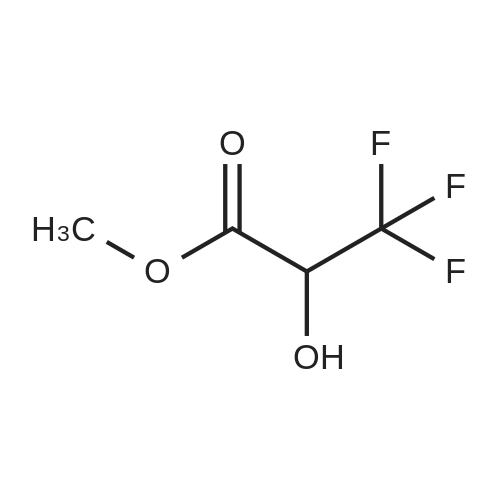
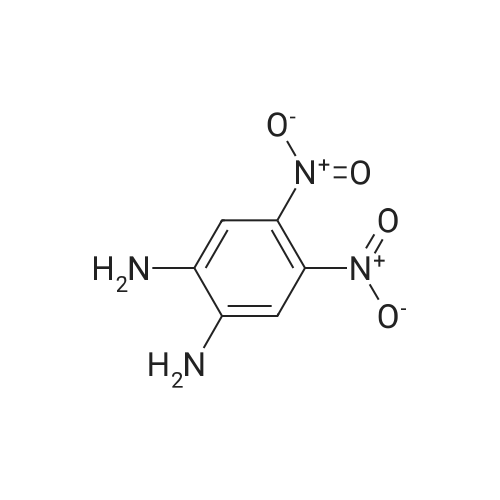
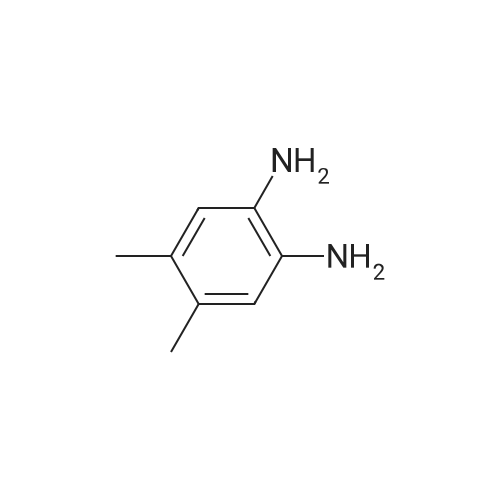
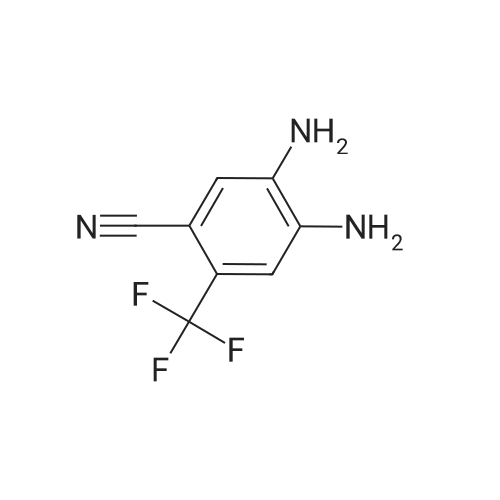
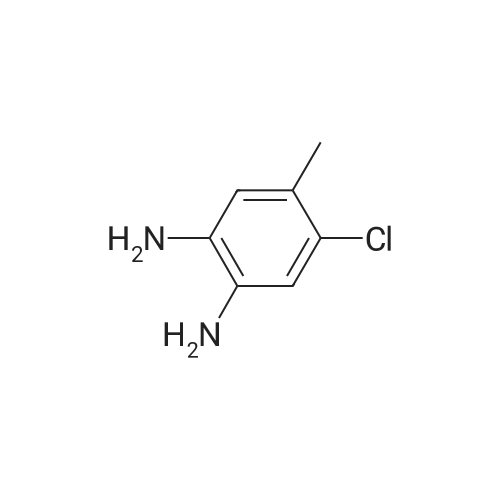
| 12.9 mg |
With N-ethyl-N,N-diisopropylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate In dichloromethane at 20℃; for 4h; |
59 Example 59: A-(5-(l-(4-ethylphenyl)-l//-pyrazol-4-yl)-l//-indol-3-yl)-3,3,3- trifluoro-2-hydroxypropanamide (Compound 166)
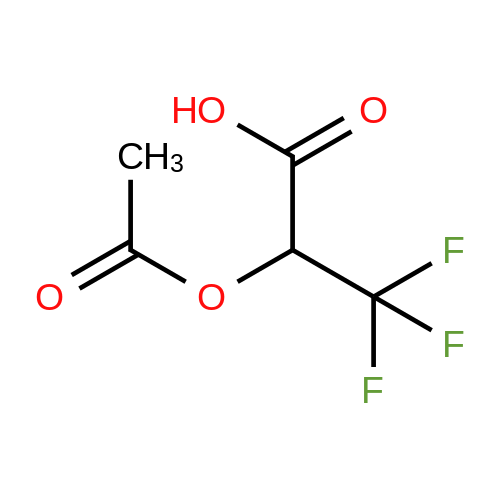
3,3,3-Trifluoro-2-hydroxypropanoic acid (114.3 mg, 0.8 mmol, 1.2 equiv.) was dissolved in DCM (10 mL), then DIEA (0.4 mL, 2.6 mmol, 4.0 equiv.), HATU (377.2 mg, 1.0 mmol, 1.5 equiv.) and 5-(l-(4-ethylphenyl)-li7-pyrazol-4-yl)-li7-indol-3-amine hydrogen chloride (224.1 mg, 0.7 mmol, 1.0 equiv.) were added. The reaction mixture was stirred for 4 hours at ambient temperature, then quenched by the addition of water. The resulting solution was extracted with DCM, dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by Flash-Prep-HPLC with the following conditions: Column: XBridge Prep OBD C18 Column, 30x 150mm, 5pm; Mobile Phase A: Water (10 mM NH4HCC>3+0.1% NH4OH), Mobile Phase B: ACN; Flow rate: 60 mL/min; Gradient: 45%B to 75%B in 8 min; 254 nm; RT1: 7.23 min. This resulted in A-(5 -( 1 -(4-ethylphenyl)- liT-pyrazol-4-yl)- liT-indol-3 -yl)-3 ,3 , 3 -trifluoro-2- hydroxypropanamide (12.9 mg) as a yellow solid. LCMS Method D: [M+H]+= 429. NMR (400 MHz, DMSO-de) d 11.09 (s, 1H), 10.07 (s, 1H), 8.82 (s, 1H), 8.10 (s, 1H), 8.04 (s, 1H), 7.82 (d, 2H), 7.77 (d, 1H), 7.52-7.49 (m, 1H), 7.42-7.36 (m, 3H), 7.30 (d,1H), 4.88-4.85 (m, 1H), 2.67 (q, 2H), 1.24 (t, 3H). |
| 12.9 mg |
With N-ethyl-N,N-diisopropylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate In dichloromethane at 20℃; for 4h; |
59 Example 59: A-(5-(l-(4-ethylphenyl)-l//-pyrazol-4-yl)-l//-indol-3-yl)-3,3,3- trifluoro-2-hydroxypropanamide (Compound 166)
3,3,3-Trifluoro-2-hydroxypropanoic acid (114.3 mg, 0.8 mmol, 1.2 equiv.) was dissolved in DCM (10 mL), then DIEA (0.4 mL, 2.6 mmol, 4.0 equiv.), HATU (377.2 mg, 1.0 mmol, 1.5 equiv.) and 5-(l-(4-ethylphenyl)-li7-pyrazol-4-yl)-li7-indol-3-amine hydrogen chloride (224.1 mg, 0.7 mmol, 1.0 equiv.) were added. The reaction mixture was stirred for 4 hours at ambient temperature, then quenched by the addition of water. The resulting solution was extracted with DCM, dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by Flash-Prep-HPLC with the following conditions: Column: XBridge Prep OBD C18 Column, 30x 150mm, 5pm; Mobile Phase A: Water (10 mM NH4HCC>3+0.1% NH4OH), Mobile Phase B: ACN; Flow rate: 60 mL/min; Gradient: 45%B to 75%B in 8 min; 254 nm; RT1: 7.23 min. This resulted in A-(5 -( 1 -(4-ethylphenyl)- liT-pyrazol-4-yl)- liT-indol-3 -yl)-3 ,3 , 3 -trifluoro-2- hydroxypropanamide (12.9 mg) as a yellow solid. LCMS Method D: [M+H]+= 429. NMR (400 MHz, DMSO-de) d 11.09 (s, 1H), 10.07 (s, 1H), 8.82 (s, 1H), 8.10 (s, 1H), 8.04 (s, 1H), 7.82 (d, 2H), 7.77 (d, 1H), 7.52-7.49 (m, 1H), 7.42-7.36 (m, 3H), 7.30 (d,1H), 4.88-4.85 (m, 1H), 2.67 (q, 2H), 1.24 (t, 3H). |
| 12.9 mg |
With N-ethyl-N,N-diisopropylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate In dichloromethane at 20℃; for 4h; |
59 Example 59: A-(5-(l-(4-ethylphenyl)-l//-pyrazol-4-yl)-l//-indol-3-yl)-3,3,3- trifluoro-2-hydroxypropanamide (Compound 166)
3,3,3-Trifluoro-2-hydroxypropanoic acid (114.3 mg, 0.8 mmol, 1.2 equiv.) was dissolved in DCM (10 mL), then DIEA (0.4 mL, 2.6 mmol, 4.0 equiv.), HATU (377.2 mg, 1.0 mmol, 1.5 equiv.) and 5-(l-(4-ethylphenyl)-li7-pyrazol-4-yl)-li7-indol-3-amine hydrogen chloride (224.1 mg, 0.7 mmol, 1.0 equiv.) were added. The reaction mixture was stirred for 4 hours at ambient temperature, then quenched by the addition of water. The resulting solution was extracted with DCM, dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by Flash-Prep-HPLC with the following conditions: Column: XBridge Prep OBD C18 Column, 30x 150mm, 5pm; Mobile Phase A: Water (10 mM NH4HCC>3+0.1% NH4OH), Mobile Phase B: ACN; Flow rate: 60 mL/min; Gradient: 45%B to 75%B in 8 min; 254 nm; RT1: 7.23 min. This resulted in A-(5 -( 1 -(4-ethylphenyl)- liT-pyrazol-4-yl)- liT-indol-3 -yl)-3 ,3 , 3 -trifluoro-2- hydroxypropanamide (12.9 mg) as a yellow solid. LCMS Method D: [M+H]+= 429. NMR (400 MHz, DMSO-de) d 11.09 (s, 1H), 10.07 (s, 1H), 8.82 (s, 1H), 8.10 (s, 1H), 8.04 (s, 1H), 7.82 (d, 2H), 7.77 (d, 1H), 7.52-7.49 (m, 1H), 7.42-7.36 (m, 3H), 7.30 (d,1H), 4.88-4.85 (m, 1H), 2.67 (q, 2H), 1.24 (t, 3H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping