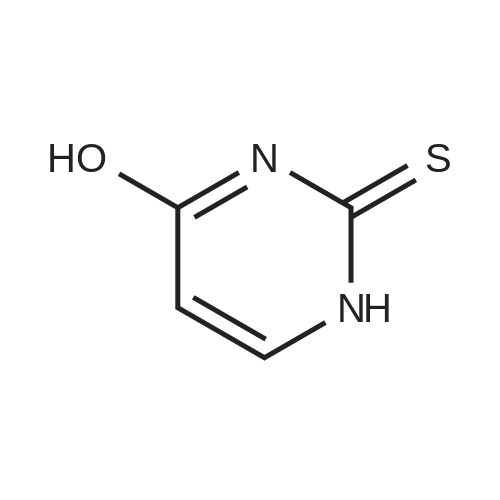
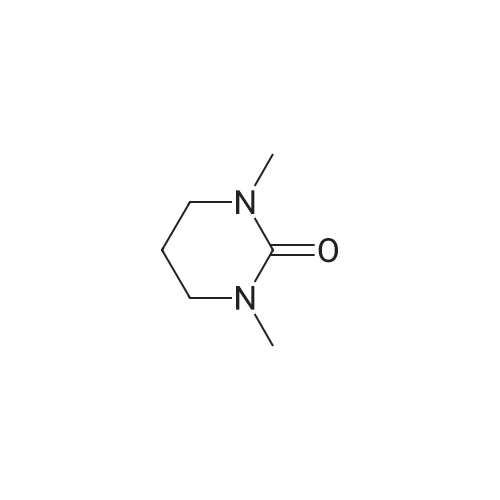
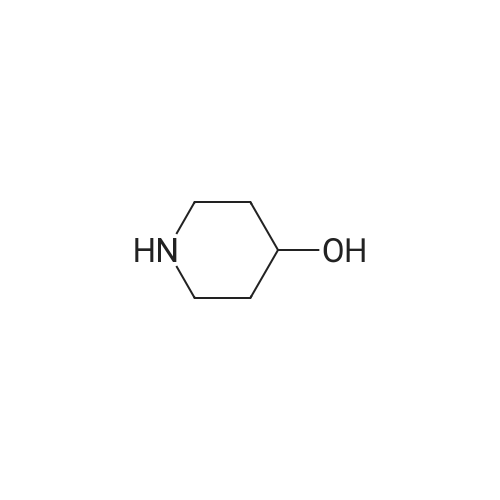
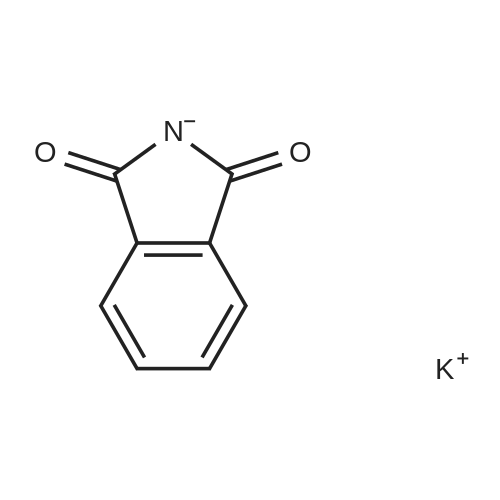
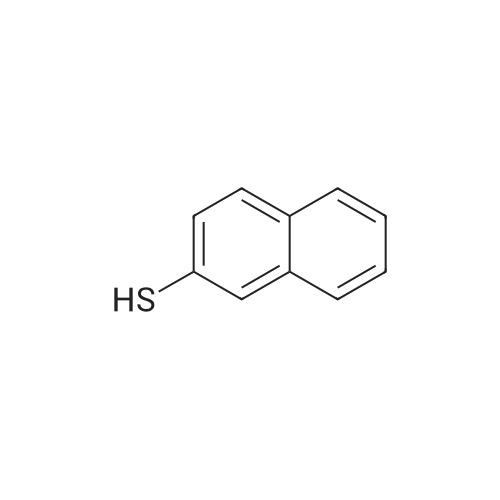
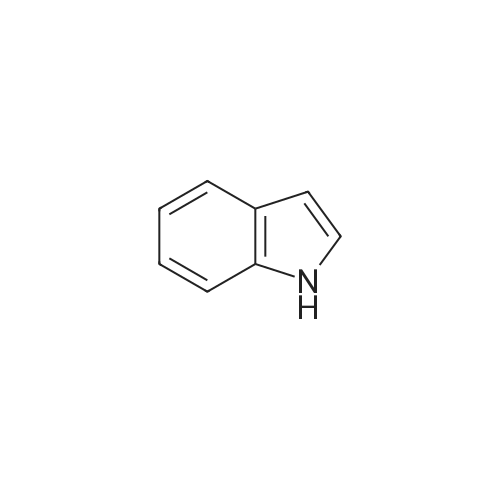
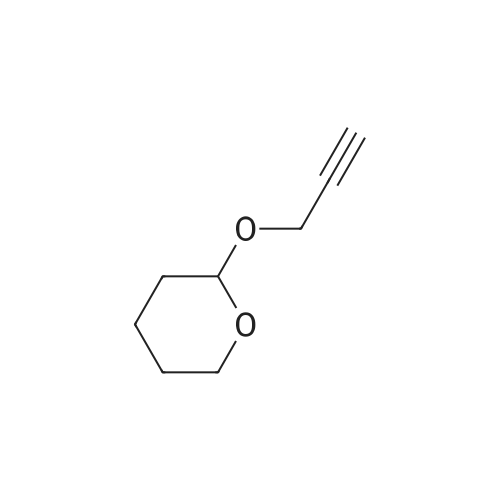
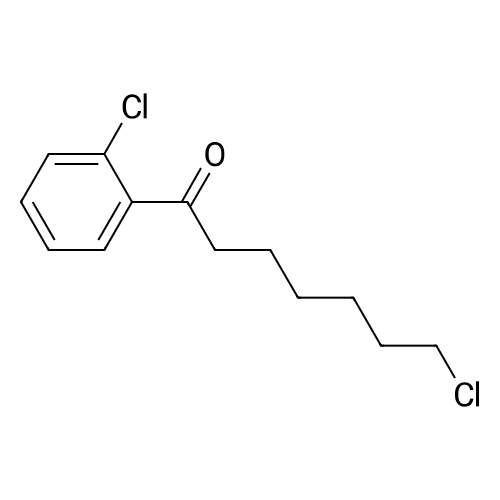
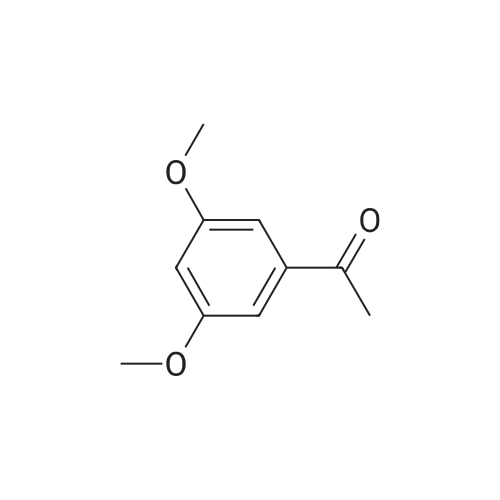
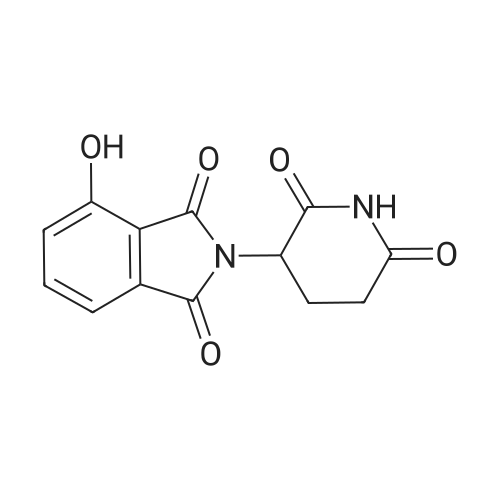
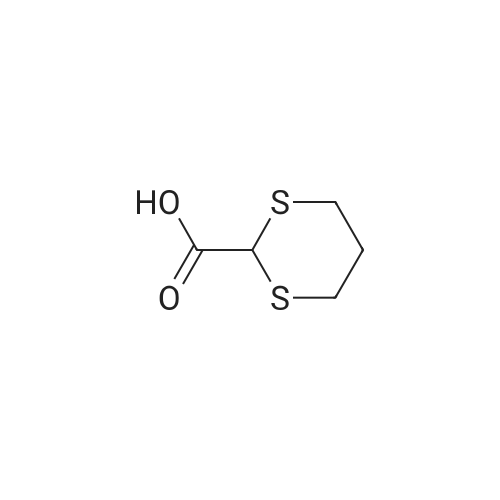
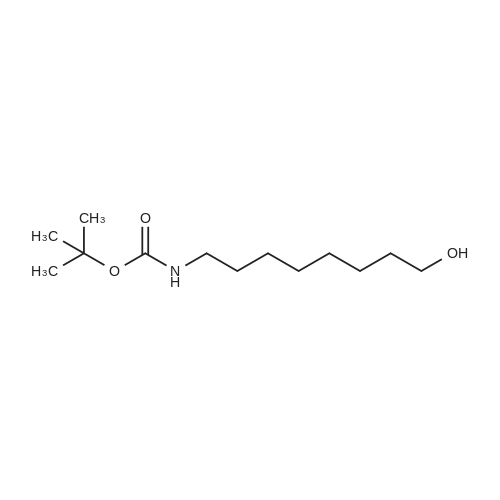
| 78% |
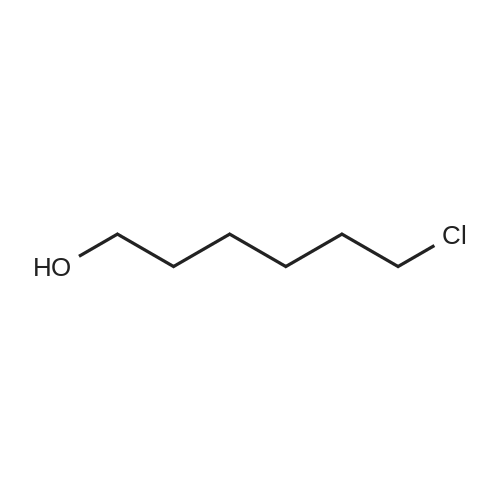
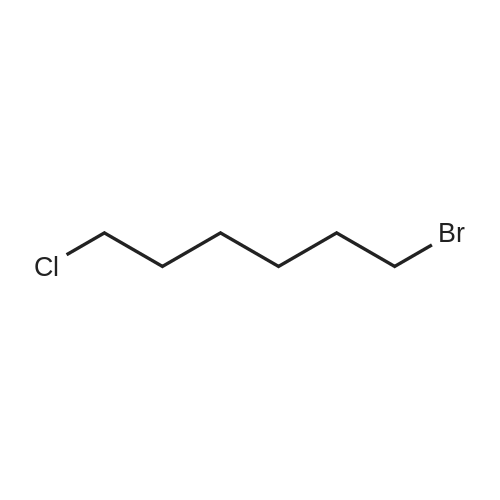
With sodium hydride In N,N-dimethyl-formamide at 20℃; for 16h; |
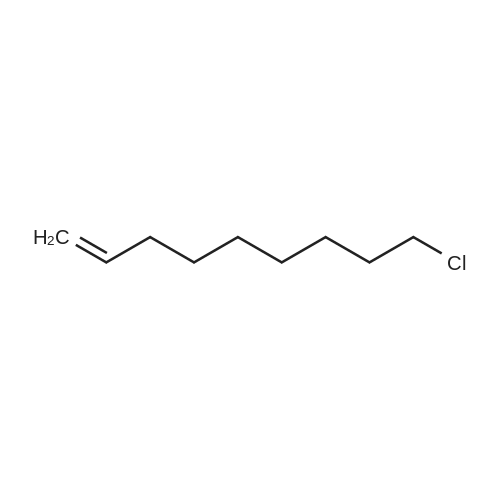
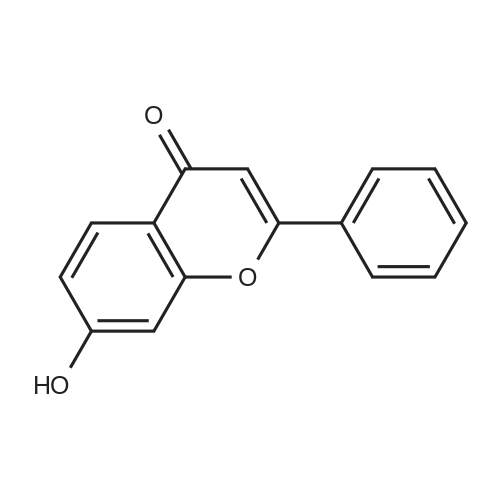
4.28 Synthesis of (2R,3R)-6,8-bis(benzyloxy)-3-((6-chlorohexyl)oxy)-2-(3,4,5-tris(benzyloxy) phenyl)chromane (16a)
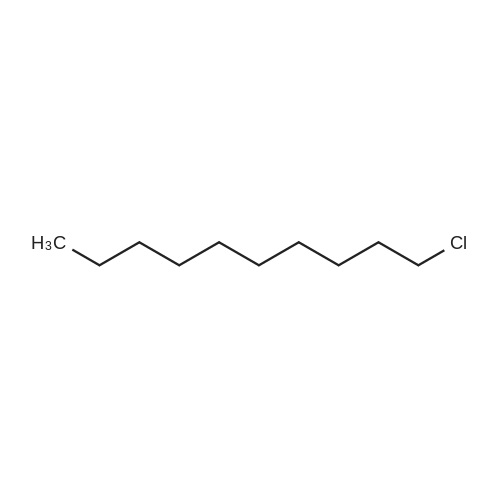
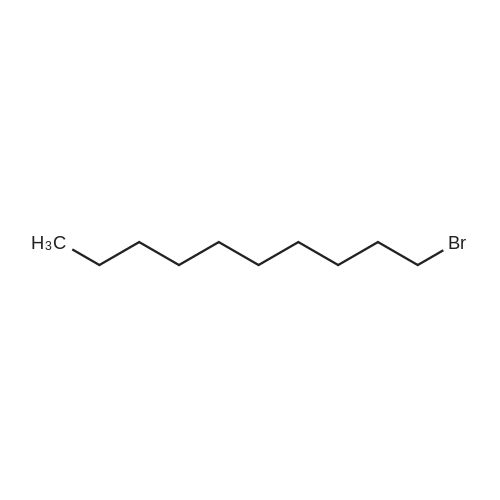
Compound 15 (2.0g, 2.6mmol) was dissolved in anhydrous DMF (30mL), and then NaH (530mg, 22.1mmol) was added in batches, and finally 1-bromo-6-chlorohexane (1.0g, 5.0mmol) was introduced dropwise. The mixture was stirred for 16h at room temperature. After all the starting materials were consumed, the addition of water (10.0mL) was used to carefully quench the mixture. Ethyl acetate (100mL) was added to extract the target compound. The organic layer was washed with water and brine, and dried with anhydrous Na2SO4, and filtered, and then condensed under the diminished pressure to afford the crude residue, which was further purified by a silica-gel column chromatogram with a mixture of petrol ether and ethyl acetate (V/V=5/1) as an eluent to afford 1.8g of compound 16a as a yellow solid. Yield, 78%, 1H NMR (400MHz, CDCl3) δ 7.42-6.95 (m, 25H), 6.74 (s, 2H), 6.22-6.11 (m, 2H), 5.07-4.77 (m, 10H), 4.17-3.92 (m, 1H), 3.69 (s, 1H), 3.37-2.99 (m, 4H), 2.99-2.56 (m, 2H), 1.63-1.51 (m, 2H), 1.27-1.17 (m, 3H), 1.09-0.94 (m, 2H), 0.85-0.75 (m, 1H); 13C NMR (100MHz, CDCl3) δ 162.58, 158.65, 158.04, 155.50, 152.53, 137.94, 137.21, 137.17, 137.13, 137.02, 134.57, 129.77, 129.02, 128.79, 128.62, 128.57, 128.54, 128.52, 128.50, 128.47, 128.44, 128.41, 128.24, 128.21, 128.18, 128.15, 128.00, 127.92, 127.87, 127.84, 127.80, 127.78, 127.70, 127.60, 127.57, 127.54, 127.50, 127.46, 127.26, 106.70, 106.18, 105.57, 101.62, 78.28, 75.26, 73.90, 71.24, 70.12, 69.96, 69.66, 51.27, 29.75, 29.21, 28.56, 24.99, 23.21. |
| 78% |
With sodium hydride In N,N-dimethyl-formamide at 20℃; for 16h; |
4.28 Synthesis of (2R,3R)-6,8-bis(benzyloxy)-3-((6-chlorohexyl)oxy)-2-(3,4,5-tris(benzyloxy) phenyl)chromane (16a)
Compound 15 (2.0g, 2.6mmol) was dissolved in anhydrous DMF (30mL), and then NaH (530mg, 22.1mmol) was added in batches, and finally 1-bromo-6-chlorohexane (1.0g, 5.0mmol) was introduced dropwise. The mixture was stirred for 16h at room temperature. After all the starting materials were consumed, the addition of water (10.0mL) was used to carefully quench the mixture. Ethyl acetate (100mL) was added to extract the target compound. The organic layer was washed with water and brine, and dried with anhydrous Na2SO4, and filtered, and then condensed under the diminished pressure to afford the crude residue, which was further purified by a silica-gel column chromatogram with a mixture of petrol ether and ethyl acetate (V/V=5/1) as an eluent to afford 1.8g of compound 16a as a yellow solid. Yield, 78%, 1H NMR (400MHz, CDCl3) δ 7.42-6.95 (m, 25H), 6.74 (s, 2H), 6.22-6.11 (m, 2H), 5.07-4.77 (m, 10H), 4.17-3.92 (m, 1H), 3.69 (s, 1H), 3.37-2.99 (m, 4H), 2.99-2.56 (m, 2H), 1.63-1.51 (m, 2H), 1.27-1.17 (m, 3H), 1.09-0.94 (m, 2H), 0.85-0.75 (m, 1H); 13C NMR (100MHz, CDCl3) δ 162.58, 158.65, 158.04, 155.50, 152.53, 137.94, 137.21, 137.17, 137.13, 137.02, 134.57, 129.77, 129.02, 128.79, 128.62, 128.57, 128.54, 128.52, 128.50, 128.47, 128.44, 128.41, 128.24, 128.21, 128.18, 128.15, 128.00, 127.92, 127.87, 127.84, 127.80, 127.78, 127.70, 127.60, 127.57, 127.54, 127.50, 127.46, 127.26, 106.70, 106.18, 105.57, 101.62, 78.28, 75.26, 73.90, 71.24, 70.12, 69.96, 69.66, 51.27, 29.75, 29.21, 28.56, 24.99, 23.21. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping