* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: at 18℃; for 15 h; Stage #2: at 28℃; for 20 h;
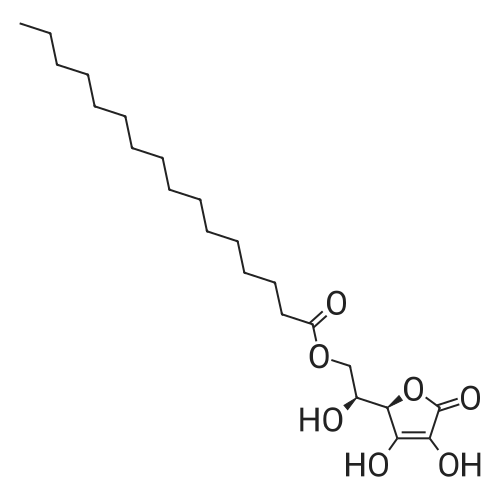
(1)500g of concentrated sulfuric acid was added to the three-necked flask, add 50g of palmitic acid, stirring to dissolve Solution in concentrated sulfuric acid was added 50gL- ascorbic acid, 18 ° C reaction 15h; (2)50g palmitic anhydride was added to the reaction mixture, the temperature was raised to 28 ° C, the reaction 20h, then After adding 10g of activated carbon and stirred for 15min; (3)The step (2) in the resulting mixture is added to 1250ml10 ° C cold water, filtered The filter cake is too crude, the crude product was rinsed with 100ml water, then washed with water after the crude product was dissolved in 750ml of butyl acetate, 50 ° C incubation decolorization 30min. Filtered and allowed to stand, stratification, the upper organic layer (Product containing layer) 50 ° C, washed twice with water, water per 500ml. After washing to the water layer, The organic layer was distilled off under reduced pressure to 400ml of butyl acetate, allowed to stand for cooling to 15 ° C, the solid was filtered off with 50ml The resulting solid was rinsed with ethyl acetate, drained, placed in a vacuum drying oven at 50 ° C. L-ascorbic acid-6-palmitate was obtained as a white flake with a purity of 98. 1percent and a yield of 91.3percent.
Reference:
[1] Patent: CN105315244, 2016, A, . Location in patent: Paragraph 0047-0050
2
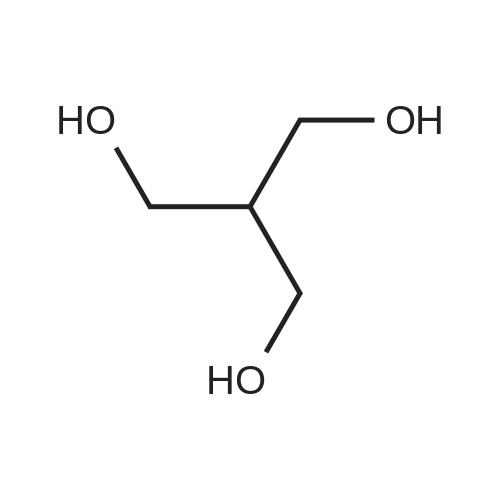
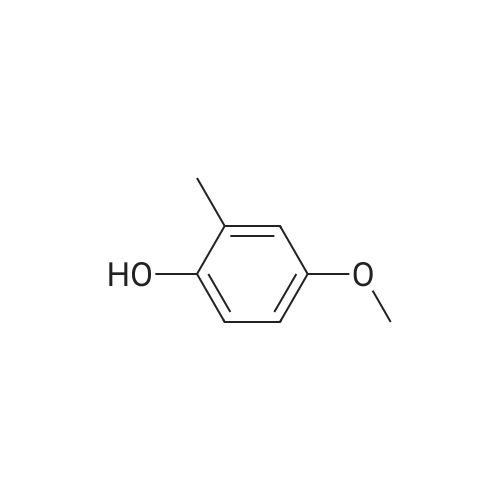
[ 57-10-3 ]
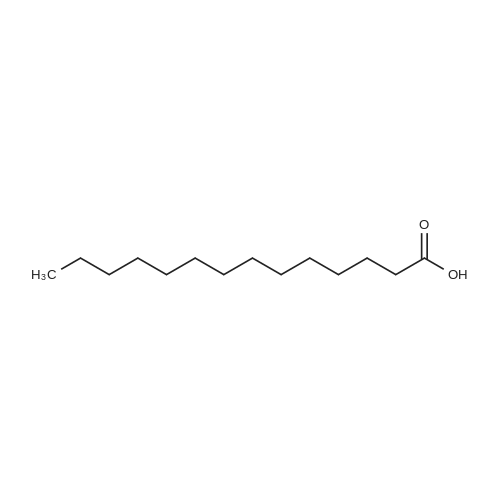
[ 50-81-7 ]
[ 137-66-6 ]
Reference:
[1] JAOCS, Journal of the American Oil Chemists' Society, 2003, vol. 80, # 8, p. 795 - 799
[2] Journal of Molecular Catalysis B: Enzymatic, 2014, vol. 102, p. 127 - 131
[3] Ultrasonics Sonochemistry, 2011, vol. 18, # 5, p. 988 - 996
[4] JAOCS, Journal of the American Oil Chemists' Society, 2011, vol. 88, # 1, p. 57 - 64
[5] Oil and Soap (Alexandria, Egypt), 1943, vol. 20, p. 224
[6] Annales Pharmaceutiques Francaises, 1957, vol. 15, p. 691,693
[7] JAOCS, Journal of the American Oil Chemists' Society, 1999, vol. 76, # 11, p. 1291 - 1295
[8] Farmaco, 2003, vol. 58, # 12, p. 1271 - 1276
[9] Journal of Physical Chemistry B, 2014, vol. 118, # 11, p. 3053 - 3062
[10] Patent: CN103254160, 2016, B, . Location in patent: Paragraph 0017-0022
Reference:
[1] Journal of Molecular Catalysis B: Enzymatic, 2014, vol. 102, p. 16 - 24
5
[ 141-43-5 ]
[ 57-10-3 ]
[ 544-31-0 ]
Yield
Reaction Conditions
Operation in experiment
95.7%
Stage #1: With triethylamine; diisopropyl-carbodiimide In dichloromethane at 20℃; for 4 h; Stage #2: at 20℃; for 0.5 h; Inert atmosphere
1) under nitrogen at room temperature,Triethylamine (2 ml) was added dropwise to mercaptomethyl resin (2 g,MATRIX-INN), N-hydroxy maleimide (1.1 g, 9.7 mmol) and DMF (40 ml)In the reaction vessel.After stirring at room temperature for 24 hours,Afterwards, stirring was continued for 4 hours at 55 degrees,After cooling to room temperature and filtered to give NHS resin,Using DMF,Distilled water and isopropyl alcohol were washed twice,NHS resin was obtained after vacuum drying.2) palmitic acid (1.465 g, 5.72 mmol) and the above NHS resin (1.50 g),DIC (diisopropylcarbodiimidediisoprpylcarbodiimide, 0.72 g, 5.72 mmol),Triethylamine (2 ml) was suspended in 15 ml of dichloromethane.The mixture was stirred at room temperature for 4 hours.Then filtered (filtrate retained,Appropriate amount of palmitic acid was detected by HPLC,DIC and solvent,For the next batch of reactions),The collected resin using DMF,Water, isopropanol, and dichloromethane were separately washed twice and dried in vacuo to give 1.70 g of dry resin to obtain an immobilized palmitic acid active ester with a loading of -1.0 mmol / g.3) Ethanolamine (93.2 mg, 1.58 mmol) was added to a flask containing active ester (1.75 g) and 50 ml of BAlcohol suspension,For 0.5 hour,Centrifugal removal of solid resin,The resin was washed twice with ethanol (the resin was left after vacuum drying)The combined liquid phase was concentrated under reduced pressure,453 mg of palmitic monoethanolamide (96.6percent yield for ethanolamine) was obtained,Purity> 99.5percent (HPLC).
90%
Stage #1: With 1-[(1-(cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino)]-uronium hexafluorophosphate; N-ethyl-N,N-diisopropylamine In dichloromethane; acetonitrile at 20℃; for 0.166667 h; Inert atmosphere Stage #2: at 20℃; Inert atmosphere
General procedure: These compounds were synthesized according to the procedure described previously with slight modifications (El-Faham and Albericio, 2010) The appropriate acid (0.15 mmol), (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluoro phosphate (COMU, 64.2 mg, 0.15 mmol), and DIPEA (0.05 ml, 0.30 mmol) were dissolved in anhydrous CH2Cl2 (0.5 ml) and CH3CN (2.5 ml) and the resulting orange-red solution was stirred at rt for 10 min under a nitrogen atmosphere. Ethanolamine (3) (0.15 mmol) in CH3CN (0.2 ml) was then injected into the reaction mixture and vigorous stirring at rt was continued until TLC (CH2Cl2/MeOH 98:2) confirmed the completion of the reaction (3–6 h). The reaction mixture was diluted with CH2Cl2 (3 ml) and the resulting mixture was washed with 5percent HCl, saturated NaHCO3 and brine. The organic layer was collected, dried over anhydrous Na2SO4, filtered. The solvent was evaporated under reduced pressure and crude purified by flash chromatography.
72%
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride In dichloromethane at 0 - 20℃; for 4 h; Inert atmosphere
Stepl . Palmitoylethanolamide, 2: EDCl (673 mg, 3.51 mmol), DMAP (44 mg, 0.35 mmol), and ethanolamine (0.141 mL, 2.34 mmol) were added to a solution palmitic acid 1 (300 mg, 1.17 mmol) was stirred in 10 mL of anhydrous ΟΗ2( at 0 °C. The reaction was allowed to stir under argon for 4 hours while warming to room temperature. Upon completion the reaction mixture was diluted with CH2C, washed with water and brine. The organic layer was collected and concentrated. The resulting residue was chromatographed on silica to yield 2 (312 mg, 72percent) as a white solid. XH NMR (500 MHz, CDC13) δ 3.70 - 3.76 (m, 2H), 3.43 (q, J = 5.37 Hz, 2H), 2.17 - 2.24 (m, 2H), 1.64 (quin, J = 7.45 Hz, 2H), 1.52 (d, J = 1.0 Hz, 1H), 1.19 - 1.35 (m, 26H).
Reference:
[1] Beilstein Journal of Organic Chemistry, 2009, vol. 5,
[2] Patent: CN107188816, 2017, A, . Location in patent: Paragraph 0048-0052
[3] Chemistry and Physics of Lipids, 2012, vol. 165, # 7, p. 705 - 711
[4] Patent: WO2015/179190, 2015, A1, . Location in patent: Paragraph 0137
[5] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 11, p. 3231 - 3234
[6] Patent: US5925678, 1999, A,
[7] Bioorganic and Medicinal Chemistry, 2011, vol. 19, # 4, p. 1520 - 1527
[8] Bioscience, Biotechnology and Biochemistry, 2011, vol. 75, # 4, p. 768 - 770
[9] ChemSusChem, 2015, vol. 8, # 16, p. 2670 - 2680
[10] Green Chemistry, 2018, vol. 20, # 2, p. 375 - 381
6
[ 57-10-3 ]
[ 544-31-0 ]
Reference:
[1] Tetrahedron Letters, 1980, vol. 21, p. 841 - 844
[2] Bioorganic and Medicinal Chemistry, 2015, vol. 23, # 2, p. 340 - 347
[3] Medicinal Chemistry Research, 2017, vol. 26, # 11, p. 2951 - 2966
7
[ 36653-82-4 ]
[ 57-10-3 ]
[ 540-10-3 ]
Yield
Reaction Conditions
Operation in experiment
98.3 %Chromat.
With tungsten oxide impregnated Zr incorporated mesoporous silica SBA-15 In 1,3,5-trimethyl-benzene at 162℃; for 6 h; Inert atmosphere; Dean-Stark
Cetyl alcohol (CA) and palmitic acid (PA) esterification reactions were performed under N2 atmosphere in a four necked round bot-tom flask (250 ml) equipped with a Teflon coated magnetic stirring bar with a stirring rate of 520 rpm and a Dean Stark apparatus surmounted with a reflux condenser. In a typical experiment, 160 mgof catalyst was added into 25 ml of mesitylene and heated up to reaction temperature of 162 C. An equimolar solution of palmitic acid and cetyl alcohol (6 mmol) in 15 ml of mesitylene at room temperature was added into the reactor. All the reactions were carried out for a reaction time of 6 h. In a preliminary set of experiments (Table 1) it was found that the reaction was not controlled by external diffusion at 520 rpm. Samples taken at regular intervals were analyzed by Agilent 6890 gas chromatography using Ultra 1(25 m × 0.3 mm) capillary column equipped with FID. The injector temperature was 280C and the detector temperature was 320 C.The GC oven temperature was changed from 50C at 12C/min to 300C where it was kept for 35 min. Helium was used as the car-rier gas at a flow rate of 37.3 ml/min The split ratio was 24.9:1. Conversion of cetyl alcohol (CA), yield of cetyl palmitate (CP) and the selectivity to CP were defined as below. Conversion(percent)= (CAin −CAout )CAin×100 Yield(percent)= CPoutCAin×100 Selectivity to CP(percent) = CPout(CAin −CAout )×100
Reference:
[1] Synthesis, 1990, # 9, p. 853 - 854
[2] Chemistry - A European Journal, 2013, vol. 19, # 15, p. 4732 - 4741
13
[ 57-10-3 ]
[ 629-62-9 ]
[ 36653-82-4 ]
[ 540-10-3 ]
Reference:
[1] Chemistry - A European Journal, 2013, vol. 19, # 15, p. 4732 - 4741
14
[ 57-10-3 ]
[ 629-62-9 ]
[ 544-76-3 ]
[ 36653-82-4 ]
[ 540-10-3 ]
Reference:
[1] Chemistry - A European Journal, 2013, vol. 19, # 15, p. 4732 - 4741
15
[ 112-39-0 ]
[ 36653-82-4 ]
[ 629-80-1 ]
[ 502-73-8 ]
[ 540-10-3 ]
[ 57-10-3 ]
Reference:
[1] Applied Catalysis A: General, 2016, vol. 526, p. 183 - 190
16
[ 127-47-9 ]
[ 57-10-3 ]
[ 79-81-2 ]
Reference:
[1] Journal of Nanoscience and Nanotechnology, 2011, vol. 11, # 9, p. 7593 - 7602
[2] Journal of Lipid Research, 2014, vol. 55, # 2, p. 319 - 328
17
[ 127-47-9 ]
[ 57-10-3 ]
[ 79-81-2 ]
[ 68-26-8 ]
Yield
Reaction Conditions
Operation in experiment
78%
With Novozyme 435 (from Candida antarctica immobilized on acrylic resin); Amberlyst A-21 In toluene at 20℃; for 15 h; Enzymatic reaction
Example 28; Preparation of Retinyl Palmitate in the Presence of Amberlyst A-21; Retinyl acetate (1.00 g; 3.04 mmol) was dissolved in 8.5 mL of toluene and palmitic acid (1.56 g; 6.09 mmol; 2.0 equiv) was added followed by 120 mg of Novozyme 435 and 0.5g of dried Amberlyst A-21. The reaction mixture was stirred at RT for 15 h, at which point a sample was removed and analyzed by HPLC, indicating 89.2percent conversion to retinyl palmitate with 9.1percent retinyl acetate and 1.7percent retinol. The reaction mixture was filtered and concentrated, then concentrated twice with heptane (10 mL each). The residue was dissolved in heptane (15 mL) and washed with 2.x.20 mL with a 1:1 mixture of 10percent aqueous potassium carbonate and methanol. The organic layer was washed further with a mixture of saturated sodium bicarbonate (2.5 mL), water (7.5 mL), and methanol (10 mL), dried (sodium sulfate) and concentrated to afford 1.25 g (78percent) of a yellow oil. Analysis of this product indicated 91.2percent retinyl palmitate (HPLC area percent), 0.4 wt percent palmitic acid, and 0.2percent retinol. HPLC (4.6.x.150 mm Zorbax SB-C8 column [Agilent], 3.5μ thickness, methanol eluent, detection at 350 nm): tR 5.52 min (retinyl palmitate); tR 2.32 min (retinyl acetate); tR 2.08 min (retinol).
With alumina methanesulfonic acid; at 120℃; for 0.333333h;Microwave irradiation;
General procedure: In a typical reaction, AMA 2:3 (332 g, 0.6 mol), the corresponding carboxylicacid (1 mol), and alcohol (1.5-2 mol) were mixed in the provided reaction glass tubeequipped with a screw cap and magnetic agitation until a wet mixture was achieved.The reaction mixture was irradiated with microwaves (Anton Parr Monowave 300reactor) at 80 C for 8 min or 120 C for 20 min. On cooling, the mixture was diluted with dichloromethane (41 mL), filtered under gravity, and washed with dichloromethane;then the filtrate was washed with Na2CO3 (ss) and water. The organic layerwas dried over Na2SO4, filtered, and concentrated under reduced pressure to give theester.
95.5%
With 8-hydroxyquinoline sulphate; at 85℃; for 4h;
In a 100 mL three-necked flask, 0.05 mol of palmitic acid, 0.35 mol of ethanol and 0.03 mol of 8-hydroxyQuinoline bisulfate ionic liquid, to maintain the reaction temperature of about 85 , equipped with reflux condenser, stirring at constant temperature 4h. anti-At the end of this step, the unreacted excess ethanol was removed under reduced pressure, the mixture was transferred to a separatory funnel, and an appropriate amount of saturated brine was addedPolyester, shaking, to be layered, discard the lower water, keep the upper oil, repeated washing 2 to 3 times, the final oilThe product was the product palmitic acid ethyl ester in a yield of 95.5%.
84.9%
With glycine ethyl ester hydrochloride; In cyclohexane; at 78℃; for 8h;
Into a 500 mL three-necked flask equipped with a thermometer and a water separator, add 150 mL of absolute ethanol, 51.3 g (0.2 mol) of palmitic acid, stir and mix well, and add 50 mL of cyclohexane and 2.52 g (0.02 mol) of glycine methyl ester hydrochloride. The temperature was raised to reflux (temperature: 78 C) and the reaction was performed for 8 hours. After the temperature was lowered to room temperature, the solvent was recovered by concentration, and 50 mL of water was added to the concentrate, followed by extraction with 150 mL of ethyl acetate. The ethyl acetate layer was washed twice with 50% aqueous 5% sodium chloride, each 50 mL. The ethyl acetate layer was concentrated to obtain 55.4 g of a crude product as a final product, which was distilled under reduced pressure, and collected 171-175oC / 0.02 MPa to obtain 48.3 g of ethyl palmitate,The final product was a light yellow oily liquid, which became white crystals upon standing.The purity by gas chromatography was 98.6% and the reaction yield was 84.9%. The structure was confirmed by nuclear magnetic characterization.
91.7%Chromat.
at 300℃; under 112511 Torr; for 0.233333h;
An esterification reaction of a fatty acid and alcohol or a transesterification of rapeseed oil and alcohol was conducted using, as a raw material, fats and oils and alcohols shown in Table 7, under conditions of mole ratio, temperature, pressure and reaction time shown in Table 7. Since about 98.5% of rapeseed oil is composed of a tri-glyceride, the reaction from rapeseed oil can be judged to be a transesterification. The reaction product was subjected to HPLC analysis in the same manner as in Example 1, from the HPLC analysis result, conversion into a fatty acid alkyl ester from a fatty acid or rapeseed oil (=yield of alkyl ester) was obtained. The results are shown in Table 7 together with the reaction conditions. TABLE 7 Alcohol/fats and oils Temperature Pressure Reaction Example (mole ratio) Fats and oils Alcohol ( C.) (Mpa) time (min) Yield (%) Example 42/1C18-3 Methanol 300 20 8 96.2 5-1 Example 42/1C18-2 300 20 8 95.1 5-2 Example 42/1C18-1 300 20 8 95.8 5-3 Example 42/1C18-0 300 20 8 94.7 5-4 Example 42/1C16-0 300 20 8 94.0 5-5 Example 42/1 Rapeseed 300 20 15 98.0 5-6 oil Example 42/1 Rapeseed 350 43 4 98.0 5-7 oil Example 42/1C18-3 Ethanol 300 15 12 94.6 5-8 Example 42/1C18-2 300 15 14 97.4 5-9 Example 42/1C18-1 300 15 14 95.9 5-10 Example 42/1C18-0 300 15 15 91.2 5-11 Example 42/1C16-0 300 15 14 91.7 5-12 Example 42/1 Rapeseed 300 15 45 96.7 5-13 oil Example 42/1 Rapeseed 350 25 10 97.1 5-14 oil Example 42/1C18-3 1-propanol 300 10 15 97.0 5-15 Example 42/1C18-2 300 10 14 92.7 5-16 Example 42/1C18-1 300 10 14 92.3 5-17 Example 42/1C18-0 300 10 14 89.6 5-18 Example 42/1C16-0 300 10 14 90.1 5-19 Example 42/1 Rapeseed 300 10 45 96.1 5-20 oil Example 42/1 Rapeseed 350 23 14 98.8 5-21 oil Example 42/1C18-3 1-butonal 300 9 15 97.3 5-22 Example 42/1C18-2 300 9 14 92.4 5-23 Example 42/1C18-1 300 9 14 86.1 5-24 Example 42/1C18-0 300 9 14 82.5 5-25 Example 42/1C16-0 300 9 14 81.1 5-26 Example 42/1 Rapeseed 300 9 45 87.1 5-27 oil Example 42/1 Rapeseed 350 23 14 95.3 5-28 oil Example 42/1 Rapeseed 1-octanol 300 6 45 68.7 5-29 oil Example 42/1 Rapeseed 350 19 20 90.7 5-30 oil C16-0: palmitic acid, C18-0: stearic acid, C18-1: oleic acid, C18-2: linoleic acid, C18-3: linolenic acid
amberlyst-15; at 80 - 120℃; under 3620.13 Torr; for 1 - 6h;Conversion of starting material;
EXAMPLE 1; Palmitic acid (90%, Sigina-Aldrichi) was combined with ethanol at a composition of 1 mole palmitic acid to 5 moles ethanol. Palmitic acid is a solid at room temperature and must be heated to about 60 C. to first melt it prior to mixing with hot ethanol, or alternatively the mixture can be combined and heated to about 50 C., at which point this amount of palmitic acid will dissolve in ethanol. The reactants were then heated and pumped through a packed bed catalytic reactor containing Amberlyst-15. The reactants were maintained at a temperature above 60 C. prior to entering the catalytic reactor. The temperature of the reactor was kept at a temperature of between 60 C. and 120 C. For each run an elevated pressure was maintained on the reactor and pre-reactor feed lines suitable to keep the ethanol and reaction by-product water in a liquid state. Run 1 used a pre-reactor temperature of 80 C. and a reactor temperature of 120 C. (reactor contained a packed bed of Amberlyst-15) and a residence time in the packed bed reactor of 2 hours. The pressure was maintained at 70 psia (the bubble point pressure of ethanol at 120 C. is 62 psia). The acid value of the product that exited the catalytic reactor was determined by ASTM method D1980-87. The acid value (AV) of the product was 10.8 (mg KOH/g sample). The original acid value of the feedstock was 115.0, demonstrating that a majority of the free fatty acid was converted to the ethyl ester product in the reactor. The AV of 10.8 is very near the reported equilibrium concentration of free fatty acid for this esterification reaction. The product was a clear liquid at room temperature. The product from the reactor contained the ethyl palmitate ester, a small amount of palmitic acid, water and excess ethanol at a concentration approximately at equilibrium. Run 2 was performed using the same palmitic acid and ethanol (1:5 mole:mole) feed and the same reactor and catalyst (Amberlyst-15) as run 1, but the residence time was 1 hour (at 120 C. and at a pressure of 70 psia). The product from run 2 had an acid value of 13.9, slightly above the equilibrium value. Run 3 was performed using the same palmitic acid and ethanol (1:5 mole:mole) feed and the same reactor and catalyst (Amberlyst-15) as run 1, but the residence time was 6 hours at 110 C. and at a pressure of 70 psia. The product from run 3 had an acid value of 16.6. Thus a large fraction of the free fatty acids were converted to alkyl ester in one pass at these conditions. Run 4 was performed using the same palmitic acid and ethanol (1:5 mole:mole) feed and the same reactor and catalyst (Amberlyst-15) as run 1, but the residence time was 4 hours (at 120 C. and at a pressure of 70 psia). The product from run 4 had an acid value of 10.8, indicating that the product from Run 1 (with a 2 hour residence time) is essentially at equilibrium.
95%Chromat.
With sulfuric acid; for 4h;Reflux;
General procedure: General procedure for the synthesis of compounds (6a-p); organic acid (0.40 mmol.), and catalyst (0.0005 mmol.) was combined with 20 mL ethanol in a 50 mL round bottomed flask equipped with a stir bar. Reaction was allowed to stir at reflux temperature for the appropriate amount of time (4 h). After completion of reaction, the reaction mixture was concentrated in vacuum to give a crude product which was analyzed by 1H NMR and GC-MS.
With zirconium containing 2-aminoterephthalate metal organic framework; at 78℃;Kinetics;
General procedure: In view of the good catalytic activity and recyclability of UiO-type MOFs for the esterification of lauric acid with MeOH and EtOH,we wanted to investigate the applicability of the MOFs to otherbiomass derived free fatty acids with longer chain lengths, bothsaturated and unsaturated. Thus, we extended our study to theesterification with MeOH and EtOH of palmitic (hexadecanoic acid,C16), Stearic (octadecanoic acid, C18), Oleic (cis-9-octadecenoicacid C18:1), linoleic (cis,cis-9,12-octadecadienoic acid, C18:2) and-linolenic acids (cis,cis,cis-9,12,15-octadecatrienoic acid, C18:3).For the sake of brevity, the complete catalytic data obtained for eachfatty acid and the comparison with other acid catalysts from theliterature is provided as Supporting Information (Tables S1-S10).In order to illustrate the dependence of the chain length andunsaturation degree of the fatty acid on reaction rate, Fig. 3 showsthe calculated pseudo-first order reaction rate constants, k, of ester-ification of various fatty acids with ethanol over UiO-66-NH2. Thesame tendency was also observed for UiO-66, although this mate-rial was in general less active than UiO-66-NH2(as already observed C12 for C12 esterification commented above). As it can be observed, thereaction rate decreases as the chain length and the degree of unsa-turation of the fatty acid increases. This is probably due to higheradsorption of the unsaturated fatty acid (or fatty ester) on the sur-face of the solid, which causes the progressive deactivativation ofthe catalyst. However, it is worth mentioning that this deactivationdue to product adsorption is fully reversible, and the activity of thecatalysts is completely recovered by simply washing with EtOH.In conclusion, the above experiments demonstrates that bothZr-containing UiOs can efficiently catalyze the esterification of var-ious fatty acids with MeOH and EtOH, being less active as the alkylchain length and degree of unsaturation of the acid increases. It isalso worth mentioning that in all the reactions tested, the Zr-MOFswere found to be stable and reusable without significant loss ofactivity, as we have previously demonstrated for the esterificationof C12 with EtOH over UiO-66-NH2.
With trimethylcyclohexylammonium methanesulfonate; toluene-4-sulfonic acid; at 60℃; under 760.051 Torr; for 5h;
According to the flow of Figure 2, the following processing is performed:2.5 mol of hexadecanoic acid, 2.5 mol of ethanol and 0.75 mol of octyltrimethylammonium methanesulfonate-p-toluenesulfonateThe molten solvent (the molar ratio of trimethylcyclohexylammonium methanesulfonate to p-toluenesulfonic acid is 1:2) is added to the esterification reactor to form the ester.The reaction vessel was heated to 60 C, and the reaction was stirred at normal pressure for 5 hours, and the stirring speed was 1000 rpm. After the reaction, the reaction solution is introduced into the decantationThe device was allowed to stand for phase separation and the rest time was 5 h. The upper liquid (ester phase) and the lower liquid (water) obtained after phase separation in the decanterThe phase is introduced into the washing tank and the flash tank respectively to carry out product ester purification and raw materials (mainly eutectic solvents, carboxylic acids and alcohols).Received. The working pressure of the washing tank is normal pressure, the operating temperature is room temperature, and the mass fraction is taken from the top of the washing tank.93% product ethyl hexadecanate, ie high purity ester, a mixture of eutectic solvent and water at the bottom, introduced into the flash tank; flashingThe tank has an operating pressure of 0.01 bar and an operating temperature of 200 C. The unreacted raw material is taken from the top of the flash tank and containsA mixture of water and ester with a eutectic solvent having a mass fraction of 99.99% at the bottom. Low eutectic solution obtained at the bottom of the flash tankThe agent is respectively exchanged to a temperature of 60 C through a heat exchanger and returned to the esterification reactor for recycling. Mixture produced at the top of the flash tankThe alcohol recovery tower is introduced, the actual number of trays of the alcohol recovery tower is 50, the operating pressure is normal pressure, the operation reflux ratio is 5.5, and the alcohol recovery towerThe unreacted alcohol was obtained from the top of the column, and was cooled to 60 C by a heat exchanger, and then returned to the esterification reactor for recycling. Recycling alcoholThe material of the Tata kettle is introduced into the carboxylic acid recovery tower. The actual number of plates in the carboxylic acid recovery column is 55, the operating pressure is normal pressure, and the operation is refluxed.The ratio is 4.2, the by-product water is obtained at the top of the acid recovery tower, and finally the water is removed from the esterification reaction system, and the acid recovery tower is obtained.The column kettle is obtained as a mixture containing unreacted carboxylic acid and a part of the product, and is cooled to 60 C by a heat exchanger to return to the esterification reaction.The kettle should be recycled. The yield of ethyl hexadecanate in Example 18 was 99.0%, and the purity was 93%.
sulfuric acid; In water; at 70 - 75℃; for 6.0h;Product distribution / selectivity;
COMPARATIVE EXAMPLE 1; This example demonstrates a typical prior art process of using a conventional acid catalyst. A reactor was charged with palmitic acid (7.79 parts), cetyl alcohol (14.71 parts) and water (6.47 parts). After warming under agitation to 70-75 C., sulphuric acid (10% aqueous solution, 1.15 parts) was added and the mixing temperature maintained for six hours. The reaction mixture was biphasic. A sample of the mixture revealed that 8.2 mole % of the palmitic acid was converted into the ester.
sulfuric acid; 1-butanesulfonic acid sodium salt; In water; at 70 - 75℃; for 6.0h;Product distribution / selectivity;
EXAMPLES 14-33; Experiments were conducted to study the effect of the amount and type of hydrolyzable catalyst. The reactor was charged with palmitic acid, cetyl alcohol, the sodium salt of the hydrolyzable catalyst, and water. After warming under agitation to 70-75 C., sulphuric acid is added and the mixing and temperature maintained for 6 hours. Samples of the mixture are taken to determine the mole conversion of palmitic acid into ester. The results are shown in Table 3 below:
sulfuric acid; sodium hexyl sulfate; In water; at 70 - 75℃; for 6.0h;Product distribution / selectivity;
EXAMPLES 14-33; Experiments were conducted to study the effect of the amount and type of hydrolyzable catalyst. The reactor was charged with palmitic acid, cetyl alcohol, the sodium salt of the hydrolyzable catalyst, and water. After warming under agitation to 70-75 C., sulphuric acid is added and the mixing and temperature maintained for 6 hours. Samples of the mixture are taken to determine the mole conversion of palmitic acid into ester. The results are shown in Table 3 below:
sulfuric acid; sodium dodecyl-sulfate; In water; at 70 - 75℃; for 6.0h;Product distribution / selectivity;
EXAMPLES 3-7; Experiments were conducted to determine the preferred ratio of carboxylic acid to alcohol. A reactor was charged with various amounts of palmitic acid, cetyl alcohol, sodium lauryl sulphate, (10% aqueous solution, 6.75 parts), and water (0.29 parts). After warming under agitation to 70-75 C., sulphuric acid (10% aqueous solution, 1.26 parts) was added and the mixing and temperature maintained for six hours. Samples of the reaction products were taken to determine the mole % conversion of palmitic acid into ester. The results are shown in Table 1 below:; Experiments were conducted to study the amount and type of strong acid. A reactor was charged with palmitic acid (1.56 parts), cetyl alcohol (2.94 parts), sodium lauryl sulphate (0.14 parts), and water. After warming under agitation at 70-75 C., the strong acid was added and the mixing and temperature maintained for 6 hours. Samples of the mixture were taken to determine the mole conversion of the palmitic acid into ester. The results are shown in Table 2 below:; Experiments were conducted to study the effect of the amount and type of hydrolyzable catalyst. The reactor was charged with palmitic acid, cetyl alcohol, the sodium salt of the hydrolyzable catalyst, and water. After warming under agitation to 70-75 C., sulphuric acid is added and the mixing and temperature maintained for 6 hours. Samples of the mixture are taken to determine the mole conversion of palmitic acid into ester. The results are shown in Table 3 below:
sulfuric acid; In water; at 70 - 75℃; for 6.0h;Product distribution / selectivity;
EXAMPLES 14-33; Experiments were conducted to study the effect of the amount and type of hydrolyzable catalyst. The reactor was charged with palmitic acid, cetyl alcohol, the sodium salt of the hydrolyzable catalyst, and water. After warming under agitation to 70-75 C., sulphuric acid is added and the mixing and temperature maintained for 6 hours. Samples of the mixture are taken to determine the mole conversion of palmitic acid into ester. The results are shown in Table 3 below:
sulfuric acid; In water; at 70 - 75℃; for 6.0h;Product distribution / selectivity;
EXAMPLES 14-33; Experiments were conducted to study the effect of the amount and type of hydrolyzable catalyst. The reactor was charged with palmitic acid, cetyl alcohol, the sodium salt of the hydrolyzable catalyst, and water. After warming under agitation to 70-75 C., sulphuric acid is added and the mixing and temperature maintained for 6 hours. Samples of the mixture are taken to determine the mole conversion of palmitic acid into ester. The results are shown in Table 3 below:
sulfuric acid; In water; at 70 - 75℃; for 6.0h;Product distribution / selectivity;
EXAMPLES 14-33; Experiments were conducted to study the effect of the amount and type of hydrolyzable catalyst. The reactor was charged with palmitic acid, cetyl alcohol, the sodium salt of the hydrolyzable catalyst, and water. After warming under agitation to 70-75 C., sulphuric acid is added and the mixing and temperature maintained for 6 hours. Samples of the mixture are taken to determine the mole conversion of palmitic acid into ester. The results are shown in Table 3 below:
sulfuric acid; In water; at 70 - 75℃; for 6.0h;Product distribution / selectivity;
EXAMPLES 14-33; Experiments were conducted to study the effect of the amount and type of hydrolyzable catalyst. The reactor was charged with palmitic acid, cetyl alcohol, the sodium salt of the hydrolyzable catalyst, and water. After warming under agitation to 70-75 C., sulphuric acid is added and the mixing and temperature maintained for 6 hours. Samples of the mixture are taken to determine the mole conversion of palmitic acid into ester. The results are shown in Table 3 below:
sulfuric acid; In water; at 70 - 75℃; for 6.0h;Product distribution / selectivity;
EXAMPLES 14-33; Experiments were conducted to study the effect of the amount and type of hydrolyzable catalyst. The reactor was charged with palmitic acid, cetyl alcohol, the sodium salt of the hydrolyzable catalyst, and water. After warming under agitation to 70-75 C., sulphuric acid is added and the mixing and temperature maintained for 6 hours. Samples of the mixture are taken to determine the mole conversion of palmitic acid into ester. The results are shown in Table 3 below:
sulfuric acid; sodium octylsulfonate; In water; at 70 - 75℃; for 6.0h;Product distribution / selectivity;
EXAMPLES 14-33; Experiments were conducted to study the effect of the amount and type of hydrolyzable catalyst. The reactor was charged with palmitic acid, cetyl alcohol, the sodium salt of the hydrolyzable catalyst, and water. After warming under agitation to 70-75 C., sulphuric acid is added and the mixing and temperature maintained for 6 hours. Samples of the mixture are taken to determine the mole conversion of palmitic acid into ester. The results are shown in Table 3 below:
sodium lauryl sulfate, mixed alcohol sulfates; mixture of; sulfuric acid; In water; at 70 - 75℃; for 6.0h;Product distribution / selectivity;
EXAMPLES 14-33; Experiments were conducted to study the effect of the amount and type of hydrolyzable catalyst. The reactor was charged with palmitic acid, cetyl alcohol, the sodium salt of the hydrolyzable catalyst, and water. After warming under agitation to 70-75 C., sulphuric acid is added and the mixing and temperature maintained for 6 hours. Samples of the mixture are taken to determine the mole conversion of palmitic acid into ester. The results are shown in Table 3 below:
With sodium dodecyl-sulfate;hydrogenchloride; In water; at 70 - 75℃; for 6.0h;Product distribution / selectivity;
EXAMPLES 8-13; Experiments were conducted to study the amount and type of strong acid. A reactor was charged with palmitic acid (1.56 parts), cetyl alcohol (2.94 parts), sodium lauryl sulphate (0.14 parts), and water. After warming under agitation at 70-75 C., the strong acid was added and the mixing and temperature maintained for 6 hours. Samples of the mixture were taken to determine the mole conversion of the palmitic acid into ester. The results are shown in Table 2 below:
With sodium dodecyl-sulfate;nitric acid; In water; at 70 - 75℃; for 6.0h;Product distribution / selectivity;
EXAMPLES 8-13; Experiments were conducted to study the amount and type of strong acid. A reactor was charged with palmitic acid (1.56 parts), cetyl alcohol (2.94 parts), sodium lauryl sulphate (0.14 parts), and water. After warming under agitation at 70-75 C., the strong acid was added and the mixing and temperature maintained for 6 hours. Samples of the mixture were taken to determine the mole conversion of the palmitic acid into ester. The results are shown in Table 2 below:
98.3%Chromat.
With tungsten oxide impregnated Zr incorporated mesoporous silica SBA-15; In 1,3,5-trimethyl-benzene; at 162℃; for 6.0h;Inert atmosphere; Dean-Stark;
Cetyl alcohol (CA) and palmitic acid (PA) esterification reactions were performed under N2 atmosphere in a four necked round bot-tom flask (250 ml) equipped with a Teflon coated magnetic stirring bar with a stirring rate of 520 rpm and a Dean Stark apparatus surmounted with a reflux condenser. In a typical experiment, 160 mgof catalyst was added into 25 ml of mesitylene and heated up to reaction temperature of 162 C. An equimolar solution of palmitic acid and cetyl alcohol (6 mmol) in 15 ml of mesitylene at room temperature was added into the reactor. All the reactions were carried out for a reaction time of 6 h. In a preliminary set of experiments (Table 1) it was found that the reaction was not controlled by external diffusion at 520 rpm. Samples taken at regular intervals were analyzed by Agilent 6890 gas chromatography using Ultra 1(25 m × 0.3 mm) capillary column equipped with FID. The injector temperature was 280C and the detector temperature was 320 C.The GC oven temperature was changed from 50C at 12C/min to 300C where it was kept for 35 min. Helium was used as the car-rier gas at a flow rate of 37.3 ml/min The split ratio was 24.9:1. Conversion of cetyl alcohol (CA), yield of cetyl palmitate (CP) and the selectivity to CP were defined as below. Conversion(%)= (CAin -CAout )CAin×100 Yield(%)= CPoutCAin×100 Selectivity to CP(%) = CPout(CAin -CAout )×100
With C28H60O3PS(1+)*C2F6NO4S2(1-); at 60℃; for 6.0h;Sealed tube;
General procedure: In a 15 mL vial containing a stirring bar,0.3 mmol of the ionic liquid catalyst shown in Table 11 mmol of carboxylic acid,And 1 mmol of alcohol was added.Seal the vial with a cap,Immerse the vial in a water bath or oil bath adjusted to the temperature shown in Table 1,Stir for 6 hoursCarboxylic acid and alcohol were reacted.After completion of the reaction, in all examples,The liquid phase containing the ester compound (hereinafter sometimes referred to as ?product phase?) was separated from the ionic liquid phase.
With ammonia; zircornium(IV) n-propoxide; at 165℃; for 7h;
General procedure: According to the embodiment of the present invention described above, stearic acid amide, which is a kind of carboxylic acid amide compound, was prepared in Example 1 as follows. First, the carboxylic acid injector 250 injects 1000 g of stearic acid into the heater 100, and the heater 100 heats 1000 g of stearic acid to 120 C. Subsequently, when stearic acid is injected into the first reaction tank 210, the first catalyst injector 261 injects 10 g of tetraisopropyl titanium, which is a metal catalyst, into the first reaction tank 210, Was heated by the heater attached to the first reaction tank 210. When 150 g of stearic acid was charged into the first reaction tank 210, the first ammonia injector 281 started to feed the ammonia gas through the ammonia pipe 283 at a rate of 100 L / hr. When 500 g of stearic acid was charged into the first reaction tank 210, the introduction of stearic acid into the first reaction tank 210 was stopped. The propeller in the first reaction tank 210 was mixed with stearic acid and ammonia while maintaining the reaction temperature at 165 C in the first reaction tank 210.Next, when the supply of the stearic acid to the first reaction tank 210 is stopped, the 500 g of stearic acid remaining in the heater 100 through the valve is changed to be supplied to the second reaction tank 220. When stearic acid is injected into the second reaction tank 220, the second catalyst injector 262 injects 10 g of tetraisopropyl titanium as a metal catalyst into the second reaction tank 220 and starts heating the second reaction tank 220 . When 150 g of stearic acid was charged into the second reaction tank 220, the second ammonia feeder 282 started to feed the ammonia gas through the ammonia pipe 283 at a rate of 100 L / hr. When all 500 g of stearic acid was fed to the second reaction tank, the addition of stearic acid to the second reaction tank was stopped. The propeller in the second reaction tank 220 was mixed with stearic acid and ammonia while maintaining the reaction temperature of 165 C in the second reaction tank 220.In Examples 2 to 10, other carboxylic acid amide compounds were prepared in the same manner as in Example 1, except that stearic acid was used instead of stearic acid and other carboxylic acids as shown in the following Table 1 as "acid".
25%
General procedure: To a stirred solution of the fatty acid (1.0 mmol, 1.0 equiv.) inCH2Cl2 (5 mL) was added CDI (0.178 g, 1.1 mmol, 1.1 equiv.).After 30 min at room temperature, the amine (1.1 mmol, 1.1equiv.) was added. After 12 h, CH2Cl2 (25 mL) was added, followedby saturated aqueous NH4Cl. The mixture was acidified topH 2 by addition of HCl, the organic phase was separated, andthe aqueous layer was further extracted with CH2Cl2 (3 × 10mL). The organic phases were combined, dried over Na2SO4, filtered,and concentrated in vacuo, to give the amide.
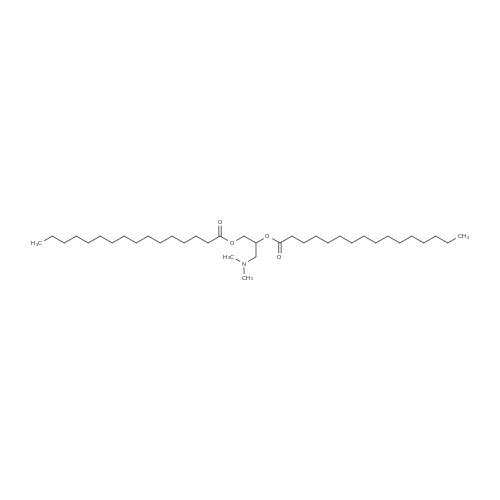
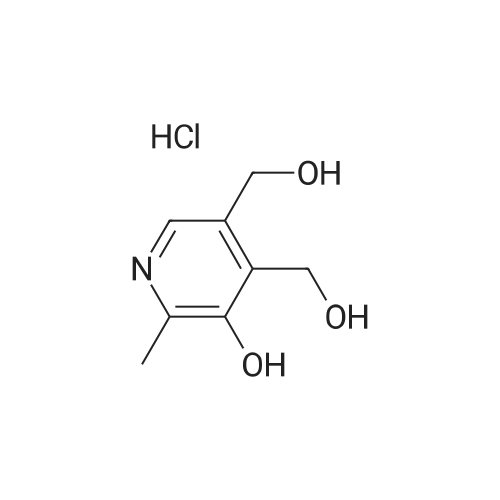
(1) 100 ml (about 184 g) 99-99.5% sulfuric acid was added to 250 ml four-neck flask with stirring, 35 g of palmitic acid was added at 30-35C dissolved with stirring for 0.5-1 hour. The temperature was dropped to 20-25C under protection of nitrogen, 17.6 grams of vitamin C was added with stirring, and stirring was stopped until completely dissolved, and insulated at 18-20C for 48 hours to obtain reaction liquid vitamin C palmitate. (2) The temperature was controlled to below 40C , the reaction solution was slowly poured into 200 ml 4-neck flask placed in ice-bath containing 200 ml cooling water. 700 ml of toluene was added and then subjected to extraction, heated to 60C and cooled naturally with stirring, when the temperature of the system was reduced to about 40C, stratified, and lower waste acid was separated leaving the toluene layer. (3) To the toluene layer 30-35 200 ml of water was added, stirred at 30-35C for 3 hours, crystallized at 30-35C for three hours, cooled in ice-water at 10-15C, stirred for one hour and filtered. The filter cake was washed with 50 ml of toluene, and then washed with 500 ml of water at 30-35C. (4) The filter cake was added to 500 ml water at 30-35C for 1 hour, filtered, and then washed with 200 ml water at 30-35C and the effluent water PH =7 was drained, discharged, and air-dried at room temperature for one day to constant weight. The crude product of vitamin C palmitate 33-34 g was obtained. Yield of 79.7-82.1%. (5) Take the above crude vitamin C palmitate, dissolved in 7.5 times the volume of 95% ethanol at 35-40 deg. C, filtered, washed with 0.5 volume of 95% ethanol and filtrate had crystallized, heated at 35-40C until fully dissolved and transferred to 500 ml 4-neck flask, rapidly cooled to -25--20C, crystallized by stirring for 3 hours, filtered, the filter cake was washed with 95% cold ethanol and vacuum-dried at 40-50C for 8 hours to obtain 30-31 g of product. Yield 90-92%.
ethyl 6-O-hexadecanoyl-D-glucopyranoside[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91%
lipase;
EXAMPLE 4 Preparation of ethyl 6-O-hexadecanoyl-D-glucopyranoside The title compound was obtained as a crude product (1220 g, 91% monoester, 7% <strong>[3198-49-0]ethyl D-glucopyranoside</strong>, 2% diesters) according to example 1 using <strong>[3198-49-0]ethyl D-glucopyranoside</strong> (603 g, 2.9 mol), hexadecanoic acid (1001 g, 3.91 mol) and immobilized lipase (30.5 g). The reaction was complete in 48 hours. NMR-spectra of the chromatographically purified product are in accordance with the 1 H and 13 C NMR-spectra given for the pure alpha and beta anomers in tables 4 a/4 b.
With alumina methanesulfonic acid; at 120℃; for 0.333333h;Microwave irradiation;
General procedure: In a typical reaction, AMA 2:3 (332 g, 0.6 mol), the corresponding carboxylicacid (1 mol), and alcohol (1.5-2 mol) were mixed in the provided reaction glass tubeequipped with a screw cap and magnetic agitation until a wet mixture was achieved.The reaction mixture was irradiated with microwaves (Anton Parr Monowave 300reactor) at 80 C for 8 min or 120 C for 20 min. On cooling, the mixture was diluted with dichloromethane (41 mL), filtered under gravity, and washed with dichloromethane;then the filtrate was washed with Na2CO3 (ss) and water. The organic layerwas dried over Na2SO4, filtered, and concentrated under reduced pressure to give theester.
76%
With glycine ethyl ester hydrochloride; In cyclohexane; at 82℃; for 10h;
In a 500 mL three-necked flask equipped with a thermometer and a water separator, add 150 mL of isopropanol, 51.3 g (0.2 mol) of palmitic acid, stir and mix well and add 50 mL of cyclohexane and 2.52 g (0.02 mol) of glycine methyl ester hydrochloride. The temperature was raised to reflux (temperature: 82 C) and the reaction was carried out for 10 hours. After the temperature was lowered to room temperature, the solvent was recovered by concentration, and 50 mL of water was added to the concentrate, followed by extraction with 150 mL of ethyl acetate. The ethyl acetate layer was washed twice with 50% aqueous 5% sodium chloride, each 50 mL. After concentrating the ethyl acetate layer, 51.3 g of a crude product of the final product was obtained. The product was distilled under reduced pressure, and 174-179oC / 0.01 MPa product was collected to obtain 45.4 g of methyl palmitate as a pale yellow oily liquid. The purity by gas chromatography was 97.3% and the reaction yield was 76.0%. The structure was confirmed by nuclear magnetic characterization.
With SBA-15 silica functionalized with 15 molar percentage of phenolsulfonic acid (15PholSO3H-SBA-15-p); for 12h;Reflux;Catalytic behavior;
Example 7 PholSO3H-SBA-15 Catalyzed Esterification of Palmitic AcidThe phenolsulfonic acid-functionalized SBA-15 (15PholSO3H-SBA-15-p) was used as the solid acid catalyst in the liquid phase esterification of palmitic acid (PA) with methanol (MeOH) and iso-propanol (iPrOH) to form methylpalmitate and iso-propylpalmitate as the products, respectively. The reactions were carried out at the reflux temperatures of the alcohols. The esters were found to be the only products in the present reaction condition based on the GC and GC-MS analyses. (0084) FIG. 7A and FIG. 7B show the conversions of palmitic acid in the esterification of palmitic acid with MeOH and iPrOH, respectively. In FIGS. 7A and 7B, the conversions of palmitic acid are shown as a function of reaction period over 15PholSO3H-SBA-15-p and commercially available Amberlyst-15 resin. It is clearly shown that the conversions of palmitic acid over 15PholSO3H-SBA-15-p increase much faster than those over Amberlyst-15 resin. After 12 hours reaction, significantly larger amounts of esters are obtained over 15PholSO3H-SBA-15-p than Amberlyst-15 resin. (0085) Table 5 demonstrates the recyclability of the 15PholSO3H-SBA-15-p catalyst. The used catalyst was regenerated by simple filtration and drying at 100 C. The catalytic activities of 15PholSO3H-SBA-15-p were well retained in comparison to that of the fresh catalyst after recycling for two times.
With silica supported tetrasulfonic acid functionalized ionic liquid; at 85℃; under 760.051 Torr; for 6h;Green chemistry;
Add 0.03 mol (7.69 g) of palmitic acid to a 100 mL one-neck flask. 0.15 mol (9.01 g) of isopropanol, Catalyst ionic liquid (j) 0.34g, It is used in an amount of 2% of the total mass of the raw materials palmitic acid and isopropanol. The separator is used to keep the generated water separated and removed in time during the reaction. The reaction was carried out at 85 C for 6 h under normal pressure, and the reaction was completed to filter out the solid catalyst. Allow to stand at room temperature, Rotary evaporation under reduced pressure, Excess isopropyl alcohol and moisture are removed.The ester layer filtrate is washed with alkali, After washing to neutral, Dry to get no (low) color, High quality isopropyl palmitate, The esterification rate is about 98%. The filtered solid catalyst is vacuum-dried with ethyl acetate to obtain a recovered ionic liquid. And it was used in the investigation of the catalytic cycle.
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride;dmap; In dichloromethane; at 20℃; for 7h;
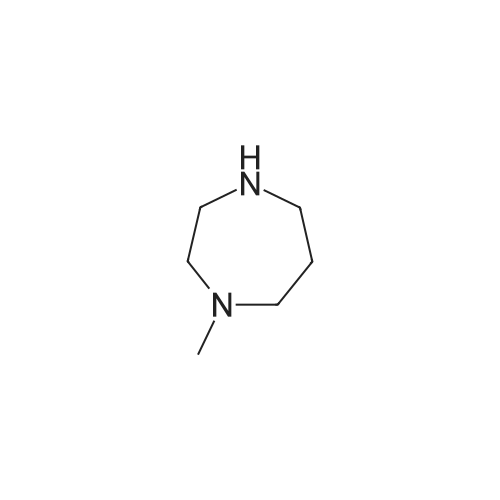
[249] Example 116 : Preparation of l-ethyl-l-methyl-4-palmitoyl-l,4-diazepan-l-ium iodide according to Reaction Scheme 3; [250] 1. Preparation of l-(4-methyl-1.4-diazepan-l-yl)hexadecan-l-one; [251][252] <strong>[4318-37-0]1-methylhomopiperazine</strong> (0.9 D, 7.54 mmol), l-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (1.4 g, 7.54 mmol), and 4-dimethylaminopyridine (0.2 g, 1.74 mmol) were added to a 0.1 M methylene chloride solution of palmitic acid ( 1.5 g, 5.80 mmol) under stirring and anhydrous conditions, and stirred at room temperature for 7 hours. The produced mixture was diluted with chloroform, washed with the saturated ammonium chloride solution three times, and then with the saturated brine solution. Then, the organic layer was dried over magnesium sulfate, and distilled off under reduced pressure. The resulting primary compound was purified by a silica gel column chromatography (eluent: 5% methanol/chloroform) to obtain the target compound in 92.4% yield (1.89 g).[253] 1H-NMR (300MHz, DMSO) delta 3.48-3.41 (m, 4H), 2.45-2.40(m, 4H), 2.28-2.22 (m, 5H), 1.81-1.71(m, 2H), 1.47(br s, 2H), 1.24(br s, 24H), 0.85(t, 3H, J=6.3 Hz)
92.4%
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 20℃; for 7h;
Example 116; Preparation of 1-ethyl-1-methyl-4-palmitoyl-1,4-diazepan-1-ium iodide according to Reaction Scheme 3; 1. Preparation of 1-(4-methyl-1,4-diazepan-1-yl)hexadecan-1-one; <strong>[4318-37-0]1-methylhomopiperazine</strong> (0.9, 7.54 mmol), 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (1.4 g, 7.54 mmol), and 4-dimethylaminopyridine (0.2 g, 1.74 mmol) were added to a 0.1 M methylene chloride solution of palmitic acid (1.5 g, 5.80 mmol) under stirring and anhydrous conditions, and stirred at room temperature for 7 hours. The produced mixture was diluted with chloroform, washed with the saturated ammonium chloride solution three times, and then with the saturated brine solution. Then, the organic layer was dried over magnesium sulfate, and distilled off under reduced pressure. The resulting primary compound was purified by a silica gel column chromatography (eluent: 5% methanol/chloroform) to obtain the target compound in 92.4% yield (1.89 g).1H-NMR (300 MHz, DMSO) delta 3.48-3.41 (m, 4H), 2.45-2.40 (m, 4H), 2.28-2.22 (m, 5H), 1.81-1.71 (m, 2H), 1.47 (br s, 2H), 1.24 (br s, 24H), 0.85 (t, 3H, J=6.3 Hz)
0059] the same construction as in Example 1 was realised in a pressure column. In this, an already pre-activated feed was used, with which a pre-reactor could be simulated. By means of slight excess pressure, a temperature in the range of the reaction zone of approximately 140 C. was set in the reaction zone. The following operating conditions led to a significantly higher reaction conversion. [0060] Feed streams: [0061] feed 1: 2.3 kg/h with the following composition (in % by weight): [TABLE-US-00002] isopropylpalmitate: 24.3 isopropanol: 21.6 palmitic acid: 52.1 water: 2.0 feed 2: isopropanol: 1.84 kg/h [0062] Product streams: [0063] head product: 1-43 kg/h-sump product: 2.71 kg/h [0064] Operating conditions: [0065] head pressure: 5100 mbar-return ratio: 0.2 [0066] temperature in the reaction zone: approximately 140 C. [0067] sump temperature: 192 C. [0068] Measure sump concentration (in % by weight): [TABLE-US-00003] isopropylpalmitate: 64.0 isopropanol: 7.7 palmitic acid: 28.3 water: 0.06
14.04%; 0.83%
at 98℃; under 759.826 Torr;
[0044] The exterification of palmitic acid with isopropanol was continually carried out in a column at atmospheric pressure under the following conditions: [0045] Reaction column: [0046] inner diameter: 50 mm. [0047] Enrichment section and stripping section: Sulzer packing BX [0048] Reaction zone: Katapak-S/filled with strongly acidic ion exchanger, which is stable at temperatures of up to 140 C. [0049] Feed streams: [0050] palmitic acid (feed 1): 2.16 kg/h [0051] isopropanol (feed 2): 1.44 kg/h [0052] Product streams: [0053] head product: 1.2 kg/h-sump product: 2.4 kg/h [0054] Operating conditions: [0055] head pressure: 1013 mbar-return ratio: 0.2 [0056] temperature in the reaction zone: approximately 98 C. [0057] sump temperature: 157.9 C. [0058] Measured sump concentrations (in % by weight): [TABLE-US-00001] isopropylpalmitate 14.04 isopropanol: 10.61 palmitic acid: 74.5 water: 0.83
To a stirred solution [OF RACEMIC 4- [4-DIMETHYLAMINO-1- (4-FLUORO-PHENYL)-1-HYDROXY-] [BUTYL]-3-HYDROXYMETHYL-BENZONITRILE] (0,29 mmol, 100 mg) and vinylbutyrate (0,29 mmol, [37 L)] in anhydrous toluene (2,925 ml) is added Novozymes 435, (0,2 mg) and 1,1 eq. Carboxylic acid. The reaction is heated to 40 degrees celcius and followed by HPLC. The enzyme is filtered off and washed with a small amount of toluene. The combined organic phases are evaporated in vacuo and subsequently analyzed on super critical fluid chromatography. Result is shown in table 19.
With phosphoric acid; In hexane; water; for 8.5h;Heating / reflux;Product distribution / selectivity;
Experiment A.-Hexane Solvent; Myristic acid/palmitic acid, 200 cc. of 85% phosphoric acid and 1800 ml. of hexane were mixed, heated to reflux and then 251 grams of cetyl alcohol added in 30 min. The mixture was refluxed further for 8 hours. Then the hot mixture consisted of a muddy acid layer and a opaque solvent layer which could not be separated by decantation or filtration. The mixture was further diluted with three volumes of hexane causing the slushy hexane layer to further soften enough to be separated from aqueous layer. The hexane layer was then cooled to bring about crystallization of fatty ester. The weight of cetyl myristate isolated was 294 grams which had a melting point of 54-59 C. The conversion, based on the cetyl alcohol used, was 63.71%
With phosphoric acid; In n-heptane; water; for 18.0h;Heating / reflux;Product distribution / selectivity;
Experiment B.-Heptane Solvent; Myristic acid/palmitic acid, 200 cc. of phosphoric acid, and 1800 ml. of heptane were mixed, heated to reflux and then 251 grams of cetyl alcohol refluxed further for 18 hours and separated as in example A. On crystallization, the cetyl myristate obtained was much darker in colour then in Experiment-A. [0030] It is evident that this process as exemplified by Experiment B is even less satisfactory than that set forth in Experiment-A
With phosphoric acid; In water; toluene; at 92℃; for 38.5h;Heating / reflux;Product distribution / selectivity;
EXAMPLE 1; Toluene Solvent; 1800 cc. of toluene, myristic acid/palmitic acid and 400 cc. of 85% phosphoric acid were mixed, heated to 92 C. and 251 grams of cetyl alcohol was introduced over a 30-minute period. When the addition was complete, the reaction mixture was further refluxed for 38 hours. The hot reaction mixture was a two phase system consisting of a toluene layer and an aqueous phosphoric acid layer. No solid material was present. The hot toluene layer was separated and mixed with charcoal to remove the undesired colouring matter. [0037] The filtrate was cooled to bring about crystallization of cetyl myristate which was isolated by filtration. The weight of cetyl myristate isolated was 436 grams which had a melting point of 54-58 C. The percentage conversion based on the cetyl alcohol employed was 92.3 percent
With phosphoric acid; In water; xylene; at 105℃; for 1.0h;Product distribution / selectivity;
EXAMPLE 2; Xylene Solvent; Myristic acid/palmitic acid, 250 grams of 85% phosphoric acid and 1000 cc. of xylene were mixed in a three neck flask provided with thermometer, agitator and reflux condenser. The temperature was increased to 105 with good agitation and 55 grams of cetyl alcohol was introduced over a one-hour period. After the reaction the supernatant xylene layer was drawn off, and the lower phosphoric acid layer was preserved for use in the following run. [0040] The xylene layer on cooling deposited a crystalline solid which weighed 154 gms. This material consisted of cetyl myristate and any unreacted fatty acid. The crude product was easily purified by recrystallization from hot xylene to yield pure cetyl myristate M.P.=54-56 C; EXAMPLE 3; Xylene Solvent; Myristic acid/palmitic acid, 400 cc. of 85% phosphoric acid and 2400 cc. of xylene were mixed in a three neck flask provided with a thermometer, agitator and reflux condenser. The temperature was raised to 105 C. with good agitation and 251 grams of cetyl alcohol was introduced with good agitation over a 1-hour period. The mixture reflux for 36 hour. Next, the supernatant xylene layer was drawn off, and the lower phosphoric acid layer was preserved for use in a subsequent run. The xylene layer on cooling deposited a crystalline solid which weighed 438 grams. This crude material was substantially cetyl myristate and was purified by recrystallization from hot xylene so as to yield pure cetyl myristate having a melting point of 54-56 C. [0045] The water which is formed by the employment of cetyl alcohol in the course of the reaction as in Example 2 dilutes the reaction mixture but can be readily removed by azeotropic distillation of the reaction mixture
With phosphoric acid; In water; at 95℃; for 0.5h;Product distribution / selectivity;
Experiment C.-Alkylation in Absence of a Solvent; Myristic acid/palmitic acid, 400 cc. of 85% phosphoric acid were mixed, heated to 95 C., and 251 grams of cetyl alcohol was added over a period of 30 minutes. The mixture further heated in vacuum and then on cooling. The reaction mixture, which contained a finely divided white solid, was diluted to 3000 ml. with water cooled to 25 C. and filtered. The white product was treated with hot water, and the mixture filtered hot to remove any alcohol. [0033] The unreacted fatty acid was present in a large quantity. The reaction was not complete
PREPARATIVE EXAMPLE 1; Synthesis of CLA by Alcaline Isomerization of Grape Seed Oil in Glycerol (The following synthesis makes the object of a co-pending application); 1 kg glycerol, 235 g potassium hydroxide (KOH) and 1000 g of grape seed oil were added into a 4-neck round bottom flask (5000 ml) equipped with a mechanical stirrer, a thermometer, a reflux condenser, and a nitrogen inlet, the nitrogen being introduced in first run through two oxygen traps.Nitrogen was bubbled into the reaction mixture for 20 min and the temperature was then raised to 90-100 C., and kept under mechanical stirring for about 20 minutes to convert the trigliceride in the corresponding potassium salts. The double phase system disappears to form a glyceric soap suspension, then heated at 230 C. under inert atmosphere and stirred for 4 hours.The reaction mixture was cooled to about 100 C., and the stirring stopped as the reaction mixture tend to reach very high viscosity during cooling. 2 l of water was then slowly added, and the mixture kept at 95 C. for 2 hour. This operation becomes necessary because of the neglegible presence of water and high content of glycerol causing fatty acids to be present as mono- and diglyceride from 5% to 10% by weight of the total lipid content. As partial glyceride esters tend to form W/O emulsion, the water addition and re-heating provides full saponification of the residual esterified fatty acid.The mixture was transferred into a becker, then cooled to room temperature and 50% w/v sulfuric acid was added to the mixture which was stirred for 1 hour until the pH stabilized at about 3.The acidulated oil phase formed a lower hydroglyceric layer and an upper fatty acid oil layer containing CLA, which was separated by decantating. Noteworthy, in industrial operation the separation could be carried out by centrifugation.The CLA was washed with water and finally it was made anhydrous with sodium sulphate and filtered, then it is stored in a dark bottle at 4 C. until time of use. Total yield about 770 g af an amber oil, whose GC-analysis is shown in Table 1; The composition of CLA appears to be a complex mixture, i.e. 9c, 11t- and 8c, 10t-octadecadienoic acids at 30.90%, 11c, 13t-10t, 12c-octadecadienoic acids at 32.05%, 11t, 13c-8c, 10c-9c, 11c-octadecadienoic acid at 1.55%, 10c, 12c-11c, 13c-11t, 13t, 9t, 11t-10t, 12t-8t,10t-octadecadienoic acids making the remaining part.
EXAMPLE 10; Preparation of Various Complexes Comprising Brimonidine and Selected CounterionsIn this experiment, various complexes comprising Brimonidine and counterions of one of the following acids were prepared: pamoic acid, capric acid, sebacic acid, hippuric acid, naproxen, 1-hydroxy-2-naphthoic acid, palmitic acid, and stearic acid. Variations of the procedure described in the following disclosure may be made within the skill of a person of ordinary skill in the art without departing from the scope of the present invention. Brimonidine free base in a preselected solvent was heated to about 60-70 C. The organic acid in another portion of the solvent was added into the heated mixture or was included in the original mixture before heating. The heating of the combined mixture was continued for an additional period, which was not critical. In certain embodiments, an antisolvent was added to the combined mixture, preferably at a lower temperature, to effect a precipitation of the complex of brimonidine and the counterion. It may be advantageous to remove a portion of the solvent and antisolvent to assist the precipitation. In certain other embodiments, the heated combined mixture was cooled down to a lower temperature, such as room temperature (or below) to effect the precipitation of the complex of brimonidine and the counterion. The precipitate was then filtered and dried to yield the final complex. The solubility of various complexes in water at the resulting pH is shown in Table 9.
With Novozyme 435 (from Candida antarctica immobilized on acrylic resin); Amberlyst A-21; In toluene; at 20℃; for 15.0h;Enzymatic reaction;Product distribution / selectivity;
Example 28; Preparation of Retinyl Palmitate in the Presence of Amberlyst A-21; Retinyl acetate (1.00 g; 3.04 mmol) was dissolved in 8.5 mL of toluene and palmitic acid (1.56 g; 6.09 mmol; 2.0 equiv) was added followed by 120 mg of Novozyme 435 and 0.5g of dried Amberlyst A-21. The reaction mixture was stirred at RT for 15 h, at which point a sample was removed and analyzed by HPLC, indicating 89.2% conversion to retinyl palmitate with 9.1% retinyl acetate and 1.7% retinol. The reaction mixture was filtered and concentrated, then concentrated twice with heptane (10 mL each). The residue was dissolved in heptane (15 mL) and washed with 2×20 mL with a 1:1 mixture of 10% aqueous potassium carbonate and methanol. The organic layer was washed further with a mixture of saturated sodium bicarbonate (2.5 mL), water (7.5 mL), and methanol (10 mL), dried (sodium sulfate) and concentrated to afford 1.25 g (78%) of a yellow oil. Analysis of this product indicated 91.2% retinyl palmitate (HPLC area percent), 0.4 wt % palmitic acid, and 0.2% retinol. HPLC (4.6×150 mm Zorbax SB-C8 column [Agilent], 3.5mu thickness, methanol eluent, detection at 350 nm): tR 5.52 min (retinyl palmitate); tR 2.32 min (retinyl acetate); tR 2.08 min (retinol).
4-[2-(dimethylamino)-1-(1-hydroxycyclohexyl)ethyl]phenol palmitate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In ethanol; at 20℃;Reflux;
Example 22[00168] A 100 ml one necked flask equipped with a magnetic stirrer was charged with ODV base (3 g, 11.39 mmol) and EtOH 95% (15ml), the suspension being stirred at reflux. Palmitic acid (3g 11.7 mmol) was added and a turbid solution appeared. A slurry was obtained after overnight stirring at room temperature. The mixture was filtered and dried at 5O0C under vacuum to get a mixture ODV base and ODV palmitate salt.
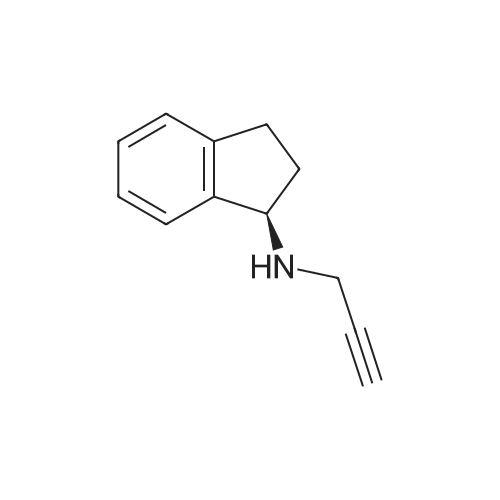
Example 24 : Preparation of <strong>[136236-51-6](1R)-2,3-dihydro-N-2-propynyl-1H-indane-1-amine</strong> palmitate (Rasagiline palmitate): Rasagiline base (81 g, 0.47 moles) was dissolved in IPA (810 ml) under stirring to obtain a solution. The obtained solution was cooled to 5C. A solution of palmitic acid (0.49 moles, 125.6 g) in IPA (200 ml) was added dropwise to the cooled solution. After complete addition, the solution was stirred for 30 min at 5-10C. The obtained solid was filtered and dried to get (172 g, 85%) of the title compound.
85%
In isopropyl alcohol; at 5 - 10℃; for 0.5h;
Example 24 Preparation of <strong>[136236-51-6](1R)-2,3-dihydro-N-2-propynyl-1H-indane-1-amine</strong> palmitate (Rasagiline Palmitate) Rasagiline base (81 g, 0.47 moles) was dissolved in IPA (810 ml) under stirring to obtain a solution. The obtained solution was cooled to 5 C. A solution of palmitic acid (0.49 moles, 125.6 g) in IPA (200 ml) was added dropwise to the cooled solution. After complete addition, the solution was stirred for 30 min at 5-10 C. The obtained solid was filtered and dried to get (172 g, 85%) of the title compound.
With novozyme 435; In toluene; at 20℃; for 20.0h;Schlenk technique; Inert atmosphere; Enzymatic reaction;
General procedure: Retinyl esters were synthesized via an enzyme-catalyzed transesterification (19) as follows. Into a dry Schlenk flask, retinyl acetate (33 mg, 0.10 mmol), Novozyme 435 (120 mg), and AberlystA-21 (50 mg) were suspended in dry toluene (5 ml). The reaction mixture was stirred under an atmosphere of N2 , and five equivalents (0.50 mmol) of the appropriate acid (palmitic, stearic, linoleic, or oleic) were added. After 20 h at room temperature, the reaction mixture was filtered and the solvent was removed under reduced pressure to give a mixture (approximately 1:4) of the desired retinyl ester and unreacted acid. The resulting mixtures were used without further purification as LC/MS/MS standards for the corresponding retinyl esters.
A mixture of methylene chloride, 3-(2-chloroethyl)-6,7,8,9-tetrahydro-2- methyl-9-hydroxy-4H-pyrido[ l ,2-a] pyrimidine-4-one, palmitic acid , 4-dimethyl amino pyridine and triethylamine was stirred for 15 min at 25-30 C, pivaloyl ' chloride was added slowly to above reaction mass at 25-40 C and the reaction was maintained for 2 hours at 30-35 C then the reaction mass was cooled to 25 C, charged purified water, separated the organic layer, washed the organic layer with dilute hydrochloric acid followed by water, distilled off methylene chloride completely and co distilled with acetonitrile. The resulted residue was dissolved in acetonitrile and added purified water slowly at 25-30 C, stirred the contents for 30 minutes at the same temperature and cooled the mass to 10-15 C stirred for 2 hours at the same temperature, filtered the isolated compound, washed the wet cake with chilled acetonitrile and dried the compound under vacuum.Yield: 95% (Theoretical).
95%
A mixture of methylene chloride, <strong>[130049-82-0]3-(2-chloroethyl)-6,7,8,9-tetrahydro-2-methyl-9-hydroxy-4H-pyrido[1,2-a]pyrimidine-4-one</strong>, palmitic acid, 4-dimethyl amino pyridine and triethylamine was stirred for 15 min at 25-30 C., pivaloyl chloride was added slowly to above reaction mass at 25-40 C. and the reaction was maintained for 2 hours at 30-35 C. then the reaction mass was cooled to 25 C., charged purified water, separated the organic layer, washed the organic layer with dilute hydrochloric acid followed by water, distilled off methylene chloride completely and co distilled with acetonitrile. The resulted residue was dissolved in acetonitrile and added purified water slowly at 25-30 C., stirred the contents for 30 minutes at the same temperature and cooled the mass to 10-15 C. stirred for 2 hours at the same temperature, filtered the isolated compound, washed the wet cake with chilled acetonitrile and dried the compound under vacuum. Yield: 95% (Theoretical).
With lipase from Pseudomonas stutzeri PS59; In aq. phosphate buffer; isopropyl alcohol; at 30℃; for 0.25h;pH 8.0;Enzymatic reaction;
General procedure: An assay mixture consisting of 1 ml of liquid ester or 1 g of solid ester, 3 ml of isopropanol, 5 ml of phosphate buffer (pH=8.0), and 1 ml of the lipase solution was incubated for 15 min at 30C with stirring at 180 rpm. The reaction was terminated by the addition of 95% ethanol, and the amount of liberated fatty acids after incubation was determined by titrating with 50 mM NaOH in the presence of two drops of phenolphthalein solution as the indicator. The control experiment was performed under the same conditions with the addition of 95% ethanol prior to the reaction. One unit of lipase activity was defined as the amount of enzyme required to liberate 1 mumol of free fatty acid per minute under the experimental conditions.
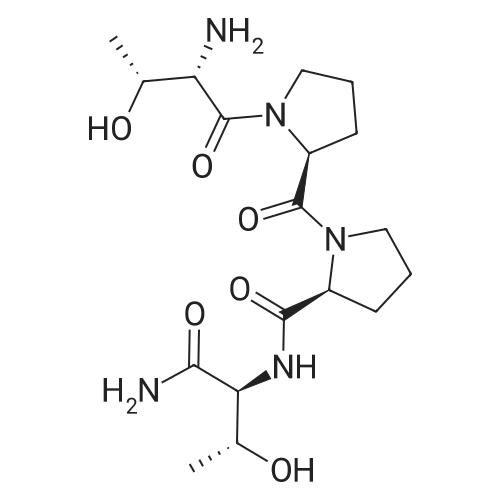
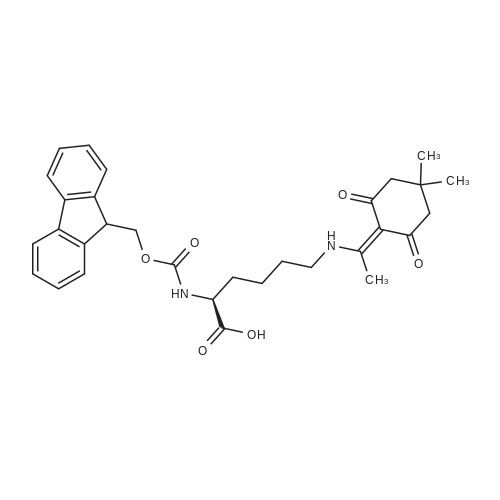
(S)-6-[(Diphenyl-p-tolyl-methyl)-amino]-2-(9H-fluoren-9-ylmethoxycarbonylamino)-hexanoic acid[ No CAS ]
C100H183N27O26[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
PACA?s 2 and 3 were synthesized using orthogonal protection group chemistry for the amines at the a and ? positionsof the lysine residue. Fmoc-Lys(Mtt)-OH was coupled to MBHA Rink Amide resin, followed by cleavage of the Mtt 10protecting group on the c-amine for palmitic acid coupling without affecting the Fmoc protection. This was followed by Fmoc removal on the a-amine to extend the peptide segment of the PA. The branching point in the PA?s was introduced ata lysine dendron using Fmoc-Lys(Mtt)-OH. To extend the first arm of PA?s, Fmoc on the a-amine was removed before Mtt. For molecule 2, Boc-Lys(Boc)-OH was coupled at the end of the first branch. Both a and c-amine positions of the lysine were blocked with Boc protection as it is more stableunder the cleavage conditions employed for the removal of Fmoc and Mu. For 3, <strong>[84624-27-1]Boc-Lys(Fmoc)-OH</strong> was coupled as the first branch. Fmoc was deprotected and tris-tert-butyl-protected 1,4,7,1 0-tetraazacyclododecane- 1,4,7,10 tetraacetic acid (DOTA) (purchased from Macrocyclics) was coupled atthe end of the first branch. Mtt was removed, and the other branch was grown using <strong>[84624-27-1]Boc-Lys(Fmoc)-OH</strong>. The peptide sequence RGDS was coupled to the c-amine in order to combine bioactivity and MR thnctionality. For 2, the DOTA moiety was coupled on SPPS to the N-terminus of the peptidesequence by using a tert-butyl ester-protected DOTA molecule. Compounds 2 and 3 were cleaved from the resin in 95:2.5:2.5 TFA:TIS:H20. Excess TFA was evaporated under reduced pressure, and crude PA solutions were triturated using cold ethet Compounds 2 and 3 were purified by reversephase HPLC, then dried under vacuum and characterized by MALDI-MS with a single peak found at 2308.66 (calcd 2308.87) and 2180.02 (calcd 2179.69), respectively. The final product was obtained by the addition of GdC13 with stirring at pH 6.5 for 72 h and purified by dialysis in deionized water(1000MWC Spectrum Laboratories Inc.). Samples were then lyophilized into a white powdet MALDI-MS showed peaks at 2464.40 (calcd 2463.09) and 2336.08 (calcd 2335.61) corresponding to 2 and 3 M+H, respectively.
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In chloroform; at 20℃; for 6h;
Into 10 mL of chloroform were dissolved 75 mg (0.2 mmol) of <strong>[26687-82-1]arctigenin</strong> and 48.5 mg (0.2 mmol) of palmitic acid. Thereto were further added 76.68 mg (0.4 mmol) of the water-soluble carbodiimide and 48 mg (0.4 mmol) of DMAP to cause the reactive components to react with each other in chloroform at room temperature for 6 hours. Water was added to the reaction liquid, and this system was stirred. The organic layer was then washed with 1 N HCL and saturated NaHCO3 solution in water. The chloroform layer was distilled off under a reduced pressure to yield 90.6 mg of palmitate <strong>[26687-82-1]arctigenin</strong>.; Molecular formula: C37H54O7; 6.92 (d, 1H), 6.76 (d, 1H), 6.74 (d, 1H), 6.66 (dd, 1H), 6.53 (dd, 1H), 6.49 (d, 1H), 4.17 (dd, 1H), 3.90 (dd, 1H), 3.85 (s, 3H), 3.82 (s, 3H), 3.75 (S, 3H), 2.97 (m, 2H), 2.54-2.56 (m, 6H), 1.75 (q, 2H), 1.25 (m, 24H), 0.88 (t, 3H)
With N-ethyl-N,N-diisopropylamine; HATU; In N,N-dimethyl-formamide; at 0 - 20℃; for 12.0h;
j00172j <strong>[117928-94-6]GLYX-13</strong> (58 mg, 1.4 mmol), palmitic acid (30 mg, 1.17 mmol), and DIPEA (45.7 mg, 3.5 mmol) were dissolved in DMF (5 mL) and cooled to 0 C. HATU (111 mg, 2.93 mmol) was added to the above reaction mixture at 0 C. The reaction mixture was stirred at rt for 12 h. After the completion of starting materials, the reaction mixture was diluted with water (2 x 5 mL). The aqueous layer was extracted with ethyl acetate (2 x 25 mL), washed with brinesolution (25 mL) and dried over anhydrous Na2504. Evaporation of the solvent furnished thecrude product, which was purified by column chromatography (100-200 mesh silica gel, 20%EtOAc in hexane) to yield 20 mg of the palmitic acid coupled <strong>[117928-94-6]GLYX-13</strong> NRX-7005 as agummy solid.
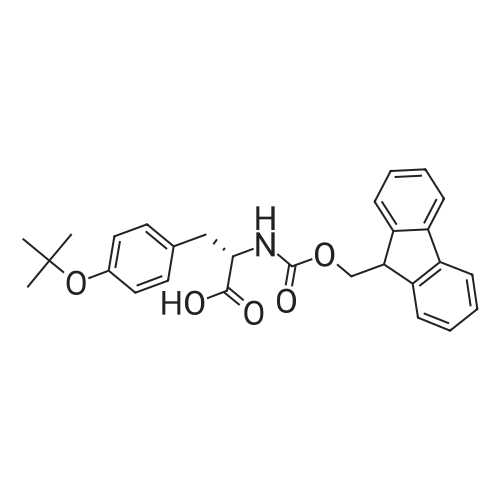
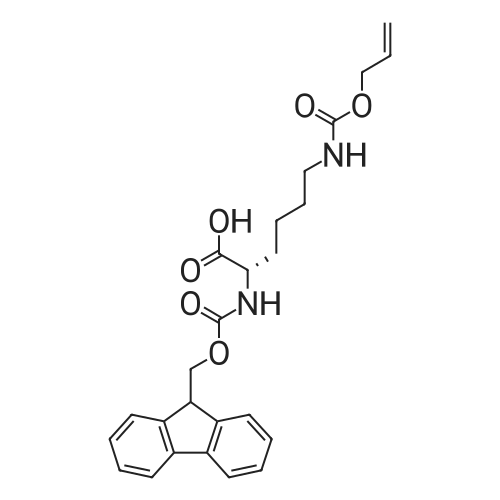
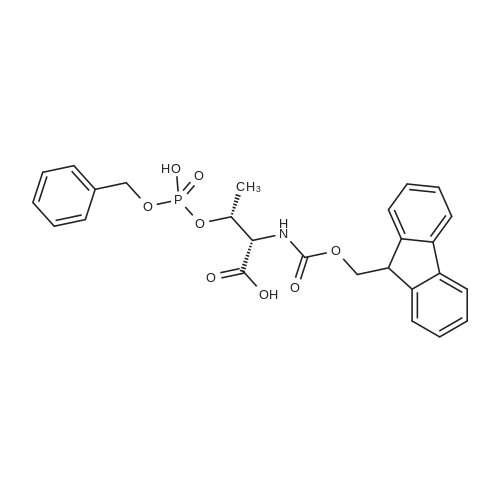
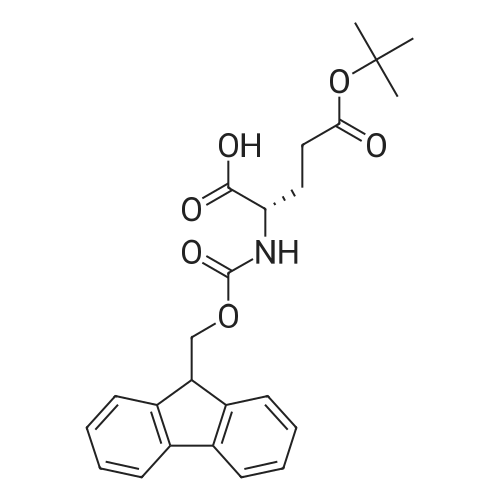
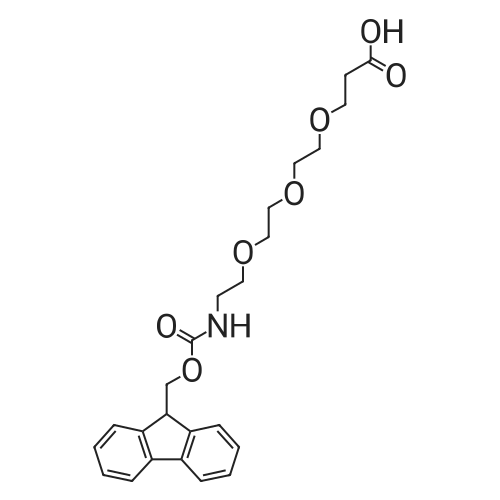
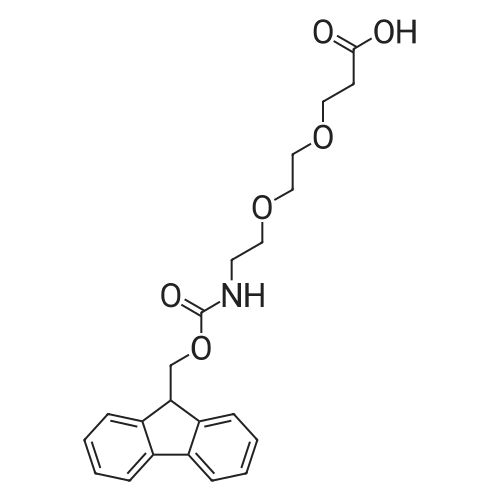
5,8,11,14,17,20,23,26-octaoxa-2-azanonacosanedioic acid, 1-(9H-fluoren-9-ylmethyl) ester[ No CAS ]
[ 4530-20-5 ]
[ 2488-15-5 ]
[ 7536-58-5 ]
[ 13734-34-4 ]
[ 47355-10-2 ]
[ 57-10-3 ]
C70H111N9O20S[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: The second portion of the CCK4 peptide and the SubP peptide were modified on resin as follows to yield test lipidated peptides (1-SubP-COOH and 1-CCK4-Gly-COOH). Spacers (these are AA's used between the peg linker and the peptide of interest) were introduced on the peptides before pegylation (KGG for SubP and GG for CCK4). The free N-terminus of the peptide on resin was first pegylated with N-Fmoc-PEG8-propionic acid using standard HBTU coupling conditions. The N-Fmoc protecting group was removed by treatment with 10% piperidine in DMF (N,N-Dimethylformamide) for 5 min. Palmitic acid was subsequently coupled with the N-terminal free amine of the pegylated peptide. Peptides were cleaved from the resin using high HF conditions with minor modifications to the usual procedure. For the SubP peptide, longer times were used to ensure removal of Arg(Tos) protecting group (90% anhydrous HF/10% anisole at 0 C. for 2 h). For the CCK-4 peptides, 1,3-propanedithiol was used in the HF cleavage mixture to ensure deprotection of the formyl protecting group and prevent oxidation of methionine to its sulfoxide derivative: 85% anhydrous HF/10% anisole/5% PDT (1,3-propaneithiol) at 0 C. for 2 h) (Matsueda, 1982). Following cleavage from resin, peptides were precipitated with cold Et2O. Unmodified peptides were extracted using 10% AcOH in water and the lipidated peptides were extracted using 10% AcOH in H2O followed by 10% AcOH in 50% EtOH/H2O. Crude peptides were purified by RP-HPLC [Vydac C18, 10 m, 22 mm×250 mm]. The purities of the peptides were assessed by analytical RP-HPLC [Vydac C18, 5 m, 4 mm×250 mm].
5,8,11,14,17,20,23,26-octaoxa-2-azanonacosanedioic acid, 1-(9H-fluoren-9-ylmethyl) ester[ No CAS ]
[ 4530-20-5 ]
[ 15761-39-4 ]
[ 13139-15-6 ]
[ 2488-15-5 ]
[ 13734-34-4 ]
[ 13836-37-8 ]
[ 84624-27-1 ]
[ 54613-99-9 ]
[ 55260-24-7 ]
[ 57-10-3 ]
C111H185N21O28S[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: The second portion of the CCK4 peptide and the SubP peptide were modified on resin as follows to yield test lipidated peptides (1-SubP-COOH and 1-CCK4-Gly-COOH). Spacers (these are AA's used between the peg linker and the peptide of interest) were introduced on the peptides before pegylation (KGG for SubP and GG for CCK4). The free N-terminus of the peptide on resin was first pegylated with N-Fmoc-PEG8-propionic acid using standard HBTU coupling conditions. The N-Fmoc protecting group was removed by treatment with 10% piperidine in DMF (N,N-Dimethylformamide) for 5 min. Palmitic acid was subsequently coupled with the N-terminal free amine of the pegylated peptide. Peptides were cleaved from the resin using high HF conditions with minor modifications to the usual procedure. For the SubP peptide, longer times were used to ensure removal of Arg(Tos) protecting group (90% anhydrous HF/10% anisole at 0 C. for 2 h). For the CCK-4 peptides, 1,3-propanedithiol was used in the HF cleavage mixture to ensure deprotection of the formyl protecting group and prevent oxidation of methionine to its sulfoxide derivative: 85% anhydrous HF/10% anisole/5% PDT (1,3-propaneithiol) at 0 C. for 2 h) (Matsueda, 1982). Following cleavage from resin, peptides were precipitated with cold Et2O. Unmodified peptides were extracted using 10% AcOH in water and the lipidated peptides were extracted using 10% AcOH in H2O followed by 10% AcOH in 50% EtOH/H2O. Crude peptides were purified by RP-HPLC [Vydac C18, 10 m, 22 mm×250 mm]. The purities of the peptides were assessed by analytical RP-HPLC [Vydac C18, 5 m, 4 mm×250 mm].
With toluene-4-sulfonic acid; In neat (no solvent); at 60℃; for 4.0h;
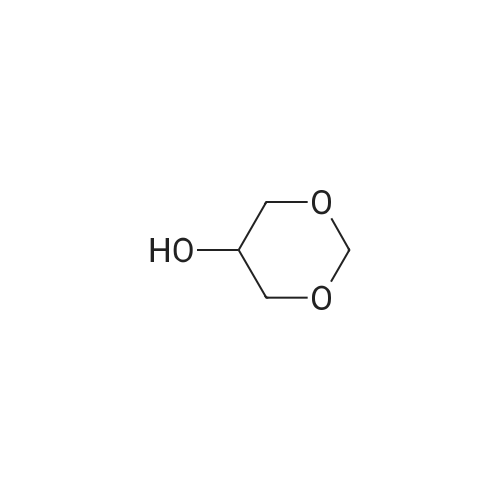
General procedure: The FFA (2.0 g), glycerol formal (GlyF: 1.5 molar equivalent respect to the FFA) andp-toluene-sulfonic acid (5percent w/w respect of the weight of the FFA) were charged in a conventionalround bottom flask with a magnetic stirrer. The mixture was heated at 60 C and stirred vigorously for4 h. The reaction was quenched by neutralizing the acid catalyst with saturated solution of sodiumcarbonate then filtered. The liquid phase was collected in a separatory funnel and partitioned betweenchloroform and water (3 x 20 mL). The organic layer was dried over sodium sulfate, filtered and thesolvent eliminated using a rotary evaporator. The sample was stored in a glass vial Table 2 summarizesthe experimental conditions and the results of reactions between GlyF and the FFAs.
(S)-6-[(Diphenyl-p-tolyl-methyl)-amino]-2-(9H-fluoren-9-ylmethoxycarbonylamino)-hexanoic acid[ No CAS ]
Fmoc-Arg(pg)[ No CAS ]
Fmoc-Ser(pg)[ No CAS ]
Fmoc-Tyr(pg)[ No CAS ]
H-K(Pal)-K(Pal)-GYSSPGS(p)PGT(p)PGSRSR-K(Pal)-K(Pal)-NH2[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
2.5: Synthesis of peptide antigen T9 The orthogonally protected amino acid Fmoc-Lys(Mtt)-OH (3 eq) was manually loaded to an amide resin (Rink amide MBHA resin, 1 eq, 0.4 mmol) in the presence of PyBOP/HOBt/DIEA in DMF. The resin was then washed with DMF (3 x 1 min). After removing the N-terminal Fmoc group with 25% piperidine in DMF (1 x 1 min and 2 x 15 min), the second residue of Fmoc-Lys(Mtt)-OH (3 eq), was coupled using the same loading conditions. The following 16 amino acids bearing the Fmoc standard side-chain protecting groups were incorporated applying the previously described coupling protocol. The phosphoaminoacids were introduced as monobenzyl esters at the phosphate group. The coupling time was determined by TNBT test or chloranyl test after a Proline. If necessary, a second coupling was performed with 2 eq of Fmoc-aminoacid in the presence of DIC/HOBt or HATU/DIEA. Each coupling step was followed by a wash step with DMF (3 x 1 min), Fmoc removal step with 25% piperidine in DMF (1 x 1 min and 2 x 15 min) and a second wash step with DMF (7 x 1 min). After the coupling of the Thr(PO(OBzI)OH), 0.5% DBU in DMF was used for the Fmoc-deprotection step. The assembly of the peptide sequence finished with the addition of the last two Fmoc-Lys(Mtt)-OH. Then, the Mtt-groups of the terminal lysine residues were selectively cleaved by treatment of the resin (1 eq, 650 mg, 0.156 mmol) with 10 mL of TtPS/TFA/DCM (1:1:98) during several cycles of 10 min. After washing with DCM (x3) and DMF (x3), Palmitic acid (20 eq, 1.01 g, 3.15 mmol) was coupled to those deprotected amino groups using TBTU (20 eq, 814 mg, 3.15 mmol) and DIEA (40 eq, 1.1 mL, 6.30 mmol) in DCM/DMF (1:1) (6 mL). The resin was washed thoroughly with DCM (x5) and DMF (x5). Then the N-terminal Fmoc group was removed with 20% piperidine in DMF (3 x 10 min) and the resin was washed again with DMF (x3) and DCM (x5). Finally simultaneous resin cleavage and side-chain deprotections were carried out using a mixture of TFA/TIPS/H2O (95:2.5:2.5) (9 mL) during 3 h. Trituration from cold diethyl ether gave the crude product T9 as a white solid (291 mg, 59% yield) with a purity of 69% (from HPLC analysis). MALDI-TOF mass spectrometry confirmed the identity of the major product (m/z expected: 3172.98 [MH+], found: 3172.90).
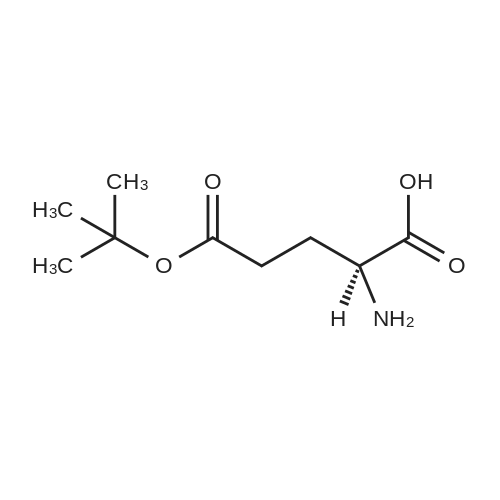
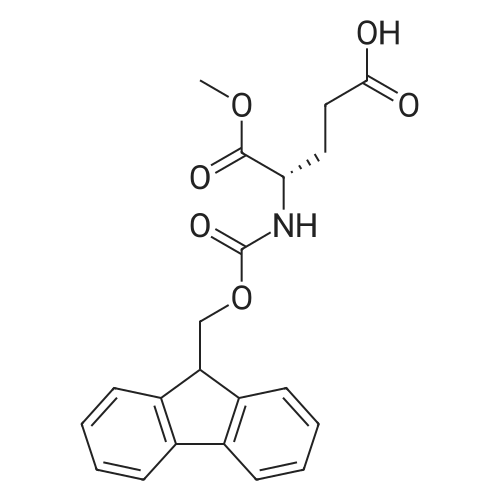
N-hexadecanoyl-L-glutamic acid 5-tert-butyl-1-methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
99.7%
In a 250 ml dry bottom flask, palmitic acid (3.0 g, 11.70 mmol) was dissolved in 50 ml of dichloromethane at room temperature under argon atmosphere. Triethylamine (3.3 ml, 23.64 mmol) was then added drop wise and the mixture was stirred for 5 min. 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluranium hexafluorophosphate (HATU) (6.74 g, 17.7 mmol) was added and allowed to stir for additional 10 min. In a separate round bottom flask, L-Glutamic acid tert-butyl ester (3.0 g, 11.82 mmol) was taken in 30 ml of dichloromethane and then added dropwise to the above activated palmitic acid in dichloromethane solution. The reaction mixture was allowed to stir at room temperature overnight. The suspension was filtered, quenched with water, extracted with dichloromethane and dried using sodium sulphate. It is then purified by silica gel column chromatography using methanol and dichloromethane to yield 5-t-butyl-1-methyl palmityl glutamate as a white solid. (5.37g, 99.7% yield). ESI-MS (+ve) m/z: 456.63(MH+);
(1)500g of concentrated sulfuric acid was added to the three-necked flask, add 50g of palmitic acid, stirring to dissolve Solution in concentrated sulfuric acid was added 50gL- ascorbic acid, 18 C reaction 15h; (2)50g palmitic anhydride was added to the reaction mixture, the temperature was raised to 28 C, the reaction 20h, then After adding 10g of activated carbon and stirred for 15min; (3)The step (2) in the resulting mixture is added to 1250ml10 C cold water, filtered The filter cake is too crude, the crude product was rinsed with 100ml water, then washed with water after the crude product was dissolved in 750ml of butyl acetate, 50 C incubation decolorization 30min. Filtered and allowed to stand, stratification, the upper organic layer (Product containing layer) 50 C, washed twice with water, water per 500ml. After washing to the water layer, The organic layer was distilled off under reduced pressure to 400ml of butyl acetate, allowed to stand for cooling to 15 C, the solid was filtered off with 50ml The resulting solid was rinsed with ethyl acetate, drained, placed in a vacuum drying oven at 50 C. L-ascorbic acid-6-palmitate was obtained as a white flake with a purity of 98. 1% and a yield of 91.3%.
N-hexadecanoyl-L-glutamic acid 5-tert-butyl-1-methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; triethylamine; In N,N-dimethyl-formamide; at 20℃; for 22h;Cooling with ice;
To L-glutamic acid 5-tert-butyl-l-methyl ester (1079) hydrochloride (132 mg, 0.52 mmol) were added N,N- dimethylformamide (5.2 ml), and palmitic acid (200 mg, 0.78 mmol) at room temperature, and the mixture was cooled in an ice bath. Then, l-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (150 mg, 0.78 mmol), 1-hydroxybenzotriazole (105 mg, 0.78 mmol), and triethylamine (0.22 ml, 1.56 mmol) were added, and the mixture was removed from the ice bath and stirred at room temperature for 22 hr. Silica gel was added to the reaction solution to evaporate the solvent, and the residue was purified by silica gel column chromatography (hexane/ethyl acetate) to give N-hexadecanoyl-L-glutamic acid 5-tert-butyl-l- methyl ester (214 mg, 0.47 mmol, yield 90%) as a white solid. FontWeight="Bold" FontSize="10" H-NMR (400 MHz, CDC13)5: 0.88 (t, 3H, J=6.8 Hz), 1.25-1.29 (m, 24H) , 1.44(s, 9H) , 1.63(m, 2H) , 1.95(m, 1H) , 2.13 (m, 1H) , (1080) 2.21(t, 2H, J=8.0 Hz), 2.31(m, 2H) , 3.75(s, 3H) , 4.60(dt, 1H, J=5.2 Hz, 8.0 Hz), 6.17(d, 1H, J=7.6 Hz). ESIMS(m/z): 456.4 ( [M+H] +) , 911.8 ( [2 M+H] +) .
3 mmol of palmitic acid, 3 mmol of DCC, 3 mmol of DMAP were added to the reaction vessel, 20 mL of anhydrous dichloromethaneAnd the mixture was stirred under ice-cooling for 30 minutes; 1 mmol of <strong>[38748-32-2]TP</strong> was dissolved in an appropriate amount of anhydrous dichloromethane and slowly added dropwiseThe reaction was carried out under ice-cooling for 45 minutes and the reaction was continued overnight at room temperature. The reaction was purified by silica gel column and purifiedTriptolylate palmitate 504.7 mg. Yield 84.3%.
As in Reaction Scheme below, <strong>[529-44-2]myricetin</strong> derivatives were prepared by subjecting <strong>[529-44-2]myricetin</strong> to acylation usinga fatty acid by using a base and oxalyl chloride. [0072] In particular, each of a plurality of types of acyl donors (2844.8 mg of stearic acid, 2564.2 mg of palmitic acid,and 2003.1 mg of lauric acid) was first allowed to react with oxalyl chloride to prepare a highly reactive fatty acid.Subsequently, 318.24 mg of <strong>[529-44-2]myricetin</strong> was mixed with a reaction product obtained from the reaction and 50 ml of pyridinebase. The resulting mixed solution was slowly heated to a temperature equal to room temperature or higher (40 C) toallow a reaction to occur and last for 16 hours.[0073] Next, the reaction solution was developed in thin layer chromatography to identify a spot corresponding to thecompound. Thereafter, column chromatography (Product Name: "M.S.GEL", AGC Si-Tech CO., INC.) was performedto remove unreacted <strong>[529-44-2]myricetin</strong>. Thereafter, the corresponding compound was dissolved in water and then extracted withchloroform, and this process was repeated several times to obtain a compound with high purity.[0074] As described above, organic compounds, i.e., <strong>[529-44-2]myricetin</strong> derivatives, prepared by acylation using a fatty acidand oxalyl chloride in the presence of a base catalyst were obtained. 1H NMR analysis results of a <strong>[529-44-2]myricetin</strong> derivativeobtained using palmitic acid as a fatty acid are illustrated in FIG. 4. In addition, 1H NMR analysis results of <strong>[529-44-2]myricetin</strong>derivatives obtained using acetic acid, stearic acid, and lauric acid as fatty acids instead of palmitic acid are illustratedin FIGS. 5, 6, and 7, respectively
(S)-6-[(Diphenyl-p-tolyl-methyl)-amino]-2-(9H-fluoren-9-ylmethoxycarbonylamino)-hexanoic acid[ No CAS ]
Nα-(9-fluorenylmethyloxycarbonyl)-Nγ-2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl-L-arginine[ No CAS ]
H-His-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys(γ-Glu-palmitoyl)-Glu-Phe-Ile-Ala-Trp-Leu-Val-Arg-Gly-Arg-Gly-OH[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: The Wang resin (0.3 -0.6 mmol/g, loading capacity) was loaded to peptide synthesis vessel, washed twice with 10 v of MDC, decanted the washings, added 10 v of MDC and kept for swelling for 1 h. Fmoc-Gly-OH (3.0 - 5.0 eq.) was dissolved in MDC, added minimum quantity of DMF to obtain clear solution and the mixture was transferred to reaction vessel. Added DIPC (3.0 - 6.0 eq.) followed by DMAP (0.01- 0.1 eq.) to the reaction vessel and stirred for 1.0? 3.0 h, at rt. Drained the reaction mass and washed the amino acid loaded resin twice with MDC followed by DMF. Capping of the unreacted functional sites were carried out using acetic anhydride and DIPEA. Fmoc-deprotection of the loaded amino acid was carried out by washing the resin using 15-25 percent piperidine in DMF two times for 5 and 10 min. followed by the resin was washed with 3-5*8 v 0.01? 0.1 M HOBt in DMF. The Fmoc-Arg(Pbf)-OH (2.0? 4.0 eq.), was coupled using coupling agents such as HBTU, COMU, DEPBT, and DIC, preferably DEPBT (2.0 - 4.0 eq.) and oxymapure, HOBt, preferably oxymapure (2.0? 4.0 eq.) and DIPEA, NMM, TMP, preferably DIPEA (5.0 -8.0 eq.) and MgCl2, ZnCl2, preferably MgCl2 (0.01? 0.1 eq) and DMF/NMP mixture as solvent. The reaction was performed in nitrogen atmosphere and r.t. Upon completion of coupling of the amino acid confirmed by Kaiser Test, the excess reagents were drained and washed the peptidyl resin with 3 x 10 v DMF
9-fluorenylmethoxycarbonyl-Nim-trityl-L-histidine[ No CAS ]
N-α-[(9H-fluoren-9-ylmethoxy)carbonyl]-NG-(2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl)-D-arginine[ No CAS ]
C88H140N22O20S2[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
The synthesis of Example 33 (Ac-C+-DTHFPr-C+-rF-PEG2-K(Palmitoyl)-NH2) is representative: The following reagents were used:MB HA rink amide resin (1 equiv)Fmoc_Lys(Dde)-OH (3 equiv)Fmoc-Phe-OH (3 equiv)Fmoc-D-Arg(Pbf)-OH (3 equiv)Fmoc-Cys(Trt)-OH (3 equiv)Fmoc-D-Arg(Pbf)-OH (3 equiv)Fmoc-Pro-OH (3 equiv)Fmoc-Phe-OH (3 equiv)Fmoc-His(Trt)-OH (3 equiv)Fmoc-Thr(OtBu)-OH (3 equiv)Fmoc-Asp(OtBu)-OH (3 equiv)Fmoc-Cys(Trt)-OH (3 equiv)<strong>[867062-95-1]Fmoc-PEG2-OH</strong> (2.5 equiv)Palmitic acid (3 equiv)The peptide was synthesized using standard Fmoc chemistry by manual synthesis. 1. Swell the resin for 30 min in DMF and push out the DMF out of column with nitrogen.2. The Fmoc group was cleaved from the resin by adding 20% (v/v) piperidine in DMF.The resin was allowed to react with the 20% piperidine solution for 20 min. 3. After the Fmoc cleavage from the resin is complete, the 20% piperidine solution is pushed out of the column. The resin is washed 3 times with DMF:4. Preparing (or activating) the amino acid: 3 eq of the amino acid and 2.95 eq of HBTU were weighed and were then dissolved in DMF. 6 eq of DIEA was added to the above solution. The activated solution was then added to the column containing the resin and reacted for about 2 h.5. The resin was drained and the loaded resin was wasged 3 times with DMF. 6. Steps 2-5 were repeated for each amino acid coupling.7. The acetyl group protection was conducted by adding the cocktail of 5% Ac2O/10% NMM 85% DMF. The solution was reacted for about 0.5 h. 8. Boc group protection when necessary was conducted by adding the 3 eq of (Boc)20 and 6 eq of DIEA to the resin in DMF. The reaction mixture was reacted for about 0.5 h.9. The resin was drained and washed with DMF 3 times and with MeOH 3 times. Cleavage and disulfide bond formation:The resulting residue was treated with cocktail of 90%TFA/5%TIPS/2.5%H2O/2.5%EDT (10 mL) and swelled for about 2 h. The crude peptide was precipitated out by ether. Cleavage:The resulting residue was treated with cocktail of 90%TFA/5%TIPS/2.5%H2O/2.5%EDT (10 mL) and swelled for about 2 h. The crude peptide was precipitated out by ether. If no disulfide bond formation was required, the crude peptide was purified by reversed-phase HPLC.Optional Cyclization (disulfide bond formation):The crude peptide was dissolved in H20/ACN (1:1) to adjust the concentration to 1 mM. Then 1 M NH4HC03was added to the above solution to adjust the pH to about 8-9. The solution was allowed to react for about 8 h at room temperature. The reaction was monitored by LCMS. After the reaction was completed, the reaction was quenched by acetic acid to adjust the pH to about 6. The reaction mixure was lyophilized and the resulting solid was purified by reversed- phase HPLC.
9-fluorenylmethoxycarbonyl-Nim-trityl-L-histidine[ No CAS ]
N-α-[(9H-fluoren-9-ylmethoxy)carbonyl]-NG-(2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl)-D-arginine[ No CAS ]
Fmoc-Ser(*)-OH[ No CAS ]
C83H135N21O19[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: The synthesis of Example 33 (Ac-C+-DTHFPr-C+-rF-PEG2-K(Palmitoyl)-NH2) is representative: The following reagents were used:MB HA rink amide resin (1 equiv)Fmoc_Lys(Dde)-OH (3 equiv)Fmoc-Phe-OH (3 equiv)Fmoc-D-Arg(Pbf)-OH (3 equiv)Fmoc-Cys(Trt)-OH (3 equiv)Fmoc-D-Arg(Pbf)-OH (3 equiv)Fmoc-Pro-OH (3 equiv)Fmoc-Phe-OH (3 equiv)Fmoc-His(Trt)-OH (3 equiv)Fmoc-Thr(OtBu)-OH (3 equiv)Fmoc-Asp(OtBu)-OH (3 equiv)Fmoc-Cys(Trt)-OH (3 equiv)<strong>[867062-95-1]Fmoc-PEG2-OH</strong> (2.5 equiv)Palmitic acid (3 equiv)The peptide was synthesized using standard Fmoc chemistry by manual synthesis. 1. Swell the resin for 30 min in DMF and push out the DMF out of column with nitrogen.2. The Fmoc group was cleaved from the resin by adding 20% (v/v) piperidine in DMF.The resin was allowed to react with the 20% piperidine solution for 20 min. 3. After the Fmoc cleavage from the resin is complete, the 20% piperidine solution is pushed out of the column. The resin is washed 3 times with DMF:4. Preparing (or activating) the amino acid: 3 eq of the amino acid and 2.95 eq of HBTU were weighed and were then dissolved in DMF. 6 eq of DIEA was added to the above solution. The activated solution was then added to the column containing the resin and reacted for about 2 h.5. The resin was drained and the loaded resin was wasged 3 times with DMF. 6. Steps 2-5 were repeated for each amino acid coupling.7. The acetyl group protection was conducted by adding the cocktail of 5% Ac2O/10% NMM 85% DMF. The solution was reacted for about 0.5 h. 8. Boc group protection when necessary was conducted by adding the 3 eq of (Boc)20 and 6 eq of DIEA to the resin in DMF. The reaction mixture was reacted for about 0.5 h.9. The resin was drained and washed with DMF 3 times and with MeOH 3 times. Cleavage and disulfide bond formation:The resulting residue was treated with cocktail of 90%TFA/5%TIPS/2.5%H2O/2.5%EDT (10 mL) and swelled for about 2 h. The crude peptide was precipitated out by ether. Cleavage:The resulting residue was treated with cocktail of 90%TFA/5%TIPS/2.5%H2O/2.5%EDT (10 mL) and swelled for about 2 h. The crude peptide was precipitated out by ether. If no disulfide bond formation was required, the crude peptide was purified by reversed-phase HPLC.Optional Cyclization (disulfide bond formation):The crude peptide was dissolved in H20/ACN (1:1) to adjust the concentration to 1 mM. Then 1 M NH4HC03was added to the above solution to adjust the pH to about 8-9. The solution was allowed to react for about 8 h at room temperature. The reaction was monitored by LCMS. After the reaction was completed, the reaction was quenched by acetic acid to adjust the pH to about 6. The reaction mixure was lyophilized and the resulting solid was purified by reversed- phase HPLC.
160 ml of dimethylformamide and 32 g of palmitic acid were sequentially added to the reaction flask.The temperature was raised to 40-50 C and stirred to dissolve. Then, 19 g of carbonyl diimidazole was added to the temperature control.The reaction was stirred for 3 hours. TLC showed the reaction was complete. Add 7.7g of pyridine under heat preservation conditions,Slowly add 10 g of <strong>[58-56-0]pyridoxine hydrochloride</strong>, and then slowly heat up to 90 C for 3 hours.Naturally cool down to room temperature, quench with water,Adding organic solvent ethyl acetate 1000ml dissolution system,Wash the organic phase 2-5 times with 0.1% dilute hydrochloric acid 300ml.Wash 300ml of saturated saline solution 1-2 times,Dry 5 g of anhydrous sodium sulfate for 1-2 hours, concentrate to dryness,Add 400 ml of ethyl acetate to dissolve and decolorize, and cool to 0 C to crystallize,Filter and dry to obtain 25.4gPyridoxine dipalmitate,HPLC purity ?99%, content ?99%,The molar yield was 78.6%.
(S)-6-[(Diphenyl-p-tolyl-methyl)-amino]-2-(9H-fluoren-9-ylmethoxycarbonylamino)-hexanoic acid[ No CAS ]
Nα-(9-fluorenylmethyloxycarbonyl)-Nγ-2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl-L-arginine[ No CAS ]
palmitic acid-Lys(Ser-Pro-Pro-Pro-Ala-Gly-Ser-Ser-Pro-Gly-Gly-Arg-Val-Leu-Trp-Ala-Ile-Phe-Glu-Lys-Ala-Ala-Glu-Glu-Glu-Leu-Tyr-Ser-Ser-Val-Asp-Ser-Thr-Phe-Thr-Gly-Glu-Aib-His)[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
22.5%
General procedure: 1. Preparation of Fmoc-Lys (Mtt)-HMP-AM Resin (1) drying and swelling of HMP-AM resin 50g (30mmol) HMP-AM resin (0.6mmol/g) dried for 24h in vacuum are placed into a 2L bubblling bottle, resinsare swelled with 500mL N, N-dimethylformamide (DMF) for 30 minutes, the DMF is drawn-off, the resins are washed with DMF for 1 min, the washing step is repeated twice. (2)Preparation of Fmoc-Lys (Mtt)-HMP-AM Resin Coupling of Fmoc-Lys (Mtt)-OH with HMP-AM resin The resins are washed with 500mL DCM, and then the washing step is repeated twice. 56.2g (90mmol) Fmoc-Lys (Mtt)-OH and 11.4g (90mmol) DIC are dissolved in 1L DCM, then are added into the swelled HMP-AM resin, then366mg (3mmol) DMAP are added to react for 24h; Washing of the resin After the reaction, the resin is washed alternately with DMF and IPA twice, washed with DMF for 3 times; Capping of hydroxyl 15.3g (150mmol) acetic anhydride and 19.4g (150mmol) DIEA are dissolved in 1L DMF and are added into theresin to react for 10min. washing of the resin The resin is washed twice with 1 L 50percent MeOH/DMF, 50percent DCM/DMF, and then washed with DCM for threetimes and with dehydrated ethanol for three times in turn. After dried under vacuum, the Fmoc-Lys (Mtt)-HMP-AM resinis obtained. (3) Loading assays of Fmoc-Lys(Mtt)-HMP-AM resin Accurate 5 ? 10 mg resin are put into 1mL 20percent Hexahydropyridine / DMF solution, stirred for 20min, then 50uLsupernatant is taken with a pipet and is diluted in 2.5ml DMF; Blank samples: 50uL 20percent Hexahydropyridine/DMF is taken with a pipet and is diluted in 2.5ml DMF;Degree of substitution is calculated as follows: wherein A is the absorption value of UV at 301nm; m is the weight of the resin, the unit is mg 2. Drying and swelling of the solid-phase synthesized resin 50g (20mmol) Fmoc-Lys (Mtt)-HMPA-AM resin (0.4mmol/g) dried in vacuum for 24h are placed into a 2Lbubbling bottle, then 500ml N, N-dimethylformamide (DMF) are added to swell the resin for 30min, then the DMF solutionis drawn-off. 3. Removal of 4 - methyl triphenylmethyl (Mtt) protecting group of Fmoc-Lys (Mtt)-HMPA-AM resin The resin is washed with 200ml DCM twice, then 1200mL 1percent TFA / DCM (TFA is about 8-fold excess) areadded to remove Mtt protecting group for 1h, the resin is alternately washed with 200mL 5percent N, N-diisopropyl ethylamine(DIEA) / DMF and DMF for three times, then washed with DMF for 3 times; 4. Palmitic acid condensation 50mmol palmitic acid and 50mmol 3 - (diethoxyphosphoryloxy) -1,2,3 - phentriazine -4- ketone (DEPBT) aredissolved in 400ml DMF, then 100mmol DIEA are added and are stirred for 3min at room temperature, said solution isadded to the resin, reacted in 37°C water baths for 2h under N2. After the reaction, the reaction solution is drawn-off,the resin is washed with DMF, isopropyl alcohol (IPA) and DMF in turn. 5. Removal of 9- Fmoc (fluorenylmethyloxycarbonyl) protecting group of Fmoc-Lys (N-epsilon-palmitic acid)-HMPA-AM resin 200mL 20percent piperidine/DMF solution are placed into a bubbling bottle filled with Fmoc-Lys (Mtt)-HMPA-AMresin, reacted for 5min and then is drawn out, then 200mL 20percent piperidine/DMF solution are added to react for 20minat room temperature. After the reaction, the resin is washed with 200mL DMF for four times. 6. Solid phase synthesis of peptide chain part of HS-20001 Condensation of Fmoc-Ser (tBu)-OH 50mmol Fmoc-Ser (tBu)-OH are dissolved in 125mL 0.4M 1-hydroxybenzo triazole (HOBt) / DMF, then 125ml0.4M N, N?-diisopropyl carbodiimide (DIC) / DCM are added to activate and react for 10min at room temperature; saidsolution is added into the resin, reacted under protection of nitrogen at room temperature, and ninhydrin is used to detectand control the degree of the reaction. After the reaction, the reaction solution is removed; the resin is washed with DMF,IPA and DMF in turn. Extension of the peptide chain HS-20001resin peptide is synthesized according to the sequence of the peptide chain of HS-20001 from theamino terminal (N-terminal) to the carboxy-terminal (C-terminal) (His-(D)-Ala-Glu-Gly-Thr-Phe-Thr-Ser- Asp-Leu-Ser-Lys-Gln-Nle-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Gln-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser), wherein the amounts of amino acids and condensation reagents are the same as the amounts for Fmoc-Ser(tBu)-OH, protected amino acids are Fmoc-Ser(tBu)-OH, Fmoc-Pro-OH, Fmoc-Ala-OH, Fmoc-Gly-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Trp(Boc)-OH, Fmoc-Glu(OtBu)-OH, Fmoc-Ile-OH, Fmoc-Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Nle-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Thr(tBu)-OH, Fmoc-D-Ala-OH andFmoc-His(Trt)-OH respectively, and condensation and deprotection reactions are repeated. Post-processing of HS-20001 resin peptide Said HS-20001 resin peptide obtained in step P is washed with DMF, IPA and DMF in turn, washed withabsolute ether twice, then dried under vacuum, and the HS-20001 resin peptide is obtained Preparation of HS-20001 crude...
His-(D-Ser)-Gln-Gly-Thr-Phe-Thr-Ser-Asp-Tyr-Ser-Lys-Tyr-Leu-Asp-Lys(PEG<SUB>2</SUB>-PEG<SUB>2</SUB>-γGlu-CO(CH<SUB>2</SUB>)<SUB>14</SUB>CH<SUB>3</SUB>)-Arg-Arg-Ala-Gln-Asp-Phe-Val-Gln-Trp-Leu-Met-Asn-Thr-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser-NH<SUB>2</SUB>[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
All amino acids were purchased from NovaBiochem Company. Unless otherwise specified, all other reagents were analytically pure and purchased from Sigma Company. Protein Technologies PRELUDE 6-channel polypeptide synthesizer was used. Phenomenex Luna C18 preparative column (46 mm×250 mm) was used for purification of the polypeptides. High performance liquid chromatograph was manufactured by Waters Company. MS analysis was determined using Agilent mass spectrometer. Synthetic method of polypeptide compounds of the invention is illustrated by taking the polypeptide compound 6 as an example: Structure Sequence: His-(D-Ser)-Gln-Gly-Thr-Phe-Thr-Ser-Asp-Tyr-Ser-Lys-Tyr-Leu-Asp-Lys(PEG2-PEG2-gammaGlu-CO(CH2)14CH3)-Arg-Arg-Ala-Gln-Asp-Phe-Val-Gln-Trp-Leu-Met-As n-Thr-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser-NH2 a) Main peptide chain assembly: The following polypeptide in a scale of 0.25 mmol was synthesized on a CS336X peptide synthesizer (CS Bio American Company) according to Fmoc/t-Bu strategy: Boc-His(Boc)-D-Ser(t-Bu)-Gln(OtBu)-Gly-Thr(t-Bu)-Phe-Thr(t-Bu)-Ser(tBu)-Asp(OtBu)-Tyr(t-Bu)-Ser(t-Bu)-Lys(Boc)-Tyr(t-Bu)-Leu-Asp(OtBu)-Lys(ivDde)-Arg(Pbf)-Arg(Pbf)-Ala-Gln(Trt)-Asp(OtBu)-Phe-Val-Gln(Trt)-Trp(Boc)-Leu-Met- n(Trt)-Thr(t-Bu)-Gly-Gly-Pro-Ser(t-Bu)-Ser(t-Bu)-Gly-Ala-Pro-Pro-Pro-Ser(t-Bu)-rink amide resin (1) Step 1: 0.75 g of Rink amide MBHA-LL resin (Novabiochem, loading 0.34 mmol/g) was swelled in dichloromethane (DCM) for 1 hour, and the resin was fully washed with N,N-dimethylformamide (DMF) for three times (2) Step 2: The procedure reaction was performed using Rink amide resin as carrier, the mixture of 6-chloro-benzotriazole-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU), organic base N,N-diisopropylethylamine (DIEPA) at a molar ratio of 1:1 as coupling agent, and N,N-dimethylformamide (DMF) as solvent, the condensation reactions were performed to successively link. Fmoc-Ser(t-Bu)-OH, Fmoc-Pro-OH (3x), Fmoc-Ala-OH, Fmoc-Gly-OH, Fmoc-Ser(t-Bu)-OH (2x), Fmoc-Pro-OH, Fmoc-Gly-OH (2x), Fmoc-Thr(t-Bu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Met-OH, Fmoc-Leu-OH, Fmoc-Trp(Boc)-OH, Fmoc-Glu(OtBu)-OH, Fmoc-Val-OH, Fmoc-Phe-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ala-OH, Fmoc-Arg(Pbf)-OH (2x), Fmoc-Lys(ivDde)-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Leu-OH, Fmoc-Tyr(t-Bu)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Ser(t-Bu)-OH, Fmoc-Tyr(t-Bu)-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Ser(t-Bu)-OH, Fmoc-Thr(t-Bu)-OH, Fmoc-Phe-OH, Thr(t-Bu)-OH, Fmoc-Gly-OH, Fmoc-Gln(Trt)-OH, Fmoc-D-Ser(t-Bu)-OH, Boc-His(Boc)-OH to obtain: Boc-His(Boc)-D-Ser(t-Bu)-Gln(OtBu)-Gly-Thr(t-Bu)-Phe-Thr(t-Bu)-Ser(tBu)-Asp(OtBu)-Tyr(t-Bu)-Ser(t-Bu)-Lys(Boc)-Tyr(t-Bu)-Leu-Asp(OtBu)-Lys(ivDde)-Arg(Pbf)-Arg(Pbf)-Ala-Gln(Trt)-Asp(OtBu)-Phe-Val-Gln(Trt)-Trp(Boc)-Leu-Met- Asn(Trt)-Thr(t-Bu)-Gly-Gly-Pro-Ser(t-Bu)-Ser(t-Bu)-Gly-Ala-Pro-Pro-Pro-Ser(t-Bu)-rink amide resin. Subsequently, the resin was fully washed with N,N-dimethylformamide (DMF), dichloromethane (DCM), Methanol, dichloromethane (DCM), and N,N-dimethylformamide (DMF) in sequence for three times respectively. In the reaction, 1) the amount of the first amino acid Fmoc-Ser(t-Bu)-OH and the amount of the resin was at a ratio of 1:16:1; and 2) in each of the subsequent condensation reactions, each of the amount of Fmoc protected amino acid, 6 -chloro-benzotriazole-1,1,3,3 -tetram ethyluronium hexafluorophosphate (HCTU), organic base N,N-diisopropylethylamine (DIEPA) was excess by 2-8 times, the reaction time was 1-5 hours. b) Removal of 1-(4,4-dimethyl-2,6-dioxo-cyclohexylidene)-3-methyl-butyl (ivDde) and introduction of lipophilic substituent: The resin was washed twice in the solution of N,N-dimethylformamide (DMF)/dichloromethane (DCM)=1:1 (volume ratio), and added with freshly prepared 3.0% hydrazine hydrate in N,N-dimethylformamide (DMF). The reaction mixture was shaken at room temperature for 10-30 minutes, and then filtered. The hydrazine treatment step was repeated five times to obtain: Boc-His(Boc)-D-Ser(t-Bu)-Gln(OtBu)-Gly-Thr(t-Bu)-Phe-Thr(t-Bu)-Ser(tBu)-Asp(OtBu)-Tyr(t-Bu)-Ser(t-Bu)-Lys(Boc)-Tyr(t-Bu)-Leu-Asp(OtBu)-Lys-Arg(Pbf)-Arg(Pbf)-Ala-Gln(Trt)-Asp(OtBu)-Phe-Val-Gln(Trt)-Trp(Boc)-Leu-Met- Asn(Trt)Thr(t-Bu)-Gly-Gly-Pro-Ser(t-Bu)-Ser(t-Bu)-Gly-Ala-Pro-Pro-Pro-Ser(t-Bu)-rink amide resin. Subsequently, the resin was fully washed with N,N-dimethylformamide (DMF), dichloromethane (DCM), Methanol, dichloromethane (DCM), N,N-dimethylformamide (DMF) in sequence for three times respectively. Thereto was added an N,N-dimethylformamide (DMF) mixed coupling solution (5 times excess of each) of FmocNH-PEG2-OH (Quanta BioDesign), 2-(7-azo BTA)-N,N,N?,N?-tetramethyluronium hexafluorophosphate (HATU) and diisopropylethyl amine (DIEPA), shaken for 2 hours, and filtrated. Subsequently, the resin was fully washed with N,N-dimethylformamide (DMF), dichloromethane (DCM), methanol, dichloromethane (DCM), and N,N-dimethylformamide (DMF) in sequence for three times respectively to obtain: Boc-His(Boc)-D-Ser(t-Bu)-Gln(OtBu)-Gly-Thr(t-Bu)-Ph...
13-<strong>[1094-61-7]nicotinamide mononucleotide</strong> (13-NMN, 0.133 g, 0.40 mmol) was added to sulfuric acid (H2504, 8.0 mE, pKa was unable to be measured in water due to the acid being excessively acidic), which is a strongly acidic liquid, and completely dissolved therein followed by the addition of palmitic acid (PaOH, 0.410 g, 1.60 mmol) as an acylating agent, stirring for 12 hours and allowing to undergo acylation. Following completion of the acylation reaction, a portion of the reaction liquid was sampled followed by quanti1,?ing the product under HPEC Analysis Conditions 1 and then quantifying the raw material under HPEC Analysis Conditions 2 to determine the conversion rate and product yield. The results are shown in Table 1. Table 1 indicates the acylating agent used, molar ratio of acylating agent to NMN (acylating agent/NMN) and solvent in Example 9. In addition, the structure of the product of Example9 was confirmed by carrying out ?H-NMR and ?3C-NMR measurements. As a result, the main component was 3?-palmitoyl-3-NMN. As shown in Table 1, 3?-palmitoyl-(3-NMN was obtained as the main product by using sulthric acid as the strongly acidic liquid, using palmitic acid as the acylating agent, and acylating NMN at the molar ratio of acylating agent to NMN shown in Table 1.
Molecule A27 is obtained by means of the conventional solid phase peptide synthesis (SPPS) method on 2-chlorotrityl chloride (CTC) resin (24.00 g, 1.37 mmol/g). (1288) The grafting of the first amino acid Fmoc-6-aminohexanoic acid (1.5 equivalents) is performed in DCM (10 V), in the presence of DIPEA (2.5 equivalents). The unreacted sites are capped with methanol (0.8 mL/g resin) at the end of the reaction. (1289) The protected amino acid <strong>[145038-49-9]Fmoc-Glu-OMe</strong> (1.5 equivalents) and palmitic acid (1.5 equivalents) are coupled in DMF (10 V), in the presence of HATU (1.0 equivalent with respect to the acid) and DIPEA (1.5 equivalents with respect to the acid). (1290) The protecting groups Fmoc are removed using an 80:20 DMF/piperidine solution (10 V). (1291) The product is cleaved from the resin using an 80:20 DCM/HFIP solution (10 V). (1292) After concentration at reduced pressure, two co-evaporations are performed on the residue with dichloromethane followed by toluene. The product is purified by recrystallization in ethyl acetate. A white solid of molecule A27 is obtained. (1293) Yield: 11.54 g (68% in 6 stages) (1294) 1H NMR (CDCl3, ppm): 0.88 (3H); 1.19-1.35 (24H); 1.35-1.44 (2H); 1.50-1.70 (6H); 1.91-2.01 (1H); 2.14-2.40 (7H); 3.14-3.34 (2H); 3.75 (3H); 4.51-4.59 (1H); 6.53 (1H); 6.70 (1H). (1295) LC/MS (ESI+): 513.4 (calculated ([M+H]+): 513.4).
Molecule A27 is obtained by means of the conventional solid phase peptide synthesis (SPPS) method on 2-chlorotrityl chloride (CTC) resin (24.00 g, 1.37 mmol/g). The grafting of the first amino acid Fmoc-6-aminohexanoic acid (1.5 equivalents) is performed in DCM (10 V), in the presence of DIPEA (2.5 equivalents). The unreacted sites are capped with methanol (0.8 mL/g resin) at the end of the reaction. The protected amino acid <strong>[145038-49-9]Fmoc-Glu-OMe</strong> (1.5 equivalents) and palmitic acid (1.5 equivalents) are coupled in DMF (10 V), in the presence of HATU (1.0 equivalent with respect to the acid) and DIPEA (1.5 equivalents with respect to the acid). The protecting groups Fmoc are removed using an 80:20 DMF/piperidine solution (10 V). The product is cleaved from the resin using an 80:20 DCM/HFIP solution (10 V). After concentration under reduced pressure, two co-evaporations are performed on the residue with dichloromethane followed by toluene. The product is purified by recrystallization in ethyl acetate. A white solid of molecule A27 is obtained. Yield: 11.54 g (68% in 6 stages) 1H NMR (CDCl3, ppm): 0.88 (3H); 1.19-1.35 (24H); 1.35-1.44 (2H); 1.50-1.70 (6H); 1.91-2.01 (1H); 2.14-2.40 (7H); 3.14-3.34 (2H); 3.75 (3H); 4.51-4.59 (1H); 6.53 (1H); 6.70 (1H). LC/MS (ESI+): 513.4 (calculated ([M+H]+): 513.4).
L-Glutamic acid alpha-tert-butyl ester (H-Glu-OtBu) was reacted with palmitic acid in presence of IBCF and NMM to yield CH3-(CH2)14-C(O)-Glu-OtBu, which was then reacted with HOSu in the presence of IBCF and NMM to yield CH3-(CH2)14-C(O)-Glu(OSu)-OtBu, which was then de-protected with trifluoroacetic acid to yield Moiety B-OSu.
56g of hexadecanoic acid (0.22mol),19.1 g of N-Boc-2,3-dihydroxypropylamine (0.1 mol) was added to a 500 mL reaction flask, 300 mL of dichloromethane was added, and after stirring well, 27.8 g of N, N'-diisopropylcarbodiimide ( DIC) (0.22mol) and 0.2g of 4-dimethylaminopyridine (DMAP). After stirring at room temperature for 24 hours, it was filtered. The solid obtained after rotary distillation of the filtrate was further recrystallized and purified to obtain 62 g of intermediate product. This step The yield was 92.8%.Add all the obtained intermediate products to a 250 mL reaction flask, add 150 mL of dichloromethane, stir well and add 20% by volume of trifluoroacetic acid, react at room temperature for 5 hours, and then recrystallize after removing the solvent by rotary distillation to obtain 51 g of the final product. . The yield in this step is 96.8%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping