HazMat Fee +
HazMat Fee + There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
| Type | HazMat fee for 500 gram (Estimated) |
| Excepted Quantity | USD 0.00 |
| Limited Quantity | USD 15-60 |
| Inaccessible (Haz class 6.1), Domestic | USD 80+ |
| Inaccessible (Haz class 6.1), International | USD 150+ |
| Accessible (Haz class 3, 4, 5 or 8), Domestic | USD 100+ |
| Accessible (Haz class 3, 4, 5 or 8), International | USD 200+ |
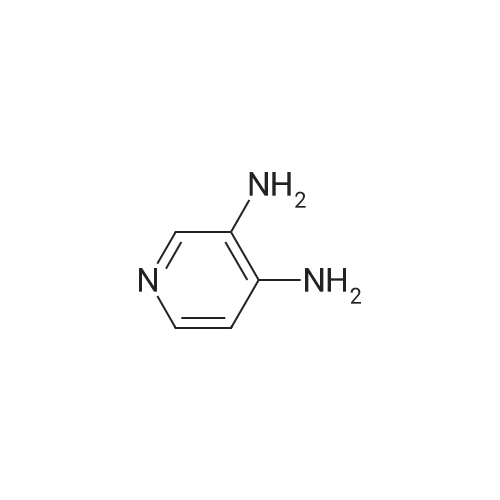

*Storage: Keep in dark place,Inert atmosphere,Room temperature.
Amifampridine is a quaternary ammonium compound that blocks presynaptic potassium channels, subsequently prolongs the action potential and increases presynaptic calcium concentrations, it was approved by the US FDA for the treatment of adults with LEMS.
Synonyms: 3,4-Diaminopyridine
4.5 
 *For Research Use Only !
*For Research Use Only !
Change View
| Size | Price | USA Stock *0-1 Day | Global Stock *5-7 Days | In Stock |
| 100mg | łÇʶÊÊ | Inquiry | Inquiry | Login |
| 1g | łÇͶÊÊ | Inquiry | Inquiry | Login |
| 5g | ł§ď¶ÊÊ | Inquiry | Inquiry | Login |
| 10g | łòʶÊÊ | Inquiry | Inquiry | Login |
Please Login or Create an Account to: See VIP prices and availability
łÇʶÊÊ
łÇͶÊÊ
ł§ď¶ÊÊ
łòʶÊÊ
In Stock
- +
Please Login or Create an Account to: See VIP prices and availability
Search for reports by entering the product batch number.
Batch number can be found on the product's label following the word 'Batch'.
Search for reports by entering the product batch number.
Batch number can be found on the product's label following the word 'Batch'.
Search for reports by entering the product batch number.
Batch number can be found on the product's label following the word 'Batch'.
Search for reports by entering the product batch number.
Batch number can be found on the product's label following the word 'Batch'.
Search for reports by entering the product batch number.
Batch number can be found on the product's label following the word 'Batch'.

| CAS No. : | 54-96-6 |
| Formula : | C5H7N3 |
| M.W : | 109.13 |
| SMILES Code : | C1=CN=CC(=C1N)N |
| Synonyms : |
3,4-Diaminopyridine
|
| MDL No. : | MFCD00006401 |
| InChI Key : | OYTKINVCDFNREN-UHFFFAOYSA-N |
| Pubchem ID : | 5918 |
| GHS Pictogram: |  |
| Signal Word: | Danger |
| Hazard Statements: | H300+H330-H311-H315-H319-H335 |
| Precautionary Statements: | P260-P264-P280-P284-P301+P310-P305+P351+P338 |
| Class: | 6.1 |
| UN#: | 2811 |
| Packing Group: | Ⅱ |
| Num. heavy atoms | 8 |
| Num. arom. heavy atoms | 6 |
| Fraction Csp3 | 0.0 |
| Num. rotatable bonds | 0 |
| Num. H-bond acceptors | 1.0 |
| Num. H-bond donors | 2.0 |
| Molar Refractivity | 33.05 |
| TPSA ? Topological Polar Surface Area: Calculated from | 64.93 Ų |
| Log Po/w (iLOGP)? iLOGP: in-house physics-based method implemented from | 0.57 |
| Log Po/w (XLOGP3)? XLOGP3: Atomistic and knowledge-based method calculated by | -0.51 |
| Log Po/w (WLOGP)? WLOGP: Atomistic method implemented from | 0.26 |
| Log Po/w (MLOGP)? MLOGP: Topological method implemented from | -0.86 |
| Log Po/w (SILICOS-IT)? SILICOS-IT: Hybrid fragmental/topological method calculated by | 0.01 |
| Consensus Log Po/w? Consensus Log Po/w: Average of all five predictions | -0.1 |
| Log S (ESOL):? ESOL: Topological method implemented from | -0.75 |
| Solubility | 19.4 mg/ml ; 0.178 mol/l |
| Class? Solubility class: Log S scale | Very soluble |
| Log S (Ali)? Ali: Topological method implemented from | -0.39 |
| Solubility | 44.9 mg/ml ; 0.412 mol/l |
| Class? Solubility class: Log S scale | Very soluble |
| Log S (SILICOS-IT)? SILICOS-IT: Fragmental method calculated by | -1.24 |
| Solubility | 6.25 mg/ml ; 0.0573 mol/l |
| Class? Solubility class: Log S scale | Soluble |
| GI absorption? Gatrointestinal absorption: according to the white of the BOILED-Egg | High |
| BBB permeant? BBB permeation: according to the yolk of the BOILED-Egg | No |
| P-gp substrate? P-glycoprotein substrate: SVM model built on 1033 molecules (training set) | No |
| CYP1A2 inhibitor? Cytochrome P450 1A2 inhibitor: SVM model built on 9145 molecules (training set) | No |
| CYP2C19 inhibitor? Cytochrome P450 2C19 inhibitor: SVM model built on 9272 molecules (training set) | No |
| CYP2C9 inhibitor? Cytochrome P450 2C9 inhibitor: SVM model built on 5940 molecules (training set) | No |
| CYP2D6 inhibitor? Cytochrome P450 2D6 inhibitor: SVM model built on 3664 molecules (training set) | No |
| CYP3A4 inhibitor? Cytochrome P450 3A4 inhibitor: SVM model built on 7518 molecules (training set) | No |
| Log Kp (skin permeation)? Skin permeation: QSPR model implemented from | -7.33 cm/s |
| Lipinski? Lipinski (Pfizer) filter: implemented from | 0.0 |
| Ghose? Ghose filter: implemented from | None |
| Veber? Veber (GSK) filter: implemented from | 0.0 |
| Egan? Egan (Pharmacia) filter: implemented from | 0.0 |
| Muegge? Muegge (Bayer) filter: implemented from | 1.0 |
| Bioavailability Score? Abbott Bioavailability Score: Probability of F > 10% in rat | 0.55 |
| PAINS? Pan Assay Interference Structures: implemented from | 0.0 alert |
| Brenk? Structural Alert: implemented from | 0.0 alert: heavy_metal |
| Leadlikeness? Leadlikeness: implemented from | No; 1 violation:MW<1.0 |
| Synthetic accessibility? Synthetic accessibility score: from 1 (very easy) to 10 (very difficult) | 1.33 |
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
 [ 54-96-6 ]
[ 54-96-6 ]
 [ 1590409-73-6 ]
[ 1590409-73-6 ]