| 92% |
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; potassium carbonate In tetrahydrofuran at 20℃; for 48h; |
|
| 92% |
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; potassium carbonate In tetrahydrofuran at 20℃; for 48h; |
|
| 92% |
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; potassium carbonate In tetrahydrofuran at 0 - 50℃; Inert atmosphere; |
|
| 92% |
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; Cs2CO3 In tetrahydrofuran at 20℃; for 48h; Inert atmosphere; |
|
| 90% |
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; triethylamine In tetrahydrofuran for 16h; Heating; |
|
| 90% |
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; triethylamine In tetrahydrofuran for 15h; Heating; |
|
| 90% |
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; triethylamine In tetrahydrofuran for 16h; Reflux; |
|
| 89% |
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; potassium carbonate In tetrahydrofuran for 24h; Inert atmosphere; Cooling with ice; Reflux; |
Synthesis of HAPCP
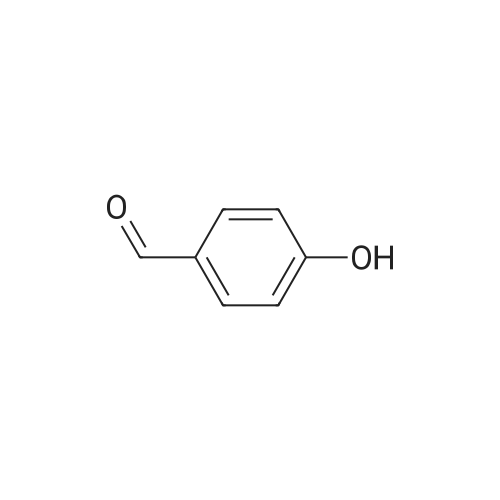
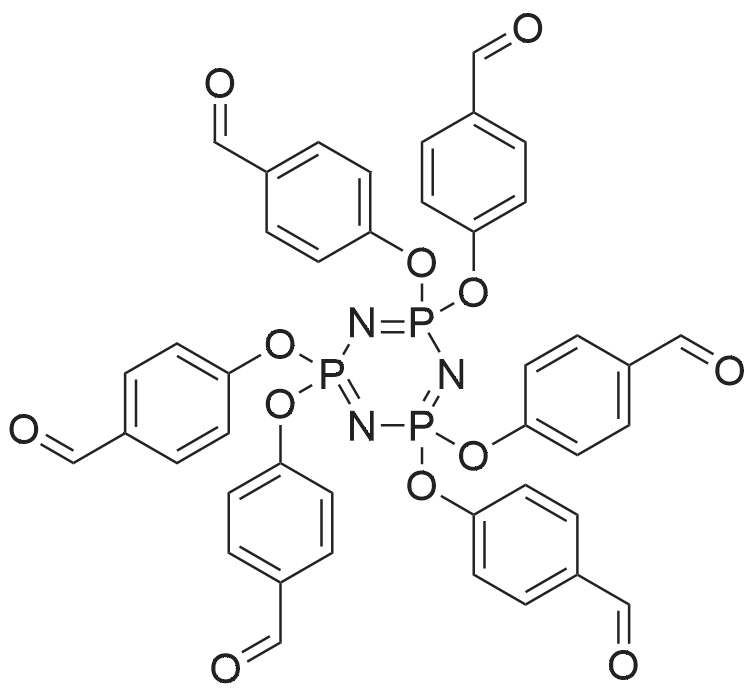
In a three-neck round-bottom flask equipped with a reflux condenser and a constant drip funnel, HCCP (5.0 g, 14.4 mmol) and K2CO3 (25.8 g, 187.0 mmol) were stirred in THF (100 mL) in an ice bath under nitrogen atmosphere. Simultaneously, p-hydroxybenzaldehyde (12.3 g, 10.1 mmol) in THF (50 mL) was added dropwise into the above mixture. Then the reaction mixture was heated to reflux, and stirred vigorously for 24 h. After that the mixture was filtered and the filtration was concentrated on a rotary evaporator. The obtained crude product was recrystallized from chloroform/petroleum ether, then washed by ethanol and dried in vacuum. Finally, HAPCP was obtained as a white solid powder. Yield: 89%. 1H NMR (400 MHz, CDCl3) ä 9.94 (s, 1H), 7.74(d, J=8.6Hz, 2H), 7.15 (d, J=8.5Hz, 2H). 13CNMR(101MHz,CDCl3) ä 190.5 (-CHO), 154.5, 133.8, 131.5, 121.3(-C6H4). 31PNMR (162 MHz, CDCl3) ä 7.3. |
| 87% |
Stage #1: 4-hydroxy-benzaldehyde With potassium carbonate In tetrahydrofuran for 2.5h; Cooling with ice;
Stage #2: With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine In tetrahydrofuran at 20℃; for 48h; |
|
| 85% |
With triethylamine In tetrahydrofuran for 48h; Reflux; |
|
| 85% |
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; sodium hydride In tetrahydrofuran; mineral oil |
|
| 85% |
Stage #1: 4-hydroxy-benzaldehyde With potassium carbonate In tetrahydrofuran for 0.5h; Cooling with ice;
Stage #2: With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine In tetrahydrofuran at 65℃; for 24h; |
|
| 81% |
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; potassium carbonate In acetonitrile at 20℃; for 24h; |
2.3.1. Hexa(4-formyl-phenoxy)cyclotriphosphazene
4-hydroxybenzaldehyde (17.56 g, 103.55 mmol) was added to amixture of N3P3Cl6 (1) (3.00 g, 8.63 mmol) and K2CO3 (14.40 g,103.55 mmol) in acetonitrile (300 mL) at room conditions. Thereactionwas stirred for 24 h at room temperature. After acetonitrileused as solvent was removed under vacuum, CH2Cl2 (350 mL) waspoured into the residual solid. The mixture was filtered and thesolvent was evaporated to about 30 mL. Then solution of themixture was slowly added to ethanol (400 mL) and a white solidprecipitated out. The resulting white solid was filtered, washed with hexane and dried at room temperature. The yield of thecompound 2 was 6.02 g (81%): mp:157 C. FT-IR (cm1): 1704 (HC]O), 1206, 1180 and 1156 (P]N), 960 (P-O-Aryl). 1H NMR d (ppm,(CD3)2CO): 10.0 (H-C]O), 8.0e7.0 (Aryl H). 31P NMR d (ppm): 7.95. |
| 80% |
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; potassium carbonate In acetone for 12h; |
|
| 77.5% |
Stage #1: 4-hydroxy-benzaldehyde With potassium carbonate In tetrahydrofuran at 20℃; for 0.5h;
Stage #2: With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine In tetrahydrofuran for 48h; Reflux; |
N3P3(OC6H4-p-CHO)6
N3P3(OC6H4-p CHO)6 was prepared according to the literature29 with minor modification. 4-Hydroxybenzaldehyde (25.20 g, 0.206 mol) was dissolved in dry THF (300 mL).To this solution was added K2CO3 (45.50 g, 0.33 mol). The mixture was stirred at room temperature for 30 min. A solution of hexachlorocyclotriphosphazene (10.00 g, 0.0288 mol) in THF (50 mL) was added drop-wise to the reaction mixture over 1 h. After stirring at refluxing temperature for 48 h, the reaction mixture was concentrated in a rotary evaporator and washed with 800 mL of water. The crude product was then recrystallized from ethyl acetate to yield white crystalline N3P3(OC6H4-p CHO)6. Yield 77.5%, mp 160-162 C. 1HNMR(DMSO-d6, TMS, ppm): 9.95(1H, CHO), 7.19-7.82 (4H, dd, ArH). 13C NMR (DMSO-d6,TMS, ppm): 192.0 (C O), 154.1 (C O), 134.1 (C C), 131.9 (CH), 121.5(CH). 31P NMR(DMSO-d6, ppm): 7.64. |
| 71% |
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; triethylamine In tetrahydrofuran for 8h; |
1.1; 2.1; 3.1
In a 250 mL three-necked flask, 14.76 g of p-hydroxybenzaldehyde, 60 mL of tetrahydrofuran and 20 mL of triethylamine were successively added and stirred to dissolve. 6.95 g of hexachlorocyclotriphosphazene was dissolved in 40 mL of tetrahydrofuran and added dropwise to the flask And the reaction was carried out for 8 hours. After the reaction was completed, the product was filtered and the filtrate was taken from the vials. The solvent was removed by steaming, then the cold ethanol was added, and the upper layer was collected by suctioning. The solid was recrystallized from ethyl acetate. Hexahydrophthalazine (HCCP-PHBA) in 71% yield. |
| 70% |
With tripotassium phosphate tribasic; 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine In acetonitrile for 3h; Heating; |
|
| 63% |
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; triethylamine In tetrahydrofuran for 48h; Reflux; |
2.2 Synthesis of hexakis(4-formylphenoxy)cyclophosphazene (HAPCP)
The HAPCP was prepared according to a reference [41]. (0008) The hexachlorocyclotriphosphazene (5.1g, 0.015mol) and triethylamine (12.0g, 0.119mol) were dissolved in THF (34mL). 4-Hydroxybenzaldehyde (11.8g, 0.096mol) was added to the above solution, and the mixture was stirred under reflux for 48h. After reaction, the mixture was cooled to room temperature, filtered to remove solids, and then recrystallized with ethyl acetate. The product was dried under vacuum at 50°C for 12h to obtain yellow solid. Yield: 7.9g (63%). 1H NMR (Fig. S1(a), 300.53MHz, CDCl3, δ): 9.9 (s, 6H), 7.7 (d, 12H), 7.2 (d, 12H). 13C NMR (Fig. S1(b), 75.57MHz, CDCl3, δ): 190.4, 154.5, 133.8, 131.4, 121.2. 31P NMR (Fig. S1(c), CDCl3, δ): 6.96 (s). |
|
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; sodium hydride In tetrahydrofuran at 20℃; for 6h; |
|
|
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; sodium hydride In tetrahydrofuran for 6h; Heating; |
|
|
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; sodium hydride In tetrahydrofuran; mineral oil Reflux; |
|
|
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; potassium carbonate In toluene for 24h; Reflux; |
|
|
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine Alkaline conditions; |
|
| 37.3 g |
Stage #1: 4-hydroxy-benzaldehyde With potassium carbonate In tetrahydrofuran for 0.666667h;
Stage #2: With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine In tetrahydrofuran at 70 - 75℃; for 24h; |
4
In a 500 ml dry three-necked flask equipped with a magnet, a thermometer, a constant pressure dropping funnel and a condensing reflux, a pulverized and dried k2CO3Powder 44.23 g, then 43.92 g of 4-hydroxybenzaldehyde was dissolved in 120 ml of dry THF and added dropwise to a three-necked flask. The reaction was carried out under stirring for 40 min. The system was heated to 75C while 17.38 g of HCCP was dissolved in 100 ml of anhydrous THF, the solid was completely dissolved, the dropwise addition to the reaction system, dropping about 2h, the system at 70 reflux reaction 22h.After the reaction, the white turbid solution was removed and the solid KCl and K were removed by suction filtration2CO3Etc., and 1/2 volume of tetrahydrofuran was distilled off under reduced pressure. The remaining solid and liquid were poured into 400 ml of distilled water. A white precipitate appeared immediately, filtered and the product was washed three times with deionized water.The precipitate was finally recrystallized twice from ethanol and dried at 60 ° C for 12 h to give 37.3 g of intermediate HAPCP |
|
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; potassium carbonate In tetrahydrofuran at 66℃; for 48h; |
1 Example 1: Preparation of hexa(4-carboxyphenoxy)cyclophosphazene (CTP-CHO)
In a 250 ml single-necked flask, 100 ml of tetrahydrofuran, 2.50 g (7.19 mmol) of hexachlorocyclotriphosphazene and 6.30 g (51.59 mmol) of p-hydroxybenzaldehyde, and 11.38 g (82.30 mmol) of potassium carbonate were sequentially added, stirred under heating at 66 ° C, and refluxed under reflux for 48 h. After the completion of the reaction was changed to distillation apparatus, the solvent tetrahydrofuran was distilled off, and then the product was added to 1000ml of distilled water, stirred for half an hour, still get a white precipitate, suction filtration, washed three times with distilled water, placed in a vacuum oven dried 60 degrees. Recrystallization from ethyl acetate gave the product hexa (4-carboxyphenoxy) cyclophosphazene (CTP-CHO). The reaction equation is as follows: |
|
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; triethylamine In tetrahydrofuran for 24h; Inert atmosphere; Reflux; |
1.1 Example 1
Step one, 24.6g of p-hydroxybenzaldehyde,20.4 g of triethylamine was dissolved in 150 mL of tetrahydrofuran,Under ice bath and inert gas, 10 g of hexachlorocyclotriphosphazene (HCCP) dissolved in 50 mL of tetrahydrofuran was added dropwise. After the addition is completed,Gradually warmed to reflux temperature, the reaction 24h. After the reaction,Suction filtration, triethylamine hydrochloride was removed, the solvent was removed by rotary evaporation,The crude product was precipitated by addition of 200 mL of water and finally recrystallized from ethyl acetate. The pure compound HAPCP was dried in vacuo to give a white crystalline solid with the structural formula: |
|
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; potassium carbonate In tetrahydrofuran at 20℃; for 48h; Inert atmosphere; |
2.1. Synthesis of compound-I
A common precursor ‘compound-I’ was used for the synthesis ofboth HNM-1 and CHNM-1 following previous reports [29,30]. In a typicalsynthesis, 10 mmol of phosphonitrilic chloride trimer (PNC, 99%,Sigma Aldrich India) dissolved in 50ml of THF (AR, SRL India)was condensedwith 61 mmol of 4-hydroxybenzaldehyde (99%, Sigma AldrichIndia) dissolved in 100 ml THF in presence of 121 mmol of K2CO3 (AR,Fisher Scientific India) under argon atmosphere. The reaction wasfound to be completed after stirring of reaction mixture for 48 h at RT.The detailed synthesis condition and processing/purification ofcompound-I has been reported elsewhere [29]. The formation ofcompound-I was confirmed by 1H, 13C and 31P NMR spectra [29]. 1HNMR (CDCl3): δ 9.95 (s), 7.75 (d), 7.17 (d). 13C NMR (CDCl3): δ 190,155, 134, 132, 122 and 31P NMR (CDCl3): δ 7.04. |
|
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; Cs2CO3 In tetrahydrofuran at 20℃; |
|
|
With 2,2,4,4,6,6-hexachloro-1,3,5-triaza-2,4,6-triphosphorine; potassium carbonate In tetrahydrofuran at 74℃; for 24h; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping