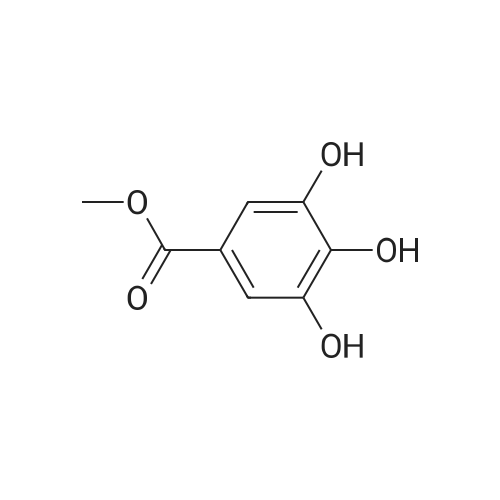
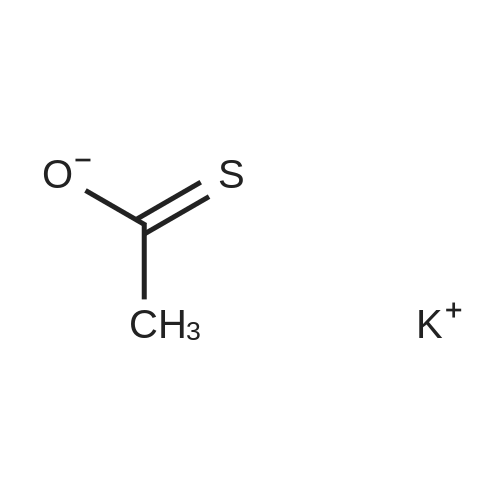
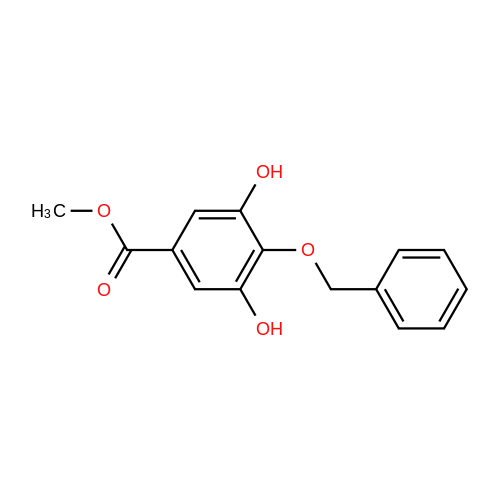
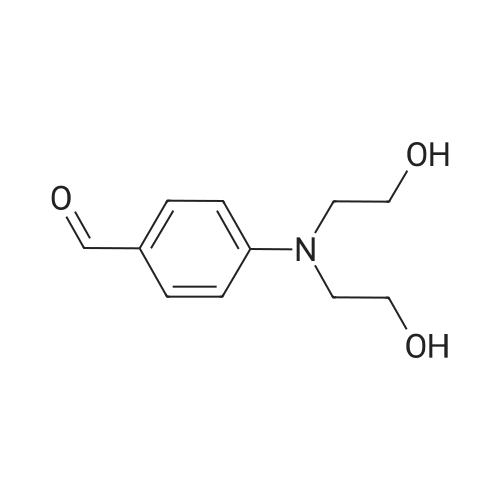
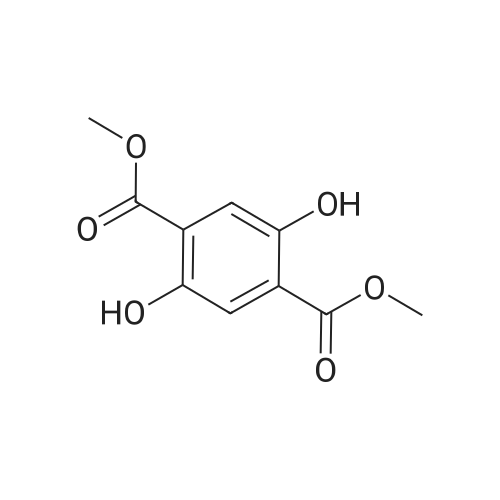
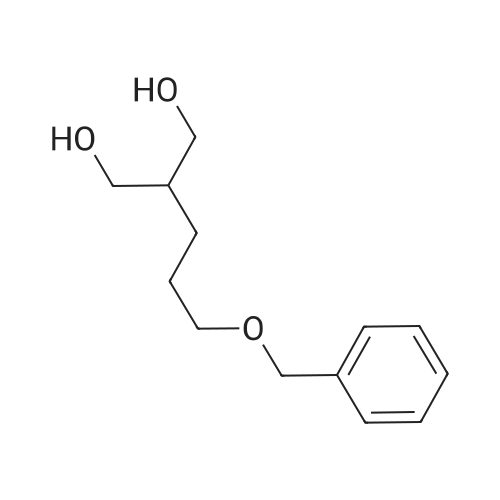
| 95% |
|
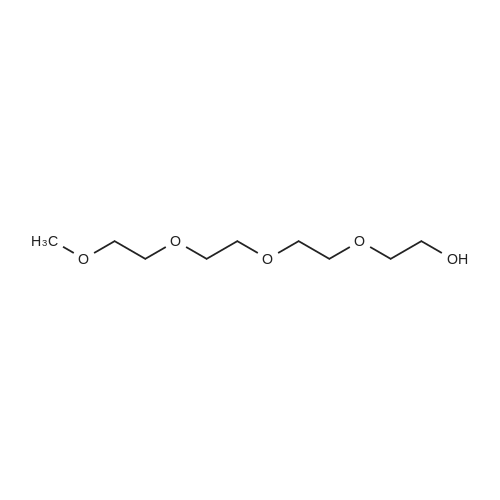
A solution of tetra (ethylene glycol) ethyl ether (1.96 g, 9.41 mmol, 1 eq.) Dissolved in THF (5 mL) was placed in a 30 mL flask and cooled to 0 C. in an ice bath.Then, a solution of sodium hydroxide (0.56 g, 14 mmol, 1.5 eq.) Dissolved in water (2 mL) was added and reacted with stirring for 15 minutes.Subsequently, p-toluenesulfonyl chloride (1.97 g, 10.35 mmol, 1.1 eq.) Was added dropwise over 10 minutes.The mixture was then reacted vigorously with stirring at 0 C. for 3 hours, then allowed to warm to room temperature and reacted with continuous stirring for a further 3 hours.The mixture was then diluted with ethyl acetate (30 mL), washed twice with water and brine, and the organic layer was dried over anhydrous sodium sulfate.Subsequently, the solvent was removed under reduced pressure to obtain compound (C-1) as a colorless oil (yield 3.231 g, 8.91 mmol, yield 95%). |
| 94% |
With pyridine; In dichloromethane; at -20℃; for 48h;Inert atmosphere; |
To a solution of tetraethyleneglycol monomethyl ether (2) (10.0 g, 48.0 mmol) and pyridine (84 mL) in CH2Cl2 (170 mL), solid p-toluenesulfonyl chloride (22.0 g, 115.4 mmol) was added portion-wise at -20 C under nitrogen. The resulting reaction mixture was stirred for 2 days at -20 C. Then, the reaction mixture was allowed to warm to room temperature and water (200 mL) was added. The aqueous layer was extracted with CH2Cl2 (150 mL × 3). The combined organic fractions were dried over MgSO4 and the solvent was removed under reduced pressure. The crude product was purified by chromatography on silica (1:1 EtOAc:hexanes; Rf = 0.3) to yield 3 as a colorless oil, 16.4 g, 94%.1H NMR (500 MHz, CDCl3): delta = 7.80 (d, Ar, 2H), 7.34 (d, Ar, 2H), 4.16 (t, 2H), 3.66 (t, 2H), 3.62-3.65 (m, 6H), 3.58 (s, 4H), 3.532-3.56 (m, 2H), 3.34 (s, 3H), 2.43 (s, 3H). 13C NMR (125 MHz, CDCl3): delta = 144.71 (s, CSO2O), 132.94 (s, CH3CCH), 129.74 (s, CHCHCSO2), 127.89 (s, CCHCH), 71.79, 70.57, 70.46, 70.44, 70.38, 70.36, 69.20, 68.52, 58.94 (CH3OCH2), 21.56 (CH3CHCH). Data was consistent with a previously reported compound [5]. |
| 94% |
|
General procedure: Pyridine (1.09 mL, 13.5 mmol, 2.0 eq.) and DMAP (1.65 g, 13.5 mmol, 2.0 eq.) were added to a solution of methoxy-oligo(ethylene glycol) (6.75 mmol, 1.0 eq.) in DCM (10 mL), and the mixture was stirred at 0 C for 15 min. A solution of tosyl chloride (1.93 g, 10.1 mmol, 1.5 eq.) in DCM (10 mL) was then added dropwise and the reaction mixture was stirred at room temperature for 6 h. The resulting mixture was diluted in DCM (150 mL) and washed with 0.5 N HCl (100 mL). The aqueous layer was washed with DCM (5 × 75 mL) and all the organic fractions were collected, dried over MgSO4 and filtered.The solvent was evaporated under reduced pressure and the residue was purified by flash chromatography. |
| 94% |
With sodium hydroxide; In tetrahydrofuran; water; at 0 - 20℃; for 21h; |
A solution of /?ara-toluenesulfonyl chloride (22.3 g, 105 mmol) in THF (35 mL) was added dropwise to a solution of tetraethyleneglycol methyl ether (20.0 g, 96 mmol) and NaOH (6.7 g, 166 mmol) in a mixture of THF/H20 (135 mL/45 mL) at 0C. After 1 hour stirring at 0C, the reaction was allowed to warm at room temperature and was stirred 20 additional hours. The solution was then poured into 200 mL of brine and the volatiles were evaporated. The resulting mixture was extracted several times with dichloromethane and the combined organic layers were washed with brine, dried over MgSC>4 and filtered. The solvent was evaporated under reduced pressure and the oil and was purified by column chromatography on silica gel eluting with dichloromethane/methanol (98/2). Compound 2 was obtained as a pale yellow oil in 94% yield. FontWeight="Bold" FontSize="10" H NMR (300 MHz, CDC13) delta 7.73 (d, J = 1.5 Hz, 2H, Ar-2,6-H), 7.28 (d, J = 1.5 Hz, 2H, Ar-3,5-H), 4.11-4.08 (m, 2Eta, ArS02OCH2), 3.64-3.47 (m, 14Eta, OCH2CH20), 3.31 (s, 3H, OC), 2.39 (s, 3H, ArC); 1 C NMR (75 MHz, CDC13) delta 144.9, 133.2, 130.0, 72.1, 70.9, 70.7, 70.6, 69.5, 68.8, 59.1, 28.1 , 21.8. |
| 94% |
With sodium hydroxide; In tetrahydrofuran; water; at 0 - 10℃; for 6h; |
A method of synthesizing the silybin phenolic ether derivatives as shown in Formula I-d is as follows:1) Add 8.00 g (0.20 mol) NaOH, 80 ml water to a 250 ml three-necked flask.Stir to dissolve it evenly. 24.96 g (0.12 mol) of tetraethylene glycol monomethyl ether was added to 50 ml of THF to be uniformly dissolved, then added to the above-mentioned three-necked flask and uniformly mixed with the NaOH solution.The mixture was stirred in an ice bath at 0 55 C. and thoroughly deoxygenated with nitrogen. 22.88 g (0.12 mol) of p-toluenesulfonyl chloride and 40 ml of THF were mixed well, and then slowly added dropwise to the above three-necked flask. The dropping process kept the temperature of the reaction liquid. After exceeding 10C, the reaction was continued for 6 hours after completion of the dropwise addition to stop the reaction.The reaction solution was extracted three times with diethyl ether, and the ether extract was washed with water until neutral and then dried over anhydrous magnesium sulfate. After filtration and rotary evaporation, the ether was removed.This gave 40.83 g (yield: 94%) of the corresponding p-toluenesulfonate of formula III-b, |
| 90% |
With dmap; triethylamine; In dichloromethane; at 20℃; for 20h; |
(JACS, 2007, 129, 13364) To a solution of 2,5,8,11-tetraoxamidecan-13-ol (7 g, 33.6 mmol) and triethylamine (4.9 ml, 35.3 mmol) in dry CH2Cl2 (100 ml), 4-toluenesulfonyl chloride (6.7 g, 35.3 mmol) and DMAP (120 mg) were added. The mixture was stirred at room temperature for 20 h. The reaction mixture was washed with 80 mL of HCl (1M) and then water. The extract was dried over anhydrous MgSO4, filtrated, and the filtrate was evaporated. The residue was used in the next step without further purification. Yield: 11.0 g (90%) NMR: 1H NMR (400 MHz, CDCl3) delta 7.75-7.64 (m, 2H), 7.31-7.26 (m, 2H), 4.16-4.06 (m, 2H), 3.62 (m 2H), 3.59-3.40 (m, 10H), 3.30 (s, 3H), 2.38 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3) delta 144.75 (s), 132.90 (s), 129.77 (s), 127.8 (s), 71.82 (s), 70.60 (s), 70.48 (s), 70.47 (s), 70.41 (s), 70.39 (s), 69.23 (s), 68.55 (s), 58.90 (s), 21.53 (s). |
| 90% |
With dmap; triethylamine; In dichloromethane; at 20℃; for 20h; |
Procedure: (JACS, 2007, 129, 13364) To a solution of 2,5,8,11-tetraoxatridecan- 13-ol (7 g, 33.6 mmol) and triethylamine (4.9 ml, 35.3 mmol) in dry CH2C12 (100 ml), 4-toluenesulfonyl chloride (6.7 g, 35.3 mmol) and DMAP (120 mg) were added. The mixture was stirred at room temperature for 20 h. The reaction mixture was washed with 80 mL of HCl (1M) and then water. The extract was dried over anhydrous MgS04, filtrated, and the filtrate was evaporated. The residue was used in the next step without further purification. Yield: 11.0 g (90%) NMR: NMR (400 MHz, CDC13) delta 7.75 - 7.64 (m, 2H), 7.31 - 7.26 (m, 2H), 4.16 - 4.06 (m, 2H), 3.62 (m 2H), 3.59 - 3.40 (m, 10H), 3.30 (s, 3H), 2.38 (s, 3H). 13C{1H} NMR (101 MHz, CDC13) delta 144.75 (s), 132.90 (s), 129.77 (s), 127.8 (s), 71.82 (s), 70.60 (s), 70.48 (s), 70.47 (s), 70.41 (s), 70.39 (s), 69.23 (s), 68.55 (s), 58.90 (s), 21.53 (s). |
| 90% |
With dmap; triethylamine; In dichloromethane; at 20℃; for 20h; |
Procedure: (JACS, 2007, 129, 13364) To a solution of 2,5,8,11-tetraoxatridecan- 13-ol (7 g, 33.6 mmol) and triethylamine (4.9 ml, 35.3 mmol) in dry CH2C12 (100 ml), 4-toluenesulfonyl chloride (6.7 g, 35.3 mmol) and DMAP (120 mg) were added. The mixture was stirred at room temperature for 20 h. The reaction mixture was washed with 80 mL of HCl (1M) and then water. The extract was dried over anhydrous MgS04, filtrated, and the filtrate was evaporated. The residue was used in the next step without further purification. Yield: 11.0 g (90%) NMR: NMR (400 MHz, CDC13) delta 7.75 - 7.64 (m, 2H), 7.31 - 7.26 (m, 2H), 4.16 - 4.06 (m, 2H), 3.62 (m 2H), 3.59 - 3.40 (m, 10H), 3.30 (s, 3H), 2.38 (s, 3H). 13C{1H} NMR (101 MHz, CDC13) delta 144.75 (s), 132.90 (s), 129.77 (s), 127.8 (s), 71.82 (s), 70.60 (s), 70.48 (s), 70.47 (s), 70.41 (s), 70.39 (s), 69.23 (s), 68.55 (s), 58.90 (s), 21.53 (s). |
| 90% |
With dmap; triethylamine; In dichloromethane; at 20℃; for 20h; |
Procedure: (JACS, 2007, 129, 13364) To a solution of 2,5,8,11-tetraoxatridecan- 13-ol (7 g, 33.6 mmol) and triethylamine (4.9 ml, 35.3 mmol) in dry CH2C12 (100 ml), 4-toluenesulfonyl chloride (6.7 g, 35.3 mmol) and DMAP (120 mg) were added. The mixture was stirred at room temperature for 20 h. The reaction mixture was washed with 80 mL of HCl (1M) and then water. The extract was dried over anhydrous MgS04, filtrated, and the filtrate was evaporated. The residue was used in the next step without further purification. Yield: 11.0 g (90%) NMR: NMR (400 MHz, CDC13) delta 7.75 - 7.64 (m, 2H), 7.31 - 7.26 (m, 2H), 4.16 - 4.06 (m, 2H), 3.62 (m 2H), 3.59 - 3.40 (m, 10H), 3.30 (s, 3H), 2.38 (s, 3H). 13C{1H} NMR (101 MHz, CDC13) delta 144.75 (s), 132.90 (s), 129.77 (s), 127.8 (s), 71.82 (s), 70.60 (s), 70.48 (s), 70.47 (s), 70.41 (s), 70.39 (s), 69.23 (s), 68.55 (s), 58.90 (s), 21.53 (s). |
| 90% |
With dmap; triethylamine; In dichloromethane; at 20℃; for 20h; |
(JACS, 2007, 129, 13364) To a solution of 2,5,8,11-tetraoxatridecan-13-ol (7 g, 33.6 mmol) and triethylamine(4.9 ml, 35.3 mmol) in dry CH2C12 (100 ml), 4-toluenesulfo-nyl chloride (6.7 g, 35.3 mmol) and DMAP (120 mg) wereadded. The mixture was stirred at room temperature for 20 h.The reaction mixture was washed with 80 mE of HC1 (1M)and then water. The extract was dried over anhydrous Mg504,filtrated, and the filtrate was evaporated. The residue was usedin the next step without further purification.10137] Yield:10138] 11.0 g (90%)10139] NMR:10140] ?H NMR (400 MHz, CDC13) oe 7.75-7.64 (m, 2H),7.3 1-7.26 (m, 2H), 4.16-4.06 (m, 2H), 3.62 (m2H), 3.59-3.40(m, 1OH), 3.30 (s, 3H), 2.38 (s, 3H).10141] ?3C{?H} NMR (101 MHz, CDC13) oe 144.75 (s),132.90 (s), 129.77 (s), 127.8 (s), 71.82(s), 70.60 (s), 70.48 (s),70.47 (s), 70.41 (s), 70.39 (s), 69.23 (s), 68.55 (s), 58.90 (s), |
| 86% |
With dmap; triethylamine; In dichloromethane; at 20℃;Inert atmosphere; |
To a solution of 2,5,8,11-tetraoxatridecan-13-ol (1.50 g, 7.21 mmol), triethylamine (2.18 g, 21.6 mmol) and N,N-dimethylpyridin-4-amine (88 mg, 0.720 mmol) in dichloromethane (30 mL) was added 4-methylbenzene-1-sulfonyl chloride (1.37 g, 7.19 mmol) under nitrogen atmosphere. After stirred at room temperature overnight, the reaction mixture was washed with water (30 mL) for three times, dried over Na2 SO4()and filtered. The filtrate was concentrated under reduced pressure to afford the title compound (1.80 g, 86% yield) as colorless oil. ?HNIVIR (300 1VIFIz, CDC13) 7.82- 7.79 (m, 2H), 7.36-7.27 (m, 2H), 4.16 (s, 2H), 3.70 - 3.56 (m, 14H), 3.38 (s, 3H), 2.45 (s, 3H). |
| 82% |
With sodium carbonate; In tetrahydrofuran; water; at 0℃; for 3h; |
To a mixture of tetraethylene glycol monomethyl ether (3.00 g, 14.406 mmol) and soda (0.865 g, 21.6 mmol) diluted in THF (33 ml) and water (4 ml) cooled to 0 C., a solution of p-toluenesulfonic acid chloride (3.021 g, 15.8 mmol) in THF (4 ml) is slowly added. After 3 hours of stirring at 0 C., the mixture is poured into iced water (10 ml) and is diluted by dichloromethane. The aqueous phase is extracted with dichloromethane and the recombined organic phases are washed with water then with a NaCl-saturated solution, dried on MgSO4, filtered and concentrated under reduced pressure. The residue is purified by flash chromatography on a silica gel (petroleum ether/ethyl acetate 1/1 to 1/4) to yield 4.234 g (82%) of a colorless oil. deltaH (300 MHz, CDCl3) identical to the literature. |
| 70% |
With pyridine; In dichloromethane; water; |
O-Tosyltetraethyleneglycol methyl ether (109) Tetraethyleneglycol monomethyl ether (2.0 g, 9.6 mmol) was combined with pyridine (0.93 mL, 11.5 mmol) in methylene chloride (20 mL) at 0 C. p-Toluenesulfonyl chloride (2.2 g, 11.5 mmol) dissolved in methylene chloride (10 mL) was added dropwise and the reaction was allowed to warm to room temperature as the ice bath thawed. After stirring nine days, the product mixture was transferred to a separatory funnel with water (70 mL). The layers were separated and the aqueous layer was extracted with methylene chloride (3*20 mL). The extracts were combined and evaporated. The residue (3.3 g) was chromatographed (silica gel, 4:1 to 3:1 gradient elution, methylene chloride/ethyl acetate) to afford 2.5 g (70%) of compound 109 as an oil. 1H NMR (300 MHz, DMSO-d6) delta 1.56 (br s, 4H), 2.42 (br s, 2H), 3.34 (br s, 4H), 6.05 (s, 3H), 7.09 (s, 0.5H), 7.26 (s, 0.5H), 7.42 (br s, 2H), 7.70 (br s, 2H), 8.87 (br d, 2H), 9.07 (s, 2H), 9.22 (br s, 1H), 10.51 (s, 1H). CI MS m/z=363 [C16H26O7S+H]+. |
| 47.3% |
With pyridine; In tetrahydrofuran; at 20℃; |
2,5,8,1 1-Tetraoxatridecan-13-ol (5.0 g, 24.01 mmol) was dissolved in THF(20.01 ml). pyridine (5.83 ml, 72.0 mmol) was added to the mixture followed by 4-methylbenzene-1-sulfonyl chloride (5.49 g, 28.8 mmol). The mixture stirred at roomtemperature overnight. Mixture was concentrated by roto-vap. Residue was dissolved in dichloromethane. Material was washed twice with saturated aqueous sodium bicarbonate. Combined aqueous layers were back-extracted with dichloromethane. Combined organics were washed twice with iN hydrochloric acidand once with brine. Organics were dried MgSO4, filtered and then concentrated todryness to get 2,5,8,11-tetraoxatridecan-13-yl 4-methylbenzenesulfonate (4.12 g, 11.37 mmol, 47.3 % yield) which was used as is in the next step. |
| 39.6% |
With triethylamine; In dichloromethane; at 0 - 20℃; for 3h; |
150.41 g (0.79 mol) tosyl chloride was dissolved in 400 ml methylene chloride. 156.41 g (0.752 mol) tetraethylene glycol monomethyl ether was added and the mixture was cooled to 0C. 79.74 g (0.79 mol) triethyl amine in 100 ml methylene chloride was added while the temperature was kept below 10 C. The reaction was allowed to continue for three hours at room temperature. The reaction mixture was extracted twice with 300 ml 5N NaOH and twice with 300 ml water. The organic fraction was dried over Na2S04 and evaporated under reduced pressure. The crude product was purified using preparative column chromatography on a Prochrom LC80 column, using Kromasil Si60A 10 mutaueta as statoionary phase and methylene (0239) chloride/ethyl acetate 35/65 as eluent. 108 g of the tosylated tertaethylene glycol monomethyl ether was isolated as a slightly colored oil (y : 39.6 %) (TLC analysis on TLC Silicagel 60F254 supplied by Merck, methylene chloride/ethyl acetate 35/65 as eluent : Rf = 0.45) . |
| 2.5 g (70%) |
With pyridine; In dichloromethane; water; |
O-Tosyltetraethyleneglycol Methyl Ether (109) Tetraethyleneglycol monomethyl ether (2.0 g, 9.6 mmol) was combined with pyridine (0.93 mL, 11.5 mmol) in methylene chloride (20 mL) at 0 C. p-Toluenesulfonyl chloride (2.2 g, 11.5 mimol) dissolved in methylene chloride (10 mL) was added dropwise and the reaction was allowed to warm to room temperature as the ice bath thawed. After stirring nine days, the product mixture was transferred to a separatory funnel with water (70 mL). The layers were separated and the aqueous layer was extracted with methylene chloride (3*20 mL). The extracts were combined and evaporated. The residue (3.3 g) was chromatographed (silica gel, 4:1 to 3:1 gradient elution, methylene chloride/ethyl acetate) to afford 2.5 g (70%) of compound 109 as an oil. 1H NMR (300 MHz, DMSO-d6) delta 1.56 (br s, 411), 2.42 (br s, 2H), 3.34 (br s, 4H), 6.05 (s, 3H), 7.09 (s, 0.5H), 7.26 (s, 0.5H), 7.42 (br s, 2H), 7.70 (br s, 2H), 8.87 (br d, 2H), 9.07 (s, 2H), 9.22 (br s, 1H), 10.51 (s, 1H). CI MS m/z=363 [C16H26O7S+H]+. |
|
With dmap; triethylamine; In dichloromethane; at 20℃; for 20h; |
To a solution of 2,5,8,11-tetraoxatridecan- 13-ol (7 g, 33.6 mmol) and triethylamine (4.9 ml, 35.3 mmol) in dry CH2C12 (100 ml), 4-toluenesulfonyl chloride (6.7 g, 35.3 mmol) and DMAP (120 mg) were added. The mixture was stirred at room temperature for 20 h. The reaction mixture was washed with 80 mL of HCl (1M) and then water. The extract was dried over anhydrous MgS04, filtrated, and the filtrate was evaporated. The residue was used in the next step without further purification.Yield: 11.0 g (90%)NMR:1H NMR (400 MHz, CDC13) delta 7.75 - 7.64 (m, 2H), 7.31 - 7.26 (m, 2H), 4.16 -4.06 (m, 2H), 3.62 (m 2H), 3.59 - 3.40 (m, 10H), 3.30 (s, 3H), 2.38 (s, 3H).13C{1H} NMR (101 MHz, CDC13) delta 144.75 (s), 132.90 (s), 129.77 (s), 127.8 (s), 71.82 (s), 70.60 (s), 70.48 (s), 70.47 (s), 70.41 (s), 70.39 (s), 69.23 (s), 68.55 (s), 58.90 (s), 21.53 (s) |
|
With dmap; triethylamine; In dichloromethane; at 20℃; for 20h; |
Synthesis of 2,5,8,11-tetraoxatridecan-13-ol Tosylate Procedure: (JACS, 2007, 129, 13364) To a solution of 2,5,8,11-tetraoxatridecan-13-ol (7 g, 33.6 mmol) and triethylamine (4.9 ml, 35.3 mmol) in dry CH2Cl2 (100 ml), 4-toluenesulfonyl chloride (6.7 g, 35.3 mmol) and DMAP (120 mg) were added. The mixture was stirred at room temperature for 20 h. The reaction mixture was washed with 80 mL of HCl (1M) and then water. The extract was dried over anhydrous MgSO4, filtrated, and the filtrate was evaporated. The residue was used in the next step without further purification. Yield: 11.0 g (90%) NMR: 1H NMR (400 MHz, CDCl3) delta 7.75-7.64 (m, 2H), 7.31-7.26 (m, 2H), 4.16-4.06 (m, 2H), 3.62 (m 2H), 3.59-3.40 (m, 10H), 3.30 (s, 3H), 2.38 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3) delta 144.75 (s), 132.90 (s), 129.77 (s), 127.8 (s), 71.82 (s), 70.60 (s), 70.48 (s), 70.47 (s), 70.41 (s), 70.39 (s), 69.23 (s), 68.55 (s), 58.90 (s), 21.53 (s). |
|
With dmap; triethylamine; In dichloromethane; at 20℃; for 20h; |
Synthesis of 2,5,8,11-tetraoxatridecan-13-ol Tosylate Procedure: (JACS, 2007, 129, 13364) To a solution of 2,5,8,11-tetraoxatridecan-13-ol (7 g, 33.6 mmol) and triethylamine (4.9 ml, 35.3 mmol) in dry CH2Cl2 (100 ml), 4-toluenesulfonyl chloride (6.7 g, 35.3 mmol) and DMAP (120 mg) were added. The mixture was stirred at room temperature for 20 h. The reaction mixture was washed with 80 mL of HCl (1M) and then water. The extract was dried over anhydrous MgSO4, filtrated, and the filtrate was evaporated. The residue was used in the next step without further purification. Yield: 11.0 g (90%) NMR: 1H NMR (400 MHz, CDCl3) delta 7.75-7.64 (m, 2H), 7.31-7.26 (m, 2H), 4.16-4.06 (m, 2H), 3.62 (m 2H), 3.59-3.40 (m, 10H), 3.30 (s, 3H), 2.38 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3) delta 144.75 (s), 132.90 (s), 129.77 (s), 127.8 (s), 71.82 (s), 70.60 (s), 70.48 (s), 70.47 (s), 70.41 (s), 70.39 (s), 69.23 (s), 68.55 (s), 58.90 (s), 21.53 (s). |
|
With dmap; triethylamine; In dichloromethane; at 20℃; for 20h; |
Synthesis of 2,5,8,11-tetraoxatridecan-13-ol tosylate Procedure: [0187] (JACS, 2007, 129, 13364) To a solution of 2,5,8,11-tetraoxatridecan-13-ol (7 g, 33.6 mmol) and triethylamine (4.9 ml, 35.3 mmol) in dry CH2Cl2 (100 ml), 4-toluenesulfonyl chloride (6.7 g, 35.3 mmol) and DMAP (120 mg) were added. The mixture was stirred at room temperature for 20 h. The reaction mixture was washed with 80 mL of HCl (1M) and then water. The extract was dried over anhydrous MgSO4, filtrated, and the filtrate was evaporated. The residue was used in the next step without further purification. Yield: [0188] 11.0 g (90%) NMR: [0189] 1H NMR (400 MHz, CDCl3) delta 7.75-7.64 (m, 2H), 7.31-7.26 (m, 2H), 4.16-4.06 (m, 2H), 3.62 (m 2H), 3.59-3.40 (m, 10H), 3.30 (s, 3H), 2.38 (s, 3H). [0190] 13C{1H} NMR (101 MHz, CDCl3) delta 144.75 (s), 132.90 (s), 129.77 (s), 127.8 (s), 71.82 (s), 70.60 (s), 70.48 (s), 70.47 (s), 70.41 (s), 70.39 (s), 69.23 (s), 68.55 (s), 58.90 (s), 21.53 (s). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping