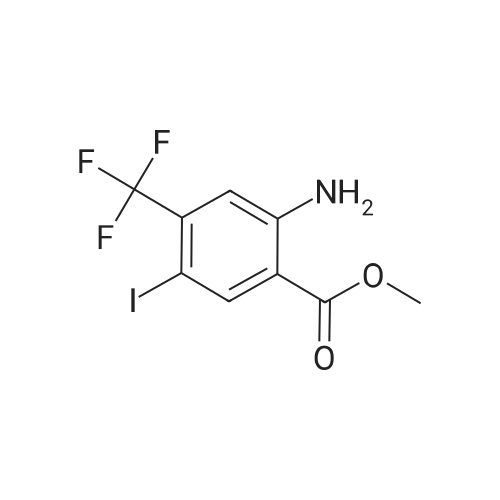
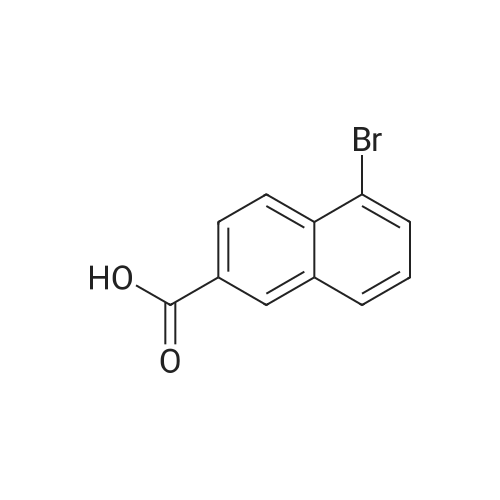
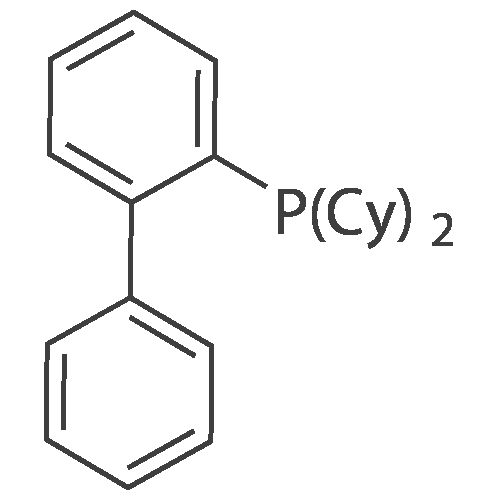
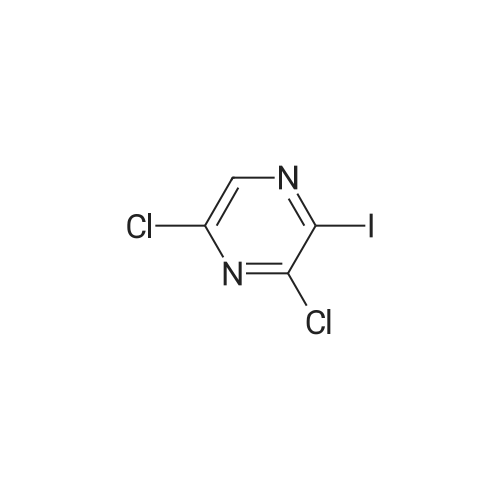
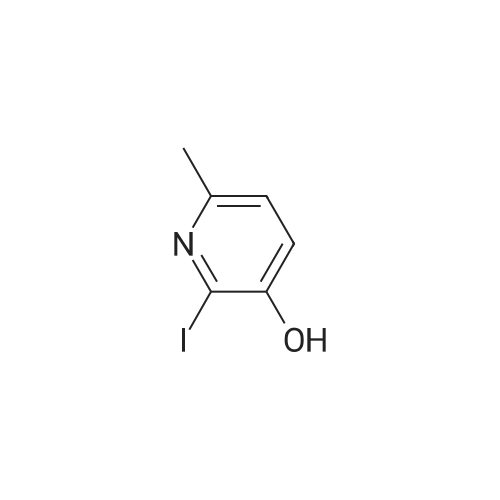
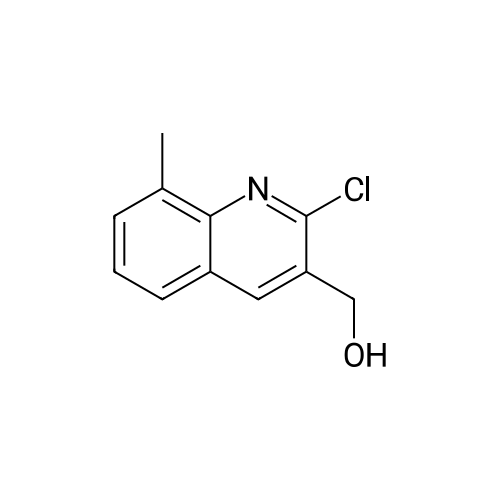
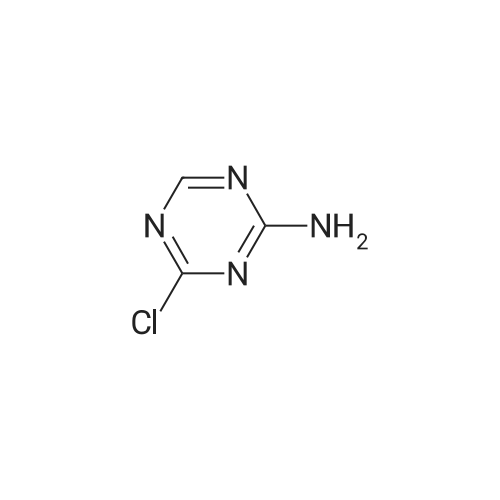
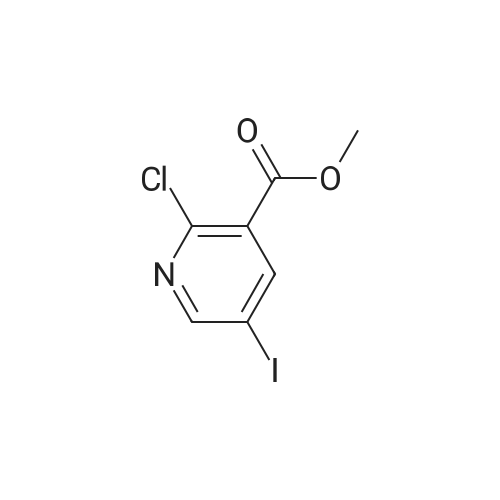
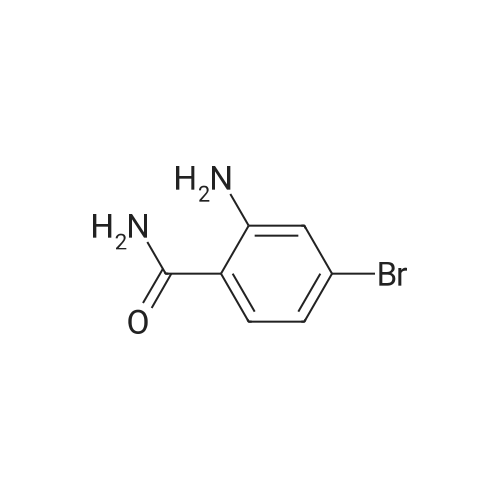
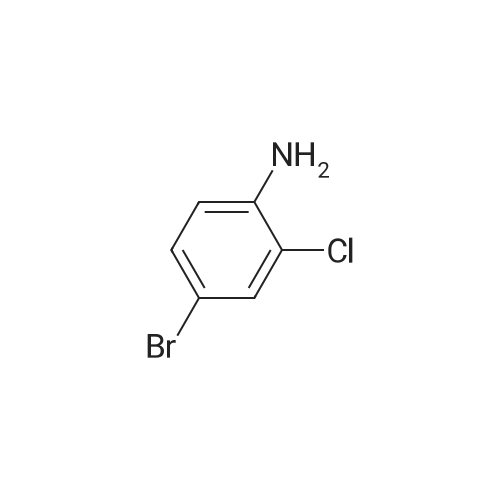
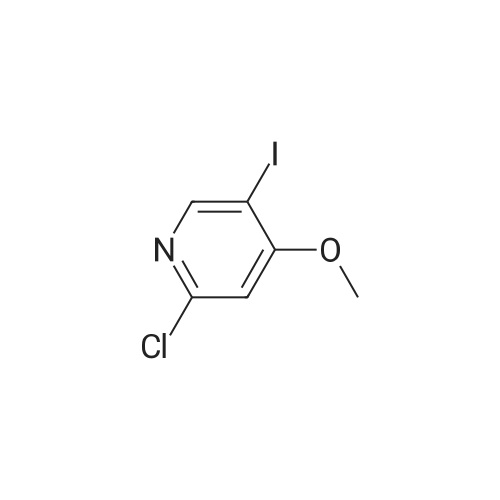
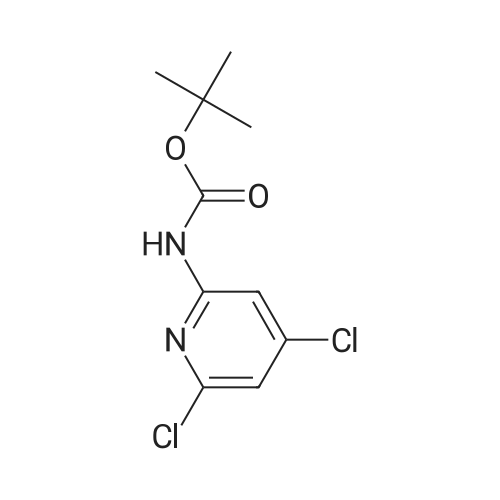
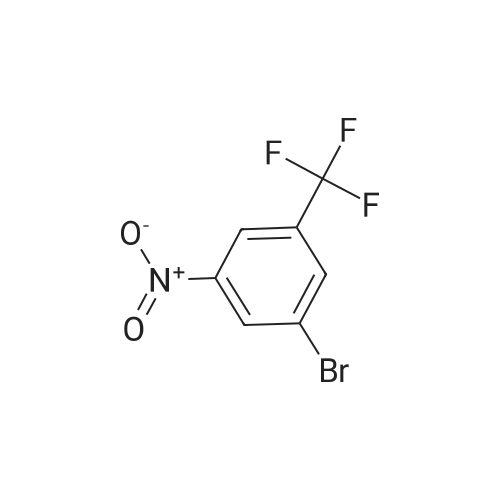
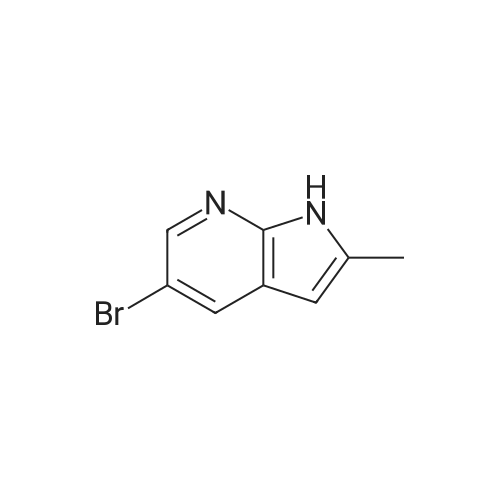
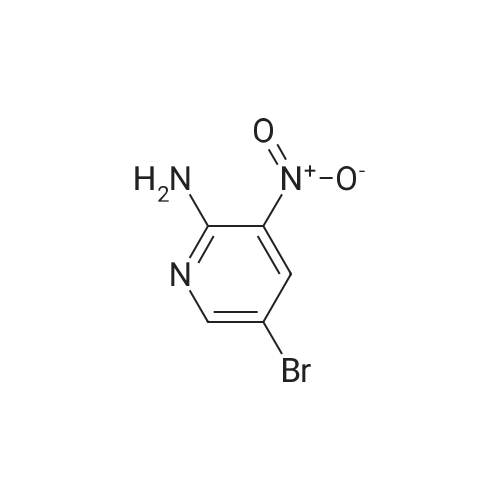
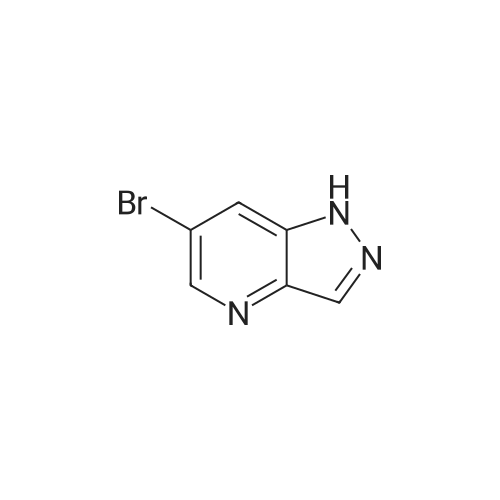
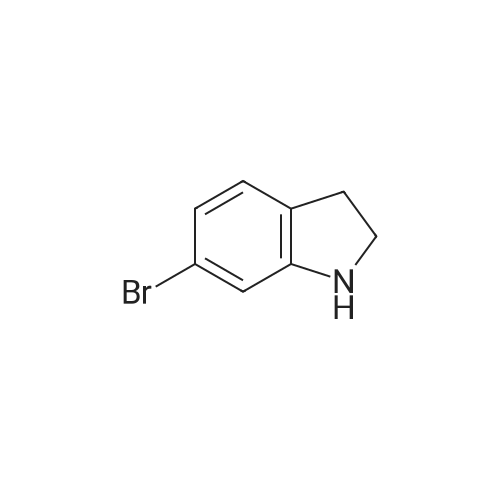
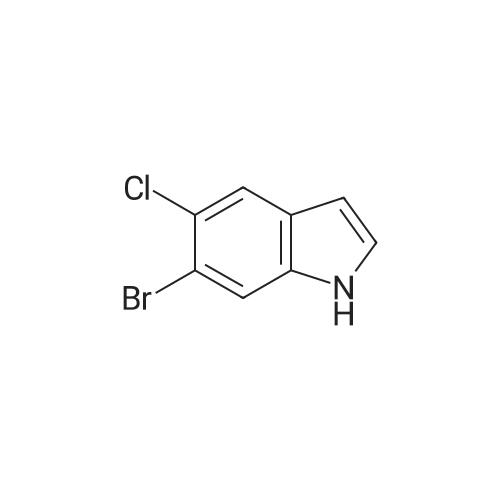
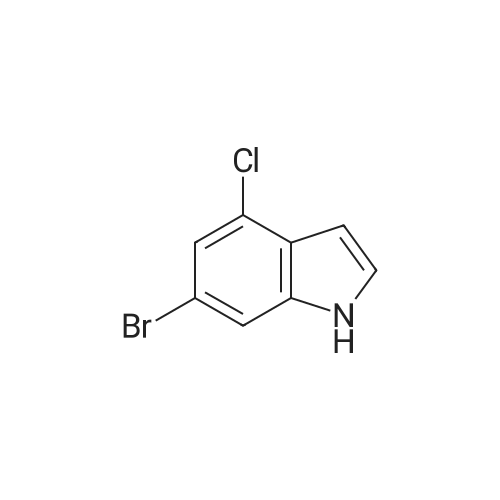
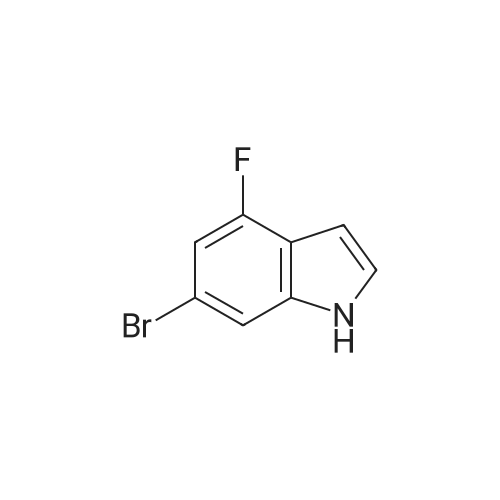
|
|
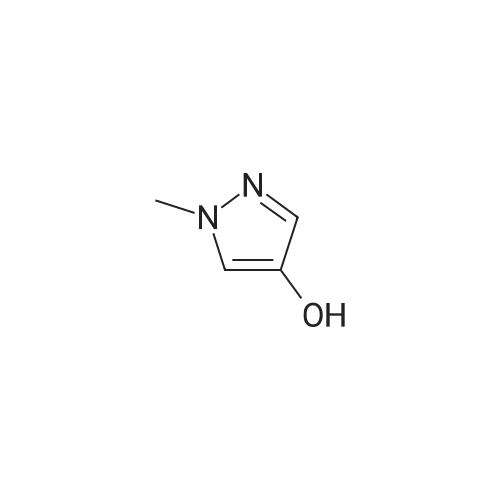
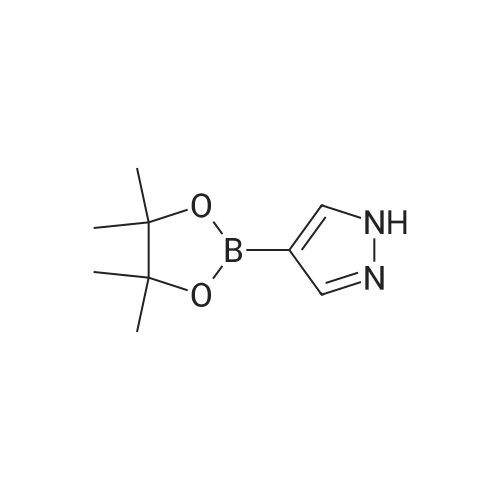
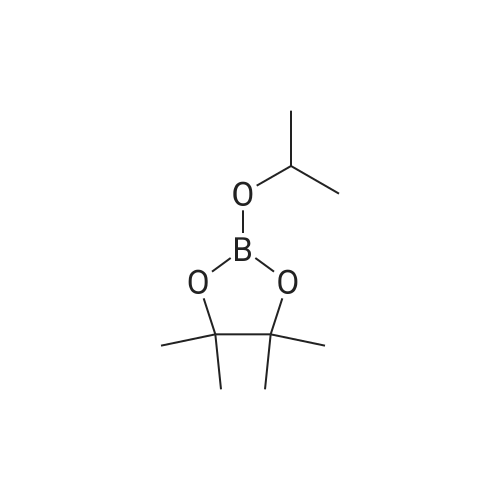
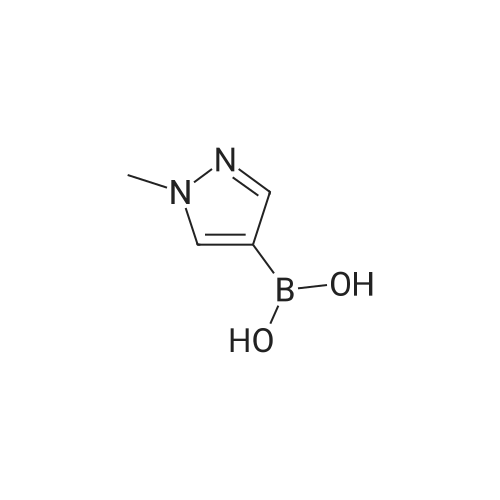
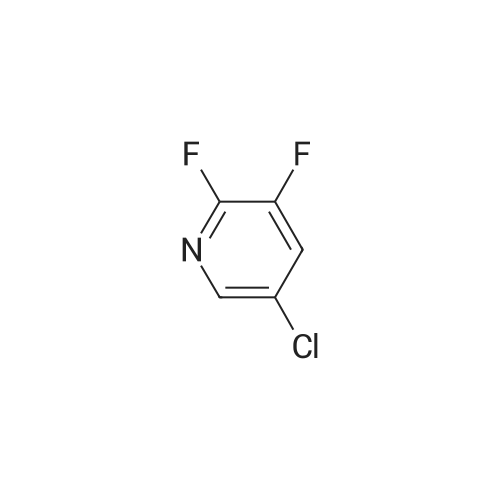
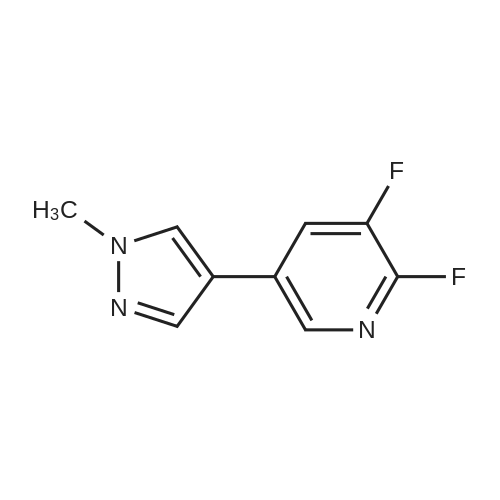
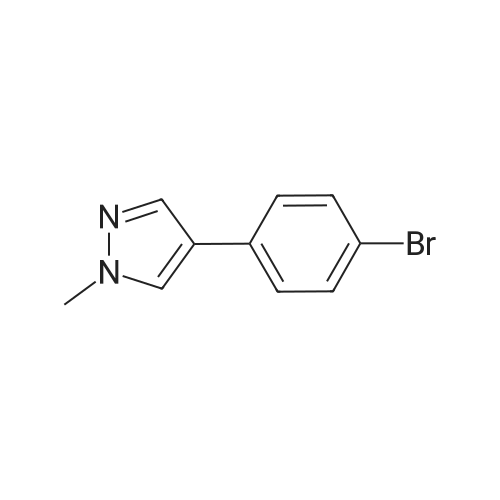
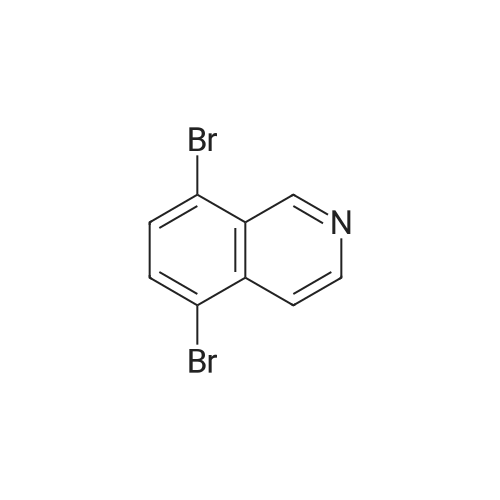
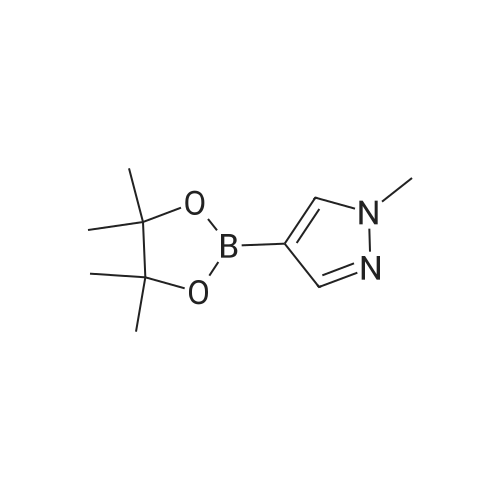
A suspension of 573 mg (2.0 mmol) <strong>[81045-39-8]5,8-dibromoisoquinoline</strong>, 458 mg (2.2 mmol) 1- methyM^^.S.S-tetramethyl-fl .S^ldioxaborolan^-y -I H-pyrazole, 849 mg (4.0 mmol) tri-potassium-phosphate trihydrate and 140 mg (0.20 mmol) bis- (triphenylphosphine)-palladium(ll)-chloride in 4 ml 1 ,2-dimethoxyethane were stirred for 18 hours at 80 C under nitrogen. The reaction mixture was cooled to room temperature, diluted with THF and filtered. The filtrate was evaporated and the residue was chromatographed on a silica gel column with ethylacetate/methanol as eluent. The two isomers were obtained separately.First eluted isomer: 8-bromo-5-(1-methyl-1 H-pyrazol-4-yl)-isoquinoline as colourless crystals; HPLC/MS 1.90 min, [M+H] = 288/290.Second eluted isomer: 5-bromo-8-(1-methyl-1 H-pyrazol-4-yl)-isoquinoline as yellow crystals; HPLC/MS (A) 2.02 min, [M+H] = 288/290.; A suspension of 135 mg (0.47 mmol) 8-bromo-5-(1-methyl-1H-pyrazol-4-yl)- isoquinoline, 126 mg (0.47 mmol) 2-(2-fluoro-phenyl)-[1,8]naphthyridine-4-boronic acid and 47.4 mg (0.56 mmol) sodium hydrogen carbonate in 1.2 ml DMF and 0.6 ml water was heated to 80 C under nitrogen. Then 6.6 mg (0.009 mmol) bis- (triphenylphosphine)-palladium(ll)-chloride were added. The reaction mixture was stirred for 18 hours at 80 C. The reaction mixture was cooled to room temperature and partitioned between water and dichloromethane. The organic phase was dried over sodium sulfate and evaporated. The residue was chromatographed on a silica gel column with ethyl acetate/methanol as eluent yielding 2-(2-fluoro-phenyl)-4-[5- (1-methyl-1H-pyrazol-4-yl)-isoquinolin-8-yl]-[1,8]naphthyridine as colourless solid; HPLC/MS (A): 1.87 min, [M+H] 432.1H NMR (400 MHz, DMSO) delta = 9.19 (dd, J=4.1, 1.9, 1H), 8.83 (d, J=0.9, 1H), 8.60 (d, J=6.0, 1H), 8.27 (s, 1H), 8.23 (td, J=7.9, 1.8, 1H), 8.17 (dd, J=6.0, 0.9, 1H), 8.11 (d, J=2A, 1H), 7.95 (d, J=7.4, 1H), 7.91 (m, 2H), 7.80 (d, J=7A, 1H), 7.62 (m, 1H), 7.57 (dd, J=8.4, 4.1, 1H), 7.47 (td, J=7.6, 1.1, 1H), 7.41 (ddd, J=11.6, 8.3, 1.0, 1H), 4.01 (s, 3H). Similarly was prepared: 2-(2-Fluoro-phenyl)-4-[8-(1-methyl-1H-pyrazol-4-yl)- isoquinolin-5-yl]-[1,8]naphthyridine as yellow solid; HPLC/MS (A): 1.84 min, [M+H] 432.1H NMR (400 MHz, DMSO) delta = 9.66 (d, J=0.9, 1H), 9.18 (dd, J=4.1, 1.9, 1H), 8.45 (d, J=5.9, 1H), 8.33 (s, 1H), 8.22 (td, J=7.9, 1.8, 1H), 8.06 (d, J=2.3, 1H), 7.95 (d, J=0.8, 1H), 7.93 (d, J=7.4, 1H), 7.87 (dd, J=8.4, 1.9, 1H), 7.83 (d, J=7.4, 1H), 7.62 (dddd, J=8.2, 7.2, 5.1, 1.9, 1H), 7.56 (dd, J=8A, 4.1, 1H), 7.47 (td, J=7.6, 1.1, 1H), 7.41 (ddd, .7=11.7, 8.3, 1.0, 1H), 7.29 (dd, .7=5.9, 0.9, 1H), 4.02 (s, 3H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping