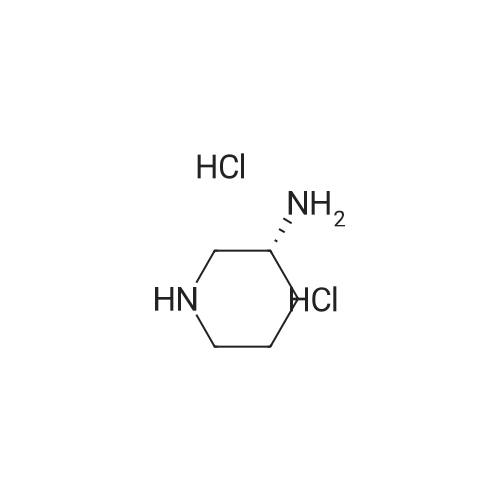
| 92.8% |
With methanol; water Inert atmosphere; Reflux; |
1.4 (4) Synthesis of linagliptin
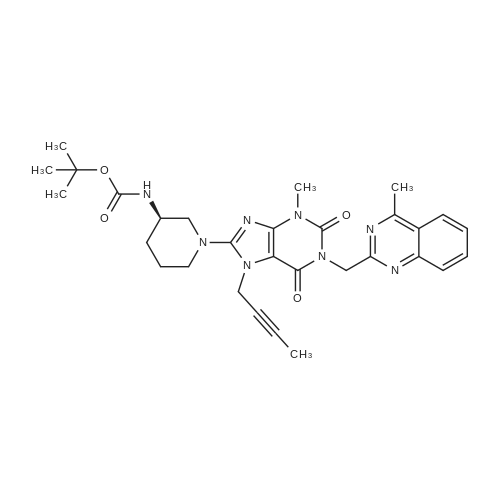
After deprotection of intermediate (g)The final product of glibenclamide.Steps:Into a 5 L reaction flask was charged 792.7 g of intermediate (g)2.4 L of methanol,1.5 L of water,After substitution with nitrogen three times,System confined,Turn on agitation,Heated to reflux.12 ~ 18h after the end of the reaction.Cooled to room temperature,Precipitation of solid.filter,The filter cake was washed with a small amount of methanol,The product was dried to give 586.5 g.The yield was 92.8%Purity 99.6%. |
| 92.8% |
With methanol; water Reflux; Inert atmosphere; |
1.4 (4) Synthesis of linagliptin
After deprotection of the intermediate (g), the final product, linagliptin, is obtained. Steps: Into a 5 L reaction vessel, 792.7 g of intermediate (g), 2.4 L of methanol and 1.5 L of water were added and replaced with nitrogen three times, closed system, turn on agitation, heated to reflux. 12 ~ 18h after the end of the reaction. Cooled to room temperature, precipitation of solid. Filtered, the filter cake was washed with a small amount of methanol, the product was dried to give 586.5 g. The yield was 92.8% and the purity was 99.6%. |
| 92.8% |
With methanol; water Inert atmosphere; Reflux; |
1.4 (4) Synthesis of linagliptin
After deprotection of intermediate (g)The final product of glibenclamide.Steps:Into a 5 L reaction flask was charged 792.7 g of intermediate (g)2.4 L of methanol,1.5 L of water,After substitution with nitrogen three times,System confined,Turn on agitation,Heated to reflux.12 ~ 18h after the end of the reaction.Cooled to room temperature,Precipitation of solid.filter,The filter cake was washed with a small amount of methanol,The product was dried to give 586.5 g.The yield was 92.8%Purity 99.6%. |
| 91% |
With trifluoroacetic acid In dichloromethane at 20℃; for 1h; |
|
| 91% |
With trifluoroacetic acid In dichloromethane at 5 - 20℃; for 5h; |
7 7. Preparation of linagliptin
Into a 1L four-neck reaction flask, compound formula VII (30.0g, 52.4mmol) and CH2Cl2 (200mL) were added. After the system was stirred, the ice-salt bath was cooled to 5 ° C. Then, a solution of TFA (75 mL) in CH2CI2 (100 mL) was slowly added dropwise to the system. After the dropwise addition, the system was naturally warmed to room temperature and reacted for 5h. Then add CH2CI2 (100mL) and H2O (300mL) to the system. After the system was stirred for 30min, the ice salt bath was cooled to 0 ° C. K2CO3 was slowly added to the system to adjust the pH value of the aqueous phase of the system to 9-10, and the organic phase was separated. The organic phase was washed twice with H20 (2X 150 mL). The organic phase solvent was removed under reduced pressure, and then heptane (500 mL) was added to the residue and stirred vigorously for 10 h. A large amount of solid was generated in the system. Filter by Büchner funnel, filter cake is blown to dry at 30C, light yellow Solid (linagliptin) (22.5g, 91%). |
| 90% |
With trifluoroacetic acid In dichloromethane at 0 - 30℃; for 12h; |
8 Example 8:
Compound D was added to 400 ml of DCM and dissolved by stirring.At 0-10 ° C, 180 ml of TFA was added dropwise, and after the addition was completed, the temperature was raised to 20-30 ° C for 12 h, and the reaction was completed by TLC.Post-treatment: control the temperature 0-10 ° C, add concentrated ammonia water, adjust PH = 8.The layers were separated and the aqueous layer was extracted with 200 ml*2DCM.The oil was evaporated to dryness under reduced pressure, and then 50 ml of anhydrous ethanol was evaporated.Add 200 ml of absolute ethanol again, stir at 0-10 °C for 2 h.After suction filtration, the filter cake was rinsed with 50 ml of absolute ethanol to obtain 69 g of linagliptin, the yield was 90%, and the HPLC purity was 99.5%. |
| 88% |
Stage #1: 1-[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-[(R)-3-(tert-butoxycarbonylamino)-piperidin-1-yl]-2,6-dioxo-2,3,6,7-tetrahydro-1H-purine With trifluoroacetic acid In dichloromethane at 20℃; Inert atmosphere;
Stage #2: With water In 1,2-dichloro-ethane |
24.I Process I
Under nitrogen protection, 1-[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-(3-(R)-tert-butoxycarbonylaminopiperidin-1-yl)xanthine (5.0 g, 0.0087 mol) was added to 50 mL of dichloromethane, and stirred for dissolution. Trifluoroacetic acid (20 mL) was slowly added, and stirred at room temperature for 1-2 hours. 5 mL of water was added to the reaction mixture, and the organic phase was separated. The aqueous phase was washed with dichloromethane. The organic phase was combined, and washed triply with saturated sodium chloride solution. The dichloromethane solution was concentrated to give a crude 1-[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-(3-(R)-aminopiperidin-1-yl)xanthine. (0226) The above crude 1-[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-(3-(R)-amino piperidin-1-yl)xanthine was dissolved in 20 times volume of methanol, added with activated carbon (10 to 25% by weight), and heated to reflux for 1 hour. The activated carbon was removed by hot filtration, and the filtrate was concentrated to obtain a residue. A mixed solvent of methylene chloride:methyl t-butyl ether=1:10 (v/v) was added to the concentrated residue, and the obtained mixture was stirred for 1 hour and filtered to obtain a filter cake, which was dried at 45° C. in a drying oven to give 1-[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-(3-(R)aminopiperidin-1-yl)xanthine (Compound I) with an optical purity of 100%. (0227) Yield: 3.61 g (88% of theoretical value) (0228) MS: [M+H]+=473.3 |
| 87.2% |
With trifluoroacetic acid In dichloromethane at 10 - 20℃; for 5h; |
1
(1- [(4- methyl - quinazolin-2-yl) methyl] -3-methyl-7- (2-butyn-1-yl) -8 - [(R) -3- (t-butoxycarbonyl amino) -Piperidin-1-yl] -2,3,6,7-tetrahydro-2,6-dione --1H- purine)17.7g, 91.8% yield, 99.6% purity.Intermediate E (15g, 26.2mmol) was dissolved in 150ml of methylene chloride, cooled to 10 , dropwise 37.5mlTrifluoroacetic acid, dropwise was completed, the reaction for 5 hours at room temperature, was added dropwise 20% mass percentage of aqueous potassium carbonate solution to pH = 10 ~ 11, the organic layer was washed with water to pH = 6 ~ 7, dried over anhydrous sodium sulfate, filtered, and the organic layer evaporated to dryness,With ethanol and methyl tert-butyl ether to obtain crystals linagliptin 10.8g, 87.2% yield, 99.9% purity. |
| 87.3% |
With formic acid; trifluoroacetic acid at 10 - 20℃; |
1 Embodiment 1: [(3R) -3 - amino -1 - piperidinyl] -7 - (2 - ethyl-acetylene base) - 3, 7 - dihydro -3 - methyl -1 - [(4 - methyl -2 - pyridinyloxy) methyl] - 1H - purine - 2, 6 - dione (advantage Geleg sandbank) preparation of
In the four neck bottles, taking intermediate II (100g, 1eq), adding formic acid (80.4g, 10eq), 10 - 20 °C adds by drops three fluorine acetic acid under (99.7g, 5eq), dropped 10 - 20 °C reaction 2 - 3 hours, TLC display of reaction, the reaction liquid concentrated steam 200 - 300 ml carboxylic acid, water (2.0L) in, adding dichloromethane 1L, adding NaOH solution (a concentration of 10 mol/L, referred to as 10N) adjusted to pH 9 - 10, separating the organic phase, dichloromethane again 2 time, combined with the organic phase, washed 1 time, saturated salt wash 1 time, dried with anhydrous sodium sulfate, concentrated, toluene (600 ml) re-crystallization, crystallization overnight at room temperature, filtered, 90 °C drying by blowing 2h, shall 72g advantage Geleg sandbank solid, yield 87.3%. Through test, HPLC purity _AOMARKENCODEGTX0AO _ 99.5%, ee % _AOMARKENCODEGTX0AO _ 99.5%, trifluoro acetylation impurity (type 2) _AOMARKENCODELTX0AO _ 0.05% |
| 87.21% |
With trifluoroacetic acid In dichloromethane at 10℃; for 3h; |
1.2; 2-7; 1-4 2) Preparation of linagliptin
At room temperature, compound D (50.00 g, 87.3 mmol) was added to the reaction flask,Add 500 ml of dichloromethane and stir to dissolve, lower the temperature to below 10 ° C, and slowly add 125 ml of trifluoroacetic acid dropwise. After the dropwise addition, the reaction is performed for 3 hours.Post-treatment: Add 600ml of purified water to another reaction bottle, add sodium hydroxide (67.00g, 1675.0mmol), stir and dissolve to obtain a sodium hydroxide solution, lower the temperature to below 5 , and cool the reaction liquid in the reaction bottle When the temperature is below 10 , slowly add dropwise to the sodium hydroxide solution to control the temperature below 15 . After the dropwise addition, the solution pH = 9 10, stir for 15min, let stand for separation, the organic layer is extracted with water 250g * 2, the pH of the water layer = 7-8, the aqueous layer was extracted with 150 ml of dichloromethane * 2.The organic layers were combined, dried over anhydrous sodium sulfate, 2.5 g of activated carbon was added, stirred for 3 hours, filtered and concentrated to dryness to obtain crude E.To the crude product of E was added 200ml of absolute ethanol, and the temperature was raised to 80-85 ° C and stirred and dissolved.Slowly lower the temperature to 30 , keep warm and stir for 3 hours, slowly add 200ml of methyl tert-butyl ether. After the addition is completed, keep stirring at 30 for 2 hours, slowly cool to 15-20 and filter with methyl tert-butyl ether 140ml rinse.Drain until there is no filter drop.After drying, a white solid E (35.98 g, 76.1 mmol) was obtained with a yield of 87.21% and a purity of 99.992%. |
| 82% |
With trifluoroacetic acid In dichloromethane at 5 - 25℃; for 4h; Large scale; |
1.3 Li Gelitinepreparation:
The reaction vessel was charged with 42 kg of dichloromethane,Trifluoroacetic acid 24 kg,Stir and cool to 5 ° C,Another 50L drop cans by adding methylene chloride 42kg,Intermediate II5.3kg stirred and dissolved,The solution was slowly added dropwise to the above reaction vessel,Keep the system temperature does not exceed 10 ,About 1hr drops finished,Heated to 25 temperature stirring 3hr,Detection of raw material intermediates II residues no more than 0.5% stop,The reaction solution was added to ice water to quench,Static sub-water phase,The organic phase was extracted three times with 25 kg of purified water,Consolidate the aqueous phase,Plus tetrahydrofuran 30kg,Temperature control at 50 plus 20% aqueous solution of sodium hydroxide 1.6Kg,Stir for 1 hr,Plus toluene 50kg stirring stratification,Organic addition of purified water 10kg washed 1 times,The tetrahydrofuran was distilled off under reduced pressure,10 kg of methyl tert-butyl ether was added,Cooling to 5 ° C stirring crystallization 2hr,filter,Methyl tert-butyl ether 2kg leaching to get crude,Crude plus anhydrous ethanol 20Kg,Stir to warm up to dissolve,Add activated carbon 0.5Kg, stirring and decolorizing for 30 minutes, precision filtration to the clean area crystallization kettle, slowly cooled to 5 , insulation crystallization 2hr filtration, filter cake with methyl tert-butyl ether 3kg washed in light yellow solid, wet The product is dried under reduced pressureDry weight loss does not exceed 0.5%A pale yellow dry product was used in the presence of 3.58 kg of the product, the yield was 82% and the purity was 99.87%. |
| 79% |
With trifluoroacetic acid In dichloromethane at 1 - 15℃; for 20h; |
3 Preparation of Linagliptin (Formula I)
tert-Butyloxycarbonyl protected intermediate (Formula II; 35 g; prepared according to the process of Working Example 2 and dichloromethane (525 mL) were added into a reaction vessel at ambient temperature. The moisture content was adjusted to less than 0.1% by azeotropically distilling dichloromethane from the reaction mixture. The reaction mixture was cooled to 1°C, and trifluroacetic acid (139.6 g) was added slowly into the reaction mixture at 1°C to 4°C. The reaction mixture was stirred at 10°C to 15 °C for 20 hours. Progress of the reaction was monitored by HPLC. The reaction mixture was cooled to 2°C, and aqueous sodium hydroxide solution (56 g in 560 mL water) was slowly added at 2°C to 13°C. The reaction mixture was stirred at 26°C to 28°C for 4 hours followed by layer separation. The organic layer was washed with water (2 x 175 mL). Activated carbon (3.5g) was added to the organic layer, and the contents were stirred for 30 minutes at 27°C to 28°C. The contents were filtered through a Hyflo bed and washed with dichloromethane (2 x 35 mL). The organic layer was concentrated at 30°C under reduced pressure. Iso-propanol (105 mL) was added, and the contents were stirred at about 28°C for 2 hours and 30 minutes. The contents were filtered, washed with iso-propanol (35 mL), and dried under reduced pressure at 60°C to 65°C for 14 hours to obtain linagliptin. Yield: 79% HPLC Purity: 99.83% |
| 70% |
Stage #1: 1-[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-[(R)-3-(tert-butoxycarbonylamino)-piperidin-1-yl]-2,6-dioxo-2,3,6,7-tetrahydro-1H-purine With hydrogenchloride In methanol; water for 4h; Reflux;
Stage #2: With sodium hydroxide In methanol; water for 5h; Reflux; |
1A; 1B
Methanol (8.6 mL) was added to compound 4 (3.3 g, 5.7 mmol) and dissolved under heating under reflux, and then concentrated hydrochloric acid (concentration 37% (w/v))/methanol (1/3). (2.9 mL) was added, and the mixture was stirred at the same temperature for 4 hours (water content in the reaction was 3.9%). After the reaction was completed, deionized water (10 mL) was added, and methanol was distilled off under reduced pressure. Subsequently, extraction with ethyl acetate/deionized water was performed, and the aqueous layer was collected.[0064]Next, methanol (10 mL) and aqueous sodium hydroxide solution (6.0 mol/L, 8.1 mL) were sequentially added to the recovered aqueous layer to make it basic conditions, and then the mixture was stirred under heating under reflux for 5 hours. After adding sodium hydroxide, it was confirmed by pH test paper that the pH was 11 or more. After completion of the reaction, extraction with ethyl acetate/deionized water was performed, and the organic layer was collected.; As a result of HPLC analysis of the liquid, the impurity 1 was 0.101% and the impurity 2 was below the detection limit in the area area (%) of the compound 1, and the formation of the impurities 1 and 2 was certainly suppressed sufficiently. It became clear that there is.[0066]After the solvent was distilled off under reduced pressure, ethanol (11 mL) was added to the residue, and the mixture was stirred at 80°C. After the residue was completely dissolved, the temperature of the solution was lowered to 40° C. to precipitate crystals. The precipitated crystals were collected to obtain compound 1 (1.9 g, 4.0 mmol, 70%). The HPLC purity of compound 1 in the obtained crystals was 99.6%. |
| 44% |
With trifluoroacetic acid In dichloromethane at 0 - 25℃; for 24h; Inert atmosphere; |
1 Synthesis of Linagliptin from tert-butyl (R)-(l-(7-(but-2-yn-l-yl)-3-methyl-l-((4- methylquinazolin-2-yl)methyl)-2, 6-dioxo-2,3, 6, 7-tetrahydro-lH-purin-8-yl)piperidin-3- yl)carbamate (BOC-protected Linagliptin)
Under inert atmosphere, 20 g (34.5 mmol) of (R)-l-(4-methyl-quinazolin-2- ylmethyl)-7-(but-2-ynyl)-8-(3 -tert-butoxycarbonylamino-piperidin- 1 -yl)-xanthine and (0103) 140 ml of methylene chloride are charged in a round bottom flask. 26.5 ml (345 mmol) of trifluoroacetic acid are slowly added at 0-5°C. The temperature is raised to 20-25°C and the mixture is kept under stirring, at the same temperature, for 24 hours. The mixture is cooled to 0-5°C and 200 ml of water are added. After phase separation, to the aqueous phase, maintaining the temperature at 5- 10°C, 100 ml of methylene chloride and 27 ml of an aqueous solution of 30% sodium hydroxide are then added (up to a pH of about 10). After separation of the aqueous phase, the organic phase is concentrated under vacuum. To the obtained residue, 83 ml of ethanol are added and the mixture is heated to reflux. The solution is cooled to 20-25°C, and 83 ml of methyl-t-butyl ether are added to the obtained suspension. The mixture is kept under stirring at 20-25°C for 1 h, then it is cooled at 0-5°C for 2 h. The solid is filtered, washed with 33 ml of methyl-t-butyl ether at 0-5°C and dried under vacuum at 45°C for 16 h to give 7 g of Linagliptin (I) (yield 44% HPLC purity 99.4%). |
| 69 g |
With trifluoroacetic acid In dichloromethane; water at 20℃; for 3h; |
11 Preparation of Linagliptin Form VIII
Example 11 Preparation of Linagliptin Form VIII 1-[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-[(R)-3-(tert-butoxycarbonylamino)-piperidin-1-yl]-2,6-dioxo-2,3,6,7-tetrahydro-1H-purine (80 g, 0.14 mol) was mixed with dichloromethane (1.6 l, 20V), and trifluoroacetic acid (400 ml, 5V) to obtain a brown solution. The solution was stirred at room temperature for 3h. Then, water (400m1, 5V) was added, and the pH was adjusted to 8 with NH4OH 25%. The organic phase was then separated, and concentrated to dryness to provide a yellow solid (92 g). The yellow solid was then mixed with ethanol (400 ml) at room temperature. The resulting brown solution turned into an off-white suspension after 5-10 min. The suspension was stirred for 1.5 h, and then filtered to provide a wet solid (143 g). The wet solid was dried for 15 h at 60° C. under vacuum to provide 8-[3(R)-Aminopiperidin-1-yl]-7-(2-butynyl)-3-methyl-1-(4-methylquinazolin-2-ylmethyl)xanthine Form VIII (69 g). |
|
With trifluoroacetic acid In dichloromethane at 25℃; for 2h; |
7 EXAMPLE-7: Preparation of the amorphous form of linagliptin
EXAMPLE-7: Preparation of the amorphous form of linagliptin 19 g compound (2) and 190 mL methylene dichloride were stirred at 25°C in round bottom flask. 70.3 g triflouro acetic acid and 95 mL methylene dichloride were added to the reaction mixture and stirred for 2 hours. 76 g aqueous potassium carbonate solution was cooled at 10°C and added to the reaction mixture. The reaction mixture was stirred for 30 min at 25°C and settled to separate the organic layer. The organic layer was treated with 3.68 g oxalic acid aqueous solution and stirred. 190 mL methylene dichloride was added to the separated aqueous layer and cooled to 20°C. 10% sodium hydroxide solution was added to the reaction mixture thereby to adjust the pH of 8-9. The separated organic layer was dried over sodium sulfate and filtered. The filtrate was distilled to remove methylene dichloride atmospherically at 40-45°C. 152 mL isopropanol was added to the residue and partially removed by distillation under vacuum at 50-55°C followed by cooling to 0-5°C. The reaction mixture was filtered and washed with methyl tert-butyl ether. The wet-cake was dissolved in 190 mL methylene dichloride and stirred for 30 min. The methylene dichloride was distilled under atmospheric pressure and degassed. The reaction mixture was cooled at ambient temperature and 76 mL methyl tert-butyl ether was added. The precipitated product was stirred for 30 min and filtered. The wet-cake was washed with methyl tert-butyl ether and dried under vacuum at 60°C for 6 hours to obtain amorphous linagliptin characterized by x-ray powder diffraction as depicted in FIGURE-2. |
|
With trifluoroacetic acid In dichloromethane at 25 - 40℃; |
6 Example 6: Preparation of 1-[(4-methyl-quinazolin-2-yl)methyl]-3-methyI-7-(2- butyn-1-yI)-8-[3-(R)-amino-piperidin-1-yl]xanthine (Linagliptin)
500 ml of Dichloromethane and 100 g of l-[(4-methyl-quinazolin-2yl) methyl]-3- methyl-7-(2-butyn-l-yl)-8-[(R)-3-(tert-butyloxycarbonylamino)-piperidin-l-yl]- xanthine (0.1746 moles prepared in example-4) were added into 1 lit round bottom flask equipped with overhead stirrer, thermo pocket and dropping funnel at 25-30°C. To the reaction mixture was slowly added 200 ml of trifluoroacetic acid at 25-30 for about 30-60 min. The reaction mixture temperature was raised to 35-40°C and maintained for lhr. After completion of the reaction, in another flask 4000 ml DM water was charged and cooled to 10-15°C and slowly added to the above reaction. The reaction mixture temperature was raised to 25-30°C and maintained for 1 hr at same temperature; and the layers were separated. The aqueous layer was washed with 300 ml of dichloromethane and charged aqueous layer into RB flask and adjusted the pH 8.5-9.0 with 30% potassium carbonate solution. 800 ml of dichloromethane was charged and stirred for 15min and separated layers. The aqueous layer was again extracted with 300 ml of dichloromethane. The organic layers were combined and washed with brine solution. The solvent was distilled out completely U/V at 35-40°C and charged 350 ml of ethanol into the residue and the temperature was raised to 70- 75°C. The reaction mixture was maintained for 30 minutes at 75-80°C. The reaction mixture was slowly cooled to 25-35°C and stirred for 2-4 hrs. To the reaction mixture was charged 350 ml of methyl tertiary butyl ether, cooled to 0- 5°C and maintained 2hrs at same temperature. The solid was filtered and washed with 100 ml tertiary butyl ether. The wet material was dissolved in 600 ml of methanol and the methanol was evaporated using spray drier at below 50°C to obtain amorphous Linagliptin. |
| 2.8 g |
With trifluoroacetic acid In dichloromethane at 20℃; for 3h; |
8 (S)-8-(3-aminopiperidin-1-yl)-3-methyl-1-[(4-methylquinazolin-2-yl)methyl]-7-(prop-2-ynyl)-1H-purine-2,6(3H,7H)-dione (1i)
General procedure: A mixture of 4b (88 mg, 0.2 mmol), (S)-3-(N-Boc-amino)piperidine (44 mg, 0.22 mmol) and K2CO3 (55 mg, 0.4 mmol) in DMF (6 mL) was stirred at 75 °C for 6 h. After cooling to r.t., the mixture was poured into water (12 mL) and extracted with DCM (3 * 10 mL). The combined organic layer was washed with saturated brine, dried over anhydrous Na2SO4, and concentrated. The crude product was purified by flash chromatography (petroleum ether/ethyl acetate, 1:1) to give the Boc precursor of 1i as a colorless syrup (80 mg, 72%), which was dissolved in DCM (2 mL), and TFA (390 μL) was added. The solution was stirred at room temperature for 3 h and then poured into ice-cold water (4 mL). The organic phase was separated, and the aqueous phase was basified with K2CO3 and extracted with DCM (2 * 10 mL). The organic layers were combined and washed with saturated brine, dried over anhydrous Na2SO4, and concentrated. The crude product was purified by flash chromatography (DCM/MeOH/TEA, 100:0.5:0.5) to give pure 1i as a white solid (51 mg, 85%). |
| 23 g |
With trifluoroacetic acid In dichloromethane at 15 - 30℃; for 3h; |
3 Example-3: Preparation of Linagliptin-Crude
To the stirring solution of Boc-Linagliptin (35 gm) in dichloromethane (350 ml) at 15 °C was added trifluoroacetic acid (140g) slowly. After addition of trifluoro acetic acid, reaction mixture was heated to 25-30 °C and maintained for 3 hour. Reaction mixture was cooled to 5°C and water was added (200 ml) and adjust ed the pH of the reaction mixture 9-10 using ammonia solution (1 10 g). Dichloromethane layer Separated and concentrated. The reaction mass is treated with Isopropyl Acetate (490 ml) and its partial removal by distillation and cooling to 10-15°C yields crude Linagliptin (23 g). HPLC Purity: 98.43% |
|
With trifluoroacetic acid In dichloromethane for 2h; |
7 Example-7 Preparation of the Amorphous Form of Linagliptin
19 g compound (2) and 190 mL methylene dichloride were stirred at 25° C. in round bottom flask. 70.3 g triflouro acetic acid and 95 mL methylene dichloride were added to the reaction mixture and stirred for 2 hours. 76 g aqueous potassium carbonate solution was cooled at 10° C. and added to the reaction mixture. The reaction mixture was stirred for 30 min at 25° C. and settled to separate the organic layer. The organic layer was treated with 3.68 g oxalic acid aqueous solution and stirred. 190 mL methylene dichloride was added to the separated aqueous layer and cooled to 20° C. 10% sodium hydroxide solution was added to the reaction mixture thereby to adjust the pH of 8-9. The separated organic layer was dried over sodium sulfate and filtered. The filtrate was distilled to remove methylene dichloride atmospherically at 40-45° C. 152 mL isopropanol was added to the residue and partially removed by distillation under vacuum at 50-55° C. followed by cooling to 0-5° C. The reaction mixture was filtered and washed with methyl tert-butyl ether. The wet-cake was dissolved in 190 mL methylene dichloride and stirred for 30 min. The methylene dichloride was distilled under atmospheric pressure and degassed. The reaction mixture was cooled at ambient temperature and 76 mL methyl tert-butyl ether was added. The precipitated product was stirred for 30 min and filtered. The wet-cake was washed with methyl tert-butyl ether and dried under vacuum at 60° C. for 6 hours to obtain amorphous linagliptin characterized by x-ray powder diffraction as depicted in FIG. 2. |
|
With trifluoroacetic acid In dichloromethane at 20℃; for 1.5h; |
4 Example 4 Preparation of a new crystal form of linagliptin
The 8-[(3R)-3-Boc-amino-1-piperidinyl]-7- (2-butynyl) -3, 7-dihydro-3-methyl -l - [( methyl-2-quinazolinyl) methyl] lH-purine-2,6-dione 60mL of dichloromethane was added 6g, 24. 3g of trifluoroacetic acid was added, the reaction stirred for 1. 5h at room temperature, the the reaction was added saturated aqueous sodium carbonate solution adjusted to pH 6-8, the organic phase was separated, the solvent was distilled off under reduced pressure to give an oil; 48ml of ethanol was added, heated to reflux to dissolve, was added 6mL of purified water, 60 ° C incubation with stirring to precipitate crystals, 120ml of isopropyl ether was added and stirred cooling to 20 ~ 30 ° C, filtered, and dried to give a white crystalline solid 3. 81g, yield 60.9%, purity 99.88%, water content 10.3%; was measured whose X-RPD spectrum consistent with 1, 2 which is consistent with the spectrum in FIG DSC-TGA, infrared absorption spectrum consistent with FIG. |
|
With trifluoroacetic acid In dichloromethane at 20 - 25℃; |
3 Example 3: Preparation of Linagliptin:
To a 3000 mL glass vessel equipped with a stirrer, condenser and a thermometer probe were added Formula V (100.0 g, 0.17 mol) and MDC (600 mL, 6.0 vol), stirred to dissolveat 25±5 °C. The reaction mixture was cooled to 20 ±5 °C and TFA (200 mL, 2.0 vol) was added slowly and warmed to 25±5 °C and stirred for 6-8h. After completion of the reaction MDC (500 mL) was added and cooled the reaction mass to 3±3 °C, water (500 mL) prechilled to 5±3 °C was added and adjusted pH of the reaction mass to 9 to 11 using aq. Ammonia maintaining the reaction temperature at 5±3 °C. The reaction mass was warmed to 25±5 °C and stirred for 2 h. Layers were separated and MDC layer was preserved. The aqueous layer was re-extracted with MDC (300 mL). Combined MDC layers were treated with activated charcoal and stirred for 30 mm. The reaction mass was filtered over celitebed and washed the celite bed with MDC (200 mL). Filtrate as obtained was concentrated at a temperature below 45 °C up to 3.0 vol. with respect to weight of Formula V used as input. MTBE (1200 mL) was added dropwise at 25±5 °C to the partially concentrated product and stirred for 1 h. The reaction mass was further cooled to 5±3 °C and stirred for 2 h. The product as obtained was filtered off, washed with MTBE (200 mL) and suck dried.The product was dried at 45±5 °C under vacuum for lOh to obtain Linagliptin as a pale yellow solid. The product was kept at -5±5°C for 36 h, raised the temperature to 25±5°C and hold it for 4-5 h to obtain anhydrous crystalline form A/B of Linagliptin. The anhydrous crystalline form A/B of Linagliptin which is prepared as per Example-3 ischaracterized by DSC as represented in Figure-6. |
| 4.99g |
With hydrogenchloride In methanol; dichloromethane at 45℃; for 4h; |
8-9 Example 8: Preparation of linagliptin
In 200 mL of a dichloromethane and methanol mixed solvent (the volume ratio of dichloromethane and methanol is 1: 4), 8.59 g (15 mmol) of Compound 7 was added, and 7.2 mL of concentrated hydrochloric acid was added dropwise. After the dropwise addition was completed, the mixture was stirred at 45 ° C. for 4 hours. TLC monitors the progress of the reaction. After the reaction is completed, cool to room temperature, remove the solution under reduced pressure, add 60 mL of distilled water, adjust the pH value of the system with a saturated sodium carbonate aqueous solution, and precipitate a solid. Crystallize at 0 ° C for 4 hours, filter, wash, and dry. 5.81 g (12.3 mmol) of crude liraritin was obtained with a yield of 82% and a purity of 97.7% by HPLC. Example 9: Refining of liraliptinDissolve 5.01 g of crude liraritin in 20 mL of ethanol under reflux, add methyl tert-butyl ether dropwise under stirring, stop solids in the system, stop heating, slowly cool to 0 ° C, crystallize for 2 hours, filter, After drying, 4.99 g of pure liraliptin was obtained, and the purity by HPLC was 99.9%. |
|
With trifluoroacetic acid In dichloromethane at 40 - 45℃; |
1 Example 1 Synthesis of Linagliptin Picolinate Salt
150 ml of methylene chloride (methylene chloride, MC) is added to 30 g of a compound of Formula 1 (hereinafter, LGT-1), and 65.7 g of trifluoroacetic acid (TFA) is slowly injected. The temperature inside the reactor is raised to 40 ° C to 45 ° C and stirred under reflux for 2 to 4 hours. After cooling to room temperature, 210 ml of H2O. Inject 60 ml of MC and stir at room temperature for 30 minutes. After stirring at room temperature, the aqueous layer is taken and stirred at room temperature for 30 minutes by injecting 300 ml of H 2 O into the separated organic layer. After stirring at room temperature, the aqueous layer is taken, and 300 ml of H 2 O is injected again into the separated organic layer and stirred at room temperature for 30 minutes. Thus, the repetition of the same process can raise the purity. Take water layer after stirring at room temperature and collect all separated water layers. The aqueous layer is washed twice with 60 ml of H 2 O under celite filtration. 300 ml of MC and 24 g of NaOH were added to the filtrate and stirred for 30 minutes. After stirring, the layers were separated to obtain an organic layer. Inject 150 ml of MC into the separated aqueous layer and stir for 30 minutes. After stirring, the layers were separated to obtain an organic layer. Collect all the organic layers and concentrate under reduced pressure. 300 ml of MeOH is added to a concentrated residue compound of Formula 2 (hereinafter referred to as Crude LGT in Scheme 1), and 6.45 g of picolinic acid is added thereto. The reaction solution was stirred under reflux for 30 minutes, cooled slowly to room temperature, stirred for 3 hours, and dehydrated. Washing twice with 30 ml of MeOH gave 21.84 g of novel linagliptin picolinate, a compound of formula 3 (LGT Picolinate in Scheme 1 below). (Yield 77.71%, Purity 98.82%) |
| 68.6 g |
With zinc(II) chloride In dichloromethane at 30℃; for 5h; |
1; 2 Example 1
The compound of formula II (100.0 g, 174.6 mmol) was added to dichloromethane (600 mL) with stirring, and ZnCl2 (95.2 g, 698.5 mmol) was added to the reaction solution. The temperature was raised to 30 ° C, and the reaction was stirred for 5 hours, and the reaction was monitored by TLC for completion. Cool to room temperature and filter. Add isopropyl alcohol (600 mL) to the filtrate, and a solid precipitates. Stir and filter at room temperature for 2 hours.The filter cake was dried under vacuum at 55 ± 5 ° C to obtain 68.6 g of ligagliptin, and the purity by HPLC (area normalization method) was 99.5%. |
| 3.4 kg |
With trifluoroacetic acid In dichloromethane at 25℃; for 2h; Large scale; |
1-3; 1
S4. Take another reactor, add 110kg of dichloromethane and 4.6kg of intermediate II obtained in step S3, add 25kg of trifluoroacetic acid to the above system at 25 ° C, and stir at 300rpm for 2h After the reaction is monitored by HPLC, 15% sodium carbonate is added dropwise, the pH is adjusted to 9.5, the layer is left standing, the organic layer is collected, acetic acid with a volume fraction of 5% is added dropwise, stirred for 30 minutes, and the water is collected standing Layer, add sodium carbonate with a mass fraction of 15%, adjust the pH to 9.5, let stand for layering, collect the organic layer, add 40 kg of dichloromethane to the water layer, stir and stand for layering, collect the organic layer, combine the organic layers , Wash with saturated sodium chloride solution 30kg, stand still and layer, collect the organic layer and concentrate to get the concentrated product; S5, take a new reaction kettle, add the concentrated product obtained in step S4 and 15.2kg of absolute ethanol to it, start Stir, heat and reflux until the solid is completely dissolved, add 0.1kg of activated carbon, filter while hot, then cool to 5 ° C, add 15kg of methyl tert-butyl ether dropwise, keep warm, stir at 450rpm for 1.0h, filter, filter cake With methyl tert-butyl ether 0.9kg washed once filter cake was collected, dried, to obtain. |
| 153.5 g |
With trifluoroacetic acid at 30 - 40℃; for 3h; |
1.4; 2-6 Step 4: Preparation of Linagliptin
The above Boc-linagliptin was added to 400ml of trifluoroacetic acid, and the reaction was stirred at 30-40 °C. The reaction was detected by TLC. The reaction was completed in 3 hours, and the solvent of the reaction solution was evaporated to dryness. Add 300ml of water, 300ml of ethyl acetate, add dropwise 2mol/L hydrochloric acid pH 2 to 3, and separate the liquids. Add 100ml*2 water to the organic phase, extract and separate. Combine the water phases, add 300 ml of ethyl acetate, add dropwise a 1 mol/L sodium hydroxide solution pH 8-9, and separate the liquids. Add 100ml*2 ethyl acetate to the water phase, separate the liquids, combine the organic phases, dry with anhydrous sodium sulfate, and filter.The filtrate was evaporated to dryness under reduced pressure at 40 °C to obtain 153.5 g of off-white solid linagliptin. The total yield of the three-step reaction is 79.6%, and the HPLC purity is 99.96% |
| 88 % |
Stage #1: 1-[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-[(R)-3-(tert-butoxycarbonylamino)-piperidin-1-yl]-2,6-dioxo-2,3,6,7-tetrahydro-1H-purine With hydrogenchloride In dichloromethane; water at -15℃;
Stage #2: With tetrabutylammomium bromide; sodium hydroxide In water; toluene at 60 - 65℃; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping