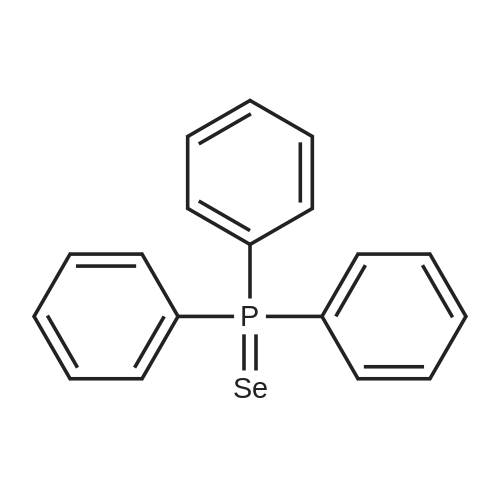
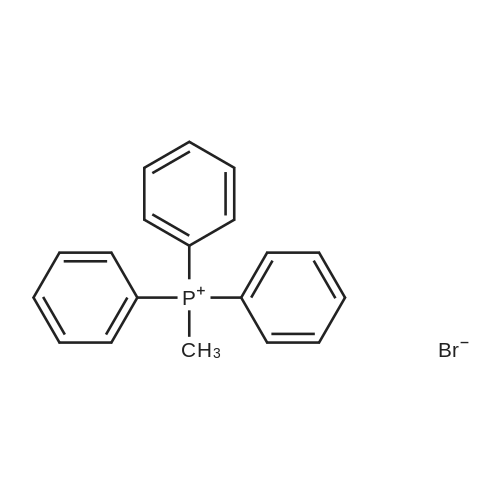
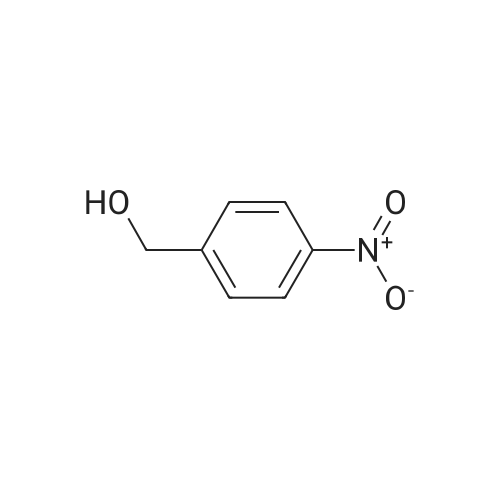
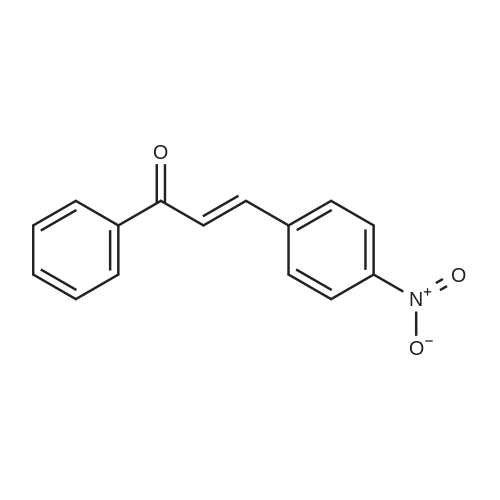
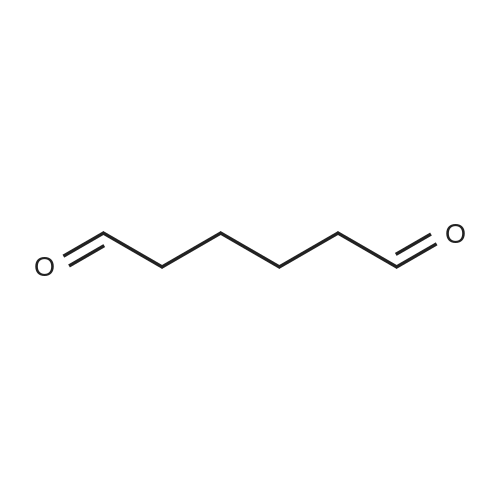
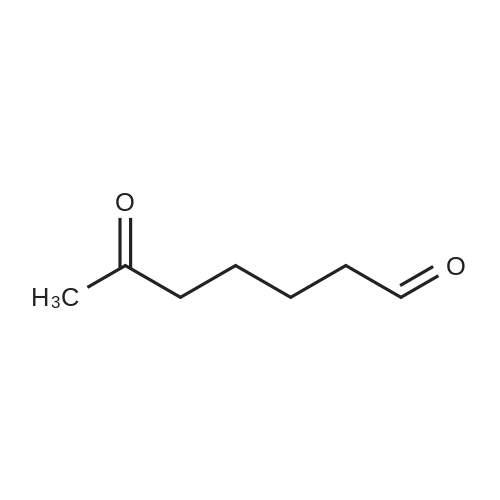
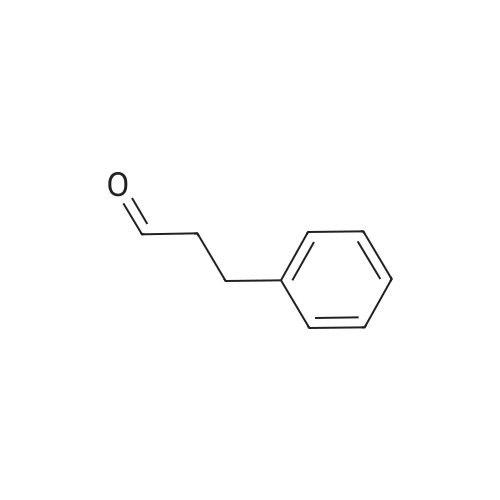
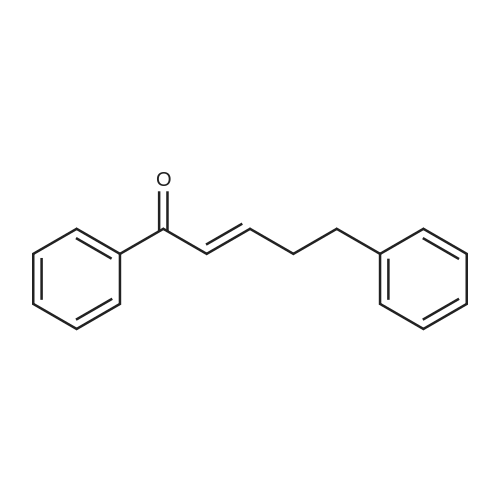
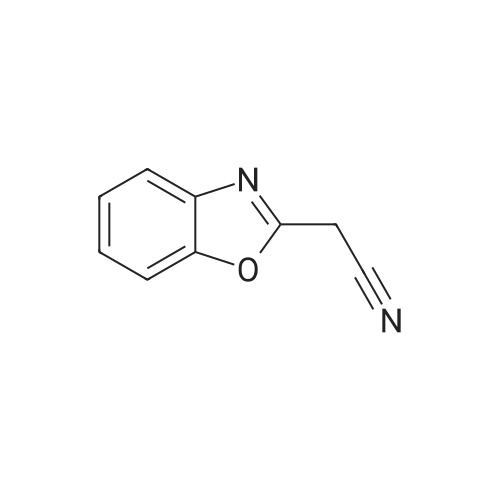
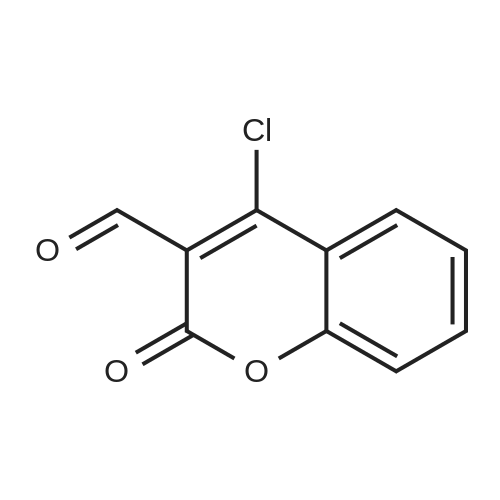
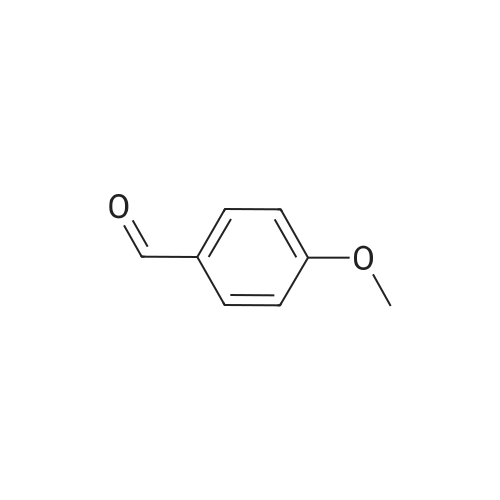
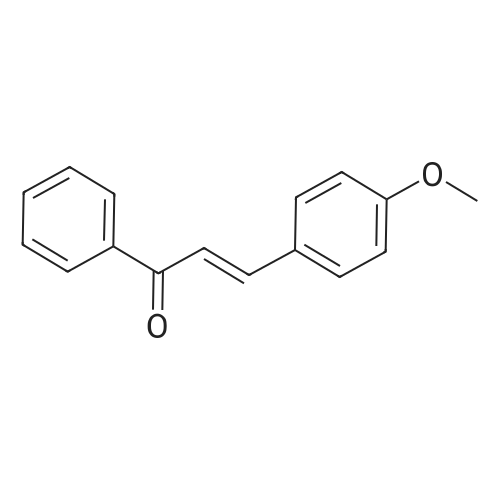
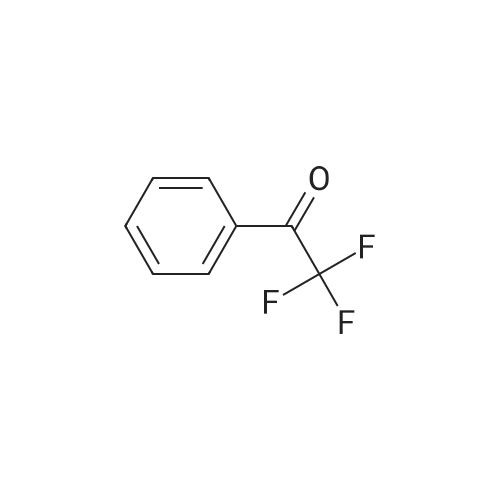
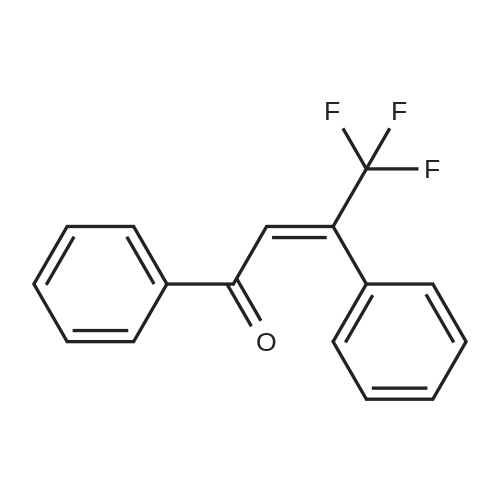
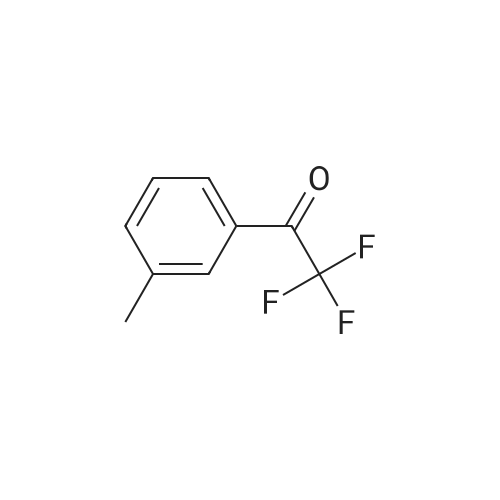
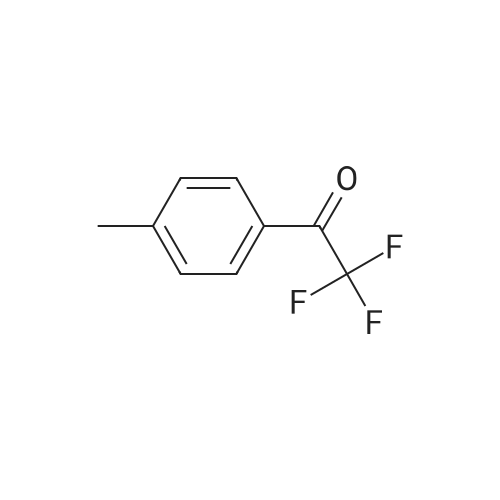
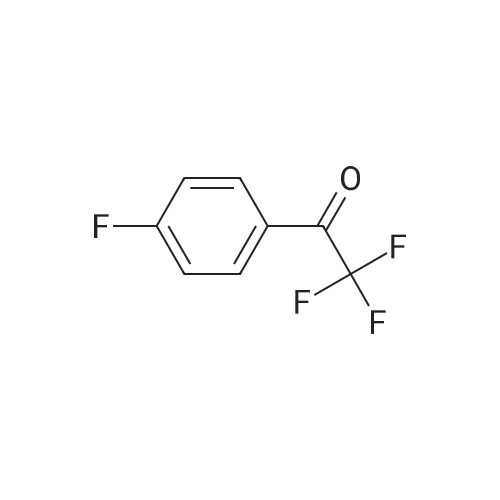
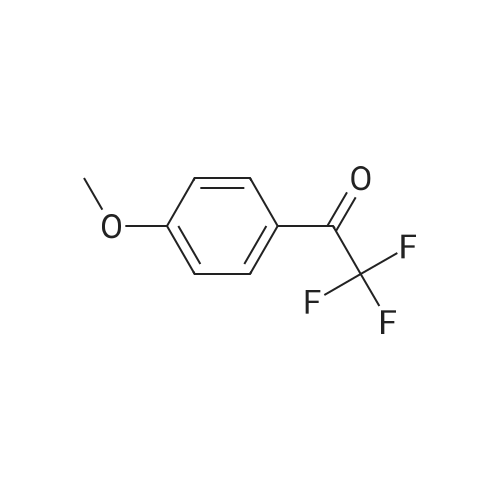
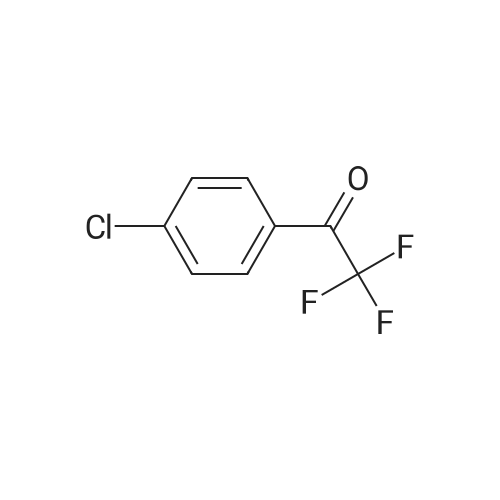
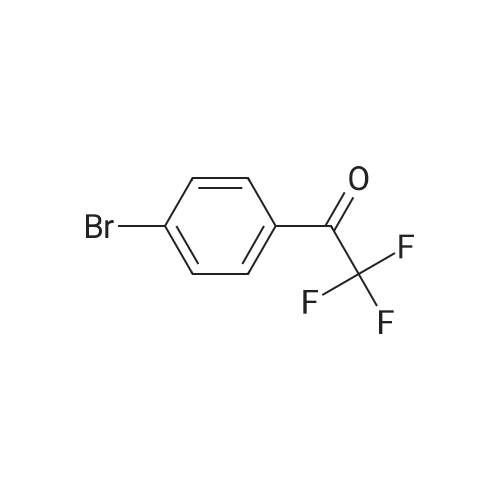
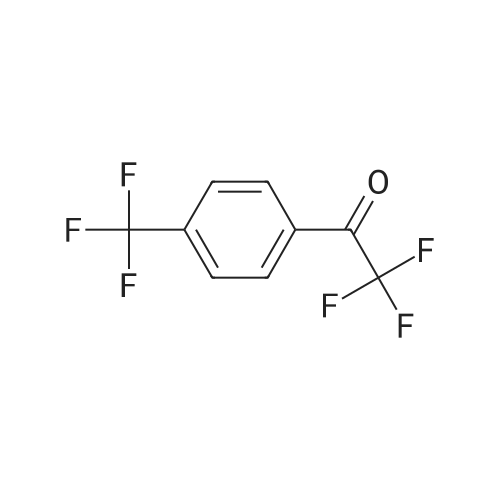
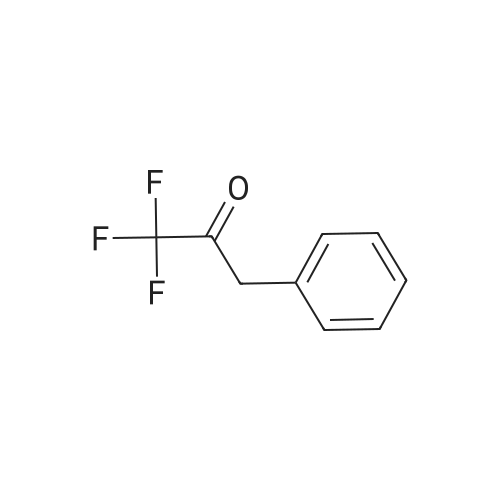
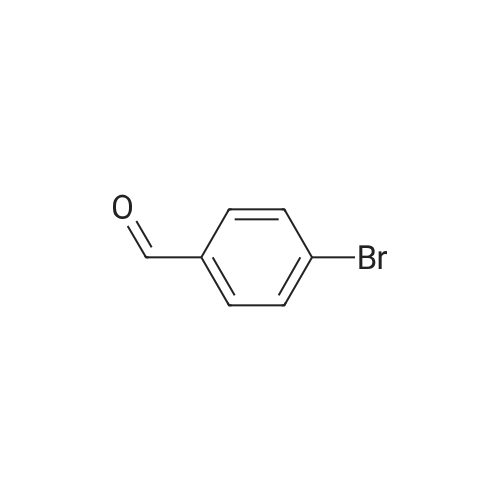
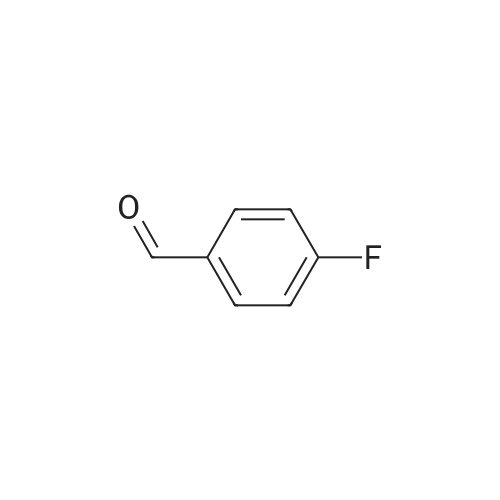
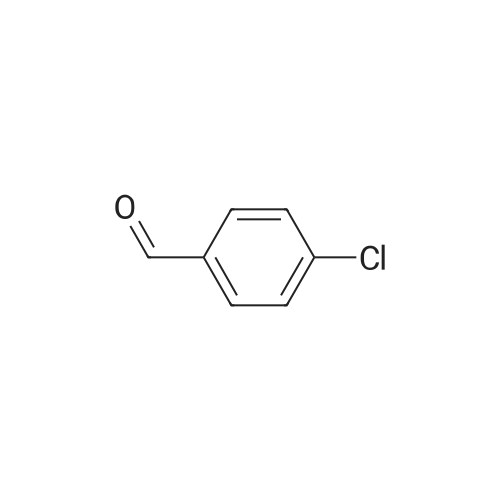
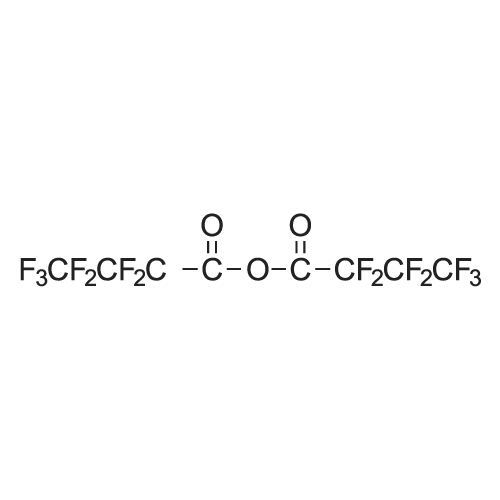
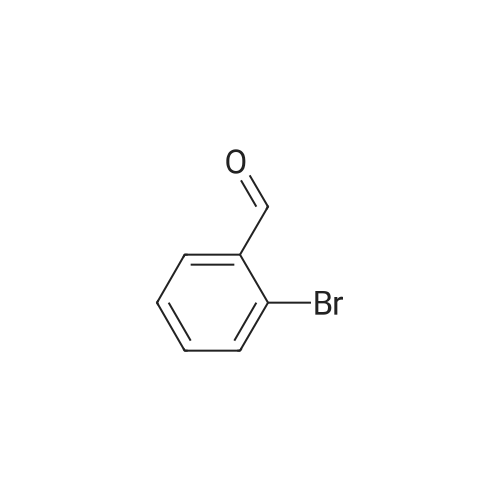
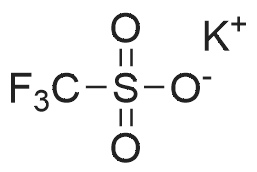
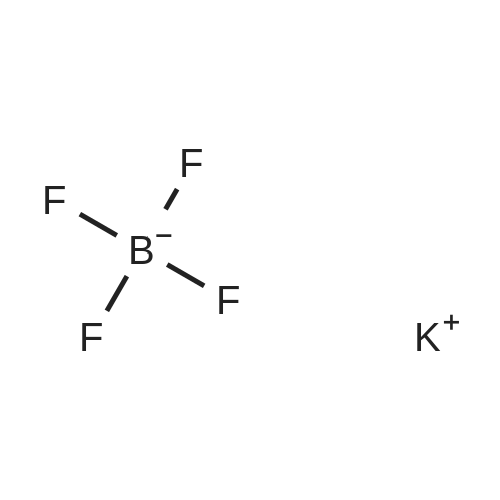
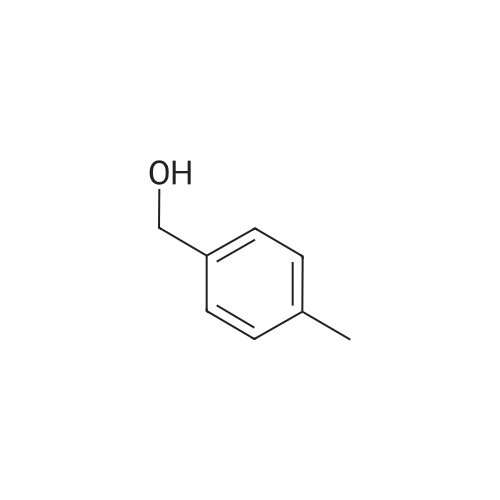
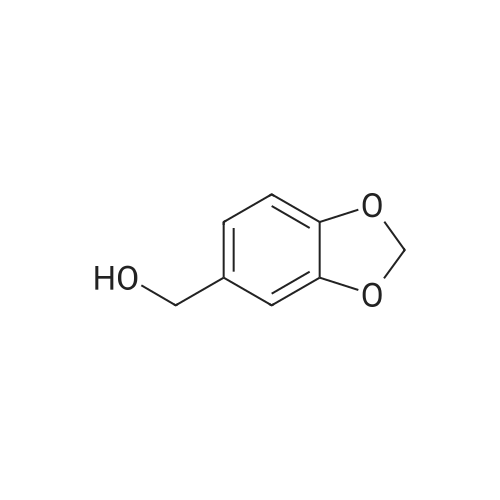
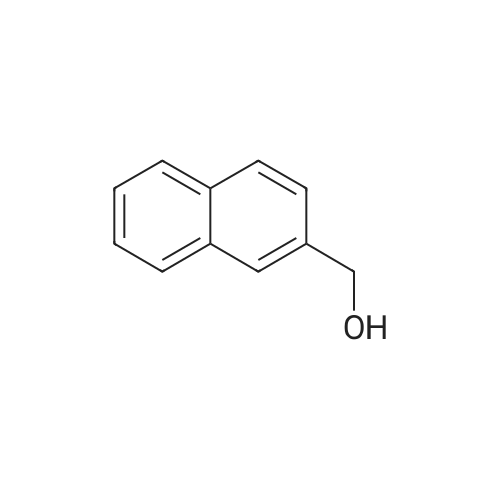
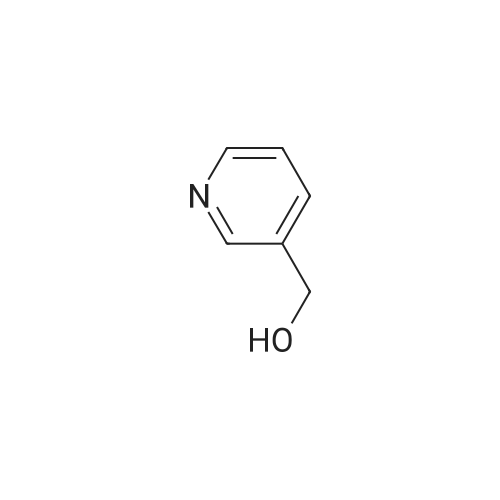
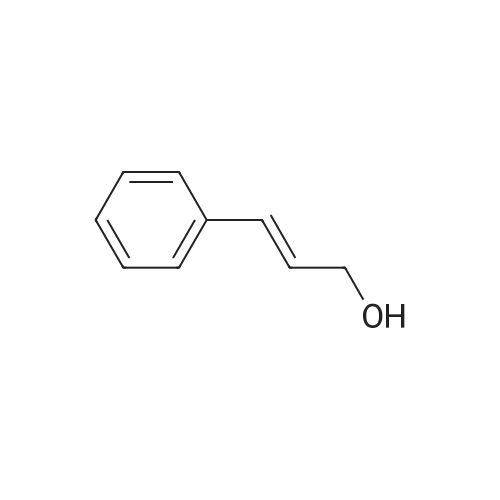
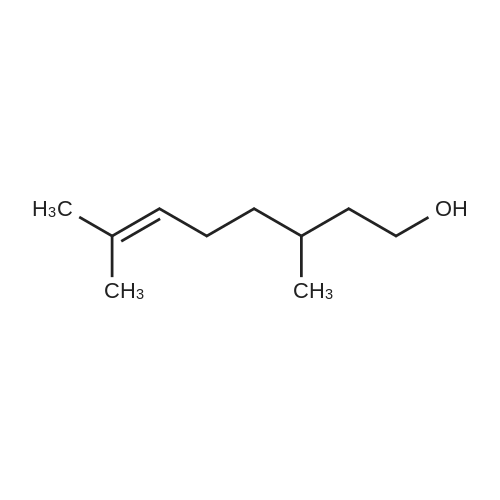
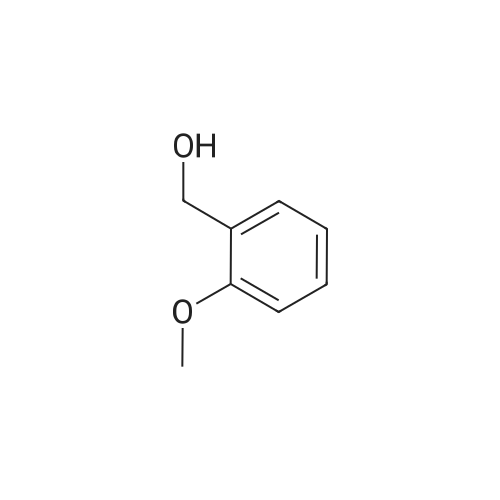
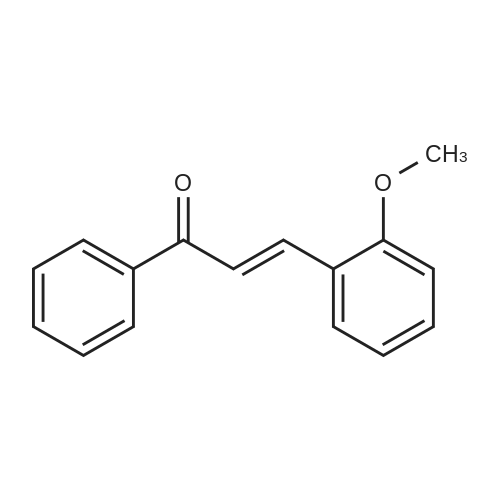
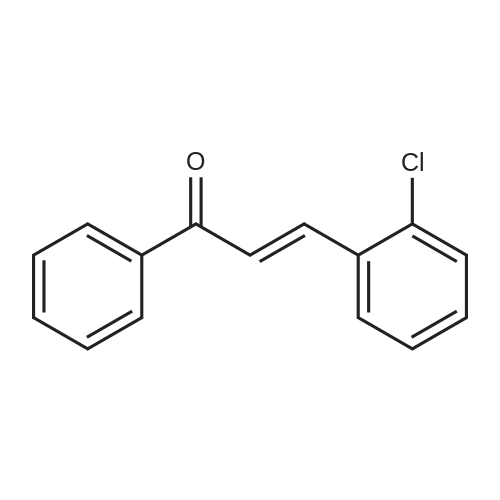
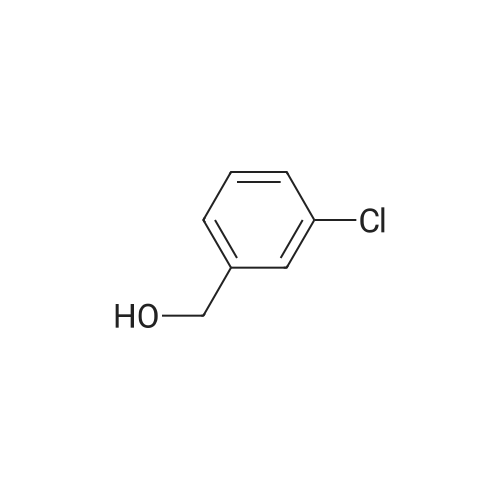
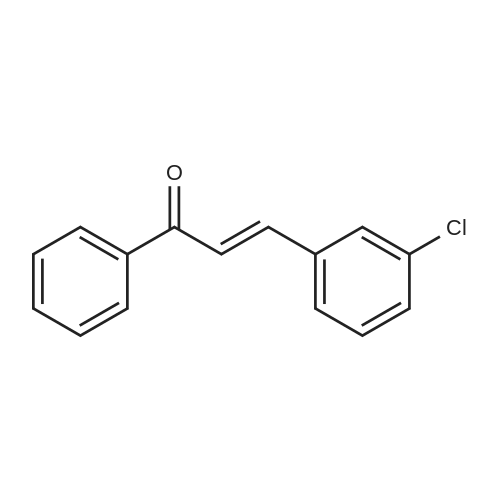
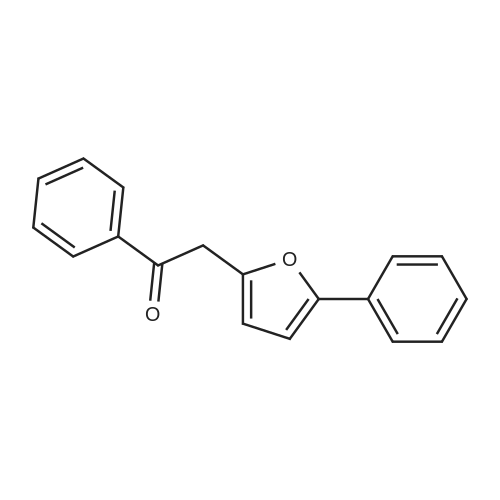
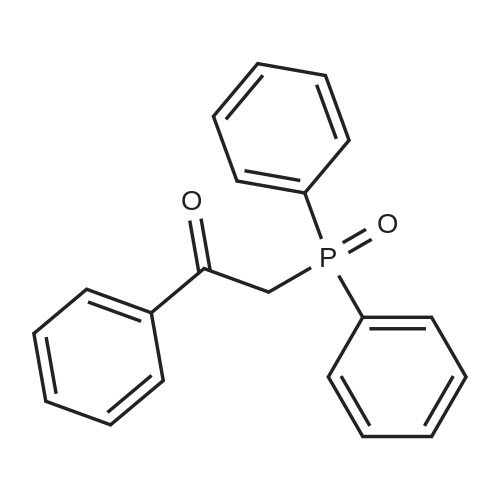
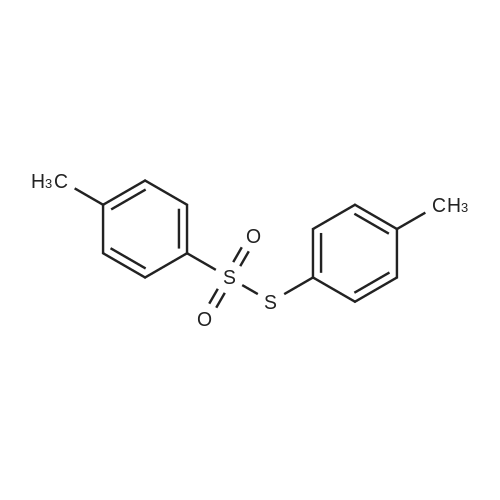
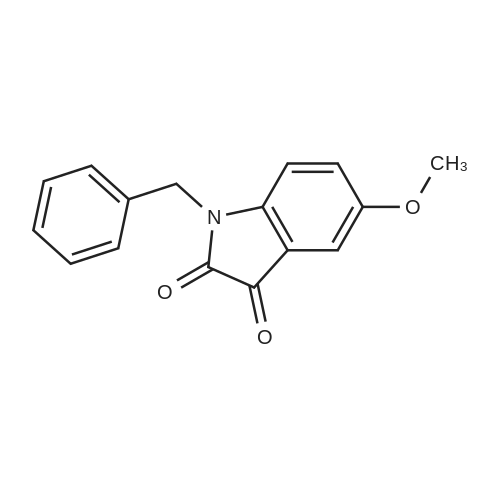
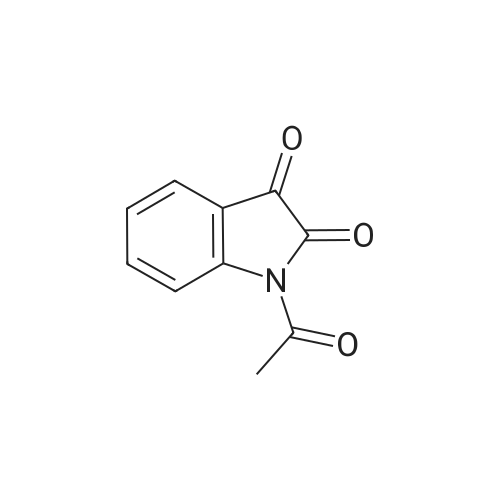
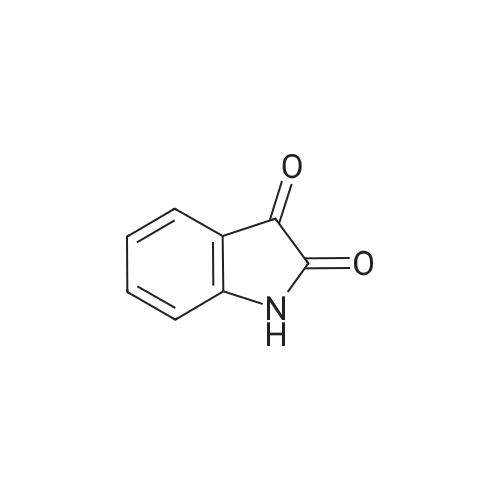
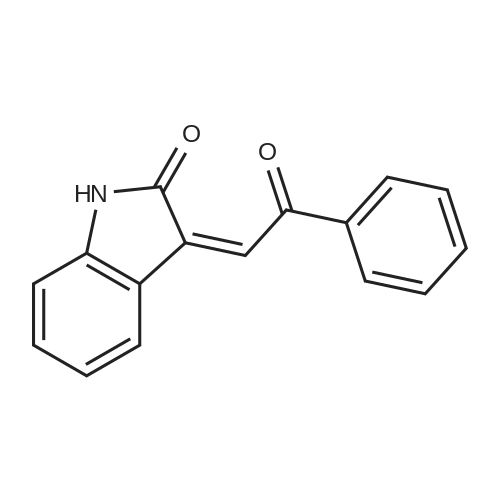
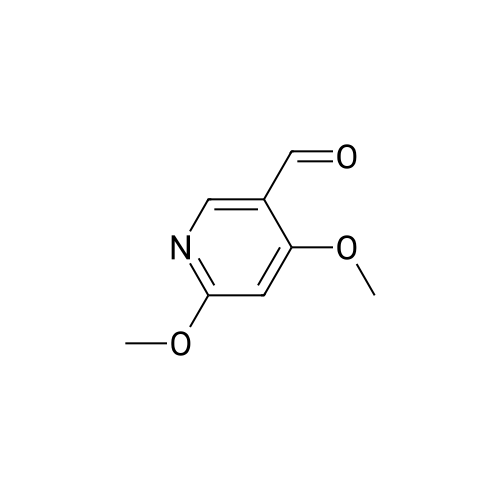
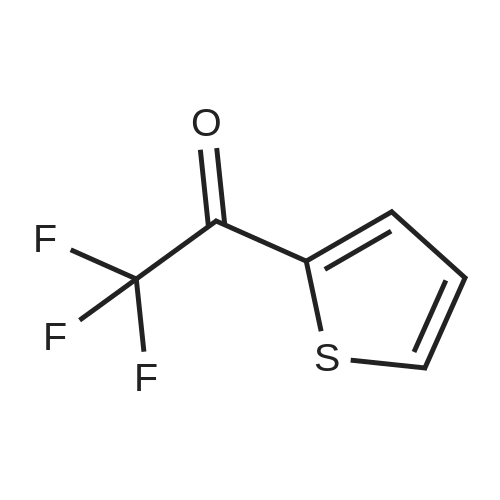
| 90% |
With sodium hydroxide In methanol at 20℃; |
|
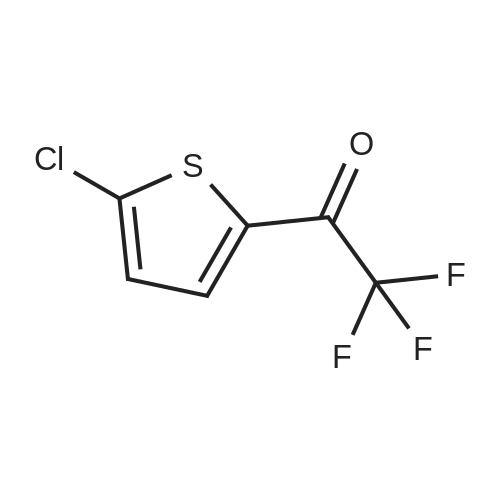
| 73% |
With lithium hydroxide monohydrate; sodium hydroxide at 20℃; |
General procedure for alkaline hydrolysis of phosphonium salts or phosphorus ylides in 3 M NaOH
General procedure: To a round-bottom flask (5 mL) containing phosphonium salt 1 or ylide 3 or 4 (0.5 mmol), 3 M NaOH (2 mL) was added. The resulting mixture was stirred at room temperature (1a, 1h, 1i, overnight; 1c, 3 h) or refluxed for 3-10 h (1b, 1d, 1e, 4, 3 h; 1f, 5 h; 1g, 10 h) till the phosphonium salt or ylide was consumed as monitored by TLC. Then the mixture was extracted with EtOAc (10 mL x 3), and the combined extract was evaporated to remove the solvent. The residue was dried in vacuo or further isolated by column chromatography or preparative TLC to give phosphine oxides 2 or Ph3PO. |
| 56% |
With sodium hydroxide In lithium hydroxide monohydrate |
|
| 5.8% |
With anhydrous sodium carbonate for 15h; |
|
|
With anhydrous sodium carbonate In lithium hydroxide monohydrate |
|
| 7.39 g |
With sodium hydroxide In methanol; lithium hydroxide monohydrate for 1h; |
|
|
With anhydrous sodium carbonate In dichloromethane; lithium hydroxide monohydrate |
|
|
With anhydrous sodium carbonate In dichloromethane; lithium hydroxide monohydrate at 20℃; for 16h; |
|
|
With sodium hydroxide In methanol; lithium hydroxide monohydrate at 20℃; for 1h; |
|
|
In methanol; lithium hydroxide monohydrate at 20℃; |
|
|
With sodium hydroxide In methanol; lithium hydroxide monohydrate for 5h; |
7.1.2 Procedure for the preparation of compounds 9a, 9d-l [24,25]
General procedure: Solution of substituted bromoacetophenone (1.0 equivalent) in methylene chloride (10 mL) was added to a solution of triphenylphosphine (1.2 equivalent) in methylene chloride (10 mL) under nitrogen. The mixture was stirred overnight and then added 100 mL ether. After stirred for 1 h, the resulting phosphonium salt was filtered and the precipitate was washed with ether and dried under vacuum. The dried phosphonium salt was suspended in a mixture of water (50 mL) and methanol (50 mL), and the mixture was stirred for 1 h. Aqueous sodium hydroxide (2 M) was added to the mixture until pH reached between 7 and 8. The mixture was then stirred vigorously for 5 h. After evaporated methanol, the aqueous layer was extracted with methylene chloride. Organic layer was dried over Na2SO4 and evaporated to obtain the compounds 9a, 9d-l. |
|
With sodium hydroxide In methanol; lithium hydroxide monohydrate |
|
| 2.8 g |
With sodium hydroxide In dichloromethane; lithium hydroxide monohydrate for 2h; |
|
|
With sodium hydroxide In lithium hydroxide monohydrate |
Typical procedure forsynthesis of (4-(Alkyl)-1H-pyrrol-3-yl)(aryl)methanone
General procedure: A mixture of phenacyl bromide (5.0 mmol), triphenylphosphine (5.0 mmol) in toluene (20 mL) was stirred at room temperature. The precipitate formed was filtered and washed with toluene. It was dissolved in water and the aqueous layer was treated with aqueous sodium hydroxide (1N). It was then extracted with DCM (2x20 mL). The combined organic layer was dried using sodium sulfate |
| 9.03 g |
With sodium hydroxide In methanol; lithium hydroxide monohydrate at 20℃; for 5h; |
|
|
With potassium carbonate In acetonitrile for 3h; |
|
|
With sodium hydroxide In dichloromethane; lithium hydroxide monohydrate; ethyl acetate |
General procedure for the syntheses of trifluoromethyl-/cyclopropyl-substituted enones 2
General procedure: α-Bromo ketone (12 mmol) was added to the solution of PPh3 (14.4 mmol) in toluene (24 mL) in one portion. Then the mixture was stirred at room temperature for 12h. After filtration, the precipitated phosphonium bromide was mixed with CH2Cl2/EtOAc (20 mL, 1:3) and stirred. To this stirred suspensions, NaOH (2N, aq.) was added slowly until all solids disappeared. Then the reaction mixture was diluted with CH2Cl2 (30 mL), washed with water (3×10 mL) and dried over anhydrous magnesium sulfate. The solvent was evaporated and phosphorus ylide reagent was obtained. After that, trifluoromethyl cyclopropyl ketone (10 mol) was mixed with phosphorus ylide reagent (12 mol) in CH2Cl2 (10 mL) under refluxing. The completion of the reaction was monitored by 19F NMR. After completion of the reaction, solvent was evaporated and the residue was purified by column chromatography on silica gel to afford the desired α, β-unsaturatedketone 2 as product. Both E- and Z-type α, β-unsaturated ketones were formed in the reaction. |
| 15.67 g |
With sodium hydroxide In dichloromethane; lithium hydroxide monohydrate for 0.166667h; |
|
|
With sodium hydroxide In lithium hydroxide monohydrate for 0.5h; Inert atmosphere; Schlenk technique; |
|
|
With sodium hydroxide In dichloromethane; lithium hydroxide monohydrate at 20℃; for 16h; |
|
|
With sodium hydroxide In dichloromethane; lithium hydroxide monohydrate at 20℃; Inert atmosphere; |
|
|
With potassium-t-butoxide In tetrahydrofuran at 20℃; for 0.5h; |
62.2 Step 2: Compound 62-c
At 20°C., potassium tert-butoxide (3.65 g, 32.52 mmol, 1.50 eq) was added into a solution of Compound 62-b (10.00 g, 21.68 mmol, 1.00 eq) in tetrahydrofuran (100 mL). The mixture was stirred at 20°C. for 0.5 h. The reaction solution was filtered, and the filtrate was extracted with ethyl acetate (100 mL3). The combined organic phase was washed with water (100 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to give Compound 62-c . MS m/z (ESI): 381.1 [M+1]. 1H NMR (400 MHz, CDCl3) δ ppm 8.01 (d, J=7.2 Hz, 2H), 7.83 (t, J=5.2 Hz, 4H), 7.51-7.49 (m, 2H), 7.48-7.38 (m, 13H). |
|
With sodium hydroxide In dichloromethane at 20℃; for 0.5h; |
|
|
With anhydrous sodium carbonate In lithium hydroxide monohydrate |
6.2. Common procedure for the synthesis of acenaphthenone-2-ylidene ketones (3a-f) by Wittig’s reaction
General procedure: A mixture of corresponding triphenylphosphonium bromide ( 8a-f , 7.0 g) and 10% aqueous sodium carbonate (250 mL) was well mixed for 15h. The mixture was filtered and insoluble portion was taken up in hot benzene (200 mL). Some unreacted bromide was removed by filtration; addition of petroleum ether to the benzene filtrate afforded the compound 9a-f (58-65%) as white powder. |
|
With sodium hydroxide In dichloromethane; lithium hydroxide monohydrate |
4.4. General procedure for the preparation of 3h-3j
General procedure: The synthesis of 3h, 3i, 3j was accomplished via the following route according to reported procedures [10]. A solution of aryl-substituted 2-bromoethanone (2.0 mmol) and PPh3 (2.2 mmol) in THF (6 mL) was refluxed for 4 h. Upon cooling, volatiles were removed, the solid was re-dissolved in DCM and extracted with aq. NaOH (20% w/w in H2O), washed with water, dried over MgSO4, filtered and evaporated to dryness. The crude product was purified by silica gel chromatography (EtOAc/n-hexane=2/1) to afford the pure product. |
|
With sodium hydroxide In methanol; lithium hydroxide monohydrate for 3h; |
|
|
With sodium hydroxide In dichloromethane; lithium hydroxide monohydrate at 20℃; for 3h; |
|
|
With lithium hydroxide monohydrate; anhydrous sodium carbonate In dichloromethane at 20℃; Inert atmosphere; |
|
|
In lithium hydroxide monohydrate Cooling; Alkaline conditions; |
3-Methyl-1-(triphenyl-λ6-phosphonylidene)butan-2-one (5a).
General procedure: A solution of 1-bromo-3-methylbutan-2-one (3a) [46] was added to a stirred solution of 2.63 g(0.01 mol) of triphenylphenylphosphine in 15 mLof benzene. The mixture was left to stand at roomtemperature until a precipitate formed, after which itwas heated for 1 h, and the precipitate of phosphoniumsalt 4a was fi ltered off. (3-Methyl-2-oxobutyl)triphenylphosphoniumbromide was washed with a little ofbenzene and dried. 1H NMR spectrum (400 MHz,DMSO-d6), δ, ppm: 1.09 d (6H, CH3, J 6.9 Hz),2.92 heptet (1H, CH, J 6.7 Hz), 5.83 d (2H, CH2, J12.7 Hz), 7.65-7.94 m (15Harom). Salt 4a was thendissolved in water, and the solution was brought to pH8 by adding 10% KOH on cooling. Compound 5 thatprecipitated was fi ltered off and dried in a vacuum. |
|
With sodium methoxide In methanol at 0℃; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping