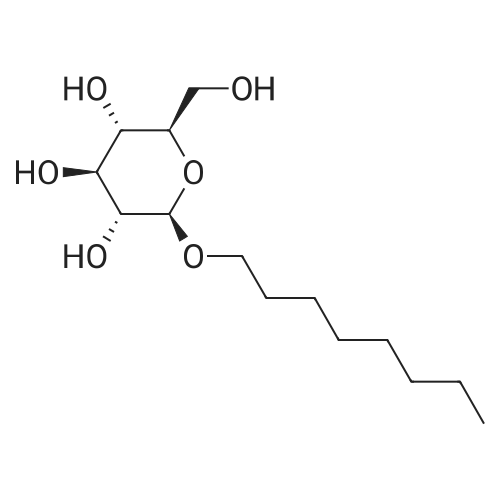
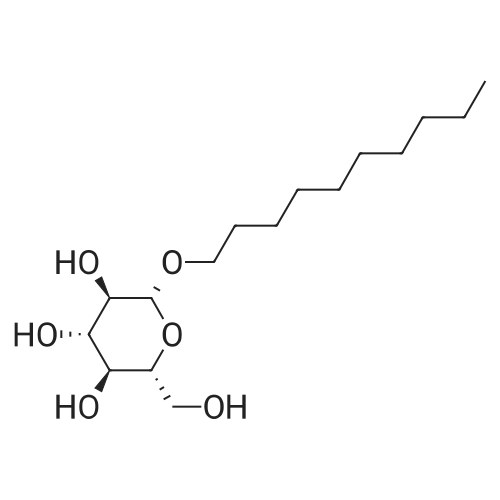
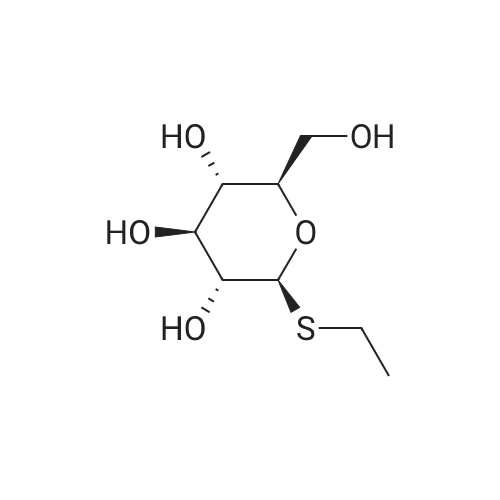
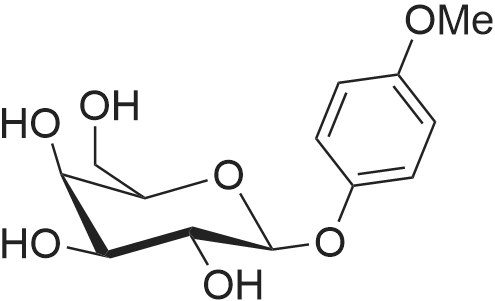
| 100% |
With hydrogen bromide In acetic acid at 20℃; for 4h; |
|
| 100% |
With hydrogen bromide; acetic acid at 0 - 20℃; |
|
| 100% |
With hydrogen bromide-acetic acid In chloroform at 20℃; for 4.5h; Inert atmosphere; |
|
| 99% |
With hydrogen bromide; acetic acid at 20℃; for 0.5h; |
|
| 98.6% |
With hydrogen bromide; acetic acid at 0 - 20℃; |
3 Example 3 Preparation of Compound 5
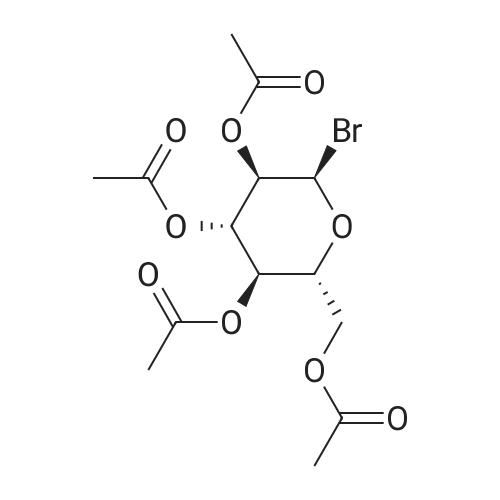
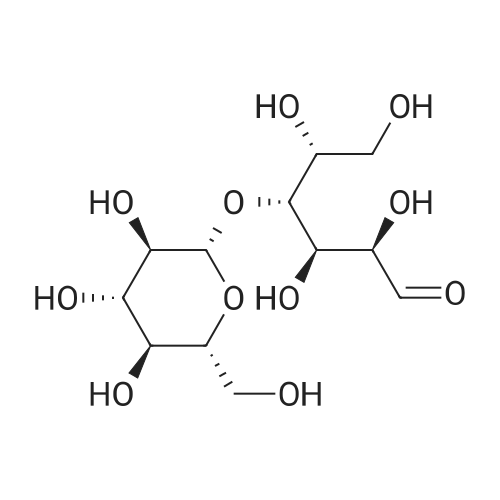
Dissolve pentaacetyl-β-D-glucose in dry dichloromethane and add HBr-AcOH (33% W/0°C)W) solution, the final mixture was stirred at 0°C for 30 min, allowed to stir at room temperature overnight, and the reaction mixture was poured into ice water.After extraction, the organic layer was extracted with aqueous NaHCO 3 solution and brine, dried, and concentrated to give compound 5 in a yield of 98.6%. |
| 97% |
With hydrogen bromide; acetic acid In dichloromethane at 0 - 20℃; for 3h; |
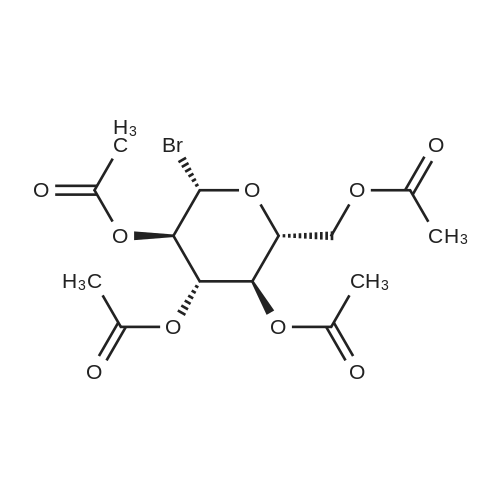
To a 500 ml flask was added 50 g of glucose pentaacetate (C6H22O11) and 80 ml of methylene chloride. The mixture was stirred at ice-water bath for 20 min HBr in HOAc (33%, 50 ml) was added to the reaction mixture. After stirring for 2.5 hr another 5 ml of HBr was added to the mixture. After another 30 min, the mixture was added 600 ml of methylene chloride. The organic mixture was washed with cold water (200 ml ×2), Saturated NaHCO3(200 ml×2), water (200 ml) and brine (200 ml ×2). The organic layer was dried over Na2SO4 and the mixture was evaporated at RT to give white solid as final product, bromide derivative, IntD1 (95% yield). C14H19BrO9, TLC Rf=0.49, SiO2, 40% ethyl acetate /60% hexanes ; Exact Mass 410.02. |
| 96% |
With hydrogen bromide In acetic acid at 0 - 20℃; for 18h; |
1.a
p-D-glucose pentaacetate (33.0 g, 84.5 mmol, ALDRICH)was reacted under nitrogen with a 30% solution of HBr inacetic acid (70 mL, 351 mmol ) at 0°C. After completion ofaddition, the reaction mixture was warmed to roomtemperature and stirred for 18 h. TLC (40% EtOAc/Petrol)indicated the disappearance of starting material. Thereaction mixture was then poured into ice and extractedthree times with CH2C12. The combined organic extracts werecarefully washed with saturated aqueous NaHCOa (3x), brine(2x) , dried (MgS04) , filtered and concentrated to give the a-bromide 6 as a white solid (34.7 g, 96%) .1H NMR (300 MHz) § 2.04 (s, 3H, OAc), 2.05 (s, 3H, OAc), 2.10(s, 3H, OAc), 2.10 (s, 3H, OAc), 4.09-4.36 (m, 3H, H5-H6),4.84 (dd, J = 3.9 Hz, 1H, H2), 5.16 (t, J= 9.6 Hz, 1H, H3),5.56 (t, J= 9.9 Hz, 1H, H4), 6.61 (d, J= 4.2 Hz, 1H, Hi) |
| 96% |
With hydrogen bromide; acetic acid In dichloromethane at 0 - 20℃; for 6h; |
4.2.1. 3,4,6-Tri-O-allyloxycarbonyl-D-glucal (7)
General procedure: β-D-Glucose pentaacetate 5 (10 g, 25.6 mmol) was dissolved in dryCH2Cl2 (60 mL) and hydrogen bromide, 33% w/w (45% w/v) solutionin acetic acid (4.9 mL) was added dropwise at 0 °C. The mixture wasthen allowed to warm to room temperature and stirred for 6 h. Thereaction mixture was diluted with CH2Cl2 (150 mL) and washed successivelywith saturated aqueous solution of NaHCO3 (80 mL). Theorganic layer was drying over Na2SO4 and concentrated to give 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide (10.1 g, 96%) as a colorlesssyrup. Rf = 0.60 (PE: EtOAc, 2:1). To a solution of 2,3,4,6-tetra-Oacetyl-α-D-glucopyranosyl bromide (5.0 g, 12.16 mmol) dissolved inacetone was added zinc power (7.63 g, 117 mmol) and saturatedNaH2PO4 solution (30 mL). The mixture was stirred overnight at roomtemperature and the mixture was filtered and concentrated. The resultingmixture was diluted with CH2Cl2 and washed successively withsaturated aqueous solution of NaHCO3 and then dried over Na2SO4 andconcentrated to afford 3,4,6-tri-O-acetyl-D-glucal (3.1g, 94%).Rf = 0.30 (petroleum ether: EtOAc, 5:1). To a solution of 3,4,6-tri-Oacetyl-D-glucal (2 g, 7.35 mmol) in dry MeOH (15 mL) was addedNaOMe (159 mg, 2.94 mmol) and stirred for 2 h. The reaction mixturewas neutralized with amberlite IR120 resin, filtered and concentratedin vacuo to obtain D-glucal 6 (1 g, 93%). Rf = 0.25 (CH2Cl2: MeOH,10:1). To a solution of D-glucal 6 (2 g, 13.69 mmol) in anhydrouspyridine (20 mL), allyl chloroformate (17.5 mL, 12 equiv.) was addeddropwise at 0 °C. The mixture was stirred for 12 h at room temperature.The residue was diluted with CH2Cl2 and washed first with 1.0 M HCland then saturated aqueous NaHCO3. The organic layer was dried overNa2SO4 and concentrated. The residue was purified by flash chromatography(petroleum ether: EtOAc, 20:1) to give compound 7 (2.2 g,40%) as yellow syrup. Rf = 0.8 (petroleum ether: EtOAc, 2:1); [α][20]D-23.3 (c 0.24, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.46 (d, 1H,J = 5.6 Hz), 5.96-5.82 (m, 3H), 5.38-5.28 (d, 3H, J = 17.2 Hz),5.28-5.15 (m, 4H), 5.07 (s, 1H), 4.92 (s, 1H), 4.65-4.55 (m, 6H),4.49-4.41 (m, 1H), 4.38-4.32 (s, 1H), 4.32-4.24 (m, 1H). 13C NMR(101 MHz, CDCl3) δ 154.48, 154.01, 153.60, 145.86, 131.27, 131.23,130.97, 119.29, 118.97, 118.90, 97.79, 73.27, 70.62, 69.94, 69.07,68.68, 68.64, 64.56. HRMS m/z calcd for C18H22O10Na [M+Na]+:421.1111, found: 421.1086. |
| 94% |
With hydrogen bromide; acetic acid In dichloromethane at 20℃; for 2h; Inert atmosphere; |
|
| 94% |
With hydrogen bromide In dichloromethane; acetic acid at 0 - 20℃; Inert atmosphere; |
2,3,4,6-Tetra-O-acetyl-α-D-glucopyranosyl bromide 8
Hydrogen bromide (33% in acetic acid, 25 mL) was added dropwise to a solution of β-d-glucose pentaacetate 6 (4.90 g, 12.6 mmol) in dry dichloromethane (50 mL) at 0 °C under argon. The reaction was allowed to warm to RT and stirred for 2 h, after which TLC analysis (1:1, cyclohexane/ethyl acetate) indicated the complete consumption of the starting material (Rf 0.30) and the formation of a single product (Rf 0.60). The reaction mixture was poured onto ice-water (200 mL) and extracted with cold dichloromethane (2 * 100 mL). The organic fractions were combined, washed with a saturated sodium bicarbonate solution (3 * 150 mL), a saturated brine solution (150 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was recrystallized (diethyl ether/cyclohexane) to give the bromide 8 as a white crystalline solid (4.75 g, 94%); m.p. 86-89 °C (diethyl ether/cyclohexane) {Lit. ( Weygand et al., 1958 ) 88-89 °C (diethyl ether/pet. ether)}; +186.1° (c 2.0, CHCl3) {Lit. (Weygand et al., 1958) +194° (c 3.94, CHCl3)}; ν cm-1: 1759 (s, >C=O); 1H NMR spectral data (400 MHz, CDCl3): δ 2.04, 2.05, 2.10, 2.10 (4 * 3H, s, COCH3), 4.14 (1H, dd, H-6a, J6a,5 = 2.0, J6a,6b = 12.5), 4.29 (1H, ddd, H-5, J5,6a = 2.0, J5,6b = 4.2, J5,4 = 9.4), 4.34 (1H, dd, H-6b, J6b,5 = 4.2, J6b,6a = 12.5), 4.84 (1H, dd, H-2, J2,1 = 4.0, J2,3 = 9.9), 5.16 (1H, a-t, H-4, J4,5 = J4,3 = 9.7), 5.56 (1H, a-t, H-3, J3,4 = J3,2 = 9.7), 6.61 (1H, d, H-1, J1,2 = 4.0); 13C NMR spectral data (100 MHz, CDCl3): δ 20.5, 20.6, 20.6, 20.6 (COCH3), 60.9 (C-6), 67.1 (C-4), 70.1 (C-3), 70.6 (C-2), 72.1 (C-5), 86.5 (C-1), 169.4, 169.8, 169.8, 170.5 (COCH3); HRMS (ESI): found 433.0110 [M+Na]+; C14H19BrNaO9+ requires 433.0110; EIMS (probe) eV, m/z (rel. int.): 433 (100% [M+Na]+). |
| 93.5% |
With water monomer; phosphorus tribromide In dichloromethane at 30℃; for 10h; |
|
| 93% |
With black phosphorus; bromine In dichloromethane at 20℃; |
|
| 93.7% |
With hydrogen bromide; acetic acid In dichloromethane at 5 - 25℃; for 6h; |
1-3
Add 100.0g of β-D-glucose pentaacetate to 400ml of dichloromethane and reduce the temperature to below 5°C; when the temperature is controlled not to exceed 5°C, add 125.6g of 33% hydrobromic acid acetic acid and heat up to Stir at 15~25°C for 6h.After TLC monitors the raw material SM1 reaction is complete.The reaction solution was added to 800 mL of saturated aqueous sodium bicarbonate solution at 5°C and stirred for 10 min, and the layers were separated.The aqueous phase was extracted once with 400 ml of DCM, and the organic phases were combined and washed with 400 ml of aqueous solution.The organic phase was dried with 40 g of anhydrous sodium sulfate, filtered, and the filtrate was distilled under reduced pressure to obtain a light yellow transparent oil.200ml of ethanol was added to the oil, stirred at room temperature for 20min, filtered, and the filter cake was washed with 100ml of n-hexane, and the filter cake was dried under reduced pressure at 20°C for 3h to obtain 98.7g of a white solid.The molar yield is 93.7%. |
| 93% |
With triethylsilane; bromine In dichloromethane at 0℃; for 2h; |
|
| 92% |
With hydrogen bromide; acetic acid In dichloromethane at 0℃; for 3h; |
1 Intermediate 1-7:
To a solution of b-D-glucose pentaacetate (5.0 g, 12.8 mmol) in DCM (30 mL) at 0 °C, was added hydrobromic acid solution in acetic acid (8 mL). Stirring was continued at 0 °C until complete conversion of starting material (about 3 h). The reaction mixture was quenched with ice water (200 mL), and extracted with DCM (3x80 mL). The organic layer was combined and washed with ice water (3x80 mL), saturated NaHCCL, and brine, dried over NaiSCri. The mixture was filtered and concentrated to provide 2,3,4,6-Tetra-O-acetyl-a-D- glucopyranosyl bromide as intermediate 1-7 (4.85 g, 11.8 mmol, 92%) as a white solid. |
| 91% |
With bromine In ethyl acetate at 30℃; for 23h; Irradiation; Green chemistry; |
2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide (2a)
General procedure: Bromine (1.5 mmol, 0.08 mL) was added slowly to a magnetic stirring barand perfluorohexanes (4.0 mL) in a test tube (14 mmφ x 105 mm) with a septum and then 1-O-acetylsugar 1a (1 mmol, 392 mg) in ethyl acetate (2.0 mL) wasadded slowly, forming three layers. The test tube was stirring upon irradiationwith 15 W black light (at 352 nm, TOSHIBA EFD15BLB-T) at 30 C. The light source wasplaced away from the test tube. After 23 hours, the bromine layer disappearedand the fluorous layer recovered transparency. The ethyl acetate layer wastaken up with a pipette. Then, additional ethyl acetate (2 mL x 4) was placedon the residual FC-72 layer, followed by decanting off. The combined ethylacetate layer was washed with water (15 mL), aqueous sat. NaHCO3 (20mL), brine (20 mL) and, dried over Na2SO4, andconcentrated. Purification by chromatography on silica gel with hexane/AcOEt = 2/1gave glycosyl bromide 2a (0.91 mmol,374 mg) in 91% yield |
| 91% |
With hydrogen bromide; acetic acid at 20℃; for 1h; |
|
| 91% |
With hydrogen bromide; acetic acid at 0 - 20℃; for 1h; |
|
| 91% |
With hydrogen bromide; acetic acid In dichloromethane at 0 - 20℃; for 2h; |
2,3,4,6-Tetra-O-acetyl-alpha-d-glucopyranosyl bromide (II)
Compound I (24mmol) was dissolved in 25mL of dichloromethane (DCM) and the solution was cooled down to 0°C. A solution of HBr (33%) in glacial acetic acid (25mL) was added to the cooled solution and the reaction mixture was warmed up to ambient temperature under magnetic stirring. After consumption of starting materials which was indicated by TLC test, the mixture was quenched with ice water (50mL), extracted with DCM (2×60mL), washed whit saturated solution of NaHCO3 (2×50mL), dried over sodium sulfate, filtered and concentrated in vacuum. The product was recrystallized (Hexane: ether 1:1) to obtain compound (II) as white needles, (8.98g, yield 91%). (0017) FT-IR bands in cm-1 (KBr): 1744 (C=O acetate), 550 (C-Br). 1HNMR (500MHz, CDCl3): δH 2.04, 2.05, 2.1, 2.11 (S, CH3, 12H), 4.13ppm (dd, AcOC(6)H2, 1H, J=12.2, 1.8Hz), 4.27-4.37 (m, AcOC(6)H2,C(5)H, 2H), 4.84ppm (dd, C(2)H, 1H, J=10, 3.9Hz, axial), 5.17ppm (t, C(4)H, 1H, J=9.8Hz, axial), 5.56ppm (t, C(3)H, 1H, J=9.7Hz, axial), 6.62ppm (d, C(1)H, 1H J=4Hz, equatorial). 13CNMR (500MHz, CDCl3): δC 20.5-20.7ppm (4×CH3), 60.94ppm (AcOCH2), 67.16, 70.15, 70.59, 72.13ppm (C(4), C(3), C(2), C(5)), 86.55ppm (C(1)), 169.43, 169.76, 169.82, 170.47ppm (4×CO). |
| 90% |
With hydrogen bromide In acetic acid for 12h; Ambient temperature; |
|
| 90% |
With hydrogen bromide; acetic acid at 30℃; for 2h; |
|
| 90% |
With hydrogen bromide; acetic acid In dichloromethane at 20℃; for 12h; Darkness; |
1.2.2 α-acetobromoglucose (17)
To a solution of 15 (13.02 g, 0.0334 mol) in 65ml CH2Cl2 was added 25 ml of 30 % hydrogen bromide in acetic acid. The reactionwas stirred under darkness at room temperature. After 12 hours the reaction wasdiluted with 100 ml CH2Cl2 and poured on ice. After the ice had melted, theorganic and aqueous layers were separated and the organic layer was washed withtwo portions of 50 ml cold distilled water, two portions of 75 ml coldsaturated sodium bicarbonate, then with two portions of 50 ml brine. Theorganic layer was dried over sodium sulfate and concentrated in-vacuo toobtain 17 as a pale yellow syrup (12.35 g, 0.0300 mol, 90 % yield).1H NMR (400 MHz, CHLOROFORM-d) δ ppm2.01 (s, 3 H), 2.03 (s, 3 H), 2.07 (s, 3 H), 2.08 (s, 3 H), 4.04 - 4.14 (m, 1H), 4.22 - 4.36 (m, 2 H), 4.81 (dd, J=9.96, 4.10 Hz, 1 H), 5.13 (t, J=9.96Hz, 1 H), 5.28 (s, 1 H), 5.45 - 5.59 (m, 1 H), 6.58 (d, J=3.91 Hz, 1 H).13C NMR (300 MHz, CHLOROFORM-d) δ ppm20.74 (s, 1 C), 20.80 (s, 1 C), 20.83 (s, 1 C), 20.85 (s, 1 C), 61.15 (s, 1 C),67.35 (s, 1 C), 70.36 (s, 1 C), 70.78 (s, 1 C), 72.34 (s, 1 C), 86.80 (s, 1 C),169.64 (s, 1 C), 169.96 (s, 1 C), 170.02 (s,1C), 170.68 (s, 1 C). |
| 90% |
With hydrogen bromide; acetic acid In dichloromethane at 0℃; for 2h; |
Synthesis of S2.
S1 (2.00 g, 4.86 mmol) was dissolved in DCM (2 mL) at 0 °C for 15 min. Then hydrogen bromide (HBr/AcOH, 33 wt.% solution in acetic acid, 20 mL) was dropped into this mixture, stirring 2 h. After this reaction the mixture was added DCM (50 mL) then washed by NaHCO3 saturated solution and brine three times respectively. The organic phase was dried by MgSO4 and condensed to get S2 (1.97 g, 90%). |
| 86% |
With hydrogen bromide; acetic acid at 0 - 20℃; for 0.75h; |
|
| 86% |
With hydrogen bromide; acetic acid In dichloromethane at 20℃; for 0.125h; Flow reactor; |
Flow synthesis of 2,3,4,6-tetra-O-acetyl--D-glucopyranosyl bromide (2)
A solution of β-D-glucose pentaacetate (1, 1 M in dry CH2Cl2, 1 equiv, 107 L/min) and asolution of HBr (33% in glacial acetic acid, 5 equiv, 93 L/min) were mixed and pumped through flow reactor R3 with a total reactor volume of 1.5 mL at rt, resulting in a total flow rate of 200 L/min and a residence time of 7.5 min. The reaction was quenched by pumping water through a third inlet with a flow rate of 200 L/min. The biphasic mixture was passed through a CSTR (V = 1 mL) with an in-printed stirring bar. The phases were separated under flow conditions in a 10 mL syringe and the organic phase was passed through a second syringe containing a NaHCO3-solution for neutralization. Through an outlet in the cap, the excess water and formed CO2 was removed. The organic phase was collected, dried over Na2SO4, filtered and concentrated in vacuo. The yellowish residue was washed with a mixture of PE and EtOAc (1:1) to afford glucopyranosyl bromide 2 as a white solid with a yield of 86% based on the steady state. Rf = 0.71 (PE:EtOAc, 1:1). 1H NMR (400 MHz, CDCl3): 6.61 (d, J1,2 = 4.0 Hz, 1H, 1-H), 5.57 (app. t, J = 9.7 Hz, 1H,3-H), 5.17 (app. t, J = 10.3 Hz, 1H, 4-H), 4.85 (dd, J2,3 = 10.0 Hz, J1,2 = 4.0 Hz, 1H, 2-H) 4.36- 4.28 (m, 2H, 5-H, 6-Ha), 4.13 (d, J6b,6a = 10.0 Hz, 6-Hb), 2.11, 2.10, 2.06, 2.04 (4 s, 12 H, 4× COCH3) ppm.13C NMR (100 MHz, CDCl3): 170.5, 169.8, 169.8, 169.4 (4 × COCH3), 86.5 (C1), 72.1(C5), 70.6 (C2), 70.1 (C3), 67.1 (C4), 60.9 (C6), 20.6, 20.6, 20.6, 20.5 (4 × COCH3) ppm.Spectroscopic data are in agreement with reported literature.[1] |
| 85% |
With hydrogen bromide In acetic acid |
|
| 85% |
With water monomer; hydrogen bromide; acetic acid at 0℃; for 4h; |
|
| 85% |
With hydrogen bromide; acetic acid at 20℃; for 2h; |
1.2
(Example 1-2) Synthesis of intermediate 4 for glucose derivative () Acetobromo-α-D-glucose (4) β-D-Glucose pentaacetate (2.5 g, 6.41 mmol) was added toHBr-AcOH (30%) with penetrating. The reactionmixture was stirred at room temperature for 2 hours. The resultant reaction mixture was diluted with CHCl3 (50 mL), and the mixture was poured into ice water (15 mL). The chloroform layer was separated into an aqueous layer and a chloroform extraction layer (2*20 mL of CHCl3). The chloroform extraction layer was combined with the chloroform layer, and the mixture was then washed with water and dried over MgSO4, to thereby obtain acetobromo-a-D-glucose (4). The solvent was removed under reduced pressure, and the residue was recrystallized from a mixed solution of AcOEt and n-hexane, to thereby obtain a colorless needles 4 (2.23 g, 85%). Melting point (m.p.) 89-90°C (lit. 88-89°C). Rf=0.25 (AcOEt:n-hexane, 1:2, v/v). 1HNMR (CDCl3, 300 MHz) δ 2.04, 2.06, 2.10, and 2.11 (each s, 12H, 4*COCH3), 4.13 (br d, Jgem=10.5 Hz, 1H, 6'-Hb), 4.27-4.37 (m, 2H, 5',6'-Ha), 4.84 (dd, J3',4'=9.6 Hz, J2',3'=9.9 Hz, 1H, 3'-H), 5.16 (t, J2',3,=9.9 Hz, 1H, 2'-H), 5.56 (t, J3',4'=9.6 Hz, 1H, 4'-H), 6.61 (d, J1',2'=4.2 Hz, 1H, 1'-H). |
| 84% |
With water monomer; phosphorus tribromide In chloroform at 20℃; for 2h; |
|
| 84% |
With hydrogen bromide; acetic acid In dichloromethane at 20℃; for 2.5h; Inert atmosphere; |
|
| 83% |
With hydrogen bromide; acetic anhydride; acetic acid Inert atmosphere; |
|
| 80% |
With hydrogen bromide; acetic acid In dichloromethane at 20℃; for 2h; Inert atmosphere; |
|
| 80% |
With Acetyl bromide; acetic acid In methanol at 0 - 20℃; for 1h; |
Synthesis of acetobromo-a-D-glycosides [14] 1 and 2
General procedure: To a solution of β-D-glucose pentaacetate (5.00 g, 0.0128 mol) or β-D-galactose pentaacetate (5.00 g, 0.0128 mol) in acetic acid (30.0 mL) was cooled to 0 °C. Then, methanol (0.68 mL, 0.0166 mol) and acetyl bromide (2.84 mL, 0.0384 mol) were added dropwise, respectively. The resulting mixture was stirred at room temperature for 1.0 h. After TLC (30% EtOAc/n-hexane) showed the completed conversion. The reaction mixture was quenched with cooled aq. NaHCO3 (20 mL) and extracted with EtOAc (3 100 mL). The combined organic layer was washed with brine, then dried with anh. Na2SO4 and then concentrated under reduce pressure. The crude product was purified by column chromatography (30% EtOAc/n-hexane) to give acetobromo-α-D-glucose 1 (4.2138 g, 80%) as a white solid or acetobromo-α-D-galactose 2 (4.5158 g, 86%) as a white solid, respectively. Acetobromo-α-D-glucose (1) [14] 80% yield (4.2138 g) as a white solid; CAS No. 572-09-8; Rf = 0.54 (40% EtOAc/n-hexane); 1H NMR (400 MHz, CDCl3): δ 6.61 (d, 1H, J = 4.0 Hz), 5.56 (t, 1H, J = 9.6 Hz), 5.16 (d, 1H, J = 10.0 Hz), 4.84 (dd, 1H, J = 10.0, 4.4 Hz), 4.30 (td, 2H, J = 12.4, 4.0 Hz), 4.13 (d, 1H, J = 10.8 Hz), 2.10 (s, 3H), 2.09 (s, 3H), 2.05 (s, 3H), 2.03 (s, 3H). |
| 78% |
With hydrogen bromide; acetic acid at 0 - 20℃; for 1.5h; |
|
| 75% |
With hydrogen bromide In dichloromethane; acetic acid at 0 - 20℃; Inert atmosphere; |
|
| 72.3% |
With black phosphorus; bromine; acetic acid at 20℃; |
|
| 70% |
With hydrogen bromide; acetic acid at 20℃; |
|
| 70% |
With hydrogen bromide; acetic acid at 20℃; Inert atmosphere; |
|
| 65% |
With hydrogen bromide In chloroform for 3h; Ambient temperature; |
|
| 55% |
With hydrogen bromide; acetic acid In dichloromethane at 20℃; for 4h; |
2 Example 2 Preparation of α-D-bromotetraacetylpyranose
Add 10 g of peracetylglucose to a 250 ml single-mouth flask.Using 100 ml of CH2Cl2 as a solvent,Add 40 ml of 33% hydrogen bromide acetic acid solution under ice bath.After reacting at room temperature for 4 h, after the reaction is completed, ice water is added, stirred, and allowed to stand for stratification.The lower organic layer was separated using a separating funnel, and the aqueous layer was extracted twice with CH2Cl2.The CH2Cl2 layer was combined and washed successively with saturated NaHCO3 solution and water.The solvent was evaporated, and the crude material was crystallised from diethyl ether. Yield 55% |
| 55% |
With hydrogen bromide; acetic acid In dichloromethane at 20℃; for 4h; Cooling with ice; |
2 Example 2 Preparation of α-D-Bromotetraacetylpyranose
Add 10 g of peracetylglucose to a 250 ml single-mouth flask.Using 100 ml of CH2Cl2 as a solvent,40 ml of 33% hydrogen bromide acetic acid solution was added under ice bath, and reacted at room temperature for 4 h.After the reaction is completed, ice water is added, stirred, and allowed to stand for stratification.The lower organic layer was separated by a separating funnel, and the aqueous layer was extracted twice with CH2Cl2, and the CH2Cl2 layer was combined.Wash with saturated NaHCO3 solution and water in turn,The solvent was evaporated, and the crude material was crystallised from diethyl ether.The yield was 55%. |
|
With Acetyl bromide; acetic acid |
|
|
With hydrogen bromide |
|
|
With hydrogen bromide In acetic acid |
|
|
With triethylsilane; carbon tetrabromide; palladium (II) chloride In dichloromethane at 20℃; for 2h; |
|
|
With hydrogen bromide; acetic acid at 0℃; for 2.4h; |
|
|
With hydrogen bromide; acetic acid |
|
|
With hydrogen bromide; acetic acid |
|
|
With hydrogen bromide; acetic anhydride; acetic acid at 20℃; for 1.5h; |
|
|
With hydrogen bromide; acetic acid at 20℃; |
|
|
Multi-step reaction with 2 steps
1: 89 percent / BF3*Et2O / CH2Cl2 / 7 h / 0 °C
2: 97 percent / AcBr, ZnBr2 / CH2Cl2 / 24 h / 22 °C |
|
|
With hydrogen bromide In acetic acid at 20℃; for 0.5h; |
1.1
To (?)-D-glucose penta-acetate (24.6 g, 63.0 mmol) was added a solution of hydrogen bromide in acetic acid (33 wt %, 100 ml). A dark brown color immediately appears. The reaction mixture was stirred at room temperature for 30 minutes under argon atmosphere. Subsequently the solvent was removed by azeotropic distillation in vacuo with toluene (4?50 ml), yielding a green-brown solid Compound 1.1. The crude product was used in the next reaction step without further purification. Formula: C14H19O9Br Molecular weight: 411.20 Rf: 0.46 (cyclohexane/ethyl acetate 1:1) IR (KBr): 2962, 2360, 2342, 1748, 1435, 1369, 1218, 1162, 1112, 1079, 1042, 911, 752, 668, 601, 563 cm?1 ES-MS: 433=[410+Na]+, 435=[412+Na+] 1H-NMR (500 MHz, CDCl3): ? 6.61 (1H, d, J=4.0 Hz), 5.56 (1H, dd, app. t, J=9.7 Hz), 5.16 (1H, dd, app. t, J=9.7 Hz), 4.84 (1H, dd, J=10.0, 4.0 Hz), 4.33 (1H, m), 4.30 (1H, m), 4.13 (1H, dd, J=12.3, 1.5 Hz), 2.11 (3H, s), 2.10 (3H, s), 2.05 (3H, s), 2.03 (3H, s) 13C-NMR (125 MHz, CDCl3): ? 170.37, 169.70, 169.64, 169.31, 86.34, 71.91, 70.39, 69.94, 66.94, 60.76, 20.48, 20.48, 20.38, 20.38 |
|
With hydrogen bromide; acetic anhydride; acetic acid In toluene at 18℃; for 0.5h; |
25
Example 25; Synthesis of 2,3,4,6-Tetra-O-acetyl-α-D-glucopyranosyl bromide (11.1); 1 ,2,3,4,6-Penta-O-acetyl-β-D-glucopyranose (100g, 0,251 mol) is suspended in toluene (210 ml). Acetic anhydride (9,5 ml; 0,1 mol) is added followed by hydrobromic acid 30% in acetic acid (200 ml, 1 mol). The mixture is stirred at 18°C for 30 min. A mixture of ice/water (300 ml) EPO and brine (100 ml) is then added with stirring. The phases are separated and the aqueous phase is extracted with toluene (100 ml). The organic phases are combined and washed with aqueous sodium hydrogencarbonate (100 ml) and brine (100 ml). Dyring and evaporation of the solvent under reduced pressure yields an oil which is crystallised by addition of methyl-t- butylether (150 ml) and methylcyclohexane (300 ml). The product is isolated by filtration, washed with methylcyclohexane and dried under vacuo at 45°C. |
|
With hydrogen bromide; acetic anhydride; acetic acid In toluene at 18℃; for 0.5h; |
1.1.7
Example 1.7: 2,3,4,6-Tetra-O-acetyl-α-D-glucopyranosyl bromide (11.1); 1 ,2,3,4,6-Penta-O-acetyl-β-D-glucopyranose (100g, 0,251 mol) is suspended in toluene (210 ml). Acetic anhydride (9,5 ml; 0,1 mol) is added followed by hydrobromic acid 30% in acetic acid (200 ml, 1 mol). The mixture is stirred at 18°C for 30 min. A mixture of ice/water (300 ml) and brine (100 ml) is then added with stirring. The phases are separated and the aqueous phase is extracted with toluene (100 ml). The organic phases are combined and washed with aqueous sodium hydrogencarbonate (100 ml) and brine (100 ml). Dyring and evaporation of the solvent under reduced pressure yields an oil which is crystallised by addition of methyl-t-butylether (150 ml) and methylcyclohexane (300 ml). The product is isolated by filtration, washed with methylcyclohexane and dried under vacuo at 45°C. |
|
With hydrogen bromide; acetic anhydride In acetic acid; toluene at 18℃; for 0.5h; |
25
EXAMPLE 25Synthesis of 2,3,4,6-Tetra-O-acetyl-α-D-glucopyranosyl bromide (II.1) 1,2,3,4,6-Penta-O-acetyl-α-D-glucopyranose (100 g, 0,251 mol) is suspended in toluene (210 ml). Acetic anhydride (9,5 ml; 0,1 mol) is added followed by hydrobromic acid 30% in acetic acid (200 ml, 1 mol). The mixture is stirred at 18° C. for 30 min. A mixture of ice/water (300 ml) and brine (100 ml) is then added with stirring. The phases are separated and the aqueous phase is extracted with toluene (100 ml). The organic phases are combined and washed with aqueous sodium hydrogencarbonate (100 ml) and brine (100 ml). Dyring and evaporation of the solvent under reduced pressure yields an oil which is crystallised by addition of methyl-t-butylether (150 ml) and methylcyclohexane (300 ml). The product is isolated by filtration, washed with methylcyclohexane and dried under vacuo at 45° C. |
|
With hydrogen bromide; acetic acid In dichloromethane at 0℃; for 2h; |
|
|
With hydrogen bromide; acetic acid at 20℃; Inert atmosphere; |
|
| 1.03 g |
With hydrogen bromide; acetic acid In water monomer at 20℃; for 3h; |
|
|
With bromine; acetic acid at 20℃; Cooling with ice; |
|
|
With black phosphorus; bromine; acetic acid at 20℃; |
|
| 31 g |
With black phosphorus; bromine; acetic anhydride In water monomer for 2.5h; |
|
|
With water monomer; phosphorus tribromide at 15 - 25℃; |
|
|
With Acetyl bromide; acetic acid In methanol at 20℃; for 24h; Inert atmosphere; |
|
|
With black phosphorus; bromine |
|
|
With hydrogen bromide; acetic acid |
|
|
With red phosphorus; bromine In water monomer |
|
|
With hydrogen bromide; acetic acid at 0℃; for 2h; Inert atmosphere; |
General procedure for synthesis of bromo-sugars (2f-j)
General procedure: The stirring solutions of compounds 1f-j at 0 °C were treated with 33% HBr solution in glacial acetic acid under anhydous condition. The reation mixture was further stirred for 2 h at 0 °C. After completion of reaction (monitored by TLC), the reaction mixtures were neutralized with saturated NaHCO3 solution followed by extraction in dichloromethane. The organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford compounds 2f-j. Because of low stability at room temperature, all the developed bromo-sugars 2f-j was subsequently utilized for the synthesis of sugar azides 3f-j without further purification. |
|
With hydrogen bromide; acetic acid |
|
|
With hydrogen bromide; acetic acid at 0℃; |
|
|
With hydrogen bromide In acetic acid for 3h; |
2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl azide (3)
Adapted from the method of Tropper et al.4 1,2,3,4,6-Penta-O-acetyl-β-D-glucopyranose (S1) (16.9 g, 43.3 mmol) was dissolved in 33% w/w HBr in AcOH solution (70 mL) and stirred for 3 h. The solution was diluted with DCM (400 mL), and washed with ice-cold water (3 × 400 mL), saturated aqueous sodium bicarbonate solution (2 × 200 mL), and brine (100 mL). The organic layer was concentrated to 75 mL by evaporation in vacuo, then tetrabutylammonium hydrogen sulfate (3.67 g, 10.8 mmol), sodium azide (14.0 g, 215 mmol) and saturated aqueous sodium bicarbonate solution (75 mL) were added and the mixture was stirred vigorously for 18 h. The mixture was extracted with ethyl acetate (2 × 250 mL), and the combined organic extracts were washed with saturated aqueous sodium bicarbonate solution (2 × 200 mL), water (2 × 250 mL), and brine (100 mL), then dried (MgSO4), filtered and evaporated in vacuo to obtain compound 3, a white powder (14.0 g, 87%) |
|
With hydrogen bromide; acetic anhydride; acetic acid In dichloromethane at 0℃; |
3.2 4.2.1. Bromination of glycosyl per-O-acetate14
General procedure: The per-acetylated sugar (1 mmol) was dissolved in DCM (1 mL),acetic anhydride (0.1 mL) and HBr/AcOH (0.8 mL, 30% w/w), and stirred overnight at 0 °C. The reaction mixture was diluted with DCM (10 mL), washed with cold water (2 × 5 mL), cold NaHCO3 (2 × 5 mL) and cold brine (5 mL), and dried over MgSO4. After evaporation, theα-D-glycosyl bromide was obtained |
|
With hydrogen bromide; acetic acid at 20℃; for 0.666667h; |
40.A Step A: 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide
To β-D-glucose pentaacetate (5 g, 12.81 mmol) was added 33% HBr in acetic acid (30 mL, 192 mmol) at rt. After stirring for 40 min, the mixture was diluted with CH2Cl2 (150 mL) and washed with ice cold water until the washings were neutral pH. The organic layer was dried over MgSO4, filtered and concentrated to give the title compound. 1H NMR (CDCl3) δ 2.06 (s, 3H), 2.08 (s, 3H), 2.12 (s, 3H), 2.13 (s, 3H), 4.15 (d, J = 11.1, 1H), 4.31-4.37 (m, 2H), 4.86 (dd, J = 9.9, 4.0, 1H), 5.19 (t, J = 9.8, 1H), 5.58 (t, J = 9.8, 1H), 6.63 (d, J = 4.0, 1H). |
| 3.07 g |
With hydrogen bromide In acetic acid at 0℃; |
Two-Step Synthesis of Peracetylated Pyranosyl Bromides 1a,b,e; General Procedure A
General procedure: A suspension of dry NaOAc (1.1 equiv) in Ac2O (13.1 equiv) was heated to reflux (~140 °C). The heater was removed and the carbohydrate (1 equiv) was added in small portions to the hot solution so that themixture starts to reflux on its own. After this, the mixture was cooled to r.t., and then poured onto ice. After crystallization, the formed solid was separated by filtration and washed with ice water. The product was vacuum-dried.The acetylated carbohydrate was added to stirred 33 wt% HBr in AcOH (1 mL per 1 g acetylated carbohydrate) at 0 °C in small portions. After full conversion (45 min-4 h), the reaction was poured onto ice water and extracted with CH2Cl2 (3 ×). The combined organic phase was washed with sat. NaHCO3 solution and dried (anhyd MgSO4). The solvent was removed under reduced pressure.12 |
|
With hydrogen bromide; acetic acid at 0 - 20℃; for 0.75h; |
4.3. general procedure for preparation of glucosyl bromide (6) and maltosyl bromide (11)
General procedure: To glucose/maltose peracetate (3 g) was added 15 mL of 33% HBr in AcOH at 0 °C, slowly allowed the reaction to come to room temperature under stirring and maintained for 45 min. DCM (80 mL) was added to the reaction mixture and the solution was dropped into ice-cold water. Separated the organic layer and then washed with solution of NaHCO3, water and dried over Na2SO4. Organic layer was evaporated under vacuum to obtain required compounds, which were stored in refrigerator. |
|
With hydrogen bromide; acetic acid In dichloromethane |
|
|
With red phosphorus; bromine; acetic acid |
|
|
With hydrogen bromide; acetic acid In dichloromethane |
|
|
With hydrogen bromide In 4-(dicyanomethylene)-2-methyl-6-(4-dimethylaminostyryl)-4H-pyran at 0℃; for 6h; |
4.4 General synthetic procedures for 5-6 and 7a-7d
The 30% 81 HBr (8mL) was added to the solution of 82 pentalacetylated glucose (3.8mmol) in anhydrous 83 CH2Cl2 (10mL) and stirred at 0°C for 6h until the starting material was not observed by TLC check. The crude solution was filtered and washed with CH2Cl2 (2×20mL), and the CH2Cl2 solution was further washed with distilled water (3×30mL). Then the organic layer was dried over anhydrous Na2SO4 and concentrated to dryness under reduced pressure to yield the 84 α-d-glucopyranosyl bromide tetra-acetate. Magnolol was dissolved in 10mL NaOH solution (0.8N) and stirred for 30min and then added to the solution of α-d-glucopyranosyl bromide tetra-acetate (1 or 2 equiv.) and tetrabutyl ammonium bromide (TBAB, 1 or 2 equiv.) in 10mL CHCl3. After stirring for 3h, the reaction mixture was filtered and washed with CHCl3 (3×20mL). Subsequently, the CHCl3 solution was washed by 5% HCl (3×50mL), saturated NaHCO3 (3×50mL) and saturated NaCl (3×50mL) respectively and further concentrated to dryness in vacuo [18]. Purification by column chromatography on silica gel to get the target compounds 5 and 6. The obtained compounds 5 or 6 was further deacetylated in sodium methoxide-methanol solution (0.8 equiv.) for different hours (4-8h), and then purified by column chromatography over silica gel to afford 7a-7d. |
|
With hydrogen bromide; acetic acid at 20℃; for 1h; |
|
|
With hydrogen bromide; acetic acid In dichloromethane at 0 - 20℃; for 2h; |
|
| 4.4 g |
With hydrogen bromide; acetic acid In dichloromethane at 20℃; |
|
| 72.5 g |
With phosphorus tribromide In water monomer; acetic anhydride at 0 - 60℃; for 1h; |
5
80 g of the β-D-glucose pentaacetate obtained above was added to 400 ml of acetic anhydride and 120 ml of phosphorus tribromide.Reduce the temperature to below 0 ° C. Add 175g of water drop at controlled temperature and add 60 ° C for 60min.The reaction was detected by TLC. The reaction solution was poured into ice water. Extracted with dichloromethane, washed the organic layer with water, washed with sodium bicarbonate solution, washed with saturated brine, dried and concentrated to obtain the residue, which is a white solid compound refined by isopropyl ether, 72.5 g. |
|
With hydrogen bromide; acetic acid In dichloromethane for 2h; |
|
|
With hydrogen bromide; acetic acid In dichloromethane at 0 - 20℃; for 3.5h; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping