| 85% |
With dmap; triethylamine; In dichloromethane; for 4h;Inert atmosphere; |
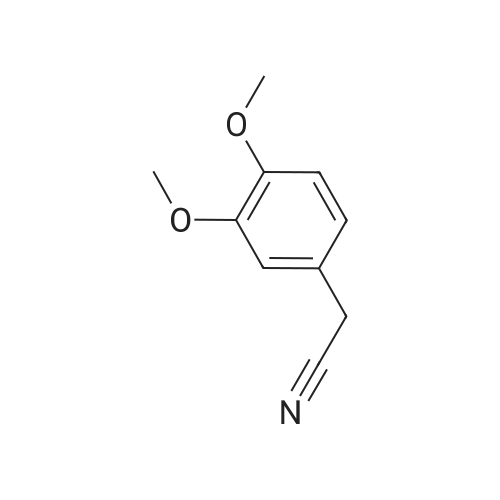
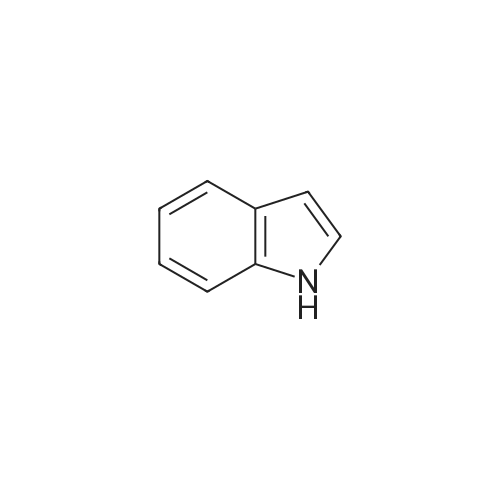
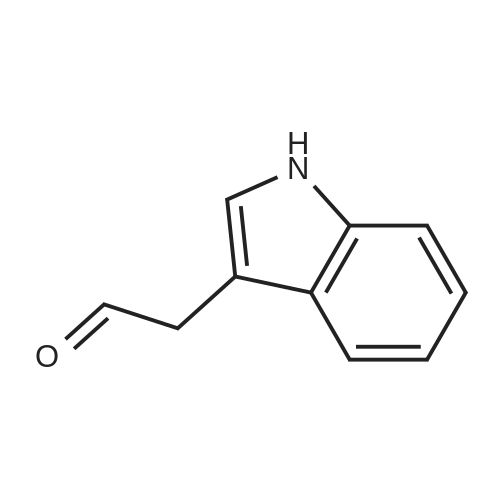
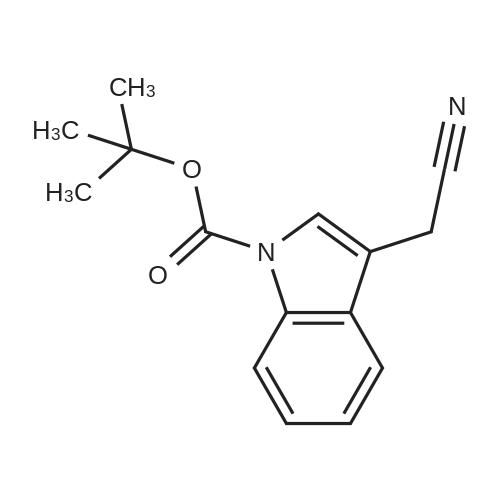
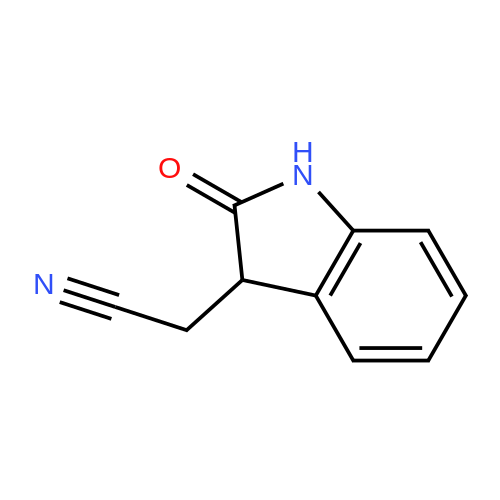
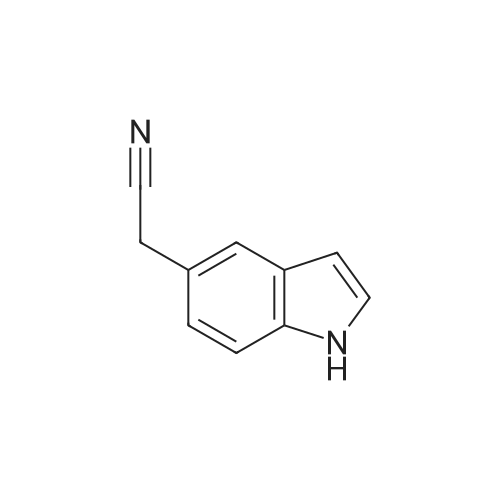
To a stirred solution of 2-(1H-indol-3-yl)acetonitrile 1 (5.0 g, 32.0 mmol) in CH2Cl2 (100 mL) were added Et3N (5.8 g, 57.6 mmol), DMAP (234 mg, 1.92 mmol) and Boc-anhydride (8.3 g, 38.4 mmol) at RT under inert atmosphere. The reaction was stirred for 4 h and monitored by TLC. The reaction mixture was diluted with water (100 mL) and extracted with CH2Cl2 (3 x 75 mL). The combined organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure to obtain the crude. The crude product was triturated with n-pentane (2 x 20 mL) and dried under reduced pressure to afford compound 2 (7.0 g, 85%) as an off-white solid. 1H NMR (500 MHz, DMSO-d6): delta 8.08 (d, J= 8.0 Hz, 1H), 7.70-7.67 (m, 2H), 7.40 (t, J= 8.0 Hz, 1H), 7.32 (t, J= 8.0 Hz, 1H), 4.12 (s, 2H), 1.63 (s, 9H). |
| 84% |
With dmap; triethanolamine; In 4-(dicyanomethylene)-2-methyl-6-(p-dimethylaminostyryl)-4H-pyran; ethyl acetate; |
A. To a stirred solution of 3-indolylacetonitrile (3.9 g, 25 mmol) in DCM (100 mL, anhyd) was added BOC anhydride (6.5 g, 30 mmol), DMAP (3.6 g, 30 mmol) and TEA (4.2 mL, 30 mmol). After 2 hours, the reaction mixture was diluted with DCM (100 mL), washed with 1N HCl (2*50 mL) and brine, then dried (Na2SO4), concentrated and chromatographed (silica gel, 6% EtOAc/Hex) to yield (1-tert-butoxycarbonylindol-3-yl)acetonitrile (5.4 g, 84%) as a pale yellow solid; 1H-NMR (CDCl3): delta 8.17 (1H, br d), 7.64 (1H, br s), 7.52 (1H, br d), 7.38 (1H, app t), 7.30 (1H, app t), 3.78 (2H, s), 1.68 (9H, s). |
| 84% |
With dmap; triethylamine; In dichloromethane; for 2h; |
A. To a stirred solution of 3-indolylacetonitrile (3.9 g, 25 mmol) in DCM (100 mL, anhyd) was added BOC anhydride (6.5 g, 30 mmol), DMAP (3.6 g, 30 mmol) and TEA (4.2 mL, 30 mmol). After 2 hours, the reaction mixture was diluted with DCM (100 mL), washed with 1 N HCI (2 x 50 mL) and brine, then dried (NaPS04), concentrated and chromatographed (silica gel, 6% EtOAc/Hex) to yield (1-tert- butoxycarbonylindol-3-yl) acetonitrile (5.4 g, 84%) as a pale yellow solid ;'H-NMR (CDCI3) : 8 8.17 (1 H, br d), 7.64 (1 H, br s), 7.52 (1H, br d), 7.38 (1 H, app t), 7.30 (1 H, app t), 3.78 (2H, s), 1.68 (9H, s). |
| 44% |
With dmap; In dichloromethane; at 25℃; for 8h; |
At 25C the 2- (1H- indol-3-yl) acetonitrile (10.0g, 64.1mmol) and DMAP (500mg, 4.1mmol) was added to a dichloromethane (50 mL), and then slowly added Boc2O (21.0g , 96.2mmol). Reaction was continued for 8 hours, then washed with a saturated sodium chloride solution (40 mL) with, liquid-separation the organic phase was dried over anhydrous sodium sulfate. Filtered, the filtrate was spin dry, purified by column chromatography (petroleum ether / ethyl acetate (v / v) = 10/1) to give the title compound as a pale yellow solid (7.2g, 44.0%). |
| 44% |
With dmap; In dichloromethane; at 25℃; for 8h; |
2- (1H-indol-3-yl) acetonitrile (10.0 g, 64.1 mmol) and DMAP (500 mg, 4.1 mmol) were added to dichloromethane (50 mL) at 25 C followed by slow addition of Boc20 , 96.2 mmol).The reaction was continued for 8 hours, then washed with saturated sodium chloride solution (40 mL) and the organic phase separated on drying with anhydrous sodium sulfate.Filtration and the filtrate was evaporated to dryness and purified by column chromatography (petroleum ether / ethyl acetate = 10/1) to give the title compound as a pale yellow solid (7.2 g, 44.0%). |
|
In acetonitrile; at 20℃; |
A solution of 1H-indol-3-ylacetonitrile (8.0g) in acetonitrile (150mL) was added by di-tert-butyl dicarbonate (13.5g) and dimethylaminopyridine (938mg) successively and stirred at the room temperature. The reaction mixture was concentrated and the obtained residue was purified by column chromatography on silica gel (hexane: ethyl acetate = from 10: 1 to 5: 1) to give the title compounds (10.5mg) having the following physical data. TLC: Rf 0.36 (hexane: ethyl acetate =5:1); NMR(CDCl3):delta 1.68 (s, 9H), 3.78 (d, J=1.28 Hz, 2H), 7.26-7.33 (m, 1H), 7.34-7.44 (m, 1H), 7.49-7.57 (m, 1H), 7.64 (s, 1H), 8.17 (d, J=8.06 Hz, 1H). |
|
With potassium carbonate; In DMF (N,N-dimethyl-formamide); at 20℃; for 12h; |
INTERMEDIATE 46 : 3-cyanomethyl-indole-l-carboxylic acid tert-butyl ester [00183] A solution of <strong>[771-51-7]3-indoleacetonitrile</strong> (10 g, 64 mmol) in DMF (160 ml) was stirred at RT. K2CO3 (13.3 g, 96 mmol) and di-tert-butyl dicarbonate (15.35 g, 70 mmol) were added thereto and the reaction mixture was stirred at RT for 12 hr. H20 (100 ml) was added to the reaction mixture and the resulting precipitate was captured by filtration. The solids were dissolved in hot methanol (20 ml) and the solution was allowed to cool slowly, producing light orange solids that were isolated by filtration to give 3- cyanomethyl-indole-1-carboxylic acid tert-butyl ester (9.2 g). 1H-NMR (300 MHz, DMSO-d6) : No. 8.08 (d, 1); 7.70 - 7.66 (m, 2); 7.42-7. 29 (m, 2); 4.12 (s, 2); 1.63 (3,9). NH40Ac standard conditions. DAD Rf = 3.31 min. M+H = 257. |
|
With potassium carbonate; In DMF (N,N-dimethyl-formamide); at 20℃; for 12h; |
Intermediate 46: 3-cyanomethyl-indole-1-carboxylic acid tert-butyl ester A solution of <strong>[771-51-7]3-indoleacetonitrile</strong> (10 g, 64 mmol) in DMF (160 ml) was stirred at RT. K2CO3 (13.3 g, 96 mmol) and di-tert-butyl dicarbonate (15.35 g, 70 mmol) were added thereto and the reaction mixture was stirred at RT for 12 hr. H2O (100 ml) was added to the reaction mixture and the resulting precipitate was captured by filtration. The solids were dissolved in hot methanol (20 ml) and the solution was allowed to cool slowly, producing light orange solids that were isolated by filtration to give 3-cyanomethyl-indole-1-carboxylic acid tert-butyl ester (9.2 g). 1H-NMR (300 MHz, DMSO-d6): delta 8.08 (d, 1); 7.70-7.66 (m, 2); 7.42-7.29 (m, 2); 4.12 (s, 2); 1.63 (3, 9). NH4OAc standard conditions. DAD Rf=3.31 min. M+H=257. |
| 68 g |
With dmap; In dichloromethane; at 20℃; |
(f) . tert-butyl 3-(cyanomethyl)-1 H-indole-1 -carboxylate <strong>[771-51-7]3-indoleacetonitrile</strong> (50 g), di-ie f-butyl dicarbonate (76.8 g) and 4-dimethylaminopyridine (1 .96 g) were added to DCM (200 ml). The mixture was stirred at room temperature overnight. The solution was washed with brine and water and then dried (Na2S04). The organic layer was concentrated and the residue was purified by chromatography on silica gel eluting with heptane and increasing amounts of ethyl acetate. Yield: 68 g |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping