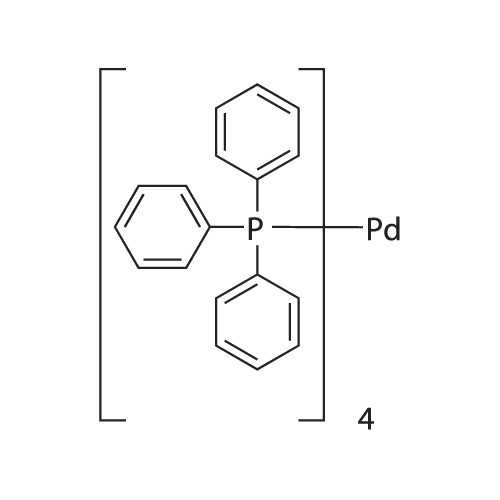
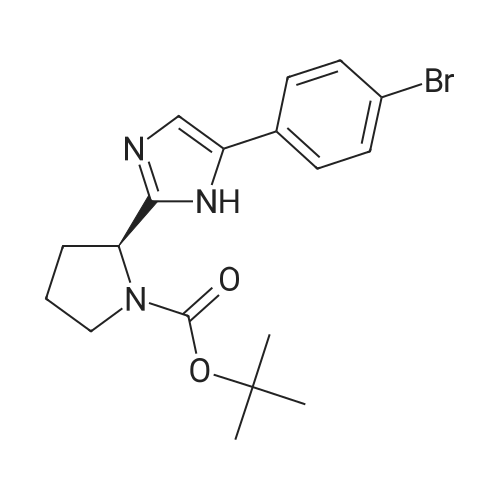
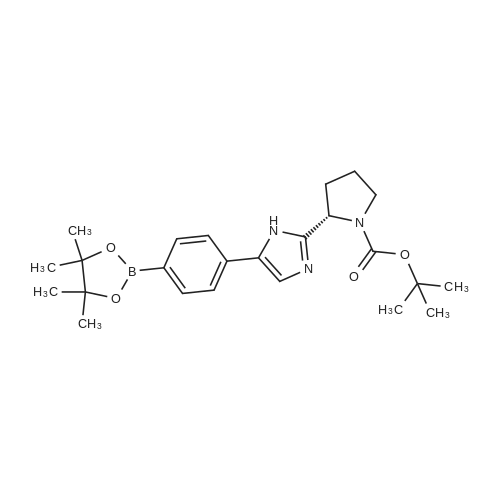
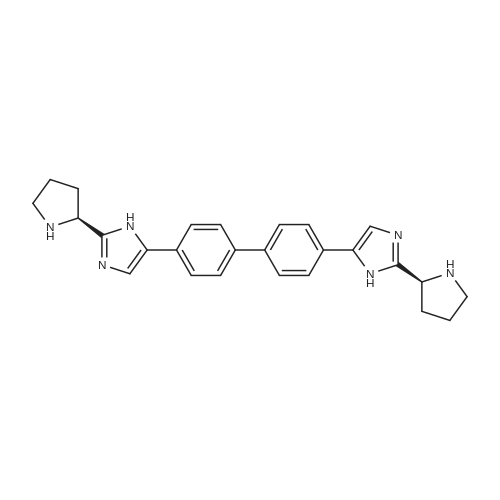
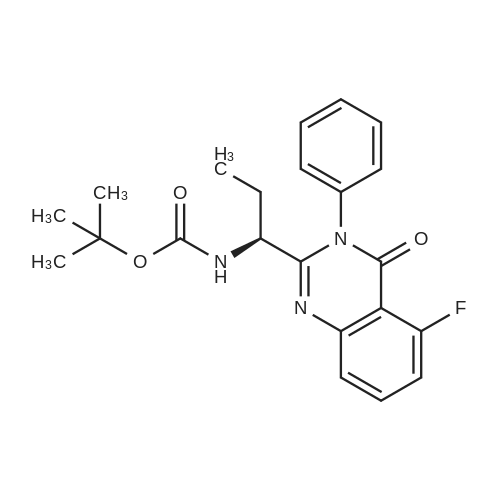
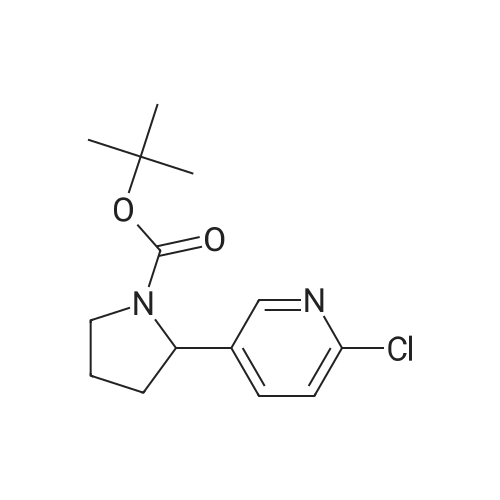
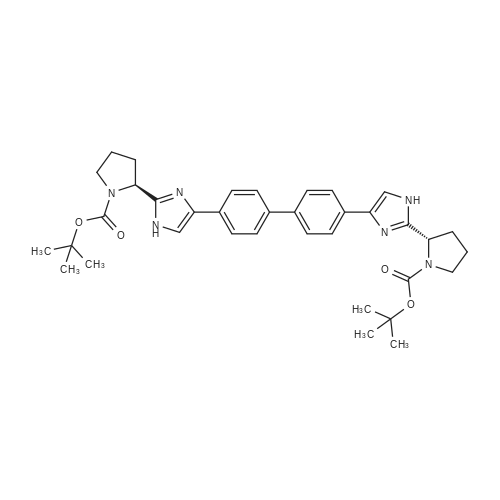
| 63% | With ammonium acetate; In toluene; at 95 - 100℃; for 15h; | To the toluene solution of compound 3 was added 78 g (1.011 mole, 20 eq.) Of ammonium acetate and heated to 95-100 . The mixture was stirred at 95-100 & lt; 0 & gt; C for 15 h. After completion of the reaction, the mixture was cooled to 70-80 , and 7 mL acetic acid, 40 mL n-butanol, and 80 mL of 5 vol% aqueous acetic acid were added. The resulting biphasic solution was separated while maintaining the temperature above 50 & lt; 0 & gt; C. 80 mL of 5 vol% aqueous acetic acid, 30 mL acetic acid and 20 mL n-butanol were added to the rich organic phase while maintaining the temperature above 50 & lt; 0 & gt; C. The resulting biphasic solution was separated while maintaining the temperature above 50 & lt; 0 & gt; C, The rich organic phase was washed with an additional 80 mL of 5 vol% aqueous acetic acid. The rich organic phase was then solvent exchanged to toluene with a target volume of 215 mL by vacuum distillation. 64 mL of methanol was added while maintaining the temperature above 60 & lt; 0 & gt; C. The resulting slurry was heated to 70-75 & lt; 0 & gt; C and aged for 1 hour. The slurry was cooled to 20-25 [deg.] C over 1 hour and aged at this temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL 10: 3 toluene: methanol. The product was dried under vacuum at 70 & lt; 0 & gt; C to give 19.8 g (31.7 mmol, 63%) of the desired product: |
| 63.5% | With ammonium acetate; In toluene; at 95 - 100℃; for 15h;Large scale; | To a solution of dakatatvein IV (8.5 kg) in toluene (45.5 kg) (51 liters), ammonium acetate (14.59 kg) was added and the temperature was raised to 95 to 100 C. After stirring for 15 hours, the temperature was lowered to 20-25 C, 1.75 Kg of glacial acetic acid, 8.14 Kg of n-butanol and 20.1 Kg of 13% by mass sodium chloride aqueous solution (The mass percentage refers to the percentage of the mass of sodium chloride as a percentage of the total mass of sodium chloride aqueous solution) to form a mixed solution, and the mixture is stirred and left to stand for delamination. The organic phase was further added with 20.1 Kg of 13% by mass sodium chloride aqueous solution (Said mass percentage refers to the percentage of the mass of sodium chloride as a percentage of the total mass of sodium chloride aqueous solution), and the mixture is stirred for standing and stratification. The organic phase was decompressed and swirled with 30 liters of solvent, and 4.74 Kg of methanol was added at a temperature higher than 50 C. The mixture was heated to 60 C.-65 C. for 1 hour, cooled to 10 C.-15 C., stirred for 2 hours, filtered, Washed with 4.26Kg mixed solvent of toluene and methanol (the mass ratio of toluene and methanol in the mixed solvent is 10: 3), and dried at 50 C under vacuum for 16 hours to obtain 5.00 kg of intermediate product of daclatasvir with a yield of 63.5%. HPLC : 97.46%. |
| 63% | With ammonium acetate; In toluene; at 95 - 100℃; for 15h;Product distribution / selectivity; | The above toluene solution of Example A-1e-2 was charged with 78 g (1.011 moles, 20 equiv) ammonium acetate and heated to 95-100 C. The mixture was allowed to stir at 95-100 C. for 15 h. After reaction completion, the mixture was cooled to 70-80 C. and charged with 7 mL acetic acid, 40 mL n-butanol, and 80 mL of 5 vol % aqueous acetic acid. The resulting biphasic solution was split while maintaining a temperature>50 C. The rich organic phase was charged with 80 mL of 5 vol % aqueous acetic acid, 30 mL acetic acid and 20 mL n-butanol while maintaining a temperature>50 C. The resulting biphasic solution was split while maintaining a temperature>50 C. and the rich organic phase was washed with an additional 80 mL of 5 vol % aqueous acetic acid. The rich organic phase was then solvent exchanged into toluene to a target volume of 215 mL by vacuum distillation. While maintaining a temperature>60 C., 64 mL MeOH was charged. The resulting slurry was heated to 70-75 C. and aged for 1 h. The slurry was cooled to 20-25 C. over 1 h and aged at that temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL 10:3 toluene:MeOH. The product was dried under vacuum at 70 C., resulting in 19.8 g (31.7 mmol, 63%) of the desired product: 1H NMR (400 MHz, DMSO-d6) delta 13.00-11.00 (s, 2H), 7.90-7.75 (m, 4H), 7.75-7.60 (m, 4H), 7.60-7.30 (s, 2H), 4.92-4.72 (m, 2H), 3.65-3.49 (m, 2H), 3.49-3.28 (m, 2H), 2.39-2.1 (m, 2H), 2.10-1.87 (m, 6H), 1.60-1.33 (s, 8H), 1.33-1.07 (s, 10H); 13C NMR (100 MHz, DMSO-d6) delta 154.1, 153.8, 137.5, 126.6, 125.0, 78.9, 78.5, 55.6, 55.0, 47.0, 46.7, 33.7, 32.2, 28.5, 28.2, 24.2, 23.5; IR (KBr, cm-1) 2975, 2876, 1663, 1407, 1156, 1125; HRMS calcd for C36H45N6O4 (M+H; ESI+): 625.3502. Found: 625.3502. mp 190-195 C. (decomposed). |
| 62% | | Alternative Preparation of Compound (6); The toluene solution of Compound 5 was charged with 78 g (1.011 moles, 20 equiv) ammonium acetate and heated to 95-100 C. The mixture was allowed to stir at 95-100 C. for 15 hours. After reaction completion, the mixture was cooled to 50-60 C. and charged with 140 mL of 2:1 acetic acid:water. The resulting biphasic solution was split while maintaining a temperature >50 C. The organic layer was washed with 70 mL 1:1 acetic acid:water. The rich aqueous layers were combined and the residual toluene removed via vacuum distillation. While maintaining a temperature of 50-60 C., 50 mL methanol was charged followed by 68 mL 10 N NaOH. The resulting slurry was cooled to 20-25 C. over 1 hour and aged at that temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL water followed by 75 mL MeOH. The product was dried under vacuum at 70 C., resulting in 27.4 g of crude product. A 1 L jacketed flask, equipped with a nitrogen line, overhead stirrer and thermocouple was charged with 63 mL NMP and 25 g of the above crude product. The mixture was heated to 50-60 C. and charged with 83 mL MeOH. The resulting slurry was allowed to stir at 50-60 C. for 18 hours. The slurry was then charged with 208 mL MeOH while maintaining a temperature >50 C. The slurry was cooled to ambient temperature over 1.5 hours and stirred for an additional 2 hours. The solids were filtered, washed with 75 mL MeOH and dried under vacuum (at)70 C. to give 18.0 g (28.8 mmol, 62% adjusted) of the desired product. |
| 62% | With ammonium acetate; In toluene; at 95 - 100℃; for 15h; | Ester (15.6 g, 23.5 mmol) and 95 mL of toluene were added into the reaction flask,then ammonium acetate (30 g, 389 mmol) was added and the temperature was raised to 98 C (temperature range 95 to 100 C) for 15 hours; ] Cooled to 60 C, slowly add saturated aqueous sodium bicarbonate solution to adjust the pH to 8 (PH range of 7-8); at about 60 C carried out stratification , then add 10 g of methanol into to the toluene layer, slowly drop temperature to 14 C (temperature range 10 to 15 C), precipitate the solid and filter to give 9. 1g imidazole product (33. 1g, yield 62 %) |
| 19.8 g | With ammonium acetate; In toluene; at 95 - 100℃; for 15h; | To the above toluene solution was added 78g of ammonium acetate and heated to 95-100 C and kept at this temperature for 15 hours. When the reaction is complete, the reaction system was cooled to 70-80 degrees, added to the system 7mL of acetic acid, 40mL of n-butanol and 80mL 5% aqueous acetic acid. 50 degrees phase. The organic phase was washed with 5% acetic acid solution, the organic phase was poured into a methanol solution, filtered, the cake washed with a volume ratio of 10: 3 methanol - toluene solution, and dried under 70 degrees, to give 19.8g product, 63% yield. |
| 100 g | With ammonium acetate; In toluene; for 18h;Reflux; | A reaction vessel was charged with 2,2'-[biphenyl-4,4'-diylbis(2-oxoethane-2,l-diyl)] 1, l'-di- tert-butyl dipyrrolidine- 1 ,2-dicarboxylate (formula IVa, 50.0 g), ammonium acetate (115.9 g), and toluene (750 mL). The reaction mass was stirred and the temperature was raised to reflux and stirring continued for about 18 hours. Upon completion of the reaction (as monitored by TLC), the reaction mass was cooled to below about 60 C and toluene (-400 mL) was distilled off under vacuum. After distillation, ethyl acetate (600 mL) and 5% aqueous acetic acid (600 mL) were added. Stirring was continued for an additional 3 hours. The solution was filtered and the resulting solid washed with ethyl acetate (200 mL) to afford di-tert-butyl (2S,2'S)-2,2'-[biphenyl-4,4'-diylbis(lH-imidazole-5,2-diyl)]dipyrrolidine-l-carboxylate. Yield: 100 g |
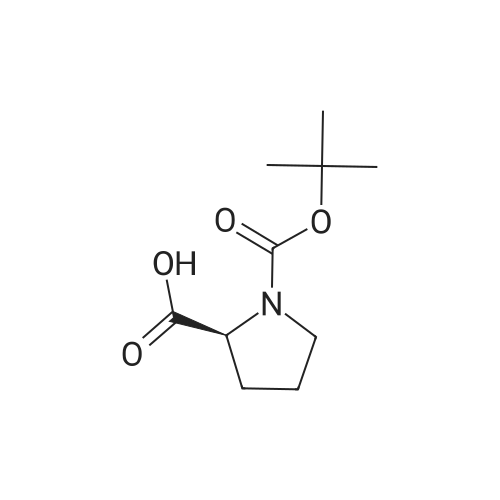
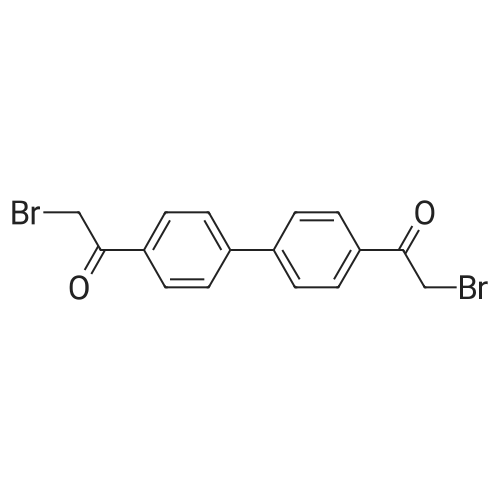
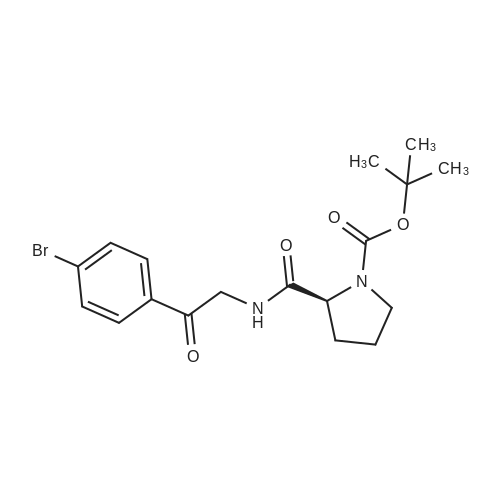
| 70.2 g | With ammonium acetate; In toluene; at 95℃; for 20h; | In 2 lit three necked round bottom flask equipped with mechanical stiner, 50 g of 1,1- biphenyl-4,4-diylbis(2-chloroethanone) of Formula V, 400 ml toluene, 80.51 g of N-bocproline and 63 g diisopropylethylamine were added at 25-30C and stined for 15 mm at same temperature. Reaction mass was heated to 70-75C and stined for 4 hr. After completion of the reaction, filtered the reaction mass and washed with 100 ml toluene. To the filterate, 250.5 g ammonium acetate was added and heated to 95C and stined for about 2Ohr at same temperature. After completion of the reaction, reaction mass was allowed to cool to 60C and 500 ml 10% methanol in water was added slowly at 50-55C. The precipitated solids was filtered and washed with water. The obtained solid compound was stined in 250 ml methanol at 60-65C for 1 hr and the reaction mass was allowed to cool to 25-30C, filtered the solids and washed with 50 ml methanol followed by dried at 60-65C to get title compound. Yield: 70.2 g. |
| With ammonium acetate; In toluene; at 55℃; for 18h; | To 1-1 was added 600 g of toluene and 32 g of ammonium acetate,Heated to 55 C and stirred for 18h,A solid precipitation.The solid was filtered to dry at 70 C to a moisture of less than 1.0%Intermediate N-3 was obtained |
| 130 g | With ammonium acetate; In toluene; at 0.105 - 0.95℃; | Ammonium acetate (390g) was added to the toluene solution of example 1 and the reaction mixture was stirred, heated to about 95C to about 105C and maintained for about 1 Oh to about 1 5h. The reaction mixture was cooled to about 75C to about 80C and water (400mL) was added to it while maintaining the temperature at about above75C. The reaction mixture was stirred and the two layers were separated at about the same temperature. The organic layer was cooled to about room temperature and stirred for about 2h to about 3h. The solid obtained was filtered, washed with toluene and dried under vacuum at about 45C to about 55C. Yield: 130g |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping