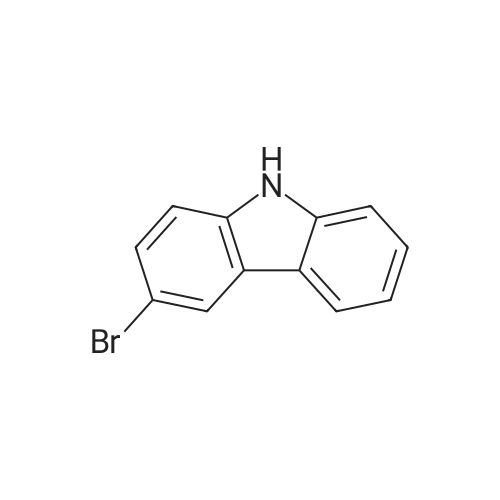
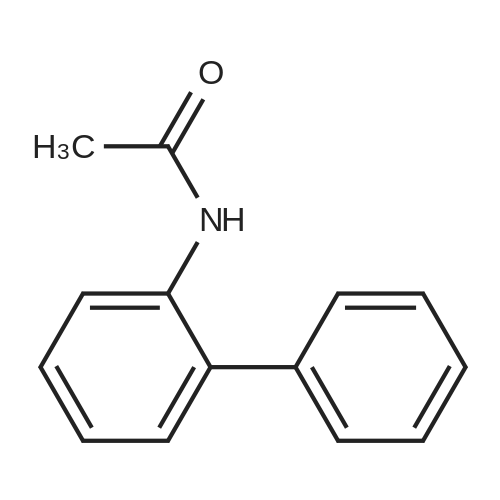
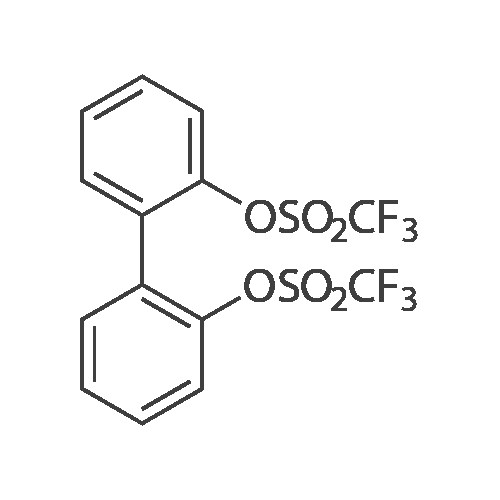
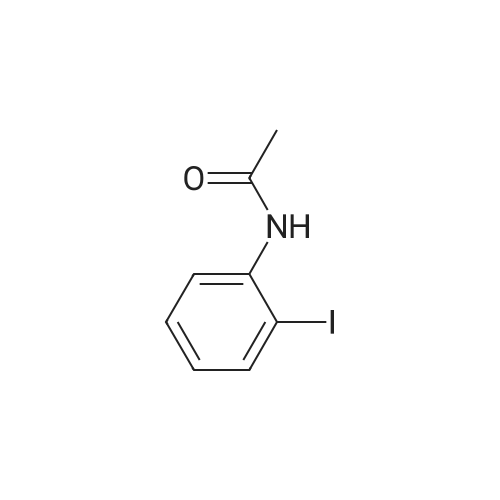
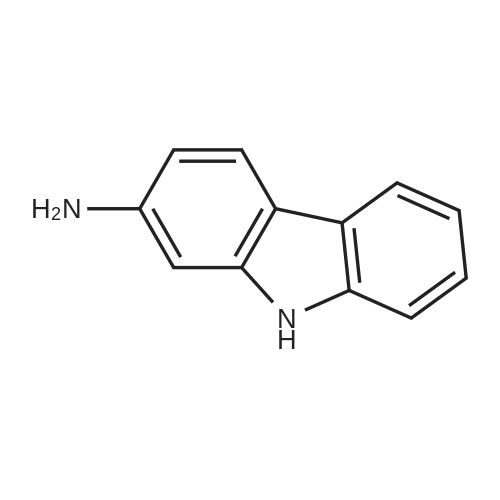
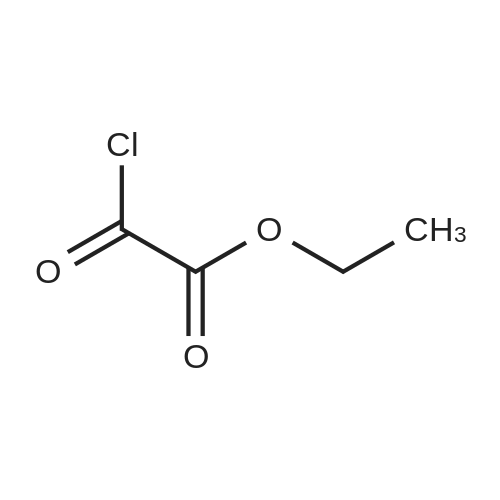
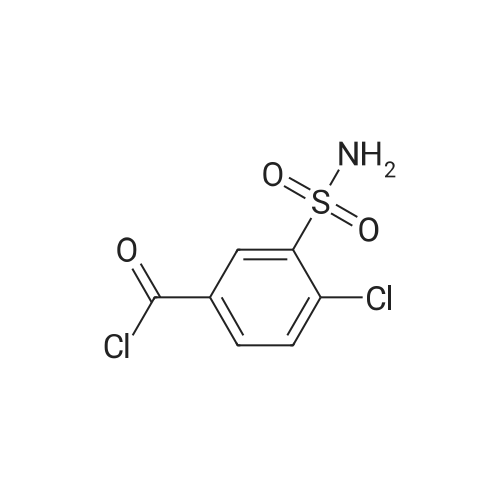
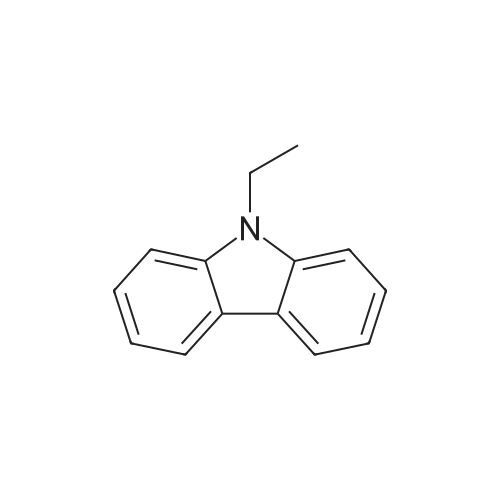
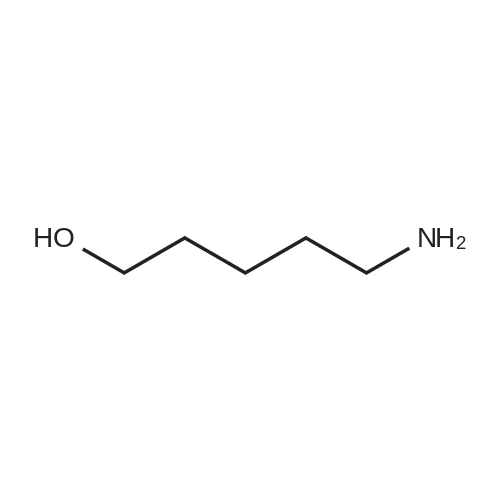
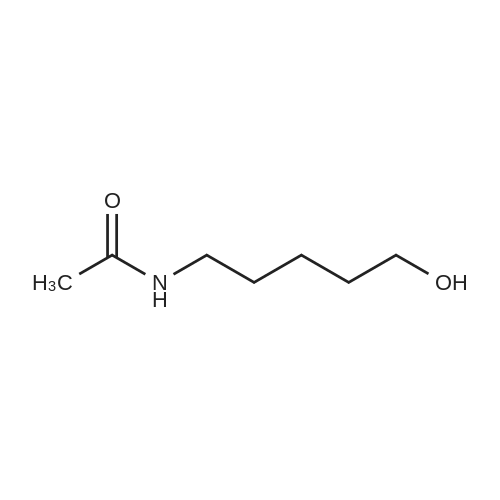
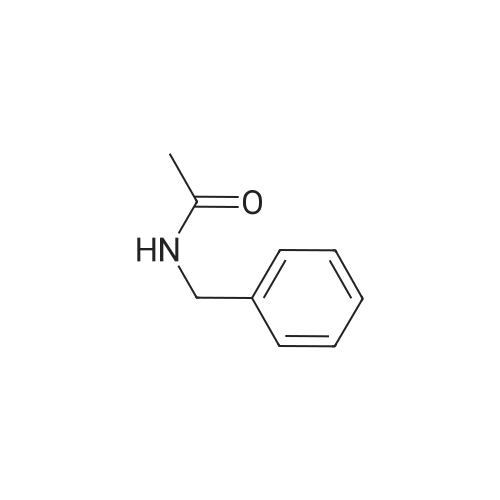
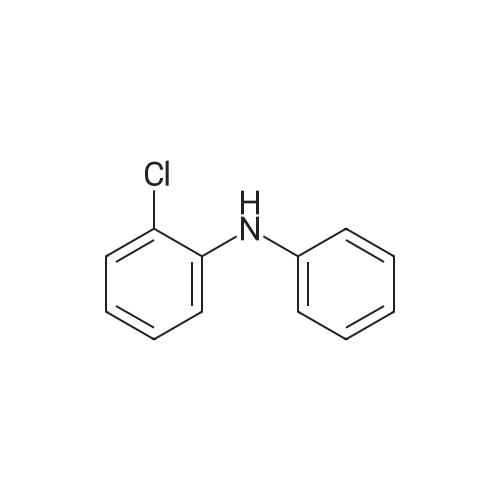
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
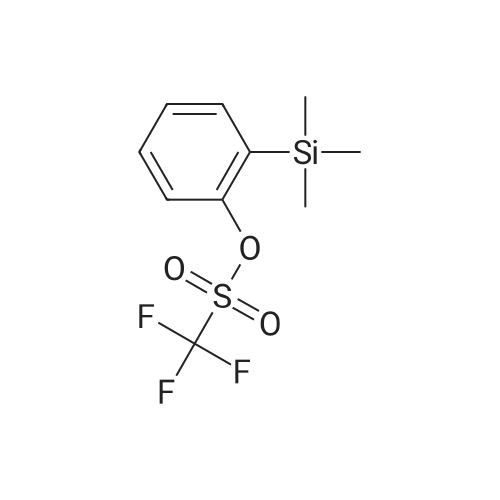
With bis(1,5-cyclooctadiene)diiridium(I) dichloride; sodium carbonate In toluene at 100℃; for 15 h; Inert atmosphere
General procedure: A mixture of [Ir(Cl)(cod)]2 (6.7 mg, 0.01 mmol), Na2CO3 (127 mg, 1.2 mmol), 1a (167 mg, 1 mmol) and 2 (430 mg, 5mmol) in Toluene (1 mL) was stirred at 100 °C for 15 h under Ar. The conversions and yields of products were estimated from peak areas based on an internal standard using GC and the product 3a was obtained in 96percent. The product 3a was isolated by column chromatography (230-400 mesh silica gel neutralized with 5percent Et3N in n-hexane) and in94percent yield (181 mg). The product 3e was only isolated by column chromatography (70-230 mesh aluminium oxide, n-hexane).
Reference:
[1] Chemistry of Heterocyclic Compounds (New York, NY, United States), 1983, vol. 19, # 11, p. 1192 - 1197[2] Khimiya Geterotsiklicheskikh Soedinenii, 1983, # 11, p. 1504 - 1509
[3] Synlett, 2017, vol. 28, # 6, p. 719 - 723
5
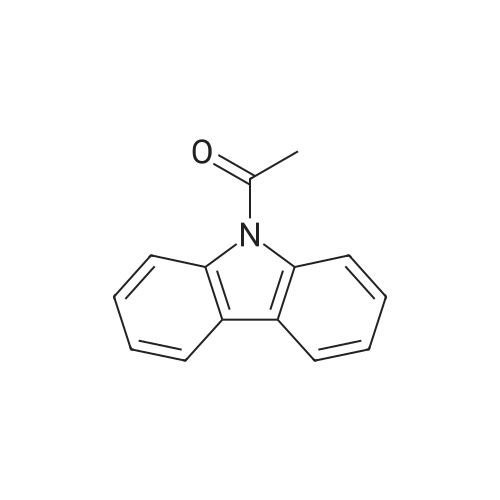
[ 60-35-5 ]
[ 17763-95-0 ]
[ 574-39-0 ]
[ 86-74-8 ]
Reference:
[1] Journal of Organic Chemistry, 2005, vol. 70, # 2, p. 413 - 419
6
[ 19591-17-4 ]
[ 88284-48-4 ]
[ 217-59-4 ]
[ 574-39-0 ]
Reference:
[1] Journal of Organic Chemistry, 2012, vol. 77, # 24, p. 11153 - 11160
Reference:
[1] Journal of Organometallic Chemistry, 1988, vol. 350, p. 217 - 226
9
[ 90-41-5 ]
[ 574-39-0 ]
Reference:
[1] Organic Letters, 2016, vol. 18, # 22, p. 5896 - 5899
[2] European Journal of Organic Chemistry, 2016, vol. 2016, # 33, p. 5474 - 5479
[3] Tetrahedron, 2019, vol. 75, # 3, p. 387 - 397
10
[ 108-24-7 ]
[ 86-74-8 ]
[ 574-39-0 ]
Reference:
[1] Photochemistry and Photobiology, 1998, vol. 68, # 5, p. 640 - 645
[2] Tetrahedron, 1980, vol. 36, # 20-21, p. 3017 - 3019
[3] Justus Liebigs Annalen der Chemie, 1872, vol. 163, p. 352
[4] Justus Liebigs Annalen der Chemie, 1872, vol. 163, p. 352
[5] Recueil des Travaux Chimiques des Pays-Bas, 1912, vol. 31, p. 364
[6] Recueil des Travaux Chimiques des Pays-Bas, 1912, vol. 31, p. 364
[7] Diss.<Braunschweig 1926> S.30,
[8] Zhurnal Obshchei Khimii, 1944, vol. 14, p. 438,441,445[9] Chem.Abstr., 1945, p. 4605
[10] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 1996, vol. 35, # 6, p. 617 - 620
11
[ 33650-07-6 ]
[ 75-36-5 ]
[ 574-39-0 ]
Reference:
[1] Bulletin of the Chemical Society of Japan, 1981, vol. 54, # 1, p. 25 - 30
12
[ 103-84-4 ]
[ 98-80-6 ]
[ 574-39-0 ]
Reference:
[1] Green Chemistry, 2016, vol. 18, # 11, p. 3295 - 3301
Reference:
[1] European Journal of Organic Chemistry, 2016, vol. 2016, # 33, p. 5474 - 5479
17
[ 33650-07-6 ]
[ 574-39-0 ]
Reference:
[1] Journal of Organometallic Chemistry, 1988, vol. 350, p. 217 - 226
18
[ 13390-92-6 ]
[ 574-39-0 ]
Reference:
[1] Journal of Organometallic Chemistry, 1988, vol. 350, p. 217 - 226
19
[ 103-84-4 ]
[ 574-39-0 ]
Reference:
[1] Chemical Communications, 2014, vol. 50, # 94, p. 14862 - 14865
20
[ 108-05-4 ]
[ 86-74-8 ]
[ 574-39-0 ]
[ 4901-29-5 ]
Reference:
[1] Chemistry of Heterocyclic Compounds (New York, NY, United States), 1983, vol. 19, # 11, p. 1192 - 1197[2] Khimiya Geterotsiklicheskikh Soedinenii, 1983, # 11, p. 1504 - 1509
21
[ 7664-93-9 ]
[ 108-24-7 ]
[ 86-74-8 ]
[ 574-39-0 ]
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1912, vol. 31, p. 364
22
[ 108-24-7 ]
[ 7705-08-0 ]
[ 86-74-8 ]
[ 574-39-0 ]
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1912, vol. 31, p. 364
23
[ 21865-49-6 ]
[ 108-24-7 ]
[ 574-39-0 ]
Reference:
[1] Chemische Berichte, 1907, vol. 40, p. 3227
24
[ 574-39-0 ]
[ 51035-17-7 ]
Reference:
[1] Journal of the Chemical Society, 1934, p. 1142
With potassium hydroxide In acetone at 30℃; for 0.5h;
8 %Chromat.
With bis(1,5-cyclooctadiene)diiridium(I) dichloride; sodium carbonate In toluene at 100℃; for 15h; Inert atmosphere;
A typical reaction was carried out as follows (Table 1, entry 1):
General procedure: A mixture of [Ir(Cl)(cod)]2 (6.7 mg, 0.01 mmol), Na2CO3 (127 mg, 1.2 mmol), 1a (167 mg, 1 mmol) and 2 (430 mg, 5mmol) in Toluene (1 mL) was stirred at 100 °C for 15 h under Ar. The conversions and yields of products were estimated from peak areas based on an internal standard using GC and the product 3a was obtained in 96%. The product 3a was isolated by column chromatography (230-400 mesh silica gel neutralized with 5% Et3N in n-hexane) and in94% yield (181 mg). The product 3e was only isolated by column chromatography (70-230 mesh aluminium oxide, n-hexane).
With 1,8-diazabicyclo[5.4.0]undec-7-ene In methanol for 0.5h; Heating;
97%
With sulfuric acid In methanol at 60℃; for 0.25h;
71%
With triethyl borane; sodium hydroxide In tert-butyl methyl ether at 80℃; for 6h; Inert atmosphere; Sealed tube;
8 Example 8, wherein the amide substrate is as follows:
Under argon atmosphere,Firstly, NaOH and triethylboron are stirred to form a transparent clear solution at room temperature.The concentration is 1M/L;Then,The above triethylboron solution was successively treated with 10umol (2mol%),Amide substrate 5mmol,Triethoxysilane or polymethylhydrogensiloxane 15mmol,MTBE 2mL added to the 10mL sealed tube,It is heated and stirred in an 80°C oil bath for 6 hours.The reaction is over,Exposing the reaction to air quenching,The amine product is then separated directly by column chromatography.According to the results of column chromatography separation, when using triethoxysilane or polymethylhydrogensiloxane (PMHS),The yield of the target product was:
70%
With sodium hydroxide; dipotassium hydrogenphosphate; potassium dihydrogenphosphate; D-glucose; Pseudomonas aeruginosa; magnesium sulfate; potassium nitrate; sodium chloride; calcium chloride In water for 72h; pH 7.0;
65%
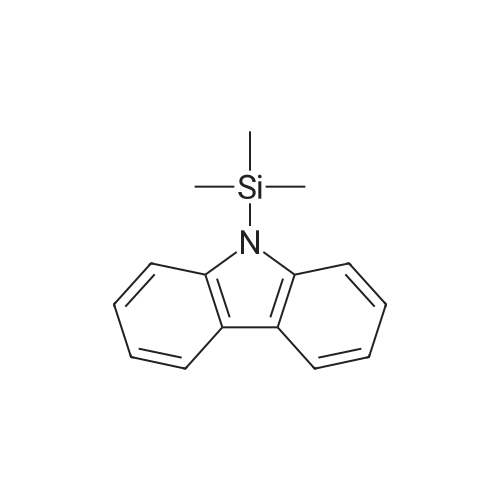
Stage #1: 9-acetylcarbazole With Triethoxysilane; sodium triethylborohydride In tert-butyl methyl ether at 80℃; for 6h;
Stage #2: With hydrogenchloride In tert-butyl methyl ether; water at 20℃; for 1h; chemoselective reaction;
With palladium diacetate; copper diacetate; 3 A molecular sieve In toluene at 120℃; for 12h;
95%
With pyridine N-oxide; 10% Pd/C; oxygen In dimethyl sulfoxide at 120℃; for 24h; Sealed tube;
1
General procedure: Palladium carbon catalyst containing 10 wt% palladium in terms of metal as a catalyst with respect to 0.25 mmol of each substrate shown in Table 1 below (N.E. Chemcat Co., Ltd. [K-type catalyst]: Palladium content; 10 wt%, specific surface area Value; 1,100 m2 / g) in terms of palladium metal 10 mol% with respect to the substrate, pyridine N-oxide as an oxidizing agent with 20 mol% with respect to the substrate, and 1 ml of anhydrous dimethyl sulfoxide (DMSO) as a solvent in a closed silicon container. After sealing with (Septum), the mixture was degassed, replaced with oxygen, heated to a temperature of 120 ° C., and stirred.After the reaction, the catalyst was filtered, the filtrate was extracted with 10 ml of ethyl acetate and 10 ml of water, and the aqueous phase was further extracted twice with 10 ml of ethyl acetate. The ethyl acetate phase was washed with 20 ml of saturated brine, dried over anhydrous magnesium sulfate, the solvent was further evaporated under reduced pressure, and the residue was purified by silica gel column chromatography (dichloromethane: methanol = 1000: 1). The outline of this reaction is shown below, and the results are shown in Table 1. Each substrate is a modification of R1 in Formula I, and R1 is also shown in Table 1 below.
94%
With copper diacetate; air; 3 A molecular sieve In toluene at 120℃; for 24h;
93%
With sodium persulfate; tetrabutylammomium bromide In water for 1.5h; Schlenk technique; Inert atmosphere; Sealed tube; Reflux;
92%
With potassium phosphate; Pd/C; oxygen In dimethyl sulfoxide at 120℃; for 37h; Molecular sieve;
90%
With 5,7-bis(acetyloxy)-5,7-dihydro-1,3,9,11-tetramethydibenzo[d,f][1,3,2]diiodoxepin In dichloromethane; 1,1,1,3',3',3'-hexafluoro-propanol at 20℃; for 6h;
89%
With 1,1,1,3',3',3'-hexafluoro-propanol at 20℃;
85%
With 1,3-bis(2,4,6-trimethylphenyl)-1H-imidazolium chloride; dihydrogen peroxide; palladium diacetate; acetic acid at 120℃; for 3h; Microwave irradiation; Sealed tube;
81%
With [Ir(dfppy)2(phen)](PF6); oxygen; palladium diacetate In dimethyl sulfoxide at 80℃; for 15h; Irradiation;
77%
With peracetic acid; 1,1,1,3',3',3'-hexafluoro-propanol; 2-(2-iodo-3-methoxyphenyl)benzo[d]oxazole In dichloromethane at 20℃; for 24h; Schlenk technique; Inert atmosphere;
73%
With phenylbis(2,2,2-trifluoroethoxy)- λ3-iodane In 2,2,2-trifluoroethanol for 48h; Inert atmosphere; Molecular sieve;
63%
With sodium ethanolate In ethanol at 25℃; for 0.5h; Electrolysis;
53%
With lithium perchlorate; palladium diacetate; toluene-4-sulfonic acid; trifluoroacetic acid In acetonitrile at 20℃; for 3h; Electrolysis; Inert atmosphere;
48%
With phenylbis(2,2,2-trifluoroethoxy)- λ3-iodane In 2,2,2-trifluoroethanol at 50℃; for 1h; Flow reactor;
36%
With tetra-(n-butyl)ammonium iodide; tert-butylammonium hexafluorophosphate(V) In dichloromethane; 2,2,2-trifluoroethanol at 25℃; for 12h; Inert atmosphere; Electrolysis;
10%
With [bis(acetoxy)iodo]benzene; palladium diacetate In dichloromethane at 20℃;
63 %Spectr.
With [bis(acetoxy)iodo]benzene; copper(II) bis(trifluoromethanesulfonate); trifluoroacetic acid In 1,2-dichloro-ethane at 50℃; for 0.25h;
10 %Spectr.
With Oxone; palladium diacetate; toluene-4-sulfonic acid; Trimethylacetic acid In N,N-dimethyl-formamide at 25℃; for 12h;
With copper diacetate; palladium diacetate
With pyridine N-oxide; 10% Pd/C; oxygen; dimethyl sulfoxide at 120℃; for 24h;
With sodium acetate; palladium diacetate; silver carbonate; benzoquinone In tert-Amyl alcohol; 1,2-dichloro-ethane at 120℃; for 12h; Schlenk technique;
4.1.3. General procedures for the formations of N-(2'-(di-ptolylphosphoryl)-[1,10-biphenyl]-2-yl)-2,2,2-trifluoroacetamide (3_caab)
General procedure: Into a 20 ml Schlenk tube was charged with a stir bar. Then,reactants such as 1_eaa (72.6 mg, 0.20 mmol), Pd(OAc)2 (4.5 mg, 0.02 mmol), Ag2CO3 (110 mg, 0.40 mmol), BQ (10.8 mg, 0.10 mmol) and NaOAc (33 mg, 0.40 mmol) were added to the tube with 1.5mL t-AmylOH (2-methylbutan-2-ol). The solution was heated at 120 °C for 10 minutes with cap opened. Then, the cap is closed again. Another flask containing 2_a (97.1 mg, 0.48 mmol), which was dissolved in dichloroethane (1.0 mL) beforehand, was placed in a Syringe before mounted on a Syringe Pump (KDS100). The solution was then injected into the Schlenk tube rather slowly (0.1mL per hour) for totally about 10 hours. After the completion of the reaction, the solution was cooled down and later purified by column chromatography packed with silica gel and eluted firstly by hexanes. A colorless solid 4_eaa (4.8 mg, 0.013 mmol) in the yield of 6.6% was obtained. Further separation using mixed solvent (Hexanes/ethyl acetate 4/1). A pale yellow solid powder 3_eaaa in the yield of 58.0% (65.4 mg, 0.116 mmol) was obtained.
92 %Spectr.
With 1,2-diiodo-4,5-dimethoxybenzene; tetramethyl ammonium acetate; tert-butylammonium hexafluorophosphate(V) Electrolysis;
2 Synthesis of 2-amino Carbazole
EXAMPLE 2 Synthesis of 2-amino Carbazole Carbazole (100.24 g) was suspended in acetic anhydride (300 mL) with catalytic boron trifluoride etherate (0.65 mL) and the solution refluxed for 25 min. then cooled to 0° C. and the solid was then collected and recrystallized from hexane to give 82.65 g 9-acetyl-carbazole. 9-Acetyl-carbazole (41.85 g, 0.2 mol) was dissolved in 1 L methylene chloride and 28.5 mL acetylchloride (0.4 mol) and 120 g aluminum chloride (0.9 mol) were added and the solution refluxed for 1.5 h. The solution was cooled to -78° C. and 6N HCl was added slowly while stirring and allowed to warm to room temperature. Methylene chloride was added to dissolve the precipitate and the solution was extracted, and the organics dried over Na2SO4, decolorized with charcoal, filtered and concentrated. Recrystallization from benzene/hexane gave 27.9 g 2,9-diacetylcarbazole. 2,9-Diacetylcarbazole, (25.1 g, 0.1 mol) was dissolved in 100 mL pyridine. Hydroxylamine hydrochloride (10.42 g, 0.15 mol) was added and the solution refluxed for 20 min. The mixture was cooled and poured into 70 mL conc. HCl in ice. The precipitate was collected by filtration, washed with a large volume of water and dried in vacuum over KOH to yield 24 g white solid. 5 g of this solid (18.8 mmol) was dissolved in 17 mL TFA and 0.3 mL trifluoroacetic anhydride (2.1 mmol) was added. The mixture was refluxed for 20 min then concentrated to give the crude 2,9-diacetylcarbazole amine. Both acetyl groups were removed by suspending the crude material in 400 mL 10% aq. KOH and refluxing for 1 day. The solution was cooled and ethyl acetate added and the product 2-aminocarbazole was extracted, washed with brine, dried over Na2SO4 and concentrated.
With hydrogenchloride; sulfuric acid; acetic anhydride; sodium hydrogencarbonate In ethanol; dichloromethane; water
1 Preparation of ethyl carbazole-2-oxalate
EXAMPLE 1 Preparation of ethyl carbazole-2-oxalate To a stirred suspension of 167.2 g. of carbazole in 400 ml. of methylene chloride was added 103 ml. of acetic anhydride and 2.0 ml. of concentrated sulfuric acid. The mixture was degassed, placed under nitrogen and heated at reflux for 20 hours. The violet-colored solution was poured, after cooling to 20°, in a steady stream into a vigorously stirred solution of 175 g. of sodium bicarbonate in 1600 ml. of water. A color change to yellow-brown was accompanied by copious evolution of carbon dioxide. The mixture was stirred for 30 minutes and then the aqueous layer was extracted with 200 ml. of methylene chloride. The combined organic solutions were washed with 200 ml. of saturated sodium bicarbonate solution and dried over sodium sulfate. The sodium sulfate was removed by filtration and the filtrate was stirred for 1.0 hour with calcium chloride and 10 g. of Norite A. The mixture was filtered through Celite on medium sintered glass. The flask and funnel were washed with about 400 ml. of fresh methylene chloride to give a total filtrate of 1000 ml. of a 1 M solution of 9-acetylcarbazole. Such solutions appear to be stable indefinitely. A mechanically-stirred suspension of 600 g. of aluminum chloride in 2000 ml. of methylene chloride was degassed, placed under nitrogen and cooled to 3° in an ice bath. A mixture of the 9-acetylcarbazole solution and 134 ml. of ethyl oxalyl chloride was added at a rate such that an internal temperature of 5° was maintained. This took about 2.0 hours and led to a deep red viscous mixture. The nitrogen inlet was disconnected and the system was vented to a gas scrubber to remove the hydrogen chloride gas. The ice bath was replaced by a 25° water bath. The bath temperature was raised to 45° over a 15 minute period as vigorous stirring was maintained to ensure a smooth evolution of the hydrogen chloride. The internal temperature rose to 35°--reflux-- which was maintained for 1.0 hour. The heating bath was removed and the mixture was stirred as 2000 ml. of ethanol was added at a rate such that a gentle reflux was maintained. This required 1.0 hour. The deep red mixture was then treated with 4000 ml. of 6 N hydrochloric acid at a rate such that gentle reflux continued. This required 1.0 hour, although the initial addition must be slow to control the exotherm. The two phase mixture was heated at reflux for 1.0 hour and cooled. The aqueous layer was extracted with 500 ml. of methylene chloride and the combined organic solutions were filtered through a layer of sodium sulfate. Solvent removal on a rotary evaporator at 50° gave crude ethyl 9acetylcarbazole-2-oxalate as a dark greenish crystalline solid.
322.1 322.1
322.1 9-acetyl-9H-carbazole This compound is obtained according to Tetrahedron (1980), 36, 3017-3019. The carbazole (10 g; 60 mmol) is suspended in 150 ml of acetic anhydride. 70% perchloric acid (0.5 ml) is added. After stirring for 30 minutes at ambient temperature, the mixture is poured into ice and the precipitate formed is filtered. After drying under vacuum, redissolving in dichloromethane and treatment with bone charcoal, the suspension is filtered on celite, the solvents are evaporated off and the product recrystallized from heptane. 12 g of brown crystals (yield of 90%) is obtained in this way. Melting point: 70-71° C. (literature: 72-74° C.).
With acetic anhydride In (2S)-N-methyl-1-phenylpropan-2-amine hydrate; ethanol
1.A A.
A. Preparation of N-acetylcarbazole A 500 ml, 3-necked, round bottom flask was fitted with mechanical stirrer, reflux condenser and means for providing nitrogen atmosphere. To the flask was added 30 g (0.18 mole) of carbazole, 24.6 g (0.18 mole) of finely powdered zinc chloride, and 55 g (.054 mole) of acetic anhydride. The resulting mixture was stirred and heated with an oil bath of 80° C for 10 minutes. The reaction mixture was then poured into 1200 ml of ice water. After stirring for 2 hrs, the precipitate was collected by filtration, washed five times with 100 ml portions of water and dried, resulting in 37 g (99% yield) of solid. Recrystallization of the crude product (37 g) from ethanol (185 ml) gave 22 g (59% yield) of the desired product, m.p. 71.2°-72.2° C. IR (KBr pellet): 1680 cm-1 (C=O).
With sulfuric acid; acetic anhydride In chloroform
5 Preparation of 9-acetylcarbazole
EXAMPLE 5 Preparation of 9-acetylcarbazole A stirred solution of 5 g. of carbazole, 50 ml. of chloroform, 5 ml. of acetic anhydride and 3 drops of concentrated sulfuric acid was heated under reflux (dry nitrogen atmosphere) for 5 hours. The reaction mixture was concentrated to dryness under reduced pressure and the residue was partitioned between ether and water. After the water was separated, the organic phase was washed by extraction with water (three times), dilute sodium bicarbonate (once) and water again (twice). Following drying of the ether solution over anhydrous magnesium sulfate, the desiccant was removed by filtration and the ether was evaporated; yielding 6.1 g., mp 76°-77°, of 9-acetylcarbazole.
33
Preparation of 5-(9-acetyl-9H-carbazole-2-carbonyl)-2-chloro-benzenesulfonamide; 52419ATo a solution of 1 g of 1-carbazol-9-yl-ethanone and 2.43 g of 4-chloro-3-sulfamoyl- benzoyl chloride in methylene chloride (20 mL) is added 2.55 g of aluminum chloride. The mixture is stirred at 5O0C overnight. The solution is then cooled at -78 "C, quenched with a 6 N HCI solution and allowed to warm up to room temperature. Methylene chloride is added to dissolve the precipitate and the solution is extracted, washed with a saturated sodium chloride solution, dried over sodium sulfate, filtered and concentrated. The residue is loaded on Celite and purified by silica gel chromatography (1 :1 hexanes/ethyl acetate) to afford 5- (9-acetyl-9H-carbazole-2-carbonyl)-2-chloro-benzenesulfonamide as yellow foam. MS (m/z): 427 (M+1). HPLC Reverse Phase (Nucleosil 100-5 C18, gradient 10->100% CH3CN in 5 min) room temperature = 5.35 minutes.
Stage #1: 9-acetylcarbazole With sodium triethylborohydride; benzo[1,3,2]dioxaborole In tetrahydrofuran at 80℃; for 12h; Inert atmosphere; Sealed tube;
Stage #2: With hydrogenchloride In tetrahydrofuran; water at 20℃; for 2h;
3 Embodiment 3, wherein the amide substrate is as follows:
Under an argon atmosphere, the amide substrate (0.5 mmol), borane (1.5 mmol, 3.0 equiv.),THF (2 mL) and sodium triethylborohydride (4 mol%) were added to a 10 mL sealed tube and placed in an oil bath at 80 ° C. and stirred for 12 hours.After the reaction was completed, a 1M aqueous hydrochloric acid solution (4 mL) was added to the reaction solution under an air environment, and the mixture was stirred at room temperature for 2 hours.The organic phase was extracted and collected, and the product was separated by column chromatography in a yield of 72%.
In tetrahydrofuran at 30℃; for 24h; chemoselective reaction;
The General Procedure of N-Acylation in Scheme 5
General procedure: To a solution of NAC 1a or 1b (1.22.5eq.) in THF (0.5 M)was added amine (0.3mmol, 1.0 eq.) at 30 °C. After stirring the mixture for 24 h, Ac2O and pyridine (0.6mmol, 2.0 eq. each) orBoc2O (0.6mmol, 2.0 eq.) was added. The mixture was stirred for 1 h, then diluted with dichloromethane. The whole mixture was purified by column chromatography on silica gel (CH2Cl2 then EtOAc) to give the corresponding N-Bz or N-Ac amides 3.
In tetrahydrofuran at 30℃; for 24h; chemoselective reaction;
The General Procedure of N-Acylation in Scheme 3,4, and 6.
General procedure: To a solution of NAC 1a or 1b12 (0.3mmol) in THF(0.5 M) was added amine (0.9mmol) at 30 °C. The mixture was stirred for 24 h, quenched with 1N HCl, and extracted with EtOAc (3). The combined organic layers were washed with brine, dried over anhydrous MgSO4, and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel to give the corresponding N-Bz or N-Ac amides 3.
N-Acylation of 2a, 2o, and 2s with NAC on 3mmol Scale.
General procedure: To a solution of NAC 1a or 1b (3.6mmol) in THF(0.5 M) was added amine (3.0mmol) at 30 °C. The mixture was stirred for 24 h, quenched with 1N HCl, extracted with EtOAc(*3). The combined organic layers were washed with brine,dried over anhydrous MgSO4, and filtered. The filtrate was concentrated under reduced pressure. The residue was purifiedby column chromatography on silica gel to give 3aa, 3ao, or 3bs.
The General Procedure of N-Acylation in Scheme 3,4, and 6.
General procedure: To a solution of NAC 1a or 1b12 (0.3mmol) in THF(0.5 M) was added amine (0.9mmol) at 30 °C. The mixture was stirred for 24 h, quenched with 1N HCl, and extracted with EtOAc (3). The combined organic layers were washed with brine, dried over anhydrous MgSO4, and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel to give the corresponding N-Bz or N-Ac amides 3.
Stage #1: 9-acetylcarbazole With n-butyllithium; N,N,N,N,-tetramethylethylenediamine In hexane at -40 - 20℃; for 4.5h; Inert atmosphere;
Stage #2: With ethylene dibromide In hexane at -60 - 20℃; for 7h;
1.2; 2.2; 3.2; 4.2; 5.2; 6.2; 7.2
(2) Under the protection of a nitrogen atmosphere, the N-acetylcarbazole prepared in step (1), TMEDA (98%, 23.2g, 0.20mol),400mL of n-hexane is added to the dropping funnel with stirring blade, constant pressure,In the four-necked reaction flask of the thermometer, reduce the temperature of the system to -40°C,Add 1.6mol/L n-BuLi (130mL, 0.21mol) n-hexane solution dropwise through a constant pressure dropping funnel, keep the temperature for 30 minutes at the end of the dropwise addition, and warm up to room temperature to react for 4 hours;Then, the temperature of the reaction system was reduced to -60°C,Add 100 mL of a n-hexane solution of 1,2-dibromoethane (45.0g, 0.24mol) through a constant pressure dropping funnel, and keep it for 1 hour after the addition.Warm up to room temperature to react for 6 hours, and the liquid phase detection reaction conversion rate reaches 99.5% or more to stop the reaction;
Multi-step reaction with 2 steps
1.1: sodium t-butanolate; palladium diacetate; tri tert-butylphosphoniumtetrafluoroborate / 1,4-dioxane / 18 h / 110 °C / Inert atmosphere
2.1: sodium hydride / tetrahydrofuran; mineral oil / 0.5 h / 20 °C
2.2: 5 h / 0 - 20 °C
Stage #1: 9H-carbazole With sodium hydride In tetrahydrofuran; mineral oil at 20℃; for 0.5h;
Stage #2: acetyl chloride In tetrahydrofuran; mineral oil at 0 - 20℃; for 5h;
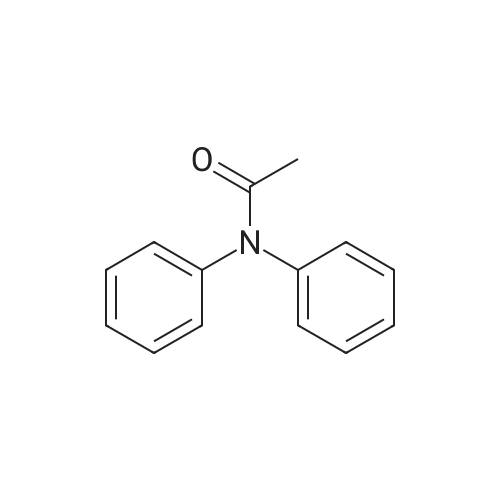
With oxygen; palladium diacetate; acetic acid at 120℃; chemoselective reaction;
General procedure for the intramolecular Pd(II)-catalyzed biaryl coupling reaction
General procedure: In a dry 100 mL round-bottom flask, diphenylamine (0.3 mmol, 1 equiv) and Pd(OAc)2 (0.05 equiv) were dissolved in 2 mL of AcOH, the resulting mixture was heated to 120°C under 1 atm of O2 for 14 h. The solution was then cooled to rt, evaporated under reduced pressure, diluted with CH3COOC2H5, washed with a saturated aqueous solution of NaCl, dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography to afford the corresponding carbazole.
With oxygen; palladium diacetate; acetic acid at 120℃; chemoselective reaction;
General procedure for the intramolecular Pd(II)-catalyzed biaryl coupling reaction
General procedure: In a dry 100 mL round-bottom flask, diphenylamine (0.3 mmol, 1 equiv) and Pd(OAc)2 (0.05 equiv) were dissolved in 2 mL of AcOH, the resulting mixture was heated to 120°C under 1 atm of O2 for 14 h. The solution was then cooled to rt, evaporated under reduced pressure, diluted with CH3COOC2H5, washed with a saturated aqueous solution of NaCl, dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography to afford the corresponding carbazole.
With oxygen; palladium diacetate; acetic acid at 120℃; chemoselective reaction;
General procedure for the intramolecular Pd(II)-catalyzed biaryl coupling reaction
General procedure: In a dry 100 mL round-bottom flask, diphenylamine (0.3 mmol, 1 equiv) and Pd(OAc)2 (0.05 equiv) were dissolved in 2 mL of AcOH, the resulting mixture was heated to 120°C under 1 atm of O2 for 14 h. The solution was then cooled to rt, evaporated under reduced pressure, diluted with CH3COOC2H5, washed with a saturated aqueous solution of NaCl, dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography to afford the corresponding carbazole.
With oxygen; palladium diacetate; acetic acid at 120℃; chemoselective reaction;
General procedure for the intramolecular Pd(II)-catalyzed biaryl coupling reaction
General procedure: In a dry 100 mL round-bottom flask, diphenylamine (0.3 mmol, 1 equiv) and Pd(OAc)2 (0.05 equiv) were dissolved in 2 mL of AcOH, the resulting mixture was heated to 120°C under 1 atm of O2 for 14 h. The solution was then cooled to rt, evaporated under reduced pressure, diluted with CH3COOC2H5, washed with a saturated aqueous solution of NaCl, dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography to afford the corresponding carbazole.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping