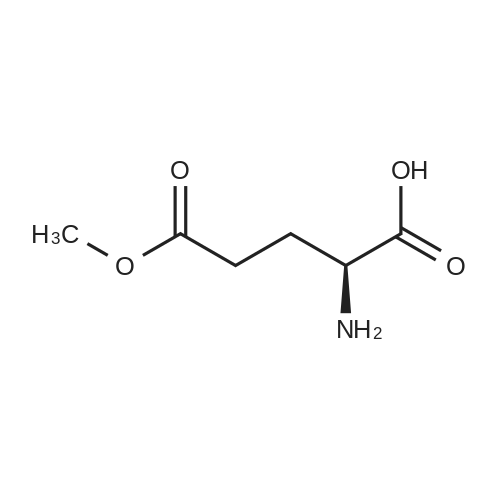
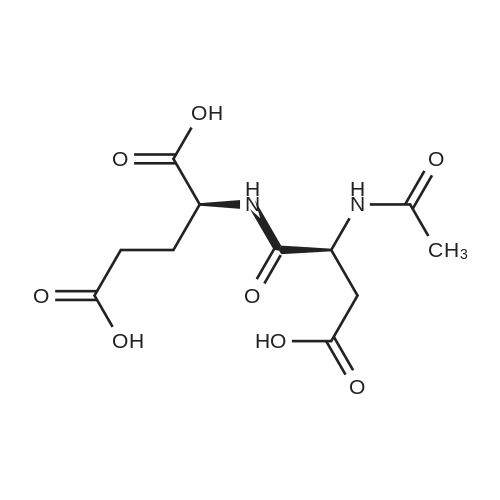
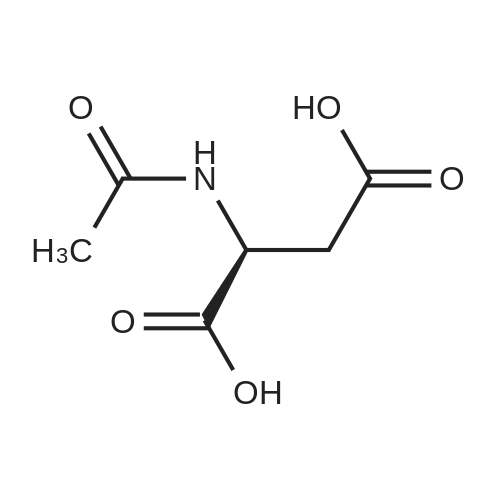
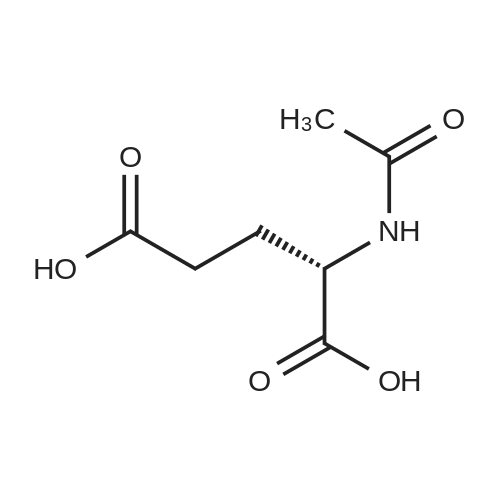
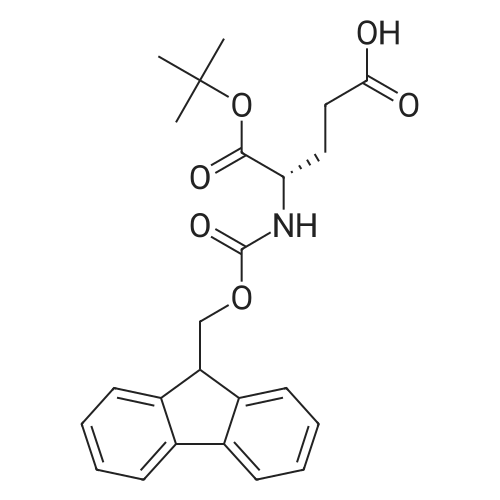
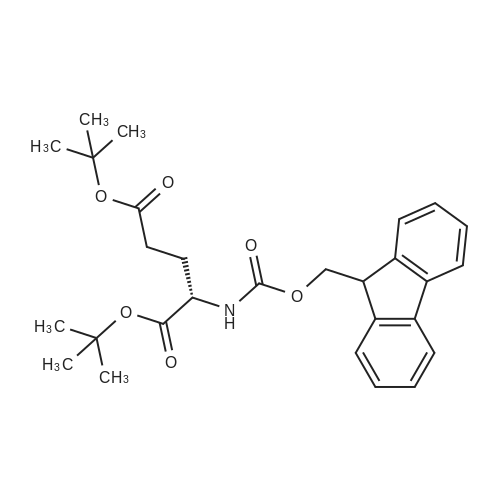
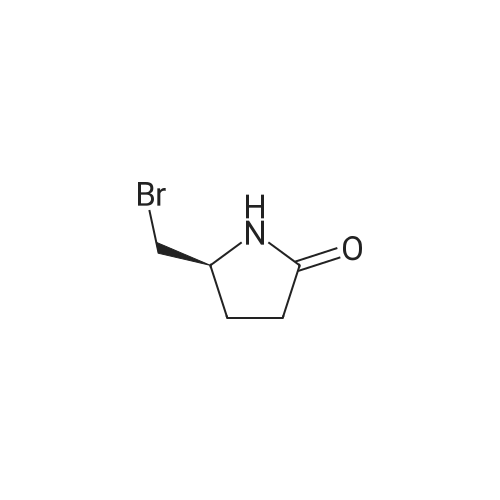
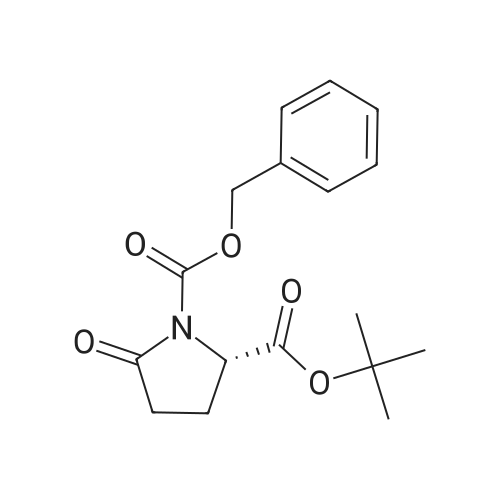
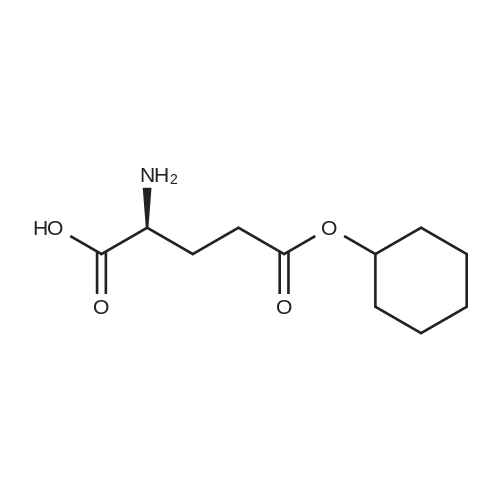
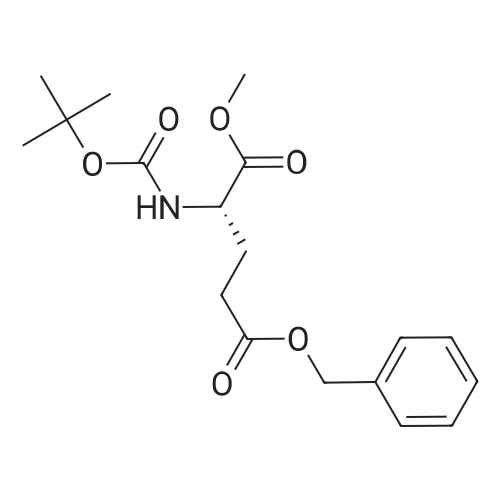
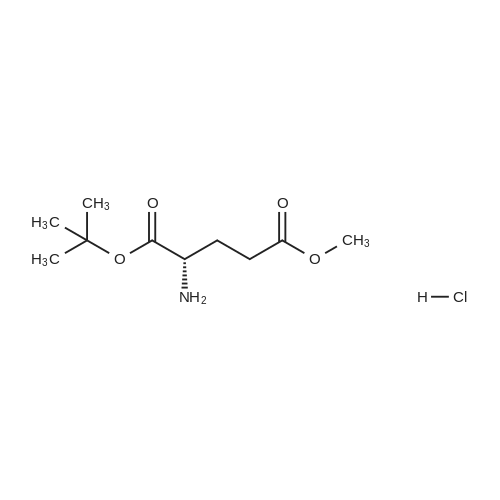
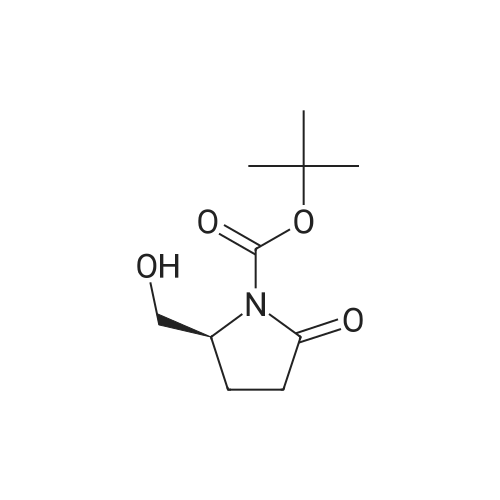
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of the American Chemical Society, 1997, vol. 119, # 42, p. 10004 - 10013
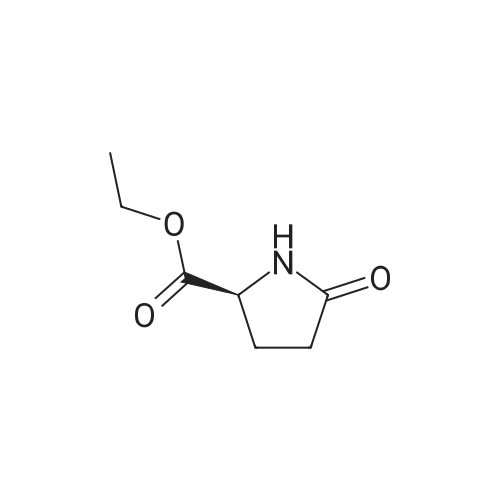
2
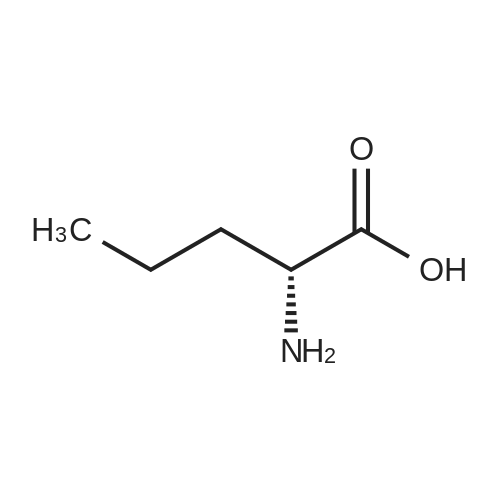
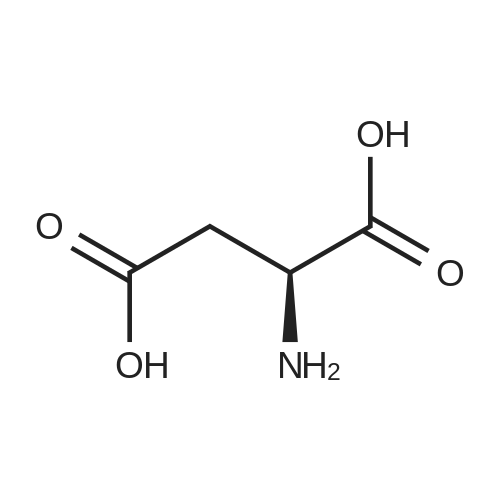
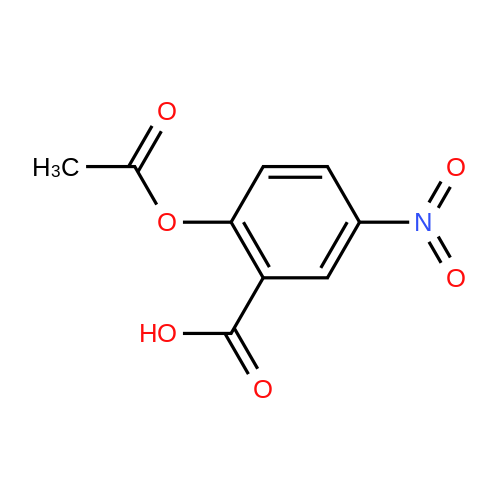
[ 56-86-0 ]
[ 17342-08-4 ]
Reference:
[1] Tetrahedron Letters, 1986, vol. 27, # 3, p. 369 - 372
[2] Heterocycles, 2001, vol. 55, # 11, p. 2099 - 2108
[3] Synthetic Communications, 2000, vol. 30, # 12, p. 2143 - 2159
[4] Journal of the American Chemical Society, 1999, vol. 121, # 45, p. 10478 - 10486
[5] Journal of Organic Chemistry, 1999, vol. 64, # 16, p. 6005 - 6018
[6] Journal of Organic Chemistry, 1980, vol. 45, # 5, p. 815 - 818
[7] Journal of Organic Chemistry, 2011, vol. 76, # 14, p. 5574 - 5583
[8] Organic and Biomolecular Chemistry, 2013, vol. 11, # 31, p. 5173 - 5183
[9] Patent: CN105837486, 2016, A,
3
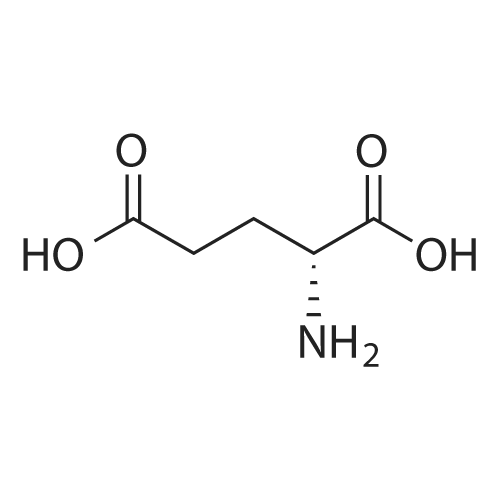
[ 56-86-0 ]
[ 5626-52-8 ]
Reference:
[1] Gazzetta Chimica Italiana, 1894, vol. 24 I, p. 383[2] Atti della Accademia Nazionale dei Lincei, Classe di Scienze Fisiche, Matematiche e Naturali, Rendiconti, 1891, vol. <4> 7 I, p. 39
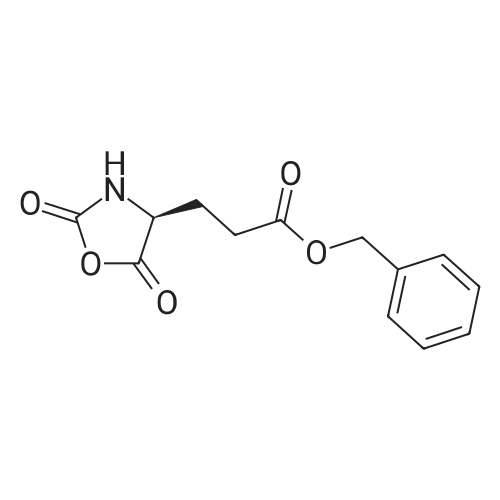
4
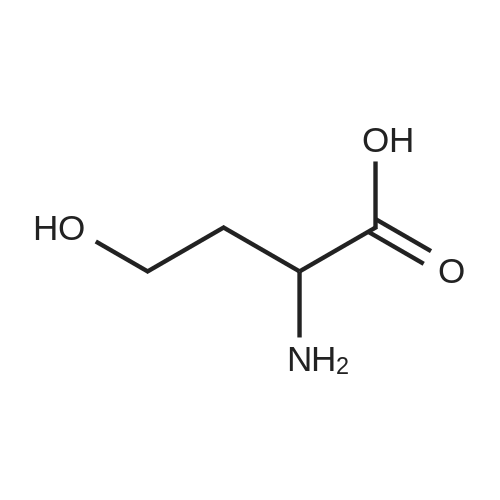
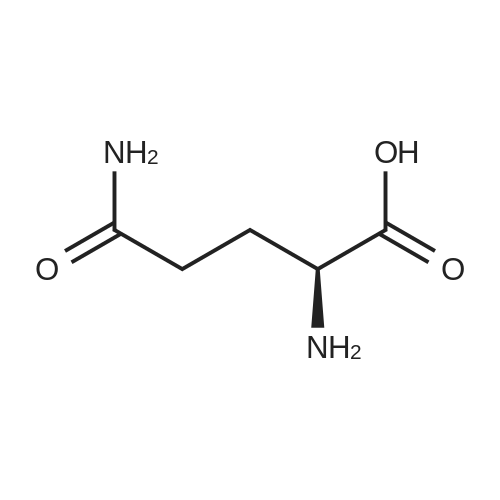
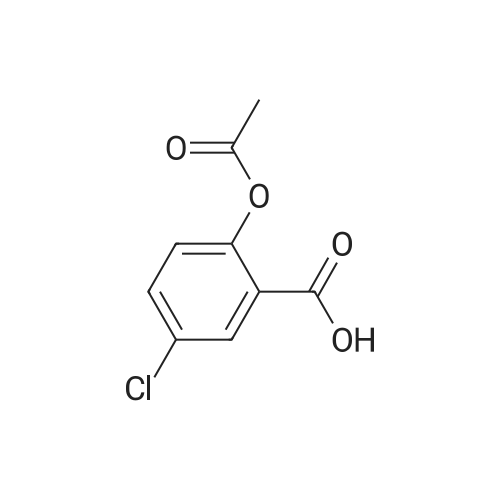
[ 56-86-0 ]
[ 16395-57-6 ]
Reference:
[1] Heterocycles, 2003, vol. 61, p. 259 - 269
5
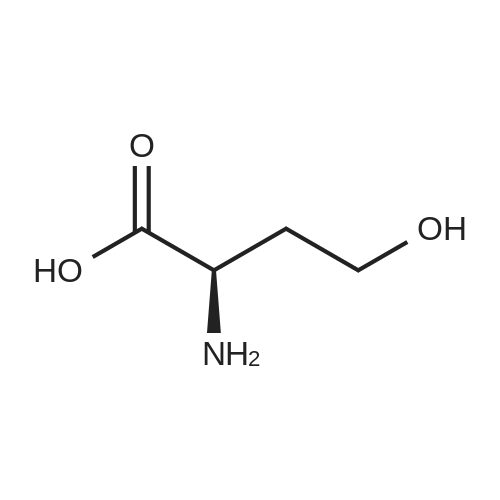
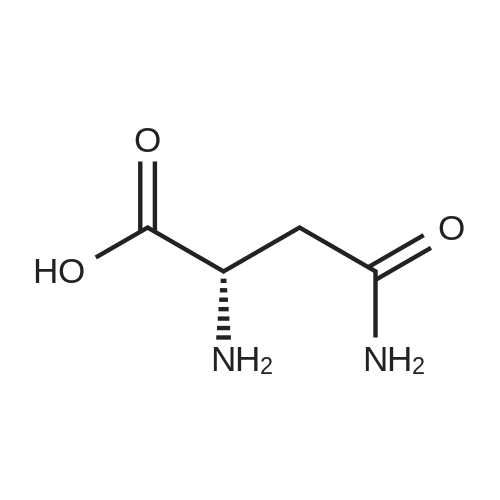
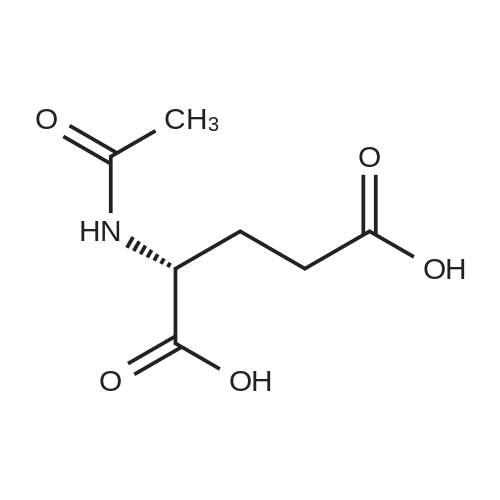
[ 56-86-0 ]
[ 7149-65-7 ]
Reference:
[1] Journal of Organic Chemistry, 1980, vol. 45, # 5, p. 815 - 818
[2] Patent: US4621145, 1986, A,
[3] Patent: CN105837486, 2016, A,
6
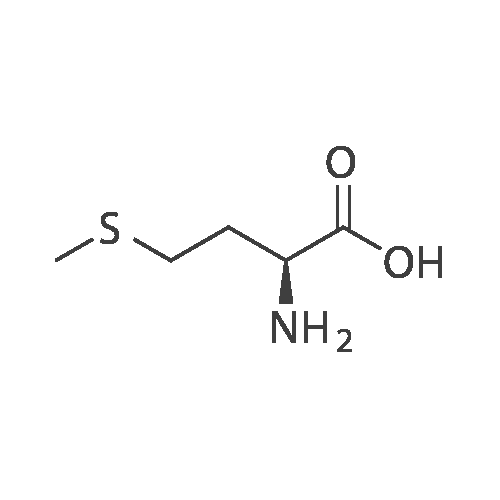
[ 64-17-5 ]
[ 56-86-0 ]
[ 7149-65-7 ]
Reference:
[1] Synlett, 2006, # 6, p. 893 - 896
[2] Tetrahedron, 1992, vol. 48, # 15, p. 2977 - 2992
[3] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1994, # 17, p. 2485 - 2492
[4] Journal of Organic Chemistry, 1994, vol. 59, # 7, p. 1719 - 1725
[5] Journal of Organic Chemistry, 2011, vol. 76, # 14, p. 5574 - 5583
[6] Tetrahedron Letters, 2002, vol. 43, # 50, p. 9191 - 9194
[7] Organic and Biomolecular Chemistry, 2004, vol. 2, # 14, p. 2003 - 2011
[8] Tetrahedron, 2010, vol. 66, # 25, p. 4462 - 4468
[9] Journal of Organic Chemistry, 1992, vol. 57, # 23, p. 6169 - 6173
7
[ 56-86-0 ]
[ 3190-71-4 ]
Reference:
[1] Nippon Kagaku Zasshi, 1956, vol. 77, p. 44,46[2] Chem.Abstr., 1958, p. 258
[3] Bulletin of the Chemical Society of Japan, 1956, vol. 29, p. 654,658[4] Chem.Abstr., 1958, p. 258
[5] Journal of the Chemical Society, 1950, p. 3239,3243
Reference:
[1] Bulletin de la Societe Chimique de France, 1989, # 1, p. 88 - 94
10
[ 56-87-1 ]
[ 56-86-0 ]
[ 78-98-8 ]
[ 123-32-0 ]
[ 5910-89-4 ]
[ 108-50-9 ]
[ 55138-72-2 ]
[ 55138-74-4 ]
[ 14667-55-1 ]
[ 17398-16-2 ]
[ 22047-27-4 ]
[ 15707-34-3 ]
[ 18433-97-1 ]
Reference:
[1] Journal of Agricultural and Food Chemistry, 2010, vol. 58, # 4, p. 2470 - 2478
11
[ 56-86-0 ]
[ 541-59-3 ]
[ 123-56-8 ]
[ 109-97-7 ]
[ 57-57-8 ]
[ 108-99-6 ]
[ 64-17-5 ]
[ 64-19-7 ]
[ 107-13-1 ]
[ 3680-71-5 ]
[ 75-05-8 ]
[ 107-12-0 ]
Reference:
[1] Journal of Agricultural and Food Chemistry, 2013, vol. 61, # 32, p. 7696 - 7704
12
[ 56-86-0 ]
[ 636-41-9 ]
[ 616-45-5 ]
[ 142-08-5 ]
[ 541-59-3 ]
[ 123-56-8 ]
[ 109-97-7 ]
[ 3680-71-5 ]
[ 75-05-8 ]
Reference:
[1] Journal of Agricultural and Food Chemistry, 2013, vol. 61, # 32, p. 7696 - 7704
13
[ 56-87-1 ]
[ 56-86-0 ]
[ 50-99-7 ]
[ 290-37-9 ]
[ 109-08-0 ]
[ 123-32-0 ]
[ 5910-89-4 ]
[ 13360-65-1 ]
[ 108-50-9 ]
[ 14667-55-1 ]
[ 13925-03-6 ]
[ 13925-07-0 ]
[ 13360-64-0 ]
Reference:
[1] Journal of Agricultural and Food Chemistry, 2010, vol. 58, # 4, p. 2470 - 2478
14
[ 56-86-0 ]
[ 72886-97-6 ]
Reference:
[1] Tetrahedron Letters, 2017, vol. 58, # 42, p. 3966 - 3969
[2] Patent: CN107253939, 2017, A,
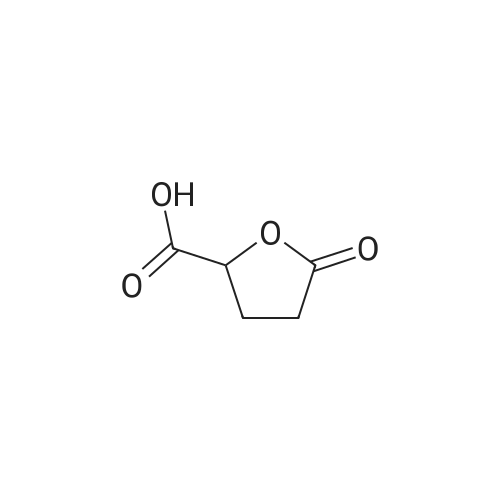
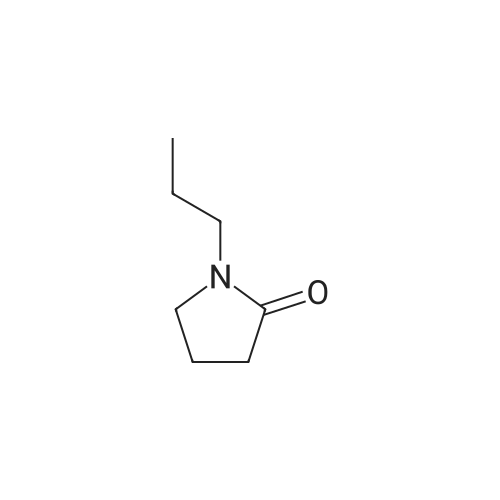
15
[ 56-86-0 ]
[ 21461-84-7 ]
Yield
Reaction Conditions
Operation in experiment
91.6%
With hydrogenchloride; sodium nitrite In water at -5 - 20℃; Inert atmosphere
To a solution of L-glutamic acid (10.07 g, 0.068 mol, JK CHEMICA) in 20 ml of coned. HCl and 40 mL H&2O was added a solution OfNaNO2 (7.0 g, 0.102 mol, Shantou Xilong chemical factory) in H2O (20 mL) slowly at -5 0C. The mixture was continued to stir for 12 hrs at room temperature. The reaction mixture was evaporated in vacuo below 50°C to give yellow oil, which was dissolved in EtOAc. The solid formed was filtered and washed with EtOAc. The filtrate and washing solution were combined, dried over Na2SO4. The solvent was concentrated in vacuo to give (S)-tetrahydro-5- oxofuran-2-carboxylic acid as pale yellow oil (8.1 g, 91.6 percent)\\ MS (ESI, pos. ion) m/z: 130.9 (M+ 1);1H NMR (400MHz, CDCl3): δ2.27 - 2.41 (m, IH), 2.44 - 2.65 (m, 3H), 5.09 (m, IH), 9.12- 9.55 (m, IH).
78%
With hydrogenchloride; sodium nitrite In 1,4-dioxane; water at 0 - 20℃; for 24 h;
l-glutamic acid (30.0g, 200mmol) was suspended in a water/ dioxane mixture (75/25mL) and stirred at 0°C for 30min. The white slurry became clear after 40mL of concentrated HCl (37percent) was added, followed by drop-wise addition of a solution of NaNO2 (21.0g, 300mmol) in 50mL of water. The reaction temperature was maintained around 0°C during the 4h of addition. The reaction mixture was then left stirring at room temperature for 20h. Upon completion, the solvent was evaporated under reduced pressure to provide a white solid, which was then treated with EtOAc (300mL) and Na2SO4 for 30min. The solution was filtered and the solvent was evaporated to yield S-2 as a white solid (21.50g, 78percent). 1H NMR (400MHz, CDCl3) δ 11.08 (bs, 1H), 5.01 (m, 1H), 2.71–2.55 (m, 3H), 2.45–2.37 (m, 1H); 13C NMR (100MHz, CDCl3) δ 176.8, 174.5, 75.4, 26.8, 25.7.
66%
With sulfuric acid; sodium nitrite In water at 20℃; for 15 h;
To a solution of L-glutamic acid in water, an aqueous solution of NaNO2 (1.2 molar equiv) and 2N H2SO4 (1.2 molar eqiv) were added simultaneously drop by drop. After the addition was over, the solution was stirred at room temperature for an additional 15 h. The water was removed in a rotary evaporator under reduced pressure by heating below 50° C. The gummy solid was triturated with 150 mL of boiling acetone and the hot solution was filtered and set aside to cool. This operation is repeated four times. Removal of solvent in a rotary evaporator afforded crude (5S)-5-carboxyl-2-oxo-tetrahydrofuran, which was purified by vacuum distillation to afford pure compound in 66percent yield as an oil.
93.5 % ee
With hydrogenchloride; sodium nitrite In 1,4-dioxane; water at 0 - 20℃; for 6 h;
A solution of sodium nitrite (140 g, 2.03 mol) in water (320 mL) was added over 4 h to a mixture of L-glutamic acid (200 g, 1.36 mol), dioxane (150 mL) and HCl (280 mL) in water (530 mL) and maintained at an internal temperature of 0-5° C. Some evolution of nitric oxide (brown gas) was observed. HPLC monitoring indicated that the reaction was complete once the addition of a stoichiometric amount of aqueous sodium nitrite was added (e.g., after approx. 30 min. for a reagent addition step requiring 45 minutes to complete). Further investigation using an in situ ReactIR probe indicated that the reaction required 2 h reaction time after the addition of the aqueous sodium nitrite solution. On completion of the addition, the mixture was warmed to r.t. and stirred 2 h, then solvents were removed in vacuo at 50-55° C. The residue was coevaporated with toluene (2.x.500 mL) to remove additional water, then ethyl acetate (1 L) was added, followed by sodium sulfate (100 g), and the mixture was stirred 0.5 h. The ethyl acetate solution was decanted and filtered, and the solids were washed and stirred with additional ethyl acetate (1 L). The combined filtrates were concentrated in vacuo, and the resulting residue was further dried under high vacuum (1 torr). The mass of crude residue containing EP2104-01 was 184 g, exceeding the mass of the theoretical yield by 7 g, attributable to solvent trapped by the viscous, syrupy residue. A sample of this residue was subjected to chiral GC analysis, which indicated the optical purity of this material to be 93.5percent e.e.
1.5 kg
With hydrogenchloride; sodium nitrite In water at -5 - 28℃; for 16 h; Large scale
(2S-2-Arninopentanedioic acid (2.50 kg, 16.99 mol) was dissolved in H20 (6 L) andconcentrated HC1 (3.5 L), then a solution ofNaNO2 (1.76 kg, 25.49 mol) in H20 (5 L) was addedslowly at -5 °C to 0 °C. After being stirred at 28 °C for 16 hours, the reaction mixture was concentrated below 50 °C to give a residue, which was treated with EtOAc (5 L). After being filtered , the filtrate was dried over Na2SO4 and concentrated in vacuo to give 1.5 kg of(2S-5-oxotetrahydrofuran-2-carboxylic acid as a colorless oil which was used for the next step without further purification.
Reference:
[1] Journal of Organic Chemistry, 2010, vol. 75, # 2, p. 390 - 398
[2] Patent: WO2010/45095, 2010, A1, . Location in patent: Page/Page column 69; 70
[3] Synthesis, 2007, # 15, p. 2279 - 2286
[4] Tetrahedron, 2009, vol. 65, # 48, p. 9997 - 10001
[5] Carbohydrate Research, 1983, vol. 124, p. 35 - 42
[6] Angewandte Chemie - International Edition, 2018, vol. 57, # 33, p. 10712 - 10717[7] Angew. Chem., 2018, vol. 130, # 33, p. 10872 - 10877,6
[8] European Journal of Medicinal Chemistry, 2014, vol. 80, p. 308 - 315
[9] Journal of the Chemical Society, Chemical Communications, 1994, # 9, p. 1139 - 1142
[10] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1995, # 21, p. 2695 - 2708
[11] Heterocycles, 1993, vol. 35, # 1, p. 115 - 120
[12] Tetrahedron, 1987, vol. 43, # 21, p. 5055 - 5072
[13] Journal of Organic Chemistry, 1984, vol. 49, # 26, p. 5041 - 5050
[14] Tetrahedron Letters, 1995, vol. 36, # 16, p. 2757 - 2760
[15] Organic Syntheses, 1985, vol. 63, p. 121 - 121
[16] Tetrahedron, 1995, vol. 51, # 21, p. 5935 - 5950
[17] Tetrahedron Letters, 1996, vol. 37, # 26, p. 4529 - 4532
[18] Journal of Medicinal Chemistry, 2017, vol. 60, # 12, p. 4949 - 4962
[19] Patent: US2005/32707, 2005, A1, . Location in patent: Page/Page column 5
[20] Journal of the Chemical Society, Chemical Communications, 1994, # 11, p. 1331 - 1332
[21] Helvetica Chimica Acta, 2003, vol. 86, # 5, p. 1397 - 1409
[22] Liebigs Annalen der Chemie, 1988, p. 1149 - 1154
[23] Tetrahedron Asymmetry, 1992, vol. 3, # 12, p. 1537 - 1538
[24] Journal of Organic Chemistry, 1988, vol. 53, # 20, p. 4780 - 4786
[25] Chemical Communications, 2007, # 5, p. 504 - 506
[26] Gazzetta Chimica Italiana, 1986, vol. 116, # 1, p. 29 - 34
[27] Journal of the American Chemical Society, 2011, vol. 133, # 6, p. 1793 - 1798
[28] Heterocycles, 2001, vol. 55, # 11, p. 2099 - 2108
[29] Organic and Biomolecular Chemistry, 2011, vol. 9, # 2, p. 531 - 537
[30] Organic and Biomolecular Chemistry, 2006, vol. 4, # 11, p. 2119 - 2157
[31] Journal of Organic Chemistry, 2008, vol. 73, # 16, p. 6330 - 6340
[32] Tetrahedron Letters, 1982, vol. 23, # 48, p. 5051 - 5054
[33] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1986, p. 1565 - 1580
[34] Bulletin de la Societe Chimique de France, 1987, vol. No. 1, p. 116 - 122
[35] Tetrahedron: Asymmetry, 1993, vol. 4, # 6, p. 1391 - 1396
[36] Tetrahedron Letters, 1991, vol. 32, # 51, p. 7539 - 7542
[37] European Journal of Medicinal Chemistry, 1997, vol. 32, # 7-8, p. 617 - 623
[38] Journal of Heterocyclic Chemistry, 2001, vol. 38, # 6, p. 1307 - 1312
[39] Polish Journal of Chemistry, 2004, vol. 78, # 2, p. 315 - 318
[40] Journal of the American Chemical Society, 2005, vol. 127, # 43, p. 15026 - 15027
[41] Synthesis, 1986, vol. NO. 3, p. 232 - 233
[42] Tetrahedron, 1984, vol. 40, # 18, p. 3521 - 3530
[43] Patent: US2006/155120, 2006, A1, . Location in patent: Page/Page column 13; 14; sheet 1; 2; 4; 5
[44] Patent: US5149462, 1992, A,
[45] Organic Process Research and Development, 2009, vol. 13, # 3, p. 535 - 542
[46] Journal of Organic Chemistry, 2010, vol. 75, # 9, p. 2820 - 2835
[47] Nucleosides, Nucleotides and Nucleic Acids, 2011, vol. 30, # 3, p. 210 - 226
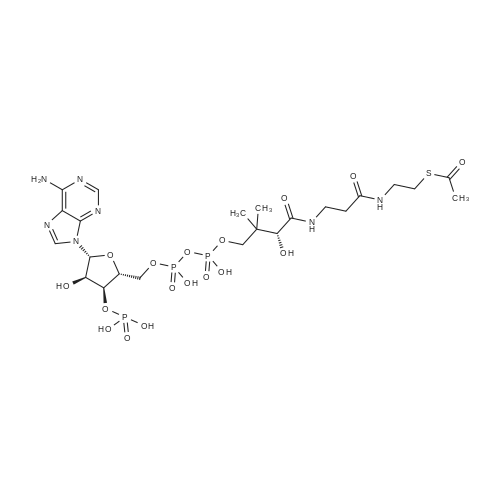
[48] Beilstein Journal of Organic Chemistry, 2013, vol. 9, p. 983 - 990
[49] Organic Process Research and Development, 1999, vol. 3, # 1, p. 73 - 76
[50] Patent: WO2014/124430, 2014, A1, . Location in patent: Page/Page column 143-144
[51] Patent: WO2015/38596, 2015, A1, . Location in patent: Page/Page column 195; 196
[52] Molecular BioSystems, 2015, vol. 11, # 5, p. 1389 - 1399
[53] Nucleosides, Nucleotides and Nucleic Acids, 2014, vol. 33, # 12, p. 774 - 785
[54] Patent: US2016/194350, 2016, A1, . Location in patent: Paragraph 0486; 0487; 0488
[55] Patent: WO2016/180743, 2016, A1, . Location in patent: Page/Page column 37; 38
[56] Patent: WO2017/1307, 2017, A1, . Location in patent: Page/Page column 19-20
[57] Angewandte Chemie - International Edition, 2018, vol. 57, # 10, p. 2712 - 2715[58] Angew. Chem., 2018, vol. 130, p. 2742 - 2745,4
Reference:
[1] Chemistry - A European Journal, 2013, vol. 19, # 21, p. 6824 - 6830
[2] Chemical Biology and Drug Design, 2018, vol. 91, # 4, p. 893 - 901
Reference:
[1] Biochemical Journal, 1959, vol. 71, p. 400,403, 404
27
[ 67-56-1 ]
[ 56-86-0 ]
[ 1499-55-4 ]
Yield
Reaction Conditions
Operation in experiment
100%
at 20℃; for 0.25 h; Inert atmosphere
Glutamic acid (22.1 g, 0.15 mol) was suspended in methanol (450 mL), under nitrogen at room temperature. TMS chloride (35.9 g, 0.33 mol) was then added dropwise over 5 minutes (note: by the end of addition most of solids dissolved). The mixture was stirred for additional 5 min and tested by TLC (15percent ammonia in methanol, ninhydrin visualization)—traces of starting material and no bis-methyl ester were visible on TLC. The mixture was stirred for additional 5 min, concentrated on a roto-vap and then dried in a vacuum oven (40° C.) to give pure by TLC and NMR product as a white solid. Yield: 30.0 g (100percent). 1H NMR (300 MHz, CD3OD, ppm) 2.2 (2H, m), 2.6 (2H, m), 3.7 (3H, s), 4.1 (1H, t)13C NMR (75 MHz, CD3OD, ppm) 26.6, 30.4, 52.5, 53.2, 171.4, 174.3.
50%
Stage #1: at 0 - 20℃; for 20 h; Stage #2: With pyridine In acetyl chloride at 20℃; for 16 h;
Acetyl chloride (20 ml) was added to methanol (300 ml) and the solution was cooled in an ice bath then L-glutamic acid (36 g) was added and the solution was stirred until all the solid has dissolved, and the reaction was kept at room temperature for 20 hours. Dry pyridine (40 ml) was added, and the reaction mixture was kept at room temperature for another 16 hours. The precipitated product (20 g, 50percent) was filtered and, washed successively with ether and air dried. The product, glutamic acid 5-methyl ester, can be used directly for the next step. An analytical sample can be obtained by recrystallization in 70percent aqueous methanol.
Reference:
[1] Patent: US2016/16890, 2016, A1, . Location in patent: Paragraph 0163
[2] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 13, p. 3793 - 3797
[3] Tetrahedron Letters, 2010, vol. 51, # 9, p. 1333 - 1335
[4] Synthesis, 1987, # 7, p. 635 - 637
[5] Organic Process Research and Development, 2004, vol. 8, # 1, p. 72 - 85
[6] Patent: WO2004/50084, 2004, A2, . Location in patent: Page/Page column 57; 58
[7] Helvetica Chimica Acta, 1955, vol. 38, p. 1491,1499
[8] Tetrahedron Letters, 2003, vol. 44, # 28, p. 5251 - 5253
[9] Journal of the Chemical Society, 1951, p. 2294
[10] Biochemical Preparations, 1957, vol. 5, p. 79,82
[11] Journal of the Chemical Society, Chemical Communications, 1988, # 12, p. 828 - 829
[12] Journal of the Chemical Society, 1950, p. 3239,3243
[13] Tetrahedron Letters, 2000, vol. 41, # 32, p. 6191 - 6194
[14] Tetrahedron Asymmetry, 2000, vol. 11, # 24, p. 4965 - 4973
[15] Organic and Biomolecular Chemistry, 2006, vol. 4, # 9, p. 1796 - 1805
[16] ChemSusChem, 2011, vol. 4, # 6, p. 785 - 791
[17] Chemistry - A European Journal, 2013, vol. 19, # 21, p. 6824 - 6830
[18] Organic Process Research and Development, 2004, vol. 8, # 1, p. 72 - 85
[19] Patent: US2561323, 1948, ,
Reference:
[1] Angewandte Chemie - International Edition, 2011, vol. 50, # 10, p. 2307 - 2312
31
[ 463-51-4 ]
[ 56-86-0 ]
[ 5817-08-3 ]
Reference:
[1] Journal of Biological Chemistry, 1940, vol. 132, p. 165
32
[ 463-51-4 ]
[ 56-86-0 ]
[ 623-11-0 ]
[ 5817-08-3 ]
[ 1188-37-0 ]
Reference:
[1] Chemische Berichte, 1930, vol. 63, p. 438
[2] Journal of Biological Chemistry, 1940, vol. 132, p. 165
33
[ 56-86-0 ]
[ 108-24-7 ]
[ 1188-37-0 ]
Reference:
[1] Synthetic Communications, 1992, vol. 22, # 2, p. 257 - 264
[2] Chemistry of Heterocyclic Compounds (New York, NY, United States), 1995, vol. 31, # 1, p. 33 - 34[3] Khimiya Geterotsiklicheskikh Soedinenii, 1995, # 1, p. 39 - 41
[4] Chemical Papers, 2011, vol. 65, # 1, p. 70 - 76
34
[ 56-86-0 ]
[ 72-89-9 ]
[ 1188-37-0 ]
Reference:
[1] Cell Chemical Biology, 2016, vol. 23, # 8, p. 935 - 945
35
[ 463-51-4 ]
[ 56-86-0 ]
[ 1188-37-0 ]
Reference:
[1] Chemische Berichte, 1930, vol. 63, p. 438
[2] Journal of Biological Chemistry, 1940, vol. 132, p. 165
36
[ 56-86-0 ]
[ 17336-14-0 ]
[ 1188-37-0 ]
Reference:
[1] Journal of Physical Organic Chemistry, 2012, vol. 25, # 11, p. 939 - 945
37
[ 56-86-0 ]
[ 1734-62-9 ]
[ 1188-37-0 ]
Reference:
[1] Journal of Physical Organic Chemistry, 2012, vol. 25, # 11, p. 939 - 945
38
[ 56-86-0 ]
[ 50-78-2 ]
[ 1188-37-0 ]
Reference:
[1] Journal of Physical Organic Chemistry, 2012, vol. 25, # 11, p. 939 - 945
39
[ 463-51-4 ]
[ 56-86-0 ]
[ 623-11-0 ]
[ 5817-08-3 ]
[ 1188-37-0 ]
Reference:
[1] Chemische Berichte, 1930, vol. 63, p. 438
[2] Journal of Biological Chemistry, 1940, vol. 132, p. 165
40
[ 5817-08-3 ]
[ 56-86-0 ]
[ 19146-55-5 ]
Reference:
[1] Journal of the American Chemical Society, 1989, vol. 111, # 16, p. 6354 - 6364
41
[ 590-28-3 ]
[ 56-86-0 ]
[ 1188-38-1 ]
Yield
Reaction Conditions
Operation in experiment
62%
With hydrogenchloride In water at 59.84℃; for 4 h;
4mmol of L-glutamic acid was dissolved in 20mL of water and the solution was acidified withconcentratedHCl (37percentv/v).Then, 12mmol of potassiumthiocyanate (KOCN) was added tothis solution. The mixture was warmed up, with agitation, to 333 K, during 4 hr. The resultantsolution was cooled at roomtemperature into a glass vial sealed with parafilm, which was perforatedwitha needle to allowslowevaporation until the precipitation of awhite solid.Crystalsof (I) suitable for X-ray analysis were obtained by slow evaporation of an ethanol solution atroom temperature (Scheme 1). Yield 62percent, mp 170°C–171°C. This experimental procedurecorrespond with a modified and improved methodology previously reported
Reference:
[1] Molecular Crystals and Liquid Crystals, 2016, vol. 625, # 1, p. 225 - 232
[2] Pharmaceutical Chemistry Journal, 1978, vol. 12, # 5, p. 601 - 606[3] Khimiko-Farmatsevticheskii Zhurnal, 1978, vol. 12, # 5, p. 53 - 59
42
[ 462-88-4 ]
[ 56-86-0 ]
[ 1188-38-1 ]
Yield
Reaction Conditions
Operation in experiment
58%
Stage #1: With potassium cyanate; potassium hydroxide In water at 20 - 80℃; Stage #2: With hydrogenchloride In water
In a 2 L reaction vessel, glutamic acid (100 g, 0.68 mol), potassium cyanate (65.3 g, 0.816 mol),Potassium hydroxide (40 g, 0.71 mol) and 1 L of water,After slowly heating to 30-50 ° C to completely dissolve the solid,Continue to heat up to 60-80 ° C reaction 2-2.5h,Slowly cool to room temperature and let stand at room temperature for 10-15h.Acidified with concentrated hydrochloric acid to adjust pH=7, then add the catalyst urea-propionic acid (100mL), let stand for 2-3h, precipitate a large amount of solid,After filtration, 86 g of crude product was obtained, and N-carbamoylglutamic acid was crystallized from water, filtered, and dried at 80 ° C to obtain 76 g of fine product.The molar yield is 58percent, the content is 92percent,
Reference:
[1] Patent: CN108484448, 2018, A, . Location in patent: Paragraph 0013; 0014; 0015; 0016; 0017
Reference:
[1] Synlett, 2004, # 12, p. 2180 - 2184
[2] Green Chemistry, 2011, vol. 13, # 4, p. 807 - 809
[3] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1987, p. 1569 - 1574
[4] Synlett, 2003, # 4, p. 542 - 546
[5] Green Chemistry, 2017, vol. 19, # 21, p. 5178 - 5186
[6] Molecular Catalysis, 2017, vol. 443, p. 92 - 100
[7] Green Chemistry, 2018, vol. 20, # 18, p. 4217 - 4223
45
[ 56-86-0 ]
[ 3515-93-3 ]
[ 16051-87-9 ]
Reference:
[1] Green Chemistry, 2011, vol. 13, # 4, p. 807 - 809
46
[ 56-86-0 ]
[ 16051-87-9 ]
[ 692-29-5 ]
Reference:
[1] Journal of the Indian Chemical Society, 2000, vol. 77, # 9, p. 413 - 420
[2] Indian Journal of Chemistry - Section A Inorganic, Physical, Theoretical and Analytical Chemistry, 2004, vol. 43, # 6, p. 1186 - 1192
47
[ 56-86-0 ]
[ 16051-87-9 ]
[ 124-38-9 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1983, p. 323 - 334
48
[ 56-86-0 ]
[ 5680-86-4 ]
Reference:
[1] Journal of the Chemical Society, 1950, p. 3239,3243
[2] Journal of Organic Chemistry, 2011, vol. 76, # 20, p. 8513 - 8517
49
[ 56-86-0 ]
[ 35726-62-6 ]
Reference:
[1] Helvetica Chimica Acta, 1948, vol. 31, p. 737,743
[2] Hoppe-Seyler's Zeitschrift fuer Physiologische Chemie, 1933, vol. 219, p. 155
[3] European Journal of Organic Chemistry, 2013, # 25, p. 5555 - 5560
Reference:
[1] Tetrahedron Letters, 2003, vol. 44, # 28, p. 5251 - 5253
[2] Journal of the Chemical Society, 1950, p. 3239,3243
54
[ 56-86-0 ]
[ 13574-13-5 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2007, vol. 5, # 12, p. 1915 - 1923
[2] Doklady Chemistry, 2006, vol. 408, # 1, p. 57 - 60
[3] Synthetic Communications, 1990, vol. 20, # 15, p. 2235 - 2249
55
[ 67-56-1 ]
[ 56-86-0 ]
[ 24424-99-5 ]
[ 59279-60-6 ]
Yield
Reaction Conditions
Operation in experiment
95%
Stage #1: at 20℃; for 6 h; Stage #2: at 0 - 20℃; for 48 h;
7.4 g L-glutamic acid(6,50.0 mmol) and 100 ml of methanol were added to the reaction flask. To the flask was added dropwise 32 ml of trimethylchlorosilane (TMSC1, 250.0 mmol) at room temperature and the mixture was stirred at room temperature for 6 hours. The esterification reaction was terminated by TLC. To the reaction system was added 49 ml of triethylamine (Et3N, 350.0 mmol) and 13.2 g of Boc20 (60. 0 mmol) at 0 ° C, and the mixture was stirred at room temperature for 48 hours. Ethyl acetate (200 ml) and water (100 ml) were added to the residue, and the mixture was partitioned by shaking. The aqueous phase was extracted with ethyl acetate (100 mL χ 3). The combined organic phases were dried over anhydrous sodium sulfate, filtered, Column chromatography to obtain 13.1 g of glutamic acid ester (7). The overall yield in 2 steps was 95percent.
95.2%
Stage #1: at 0℃; for 0.0833333 h; Stage #2: for 2 h; Reflux Stage #3: With triethylamine In tetrahydrofuran at 0 - 20℃; for 2.58333 h;
Acetyl chloride (5 mL) was slowly added dropwise to methanol (100 mL) at 0° C., stirred for 5 minutes, then glutamic acid (10 g, 67.9 mmol) was added, stirring was continued and the mixture was heated to reflux for 2 hours while maintaining the reflux temperature. .The reaction was stopped and the solvent was removed under reduced pressure and recrystallized from ether. The resulting oil was dissolved in THF (150 mL) and TEA (28.5 mL, 203.7 mmol) was added dropwise at 0°C.After stirring at 0° C. for 5 minutes, di-tert-butyl dicarbonate (17.8 g, 81.5 mmol) dissolved in THF (30 mL) was continuously added dropwise, and the mixture was stirred at room temperature for 2.5 hours.After the reaction was completed, the solvent was evaporated under reduced pressure, and the residue was added with water (200 mL), extracted with DCM (2×200 mL) from the aqueous phase, and the combined organic phases were dried over anhydrous sodium sulfate.After concentration, the resulting crude product was purified by flash column (PE:EA=5:1) to give N-Boc-L-(+)-glutamic acid dimethyl ester (17.7 g, yield 95.2percent) as none Oily liquid
1.30 kg
Stage #1: at 0 - 20℃; Stage #2: With triethylamine In methanol at 25℃;
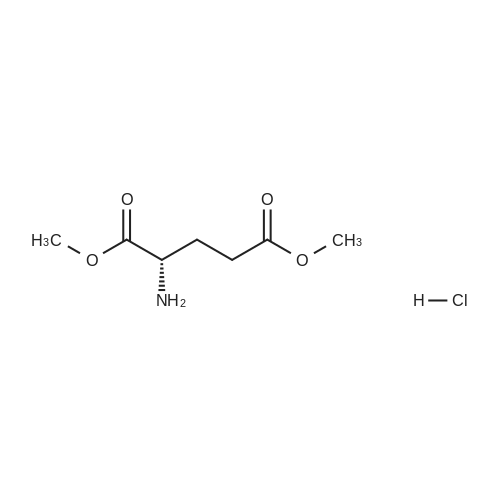
H-CI(S)-(Tetrahydro-pyran-3-yl)amine hydrochloride (S)-Dimethyl 2-(tert-butoxycarbonylamino)pentanedioateTo MeOH (7L) was added TMSC1 slowly at 0 °C, and the mixture was stirred for 30 min, then L-glutamic acid (700 g, 4.76 mol) was added to the mixture. The mixture was stirred at room temperature until complete reaction was observed (monitored by TLC). After cooling to 0 °C, triethylamine (313 g, 31.0 mol) and Boc20 (1.14 Kg, 5.23 mol) were added slowly to the reaction solution successively while keeping the internal temperature below 25 °C, and the resultant solution was stirred for 16 hours. After concentration, the residue was poured into water (5 L) and extracted with ethyl acetate (10 L). The organic phase was washed with 4L of 20percent citric acid and brine, and dried over sodium sulfate. After filtration and concentration, the crude (S)-dimethyl 2-(tert-butoxycarbonylamino)pentanedioate (1.30 Kg) was obtained as pale yellow oil.
Reference:
[1] Angewandte Chemie - International Edition, 2018, vol. 57, # 25, p. 7528 - 7532[2] Angew. Chem., 2018, vol. 130, # 25, p. 7650 - 7654,5
[3] Patent: CN105294519, 2016, A, . Location in patent: Paragraph 0011
[4] Patent: CN107459511, 2017, A, . Location in patent: Paragraph 0084; 0085; 0086
[5] Angewandte Chemie - International Edition, 2016, vol. 55, # 6, p. 2191 - 2194[6] Angew. Chem., 2016, vol. 128, # 6, p. 2231 - 2234,4
[7] Organic Letters, 2008, vol. 10, # 11, p. 2175 - 2178
[8] Chemical Communications, 2018, vol. 54, # 23, p. 2890 - 2893
[9] Journal of Organic Chemistry, 1998, vol. 63, # 11, p. 3741 - 3744
[10] Science China Chemistry, 2012, vol. 55, # 6, p. 1101 - 1107
[11] Patent: WO2013/7768, 2013, A1, . Location in patent: Page/Page column 109; 110
[12] Synlett, 2013, vol. 24, # 8, p. 987 - 990
[13] Journal of the American Chemical Society, 2018, vol. 140, # 23, p. 7116 - 7126
56
[ 56-86-0 ]
[ 24424-99-5 ]
[ 59279-60-6 ]
Yield
Reaction Conditions
Operation in experiment
85%
Stage #1: With thionyl chloride In methanol at 20℃; for 10 h; Reflux; Inert atmosphere Stage #2: With triethylamine In tetrahydrofuran at 0 - 20℃; for 8 h; Inert atmosphere
Freshly distilled thionyl chloride (0.60 mL, 8.16 mmol) was added dropwise with stirring to a cooled suspension of l-glutamic acid (1.0 g, 6.8 mmol) in anhydrous methanol (7 mL). The reaction mixture was stirred at rt for 2 h and was refluxed for 8 h. The solvent was removed under reduced pressure and co-evaporated with methanol (3.x.7 mL) to afford crude product as sticky liquid. To a solution of this crude product in THF (20 mL) were added triethylamine (2.0 mL, 14.6 mmol) and a solution of di-tert-butyl dicarbonate (1.78 g, 8.16 mmol) in THF (10 mL) at 0 °C. The reaction mixture was stirred at rt for 8 h. After completion of the reaction, solvent was removed in vacuo and the residue partitioned between AcOEt (20 mL) and water (20 mL). The aqueous phase was extracted with AcOEt (2.x.15 mL) and the combined organic layers were washed with 3percent HCl (15 mL), saturated NaHCO3 (15 mL), and brine (20 mL), dried (Na2SO4), and evaporation of the solvent gave 16 (1.5 g, 85percent) as a colorless liquid.Comment[α]D26 +13.7 (c 0.89, CHCl3), lit.refPreviewPlaceHolder20 [α]D25 +12.5 (c 2, CHCl3); 1H NMR (300 MHz, CDCl3) δ 5.16 (br d, 1H, J=6.4 Hz), 4.28 (br d, 1H, J=4.6 Hz), 3.69 (s, 3H), 3.63 (s, 3H), 2.33-2.42 (m, 2H), 2.10-2.16 (m, 1H), 1.86-2.00 (m, 1H), 1.38 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 173.2, 172.7, 155.4, 80.0, 52.9, 52.4, 51.8, 30.1, 28.3, 27.7; HRMS (ESI) (M+Na)+ calculated for C12H21NO6Na+=298.1267, found 298.1266.
Stage #1: With triethylamine In methanol at 0 - 20℃; Stage #2: at 20℃; Cooling with ice
[0027] To a stirred solution of (S)-Glutamic acid (100 g, 0.68 mol) in dry methanol (1740 ml) was added TMSC1(375 ml, 2.9 mol) at 0° C. After addition, the mixture was allowed to reach room temperature and stirred overnight. NEt3 (446 g, 4.42 mol) was added to the solution under ice-bath and followed by addition of Boc2O (163 g, 0.75 mol). After the evolution of gas had ceased, the mixture became clear and was allowed to warm to room temperature. The solvent was evaporated under reduced pressure, and the residue was triturated and washed with ether (3×500 ml). The combined filtrates were concentrated to provide crude product which was purified by column chromatography on silica gel to yield TA-101 (173 g, 92.5percent yield) as an oil. 1H NMR (CDCl3): 5.16-5.14 (d, 1H), 4.34-4.33 (d, 1H), 3.74 (s, 3H), 3.68 (s, 3H), 2.48-2.34 (m, 2H), 2.23-2.16 (m, 1H), 2.16-1.92 (m, 1H), 1.43 (s, 9H);
Reference:
[1] Journal of Medicinal Chemistry, 2006, vol. 49, # 16, p. 4971 - 4980
[2] Bioorganic and Medicinal Chemistry, 2005, vol. 13, # 17, p. 5240 - 5252
[3] Journal of Medicinal Chemistry, 2005, vol. 48, # 22, p. 6767 - 6771
[4] Journal of Organic Chemistry, 2003, vol. 68, # 3, p. 743 - 746
[5] Tetrahedron Letters, 2001, vol. 42, # 43, p. 7599 - 7603
[6] Tetrahedron Letters, 2011, vol. 52, # 19, p. 2488 - 2491
[7] Patent: EP2319850, 2011, A1,
[8] Organic Letters, 2012, vol. 14, # 17, p. 4518 - 4521
[9] Organic Process Research and Development, 2005, vol. 9, # 6, p. 853 - 856
[10] Organic Letters, 2013, vol. 15, # 22, p. 5670 - 5673
[11] Chemistry - An Asian Journal, 2015, vol. 10, # 2, p. 474 - 482
[12] Journal of Medicinal Chemistry, 2015, vol. 58, # 23, p. 9414 - 9420
[13] Patent: WO2017/156074, 2017, A1,
59
[ 75-77-4 ]
[ 56-86-0 ]
[ 24424-99-5 ]
[ 72086-72-7 ]
Yield
Reaction Conditions
Operation in experiment
95%
Stage #1: at 20℃; Cooling with ice Stage #2: at 20℃; for 3 h;
L-glutamic acid (38 g, 0.26 mol) was dissolved in anhydrous methanol (450 mL).Me3SiCl was added dropwise on ice bath (120 mL,0.94mmol),Stir at room temperature overnight.Add Et3N (230 mL, 1.66 mol) and (Boc)2O (63.5 g, 0.3 mol) stirring at room temperature For an 3 hours, the concentrated column was evaporated to dryness to give compound L-1 (67.6 g, 95percent).
The reaction takes place via typical condition, that is 6.8 mmol L-glutamic acid, 10.2 mmol benzyl alcohol (L-glutamic acid/benzyl alcohol molar ratio = 1:1.5) and 0.68 mmol CuCl2 are loaded into a 50 ml single-port reaction flask, and the mixture is stirred at 60 °C for 2 h. After the reaction, the reaction mixture was cooled to be analyzed.
Reference:
[1] Journal of the Chemical Society - Perkin Transactions 1, 1999, # 10, p. 1311 - 1323
[2] Journal of the Chemical Society, 1949, p. 1401,1404
72
[ 56-86-0 ]
[ 45214-91-3 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 13, p. 3793 - 3797
[2] Organic and Biomolecular Chemistry, 2006, vol. 4, # 9, p. 1796 - 1805
[3] Tetrahedron Letters, 2003, vol. 44, # 28, p. 5251 - 5253
[4] Angewandte Chemie - International Edition, 2013, vol. 52, # 36, p. 9558 - 9562[5] Angew. Chem., 2013, vol. 125, # 36, p. 9737 - 9741,5
[6] Angewandte Chemie - International Edition, 2015, vol. 54, # 45, p. 13366 - 13369[7] Angew. Chem., 2015, vol. 54, # 45, p. 13564 - 13567
[8] Patent: US2016/16890, 2016, A1,
[9] Patent: US2016/16890, 2016, A1,
73
[ 2280-44-6 ]
[ 73-32-5 ]
[ 56-86-0 ]
[ 657-27-2 ]
[ 60-18-4 ]
Reference:
[1] Agricultural and Biological Chemistry, 1990, vol. 54, # 12, p. 3275 - 3282
74
[ 56-86-0 ]
[ 102774-86-7 ]
[ 115-11-7 ]
[ 84793-07-7 ]
[ 71989-18-9 ]
[ 129460-14-6 ]
Reference:
[1] Synthesis, 1990, # 7, p. 571 - 572
75
[ 56-86-0 ]
[ 72479-05-1 ]
Reference:
[1] Journal of Organic Chemistry, 1980, vol. 45, # 5, p. 815 - 818
76
[ 540-88-5 ]
[ 56-86-0 ]
[ 501-53-1 ]
[ 81470-51-1 ]
Reference:
[1] Journal of Organic Chemistry, 1990, vol. 55, # 6, p. 1711 - 1721
77
[ 56-86-0 ]
[ 73821-97-3 ]
Reference:
[1] Synthesis, 1992, # 4, p. 361 - 362
78
[ 64-17-5 ]
[ 56-86-0 ]
[ 1118-89-4 ]
Yield
Reaction Conditions
Operation in experiment
98%
Stage #1: at 70 - 75℃; for 5 h; Stage #2: at 20 - 25℃; for 0.5 h;
To a 500 ml four-necked flask equipped with a stirrer, a thermometer and a reflux condenser (connected with 30percent aqueous sodium hydroxide solution), 300 g of ethanol was added.14.7 g (0.10 mol) L-glutamic acid, 25.0 g (0.08 mol) triphosgene, Heat, react at 70-75°C for 5 hours, cool to 20-25°C,Hydrogen chloride gas in the nitrogen substitution system was replaced 30 minutes later.Distillation recovers excess triphosgene and ethanol, then adds 100 to the residueMethyl tert-butyl ether, beaten, filtered and dried to give 23.5 g of a white solidL-glutamic acid diethyl ester hydrochloride, liquid purity 99.7percent, yield 98.0percent.
77.76%
Stage #1: at -10 - 10℃; for 1 h; Stage #2: at 20 - 80℃; for 2 h;
Add 320 mL (5.48 mol, 40 eq) of absolute ethanol to a 1000 mL single-mouth bottle and cool to minus 10 ° C.To the reaction system, 28 mL (378.92 mmol, 2.8 eq) of thionyl chloride was slowly added dropwise.After the dropwise addition was completed, the reaction solution was stirred at a constant temperature of 10 ° C for 1 hour.Further, 20.08 g (136.48 mmol, 1 eq) of L-glutamic acid was added to the reaction system.The reaction solution was stirred at room temperature for 2 hours, then warmed to 80 ° C and the reaction was monitored by TLC.The reaction solution was concentrated under reduced pressure to remove the solvent, which was cooled and crystallized in a refrigerator.Add ethyl acetate (150 mL × 3), wash and filter by suction.The product obtained by drying (L-glutamic acid diethyl ester hydrochloride) was 25.44 g of a white solid, yield 77.76percent.
Reference:
[1] Synthetic Communications, 2010, vol. 40, # 8, p. 1161 - 1179
[2] Patent: CN107602436, 2018, A, . Location in patent: Paragraph 0051; 0052
[3] Patent: CN108218739, 2018, A, . Location in patent: Paragraph 0116; 0117
[4] Chemistry of Natural Compounds, 1994, vol. 30, # 2, p. 238 - 244[5] Khimiya Prirodnykh Soedinenii, 1994, # 2, p. 261 - 268
[6] Synthetic Communications, 1989, vol. 19, # 20, p. 3485 - 3496
[7] European Journal of Medicinal Chemistry, 2011, vol. 46, # 1, p. 11 - 20
79
[ 56-86-0 ]
[ 75-36-5 ]
[ 1118-89-4 ]
Yield
Reaction Conditions
Operation in experiment
99%
at 0℃; for 4 h; Reflux
EtOH (42 mL) was placed in a round-bottomed flask and cooled to 0 °C in an ice bath. Acetylchloride (3.6 mL, 50.0 mmol) was then slowly added to the EtOH with keeping thetemperature and magnetically string. After the reaction solution was starred at 0 °C for 30 min,L-glutamic acid (3.68 g, 25.0 mmol) was added to the mixture solution. The reaction solutionwas stirred under a reflux condition for 4 h. The resulting solution was evaluated undervacuum to obtain a colorless oil of 11′.
Reference:
[1] Molecules, 2017, vol. 22, # 3,
80
[ 56-86-0 ]
[ 1118-89-4 ]
Reference:
[1] Chemische Berichte, 1933, vol. 66, p. 151,158[2] Nippon Kagaku Kaishi, 1931, vol. 52, p. 844,850
81
[ 56-86-0 ]
[ 104091-09-0 ]
Yield
Reaction Conditions
Operation in experiment
76%
With potassium carbonate In acetonitrile at 20℃; for 2 h;
General procedure: To a solution of H-Phe-OH (100 mg, 60.5 mmol) in 50 percent MeCN (6.1 mL)were added Fmoc-OPhth (233 mg, 60.5 mmol) and K2CO3 (167 mg, 121 mmol) and stirred at room temperature. After 2 h of stirring saturated sodium bicarbonate solution and H2O were added and the resulting solution was washed with diethyl ether. The aqueous phase is acidified to pH 1 with 1M HCl and extracted with diethyl ether. The organic phase was washed with 1 M HCl, H2O, brine, dried over MgSO4. The filtrate was evaporatedevaporated under reduced pressure to give yellow solid as crude product.
Reference:
[1] Synlett, 2011, # 14, p. 2013 - 2016
83
[ 56-86-0 ]
[ 28920-43-6 ]
[ 104091-09-0 ]
Reference:
[1] Journal of the Chinese Chemical Society, 2011, vol. 58, # 4, p. 509 - 515
[2] Soft Matter, 2011, vol. 7, # 19, p. 8913 - 8922
84
[ 56-86-0 ]
[ 102774-86-7 ]
[ 115-11-7 ]
[ 84793-07-7 ]
[ 71989-18-9 ]
[ 129460-14-6 ]
Reference:
[1] Synthesis, 1990, # 7, p. 571 - 572
85
[ 56-86-0 ]
[ 24277-39-2 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 13, p. 3793 - 3797
[2] Tetrahedron Letters, 2003, vol. 44, # 28, p. 5251 - 5253
[3] Journal of the Chemical Society - Perkin Transactions 1, 1999, # 8, p. 855 - 866
[4] Angewandte Chemie - International Edition, 2013, vol. 52, # 36, p. 9558 - 9562[5] Angew. Chem., 2013, vol. 125, # 36, p. 9737 - 9741,5
[6] Patent: US2016/16890, 2016, A1,
[7] Patent: US2016/16890, 2016, A1,
[8] Patent: US2016/16890, 2016, A1,
[9] Patent: US2016/16890, 2016, A1,
86
[ 56-86-0 ]
[ 104-15-4 ]
[ 100-51-6 ]
[ 2791-84-6 ]
Yield
Reaction Conditions
Operation in experiment
61 g
Inert atmosphere
10135] 29.4 g (0.2 mol) L-(+)-glutamic acid, 40 g (0.23 mol) p-toluenesulfonic acid, and 80 mE benzyl alcohol were dissolved in 500 mE methylbenzene. 11 mE water was separated out by backflow under the protection of nitrogen gas. The backflow was continued for 3 h, and 150 mE liquid was evaporated and removed. The solution was cooled down to 50° C., and then the reaction solution was poured into a beaker containing 600 mE petroleum ether for stirring 1 h. The precipitation was collected by filtration. The filter cake was dissolved in 280 mE of 95percent ethanol by heating, then the heating was stopped and the solution was cooled overnight. The precipitation was collected by filtration and dried in vacuum to produce 61 g E-(+)-glutamic acid dibenzyl ester p-toluenesulfonate (compound 1).
Reference:
[1] European Journal of Medicinal Chemistry, 2008, vol. 43, # 12, p. 2699 - 2716
[2] Journal of Chemical Research, 2013, vol. 37, # 3, p. 177 - 180
[3] Journal of Chemical Research, 2013, vol. 37, # 3, p. 181 - 185
[4] Journal of the Chemical Society - Perkin Transactions 1, 1999, # 8, p. 855 - 866
[5] Patent: US2015/359900, 2015, A1, . Location in patent: Paragraph 0134; 0135
Stage #1: at 80 - 85℃; for 5 h; Stage #2: at 20 - 25℃; for 0.5 h;
To a 500 ml four-necked flask equipped with a stirrer, a thermometer and a reflux condenser (connected with a 30percent aqueous sodium hydroxide absorber) was added 280 g of benzyl alcohol.14.7 g (0.10 mol)L-glutamic acid, 30.0 g (0.25 mole) of thionyl chloride,Heat, react at 80-85°C for 5 hours, cool to 20-25°C,Hydrogen chloride gas in the nitrogen displacement system, after 30 minutes of replacement,Distillation recovers excess thionyl chloride and benzyl alcohol,Then add 120 grams methyl tert-butyl ether to the residue and beat it.Filter and dry to give 35.9 g of a white solidDibenzyl L-glutamate hydrochloride,The liquid phase purity was 99.8percent and the yield was 98.7percent.
Reference:
[1] Patent: CN107602436, 2018, A, . Location in patent: Paragraph 0047; 0048; 0049; 0050
[2] Journal of the Chinese Chemical Society, 2009, vol. 56, # 5, p. 1010 - 1017
[3] Bioorganic and Medicinal Chemistry, 2010, vol. 18, # 17, p. 6220 - 6229
90
[ 56-86-0 ]
[ 4561-10-8 ]
Reference:
[1] Patent: WO2018/26763, 2018, A1,
91
[ 67-56-1 ]
[ 56-86-0 ]
[ 23150-65-4 ]
Yield
Reaction Conditions
Operation in experiment
98.1%
Stage #1: at 60 - 63℃; for 7 h; Stage #2: at 20 - 25℃; for 0.5 h;
To a 500 ml four-necked flask equipped with a stirrer, a thermometer and a reflux condenser (connected with a 30percent aqueous sodium hydroxide absorber) was added 300 g of methanol.14.7 g (0.10 mol) of L-glutamic acid, 30.0 g (0.25 mol) of thionyl chloride,Heating, reaction at 60-63°C for 7 hours, cooling to 20-25°C,Hydrogen chloride gas in the nitrogen displacement system, after 30 minutes of replacement,Distillation recovers excess thionyl chloride and methanol,Then 100 g methyl tert-butyl ether was added to the residue, which was beaten and filtered.Dry to obtain 20.8 g of L-glutamic acid dimethyl ester hydrochloride as a white solid.The liquid purity was 99.5percent and the yield was 98.1percent.
47.5 g
at 0 - 20℃; for 40 h;
50.0 g L-Glutamic acid was taken in assembly containing 300 ml methanol, chilled the reaction mass up to 0 to 5 °C. Added dropwise 40.5 g Thionyl chloride into the reaction mass. The reaction mixture turned into a clear solution that was stirred for 40 hrs at room temperature. The solvent was evaporated to give crude L-glutamic acid dimethyl ester hydrochloride as residue. Added Methanol (150 ml) and distilled out to remove residual thionyl chloride. Added Ethyl Acetate (250 ml) to precipitate solid. Filter the product and washed with ethyl acetate (50 ml) to get 47.5 g L-glutamic acid dimethyl ester hydrochloride.
Reference:
[1] Journal of Medicinal Chemistry, 2007, vol. 50, # 24, p. 6144 - 6153
[2] Archiv der Pharmazie, 2005, vol. 338, # 5-6, p. 281 - 290
[3] Bioorganic and Medicinal Chemistry, 2007, vol. 15, # 18, p. 6273 - 6290
[4] Bioorganic and Medicinal Chemistry, 2009, vol. 17, # 2, p. 777 - 793
[5] Patent: CN107602436, 2018, A, . Location in patent: Paragraph 0053; 0054
[6] Phytochemistry, 1997, vol. 45, # 1, p. 37 - 40
[7] Monatshefte fur Chemie, 2007, vol. 138, # 12, p. 1283 - 1287
[8] Journal of Organic Chemistry, 2007, vol. 72, # 6, p. 2106 - 2117
[9] Journal of the Chemical Society - Perkin Transactions 1, 1999, # 10, p. 1311 - 1323
[10] Patent: EP1229041, 2002, A1, . Location in patent: Example 1
[11] Patent: WO2004/14893, 2004, A2, . Location in patent: Page 103; 105
[12] Patent: US2010/152452, 2010, A1, . Location in patent: Page/Page column 9
[13] Patent: US2008/26017, 2008, A1, . Location in patent: Page/Page column 11
[14] Tetrahedron Letters, 2011, vol. 52, # 19, p. 2488 - 2491
[15] Patent: EP2319850, 2011, A1, . Location in patent: Page/Page column 8
[16] Organic Letters, 2012, vol. 14, # 17, p. 4518 - 4521
[17] Journal of Medicinal Chemistry, 2016, vol. 59, # 11, p. 5505 - 5519
[18] Bioorganic and Medicinal Chemistry, 2017, vol. 25, # 3, p. 870 - 885
[19] Russian Journal of Organic Chemistry, 2017, vol. 53, # 5, p. 769 - 776[20] Zh. Org. Khim., 2017, vol. 53, # 5, p. 756 - 762,7
[21] Patent: WO2017/168442, 2017, A1, . Location in patent: Page/Page column 8; 13
[22] Journal of Medicinal Chemistry, 2018, vol. 61, # 8, p. 3503 - 3515
With hydrogenchloride; In water; at 59.84℃; for 4h;
4mmol of L-glutamic acid was dissolved in 20mL of water and the solution was acidified withconcentratedHCl (37%v/v).Then, 12mmol of potassiumthiocyanate (KOCN) was added tothis solution. The mixture was warmed up, with agitation, to 333 K, during 4 hr. The resultantsolution was cooled at roomtemperature into a glass vial sealed with parafilm, which was perforatedwitha needle to allowslowevaporation until the precipitation of awhite solid.Crystalsof (I) suitable for X-ray analysis were obtained by slow evaporation of an ethanol solution atroom temperature (Scheme 1). Yield 62%, mp 170C-171C. This experimental procedurecorrespond with a modified and improved methodology previously reported
With hydrogenchloride; sodium nitrite; In water; at -5 - 15℃; for 12h;
To a solution of 2-aminopentanedioic acid (210 g, 1.43 mol, CAS617-65-2) in H2O (800 mL) and HCl (12 M, 210 mL) was added a solution of NaNO2 (147 g, 2.13 mol) in H2O (400 mL) at -5 C. The mixture was stirred at 15 C. for 12 hrs. On completion, the mixture was concentrated and then dissolved in EA (500 mL) and filtered and washed with EA (3×100 mL). The filtrate and washed solution were dried over Na2SO4, filtered and concentrated in vacuo to give the title compound (200 g, crude) as yellow oil. 1H NMR (400 MHz, CDCl3) delta 6.43 (s, 1H), 5.02-4.95 (m, 1H), 2.67-2.38 (m, 4H)
L-glutamic acid (10.0 g, 67.97 mmol) was dissolved in 200 mL of methanol, and SOCl2(10.85 mL, 149.53 mmol)was slowly added dropwise under an ice bath. After the addition, reflux was performed at 65 C. After 2 hours, the reaction system was cooled to room temperature, and methanol was evaporated to dryness under reduced pressure to obtain a colorless viscous substance.This colorless viscous substance was dissolved in 200 mL of THF, (Boc)2O (22.25 g, 101.95 mmol) was addedunder an ice bath, and triethylamine (14.13 mL, 101.95 mmol) was slowly added dropwise.After the addition was complete, the reaction was allowed to proceed at room temperature overnight.After completion of the reaction, the solvent was distilled off under reduced pressure.The residue was dissolved in 200 mL of dichloromethane.The organic phase was washed with water (200 mL * 2), saturated citric acid solution (200 mL * 2), saturated sodium bicarbonate solution (200 mL * 2), and saturated brine (200 mL * 2) in that order.The organic phase was dried over anhydrous Na2SO4. Thefiltrate was collected by filtration, and the solvent was evaporated under reduced pressure. The residue was purified by column chromatography (petroleum ether: ethyl acetate = 4: 1 v / v) to give a colorless oily product 2 (18.34 g, yield 98%)
95%
7.4 g L-glutamic acid(6,50.0 mmol) and 100 ml of methanol were added to the reaction flask. To the flask was added dropwise 32 ml of trimethylchlorosilane (TMSC1, 250.0 mmol) at room temperature and the mixture was stirred at room temperature for 6 hours. The esterification reaction was terminated by TLC. To the reaction system was added 49 ml of triethylamine (Et3N, 350.0 mmol) and 13.2 g of Boc20 (60. 0 mmol) at 0 C, and the mixture was stirred at room temperature for 48 hours. Ethyl acetate (200 ml) and water (100 ml) were added to the residue, and the mixture was partitioned by shaking. The aqueous phase was extracted with ethyl acetate (100 mL chi 3). The combined organic phases were dried over anhydrous sodium sulfate, filtered, Column chromatography to obtain 13.1 g of glutamic acid ester (7). The overall yield in 2 steps was 95%.
95.2%
Acetyl chloride (5 mL) was slowly added dropwise to methanol (100 mL) at 0 C., stirred for 5 minutes, then glutamic acid (10 g, 67.9 mmol) was added, stirring was continued and the mixture was heated to reflux for 2 hours while maintaining the reflux temperature. .The reaction was stopped and the solvent was removed under reduced pressure and recrystallized from ether. The resulting oil was dissolved in THF (150 mL) and TEA (28.5 mL, 203.7 mmol) was added dropwise at 0C.After stirring at 0 C. for 5 minutes, di-tert-butyl dicarbonate (17.8 g, 81.5 mmol) dissolved in THF (30 mL) was continuously added dropwise, and the mixture was stirred at room temperature for 2.5 hours.After the reaction was completed, the solvent was evaporated under reduced pressure, and the residue was added with water (200 mL), extracted with DCM (2×200 mL) from the aqueous phase, and the combined organic phases were dried over anhydrous sodium sulfate.After concentration, the resulting crude product was purified by flash column (PE:EA=5:1) to give N-Boc-L-(+)-glutamic acid dimethyl ester (17.7 g, yield 95.2%) as none Oily liquid
95.2%
Acetyl chloride (5 mL) was slowly added dropwise to methanol (100 mL) at 0 C.Stir for 5 minutes, then add glutamic acid (10 g, 67.9 mmol).Stirring was continued and heated to reflux, and the reaction was maintained at reflux temperature for 2 hours.After the reaction was completed, the solvent was evaporated under reduced pressure and crystallised from diethyl ether.The obtained oil was dissolved in THF (150 mL). EtOAc (2.After stirring at 0 C for 5 minutes, di-tert-butyl dicarbonate (17.8 g, 81.5 mmol) dissolved in THF (30 mL) was added dropwise.Stir to room temperature and stir for 2.5 hours.After the reaction was completed, the solvent was evaporated under reduced pressure and water (200 mL)Extracted from the aqueous phase with DCM (2×200 mL).It is then concentrated and the crude product obtained is purified by flash column (PE: EA = 5:1).Compound 1-2 was obtained as a colorless oily liquid (17.7 g, yield 95.2%).
1.30 kg
H-CI(S)-(Tetrahydro-pyran-3-yl)amine hydrochloride (S)-Dimethyl 2-(tert-butoxycarbonylamino)pentanedioateTo MeOH (7L) was added TMSC1 slowly at 0 C, and the mixture was stirred for 30 min, then L-glutamic acid (700 g, 4.76 mol) was added to the mixture. The mixture was stirred at room temperature until complete reaction was observed (monitored by TLC). After cooling to 0 C, triethylamine (313 g, 31.0 mol) and Boc20 (1.14 Kg, 5.23 mol) were added slowly to the reaction solution successively while keeping the internal temperature below 25 C, and the resultant solution was stirred for 16 hours. After concentration, the residue was poured into water (5 L) and extracted with ethyl acetate (10 L). The organic phase was washed with 4L of 20% citric acid and brine, and dried over sodium sulfate. After filtration and concentration, the crude (S)-dimethyl 2-(tert-butoxycarbonylamino)pentanedioate (1.30 Kg) was obtained as pale yellow oil.
10135] 29.4 g (0.2 mol) L-(+)-glutamic acid, 40 g (0.23 mol) p-toluenesulfonic acid, and 80 mE benzyl alcohol were dissolved in 500 mE methylbenzene. 11 mE water was separated out by backflow under the protection of nitrogen gas. The backflow was continued for 3 h, and 150 mE liquid was evaporated and removed. The solution was cooled down to 50 C., and then the reaction solution was poured into a beaker containing 600 mE petroleum ether for stirring 1 h. The precipitation was collected by filtration. The filter cake was dissolved in 280 mE of 95% ethanol by heating, then the heating was stopped and the solution was cooled overnight. The precipitation was collected by filtration and dried in vacuum to produce 61 g E-(+)-glutamic acid dibenzyl ester p-toluenesulfonate (compound 1).
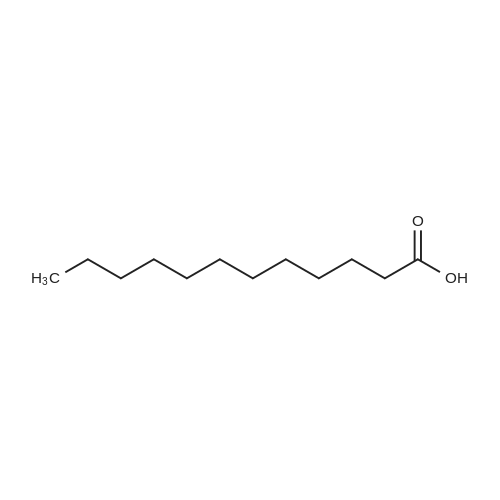
20.44 g of lauric acid (content of 98%) and 20.84 g of N,N'-dicyclohexylcarbodiimide (DCC) (content: 99%) were added to a ball mill and ground for 30 min. Add 14.86 g of glutamic acid (99%) and 8.42 g of sodium bicarbonate. After continuing to grind for 45 min, a white solid product was obtained which was the target collector product. After analysis and testing, The collector product 2-lauroamide glutaric acid content is 52.12%, The yield of 2-lauroyl glutaric acid based on lauric acid was 96.28%.
Freshly distilled thionyl chloride (0.60 mL, 8.16 mmol) was added dropwise with stirring to a cooled suspension of l-glutamic acid (1.0 g, 6.8 mmol) in anhydrous methanol (7 mL). The reaction mixture was stirred at rt for 2 h and was refluxed for 8 h. The solvent was removed under reduced pressure and co-evaporated with methanol (3×7 mL) to afford crude product as sticky liquid. To a solution of this crude product in THF (20 mL) were added triethylamine (2.0 mL, 14.6 mmol) and a solution of di-tert-butyl dicarbonate (1.78 g, 8.16 mmol) in THF (10 mL) at 0 C. The reaction mixture was stirred at rt for 8 h. After completion of the reaction, solvent was removed in vacuo and the residue partitioned between AcOEt (20 mL) and water (20 mL). The aqueous phase was extracted with AcOEt (2×15 mL) and the combined organic layers were washed with 3% HCl (15 mL), saturated NaHCO3 (15 mL), and brine (20 mL), dried (Na2SO4), and evaporation of the solvent gave 16 (1.5 g, 85%) as a colorless liquid.Comment[alpha]D26 +13.7 (c 0.89, CHCl3), lit.refPreviewPlaceHolder20 [alpha]D25 +12.5 (c 2, CHCl3); 1H NMR (300 MHz, CDCl3) delta 5.16 (br d, 1H, J=6.4 Hz), 4.28 (br d, 1H, J=4.6 Hz), 3.69 (s, 3H), 3.63 (s, 3H), 2.33-2.42 (m, 2H), 2.10-2.16 (m, 1H), 1.86-2.00 (m, 1H), 1.38 (s, 9H); 13C NMR (75 MHz, CDCl3) delta 173.2, 172.7, 155.4, 80.0, 52.9, 52.4, 51.8, 30.1, 28.3, 27.7; HRMS (ESI) (M+Na)+ calculated for C12H21NO6Na+=298.1267, found 298.1266.
With triethylamine; In methanol;
EXAMPLE 1 Preparation of Compound 1 TMSCl (190 mL, 4.4 eq) was slowly added to a stirred suspension of L-glutamic acid (50.0 g, 1 eq.) in dry MeOH (1100 mL, 0.3 M) surrounded by an ice-cold bath. After the addition was complete, the ice-clod bath was removed and the reaction was stirred overnight until TLC showed completed conversion. Then, Et3N (306 mL, 6.5 eq) and (Boc)2O (82 g, 1.1 eq) were added sequentially to the above reaction mixture. The reaction mixture was then stirred until TLC showed complete protection. The solvent was removed under reduced pressure. The residue was then filtered and washed with Et2O using a pad of celite. The organic layers were combined and concentrated. The resulting crude product was purified by silica gel column chromatography to afford intermediate 1, N-Boc-L-(+)-glutamic acid dimethyl ester, (88 g, 95%) as an oil: 1H NMR (CDCl3) delta 1.40 (s, 9H), 1.91 (m, 1H), 2.14 (m, 2H), 2.37 (m, 2H), 3.64 (s, 3H), 3.70 (s, 3H), 4.29 (br, s, 1H). ESI-MS (M+H+)=276.
With triethylamine; at 20℃; for 3h;Inert atmosphere;
General procedure: L-Glutamic acid (20.4 mmol, 1equiv) is dissolved in dry methanol (50 ml) under N2. At 0 C, TMSCl (89.8 mmol, 4.4 equiv) is addeddropwise via a syringe. The ice bath is removed and the mixture stirred at rt until LC/MS or TLCshows the reaction is complete. The crude is cooled at 0 C and TEA (132.6 mmol, 6.5 equiv) is addeddropwise. Boc2O (22.4 mmol, 1.1 equiv) is then added in one portion. After 3 h at rt, 0.5 equiv ofBoc2O is added to allow the full conversion of the primary amine to the monoprotected amino acid.The mixture is stirred at rt overnight. Filtration of the salts over a pad of celite, concentration in vacuoand classical aqueous work up gave compound 1 as a colorless oil. 1 is then diluted in dry acetonitrile(60 ml) under N2. DMAP (4.08 mmol, 0.2 equiv) and Boc2O (22.4 mmol, 1.1 equiv) are added in oneportion. After 2 h at rt, 0.5 equiv of Boc2O is added. When TLC shows the complete formation ofdiBoc compound, the crude is concentrated in vacuo and purified by flash chromatography over silicagel (Petroleum Ether/Ethyl Acetate) to afford 2 (7.005 g, 18.6 mmol, 92 %) as white crystals. 1H NMR(CDCl3, 300 MHz) 4.93 (dd, 1H, J = 9 Hz & 4.2 Hz), 3.71 (s, 3H), 3.67 (s, 3H), 2.492.37 (m, 3H),2.19 (m, 1H), 1.49 (s, 18H); 13C NMR (CDCl3, 75.45 MHz) 173.0, 170.8, 151.8, 83.2 (C), 57.3(CH), 52.2, 51.6 (CH3), 30.5 (CH2), 27.9 (6 CH3), 25.1 (CH2). LC/MS (ESI+): tR = 17.20 min, m/z376.17 ([M + H]+). []20D 36.1 (c 2.38, CHCl3). HRMS (ESI+) m/z calcd for C17H29NO8Na ([M +Na]+) 398.1791, found 398.1791.
With water; sodium acetate; at 50℃; for 25h;pH 4.9;Conversion of starting material;
[0036] A reaction mixture containing 670 mM of L-glu-di-tert-butyl ester HCl and 2 mole equivalents of sodium acetate, pH 4.9, was stirred in a round bottomed flask fitted with a condenser and Therm-o-watch [12R, Cheltenhana, Pa.], at 50 C. Samples were taken over time intervals and analyzed by HPLC for L-glu-alpha-tert-butyl ester alpha-monoester), L-glu-gamma-tert-butyl ester (gamma-monoester), L-glu-di-tert-butyl esters (diester) and L-glutamic acid (diacid). [0037] The results which are present in TABLE I show that under the reaction condition set forth above, approximately half of the substrate (diester) was hydrolyzed within 25 hours. The product of the hydrolytic reaction included alpha-monoester, gamma-monoester, and diacid. However, gamma-monoester was predominant, indicating that the hydrolysis was selective for the alpha-ester group.
With hydrogenchloride; water; sodium acetate; for 20 - 24h;pH 4.7 - 5.0;Conversion of starting material;
[0038] Two reaction mixtures were prepared by adding 2.0 mL of 200 g/L of L-glu-di-tert-butyl ester free base as substrate, to a reaction vessel. Then 2.10 g of solid sodium acetate (NaOAc.3H2O) was added with about 5.0 mL of water. The pH of the first reaction mixture was adjusted to a pH of about 4.7 to about 5.0 using concentrated hydrochloric acid (HCl). The pH of the second reaction mixture was adjusted to a pH of about 4.8 with concentrated sulfuric acid (H2SO4). The volume of each mixture was adjusted to 10 mL. At about 20 to 24 hours, the samples of the reaction mixture were analyzed. The results shown in TABLE II indicate that L-glu-di-tert-butyl ester free base could be used as the substrate for the non-enzyme hydrolysis method. Although at 20 to 24 hours, the conversion of the diester was not complete, the conversion resulted mainly in gamma-monoester. The percent conversion to gamma-monoester reached 42.32% for the first reaction mixture and 42.55% for the second mixture.
With potassium phosphate; water; at 40℃; for 46h;pH 5.5 - 6.5;Conversion of starting material;
[0042] Four sets of reactions were initiated by mixing 30 mM of L-glu-di-tert-butyl ester with 100 mM of potassium phosphate (3 sets) or sodium acetate (1 set). The pH of the first 3 sets of the reaction was adjusted to 6.5, 6.0, and 5.5, while the pH of the last set was adjusted to 5.5. The reactions were run at a temperature of 40 C. for about 46 hours. Samples were taken at various intervals and analyzed. The results which are presented in TABLE VI, generally indicate that under the specified reaction condition, L-glu-di-tert-butyl ester was converted to mainly gamma-monoester and a small amount of alpha-monoester, and L-glu (diacid). The data further indicates that in the presence of potassium phosphate, the percent conversion of the diester to gamma-monoester increased from 48.56% to 92.45% with the increased pH from pH 5.5 to 6.5. However, when sodium acetate was used in place of potassium phosphate, the percent conversion was high (93.53%) at a lower pH (5.5).
With sulfuric acid; water; sodium acetate; for 20 - 24h;pH 4.8;Conversion of starting material;
[0038] Two reaction mixtures were prepared by adding 2.0 mL of 200 g/L of L-glu-di-tert-butyl ester free base as substrate, to a reaction vessel. Then 2.10 g of solid sodium acetate (NaOAc.3H2O) was added with about 5.0 mL of water. The pH of the first reaction mixture was adjusted to a pH of about 4.7 to about 5.0 using concentrated hydrochloric acid (HCl). The pH of the second reaction mixture was adjusted to a pH of about 4.8 with concentrated sulfuric acid (H2SO4). The volume of each mixture was adjusted to 10 mL. At about 20 to 24 hours, the samples of the reaction mixture were analyzed. The results shown in TABLE II indicate that L-glu-di-tert-butyl ester free base could be used as the substrate for the non-enzyme hydrolysis method. Although at 20 to 24 hours, the conversion of the diester was not complete, the conversion resulted mainly in gamma-monoester. The percent conversion to gamma-monoester reached 42.32% for the first reaction mixture and 42.55% for the second mixture.
With water; sodium acetate; at 50℃; for 43 - 65h;pH 5.0;Conversion of starting material;
[0041] Three reaction mixtures were prepared with 340 mM of L-glu-di-tert-butyl ester free base as substrate. The first mixture further contained 1×(=340 mM) sodium acetate, the second mixture contained 1.5×(=510 mM) sodium acetate, and the third mixture contained 2×(=680 mM) sodium acetate. The reactions were run with the pH adjusted to 5.0 and the reaction temperature was maintained at 50 C. The reactions were run for about 43 to about 65 hours. The results shown in TABLE V indicate that the conversion of the diester to gamma-monoester depended on the concentration of sodium acetate. The addition of 1× concentration of sodium acetate resulted in the conversion of diester to mainly diacid and gamma-monoester. The amount of diacid recovered was about twice as much as the amount of gamma-monoester. The addition of 1.5× concentration of sodium acetate resulted in a higher percent conversion to gamma-monoester, and the addition of 2× concentration of sodium acetate resulted in the a highest percent conversion to gamma-monoester, (83.47%).
With water; sodium acetate; at 40℃; for 46h;pH 5.5;Conversion of starting material;
[0042] Four sets of reactions were initiated by mixing 30 mM of L-glu-di-tert-butyl ester with 100 mM of potassium phosphate (3 sets) or sodium acetate (1 set). The pH of the first 3 sets of the reaction was adjusted to 6.5, 6.0, and 5.5, while the pH of the last set was adjusted to 5.5. The reactions were run at a temperature of 40 C. for about 46 hours. Samples were taken at various intervals and analyzed. The results which are presented in TABLE VI, generally indicate that under the specified reaction condition, L-glu-di-tert-butyl ester was converted to mainly gamma-monoester and a small amount of alpha-monoester, and L-glu (diacid). The data further indicates that in the presence of potassium phosphate, the percent conversion of the diester to gamma-monoester increased from 48.56% to 92.45% with the increased pH from pH 5.5 to 6.5. However, when sodium acetate was used in place of potassium phosphate, the percent conversion was high (93.53%) at a lower pH (5.5).
With water; sodium acetate; at 40℃; for 46h;Conversion of starting material;
[0039] Eight reaction mixtures were prepared with different amounts of L-glu-di-tert-butyl ester free base (diester) as substrate. The mixtures were divided into two groups. Different amounts of sodium acetate were added into the mixtures in the first group. No sodium acetate was added to the mixtures in the second group. The reactions were run at 40 C. for about 46 hours. The results in TABLE III show generally that in the absence of sodium acetate, the conversion of the diester was complete in 46 hours. However, most of the conversion resulted in the diacid. The percent conversion of the diester into gamma-monoester was relatively low, but slightly increased with the decreasing concentrations of the substrate. The data in TABLE III also show that, in the presence of sodium acetate, the conversion of the diester was almost complete at 46 hours. Only relatively small amounts of the diester remained. The conversion resulted mainly in the production of gamma-monoester indicating selective hydrolysis. The increased amounts of the substrate and sodium acetate had little effect on the percent conversion of the diester to gamma-monoester.
With water; for 24h;pH 5.5;Aqueous sodium acetate buffer;Conversion of starting material;
[0040] Four reaction mixtures were each prepared by adding 100 mg/L of L-glu-di-tert-butyl ester (diester) as substrate, to 340 mM of one of four different buffers. The substrate was first prepared by mixing 2.0 g of the diester with 10 ml of water. To each vial, 2 mL of the diester solution was added, followed by 1.36 mL of 1M buffer and 640 muL of water. The four different buffers were sodium citrate, sodium succinate, sodium formate and sodium acetate. Sodium citrate was prepared by titration of citric acid and sodium hydroxide to a pH 5.5. Sodium succinate was prepared by titration of succinic acid and sodium hydroxide to a pH 5.5. Sodium formate was prepared by titration of formic acid with sodium hydroxide to a pH of 5.5. The reaction was run for 24 hours. The results in TABLE IV indicate that all the non-enzyme additives tested had similar effects on the percent conversion of the diester gamma-monoester. The conversion to alpha-monoester was minimal when the reaction was run in the presence of sodium citrate and sodium formate. On the other hand, in the presence of sodium succinate or sodium acetate, the conversion to alpha-monoester was relatively high in this particular experiment. Nevertheless, selective hydrolysis of the alpha-ester group was evidenced by the % conversion to gamma-monoester. After 24 hours, the percent conversion ranged from 59.91% to 65.70%.
With water; for 24h;pH 5.5;Aqueous sodium citrate buffer;Conversion of starting material;
[0040] Four reaction mixtures were each prepared by adding 100 mg/L of L-glu-di-tert-butyl ester (diester) as substrate, to 340 mM of one of four different buffers. The substrate was first prepared by mixing 2.0 g of the diester with 10 ml of water. To each vial, 2 mL of the diester solution was added, followed by 1.36 mL of 1M buffer and 640 muL of water. The four different buffers were sodium citrate, sodium succinate, sodium formate and sodium acetate. Sodium citrate was prepared by titration of citric acid and sodium hydroxide to a pH 5.5. Sodium succinate was prepared by titration of succinic acid and sodium hydroxide to a pH 5.5. Sodium formate was prepared by titration of formic acid with sodium hydroxide to a pH of 5.5. The reaction was run for 24 hours. The results in TABLE IV indicate that all the non-enzyme additives tested had similar effects on the percent conversion of the diester gamma-monoester. The conversion to alpha-monoester was minimal when the reaction was run in the presence of sodium citrate and sodium formate. On the other hand, in the presence of sodium succinate or sodium acetate, the conversion to alpha-monoester was relatively high in this particular experiment. Nevertheless, selective hydrolysis of the alpha-ester group was evidenced by the % conversion to gamma-monoester. After 24 hours, the percent conversion ranged from 59.91% to 65.70%.
With water; for 24h;pH 5.5;Aqueous sodium formate buffer;Conversion of starting material;
[0040] Four reaction mixtures were each prepared by adding 100 mg/L of L-glu-di-tert-butyl ester (diester) as substrate, to 340 mM of one of four different buffers. The substrate was first prepared by mixing 2.0 g of the diester with 10 ml of water. To each vial, 2 mL of the diester solution was added, followed by 1.36 mL of 1M buffer and 640 muL of water. The four different buffers were sodium citrate, sodium succinate, sodium formate and sodium acetate. Sodium citrate was prepared by titration of citric acid and sodium hydroxide to a pH 5.5. Sodium succinate was prepared by titration of succinic acid and sodium hydroxide to a pH 5.5. Sodium formate was prepared by titration of formic acid with sodium hydroxide to a pH of 5.5. The reaction was run for 24 hours. The results in TABLE IV indicate that all the non-enzyme additives tested had similar effects on the percent conversion of the diester gamma-monoester. The conversion to alpha-monoester was minimal when the reaction was run in the presence of sodium citrate and sodium formate. On the other hand, in the presence of sodium succinate or sodium acetate, the conversion to alpha-monoester was relatively high in this particular experiment. Nevertheless, selective hydrolysis of the alpha-ester group was evidenced by the % conversion to gamma-monoester. After 24 hours, the percent conversion ranged from 59.91% to 65.70%.
With water; for 24h;pH 5.5;Aqueous sodium succinate buffer;Conversion of starting material;
[0040] Four reaction mixtures were each prepared by adding 100 mg/L of L-glu-di-tert-butyl ester (diester) as substrate, to 340 mM of one of four different buffers. The substrate was first prepared by mixing 2.0 g of the diester with 10 ml of water. To each vial, 2 mL of the diester solution was added, followed by 1.36 mL of 1M buffer and 640 muL of water. The four different buffers were sodium citrate, sodium succinate, sodium formate and sodium acetate. Sodium citrate was prepared by titration of citric acid and sodium hydroxide to a pH 5.5. Sodium succinate was prepared by titration of succinic acid and sodium hydroxide to a pH 5.5. Sodium formate was prepared by titration of formic acid with sodium hydroxide to a pH of 5.5. The reaction was run for 24 hours. The results in TABLE IV indicate that all the non-enzyme additives tested had similar effects on the percent conversion of the diester gamma-monoester. The conversion to alpha-monoester was minimal when the reaction was run in the presence of sodium citrate and sodium formate. On the other hand, in the presence of sodium succinate or sodium acetate, the conversion to alpha-monoester was relatively high in this particular experiment. Nevertheless, selective hydrolysis of the alpha-ester group was evidenced by the % conversion to gamma-monoester. After 24 hours, the percent conversion ranged from 59.91% to 65.70%.
With water; at 40℃; for 46h;Conversion of starting material;
[0039] Eight reaction mixtures were prepared with different amounts of L-glu-di-tert-butyl ester free base (diester) as substrate. The mixtures were divided into two groups. Different amounts of sodium acetate were added into the mixtures in the first group. No sodium acetate was added to the mixtures in the second group. The reactions were run at 40 C. for about 46 hours. The results in TABLE III show generally that in the absence of sodium acetate, the conversion of the diester was complete in 46 hours. However, most of the conversion resulted in the diacid. The percent conversion of the diester into gamma-monoester was relatively low, but slightly increased with the decreasing concentrations of the substrate. The data in TABLE III also show that, in the presence of sodium acetate, the conversion of the diester was almost complete at 46 hours. Only relatively small amounts of the diester remained. The conversion resulted mainly in the production of gamma-monoester indicating selective hydrolysis. The increased amounts of the substrate and sodium acetate had little effect on the percent conversion of the diester to gamma-monoester.
With boric acid;glutaminase; In water; at 30℃; for 22h;pH 11;
0.3 M glutamine and 1.5 M methylamine hydrochloride were reacted in the presence of 0.3 U glutaminase (commercially available) at 30 C. for 22 hours in a buffer solution of 0.05 M boric acid (pH 11), whereby 225 nm theanine was obtained. Reaction liquid was applied to Dowex 50×8 columnar chromatgraphy and Dowex 1×2 columnar chromatography (both made by Muromachi Chemical Co., Ltd.) thereby to be processed by ethanol, whereby an object substance is isolated from the reaction liquid. The isolated substance was applied to an amino acid analyzer (made by Hitachi Co.) and paper chromatography. Since the isolated substance behaved in the same way as a standard substance, it was recognized as L-theanine. When the isolated substance was processed by hydrolysis using hydrochloric acid or glutaminase, glutamine acid and ethylamine were produced in a ratio of 1:1. Thus, since the isolated substance was hydrolyzed by glutaminase, it was shown that ethylamine was gamma-ethylamine of glutamine acid. Furthermore, it was confirmed on the basis of glutamate dehydrogenase that glutamine acid produced by hydrolysis was L-glutamine acid. As a result, 8.5 g theanine was obtained.
With sodium hydroxide; triethylamine; In water; 1,2-dichloro-ethane; acetone;
EXAMPLE 1 Lauric acid 20 g was dissolved in 80 ml 1.2-dichloroethane and then 10 g triethylamine was added. The resulting solution was held at -2 - 5C and 8 g sulfuric anhydride was added dropwise with stirring. The reaction mixture was stirred at that temperature for 50 minutes and a solution of lauric acid-sulfuric acid mixed acid anhydride was prepared and employed in the following acylation. Sodium hydroxide 8 g was added to a suspension of 14.7 g L-glutamic acid in 72 ml water and 48 ml acetone. The resulting solution of disodium L-glutamate was cooled to 0C and 10 ml of 10 N aqueous sodium hydroxide solution and the mixed acid anhydride prepared in the above was added drop by drop under stirring while the pH was controlled to 12 - 13. This reaction solution was stirred for 1 hour at 0C. Thereafter, the reaction mixture was concentrated under reduced pressure to distill off the solvent. The residue was adjusted to a pH 1 with 6 N sulfuric acid. The crude crystals of N-lauroyl-L-glutamic acid precipitated, and were filtered and dried (25.7 g). They were washed with petroleum benzine and 23.2 g of the purified acid were recovered by filtration (70.3% yield) M.P. 102 - 105C. After recrystallization from ethanol-petroleum benzine, the elemental analysis was as follows:
With glutaminase; water; boric acid; at 30℃; for 22h;pH 11;Enzymatic reaction;
0.3 M glutamine and 1.5 M methylamine hydrochloride were reacted in the presence of 0.3 U glutaminase (commercially available) at 30°C for 22 hours in a buffer solution of 0.05 M boric acid (pH 11), whereby 225 nm theanine was obtained. Reaction liquid was applied to Dowex 50.x.8 columnar chromatography and Dowex 1.x.2 columnar chromatography (both made by Muromachi Chemical Co., Ltd.) thereby to be processed by ethanol, whereby an object substance is isolated from the reaction liquid. As a result, 8.5 g theanine was obtained. The isolated substance was applied to an amino acid analyzer (made by Hitachi Co.) and paper chromatography. Since the isolated substance behaved in the same way as a standard substance, it was recognized as L-theanine. When the isolated substance was processed by hydrolysis using hydrochloric acid or glutaminase, glutamine acid and ethylamine were produced in a ratio of 1:1. Thus, since the isolated substance was hydrolyzed by glutaminase, it was shown that ethylamine was gamma-ethylamine of glutamine acid. Furthermore, it was confirmed on the basis of glutamate dehydrogenase that glutamine acid produced by hydrolysis was L-glutamine acid.
2.00 g of desvenlafaxine base is dissolved in 50 ml of absolute ethanol at reflux conditions and 1.22 g of L-glutamic acid is added. The solution is further stirred at reflux conditions for 10 minutes and cooled down to -10 C. The mixture is further stirred for one hour at -10 C, the precipitated white crystals are filtered, washed with mother liquor and dried under vacuum at ambient temperature to yield 2.78 g (89.2%) of L-glutamic salt
To a 500 ml four-necked flask equipped with a stirrer, a thermometer and a reflux condenser (connected with a 30percent aqueous sodium hydroxide absorber) was added 280 g of benzyl alcohol.14.7 g (0.10 mol)L-glutamic acid, 30.0 g (0.25 mole) of thionyl chloride,Heat, react at 80-85°C for 5 hours, cool to 20-25°C,Hydrogen chloride gas in the nitrogen displacement system, after 30 minutes of replacement,Distillation recovers excess thionyl chloride and benzyl alcohol,Then add 120 grams methyl tert-butyl ether to the residue and beat it.Filter and dry to give 35.9 g of a white solidDibenzyl L-glutamate hydrochloride,The liquid phase purity was 99.8percent and the yield was 98.7percent.
With ammonium hydroxide; In water; at 20℃; for 6h;pH 9.7 - 9.9;Green chemistry; Enzymatic reaction;
At first, 14.7 g (100 mmol) of l-glutamic acid and 5.52 g (36.2 mmol) of d-PGA were suspended in water and the pH was adjusted to 9.7, making an overall volume of 500 mL. To this, 12.2 g of immobilized penicillin acylase was added at room temperature. Two further portions of d-PGA (4.74 g, 31.6 mmol) were added, with a 2-hour interval each, at which the pH was adjusted to 9.9 with the NH4OH. After 6 hours, the enzyme was filtered and a turbid mixture was obtained. The pH was then lowered to 3.3 with approximately 30 mL of H2SO4 (6 M). The mixture was left stirring for a period of two days, after which the formed precipitate was filtered, washed with petroleum ether 40-60, dried and 13.2 g was obtained. The mixture was kept stirring for an extra period of 10 days. More precipitate was formed and collected, following the same procedure, affording 7.28 g, which corresponds to an overall yield of 73percent. The product was isolated NMR pure. Mp: 217-219 °C. [alpha]D24 = -87.5 (c 1, 2.5 M HCl). 1H NMR (300 MHz, D2O+DCl): delta = 7.487 (m, aromatic protons), delta = 5.173 (s, CHNH2), delta = 4.485 (dd, J1 = 3.9 Hz, J2 = 9.6 Hz, CHCH2CH2COOH), delta = 2.15, 1.81 (mm, CHCH2CH2COOH); 13C NMR (75 MHz, D2O+DCl): delta = 178.1, 176.0, 170.3 (C=O), delta = 133.4, 132.0, 131.3, 129.5 (Ar); delta = 58.14, 53.44 (Calpha), delta = 30.99, 26.91 (CHCH2CH2COOH).
[0027] To a stirred solution of (S)-Glutamic acid (100 g, 0.68 mol) in dry methanol (1740 ml) was added TMSC1(375 ml, 2.9 mol) at 0 C. After addition, the mixture was allowed to reach room temperature and stirred overnight. NEt3 (446 g, 4.42 mol) was added to the solution under ice-bath and followed by addition of Boc2O (163 g, 0.75 mol). After the evolution of gas had ceased, the mixture became clear and was allowed to warm to room temperature. The solvent was evaporated under reduced pressure, and the residue was triturated and washed with ether (3×500 ml). The combined filtrates were concentrated to provide crude product which was purified by column chromatography on silica gel to yield TA-101 (173 g, 92.5% yield) as an oil. 1H NMR (CDCl3): 5.16-5.14 (d, 1H), 4.34-4.33 (d, 1H), 3.74 (s, 3H), 3.68 (s, 3H), 2.48-2.34 (m, 2H), 2.23-2.16 (m, 1H), 2.16-1.92 (m, 1H), 1.43 (s, 9H);
With C12H20CuN2O8; N,N,N',N'-tetramethylguanidine; In water; N,N-dimethyl-formamide; at 40℃; for 38h;
Synthesis of gamma-4,5-dimethoxy-2-nitrobenzyl-L-glutamate (3). In a 300-mL round bottom flask, N,N,N?,N?-tetramethylguanidine (1.1 mL, 0.86 mmol) was added slowly to a stirred mixture of L-Glutamic acid (0.64 g, 4.3 mmol) and L-glutamic acid copper(II) complex (1.05 g, 2.14 mmol) in DMF (4 mL) and distilled water (0.6 mL). The mixture turned to dark blue. After the dissolution of all solids (2 h), an additional DMF (3.1 mL) was added. Then, <strong>[53413-67-5]4,5-dimethoxy-2-nitro-1-bromomethylbenzene</strong> (DMNB, 2.5 g, 9 mmol) was added to the above solution in one portion. The reaction solution became darker and kept at 40 C. for 38 h. After that, acetone (100 mL) was added into the mixture and kept stirring until a fine precipitate was obtained (1 h). The violet solid was collected by filtration, followed by mixed with freshly prepared EDTA (2 g)/sodium bicarbonate (1 g) aqueous solution (15 mL) and kept stiffing for 24 h. The product was collected by filtration and washed with DI water. Further purification can be performed by recrystallizing from H2O/isopropanol. Freeze dry to yield the purified product as a yellow crystalline solid. Obtained 1.51 g (yield: 49%). 1H NMR [D2O/DCI (1 wt %), delta, ppm]: 7.52 (s, 1H, ArH), 6.90 (s, 1H, ArH), 5.21 (s, 2H, ArCH2-), 3.95 (t, 1H, -CHNH2), 3.74 (s, 3H, CH3O-), 3.70 (s, 3H, CH3O-), 2.55 (t, 2H, -COCH2CH2-), 2.08 (m, 2H, -COCH2CH2-).
N-[3-fluoro-4-[[6-methoxy-7-[[3-(morpholin-4-yl)propyl]oxy]quinolin-4-yl]oxy]phenyl]-N'-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide glutamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
58%
In tetrahydrofuran; water; at 0 - 20℃; for 8h;
Compound 1(N- [3-fluoro-4 - [[6-methoxy-7 - [[3- (morpholin-4-yl) propyl] oxy] quinolin-4-yl] oxy] N '- (4-fluorophenyl) cyclopropane-1,1-dicarboxamide) (632 mg, 1 mmol)Was dissolved in tetrahydrofuran / water (10 mL, 1: 1)Ice bath to the internal temperature of 0 ,L-glutamic acid (147 mg, 1 mmol) was added,Reaction at room temperature for 8 h,After completion of the reaction, the white solid was filteredN- [3-fluoro-4 - [[6-methoxy-7 - [[3- (morpholin-4-yl) propyl] oxy] quinolin-4-yl] oxy] (4-fluorophenyl) cyclopropane-1,1-dicarboxamide glutamate (Compound 3) 450 mg Yield 58%.
In a 2 L reaction vessel, glutamic acid (100 g, 0.68 mol), potassium cyanate (65.3 g, 0.816 mol),Potassium hydroxide (40 g, 0.71 mol) and 1 L of water,After slowly heating to 30-50 C to completely dissolve the solid,Continue to heat up to 60-80 C reaction 2-2.5h,Slowly cool to room temperature and let stand at room temperature for 10-15h.Acidified with concentrated hydrochloric acid to adjust pH=7, then add the catalyst urea-propionic acid (100mL), let stand for 2-3h, precipitate a large amount of solid,After filtration, 86 g of crude product was obtained, and N-carbamoylglutamic acid was crystallized from water, filtered, and dried at 80 C to obtain 76 g of fine product.The molar yield is 58%, the content is 92%,
With phosphoric acid; boron trifluoride diethyl etherate; In tetrahydrofuran;
The reaction of L-glutamic acid 2 with isobutylene in TI-IF in the presence of catalytic BF3 Et20 and H3P04 provides 5-tert-butyl ester compound 11. The formation of Ncarboxylate anhydride 12 is conducted using phosgene or N,N?-carbonyldiimidazole (CDI). The subsequent addition of ammonia (as a solution in water or a solvent such as methanol) provides free base amide 1. Lastly, formation of HC1 salt of the free base amino-amide provides the target molecule 1.
Pemetrexed (42.7 g, 0.10 mol) was added to the reaction kettle.Add 1708 ml of methanol,Heated to 55 C,Stir well,Insulation at 55 C,An aqueous solution of glutamic acid (44.2 g, 0.3 mol) was added dropwise to the reaction vessel.After the completion of the dropwise addition, stirring was continued for 1 h, the temperature was lowered to room temperature, and the mixture was decanted for 2 h, centrifuged and filtered, 45-55 C, and the vacuum was -0.080 to -0.100 Mpa, and dried under vacuum for 5-6 h.Demimethrin glutamate,Molar yield 98.0%,The HPLC purity was 99.76%






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping