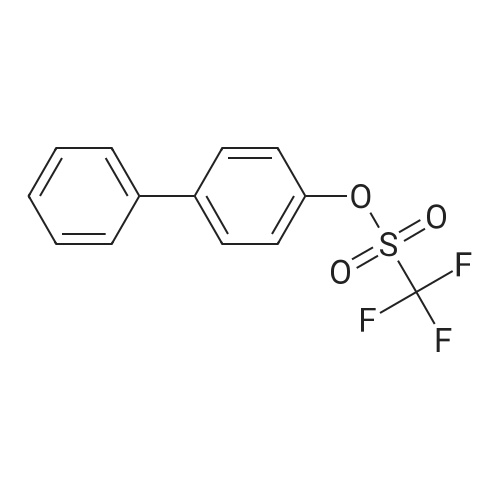
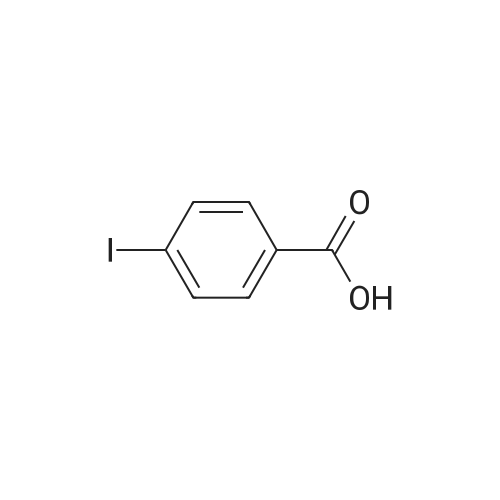
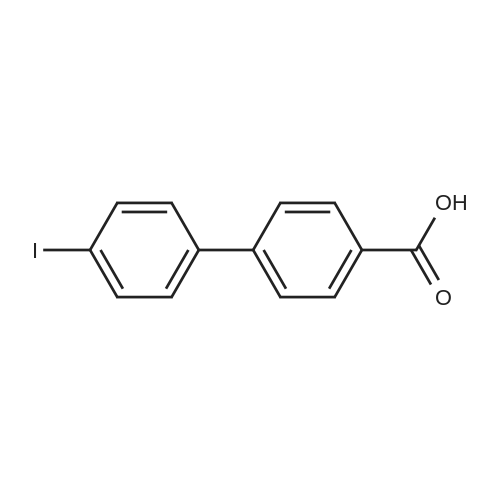
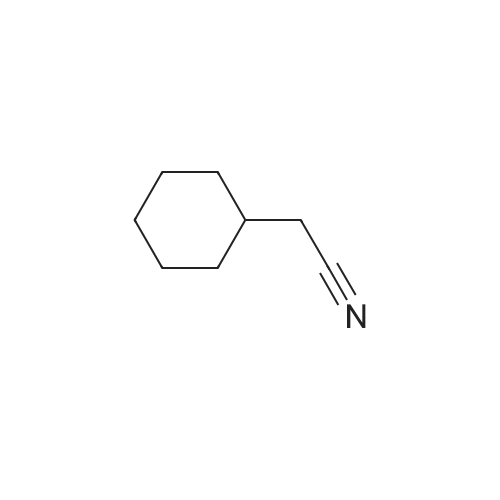
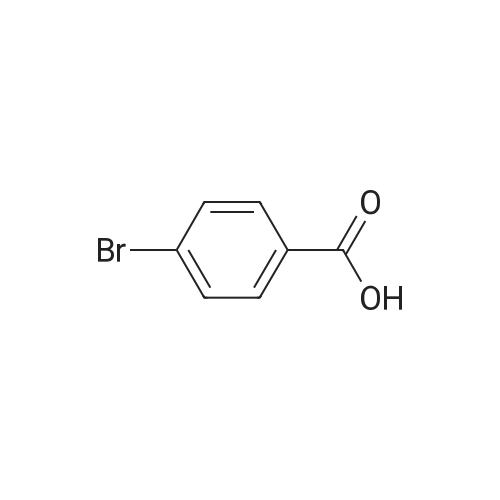
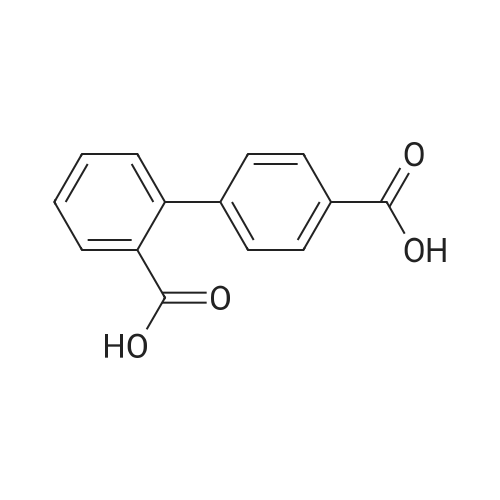
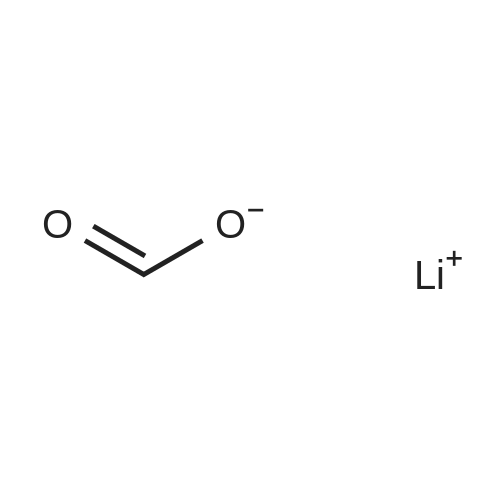
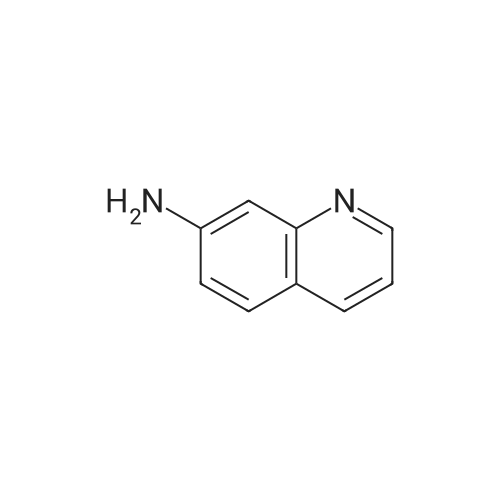
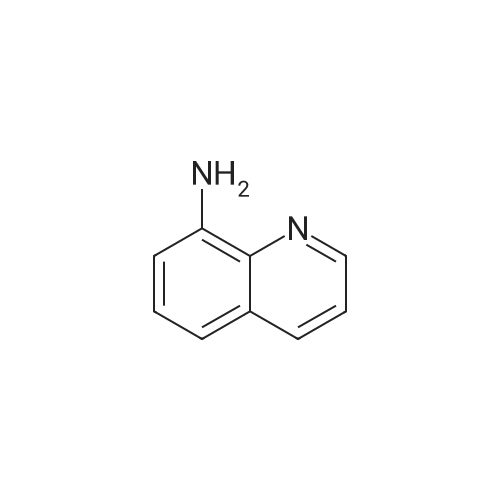
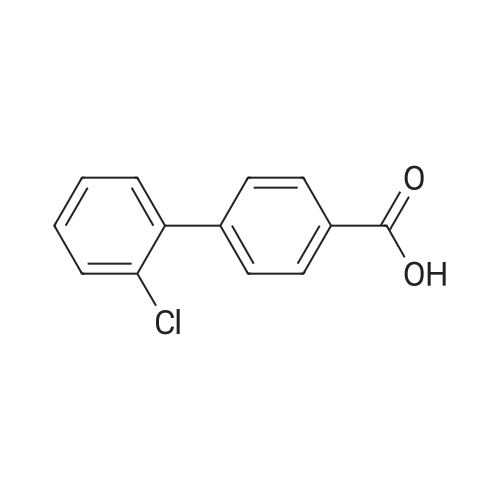
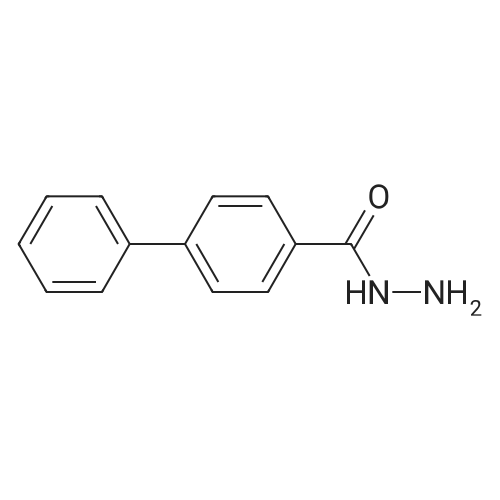
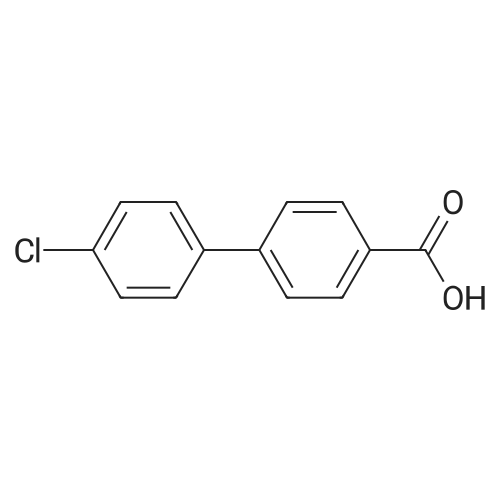
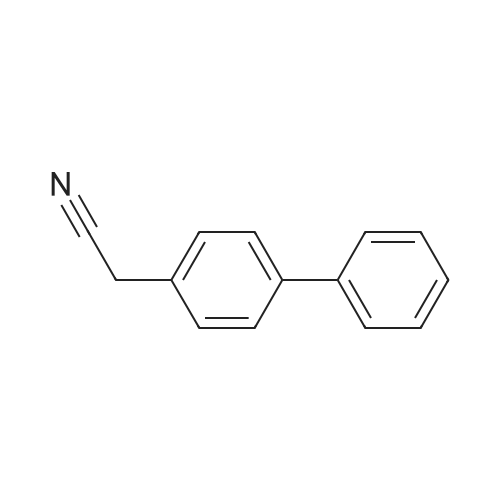
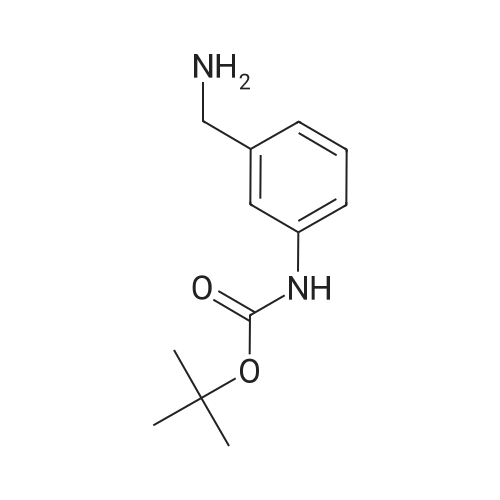
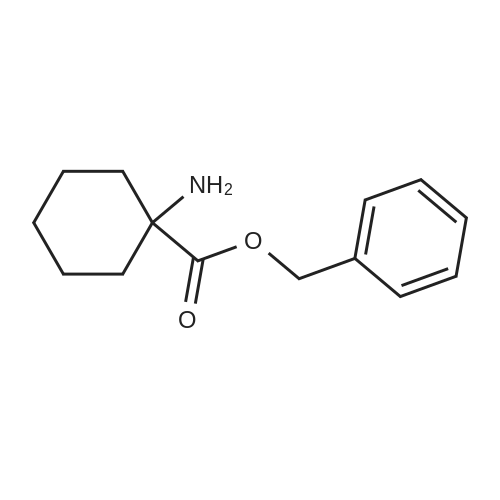
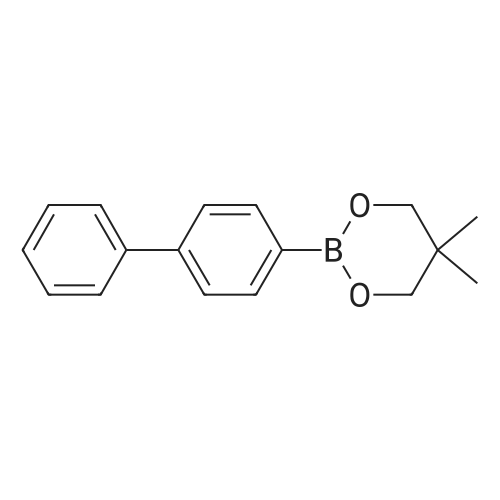
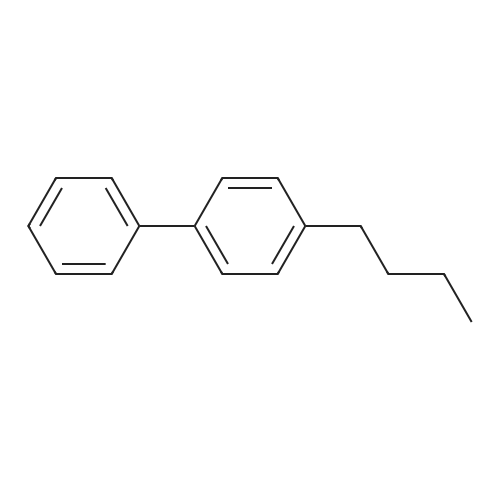
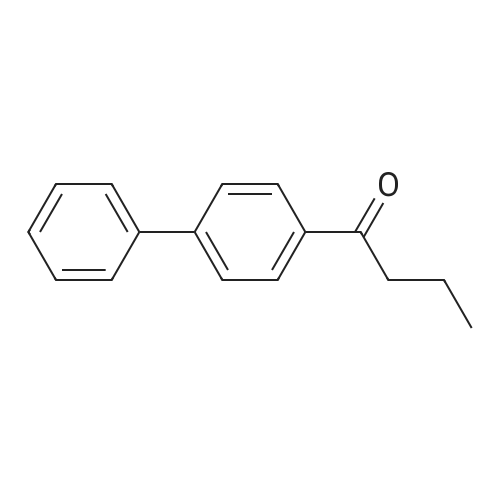
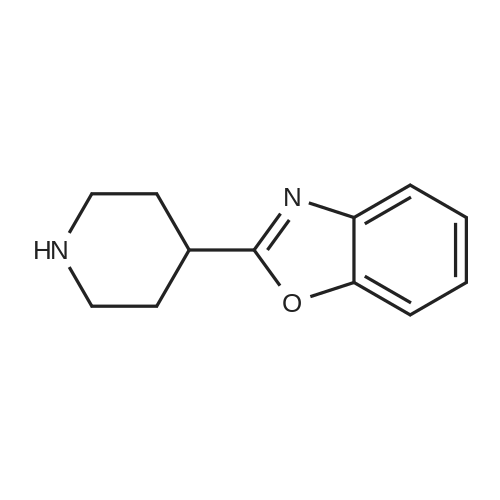
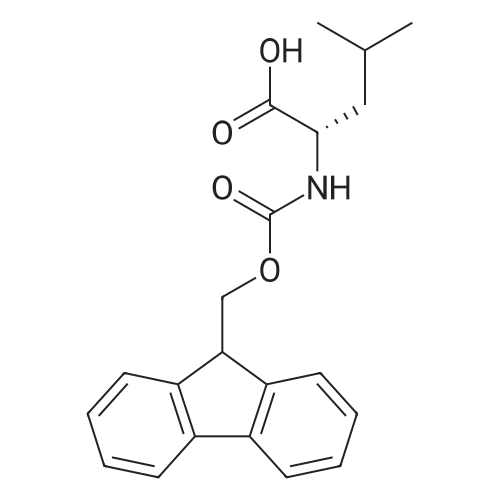
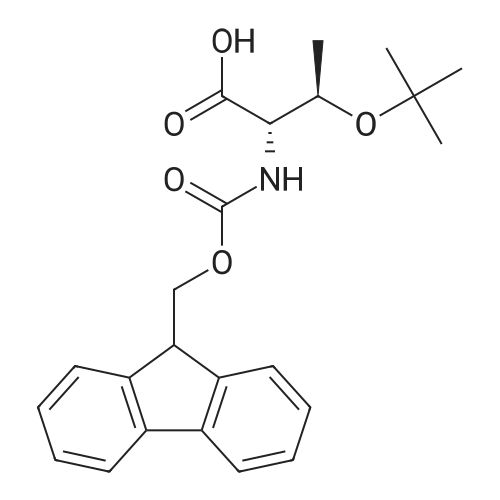
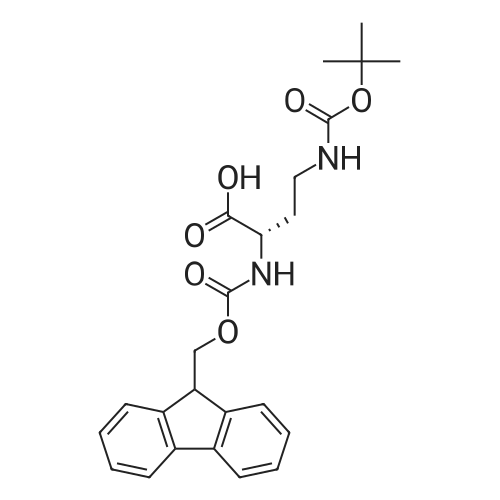
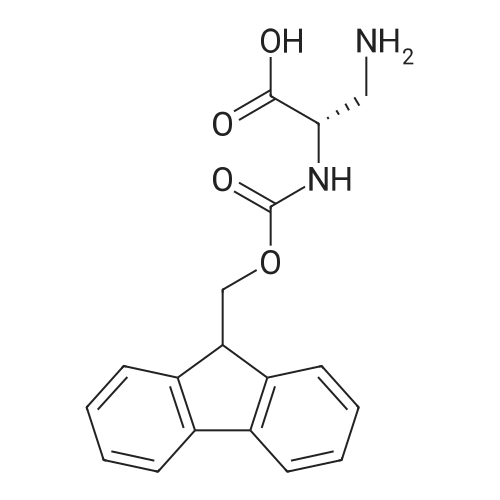
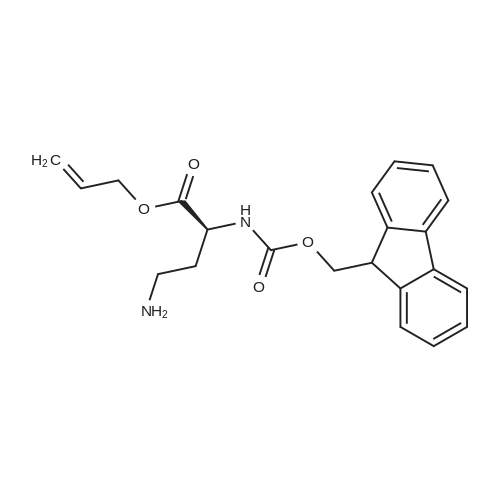
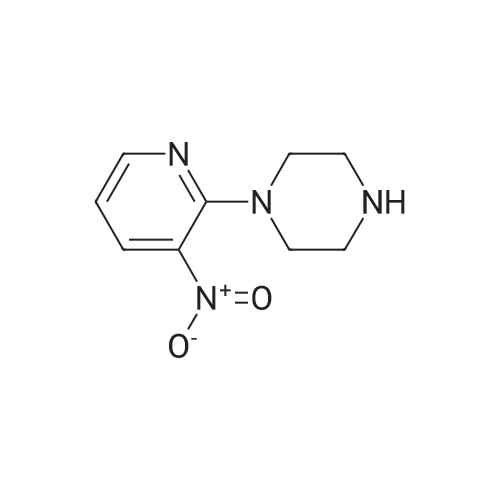
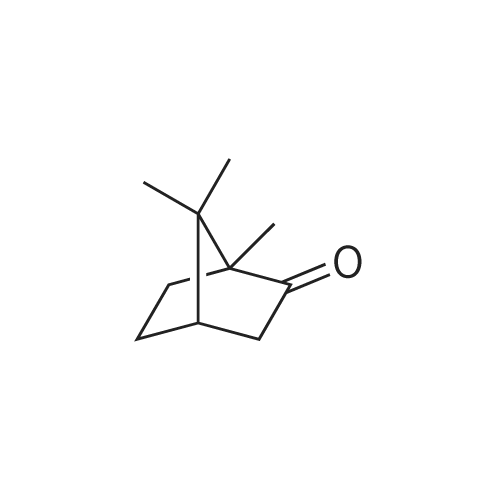
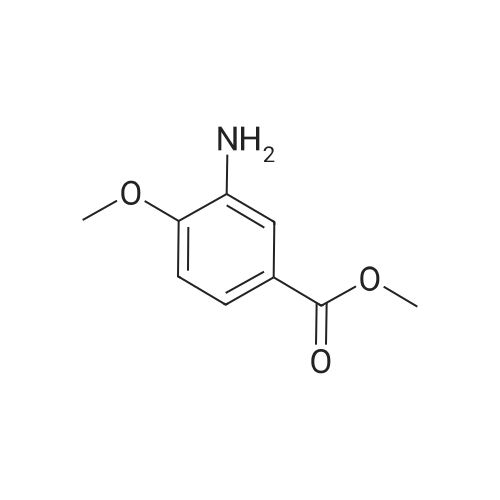
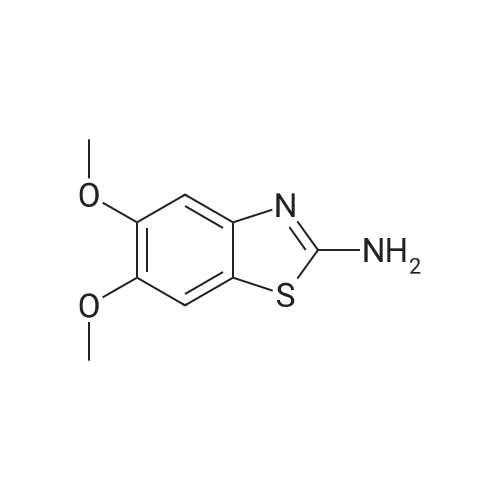
| 100% |
With potassium carbonate In lithium hydroxide monohydrate at 60℃; for 3h; |
|
| 100% |
With sodium phosphate tribasic dodecahydrate In lithium hydroxide monohydrate; isopropanol at 20℃; for 7h; Inert atmosphere; |
|
| 99% |
With tetrabutylammonium bromide; anhydrous sodium carbonate In lithium hydroxide monohydrate at 150℃; for 0.0833333h; MW irradiation; |
|
| 99% |
With tetrabutylammonium bromide; anhydrous sodium carbonate In lithium hydroxide monohydrate at 150℃; for 0.0833333h; microwave irradiation; |
|
| 99% |
With potassium carbonate In lithium hydroxide monohydrate at 80℃; for 0.5h; |
|
| 99% |
With potassium carbonate; palladium (II) chloride In lithium hydroxide monohydrate at 20 - 90℃; Inert atmosphere; |
|
| 99% |
With potassium carbonate In lithium hydroxide monohydrate at 90℃; for 0.5h; Inert atmosphere; |
|
| 99% |
With potassium phosphate tribasic trihydrate; C25H27ClN3Pd(1+)*F6P(1-) In lithium hydroxide monohydrate at 80℃; for 1h; |
|
| 99% |
With potassium carbonate; palladium (II) chloride In PEG400; lithium hydroxide monohydrate at 20℃; for 0.133333h; Air atmosphere; |
4.2. General procedure for Suzuki-Miyaura reaction
General procedure: A mixture of arylboronic acid (0.5 mmol), aryl bromides/iodides (0.6 mmol), PdCl2 (0.0025 mmol), K2CO3 (1.75 mmol), PEG400 1.5 mL, and H2O 1.5 mL were added to a 50 mL round-flask, and stirred at room temperature for the desired time until complete consumption of starting material as judged by TLC. Then, the reaction mixture was extracted with ether (10 mL×4) and the combined organic layers were dried over anhydrous MgSO4 (acidification was needed for carboxyl substituted substrates before extraction). The solvent was removed by evaporation under reduced pressure to afford the crude products, which were further purified by column chromatography on silica gel using petroleum ether and ethyl acetate as the eluent. |
| 99% |
With potassium carbonate In ethanol; lithium hydroxide monohydrate at 65℃; for 8h; |
|
| 99% |
With potassium dihydrogen orthophosphate; 4C3H6N6*Pd(2+)*2C2H3O2(1-); potassium hydroxide In lithium hydroxide monohydrate at 80℃; for 2h; Green chemistry; |
|
| 99% |
With potassium hydroxide In lithium hydroxide monohydrate at 80℃; for 2h; Green chemistry; |
1.3. General experimental procedure for Suzuki coupling
General procedure: Aryl halide (0.4 mmol), phenylboronic acid (0.48 mmol, 1.2 equiv) and a base (1.6mmol, 4eq) were added in deionized water (5 ml) and this aqueous solution was injected into a reaction vessel (20 ml). EumPd NPs catalyst (containing 0.05 mol% Pd) dispersed in water (5 ml) was added into the solution. The mixture was stirred at 80 °C for 2 h. After the reaction, the organic products were extracted with diethyl ether (10 ml). The extracts were taken to be analyzed by GC-MS (equipped with a DB-5 capillary column). |
| 99% |
With potassium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 100℃; for 6h; |
|
| 98% |
With (2-di-tert-butylphosphinoethyl)trimethylammonium chloride; anhydrous sodium carbonate In lithium hydroxide monohydrate at 20℃; |
|
| 98% |
With anhydrous sodium carbonate In N,N-dimethyl-formamide at 20℃; for 24h; |
|
| 98% |
Stage #1: 4-Bromobenzoic acid; phenylboronic acid With bis(acetonitrile)palladium(II) chloride; anhydrous sodium carbonate In lithium hydroxide monohydrate at 45℃; for 0.5h;
Stage #2: |
2.3. Procedure for Suzuki reaction
General procedure: Catalyst (2 mol%), aryl halide (1 equiv.) and Na2CO3 (1.1 equiv.) were stirred in H2O (5 mL) taken in the round bottom flask. The aryl boronic acid (1.1 equiv.) was added to the stirring solution. Stirring was continued for required time at 45 °C. After the requisite time, the reaction mixture was diluted with water and the product was extracted with ethyl acetate. The ethyl acetate extract was passed through celite bed and then analyzed by GC. Authentic samples of both reactant and product were used to verify the retention time and to confirm the product formation. The ethyl acetate extract was concentrated and chromatographed on a silica gel column using hexane and ethylacetate as eluent to afford coupled product. The products are characterized by NMR, GC MS and UPLC analyses. |
| 98% |
With C56H98O35*C23H35Cl2N5Pd; anhydrous sodium carbonate In methanol; lithium hydroxide monohydrate at 20℃; for 4h; |
|
| 98% |
With potassium carbonate In ethanol; lithium hydroxide monohydrate at 50℃; for 2.5h; Inert atmosphere; Schlenk technique; |
2.4. General procedure for Suzuki-Miyaura coupling reaction
General procedure: A Schlenk tube was charged with a suspension of aryl halide (1.0mmol), arylboronic acid (1.2 mmol), potassium carbonate (276 mg,2.0 mmol) and Pd/HCCP-DABP (20 mg, 0.3 mol%) in EtOH-H2O (3 mL,v:v=1:1), evacuated, and backfilled with nitrogen. The resulting reaction mixture was stirred at 50 °C for 2.5 h. After completion of the reaction,the mixture was cooled and extracted with CH2Cl2. The combined organic phase was dried, evaporated and purified by column chromatographyon silica gel to give the coupling product. The aqueous phasewas filtered and the solid was washed with CH2Cl2 and reused in further reactions as the recovered catalyst. |
| 98% |
With [Pd(N-(3-chloro-2-quinoxalinyl)-N'-(2,6-diisopropylphenyl)imidazolium)(PPh3)Cl2]; potassium carbonate In lithium hydroxide monohydrate at 70℃; for 3h; |
|
| 98.2% |
With potassium carbonate In lithium hydroxide monohydrate at 50℃; for 6h; |
Add 1.0 mmol of p-bromobenzoic acid to a round bottom flask.1.2mmol phenylboric acid,2.0mmol potassium carbonate,5.0 ml of pure water and 0.1 mol% of water-soluble fullerene nanopalladium catalyst,The reaction was stirred at room temperature for 6 hours in the air.Add 2mol/L hydrochloric acid solution to the pH of the solution 3-4,With the addition of hydrochloric acid solution,A large amount of white solid precipitated in the solution.Add 100 ml of distilled water to the solution and heat to 100 ° C.Keep it for 10 minutes,Then hot filtered,Washed with a large amount of water at 80 ° C,a white solid obtained by suction filtration,After drying, it was recrystallized from ethyl acetate.The calculated yield was 97.4%. |
| 98% |
With potassium carbonate In lithium hydroxide monohydrate at 20℃; for 4h; Green chemistry; |
Suzuki-Miyaura cross-coupling reactioncatalyzed by C60-TEGs/PdCl2
General procedure: A 50 mL flask was charged with an aryl halide (1.0 mmol), anaryl boronic acid (1.2 mmol), K2CO3 (2.0 mmol), nanocatalyst(0.05 mol% Pd) and deionized water (5 mL). The reactionwas stirred at room temperature for 4 h. The progress ofthe reaction was monitored by thin-layer chromatography After the reaction was completed, distilled water (25 mL) wasadded to the mixture and dilute 2 mol/L HCl was added dropwiseto pH 3.0-4.0 with stirring, and the mixture was heatedto 100 °C for 10 min. The white solid that had formed wasfiltered off and washed with hot water. After drying, it wasdissolved in ether (5 mL) and was rapidly separated using asilica gel column. Elution with ether left behind small amountsof impurities and traces of palladium black to give a crudeproduct. The ether solution was evaporated to 3-5 mL andrecrystallized to obtain a pure product. All the products 3a-uare known compounds and were characterized by comparingtheir melting points and 1H NMR spectra with those preparedrecently by using a water-soluble glycine-based Pd catalyst,PdCl2(NH2CH2CO2H)2.10 The melting points and 1H NMRdata of 3a-u are listed in the supplemental material. |
| 98% |
With potassium carbonate In lithium hydroxide monohydrate at 29 - 30℃; for 24h; |
|
| 98% |
With anhydrous sodium carbonate In lithium hydroxide monohydrate at 70℃; for 2.5h; |
Synthesis of 1
4-bromobenzoic acid (3 g, 15 mmole), phenylboronic acid (1.99 g, 16.5 mmole) and sodium carbonate (4.77 g, 45 mmole) were taken in a beaker. 120 mL water was added to this mixture. The beaker was placed on a magnetic stirrer and then it was heated at 70 oC. Within 10 minutes the whole mixture dissolved in the hot water. Then a water-soluble palladium catalyst1 (0.05 mole percent) was added to it. The reaction mixture was stirred at 70 oC for 2.5 hours. Then the mixture was acidified using 6 N HCl and pH was adjusted to 2. White colored precipitate of compound 1 was obtained. The white precipitate was filtered and washed with 200 mL distilled water. The crude compound 1 was recrystallized from a methanol-water mixture. Yield = 2.91 g (98%). 1H NMR (400 MHz, DMSO-d6, d in ppm): 12.93 (s, 1 H), 8.02 (d, 2 H, J = 8.5), 7.79 (d, 2 H, J = 7.9), 7.72 (d, 2 H, J = 7.9), 7.49 (d, 2 H, J = 7.3), 7.41 (t, 1H, J = 7.3). 13C NMR (100 MHz, DMSO-d6, d in ppm): 144.33, 139.05, 130.01, 129.67, 129.32, 126.99, 126.85. IR (KBr): 1607, 1678, 1419, 1289 cm-1. |
| 97% |
With palladium diacetate; anhydrous sodium carbonate In lithium hydroxide monohydrate at 110℃; for 0.166667h; MW irradiation; |
|
| 97% |
With potassium carbonate In lithium hydroxide monohydrate at 100℃; for 1h; |
|
| 97% |
With potassium carbonate In ethanol; lithium hydroxide monohydrate for 4h; |
|
| 97% |
With phosphoric acid disodium salt; Pd(OAc)2(2-amino-4,6-dihydroxypyrimidine disodium salt)2 In lithium hydroxide monohydrate at 90℃; for 2h; |
|
| 97% |
With palladium phosphide; potassium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 100℃; for 1h; |
|
| 97% |
With potassium carbonate In lithium hydroxide monohydrate at 80℃; for 8h; Reflux; |
|
| 97% |
With palladium diacetate; potassium carbonate In lithium hydroxide monohydrate; isopropanol at 20℃; for 0.333333h; |
Typical experimental procedure:
General procedure: In a 50 mL round bottomed flask, a mixture ofaryl halide (1 mmol), arylboronic acid (1.2 mmol), Pd(OAc)2 (1 mol %), biuret (0.01 mmol) and K2CO3 (2 mmol) in iPrOH/H2O (1:1, v/v, 4 mL) and the mixture was stirred at room temperature for a time period as mentioned in Table 2. The progress of the reaction was monitored by TLC. After completion of the reaction it was extracted with diethyl ether (3 x 10 mL) and washed with water. The combined ether extract was dried over anhydrous Na2SO4. The filtrate was concentrated under reduced pressure. The product was purified by column chromatography over silica gel using hexane/ethyl acetate (9:1 v/v) to get the desired coupling product. The products were characterized by IR, 1H NMR, 13C NMR and GC-MS. |
| 96% |
With potassium phosphate tribasic trihydrate; palladium diacetate In lithium hydroxide monohydrate; Aminol at 20℃; for 9h; |
|
| 96% |
With anhydrous sodium carbonate In lithium hydroxide monohydrate at 90℃; for 10h; Inert atmosphere; |
|
| 96% |
With Triphenylmethylamin; potassium carbonate; palladium (II) chloride In lithium hydroxide monohydrate at 20℃; for 16h; |
|
| 96% |
With trans-{PdCl2-[η1-(P)-PPh2(p-C6H4NMe2)]2}; potassium carbonate In lithium hydroxide monohydrate; isopropanol at 20℃; for 30h; |
|
| 96% |
With potassium carbonate In lithium hydroxide monohydrate at 90℃; for 1h; Inert atmosphere; Green chemistry; |
|
| 96% |
With C23H35N4O4Pd(1+)*Br(1-); potassium carbonate In lithium hydroxide monohydrate at 100℃; for 1h; |
|
| 96% |
With C23H35Cl2N5Pd; anhydrous sodium carbonate In methanol; lithium hydroxide monohydrate at 20℃; for 2h; |
|
| 96% |
With [PdCl2(2-pyridin-2-yl-benzo[b]thiophen-3-ol)]; potassium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 90℃; for 10h; |
|
| 96% |
With pentamethylbenzene,; potassium carbonate In ethanol; lithium hydroxide monohydrate at 80℃; for 0.05h; |
2.5. Suzuki cross-coupling reaction
General procedure: The Pd/PEG-PNIPAM catalyst (3 mg) was dissolved in water(3 mL). Iodobenzene (0.15 mmol), phenylboronic acid (0.3mmol), K2CO3 (0.3 mmol), and pentamethylbenzene (0.15mmol) were added to ethanol (3 mL) under stirring. The twosolutions were mixed under magnetic stirring at a speed of 800rpm and the reaction was performed at 80 °C in air. A sample ofthe reaction mixture was collected and analyzed usinghigh-performance liquid chromatography (HPLC) and gaschromatography (GC). |
| 95% |
With 2H(1+)*Cl4Pd(2-)*2H3N; poly[N-isopropylacrylamide-co-diphenyl(4'-styryl)phosphine]; anhydrous sodium carbonate In lithium hydroxide monohydrate at 100℃; for 4h; |
|
| 95% |
With anhydrous sodium carbonate In lithium hydroxide monohydrate at 100℃; for 4h; |
|
| 95% |
With potassium carbonate; palladium(0) In lithium hydroxide monohydrate at 60℃; for 5h; |
|
| 95% |
With C29H48Cl2N2PdSe; potassium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 80℃; for 2h; |
|
| 95% |
With potassium carbonate at 100℃; for 2.5h; |
|
| 95% |
With tripotassium phosphate tribasic at 80℃; for 2h; |
|
| 95% |
With C72H56N16O4Pd4S4; potassium hydroxide In methanol for 5h; Reflux; |
|
| 94% |
With polymer-based palladium; anhydrous sodium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 50℃; for 6h; |
|
| 94% |
With palladium-ruthenium; anhydrous sodium carbonate In acetonitrile at 80℃; for 2h; |
|
| 94% |
With anhydrous sodium carbonate In ethanol; lithium hydroxide monohydrate at 80℃; for 24h; |
|
| 94% |
With {2,6-bis[(di-1-piperidinylphosphino)amino]phenyl}palladium(II) chloride; potassium carbonate In 1,4-dioxane; lithium hydroxide monohydrate; butan-1-ol at 100℃; for 3h; |
|
| 94% |
With PdCl(2-HO-C6H4-CH(Ph)-NH-(CH2)3-SeC6H5); potassium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide for 4h; Heating; Aerobic conditions; |
|
| 94% |
With potassium carbonate In toluene at 80℃; for 8h; |
2.3. Procedure for the Suzuki coupling reactions catalyzed by palladium-polymer composite
General procedure: In typical experiment aryl halide (1.0 mmol), phenylboronic acid (1.5 mol), K2CO3 (1.5 mmol) and the catalyst (0.036 mol% Pd) were added to toluene (5 ml) in a small round bottom flask with a magnetic stirring bar. The reaction mixture was placed on an oil bath at 80 - 90 °C and stirred for 6 - 8 h depending on the aryl halidesused. The reaction was monitored by a thin layer chromatography (TLC) technique. Subsequently, the mixture was extracted with ethyl acetate three times. Subsequently, the reaction mixturewas cooled, diluted with Et2O, filtered through a pad of silica gel with copious washings and purified by flash chromatography on silica gel. |
| 94% |
With C22H22ClNOPdS; potassium carbonate In N,N-dimethyl-formamide at 100℃; for 15h; Inert atmosphere; |
|
| 94% |
With potassium carbonate In ethanol; lithium hydroxide monohydrate for 8h; Reflux; |
Coupling reactions
General procedure: A solution of phenylboronic acid (11 mmol), aryl iodide (10 mmol) and K2CO3 (20 mmol) in EtOH (30 mL, EtOH: H2O = 2: 1) was treated with Ni(0)-AOFs (6 mol%) at room temperature. After the mixture was stirred and refluxed for 4 h, the catalyst was separated by filtration, washed with hot ethanol and distilled water, and dried at room temperature, and reused in a next cycle. The filtrate was cooled, and products were precipitated as white scaly crystals. The the solid product was filtered, dried, and recrystallised from EtOH-H2O to afford the pure product. |
| 94% |
With C13H13NOS; potassium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 90℃; for 10h; |
|
| 94% |
With C35H34NOP2Pd(1+)*ClO4(1-); potassium carbonate In ethanol; lithium hydroxide monohydrate at 60℃; for 0.75h; |
2.4. General procedure for the Suzuki-Miyaura couplings
General procedure: To a solution of the phenylboronic acid (0.6 mmol) and the corresponding aryl halide (0.5 mmol) in a 1:1 EtOH/H2O mixture (3 mL), K2CO3 (1 mmol) and complex 2 (2 mg, 0.45 mol%) were added and the resulting suspension was stirred at 60 °C for 45 min. The reaction progress was monitored by thin-layer chromatography (TLC). At the end of the cross-coupling reactions, the catalyst was filtered, and then EtOH was evaporated (under vacuo) and the aqueous solution extracted with n-hexane. The organic layer was dried over Na2SO4. The solvent was then evaporated off and pure biphenyl derivatives were obtained in 60 to 98% yields. All obtained products are known compounds, and their 1H and 13C NMR spectra and melting points are in accordance with those reported in the literature (see Supporting Information). |
| 93% |
With tripotassium phosphate tribasic In ethanol at 20℃; for 2h; |
|
| 93% |
With C32H26NO2PPdS; potassium carbonate In ethanol; N,N-dimethyl acetamide; lithium hydroxide monohydrate at 100℃; for 12h; |
2.5 General procedure for the Suzuki-Miyaura cross coupling reaction
General procedure: To the catalyst (1.0 mol%) dissolved in 1 ml DMAc, aryl bromide (1.0 mmol), phenyl boronic acid (1.5 mmol) in 1 ml ethanol, K2CO3 (2.0 mmol) in 1 ml water and DMAc (5 ml) were all added. The mixture was heated at 100 °C for 12 h. Then, the mixture was cooled, water was added and the product was extracted with ethylacetate. The organic layer was washed with brine, dried over Na2SO4, filtered, passed through celite, and analyzed by GC. Yields were based on corresponding aryl bromides. |
| 93% |
With C49H74N4O34Pd(2+)*2C2H3O2(1-); anhydrous sodium carbonate In lithium hydroxide monohydrate for 8h; Sealed tube; Reflux; Green chemistry; |
|
| 93% |
With anhydrous sodium carbonate; palladium(0) In ethanol; lithium hydroxide monohydrate at 20 - 83℃; for 0.366667h; Microwave irradiation; |
|
| 93% |
With potassium carbonate In lithium hydroxide monohydrate at 60℃; for 5h; |
General procedure for the Suzuki-Miyaura coupling reaction
General procedure: A 10 mL round-bottom flask was charged with 4-bromoacetophenone (1j, 1 mmol, 1 eq.), phenylboronic acid (2a, 1.5 mmol, 1.5 eq.), K2CO3 (414 mg, 3 mmol, 3 eq.), H2O (2 mL), and Pd catalyst (0.01 mmol). The flask was stirred at 60°C in air. The reaction was monitored by thin layer chromatography (TLC). After the reaction was complete, the reaction mixture was cooled to room temperature and then simply filtered to recover the catalyst. It was then washed with 10 mL of H2O and ethyl acetate (EtOAc). The organic phase was separated from the aqueous phase, which was extracted three times with 30 mL EtOAc. The organic phases were collected together, dried over MgSO4, and filtered. The solvent was then evaporated under reduced pressure. The pure product was obtained via silica gel column chromatography with an eluent of EtOAc and hexane. The resulting product was analyzed by 1H NMR spectroscopy |
| 93% |
With potassium carbonate at 80℃; for 0.333333h; |
|
| 93% |
With C21H25Cl2N3PdSe; potassium carbonate In lithium hydroxide monohydrate at 100℃; for 0.5h; |
|
| 93% |
With potassium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 70℃; for 1.66667h; |
|
| 93% |
With potassium carbonate In lithium hydroxide monohydrate at 80℃; for 0.916667h; Green chemistry; |
2.5 General Procedure for the Synthesis of Biphenyl Derivatives
General procedure: A mixture of aryl halide (1.0mmol), PhB(OH)2 or Ph3SnCl (0.5mmol), base (3mmol) and Pd-Vanillin-MCM-41 were stirred in water at 80 °C for an appropriate time. The progress of reaction was monitored by TLC (eluent; n-hexane/acetone, 8:2). After completion of the reaction, the solution was allowed to cool to room temperature and was subsequently extracted with diethyl ether. Hereon, the pure product was obtained. |
| 93% |
With potassium carbonate In ethanol; lithium hydroxide monohydrate at 70℃; for 12h; |
|
| 93% |
With potassium carbonate In lithium hydroxide monohydrate at 80℃; for 4h; Schlenk technique; |
General procedure: To investigate the catalytic potential of Pd-[P(NA)] towardsSuzuki coupling reactions, an aryl halide (0.250 mmol), phenylboronic acid (0.370 mmol), K2CO3 (0.750 mmol), water (2.00 mL)and Pd-[P(NA)] (2.50 mL) were stirred in a Schlenk tube at 80 C.The reaction progress was examined by TLC. Ethyl acetate (EtOAC)was used for extraction of crude mixture. The final product was obtained by loading the crude mixture on silica gel column. TMS was used as internal standard for NMR analysis of resulting products. |
| 92% |
With PdCl2(PPh3O)2; potassium carbonate In N,N-dimethyl-formamide at 20℃; for 18h; |
|
| 92% |
With potassium phosphate tribasic trihydrate; C21H26ClN2OPd In ethanol at 60℃; for 5h; |
|
| 92% |
With Pd/SiO2; potassium hydroxide In lithium hydroxide monohydrate for 0.116667h; Microwave irradiation; Green chemistry; |
|
| 92% |
With palladium diacetate; potassium carbonate; urea In lithium hydroxide monohydrate; isopropanol at 20℃; for 0.333333h; Green chemistry; |
Typical experimental procedure:
General procedure: In a 50mL round bottomed flask, a mixture of aryl halide (1mmol), arylboronic acid (1.2mmol), Pd(OAc)2 (1mol%), urea (0.01mmol) and K2CO3 (3mmol) in iPrOH/H2O (1:1, v/v, 4mL) and the mixture was stirred at room temperature for a time period as mentioned in Table 2. The progress of the reaction was monitored by TLC. After completion of the reaction it was extracted with diethyl ether (3×10mL) and washed with water. The combined ether extract was dried over anhydrous Na2SO4. The filtrate was concentrated under reduced pressure. The product was purified by column chromatography over silica gel using hexane/ethyl acetate (9:1 v/v) to get the desired coupling product. The products were characterized by IR, 1H NMR, 13C NMR and GC-MS. |
| 92% |
With C20H18I2N4Pd; potassium-t-butoxide In ethanol at 30℃; for 3.5h; |
General procedure for Suzuki-Miyaura coupling
General procedure: A mixture of aryl halide (100 mg scale, 1.0 eq), phenylboronic acid (1.2 eq),t-BuOK (1.5 eq), complex 5 (1 mol%with respect to substrate) in EtOH (5 mL) was stirred at room temperature (30 oC)until the starting aryl halide disappeared (checked by TLC). The reactionmixture was diluted with ice cold water (10 mL) and extracted with ethylacetate. Removal of solvent under vacuumgave the crude product which was purified either by chromatography on silicagel or by simply washing with hexane. The products and their spectral data arereported in literature and they were characterized by 1H and 13CNMR spectroscopic data in the present study. |
| 92% |
With tripotassium phosphate tribasic In 1,4-dioxane at 80℃; for 2h; |
|
| 92% |
With C42H37N2O4P2Pd(1+)*ClO4(1-); potassium carbonate In ethanol; lithium hydroxide monohydrate at 60℃; for 0.5h; |
2.3. Typical procedure for the Suzuki-Miyaura reaction between aryl halides and phenylboronic acid
General procedure: Pd-catalyst 3 (2 mg, 0.44 mol%) was added to a mixture of aryl halide (0.5 mmol), phenylboronicacid (0.6 mmol), and K2CO3 (1 mmol) in a 1:1 mixture of EtOH/H2O (3 mL) ina 25mL round bottom flask. The mixture was stirred at 60 °C, and the reaction progresswas monitored by thin-layer chromatography (TLC). At the end of the cross-couplingreactions, the catalyst was filtered, and the remaining mixture was extractedwith H2O and n-hexane. The organic layer was dried over Na2SO4. Afterward, the solvent was evaporated, and pure biphenyl derivatives were found in 64% to 97% yields. |
| 91.7% |
With anhydrous sodium carbonate In lithium hydroxide monohydrate at 100℃; |
|
| 91% |
With potassium fluoride; potassium carbonate In lithium hydroxide monohydrate at 100℃; for 8h; |
|
| 91% |
With [Pd2(2,3-bis[(phenylthio)methyl]quinoxaline(-1H))2Cl2]; potassium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 90℃; for 3h; |
|
| 91% |
With anhydrous sodium carbonate at 80℃; for 5.66667h; Green chemistry; |
|
| 91% |
With Pd-BaSO4 at 100℃; for 2h; |
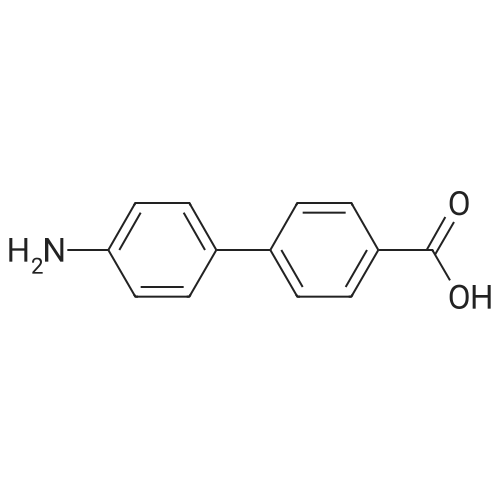
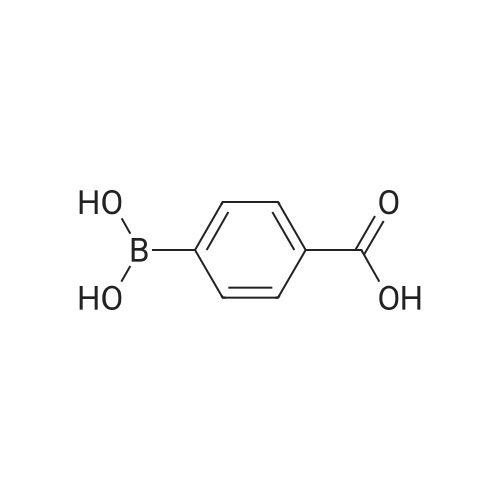
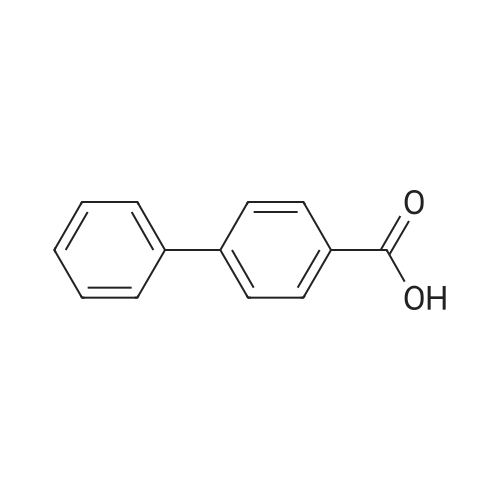
12 Example 12: Preparation of 4-carboxybiphenyl
Add 4-carboxybromobenzene (1 mmol), phenylboronic acid (1.1 mmol) and palladium barium sulfate catalyst (0.01 mmol), and Suaeda extract (4 ml) to a 10 ml round bottom flask, stir at 100°C for 2 hours under air . After the reaction, the mixture was cooled to room temperature. After filtration, the obtained filter residue was mixed with a palladium catalyst to obtain a mixture dissolved in 20 mL of ethyl acetate. The solution was collected after filtration, and the final product was obtained after evaporation to dryness. The structure of the product was identified by nuclear magnetic resonance hydrogen spectrum and carbon spectrum. The yield was 91%. |
| 90% |
With potassium carbonate In ethanol; lithium hydroxide monohydrate at 80℃; for 5h; Inert atmosphere; |
|
| 90% |
With polymer anchored Pd(II) Schiff base complex In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 80℃; for 8h; |
General experimental procedure for Suzuki coupling reaction
General procedure: A mixture of aryl halide (1.0 mmol), arylboronic acid (1.2 mmol), K2CO3 (2.0 mmol), DMF-H2O (3-3 ml), and n-dodecane (15-20 mg) as an internal GC standard and 0.5 mol% of catalyst was stirred at 80◦ C in air. Progress of the reaction was monitored by withdrawing the reaction mixtures periodically and analyzed by GC/GC-MS. GC yields were based on the amount of aryl halide employed. At the end of the reaction, the catalyst was separated by simple filtration. Filtrate was dried over Na2SO4, filtered, concentrated and the residue was purified by flash column chromatography on silica gel. The product was analyzed by GC-MS. All the prepared compounds are known and compared with authentic samples. |
| 90% |
With Pd(L-proline)<SUB>2</SUB>; potassium carbonate In lithium hydroxide monohydrate for 5h; Sealed tube; Reflux; |
|
| 90% |
With anhydrous sodium carbonate at 80℃; for 0.833333h; |
|
| 90% |
With potassium carbonate In ethanol; lithium hydroxide monohydrate at 20℃; for 2h; |
|
| 90% |
With palladium diacetate; triethylamine at 25℃; for 24h; |
10 Preparation of 4-carboxybiphenyl
In the air, 4-bromobenzoic acid, 1.0 mmol of phenylboronic acid,2.5 mmol of triethylamine, 0.005 mmol of palladium acetate was added to a 10 mL round bottom flask.The reaction was stirred at 25 ° C for 24 hours,The reaction was quenched by the addition of 15 mL of saturated brine to a round bottom flask,The mixture was extracted with 3 x 15 mL of ethyl acetate and the combined organic phases were extracted.The organic phase was concentrated under reduced pressure to give a crude product which was subjected to column chromatography using petroleum ether as an eluent,The final product was obtained. The product structure was identified by nuclear magnetic resonance spectroscopy and mass spectrometry. The yield was 90%. |
| 90% |
With C41H35N2O4P2Pd(1+)*ClO4(1-); potassium carbonate In ethanol; lithium hydroxide monohydrate at 20℃; for 1h; |
2.4. General procedure for the SuzukiMiyaura reaction
General procedure: In a general procedure, to a round-bottom flask equipped with amagnetic stirring bar a mixture of palladium catalyst 3 (0.5 mol%),phenylboronic acid (0.6 mmol), aryl bromide (0.5 mmol) and K2CO3(0.75 mmol) in 1:1 mixture of EtOH/H2O (2 mL) was added. Themixture was stirred at room temperature in the presence of air andthe progress was monitored by thin layer chromatography (TLC).After the completion of the reaction, catalyst was filtered and theremaining reaction mixture was extracted with H2O and n-hexane.The organic layer was dried over anhydrous Na2SO4 (1.5 g). Then nhexanewas evaporated and pure biphenyl derivatives were obtainedin 84e96% of yields. |
| 90% |
With C38H28N2O2Pd; potassium carbonate In ethanol at 80℃; for 2h; |
2.4. General procedure for the Suzuki coupling reaction
General procedure: In a typical run, an oven-dried 10 mL round bottom flask wascharged with a catalyst (0.01 mmol), K2CO3 (2 mmol), phenylboronicacid (1.5 mmol) and aryl halide (1 mmol) with the appropriatesolvents (5 mL). The flask was placed in a preheated oil bathat required temp. After the specified time, the flask was removedfrom the oil bath and water (20 mL) added, followed by extractionwith ether (4 10 mL). The combined organic layers were washedwith water (3 10 mL), dried over anhydrous Na2SO4, and solventwas removed under vacuum. The crude products were purified byusing silica gel column chromatography. The products were characterizedby NMR spectra. |
| 90% |
With tripotassium phosphate tribasic In 1,4-dioxane at 70℃; for 3h; |
|
| 89% |
With 2C2H5NO2*Pd(2+)*2Cl(1-); potassium carbonate In lithium hydroxide monohydrate at 20℃; for 1.5h; Green chemistry; |
|
| 89% |
With C19H16ClNPdS; potassium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 90℃; for 2h; |
|
| 89% |
With potassium carbonate In ethanol; lithium hydroxide monohydrate at 80℃; for 0.333333h; |
|
| 89% |
With sodium tetrahydridoborate; potassium carbonate; triphenylphosphine In ethanol; dichloromethane at 20℃; for 1.5h; |
2.2.4. General procedure for the synthesis of compounds 3a-3l via a Suzuki-Miyaura cross-coupling reaction catalyzed by the Fe3O4/o-PDA-Pd nanocatalyst
General procedure: In a round-bottomed flask (100 mL), Fe3O4/o-PDA-Pd MNPs (0.01)were dispersed in dichloromethane (5.0 mL) and ethanol (2.0 mL) by ultra sonication for 20 min at room temperature. Then aryl halides 1(1 mmol), phenylboronic acid 2 (1.2 mmol), NaBH4 (0.005 g, 0.1 mmol), K2CO3 (0.21 g, 1.5 mmol), and PPh3 (0.0026 g, 0.1 mmol) were added tothe flask and the mixture were stirred for 10 min at room temperature. After completion of the reaction, the magnetic nanocatalyst was conveniently isolated by use of an external magnet, and excess dichloromethane (2.0 mL) was added to the flask. Deionized water(5.0 mL) was then added, and after much stirring the organic phase wasseparated and dried with MgSO4 salt. The crude product was purified by column chromatography. Evaporation of the solvent gave the puredesired products 3a-3l. |
| 88% |
With potassium carbonate In ethanol; lithium hydroxide monohydrate at 50℃; for 19h; |
|
| 88.6% |
With tetrakis-(triphenylphosphine)-palladium; potassium carbonate In 1,4-dioxane; lithium hydroxide monohydrate at 80 - 85℃; for 5h; |
4.1.2 4.1.2 [1,1'-Biphenyl]-4-carboxylic acid (2)
In a 100 mL flask, phenylboronic acid (1.68 g, 12 mmol), K2CO3 (3.31 g, 24.0 mmol), Pd[P(C6H5)3]4 (0.2 g) and compound 1 (2 g,10 mmol) were dissolved into the mixed solution of dioxane/water (30/5 mL). Then the mixture was heated to 80-85 °C and was maintained at this temperature for 5 h. The reaction mixture was filtered, and the filtrate was adjusted to pH 2-3 with 2 N hydrochloric acid solution. A lot of white solid precipitation was filtered and dried in vacuo at 40 °C for 24 h to give a white solid (1.75 g, yield: 88.6%). |
| 88% |
Stage #1: 4-Bromobenzoic acid With acetonitrile-N<SUP>2</SUP>,N<SUP>6</SUP>-dibenzylpyridine-2,6-dicarboxamidopalladium(II); potassium carbonate In ethanol; lithium hydroxide monohydrate for 0.0833333h;
Stage #2: phenylboronic acid In ethanol; lithium hydroxide monohydrate at 82℃; for 7h; |
2.3 Suzuki-Miyaura cross-coupling reaction
General procedure: A mixture of aryl halide (1mmol), catalyst (0.005mmol) and K2CO3 (2mmol) was stirred in EtOH-H2O (4:1) (5mL) for 5min. Phenylboronic acid (1.5mmol) was added to the above mixture and stirring was continued for required time at 82°C. Then, reaction mixture was diluted with ethyl acetate and water, and the catalyst was separated by centrifugation. The centrifugate was dried over anhydrous sodium sulphate, filtered and evaporated. Then the product was analyzed by GC/GCMS. |
| 88% |
With potassium carbonate In ethanol; lithium hydroxide monohydrate at 20℃; for 1h; Green chemistry; |
2.4 General procedure for Suzuki-Miyaura cross-coupling reaction
General procedure: In a 10 mL glass vial equipped with a cap containing 5 mL of ethanol:water (1:1) mixture, aryl halide (1 equiv), phenylboronic acid (1.1 equiv), K2CO3 (2.5 equiv) were added followed by dipping of the dip catalyst into the reaction mixture which was then stirred magnetically at room temperature for required time. The progress of the reaction was monitored by thin layer chromatography (TLC). After reaction completion, the dip catalyst was simply removed from the reaction mass and washed with ethanol (1 x 5 mL) and water (1 x 5 mL) and was reused without purifying further. The product was extracted using dichloromethane (2 x 10 mL) and the combined organic layer was subjected to water wash (2 x 10 mL) followed by drying of the organic layer over Na2SO4. The dried organic layer was concentrated in vacuo, and the product was purified by column chromatography using n-hexane and ethyl acetate as eluents to afford the corresponding products in good to excellent yields. All the coupled products were known molecules and were confirmed by comparing with our previous standards (Kandathil et al., 2017; Vishal et al., 2017). |
| 88% |
With potassium carbonate In lithium hydroxide monohydrate; N,N-dimethyl-formamide at 80℃; for 0.283333h; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping