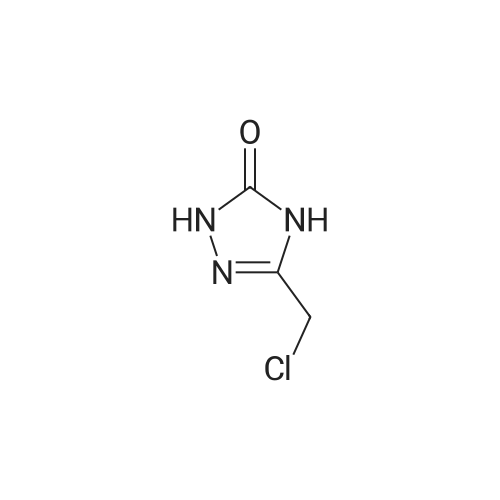
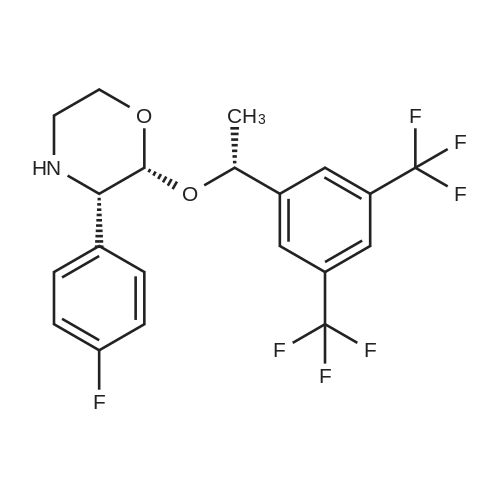
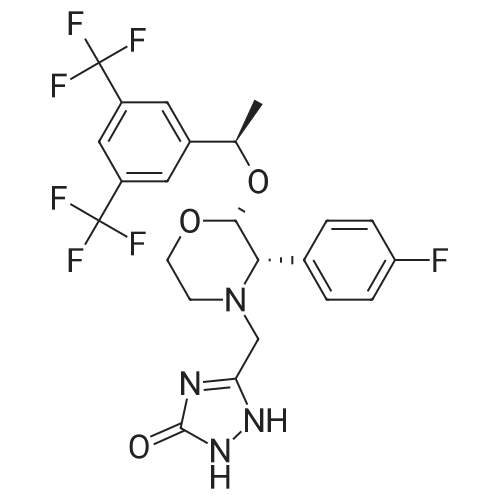
| 68% | With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 0.5h; | Example 8: Synthesis of I5-Chloromethyl-2,4-dihydro-[1 ,2,4]triazol-3-one-variant:To a solution of 0.6Og (1.48mmol, 1 equivalent) of III in 3.1 mL of DMF were added 226mg (1.64mmol, 1.1 equivalents) of potassium carbonate at ambient temperature. The mixture was stirred at 209C and a solution of 238mg (1.78mmol, 1.2 equivalents) of 5-Chloromethyl-2,4-dihydro-[1 ,2,4]triazol-3-one in 1.5ml_ of DMF was added dropwise within 15min. The reaction was stirred for 15min at 209C before 1 OmL of water were added dropwise while the product started to crystallize. The resulting suspension was stirred for 10min at 259C before it is cooled to 00C and stirred for 1 h. The crystals was collected by filtration and washed with cold water to give 416mg (68percent) of the title compound after drying under reduced pressure (400C, 10mbar) as a white crystalline product.Example 8a: Synthesis of I lambda^I -Amino^-chloro-eth-^-ylideneJ-hydrazinecarboxylic acid methyl ester-variant: To a solution of 500mg (1.14mmol, 1 equivalent) of III in 4.2mL of acetonitrile were added 546muL (3.29mmol, 2.9 equivalents) of lambda/,lambda/-diisopropylethylamine and 244mg (1.48mmol, 1.29 equivalents) of /V- [1 -Amino-2-chloro-eth-(Z)-ylidene]-hydrazinecarboxylic acid methyl ester. The resulting suspension was stirred at ambient temperature for 3h while a clear solution was formed. The reaction mixture was concentrated under reduced pressure (450C, 100mbar) and the residue was dissolved in 1 OmL of dichloromethane and washed with 1 OmL of a 26.5percent aqueous sodium chloride solution. The organic layer was concentrated under reduced pressure (459C, 100mbar). Then 4.2mL of acetonitrile were added to the residue and the mixture was transferred to a reactor where it was stirred for 55h at 1109C and 1.5bar. Then the reaction mixture was concentrated under reduced pressure (450C, 10 mbar) and the residue was dissolved in 5.6mL of methanol. The reaction mixture was heated to reflux and charcoal was added. <n="20"/>The reaction mixture was kept at reflux for 30min before it was filtered over a bed of celite and washed with methanol. The filtrate was concentrated under reduced pressure and then suspended in acetonitrile. The resulting crystalline product I was collected by filtration and washed with cold acetonitrile to give 324mg (53percent) of the title compound as a white crystalline product.Example 9: Synthesis of I without isolation of intermediates:A mixture of 33.5g (77mmol, 1 equivalent) of Xl dissolved in 496ml_ of methanol and 6.69g of Pd/C (10percent) was charged with hydrogen and stirred for 2h at ambient temperature. The catalyst was filtered off and washed three times with 5OmL of methanol. The filtrate was concentrated under reduced pressure (450C, 10mbar) to yield in 34g of III as a colourless oil. This oil was dissolved in 278ml_ of acetonitrile and 28g (218mmol, 2.9 equivalents) of lambda/,lambda/-diisopropylethylamine and 16g (97,8mmol, 1.3 equivalents) of /V-[1 - Amino-2-chloro-eth-(Z)-ylidene]-hydrazinecarboxylic acid methyl ester were added. The mixture was stirred at ambient temperature for three hours and then concentrated under reduced pressure. The residue was dissolved in 30OmL of dichloromethane and washed with 30OmL of a 26.5percent aqueous sodium chloride solution. The organic layer was concentrated under reduced pressure (45°C, 10mbar). Then 13OmL acetonitrile were added and the resulting mixture was transferred to a reactor where it was stirred for 45h at 1100C and 1.5 bar. The reaction mixture was then concentrated under reduced pressure (450C, 10mbar) and the residue was dissolved in 371 mL methanol. The reaction mixture was heated to reflux and charcoal was added. The reaction mixture was kept at reflux for 30min before it was filtered over a bed of celite and washed with methanol. The filtrate was concentrated under reduced pressure and then suspended in 278mL of acetonitrile. The resulting crystals were collected by filtration and washed with acetonitrile to give 377g (69percent) of the title compound as a white crystalline product.NMR: 1H-NMR (DMSOd6, 300MHz) delta (ppm) = 1.36 (d, CH3, 3H, J 6.5Hz), 2.39(dt, CH2, 1 H, J 11.7Hz, J 3.1 Hz), 2.75 (d, CH2, 1 H, J 14.2Hz), 2.84 (d, CH2, 1 H, J 11.7Hz), 3.38 (d, CH2, 1 H, J 13.9Hz), 3.49 (d, CH, 1 H, J 2.54), 3.62 (d, CH2, 1 H, J 10.9Hz), 4.12 (t, CH2, 1 H, J 9.9Hz), 4.33 (d, CH, 1 H, J 2.7Hz), 4.94 (q, CH, 1 H, J 6.5Hz), 7.07 (t, CH, 2H, J 8.8Hz), 7.37 (s, CH, 2H), 7.51 (t, CH, 2H, J 6.1 Hz), 7.83 (S, CH, 1 H), 11.29 (bs, NH, 2H). 13C-NMR (DMSOd6, 75.47MHz) delta (ppm) = 24.75, 50.79, 51.88, 59.07, 68.02, 71.87, 95.77, 114.79, 115.07, 121.39, 121.60, 125.22, 126.87, 128.83, 129.88, 130.31 , 130.74, 131.18, 131.37, 131.47, 133.52, 133.56, 144.24, 146.87, 156.72, 160.45, 163.68. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping