There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
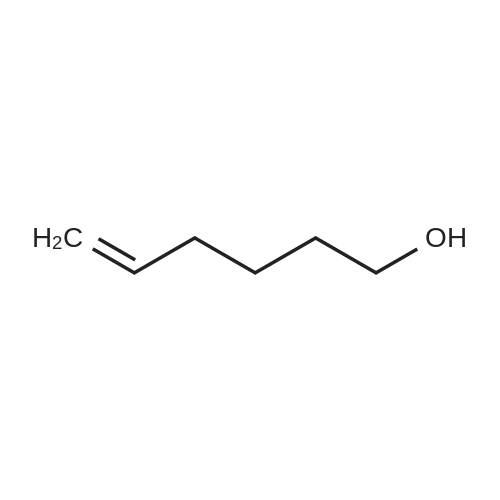
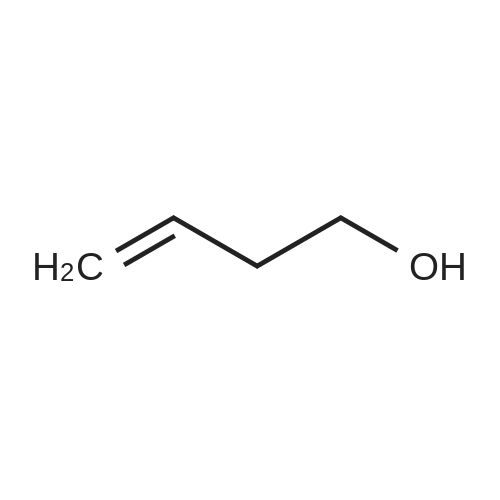
Structure of 821-09-0 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of Organic Chemistry, 1985, vol. 50, # 10, p. 1582 - 1589
2
[ 95650-50-3 ]
[ 4801-58-5 ]
[ 2937-83-9 ]
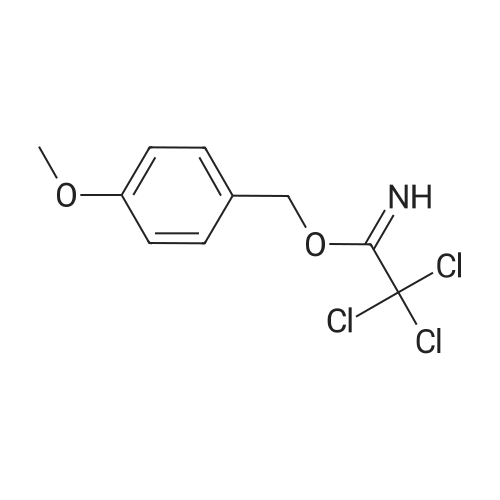
[ 821-09-0 ]
Reference:
[1] Collection of Czechoslovak Chemical Communications, 1984, vol. 49, # 10, p. 2410 - 2414
3
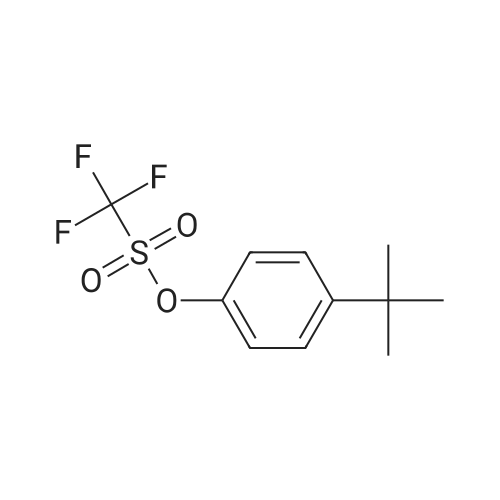
[ 821-09-0 ]
[ 1119-51-3 ]
Reference:
[1] Tetrahedron, 2004, vol. 60, # 48, p. 10943 - 10948
[2] Canadian Journal of Chemistry, 1984, vol. 62, p. 829 - 837
[3] Journal of Organic Chemistry, 1982, vol. 47, # 10, p. 1893 - 1904
[4] European Journal of Organic Chemistry, 2005, # 6, p. 1028 - 1043
[5] J. Gen. Chem. USSR (Engl. Transl.), 1981, vol. 51, # 2, p. 322 - 329[6] Zhurnal Obshchei Khimii, 1981, vol. 51, # 2, p. 396 - 404
[7] Liebigs Annalen der Chemie, 1981, vol. No. 9, p. 1705 - 1720
[8] Journal of the American Chemical Society, 1948, vol. 70, p. 3709
[9] Journal of the Chemical Society, 1937, p. 1973
[10] Chemische Berichte, 1930, vol. 63, p. 1991
[11] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1931, vol. 193, p. 599[12] Annales de Chimie (Cachan, France), 1932, vol. <10>18, p. 342
[13] Bulletin de la Societe Chimique de France, 1962, p. 177 - 182
[14] Journal of Organic Chemistry, 1960, vol. 25, p. 1628 - 1632
[15] Bulletin de la Societe Chimique de France, 1964, p. 1109 - 1116
[16] Chemische Berichte, 1974, vol. 107, p. 2887 - 2898
[17] Bulletin de la Societe Chimique de France, 1969, p. 2415 - 2427
[18] Canadian Journal of Chemistry, 1976, vol. 54, p. 2385 - 2401
[19] Journal of the Chemical Society, 1965, p. 1932 - 1939
[20] Journal of Organic Chemistry, 1978, vol. 43, p. 2361 - 2366
[21] Journal of Organic Chemistry, 1978, vol. 43, p. 2361 - 2366
[22] Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry, 1984, vol. 23, # 3, p. 238 - 240
[23] Journal of Organic Chemistry, 1991, vol. 56, # 5, p. 1874 - 1878
[24] Journal of the American Chemical Society, 1981, vol. 103, # 9, p. 2292 - 2296
[25] Synthesis, 2002, # 4, p. 479 - 482
[26] Recueil des Travaux Chimiques des Pays-Bas, 1986, vol. 105, p. 436 - 442
[27] Advanced Synthesis and Catalysis, 2013, vol. 355, # 7, p. 1315 - 1322
[28] Macromolecules, 2015, vol. 48, # 16, p. 5470 - 5473
4
[ 110-86-1 ]
[ 821-09-0 ]
[ 7789-60-8 ]
[ 1119-51-3 ]
Reference:
[1] Chemische Berichte, 1930, vol. 63, p. 1991
5
[ 821-09-0 ]
[ 928-50-7 ]
Yield
Reaction Conditions
Operation in experiment
95.22%
at 0 - 30℃; for 72 h;
In a 2000 ml four-necked flask equipped with a mechanical stirrer, a dropping funnel, a reflux condenser and a thermometer,200 g (2.33 mol) of 4-penten-1-ol, 2 g of bismuth trioxide and 700 g of pyridine were added.The temperature of the system was lowered to 0 ° C with ice-salt bath with stirring. At this point, 238 g (2 mol) of thionyl chloride was added dropwise to the system with a dropping funnel.Control the rate of dropping to ensure that the material temperature does not exceed 5 , dropping the end, remove the ice salt bath. Naturally heated to between 10-30 , the reaction was stirred for 72 hours.When the temperature exceeds 30 , use ice salt bath to cool below 30 . After the reaction was completed, the mixture was filtered through a Buchner funnel and the filter cake was taken as a catalyst and pyridine hydrochloride. The filter cake was rinsed twice with 200 ml of acetone.The combined filtrate and washings, poured into 1500ml of distilled water, allowed to stand, separate the lower reservoir.The oil layer was further washed with distilled water to neutrality, and then distilled under reduced pressure in a water pump to collect the distillate fraction (vacuum 255 mmHg) at a head temperature of 88-95 ° C.199g colorless liquid was obtained in a yield of 95.22percent. Gas chromatography detection of content of 98.27percent.
Reference:
[1] Patent: CN105837396, 2016, A, . Location in patent: Paragraph 0038; 0039
[2] Journal of Organic Chemistry, 1982, vol. 47, # 19, p. 3777 - 3779
[3] Journal of Organic Chemistry, 1980, vol. 45, # 18, p. 3578 - 3580
[4] Chemische Berichte, 1930, vol. 63, p. 1991
[5] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1931, vol. 193, p. 599[6] Annales de Chimie (Cachan, France), 1932, vol. <10>18, p. 342
[7] Journal of the Chemical Society, 1957, p. 1788,1793
[8] Journal of Organic Chemistry, 1953, vol. 18, p. 1356,1361
[9] Journal of Organometallic Chemistry, 1974, vol. 71, p. 315 - 334
[10] Journal fuer Praktische Chemie (Leipzig), 1971, vol. 313, p. 956 - 968
[11] Journal of the Chemical Society, 1964, p. 3419 - 3423
[12] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1980, p. 819 - 824
[13] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1984, # 10, p. 2245 - 2254
[14] Journal of the American Chemical Society, 2005, vol. 127, # 2, p. 528 - 529
[15] Journal of Organic Chemistry, 2014, vol. 79, # 3, p. 1511 - 1515
[16] Journal of the American Chemical Society, 2016, vol. 138, # 39, p. 12779 - 12782
6
[ 91606-67-6 ]
[ 821-09-0 ]
[ 928-50-7 ]
Reference:
[1] Journal of the Chemical Society. Perkin Transactions 2, 1998, # 6, p. 1423 - 1429
7
[ 110-86-1 ]
[ 821-09-0 ]
[ 7719-12-2 ]
[ 928-50-7 ]
Reference:
[1] Chemische Berichte, 1930, vol. 63, p. 1991
8
[ 821-09-0 ]
[ 111-45-5 ]
Reference:
[1] Tetrahedron, 1979, vol. 35, p. 1365 - 1372
With pyridine; thionyl chloride; bismuth(III) oxide; at 0 - 30℃; for 72h;
In a 2000 ml four-necked flask equipped with a mechanical stirrer, a dropping funnel, a reflux condenser and a thermometer,200 g (2.33 mol) of 4-penten-1-ol, 2 g of bismuth trioxide and 700 g of pyridine were added.The temperature of the system was lowered to 0 C with ice-salt bath with stirring. At this point, 238 g (2 mol) of thionyl chloride was added dropwise to the system with a dropping funnel.Control the rate of dropping to ensure that the material temperature does not exceed 5 , dropping the end, remove the ice salt bath. Naturally heated to between 10-30 , the reaction was stirred for 72 hours.When the temperature exceeds 30 , use ice salt bath to cool below 30 . After the reaction was completed, the mixture was filtered through a Buchner funnel and the filter cake was taken as a catalyst and pyridine hydrochloride. The filter cake was rinsed twice with 200 ml of acetone.The combined filtrate and washings, poured into 1500ml of distilled water, allowed to stand, separate the lower reservoir.The oil layer was further washed with distilled water to neutrality, and then distilled under reduced pressure in a water pump to collect the distillate fraction (vacuum 255 mmHg) at a head temperature of 88-95 C.199g colorless liquid was obtained in a yield of 95.22%. Gas chromatography detection of content of 98.27%.
2. Preparation of 2-Vinylhept-6-enoic acid (4) 2.1. Pent-4-enyl p-toluenesulfonate p-Toluenesulfonyl chloride (22.15 g, 116 mmol, 1.5 eq.) was added in small portions to a solution of pent-4-en-1-ol (6.67 g, 77.5 mmol, 1 eq.) and pyridine (abs., 12.53 ml, 12.25 g, 154.9 mmol, 2 eq.) in CH2Cl2 (80 ml) at 0 C. The reaction mixture was stirred at 0 C. for 3 hours. After addition of water (60 ml), the mixture was extracted with diethyl ether (125 ml). The organic phase was washed in succession with aqueous hydrochloric acid (2 M), an aqueous Na2CO3 solution (5%) and water, dried over MgSO4 and freed of the solvent under reduced pressure. The residue was fractionated by column chromatography (petroleum ether/diethyl ether, 8:1). Pent-4-enyl p-toluenesulfonate was obtained as a colorless oil in an amount of 17.7 g (yield: 95%).1H NMR (400.1 MHz, CDCl3): delta=1.71-1.78 (m, 2H); 2.05-2.11 (m, 2H); 2.45 (s); 4.04 (t, J=6.4 Hz, 2H); 4.93-4.98 (m, 2H); 5.64-5.74 (m, 1H); 7.35 (dm, J=8.3 Hz, 2H); 7.79 ppm (dm, J=8.3 Hz, 2H). 13C{1H}-NMR (100.6 MHz, CDCl3): delta=21.6; 28.0; 29.4; 69.8; 115.8; 127.9; 129.8; 133.2; 136.6; 144.7 ppm.
93%
With pyridine; In dichloromethane; at 0 - 20℃; for 21h;
A. To a solution of 4-penten-1-ol (4.8 mL, 4.00 g, 46.4 mmol) in dichloromethane (60 mL) was added pyridine (10mL), followed by the addition of p-toluenesulfuryl chloride (7.2 g, 37.8 mmol) at 0 C. The reaction mixture wasstirred for 21 h at ambient temperature. The reaction mixture was then diluted with diethyl ether (350 mL), washedsequentially with water, 1% HCl, water and brine. The organic layer was dried over Na2SO4 and concentrated toaffors the product in 93% yield (8.48 g) which was used for the next step reaction without further purification.
86%
With dmap; triethylamine; In dichloromethane; at 20℃;Inert atmosphere;
4.2.1. General procedure A. To a stirred solution of 4-penten-1-ol(0.6 mL, 5.81 mmol) in anhydrous CH2Cl2 (11 mL) was added triethylamine(1.21 mL, 8.71 mmol) followed by DMAP (28 mg,0.232 mmol) and TsCl (1.1 g, 5.81 mmol). The resulting mixture wasstirred at room temperature overnight. The reaction was dilutedwith water (10 mL) and extracted with chloroform (10 mL, threetimes). The combined organic layers were washed with aqueoussatd NH4Cl (10 mL), separated, dried over MgSO4, filtered, andconcentrated. Silica gel column chromatography purification (20%EtOAc/Hexanes) afforded the corresponding known tosylate20 (31)as an oil (1.19 g, 86%).
In an oven dried 50 mL round bottom flask equipped with a stir bar were added 4-penten-1-ol (7; 1.20 mL, 11.6 mmol) and dry CH2Cl2 (15 mL). The reaction was allowed to stir for 10 minutes, followed by the drop-wise addition of pyridine (1.40 mL, 17.4 mmol) with additional stirring for 1 h. The reaction mixture was then cooled to 0 C on an ice bath for 30 min followed by the drop-wise addition of benzoyl chloride (2.70 mL, 23.2 mmol). The mixture was warmed to r.t. and was stirred for 12 h. The solids were removed by filtration and the filtrate was concentrated under reduced pressure. The crude product was then purified by silica gel flash chromatography (EtOAc/hexanes 1:9) to afford 8c as a clear viscous oil (2.21 g, 86%); Rf =0.92 (EtOAc/hexanes 1:9.). 1H NMR (300 MHz, DMSO-d6): d =7.95 (d, J = 9.6 Hz, 2H), 7.65-7.47 (m, 3H), 5.89-5.73 (m, 1H), 5.06-4.95 (m, 2H), 4.24 (t, J = 6.6 Hz, 2H), 2.14 (q, J = 6.9 Hz, 2H), 1.77 (quint., J = 6.6Hz,2H); 13C NMR (300 MHz, DMSO-d6): d = 166.1, 138.1, 133.6, 130.3, 129.1, 115.7, 64.5, 30.1, 27.8.
78%
With pyridine; In dichloromethane; at 0 - 20℃; for 1h;
<strong>[821-09-0]4-Penten-1-ol</strong> (0.86 g, 10.00 mmol) and pyridine (0.95 g, 12.00 mmol) were dissolved in dry CH2Cl2 (20 mL). Benzoyl chloride (2.12 g, 12.00 mmol) was then added dropwise tothe solution at 0 C. After stirring for 1 h at r.t., the salt was filtered and the solvent was removed in vacuo. The crude material was purified by column chromatography on silica gel (Pet. Ether/EtOAc 100:1); Colorless oil; 78% yield; 1H NMR (200 MHz, CDCl3) delta: 8.06-8.02 (2H, m, ArH), 7.57-7.37 (3H, m, ArH), 5.94-5.74 (1H, m, =CH), 5.10-4.98 (2H, m, =CH2), 4.32 (2H, t, J = 6.5 Hz, OCH2), 2.26-2.15 (2H, m, CH2), 1.92-1.79 (2H, m, CH2); 13C NMR (50 MHz, CDCl3) delta: 166.4, 137.3, 132.7, 130.3, 129.4, 128.2, 115.2, 64.2, 30.0, 27.8; MS 191 [M+H]+.
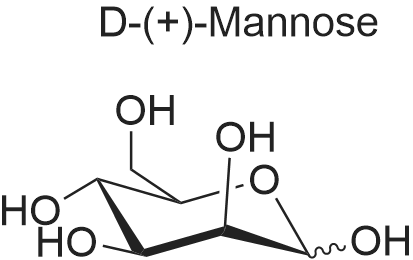
(2R,3S,4S,5S,6S)-2-(hydroxymethyl)-6-(pent-4-en-1-yloxy)tetrahydro-2H-pyran-3,4,5-triol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65%
With camphor-10-sulfonic acid In dimethyl sulfoxide at 90℃; for 24h;
With camphor-10-sulfonic acid at 100℃; for 20h;
8
4'-Pentenyl α-D-mannopyranose Camphorsulfonic acid (0.2 g) was added to stirred mixture of D-mannose (15 g) and 4-penten-1-ol (100 g) and the mixture heated to 100° C. After 20 hours the mixture was evaporated under reduced pressure and the residue chromatographed [SiO2, ethyl acetate] to give compound 8 (17.1 g).
With triethylamine; In dichloromethane; at 20℃; for 48h;Inert atmosphere;
Trityl chloride (3.88 g, 13.9 mmol) and triethylamine (3.24 mL, 23.2 mmol) were added to a solution of 4-penten-1-ol (7; 1.20 mL, 11.6 mmol) and dry CH2Cl2 (70 mL) in a 250 mL round bottom flask equipped with a stir bar under an argon atmosphere. The reaction mixture was stirred at r.t. for 48 h. The solvent was then removed under reduced pressure, the residue was dissolved in ethyl acetate (70 mL) and then washed sequentially with 1 M HCl (2 x 70 mL), saturated aqueous NaHCO3 (70 mL), and brine solution (70 mL). The organic layer was then dried (MgSO4), filtered, and concentrated under reduced pressure. The crude oil was then purified by flash silica gel chromatography (EtOAc/hexanes 1:9) to afford 8b as a clear oil (3.81 g, 84%); Rf = 0,64 (EtOAc/hexanes 1:9). 1H NMR (300 MHz, DMSO-d6): d = 7.37-7.17 (m, 15H), 5.78-5.65 (m, 1H), 4.96-4.85 (m, 2H), 2.96 (t, J = 6.3Hz, 2H), 2.06 (q, J = 6.9 Hz, 2H), 1.62 (p, J = 6.6Hz, 2H). 13C NMR (75 MHz, DMSO-d6): d = 144.5, 138.6, 128.6, 128.3, 127.4, 115.3, 86.3, 62.8, 30.4, 29.1.
With 1H-imidazole; In dichloromethane; at 20℃; for 2h;
A 1000 mL round-bottom flask was charged with penten-1-ol (TCI, 51.8 g, 0.6 mol, 1.0 equiv) and dissolved in CH2C12 (500 mL, 1.2 M). The reaction was then added sequentially with tert-butyldimethylsilyl chloride (99.5 g, 0.66 mol, 1.1 equiv) and imidazole (49.0 g, 0.72 mol, 1.2 equiv), and allowed to stir at ft for 2 h. The reaction slurry was then washed with saturated NaC1 solution (200 mL twice). The aqueous phase was extracted with CH2C12 (100 mL twice) and the combined organic layer was dried over anhydrous MgSO4. The solvent was removed under reduced pressure to afford a crude product, which was purified by house vacuum distillation at 105 C to afford the product tert-butyldimethyl(pent4-en-1-yloxy)silane as a clear oil (119.3 g, 99% yield).
98%
With 1H-imidazole; In N,N-dimethyl-formamide; at 0 - 23℃; for 8h;
To a stirred solution of 4-penten-l-ol (11.4 mL, 110 mmol) in DMF (200 mL) were added imidazole (18.7 g, 275 mmol), and TBSCl (24.9 g, 165 mmol) at 0 C. The resultant mixture was stirred for 8 h at 23 C. The reaction mixture was then quenched with H20 and extracted with ethyl acetate three times, washed by H20 and Brine, dried with MgS04, concentrated, and purified by column chromatography (silica gel, hexanes) to afford the title product (21.6 g, 98% yield). ]H NMR (300 MHz, CDC13) delta 0.05 (s, 6 H), 0.90 (s, 9 H), 1.61 (m, 2 H), 2.11 (m, 2 H), 3.62 (t, / = 6.0 Hz, 2 H), 5.00 (m, 2 H), 5.83 (m, 1H).
98%
With 1H-imidazole; In N,N-dimethyl-formamide; at 0 - 23℃; for 8h;
To a stirred solution of 4-penten-1-ol (11.4 mL, 110 mmol) in DMF (200 mL) were added imidazole (18.7 g, 275 mmol), and TBSCl (24.9 g, 165 mmol) at 0 C. The resultant mixture was stirred for 8 h at 23 C. The reaction mixture was then quenched with H2O and extracted with ethyl acetate three times, washed by H2O and Brine, dried with MgSO4, concentrated, and purified by column chromatography (silica gel, hexanes) to afford the title product (21.6 g, 98% yield). 1H NMR (300 MHz, CDCl3) delta 0.05 (s, 6H), 0.90 (s, 9H), 1.61 (m, 2H), 2.11 (m, 2H), 3.62 (t, J=6.0 Hz, 2H), 5.00 (m, 2H), 5.83 (m, 1H).
98%
With 1H-imidazole; In dichloromethane; at 25℃;Inert atmosphere;
To a solution of 4-penten-1-ol (2.15g, 25.0mmol, 1.0 equiv) in CH2Cl2 (25mL) was added imidazole (2.04g, 30.0mmol, 1.2 equiv) followed by TBSCl (4.52g, 30.0mmol, 1.2 equiv) and the reaction mixture was stirred at room temperature for 3h. After the reaction had completed, water (30mL) was added and the mixture was extracted with EtOAc (2×30mL). The organic layer was dried (MgSO4) and concentrated in vacuo. The crude product was purified by column chromatography on silica gel eluting with 1:99 Et2O/hexanes to afford the title compound 25 as a colorless oil (4.91g, 98%). The spectroscopic data of this compound matched with those in the literature.34 1H NMR (500MHz, CDCl3): delta 5.82 (ddt, J=17.0, 10.2, 6.7Hz, 1H, H-4), 5.02 (d, J=17.0Hz, 1H, H-5A), 4.95 (d, J=10.2Hz, 1H, H-5B), 3.62 (t, J=6.5Hz, 2H, H-1), 2.10 (q, J=7.3Hz, 2H, H-3), 1.61 (p, J=6.9Hz, 2H, H-2), 0.90 (s, 9H, (CH3)3CSi), 0.05 (s, 6H, 2× CH3Si).
94%
To a solution of the pent-4-en-1-ol (5 g, 58.14 mmol) in dry CH2Cl2 (50 ml) was added imidazole (5.93 g, 87.21 mmol), and the mixture was stirred for 10 min at 0C. To this solution tert-Butyl dimethyl silylchloride (10.51 g, 69.77 mmol) was added at 0C, and the mixture was stirred at rt for 4 h. After completion of the reaction, the mixture was diluted with H2O, extracted with ethyl acetate (2x50 ml), dried (anh. Na2SO4), and concentrated under reduced pressure. The crude residue was purified by Column chromatography (2% ethyl acetate hexane) to give 7 as colorless liquid (10.9 g, 94%); 1H NMR (300 MHz, CDCl3): delta = 5.92-5.74 (m, 1 H), 5.05-4.88 (m, 2 H), 3.62 (t, J = 6.6 HZ, 2 H), 2.15-2.06 (m, 2 H), 1.67-1.56 (m, 2 H), 0.89 (s, 9 H), 0.05 (s, 6 H); 13C NMR (75 MHz, CDCl3): 138.5, 114.4, 62.5, 32.0, 30.0, 29.9 (3 C), -5.2 (2 C); IR (neat): umax 2928, 2860, 1640, 1474, 1254, 1102 cm-1; ESI-MS: 202 [M+H]+.
85%
With 1H-imidazole; In N,N-dimethyl-formamide; at 0 - 20℃; for 1.25h;
To a stirred 75 mL DMF solution of alcohol 1 (5.4 g, 63 mmol) and imidazole (4.5 g, 66 mmol) at 0 C. was added TBDMSCI (10 g, 66 mmol). The reaction was stirred 15 min at 0 C. and 1 h at RT at which time the alcohol 1 could not be detected by TLC analysis. The reaction was diluted with 400 mL of hexanes and washed successively with water (4×100 mL) and saturated brine (50 mL). The organic layer was dried (MgSO4), filtered through a pad of silica gel and concentrated. The residual solvents removed in vacuo (<1 mmHg) overnight to provide 10.7 g (85%) of 2 as a thick oil.
With dmap; triethylamine; In dichloromethane; at 0 - 25℃; for 4h;
The solution of pent-3-en-l-ol (20g, 0.232 mol) in dichloromethane (200 ml) was cooled to 0C, and triethylamine (45.01 mL, 0.325 mol), followed by tert- butyldimethylsilyl chloride (50.3 g, 0.0.278 mol) and DMAP (0.56 g, 4.644 mmol) were added to the reaction mixture and stirred at 25C for 4 hours. After completion of the reaction, reaction mixture was diluted with dichloromethane (100 mL) and washed successively with water and brine solution. Organic layer was dried over Na2S04, and concentrated in vacuo at 40C to obtain (pent-3-en-l-yloxy)(tert-butyl)dimethylsilane, IIIc ( 42.06g ). The crude material was taken for the next step without any purification. 1HNMR (DMSO-d6) delta 5.76 - 5.86 (m, 1H), 4.93-5.04 (m, 2H), 3.58 (t, = 8.4 hz, 2H), 2.02-2.10 (m, 2H), 1.49-1.58 (m, 2H), 0.93 (s, 9H), 0.09 (s, 6H)
With dmap; triethylamine; In dichloromethane; at 0 - 20℃;
27A. 2, 2-DIMETHYL-1-PENT-4-ENYLOXY-1, 1-DIPHENYL-1-SILAPROPANE To a solution OF 4-PENTENE-1-OL (Lancaster Synthesis, 8.76 g, 101.76 mmol), triethylamine (12.05 g, 119.06 mmol) and 4-dimethylaminopyridine (0.623 g, 5.09 mmol) in dichloromethane (200 ML) at 0 C was added TERT-BUTYLCHLORODIPHENYLSILANE (Acros Organics, 29.37 g, 106.85 mmol) dropwise with stirring over 20 minutes. The reaction mixture was warmed to room temperature, and stirred overnight. The reaction mixture was partitioned between water and dichloromethane. The organic layer was washed with 1N HC1, water (2X) saturated sodium bicarbonate solution, brine, and dried over magnesium sulfate. The solvent was removed IN VACUO to obtain the title compound (33.03 g, 100% yield) as a pale yellow oil : 1H NMR (300 MHz, CDC13) 8 7.73-7. 70 (m, 4H), 7.47-7. 42 (m, 6H), 5.84 (m, 1H), 5.08-4. 96 (m, 2H), 3.72 (t, J = 7.3 Hz, 2H), 2.23-2. 14 (m, 2H), 1.74-1. 66 (m, 2H), 1.09 (s, 9H); MS (API-TIS) M/Z 325 (MH+), 342 (MNH4+).
97.3%
With 1H-imidazole; In N,N-dimethyl-formamide; at 0℃; for 1h;
Dissolve 4-penten-1-ol (30.0g, 0.35mol) in 150mL DMF, add imidazole (35.6g, 0.52mol), cool to 0 degree, TBDPSCl (100.5g, 0.365mol) dropwise to the reaction solution .After stirring for 1 hour, it was quenched with water, extracted with 300 mL of tert-butyl methyl ether, and washed successively with 150 mL of 5% HCl water and 150 mL of saturated brine.Concentration yielded 110 g of product XIVa as a colorless oil with a yield of 97.3%.
With triphenylphosphine; diethylazodicarboxylate In tetrahydrofuran at 20℃; for 3h; Inert atmosphere;
96%
With di-isopropyl azodicarboxylate; triphenylphosphine In tetrahydrofuran at 0 - 20℃; for 20h; Inert atmosphere;
95%
With triphenylphosphine; acetylenedicarboxylic acid diethyl ester In tetrahydrofuran for 3h; Ambient temperature;
87%
With triphenylphosphine; diethylazodicarboxylate In tetrahydrofuran; toluene at 20℃; Inert atmosphere;
86%
With di-isopropyl azodicarboxylate; triphenylphosphine In tetrahydrofuran at 0 - 20℃; Inert atmosphere;
84%
With triphenylphosphine; diethylazodicarboxylate In tetrahydrofuran at 20℃;
With di-isopropyl azodicarboxylate; triphenylphosphine In tetrahydrofuran at 0℃;
With triphenylphosphine; diethylazodicarboxylate In tetrahydrofuran at 0 - 20℃;
With triphenylphosphine; diethylazodicarboxylate In tetrahydrofuran; toluene at 20℃; for 3h;
With di-isopropyl azodicarboxylate; triphenylphosphine In tetrahydrofuran at 20℃; for 15h; Inert atmosphere;
With triphenylphosphine; diethylazodicarboxylate In tetrahydrofuran at 20℃; for 16h; Inert atmosphere; Schlenk technique;
With di-isopropyl azodicarboxylate; triphenylphosphine In tetrahydrofuran at 0 - 20℃; for 3h; Inert atmosphere;
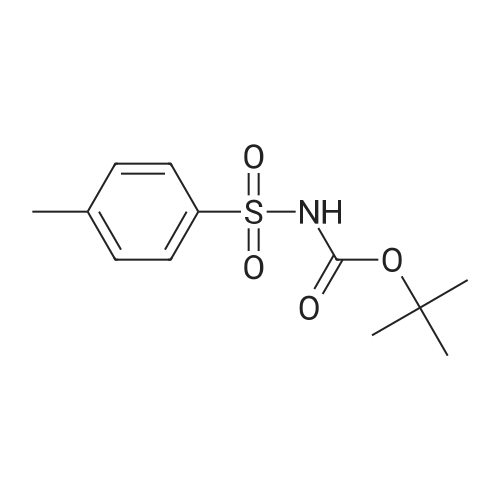
Stage #1: n-Pent-4-enyl alcohol; N-[(tert-butoxy)carbonyl]-4-methylbenzenesulfonamide With triphenylphosphine In tetrahydrofuran for 0.25h; Inert atmosphere;
Stage #2: With di-isopropyl azodicarboxylate In tetrahydrofuran at 0 - 20℃; Inert atmosphere;
With dmap; triethylamine; In dichloromethane; at 0℃;Inert atmosphere;
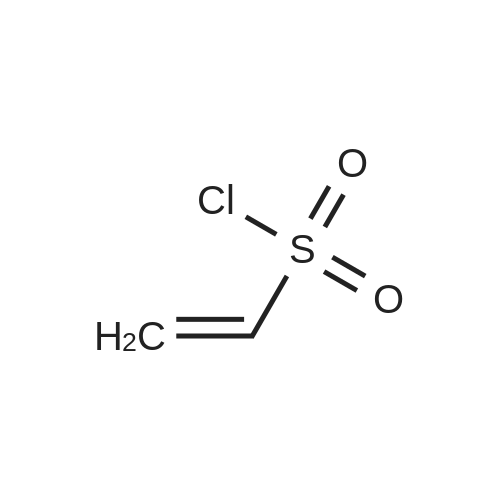
General procedure: The unsaturated alcohol (1.00 mmol) was dissolved in CH2Cl2 (10 mL), and a small amount ofN,N-dimethylaminopyridine was added. The solution was cooled to 0 C, and triethylamine (1.20mmol) was added. Then, 2-methyl<strong>[14418-84-9]allylsulfonyl chloride</strong> (0.186 g, 1.20 mmol) was slowly added withcooling and stirring. The resultant solution was stirred at 0 C until complete conversion of the alcoholsubstrate (1-2 h). In order to remove triethylammonium hydrochloride, the mixture was filteredthrough a plug of silica gel, which was eluted with Et2O. After removal of the volatiles under reducedpressure, the crude product was purified by flash chromatography on silica gel to give the sulfonates2b and 2c.
Stage #1: n-Pent-4-enyl alcohol With sodium hydride In 1,4-dioxane
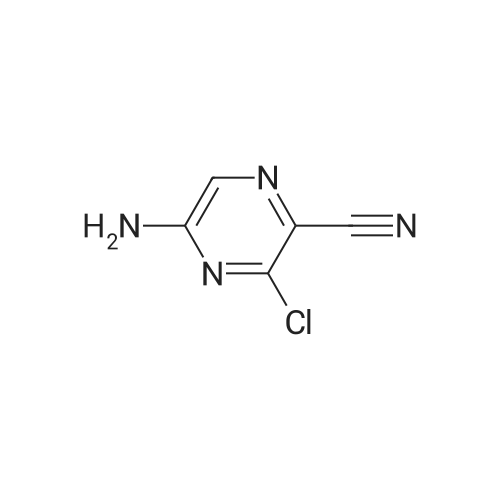
Stage #2: 5-amino-3-chloro-pyrazine-2-carbonitrile In 1,4-dioxane at 100℃; Further stages.;
General procedure: In air, Cu(OAc)2·H2O (5.0mg, 5mol%) and IPr·HCl (10.6mg, 5mol%) were placed in a screw-capped reaction vial. The vial was moved in to a glove box and t-BuOK (5.6mg, 10mol%) and solvent (1.0ml) were added. The vial was moved out of the glove box and connected to an argon line through a needle. The mixture was raised to 50C and stirred for 1h. PMHS (131mg, 4.0equiv) wasthen added dropwise with a microsyringe and the solution was stirred for an additional 30min. After the mixture was changed to the specified reaction temperature, liquid alkyne (0.5mmol) and t-BuOH (74mg, 2.0equiv) was added dropwise. The mixture was stirred for a specified period of time. The reaction mixture was subsequently hydrolyzed by adding 1M NaOH aqueous (2ml) (for substrates with no hydroxyl group) or 1M TBAF in THF (2ml) at 0C (for substrates with a hydroxyl group) for several hours. The mixture was extracted with ether (2ml×3). Crude products wereobtained after evaporation and purified by silica gel chromatography.
With diisobutylaluminium hydride; In hexane; at 20℃; for 15h;Inert atmosphere;
8m were made from corresponding terminal alkyne and iodoalkyne S87via route shown in Scheme 4. To asolution of 1.5 M DiBAL-H (27.7 mL, 41.6 mmol) in Hexanes under argon at -20 C was added slowly S7 (1 g, 11.9mmol). The solution was stirred 15 h at room temperature, after removal of volatiles under vacuo. The resultant colourless oil was dissolved in THF (20mL) and a solution of iodine (3.62 g, 14.3 mmol) in THF (20 mL) was slowly added at -78 C under argon. Then the reaction was stirred for 20 min at -78 C and warmed to room temperature. After the solution was cautiously poured into a 2N solution of HCl (10 mL) at 0 C and more HCl (8 mL) was carefully added. The aqueous layer was extracted with PE/EA 1/1 (2 *30 mL). The combined organic extracts were dried over Na2SO4, filtered and concentrated under vacuo. Purification by flash chromatography (PE/EA 1/1) gave S8 as a yellow oil (1.26 g, 50%).6 Pd(PPh3)4 (68 mg, 0.059 mmol) and copper iodide (23 mg, 0.12 mmol) were added to a seal-tube. Absorb the air andfill with nitrogen. Oct-1-yne (143 mg, 1.3 mmol) ,S8 (250 mg, 1.18 mmol), triethylamine (143 mg, 1.42 mmol) and 4 mL THF was added. The mixture was heated to 50 C and stirred for 10 h. The reaction was then cooled and diluted with Et2O (10 mL) and saturated aqueous NH4Cl (5 mL). The organics were extracted with Et2O (2 *5 mL), dried over Na2SO4, filtered, and concentrated in vacuo. Purification via flash chromatography on silica gel (PE/EA = 7/1) provided 8m (117mg, 90% yield).
4-pentenyl 5-aminolevulinate hydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
47%
Example 3 Preparation of 4-pentenyl 5-aminolevulinate hydrochloride To 1 ml of thionyl chloride (SOCl2) were added 3 drops of N,N-dimethylformamide (DMF) with stirring. Following the addition of <strong>[5451-09-2]5-aminolevulinic acid hydrochloride</strong> (ALA.HCl, 200 mg, 1.19 mmol), the solution was stirred for 12 hours at room temperature. Concentration in a vacuum was conducted before the addition of 4-pentenol. Then, the reaction mixture was stirred for 1.5 hours at room temperature, followed by purification by silica gel chromatography to afford 4-pentenyl 5-aminolevulinate hydrochloride at a yield of 47%. 1H NMR (300 MHz, DMSO-d6): delta8.27 (s, 3H), 5.84-5.73 (m, 1H), 5.06-4.94 (m, 2H), 4.0 (t, J=6.6 Hz, 2H), 3.97 (s, 2H), 2.79 (t, J=6.3 Hz, 2H), 2.54 (t, J=6.6 Hz, 2H), 2.06 (q, J=7.6 Hz, 2H), 1.65 (quintet, J=6.6 Hz, 2H); 13C NMR (75 MHz, DMSO-d6): delta172.09, 152.67, 143.18, 137.68, 115.20, 63.20, 31.91, 29.39, 28.91, 27.18.
Pentenol 3 (1.2 mL, 11.6 mmol) was added dropwise to a stirred suspension of NaH (557 mg, 60% dispersion in mineral oil, 13.9 mmol) in anhydrous THF (23 mL) at 0 C. After 1 h at the same temperature, chloroacetamide 1 (1.4 mL, 13.9 mmol) was added at 0 C and the mixture was stirred for 24 h at room temperature. The mixture was neutralized with saturated aqueous NH4Cl and THF was evaporated under reduced pressure. The residue was dissolved in EtOAc and washed with water and brine. The organic layer was dried over anhydrous MgSO4 and concentrated at reduced pressure. Purification of the residue by flash column chromatography on silica gel (hexanes/EtOAc, 1:1) afforded 6b (1.87 g, pale orange oil) in 94% yield. 1H NMR (CDCl3, 300 MHz) delta 5.89-5.74 (m, 1H), 5.07-4.93 (m, 2H), 4.13 (s, 2H), 3.52 (t, J = 6.5 Hz, 2H), 3.02 (s, 3H), 2.96 (s, 3H), 2.19-2.08 (m, 2H), 1.77-1.66 (m, 2H); 13C NMR (CDCl3, 75 MHz) delta 169.4, 138.1, 114.9, 70.9, 70.4, 36.5, 35.6, 20.3, 28.8; HRMS (EI) m/z calcd for C9H17NO2 (M+) 171.1259, found 171.1243.
94%
General procedure: To a suspension of NaH (344 mg, 60% dispersion in mineral oil) in THF (17 mL) was added allyl alcohol (2) (500 mg, 8.6 mmol) at 0 C. After 1 h at the same temperature, chloroacetamide 1 (0.97 mL, 9.5 mmol) was added and the mixture was stirred for 24 h. The mixture was quenched with saturated aqueous NH4Cl and concentrated at reduced pressure. The resulting residue was dissolved in EtOAc and washed with water and brine. The organic layer was dried over anhydrous MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (hexanes/EtOAc, 3:1) to give 6a (1.05 g, pale yellow oil) in 85% yield.
With lanthanum(lll) triflate In toluene at 80℃; for 0.0333333h;
General procedure for the preparation of the PMB ethers (General Method A)
General procedure: To a solution of the alcohol (3.0 mmol) in dry toluene (30 mL) was added 4-methoxybenzyl trichloroacetimidate (S3) (1.27 g, 4.5 mmol) and La(OTf)3 (88 mg, 0.15 mmol). The reaction mixture was heated for 2 min at 80 °C. After cooling to 21 °C the solvent was removed under reduced pressure and the residue was treated with a mixture of hexanes-ethyl acetate (5:1, V:V).The resulting precipitate was filtered off and the filtrate was evaporated. The residue was purified by flash-chromatography to yield the corresponding PMB ether.
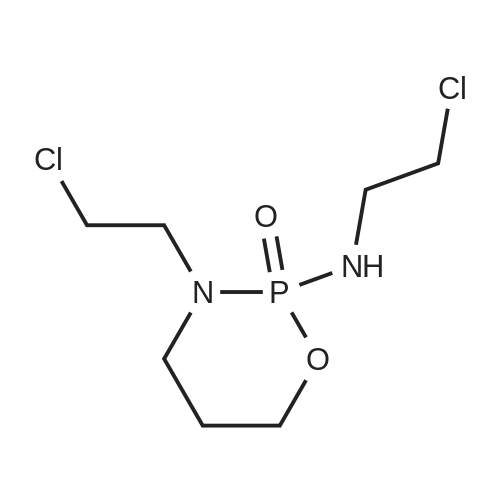
(2-chloroethyl)-[3-(2-chloroethyl)-2-oxo-4-pent-4-enyloxy-2λ5-[1,3,2]oxazaphosphinan-2-yl]amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
19%
With N,N,N,N-tetraethylammonium tetrafluoroborate; In acetonitrile;Electrochemical reaction;
IFO therefore was electrochemically oxidized in acetonitrile in the presence of various alcohols with 5 carbon atoms. The reaction was stopped when all the starting material appeared to be consumed as monitored by TLC. At the end of the reaction, sodium bicarbonate (1 Eq.) was added to the medium to neutralize electrogenerated hydrons. The proportions of the various diastereomers were determined on the crude reaction mixture by 31P-NMR before being isolated by ?flash chromatography?.
With dmap; N-ethyl-N,N-diisopropylamine; HATU In dichloromethane at 20℃; for 14h;
4.4A Step 4A
To N-Boc-L-leucine 4a-l (3.05 g, 12.234 mmol) and alcohol (1.58 g, 18.35 mmol) in DCM (20 mL) was added HATU (5.582 g, 14.681 mmol), DIPEA (4.3 mL, 24.468 mmol) and DMAP (75 mg, 0.612 mmol). The resulted mixture was stirred at RT for 14 h, and was diluted with DCM, washed with IN HC1, H20 and brine sequentially. The organic layer was dried ( a2S04) and concentrated in vacuo and the residue was purified by silica gel chromatography (hexane to 30% acetone in hexane) to afford the product 4a (3.25 g, 89%). MS (ESI): m/z 322.24 (M+Na).
4-pentenyl 3,4,6-tri-O-acetyl-2-deoxy-2-(2,2,2-trichloroethoxysulfonyl)amido-β-D-galactopyranoside[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
63%
With tetrakis(μ-trifluoroacetamidato)dirhodium(II); iodosylbenzene In chlorobenzene at 5 - 20℃; Molecular sieve; Inert atmosphere; stereoselective reaction;
63%
Stage #1: n-Pent-4-enyl alcohol; 2,2,2-trichloroethyl sulfamate; triacetyl-D-galactal With tetrakis(μ-trifluoroacetamidato)dirhodium(II) In chlorobenzene Molecular sieve; Inert atmosphere;
Stage #2: With iodosylbenzene In chlorobenzene at 5 - 20℃; Molecular sieve; Inert atmosphere; stereoselective reaction;
General procedure for one-pot amidoglycosylation
General procedure: To a mixture of glycal 1a,b,c (0.2 mmol), alcohol (0.4 mmol),TcesNH2 (80 mg, 0.35 mmol), Rh2(NHCOCF3)4 (12 mg, 0.02 mmol),and activated powdered molecular sieves 4 Å (160 mg) under nitrogen was added PhCl (3 mL), and the resulting light-purple suspension was cooled with an ice-water bath. PhIO (80 mg,0.36 mmol) was added in several portions for 1 h, and the resulting light-brown suspension was stirred at 5°C for 1 h and then at rt for 5-15 h with monitoring the reaction by TLC. The reaction mixture was filtered, washed with CH2Cl2, and the combined filtrates were concentrated under reduced pressure to remove CH2Cl2. The residue was purified by silica gel chromatography, usually eluting with hexane-EtOAc mixture
With oxygen; potassium carbonate at 60℃; for 8h; Sealed tube;
2.6. General procedure for oxidative esterification of benzylicalcohols
General procedure: A 50 ml round bottomed flask was charged with heterogeneousPd catalyst 1 (1 mol%), K2CO3(1.2 equiv.) and methanol (3 ml). Thebase was soluble in the methanol and methanol acted as bothreactant and reaction media for the present transformation. Theresulting flask was sealed with septum, evacuated and back-filledwith oxygen. After that the benzyl alcohol (1.0 equiv.) was added tothe above suspension via syringe. The reaction mixture was stirredat 60C for the time as given in Table 1 in the presence of oxy-gen balloon. After completion, the mixture was cooled to roomtemperature and catalyst was recovered by the influence of exter-nal magnet. The crude product was dissolved in ethyl acetate andwashed successively with water to remove the base. The organiclayer was dried over anhydrous MgSO4and concentrated underreduced pressure. The residue so obtained was purified by columnchromatography using eluent ethyl acetate in hexane (20:80) togive the pure desired product. The recovered catalyst was washedwith methanol, dried in vacuum and used for the subsequent runs.
(5-hydroxypentyl)(phenyl)phosphinic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
In 1,4-dioxane; at 160℃; for 1h;Microwave irradiation; Sealed tube;
General procedure: Phenyphosphinic acid (0.2 mmol), alkene (0.4 mmol) and dioxane (4 mL) were added into a 10 mL microwavevial containing a Teflon-coated stirrer bar and a septum. The vial was sealed and was heated under microwaveirradiation at 160 C for 1 hour. After cooling to room temperature the reaction mixture was purified by columnchromatography on silica gel using CH2Cl2-MeOH-CH3COOH: 9-0.5-0.5 as eluent to give the corresponding<strong>[1779-48-2]phenyl phosphinic acid</strong>s 6.
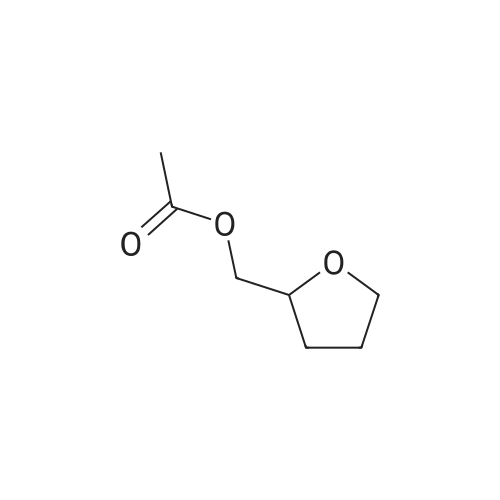
With dihydrogen peroxide; potassium iodide; at 20℃; for 4.0h;
General procedure: An alkenoic acid (0.3 mmol), H2O2 (30%, 0.6 mmol) and KI (0.06 mmol) were dissolved in AcOH (2 mL), and the mixture was stirred at room temperature for 4 h. When the reaction was complete, H2O (5 mL), sat.aq Na2S2O3 (2 mL) and sat. aq Na2CO3 (2 mL) were added. The mixture was extracted with CH2Cl2 (3×5 mL) and the combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified on silica gel plate using (4:1 hexane-ethyl acetate) as eluant to afford the pure product. 2-Acetoxymethyltetrahydrofuran (2j): Oil;20 IR (neat, cm-1): 1745, 1237, 1043; 1H NMR: delta 4.14-4.02 (m, 2H), 4.00-3.90 (m, 1H), 3.85-3.72 (m, 2H), 2.09 (s, 3H), 2.05-1.85 (m, 3H), 1.70-1.57 (m, 1H); 13C NMR: delta 170.8, 78.4, 71.5, 65.7, 27.5, 23.9, 20.1; MS (EI, m/z, %): 144 (M+, 23.3), 43 (100).
With ammonium iodide; 3-chloro-benzenecarboperoxoic acid In 2,2,2-trifluoroethanol at 20℃; for 5h;
Selenolactonization; General Procedure
General procedure: Alkenoic acid 1 (0.4 mmol), diselenide 2 (0.24 mmol), NH4I (0.04mmol) and mCPBA (0.22 mmol) were added successively to a reaction vessel containing CF3CH2OH (1.5 mL). The resulting suspension was stirred vigorously at r.t. for 5 h. Upon completion, the reaction mixture was quenched by the addition of sat. aq Na2S2O3 (2 mL) solution, and made basic by the addition of sat. aq Na2CO3(8 mL) solution and then H2O (5 mL). The mixture was extracted with CH2Cl2(3 × 5 mL) and the combined organic phase dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified on a silica gel plate (PE-EtOAc, 2:1) to furnish the corresponding selenolactone 3
With ammonium iodide; 3-chloro-benzenecarboperoxoic acid; In 2,2,2-trifluoroethanol; at 20℃; for 5h;
General procedure: Alkenoic acid 1 (0.4 mmol), diselenide 2 (0.24 mmol), NH4I (0.04mmol) and mCPBA (0.22 mmol) were added successively to a reaction vessel containing CF3CH2OH (1.5 mL). The resulting suspension was stirred vigorously at r.t. for 5 h. Upon completion, the reaction mixture was quenched by the addition of sat. aq Na2S2O3 (2 mL) solution, and made basic by the addition of sat. aq Na2CO3(8 mL) solution and then H2O (5 mL). The mixture was extracted with CH2Cl2(3 × 5 mL) and the combined organic phase dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified on a silica gel plate (PE-EtOAc, 2:1) to furnish the corresponding selenolactone 3
tetra(4-pentenyl)-1,2,3,4-cyclobutanetetracarboxylic acid ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
25 g
With toluene-4-sulfonic acid; In toluene; for 4h;Dean-Stark; Reflux;
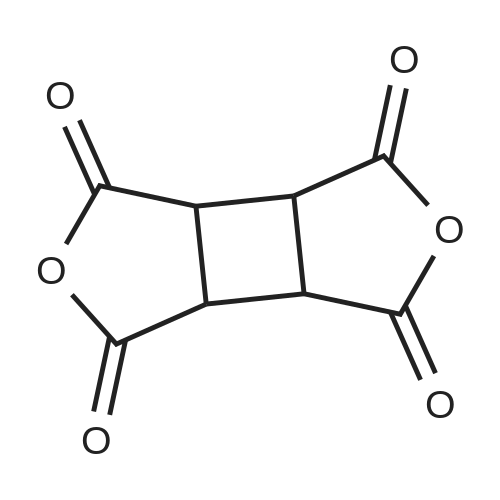
To a reactor equipped with a Dean-Stark apparatus and a condenser, 10 g of <strong>[4415-87-6]1,2,3,4-cyclobutanetetracarboxylic acid dianhydride</strong>, 1 g of p-toluenesulfonic acid monohydrate, 100 mL of toluene, and 21 g of 5-hexen-1-ol were charged, and the mixture was caused to react at a reflux temperature for 4 hours. After completion of the reaction, the mixture was washed with sodium bicarbonate water and washed with water, and then concentrated to obtain 25 g of tetra(4-pentenyl)-1,2,3,4-cyclobutanetetracarboxylic acid ester as a light yellow liquid. (0167) H-NMR (300 MHz, CDCl3): delta=5.86-5.72 (m, 4H), 5.07-4.98 (m, 8H), 4.18-4.04 (m, 8H), 3.75 (s, 4H), 2.17-2.08 (m, 8H), 1.77-1.68 (m, 8H), (0168) LC-MS (ESI): m/z=505.5 (M+H).
With dmap; diisopropyl-carbodiimide; In dichloromethane; at 20 - 30℃; for 12h;
2 - ((3-fluoro-4- (methylcarbamoyl) phenyl) amino) -2-methyl-propionic acid (500g, 2.0mol, 1eq), 4- penten-1-ol (172g, 2.0mol, 1.0eq), dimethylaminopyridine (24g, 0.2mol), 1,3- diisopropyl carbodiimide (500g, 4.0mol) and dichloromethane (2L) were sequentially added 10L reaction flask within the control temperature 20 ~ 30 , the reaction was stirred for 12 hours. 2L water was added to the reaction flask, extracted, the aqueous phase was extracted with dichloromethane 2L × 3, dichloromethane layers were combined, washed twice with saturated sodium chloride solution, 2L, dried over anhydrous sodium sulfate, filtered, and concentrated to dryness under reduced pressure, 4L of acetone was added with stirring, filtered, and added to the solution with stirring 10L hexane crystallization 2h, give crude intermediate 1 (451g, 1.4mol).
Stage #1: 3-Hydroxy-2-naphthoic acid With 1,1'-carbonyldiimidazole In N,N-dimethyl-formamide at 20℃; Inert atmosphere;
Stage #2: n-Pent-4-enyl alcohol With 1,8-diazabicyclo[5.4.0]undec-7-ene In N,N-dimethyl-formamide at 20℃; for 24h; Inert atmosphere;
General Procedure for the Synthesis of Alkenyl 3-Hydroxy-2-naphthoates (3a-3h) from Primary Alcohols
General procedure: N,N'-Carbonyldiimidazole (CDI, 80 mmol) was added to a solution of 3-hydroxy-2-naphthoic acid (80 mmol) in DMF (70 mL) for 10-15 min while stirring at 20°C in an argon atmosphere. The reaction occurred with evolution of CO2. After the whole amount of CDI was added, the stirring of the reaction mixture (solution I) was continued for another 1 h. A mixture of alkenol (53.1 mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 79.7 mmol) was stirred in DMF (60 mL) (solution II) under the same conditions as indicated above. Then solution II was added to solution I at 20°C; the resulting reaction mixture was stirred for 24 h at the same temperature. The reaction mixture was diluted with diethyl ether (150 mL) and washed in sequence with a 10% HCl solution (4 × 50 mL), H2O (3 × 50 mL) until neutral pH of the medium, and then with a saturated NaHCO3 solution (3 × 50 mL) and H2O (3 × 50 mL)until neutral pH of the medium. The organic layer was dried over MgSO4. The solvent was removed at reduced pressure.
With ethanol; sodium In hexane; mineral oil at 0℃; for 0.0833333h; Inert atmosphere;
3 Preparation of compound of formula 6
Under the condition of 0 ° C nitrogen protection and vigorous stirring,Compound of formula 10 (12.20 mmol, 1.39 g)Anhydrous n-hexane (30 mL)The solution was added to absolute ethanol (55.00 mmol, 2.50 g).Sodium mineral oil suspension (34.1 wt%, 55.00 mmol, 3.71 g) was then added.The reaction was carried out at 0 ° C for 5 min and was quenched to room temperature with water (10 mL).A saturated sodium hydroxide solution (10 mL) was added to the reaction mixture.It was refluxed at 100 ° C for 2 hours.After cooling to room temperature, the organic layer was washed with brine (3×10 mL).Dry over anhydrous magnesium sulfate, filter and store with nitrogen for use.
ethyl 1-methyl-5-(pent-4-en-1-yloxy)-1H-pyrazole-4-carboxylate[ No CAS ]
pent-4-en-1-yl 1-methyl-5-(pent-4-en-1-yloxy)-1H-pyrazole-4-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
4.1.6 1-Methyl-5-(pent-4-en-1-yloxy)-1H-pyrazole-4-carboxylic acid 10b A solution of pent-4-en-1-ol (1.45?mL, 14.0?mmol) in THF (24.0?mL) was cooled to -8?C and treated with 1.0?M sodium hexamethyldisilazane in THF (14.0?mL, 14.0?mmol). The reaction mixture was stirred at the reduced temperature for 5?min before the ice bath was removed. Stirring was continued for an additional 25?min before being treated with a solution of <strong>[105486-72-4]ethyl 5-bromo-1-methyl-1H-pyrazole-4-carboxylate</strong> (1.1?g, 4.7?mmol) in THF (24?mL). The reaction mixture was stirred for 1 hour at room temperature before being quenched with 50?mL of sat. NH4Cl. The crude product was extracted with DCM (60?mL x 3), dried with Na2SO4 and concentrated under reduced pressure. This material purified by SiO2 chromatography (ethyl acetate: petroleum ether?=?1:5) to afford 0.65?g impure product 9b mixed with pent-4-en-1-yl 1-methyl-5-(pent-4-en-1-yloxy)-1H-pyrazole-4-carboxylate (produced through transesterification reaction), the mixed esters could both be the raw materials for next hydrolysis, so the mixture was put into next step without further separation. To a solution of the mixed esters in 5?mL MeOH/THF (VMeOH:VTHF?=?1:1) at 0? was added 5?mL 2M NaOH (aq). The mixture was stirred at room temperature for 3?h and MeOH and THF were evaporated in vacuo. The residue was acidified to pH?=?2-3 with 1?N HCl and extracted with ethyl acetate (10?mL?*?3). The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure to afford compound 10b as yellow solid (0.45?g, 46%). 1 H NMR (500?MHz, CDCl3) delta 7.84 (s, 1H), 5.89-5.77 (m, 1H), 5.10-5.00 (m, 2H), 4.46 (t, J?=?6.5?Hz, 2H), 3.70 (s, 4H), 2.28-2.19 (m, 2H), 1.93-1.85 (m, 2H); ESI-MS: m/z?=?209 [M-H]-.
1-methyl-5-(pent-4-en-1-yloxy)-1H-pyrazole-4-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
46%
A solution of pent-4-en-1-ol (1.45?mL, 14.0?mmol) in THF (24.0?mL) was cooled to -8?C and treated with 1.0?M sodium hexamethyldisilazane in THF (14.0?mL, 14.0?mmol). The reaction mixture was stirred at the reduced temperature for 5?min before the ice bath was removed. Stirring was continued for an additional 25?min before being treated with a solution of <strong>[105486-72-4]ethyl 5-bromo-1-methyl-1H-pyrazole-4-carboxylate</strong> (1.1?g, 4.7?mmol) in THF (24?mL). The reaction mixture was stirred for 1 hour at room temperature before being quenched with 50?mL of sat. NH4Cl. The crude product was extracted with DCM (60?mL x 3), dried with Na2SO4 and concentrated under reduced pressure. This material purified by SiO2 chromatography (ethyl acetate: petroleum ether?=?1:5) to afford 0.65?g impure product 9b mixed with pent-4-en-1-yl 1-methyl-5-(pent-4-en-1-yloxy)-1H-pyrazole-4-carboxylate (produced through transesterification reaction), the mixed esters could both be the raw materials for next hydrolysis, so the mixture was put into next step without further separation. To a solution of the mixed esters in 5?mL MeOH/THF (VMeOH:VTHF?=?1:1) at 0? was added 5?mL 2M NaOH (aq). The mixture was stirred at room temperature for 3?h and MeOH and THF were evaporated in vacuo. The residue was acidified to pH?=?2-3 with 1?N HCl and extracted with ethyl acetate (10?mL?*?3). The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure to afford compound 10b as yellow solid (0.45?g, 46%). 1 H NMR (500?MHz, CDCl3) delta 7.84 (s, 1H), 5.89-5.77 (m, 1H), 5.10-5.00 (m, 2H), 4.46 (t, J?=?6.5?Hz, 2H), 3.70 (s, 4H), 2.28-2.19 (m, 2H), 1.93-1.85 (m, 2H); ESI-MS: m/z?=?209 [M-H]-.
ethyl 4-((ethoxycarbonothioyl)thio)-7-hydroxyheptanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
84%
With [4,4?-bis(1,1-dimethylethyl)-2,2?-bipyridine-N1,N1?]bis [3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl-N]phenyl-C]iridium(III) hexafluorophosphate; In dimethyl sulfoxide; at 30℃; for 37h;Irradiation; Sealed tube; Inert atmosphere;
General procedure: To a 10 mL sealed tube was added ethyl 2-((ethoxycarbonothioyl)thio)acetate (1a,61.3 mg, 0.294 mmol), [Ir{dF(CF3)ppy}2(dtbbpy)](PF6) (8, 1.6 mg, 0.00143 mmol),and 1-octene (2a) (90 L, 0.573 mmol). Degassed anhydrous DMSO (290 L) wasadded and the reaction was sparged with Ar. The tube was sealed and surrounded byblue LEDs for 20 h. The reaction mixture was diluted with EtOAc and water. Theaqueous layer was separated and extracted twice with EtOAc. The combined organicextracts were washed with brine, dried over MgSO4, filtered and concentrated invacuo. The resulting crude material was purified by flash column chromatography(silica gel; hexane/EtOAc 200:1) to give 3aa (84.3 mg, 0.263 mmol) in 89% yield as acolorless oil.
2-(7-hydroxyhept-3-en-1-yl)isoindoline-1,3-dione[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
45%
With tricyclohexylphosphine[1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidine][benzylidene]ruthenium(II) dichloride In dichloromethane at 40℃; for 5.5h; Inert atmosphere;
General procedure D
General procedure: Cross Metathesis Reaction Under an atmosphere of argon, 4-penten-1-ol (3 equiv) and alkene (1 equiv) are dissolved in degassed, anhydrous CH2Cl2 (0.5 M). Grubbs catalyst 2nd generation (0.05 equiv) is added and the reaction mixture is allowed to stir at 40 °C. The solvent is removed under reduced pressure and the residue is purified on silica gel to yield the title compound.
pent-4-enyl 1-(pyrimidin-2-yl)-1H-indole-2-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65%
With rhodium(III) chloride trihydrate; copper diacetate; In N,N-dimethyl-formamide; at 90℃; under 760.051 Torr; for 10h;Sealed tube;
AddN-pyrimidinylpurine 1a (0.2 mmol), RhCl3•3H2O (0.004 mmol), Cu(OAc)2(0.4 mmol) andpent-4-en-1-ol 2l (1.0 mmol) to 2.0 In mL of DMF, after replacing carbon monoxide three times in Young's tube,add carbon monoxide (1 atm), react with 90 C oil bath for 10 hours, stop the reaction, wait until the reaction solution is cooled to room temperature, addethyl acetate and saturate The saline is washed and extracted several times.The organic phase was dried over anhydrous sodium sulfate and filtered.The solvent was evaporated to dryness, andethyl acetate / petroleum ether (1:10 to 1:1) was obtained.The product was a colorless oily liquidwith a yield of 65%.
With toluene-4-sulfonic acid; hydroquinone In toluene for 18h; Reflux; Dean-Stark;
21 EXAMPLE 21 (0209) Synthesis of Di(4-pentenyl) Itaconate (DPI)
Into a 100 mL round-bottom flask, itaconic acid (3.90 grams, 30 mmol), 4-pentenol (5.17 grams, 60 mmol), p-toluenesulfonic acid (0.456 grams, 2.65 mmol), hydroquinone (0.29 grams, 2.68 mmol), and toluene (45 mL) were transferred and then fitted with a Dean-Stark trap. The mixture was refluxed under vigorous stirring for 18 hours. The reaction solution was then cooled to room temperature and washed with 90 mL of deionized water three times. The product was dried over magnesium sulfate and concentrated by rotary evaporation. The obtained crude product was purified by silica column chromatography on the Biotage using a SNAP Ultra 50 g silica column (hexanes/ethyl acetate = 9/1) to afford the final product as a colorless oil (57% yield). 1H MR (400 MHz, CDC13), d (TMS, ppm): 6.32 (s, 1H), 5.82 (m, 2H), 5.69 (s, 1H), 4.94 (m, 4H), 4.14 (t, 2H), 4.08 (t, 2H), 3.33 (s, 2H), 2.03 (m, 4H), 1.77-1.55 (m, 4H).
2-bromo-2,2-difluoroacetate allylpentyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
98.3%
With phosphotungstic acid; phosphomolybdic acid In toluene at 110℃; for 10h;
2 Example 2
Equipped with a thermometer, reflux condenser, Add 87.5 grams of 2-bromo-2,2-difluoroacetic acid, 140 grams of 4-penten-1-ol, 8.75 grams of phosphomolybdic acid and 500 grams of toluene, The reaction was refluxed at 110°C until no water was released, and the reaction time was 10 hours. After the reaction is completed, the solvent and excess 4-penten-1-ol are recovered by distillation, and then the fraction under the condition of 158159/50mmHg is collected by vacuum distillation. Obtain 2-bromo-2,2-difluoroacetate enpentyl ester 119.5 g, yield 98.3%, The purity is 99.7%.
1,2,4,5-tetrafluoro-3-(pent-4-en-1-yloxy)benzene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
With potassium <i>tert</i>-butylate In acetonitrile at 70℃; for 8h;
2. General procedure for polyfluorinated aromatic ethers
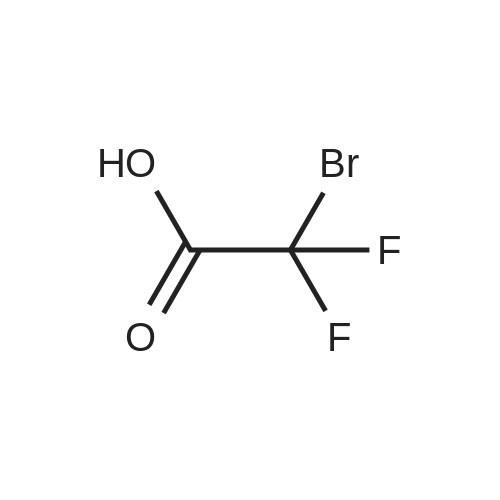
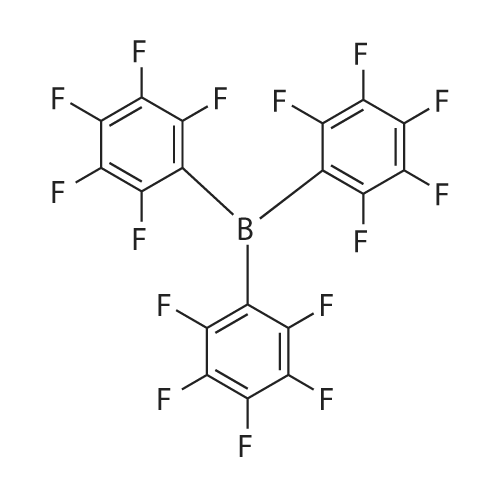
General procedure: Reaction conditions: To a 25 mL Shrek tube were sequentially added (thio)alcohol or (thio)phenol (0.2 mmol), B(C6F5)3 (35mol%, 35.8 mg), t-BuOK (0.2 mmol, 1.0 equiv, 22.4 mg) and CH3CN (2.0 mL). The formed mixture was stirred at 70 °C for 8h as monitored by TLC and GC-MS. After completion of the reaction, the crude product was concentrated and purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate) to afford the corresponding products.
With Cp*Ir(6,6'-dionato-2,2'-bipyridine)(H2O); caesium carbonate In tert-Amyl alcohol at 125℃; for 12h; Inert atmosphere;
4.2. Procedure for the a-alkylation of ketones with alkenyl alcohols catalyzed by [Cp*Ir(2,2'-bpyO)(H2O)] (cat. 10) (Tables 1-3)
General procedure: To an oven-dried, N2-purged 25 mL Schlenk tube were added ketone (1 mmol), alkenyl alcohol (1.2 mmol), cat. 10 (5.3 mg,0.01 mmol, 1 mol%), Cs2CO3 (98 mg, 0.3 mmol, 0.3 equiv) and tert amylalcohol (1 mL). The mixture of reaction was heated under 125 °C in an oil bath for 12 h. The reaction mixture was cooled to ambient temperature, concentrated in vacuo and purified by flash column chromatography with ethyl acetate/hexanes to afford the corresponding product.1-Phenylhept-6-en-1-one (3aa) [3e]. Purified by flash column chromatography on silica gel (ethyl acetate/hexanes = 1/100);Pale-yellow oil;
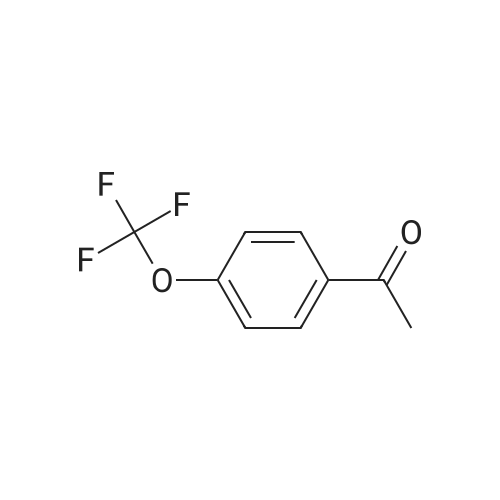
1-(4-(trifluoromethoxy)phenyl)hept-6-en-1-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
With Cp*Ir(6,6'-dionato-2,2'-bipyridine)(H2O); caesium carbonate In tert-Amyl alcohol at 125℃; for 12h; Inert atmosphere;
4.2. Procedure for the a-alkylation of ketones with alkenyl alcohols catalyzed by [Cp*Ir(2,2'-bpyO)(H2O)] (cat. 10) (Tables 1-3)
General procedure: To an oven-dried, N2-purged 25 mL Schlenk tube were added ketone (1 mmol), alkenyl alcohol (1.2 mmol), cat. 10 (5.3 mg,0.01 mmol, 1 mol%), Cs2CO3 (98 mg, 0.3 mmol, 0.3 equiv) and tert amylalcohol (1 mL). The mixture of reaction was heated under 125 °C in an oil bath for 12 h. The reaction mixture was cooled to ambient temperature, concentrated in vacuo and purified by flash column chromatography with ethyl acetate/hexanes to afford the corresponding product.1-Phenylhept-6-en-1-one (3aa) [3e]. Purified by flash column chromatography on silica gel (ethyl acetate/hexanes = 1/100);Pale-yellow oil;






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping