali Riuniti di Bergamo
Bergamo, BG, Italy, 24128
A. O. Ospedale Treviglio-Caravaggio
Treviglio, BG, Italy, 24047
A.O. Sant'Orsola Malpighi
Bologna, BO, Italy, 40138
Policlinico Sant'Orsola Malpighi
Bologna, BO, Italy, 40138
A.O. Santa Croce e Carle
Cuneo, CN, Italy, 12100
Azienda Ospedaliero-Universitaria "Policlinico-Vittorio Emanuele"
Catania, CT, Italy, 95123
Policlinico Universitario Mater Domini
Catanzaro, CZ, Italy, 88100
Azienda Ospedaliero- Universitaria Careggi - Firenze
Firenze, FI, Italy, 50141
A.O. Ospedale San Gerardo
Monza, MB, Italy, 20052
Azienda Ospedaliera di Desio e Vimercate
Vimercate, MB, Italy, 20059
Fondazione IRCCS Ospedale Maggiore Policlinico
Milano, MI, Italy, 20122
Istituto Nazionale per la Cura e lo Studio dei Tumori
Milano, MI, Italy, 20133
Istituto Oncologico Europeo
Milano, MI, Italy, 20141
Azienda Ospedaliera "San Paolo"
Milano, MI, Italy, 20142
Istituto Clinico Humanitas
Rozzano, MI, Italy, 20089
Casa di Cura MultiMedica
Sesto San Giovanni, MI, Italy, 20099
A. O. "Carlo Poma"
Mantova, MN, Italy, 46100
Ospedale "Ramazzini " di Carpi
Carpi, MO, Italy, 41012
Ospedale "Guglielmo da Saliceto"
Piacenza, PC, Italy, 29100
IRCCS Istituto Oncologico Veneto
Padova, PD, Italy, 35128
Ospedale Misericordia e Dolce - USL 4
Prato, PO, Italy, 59100
Ospedale Santa Croce Fano
Fano, PU, Italy, 61032
Azienda Ospedaliera 'San Carlo'
Potenza, PZ, Italy, 85100
Arcispedale S. Maria Nuova Azienda Ospedaliera
Reggio Emilia, RE, Italy, 42100
Università "Campus Bio-Medico"
Roma, RM, Italy, 00128
Policlinico Universitario A. Gemelli
Roma, RM, Italy, 00168
A.O. della Valtellina e della Valchiavenna - "Ospedale E. Morelli"
Sondalo, SO, Italy, 23100
IRCC/FPO -Istituto per la Ricerca e la Cura del Cancro di Candiolo
Candiolo, TO, Italy, 10060
A. O. "Ospedale di Circolo di Busto Arsizio" - Busto Arsizio (VA)
Busto Arsizio, VA, Italy, 21052
A. O. Busto Arsizio - P.O. Saronno
Saronno, VA, Italy, 21047
A.O. Ospedale di Circolo e Fondazione Macchi
Varese, VA, Italy, 21100
Fondazione "G. Pascale" Istituto Tumori di Napoli
Napoli, Italy, 80131 Less << |
|
NCT02951897
|
Lung Adenocarcinoma, Stage I
... More >> Diagnoses Diseases
Circulating Tumor Cells
Treatment Less << |
Not Applicable |
Recruiting |
December 2019 |
China
... More >> Daping hospital
Recruiting
Chongqing, China
Contact: Bo Deng +86-13637782166 deng.bo@dphospital.tmmu.edu.cn Less << |
|
NCT00567567
|
Localized Resectable Neuroblas... More >>toma
Localized Unresectable Neuroblastoma
Recurrent Neuroblastoma
Regional Neuroblastoma
Stage 4 Neuroblastoma
Stage 4S Neuroblastoma Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00567567
|
- |
|
Completed |
- |
- |
|
NCT01418859
|
- |
|
Unknown |
December 2015 |
China, Shanxi
... More >> Xi'an Jiaotong University College of Medicine
Recruiting
Xi'an, Shanxi, China, 710061
Contact: Zi Liu, M.D 086-18991232167 18991232167@189.cn
Contact: Tao Wang, M.D 086-18991232386 18991232386@189.cn
Sub-Investigator: Tao Wang, M.D Less << |
|
NCT03057106
|
Lung Cancer Metastatic |
Phase 2 |
Active, not recruiting |
December 2019 |
- |
|
NCT03126916
|
Childhood Ganglioneuroblastoma... More >>
Childhood Neuroblastoma
INRG Stage L2
INRG Stage M
INRG Stage MS
NMYC Gene Amplification
Recurrent Neuroblastoma Less << |
Phase 3 |
Recruiting |
June 30, 2026 |
- |
|
NCT00696735
|
Follicular Lymphoma |
Phase 3 |
Completed |
- |
France
... More >> Emmanuel GYAN
Tours, France, 37000
Philippe COLOMBAT
Tours, France, 37000 Less << |
|
NCT02371161
|
Non Hodgkin Lymphoma |
Phase 2 |
Recruiting |
February 2023 |
Italy
... More >> A.O. SS.Antonio e Biagio e C. Arrigo
Recruiting
Alessandria, AL, Italy, 15121
Contact: Flavia Salvi, MD 0131-206066 fsalvi@ospedale.al.it
Principal Investigator: Flavia Salvi, MD
Ospedali Riuniti
Recruiting
Ancona, AN, Italy, 60126
Contact: Attilio Olivieri, MD 071-5964226 a.olivieri@univpm.it
Principal Investigator: Attilio Olivieri, MD
Ospedale Perrino
Active, not recruiting
Brindisi, BR, Italy, 72100
Spedali Civili
Recruiting
Brescia, BS, Italy, 25123
Contact: Alessandra Tucci, MD 030-3995438 alessandra.tucci@spedalicivili.brescia.it
Principal Investigator: Alessandra Tucci, MD
Policlinico Vittorio Emanuele
Active, not recruiting
Catania, CT, Italy, 95123
Casa Sollievo della Sofferenza
Active, not recruiting
San Giovanni Rotondo, FG, Italy, 71013
IRST Meldola
Recruiting
Meldola, Forlì-Cesena, Italy, 47014
Contact: Gerardo Musuraca 0543-739291 g.musuraca@irst.emr.it
Principal Investigator: Gerardo Musuraca, MD
IRCCS San Martino - IST
Active, not recruiting
Genova, GE, Italy, 16132
ASUR Marche, Area Vasta 3
Active, not recruiting
Civitanova Marche, MC, Italy, 62012
AO Riuniti Papardo Piemonte
Recruiting
Messina, ME, Italy, 98158
Contact: Donato Mannina, MD 090-3992239 donamanni@gmail.com
Principal Investigator: Donato Mannina, MD
IRCCS Humanitas
Recruiting
Rozzano, Milano, Italy, 20089
Contact: Luca Castagna, MD 02-82244580 luca.castagna@humanitas.it
Principal Investigator: Luca Castagna, MD
AO Niguarda Ca' Granda
Recruiting
Milano, MI, Italy, 20162
Contact: Chiara Rusconi, MD 02-64442668 chiara.rusconi@ospedaleniguarda.it
Principal Investigator: Chiara Rusconi, MD
Ospedale Civile "Guglielmo da Saliceto"
Recruiting
Piacenza, PC, Italy, 29100
Contact: Daniele Vallisa, MD 0523-303719 d.vallisa@ausl.pc.it
Principal Investigator: Danilele Vallisa, MD
Centro di Riferimento Oncologico
Recruiting
Aviano, PN, Italy, 33081
Contact: Mariagrazia Michieli, MD 0434-659601 mmichieli@cro.it
Principal Investigator: Mariagrazia Michieli, MD
Azienda Ospaliero Universitaria di Parma
Recruiting
Parma, PR, Italy, 43126
Contact: Francesca Re, MD 0521-033306 fre@ao.pr.it
Principal Investigator: Francesca Re, Prof.
Ospedale San Carlo
Active, not recruiting
Potenza, PZ, Italy, 85100
AUSL di Ravenna
Recruiting
Ravenna, RA, Italy, 48100
Contact: Monica Tani, MD 0544-286223 monica.tani@ausl.ra.it
Principal Investigator: Monica Tani, MD
A.O. Bianchi - Melacrino - Morelli
Active, not recruiting
Reggio Calabria, RC, Italy, 89125
Asmn-Irccs
Recruiting
Reggio Emilia, RE, Italy, 43123
Contact: Francesco Merli, MD 0522-296618 merli.francesco@asmn.re.it
Principal Investigator: Francesco Merli, MD
Policlinico Sant'Andrea
Recruiting
Roma, RM, Italy, 00189
Contact: Maria Christina Cox, MD 338-2268017 chrisscox@gmail.com
Principal Investigator: Maria Christina Cox, MD
Ospedale Nocera- Pagani
Active, not recruiting
Nocera Inferiore, SA, Italy, 84014
AOU San Giovanni e Ruggi
Active, not recruiting
Salerno, SA, Italy, 84131
AOU Città della Salute e della Scienza
Recruiting
Torino, TO, Italy, 10126
Contact: Federica Cavallo, MD 011-6334301 f.cavallo@unito.it
Principal Investigator: Federica Cavallo, MD
Ospedale Mauriziano
Active, not recruiting
Torino, TO, Italy, 10128
AO Santa Maria
Active, not recruiting
Terni, TR, Italy, 05100
Ospedale S.Andrea
Active, not recruiting
Vercelli, VC, Italy, 13100
Ospedale degli Infermi
Recruiting
Rimini, Italy, 47923
Contact: Anna Lia Molinari 0541-705423 annalia.molinari@auslromagna.it
Principal Investigator: Anna Lia Molinari, MD Less << |
|
NCT02556762
|
Esophageal Cancer |
Phase 3 |
Recruiting |
August 2021 |
China, Guangdong
... More >>
Cancer Hospital, Shantou University Medical College
Recruiting
Shantou, Guangdong, China, 515031
Contact: Chuangzhen Chen, MD 86-13923995569 stccz@139.com
Contact: Jianzhou Chen, MD 86-13417049908 cjzeoeo@gmail.com
Principal Investigator: Chuangzhen Chen, MD Less << |
|
NCT00460551
|
Non Small Cell Lung Cancer |
Phase 2 |
Terminated(Due to consideratio... More >>ns regarding the appropriate therapeutic regimen for these patients.) Less << |
- |
United States, Maryland
... More >>
Towson, Maryland, United States, 21204
United States, Oregon
Providence Portland Medical Center
Portland, Oregon, United States, 97213-2967
Belgium
UZ Gent
Gent, Belgium, 9000
CHR La Citadelle
Liege, Belgium, 4000
CHU Sart-Tilman Domaine Universitaire du Sart-Tilman
Liege, Belgium, 4000
France
CHRU Reims, Hospital Maison Blanche
Reims, Cedex, France, 51092
Netherlands
VU Medisch Centrum (VUMC)
Amsterdam, Netherlands, 1004MB
United Kingdom
The Institute of Cancer Research and the Royal Marsden NHS Foundation Trust
Sutton, Surrey, United Kingdom, SM2 5PT Less << |
|
NCT01275664
|
- |
|
Terminated(Study terminated du... More >>e to no patient population available) Less << |
- |
- |
|
NCT00574080
|
Multiple Myeloma |
Phase 3 |
Terminated(low accrual) |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT02883543
|
Non-small Cell Lung Cancer |
Phase 3 |
Active, not recruiting |
June 2019 |
- |
|
NCT01275664
|
Nausea and Vomiting
... More >> Ovarian Brenner Tumor
Ovarian Clear Cell Cystadenocarcinoma
Ovarian Endometrioid Adenocarcinoma
Ovarian Mucinous Cystadenocarcinoma
Ovarian Seromucinous Carcinoma
Ovarian Serous Cystadenocarcinoma
Stage II Ovarian Cancer
Stage IIA Fallopian Tube Cancer
Stage IIA Ovarian Cancer
Stage IIB Fallopian Tube Cancer
Stage IIB Ovarian Cancer
Stage IIC Fallopian Tube Cancer
Stage IIC Ovarian Cancer
Stage IIIA Fallopian Tube Cancer
Stage IIIA Ovarian Cancer
Stage IIIA Primary Peritoneal Cancer
Stage IIIB Fallopian Tube Cancer
Stage IIIB Ovarian Cancer
Stage IIIB Primary Peritoneal Cancer
Stage IIIC Fallopian Tube Cancer
Stage IIIC Ovarian Cancer
Stage IIIC Primary Peritoneal Cancer
Stage IV Fallopian Tube Cancer
Stage IV Ovarian Cancer
Stage IV Primary Peritoneal Cancer
Undifferentiated Ovarian Carcinoma Less << |
Not Applicable |
Terminated(Study terminated du... More >>e to no patient population available) Less << |
- |
United States, Illinois
... More >>
Sudarshan K Sharma MD Limted-Gynecologic Oncology
Hinsdale, Illinois, United States, 60521
United States, Rhode Island
Women and Infants Hospital
Providence, Rhode Island, United States, 02905 Less << |
|
NCT00574080
|
- |
|
Terminated(low accrual) |
- |
- |
|
NCT00531960
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT01327521
|
Carcinoma, Hepatocellular |
Phase 3 |
Withdrawn(Accrual below target... More >> levels) Less << |
- |
United States, California
... More >>
Stanford University School of Medicine
Stanford, California, United States, 94305 Less << |
|
NCT00003643
|
Extragonadal Germ Cell Tumor
... More >> Teratoma
Testicular Germ Cell Tumor Less << |
Phase 2
Phase 3 |
Unknown |
- |
- |
|
NCT03294122
|
- |
|
Recruiting |
December 31, 2019 |
Italy
... More >> Fondazione IRCCS Istituto Nazionale Tumori
Recruiting
Milan, Italy, 20133
Contact: Riccardo Valdagni, MD, PhD +39 02-23903034 riccardo.valdagni@istitutotumori.mi.it
Contact: Tiziana Rancati, MS +39 02-23903786 tiziana.rancati@istitutotumori.mi.it
Principal Investigator: Riccardo Valdagni, MD, PhD Less << |
|
NCT01691625
|
Esophageal Carcinoma |
Not Applicable |
Recruiting |
September 2019 |
China, Beijing
... More >> Capital Medical University Cancer Center
Recruiting
Beijing, Beijing, China, 100038
Contact: Jun Ren, MD, PhD 86-10-63926317 renjun9688@yahoo.com
Principal Investigator: Jun Ren, MD, PhD Less << |
|
NCT03775486
|
Non-small Cell Lung Cancer NSC... More >>LC Less << |
Phase 2 |
Not yet recruiting |
May 23, 2022 |
- |
|
NCT00531960
|
- |
|
Completed |
- |
- |
|
NCT00009880
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT02716896
|
Muscle Invasive Bladder Cancer |
Not Applicable |
Active, not recruiting |
June 2019 |
United States, Texas
... More >>
University of Texas Health Science Center at San Antonio
San Antonio, Texas, United States, 78829 Less << |
|
NCT00033644
|
Sarcoma |
Phase 2 |
Terminated |
- |
- |
|
NCT00003777
|
Head and Neck Cancer |
Phase 2 |
Unknown |
- |
United States, Pennsylvania
... More >>
University of Pennsylvania Cancer Center
Philadelphia, Pennsylvania, United States, 19104-4283 Less << |
|
NCT03353675
|
NSCLC |
Phase 2 |
Recruiting |
September 2020 |
United States, North Carolina
... More >>
Charlotte
Recruiting
Charlotte, North Carolina, United States, 28204
United States, Tennessee
Nashville
Recruiting
Nashville, Tennessee, United States, 37203
Belgium
Libramont
Recruiting
Libramont, Belgium
Denmark
Herlev
Recruiting
Herlev, Denmark
France
Créteil
Recruiting
Créteil, France
Mulhouse
Recruiting
Mulhouse, France
Rennes
Recruiting
Rennes, France
Strasbourg
Recruiting
Strasbourg, France
Hungary
Budapest
Recruiting
Budapest, Hungary
Szekesfehervar
Recruiting
Szekesfehervar, Hungary Less << |
|
NCT01216527
|
Squamous Cell Esophageal Carci... More >>noma Less << |
Phase 3 |
Active, not recruiting |
December 2019 |
China, Guangdong
... More >>
Sun Yat-sen Uniersity Cancer Center
GuangZhou, Guangdong, China, 510060
Cancer Hospital of Shantou University Medical College
Shantou, Guangdong, China, 515000 Less << |
|
NCT00002884
|
Esophageal Cancer |
Phase 3 |
Unknown |
- |
France
... More >> Hopital Jules Courmont - Centre Hospitalier Lyon Sud
Pierre Benite, France, 69495 Less << |
|
NCT00002631
|
Esophageal Cancer |
Phase 3 |
Completed |
- |
United States, Arizona
... More >>
CCOP - Scottsdale Oncology Program
Scottsdale, Arizona, United States, 85259-5404
United States, Illinois
CCOP - Illinois Oncology Research Association
Peoria, Illinois, United States, 61602
CCOP - Carle Cancer Center
Urbana, Illinois, United States, 61801
United States, Iowa
CCOP - Cedar Rapids Oncology Project
Cedar Rapids, Iowa, United States, 52403-1206
CCOP - Iowa Oncology Research Association
Des Moines, Iowa, United States, 10309-1016
Siouxland Hematology-Oncology
Sioux City, Iowa, United States, 51101-1733
United States, Kansas
CCOP - Wichita
Wichita, Kansas, United States, 67214-3882
United States, Minnesota
CCOP - Duluth
Duluth, Minnesota, United States, 55805
Mayo Clinic Cancer Center
Rochester, Minnesota, United States, 55905
CentraCare Clinic
Saint Cloud, Minnesota, United States, 56303
United States, North Dakota
Quain & Ramstad Clinic, P.C.
Bismarck, North Dakota, United States, 58501
CCOP - Merit Care Hospital
Fargo, North Dakota, United States, 58122
Altru Health Systems
Grand Forks, North Dakota, United States, 58201
United States, Ohio
CCOP - Toledo Community Hospital Oncology Program
Toledo, Ohio, United States, 43623-3456
United States, Pennsylvania
CCOP - Geisinger Clinical and Medical Center
Danville, Pennsylvania, United States, 17822-2001
United States, South Dakota
Rapid City Regional Hospital
Rapid City, South Dakota, United States, 57709
CCOP - Sioux Community Cancer Consortium
Sioux Falls, South Dakota, United States, 57105-1080
Canada, Saskatchewan
Saskatchewan Cancer Agency
Regina, Saskatchewan, Canada, S4S 6X3 Less << |
|
NCT01680523
|
Cervical Cancer |
Not Applicable |
Recruiting |
July 2020 |
Korea, Republic of
... More >>
Department of Obstetrics and Gynecology, University of Ulsan College of Medicine, Asan Medical Center
Recruiting
Seoul, Korea, Republic of, 138-736
Contact: Joo-Hyun Nam, M.D., Ph.D. 82-2-3010-3633 jhnam@amc.seoul.kr Less << |
|
NCT03164616
|
Non Small Cell Lung Cancer NSC... More >>LC Less << |
Phase 3 |
Recruiting |
September 24, 2021 |
- |
|
NCT00041262
|
Esophageal Cancer |
Phase 3 |
Unknown |
- |
United Kingdom
... More >> Queen Elizabeth Hospital at University Hospital of Birmingham NHS Trust
Recruiting
Birmingham, England, United Kingdom, B15 2TH
Contact: Derek Alderson, MD 44-121-627-2276 d.alderson@bham.ac.uk Less << |
|
NCT01539785
|
First Recurrence of Ovarian Ca... More >>ncer Less << |
Not Applicable |
Unknown |
- |
Italy
... More >> Catholic University of Sacred the Hearth
Recruiting
Rome, Italy, 00100
Contact: Catholic University of Sacred the Hearth +39 063 015 627 9
Principal Investigator: Fagotti Anna, MD, PhD
Principal Investigator: Fanfani Francesco, MD
Principal Investigator: Costantini Barbara, MD
Principal Investigator: Perelli Federica, MD Less << |
|
NCT00047112
|
Esophageal Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00673361
|
Melanoma |
Phase 2 |
Terminated |
- |
- |
|
NCT00673361
|
- |
|
Terminated |
- |
- |
|
NCT00053872
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 3 |
Unknown |
- |
Belgium
... More >> U.Z. Gasthuisberg
Leuven, Belgium, B-3000
France
Institut Curie Hopital
Paris, France, 75248
Germany
Universitaets - Kinderklinik Wuerzburg
Wuerzburg, Germany, D-97080
Italy
Ospedale Infantile Regina Margherita
Turin, Italy, 10126
Netherlands
Academisch Medisch Centrum at University of Amsterdam
Amsterdam, Netherlands, 1105 AZ
Spain
Hospital de Cruces
Vizcaya, Spain, 48
Sweden
Ostra Sjukhuset
Gothenburg, Sweden, 41685
United Kingdom
Royal Liverpool Children's Hospital, Alder Hey
Liverpool, England, United Kingdom, L12 2AP Less << |
|
NCT00735345
|
Esophageal Cancer |
Phase 2 |
Terminated(because of safety c... More >>oncerns the study was terminated prematurely) Less << |
- |
Austria
... More >> Landeskrankenhaus Feldkirch
Feldkirch, Austria, A-6806
Universitätsklinikum Graz
Graz, Austria, A-8036
Universitätsklinik Innsbruck
Innsbruck, Austria, A-6020
A.ö. Landeskrankenhaus Leoben
Leoben, Austria, A-8700
Universitaetsklinik f. Innere Medizin III
Salzburg, Austria, A-5020
Krankenhaus Barmherzige Brueder St. Veit a.d. Glan
St. Veit/ Glan, Austria, A-9300
Klinikum Kreuzschwestern Wels GmbH
Wels, Austria, A-4600 Less << |
|
NCT00002858
|
Lung Cancer |
Phase 3 |
Unknown |
- |
France
... More >> Centre Hospitalier Regional et Universitaire d'Angers
Angers, France, 49033
Centre Paul Papin
Angers, France, 49036
Institut Bergonie
Bordeaux, France, 33076
Centre Regional Francois Baclesse
Caen, France, 14076
Hopital Antoine Beclere
Clamart, France, 92141
Centre Hospitalier Sud Francilien - Site Corbeil
Corbeil, France, 91100
Hopital Intercommunal De Creteil
Creteil, France, 94010
Hopital De La Trouhade
Dijon, France, 21034
Centre de Lute Contre le Cancer,Georges-Francois Leclerc
Dijon, France, 21079
C.H. General Andre Boulloche
Montbeliard, France, 25209
CRLCC Nantes - Atlantique
Nantes-Saint Herblain, France, 44805
Centre Antoine Lacassagne
Nice, France, 06189
Hopital Haut Leveque
Pessac, France, 33604
C.H. De Saumur
Saumur, France, 49403
Hopitaux Universitaire de Strasbourg
Strasbourg, France, 67091
Institut Claudius Regaud
Toulouse, France, 31052
Centre Alexis Vautrin
Vandoeuvre-les-Nancy, France, 54511
CHRU de Nancy - Hopitaux de Brabois
Vandoeuvre-Les-Nancy, France, 54511
Institut Gustave Roussy
Villejuif, France, F-94805 Less << |
|
NCT00003032
|
Breast Cancer |
Phase 3 |
Completed |
- |
Canada, Alberta
... More >>
Tom Baker Cancer Center - Calgary
Calgary, Alberta, Canada, T2N 4N2
Canada, British Columbia
BC Cancer Agency
Vancouver, British Columbia, Canada, V5Z 4E6
Canada, Manitoba
CancerCare Manitoba
Winnipeg, Manitoba, Canada, R3E 0V9
Canada, New Brunswick
Moncton Hospital
Moncton, New Brunswick, Canada, E1C 6ZB
Saint John Regional Hospital
Saint John, New Brunswick, Canada, E2L 4L2
Canada, Newfoundland and Labrador
Newfoundland Cancer Treatment and Research Foundation
St. Johns, Newfoundland and Labrador, Canada, A1B 3V6
Canada, Nova Scotia
Nova Scotia Cancer Centre
Halifax, Nova Scotia, Canada, B3H 1V7
Canada, Ontario
William Osler Health Centre
Brampton, Ontario, Canada, L6W 2Z8
Cancer Care Ontario-Hamilton Regional Cancer Centre
Hamilton, Ontario, Canada, L8V 5C2
Cancer Care Ontario-London Regional Cancer Centre
London, Ontario, Canada, N6A 4L6
Ottawa Regional Cancer Centre - General Campus
Ottawa, Ontario, Canada, K1H 1C4
Hotel Dieu Hospital - St. Catharines
St. Catharines, Ontario, Canada, L2R 5K3
Northeastern Ontario Regional Cancer Centre, Sudbury
Sudbury, Ontario, Canada, P3E 5J1
Northwestern Ontario Regional Cancer Centre, Thunder Bay
Thunder Bay, Ontario, Canada, P7A 7T1
Toronto Sunnybrook Regional Cancer Centre
Toronto, Ontario, Canada, M4N 3M5
Toronto General Hospital
Toronto, Ontario, Canada, M5G 2C4
Princess Margaret Hospital
Toronto, Ontario, Canada, M5G 2M9
Humber River Regional Hospital
Weston, Ontario, Canada, M9N 1N8
Cancer Care Ontario - Windsor Regional Cancer Centre
Windsor, Ontario, Canada, N8W 2X3
Canada, Prince Edward Island
Queen Elizabeth Hospital, PEI
Charlottetown, Prince Edward Island, Canada, C1A 8T5
Canada, Quebec
L'Hotel Dieu de Levis
Levis, Quebec, Canada, G6V 3Z1
Centre Hospitalier de l'Universite de Montreal
Montreal, Quebec, Canada, H2L-4M1
McGill University Department of Oncology
Montreal, Quebec, Canada, H2W 1S6
Hotel Dieu de Montreal
Montreal, Quebec, Canada, H2W 1T8
Centre Hospitalier Universitaire de Quebec, Pavillion de Quebec
Quebec City, Quebec, Canada, G1R 2J6 Less << |
|
NCT03661320
|
Bladder Cancer
... More >> Muscle-Invasive Bladder Cancer
BMS-986205 Less << |
Phase 3 |
Recruiting |
December 30, 2026 |
- |
|
NCT00002747
|
Head and Neck Cancer |
Phase 3 |
Unknown |
- |
Italy
... More >> Istituto Nazionale per lo Studio e la Cura dei Tumori
Milan, Italy, 20133 Less << |
|
NCT02864251
|
Non-Small-Cell Lung Carcinoma |
Phase 3 |
Recruiting |
December 31, 2023 |
- |
|
NCT02755311
|
Hepatocellular Carcinoma |
Phase 3 |
Unknown |
June 2016 |
China, Guangdong
... More >>
The First Affiliated Hospital of Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China, 510080
Contact: Zhenwei Peng, MD pengzhenwei2005@163.com Less << |
|
NCT03643133
|
Osteosarcoma |
Phase 2 |
Recruiting |
October 2028 |
France
... More >> Institut Curie
Not yet recruiting
Paris, France, 75000
Contact: Sophie MD PIPERNO-NEUMANN, PhD 33 (0)144324672 sophie.piperno-neumann@curie.fr
Gustave Roussy
Recruiting
Villejuif, France, 94800
Contact: Nathalie MD GASPAR, PhD 33 (0)142114166 nathalie.gaspar@gustaveroussy.fr Less << |
|
NCT01346267
|
Central Nervous System Tumor, ... More >>Pediatric
Chemotherapy-induced Nausea and Vomiting
Unspecified Childhood Solid Tumor, Protocol Specific Less << |
Phase 3 |
Completed |
- |
- |
|
NCT03376958
|
Relapsed /Refractory Diffuse L... More >>arge B Cell Lymphoma Less << |
Phase 4 |
Recruiting |
February 1, 2019 |
China, Henan
... More >> Oncology Department of The First Affiliated Hospital of Zhengzhou University
Recruiting
Zhengzhou, Henan, China, 450052
Contact: Mingzhi Zhang, Pro,Dr 13838565629 mingzhi_zhang@126.com Less << |
|
NCT01432314
|
Hepatocellular Carcinoma |
Phase 2 |
Withdrawn(Another phase 2 rand... More >>omized study has been started by other investigators.) Less << |
- |
- |
|
NCT02453282
|
Non-Small-Cell Lung Carcinoma ... More >>NSCLC Less << |
Phase 3 |
Active, not recruiting |
December 31, 2019 |
- |
|
NCT02089204
|
- |
|
Unknown |
December 2015 |
- |
|
NCT00967499
|
Postoperative Nausea and Vomit... More >>ing Less << |
Phase 2 |
Completed |
- |
United States, Arizona
... More >>
Precision Trials
Phoenix, Arizona, United States, 85032
United States, California
Accurate Clinical Trials, Inc
Laguna Hills, California, United States, 92653
University of California San Francisco
San Francisco, California, United States, 94115
United States, Florida
University of Miami
Miami, Florida, United States, 33136
United States, Kansas
University of Kansas Medical Center
Kansas City, Kansas, United States, 66160
United States, North Carolina
Duke University Medical Center
Durham, North Carolina, United States, 27710
United States, Ohio
Ohio State University Medical Center
Columbus, Ohio, United States, 43210
United States, Texas
Scott and White Hospital
Temple, Texas, United States, 76508 Less << |
|
NCT01480817
|
Hepatocellular Carcinoma |
Phase 2 |
Terminated(Difficulty of parti... More >>cipant enrollment.) Less << |
- |
Korea, Republic of
... More >>
Seoul National University Hospital
Seoul, Korea, Republic of, 110-744 Less << |
|
NCT01628380
|
Ovarian Neoplasms |
Phase 3 |
Unknown |
July 2018 |
Germany
... More >> Jena University Hospital
Recruiting
Jena, Germany, 07743
Contact: Ingo B. Runnebaum, MD, MBA INGO.RUNNEBAUM@med.uni-jena.de
Principal Investigator: Ingo B. Runnebaum, MD, MBA
Italy
A.O. Papa Giovanni XXIII (former Ospedali Riuniti)
Recruiting
Bergamo, Bg, Italy, 24128
Contact: Luca Ansaloni, MD +390352673477 lansaloni@hpg23.it
Contact: Marco Lotti, MD +390352673477 mlotti@hpg23.it
Principal Investigator: Luca Ansaloni, MD
A.O. Universitaria Policlinico S.Orsola-Malpighi Di Bologna - Bologna (Bo)
Recruiting
Bologna, Bo, Italy, 40138
Contact: Pierandrea Deiaco, MD +390516364426 pierandrea.deiaco@aosp.bo.it
Principal Investigator: Pierandrea Deiaco, MD
A.O. Universitaria Di Parma - Parma (Pr) Oncologia Chirurgica
Recruiting
Parma, Pr, Italy, 43100
Contact: Fausto Catena, MD +390521703940 faustocatena@gmail.com
Principal Investigator: Fausto Catena, MD
POLICLINICO UNIVERSITARIO GEMELLI DI ROMA - ROMA (RM) GINECOLOGIA E OSTETRICIA Dipartimento per la Tutela della Salute della Donna e della Vita Nascente
Recruiting
Roma, Italy, 00168
Contact: Giovanni Scambia, MD +390630156279 clinicaltrials@rm.unicatt.it
Principal Investigator: Giovanni Scambia, MD Less << |
|
NCT03237780
|
Metastatic Urothelial Carcinom... More >>a
Recurrent Bladder Urothelial Carcinoma
Recurrent Urethral Urothelial Carcinoma
Recurrent Urothelial Carcinoma of the Renal Pelvis and Ureter
Renal Pelvis Urothelial Carcinoma
Stage III Bladder Urothelial Carcinoma AJCC v6 and v7
Stage III Renal Pelvis Cancer AJCC v7
Stage III Ureter Cancer AJCC v7
Stage III Urethral Cancer AJCC v7
Stage IV Bladder Urothelial Carcinoma AJCC v7
Stage IV Renal Pelvis Cancer AJCC v7
Stage IV Ureter Cancer AJCC v7
Stage IV Urethral Cancer AJCC v7
Ureter Urothelial Carcinoma Less << |
Phase 2 |
Recruiting |
January 31, 2020 |
United States, California
... More >>
Los Angeles County-USC Medical Center
Recruiting
Los Angeles, California, United States, 90033
Contact: Site Public Contact 323-865-0451
Principal Investigator: David I. Quinn
USC / Norris Comprehensive Cancer Center
Recruiting
Los Angeles, California, United States, 90033
Contact: Site Public Contact 323-865-0451
Principal Investigator: David I. Quinn
Keck Medical Center of USC Pasadena
Recruiting
Pasadena, California, United States, 91105
Contact: Site Public Contact 323-865-0451
Principal Investigator: David I. Quinn
University of California Davis Comprehensive Cancer Center
Recruiting
Sacramento, California, United States, 95817
Contact: Site Public Contact 916-734-3089
Principal Investigator: Chong-Xian Pan
United States, Pennsylvania
University of Pittsburgh Cancer Institute (UPCI)
Recruiting
Pittsburgh, Pennsylvania, United States, 15232
Contact: Site Public Contact 412-647-8073
Principal Investigator: Leonard J. Appleman Less << |
|
NCT03001596
|
Esophageal Squamous Cell Carci... More >>noma Stage cT3-4aN0-1M0 Less << |
Phase 3 |
Not yet recruiting |
December 2023 |
China, Shanghai
... More >>
Shanghai Zhongshan Hospital
Not yet recruiting
Shanghai, Shanghai, China, 200032
Contact: Han Tang +862164041990 htang1989@163.com
Contact: Lijie Tan, MD +862164041990 tan.lijie@zs-hospital.sh.cn
Sub-Investigator: Han Tang
Principal Investigator: Lijie Tan Less << |
|
NCT01265849
|
Squamous Cell Carcinoma of the... More >> Oral Cavity
Squamous Cell Carcinoma of the Soft Palate Less << |
Phase 3 |
Completed |
- |
- |
|
NCT01349647
|
Lung Cancer |
Phase 1 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT00355199
|
- |
|
Completed |
- |
- |
|
NCT00825669
|
Hepatocellular Carcinoma |
Not Applicable |
Completed |
- |
China, Shanghai
... More >>
Eastern Hepatobiliary Surgery Hospital
Shanghai, Shanghai, China, 200438 Less << |
|
NCT03161548
|
Tongue Cancer
... More >> Chemotherapy Effect
Surgery Less << |
Phase 2 |
Terminated(Diffiult patient re... More >>cruitment) Less << |
- |
- |
|
NCT02129699
|
Lung Cancer Non-small Cell Sta... More >>ge IV Less << |
Phase 3 |
Active, not recruiting |
September 2020 |
- |
|
NCT02938195
|
Esophageal Cancer
... More >> Chemoradiation
Surgery Less << |
Phase 2 |
Unknown |
January 2018 |
China, Shanghai
... More >>
Shanghai Chest Hospital
Recruiting
Shanghai, Shanghai, China, 200030
Contact: Jun Liu, Doctor +862122200000 ext 3602 drjunliu@qq.com Less << |
|
NCT00355199
|
Diffuse Large B-Cell Lymphoma |
Phase 3 |
Completed |
- |
Italy
... More >> Clinica di Ematologia - Nuovo Ospedale Torrette
Ancona, Italy
U.O. Ematologia - Ospedali Riuniti di Bergamo
Bergamo, Italy
Divisione di Ematologia - Ospedale Centrale di Bolzano
Bolzano, Italy
CTMO - Ematologia - Ospedale "R. Binaghi"
Cagliari, Italy
Divisione di Ematologia - Ospedale Ferrarotto
Catania, Italy
S.C. Ematologia - Azienda Ospedaliera S. Croce e Carle
Cuneo, Italy
Divisione Ematologia - Istituto S. Raffaele
Milano, Italy
Oncologia Medica - Istituto Nazionale dei Tumori
Milano, Italy
U.O. Ematologia - Istituto Nazionale dei Tumori
Milano, Italy
Divisione di Ematologia - Azienda Ospedaliera
Padova, Italy
Ematologia - Azienda Ospedaliera V. Cervello
Palermo, Italy
Ematologia Clinica - Ospedale Civile di Pescara
Pescara, Italy
Ematologia e TMO - Ospedale S. Camillo
Roma, Italy
Divisione Universitaria di Ematologia - Azienda Ospedaliera S. Giovanni Battista (Molinette)
Torino, Italy
Dipartimento di Medicina Clinica e Sperimentale - Università di Verona
Verona, Italy
Divisione di Ematologia - Presidio Ospedaliero S. Bortolo
Vicenza, Italy Less << |
|
NCT02265770
|
Childhood Ependymoma |
Phase 2
Phase 3 |
Recruiting |
August 2026 |
- |
|
NCT01626664
|
Adult T-cell Leukemia-Lymphoma |
Phase 2 |
Completed |
- |
United States, California
... More >>
Cedars-Sinai Medical Center
Los Angeles, California, United States, 90048
United States, Florida
University of Miami / Sylvester Comprehensive Cancer Center
Miami, Florida, United States, 33136
United States, Illinois
Northwestern University
Chicago, Illinois, United States, 60611
United States, Maryland
National Cancer Institute
Bethesda, Maryland, United States, 20892
United States, Missouri
Washington University School of Medicine
Saint Louis, Missouri, United States, 63110
United States, New Jersey
Hackensack University Medical Center
Hackensack, New Jersey, United States, 07601
United States, New York
Montefiore Medical Center
Bronx, New York, United States, 10467
Memorial Sloan Kettering
New York, New York, United States, 10021
Columbia Presbyterian
New York, New York, United States, 10032
Weill Cornell Medical College
New York, New York, United States, 10065
Belgium
Cliniques Universitaires Saint-Luc
Bruxelles, Belgium, 1200
Brazil
Hospital Universitario Professor Edgard Santos- UFBA
Salvador, Bahia, Brazil, 40110-060
Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo
Sao Paulo- SP, Brazil, CEP 05403-000
France
CHU de Fort de France
Fort De France Cedex, France, BP 632 97261
Hospital Necker
Paris, France, 75743
Peru
Hospital Nacional Edgardo Rebagliati Martins
Lima, Peru, Lima11
Instituto Oncologico Miraflores
Lima, Peru, Lima18
United Kingdom
Guy's Hospital
London, United Kingdom, SE1 9RT
Imperial College
London, United Kingdom, W2 1PG
Sandwell General Hospital
West Midlands, United Kingdom, B71 4HJ Less << |
|
NCT00028665
|
Lymphoma |
Phase 2 |
Completed |
- |
United States, Ohio
... More >>
Ireland Cancer Center at University Hospitals of Cleveland and Case Western Reserve University
Cleveland, Ohio, United States, 44106-7284 Less << |
|
NCT00411151
|
Gastric Adenocarcinoma
... More >> Barrett Esophagus Less << |
Phase 2 |
Completed |
- |
Germany
... More >> Universität Heidelberg, Nationales Zentrum für Tumorerkrankungen
Heidelberg, Baden-Würtemberg, Germany, 69120
Universitätsklinikum Tübingen
Tübingen, Baden-Würtemberg, Germany, 72076
TU München, Klinikum rechts der Isar, II. Med. Klinik und Poliklinik
München, Bayern, Germany, 81675
Klinikum der Universität Würzburg
Würzburg, Bayern, Germany, 97070
Universitätsklinikum der GHS Essen, Innere Klinik und Poliklinik, Tumorforschung
Essen, Nordrhein-Westfalen, Germany, 45122
Universitätsklinik zu Köln, Klinik I für Innere Medizin
Köln, Nordrhein-Westfalen, Germany, 50937
Klinikum der Johannes Gutenberg-Universität Mainz
Mainz, Rheinland-Pfalz, Germany, 55131
Universitätsklinikum des Saarlandes
Homburg/Saar, Saarland, Germany, 66421
Otto-von-Guericke-Universität Magdeburg
Magdeburg, Sachsen-Anhalt, Germany, 39120
Universitätsklinikum Carl Gustav Carus, Med. Klinik I
Dresden, Sachsen, Germany, 01307
Charité, Campus Benjamin Franklin
Berlin, Germany, 12200
KH Nordwest, Abteilung für Hämatologie und Onkologie
Frankfurt, Germany, 60488 Less << |
|
NCT00411151
|
- |
|
Completed |
- |
- |
|
NCT02640898
|
Gastric Cancer |
Not Applicable |
Recruiting |
December 2019 |
China, Shanghai
... More >>
the Ethic Committee of Shanghai General Hospital
Recruiting
Shanghai, Shanghai, China, 210000
Contact: zhixiao chen, MD 021-63240090 ext 6218 chenzhixiaochen@sohu.com Less << |
|
NCT00452803
|
Lung Cancer |
Phase 2 |
Unknown |
December 2012 |
Korea, Republic of
... More >>
National Cancer Center, Korea
Recruiting
Goyang-si, Gyeonggi-do, Korea, Republic of, 411-769
Sub-Investigator: Jae-Ill Zoo, M.D.
Sub-Investigator: Jong Mog Lee, M.D.
Sub-Investigator: Moon Soo Kim, M.D.
Sub-Investigator: Hyun-Sung Lee, M.D.
Sub-Investigator: Jin Soo Lee, M.D.
Sub-Investigator: Ji-Youn Han, M.D.
Sub-Investigator: Kwan Ho Cho, M.D.
Sub-Investigator: Hong Ryull Pyo, M.D.
Sub-Investigator: Bin Hwangbo, M.D.
Sub-Investigator: Hee Seok Lee, M.D.
Sub-Investigator: Hyae Young Kim, M.D.
Sub-Investigator: Geon Kook Lee, M.D.
Sub-Investigator: Moon Woo Seong, M.D.
Sub-Investigator: Kyeong-Man Hong, M.D.
Sub-Investigator: Byung-Ho Nam, Ph.D.
Sub-Investigator: Tak Yun, M.D. Less << |
|
NCT01365156
|
Cervical Squamous Cell Carcino... More >>ma
Adenosquamous Carcinoma
Adenocarcinoma
Locally Advanced Malignant Neoplasm
Cervical Cancer Less << |
Phase 3 |
Active, not recruiting |
September 2019 |
United States, Texas
... More >>
Lyndon B. Johnson General Hospital
Houston, Texas, United States, 77026
University of Texas MD Anderson Cancer Center
Houston, Texas, United States, 77030
Belgium
Vall d'Hebron Hospital
Brussels, Belgium Less << |
|
NCT01560195
|
NSCLC
Neutrop... More >>enia
Febrile Neutropenia Less << |
Phase 3 |
Unknown |
- |
China, Shanghai
... More >>
Shanghai Pulmonary Hospital
Recruiting
Shanghai, Shanghai, China, 200433
Contact: Caicun Zhou, M. D. caicunzhou@yahoo.com.cn Less << |
|
NCT00176800
|
- |
|
Completed |
- |
- |
|
NCT01626664
|
- |
|
Completed |
- |
- |
|
NCT00176800
|
Esophageal Carcinoma |
Phase 2 |
Completed |
- |
United States, Michigan
... More >>
University of Michigan Cancer Center
Ann Arbor, Michigan, United States, 48109 Less << |
|
NCT02367443
|
Non-small Cell Lung Cancer |
Phase 1
Phase 2 |
Unknown |
December 2016 |
Poland
... More >> Independent Public Health Care Facility and Warmian & Mazurian Oncology Centre
Recruiting
Olsztyn, Poland, 10-228
Contact: Lucyna Kepka, Prof +48 506 118 893 lucynak@coi.pl Less << |
|
NCT02151981
|
- |
|
Active, not recruiting |
- |
- |
|
NCT02151981
|
Anticancer Treatment |
Phase 3 |
Active, not recruiting |
December 30, 2020 |
- |
|
NCT00765011
|
Head and Neck Cancer |
Phase 2 |
Unknown |
May 2014 |
Spain
... More >> Hospital Germans Trias i Pujol
Recruiting
Badalona, Barcelona, Spain, 08916
Principal Investigator: Beatriz Cirauqui, MD
Hospital Duran i Reynals
Recruiting
L'Hospitalet de Llobregat, Barcelona, Spain
Principal Investigator: Ricard Mesia, MD
Hospital de Manresa
Recruiting
Manresa, Barcelona, Spain
Principal Investigator: Esther Casado, MD
Hospital Clínic i Provincial de Barcelona
Recruiting
Barcelona, Spain, 08036
Principal Investigator: Juan J Grau, MD
Hospital General de Yagüe
Withdrawn
Burgos, Spain
Hospital San Pedro de Alcántara
Recruiting
Cáceres, Spain
Principal Investigator: Mª Angeles Rodríguez, MD
Hospital Dr.Josep Trueta (ICO Girona)
Recruiting
Girona, Spain, 17007
Principal Investigator: José Rubio, MD
Hospital Universitario Virgen de las Nieves
Recruiting
Granada, Spain
Principal Investigator: Antonio L Irigoyen, MD
Oncogranada
Terminated
Granada, Spain
Hospital General Ciudad de Jaén
Withdrawn
Jaén, Spain
Hospital de León
Withdrawn
Leon, Spain
Hospital Xeral Calde
Recruiting
Lugo, Spain
Principal Investigator: José R Mel, MD
Clínica Quirón
Withdrawn
Madrid, Spain
Hospital Gregorio Marañon
Recruiting
Madrid, Spain
Principal Investigator: Yolanda escobar, MD
Hospital Universitario 12 de Octubre
Terminated
Madrid, Spain
Hospital Universitario San Carlos
Recruiting
Madrid, Spain
Principal Investigator: José A García, MD
Hospital Son Llátzer
Recruiting
Mallorca, Spain
Principal Investigator: Mª Belén González, MD
Hospital General Universitario Morales Meseguer
Withdrawn
Murcia, Spain
Hospital Universitario de Salamanca
Recruiting
Salamanca, Spain
Sub-Investigator: Juan J Cruz, MD
Principal Investigator: Elvira del Barco Morillo, MD
Sub-Investigator: Juan Carlos Adansa Klain, MD
Hospital Universitario Marqués de Valdecilla
Withdrawn
Santander, Spain
Hospital Clínico de Santiago
Withdrawn
Santiago de Compostela, Spain
Hospital Universitario la Fe de Valencia
Recruiting
Valencia, Spain
Principal Investigator: Miguel Pastor, MD
Hospital Xeral Cies
Withdrawn
Vigo, Spain
Hospital Clínico Universitario de Zaragoza
Recruiting
Zaragoza, Spain
Principal Investigator: Julio Lambea, MD
Hospital Miguel Servet
Recruiting
Zaragoza, Spain
Principal Investigator: Javier Martínez Trufero, MD Less << |
|
NCT02363400
|
Nasopharyngeal Carcinoma |
Phase 3 |
Enrolling by invitation |
December 2022 |
- |
|
NCT00391586
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Terminated(PI left institution... More >>.) Less << |
- |
United States, New Mexico
... More >>
Lovelace Medical Group
Albuquerque, New Mexico, United States, 87102
Hematology Oncology Associates NM
Albuquerque, New Mexico, United States, 87106
Presbyterian Medical Group
Albuquerque, New Mexico, United States, 87110
University of New Mexico Cancer Center
Albuquerque, New Mexico, United States, 87131
New Mexico Cancer Care Associates
Santa Fe, New Mexico, United States, 87505 Less << |
|
NCT02082886
|
- |
|
Recruiting |
January 2034 |
United States, Illinois
... More >>
Elmhurst Memorial Health Care
Recruiting
Elmhurst, Illinois, United States, 60126
Contact: Kathy Seymour, BSN 630-646-6072 Kathy.Seymour@EEHealth.org
Principal Investigator: George I Salti, MD
Edward Hospital
Recruiting
Naperville, Illinois, United States, 60540
Contact: Kathy Seymour, BSN 630-646-6072 Kathy.Seymour@EEHealth.org
Principal Investigator: George I Salti, MD Less << |
|
NCT01861509
|
Metastatic Breast Cancer
... More >> Stage IV Breast Cancer Less << |
Phase 1 |
Completed |
- |
Israel
... More >> Oncology Unit. Sheba Medical Centre
Ramat-Gan, Israel, 52621
Thailand
Lampang Cancer Center
Lampang, Thailand, 52000 Less << |
|
NCT01543620
|
- |
|
Unknown |
June 2015 |
United States, California
... More >>
University of Southern California
Recruiting
Los Angeles, California, United States, 90033
Contact: Research Coordinator1
Contact: Research Coordinator2
Principal Investigator: Paul Beringer, PharmD
United States, Minnesota
University of Minnesota - Cystic Fibrosis Center
Recruiting
Minneapolis, Minnesota, United States, 55455
Contact: Research Coordinator1
Contact: Research Coordinator2
Principal Investigator: Joanne Billings, MD
United States, Utah
University of Utah
Recruiting
Salt Lake City, Utah, United States, 84132
Contact: Research Coordinator1
Contact: Research Coordinator2
Principal Investigator: Dave Young, PharmD Less << |
|
NCT03210298
|
- |
|
Recruiting |
May 2019 |
Argentina
... More >> Roffo Cancer Institute
Recruiting
Buenos Aires, Argentina
Contact: Laura Lay, MD lauralay@ymail.com
Australia
Peter MacCallum Cancer Centre
Recruiting
Melbourne, Australia
Contact: Craig Lynch, MD
Belgium
University Hospital Gent (UZ Gent)
Recruiting
Gent, Belgium, 9000
Contact: Woulter Willaert, MD wouter.willaert@ugent.be
Denmark
University Hospital Odense
Recruiting
Odense, Denmark
Contact: Martin Graversen, MD martin.graversen@rsyd.dk
France
Hospices civils de Lyon
Recruiting
Lyon, France
Contact: Olivier Glehen, MD +33 478862371
Hôpital Lariboisière
Recruiting
Paris, France, 75010
Contact: Clarisse Eveno, MD +33 1 49 95 25 80
Germany
University Hospital Tübingen
Recruiting
Tübingen, Germany, 72076
Contact: Verena Schlaich +4970712986722 pipac@med.uni-tuebingen.de
India
Fortis Hospital
Recruiting
Bengaluru, India
Contact: Aditi Bhatt, MD
Italy
IRCC Torino Candiolo
Recruiting
Turin, Italy
Contact: Marco Vaira, MD marco.vaira@ircc.it
Poland
F. Łukaszczyk Oncology Centre
Recruiting
Bydgoszcz, Poland
Contact: Maciej Nowacki, MD maciej.s.nowacki@gmail.com
Russian Federation
Gersten Institute for Cancer Research
Recruiting
Moscow, Russian Federation
Contact: Vladimir Khomyakov, MD vladimirkhom@mail.ru
Singapore
National University Hospital
Recruiting
Singapore, Singapore
Contact: Janelle Phua niam_sin_phua@nuhs.edu.sg
Spain
Hospital de Pilar
Recruiting
Barcelona, Spain
Contact: Juan Torrent, MD juanjotorrent@yahoo.es
Switzerland
University Hospital Lausanne CHUV
Recruiting
Lausanne, Switzerland, 1011
Contact: Martin Huebner, MD chv.carcinose@chuv.ch Less << |
|
NCT03627390
|
Metastatic Pancreatic Cancer
... More >> Unresectable Pancreatic Cancer Less << |
Phase 2 |
Completed |
- |
Egypt
... More >> National Liver Institute, Menoufia University
Cairo, Egypt Less << |
|
NCT00004897
|
Esophageal Cancer |
Phase 2 |
Terminated(Institutional Revie... More >>w Board requested termination - all patients deceased and no new accrual.) Less << |
- |
United States, Illinois
... More >>
Robert H. Lurie Comprehensive Cancer Center, Northwestern University
Chicago, Illinois, United States, 60611 Less << |
|
NCT00042367
|
Brain Tumors |
Not Applicable |
Completed |
- |
United States, North Carolina
... More >>
Brain Tumor Center at Duke University
Durham, North Carolina, United States, 27710
United States, Tennessee
St. Jude Children's Research Hospital
Memphis, Tennessee, United States, 38105-0318
United States, Texas
Texas Children's Hospital
Houston, Texas, United States, 77030 Less << |
|
NCT03617133
|
Uterine Cervical Neoplasms |
Phase 2 |
Recruiting |
April 2031 |
Austria
... More >> Medical University of Vienna
Recruiting
Vienna, Austria, 1090
Contact: Richard Pötter, MD 0043140400 ext 26920 richard.poetter@akhwien.at Less << |
|
NCT03603197
|
Metastatic Breast Cancer
... More >> Stage IV Breast Cancer Less << |
Phase 2 |
Completed |
- |
Thailand
... More >> Udon Thani Cancer Hospital
Udon Thani, Udon Thani Province, Thailand, 41000
Siriraj Hospital, Mahidol University
Bangkok, Thailand
Lampang Cancer Center
Lampang, Thailand, 52000
Ubon Ratchanthani Cancer Hospital
Ubon Ratchathani, Thailand Less << |
|
NCT01737502
|
Extensive Stage Small Cell Lun... More >>g Carcinoma
Lung Adenocarcinoma
Recurrent Non-Small Cell Lung Carcinoma
Recurrent Small Cell Lung Carcinoma
Squamous Cell Lung Carcinoma
Stage IIIA Non-Small Cell Lung Cancer
Stage IIIB Non-Small Cell Lung Cancer
Stage IV Non-Small Cell Lung Cancer Less << |
Phase 1
Phase 2 |
Recruiting |
November 2018 |
United States, Arizona
... More >>
Mayo Clinic in Arizona
Recruiting
Scottsdale, Arizona, United States, 85259
Contact: Clinical Trials Referral Office 855-776-0015
Principal Investigator: Helen J. Ross Less << |
|
NCT02762266
|
Child-Pugh Class A
... More >> Child-Pugh Class B
Recurrent Hepatocellular Carcinoma Less << |
Phase 3 |
Recruiting |
February 27, 2021 |
United States, California
... More >>
Stanford University, School of Medicine
Recruiting
Palo Alto, California, United States, 94304
Contact: Rachel Freiberg 650-725-0438 rachelf@stanford.edu
Principal Investigator: Daniel T. Chang
Japan
Hokkaido University Hospital
Not yet recruiting
Sapporo, Hokkaido, Japan, 060-8648
Contact: Rei Kikuchi 81-11-706-7600 reik777@huhp.hokudai.ac.jp
Principal Investigator: Hiroki Shirato Less << |
|
NCT01493544
|
- |
|
Completed |
- |
Argentina
... More >> Research Site
Escobar, Buenos Aires, Argentina
Research Site
Ezeiza, Buenos Aires, Argentina
Research Site
Los Polvorines, Buenos Aires, Argentina
Research Site
Martinez, Buenos Aires, Argentina
Research Site
Quilmes, Buenos Aires, Argentina
Research Site
Ramos Mejia, Buenos Aires, Argentina
Research Site
Capital Federal, Argentina
Colombia
Research Site
Medellin, Antioquia, Colombia
Research Site
Barranquilla, Atlantico, Colombia
Research Site
Manizales, Caldas, Colombia
Research Site
Armenia, Quindio, Colombia
Research Site
Cali, Valle Del Cauca, Colombia
Uruguay
Research Site
Montevideo, Uruguay
Venezuela
Research Site
Barquisimeto, Venezuela
Research Site
Caracas, Venezuela
Research Site
Maracaibo, Venezuela
Research Site
Porlamar, Venezuela
Research Site
Valencia, Venezuela Less << |
|
NCT01362530
|
Chemotherapy Induced Nausea an... More >>d Vomiting Less << |
Phase 3 |
Completed |
- |
- |
|
NCT02783794
|
Metastatic Breast Cancer
... More >> Stage IV Breast Cancer Less << |
Phase 2 |
Completed |
- |
Russian Federation
... More >>
Russian Oncological Research Centre n.a. N.N. Blokhin, Russian Academy of Medical Science (RAMS)
Moscow, Russian Federation
State Budgetary Institution of Nizhniy Novgorod Region "Oncology Dispensary of Nizhniy Novgorod" (Branch nr.1)
Nizhniy Novgorod, Russian Federation
St. Petersburg State Budgetary Health Organization, City Clinical Oncology Dispensary
St Petersburg, Russian Federation
Leningrad Regional Oncological Centre
St. Petersburg, Russian Federation
Thailand
Siriraj Hospital, Mahidol University
Bangkok, Thailand
Udon Thani Cancer Hospital
Udon Thani, Thailand Less << |
|
NCT03072238
|
Metastatic Prostate Cancer |
Phase 3 |
Recruiting |
November 18, 2023 |
- |
|
NCT01726582
|
Pancreatic Adenocarcinoma |
Phase 2 |
Active, not recruiting |
December 2021 |
United States, Ohio
... More >>
University of Cincinnati Cancer Center
Cincinnati, Ohio, United States, 45221
United States, Wisconsin
Froedtert and The Medical College of Wisconsin
Milwaukee, Wisconsin, United States, 53226 Less << |
|
NCT01362530
|
- |
|
Completed |
- |
- |
|
NCT02630004
|
Oral Mucositis |
Phase 1
Phase 2 |
Completed |
- |
Spain
... More >> Institut Català d'Oncologia ICO Badalona
Badalona, Barcelona, Spain, 08916
Institut Català d'Oncologia L'Hospitalet
L'Hospitalet de Llobregat, Barcelona, Spain, 08907
Hospital Clínico Universitario de Santiago
Santiago de Compostela, La Coruña, Spain, 15706
Hospital de la Santa Creu i Sant Pau
Barcelona, Spain, 08026
Hospital Universitari de la Vall d'Hebron
Barcelona, Spain, 08035
Institut Català d'Oncologia Girona
Girona, Spain, 17007
Hospital San Carlos, Madrid
Madrid, Spain, 28040
Hospital Universitario La Paz
Madrid, Spain, 28046
Hospital Universitario Virgen de la Victoria
Málaga, Spain, 29010
Hospital Universitario Marqués de Valdecilla
Santander, Spain, 39008
Hospital Miguel Servet
Zaragoza, Spain, 50009 Less << |
|
NCT03206450
|
- |
|
Recruiting |
September 5, 2021 |
- |
|
NCT02863003
|
- |
|
Not yet recruiting |
December 2020 |
Brazil
... More >> CPO - Pucrs
Not yet recruiting
Porto Alegre, Rio Grande do Sul, Brazil
Contact: Virginia Webber
Principal Investigator: André Fay, MD
Hospital Israelita Albert Einstein
Not yet recruiting
São Paulo, Brazil
Contact: Fernanda Cardoso
Principal Investigator: Oren Smaletz, MD
Hospital São José
Not yet recruiting
São Paulo, Brazil
Contact: Cristiane Ibrahim
Principal Investigator: Fábio Schutz, MD
ICESP
Not yet recruiting
São Paulo, Brazil
Contact: Bárbara Penna
Principal Investigator: Diogo Bastos, MD
Principal Investigator: Carlos Dzik, MD Less << |
|
NCT00131638
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT02723604
|
Mucositis |
Not Applicable |
Recruiting |
March 2018 |
United States, Michigan
... More >>
Wayne State University/Karmanos Cancer Institute
Recruiting
Detroit, Michigan, United States, 48201
Contact: Harold E. Kim 313-576-9543 kimh@karmanos.org
Principal Investigator: Harold E. Kim, M.D.
Sub-Investigator: Mark A. Zaki, M.D.
Sub-Investigator: Pamela Laszewski, R.N. Less << |
|
NCT02577172
|
Testicular Neoplasms |
Not Applicable |
Terminated(Unexpected events) |
- |
Norway
... More >> Oslo University Hospital
Oslo, Norway Less << |
|
NCT03170960
|
Urothelial Carcinoma
... More >> Renal Cell Carcinoma
Non-Small Cell Lung Cancer
Castration-resistant Prostate Cancer
Triple Negative Breast Cancer
Ovarian Cancer
Endometrial Cancer
Hepatocellular Carcinoma
Gastric Cancer
Gastroesophageal Junction Adenocarcinoma
Colorectal Cancer
Head and Neck Cancer
Differentiated Thyroid Cancer Less << |
Phase 1
Phase 2 |
Recruiting |
December 2020 |
- |
|
NCT03328234
|
IMRT With or Without Concurren... More >>t Chemotherapy for Esophageal Cancer Less << |
Phase 3 |
Recruiting |
December 31, 2022 |
China
... More >> Department of Radiation Oncology, Cancer Hospital, National Cancer Center, Chinese Academy of Medical Sciences (CAMS) and Peking Union Medical College (PUMC)
Recruiting
Beijing, China, 100021
Contact: Xin Wang, MD +861013311583220 beryl_wx2000@163.com Less << |
|
NCT01951586
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00558220
|
Diffuse Large B-Cell Lymphoma.... More >>
Primary Mediastinal B-Cell Lymphoma
Follicular Lymphoma Grade III Less << |
Phase 2 |
Completed |
- |
Czech Republic
... More >> University Hospital Brno-Bohunice
Brno, Czech Republic, 625 00
Hospital Chomutov
Chomutov, Czech Republic, 430 12
University Hospital Hradec Králové
Hradec Králové, Czech Republic, 500 05
University Hospital Královské Vinohrady
Prague, Czech Republic, 100 34
General University Hospital
Prague, Czech Republic, 128 08
University Hospital Motol
Prague, Czech Republic, 150 00
Hospital Ústí nad Labem
Usti nad Labem, Czech Republic, 401 13
Hospital České Budějovice
České Budějovice, Czech Republic Less << |
|
NCT02750657
|
- |
|
Recruiting |
December 2021 |
Canada, Ontario
... More >>
Kingston Health Sciences Centre
Recruiting
Kingston, Ontario, Canada, K7L2V7
Contact: Jim Biagi, M.D. 613-549-6666 ext 8105
Princess Margaret Cancer Centre
Recruiting
Toronto, Ontario, Canada, M5G 2M9
Contact: Jennifer J. Knox, M.D. 416-946-2399
Principal Investigator: Jennifer J. Knox, M.D.
Canada, Quebec
McGill University Health Centre
Recruiting
Montréal, Quebec, Canada, H4A3J1
Contact: George Zogopoulos, M.D. 514-934-1934 ext 76333 Less << |
|
NCT00827424
|
Obesity |
Not Applicable |
Completed |
- |
Israel
... More >> Clinical Research Unit, Clalit Health Services, Haifa and Western Galilee District .
Haifa,, Israel, 35024 Less << |
|
NCT00119613
|
Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00089453
|
Multiple Myeloma |
Phase 1 |
Completed |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences/MIRT
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT03034850
|
- |
|
Completed |
- |
- |
|
NCT01951586
|
- |
|
Completed |
- |
- |
|
NCT00412425
|
- |
|
Completed |
- |
- |
|
NCT00412425
|
Melanoma |
Phase 1
Phase 2 |
Completed |
- |
United States, Texas
... More >>
U.T. M.D. Anderson Cancer Center
Houston, Texas, United States, 77030 Less << |
|
NCT00083876
|
Multiple Myeloma |
Phase 3 |
Completed |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences/MIRT
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT01451853
|
Lung Cancer
H... More >>ead and Neck Cancer
Hearing Loss
Ototoxicity
Tinnitus
Neuropathy Less << |
Phase 2 |
Not yet recruiting |
September 23, 2019 |
United States, Washington
... More >>
VA Puget Sound Health Care
Not yet recruiting
Seattle, Washington, United States, 98108
Contact: Tony S. Quang, M.D. 206-768-5356
Principal Investigator: Tony S Quang, M.D. Less << |
|
NCT01037790
|
Adult Solid Tumor
... More >> Adenocarcinoma of the Colon
Adenocarcinoma of the Rectum
Adult Central Nervous System Germ Cell Tumor
Adult Teratoma
Benign Teratoma
Estrogen Receptor-negative Breast Cancer
Estrogen Receptor-positive Breast Cancer
Familial Testicular Germ Cell Tumor
HER2-negative Breast Cancer
HER2-positive Breast Cancer
Male Breast Cancer
Ovarian Immature Teratoma
Ovarian Mature Teratoma
Ovarian Monodermal and Highly Specialized Teratoma
Progesterone Receptor-negative Breast Cancer
Progesterone Receptor-positive Breast Cancer
Recurrent Breast Cancer
Recurrent Colon Cancer
Recurrent Extragonadal Germ Cell Tumor
Recurrent Extragonadal Non-seminomatous Germ Cell Tumor
Recurrent Extragonadal Seminoma
Recurrent Malignant Testicular Germ Cell Tumor
Recurrent Melanoma
Recurrent Ovarian Germ Cell Tumor
Recurrent Rectal Cancer
Stage III Extragonadal Non-seminomatous Germ Cell Tumor
Stage III Extragonadal Seminoma
Stage III Malignant Testicular Germ Cell Tumor
Stage III Ovarian Germ Cell Tumor
Stage IV Breast Cancer
Stage IV Colon Cancer
Stage IV Extragonadal Non-seminomatous Germ Cell Tumor
Stage IV Extragonadal Seminoma
Stage IV Melanoma
Stage IV Ovarian Germ Cell Tumor
Stage IV Rectal Cancer
Testicular Immature Teratoma
Testicular Mature Teratoma Less << |
Phase 2 |
Recruiting |
- |
United States, Pennsylvania
... More >>
Abramson Cancer Center of The University of Pennsylvania
Recruiting
Philadelphia, Pennsylvania, United States, 19104
Contact: Peter ODwyer 855-216-0098 PennCancerTrials@emergingmed.com
Principal Investigator: Peter ODwyer Less << |
|
NCT01887314
|
Cancer
Nausea... More >>
Vomiting Less << |
Not Applicable |
Completed |
- |
Italy
... More >> Istituto Nazionale dei Tumori
Milan, Italy, 20133
IEO -Istituto Europeo di Oncologia-
Milan, Italy, 20141
Ospedale S. Gerardo
Monza, Italy, 20052
Policlinico Umberto I
Rome, Italy, 00186
Istituti Fisioterapici Ospitalieri - Istituto Nazionale Tumori "Regina Elena"
Rome, Italy
Ospedale S. Maria
Terni, Italy, 05100 Less << |
|
NCT01016028
|
- |
|
Withdrawn(Study assessment too... More >>l development delayed.) Less << |
- |
- |
|
NCT02381782
|
- |
|
Completed |
- |
Belgium
... More >> General Hospital Groeninge
Kortrijk, Belgium Less << |
|
NCT01941680
|
- |
|
Active, not recruiting |
March 2021 |
Guadeloupe
... More >> CHU Pinteà Pitre/Abymes
Pointe à Pitre, Guadeloupe
Martinique
CHU de Martinique
Fort de France, Martinique Less << |
|
NCT02658214
|
Small Cell Lung Carcinoma
... More >> Carcinoma, Squamous Cell of Head and Neck
Stomach Neoplasms
Triple Negative Breast Neoplasms
Ovarian Neoplasms
Fallopian Tube Neoplasms
Peritoneal Neoplasms
Esophagogastric Junction Neoplasms
Carcinoma, Pancreatic Ductal
Esophageal Squamous Cell Carcinoma Less << |
Phase 1 |
Recruiting |
June 28, 2019 |
United States, Georgia
... More >>
Research Site
Withdrawn
Augusta, Georgia, United States, 30912
United States, Pennsylvania
Research Site
Withdrawn
Pittsburgh, Pennsylvania, United States, 15232
Japan
Research Site
Recruiting
Chuo-ku, Japan, 104-0045
Research Site
Recruiting
Kashiwa, Japan, 277-8577
Korea, Republic of
Research Site
Recruiting
Seoul, Korea, Republic of, 03080
Research Site
Recruiting
Seoul, Korea, Republic of, 03722
Research Site
Recruiting
Seoul, Korea, Republic of, 05505
Research Site
Recruiting
Seoul, Korea, Republic of, 135-710 Less << |
|
NCT00040872
|
Neuroblastoma |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00302003
|
Childhood Favorable Prognosis ... More >>Hodgkin Lymphoma
Childhood Lymphocyte Depletion Hodgkin Lymphoma
Childhood Mixed Cellularity Hodgkin Lymphoma
Childhood Nodular Sclerosis Hodgkin Lymphoma
Stage I Childhood Hodgkin Lymphoma
Stage II Childhood Hodgkin Lymphoma Less << |
Phase 3 |
Unknown |
- |
United States, California
... More >>
Children's Oncology Group
Arcadia, California, United States, 91006-3776 Less << |
|
NCT03689712
|
Oral Mucositis |
Phase 3 |
Recruiting |
October 2024 |
United States, Washington
... More >>
Cancer Care NW
Recruiting
Spokane, Washington, United States, 99216 Less << |
|
NCT00513383
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 1 |
Completed |
- |
United States, Massachusetts
... More >>
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115
Massachusetts General Hospital
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00003397
|
Lymphoma |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
Marlene & Stewart Greenebaum Cancer Center, University of Maryland
Baltimore, Maryland, United States, 21201 Less << |
|
NCT01336270
|
- |
|
Completed |
- |
France
... More >> Cochin Hospital
Paris, Ile de France, France, 75014 Less << |
|
NCT00653068
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00165139
|
Neuroblastoma
... More >> Ewings Sarcoma
Non-rhabdomyosarcoma Soft Tissue Sarcoma Less << |
Phase 2 |
Completed |
- |
United States, Massachusetts
... More >>
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115 Less << |
|
NCT00017368
|
Neuroblastoma |
Phase 2 |
Completed |
- |
United States, Georgia
... More >>
AFLAC Cancer Center and Blood Disorders Service of Children's Healthcare of Atlanta - Scottish RiteCampus
Atlanta, Georgia, United States, 30342
United States, Massachusetts
Floating Hospital for Children
Boston, Massachusetts, United States, 02111
Dana-Farber/Harvard Cancer Center at Dana Farber Cancer Institute
Boston, Massachusetts, United States, 02115
United States, Oregon
CCOP - Columbia River Oncology Program
Portland, Oregon, United States, 97225
United States, Pennsylvania
Children's Hospital of Philadelphia
Philadelphia, Pennsylvania, United States, 19104
United States, Texas
Baylor College of Medicine
Houston, Texas, United States, 77030
CCOP - Scott and White Hospital
Temple, Texas, United States, 76508
United States, Wisconsin
CCOP - Marshfield Clinic Research Foundation
Marshfield, Wisconsin, United States, 54449
Australia, Western Australia
Princess Margaret Hospital for Children
Perth, Western Australia, Australia, 6001 Less << |
|
NCT00002596
|
Childhood Germ Cell Tumor
... More >> Extragonadal Germ Cell Tumor
Testicular Germ Cell Tumor Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00003086
|
Breast Cancer |
Phase 1
Phase 2 |
Unknown |
- |
United States, Louisiana
... More >>
Louisiana State University School of Medicine
Shreveport, Louisiana, United States, 71130-3932 Less << |
|
NCT00006123
|
Breast Cancer |
Phase 1
Phase 2 |
Withdrawn(Never started) |
- |
United States, Massachusetts
... More >>
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02215
United States, New Jersey
Hackensack University Medical Center
Hackensack, New Jersey, United States, 07601 Less << |
|
NCT00003386
|
Ovarian Cancer |
Phase 2 |
Terminated |
- |
- |
|
NCT00302003
|
- |
|
Unknown |
- |
- |
|
NCT00004900
|
Breast Cancer |
Phase 2 |
Completed |
- |
United States, Illinois
... More >>
Robert H. Lurie Comprehensive Cancer Center, Northwestern University
Chicago, Illinois, United States, 60611 Less << |
|
NCT00012051
|
Lymphoma |
Phase 3 |
Completed |
- |
Belgium
... More >> U.Z. Gasthuisberg
Leuven, Belgium, B-3000
Netherlands
HagaZiekenhuis - Locatie Leyenburg
's-Gravenhage, Netherlands, 2545 CH
Jeroen Bosch Ziekenhuis
's-Hertogenbosch, Netherlands, 5211 NL
Meander Medisch Centrum
Amersfoort, Netherlands, 3816 CP
Netherlands Cancer Institute - Antoni van Leeuwenhoek Hospital
Amsterdam, Netherlands, 1066 BE
Vrije Universiteit Medisch Centrum
Amsterdam, Netherlands, 1081HV
Academisch Medisch Centrum at University of Amsterdam
Amsterdam, Netherlands, 1105 AZ
Medisch Spectrum Twente
Enschede, Netherlands, 7500 KA
University Medical Center Groningen
Groningen, Netherlands, 9713 EZ
Medisch Centrum Leeuwarden - Zuid
Leeuwarden, Netherlands, 8934 AD
Leiden University Medical Center
Leiden, Netherlands, 2300 CA
Academisch Ziekenhuis Maastricht
Maastricht, Netherlands, 6202 AZ
Sint Antonius Ziekenhuis
Nieuwegein, Netherlands, 3435 CM
Universitair Medisch Centrum St. Radboud - Nijmegen
Nijmegen, Netherlands, NL-6500 HB
Daniel Den Hoed Cancer Center at Erasmus Medical Center
Rotterdam, Netherlands, 3008 AE
University Medical Center Utrecht
Utrecht, Netherlands, 3584 CX
Isala Klinieken - locatie Sophia
Zwolle, Netherlands, 8000 GK Less << |
|
NCT00238368
|
Lymphoma |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Baltimore, Maryland, United States, 21231-2410 Less << |
|
NCT00007982
|
Brain and Central Nervous Syst... More >>em Tumors
Head and Neck Cancer
Lymphoma Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Herbert Irving Comprehensive Cancer Center at Columbia University Medical Center
New York, New York, United States, 10032 Less << |
|
NCT00002772
|
Breast Cancer |
Phase 3 |
Terminated(poor accrual) |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00004906
|
Breast Cancer |
Phase 2 |
Completed |
- |
United States, Illinois
... More >>
Robert H. Lurie Comprehensive Cancer Center, Northwestern University
Chicago, Illinois, United States, 60611
United States, New Jersey
Hackensack University Medical Center
Hackensack, New Jersey, United States, 07601
United States, Ohio
Ireland Cancer Center
Cleveland, Ohio, United States, 44106-5065
United States, Pennsylvania
University of Pennsylvania Cancer Center
Philadelphia, Pennsylvania, United States, 19104 Less << |
|
NCT00017225
|
Neuroblastoma |
Phase 2 |
Completed |
- |
- |
|
NCT00024193
|
Neuroblastoma |
Phase 2 |
Unknown |
- |
France
... More >> Institut Gustave Roussy
Villejuif, France, F-94805 Less << |
|
NCT00653068
|
Childhood Atypical Teratoid/Rh... More >>abdoid Tumor Less << |
Phase 3 |
Active, not recruiting |
- |
- |
|
NCT02613286
|
Uterine Cervical Neoplasms |
Phase 2 |
Unknown |
June 2018 |
- |
|
NCT00004903
|
Multiple Myeloma and Plasma Ce... More >>ll Neoplasm Less << |
Phase 2 |
Unknown |
- |
United States, Illinois
... More >>
Robert H. Lurie Comprehensive Cancer Center, Northwestern University
Chicago, Illinois, United States, 60611
University of Chicago Cancer Research Center
Chicago, Illinois, United States, 60637 Less << |
|
NCT00003273
|
Brain and Central Nervous Syst... More >>em Tumors
Neuroblastoma
Retinoblastoma
Sarcoma Less << |
Phase 2 |
Withdrawn |
- |
United States, Hawaii
... More >>
Cancer Research Center of Hawaii
Honolulu, Hawaii, United States, 96813
United States, Maine
Maine Children's Cancer Program
Scarborough, Maine, United States, 04074-9308
United States, Michigan
Spectrum Health and DeVos Children's Hospital
Grand Rapids, Michigan, United States, 49503
United States, Nebraska
Children's Hospital of Omaha
Omaha, Nebraska, United States, 68114
United States, New Jersey
Hackensack University Medical Center
Hackensack, New Jersey, United States, 07601
United States, New York
Albert Einstein Clinical Cancer Center
Bronx, New York, United States, 10461
Winthrop University Hospital
Mineola, New York, United States, 11501
NYU School of Medicine's Kaplan Comprehensive Cancer Center
New York, New York, United States, 10016
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021
Herbert Irving Comprehensive Cancer Center
New York, New York, United States, 10032
Beth Israel Hospital North
New York, New York, United States, 10128
State University of New York Health Sciences Center - Stony Brook
Stony Brook, New York, United States, 11790-7775
State University of New York - Upstate Medical University
Syracuse, New York, United States, 13210
United States, Ohio
St. Vincent Mercy Medical Center
Toledo, Ohio, United States, 43608
United States, Pennsylvania
Milton S. Hershey Medical Center
Hershey, Pennsylvania, United States, 17033-0850
Children's Hospital of Philadelphia
Philadelphia, Pennsylvania, United States, 19104
United States, Wisconsin
University of Wisconsin Comprehensive Cancer Center
Madison, Wisconsin, United States, 53792-6164
Argentina
Children's Hospital
Buenos Aires, Argentina, 1425
Canada, British Columbia
British Columbia Children's Hospital
Vancouver, British Columbia, Canada, V6H 3V4 Less << |
|
NCT00002675
|
Retinoblastoma |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00025636
|
Lymphoma |
Phase 3 |
Unknown |
- |
- |
|
NCT00002634
|
Neuroblastoma |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT00920153
|
Lymphoma |
Phase 3 |
Terminated(Other new drugs) |
- |
France
... More >> FILO French Innovative Leukemia Organization
Tours Cedex, France, 37044 Less << |
|
NCT01172912
|
Brain and Central Nervous Syst... More >>em Tumors
Extragonadal Germ Cell Tumor
Testicular Germ Cell Tumor Less << |
Phase 2 |
Unknown |
- |
Italy
... More >> Fondazione Istituto Nazionale dei Tumori
Recruiting
Milan, Italy, 20133
Contact: Contact Person 39-02-2390-2532 alessandro.gianni@unimi.it Less << |
|
NCT00025064
|
Lymphoma |
Phase 2 |
Unknown |
- |
Ireland
... More >> Our Lady's Hospital for Sick Children
Dublin, Ireland, 12
United Kingdom
Birmingham Children's Hospital
Birmingham, England, United Kingdom, B4 6NH
Bristol Royal Hospital for Children
Bristol, England, United Kingdom, BS2 8BJ
Addenbrooke's Hospital at Cambridge University Hospitals NHS Foundation Trust
Cambridge, England, United Kingdom, CB2 2QQ
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Royal Liverpool Children's Hospital, Alder Hey
Liverpool, England, United Kingdom, L12 2AP
Saint Bartholomew's Hospital
London, England, United Kingdom, EC1A 7BE
Great Ormond Street Hospital for Children NHS Trust
London, England, United Kingdom, WC1N 3JH
Meyerstein Institute of Oncology at University College of London Hospitals
London, England, United Kingdom, WIT 3AA
Central Manchester and Manchester Children's University Hospitals NHS Trust
Manchester, England, United Kingdom, M27 4HA
Newcastle Upon Tyne Hospitals NHS Trust
Newcastle-Upon-Tyne, England, United Kingdom, NE7 7DN
Queen's Medical Centre
Nottingham, England, United Kingdom, NG7 2UH
Oxford Radcliffe Hospital
Oxford, England, United Kingdom, 0X3 9DU
Children's Hospital - Sheffield
Sheffield, England, United Kingdom, S10 2TH
Southampton General Hospital
Southampton, England, United Kingdom, SO16 6YD
Royal Marsden NHS Foundation Trust - Surrey
Sutton, England, United Kingdom, SM2 5PT
Royal Belfast Hospital for Sick Children
Belfast, Northern Ireland, United Kingdom, BT12 6BE
Aberdeen Royal Infirmary
Aberdeen, Scotland, United Kingdom, AB25 2ZN
Royal Hospital for Sick Children
Edinburgh, Scotland, United Kingdom, EH9 1LF
Royal Hospital for Sick Children
Glasgow, Scotland, United Kingdom, G3 8SJ Less << |
|
NCT02551159
|
Squamous Cell Carcinoma of the... More >> Head and Neck Less << |
Phase 3 |
Active, not recruiting |
December 31, 2018 |
- |
|
NCT00003159
|
Lung Cancer |
Phase 3 |
Completed |
- |
United Kingdom
... More >> Medical Research Council Clinical Trials Unit
London, England, United Kingdom, NW1 2DA
Royal Marsden NHS Foundation Trust - Surrey
Sutton, England, United Kingdom, SM2 5PT Less << |
|
NCT00003413
|
Ovarian Cancer |
Phase 2 |
Completed |
- |
United States, Maryland
... More >>
Marlene & Stewart Greenebaum Cancer Center, University of Maryland
Baltimore, Maryland, United States, 21201 Less << |
|
NCT00003042
|
Breast Cancer |
Phase 2 |
Active, not recruiting |
March 2019 |
United States, California
... More >>
City of Hope Comprehensive Cancer Center
Duarte, California, United States, 91010-3000 Less << |
|
NCT00019864
|
Cardiac Toxicity
... More >> Sarcoma Less << |
Phase 2 |
Terminated |
- |
United States, Maryland
... More >>
Warren Grant Magnuson Clinical Center - NCI Clinical Trials Referral Office
Bethesda, Maryland, United States, 20892-1182
United States, Oklahoma
Oklahoma University Cancer Institute
Oklahoma City, Oklahoma, United States, 73104
United States, Texas
Cook Children's Medical Center - Fort Worth
Fort Worth, Texas, United States, 76104
Texas Children's Cancer Center and Hematology Service at Texas Children's Hospital
Houston, Texas, United States, 77030-2399 Less << |
|
NCT00410631
|
Neuroblastoma |
Phase 3 |
Unknown |
- |
- |
|
NCT00006258
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 2 |
Completed |
- |
United States, California
... More >>
Children's Hospital Los Angeles
Los Angeles, California, United States, 90027-0700
United States, Pennsylvania
Geisinger Medical Center
Danville, Pennsylvania, United States, 17822-1320
Canada, Alberta
Tom Baker Cancer Centre - Calgary
Calgary, Alberta, Canada, T2N 4N2 Less << |
|
NCT00055887
|
Lung Cancer |
Phase 3 |
Withdrawn(Study never started.... More >> No patients were enrolled.) Less << |
- |
United States, Arizona
... More >>
St. Joseph's Hospital and Medical Center
Phoenix, Arizona, United States, 85013
United States, Idaho
North Idaho Cancer Center
Coeur d'Alene, Idaho, United States, 83814
United States, Kentucky
Cancer Center at Lexington Clinic
Lexington, Kentucky, United States, 40504
United States, Louisiana
Willis - Knighton Cancer Center
Shreveport, Louisiana, United States, 71103-3951
United States, Maryland
St. Agnes Cancer Center
Baltimore, Maryland, United States, 21229
Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Baltimore, Maryland, United States, 21231-2410
United States, Washington
Providence Everett Medical Center - Pacific Campus
Everett, Washington, United States, 98206
United States, West Virginia
Schiffler Cancer Center
Wheeling, West Virginia, United States, 26003
Belgium
Algemeen Ziekenhuis Middelheim
Antwerp, Belgium, 2020
Canada, Alberta
Tom Baker Cancer Center - Calgary
Calgary, Alberta, Canada, T2N 4N2
Cross Cancer Institute
Edmonton, Alberta, Canada, T6G 1Z2
Canada, Ontario
Cancer Care Ontario-London Regional Cancer Centre
London, Ontario, Canada, N6A 4L6
Ottawa Regional Cancer Centre
Ottawa, Ontario, Canada, K1H 1C4
Canada, Quebec
CHUS-Hopital Fleurimont
Fleurimont, Quebec, Canada, J1H 5N4
Maisonneuve-Rosemont Hospital
Montreal, Quebec, Canada, H1T 2M4
Hopital Notre- Dame du CHUM
Montreal, Quebec, Canada, H2L 4M1
McGill University
Montreal, Quebec, Canada, H2W 1S6
Centre Hospitalier Universitaire de Quebec
Quebec City, Quebec, Canada, G1R 2J6
Israel
Soroka University Medical Center
Beer-Sheva, Israel, 84101
Rambam Medical Center
Haifa, Israel, 31096
Sheba Medical Center
Tel Hashomer, Israel, 52621
Tel-Aviv Sourasky Medical Center
Tel-Aviv, Israel, 64239 Less << |
|
NCT00274950
|
Childhood Germ Cell Tumor
... More >> Extragonadal Germ Cell Tumor
Ovarian Cancer Less << |
Phase 3 |
Unknown |
- |
Ireland
... More >> Our Lady's Hospital for Sick Children Crumlin
Dublin, Ireland, 12
United Kingdom
Birmingham Children's Hospital
Birmingham, England, United Kingdom, B4 6NH
Institute of Child Health at University of Bristol
Bristol, England, United Kingdom, BS2 8AE
Addenbrooke's Hospital
Cambridge, England, United Kingdom, CB2 2QQ
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Royal Liverpool Children's Hospital, Alder Hey
Liverpool, England, United Kingdom, L12 2AP
Royal London Hospital
London, England, United Kingdom, E1 1BB
Great Ormond Street Hospital for Children
London, England, United Kingdom, WC1N 3JH
Royal Manchester Children's Hospital
Manchester, England, United Kingdom, M27 4HA
Sir James Spence Institute of Child Health at Royal Victoria Infirmary
Newcastle-Upon-Tyne, England, United Kingdom, NE1 4LP
Queen's Medical Centre
Nottingham, England, United Kingdom, NG7 2UH
Children's Hospital - Sheffield
Sheffield, England, United Kingdom, S10 2TH
Southampton General Hospital
Southampton, England, United Kingdom, SO16 6YD
Royal Marsden - Surrey
Sutton, England, United Kingdom, SM2 5PT
Royal Belfast Hospital for Sick Children
Belfast, Northern Ireland, United Kingdom, BT12 6BE
Royal Aberdeen Children's Hospital
Aberdeen, Scotland, United Kingdom, AB25 2ZG
Royal Hospital for Sick Children
Edinburgh, Scotland, United Kingdom, EH9 1LF
Royal Hospital for Sick Children
Glasgow, Scotland, United Kingdom, G3 8SJ
Childrens Hospital for Wales
Cardiff, Wales, United Kingdom, CF14 4XW Less << |
|
NCT00024271
|
Malignant Mesothelioma |
Phase 2 |
Unknown |
- |
United States, New York
... More >>
Herbert Irving Comprehensive Cancer Center at Columbia University
New York, New York, United States, 10032 Less << |
|
NCT00023868
|
Colorectal Cancer
... More >> Metastatic Cancer Less << |
Phase 3 |
Completed |
- |
United States, Arizona
... More >>
Arizona Cancer Center
Tucson, Arizona, United States, 85724
United States, Florida
H. Lee Moffitt Cancer Center and Research Institute
Tampa, Florida, United States, 33612-9497
United States, Illinois
Robert H. Lurie Comprehensive Cancer Center, Northwestern University
Chicago, Illinois, United States, 60611-3013
United States, Maryland
Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Baltimore, Maryland, United States, 21231-2410
United States, Massachusetts
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02215
United States, New York
NYU School of Medicine's Kaplan Comprehensive Cancer Center
New York, New York, United States, 10016
State University of New York - Upstate Medical University
Syracuse, New York, United States, 13210
United States, Pennsylvania
University of Pennsylvania Cancer Center
Philadelphia, Pennsylvania, United States, 19104-4283
United States, Texas
University of Texas - MD Anderson Cancer Center
Houston, Texas, United States, 77030-4009 Less << |
|
NCT00003309
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 2 |
Completed |
- |
United States, Florida
... More >>
H. Lee Moffitt Cancer Center and Research Institute
Tampa, Florida, United States, 33612-9497
United States, Illinois
Veterans Affairs Medical Center - Lakeside Chicago
Chicago, Illinois, United States, 60611-4494
Robert H. Lurie Comprehensive Cancer Center at Northwestern University
Chicago, Illinois, United States, 60611
CCOP - Carle Cancer Center
Urbana, Illinois, United States, 61801
United States, Iowa
CCOP - Cedar Rapids Oncology Project
Cedar Rapids, Iowa, United States, 52403-1206
CCOP - Iowa Oncology Research Association
Des Moines, Iowa, United States, 50309-1016
United States, Kansas
CCOP - Wichita
Wichita, Kansas, United States, 67214-3882
United States, Michigan
CCOP - Kalamazoo
Kalamazoo, Michigan, United States, 49007-3731
West Michigan Cancer Center
Kalamazoo, Michigan, United States, 49007
United States, Minnesota
CCOP - Metro-Minnesota
Saint Louis Park, Minnesota, United States, 55416
United States, New Hampshire
Norris Cotton Cancer Center at Dartmouth-Hitchcock Medical Center
Lebanon, New Hampshire, United States, 03756-0002
United States, Tennessee
Vanderbilt-Ingram Cancer Center at Vanderbilt Medical Center
Nashville, Tennessee, United States, 37232-6307
United States, Wisconsin
CCOP - Marshfield Clinic Research Foundation
Marshfield, Wisconsin, United States, 54449
Medical College of Wisconsin Cancer Center
Milwaukee, Wisconsin, United States, 53226-3596 Less << |
|
NCT00002822
|
Lung Cancer |
Phase 3 |
Completed |
- |
United Kingdom
... More >> Medical Research Council Clinical Trials Unit
London, England, United Kingdom, NW1 2DA Less << |
|
NCT00652860
|
Metastatic Cancer
... More >> Sarcoma Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00023998
|
Metastatic Osteosarcoma |
Phase 2 |
Completed |
- |
United States, California
... More >>
Children's Oncology Group
Arcadia, California, United States, 91006-3776 Less << |
|
NCT00070200
|
Neuroblastoma |
Phase 1 |
Completed |
- |
United States, California
... More >>
UCSF Comprehensive Cancer Center
San Francisco, California, United States, 94143-0106
United States, Illinois
Children's Memorial Hospital - Chicago
Chicago, Illinois, United States, 60614
United States, Tennessee
St. Jude Children's Research Hospital
Memphis, Tennessee, United States, 38105
United States, Washington
Children's Hospital and Regional Medical Center - Seattle
Seattle, Washington, United States, 98105
Mary Bridge Children's Hospital and Health Center - Tacoma
Tacoma, Washington, United States, 98405
Australia, New South Wales
Westmead Hospital
Westmead, New South Wales, Australia, 2145 Less << |
|
NCT00526318
|
Neuroblastoma |
Not Applicable |
Unknown |
- |
- |
|
NCT00577096
|
Multiple Myeloma |
Not Applicable |
Completed |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT00577096
|
- |
|
Completed |
- |
- |
|
NCT00002802
|
Neuroblastoma |
Phase 3 |
Completed |
- |
Germany
... More >> University of Cologne
Frechen, Germany, DOH-5-0226 Less << |
|
NCT00646659
|
Head and Neck Cancer |
Phase 2 |
Terminated(Recruitment was sus... More >>pended prematurely for safety concerns and closed after IDMC review) Less << |
- |
Belgium
... More >> Universitair Ziekenhuis Antwerpen
Edegem, Belgium, B-2650 Less << |
|
NCT00004086
|
Leukemia
Lymp... More >>homa Less << |
Phase 1 |
Unknown |
- |
United States, New Jersey
... More >>
Garden State Cancer Center
Belleville, New Jersey, United States, 07103 Less << |
|
NCT00003640
|
Bladder Cancer |
Phase 2 |
Terminated(low accrual) |
- |
Italy
... More >> San Raffaele Hospital
Rome, Italy, 00144 Less << |
|
NCT00002566
|
Extragonadal Germ Cell Tumor
... More >> Testicular Germ Cell Tumor Less << |
Phase 3 |
Unknown |
- |
France
... More >> Institut Gustave Roussy
Villejuif, France, F-94805 Less << |
|
NCT00083083
|
Lung Cancer |
Phase 2 |
Unknown |
- |
- |
|
NCT00276731
|
Neuroblastoma |
Phase 3 |
Unknown |
- |
Ireland
... More >> Our Lady's Hospital for Sick Children
Dublin, Ireland, 12
United Kingdom
Birmingham Children's Hospital
Birmingham, England, United Kingdom, B4 6NH
Institute of Child Health at University of Bristol
Bristol, England, United Kingdom, BS2 8AE
Addenbrooke's Hospital at Cambridge University Hospitals NHS Foundation Trust
Cambridge, England, United Kingdom, CB2 2QQ
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Royal Liverpool Children's Hospital, Alder Hey
Liverpool, England, United Kingdom, L12 2AP
Royal London Hospital
London, England, United Kingdom, E1 1BB
Great Ormond Street Hospital for Children NHS Trust
London, England, United Kingdom, WC1N 3JH
Central Manchester and Manchester Children's University Hospitals NHS Trust
Manchester, England, United Kingdom, M27 4HA
Sir James Spence Institute of Child Health
Newcastle-Upon-Tyne, England, United Kingdom, NE1 4LP
Queen's Medical Centre
Nottingham, England, United Kingdom, NG7 2UH
Oxford Radcliffe Hospital
Oxford, England, United Kingdom, 0X3 9DU
Children's Hospital - Sheffield
Sheffield, England, United Kingdom, S10 2TH
Southampton General Hospital
Southampton, England, United Kingdom, SO16 6YD
Royal Marsden NHS Foundation Trust - Surrey
Sutton, England, United Kingdom, SM2 5PT
Royal Belfast Hospital for Sick Children
Belfast, Northern Ireland, United Kingdom, BT12 6BE
Royal Aberdeen Children's Hospital
Aberdeen, Scotland, United Kingdom, AB25 2ZG
Royal Hospital for Sick Children
Edinburgh, Scotland, United Kingdom, EH9 1LF
Beatson Oncology Centre
Glasgow, Scotland, United Kingdom, G11 6NT
Royal Hospital for Sick Children
Glasgow, Scotland, United Kingdom, G3 8SJ
Childrens Hospital for Wales
Cardiff, Wales, United Kingdom, CF14 4XW Less << |
|
NCT00003209
|
Cervical Cancer |
Phase 3 |
Completed |
- |
United Kingdom
... More >> Derbyshire Royal Infirmary
Derby, England, United Kingdom, DE1 2QY
Beatson Oncology Centre
Glasgow, Scotland, United Kingdom, G11 6NT Less << |
|
NCT00003776
|
Sarcoma |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10021 Less << |
|
NCT03668418
|
- |
|
Recruiting |
May 31, 2022 |
Italy
... More >> Azienda Ospedaliero-Universitaria Pisana
Recruiting
Pisa, Italy, 56124
Contact: Luca Morelli, Prof luca.morelli@unipi.it
Sub-Investigator: Vittoria Raffa, Prof
Sub-Investigator: Enrico Vasile, Dr
Sub-Investigator: Gregorio Di Franco, MD
Sub-Investigator: Alice Usai, MSc Less << |
|
NCT00303810
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Not Applicable |
Completed |
- |
Germany
... More >> University Medical Center Hamburg - Eppendorf
Hamburg, Germany, D-20246 Less << |
|
NCT00287950
|
Neuroblastoma |
Not Applicable |
Unknown |
- |
Ireland
... More >> Our Lady's Hospital for Sick Children
Dublin, Ireland, 12
United Kingdom
Birmingham Children's Hospital
Birmingham, England, United Kingdom, B4 6NH
Institute of Child Health at University of Bristol
Bristol, England, United Kingdom, BS2 8AE
Addenbrooke's Hospital at Cambridge University Hospitals NHS Foundation Trust
Cambridge, England, United Kingdom, CB2 2QQ
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Royal Liverpool Children's Hospital, Alder Hey
Liverpool, England, United Kingdom, L12 2AP
Royal London Hospital
London, England, United Kingdom, E1 1BB
Great Ormond Street Hospital for Children NHS Trust
London, England, United Kingdom, WC1N 3JH
Meyerstein Institute of Oncology at University College of London Hospitals
London, England, United Kingdom, WIT 3AA
Central Manchester and Manchester Children's University Hospitals NHS Trust
Manchester, England, United Kingdom, M27 4HA
Sir James Spence Institute of Child Health
Newcastle-Upon-Tyne, England, United Kingdom, NE1 4LP
Queen's Medical Centre
Nottingham, England, United Kingdom, NG7 2UH
Oxford Radcliffe Hospital
Oxford, England, United Kingdom, 0X3 9DU
Children's Hospital - Sheffield
Sheffield, England, United Kingdom, S10 2TH
Southampton General Hospital
Southampton, England, United Kingdom, SO16 6YD
Royal Marsden NHS Foundation Trust - Surrey
Sutton, England, United Kingdom, SM2 5PT
Royal Belfast Hospital for Sick Children
Belfast, Northern Ireland, United Kingdom, BT12 6BE
Royal Aberdeen Children's Hospital
Aberdeen, Scotland, United Kingdom, AB25 2ZG
Royal Hospital for Sick Children
Edinburgh, Scotland, United Kingdom, EH9 1LF
Royal Hospital for Sick Children
Glasgow, Scotland, United Kingdom, G3 8SJ
Childrens Hospital for Wales
Cardiff, Wales, United Kingdom, CF14 4XW Less << |
|
NCT03770299
|
Non-Small Cell Lung Cancer
... More >> Non-Small-Cell Lung Carcinoma
Circulating Tumor DNA Less << |
Phase 2 |
Not yet recruiting |
March 14, 2024 |
United States, Florida
... More >>
Local Institution
Not yet recruiting
Jacksonville, Florida, United States, 32224
Contact: Site 0015
United States, Kentucky
Local Institution
Not yet recruiting
Lexington, Kentucky, United States, 40503-1489
Contact: Site 0014
United States, South Carolina
Local Institution
Not yet recruiting
Charleston, South Carolina, United States, 29414
Contact: Site 0010
United States, Texas
Local Institution
Not yet recruiting
Tyler, Texas, United States, 75701
Contact: Site 0013
Germany
Local Institution
Not yet recruiting
Gauting, Germany, 82131
Contact: Site 0005
Local Institution
Not yet recruiting
Grosshansdorf, Germany, 22927
Contact: Site 0006
Local Institution
Not yet recruiting
Heidelberg, Germany, 69126
Contact: Site 0007
Switzerland
Local Institution
Not yet recruiting
Lausanne, Switzerland, 1011
Contact: Site 0009
Local Institution
Not yet recruiting
Zuerich, Switzerland, 8091
Contact: Site 0008
United Kingdom
Local Institution
Not yet recruiting
London, United Kingdom, NW1 2BU
Contact: Site 0001 Less << |
|
NCT00003862
|
Gastric Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT00683319
|
- |
|
Active, not recruiting |
- |
United Kingdom
... More >> Birmingham Children's Hospital
Birmingham, England, United Kingdom, B4 6NH
Addenbrooke's Hospital
Cambridge, England, United Kingdom, CB2 2QQ
Leeds Cancer Centre at St. James's University Hospital
Leeds, England, United Kingdom, LS9 7TF
Royal Liverpool Children's Hospital, Alder Hey
Liverpool, England, United Kingdom, L12 2AP
Great Ormond Street Hospital for Children
London, England, United Kingdom, WC1N 3JH
Royal Manchester Children's Hospital
Manchester, England, United Kingdom, M27 4HA
Queen's Medical Centre
Nottingham, England, United Kingdom, NG7 2UH
Children's Hospital - Sheffield
Sheffield, England, United Kingdom, S10 2TH
Aberdeen Royal Infirmary
Aberdeen, Scotland, United Kingdom, AB25 2ZN
Royal Hospital for Sick Children
Glasgow, Scotland, United Kingdom, G3 8SJ Less << |
|
NCT00002496
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
United States, Colorado
... More >>
CCOP - Colorado Cancer Research Program, Inc.
Denver, Colorado, United States, 80209-5031
United States, Florida
H. Lee Moffitt Cancer Center and Research Institute
Tampa, Florida, United States, 33612
United States, Georgia
Veterans Affairs Medical Center - Atlanta (Decatur)
Decatur, Georgia, United States, 30033
United States, Illinois
CCOP - Illinois Oncology Research Association
Peoria, Illinois, United States, 61602
CCOP - Carle Cancer Center
Urbana, Illinois, United States, 61801
United States, Indiana
Indiana University Cancer Center
Indianapolis, Indiana, United States, 46202-5265
Veterans Affairs Medical Center - Indianapolis (Roudebush)
Indianapolis, Indiana, United States, 46202
United States, Iowa
CCOP - Iowa Oncology Research Association
Des Moines, Iowa, United States, 50309-1016
United States, Massachusetts
New England Medical Center Hospital
Boston, Massachusetts, United States, 02111
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02215
United States, Michigan
CCOP - Kalamazoo
Kalamazoo, Michigan, United States, 49007-3731
United States, Minnesota
CCOP - Metro-Minnesota
Saint Louis Park, Minnesota, United States, 55416
United States, New Jersey
Veterans Affairs Medical Center - East Orange
East Orange, New Jersey, United States, 07018-1095
CCOP - Northern New Jersey
Hackensack, New Jersey, United States, 07601
United States, Ohio
Ireland Cancer Center
Cleveland, Ohio, United States, 44106-5065
United States, Pennsylvania
Hahnemann University Hospital
Philadelphia, Pennsylvania, United States, 19102-1192
University of Pittsburgh Cancer Institute
Pittsburgh, Pennsylvania, United States, 15213
South Africa
Pretoria Academic Hospital
Pretoria, South Africa, 0001 Less << |
|
NCT00470223
|
Sarcoma |
Phase 3 |
Terminated(IDMC decision upon ... More >>interim efficacy analysis) Less << |
April 2025 |
France
... More >> Centre Paul Papin
Angers, France, 49036
Institut Gustave Roussy
Angers, France, 49036
Centre Hospitalier Regional de Besancon - Hopital Jean Minjoz
Besancon, France, 25030
CHR de Besancon - Hopital Saint-Jacques
Besancon, France, 25030
Institut Bergonie
Bordeaux, France, 33076
CHU Hopital A. Morvan
Brest, France, 29609
CHU de Caen
Caen, France, 14033
Centre Regional Francois Baclesse
Caen, France, 14076
CHR Clermont Ferrand, Hotel Dieu
Clermont-Ferrand, France, 63003
Centre Jean Perrin
Clermont-Ferrand, France, 63011
Centre de Lutte Contre le Cancer Georges-Francois Leclerc
Dijon, France, 21079
Centre Hospitalier Universitaire de Dijon
Dijon, France, 21079
CHU de Grenoble - Hopital Michallon
Grenoble, France, 38043
Centre Oscar Lambret
Lille, France, 59020
Centre Leon Berard
Lyon, France, 69373
Hopital Edouard Herriot - Lyon
Lyon, France, 69437
Marseille Institute of Cancer - Institut J. Paoli and I. Calmettes
Marseille, France, 13273
CHU de la Timone
Marseille, France, 13385
Hopital d'Enfants de la Timone
Marseille, France, 13385
CHU Nord
Marseille, France, 13915
Hopital Arnaud de Villeneuve
Montpellier, France, 34295
Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Montpellier, France, 34298
Centre Regional Rene Gauducheau
Nantes-Saint Herblain, France, 44805
Centre Antoine Lacassagne
Nice, France, 06189
Hopital de l'Archet CHU de Nice
Nice, France, F-06202
Institut Curie Hopital
Paris, France, 75248
Hopital Jean Bernard
Poitiers, France, 86021
Centre Eugene Marquis
Rennes, France, 35042
Hopital Charles Nicolle
Rouen, France, 76031
Centre Henri Becquerel
Rouen, France, 76038
Institut de Cancerologie de la Loire
Saint Priest en Jarez, France, 42270
Hopitaux Universitaire de Strasbourg
Strasbourg, France, 67091
Hopital Universitaire Hautepierre
Strasbourg, France, 67098
Hopital des Enfants
Toulouse, France, 31059
C.H. Bastien de Clocheville
Tours, France, 3700
CHRU de Tours - Hopital Trousseau
Tours, France, 37044
Centre Alexis Vautrin
Vandoeuvre-les-Nancy, France, 54511 Less << |
|
NCT00002887
|
Lung Cancer |
Phase 1 |
Unknown |
- |
Canada, Ontario
... More >>
Ottawa Regional Cancer Centre - General Campus
Ottawa, Ontario, Canada, K1H 1C4 Less << |
|
NCT00629863
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Not Applicable |
Completed |
- |
United Kingdom
... More >> Royal Blackburn Hospital
Blackburn, England, United Kingdom, BB2 3 HH
Gloucestershire Royal Hospital
Gloucester, England, United Kingdom, GL1 3NN
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
North Tyneside Hospital
North Shields, England, United Kingdom, NE29 8NH
Southampton General Hospital
Southampton, England, United Kingdom, SO16 6YD
Aberdeen Royal Infirmary
Aberdeen, Scotland, United Kingdom, AB25 2ZN
Ninewells Hospital
Dundee, Scotland, United Kingdom, DD1 9SY
Royal Infirmary - Castle
Glasgow, Scotland, United Kingdom, G4 0SF
North Wales Organisation for Randomised Trials in Health
Bangor, Wales, United Kingdom, LL57 2PZ Less << |
|
NCT02477826
|
Non-Small Cell Lung Cancer |
Phase 3 |
Recruiting |
December 31, 2020 |
- |
|
NCT03308552
|
Esophageal Neoplasms |
Phase 3 |
Recruiting |
August 30, 2021 |
China, Beijing
... More >> Department of Radiation Oncology, Cancer Hospital, National Cancer Center, Chinese Academy of Medical Sciences (CAMS) and Peking Union Medical College (PUMC)
Recruiting
Beijing, Beijing, China, 100021
Contact: Zefen Xiao, MD Less << |
|
NCT00075699
|
Malignant Mesothelioma |
Phase 3 |
Completed |
- |
United Kingdom
... More >> Leeds General Infirmary at Leeds Teaching Hospital NHS Trust
Leeds, England, United Kingdom, LS1 3EX
Saint Bartholomew's Hospital
London, England, United Kingdom, EC1A 7BE
Royal Marsden NHS Foundation Trust - Surrey
Sutton, England, United Kingdom, SM2 5PT Less << |
|
NCT00190528
|
Cervical Neoplasms |
Phase 3 |
Terminated |
- |
Japan
... More >> National Cancer Center Hospital
Chuo-ku, Tokyo, Japan, 1040045 Less << |
|
NCT02998528
|
Non Small Cell Lung Cancer |
Phase 3 |
Recruiting |
November 8, 2028 |
- |
|
NCT03734952
|
Radiation Oncology |
Not Applicable |
Not yet recruiting |
January 1, 2029 |
- |
|
NCT00365755
|
Neuroblastoma |
Phase 3 |
Completed |
- |
- |
|
NCT03064854
|
Non-small Cell Lung Cancer |
Phase 1 |
Recruiting |
February 15, 2022 |
- |
|
NCT02772107
|
Extensive-stage Small Cell Lun... More >>g Cancer Less << |
Phase 2
Phase 3 |
Unknown |
January 2018 |
China, Beijing
... More >> Chinese PLA General Hospital
Recruiting
Beijing, Beijing, China, 100853
Contact: haitao tao, PhD +861066937875 whatyouknow@126.com Less << |
|
NCT03615326
|
Gastric Neoplasms
... More >> Gastroesophageal Junction Adenocarcinoma Less << |
Phase 3 |
Recruiting |
March 20, 2024 |
- |
|
NCT03574649
|
Non Small Cell Lung Cancer |
Phase 2 |
Not yet recruiting |
December 30, 2020 |
United States, California
... More >>
Chan Soon-Shiong Institute for Medicine
El Segundo, California, United States, 90245 Less << |
|
NCT02013050
|
Oral Mucositis |
Phase 2 |
Completed |
- |
United States, Kentucky
... More >>
Markey Cancer Center-University of Kentucky
Lexington, Kentucky, United States, 40536 Less << |
|
NCT03161522
|
Malignant Neoplasms of Digesti... More >>ve Organs
Esophageal Cancer
Gastric Cancer Less << |
Phase 2 |
Recruiting |
February 2025 |
United States, Texas
... More >>
University of Texas MD Anderson Cancer Center
Recruiting
Houston, Texas, United States, 77030
Contact: Clinical Research Operations CR_Study_Registration@mdanderson.org Less << |
|
NCT03403049
|
Pancreatic Cancer
... More >> Cancer Less << |
Phase 1 |
Recruiting |
December 2019 |
United States, New York
... More >>
Albert Einstein College of Medicine
Not yet recruiting
Bronx, New York, United States, 10461
Contact: Ravindran S. Kathirithamby, MBA 718-430-3697 ravindran.kathirithamby@einstein.yu.edu
China, Shanghai
Shanghai Proton and Heavy Ion Center
Recruiting
Shanghai, Shanghai, China
Contact: Guo-Liang Jiang, M.D. +86-021-38296666-53609 guoliang.jiang@sphic.org.cn Less << |
|
NCT00028756
|
Stage III Bladder Cancer
... More >> Stage IV Bladder Cancer
Transitional Cell Carcinoma of the Bladder Less << |
Phase 3 |
Completed |
- |
- |
|
NCT02013050
|
- |
|
Completed |
- |
- |
|
NCT00436774
|
- |
|
Unknown |
- |
Ireland
... More >> Our Lady's Hospital for Sick Children Crumlin
Recruiting
Dublin, Ireland, 12
Contact: Contact Person 353-1-409-6653
United Kingdom
Institute of Child Health at University of Bristol
Recruiting
Bristol, England, United Kingdom, BS2 8AE
Contact: Contact Person 44-117-342-8811
Addenbrooke's Hospital
Recruiting
Cambridge, England, United Kingdom, CB2 2QQ
Contact: Contact Person 44-1223-256-298
Leeds Cancer Centre at St. James's University Hospital
Recruiting
Leeds, England, United Kingdom, LS9 7TF
Contact: Adam Glaser, MD 44-113-206-4984 adam.glaser@leedsth.nhs.uk
Great Ormond Street Hospital for Children
Recruiting
London, England, United Kingdom, WC1N 3JH
Contact: Gill Levitt, MD 44-20-7405-9200 ext. 0073
Sir James Spence Institute of Child Health at Royal Victoria Infirmary
Recruiting
Newcastle-Upon-Tyne, England, United Kingdom, NE1 4LP
Contact: Juliet Hale, MD 44-191-282-4101 j.p.hale@ncl.ac.uk
Children's Hospital - Sheffield
Recruiting
Sheffield, England, United Kingdom, S10 2TH
Contact: Mary P. Gerrard, MBChB, FRCP, FRCPCH 44-114-271-7366 mary.gerrard@sch.nhs.uk
Southampton General Hospital
Recruiting
Southampton, England, United Kingdom, SO16 6YD
Contact: Janice A. Kohler, MD, FRCP 44-23-8079-6942
Royal Marsden - Surrey
Recruiting
Sutton, England, United Kingdom, SM2 5PT
Contact: Mary Taj, MD 44-20-8642-6011 ext. 1307
Royal Aberdeen Children's Hospital
Recruiting
Aberdeen, Scotland, United Kingdom, AB25 2ZG
Contact: Veronica Neefjes 44-1224-550-217
Royal Hospital for Sick Children
Recruiting
Edinburgh, Scotland, United Kingdom, EH9 1LF
Contact: W. Hamish Wallace, MD 44-131-536-0426
Royal Hospital for Sick Children
Recruiting
Glasgow, Scotland, United Kingdom, G3 8SJ
Contact: Milind D. Ronghe, MD 44-141-201-9309
Childrens Hospital for Wales
Recruiting
Cardiff, Wales, United Kingdom, CF14 4XW
Contact: Heidi Traunecker, MD, PhD 44-29-2074-2285 heidi.traunecker@cardiffandvale.wales.nhs.uk Less << |
|
NCT00004192
|
Lymphoma
Neut... More >>ropenia Less << |
Phase 2 |
Completed |
- |
United States, California
... More >>
Jonsson Comprehensive Cancer Center, UCLA
Los Angeles, California, United States, 90095-1781
United States, Nebraska
University of Nebraska Medical Center
Omaha, Nebraska, United States, 68198-3330
United States, Ohio
Ireland Cancer Center
Cleveland, Ohio, United States, 44106-5065 Less << |
|
NCT03322566
|
Lung Cancer |
Phase 2 |
Active, not recruiting |
September 2021 |
- |
|
NCT00148395
|
Non-Small-Cell Lung Carcinoma |
Phase 3 |
Completed |
- |
Germany
... More >> Klinikum Kassel GmbH
Kassel, Germany, 34125 Less << |
|
NCT02734524
|
Non-small Cell Lung Cancer |
Phase 2 |
Recruiting |
March 2019 |
China, Chongqing
... More >>
Southwest Hospital of Third Millitary Medical University
Recruiting
Chongqing, Chongqing, China, 400000
Contact: Cheng Qian, PhD 008615086883400 cqian3184@163.com
Contact: Zhi Yang, PhD 008613206140093
Principal Investigator: Cheng Qian, PhD Less << |
|
NCT00983541
|
- |
|
Terminated(Low enrollment) |
- |
- |
|
NCT00856791
|
Ovarian Cancer |
Phase 2 |
Completed |
- |
United States, Indiana
... More >>
St. Vincent Gynecologic Oncology
Indianapolis, Indiana, United States, 46260 Less << |
|
NCT02560441
|
Extranodal Natural Killer/T-Ce... More >>ll Lymphoma, Nasal Type Less << |
Phase 2 |
Recruiting |
- |
China
... More >> Cancer Institute/Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College
Recruiting
Beijing, China
Contact: Yuankai Shi
People's Hospital of Guangxi
Recruiting
Nanning, China
Contact: Jiaxin Chen
Fourth Hospital of Hebei Medical University
Recruiting
Shijiazhuang, China
Contact: Yuhuan Gao
Shanxi Dayi Hospital
Recruiting
Taiyuan, China
Contact: Qiaohua Zhang
Shanxi Province Cancer Hospital
Recruiting
Taiyuan, China
Contact: Liping Su Less << |
|
NCT00391586
|
- |
|
Terminated(PI left institution... More >>.) Less << |
- |
- |
|
NCT03179579
|
Gastric Cancer
... More >> Peritoneal Carcinomatosis Less << |
Phase 3 |
Not yet recruiting |
August 1, 2022 |
China, Guangdong
... More >>
Department of Abdominal Surgery (Section 2), Affiliated Tumor Hospital of Guangzhou Medical University
Not yet recruiting
Guangzhou, Guangdong, China, 510095
Contact: Xianzi Yang, M.D 8602066673666 7097359@qq.com
Principal Investigator: shuzhong cui, M.D Less << |
|
NCT00983541
|
Cancer |
Phase 2 |
Terminated(Low enrollment) |
- |
United States, Utah
... More >>
Huntsman Cancer Institute
Salt Lake City, Utah, United States, 84112 Less << |
|
NCT01510990
|
Non-small Cell Lung Cancer |
Phase 2 |
Unknown |
November 2015 |
Korea, Republic of
... More >>
Asan Medical Center
Recruiting
Seoul, Korea, Republic of, 138-736
Contact: Sang-We Kim, M.D. 82-2-3010-3215 swkim@amc.seoul.kr
Principal Investigator: Sang-We Kim, M.D. Less << |
|
NCT01091168
|
Breast Cancer
... More >> Metastases Less << |
Phase 3 |
Completed |
- |
- |
|
NCT01538641
|
Refractory Aggressive Non-Hodg... More >>kin's Lymphoma
Relapsing Aggressive Non-Hodgkin's Lymphoma Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00856791
|
- |
|
Completed |
- |
- |
|
NCT03141359
|
Lung |
Phase 2 |
Recruiting |
June 2020 |
United States, Maryland
... More >>
University of Maryland Medical Center
Recruiting
Baltimore, Maryland, United States, 21201
Contact: Caroline Poff, RN, MSN 410-369-5264 caroline.poff@umm.edu
Principal Investigator: Charles Simone, MD
United States, North Carolina
Levine Cancer Institute
Recruiting
Charlotte, North Carolina, United States, 28204
Contact: Jenna Gregory, RN, BSN, OCN 704-403-1520 jenna.gregory@carolinashealthcare.org
Principal Investigator: John H Heinzerling, MD Less << |
|
NCT00807300
|
Hepatocellular Carcinoma |
Phase 2 |
Completed |
- |
Germany
... More >> Clinic of Diagnostic Radiology and Nuclear Medicine, Medical Faculty, University Magdeburg
Magdeburg, Saxony-anhalt, Germany, 39120 Less << |
|
NCT01117402
|
Postsurgery Recurrent Carcinom... More >>a Cervix Less << |
Phase 2 |
Unknown |
December 2014 |
India
... More >> Tata Memorial Centre
Recruiting
Mumbai, Maharashtra, India, 400012
Contact: Reena Engineer, MD +912224177165 reena_engineer@rediffmail.com
Contact: ShyamKishore Shrivastava, MD +912224177163 shrivastavask@tmc.gov.in
Principal Investigator: Reena Engineer, MD Less << |
|
NCT03671265
|
Esophageal Cancer |
Not Applicable |
Not yet recruiting |
September 1, 2021 |
China, Tianjin
... More >> Department of Radiation Oncology, Tianjin Medical University Cancer Hospital
Tianjin, Tianjin, China, 300060 Less << |
|
NCT02374424
|
Diffuse, Large B-Cell, Lymphom... More >>a Less << |
Phase 2 |
Terminated(interim analysis pl... More >>anned as per protocol didn't provide positive outcome) Less << |
- |
Italy
... More >> Azienda Ospedaliera S. Gerardo Di Monza
Monza, Monza Brianza, Italy, 20900
Onco-Ematologia I.R.C.C.
Candiolo, TO, Italy
Ospedale Mauriziano
Torino, TO, Italy, 10128
Ospedale San Bortolo
Vicenza, VI, Italy, 36100
Ospedale "C. e G. Mazzoni", UOC Ematologia
Ascoli Piceno, Italy, 63100
Ospedale di Bolzano, Reparto di Ematologia & CTMO
Bolzano, Italy, 39100
A.O. Universitaria Careggi Di Firenze
Firenze, Italy, 50139
USL 6 Livorno Centro Aziendale Ematologia
Livorno, Italy, 57124
SC Ematologia AO Riuniti Papardo Piemonte
Messina, Italy
Ospedale dell'Angelo, U.O. Ematologia
Mestre, Italy, 30174
A.O. Universitaria Policlinico Di Modena
Modena, Italy, 41124
AUO Padova-Ematologia e Immunologia Clinica
Padova, Italy, 35121
A.O. Ospedali Riuniti Villa Sofia-Cervello, U.O. Ematologia I
Palermo, Italy, 90146
Ematologia Policlinico San Matteo
Pavia, Italy, 27100
AO di Perugia S. Maria della misericordia
Perugia, Italy, 06132
A.O. Universitaria Pisana
Pisa, Italy, 56126
Ematologia Ospedale S.Camillo Forlanini
Roma, Italy, 00149
Ematologia e Trapianto Istituto Regina Elena IFO
Roma, Italy
Policlinico Universitario Campus biomedico
Roma, Italy Less << |
|
NCT00593723
|
Esophagus Cancer |
Phase 1 |
Completed |
- |
United States, Missouri
... More >>
Washington University School of Medicine
St. Louis, Missouri, United States, 63110 Less << |
|
NCT01124812
|
Non Small Cell Lung Cancer |
Phase 1 |
Terminated |
- |
Italy
... More >> University Hospital Pisa
Pisa, Tuscany, Italy, 56126
Irst - Istituto Scientifico Romagnolo Per Lo Studio E La Cura Dei Tumori - Meldola (Fc)
Meldola, Italy Less << |
|
NCT00522405
|
Hepatocellular Carcinoma |
Phase 2
Phase 3 |
Unknown |
October 2014 |
India
... More >> All India Institute of Medical Sciences
Recruiting
New Delhi, Delhi, India, 110029
Sub-Investigator: Shashi B Paul, Ph.D
Sub-Investigator: Shivanand Gamanagatti, MD
Sub-Investigator: Kaushal Madan, DM
Sub-Investigator: Sreenivasa B Chalamalasetty, DM
Sub-Investigator: Sreenivas Vishnubhatla, Ph.D Less << |
|
NCT03058432
|
Nasopharyngeal Carcinoma |
Phase 2 |
Recruiting |
December 2019 |
China, Zhejiang
... More >>
Xiaozhong Chen
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: Xiaozhong Chen, MD +86-571-88128202 cxzfyun@sina.com Less << |
|
NCT00602667
|
Brain and Central Nervous Syst... More >>em Tumors Less << |
Phase 2 |
Active, not recruiting |
April 30, 2023 |
United States, California
... More >>
Lucile Packard Children's Hospital at Stanford University Medical Center
Palo Alto, California, United States, 94304
Rady Children's Hospital
San Diego, California, United States, 92123
United States, Minnesota
Children's Hospitals and Clinics of Minnesota - St. Paul
Saint Paul, Minnesota, United States, 55102
United States, Tennessee
St. Jude Children's Research Hospital
Memphis, Tennessee, United States, 38105
United States, Texas
University of Texas Southwestern Medical Center at Dallas
Dallas, Texas, United States, 75390
Australia, Queensland
Lady Cilento Children's Hospital, Brisbane
Brisbane, Queensland, Australia, 4029 Less << |
|
NCT03007212
|
Hepatocellular Carcinoma (HCC) |
Phase 4 |
Completed |
- |
- |
|
NCT03220932
|
Ovarian Epithelial Cancer |
Phase 3 |
Not yet recruiting |
December 31, 2022 |
France
... More >> Centre Hospitalier Lyon Sud
Not yet recruiting
Pierre-Bénite, France
Contact: Naoual BAKRIN Less << |
|
NCT03402737
|
Head and Neck Neoplasm |
Not Applicable |
Recruiting |
January 31, 2021 |
Belgium
... More >> Radiotherapy department, University Hospital Ghent
Recruiting
Ghent, Oost-Vlaanderen, Belgium, 9000
Contact: Wilfried De Neve, MD, PhD +32 9 332 30 15 wilfried.deneve@uzgent.be
Contact: Fréderic Duprez, MD, PhD +32 9 332 30 15 frederic.duprez@uzgent.be
Principal Investigator: Wilfried De Neve, MD, PhD
Sub-Investigator: Fréderic Duprez, MD, PhD
Sub-Investigator: Katrien Vandecasteele, MD, PhD
Sub-Investigator: Sylvie Rottey, MD, PhD
UZ Leuven
Recruiting
Leuven, Belgium
Contact: Sandra Nuyts, MD, PhD
CHU Namur
Recruiting
Namur, Belgium
Contact: Jean-François Daisne, MD Less << |
|
NCT00797719
|
Malignant Pleural Mesothelioma |
Phase 1
Phase 2 |
Recruiting |
July 2019 |
Canada, Ontario
... More >>
University Health Network
Recruiting
Toronto, Ontario, Canada, M5G 2M9
Contact: John Cho, MD 416 946 2124 john.cho@rmp.uhn.on.ca
Principal Investigator: John Cho, MD Less << |
|
NCT02299999
|
Metastatic Breast Cancer |
Phase 2 |
Recruiting |
December 2022 |
France
... More >> Institut de Cancérologie de l'Ouest/Paul Papin
Recruiting
Angers, France
Contact: Mario CAMPONE, MD mario.campone@ico.unicancer.fr
Principal Investigator: Mario CAMPONE, MD
Institut Sainte-Catherine
Recruiting
Avignon, France
Contact: Alice Mege, MD a.mege@isc84.org
Principal Investigator: Alice Mege, MD
Polyclinique Bordeaux Nord Aquitaine
Recruiting
Bordeaux, France, 33077
Contact: Nadine DOHOLLOU, MD n.dohollou@bordeauxnord.com
Contact: Nadine DOHOLLOU, MD
Institut Bergonié
Recruiting
Bordeaux, France
Contact: Hervé Bonnefoi, MD h.bonnefoi@bordeaux.unicancer.fr
Principal Investigator: Hervé Bonnefoi, MD
Centre François Baclesse
Recruiting
Caen, France
Contact: Christelle Lévy, MD c.levy@baclesse.unicancer.fr
Principal Investigator: Christelle Lévy, MD
Centre Jean Perrin
Recruiting
Clermont-Ferrand, France
Contact: Marie-Ange Mouret Reynier, MD marie-ange.mouret-reynier@cjp.fr
Principal Investigator: Marie-Ange Mouret Reynier, MD
Ch Alpes Leman
Not yet recruiting
Contamine Sur Arve, France, 74130
Contact: Carol ALLIOT, MD calliot@ch-alpes-leman.fr
Contact: Carol ALLIOT, MD
Centre Georges François Leclerc
Recruiting
Dijon, France, 21079
Contact: Nicolas Isambert, MD nisambert@cgfl.fr
Principal Investigator: Nicolas Isambert, MD
Chd Vendee
Recruiting
La Roche-sur-Yon, France, 85925
Contact: Tifenn L'Haridon, MD tifenn.lharidon@chd-vendee.fr
Contact: Tifenn L'Haridon, MD
Centre Oscar Lambret
Recruiting
Lille, France
Contact: Nuria KOTECKI, MD
Principal Investigator: Nuria KOTECKI, MD
Chu Dupuytren
Not yet recruiting
Limoges, France, 87000
Contact: Laurence VENAT-BOUVET, MD laurence.venat-bouvet@chu-limoges.fr
Contact: Laurence VENAT-BOUVET, MD
Hopital Privé Jean Mermoz
Not yet recruiting
Lyon, France, 69008
Contact: Olfa DERBEL, MD o.derbelmermoz@gmail.com
Contact: Olfa DERBEL, MD
Centre Hospitalier Lyon Sud
Recruiting
Lyon, France
Contact: Benoit You, MD
Principal Investigator: Benoit You, MD
Centre Léon Bérard
Recruiting
Lyon, France
Contact: Thomas Bachelot, MD thomas.bachelot@lyon.unicancer.fr
Principal Investigator: Thomas Bachelot, MD
Institut Paoli Calmettes
Recruiting
Marseille, France
Contact: Anthony Gonçalves, MD goncalvesa@ipc.unicancer.fr
Principal Investigator: Anthony Gonçalves, MD
Institut Régional du Cancer Montpellier Val d'Aurelle
Recruiting
Montpellier, France
Contact: William Jacot, MD william.jacot@icm.unicancer.fr
Principal Investigator: William Jacot, MD
Centre Alexis Vautrin
Recruiting
Nancy, France
Contact: Elisabeth Luporsi, MD e.luporsi@nancy.unicancer.fr
Principal Investigator: Elisabeth Luporsi, MD
Institut de Cancérologie de l'Ouest/ René Gauducheau
Recruiting
Nantes, France
Contact: Mario Campone, MD mario.campone@ico.unicancer.fr
Principal Investigator: Mario Campone, MD
Centre Antoine Lacassagne
Recruiting
Nice, France
Contact: Jean-Marc Ferrero, MD jean-marc.ferrero@nice.unicancer.fr
Principal Investigator: Jean-Marc Ferrero, MD
Institut Curie
Recruiting
Paris, France
Contact: Marie-Paule Sablin, MD mariepaule.sablin@curie.fr
Principal Investigator: Marie-Paule Sablin, MD
Centre Eugène Marquis
Recruiting
Rennes, France
Contact: Claudia Lefeuvre-Plesse, MD c.lefeuvre@rennes.unicancer.fr
Principal Investigator: Claudia Lefeuvre-Plesse, MD
Centre Henri Becquerel
Recruiting
Rouen, France
Contact: Jean-Christophe Théry, MD
Principal Investigator: Jean-Christophe Théry, MD
Institut Curie
Recruiting
Saint-Cloud, France
Contact: Florence COUSSY, MD florence.coussy@curie.fr
Principal Investigator: Florence COUSSY, MD
Hopitaux Universitaire de Strasbourg - Hopital Civil
Recruiting
Strasbourg, France
Contact: Philippe BARTHELEMY, MD philippe.barthelemy@chru-strasbourg.fr
Principal Investigator: Philippe BARTHELEMY, MD
Hopitaux Du Leman
Recruiting
Thonon-les-Bains, France, 74200
Contact: Francesco DEL PIANO, MD f-delpiano@ch-hopitauxduleman.fr
Contact: Francesco DEL PIANO, MD
Institut Claudius Regaud
Recruiting
Toulouse, France
Contact: Florence Dalenc, MD dalenc.florence@iuct-oncopole.fr
Principal Investigator: Florence Dalenc, MD
Gustave Roussy
Recruiting
Villejuif, France
Contact: Monica Arnedos, MD Monica.ARNEDOS@gustaveroussy.fr
Principal Investigator: Monica Arnedos, MD
Sub-Investigator: Fabrice André, MD Less << |
|
NCT02918955
|
Head and Neck Neoplasms |
Phase 3 |
Recruiting |
June 2022 |
Switzerland
... More >> Inselspital Bern
Recruiting
Bern, Switzerland, 3010
Contact: OLGUN ELICIN, MD +41 31 632 26 32 olgun.elicin@insel.ch
Genève University Hospital
Recruiting
Geneva, Switzerland
Contact: Francesca Caparrotti, MD +41 79 553 56 12 francesca.caparrotti@hcuge.ch
Contact: Laurence Zulianello Laurence.Zulianello@hcuge.ch Less << |
|
NCT00083538
|
Multiple Myeloma |
Phase 2 |
Completed |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences/MIRT
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT02873416
|
Precision Cell Immunotherapy
... More >> Chemotherapy
Advanced Lung Cancer Less << |
Phase 1
Phase 2 |
Unknown |
August 2018 |
China, Zhejiang
... More >>
Ningbo No.5 Hospital (Ningbo Cancer Hospital)
Recruiting
Ningbo, Zhejiang, China
Contact: Bi Wan 86-13310088259 biwang0218@126.com
Contact: Jiangtao Wang 86-15888102792 Less << |
|
NCT00415818
|
Carcinoma, Non-Small-Cell Lung |
Phase 2
Phase 3 |
Completed |
- |
France
... More >> Centre Hospitalier Belfort-Montbeliard
Belfort, France, 90016
CHU, Service de Pneumologie
Besancon, France, 25000
Centre Hospitalier Général, Service de Pneumologie
Briey, France, 54151
Centre François Baclesse
Caen, France, 14076
Hôpital Pasteur - Service de médecine F- Pavillon 43
Colmar, France, 68000
Centre Oscar Lambret
Lille, France, 59020
Institut Paoli-Calmettes, Service d'oncologie médicale
Marseille, France, 13273
Centre Hospitalier Lyon Sud
Pierre-Bénite, France, 69495
CHRU Hôpital de Pontchaillou
Rennes, France, 35033
Hôpital Lyautey - Service de Pneumologie
Strasbourg, France, 67100
Centre Hospitalier de la Haute Saône, Service de Pneumologie - Allergologie
Vesoul, France, 70014
Germany
Asklepios Fachkliniken, Zentrum für Pneumonologie
Gauting, Germany, 82131
Thoraxklinik Heidelberg
Heidelberg, Germany, 69126
Lungenklinik Krankenhaus Merheim
Köln, Germany, 51109
Klinikum Mannheim der Ruprecht-Karls
Mannheim, Germany, 68167
Klinikum Offenburg, Medizinische Klinik II
Offenburg, Germany, 77654
Hungary
Fejér Megyei Szent György Kórház
Székesfehérvár, Hungary, 8000
Poland
Oddział II Chorób Płuc i Gruźlicy
Bialystok, Poland, 15-540
Kujawsko-Pomorskie Centrum Pulmonologii
Bydgoszcz, Poland, 85-326
Oddział Chemioterapii
Krakow, Poland, 31-826
Oddział III Chorób Płuc i Gruźlicy
Otwock, Poland, 05-400
Oddział Onkologii Klinicznej
Poznan, Poland, 60-569
Dolnośląskie Centrum
Wrocław, Poland, 53-439 Less << |
|
NCT01655225
|
Advanced Cancer
... More >> Metastatic Cancer
Non-Hodgkin's Lymphoma
Metastatic Breast Cancer
Malignant Mesothelioma
Non-small Cell Lung Cancer Less << |
Phase 1 |
Recruiting |
March 1, 2021 |
United States, California
... More >>
UCLA Medical Center
Recruiting
Los Angeles, California, United States, 90095
Contact 3103258252
Principal Investigator: Parvin Fatheddin Peddi
United States, New York
Memorial Sloan Kettering Cancer Center
Recruiting
New York, New York, United States, 10065
Contact 646-888-4226
Principal Investigator: Anna M Varghese
United States, Oklahoma
Peggy and Charles Stephenson Oklahoma Cancer Center
Recruiting
Oklahoma City, Oklahoma, United States, 73104
Contact 405-271-8777
Principal Investigator: Raid Aljumaily
United States, Pennsylvania
Penn Presbyterian Medical Center
Completed
Philadelphia, Pennsylvania, United States, 19104
United States, Tennessee
Sarah Cannon Cancer Center
Recruiting
Nashville, Tennessee, United States, 37203
Contact 615-239-7615
Principal Investigator: SMO Sarah Cannon Research Inst.
Tennessee Oncology PLLC
Recruiting
Nashville, Tennessee, United States, 37203
Contact 6153297274
Principal Investigator: Johanna Chock Bendell
Italy
Azienda Ospedaliero - Universitaria S. Luigi Gonzaga
Completed
Orbassano, Torino, Italy, 10043
Puerto Rico
Fundacion de Investigacion de Diego
Recruiting
San Juan, Puerto Rico, 00927
Contact 7877221248
Principal Investigator: Mirelis Acosta-Rivera Less << |
|
NCT02862561
|
Precision Cell Immunotherapy
... More >> Chemotherapy
Advanced Malignancies Less << |
Phase 1
Phase 2 |
Unknown |
July 2018 |
China, Shanghai
... More >>
Shanghai International Medical Center
Recruiting
Shanghai, Shanghai, China, 201318
Contact: Naiyan Han +86 (0) 182 1766 2469 naiyan.han@simcgroup.com Less << |
|
NCT02862587
|
Precision Cells
... More >> Chemotherapy
Advanced Lung Cancer Less << |
Phase 1
Phase 2 |
Unknown |
August 2018 |
China, Shanghai
... More >>
Shanghai International Medical Center
Recruiting
Shanghai, Shanghai, China, 201318
Contact: Naiyan Han +86 (0) 182 1766 2469 naiyan.han@simcgroup.com Less << |
|
NCT00464893
|
Gastric Cancer
... More >> Gastric Adenocarcinoma Less << |
Phase 2 |
Completed |
- |
Austria
... More >>
Innsbruck, Austria
Germany
Hamburg, Berlin, Heidelberg, Köln, Halle, Germany
Spain
Terrassa, Spain
United Kingdom
Nottingham, United Kingdom Less << |
|
NCT03058289
|
Melanoma
Head... More >> and Neck Cancer
Lymphoma
Breast Cancer
Pancreatic Cancer
Liver Cancer
Colon Cancer
Lung Cancer
Glioblastoma Less << |
Phase 1
Phase 2 |
Recruiting |
August 2020 |
United States, California
... More >>
USC Norris
Recruiting
Los Angeles, California, United States, 90033
Contact: Xiomara Menendez 323-409-4368 xiomara.menendez@med.usc.edu
Contact: Jubilee Acap 323-865-0451 jubilee.acap@med.usc.edu
USC HOAG
Recruiting
Newport Beach, California, United States, 92663
Contact: Alicia Bogardus 949-764-6755 Alicia.bogardus@hoag.org
Contact: Cristina de Leon 9497645543 cristina.deleon@hoag.org
United States, Maryland
Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins
Recruiting
Baltimore, Maryland, United States, 21205
Contact: Denise Gallagher, RN 410-502-5140 phase1trials@exchange.johnshopkins.edu
Principal Investigator: Nilofer Azad, MD
United States, Massachusetts
UMASS Memorial Medical Center
Recruiting
Worcester, Massachusetts, United States, 01655
Contact: Cancer Research Office 508-856-3216 cancer.research@umassmed.edu
United States, Pennsylvania
Fox Chase Cancer Center
Recruiting
Philadelphia, Pennsylvania, United States, 19111-2497
Contact: Daniel Sabo Daniel.Sabo@fccc.edu
Principal Investigator: Anthony J. Olszanski, MD, RPh
Canada, Ontario
Princess Margaret Cancer Center - University Health Network
Recruiting
Toronto, Ontario, Canada, M5G 2C4
Contact: Oncology Referral 416-946-4575 Less << |
|
NCT01837381
|
Hepatocellular Carcinoma |
Phase 2 |
Completed |
- |
Hong Kong
... More >> Department of Clinicl Oncology, Prince of Wales Hospital, The Chinese University of Hong Kong
Hong Kong, Hong Kong
Department of Imaging and Interventional Radiology, Prince of Wales Hospital, The Chinsese University of Hong Kong
Hong Kong, Hong Kong
Department of Surgery, Prince of Wales Hospital, The Chinese University of Hong Kong
Hong Kong, Hong Kong Less << |
|
NCT03003962
|
Non Small Cell Lung Carcinoma ... More >>NSCLC Less << |
Phase 3 |
Recruiting |
December 31, 2020 |
- |
|
NCT02873520
|
Precision Cell Immunotherapy
... More >> Chemotherapy
Advanced Gastric Cancer Less << |
Phase 1
Phase 2 |
Unknown |
August 2018 |
China, Zhejiang
... More >>
Ningbo No.5 Hospital (Ningbo Cancer Hospital)
Recruiting
Ningbo, Zhejiang, China
Contact: Bi Wan 86-13310088259 biwang0218@126.com
Contact: Jiangtao Wang 86-15888102792 Less << |
|
NCT01997775
|
Non-small Cell Lung Cancer |
Phase 2 |
Terminated(Difficult enrollmen... More >>t) Less << |
- |
Taiwan
... More >> National Cheng-Kung Uni. Hosp.
Tainan, Taiwan, 704 Less << |
|
NCT03393507
|
Ovarian Cancer
... More >> Chemotherapeutic Toxicity Less << |
Phase 2 |
Recruiting |
December 1, 2019 |
China, Sichuan
... More >> Zhang xuan
Recruiting
Leshan, Sichuan, China, 600000
Contact: Zhang Xuan, MD 861890631232 79916363@qq.com Less << |
|
NCT00424255
|
- |
|
Completed |
- |
- |
|
NCT02649829
|
Malignant Pleural Mesothelioma |
Phase 1
Phase 2 |
Recruiting |
November 2021 |
Belgium
... More >> Antwerp University Hospital
Recruiting
Edegem, Antwerp, Belgium, 2650
Contact: Zwi N Berneman, MD, PhD zwi.berneman@uza.be
Contact: Sébastien Anguille, MD, PhD sebastien.anguille@uza.be
Sub-Investigator: Paul Germonpré, MD
Sub-Investigator: Koen Deschepper, MD
AZ Middelares
Recruiting
Ghent, Belgium, 9000
Contact: Local investigator: Paul Germonpré, MD, PhD
AZ Nikolaas
Recruiting
Sint-Niklaas, Belgium, 9100
Contact: Local investigator: Koen Deschepper, MD Less << |
|
NCT00424255
|
Neoplasms, Head and Neck |
Phase 3 |
Completed |
- |
- |
|
NCT02879214
|
Squamous Cell Carcinoma of Cer... More >>vix Less << |
Phase 2 |
Recruiting |
February 2020 |
China, Sichuan
... More >> Sichuan PPH, Departmentn of Gynecology Oncology
Recruiting
Chengdu, Sichuan, China, 610072
Contact: Lan Xie, MD 086-87398097
Contact: Yi Liu, MD 086-87398097 Less << |
|
NCT00577512
|
Multiple Myeloma |
Phase 2 |
Completed |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT01806272
|
Nasopharyngeal Cancers |
Phase 2 |
Unknown |
August 2014 |
China, Guangdong
... More >>
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Wei LUO, M.D. +862087343483 luowei2@mail.sysu.edu.cn Less << |
|
NCT01731548
|
Carcinoma of Lung
... More >> Small Cell
Limited Stage Less << |
Phase 2
Phase 3 |
Unknown |
December 2016 |
China, Guangdong
... More >>
Sun Yat-Sen University, Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Hui Liu, MD. PHD 086-020-87343033 liuhui@sysucc.org.cn
Contact: Yong Bao, MD 086-020-87343504 baoyong@sysucc.org.cn Less << |
|
NCT00577512
|
- |
|
Completed |
- |
- |
|
NCT02944708
|
Nasopharyngeal Carcinoma |
Phase 3 |
Unknown |
October 2018 |
China, Beijing
... More >> Peking University Cancer Hospital
Recruiting
Beijing, Beijing, China, 100142
Contact: Yan Sun, MD
China, Fujian
The First Affiliated Hospital of Fujian Medical University
Recruiting
Fuzhou, Fujian, China, 350005
Contact: Jinshen Hong, MD hjs703@126.com
The First Affiliated Hospital of Xiamen University
Recruiting
Xiamen, Fujian, China, 361003
Contact: Qin Lin, MD linqin05@163.com
China, Guangxi
Cancer Hospital of Guangxi Medical University
Recruiting
Nanning, Guangxi, China, 530021
Contact: Xiaodong Zhu, MD
The People's Hospital of Guangxi Zhuang Autonomous Region
Recruiting
Nanning, Guangxi, China, 530021
Contact: Heming Lu, MD
China, Hubei
Tongji Hospital, Tongji Medical College of HUST
Recruiting
Wuhan, Hubei, China, 430030
Contact: Guoqing Hu, MD
China, Jiangxi
Jiangxi Province Tumor Hospital
Recruiting
Nanchang, Jiangxi, China, 330029
Contact: Xiaochang Gong, MD gxcanddw@163.com
Jiangxi Provincial Cancer Hospital
Recruiting
Nanchang, Jiangxi, China, 330029
Contact: Xiaochang Gong, MD gxcanddw@163.com
China, Shanghai
Fudan University Shanghai Cancer center
Recruiting
Shanghai, Shanghai, China, 201315
Contact: Chaosu Hu, MD hucsu62@yahoo.com
Shanghai Proton and Heavy Ion Center
Recruiting
Shanghai, Shanghai, China, 201315
Contact: Lin Kong, MD lin.kong@sphic.org.cn
Contact: Jiyi Hu, MD jiyi.hu@sphic.org.cn
Principal Investigator: Jiade J Lu, MD
Eye & ENT Hospital of Fudan University
Recruiting
Shanghai, Shanghai, China
Contact: Shengzi Wang, MD shengziwang@fudan.edu.cn
China, Sichuan
West China Hospital
Recruiting
Chengdu, Sichuan, China, 610041
Contact: Ping Li, MD
Singapore
National Cancer Centre, Singapore
Recruiting
Singapore, Singapore, 119082
Contact: Boon Cher Goh, MD
Taiwan
Taichung Veterans General Hospital
Recruiting
Taichung, Taiwan, 40705
Contact: Jin-Ching Lin, MD jclin@vghtc.gov.tw Less << |
|
NCT01020864
|
Head and Neck Neoplasms |
Phase 2 |
Unknown |
January 2018 |
Denmark
... More >> Department of Oncology, Odense University Hospital
Recruiting
Odense, Denmark, 5000
Contact: Jorgen Johansen, MD, PhD +45 6541 4568 jorgen.johansen@ouh.regionsyddanmark.dk
Principal Investigator: Jorgen Johansen, MD, PhD Less << |
|
NCT02145078
|
Recurrent Non-small Cell Lung ... More >>Cancer
Stage IV Non-small Cell Lung Cancer Less << |
Not Applicable |
Terminated(Slow accrual.) |
- |
United States, Michigan
... More >>
Barbara Ann Karmanos Cancer Institute
Detroit, Michigan, United States, 48201 Less << |
|
NCT00083551
|
Multiple Myeloma |
Phase 3 |
Completed |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences/MIRT
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT03455205
|
Digestive Tract |
Not Applicable |
Recruiting |
December 30, 2019 |
China, Guangdong
... More >>
First affiliated hospital of guangzhou university of traditional Chinese medicine
Recruiting
Guangzhou, Guangdong, China
Contact: lin lizhu, Dr. 13501505588 13501505588@163.com Less << |
|
NCT00083551
|
- |
|
Completed |
- |
- |
|
NCT00959582
|
Ovarian Cancer
... More >> Primary Peritoneal Cancer
Fallopian Tube Cancer Less << |
Phase 1 |
Completed |
- |
- |
|
NCT03215706
|
Non-Small Cell Lung Cancer |
Phase 3 |
Recruiting |
May 20, 2020 |
- |
|
NCT01540058
|
Neoplasms, Unknown Primary |
Phase 3 |
Unknown |
October 2017 |
Denmark
... More >> Rigshospitalet
Active, not recruiting
Copenhagen, Denmark, 2100
France
Institut Gustave Roussy
Recruiting
Villejuif, Val de Marne, France, 94805
Contact: Karim FIZAZI, MD, PhD +33 1 42 11 43 17 karim.fizazi@igr.fr
Principal Investigator: Karim FIZAZI, MD, PhD
Netherlands
Viecuri Medical Centre Venlo
Active, not recruiting
Venlo, Netherlands, 5912 BL Less << |
|
NCT01014208
|
Lymphoma, Large-Cell, Diffuse |
Phase 3 |
Completed |
- |
- |
|
NCT02369835
|
Head and Neck Carcinoma
... More >> Radiation-Induced Dermatitis Less << |
Phase 3 |
Active, not recruiting |
December 2019 |
United States, California
... More >>
Stanford Cancer Center South Bay
San Jose, California, United States, 95124
Stanford University School of Medicine
Stanford, California, United States, 94305 Less << |
|
NCT01805557
|
Diffuse Large B-cell Lymphoma ... More >>Refractory
Diffuse Large B-cell Lymphoma Recurrent Less << |
Phase 2
Phase 3 |
Unknown |
February 2018 |
Italy
... More >> AOU Careggi
Terminated
Firenze, FI, Italy, 50134
A.O. Niguarda
Recruiting
Milano, MI, Italy, 20162
Contact: Chiara Rusconi, MD +39 02 - 64442668 chiara.rusconi@ospedaleniguarda.it
Principal Investigator: Chiara Rusconi, MD
Osp. San Gerardo
Not yet recruiting
Monza, MI, Italy, 20052
Contact: Enrico Pogliani, MD 039-2339435 enrico.pogliani@unimib.it; marziaborsetto@yahoo.it
Principal Investigator: Enrico Pogliani, MD
Ospedale Civile Guglielmo da Saliceto
Recruiting
Piacenza, PC, Italy, 29121
Contact: Daniele Vallisa, MD +39 0523-303701/719 d.vallisa@auslpc.it
Principal Investigator: Daniele Vallisa, MD
CRO Aviano
Recruiting
Aviano, Pordenone, Italy, 33081
Contact: Mariagrazia Michieli, M.D. mmichieli@cro.it
Principal Investigator: Mariagrazia Michieli, M.D.
Osp. Umberto I
Recruiting
Nocera Inferiore, SA, Italy, 84014
Contact: Alfonso M. D'Arco, MD +39 081-9213370 medint.nocera@libero.it
Principal Investigator: Alfonso M. D'Arco, MD
Medicina Interna 2 ad indirizzo Ematologico AOU San Luigi Gonzaga
Not yet recruiting
Orbassano, Torino, Italy, 10043
Contact: Giuseppe Saglio, Prof. 00390119026721 giuseppe.saglio@unito.it
Principal Investigator: Giuseppe Saglio, Prof.
AOU San Giovanni Battista-Ematologia 1
Recruiting
Torino, TO, Italy, 10126
Contact: Marco Ladetto, MD +39 011-6334264 marco.ladetto@unito.it;antonella.fiorillo@yahoo.it
Principal Investigator: Marco Ladetto, MD
Azienda Ospedaliero - Universitaria di Udine
Recruiting
Udine, UD, Italy, 33100
Contact: Francesco Zaja, MD +39 0432-559662 zaja.francesco@aoud.sanita.fvg.it;d'odorico.cristina@aoud.sanita.fvg.it
Principal Investigator: Francesco Zaja, MD
Osp. S. Antonio Abate Gallarate
Recruiting
Gallarate, Varese, Italy
Principal Investigator: Fabrizio Ciambelli, MD
Ospedale di Circolo e Fondazione Macchi - Ematologia
Not yet recruiting
Varese, VA, Italy, 21100
Contact: Francesco Passamonti +39 0332-393648 francesco.passamonti@ospedale.varese.it
Principal Investigator: Francesco Passamonti, MD
A.O. SS. Antonio e Biagio e C. Arrigo
Recruiting
Alessandria, Italy, 15121
Contact: Flavia Salvi, MD +39 0131206294 segreteria@filinf.it
Principal Investigator: Flavia Salvi, MD
Clinica di ematologia AOU Umberto I Ospedali Riuniti
Recruiting
Ancona, Italy, 60100
Contact: Guido Gini, M.D 00390713580384 g.gini@ospedaliriuniti.marche.it
Principal Investigator: Guido Gini, M.D.
Divisione di Ematologia Spedali Civili
Recruiting
Brescia, Italy
Principal Investigator: Alessandra Tucci, MD
Presidio Ospedaliero A.Perrino - Divisione di Ematologia
Terminated
Brindisi, Italy
Divisione di Ematologia Osp. Businco
Recruiting
Cagliari, Italy
Principal Investigator: Giuseppina Cabras, MD
Ospedale Roberto Binaghi
Not yet recruiting
Cagliari, Italy
Principal Investigator: Giorgio La Nasa, DM
Università Cattolica Campobasso
Terminated
Campobasso, Italy
A.O. Universitaria Ospedali Riuniti di Foggia
Not yet recruiting
Foggia, Italy
Principal Investigator: Silvana Capalbo, MD
Ematologia 1 Ospedale S. Martino
Not yet recruiting
Genova, Italy, 16132
Principal Investigator: Angelo Michele Carella, PROF
Azienda Universitaria San Martino
Not yet recruiting
Genova, Italy
Principal Investigator: Marco Gobbi, MD
Ospedale Santa Maria Goretti
Not yet recruiting
Latina, Italy
Principal Investigator: Angelo De Blasio, MD
Ospedale Vito Fazzi
Not yet recruiting
Lecce, Italy
Principal Investigator: Nicola Di Renzo, MD
SC Ematologia ‐ Trapianto di midollo osseo Fond. IRCCS Istituto Nazionale Tumori
Recruiting
Milano, Italy, 20133
Contact: Paolo Corradini, Prof. 00390223902950 paolo.corradini@unimi.it
Principal Investigator: Paolo Corradini, Prof.
Ospedale Maggiore IRCCS-Dipartimento di Ematologia
Not yet recruiting
Milano, Italy
Principal Investigator: Luca Baldini, MD
AORN Cardarelli U.O.Ematologia
Not yet recruiting
Napoli, Italy
Principal Investigator: Vincenzo Mettivier, MD
SCDU Ematologia ‐ Università del Piemonte Orientale
Recruiting
Novara, Italy, 28100
Contact: Gianluca Gaidano, Prof. 00390321660663
Principal Investigator: Gianluca Gaidano, Prof.
Ematologia e Immunologia Clinica, Dipartimento di Medicina Università di Padova
Recruiting
Padova, Italy, 35100
Contact: Giampiero Semenzato, Prof. 00390498212298 g.semenzato@unipd.it
Principal Investigator: Giampiero Semenzato, Prof.
Ospedale S. Antonio
Not yet recruiting
Padova, Italy
Principal Investigator: Dario Marino, MD
Oncoematologia Dipartimento oncologico La Maddalena
Not yet recruiting
Palermo, Italy, 90146
Contact: Maurizio Musso, M.D. 00390916806801 mamusso@libero.it
Principal Investigator: Maurizio Musso, M.D.
U.O. Complessa di Ematologia Ospedale di Parma
Recruiting
Parma, Italy, 43100
Contact: Franco Aversa, Prof. 00390521903263 franco.aversa@unipr.it
Principal Investigator: Franco Aversa, Prof.
Fond. Maugeri - Centro medico
Not yet recruiting
Pavia, Italy
Principal Investigator: Vittorio Fregoni, MD
Osp. S. Maria delle Croci
Recruiting
Ravenna, Italy
Principal Investigator: Alfonso Zaccaria, MD
AO Bianchi Melacrino Morelli UO Ematologia
Recruiting
Reggio Calabria, Italy
Principal Investigator: Caterina Stelitano, MD
AO Arcispedale S.Maria Nuova Ematologia
Recruiting
Reggio Emilia, Italy
Principal Investigator: Francesco Merli, MD
Osp. degli Infermi Divisione di Oncologia
Not yet recruiting
Rimini, Italy
Principal Investigator: Annalisa Molinari, MD
Ospedale Oncologico regionale CROB
Not yet recruiting
Rionero in Vulture (PZ), Italy
Principal Investigator: Pellegrino Musto, MD
Ematologia Ospedale S.Camillo Forlanini
Not yet recruiting
Roma, Italy, 00149
Contact: Roberta Battistini, M.D. rbattistini@scamilloforlanini.rm.it
Principal Investigator: Roberta Battistini, M.D.
A.O. Universitaria S. Andrea
Recruiting
Roma, Italy
Principal Investigator: Maria Cristina, Cox
Osp.Sant'Eugenio Divisione di Ematologia
Not yet recruiting
Roma, Italy
Principal Investigator: Elisabetta Abruzzese, MD
Policlinico Universitario Tor Vergata
Not yet recruiting
Roma, Italy
Principal Investigator: Maria Cantonetti, MD
Universita' La Sapienza
Not yet recruiting
Roma, Italy
Principal Investigator: Maurizio Martelli, MD
Clinica Humanitas
Recruiting
Rozzano (MI), Italy
Sub-Investigator: Balzarotti Monica, MD
Policlinico Le Scotte Clinica Ematologica
Not yet recruiting
Siena, Italy
Principal Investigator: Alberto Fabbri, MD
SC Oncoematologia con autotrapianto AO Santa Maria
Recruiting
Terni, Italy, 05100
Contact: Anna Marina Liberati, Prof 0744/205971 marinal@unipg.it
Principal Investigator: Anna Marina Liberati, M.D.
Osp. San Giovanni Battista_Molinette Ematologia 2
Recruiting
Torino, Italy
Principal Investigator: Umberto Vitolo, MD
Divisione di Ematologia Ospedale S. Nicola
Not yet recruiting
Trani, Italy
Principal Investigator: Gaetano De Santis, MD Less << |
|
NCT00113919
|
Multiple Myeloma |
Phase 1
Phase 2 |
Terminated(Due to poor accrual... More >>) Less << |
- |
United States, Arkansas
... More >>
University of Arkansas for Medical Sciences, Myeloma Institute for Research and Therapy
Little Rock, Arkansas, United States, 72205
University of Arkansas for Medical Sciences
Little Rock, Arkansas, United States, 72205 Less << |
|
NCT01745445
|
Small Cell Lung Cancer |
Phase 2 |
Unknown |
December 2016 |
China, Tianjin
... More >> Department of Radiation Oncology, Tianjin Medical University Cancer Hospital
Recruiting
Tianjin, Tianjin, China, 300060
Contact: QING SONG PANG, M.D +86-22-23340123-1314 pangqingsong@yahoo.com.cn Less << |
|
NCT01664663
|
Non Small Cell Lung Cancer
... More >> Locally Advanced Disease Less << |
Phase 2 |
Terminated(Safety analysis sho... More >>wed increased grade 5 toxicity in experimental arm.) Less << |
October 2015 |
Sweden
... More >> Department of Oncology, Norrlands Universitetssjukhus
Umeå, Norrland, Sweden, 901 85
Department of Oncology, Karolinska University Hospital
Stockholm, Stockholm county, Sweden, 171 76
Department of Oncology, Sahlgrenska University Hospital
Gothenburg, Västra Götaland, Sweden, 413 45 Less << |
|
NCT00620139
|
- |
|
Completed |
- |
United States, New York
... More >>
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT01014208
|
- |
|
Completed |
- |
- |
|
NCT01627197
|
Bladder Cancer |
Phase 3 |
Recruiting |
December 2021 |
China, Guangdong
... More >>
Cancer Center, Sun Yat-sen University
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Fang-Jian Zhou, M.D Ph.D zhoufj@sysucc.org.cn
Contact: Zhuo-wei Liu liuzhw@sysucc.org.cn
Principal Investigator: Fang-Jian Zhou, M.D Ph.D
Sub-Investigator: Zhuo-Wei Liu, M.D Ph.D Less << |
|
NCT03513042
|
- |
|
Not yet recruiting |
December 2022 |
- |
|
NCT00718354
|
Gastric Cancer
... More >> Gastroesophageal Cancer Less << |
Phase 3 |
Completed |
- |
India
... More >> Mahatma Gandhi Cancer Hospital & Research Institute ,1/7 M.V.P. Colony, - ,, .
Vishakhapattanam, Andhra Pradesh, India, 530017
Mahavir Cancer Sansthan,Phulwari Sharif
Patna, Bihar, India, 801505
Gujarat Cancer Research Institute, Civil Hospital Campus,Asarwa, ,
Ahmedabad, Gujarat,, India, 380016
Department Of Radiotherapy,S.S.G. Hospital, -
Baroda,Vadodara, Gujarat,, India, 390 001
Gokula Curie Cancer Centre,M.S.Ramaiah Memorial Hospital,MSR Nagar, MSRIT Post
Bangalore, Karnataka, India, 560054
Madhavan J.P.
Trivandrum, Kerala,, India, 695011
MGM Medical College & MY Hospital,
Indore, M.P, India, 452005
Curie Manavta Cancer Centre, Opp.Hotel Sandeep Naka,Nashik
Mumbai, Maharashtra,, India, 425004
Ruby Hall Clinic,Cancer Building,40 sassoon Road, , ,
Pune, Maharashtra, India, 411001
Cancer Hospital & Research Institute, Cancer Hill
Gwalior, MP, India, 474009
Acharya Tulsi Regional Cancer Treatment & Research Institute
Bikaner, UP, India
B.P.Poddar Hospital & Medical Research Ltd,71/1, Humayun Kabir Sarani,, Block-G, New Alipore
Kolkata, west Bangol, India, 700053
Biswajit Sanyal
Calcutta, West Bengal, India
Chittaranjan National Cancer Institute,37, S.P.Mukhurjee Road
Kolkata, West Bengal, India, 700026
Dr. BRA IRCH,all India Institute of Medical Sciences,Ansari Nagar,
New Delhi, India, 110029. Less << |
|
NCT00113919
|
- |
|
Terminated(Due to poor accrual... More >>) Less << |
- |
- |
|
NCT01472250
|
- |
|
Completed |
- |
China, Beijing
... More >> Lin Shen
Beijing, Beijing, China, 100142 Less << |
|
NCT00921739
|
Non Small Cell Lung Cancer
... More >> Small Cell Lung Cancer
Thymoma
Thymus Neoplasms Less << |
Phase 1 |
Active, not recruiting |
December 2018 |
United States, North Carolina
... More >>
Duke University Medical Center
Durham, North Carolina, United States, 27710 Less << |
|
NCT00230451
|
Esophageal Cancer |
Phase 2 |
Completed |
- |
United States, Michigan
... More >>
University of Michigan Cancer Center
Ann Arbor, Michigan, United States, 48197 Less << |
|
NCT02786719
|
Neuroblastoma |
Not Applicable |
Recruiting |
June 2019 |
United States, California
... More >>
Rady Children's Hospital
Recruiting
San Diego, California, United States, 92123
Contact: Laura Conner 858-966-5983 LConner@rchsd.org
Principal Investigator: Peter Zage, MD
United States, Texas
Texas Children's Hospital
Recruiting
Houston, Texas, United States, 77030
Contact: Sarah Whittle, MD, BA 832-822-4242 sbwhittl@txch.org Less << |
|
NCT00333294
|
Non-Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
France
... More >> Research Site
Clermont Ferrand, France
Research Site
Grenoble, France
Research Site
Nantes, France
Research SIte
Paris, France Less << |
|
NCT03562897
|
Carcinoma
Ova... More >>rian Neoplasm
Endocrine Gland Neoplasm
Urogenital Neoplasms
Ovarian Diseases
Adnexal Diseases
Genital Diseases, Female
Female Urogenital Diseases
Female Urogenital Diseases and Pregnancy Complications
Endocrine System Diseases
Gonadal Disorders
Genital Neoplasm, Female
Neoplasms, Glandular and Epithelial Less << |
Phase 2 |
Not yet recruiting |
March 1, 2020 |
Cuba
... More >> National Institute of Oncology and Radiobiology (INOR)
Not yet recruiting
Havana, Cuba, 10400
Contact: Karen Lopez Miguel, Dr. +53-78388589 karenlopez@infomed.sld.cu
Principal Investigator: Karen López Miguel, Dr.
Sub-Investigator: Yaniurka Cruz Camejo, Dr.
Sub-Investigator: Laura Selis Pomar, Dr.
Sub-Investigator: Alejandro Linchenat Lambert, Dr.
Sub-Investigator: Juan M. Silveira Pablo, Dr.
Sub-Investigator: Roberto Esperón Noa, Dr.
Sub-Investigator: Karelia Silvera Candó, BSc
Sub-Investigator: Vivian Diéguez Rodríguez, BSc
Sub-Investigator: Zaida Lastres Sosa, BSc
Sub-Investigator: Idelmis Curbelo Heredia, MSc
Sub-Investigator: Mircea Betancourt Cabeza, MSc
Sub-Investigator: Juan J. Lence Anta, Dr. Less << |
|
NCT00002539
|
Sarcoma |
Phase 3 |
Completed |
- |
Belgium
... More >> Institut Jules Bordet
Brussels (Bruxelles), Belgium, 1000
Cliniques Universitaires Saint-Luc
Brussels (Bruxelles), Belgium, 1200
Universitair Ziekenhuis Antwerpen
Edegem, Belgium, B-2650
U.Z. Gasthuisberg
Leuven, Belgium, B-3000
Denmark
Aarhus Kommunehospital
Aarhus, Denmark, DK-8000
Rigshospitalet
Copenhagen, Denmark, 2100
France
Centre Eugene Marquis
Rennes, France, 35064
Netherlands
Onze Lieve Vrouwe Gasthuis
Amsterdam, Netherlands, 1091 HA
Emma Kinderziekenhuis
Amsterdam, Netherlands, NL-1100 DE
Leiden University Medical Center
Leiden, Netherlands, 2300 CA
University Medical Center Nijmegen
Nijmegen, Netherlands, NL-6500 HB
Academisch Ziekenhuis Utrecht
Utrecht, Netherlands, 3584 CX
Portugal
Instituto Portugues de Oncologia de Francisco Gentil-Centro de Lisboa
Lisbon, Portugal, 1099-023 Codex
Saudi Arabia
King Faisal Specialist Hospital and Research Centre
Riyadh, Saudi Arabia, 11211
Slovenia
Institute of Oncology, Ljubljana
Ljubljana, Slovenia, Sl-1000
United Kingdom
St. James's Hospital
Leeds, England, United Kingdom, LS9 7TF Less << |
|
NCT00002669
|
Melanoma (Skin) |
Phase 2 |
Completed |
- |
Austria
... More >> Landeskrankenanstalten - Salzburg
Salzburg, Austria, A-5020
Belgium
Institut Jules Bordet
Brussels (Bruxelles), Belgium, 1000
Hopital Universitaire Erasme
Brussels, Belgium, 1070
Universitair Ziekenhuis Antwerpen
Edegem, Belgium, B-2650
U.Z. Gasthuisberg
Leuven, Belgium, B-3000
France
CHR de Besancon - Hopital Saint-Jacques
Besancon, France, 25030
Centre Leon Berard
Lyon, France, 69373
CHU Pitie-Salpetriere
Paris, France, 75651
Germany
Universitaetsklinikum Charite
Berlin, Germany, D-10117
Universitaetsklinikum Benjamin Franklin
Berlin, Germany, D-12200
Robert Roessle Klinik
Berlin, Germany, D-13122
Haematologisch-Onkologische Praxis Altona
Hamburg, Germany, D-22765
Johannes Gutenberg University
Mainz, Germany, D-55101
III Medizinische Klinik Mannheim
Mannheim, Germany, D-68135
Italy
Istituto Europeo Di Oncologia
Milano, Italy, 20141
Netherlands
University Medical Center Nijmegen
Nijmegen, Netherlands, NL-6500 HB
Rotterdam Cancer Institute
Rotterdam, Netherlands, 3075 EA
Portugal
Instituto Portugues de Oncologia do Porto
Porto, Portugal, 4200
Switzerland
Centre Hospitalier Universitaire Vaudois
Lausanne, Switzerland, CH-1011
Universitaetsspital
Zurich, Switzerland, CH-8091
United Kingdom
St. James's Hospital
Leeds, England, United Kingdom, LS9 7TF
Royal Marsden NHS Trust
London, England, United Kingdom, SW3 6JJ
Southend NHS Trust Hospital
Westcliff-On-Sea, England, United Kingdom
Royal Bournemouth Hospital
Bournemouth, United Kingdom, BH7 7DW Less << |
|
NCT00563862
|
Nasopharyngeal Neoplasms
... More >> Carcinoma, Squamous Cell Less << |
Not Applicable |
Terminated |
- |
China
... More >> Pamela Youde Nethersole Eastern Hospital
Hong Kong, China
Prince of Wales Hospital
Hong Kong, China
Queen Elizabeth Hospital
Hong Kong, China
Queen Mary Hospital
Hong Kong, China
Tuen Mun Hospital
Hong Kong, China Less << |
|
NCT03656393
|
NSCLC
EGFR |
Phase 3 |
Recruiting |
September 30, 2020 |
China, Guangdong
... More >>
Shenzhen People's Hospital
Recruiting
Shenzhen, Guangdong, China, 518000
Contact: wang wei, Doctor 15914030296 limey@sina.com
Contact: wang suo, Doctor 13590437796 908611104@qq.com Less << |
|
NCT00003583
|
Drug/Agent Toxicity by Tissue/... More >>Organ
Lung Cancer
Radiation Toxicity Less << |
Phase 2 |
Unknown |
- |
United States, Florida
... More >>
University of Florida - Gainesville
Gainesville, Florida, United States, 32610-0277 Less << |
|
NCT00002477
|
Ovarian Cancer |
Phase 3 |
Unknown |
- |
United Kingdom
... More >> Cochrane Cancer Network
Oxford, England, United Kingdom, OX3 7LF Less << |
|
NCT00003636
|
Fallopian Tube Cancer
... More >> Ovarian Cancer
Primary Peritoneal Cavity Cancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT03100708
|
- |
|
Recruiting |
April 2021 |
Germany
... More >> Universitätsklinikum Leipzig - AöR
Recruiting
Leipzig, Sachsen, Germany, 04103 Less << |
|
NCT00002623
|
Lung Cancer |
Phase 3 |
Completed |
- |
Belgium
... More >> Algemeen Ziekenhuis Middelheim
Antwerp, Belgium, 2020
A.Z. St. Jan
Brugge, Belgium, 8000
Universitair Ziekenhuis Antwerpen
Edegem, Belgium, B-2650
Hopital de Jolimont
Haine Saint Paul, Belgium, 7100
U.Z. Gasthuisberg
Leuven, Belgium, B-3000
CHR - Clinique Saint Joseph - Hopital de Warqueguies
Mons, Belgium, B-7000
Clinique Universitaire De Mont-Godinne
Mont-Godinne Yvoir, Belgium, 5530
Stedelijk Ziekenhuis
Roesclare, Belgium, 8800
France
Academisch Ziekenhuis Utrecht
Vandoeuvre-les-Nancy, France, 54511
Italy
Istituto Nazionale per la Ricerca sul Cancro
Genoa (Genova), Italy, 16132
Netherlands
Groot Ziekengasthuis 's-Hertogenbosch
's-Hertogenbosch, Netherlands, 5211 NL
Vrije Universiteit Medisch Centrum
Amsterdam, Netherlands, 1001HV
Antoni van Leeuwenhoekhuis
Amsterdam, Netherlands, 1066 CX
Arnhems Radiotherapeutisch Instituut
Arnhem, Netherlands, 6815 AD
Ziekenhuis St Jansdal
Harderwijk, Netherlands, 3840 AC
Atrium Medical Centre
Heerlen, Netherlands, 6419 PC
Elkerliek Ziekenhuis
Helmond, Netherlands, 5707-HA
Leiden University Medical Center
Leiden, Netherlands, 2300 CA
Sint Antonius Ziekenhuis
Nieuwegein, Netherlands, 3435 CM
Canisius-Wilhelmina Ziekenhuis
Nijmegen, Netherlands, 6532 SZ
University Medical Center Nijmegen
Nijmegen, Netherlands, NL-6500 HB
Saint Franciscus Ziekenhuis
Roosendaal, Netherlands, 4708 AE
University Hospital - Rotterdam Dijkzigt
Rotterdam, Netherlands, 3000 CA
Erasmus Medical Center
Rotterdam, Netherlands, 3075 EA
Twee Steden Ziekenhuis Vestiging Tilburg
Tilburg, Netherlands, 5042 AD
Diakonessenhuis Utrecht
Utrecht, Netherlands, 3508 TG
Sophia Ziekehuis
Zwolle, Netherlands, 8000 GK
United Kingdom
Leicester Royal Infirmary
Leicester, England, United Kingdom, LE1 5WW
Royal Marsden Hospital
Sutton, England, United Kingdom, SM2 5PT
Royal Victoria Hospital
Belfast, Northern Ireland, United Kingdom, BT12 6BA Less << |
|
NCT00004176
|
Drug/Agent Toxicity by Tissue/... More >>Organ
Lung Cancer
Radiation Toxicity Less << |
Phase 2 |
Completed |
- |
United States, Colorado
... More >>
Rocky Mountain Cancer Center
Denver, Colorado, United States, 80218
United States, Missouri
Washington University Barnard Cancer Center
Saint Louis, Missouri, United States, 63110
United States, North Carolina
Lineberger Comprehensive Cancer Center, UNC
Chapel Hill, North Carolina, United States, 27599-7295
United States, Pennsylvania
University of Pittsburgh Cancer Institute
Pittsburgh, Pennsylvania, United States, 15213 Less << |
|
NCT00003592
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
United States, Georgia
... More >>
Emory University Hospital - Atlanta
Atlanta, Georgia, United States, 30322
Veterans Affairs Medical Center - Atlanta (Decatur)
Decatur, Georgia, United States, 30033
United States, Illinois
Robert H. Lurie Comprehensive Cancer Center, Northwestern University
Chicago, Illinois, United States, 60611
Veterans Affairs Medical Center - Chicago (Lakeside)
Chicago, Illinois, United States, 60611
CCOP - Illinois Oncology Research Association
Peoria, Illinois, United States, 61602
CCOP - Carle Cancer Center
Urbana, Illinois, United States, 61801
United States, Michigan
CCOP - Kalamazoo
Kalamazoo, Michigan, United States, 49007-3731
United States, Minnesota
CCOP - Metro-Minnesota
Saint Louis Park, Minnesota, United States, 55416
United States, Nebraska
CCOP - Missouri Valley Cancer Consortium
Omaha, Nebraska, United States, 68131
United States, New Jersey
Veterans Affairs Medical Center - East Orange
East Orange, New Jersey, United States, 07018-1095
United States, New York
Albert Einstein Comprehensive Cancer Center
Bronx, New York, United States, 10461
University of Rochester Cancer Center
Rochester, New York, United States, 14642
United States, Ohio
Ireland Cancer Center
Cleveland, Ohio, United States, 44106-5065
CCOP - Toledo Community Hospital Oncology Program
Toledo, Ohio, United States, 43623-3456
United States, Pennsylvania
Fox Chase Cancer Center
Philadelphia, Pennsylvania, United States, 19111
United States, Tennessee
Veterans Affairs Medical Center - Nashville
Nashville, Tennessee, United States, 37212
Vanderbilt Cancer Center
Nashville, Tennessee, United States, 37232-6838
United States, Wisconsin
Medical College of Wisconsin
Milwaukee, Wisconsin, United States, 53226
Veterans Affairs Medical Center - Milwaukee (Zablocki)
Milwaukee, Wisconsin, United States, 53295
South Africa
Pretoria Academic Hospital
Pretoria, South Africa, 0001 Less << |
|
NCT00791141
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
Germany
... More >> Department of Radiotherapy and Radiological Oncology, University Hospital Munich
Munich, Bavaria, Germany, 81377
Department of Radiotherapeutics of the University Hospital Freiburg
Freiburg, BW, Germany, 79106
Department of Radiological Oncology University Hospital Heidelberg
Heidelberg, BW, Germany, 69120
Department of Radiotherapy and Radiological Oncology University Hospital Tuebingen
Tuebingen, BW, Germany, 72076
Department of Radiotherapy and Radiological Oncology University Hospital Ulm
Ulm, BW, Germany, 89091
Department of Radiotherapy an Radiological Oncology University Hospital Essen
Essen, NW, Germany, 45122
Department of Radiotherapy and Radiological Oncology University Hospital Mainz
Mainz, Rheinland-Pfalz, Germany, 55131
Department of Radiotherapy, University Hospital Schleswig Holstein, Campus Lübeck
Lübeck, Schleswig Hostein, Germany, 23538
Department of Radiotherapy and Radiological Oncology Universität Hospital Jena
Jena, Thueringen, Germany, 07743
Charité University Medicine, Department of Radiotherapy and Radiological Oncology
Berlin, Germany, 13353 Less << |
|
NCT02009449
|
Melanoma
Pros... More >>tate Cancer
Ovarian Cancer
Renal Cell Carcinoma
Colorectal Carcinoma
Pancreatic Carcinoma
Non-small Cell Lung Carcinoma
Solid Tumors
Breast Cancer Less << |
Phase 1 |
Active, not recruiting |
March 2020 |
United States, California
... More >>
UCLA Medical Hematology & Oncology
Los Angeles, California, United States, 90024
UCSF
San Francisco, California, United States
United States, Colorado
Sarah Cannon Research Institute at HealthONE
Denver, Colorado, United States, 80218
United States, Florida
Florida Cancer Specialists & Research Institute
Sarasota, Florida, United States, 34232
United States, Massachusetts
Dana Farber Cancer Institute
Boston, Massachusetts, United States, 02215
United States, New York
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
United States, Oklahoma
Stephenson Cancer Center at Oklahoma University TSET Phase 1 Program
Oklahoma City, Oklahoma, United States, 73104
United States, Tennessee
Sarah Cannon Research Institute
Nashville, Tennessee, United States, 37203
United States, Texas
The University of Texas M.D. Anderson Cancer Center
Houston, Texas, United States, 77030
South Texas Accelerated Research Therapeutics
San Antonio, Texas, United States, 78229 Less << |
|
NCT02999087
|
HNSCC |
Phase 3 |
Recruiting |
January 2021 |
France
... More >> Centre Hospitalier Bretagne Sud
Recruiting
Lorient, France, 56322
Contact: Christian SIRE, MD (0)2 97 06 96 84 ext +33 c.sire@ch-bretagne-sud.fr
Contact: Guillaume BERA, MD (0)2 97 06 96 84 ext +33 g.bera@ch-bretagne-sud.fr Less << |
|
NCT01843647
|
Non-small-cell Lung Cancer |
Phase 2 |
Unknown |
February 2018 |
China, Zhejiang
... More >>
Zhejiang cancer hospital
Recruiting
Hangzhou, Zhejiang, China, 310022
Contact: Weimin Mao, MD 86-571-88122182
Principal Investigator: Weimin Mao, MD Less << |
|
NCT00300586
|
Stage IV Non-small Cell Lung C... More >>ancer Less << |
Phase 3 |
Completed |
- |
France
... More >> Maurice PEROL
Lyon, France, 69317 Less << |
|
NCT01260103
|
Optic Nerve Glioma |
Phase 3 |
Withdrawn(Withdrawn study halt... More >>ed prematurely, prior to enrollment of first participant) Less << |
December 2018 |
- |
|
NCT00806117
|
Cervical Cancer |
Phase 3 |
Active, not recruiting |
December 2018 |
China, Guangdong
... More >>
Department of Gynecologic Oncology, Sun Yat-sen University Cancer Center
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT00533299
|
Ovarian Cancer |
Phase 3 |
Unknown |
December 2009 |
Mexico
... More >> Instituto Nacional de Cancerologia
Recruiting
Mexico City, Tlalpan, Mexico, 14080
Principal Investigator: Dolores Gallardo, MD Less << |
|
NCT03717610
|
Ovarian Cancer Recurrent |
Phase 3 |
Recruiting |
December 31, 2020 |
Sweden
... More >> Uppsala University Hopsital
Recruiting
Uppsala, Sweden, 75185
Contact: Marta Lomnytska, PhD 018-611 00 00 ext +46 marta.lomnytska@akademiska.se
Contact: Ilvars Silins, PhD 018-611 00 00 ext +46 ilvars.silins@akademiska.se
Sub-Investigator: Wilhelm Graf, Professor Less << |
|
NCT02495896
|
Head and Neck Squamous Cell Ca... More >>rcinoma
Metastatic Pancreatic Adenocarcinoma
Non-Resectable Cholangiocarcinoma
Pancreatic Adenocarcinoma
Recurrent Gallbladder Carcinoma
Recurrent Non-Small Cell Lung Carcinoma
Stage III Pancreatic Cancer
Stage IIIA Gallbladder Cancer
Stage IIIA Non-Small Cell Lung Cancer
Stage IIIB Gallbladder Cancer
Stage IIIB Non-Small Cell Lung Cancer
Stage IV Gallbladder Cancer
Stage IV Non-Small Cell Lung Cancer
Stage IV Pancreatic Cancer
Unresectable Gallbladder Carcinoma
Unresectable Pancreatic Cancer Less << |
Phase 1 |
Recruiting |
December 3, 2020 |
United States, California
... More >>
USC / Norris Comprehensive Cancer Center
Recruiting
Los Angeles, California, United States, 90033
Contact: Anthony El-Khoueiry 323-865-3967 elkhouei@usc.edu
Principal Investigator: Anthony El-Khoueiry, MD
Hoag Memorial Hospital Presbyterian
Recruiting
Newport Beach, California, United States, 92663
Contact: Alicia Bogardus, MA 949-764-6755 Alicia.bogardus@hoag.org
Contact: Cristina De Leon, RN 949-764-5543 Cristina.deleon@hoag.org
Sub-Investigator: Diana Hanna, MD Less << |
|
NCT00404703
|
Non-Small Cell Lung Cancer |
Phase 2 |
Terminated(Primary (safety) en... More >>dpoint reached) Less << |
- |
Australia
... More >>
Tugun, Australia
Wollongong, Australia
Belgium
Liege, Belgium
Czech Republic
Ostrava, Czech Republic
Usti nad Labem, Czech Republic
France
Bobigny, France
Marseille, France
Nantes, France
Hungary
Szekesfehervar, Hungary
Szombathely, Hungary
Israel
Kfar Saba, Israel
Ramat Gan, Israel
Poland
Lublin, Poland
Poznan, Poland
Szczecin, Poland
Warszawa, Poland
Russian Federation
Balashikha, Russian Federation
Moscow, Russian Federation
St. Petersburg, Russian Federation
Spain
Madrid, Spain
Sevilla, Spain
Taiwan
Kueishan, Taiwan
Taichung, Taiwan
Taipei, Taiwan
Taoyuan, Taiwan Less << |
|
NCT00005051
|
Ovarian Cancer |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Albert Einstein Clinical Cancer Center
Bronx, New York, United States, 10461
New York Hospital Medical Center of Queens
Fresh Meadows, New York, United States, 11365
Saint Vincent Catholic Medical Center of New York
New York, New York, United States, 10011
NYU School of Medicine's Kaplan Comprehensive Cancer Center
New York, New York, United States, 10016
St. Luke's-Roosevelt Hospital Center - Roosevelt Division
New York, New York, United States, 10019
New York Weill Cornell Cancer Center at Cornell University
New York, New York, United States, 10021
Mount Sinai Medical Center, NY
New York, New York, United States, 10029
Herbert Irving Comprehensive Cancer Center at Columbia University
New York, New York, United States, 10032
New York Medical College
Valhalla, New York, United States, 10595
United States, Wisconsin
University of Wisconsin Comprehensive Cancer Center
Madison, Wisconsin, United States, 53792-3236 Less << |
|
NCT01674738
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Withdrawn(Decision of the Spon... More >>sor, as the funding of the study was no longer guaranteed.) Less << |
- |
- |
|
NCT00806923
|
Non-Squamous Non-Small Cell Lu... More >>ng Cancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT01232374
|
Squamous Cell Carcinoma |
Phase 2 |
Unknown |
January 2016 |
China, Beijing
... More >> Chinese Academy of Medical Sciences Cancer Hospital
Beijing, Beijing, China, 266000
China, Fujian
Fujian Provincial Tumor Hospital
Fuzhou, Fujian, China, 350000
China, Guangdong
Cancer Center of Sun Yat-sen
Guangzhou, Guangdong, China, 510000
China, Henan
First Affiliated Hospital of Zhengzhou University
Zhengzhou, Henan, China, 450052
China, Hubei
Huazhong University of Science and Technology, Union Hospital, Tongji Medical College
Wuhan, Hubei, China, 430000
China, Jiangsu
Cancer Hospital of Jiangsu Province
Nanjing, Jiangsu, China, 210000
The First Affiliated Hospital of Soochow University,Suzhou First People's Hospital
Suzhou, Jiangsu, China, 215006
Northern Jiangsu People's Hospital
Yangzhou, Jiangsu, China, 225001
China, Jilin
The first bethune hospital of Jilin university
Changchun, Jilin, China
China, Shandong
Affiliated Hospital of Qingdao University Medical College
Qingdao, Shandong, China, 266000
China, Shanghai
Fudan University Shanghai Cancer Center
Shanghai, Shanghai, China, 200000
Shanghai Chest Hospital
Shanghai, Shanghai, China, 201100
Shanghai First People's Hospital
Shanghai, Shanghai, China, 201100
China, Shanxi
Fourth Military Medical University Xijing Hospital
Xian, Shanxi, China, 710000
China, Tianjin
Tianjin Cancer Hospital
Tianjin, Tianjin, China, 300000
China, Zhejiang
Zhejiang Cancer Hospital
Hangzhou, Zhejiang, China, 310000
The First Affiliated Hospital of College of Medicine, Zhejiang University
Hangzhou, Zhejiang, China, 310003 Less << |
|
NCT00176137
|
Non-Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT01014767
|
Brain Cancer
... More >>Choroid Plexus Tumors Less << |
Phase 3 |
Suspended(PI departure from co... More >>ordinating institution) Less << |
- |
United States, Massachusetts
... More >>
Tufts Medical Center
Boston, Massachusetts, United States, 02111
United States, Texas
Children's Cancer Hospital at UT MD Anderson Cancer Center
Houston, Texas, United States, 77030
Germany
St. Hedwig Children's Hospital, University of Regensburg (International Study Center)
Regensburg, Germany
Hungary
Semmelweis University
Budapest, Hungary
New Zealand
Christchurch Hospital
Christchurch, New Zealand Less << |
|
NCT00012220
|
Pancreatic Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT02368990
|
- |
|
Withdrawn(Since the conception... More >> of the study, the competitive environment has developed and as a result this study is no longer able to generate the necessary information.) Less << |
June 2018 |
- |
|
NCT00573131
|
Esophageal Cancer
... More >> Adenocarcinoma of the Esophagus
Squamous Cell Carcinoma Less << |
Phase 2 |
Terminated(OncoGel did not sho... More >>w any impact on overall tumor response) Less << |
- |
United States, California
... More >>
University of California San Diego
San Diego, California, United States, 92093
United States, Illinois
Rush University Medical Center
Chicago, Illinois, United States, 60612
University of Chicago Medical Center
Chicago, Illinois, United States, 60637
United States, Indiana
Indiana University Medical Center
Indianapolis, Indiana, United States, 46202
United States, Kansas
University of Kansas Medical Center
Kansas City, Kansas, United States, 66160
United States, Pennsylvania
Thomas Jefferson University
Philadelphia, Pennsylvania, United States, 19107
United States, Texas
Baylor University Medical Center
Dallas, Texas, United States, 75246
Digestive Health Specialists of Tyler, Texas
Tyler, Texas, United States, 75701
Czech Republic
University Hospital Brno
Brno, Czech Republic, 62500
Hospital Jablonec nad Nisou
Jablonec nad Nisou, Czech Republic, 466 60
University Hospital Olomouc
Olomouc, Czech Republic, 775 20
University Hospital Motol
Praha, Czech Republic, 150 06
Massaryk's Hospital in Usti nad Labem
Usti nad Labem, Czech Republic, 401 13
India
Kidwai Memorial Institute of Oncology
Bangalore, Karnataka, India, 560029
Amrita Institute of Medical Sciences
Kochi, Kerala, India, 682026
Bombay Hospital & Medical Research Centre
Mumbai, Maharashtra, India, 400020
Deenanath Mangeshkar Hospital
Erandwane, Pune, India, 411004
Meenakshi Mission Hospital and Research Centre
Madurai, Tamil Nadu, India, 625107
Poland
Samodzielny Publiczny Szpital Kliniczny
Lublin, Poland, 20-081
Samodzielnego Publicznego Szpitala Klinicznego
Szczecin, Poland, 70-111 Less << |
|
NCT02001168
|
Non-small Cell Lung Cancer |
Phase 3 |
Active, not recruiting |
October 2022 |
China, Xinjiang
... More >>
Cancer Hospital of Xinjiang Medical University
Urumqi, Xinjiang, China, 830011 Less << |
|
NCT00263016
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Terminated(Slow recruitment) |
- |
Hong Kong
... More >> Sanofi-aventis
Hong Kong, Hong Kong Less << |
|
NCT02844114
|
Carcinoma, Non-Small-Cell Lung |
Phase 2 |
Recruiting |
January 2018 |
China, Liaoning
... More >>
First Hospital of Dalian Medical University
Recruiting
Dalian, Liaoning, China, 116000
Contact: Yajie Gao, Professor 0411-83634153 gaoyajie@126.com
Contact: Yan Dong, Physician 0411-83634153 dongyan979828@hotmail.com
Principal Investigator: Yajie Gao, Professor
Sub-Investigator: Yan Dong, Physician
Sub-Investigator: Bin Zhang, Physician
Sub-Investigator: Hui Mi, Postgraduate Less << |
|
NCT02365935
|
Uterine Cervical Neoplasms |
Not Applicable |
Completed |
- |
Italy
... More >> campus bio-medico of Rome
Rome, Italy, 00128 Less << |
|
NCT01868022
|
Neoplasms |
Phase 1 |
Completed |
- |
- |
|
NCT00006004
|
Lung Cancer |
Phase 2 |
Completed |
- |
United States, Georgia
... More >>
Emory University Hospital - Atlanta
Atlanta, Georgia, United States, 30322
United States, Iowa
Iowa Methodist Medical Center
Des Moines, Iowa, United States, 50309
Mercy Medical Center
Des Moines, Iowa, United States, 50314
Iowa Lutheran Hospital
Des Moines, Iowa, United States, 50316-2301
United States, Nebraska
Alegent Health-Midlands Community Hospital
Papillion, Nebraska, United States, 68128-4157
United States, New Jersey
CCOP - Northern New Jersey
Hackensack, New Jersey, United States, 07601
Cancer Institute of New Jersey
New Brunswick, New Jersey, United States, 08903
United States, New Mexico
MBCCOP - University of New Mexico HSC
Albuquerque, New Mexico, United States, 87131
United States, New York
James P. Wilmot Cancer Center
Rochester, New York, United States, 14642
United States, Oklahoma
CCOP - Oklahoma
Tulsa, Oklahoma, United States, 74136
United States, Pennsylvania
Milton S. Hershey Medical Center
Hershey, Pennsylvania, United States, 17033-0850
United States, Texas
CCOP - Scott and White Hospital
Temple, Texas, United States, 76508
United States, Wisconsin
CCOP - St. Vincent Hospital Cancer Center, Green Bay
Green Bay, Wisconsin, United States, 54307-3453
Australia, New South Wales
Westmead Hospital
Westmead, New South Wales, Australia, 2145
Peru
Instituto de Enfermedades Neoplasicas
Lima, Peru, 34
Puerto Rico
San Juan City Hospital
San Juan, Puerto Rico, 00936-7344 Less << |
|
NCT02644863
|
Esophageal Neoplasms
... More >> Neoplasms
Digestive System Neoplasms
Esophageal Cancer Less << |
Phase 2 |
Recruiting |
May 2019 |
China, Guangdong
... More >>
Affiliated Tumor Hospital of Guangzhou Medical University
Recruiting
Guangzhou, Guangdong, China, 510000
Contact: Zhiyuan Wang wangzhiyuan@hornetcorn.com Less << |
|
NCT02899195
|
Mesothelioma
... More >>Pleural Mesothelioma Less << |
Phase 2 |
Active, not recruiting |
December 2020 |
- |
|
NCT02161692
|
Testicular Neoplasms
... More >> Germ Cell Tumors Less << |
Phase 2 |
Completed |
- |
Italy
... More >> Istituto Nazionale dei Tumori
Milano, Italy, 20133 Less << |
|
NCT00005047
|
Bladder Cancer |
Phase 3 |
Terminated(Accrual was halted ... More >>on the basis of the Data and Safety Monitoring Board review of a futility analysis.) Less << |
- |
- |
|
NCT00005047
|
- |
|
Terminated(Accrual was halted ... More >>on the basis of the Data and Safety Monitoring Board review of a futility analysis.) Less << |
- |
- |
|
NCT02624531
|
Cervical Cancer |
Not Applicable |
Recruiting |
November 2020 |
China, Guangdong
... More >>
Clinical Trial Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Jihong Liu, Ph.D. 86-20-87343102 Liujih@mail.sysu.edu.cn
Contact: Yanling Feng, Ph.D. 86-20-87343104 fengyl@sysucc.org.cn
Sun Yat-sen University Cancer Center
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Jihong Liu, Ph.D. 86-20-87343102 Liujih@mail.sysu.edu.cn
Contact: Yanling Feng, Ph.D. 86-20-87343104 fengyl@sysucc.org.cn Less << |
|
NCT00573131
|
- |
|
Terminated(OncoGel did not sho... More >>w any impact on overall tumor response) Less << |
- |
- |
|
NCT02766348
|
NSCLC |
Phase 2 |
Unknown |
December 2018 |
China, Hubei
... More >> Jingzhou Central hospital Immunotherapy center
Not yet recruiting
Jingzhou, Hubei, China, 434020
Contact: Fan Fu, Researcher +86 15027082046 Fufan@hornetcorn.com
Contact: Fan Fu Less << |
|
NCT01363479
|
- |
|
Completed |
- |
- |
|
NCT00572572
|
Germ Cell Tumors |
Phase 3 |
Completed |
- |
United States, Indiana
... More >>
Indiana University Simon Cancer Center
Indianapolis, Indiana, United States, 46202
Medical Consultants, P.C.
Muncie, Indiana, United States, 47303
United States, Missouri
Siteman Cancer Center
St. Louis, Missouri, United States, 63110
United States, Oregon
Providence Portland Medical Center
Portland, Oregon, United States, 97213
United States, Pennsylvania
University of Pennsylvania
Philadelphia, Pennsylvania, United States, 19104
United States, Wisconsin
Froedtert/Medical College of Wisconsin
Milwaukee, Wisconsin, United States, 53226 Less << |
|
NCT00572572
|
- |
|
Completed |
- |
- |
|
NCT01363479
|
Chemotherapy-Induced Nausea an... More >>d Vomiting Less << |
Phase 3 |
Completed |
- |
- |
|
NCT00577707
|
Non Small Cell Lung Cancer
... More >> Lung Cancer Less << |
Phase 2 |
Completed |
- |
United States, New York
... More >>
Memorial Sloan-Kettering Cancer Center
New York, New York, United States, 10065 Less << |
|
NCT02915965
|
Stage II and III Esophageal Sq... More >>uamous Cell Carcinoma Less << |
Phase 2 |
Unknown |
- |
China, Guangdong
... More >>
The Sixth Affiliated Hospital of Sun Yat-sen University
Guangzhou, Guangdong, China, 510655 Less << |
|
NCT01055496
|
Lymphoma, B-Cell |
Phase 1 |
Completed |
- |
- |
|
NCT02819999
|
Small Cell Lung Cancer |
Phase 1 |
Recruiting |
December 2020 |
United States, Colorado
... More >>
University of Colorado
Recruiting
Aurora, Colorado, United States, 80010
Principal Investigator: David Camidge, MD
Rocky Mountain Cancer Centers
Recruiting
Denver, Colorado, United States, 80218
Principal Investigator: Robert Jotte, MD
United States, Florida
Cancer Institute of Florida
Recruiting
Orlando, Florida, United States, 32804
Principal Investigator: Tarek Mekhail, MD
United States, Maryland
Johns Hopkins Sidney Kimmel Comprehensive Cancer Center
Recruiting
Baltimore, Maryland, United States, 21231
Principal Investigator: Christine Hann, MD
United States, Missouri
Washington University
Recruiting
Saint Louis, Missouri, United States, 63110
Principal Investigator: Daniel Morganzstern, MD
United States, Ohio
Oncology Hematology Care
Recruiting
Cincinnati, Ohio, United States, 45242
Principal Investigator: Patrick Ward, MD
University Hospital of Cleveland
Recruiting
Cleveland, Ohio, United States, 44106
Principal Investigator: Afshin Dowlati, MD
Cleveland Clinic
Recruiting
Cleveland, Ohio, United States, 44195
Principal Investigator: Nathan Pennell, MD, PhD
United States, Pennsylvania
University of Pittsburgh
Recruiting
Pittsburgh, Pennsylvania, United States, 15232
Principal Investigator: Timothy Burns, MD
United States, Texas
Texas Oncology
Recruiting
Fort Worth, Texas, United States, 76104
Principal Investigator: Stephen Richey, MD
Texas Oncology
Recruiting
San Antonio, Texas, United States, 78258
Contact 800-381-2637
Principal Investigator: Sridhar Beeram, MD Less << |
|
NCT02135042
|
Epstein-Barr Virus Infection
... More >> Stage II Nasopharyngeal Carcinoma
Stage III Nasopharyngeal Carcinoma
Stage IVA Nasopharyngeal Carcinoma
Stage IVB Nasopharyngeal Carcinoma Less << |
Phase 2
Phase 3 |
Recruiting |
July 2026 |
- |
|
NCT00446225
|
Non-Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00002883
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 3 |
Unknown |
- |
France
... More >> Centre Regional de Lutte Contre le Cancer - Centre Val d'Aurelle
Montpellier, France, 34298 Less << |
|
NCT01774786
|
Gastric Cancer |
Phase 3 |
Active, not recruiting |
October 6, 2021 |
- |
|
NCT00403403
|
Small Cell Lung Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT01774786
|
- |
|
Active, not recruiting |
- |
- |
|
NCT00632333
|
Esophageal Cancer |
Phase 1 |
Unknown |
March 2012 |
Japan
... More >> Teikyo University
Recruiting
2-11-1 Kaga Itabashi-ku, Tokyo, Japan, 173-0003
Contact: Hisae Iinuma, PhD +81-33-964-1211 iinuma@med.teikyo-u.ac.jp
Principal Investigator: Hisae Iinuma, PhD Less << |
|
NCT00577707
|
- |
|
Completed |
- |
- |
|
NCT00001332
|
Peritoneal Neoplasms
... More >> Stomach Neoplasms Less << |
Phase 1 |
Completed |
- |
United States, Maryland
... More >>
National Cancer Institute (NCI)
Bethesda, Maryland, United States, 20892 Less << |
|
NCT01215266
|
Bladder Cancer |
Phase 2 |
Terminated(Slow recruitment) |
- |
- |
|
NCT00142350
|
Gastric Cancer |
Phase 3 |
Completed |
- |
Japan
... More >> Aichi Cancer Center Hospital
Nagoya,Chikusa-ku,Kanokoden,1-1, Aichi, Japan, 464-8681
Nagoya Medical Center
Nagoya,Naka-ku,Sannomaru,4-1-1, Aichi, Japan, 460-0001
Aichi Cancer Center,Aichi Hospital
Okazaki,Kake-machi,Kuriyado,18, Aichi, Japan, 444-0011
Aomori Prefectural Central Hospital
Higashitsukurimiti,2-1-1,Aomori, Aomori, Japan, 030-0911
Chiba Cancer Center Hospital
Chiba,Chuo-ku,Nitona-cho,666-2, Chiba, Japan, 260-8717
Asahi General Hospital
I-1326,Asahi, Chiba, Japan, 289-2511
National Cancer Center Hospital East
Kashiwa,Kashiwanoha,6-5-1, Chiba, Japan, 277-8577
National Hospital Organization Shikoku Cancer Center
Matsuyama,Horinouchi,13, Ehime, Japan, 790-0007
Kyushu University Hospital
Fukuoka,Higashi-ku,Maidashi,3-1-1, Fukuoka, Japan, 812-8582
National Kyushu Cancer Center
Fukuoka,Minami-ku,Notame,3-1-1, Fukuoka, Japan, 811-1395
Federation of national public service personnel mutual aid associations Tonan Hospital
kita 1 nishi 6,Chuo-ku,Sapporo, Hokkaido, Japan, 060-0001
Hokkaido University Hospital
North-14 West-5 Kita-ku,Sapporo, Hokkaido, Japan, 060-8648
Hyogo Medical Center for Adults
Akashi,Kitaouji-cho,13-70, Hyogo, Japan, 673-8558
Kobe University Graduate School of Medicine
Kobe,Chuo-ku,Kusunoki-cho,7-5-2, Hyogo, Japan, 650-0017
Ibaraki Kenritsu Chuo Hospital & Cancer Center
Nishi-ibarakigun,Tomobemachi,Koibuchi,6528, Ibaraki, Japan, 309-1793
Iwate Prefectural Central Hospital
Morioka,Ueda,1-4-1, Iwate, Japan, 020-0066
Kitasato University East Hospital
Sagamihara,Asamizodai,2-1-1, Kanagawa, Japan, 228-8520
Kanagawa Cancer Center
Yokohama,Asahi-ku,Nakao,1-1-2, Kanagawa, Japan, 241-0815
Yokohama Mucipical Citizen's Hospital
Yokohama,Hodogaya-ku,Okazawa-cho,56, Kanagawa, Japan, 240-8555
Yokohama City University Medical Center
Yokohama,Minami-ku,Urafunecho,4-57, Kanagawa, Japan, 232-0024
Kochi Health Science Center
Kochi,Ike,2125-1, Kochi, Japan, 781-8555
Kumamoto Regional Medical Center Hospital
Kumamoto,Honjo,5-16-10, Kumamoto, Japan, 860-0811
Kyoto University Hospital
Kyoto,Sakyo-ku,Syogoinkawara,54, Kyoto, Japan, 606-8507
Tohoku University Hospital
Sendai,Aoba-ku,Seiryo-machi,1-1, Miyagi, Japan, 980-8574
Saku Central Hospital
Saku,Usuda,197, Nagano, Japan, 384-0301
Osaka Medical College
Takatsuki,Daigakucho,2-7, Osaka, Japan, 569-0801
Saitama Medical School Hospital
Iruma-gun,Moroyama-machi,Morohongo,38, Saitama, Japan, 350-0495
Saitama Cancer Center
Kita-adachi,Ina,Komuro,818, Saitama, Japan, 362-0806
Div. of Gastrointestinal Oncology, Shizuoka Cancer Center
Sunto-gun, Nagaizumi-cho, Shimonagakubo,1007, Shizuoka, Japan, 411-8777
Tochigi Cancer Center
Utsunomiya,Yohnan,4-9-13, Tochigi, Japan, 320-0834
National Cancer Center Hospital
Chuo-ku,Tsukiji,5-1-1, Tokyo, Japan, 104-0045
Cancer Institute Hospital
Koto-ku,Ariake,3-10-6, Tokyo, Japan, 135-8550
Showa University School of Medicine
Shinagawa-ku,Hatanodai,1-5-8, Tokyo, Japan, 142-8666
Yamagata Prefectural Central Hospital
Yamagata,Aoyagi,1800, Yamagata, Japan, 990-2292 Less << |
|
NCT01625702
|
Gastric Cancer |
Not Applicable |
Completed |
- |
China, Beijing
... More >> Department of GI Oncology, Peking University Cancer Hospital
Beijing, Beijing, China, 100142 Less << |
|
NCT01958372
|
Adenocarcinoma of the Lung
... More >> Adenosquamous Cell Lung Cancer
Bronchoalveolar Cell Lung Cancer
Large Cell Lung Cancer
Recurrent Non-small Cell Lung Cancer
Squamous Cell Lung Cancer
Stage IIIA Non-small Cell Lung Cancer
Stage IIIB Non-small Cell Lung Cancer Less << |
Phase 1 |
Active, not recruiting |
June 30, 2018 |
United States, Michigan
... More >>
Karmanos Cancer Institute at McLaren Bay Region
Bay City, Michigan, United States, 48706
Barbara Ann Karmanos Cancer Institute
Detroit, Michigan, United States, 48201
Karmanos Cancer Institute at McLaren Lapeer Region
Lapeer, Michigan, United States, 48446
Karmanos Cancer Institute at McLaren Central Michigan
Mount Pleasant, Michigan, United States, 48858
Karmanos Cancer Institute at McLaren Northern
Petoskey, Michigan, United States, 49770 Less << |
|
NCT00403403
|
- |
|
Completed |
- |
- |
|
NCT02921984
|
Salivary Gland Tumors
... More >> Head and Neck Cancer Less << |
Phase 1 |
Completed |
- |
China, Shanghai
... More >>
Shanghai ninth people's hospital
Shanghai, Shanghai, China, 200011 Less << |
|
NCT02938273
|
Carcinoma, Squamous Cell of He... More >>ad and Neck Less << |
Phase 1 |
Recruiting |
October 2018 |
Netherlands
... More >> Antoni van Leeuwenhoek
Recruiting
Amsterdam, Netherlands, 1066CX
Contact: Jan Paul de Boer, MD,PhD +31 20 512 9111
Principal Investigator: Jan Paul de Boer, MD,PhD Less << |
|
NCT00808639
|
Bladder Cancer
... More >> Muscle-invasive Bladder Cancer Less << |
Phase 2 |
Completed |
- |
United States, Massachusetts
... More >>
Beth Israel Deaconess Medical Center
Boston, Massachusetts, United States, 02115
Dana-Farber Cancer Institute
Boston, Massachusetts, United States, 02115
Lahey Clinic
Burlington, Massachusetts, United States, 01805
United States, Pennsylvania
University of Pittsburgh Cancer Institute
Pittsburgh, Pennsylvania, United States, 15232 Less << |
|
NCT01343264
|
- |
|
Unknown |
- |
Germany
... More >> Dr. Horst Schmidt Klinik, Department of Thoracic Surgery
Recruiting
Wiesbaden, Germany
Contact: Joachim Schirren, MD, PhD +49 611 433132 joachim.schirren@hsk-wiesbaden.de
Principal Investigator: Joachim Schirren, MD, PhD
Sub-Investigator: Servet Bölükbas, MD, PhD Less << |
|
NCT01719835
|
Peripheral T-cell Lymphoma NOS... More >>
Anaplastic Large Cell Lymphoma, ALK-Negative
Angioimmunoblastic T-cell Lymphoma
Hepatosplenic Gamma/ Delta T-cell Lymphoma
Enteropathy-Associated T-Cell Lymphoma Less << |
Phase 2 |
Active, not recruiting |
August 2022 |
United Kingdom
... More >> Royal Marsden NHS Foundation Trust - London and Surrey
London, United Kingdom, SM2 5PT Less << |
|
NCT00006012
|
Drug/Agent Toxicity by Tissue/... More >>Organ
Lung Cancer
Radiation Toxicity Less << |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT00612495
|
Endometrial Cancer |
Phase 2 |
Completed |
- |
France
... More >> Sanofi-Aventis
Paris, France Less << |
|
NCT00808639
|
- |
|
Completed |
- |
- |
|
NCT00454779
|
Metastatic or Recurrent Squamo... More >>us Cell Carcinoma of Head and Neck Less << |
Phase 2 |
Completed |
- |
- |
|
NCT00454779
|
- |
|
Completed |
- |
- |
|
NCT01086826
|
Head and Neck Squamous Cell Ca... More >>rcinoma Less << |
Phase 3 |
Completed |
- |
- |
|
NCT02215356
|
Non-small Cell Lung Cancer |
Phase 2 |
Active, not recruiting |
February 2019 |
- |
|
NCT01640782
|
Adenocarcinoma of the Stomach
... More >>
Adenocarcinoma of the Gastroesophageal Junction Less << |
Phase 3 |
Completed |
- |
- |
|
NCT03308604
|
Gynecologic Cancer |
Phase 1 |
Recruiting |
May 2021 |
France
... More >> Gustave Roussy
Recruiting
Villejuif, Val De Marne, France, 94805
Contact: CHARGARI Cyrus, MD 0142113365 ext +33 cyrus.chargari@gustaveroussy.fr
Contact: PAOLETTI Xavier, MD 0142116564 ext +33 xavier.paoletti@gustaveroussy.fr
Principal Investigator: CHARGARI Cyrus, MD Less << |
|
NCT01817023
|
Nasopharyngeal Carcinoma |
Phase 3 |
Unknown |
April 2018 |
China, Guangdong
... More >>
Department of nasopharyngeal carcinoma, Cancer hospital, Sun Yat-Sen University
Not yet recruiting
Guangzhou, Guangdong, China, 510060
Contact: Xiang Guo, MD 8613902251681 guoxiang@sysucc.org.cn
Department of Radiation oncology, Cancer hospital, Sun Yat-Sen University
Recruiting
Guangzhou, Guangdong, China, 510060
Contact: Chong Zhao, MD 8613902206160 zhaochong@sysucc.org.cn
China, Hubei
Zhongnan Hospital of Wuhan University
Not yet recruiting
Wuhan, Hubei, China, 430030
Contact: Conghua Xie, MD 8613638607566 chxie_65@hotmail.com
Tongji hospital, Huazhong University of Science & Technology
Recruiting
Wuhan, Hubei, China, 430032
Contact: Guoqing Hu, MD 8613707189803 gqhu@tjh.tjmu.edu.cn
China, Jiangxi
Jiangxi province cancer hospital
Not yet recruiting
Nanchang, Jiangxi, China, 330029
Contact: Jingao Li, MD 8613970866296 lijingao@hotmail.com
China
Cancer hospital, Chinese Academy of Medical Sciences
Recruiting
Beijing, China, 100021
Contact: Junlin YI, MD 861087788504 junlinyi@sohu.com Less << |
|
NCT00454649
|
Neoplasms |
Phase 1 |
Completed |
- |
United States, Georgia
... More >>
Pfizer Investigational Site
Augusta, Georgia, United States, 30909
Pfizer Investigational Site
Augusta, Georgia, United States, 30912
United States, Illinois
Pfizer Investigational Site
Harvey, Illinois, United States, 60426-4265
Pfizer Investigational Site
Harvey, Illinois, United States, 60426
Pfizer Investigational Site
Tinley Park, Illinois, United States, 60477
United States, Indiana
Pfizer Investigational Site
Hobart, Indiana, United States, 46342
Pfizer Investigational Site
Munster, Indiana, United States, 46321
United States, Pennsylvania
Pfizer Investigational Site
Philadelphia, Pennsylvania, United States, 19111
United States, Texas
Pfizer Investigational Site
Houston, Texas, United States, 77030-4009
United States, Washington
Pfizer Investigational Site
Kennewick, Washington, United States, 99336
Poland
Pfizer Investigational Site
Warszawa, Poland, 02-781
Spain
Pfizer Investigational Site
Barcelona, Spain, 08003
Pfizer Investigational Site
Madrid, Spain, 28046 Less << |
|
NCT01519804
|
Non-Squamous Non-Small Cell Lu... More >>ng Cancer Less << |
Phase 2 |
Completed |
- |
- |
|
NCT02624999
|
Squamous Cell Carcinoma Head a... More >>nd Neck Less << |
Phase 2 |
Not yet recruiting |
May 2019 |
Thailand
... More >> National Cancer Institute of Thailand
Not yet recruiting
Bangkok, Thailand, 10400
Contact: Chindavijak Somjin, MD +66 89 6937679 jksomjin@hotmail.com
Contact: Nitiya Ritthidechratn +66261913356 nitiya@immunovative.com
Principal Investigator: Chindavijak Somjin, MD Less << |
|
NCT00014313
|
Sarcoma |
Phase 2 |
Terminated(low accrual) |
- |
United Kingdom
... More >> Institute of Cancer Research - UK
Sutton, England, United Kingdom, SM2 5NG Less << |
|
NCT00871403
|
Lung Cancer, Non-Small Cell |
Phase 2 |
Completed |
- |
Denmark
... More >> GSK Investigational Site
Herlev, Denmark, 2730
United Kingdom
GSK Investigational Site
Sutton, Surrey, United Kingdom, SM2 5PT Less << |
|
NCT01467115
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
United States, Pennsylvania
... More >>
Hahnemann University Hospital
Philadelphia, Pennsylvania, United States, 19102 Less << |
|
NCT03199300
|
- |
|
Recruiting |
January 2021 |
Netherlands
... More >> University Medical Center Groningen
Recruiting
Groningen, Netherlands, 9713 GZ
Contact: J.A. Gietema, MD, PhD +31 50 3612821 j.a.gietema@umcg.nl
Contact: L.C. Steggink, MD +31 50 3612821 l.c.steggink@umcg.nl
Principal Investigator: J.A. Gietema, MD, PhD Less << |
|
NCT00014534
|
Bladder Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00454649
|
- |
|
Completed |
- |
- |
|
NCT00737438
|
- |
|
Completed |
- |
- |
|
NCT00011999
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
- |
|
NCT01101451
|
Cervical Adenocarcinoma
... More >> Cervical Adenosquamous Carcinoma
Cervical Squamous Cell Carcinoma, Not Otherwise Specified
Stage IA Cervical Cancer
Stage IB Cervical Cancer
Stage IIA Cervical Cancer Less << |
Phase 3 |
Recruiting |
- |
- |
|
NCT00737438
|
Esophageal Cancer
... More >> Gastric Cancer Less << |
Phase 2 |
Completed |
- |
United States, New Jersey
... More >>
Memorial Sloan Kettering Cancer Center
Basking Ridge, New Jersey, United States
United States, New York
Memorial Sloan Kettering Cancer Center
Commack, New York, United States
Memorial Sloan Kettering Cancer Center
New York, New York, United States, 10065
Memorial Sloan Kettering Cancer Center
Rockville Centre, New York, United States
Memoral Sloan Kettering Cancer Center@Phelps
Sleepy Hollow, New York, United States Less << |
|
NCT01736917
|
- |
|
Completed |
- |
- |
|
NCT00292695
|
Lymphoma |
Phase 2 |
Completed |
- |
Taiwan
... More >> National Health Research Institutes, Lymphoma Disease Committee
Taipei, Taiwan Less << |
|
NCT00516750
|
Bladder Cancer |
Phase 2 |
Terminated(Withdrawn due to la... More >>ck of enrollment.) Less << |
- |
Japan
... More >> Nagoya University Hospital
Nagoya, Aichi, Japan, 466-8560
Shiga Medical Center for Adults
Moriyama, Shiga, Japan, 524-8524
Kyoto University Hospital
Kyoto, Japan, 606-8507
National Hospital Organization - Kyoto Medical Center
Kyoto, Japan, 612-0861
Osaka Red Cross Hospital
Osaka, Japan, 543-8555 Less << |
|
NCT00871403
|
- |
|
Completed |
- |
- |
|
NCT01672671
|
Early Triple Negative Breast C... More >>ancer Less << |
Phase 2 |
Completed |
- |
Russian Federation
... More >>
Russian Cancer Research Center named after N.N.Blokhin RAMS
Moscow, Russian Federation, 115478 Less << |
|
NCT00006237
|
Melanoma (Skin) |
Phase 3 |
Completed |
- |
- |
|
NCT01736917
|
Chemotherapy-Induced Nausea an... More >>d Vomiting Less << |
Phase 2 |
Completed |
- |
United States, Indiana
... More >>
Indiana University Melvin and Bren Simon Cancer Center
Indianapolis, Indiana, United States, 46202
United States, Missouri
Siteman Cancer Center
St. Louis, Missouri, United States, 63110
United States, Nebraska
Nebraska Cancer Specialists
Omaha, Nebraska, United States, 68114
United States, South Carolina
MUSC Hollings Cancer Center
Charleston, South Carolina, United States, 29425 Less << |
|
NCT00006237
|
- |
|
Completed |
- |
- |
|
NCT02375204
|
Germ Cell Tumor
... More >> Teratoma
Choriocarcinoma
Germinoma
Mixed Germ Cell Tumor
Yolk Sac Tumor
Childhood Teratoma
Malignant Germ Cell Neoplasm
Extragonadal Seminoma
Non-seminomatous Germ Cell Tumor
Seminoma Less << |
Phase 3 |
Recruiting |
- |
- |
|
NCT00589056
|
Lung Cancer |
Phase 1
Phase 2 |
Unknown |
- |
United States, Pennsylvania
... More >>
Abramson Cancer Center of the University of Pennsylvania
Philadelphia, Pennsylvania, United States, 19104-4283 Less << |
|
NCT02500940
|
Nasopharyngeal Carcinoma |
Phase 3 |
Recruiting |
May 2021 |
Taiwan
... More >> Taichung Vaterans General Hospital
Recruiting
Taichung, Taiwan, 40705
Contact: Jin Ching Lin, Professor +886 4 23592525 ext 5613 jclin@vghtc.gov.tw Less << |
|
NCT00003251
|
Drug/Agent Toxicity by Tissue/... More >>Organ
Head and Neck Cancer
Radiation Toxicity Less << |
Phase 1
Phase 2 |
Unknown |
- |
United States, Illinois
... More >>
University of Illinois at Chicago Health Sciences Center
Chicago, Illinois, United States, 60612 Less << |
|
NCT01345084
|
Carcinoma
Hea... More >>d and Neck Cancer Less << |
Phase 3 |
Withdrawn(change company strat... More >>egy) Less << |
November 2017 |
Brazil
... More >> Hospital Erasto Gaetner
Curitiba, Paraná, Brazil, 81520-060
Hospital de Clínicas de Porto Alegre
porto Alegre, Rio Grande do Sul, Brazil, 90035-903
Fundação Pio XII - Hospital de Câncer de Barretos
Barretos, São Paulo, Brazil, 14784-400
Hospital Amaral Carvalho
Jau, São Paulo, Brazil, 17210-120
Centro Oncológico de Mogi das Cruzes
Mogi das Cruzes, São Paulo, Brazil, 08730-500
Hospital de Base São José do Rio Preto
São José do Rio Preto, São Paulo, Brazil, 15090-000
Hospital Federal de Bonsucesso
Rio de Janeiro, Brazil, 210041-030
Instituto do Câncer de São Paulo
São Paulo, Brazil, 01246-000 Less << |
|
NCT01529411
|
Carcinoma, Transitional Cell |
Phase 2 |
Completed |
- |
Spain
... More >> Hospital General de Elda Virgen de la Salud
Elda, Alicante, Spain, 03600
ICO-Hospital Universitari Germans Trias i Pujol
Badalona, Barcelona, Spain, 08916
ICO-Hospital Duran i Reynals
L'Hospitalet de Llobregat, Barcelona, Spain, 08908
Hospital Fundació Althaia
Manresa, Barcelona, Spain, 08243
Corporació Sanitaria Parc Taulí
Sabadell, Barcelona, Spain, 08208
Complejo Hosp. Univ. de Santiago de Compostela
Santiago de Compostela, Galicia, Spain, 15706
Hospital Universitario Fundación Alcorcón
Alcorcon, Madrid, Spain, 28922
H. del Mar (Fundació Institut Mar d´Investigacions Mèdiques - FIMIM)
Barcelona, Spain, 08003
H. Universitari Vall d'Hebrón
Barcelona, Spain, 08035
Hospital Clínic i Provincial de Barcelona
Barcelona, Spain, 08036
H. General Universitario de Ciudad Real
Ciudad Real, Spain, 13005
Hospital General Universitario Gregorio Marañon
Madrid, Spain, 28007
Hospital Clínico San Carlos
Madrid, Spain, 28040
Hospital Universitario 12 de Octubre
Madrid, Spain, 28041
Hospital Universitari Son Espases
Palma de Mallorca, Spain, 07010
Clínica Universitaria de Navarra (CUN)
Pamplona, Spain, 31002
Complejo Hospitalario de Navarra
Pamplona, Spain, 31008
H. Universitario Virgen de la Macarena
Sevilla, Spain, 41009
H. Universitario Virgen del Rocío
Sevilla, Spain, 41013
IVO
Valencia, Spain, 46009 Less << |
|
NCT00665392
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
France
... More >> Hôpital Simone Veil
Montmorency, France, 95160
Hôpital Privé St Joseph
Paris, France, 75014
Hopital Europeen Georges Pompidou
Paris, France, 75015
Hopital Bichat - Claude Bernard
Paris, France, 75018
Hopital Tenon
Paris, France, 75970
centre Hospitalier Lyon Sud
Pierre Benite, France, 69495
Centre René Huguenin
Saint Cloud, France, 92100
Hopital Foch
Suresnes, France, 92151 Less << |
|
NCT01812369
|
Bladder Cancer |
Phase 3 |
Recruiting |
August 2021 |
France
... More >> UHRouen
Recruiting
Rouen, France, 76031
Contact: Christian PFISTER, MD 00332888163 christian.fister@chu-rouen.fr
Principal Investigator: Christian PFISTER, MD Less << |
|
NCT01015339
|
Advanced Gastric Cancer |
Phase 3 |
Unknown |
August 2015 |
China, Beijing
... More >> Department of GI Oncology, Peking University, School of Oncology
Recruiting
Beijing, Beijing, China, 100142
Contact: Zhihao Lu, MD 86-10-88196561 pppeirain@126.com
Principal Investigator: Shen Lin, MD Less << |
|
NCT00023660
|
Cervical Cancer |
Phase 1
Phase 2 |
Completed |
- |
- |
|
NCT02754726
|
Untreated Metastatic Pancreati... More >>c Ductal Adenocarcinoma Less << |
Phase 2 |
Recruiting |
July 2020 |
United States, Arizona
... More >>
HonorHealth Research Institute
Recruiting
Scottsdale, Arizona, United States, 85258
Contact: Joyce Schaffer, MSN, AOCNS 480-323-1339 ext option #2 Joyce.Schaffer@HonorHealth.com
Principal Investigator: Erkut Borazanci, MD Less << |
|
NCT00910117
|
Head and Neck Cancer |
Phase 2 |
Completed |
- |
China, Shanghai
... More >>
Fudan University Cancer Hospital
Shanghai, Shanghai, China, 200032 Less << |
|
NCT00455936
|
Lung Cancer |
Phase 3 |
Completed |
- |
Korea, Republic of
... More >>
National Cancer Center, Korea
Goyang-si, Gyenggi-do, Korea, Republic of, 411-769
Samsung Medical Center
Seoul, Korea, Republic of, 135-710
Asan Medical Center
Seoul, Korea, Republic of, 138-736 Less << |
|
NCT02947113
|
Non-Small Cell Lung Cancer |
Phase 2 |
Withdrawn(New studies availabl... More >>e with immunotherapy so recruitment is no longer acceptable.) Less << |
November 2017 |
- |
|
NCT00154687
|
Transitional Cell Carcinoma |
Phase 2 |
Completed |
- |
Taiwan
... More >> Department of Oncology, National Taiwan University Hospital
Taipei, Taiwan, 100 Less << |
|
NCT00349219
|
Advanced Non-Small Cell Lung C... More >>ancer Less << |
Phase 3 |
Completed |
- |
- |
|
NCT01019798
|
Non Small Cell Lung Cancer |
Phase 2 |
Unknown |
December 2011 |
Taiwan
... More >> Section of Hematology, Department of Medicine,Taipei Medical University Hospital
Recruiting
Taipei, Taiwan, 110
Contact: Cheng-Jeng Tai, M.D. 886-2-27372181 ext 3903 cjtai@tmu.edu.tw
Principal Investigator: Cheng-Jeng Tai, M.D. Less << |
|
NCT00002670
|
Head and Neck Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT00758381
|
Locally Advanced Pancreatic Ca... More >>ncer Less << |
Phase 2 |
Unknown |
August 2009 |
Italy
... More >> A.O. Treviglio-Caravaggio, P.le Ospedale n1
Recruiting
Treviglio, Bergamo, Italy, 24047
Sub-Investigator: sandro Barni, MD
Ospedale S.Orsola Fatebenefratelli
Active, not recruiting
Brescia, BS, Italy, 15100
A.O. Ospedale S.Martino
Recruiting
Genova, GE, Italy, 16132
Sub-Investigator: Alberto Sobrero, MD
A.O. san Paolo
Recruiting
Milano, MI, Italy, 20100
Sub-Investigator: Paolo Foa, MD
Casa di Cura Igea
Not yet recruiting
Milano, MI, Italy, 20100
Sub-Investigator: Gianfranco Pancera, MD
Ospedale S.Carlo Borromeo
Not yet recruiting
Milano, MI, Italy, 20123
Sub-Investigator: Donata Tabiadon, MD
A.O. S.Gerardo
Recruiting
Monza, MI, Italy, 20052
Sub-Investigator: Paolo Bidoli, MD
A.O. Universitaria Ospedali Riuniti Umberto I
Recruiting
Ancona, Italy, 60020
Principal Investigator: Stefano Cascinu, M.Pr
Ospedali Riuniti, Largo Barozzi, 1
Recruiting
Bergamo, Italy, 24128
Principal Investigator: Roberto Labianca, MD
A.O.Policlinico S.Orsola Malpighi
Active, not recruiting
Bologna, Italy, 40138
A.O. Careggi-Università, Viale Pieraccini, 17
Recruiting
Firenze, Italy, 50139
Sub-Investigator: Francesco Di Costanzo, MD
Ospedale Galliera
Active, not recruiting
Genova, Italy, 16132
A.O. Carlo Poma - Via Albertoni, 1
Recruiting
Mantova, Italy, 46100
Sub-Investigator: Enrico Aitini, MD
A.O. Cà Granda, Piazza Ospedale Maggiore, 3
Recruiting
Milano, Italy, 20162
Sub-Investigator: Salvatore Siena, MPr
Policlinico di modena
Active, not recruiting
Modena, Italy, 41100
Università Campus Biomedico, Via Emilio Longoni, 83
Active, not recruiting
Roma, Italy, 00155
A.O. S.Giovanni Calabita Fatebenefratelli
Active, not recruiting
Roma, Italy, 00186 Less << |
|
NCT02497118
|
Non Small Cell Lung Cancer |
Phase 4 |
Completed |
- |
China, Tianjin
... More >> Tianjin Medical University Cancer Institute and Hospital
Tianjin, Tianjin, China, 300060 Less << |
|
NCT03342378
|
- |
|
Recruiting |
October 2022 |
United States, Wisconsin
... More >>
University of Wisconsin Carbone Cancer Center
Recruiting
Madison, Wisconsin, United States, 53792
Contact: Cancer Connect 800-622-8922 cancerconnect@uwcarbone.wisc.edu
Principal Investigator: Matthew E Witek, MD Less << |
|
NCT00002608
|
Adrenocortical Carcinoma
... More >> Brain and Central Nervous System Tumors
Head and Neck Cancer
Liver Cancer
Malignant Mesothelioma
Pheochromocytoma
Sarcoma Less << |
Phase 2 |
Completed |
- |
Canada, Ontario
... More >>
Ottawa Regional Cancer Centre at Ottawa Hospital - General Campus
Ottawa, Ontario, Canada, K1H 1C4 Less << |
|
NCT03214003
|
Small Cell Lung Cancer Limited... More >> Stage
Radiotherapy Less << |
Not Applicable |
Enrolling by invitation |
August 2025 |
China
... More >> Beijing Cancer Hospital
Beijing, China Less << |
|
NCT00566540
|
Head and Neck Cancer |
Phase 2 |
Unknown |
- |
United States, Ohio
... More >>
Ohio State University Medical Center
Columbus, Ohio, United States, 43210 Less << |
|
NCT00387868
|
Thymoma |
Phase 2 |
Completed |
- |
United States, New Jersey
... More >>
Valley Health System - The Valley Hospital
Ridgewood, New Jersey, United States, 07450
Canada, Ontario
University of Toronto
Toronto, Ontario, Canada, M5G2C4 Less << |
|
NCT00462397
|
Cervical Cancer |
Phase 2 |
Unknown |
- |
United Kingdom
... More >> Leicester Royal Infirmary
Recruiting
Leicester, England, United Kingdom, LE1 5WW
Contact: R. Paul Symonds, MD, FRCP, FRCR 44-116-258-6296
Royal Marsden - London
Recruiting
London, England, United Kingdom, SW3 6JJ
Contact: Peter R. Blake, MD 44-20-7808-2581 peter.blake@rmh.nthames.nhs.uk
University College of London Hospitals
Recruiting
London, England, United Kingdom, WIT 3AA
Contact: Mary McCormack, MD 44-20-7380-9302 Less << |
|
NCT03240835
|
- |
|
Active, not recruiting |
September 2018 |
China, Guangdong
... More >>
Cancer Center, Sun Yat-sen University
Guangzhou, Guangdong, China, 510060 Less << |
|
NCT00003945
|
Cervical Cancer |
Phase 3 |
Completed |
- |
United States, Indiana
... More >>
Indiana University Cancer Center
Indianapolis, Indiana, United States, 46202-5289
United States, Iowa
John Stoddard Cancer Center at Iowa Methodist Medical Center
Des Moines, Iowa, United States, 50309
Mercy Cancer Center at Mercy Medical Center-Des Moines
Des Moines, Iowa, United States, 50314
Iowa Lutheran Hospital
Des Moines, Iowa, United States, 50316-2301
United States, Nebraska
Midlands Cancer Center at Midlands Community Hospital
Papillion, Nebraska, United States, 68128-4157
United States, New Mexico
MBCCOP - University of New Mexico HSC
Albuquerque, New Mexico, United States, 87131
United States, Pennsylvania
Penn State Cancer Institute at Milton S. Hershey Medical Center
Hershey, Pennsylvania, United States, 17033-0850
United States, Wisconsin
CCOP - St. Vincent Hospital Cancer Center, Green Bay
Green Bay, Wisconsin, United States, 54307-3453
Australia, New South Wales
Westmead Hospital
Westmead, New South Wales, Australia, 2145
Peru
Instituto de Enfermedades Neoplasicas
Lima, Peru, 34
Puerto Rico
San Juan City Hospital
San Juan, Puerto Rico, 00936-7344 Less << |
|
NCT00078949
|
Lymphoma |
Phase 3 |
Active, not recruiting |
December 2018 |
- |
|
NCT00043927
|
Small Cell Lung Cancer |
Phase 3 |
Completed |
- |
- |
|
NCT03196869
|
Locally Advanced Head and Neck... More >> Squamous Cell Carcinoma Less << |
Phase 2 |
Recruiting |
August 12, 2022 |
China, Guizhou
... More >> Cancer Hospital of Guizhou Medical University
Recruiting
Guiyang, Guizhou, China, 550000
Contact: Feng Jin, Bachelor 0851-86512802 jinf8865@yahoo.com.cn
Sub-Investigator: Weili Wu, master
Sub-Investigator: Jinhua Long, master
Sub-Investigator: Yuanyuan Li, master
Sub-Investigator: Xiuyun Gong, Bachelor
Sub-Investigator: Xiaoxiao Chen, Bachelor
Sub-Investigator: Juan Li, Bachelor Less << |
|
NCT01291095
|
HEAD AND NECK CANCER |
Phase 2 |
Unknown |
December 2012 |
India
... More >> Dr B.R.A.I.R.C.H, ALL INDIA INSTITUTE OF MEDICAL SCIENCES, NEW DELHI
Recruiting
New Delhi, Other state, India, 110029
Contact: Dr SUDEEP DAS, MBBS 919350114969 sudeepdas1981@gmail.com
Contact: Dr BIDHU K MOHANTI, MD 00911126173045 drbkmohanti@rediffmail.com Less << |
|
NCT03493425
|
Stage III Nasal Cavity and Par... More >>anasal Sinus Squamous Cell Carcinoma AJCC v6 and v7
Stage IVA Nasal Cavity and Paranasal Sinus Squamous Cell Carcinoma AJCC v7 Less << |
Phase 2 |
Recruiting |
February 28, 2028 |
United States, Pennsylvania
... More >>
ECOG-ACRIN Cancer Research Group
Recruiting
Philadelphia, Pennsylvania, United States, 19103
Contact: Nabil F. Saba 404-778-1868
Principal Investigator: Nabil F. Saba Less << |
|
NCT00078949
|
- |
|
Active, not recruiting |
- |
- |
|
NCT03909776
|
Event-free Survival|Overall Su... More >>rvival|Local-recurrence Free Survival|Pathological Response Less << |
|
COMPLETED |
2019-04-08 |
Peking University People's Hos... More >>pital, Beijing, Beijing, China Less << |
|
NCT05827523
|
Ovarian Cancer |
PHASE3 |
RECRUITING |
2030-12-31 |
National Cancer Center, Goyang... More >>, Gyeonggi, Korea, Republic of Less << |
|
NCT06445114
|
Oropharyngeal Cancer|Carcinoma |
PHASE2 |
NOT_YET_RECRUITING |
1932-03-01 |
Cedars-Sinai Cancer at Beverly... More >> Hills (THO), Beverly Hills, California, 90211, United States|Cedars Sinai Medical Center, Los Angeles, California, 90048, United States|CS Cancer at Valley Oncology Medical Group, Tarzana, California, 91356, United States|CS Cancer at the Hunt Cancer Center, Torrance, California, 90505, United States Less << |
|
NCT04595981
|
SCCHN|Squamous Cell Carcinoma |
PHASE2 |
NOT_YET_RECRUITING |
2025-01-29 |
University of Alabama at Birmi... More >>ngham, Birmingham, Alabama, 35233, United States Less << |
|
NCT00846508
|
Cervical Cancer |
PHASE3 |
UNKNOWN |
2025-01-13 |
Chang Gung Memory Hpspital, Ka... More >>ohsiung, Taiwan|Chang Gung Memory Hpspital, Keelung, Taiwan Less << |
|
NCT06095141
|
Pancreatic Adenocarcinoma|Homo... More >>logous Recombination Deficiency Less << |
PHASE2|PHASE3 |
RECRUITING |
2026-10-31 |
Shanghai Cancer Center, Shangh... More >>ai, China Less << |
|
NCT06358599
|
Bladder Cancer|Bladder Cancer ... More >>Stage I|Bladder Cancer Stage II Less << |
|
COMPLETED |
2024-01-15 |
Ankara Etlik City Hospital, An... More >>kara, 06170, Turkey Less << |
 HazMat Fee +
HazMat Fee +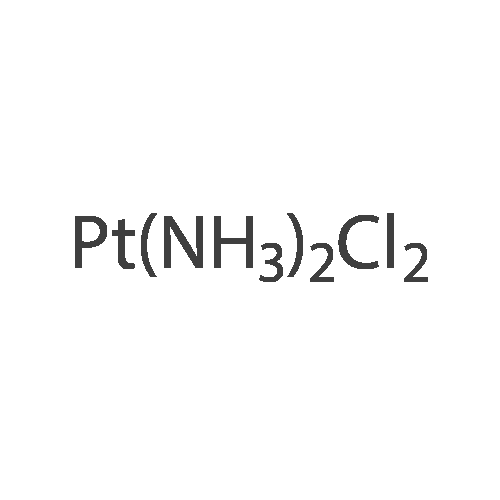





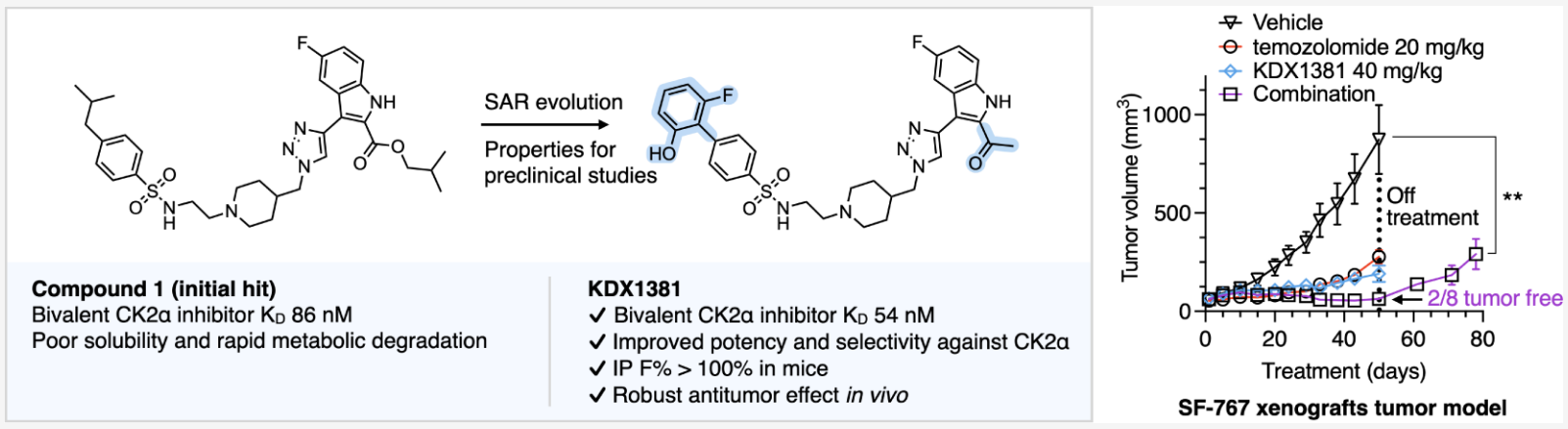

 Sulfide Complexes Suitable as Intramuscular Cyanide Countermeasures.png)


