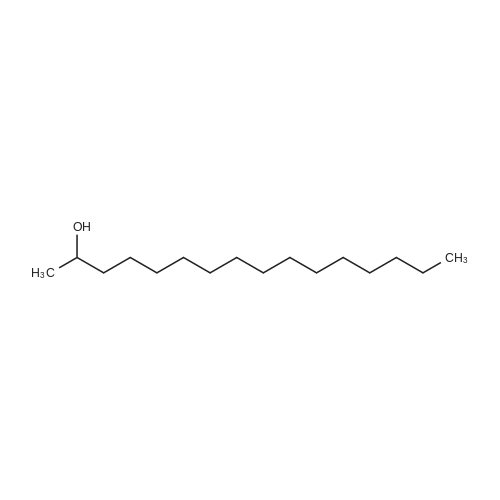
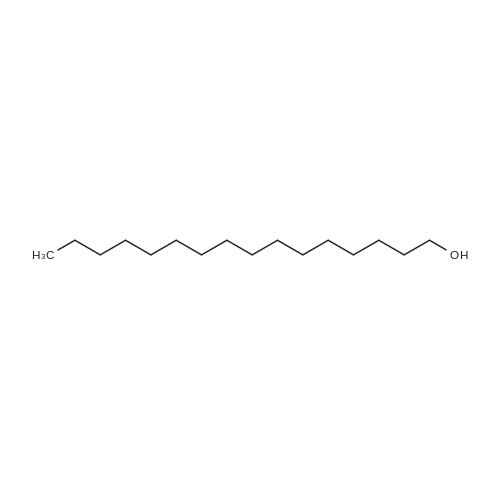
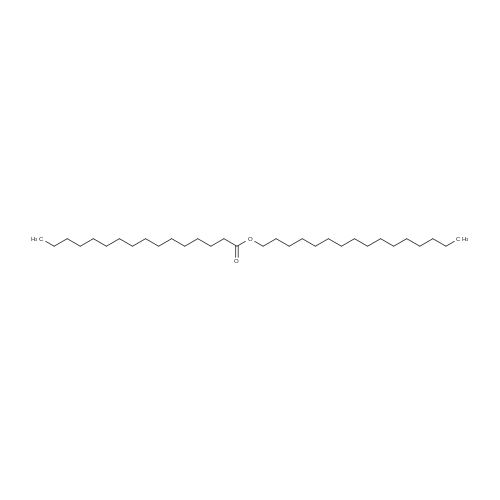
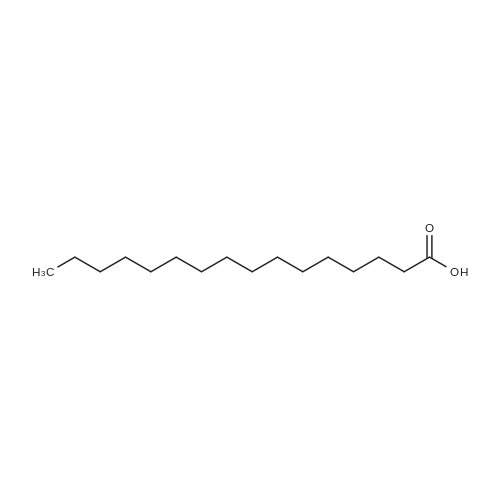
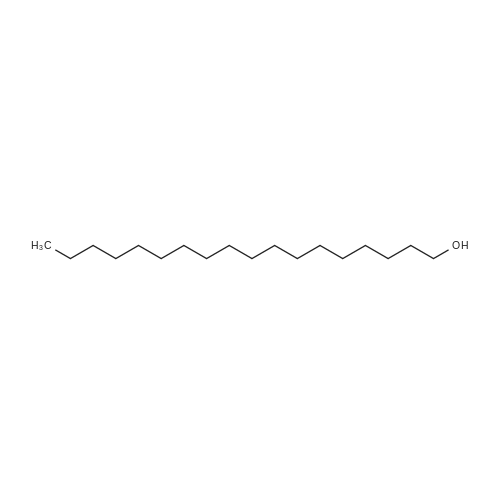
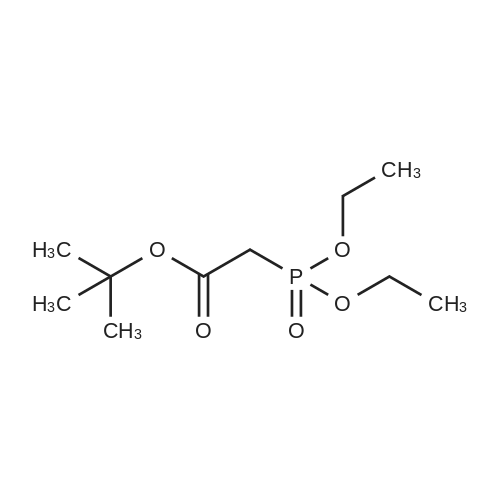
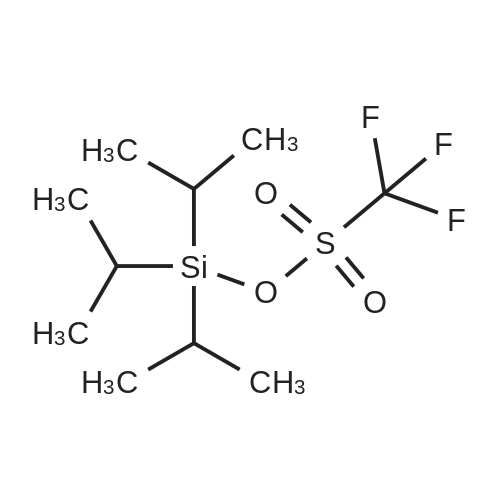
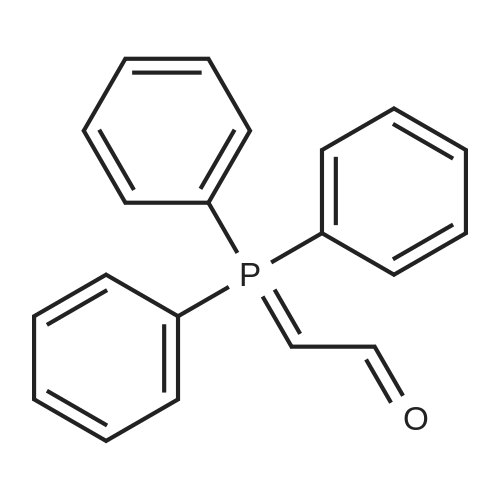
| 96% |
With oxalyl dichloride; dimethyl sulfoxide; triethylamine In dichloromethane for 0.25h; cooling; |
|
| 96% |
With sodium chlorine monoxide; Sodium hydrogenocarbonate; potassium bromide In dichloromethane at 0 - 20℃; for 0.25h; |
|
| 96% |
With sulphur trioxide pyridine complex; dimethyl sulfoxide; triethylamine In dichloromethane at 0 - 20℃; Inert atmosphere; |
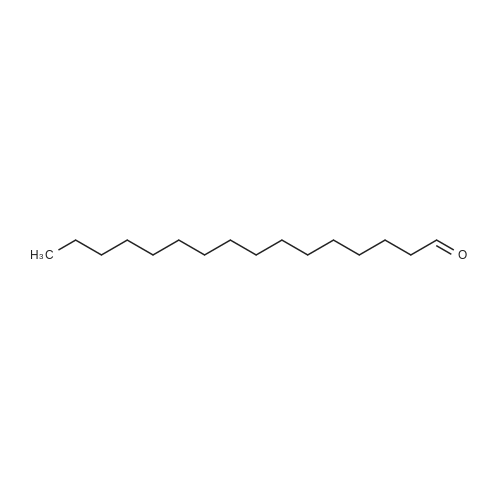
3,3-Difluorononadec-1-en-4-ol (1i)
To a mixture of dimethylsulfoxide (19.5 mL, 275 mmol), triethylamine (19.6 mL, 142 mmol) and CH2Cl2 (20 mL), hexadecan-1-ol (2.39 g, 9.8 mmol) was added and stirred at room temperature for 5 min. To a resulting clear solution, pyridine sulfur trioxide complex (13.1 g, 82.2 mmol) was slowly added at 0 °C. After being stirred at room temperature for 40 min, the reaction mixture was extracted with hexane-Et2O (1 : 1 v/v, 100 mL x 3). The combined organic layer was washed with 1 M HCl solution (50 mL) and 1 M NaHCO3 solution (100 mL), dried over anhydrous MgSO4 and concentrated under reduced pressure. Purification of the residue by column chromatography on silica gel (hexane/EtOAc = 20 : 1) gave palmitaldehyde in 96% yield (2.30 g, 9.6 mmol). According to the general procedure, difluorohomoallyl alchol 1i was prepared in 93% from this aldehyde. |
| 96% |
With sulfur(VI) fluoride; potassium carbonate; dimethyl sulfoxide at 20℃; for 21h; Sealed tube; chemoselective reaction; |
|
| 95% |
With pyridinium chlorochromate In dichloromethane at 20℃; for 3h; |
|
| 95% |
With pyridinium chlorochromate In dichloromethane for 2h; Reflux; |
|
| 95% |
With pyridinium chlorochromate In dichloromethane Reflux; |
|
| 95% |
With 1-methyl-1H-imidazole; [2,2]bipyridinyl; 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; copper(I) tetrakis(acetonitrile)tetrafluoroborate; oxygen In acetonitrile at 60℃; for 1.5h; Schlenk technique; |
|
| 94% |
With 1-(chloromethyl)-4-fluoro-1,4-diazoniabicyclo-[2.2.2]octane bis(tetrafluoroborate) In acetonitrile for 3h; Heating; |
|
| 94% |
With 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione In ethyl acetate at 70℃; for 24h; |
tert-Butyl (E)-2-octadecenoate 1. Preparation of hexadecanal
IBX (1.75 g, 6.24 mmol) was added to a stirred solution of 1-hexadecanol (500 mg, 2.08 mmol) in EtOAc (6 mL) and the suspension was heated to 70 °C for 24 h. After cooling to rt, the mixture was filtered and concentrated in vacuo to give hexadecanal as a white solid (467 mg, 94%); δH(400 MHz, CDCl3) 0.89 (3H, t, J 6.9, C(16)H3), 1.23-1.39 (24H, m, C(4)H2-C(15)H2), 1.51-1.69 (2H, m, C(3)H2), 2.43 (2H, td, J 1.7, 7.3, C(2)H2). |
| 93% |
With 2,2,6,6-tetramethyl-1-piperidinyloxy free radical; Amberlite IRA 900 bisacetoxybromate(I) In dichloromethane at 20℃; for 3.5h; |
|
| 93% |
With Burgess Reagent; dimethyl sulfoxide at 20℃; for 0.0833333h; Schlenk technique; Inert atmosphere; |
|
| 92% |
With pyridine; 2,2,6,6-tetramethyl-1-piperidinyloxy free radical; 1,2-Dichloro-3-iodobenzene In chloroform at 50℃; for 4.5h; |
|
| 92% |
With pivaloyl chloride; dimethyl sulfoxide; triethylamine In dichloromethane at -78 - 20℃; for 1h; |
|
| 92% |
With 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione In tetrahydrofuran; dimethyl sulfoxide at 20℃; Inert atmosphere; |
|
| 92% |
With 1-hydroxy-3H-benz[d][1,2]iodoxole-1,3-dione for 12h; Reflux; |
|
| 92% |
With oxalyl dichloride In dichloromethane; dimethyl sulfoxide at -41℃; for 5.5h; Inert atmosphere; |
Hexadecanal (39)[8]
To the stirring solution of DMSO (700 μL, 10.0 mmol) in CH2Cl2 (40 mL) at -78 °C was added oxalylchloride (550 μL, 6.0 mmol) under an inert atmosphere. After 20 min, 1-hexadecanol (486 mg,2.0 mmol) in CH2Cl2 (6 mL) was added and the reaction was stirred for 5.5 h at -41 °C. The reactionwas quenched with Et3N (3 mL, 21.5 mmol) and slowly warmed to ambient temperature over 30 min.The reaction mixture was washed with sat. NaHCO3 (50 mL) and extracted with CH2Cl2 (3 × 30 mL).The combined organic phase was washed with 2M HCl (2 × 20 mL), sat. NaHCO3 (20 mL), brine(20 mL), dried (MgSO4), filtered and concentrated in vacuo to afford a yellow solid (445 mg, 92%). |
| 92% |
With Dess-Martin periodane In dichloromethane at 0℃; for 3h; |
|
| 90% |
With Sodium hydrogenocarbonate; Dess-Martin periodane In dichloromethane at 0℃; Inert atmosphere; |
|
| 90% |
Stage #1: 1-Hexadecanol With copper(II) bromide In acetonitrile at 20℃; for 0.05h; Inert atmosphere;
Stage #2: With N,N'-di-tert-butyldiaziridin-3-one In acetonitrile at 60℃; for 4h; |
|
| 90% |
With pyridinium chlorochromate In dichloromethane at 20℃; for 4h; |
|
| 88% |
With pyridinium chlorochromate In dichloromethane for 1.5h; Ambient temperature; |
|
| 88% |
With 4 A molecular sieve; pyridinium chlorochromate In dichloromethane at 20℃; for 2h; |
|
| 88% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; iodosylbenzene; ytterbium(III) tris(trifluoromethanesulfonate) In dichloromethane at 0℃; for 0.833333h; Inert atmosphere; |
|
| 88% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; iodosylbenzene; ytterbium(III) tris(trifluoromethanesulfonate) In dichloromethane at 0℃; for 0.833333h; Inert atmosphere; |
|
| 87% |
With iron (ΙΙΙ) nitrate nonahydrate; 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl; oxygen; sodium chloride In 1,2-dichloro-ethane at 25℃; for 16h; chemoselective reaction; |
|
| 87% |
With 1,1,1,3',3',3'-hexafluoro-propanol; 5-trimethylammonio-1,3-dioxo-1,3-dihydro-1λ5-benzo-[d][1,2]iodoxol-1-ol anion; trifluoroacetic acid at 20℃; for 5h; |
|
| 86% |
With pyridinium chlorochromate In dichloromethane for 5h; Ambient temperature; |
|
| 85% |
Stage #1: 1-Hexadecanol With oxalyl dichloride; dimethyl sulfoxide In dichloromethane at -10℃; for 0.0833333h; Inert atmosphere;
Stage #2: In dichloromethane at -10℃; for 0.25h; Inert atmosphere; |
|
| 85% |
With pyridinium chlorochromate In dichloromethane at 20℃; for 3h; Molecular sieve; |
General procedure for oxidation of hexadecanol and octadecanol to 24 and 30:
General procedure: Hexadecanol(20 g, 82.6 mmol) was dissolved in 200 mL of anhydrous dichloromethane. The mixturewas added to a stirred suspension consisting of pyridinium chlorochromate (PCC) (26.8g, 124 mmol) and Celite (26.8 g) in 250 mL of anhydrous dichloromethane. The resultingsuspension was stirred for 3 h at rt and the progress of the reaction was monitored by TLC(Silica Gel 60, 9:1 petroleum ether/ethyl acetate (PE/EtOAc). Upon the disappearance ofthe starting material, the suspension was filtered twice through a filter paper (Whatman2). The solvent was evaporated and the residue dissolved in 500 mL of PE. The resultingsuspension was filtered through a thin layer of silica, the filter cake was rinsed with anadditional 200 mL of PE and the filtrates were combined. The solvent was removed underreduced pressure to afford hexadecanal (24) (17.08 g, 71.1 mmol, 85%) as an amorphouswhite solid. The same procedure was used for the synthesis of octadecanal (30), from octadecanol,in an 83% yield.Hexadecanal (24): Amorphous white solid; 1H NMR (400 MHz, CDCl3) δ 9.71 (1H, bs,CHO), 2.37 (2H, m, CH2CHO), 1.58 (2H, m, CH2CH2CHO), 1.23 (24 × H, bm), 0.84 (3H, t,J = 6.5 Hz, CH3). |
| 85% |
With pyridinium chlorochromate In dichloromethane at 20℃; for 3h; Molecular sieve; |
General procedure for oxidation of hexadecanol and octadecanol to 24 and 30:
General procedure: Hexadecanol(20 g, 82.6 mmol) was dissolved in 200 mL of anhydrous dichloromethane. The mixturewas added to a stirred suspension consisting of pyridinium chlorochromate (PCC) (26.8g, 124 mmol) and Celite (26.8 g) in 250 mL of anhydrous dichloromethane. The resultingsuspension was stirred for 3 h at rt and the progress of the reaction was monitored by TLC(Silica Gel 60, 9:1 petroleum ether/ethyl acetate (PE/EtOAc). Upon the disappearance ofthe starting material, the suspension was filtered twice through a filter paper (Whatman2). The solvent was evaporated and the residue dissolved in 500 mL of PE. The resultingsuspension was filtered through a thin layer of silica, the filter cake was rinsed with anadditional 200 mL of PE and the filtrates were combined. The solvent was removed underreduced pressure to afford hexadecanal (24) (17.08 g, 71.1 mmol, 85%) as an amorphouswhite solid. The same procedure was used for the synthesis of octadecanal (30), from octadecanol,in an 83% yield.Hexadecanal (24): Amorphous white solid; 1H NMR (400 MHz, CDCl3) δ 9.71 (1H, bs,CHO), 2.37 (2H, m, CH2CHO), 1.58 (2H, m, CH2CH2CHO), 1.23 (24 × H, bm), 0.84 (3H, t,J = 6.5 Hz, CH3). |
| 83% |
Stage #1: 1-Hexadecanol With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; tetrabutylammonium bromide; Sodium hydrogenocarbonate; potassium carbonate In dichloromethane; lithium hydroxide monohydrate at 20℃;
Stage #2: With N-chloro-succinimide In dichloromethane; lithium hydroxide monohydrate at 20℃; for 1h; |
1 Synthesis of Compound [2]
A solution of 200 mL of pure water in which sodium hydrogen carbonate (8.47 g, 100 mmol) and potassium carbonate (1.42 g, 10.3 mmol) were dissolved was added to a solution of 1-hexadecanol (24.4 g, 100 mmol), tetrabutylammonium bromide (TBAB) (1.67 g, 5.2 mmol), and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) (0.79 g, 5.1 mmol) in 200 mL of dichloromethane and the resultant mixture was stirred at room temperature. To this solution, N-chlorosuccinimide (NCS) (16.1 g, 120 mmol) was added and the resultant mixture was stirred at room temperature for 1 hour. After stirring, an organic phase was separated and the organic phase was washed three times with 100 mL of pure water. After washing, the organic phase was separated and sodium sulfate was added to dry the organic phase. Thereafter, sodium sulfate was removed by filtration and the filtrate was concentrated. The residue was purified by column chromatography (silica gel, hexane:ethyl acetate=100:0 to 95:5 (v/v)) to give the target product 1-hexadecanal (Compound [2]): Yield 83% (20.0 g), 1H NMR (400 MHz, CDCl3): δ 9.76 (1H, t, J=1.8 Hz), 2.42 (2H, dt, J=1.8, 7.3 Hz), 1.63 (2H, quin, J=7.3 Hz), 1.37-1.19 (24H, m), 0.88 (3H, t, J=6.9 Hz). |
| 81% |
Stage #1: 1-Hexadecanol With oxalyl dichloride; dimethyl sulfoxide In dichloromethane at -41℃; for 5.5h; Inert atmosphere; Sealed tube;
Stage #2: With triethylamine In dichloromethane at -41 - 21℃; for 0.5h; Inert atmosphere; Sealed tube; |
|
| 80% |
With TEMPOL; Cu(NO<SUB>3</SUB>)<SUB>2</SUB>3H<SUB>2</SUB>O; oxygen In 1,2-dichloro-ethane at 25℃; for 36h; |
44; 54 Example 1
General procedure: Insert an oxygen balloon on the dry reaction tube, ventilate 3 times, add Cu(NO3)2·3H2O (24.6mg, 0.1mmol), TEMPO (16.2mg, 0.1mmol), 1b (146.4mg, 1.0mmol) in sequenceMeCN (4mL) solution.The reaction tube was stirred at 25°C for 6 hours. The reaction solution was filtered with a short silica gel column (2cm),Wash with ether and remove the solvent by rotary evaporation. Use silica gel column chromatography for separation and purification (eluent:Petroleum ether/diethyl ether=60/1) to obtain product 2b (130.7 mg, 91%): a white solid. |
| 76% |
With iron (ΙΙΙ) nitrate nonahydrate; 2,2,6,6-tetramethyl-1-piperidinyloxy free radical; oxygen; sodium chloride In 1,2-dichloro-ethane at 25℃; for 8.5h; |
|
| 72% |
With dimethyl sulfoxide; triethylamine; trichloromethyl chloroformate In dichloromethane |
|
| 64% |
With 1-methyl-3-(2-oxo-2-(2,2,6,6-tetramethyl-1-ylooxy-4-piperidoxyl)ethyl)imidazolium chloride; carbon dioxide; lithium hydroxide monohydrate; oxygen; NaNO2 at 99.84℃; for 12h; Autoclave; |
|
| 62% |
With piridinium dichromate In dichloromethane |
|
| 62% |
With 2,2,6,6-Tetramethyl-1-piperidinyloxy free radical; caesium hydroxide; [(2-(sulfonylquinlium-8-yloxy)phthalic acid-2H)2Cu4(4,4′-bipyridine)4]·2H2O}n·2nH2O In acetonitrile at 80℃; for 12h; |
|
| 61% |
With tetrabutylammonium bromide; dihydrogen peroxide In dichloromethane at 20℃; for 24h; |
|
| 50% |
With tetrabutylammonium chlorochromate In chloroform for 24h; Heating; |
|
| 45% |
With piridinium dichromate; adogen 464; dihydrogen peroxide; anhydrous sodium carbonate In various solvent(s) for 24h; Heating; |
|
| 45% |
With titanium(IV) dioxide; oxygen at 29.84℃; for 24h; Sealed tube; Irradiation; |
|
| 41% |
With 1-methyl-3-(2-oxo-2-(2,2,6,6-tetramethyl-1-ylooxy-4-piperidoxyl)ethyl)imidazolium chloride; 1-(carboxymethyl)-3-methylimidazolium chloride; oxygen; NaNO2 In lithium hydroxide monohydrate at 59.84℃; for 12h; Inert atmosphere; |
|
| 31 % Turnov. |
With pentanal at 450℃; for 0.0833333h; var. of temp.; |
|
|
With N-Methylmorpholine N-oxide In acetonitrile at 25℃; |
|
|
With copper(II) oxide |
|
|
With copper oxide-chromium oxide at 250℃; |
|
|
With piridinium dichromate In dichloromethane |
|
|
With sodium chlorine monoxide In Carbon tetrachloride; lithium hydroxide monohydrate at 60 - 65℃; Yield given; |
|
|
With diphosphorus pentoxide; dimethyl sulfoxide; triethylamine 1.) CH2Cl2, from 0 deg C to RT, 30 min, 2.) CH2Cl2, 0 deg C, 30 min; Yield given. Multistep reaction; |
|
|
With pyridinium chlorochromate In dichloromethane for 2h; Ambient temperature; Yield given; |
|
|
With pyridinium chlorochromate In dichloromethane for 1.5h; Ambient temperature; |
|
| 98 % Turnov. |
With piridinium dichromate; dihydrogen peroxide; anhydrous sodium carbonate In 1,2-dichloro-ethane at 80℃; for 24h; |
|
|
With pyridinium chlorochromate |
|
|
With pyridinium chlorochromate In dichloromethane |
|
|
With diphosphorus pentoxide; dimethyl sulfoxide |
|
|
With diphosphorus pentoxide; dimethyl sulfoxide; triethylamine In dichloromethane at 0 - 20℃; |
|
|
With Celite; pyridinium chlorochromate In dichloromethane at 20℃; for 4h; |
|
|
With air; RuO2 incorporated in ZSM-5 zeolite In toluene at 80℃; for 36h; |
|
|
With oxalyl dichloride; dimethyl sulfoxide; triethylamine In dichloromethane at -78 - 20℃; |
|
|
Multi-step reaction with 4 steps
1: triethylamine / CH2Cl2 / 0.25 h / 0 °C
2: NaI / acetone / 2 h
3: 71 percent / AgNO2 / diethyl ether / 0 deg C, 15 h, next room temperature, 8 h
4: 1.) NaH, tert-butyl alcohol, 2.) KMnO4, H3BO3 / 1.) pentane, 20 min, 2.) ethyl acetate, water, 10 min |
|
|
Multi-step reaction with 2 steps
1: selenium dioxide
2: 177 °C / 0.1 Torr / Irradiation.im UV-Licht |
|
|
With diphosphorus pentoxide; dimethyl sulfoxide; triethylamine In dichloromethane at 20℃; for 2h; |
Synthesis of hexadecanal 7
To a cooled solution of hexadecanol (10.0 g, 41 mmol) in dried dichloromethane was added dimethyl sulfoxide (6.43 g, 82 mmol) and P2O5 (11.7 g, 82 mmol). After stirring 45 min at room temperature the mixture was cooled to 0 C and triethylamine (14.52g, 143.5 mmol) was added. The reaction mixture was allowed to warm to room temperature and stirred for 2 h. After addition of 50 ml HCl (10 %) the reaction mixture was extracted 3 times with dichloromethane. The combined organic layers were extracted with saturated NaCl-solution and dried with NaSO4. After evaporation a colourless solid (6.3 g) was obtained which was purified by column chromatography over silica gel with cyclohexane/ethyl acetate (4:1).1H-NMR (200 MHz, CDCl3): d (ppm) = 0.9 (t, 3H, J = 6.6 Hz, CH3); 1.3 - 1.5 (b, 26 H, -CH2-); 2.4 (td, 2H, CH2CHO); 9.7 (t, 1H, CH2CHO). |
|
With anhydrous Sodium acetate; pyridinium chlorochromate In diethyl ether; dichloromethane |
1 Hexadecanal (2)
EXAMPLE 1 Hexadecanal (2) A solution comprising 10 g (85 mmole) of 1-hexadecanol in 100 ml of methylene chloride was added to a mixture comprising 10 g (139 mmole) of pyridinium chlorochromate, 30 g of celite and 12.0 g (146 mmole) of anhydrous sodium acetate in 400 ml of methylene chloride in a dropwise fashion. Following addition, the reaction mixture was stirred at room temperature for three hours then 300 ml of diethyl ether was added. The reaction mixture was filtered and a precipitate was collected and washed with diethyl ether until clear. The solvent was removed under reduced pressure and the residue passed through Florisil with diethyl ether as the eluent. The solvent was removed under reduced pressure and the residue purified by flash chromatography on silica (230 to 400 mesh) with hexanes and ethyl acetate at a ratio of approximately 39 to 1. The synthesis yielded hexadecanal having a mass of approximately 17.5 g and of approximately 88.4% purity. Hexadecanal is a white wax having a melting point of approximately 33°-34 C. The 1 H NMR spectrum of the compound at 300 MHz using CDCl3 comprised the following peaks: δ 0.892 (3H, t, J=6.5Hz), 1.267 (24H, bs), 1.61 to 1.66 (2H, m), 2.430 (2H, dt, J=1.7 and 7.3 Hz), and 9.776 (1H, s). |
|
|
9
In this example, EVK-203 was prepared. Following a literature procedure of Haldar et al., Incorporation of Multiple Head Groups Leads to Impressive Antibacterial Activity. J. Med. Chem. 48:3823-3831 (2005), hexadecanal (1) was prepared by oxidation of commercially available 1-hexadecanol, while the tetra-Boc-polyamine (3) was synthesized following procedure from Example 1. |
|
Stage #1: 1-Hexadecanol With pivaloyl chloride; dimethyl sulfoxide In dichloromethane at -78℃;
Stage #2: With triethylamine In dichloromethane at -78 - 20℃; |
|
|
Stage #1: 1-Hexadecanol With oxalyl dichloride; dimethyl sulfoxide In dichloromethane at -78℃; for 1.16667h; Inert atmosphere;
Stage #2: With triethylamine In dichloromethane at -60 - 20℃; for 0.5h; Inert atmosphere; |
|
|
With pyridinium chlorochromate Inert atmosphere; |
|
|
With Sodium hydrogenocarbonate; Dess-Martin periodane In dichloromethane at 0 - 20℃; Inert atmosphere; Cooling with ice; |
|
|
With pyridinium chlorochromate |
|
|
With manganese(IV) oxide In n-Pentane |
|
| 7 %Chromat. |
With sulfuric acid; oxygen; NaNO2 In acetonitrile at 80℃; for 24h; |
Typical procedure for the oxidation process:
General procedure: In a three-neck round bottom flask, KCC-1/TEMPO (0.05 g, about 0.02 mmol) was dispersed in acetonitrile (15 mL) and sonicated for 15 min followed by the addition of alcohol (2 mmol), NaNO2 (6.9 mg, 0.1 mmol). Oxygen was introduced to the flask with a rate of 0.004 m3/h and the flask was placed in an oil bath thermostated at 80 °C. The reaction was initiated by the addition of 10% H2SO4 solution (0.1 mL). When the reaction was finished, the catalyst was recovered by filtration and used after drying without further treatment. After extraction with CH2Cl2 and drying over MgSO4, the crude product was analyzed by GC. |
| 68 %Chromat. |
With pyridine; Tributylphosphine oxide; oxygen; palladium diacetate In toluene at 80℃; Molecular sieve; |
|
|
With dihydrogen peroxide; N-hexadecyl-N,N,N-trimethylammonium bromide In lithium hydroxide monohydrate at 80℃; for 24h; Green chemistry; |
|
|
Stage #1: 1-Hexadecanol With oxalyl dichloride; dimethyl sulfoxide In dichloromethane at -78℃;
Stage #2: With triethylamine In dichloromethane at -78 - 20℃; |
4.29. Compatibility of AzulE with Swern oxidation
DMSO (0.5 mL), (COCl)2 (0.2 mL) and CH2Cl2 (5 mL) were added together at 78 C and left for 30 min. For the control reaction, cetyl alcohol (54 mg, 0.22 mmol, 1 eq.) was dissolved in THF (4 mL)at 78 C, and the above oxidising mixture (2 mL, containing a calculated 4.2 eq. chlorodimethylsulfonium chloride) was added.The solution became cloudy. After two hours, NEt3 (0.3 mL,2.15 mmol, 9.7 eq.) was added and the reaction was maintained at 78 C for a further 30 min before allowing it to warm up to r.t. over an hour. 1 mL 10% HCl (aq.) was added to the reaction mixture, followed by water and a separation was performed using CH2Cl2/H2O, and the organic layer was submitted for NMR analysis. |
|
With pyridinium chlorochromate In dichloromethane at 0 - 20℃; for 1h; |
|
|
With sulfur(VI) fluoride; potassium carbonate; dimethyl sulfoxide at 20℃; |
|
|
With potassium peroxymonosulfate; tetrabutylammonium hydrogensulfate In chloroform at 25℃; for 8h; |
General procedure: A typical reaction was carried out as follows: benzyl alcohol (1 equiv), Oxone (0.88 equiv), and PTC (1.5 equiv) were dissolved in 3 mL of chloroform in a 10-mL glass reactor. The mixture was stirred for 8 h, and the substrate conversion and product formation were monitored by GLC. The products were identified by comparing with authentic samples using GC or GC/MS analysis. |
|
With Sodium hydrogenocarbonate; Dess-Martin periodane In dichloromethane at 20℃; for 2h; Inert atmosphere; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping