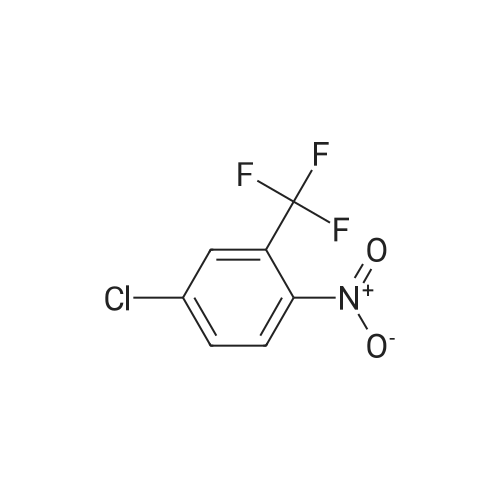
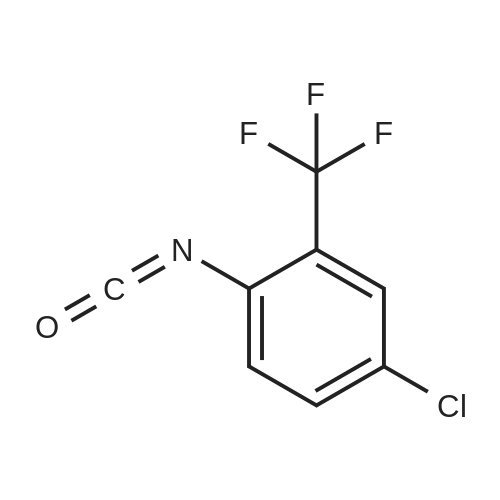
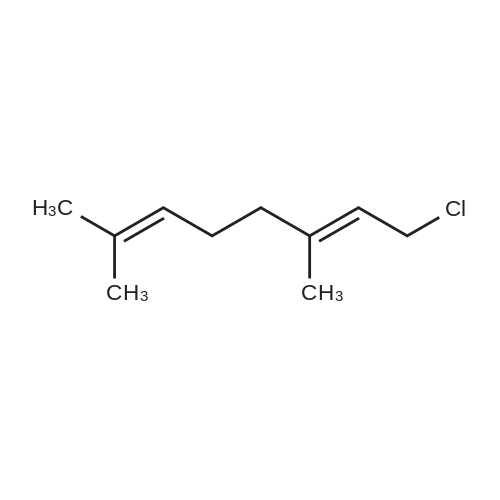
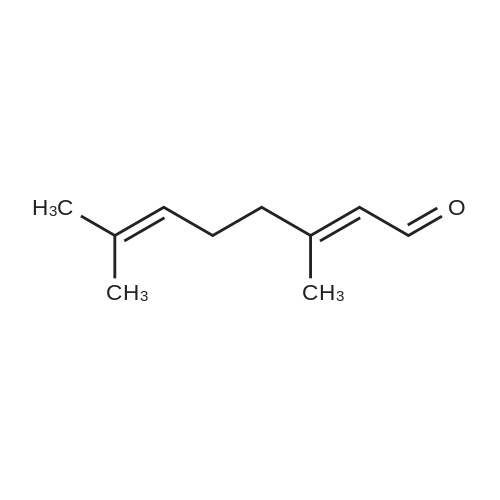
|
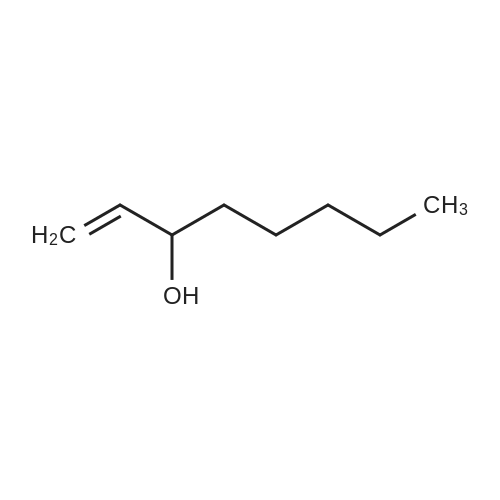
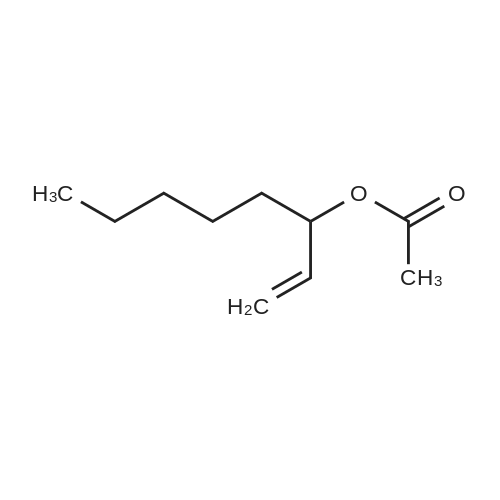
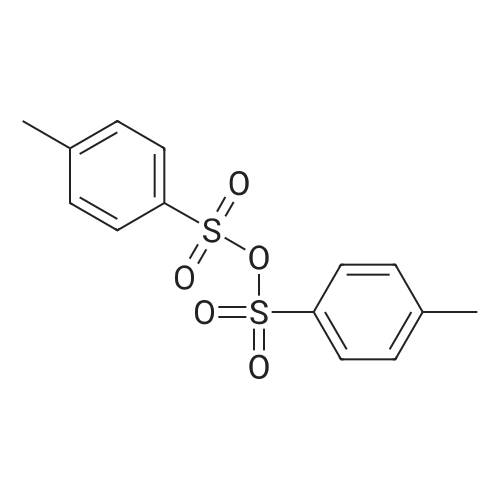
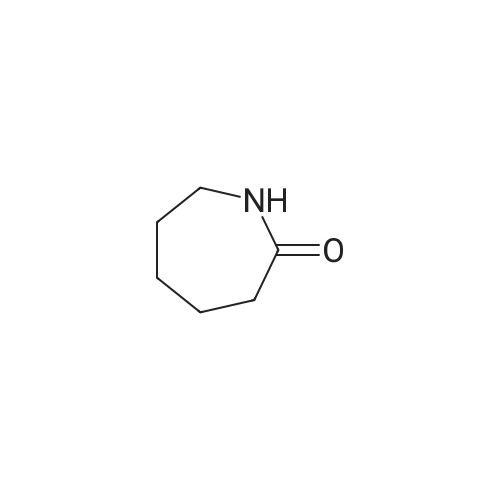
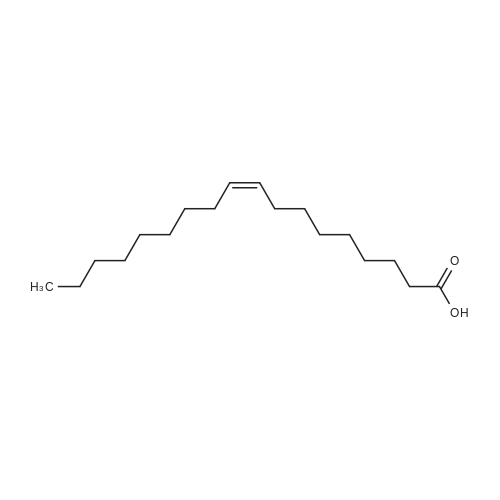
With oxygen; ozone; In water; for 0.25 - 2h; |
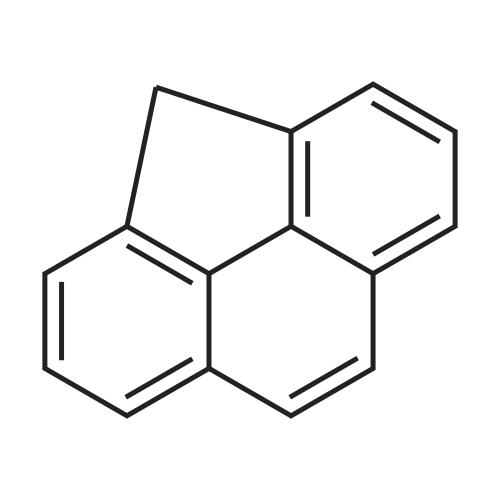
Example A; Degradation of pyrene; This example focuses on an integrated approach for the degradation of pyrene involving chemical oxidation followed by biological treatment. The objectives were to: 1) provide mechanistic details in the degradation of pyrene subject to ozone treatment, 2) test the combined technique of ozone pretreatment followed by biological degradation, and 3) test a pretreatment column to promote efficient use of chemical oxidants and biodegradability. Batch and packed column reactors were used to examine the degradation pathways of pyrene subject to ozonation in the aqueous phase. After different ozonation times, samples containing reaction intermediates and byproducts from both reactors were collected, identified for organic contents, and further biologically inoculated to determine biodegradability. The O3-pretreated samples were incubated for 5, 10, 15, and 20 days, after which biochemical oxygen demand (BOD), chemical oxygen demand (COD), and toxicity tests along with qualitative and quantitative GC/FID and GC/MS analyses of pyrene, intermediates, and products were performed. Intermediates identified at different stages included 4,5-phenanthrenedialdehyde, 2,2',6,6'-biphenyltetraaldehyde, and long-chain aliphatic hydrocarbons, which suggested that the degradation of pyrene was initiated by O3 via ring cleavage at the 4,5- and 9,10-bonds and that further oxidation ensued via reactions with both O3 and OH. until complete mineralization. Intermediates formed during chemical oxidation were biodegradable with a measured first-order rate constant (k0) of 0.243 day-. The integrated chemical-biological system appeared to be feasible for treating recalcitrant compounds, and a chemical pretreatment column was particularly useful in promoting soluble intermediates from otherwise highly insoluble, inaccessible pyrene.Materials and MethodsChemicalsOzone (1% w/w ozone in air) was generated from filtered, dry air by an ozonator (Model T-816, Polymetrics Corp.). Pyrene (99%, Aldrich Chemical Co.) was washed with distilled-deionized (DD) water three times, extracted by dichloromethane (DCM), and the solvent evaporated by a gentle stream of nitrogen gas. Stock and working indigo blue solutions were prepared from potassium indigo trisulfonate (C16H7N2O11S3K3, Aldrich Co.) per Standard Methods (APHA et al., 1992a). Polyseed (Hach Co.) was used in dilution water for biochemical oxygen demand (BOD) measurements per Standard Methods (APHA et al., 1992b). Inoculum for toxicity test was prepared according to a Hach method (HACH, 1988-1995b). COD digestion solutions (0-15,000 mg/L, 0-40 mg/L range, Hach Co.), ToxTrak reagent powder pillows, and ToxTrak accelerator solution (Hach Co.) were purchased and used according to the manufacturer's methods without further processing. Low-organic (<15 ppb as TOC), low-ion (resistivity>18 MOmega-cm), and non-pyrogenic (up to 4-log reduction with reverse osmosis pretreatment) DD water was used in all procedures (4-stage Mill-Q Plus system, Millipore Co.). Dichloromethane (Fisher Scientific) of HPLC grade was used in liquid-liquid extraction procedures. Other chemicals used in this research were of reagent grade.; Results and Discussion; Ozonation of pyrene was carried out in batch and column reactors to study: 1) the effect of reactor on intermediates and products formation, 2) the degradation pathway of pyrene under ozonation, 3) the biodegradability of intermediates, and 4) the feasibility of a combined chemical-biological treatment system for pyrene. Reaction solutions during ozonation and biodegradation processes at different stages were collected and the intermediates and byproducts identified by GC/MS techniques.1. Effects of the Reactor Type on Intermediates and Products FormationTo delineate the influence of reactor configurations on the formation of intermediates and products, ozonation experiments using aqueous and excess pyrene were carried out in batch and packed column reactors. BOD5 and COD were measured for three ozonated, filtered solutions: 1) a saturated aqueous solution of pyrene (0.13 ppm), 2) the solution after ozonation of an excess pyrene suspension (1 g/1.7 L), and 3) the effluent of a column packed with excess pyrene solid (1 g) and glass beads (7.5 in. in bed-length). The saturated pyrene solution was prepared by allowing excess pyrene solid to reach dissolution equilibrium in water overnight followed by removal of the excess solid using a 0.45-mum filter. The ozonated batch solution was obtained after 10 min of ozonation and filtered, while the effluent was collected from the packed column fed with ozonated water over a 4-hr period. Table A-I shows the results of BOD5 and COD measurements. The BOD5 for the saturated pyrene solution approximates over 80% of the COD value, suggesting that pyrene in its dissolved form is amenable to biodegradation, albeit in small quantity. The aqueous phase COD from the ozonated batch reactor increased after ozon... |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping