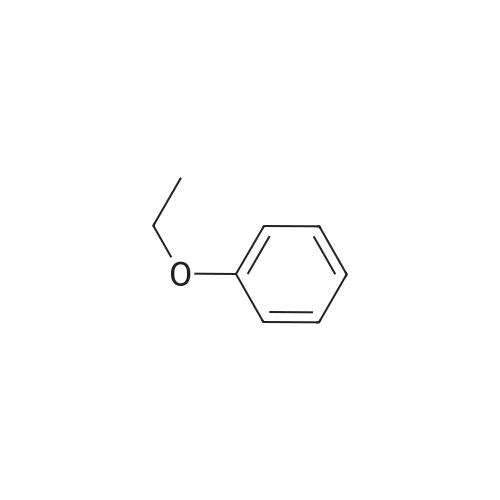
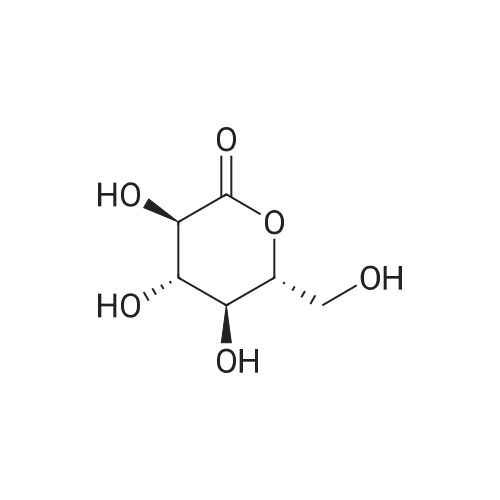
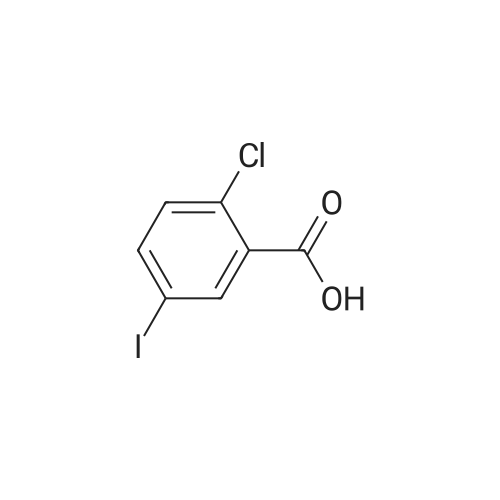
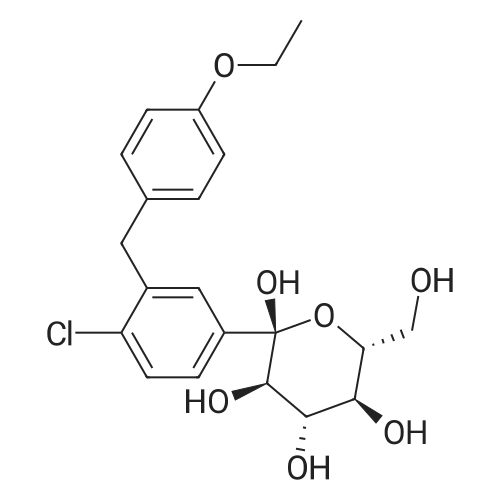
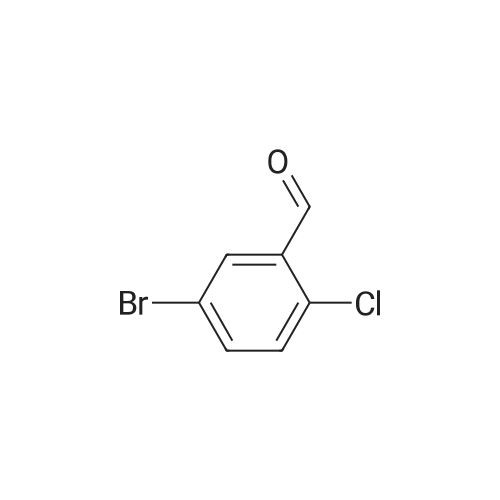
| 76% |
|
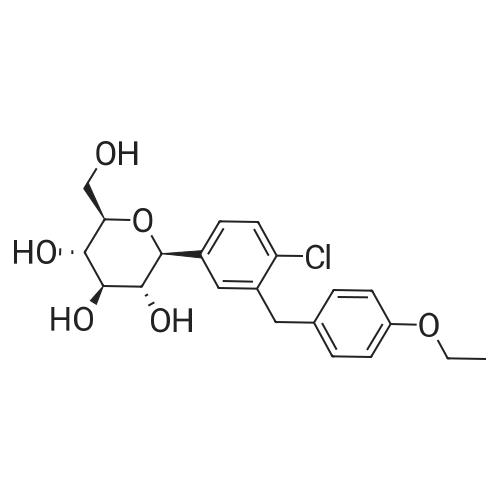
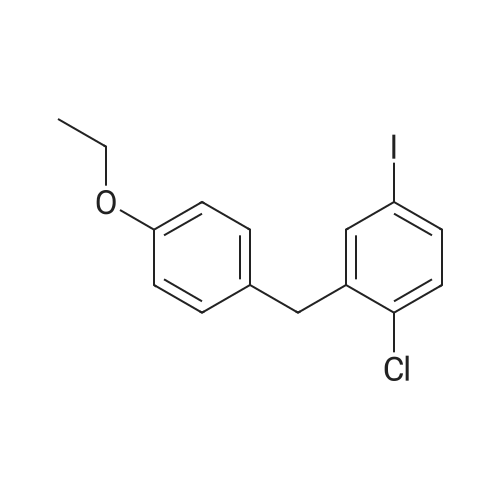
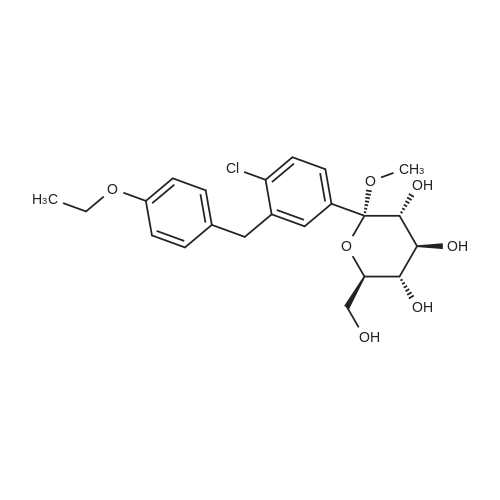
Take 1-chloro-4-(1-methoxy-D-glucopyran-1-yl)-2-(4-ethoxybenzyl)-benzene 44kg, dichloromethane 50kg and acetonitrile 150kg to add 500L reaction Stir well in the kettle. The reaction solution was cooled to -5 C, 2 kg of zinc chloride was added, and stirred for 30 min. 13 kg of Et 3 SiH was added dropwise thereto at this temperature, and the temperature was slowly raised to 10 C for 3 h.After the reaction was completed, the temperature was lowered to -5 C, and a saturated sodium hydrogencarbonate solution was added dropwise to adjust the pH to 6 to 7. Extracted with ethyl acetate (50 kg × 2), the organic phase was washed successively with saturated sodium chloride solution, water, then neutralized, then dried over anhydrous sodium sulfate, filtered, and filtrated to recover ethyl acetate under reduced pressure, 40 kg of methanol and two A mixed solution of methyl chloride (1:1) was stirred, and a large amount of solid was precipitated, and the mixture was cooled and stirred for 1 hour. Filtration, washing the solid with cold ethanol, and drying under vacuum at 50 C overnight to give a white solid 31 kg, yield 76%. The purity is 99.37%. |
| 72.1% |
With triethylsilane; boron trifluoride diethyl etherate; In dichloromethane; acetonitrile; at -20 - 0℃; |
To 100mL three-necked flask was added 4.23g of intermediate -1,20ml methylene chloride, 15ml of acetonitrile, stirred to dissolve, cooled to -15 to -20 deg.] C, was added 6.84g of triethylsilane was added dropwise 6.09g three boron trifluoride diethyl, dropping the reaction temperature is kept at -10 deg.] C, then warmed to completion of the dropwise addition -5 -0 insulation mixing the reaction, the HPLC monitoring of the reaction is complete, add saturated sodium bicarbonate ph adjusted to about 7, Save pressure distillation, liquor added ethyl acetate, dried over anhydrous sodium sulfate to afford 2.84g Dag column net yield of 72.1%. |
|
With triethylsilane; boron trifluoride diethyl etherate; water; In acetonitrile; at 5 - 10℃; for 2h;Product distribution / selectivity; |
Example 6; To acetonitrile (12 mL), at batch temperature of 8-10 C. under nitrogen atmosphere, was charged borontrifluoride diethyletherate (2.3 mL, 18.4 mmol) and water (0.82 mL, 4.6 mmol). After holding the above mixture for about 1 hour, triethylsilane (3 mL, 18.4 mmol) was added. The resulting mixture was held for about 1 hour, and then compound B (prepared as described in Example 17) in 10 mL acetonitrile was added. The batch was held at 5 to 10 C. On completion of the reaction as determined by HPLC, the reaction mixture was quenched with aqueous ammonium acetate (24 mL; 85 g) in 200 mL water. The phases were separated and product rich organic phase was dried over sodium sulfate. The product rich organic phase was concentrated under reduced pressure. Water (13 mg, 0.7 mmol, based on 0.3 g crude compound B input), (S)-propylene glycol (56 mg, 0.7 mmol), t-butylmethyl ether (5 mL, 17 mL/g compound B input), compound Ia seeds (20 mg) were mixed and held for 1 hr., to form a crystal slurry. Cyclohexane (10 mL, 33 mL/g compound B (input)) was added. The crystalline product (Ia) was isolated by filtration (4-5%) and dried in vacuo at 20-25 C. |
|
With triethylsilane; boron trifluoride diethyl etherate; In dichloromethane; acetonitrile; at -45 - -10℃; |
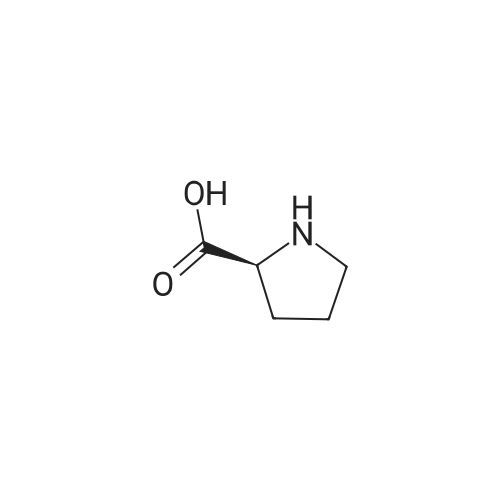
Example 1C (282 g, 0.643 mol) was dissolved in anhydrous acetonitrile /dichloromethane (3.4 L, 1 :1 (v/v)) at -45 0C stirred solution of in was added triethylsilane (299 g, 2.57 mol) followed by addition of boron trifluoride etherate (245 mL, 1.93 mol). After the addition, the mixture was stirred for another 2 h at -10 0C. The reaction was quenched with saturated aqueous bicarbonate to pH 7.5. The volatiles were removed under reduced pressure and the residues were extracted with ethyl acetate (2 x 3.0 L). The combined organic layers were washed with brine (2 x 2.0 L), dried over sodium sulfate and concentrated to give the crude product as a white solid (250 g). Purity (HPLC): 82.8% (UV).[0132] A 5 L 4-necked flask was charged with the above crude product (203 g, 82% purity) and followed by Z-proline (114 g, 0.995 mol), ethanol(1.46 L) and water (162 mL). The mixture was heated to reflux for 30 min with rapid mechanical stirring. n-Hexane (200 mL) was added dropwise to the above solution. After the addition was complete, the reaction was cooled slowly to room temperature and then further to -5C. After stirring for 3 h at -5 0C, the mixture was filtered and the filter cake was washed with cold ethano I/water (90: 10 (v/v), 2 x 100 mL) and n-hexane (2 x 500 mL), and dried under vacuum at 65C to give the desired product as a white solid (186 g). A portion of this crude product (140 g) was dissolved in ethano I/water (90:10 (v/v), 700 mL) at 75 0C with mechanical stirring. After the solution became clear, it was cooled slowly to room temperature and stirred for another 5 h. The mixture was filtered and the filter cake was washed with cold ethanol (2 x 50 mL), n-hexane (2 x 100 mL) , dried under vacuum at 65 0C to get the desired product as a white solid (130 g, yield 66%). Purity (HPLC) 99.5% (UV). 1H NMR (CD3OD, 400 MHz): delta 7.34-7.25 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.78 (d, J=8.8 Hz, 2H), 4.10 (d, J=9.2 Hz, IH), 4.06-3.95 (m, 6H), 3.88-3.85 (m, IH), 3.72-3.68 (m, IH), 3.47-3.37 (m, 5H), 3.32-3.20 (m, 3H), 2.33-2.26 (m, 2H), 2.16-2.08 (m, 2H), 2.01-1.95 (m, 4H), 1.35 (t, J=7.2Hz, 3H); MS ESI (m/z): 409 [M+ 1]+ |
|
With triethylsilane; boron trifluoride diethyl etherate; In dichloromethane; acetonitrile; at -30 - -10℃; for 1.5h;Inert atmosphere; |
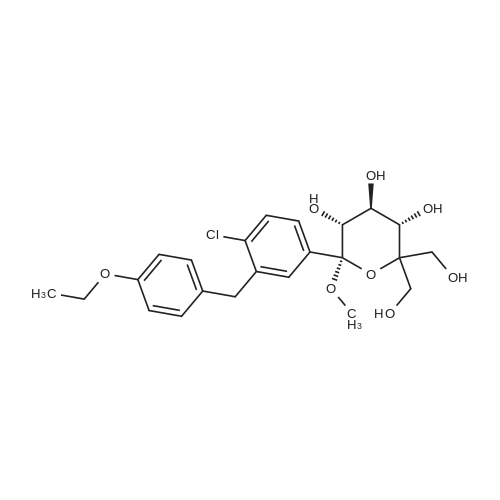
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol A solution of <strong>[714269-57-5](2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol</strong> (7.87 g, crude, ?17.9 mmol) in dichloromethane (59 mL) and acetonitrile (59 mL) was cooled to -30 C. under argon. Triethylsilane (11.5 mL, 71.6 mmole) was added to the reaction solution followed by addition of boron trifluoride etherate (6.8 mL, 53.7 mmole) so that the temperature didn't exceed -10 C. After the addition was complete the reaction solution was stirred for additional 1.5 h and then quenched with 5% sodium bicarbonate until the pH reached 7.5. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (2*80 mL). The combined organic phases were washed with brine (2*80 mL) and dried over anhydrous sodium sulfate. The sample was concentrated under reduced pressure to provide 6.8 g of the title compound as a pale solid which was used for the next step without purification. Yield: 93%. Purity (LCMS-0013) 2.9 min, 82% (UV); MS ESI (m/z) 409[M+1]+, calc. 408. |
|
With triethylsilane; boron trifluoride diethyl etherate; In dichloromethane; acetonitrile; at -20 - 20℃; for 0.333333h;Inert atmosphere; |
[0280] To a 100 mL 3-neck flask equipped with magnetic stirrer and under argon atmosphere was added dichloromethane (7.0 mL), acetonitrile (7.0 mL) and triethylsilane (5.09 mL, 31.9 mmol) successively at room temperature. The above mixture was cooled to -20 to -25 C and BF3-Et20 (3.03 mL, 23.9 mmol) was added in one portion. Another 100 mL flask was charged with crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl) phenyl)- 6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (3.5 g, 7.97 mmol), dichloromethane (7.0 mL) and acetonitrile (7.0 mL), and the resulting mixture was shaking for 20 min at ambient temperature until a clear solution was obtained. Under an atmosphere of nitrogen, the (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl) phenyl)-6- (hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol solution in dichloromethane and acetonitrile was transferred to an addition funnel and was slowly added to the solution of BF3-Et20 and triethylsilane over a period of 1 h while keeping the internal temperature between -15 to -20 C. After the addition was completed, the mixture was stirred at a temperature between -15 to -20 C. [0281] The reaction was quenched by addition of an aqueous solution of sodium bicarbonate (7.4% w/w, 25 g) via an addition funnel while keeping the internal temperature below -5 C. Additional solid sodium bicarbonate (1.7 g) was added to adjust the pH to -7.5. The volatile solvents were removed under reduced pressure at a temperature below 40 C. After cooling below room temperature, the residues were partitioned between ethyl acetate (30 mL) and water (15 mL). The organic layer was separated and the aqueous layer was extracted twice with ethyl acetate (2 x 15 mL). The combined organic layers were washed with 10% brine (2 x 20 mL). The combined extracts were concentrated under reduced pressure at a temperature below 40 C until the condensation rate slow down and almost distillation stop (not foaming). The residue was dried under oil pump (P=0.1 mm Hg) to give 3.25 g of off-white solid (99.7% yield, 89.3% pure by HPLC Method EGT-M-0001). |
|
|
Dichloromethane (1250 ml) followed by acetonitrile (1250 ml) were added to (2S ,3R,4 S,5 S ,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)-2-methoxy tetrahydro-2H-pyran-3,4,5-triol compound of formula-6 (250 gms) at 25-30 C. Cooled the reaction mixture to -20 to -25 C. under nitrogen atmosphere and stirred for 15 mins at the same temperature. Triethylsilane (132.35 gms) was slowly added to the reaction mixture at -20 to -25 C. and stirred for 15 mins. BF3-etherate (193.5 gms) was added to the reaction mixture at -25 to -20 C. and stirred for 15 mins at the same temperature. The temperature of the reaction mixture was slowly raised to -5 to 0 C. and stirred for 1 hr at the same temperature. The pH of the reaction mixture was neutralized by using 10% aqueous sodium bicarbonate solution. Ethyl acetate was added to the reaction mixture and stirred for 15 mins. Separated the both organic and aqueous layers, washed the organic layer with aqueous sodium chloride solution (50 gms of sodium chloride in 1250 ml of water) and then distilled off the solvent completely from the organic layer under reduced pressure. |
| 90 g |
With triethylsilane; boron trifluoride diethyl etherate; In dichloromethane; acetonitrile; at -5℃; for 5h; |
To a solution of Compound IV (100 gm, 0.22 mol.) in dichloromethane, acetonitrile (1 : 1, 1000 ml) mixture at -5eC was added B F3.OEt2 (0.35 mol.) and Tri ethyl si lane (0.47 mol) sequenti al ly at -5eC . T he reacti on mi xture was sti rred at the same temperature for 5 h. A f ter 5 h, reaction mixture was quenched with 10% NaHC03 solution. Reaction mass was concentrated to remove vol ati I es and added 1000ml of ethyl acetate. Layers were separated (0119) 3a and organic layer was washed with water (500 ml) followed by brine solution (500 ml). (0120) Organic layer was dried over anhydrous sodium sulphate and concentrated under vacuum to get the crude Compound V (90 g, 0.22 mol) as foamy solid. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping