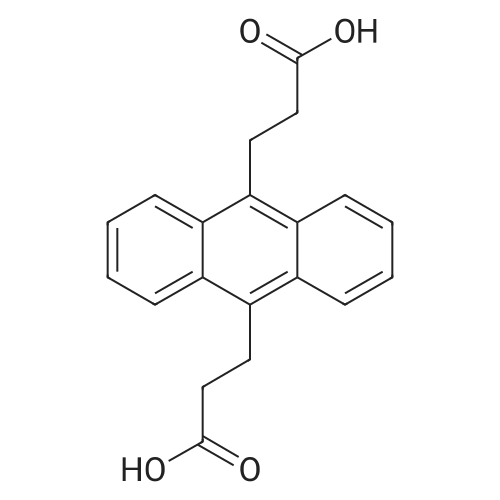
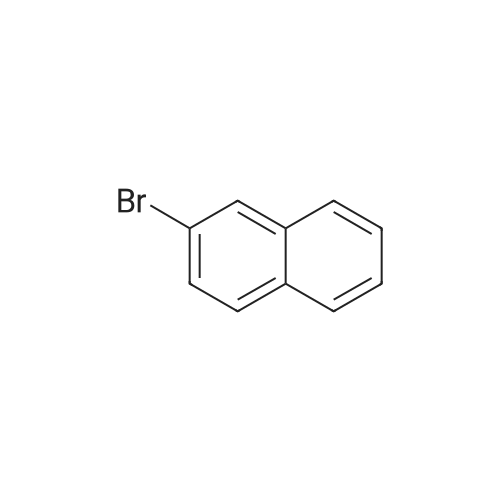
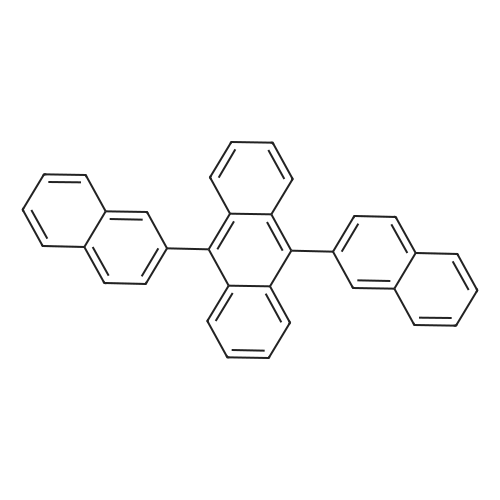
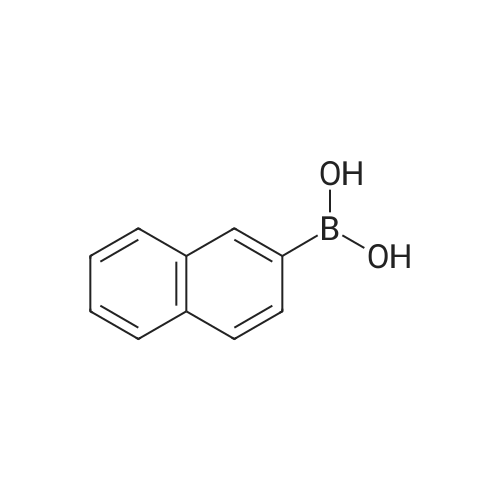
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Chem. News J. Ind. Sci., 1876, vol. 34, p. 145[2] Bulletin de la Societe Chimique de France, 1877, vol. <2> 27, p. 465
[3] Journal of the Chemical Society, 1924, vol. 125, p. 1085
[4] Recueil des Travaux Chimiques des Pays-Bas, 1925, vol. 44, p. 217,219
[5] Recueil des Travaux Chimiques des Pays-Bas, 1925, vol. 44, p. 217,219
[6] Journal of Physical Chemistry, 1984, vol. 88, # 19, p. 4380 - 4384
[7] Journal of Physical Chemistry, 1996, vol. 100, # 47, p. 18431 - 18435
[8] Molecules, 2013, vol. 18, # 1, p. 398 - 407
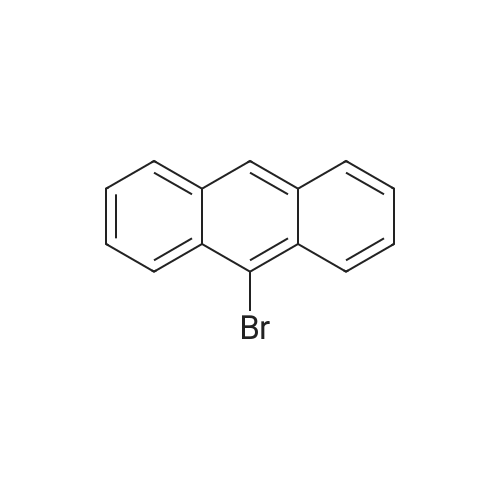
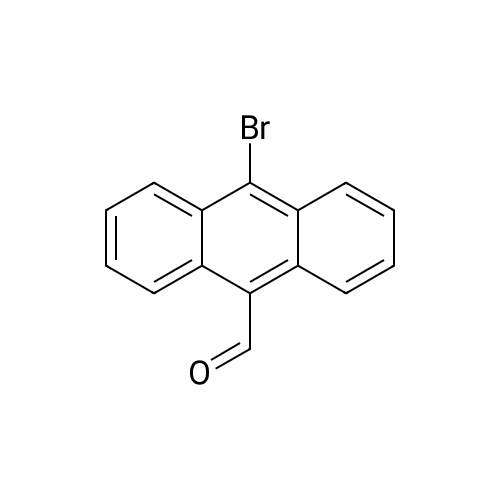
A solution of Br2 (2.90 mL, 56.60 mmol) in CHCl3 (50 mL) was added dropwise into the solution of anthracene (5.0 g, 28.05 mmol) in CHCl3 (100 mL) at room temperature. After stirring for 4 h, the solvent was removed and the crude product was purified by recrystallized in a dichloromethane to give a yellow needle in a yield 98 percent. 1H NMR (500 MHz, TMS, CDCl3): δ (ppm) = 8.59-8.56 (m, 4H), 7.64-7.61 (m, 4H) (Fig. S11). IR (KBr, cm-1): 2920, 2850, 1650, 1560, 1460, 1380, 1260, 926, 746, 579. Elemental analysis calculated for C14H8Br2: C, 54.04; H, 2.40. Found: C,53.84; H, 2.36. MS, m/z: cal: 336.0, found: 336.1.
96%
With dimethylbromosulphonium bromide In dichloromethane at 20℃; for 0.5 h;
General procedure: General procedure for the synthesis of 9,10-dibromoanthracene: A mixture of anthracene (10.0 mmol) and BDMS (24.0 mmol) in CH2Cl2 (10.0 mL) was stirred for appropriate amount of time at room temperature [CAUTION.. The reaction produces HBr and a gas trap (bubbler containing 1 mol/L NaOH solution) should be used, preferably in a fumehood]. When the reaction completed as indicated by TLC analysis, the reaction product was collected by filtration. The yellow solid was dried in vacuo for 2 h to give the compound. Mp: 222-224 °C, 1H NMR (400 MHz, CDCl3): δ 8.60 (4H), 7.66 (4H).
93%
With potassium bromide In dichloromethane; water; acetic acid at 45℃; for 5.25 h;
The anthracene (356 mg, 2 mmol), potassium bromide (309 mg, 2.6 mmol), acetic acid 9 ml, water 1 ml, dichloromethane 5 ml of the mixed solution is sequentially added with condensed tube, of the thermometer in the three-necked flask, transfer to the constant temperature heating magnetic stirring in the water bath, in 15 min by stages of ZnAl - BrO slowly added3- - LDHs (3.6 g, 3.6 mmol), control the reaction temperature 45 °C, TCL tracking reaction process, the reaction 5 h, using sodium sulfite solution to wash, then using dichloromethane (3 × 10 ml) extraction, the combined organic phase, in the dichloromethane phase bucket column chromatography silica gel by adding the two drugs (200 - 300 mesh), distilling out the organic solvent, in through the column chromatography (petroleum ether: ethyl acetate=15:1 as eluant) separation to obtain the target product 625 mg. Yellow solid, yield 93percent.
75%
With N-Bromosuccinimide In chloroform; acetic acid at 40℃; for 2 h;
The compound anthracene (1 g, 5.62 mmol) was placed in 30 ml chloroform, NBS (3 g,16.86 mmol) was dissolved in the mixture of acetic acid (30 ml) and chloroform (30 ml) and stirred for 20 min at room temperature, then added dropwise to the anthracene solution and the mixture was stirred at 40°C for 2 h. Afterwards, the mixture was neutralized with KOH solution, and the product was extracted with chloroform (4×100 mL), then the combined organic layer was dried by anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel using petroleum ether as eluent to afford compound 1 in 75percent yield as yellow solid. m.p.: 222–224°C. 1HNMR (400 MHz, CDCl3) δ 8.57 (dd, J = 6.7, 3.1 Hz, 4H), 7.62 (dd, J = 6.8,3.1 Hz, 4H).
Reference:
[1] Canadian Journal of Chemistry, 2005, vol. 83, # 2, p. 146 - 149
[2] Chemistry - An Asian Journal, 2012, vol. 7, # 10, p. 2240 - 2252
[3] Inorganic Chemistry, 2016, vol. 55, # 21, p. 10851 - 10854
[4] Chemical Communications, 2011, vol. 47, # 7, p. 2113 - 2115
[5] Tetrahedron Letters, 2013, vol. 54, # 6, p. 594 - 599
[6] Journal of Organic Chemistry, 2006, vol. 71, # 5, p. 1795 - 1801
[7] Tetrahedron Letters, 2013, vol. 54, # 4, p. 312 - 314
[8] RSC Advances, 2013, vol. 3, # 19, p. 6930 - 6938
[9] Chinese Chemical Letters, 2014, vol. 25, # 3, p. 435 - 437
[10] Journal of Organic Chemistry, 1992, vol. 57, # 9, p. 2740 - 2741
[11] Chemical Communications, 2015, vol. 51, # 62, p. 12403 - 12406
[12] Synthetic Communications, 2001, vol. 31, # 12, p. 1799 - 1802
[13] Organic Letters, 2000, vol. 2, # 3, p. 247 - 249
[14] Patent: CN107417489, 2017, A, . Location in patent: Paragraph 0054-0064
[15] European Journal of Organic Chemistry, 2016, vol. 2016, # 23, p. 4020 - 4031
[16] Organic Letters, 2005, vol. 7, # 5, p. 831 - 833
[17] Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry, 1986, vol. 25, p. 228 - 229
[18] Journal of Sulfur Chemistry, 2018, vol. 39, # 1, p. 89 - 98
[19] Tetrahedron Letters, 2010, vol. 51, # 8, p. 1161 - 1165
[20] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1989, p. 1381 - 1385
[21] Organic Letters, 2010, vol. 12, # 17, p. 3874 - 3877
[22] Journal of Chemical Research, 2006, # 3, p. 151 - 153
[23] Tetrahedron Letters, 1998, vol. 39, # 44, p. 8163 - 8166
[24] Helvetica Chimica Acta, 2003, vol. 86, # 1, p. 164 - 168
[25] Journal of Chemical Research - Part S, 1996, # 1, p. 62 - 63
[26] Organic Syntheses 3 <New York 1923>, S. 41,
[27] Journal of the American Chemical Society, 1939, vol. 61, p. 3360
[28] Kogyo Kagaku Zasshi, 1941, vol. 44, p. 731,733[29] J. Soc. chem. Ind. Japan Spl., 1941, vol. 44, p. 316
[30] Zhurnal Obshchei Khimii, 1954, vol. 24, p. 1265,1270; engl. Ausg. S. 1251, 1254
[31] Chemische Berichte, 1868, vol. 1, p. 187[32] Justus Liebigs Annalen der Chemie, 1870, vol. Suppl.7, p. 275
[33] Bulletin of the Chemical Society of Japan, 2001, vol. 74, # 6, p. 1151 - 1152
[34] Zhurnal Obshchei Khimii, 1950, vol. 20, p. 338,344; engl. Ausg. S. 359, 364
[35] Organic Letters, 2008, vol. 10, # 10, p. 2007 - 2010
[36] Dyes and Pigments, 2010, vol. 85, # 3, p. 93 - 98
[37] Tetrahedron Letters, 2010, vol. 51, # 51, p. 6730 - 6733
[38] Spectrochimica Acta - Part A: Molecular and Biomolecular Spectroscopy, 2011, vol. 78, # 1, p. 294 - 297
[39] Organic Letters, 2015, vol. 17, # 4, p. 1042 - 1045
[40] Archiv der Pharmazie, 2016, vol. 349, # 6, p. 466 - 474
[41] Tetrahedron, 2017, vol. 73, # 33, p. 4994 - 5004
[42] Photochemical and Photobiological Sciences, 2018, vol. 17, # 11, p. 1794 - 1803
15
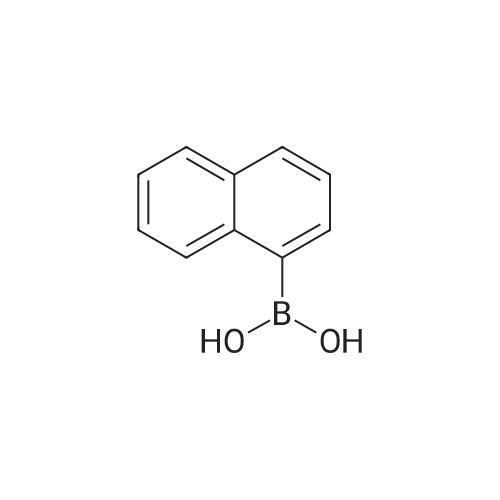
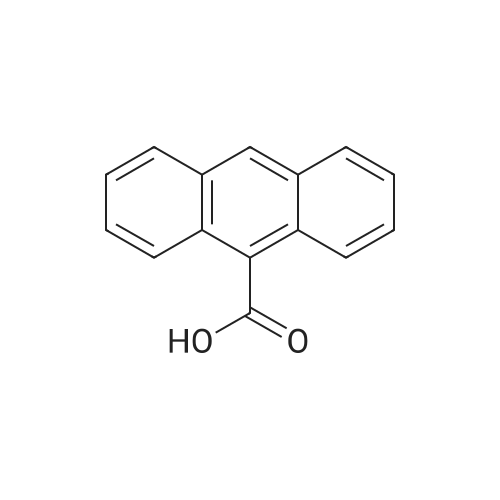
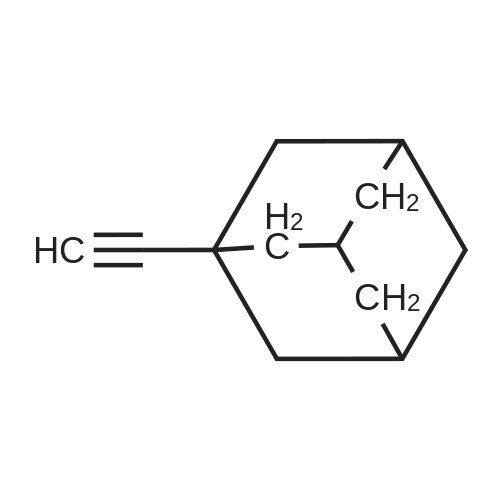
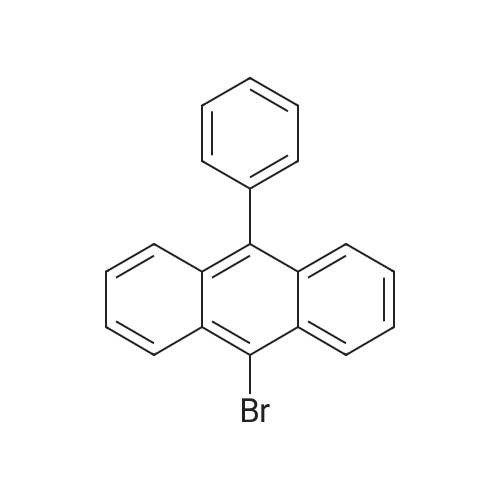
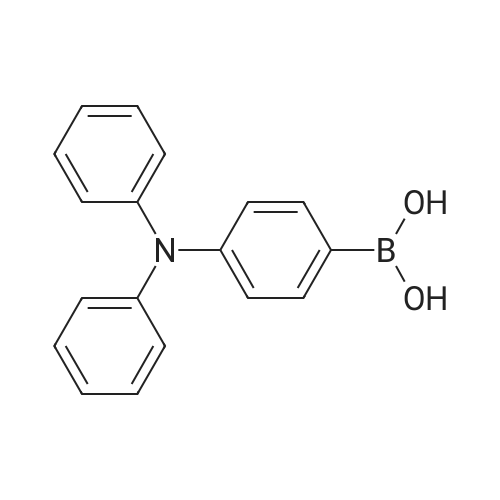
[ 120-12-7 ]
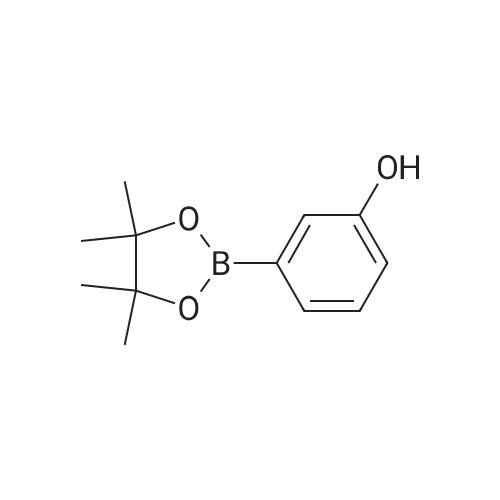
[ 7726-95-6 ]
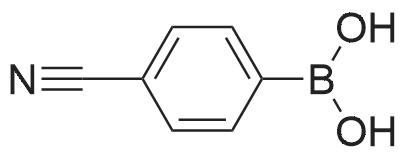
[ 523-27-3 ]
Yield
Reaction Conditions
Operation in experiment
75%
for 24 h;
(No Schlenk conditions needed). Solid anthracene (100g, 0.56 mol) was added to a 3 L round bottom flask charged with 1.5L dichloromethane. The reaction mixture was allowed to stir for 30 min until the anthracene was nearly completely dissolved. Then bromine (58.5 ml, 185g, 1.17 mol) were added dropwise over a period of 6 h. (HBr is formed: reaction flaskmust not be closed and fume cupboard requires good ventilation). The reaction mixture was left stirring for further 24 h(until no more HBr evolution was observed). The yellow precipitate was filtered using a sintered glass filter and the filtrate was washed several times with cold dichloromethane. The mother liquor was then left stirring again for 7 h and again they ellow precipitate was formed which was collected as described. The obtained yellow powder was then dried under high vacuum for 7 h.Yield: 141.6 g (75percent). Elemental analysis: Found: C = 5 1.72percent, H = 1.85percent ; Calculated: C = 50.29percent, H = 2.39percent
Reference:
[1] Zhurnal Obshchei Khimii, 1950, vol. 20, p. 338,344; engl. Ausg. S. 359, 364
[2] Journal of the Chemical Society, 1924, vol. 125, p. 1085
[3] Journal of Organic Chemistry, 1994, vol. 59, # 22, p. 6564 - 6566
Reference:
[1] Bulletin de la Societe Chimique de France, 1937, vol. <5> 4, p. 2052,2062
37
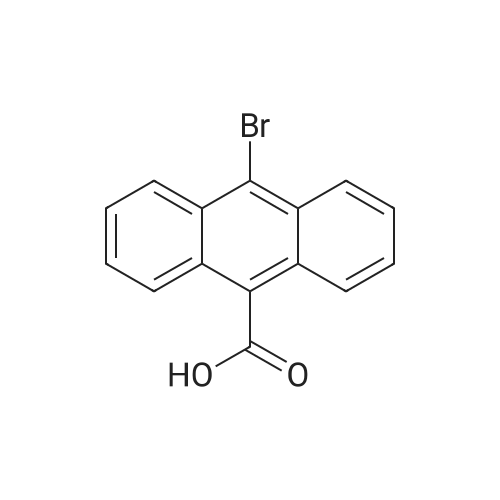
[ 613-31-0 ]
[ 7726-95-6 ]
[ 523-27-3 ]
Reference:
[1] Justus Liebigs Annalen der Chemie, 1870, vol. Suppl.7, p. 265
38
[ 10349-31-2 ]
[ 7726-95-6 ]
[ 523-27-3 ]
Reference:
[1] Chemische Berichte, 1888, vol. 21, p. 2512
39
[ 56-23-5 ]
[ 602-60-8 ]
[ 7726-95-6 ]
[ 523-27-3 ]
Reference:
[1] Journal of the Chemical Society, 1922, vol. 121, p. 2065
40
[ 723-62-6 ]
[ 7726-95-6 ]
[ 523-27-3 ]
[ 6929-81-3 ]
Reference:
[1] Chemische Berichte, 1887, vol. 20, p. 701
41
[ 75-15-0 ]
[ 1498-71-1 ]
[ 7726-95-6 ]
[ 523-27-3 ]
Reference:
[1] Journal of the Chemical Society, 1926, p. 2171[2] Journal of the Chemical Society, 1928, p. 58
42
[ 1564-53-0 ]
[ 7726-95-6 ]
[ 64-19-7 ]
[ 523-27-3 ]
Reference:
[1] Journal of the Chemical Society, 1926, p. 1285
43
[ 1564-64-3 ]
[ 7697-37-2 ]
[ 64-19-7 ]
[ 523-27-3 ]
[ 6313-44-6 ]
Reference:
[1] Journal of the Chemical Society, 1924, vol. 125, p. 1085
44
[ 523-27-3 ]
[ 71367-28-7 ]
Reference:
[1] Chemical and Pharmaceutical Bulletin, 2012, vol. 60, # 8, p. 1055 - 1062
45
[ 580-13-2 ]
[ 523-27-3 ]
[ 122648-99-1 ]
Reference:
[1] Journal of Organic Chemistry, 2008, vol. 73, # 11, p. 4326 - 4329
46
[ 523-27-3 ]
[ 32316-92-0 ]
[ 474688-73-8 ]
Yield
Reaction Conditions
Operation in experiment
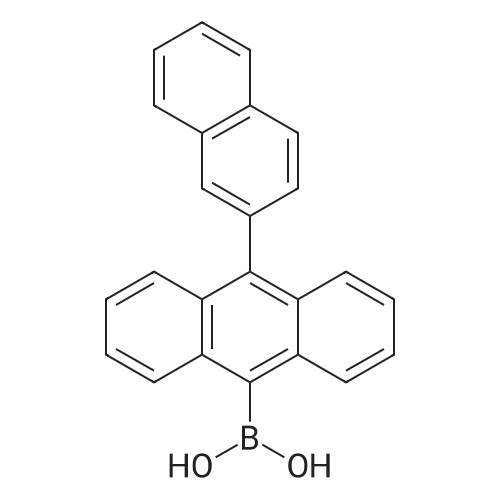
74%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In tetrahydrofuran; water at 80℃; for 72 h;
9-Bromo-10-(naphthalen-2-yl)anthracene (Compound 1) A mixture of 9,10-dibromoanthracene (16 g, 47.6 mmol), 2-naphthalenyl boronic acid (3.4 g, 19.8 mmol), Pd(PPh3)4 (1.0 g, 0.86 mmol) and sodium carbonate (8.4 g, 79 mmol) in tetrahydrofuran (THF)/water (200 ml/40 ml) was degassed and heated at about 80° C. for about 3 days. After being cooled to room temperature, the mixture was filtered, and the filtrate was extracted with dichloromethane (DCM) (250 ml), then washed with brine. The organic phase was collected and dried over Na2SO4, loaded on silica gel, purified by flash column (hexanes) to give a yellow solid (5.7 g, in 74percent yield).
64%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In toluene for 24 h; Inert atmosphere; Reflux
3.36 g (10 mmol) of 9,10-dibromoanthracene and 1.72 g (10 mmol) of Intermediate-1 were introduced under nitrogen and dissolved in 40 ml of toluene0.58 g (0.5 mmol) of Pd (PPh3) 4 and 15 ml (30 mmol) of 2M K2CO3 were added, respectively, and refluxed for 24 hours.After completion of the reaction, the temperature of the reaction mixture was cooled to room temperature, 200 ml of MC and 200 ml of H2O were added to extract the MC layer,Dried over MgSO4, concentrated, and then subjected to column chromatography with Hex: EA = 4: 1 to obtain Intermediate-7 2.45 (64percent).
61%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In ethanol; toluene at 90℃; for 24 h; Inert atmosphere
Synthesis of 9-bromo-10-(naphthalen-2-yl)anthracene A mixture of 15 g (44.6 mmol) of 9,10-dibromoanthracene, 7.7 g (44.6 mmol) of naphthalen-2-ylboronic acid, 0.52 g (0.446 mmol) of Tetrakis(triphenylphosphine)Palladium, 33 ml of 2M Na2CO3, 60 ml of EtOH and 150 ml toluene was degassed and placed under nitrogen, and then heated at 90° C. for 24 h. After the reaction finish, the mixture was allowed to cool to room temperature. The organic layer was extracted with ethyl acetate and water, dried with anhydrous magnesium sulfate, the solvent was removed and the residue was purified by column chromatography on silica (hexane-dichloromethane) to give product 10.4 g (61percent) as a yellow solid.
3 g
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In 1,2-dimethoxyethane; water at 80℃; for 3 h;
2 - 5 g of 9, 10 - 50 ml of dimethoxyethane [jiburomoantorasen[jiburomoantorasen]naphthalene boron acid 2.1 g of commercially available and dissolved, was heated to 80 °C. 50 ml distilled water and sodium carbonate 10 g was placed therein. Further thereto was 0.4 g (0) tetrakistriphenylphosphine. 3 hours after the separatory funnel by extraction with toluene, silica (SiO2 500 g) was purified. Therefore, yellowish white crystals (9 - bromo-naphthalene - yl -10 - -2 - anthracene) 3 g was obtained as thin.
Reference:
[1] Patent: US9923145, 2018, B2, . Location in patent: Page/Page column 34; 35
[2] Patent: KR2017/49295, 2017, A, . Location in patent: Paragraph 0151-0156
[3] Patent: US2014/131664, 2014, A1, . Location in patent: Paragraph 0035-0036
[4] Journal of Materials Chemistry, 2011, vol. 21, # 34, p. 12969 - 12976
[5] Patent: US2012/267615, 2012, A1, . Location in patent: Page/Page column 49
[6] Dyes and Pigments, 2013, vol. 96, # 1, p. 211 - 219
[7] Patent: JP6146001, 2017, B2, . Location in patent: Paragraph 0156-0157
[8] Patent: JP6145989, 2017, B2, . Location in patent: Paragraph 0140; 154
47
[ 523-27-3 ]
[ 13922-41-3 ]
[ 400607-04-7 ]
Yield
Reaction Conditions
Operation in experiment
80%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In tetrahydrofuran; water at 80℃; for 2 h;
9,10-Dibromo anthracene(5.04g, 15mmol), naphthalene1-boronic acid (1.72g, 10mmol), Pd (PPh3) 4 ([0344] 0.58 g, 0.5mmol), potassium carbonate (4.15g, 30mmol) and THF: H2O = 2: 1 solution was dissolved in 200ml At 80 was stirred for 2 hours under reflux. Thereafter 40ml H2O was added and the organic layer was dried 40ml ether obtained by extraction three times with magnesium sulfate, and the intermediate B-3 to the residue obtained by evaporation of the solvent purification by silica gel column chromatography (3.06g, 80percent yield ) it was obtained.
76.89%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In water; toluene at 60℃; for 0.333333 h; Inert atmosphere
In N2under the protection of the, air three times, the solvent is toluene/water = 3:1 (60 ml:20 ml), 2-naphthyl boronic acid (5.6g, 32 . 6mmol), 9,10- dibromoanthracene (11g, 32.6mmol), potassium carbonate (11.2g, 81 . 5mmol) at the same time adding to the above-mentioned three-mouth bottle, begins to stir, 30 min after, adding catalyst four triphenyl phosphine palladium (1.13g, 0 . 98mmol), air again after a time, the temperature rising to 60 °C reflux, the reaction time is 20h left and right.Processing process: TLC for detection, the diatomaceous earth ( helps filters ), DCM for washing, separating, the spin vaporization of solvent, funnel over silica gel, eluant: DCM/PE = 10:1 by the spin vaporization of the solution, after crystallization for heavy PE, filtration to obtain the product, the resulting white solid product in 50 °C bake 3h, get C-1 (9.6g, 76.8percent).
62.1%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In tetrahydrofuran at 50 - 90℃; Inert atmosphere
Under a nitrogen atmosphere, 500mlThree-neck flask 1-naphthalene boronic acid (7g, 0.0407mol) and 9,10-dibromo-anthracene (17.8g, 0.0529mol) for installation into a rear oil bath behind a 2M K2CO3 dissolved in THF and the solvent reflux sikimyeo 50 maintaining the temperature rises to Pd (PPh3) 4 catalyst 0.3g put back 80 ~ 90 temperature to sikimyeo allowed to react for about 16 to 24 hours. Extracted with dichloromethane (300ml), followed by the removal of water on the obtained organic extracts. Then, the solvent was evaporated, and to the organic phase, and separated by column chromatography using hexane-zero eluted G compound (9.7g, yield: 62.1percent) was obtained the, structure was confirmed by 1H-NMR.
62%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In toluene for 24 h; Inert atmosphere; Reflux
3.36 g (10 mmol) of 9,10-dibromoanthracene and 1.72 g (10 mmol) of Intermediate-2 were introduced under nitrogen and dissolved in 45 ml of toluene0.58 g (0.5 mmol) of Pd (PPh3) 4 and 15 ml (30 mmol) of 2M K2CO3 were added, respectively, and refluxed for 24 hours. After completion of the reaction, the temperature of the reaction mixture was cooled to room temperature, 200 ml of MC and 200 ml of H2O were added to extract the MC layer,Dried over MgSO4, concentrated and then columned with Hex: MC = 3: 1 to yield Intermediate-8 2.38 (62percent).
42%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In ethanol; toluene at 100℃; for 12 h; Inert atmosphere
Synthesis of 9-bromo-10-(naphthalen-1-yl)anthracene A mixture of 40 g(119 mmol) of 9,10-dibromoanthracene, 20.5 g(119 mmol) of naphthalen-1-ylboronic acid, 1.38 g(1.2 mmol) of Pd(PPh3)4, 120 ml of 2M Na2CO3, 200 ml of EtOH and 600 ml toluene was degassed and placed under nitrogen, and then heated at 100° C. for 12 h. After finishing the reaction, the mixture was allowed to cool to room temperature. The organic layer was extracted with ethyl acetate and water, dried with anhydrous magnesium sulfate, the solvent was removed and the residue was purified by column chromatography on silica to give product(19.2 g, 50 mmol, 42percent) as a yellow solid.
Reference:
[1] Patent: KR2016/30001, 2016, A, . Location in patent: Paragraph 0341-0344
[2] Patent: CN105384613, 2016, A, . Location in patent: Paragraph 0044; 0045
[3] Patent: KR2015/22269, 2015, A, . Location in patent: Paragraph 0322; 0323; 0327; 0328
[4] Patent: KR2017/49295, 2017, A, . Location in patent: Paragraph 0158-0163
[5] Patent: US2016/308159, 2016, A1, . Location in patent: Paragraph 0021; 0022
[6] Journal of Materials Chemistry, 2010, vol. 20, # 16, p. 3186 - 3194
[7] Patent: US2016/308157, 2016, A1, . Location in patent: Paragraph 0113
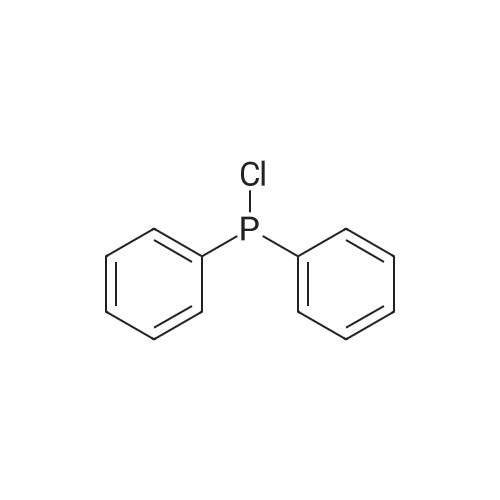
Stage #1: 9,10-Dibromoanthracene With n-butyllithium In diethyl ether for 0.166667h;
Stage #2: chloro-diphenylphosphine In diethyl ether for 12h;
73%
Stage #1: 9,10-Dibromoanthracene In tetrahydrofuran; hexane at -78℃; for 1h; Inert atmosphere;
Stage #2: chloro-diphenylphosphine In tetrahydrofuran; hexane at -78 - 20℃; Inert atmosphere;
61%
Stage #1: 9,10-Dibromoanthracene With n-butyllithium In tetrahydrofuran at -78 - -30℃; for 2h; Inert atmosphere;
Stage #2: chloro-diphenylphosphine In tetrahydrofuran at -78 - 20℃; for 12.5h; Inert atmosphere;
24.2%
Stage #1: 9,10-Dibromoanthracene With n-butyllithium In tetrahydrofuran; diethyl ether; hexane at -30℃; Inert atmosphere; Schlenk technique;
Stage #2: chloro-diphenylphosphine In tetrahydrofuran; diethyl ether; hexane at 35 - 40℃; Inert atmosphere; Schlenk technique;
Preparation of 9,10-bis-(diphenylphosphanyl)anthracene
To a 200ml Schlenk tube a mixture of 9,10-dibromoanthracene (5.0g, 15 mmol) in freshly dried diethyl ether/THF (1:5) wasadded and was allowed to stir for 15 min. The reaction mixture was subsequently cooled using a dry ice/acetone mixture. n-BuLi (15 ml 2.5M BuLi in hexane, 37.3 mmol) was then added using an oven dried syringe. After 20 min the cooling bathwas removed and the reaction mixture was allowed to reach ~-30C. The reaction was again cooled to in a dry ice/acetone bath. The chloro-diphenylphosphine (6.6 ml; 8 g, 37 mmol) was added with a syringe, then the reaction was allowed to warm up to RT and was then slowly heated to 35-40C. The reaction was quenched by addition of 100 ml water. The organic layer was separated with the separation funnel and the water phase was extracted three times with 50 ml diethylether. The combined organic layers were dried over magnesium sulfate. Then the solvent removed in vacuo. The remaining orange oil was loaded on a silica gel column (2 cm diameter). The product was eluted with ether and obtained as an orange powder after drying under high vacuum for 5h. Yield: 1.9 g (24.2%). Elemental analysis: Found: C, 84.4%, H, 5.31%. Calculated: C, 83.5% H, 5.12%.
Stage #1: 9,10-Dibromoanthracene With n-butyllithium In tetrahydrofuran at -78℃; Inert atmosphere;
Stage #2: chloro-diphenylphosphine In tetrahydrofuran at -78 - 20℃; for 12h; Inert atmosphere;
With potassium carbonate; In water; N,N-dimethyl-formamide; at 80℃; for 4h;Schlenk technique;
General procedure: An oven-dried Schlenk flask, equipped with a magnetic stir bar, septum, and a condenser was charged with aryl halide (1.0 mmol), arylboronic acid (1.2 mmol), K2CO3 (2 mmol), 4 (0.143 g, 1 mol %), and 5 mL of solvent. The flask was immersed in an oil bath and stirred at 80 C. Upon complete consumption of starting materials as determined by TLC analysis, the reaction mass was filtered and the solid washed with water (2Chi5 mL), and extracted with diethyl ether (3Chi5 mL). The combined organic layers were collected, dried over anhydrous Na2SO4, and concentrated in vacuum to afford product which was purified by silica gel column chromatography (n-hexane/EtOAc = 9:1)
80%
With C34H32Cl2FeP2Pd; In ethanol; at 80℃; for 6h;Schlenk technique;
General procedure: An oven-dried Schlenk flask, equipped with a magneticstir bar, a septum and a condenser was charged with arylhalide (1.0 mmol), arylboronic acid (1.2 mmol), the gelentrappedbase (1 g, 2 mmol), Pd(dppf)Cl2 (0.0085 g,1 mol%) and 5 mL of 95% ethanol. The flask was immersedand stirred in an oil bath at 80 8C. Upon completeconsumption of starting materials as determined by TLCanalysis, the gel was separated by filtration and water(10 mL) was added. The filtrate was extracted with diethylether (3 5 mL). The combined organic layer was collected,dried over anhydrous Na2SO4 and concentratedunder vacuum to afford the product, which was purified bysilica gel column chromatography (n-hexane:ethyl acetate9:1)
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In water; toluene; at 80℃;Inert atmosphere;
General procedure: A general Pd-catalyzed coupling reaction procedure is explained with the case of 1,4-diphenylnaphthalene 14DPN: A mixture of 1,4-dibromonaphthalene (0.57 g, 2.0 mmol), phenylboronic acid (0.55 g, 4.5 mmol), sodium carbonate (0.54 g, 5.0 mmol), water (5 mL) and toluene (32 mL) in a 100 mL two-necked round bottom flask was sealed with septa and deoxygen by bubbling with nitrogen gas for 40 minitues. Afterwards tetrakis(triphenylphosphine)palladium (47 mg, 0.045 mmol) was quickly added into the mixture by opening the septum, and the mixture was deoxygen for additional 10 minitues. The reaction was kept at 80 C uner a nitrogen atmosphere for 24 hours. After cooled down to room temperature, the reaction was extracted with ethylacetate and water for three times. The oragnic layer was collected, and the solvent was removed by a rotary evaporator. The residue was purified by silica gel chromatagraphy with hexane and dichloromethane (15:1) as the elute to afford a white solid, 1,4-diphenylnaphthalene 14DPN (0.47 g, 1.68 mmol), in a 84 % yield.
To a suspension of 9, 10-dibromoanthracene (1 eq) in THF was dropwise added a n- hexane solution of nBuLi (1.1 eq) at -78C. After stirring for 1 h, some pellets of dry ice were added under a stream of nitrogen. The yellow solution was kept at the same temperature for additional 30 min and then was allowed to warm to room temperature. A small amount of distilled water was added and solvents were concentrated under reduced pressure to obtain yellow solid. Purification by means of silica gel column using EtOAc/EtOH gradient (100%- >60%) as an eluent delivered product in 84% yield as an orange powder. Step 2. isoPropyl 10-bromo-9-anthracenecarboxylate
9,10-bis(N,N-di(4-methoxyphenyl)amino)anthracene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
84%
With tris-(dibenzylideneacetone)dipalladium(0); lithium hexamethyldisilazane; ruphos In 1,4-dioxane at 20 - 100℃; for 48h;
83%
With tri-tert-butyl phosphine; palladium diacetate; sodium t-butanolate In toluene at 100℃; for 48h; Inert atmosphere;
80%
With tris(dibenzylideneacetone)dipalladium(0) chloroform complex; tri-tert-butyl phosphine; sodium t-butanolate In hexane; toluene
70%
With tri-tert-butyl phosphine; potassium <i>tert</i>-butylate; palladium diacetate In toluene at 80℃; for 24h; Inert atmosphere;
1.1 Step 1. Synthesis of luminescent core S1CH3O
Add dibromoanthracene (5g, 14.88mmol) and 3,6-dimethoxydiphenylamine (8.5g, 37.2mmol) in a 100mL reaction flask, then add 50mL of toluene solution to dissolve, then add palladium acetate to it (0.5 g, 1.86 mmol) catalyst and (0.68 g, 3.36 mmol) tri-tert-butyl phosphine were reacted under a nitrogen atmosphere at 80° C. for 24 h. After the reaction is completed, cool to room temperature, add a large amount of water, stir and filter with suction to obtain a crude product. Then, it was purified by column chromatography to obtain a white solid of S1CH3O, and the yield of S1CH3O was 70%.
With tris(dibenzylideneacetone)dipalladium(0) chloroform complex; tris-(o-tolyl)phosphine; sodium t-butanolate In toluene Heating;
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate; In 1,4-dioxane; at 110℃; for 24h;Inert atmosphere;
Under an argon atmosphere, 9,10-dibromoanthracene (10g, 27.47mmol), bis-boronic acid ester (20.92g, 82.40mmol) and potassium acetate (13.48g, 137.33mmol), was added to 250 ml 1,4 - dioxane, was heated to 110 deg.] C, for 24 hours.The reaction mixture was poured into water and extracted with ethyl acetate, the organic layer was thoroughly washed with brine, dried over anhydrous magnesium sulfate was added.After the solution was concentrated to give crude product as a white solid, purified by silica gel column chromatography (eluent petroleum ether selecting / dichloromethane = 3/1, v / v ),the product stored in a refrigerator, to give a white solid, yield 85%.
71%
With palladium bis[bis(diphenylphosphino)ferrocene] dichloride; potassium acetate; In 1,4-dioxane; at 95℃; for 24h;
9,10-dibromoanthracene (3.4 g, 0.010 mol) in bis (pinacolato) dibron (5.0 g, 0.024 mol), PdCl2 (dppf) (0.4 g, 0.0005 mol), potassium-acetate (2.7 g, 0.020 mol) into a 100 mL 1,4-dioxane and stirred for 24 hours at 95C . The reaction was then cooled to terminate the reaction H20: After layer separation the MC column purification (n-Hexane: MC) to the intermediate 14- 3.1 g (yield:71%) was obtained.
65.9%
With sodium borate; palladium bis[bis(diphenylphosphino)ferrocene] dichloride; potassium acetate; In N,N-dimethyl-formamide; at 80℃; for 24h;Inert atmosphere;
In nitrogen atmosphere, a mixture with 3.36 g 9,10-dibromoanthracene, 5.6 g sodium borate, 6.0 g potassium acetate and 0.2 g Pd(dppf)Cl2 in 80 mL N,N-dimethylformamide was stirred at 80 C for 24 h. After cooling to room temperature, the reaction mixture was poured into icewater and rapidly stirred to precipitate crude product. The 9,10-bis(4' ,4' ,5' ,5? -tetramethyl-1' ,3' ,2? -borate) anthracene was obtained by column chromatography on silica gel with dichloromethane/petroleumether (1/5) as the eluant to obtain a faint yellow powder (yield 65.9%). 1H NMR (CDCl3, 400 MHz) delta 8.24 (d, J 3.6 Hz, 4H), 7.55 (dd, J 4.4,4.8 Hz, 4H), 1.58 (s, 24H); 13C NMR (CDCl3, 100 MHz) 25.41, 85.22,99.93, 126.35, 128.48, 134.45.
65%
With tris-(dibenzylideneacetone)dipalladium(0); sodium acetate; In N,N-dimethyl-formamide;Inert atmosphere; Reflux;
a, under the protection of nitrogen, 9,10-dibromoanthracene,Pinacol borate,Pd2 (dba) 3, sodium acetate was added to DMF,Heated to reflux overnight;After the reaction is cooled to room temperature,The combined organic phases were washed with water and extracted with ethyl acetate.Spin out the organic solvent and separate by column chromatographyTo give the compound 9,10-di-pinacol boronate,Is white powder,Yield 65%;
58%
With potassium acetate; palladium diacetate; In N,N-dimethyl-formamide; at 80℃; for 7h;Inert atmosphere;
In a nitrogen atmosphere, 9,10-dibromoanthracene (6) (0.54 g, 1.61 mmol) and of bis(pinacolato)diboron (0.98 g, 3.86 mmol) were dissolved in DMF (20 mL), followed by the addition of palladium acetate (12 mg) and potassium acetate (0.473 g, 4.82 mmol). The reaction mixture was then stirred at 80 C for 7 h under nitrogen atmosphere. After that, the mixture was cooled to room temperature and poured into water (300 mL) with stirring. The solid was collected by filtration and dried. The crude product was purified by column chromatography (silica gel) with petroleum ether/dichloromethane (v/v = 1:1) as eluent, followed by recrystallized in petroleum ether/dichloromethane to give a green powder in a yield of 58 %. Mp>250 C. 1H NMR (500 MHz, TMS, CDCl3): delta (ppm) = 8.35-8.33 (m, 4H), 7.45-7.43 (m, 4H), 1.57 (s, 24H) (Fig. S12) IR (KBr, cm-1): 2980, 2930, 1720, 1450, 1240, 1140, 980, 852, 758, 675, 579. Elemental analysis calculated for C26H32B2O4: C, 72.60; H, 7.50. Found: C,72.51; H, 7.36. MS, m/z: cal: 430.2, found: 429.7 (Fig. S13)
36.3%
With potassium acetate; palladium diacetate; In N,N-dimethyl-formamide; at 70℃; for 18h;Inert atmosphere;
Compound A (4.00 g, 11.91 mmol), bis (pinacolato) diboron (7.62 g, 30.00 mmol),Palladium (II) acetate (Palladium (II) acetate, Pd (OAc) 2) (0.15 g, 0.71 mmol), potassium acetate (Poassi acetate acetate, PoAc)(7.00 g, 71.4 mmol) and 35 mL of dimethylformamide (DMF) were placed in a flask, charged with nitrogen, and reacted at 70 C. for 18 hours.After cooling to room temperature, the product was extracted with methylene chloride (MC) and washed well with water. The extracted organics were dried over anhydrous magnesium sulfate (anhydrous MgSO 4). Then, the residue was purified by silica gel column chromatography (hexane: ethyl acetate = 20: 1 (eluent)) to obtain 1.86 g of compound A-1. (Yield 36.3%)
With potassium carbonate; palladium; In N,N-dimethyl-formamide; at 90 - 120℃;Inert atmosphere;
In actual operation, under the protection of argon, 2 mmol-15 mmol of the first reactant 4 mmol-30 mmol of the second reactant 2 mmol- 41 mmol of potassium carbonate and 0.1 mmol-1.0 mmol of palladium catalyst, 20 mL-250 mL of dimethylformamide solvent was added thereto, and the mixture was heated and refluxed at 90 C-120 C for 48 hours-96 hours The mixture including the first intermediate product is cooled to room temperature at the end of the reaction, the solvent is distilled off under reduced pressure and distillation, and the product is extracted 3 to 5 times with deionized water and CH2Cl2. Extract 3 times to 5 times, dry the obtained organic phase with anhydrous sodium sulfate, then filter, spin-dry and chromatographically separate and purify to obtain the first intermediate product
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In ethanol; water; toluene; at 100℃; for 48h;Inert atmosphere;
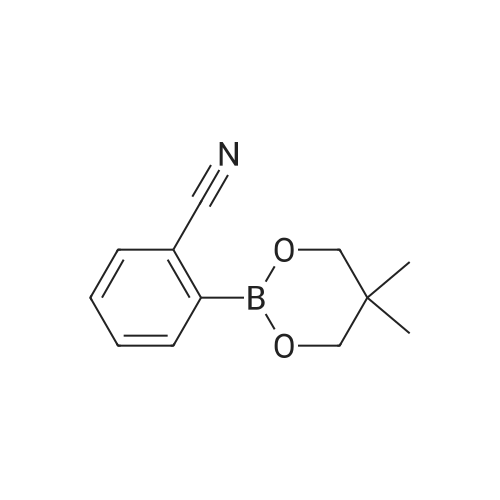
A mixture of 9,10-dibromoanthracene(3.0 g, 8.93mmol), 4-cyanophenyl boronic acid (1.44 g,9.82 mmol), Pd(PPh3)4 (0.41 g, 0.36 mmol), 50 mL toluene, and 20 mL of 2 M K2CO3was refluxed at 100C for 48 h under nitrogen, and was extracted with CH2Cl2.The organic layer was washed with brine and water anddried over magnesium sulfate, filtered and concentrated in vacuo. The crudeproduct was purified by flash column chromatography (n-hexane/DCM: 3/1). Yield: 56%. 1H-NMR (CDCl3,300MHz) [ppm]: delta 8.65 (d, 2H), 7.91 (d, 2H), 7.64-7.41 (m, 8H); FT-IR (KBr) [cm-1]2234 (-CN); EI-mass (m/z): calculated: 357.02, found: 357 (M+)
50%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In toluene; at 90℃; for 12h;Inert atmosphere;
Add 9,10-dibromoanthracene (1.67g, 5mmol), <strong>[126747-14-6]4-cyanophenylboronic acid</strong> (0.74g, 5mmol), tetratriphenylphosphine palladium (0.115 g, 0.1 mmol) was sequentially added to a 250 mL two-necked flask, the flask was evacuated under vacuum and replaced three times under dry nitrogen, then 40 mL of toluene and 10 mL of a saturated K2CO3 aqueous solution were added.The reaction was stirred under heating at 90 C for 12 hours.Extract with saturated brine and dichloromethane. Distilled under reduced pressure to obtain a yellow solid. Using silica gel as a stationary phase and petroleum ether / dichloromethane as an eluent, 0.89 g of a white powder was obtained by column chromatography (yield 50%).
40%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In water; toluene; at 90℃; for 24h;Inert atmosphere;
100mL round bottom flask,9,10-Dibromoanthraquinone (5 mmol, 1.67 g),<strong>[126747-14-6]4-cyanophenylboronic acid</strong> (5 mmol, 735 mg),Tetratriphenylphosphine Palladium (0.1 mmol, 115 mg)Dissolved in 40 mL of toluene and20mL of potassium carbonate aqueous solution (2.0mol L-1),Under a nitrogen atmosphere, the mixture was stirred and refluxed at 90C for 24 hours.After the reaction is over,Extract with dichloromethane,Rotary evaporation concentrates the extractColumn chromatographic separation (petroleum ether: dichloromethane = 3:1,Volume ratio) gives a pale yellow-green solid (710 mg,Yield: 40%).
9,10-bis(4-methyldiphenylamino)anthracene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Stage #1: 9,10-Dibromoanthracene; 4-methyldiphenylamine With tris-(o-tolyl)phosphine; sodium t-butanolate In toluene at 20℃; Heating / reflux;
Stage #2: With hydrogenchloride In water; toluene
4.A
Palladium acetate (0.27 g, 0.4 mmol) and tri-o-tolylphosphine (0.49 g, 1.6 mmol) were dissolved in toluene (150 mL). The solution was stirred at room temperature for 15 minutes, whereupon 9,10-dibromoanthracene (13.44 g, 40 mmol), 4-methyldiphenylamine (18.3 g, 100 mmol) and sodium t-butoxid (15.4 g, 160 mmol) were added. The mixture was refluxed under nitrogen overnight, then cooled to room temperature. Hydrochloric acid (1N) was added slowly to neutralize the solution and 500 mL of methanol was added. The precipitate was collected by filtration and dried in vacuo at room temperature. The solid was dissolved in 1.5 L of toluene, and the resultant solution was passed through a column of acidic alumina and eluted with toluene. The combined toluene solution was concentrated to-500 mL and poured into 500 mL of methanol to precipitate the crude product as a yellow powder. The crude product was collected by filtration and was washed with methanol. Twice re-crystallization from a mixture of toluene and ethanol afforded 14.1 g of the final product as a yellow powder
Anthracene (50 g, 0.28 mol, 1 eq) and 1,3-dibromo-5,5-dimethylhydantoin (38.15 g, 0.133 mol, 0.475 eq) were combined in a 500 mL of ethyl acetate and brought to reflux. After about 5 min of reflux, the mixture became homogeneous, and yellow, and stayed light in color throughout the 2.5 h that it was refluxed. The reaction mixture allowed to cool and then washed with water to remove the 5,5-dimethylhydantoin. When water was added, the mixture turned greenish-blue and the color persisted through additional aqueous washes. The organic layer was dried over Na2SO4 and the greenish color gradually disappeared, and the mixture became orange-brown. The liquor was concentrated to dryness, to yield a brown solid residue. The solid was dissolved in hot THF (about 50 mL) and then slowly it was precipitated again with addition of acetonitrile (about 200 mL). The yellowish solid was isolated by filtration (36.4 g) and the process was repeated twice to yield two more crops of solid (total 23.8 g). The 3 crops were combined and analyzed by high performance liquid-chromatography (HPLC). The analysis indicated that only 89% of the reaction product was the desired 9-bromoanthracene, 9.3% was unreacted anthracene and 1% was the undesirable 9,10-dibromoanthracene. The 9,10-dibromoanthracene could not be removed by recrystallization.
Example 1 In suspension of 49.8 g (2.04 mol) of Mg and 1.0 L of THF was dropwisely added 321.4 g (2.04 mol) of bromobenzene under refluxing to give Grignard reagent. Thus prepared Grignard reagent was dropwisely added to a mixed solution of 286 g (0.85 mol) of 9,10-dibromoanthracene, 566mL of THF and 0.6 g (0.00085 mol) of Pd(PPh3)2Cl2 with agitation at 30 to 60 C. in 30 minutes, followed by agitation at the same temperature for 1 hour. To the reaction solution was added 300 ml of diluted hydrochloric acid and the object substance was extracted with 3 L of toluene. The obtained organic layer was washed with saturated saline solution and concentrated to 750 mL in total. The precipitated crystal was recovered by filtration and dried at 70 C. for 1 hour to give 229.5 g of 9,10-diphenylanthracene (yield 81.7%, HPLC purity 97.7%).
73.8%
Pd(PPh3)2Cl2; In tetrahydrofuran;
Example 3 To a mixed solution of 286.6 g (0.85 mol) of 9,10-dibromoanthracene, 321.4 g (2.04 mol) of bromobenzene and 1.5 L of THF were added 49.8 g (2.04 mol) of Mg and 0.6 g (0.00085 mol) of Pd(PPh3)2Cl2, followed by heating up to refluxing and agitating at the same temperature for 1 hour. To the resultant was added 300 mL of diluted hydrochloric acid and the object substance was extracted with 3 L of toluene. The obtained organic layer was washed with saturated saline solution and concentrated to 750 ml in total. The precipitated crystal was recovered by filtration and dried at 70 C. for 1 hour to give 207.3 g of 9,10-diphenylanthracene (yield 73.8%, HPLC purity 95.2%).
Example 6 Grignard reagent prepared by the same manner as in Example 1 was dropwisely added to a mixed solution of 286 g (0.85 mol) of 9,10-dibromoanthracene, 566 mL of THF and 0.4 g (0.00085 mol) of Ni(dppe)Cl2 with agitation at 30 to 60 C. in 30 minutes, followed by agitation at the same temperature for 1 hour. To the reaction solution was added 300 mL of diluted hydrochloric acid and the object substance was extracted with 3 L of toluene. The obtained organic layer was washed with saturated saline solution and concentrated to 750 mL in total. The precipitated crystal was recovered by filtration and dried at 70 C. for 1 hour to give 229.5 g of 9,10-diphenylanthracene (Yield: 81.7%, HPLC purity:97.7%).
75.9%
Ni(AcaC)2; In tetrahydrofuran;
Example 5 Grignard reagent prepared by the same manner as in Example 1 was dropwisely added to a mixed solution of 286 g (0.85 mol) of 9,10-dibromoanthracene, 566 mL of THF and 0.24 g (0.00085 mol) of Ni(acac)2 with agitation at 30 to 60 in 30 minutes, followed by agitation at the same temperature for 1 hour. To the reaction solution was added 300 mL of diluted hydrochloric acid and the object substance was extracted with 3 L of toluene. The obtained organic layer was washed with saturated saline solution and concentrated to 750 mL in total. The precipitated crystal was recovered by filtration and dried at 70 C. for 1 hour to give 213.2 g of 9,10-diphenylanthracene (Yield: 75.9%, HPLC purity:97.0%).
9,10-bis[N-(4-diphenylaminophenyl)-N-phenylamino]anthracene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
24%
palladium diacetate; In o-xylene; water;
Example 5 (Synthesis of 9,10-bis[N-(4-diphenylaminophenyl)-N-phenylamino]-anthracene) A hexane solution of 40.0 g of N,N,N'-triphenyl-1,4-phenylenediamine, 15.9 g of 9,10-dibromoanthracene, 21.9 g of sodium t-butoxide, 0.10 mg of palladium acetate and 0.38 g of tri-t-butylphosphine were dissolved in 300 mL of o-xylene. The reaction was carried out at a temperature of 70C for 8 hours under a nitrogen atmosphere. After the reaction, water was added to the resulting reaction mixture and the resulting red precipitates were collected by filtration. The precipitates were dissolved in hot toluene, and the resulting solution was filtered while hot to remove impurities therefrom, followed by cooling, to provide 24.7 g of 9,10-bis[N-(4-diphenylaminophenyl)-N-phenylamino]anthracene as red solid. The yield was 24%.
With Pd(PCy3)2(dba); potassium carbonate; In ethanol; toluene; at 80 - 85℃;Inert atmosphere;
General procedure: The title compounds are prepared in a manner substantially similar to the proceduredescribed in reference 12 with a slight modification. A mixture of aryl dibromide or 4-bromostyrene (3 mmol), 4-vinylphenylboronic acid (6mmol or 3 mmol), 2M aq. base solution (4 mL, K2CO3), ethanol (6 mL), toluene (0.4M solution concentration of olefin), Pd(dba)(PCy3)2 1 (0.5mol%) were placed in a two-necked glass-reactor equipped with a magnetic stirring bar and reflux condenser. The suspension was degassed andheated in an oil bath at 80oC for 16h. After the reaction was completed (monitored by TLC and GCMS), the resulting mixture was concentratedand extracted with DCM and washed with water (twice). Then, the organic layer was collected and passed through by very quick ‘flash system’(glass filter, silica gel, celite) and dried over anhydrous Na2SO4 for 6h. The excess of solvent was evaporated to dryness under vacuum. Theresulting olefins were dissolved in DCM or CHCl3 (quite good or weakly soluble), isolated and purified by repeated precipitation fromDCM/hexane system. The final products were filtered off and dried under vacuum. The isolated yields were from 76 to 99% (depending on thecombination of starting dibromoarene).
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In toluene; for 48h;Heating / reflux;
SYNTHESIS EXAMPLE 1; Synthesis of Intermediate A; An intermediate A was synthesized according to Reaction Scheme 1 below: 9,10-dibromoanthracene (4.00 g, 11.90 mmol) and phenylboronic acid (1.60 g, 13.12 mmol) were dissolved in toluene (100 ml), and Pd(PPh3)4 (0.68 g, 0.59 mmol) and 2M K2CO3 (24 ml) were gradually dropwise added thereto. The reaction mixture was refluxed for 48 hours and cooled to room temperature. Then, a solvent was removed under a reduced pressure, and the residue was extracted with chloroform. The extracted solution was twice washed with a supersaturated sodium chloride solution and water (H2O), and an organic layer was collected and dried over anhydrous magnesium sulfate. Then, the solvent was evaporated to obtain a crude product, and the crude product was purified by silica gel column chromatography using as an eluant, a solution of chloroform and hexane (1:1 v/v) to give 2.27 g (yield: 57%) of a compound of Formula 1b. The compound of Formula 1b (2.20 g, 6.60 mmol) was dissolved in tetrahydrofuran (100 ml), and n-butyllithium (4.6 ml, 7.36 mmol, 1.6 M solution) and 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.48 ml, 7.25 mmol) were gradually dropwise added thereto at -78 C. The reaction mixture was gradually heated to room temperature and incubated at room temperature for 15 hours. Then, water (H2O) (50 ml) was added to the reaction solution so that the reaction was terminated, and the resultant solution was extracted with chloroform. The extracted solution was twice washed with a supersaturated sodium chloride solution and water (H2O), and an organic layer was collected and dried over anhydrous magnesium sulfate. Then, the solvent was evaporated to obtain a crude product, and the crude product was purified by silica gel column chromatography using as an eluant, a solution of chloroform and hexane (1:1 v/v) to give 1.53 g (yield: 61%) of a compound of Formula 1c. A compound of Formula 1d (1.80 g, yield: 61%) was synthesized in the same manner as in the synthesis of the compound of Formula 1b except that 3-bromoiodobenzene was used instead of 9,10-dibromoanthracene, and the compound of Formula 1c (2.75 g, 7.23 mmol) was used instead of phenylboronic acid. The intermediate A (1.70 g, yield: 56%) was synthesized in the same manner as in the synthesis of the compound of Formula 1c except that the compound of Formula 1d (2.70 g, 6.60 mmol) was used instead of the compound of Formula 1b.
57%
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In toluene;Reflux;
Synthesis Example 1 Synthesis of Intermediate AAn intermediate A was synthesized according to Reaction Scheme 1 below: 9,10-dibromoanthracene (4.00 g, 11.90 mmol) and phenylboronic acid (1.60 g, 13.12 mmol) were dissolved in toluene (100 ml), and Pd(PPh3)4 (0.68 g, 0.59 mmol) and 2M K2CO3 (24 ml) were gradually dropwise added thereto. The reaction mixture was refluxed for 48 hours and cooled to room temperature. The solvent was removed under reduced pressure, and the residue was extracted with chloroform. The extracted solution was twice washed with a supersaturated sodium chloride solution and water (H2O), and the organic layer was collected and dried over anhydrous magnesium sulfate. The solvent was evaporated to obtain a crude product, and the crude product was purified by silica gel column chromatography to yield 2.27 g (57%) of the intermediate A.
45%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; toluene; at 100℃; for 12h;Inert atmosphere;
Synthesis of 9-bromo-10-phenylanthracene A mixture of 40 g (119 mmol) of 9,10-dibromoanthracene, 16 g (131 mmol) of phenylboronic acid, 1.38 g (1.2 mmol) of Pd(PPh3)4, 120 ml of 2M Na2CO3, 150 ml of EtOH and 450 ml toluene was degassed and placed under nitrogen, and then heated at 100 C. for 12 h. After finishing the reaction, the mixture was allowed to cool to room temperature. The organic layer was extracted with ethyl acetate and water, dried with anhydrous magnesium sulfate, the solvent was removed and the residue was purified by column chromatography on silica to give product (17.8 g, 53.6 mmol, 45%) as a yellow solid.
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; at 80℃; for 3h;
Synthesis (C3-1); 2.2 g of commercially available phenylboronic acid and 6 g of 9,10-dibromoanthracene were dissolved in 100 ml of dimethoxyethane and were heated to 80 C. 50 ml of distilled water and 10 g of sodium carbonate were added thereto. Also, 0.5 g of tetrakis-triphenylphosphine palladium(0) was added thereto.After 3 hours, extraction with toluene was carried out in a separatory funnel and purification using silica gel (SiO2, 500 g) was carried out.As a result, 4 g of pale yellow crystals (9-bromo-10-phenylanthracene) was obtained.
4 g
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,2-dimethoxyethane; at 80℃; for 3h;
g 2.2 g of 9, 10 - 100 ml of phenyl boronic acid with commercially available [jiburomoantorasen[jiburomoantorasen] dissolved in dimethoxyethane, 80 C heated. 50 ml distilled water and sodium carbonate 10 g was placed therein. Further thereto was placed 0.5 g (0) tetrakistriphenylphosphine. 3 hours after the separatory funnel by extraction with toluene, silica (SiO2 500 g) was purified. Therefore, yellowish white crystals (9 - bromo - phenyl -10 - anthracene) 4 g was obtained
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In toluene; for 2h;Reflux; Inert atmosphere;
A mixture of compound 4 (33.4 g, 100 mmol), phenylboronic acid (12.2 g, 100 mmol)(5.78 g, 5 mmol) and potassium carbonate (41.4 g, 300 mmol) were added to toluene (500 ml), refluxed under nitrogen for 2 hours, cooled to room temperature and used directly in The next step.
94%Chromat.
With C30H34Cl4N4Pd2; potassium carbonate; In methanol; at 60℃; for 24h;
General procedure: [1] Reaction conditions: 0.50 mmol of halogenated aromatics, 0.60 mmol of phenylboronic acid, 1.0 mmol of K2CO3, prepared in Example 2. The prepared nitrogen heterocyclic biscarbene metal complex 0.2 mol%, solvent 5 mL, PEG400 (0.1 mL), 60 C.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; for 25h;Inert atmosphere; Reflux;
9,10-dibromoanthracene (5 mmol), phenyl boronic acid (5 mmol), Pd(PPh3)4 (0.5 mmol), Na2CO3 (2.0 M) aqueous solution (15 mL), ethanol (10 mL), and toluene (30 mL) were added to a flask and mixed. A gas was removed from the mixture, and the mixture was refluxed in a nitrogen atmosphere for 25 hours. After the mixture was cooled, a solvent was evaporated therefrom in a vacuum, and a product was extracted therefrom by using dichloromethane. CH2Cl2 solution was washed with water and dried by using MgSO4.
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; sodium carbonate; In 1,4-dioxane; water; at 110℃;
Add 9,10-dibromoanthracene and <strong>[133730-34-4]2,4-dimethoxyphenylboronic acid</strong> at a molar ratio of 1:2 into the round bottom flask,Then add anhydrous sodium carbonate and catalyst [1,1’-bis(diphenylphosphino)ferrocene] palladium dichloride 10% mol,Use 1,4-dioxane and water volume ratio 4:1 as solvent,Reflux overnight at 110C, evaporate the solvent after the reaction is complete,Extract with water and dichloromethane, dry with anhydrous sodium sulfate,The mixture was separated through a silica gel column to obtain 9,10-bis(2,4-dimethoxyphenyl)anthracene as a white solid.The yield was 32%.
palladium diacetate; In 1,4-dioxane; water; toluene;
a) Synthesis of the Atropisomer Mixture of Anthracene Derivative A1 1.83 g (6 mmol) of tris-o-tolylphosphine and then 225 mg (1 mmol) of palladium(II) acetate were added to a degassed, well-stirred suspension of 74.4 g (400 mmol) of 4-methylnaphthalene-1-boronic acid, 33.6 g (100 mmol) of 9,10-dibromoanthracene and 104.3 g (420 mmol) of potassium phosphate monohydrate in a mixture of 300 ml of toluene, 150 ml of dioxane and 400 ml of water, and the mixture was subsequently refluxed for 16 h. After the reaction mixture had been cooled to room temperature, the precipitated solid was filtered off with suction and washed three times with 200 ml of water each time and three times with 100 ml of ethanol each time. After drying in vacuo (1 mbar; 80 C., 16 h), the product was extracted with chloroform through a glass-fibre extraction thimble (pore size 0.1 pm) in a Soxhlett extractor in order to remove traces of palladium. The chloroform was concentrated to a volume of 100 ml, and 500 ml of ethanol were added. The resultant precipitate was filtered off with suction and washed with ethanol. The yield of crude product A1 was 41.3 g (90 mmol), 90.0% of theory.
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In toluene for 24h; Inert atmosphere; Reflux;
26%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In ethanol; water; toluene at 110℃; for 48h; Inert atmosphere; Schlenk technique;
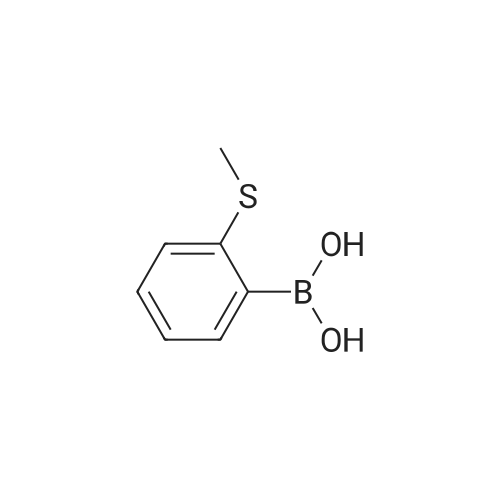
Synthesis of Ligand syn 1
A Schlenk tube was charged with 2-(methylthio)benzeneboronic acid (150 mg, 0.89mmol), 9,10-dibromoanthracene (120 mg, 0.36 mmol), tetrakis(triphenylphosphine)palladium (21 mg, 5 mol %) and potassium carbonate (248 mg, 1.8 mmol). The mixture was degassed and flushed with argon for three times. The nthe degassed solvent toluene/ethanol/water (6 mL, 4:1:1) was added. The resulting mixture was stirred at 110 °C for 48 h. After cooling to room temperature, the suspension was filtered through a pad of celite and washed with dichloromethane. The filtrate was dried over magnesium sulfate, then concentrated and purified by column chromatography on silica gel (eluent: petrol ether/ethyl acetate, 75: 1) to afford syn-atropisomer 1 as a light yellow solid (180 mg, 26% yield).
With palladium diacetate; sodium carbonate; triphenylphosphine; at 110℃; for 24h;Ionic liquid; Inert atmosphere;
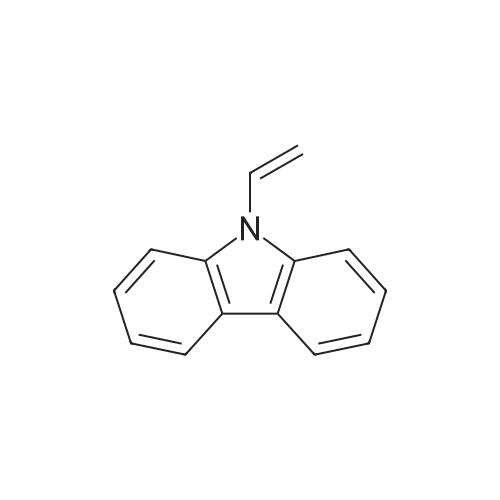
The solution of Pd(OAc)2 0.056 g (0.25 mmol) and PPh3 0.262 g (1.0 mmol) as catalyst in 20mL [BMIM]BF4 was stirred at 80 C for 30 min under nitrogen atmosphere and then cooled to room temperature. A mixture of 9,10-dibromoanthracene 0.84 g (2.5 mmol), <strong>[1484-13-5]N-vinylcarbazole</strong> 1.05 g (5 mmol), and anhydrous sodium carbonate 0.4 g (5 mmol) was added. After stirred at 110 C for 24 h, the mixture solution was extracted by 50mL ether for 5 times. The organic solution was washed with water saturated solution of NaCl and dried over MgSO4. Removal of the solvent in vacuo to give a yellow solid and purified by column chromatography on silica gel with petroleum ether:ethyl acetate (4:1) as the elutant to give slightly yellow crystals of 9,10-di-(N-carbazovinylene)anthracene (DCVA) in 58.8% yield, m.p. 167-168 C.
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In tetrahydrofuran; water at 80℃; for 2h;
4 synthesis of intermediate B-3
9,10-Dibromo anthracene(5.04g, 15mmol), naphthalene1-boronic acid (1.72g, 10mmol), Pd (PPh3) 4 ([0344] 0.58 g, 0.5mmol), potassium carbonate (4.15g, 30mmol) and THF: H2O = 2: 1 solution was dissolved in 200ml At 80 was stirred for 2 hours under reflux. Thereafter 40ml H2O was added and the organic layer was dried 40ml ether obtained by extraction three times with magnesium sulfate, and the intermediate B-3 to the residue obtained by evaporation of the solvent purification by silica gel column chromatography (3.06g, 80% yield ) it was obtained.
76.89%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In water; toluene at 60℃; for 0.333333h; Inert atmosphere;
preparation of Intermediate 9-bromo -10 - (1-naphthyl) anthracene (C-1)
In N2under the protection of the, air three times, the solvent is toluene/water = 3:1 (60 ml:20 ml), 2-naphthyl boronic acid (5.6g, 32 . 6mmol), 9,10- dibromoanthracene (11g, 32.6mmol), potassium carbonate (11.2g, 81 . 5mmol) at the same time adding to the above-mentioned three-mouth bottle, begins to stir, 30 min after, adding catalyst four triphenyl phosphine palladium (1.13g, 0 . 98mmol), air again after a time, the temperature rising to 60 °C reflux, the reaction time is 20h left and right.Processing process: TLC for detection, the diatomaceous earth ( helps filters ), DCM for washing, separating, the spin vaporization of solvent, funnel over silica gel, eluant: DCM/PE = 10:1 by the spin vaporization of the solution, after crystallization for heavy PE, filtration to obtain the product, the resulting white solid product in 50 °C bake 3h, get C-1 (9.6g, 76.8%).
62.1%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In tetrahydrofuran at 50 - 90℃; Inert atmosphere;
2 Synthesis of Compound G
Under a nitrogen atmosphere, 500mlThree-neck flask 1-naphthalene boronic acid (7g, 0.0407mol) and 9,10-dibromo-anthracene (17.8g, 0.0529mol) for installation into a rear oil bath behind a 2M K2CO3 dissolved in THF and the solvent reflux sikimyeo 50 maintaining the temperature rises to Pd (PPh3) 4 catalyst 0.3g put back 80 ~ 90 temperature to sikimyeo allowed to react for about 16 to 24 hours. Extracted with dichloromethane (300ml), followed by the removal of water on the obtained organic extracts. Then, the solvent was evaporated, and to the organic phase, and separated by column chromatography using hexane-zero eluted G compound (9.7g, yield: 62.1%) was obtained the, structure was confirmed by 1H-NMR.
62%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In toluene for 24h; Inert atmosphere; Reflux;
Synthesis of intermediate-8
3.36 g (10 mmol) of 9,10-dibromoanthracene and 1.72 g (10 mmol) of Intermediate-2 were introduced under nitrogen and dissolved in 45 ml of toluene0.58 g (0.5 mmol) of Pd (PPh3) 4 and 15 ml (30 mmol) of 2M K2CO3 were added, respectively, and refluxed for 24 hours. After completion of the reaction, the temperature of the reaction mixture was cooled to room temperature, 200 ml of MC and 200 ml of H2O were added to extract the MC layer,Dried over MgSO4, concentrated and then columned with Hex: MC = 3: 1 to yield Intermediate-8 2.38 (62%).
62%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In water; toluene for 24h; Inert atmosphere; Reflux;
3-3 (3-3) Synthesis of 9-bromo-10-(naphthalen-1-yl) PLC-3)
As shown in Figure 6a, under a nitrogen atmosphere, 9,10-dibromoanthracene (8) (3.36 g, 10.0 mmol, 1.0 equivalent), 1-naphthyl boronic acid (1.72 g, 10.0 mmol, 1.0 equivalent), Pd(PPh3)4 (0.58 g, 0.50 mmol, 0.05 eq), water (15 mL) andK2CO3 (4.14 g, 30.0 mmol, 3.0 eq) dissolved in anhydrous toluene (45 mL) was added to a dried flask (200 mL). The reaction mixture was refluxed under stirring for 24 hours. The reaction progress was monitored by TLC using hexane containing 10% dichloromethane. After completion of the reaction, the reaction mixture was cooled to room temperature, dichloromethane (100 mL) and water (100 mL) were added to the reaction mixture, and the organic solvent was separated from the reaction mixture, dried over sodium sulfate, and evaporated under reduced pressure. The white solid was purified by silica gel chromatography to obtain pure product (2.38 g, 62% yield).
42%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In ethanol; toluene at 100℃; for 12h; Inert atmosphere;
1 Synthesis of 9-bromo-10-(naphthalen-1-yl)anthracene
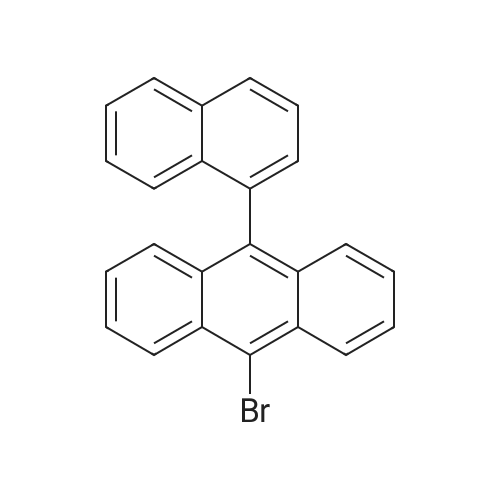
Synthesis of 9-bromo-10-(naphthalen-1-yl)anthracene A mixture of 40 g(119 mmol) of 9,10-dibromoanthracene, 20.5 g(119 mmol) of naphthalen-1-ylboronic acid, 1.38 g(1.2 mmol) of Pd(PPh3)4, 120 ml of 2M Na2CO3, 200 ml of EtOH and 600 ml toluene was degassed and placed under nitrogen, and then heated at 100° C. for 12 h. After finishing the reaction, the mixture was allowed to cool to room temperature. The organic layer was extracted with ethyl acetate and water, dried with anhydrous magnesium sulfate, the solvent was removed and the residue was purified by column chromatography on silica to give product(19.2 g, 50 mmol, 42%) as a yellow solid.
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In tetrahydrofuran; water at 80℃; for 8h; Inert atmosphere;
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In tetrahydrofuran; water
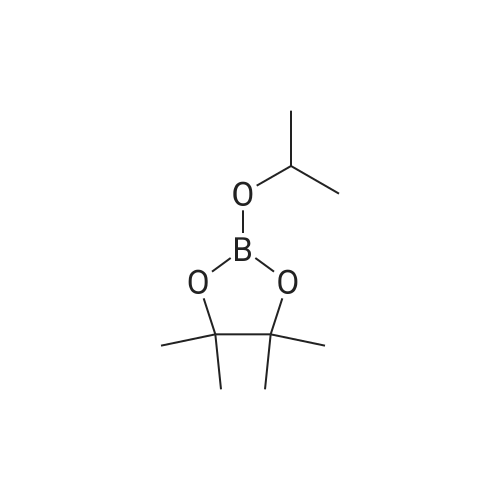
Under an argon atmosphere, 9,10-dibromoanthracene (336 mg, 1.0 mmol) was dissolved in THF (5 mL)It was cooled to -78 C. To this was added a hexane solution of butyllithium (2.78 M, 0.79 mL, 2.2 mmol). The mixture was stirred at the same temperature for 2 hours and then 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaboralone (0.60 mL, 3.0 mmol) was added.The reaction solution was warmed to room temperature and stirred for 15 hours, and then saturated aqueous ammonium chloride solution (5 mL) and chloroform (5 mL) were added. After separating the organic layer, the aqueous layer was extracted twice with chloroform and the organic layer was dried with sodium sulfate. After removing the drying agent by filtration, the low boiling point fraction was removed under reduced pressure from the filtrate to obtain a crude product. The crude product was purified by silica gel chromatography (hexane / chloroform = 80: 20 to 0: 100) to give 9,10-bis (4,4,5,5-tetramethyl-1,3-dioxaborolane- Yl) anthracene as a white solid (327 mg, 0.76 mmol, 76%).
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; at 80℃; for 72h;
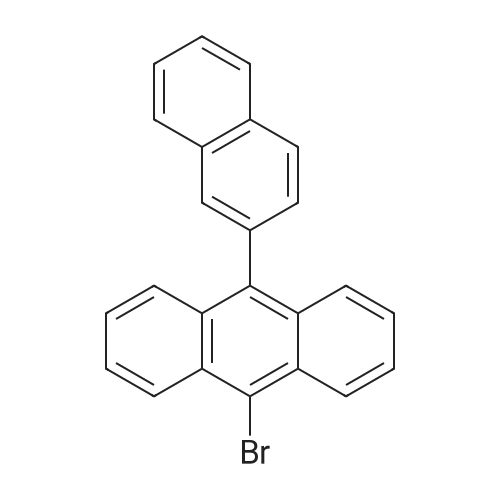
9-Bromo-10-(naphthalen-2-yl)anthracene (Compound 1) A mixture of 9,10-dibromoanthracene (16 g, 47.6 mmol), 2-naphthalenyl boronic acid (3.4 g, 19.8 mmol), Pd(PPh3)4 (1.0 g, 0.86 mmol) and sodium carbonate (8.4 g, 79 mmol) in tetrahydrofuran (THF)/water (200 ml/40 ml) was degassed and heated at about 80 C. for about 3 days. After being cooled to room temperature, the mixture was filtered, and the filtrate was extracted with dichloromethane (DCM) (250 ml), then washed with brine. The organic phase was collected and dried over Na2SO4, loaded on silica gel, purified by flash column (hexanes) to give a yellow solid (5.7 g, in 74% yield).
64%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In toluene; for 24h;Inert atmosphere; Reflux;
3.36 g (10 mmol) of 9,10-dibromoanthracene and 1.72 g (10 mmol) of Intermediate-1 were introduced under nitrogen and dissolved in 40 ml of toluene0.58 g (0.5 mmol) of Pd (PPh3) 4 and 15 ml (30 mmol) of 2M K2CO3 were added, respectively, and refluxed for 24 hours.After completion of the reaction, the temperature of the reaction mixture was cooled to room temperature, 200 ml of MC and 200 ml of H2O were added to extract the MC layer,Dried over MgSO4, concentrated, and then subjected to column chromatography with Hex: EA = 4: 1 to obtain Intermediate-7 2.45 (64%).
61%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; toluene; at 90℃; for 24h;Inert atmosphere;
Synthesis of 9-bromo-10-(naphthalen-2-yl)anthracene A mixture of 15 g (44.6 mmol) of 9,10-dibromoanthracene, 7.7 g (44.6 mmol) of naphthalen-2-ylboronic acid, 0.52 g (0.446 mmol) of Tetrakis(triphenylphosphine)Palladium, 33 ml of 2M Na2CO3, 60 ml of EtOH and 150 ml toluene was degassed and placed under nitrogen, and then heated at 90 C. for 24 h. After the reaction finish, the mixture was allowed to cool to room temperature. The organic layer was extracted with ethyl acetate and water, dried with anhydrous magnesium sulfate, the solvent was removed and the residue was purified by column chromatography on silica (hexane-dichloromethane) to give product 10.4 g (61%) as a yellow solid.
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; water; at 80℃; for 3h;
Synthesis (C1-1); 2.1 g of commercially available 2-naphthaleneboronic acid and 5 g of 9,10-dibromoanthracene were dissolved in 50 ml of dimethoxyethane and were heated to 80 C. 50 ml of distilled water and 10 g of sodium carbonate were added thereto. Also, 0.4 g of tetrakis-triphenylphosphine palladium(0) was added thereto.After 3 hours, extraction with toluene was carried out in a separatory funnel and purification using silica gel (SiO2 500 g) was carried out.As a result, 3 g of pale yellow crystals (9-bromo-10-naphthalen-2-yl-anthracene) was obtained.
3 g
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,2-dimethoxyethane; water; at 80℃; for 3h;
2 - 5 g of 9, 10 - 50 ml of dimethoxyethane [jiburomoantorasen[jiburomoantorasen]naphthalene boron acid 2.1 g of commercially available and dissolved, was heated to 80 C. 50 ml distilled water and sodium carbonate 10 g was placed therein. Further thereto was 0.4 g (0) tetrakistriphenylphosphine. 3 hours after the separatory funnel by extraction with toluene, silica (SiO2 500 g) was purified. Therefore, yellowish white crystals (9 - bromo-naphthalene - yl -10 - -2 - anthracene) 3 g was obtained as thin.
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; for 1.5h;Inert atmosphere; Reflux;
In a 1000ml three-necked flask, with mechanical stirring, Ar gas protection, 11.7g of 9,10-dibromoanthracene (molecular weight(0.35, 0.035 mol), 8.7 g of 4- (1-naphthyl) benzeneboronic acid (molecular weight: 248,0.035 mol), and Pd (PPh3)1.8 g (molecular weight 1154, 0.001556 mol), 120 ml (2M) of sodium carbonate aqueous solution, 250 ml of toluene and 150 ml of ethanol. The mixture was stirred and refluxed, and the reaction was monitored by TLC. The reaction was complete after 1.5 hrs of reaction. Cooled, separated, evaporated to dryness, the product was isolated by column chromatography, drenchedLotion with 1: 5 ethyl acetate: petroleum ether gave 15.8 g of a white solid9- (4- (1-naphthyl) phenyl) -10-bromoanthracene (AN1), product molecular weight 458, purity 98.7%, yield: 97.5%.
9,10-Dibromoanthracene (10 g, 0.03 mol), tetrakis(triphenylphosphine)palladium(0) (1.72 g, 1.49 mmol) and <strong>[870774-25-7]4-(naphthalene-1-yl)phenylboronic acid</strong> (7.4 g, 0.03 mol) were dissolved in THF (350 mL) in a double-necked flask and stirred for 30 min. Then, potassium carbonate (2 M, 200 mL) was added dropwise over 20 min. The mixture was degassed and refluxed overnight at 80 C under a nitrogen atmosphere. After being cooled, the solvent was evaporated under vacuum, and the product was extracted with ethyl acetate and water. The ethyl acetate solution was washed with water and dried with MgSO4. Evaporation of the solvent, followed by column chromatography using n-hexane on a silica gel, yielded a white yellow product
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,2-dimethoxyethane; water; for 20h;Inert atmosphere; Reflux;
To a magnetically stirred solution of 9, 10-dibromoanthracene (5.0 g; 0.015 mole) in ethylene glycol dimethyl ether (100 ml), tetrakis(triphenyl phosphine)palladium (1.9 g; 0.0016 mole) was added followed by 1-thianthrenyboronic acid (8.5 g; 0.033 mole). Potassium carbonate (12.4 g; 0.090 mole) in water (50 ml) was then added and the reaction mixture was refluxed under nitrogen atmosphere for 20 hours. After 15 minutes, the reaction mixture became yellow green in colour. The solvent was removed under reduced pressure and the residue was dissolved in dichloromethane and extracted with dilute acid. (The layers were not easily separable without the addition of acid). The organic phase was washed with water, dried over anhydrous magnesium sulphate and the solvent filtered through a pad of silica gel. After the removal of the solvent, methanol was added to the residue and stirred at room temperature overnight to give a greenish black solid which was dried under vacuum at 80C. Yield 6.0 g TLC examination showed a single product which was purified by sublimation to give a dark yellow solid that exhibited intense yellow fluorescence under UV. It was then further purified by double sublimation. The first sublimation gave 2.1 g of the product and the second sublimation gave 1.45 g of the product. M. p 381 C (DSC, onset), Tg 149 C Elemental analysis: Found: C 75.34; H 3.88, and S 21.44. C38H22S4, requires: C 75.21; H 3.65, and S 21.14 %. UV (CH2CI2): max (s/Nf 1), 259(133,155), 342(4107), 359(8929), 379(15,119) and 400(14,940). UV (Thin film): lambdaomega3chi (Abs): 196(1.47), 266(1.45), 364(0.16), 384(0.245) and 406(0.25), Film thickness: -60 nm. FL(CH2Cl2) lambdaomega3chi (em): 431, excitation wavelength: 350 nm. FL(Powder) ) max (em): 442, 465(sh), 508 and 540(sh). FL(Thin film) max (em): 422(sh), 439 and 500(sh). CV (CH2CI2): electrolyte: Tetrabutylammonium tetrafluorob orate (100 mM), analyte (ImM). Optical band gap: 2.9 ev; HOMO: -6.0 eV and LUMO: -3.1 eV. TGA/ C (% weight loss): 400 (1) and 433(5).
With palladium diacetate; potassium carbonate; trimethylpyruvic acid; tricyclohexylphosphine tetrafluoroborate In N,N-dimethyl-formamide at 150℃; for 0.0833333h; Microwave irradiation;
4.1.1. General procedure for MW-assisted direct arylation reaction.
General procedure: The MW vessel was charged with 9,10-dibromoanthracene1 (150 mg, 0.45 mmol), Pd(OAc)2 (6 mol %, 6 mg, 0.027 mmol),PCy3HBF4 (12 mol %, 20 mg, 0.054 mmol), PivOH (0.3 equiv, 14 mg,0.135 mmol), K2CO3 (1.5 equiv, 93 mg, 0.675 mmol), DMF (4 ml) andthe proper thienyl derivative (0.99 mmol for 2b,c and 3b,c;2.25 mmol in the case of 2a and 3a). The vessel was capped and themixture was irradiated with MW fixing the temperature at 150 Cfor 5 min. Then the solvent was removed under vacuum and thecrudewas dissolved in CH2Cl2,washed withwater and extracted forthree times. The combined organic phases were dried over Na2SO4and the solvent evaporated in vacuum and the so obtained solid crude was purified by flash chromatography on CombiFlash (SiO2,n-hexane with increasing amounts of DCM).
With potassium carbonate; copper(II) oxide; In N,N-dimethyl-formamide; at 100℃; for 24h;
The molar ratio of 9,10-dibromoanthracene: 1H-1,2,4-triazole: potassium carbonate: copper oxide is 2: 10-15: 30: 1In the magnet,A 50 mL three-necked round bottom flask equipped with a reflux condenser and a thermometer was charged with CuO (0.0398 mg, 0.5 mmol)Potassium carbonate (2.0731 g, 15 mmol),<strong>[288-36-8]Triazole</strong> (0.345 mg, 5 mmol),9,10-dibromoanthracene (0.3360 g, 1 mmol),20 mL DMF.Start stirring at 100 oC,Reaction for 24 hours.After the reaction,The reaction solution was cooled to room temperature,Filtration, the filtrate was added 100 mL of water,Precipitation precipitation, filtration,The filter cake was collected in a yield of 60%.
60%
With potassium carbonate; copper(II) oxide; In N,N-dimethyl-formamide; at 100℃; for 24h;
The molar ratio of 9,10-dibromoanthracene: 1H-1,2,4-triazole: potassium carbonate: copper oxide is 2: 10-15: 30: 1In the magnet,A 50 mL three-necked round bottom flask equipped with a reflux condenser and a thermometer was charged with CuO (0.0398 mg, 0.5 mmol)Potassium carbonate (2.0731 g, 15 mmol),<strong>[288-36-8]Triazole</strong> (0.345 mg, 5 mmol),9,10-dibromoanthracene (0.3360 g, 1 mmol),20 mL DMF.Start stirring at 100 oC,Reaction for 24 hours.After the reaction,The reaction solution was cooled to room temperature,filter,The filtrate was added to 100 mL of water,Precipitation precipitation,Suction filtration, collecting filter cake,Yield 60%.
With potassium carbonate; copper(II) oxide; In N,N-dimethyl-formamide; at 1500℃; for 18h;
in a polar solvent,DMF, 9,10-dibromoanthracene, triazole, potassium carbonate and copper oxide inthe preparation of the organic compound under heating; 9,10-dibromoanthracenewherein: three azole: potassium carbonate: copper oxide molar ratio of 2: 10:30: 1; The reaction temperature is1500 C, the reaction time of 18 hours
With potassium carbonate; copper(II) oxide; In N,N-dimethyl-formamide; at 150℃; for 18h;
The preparation method of 1- [9- (1H-1,2,4-triazol-1-yl) anthracene-10-yl] -1H-1,2,4-triazole is characterized by The organic compounds are prepared under heating conditions using 9,10-dibromoanthracene, triazole, potassium carbonate and copper oxide in a DMF polar solvent using a "one-pot" process, wherein 9,10-dibromoanthracene: <strong>[288-36-8]Triazole</strong>: potassium carbonate: copper oxide molar ratio of 2: 10: 30: 1;
9,10-bis(9-phenyl-9H-carbazol-2-yl)anthracene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
40%
With tetrakis(triphenylphosphine) palladium(0); In toluene; at 110℃; for 2h;Inert atmosphere; Alkaline conditions;
9,10-dibromoanthracene (1.0 g, 2.6 mmol), 2-boro-9-phenyl-9H-carbazole(2.5 g, 6.5 mmol), Pd(PPh3)4 (0.3 g, 0.2 mmol) were added to 25 mL ofanhydrous toluene under N2 atmosphere and stirred. 12 mL of tetraethylammoniumhydroxide (20 wt %) was added to the mixture andstirred for reflux at 110 C. After 2 h of reaction, the crude solution wasfiltered and the solvent of residue was vaporized. (0.4 g, Yield: 40%) 1HNMR (300 MHz, THF): delta(ppm) 8.42-8.38 (t, 1H), 8.30-8.27 (d, 1H),7.70-7.60 (m, 4H), 7.57-7.30 (m, 8H), 7.22-7.26 (q, 2H). HRMS (FABMS,m/z): calcd. for C50H32N2, 660.26; found, 660.2578 [M]+. Anal.calcd for C50H32N2: C 90.88, H 4.88, N 4.24; found: C 90.58, H 4.67 N4.19%.
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; sodium hydroxide;Inert atmosphere; Reflux;
Put 9,10-dibromoindole, triphenylamine boric acid and 4-(1-phenyl-1-H-benzimidazol-2-)benzeneboronic acid in a three-necked flask, solvent 40 ml, and install a mechanical stir bar. Nitrogen gas for 10 minutes,The catalyst PdCl2 (dppf) was added under 0.25 mol% to 3 mol%, and the 2M alkali solution was 0.018 mol, and the mixture was heated under reflux for 5-7 hours.After the reaction, suction filtration, toluene washing, and ethanol washing.After recrystallization from xylene, a powder having a purity of 99% or more was obtained, and the product yield was 78%.
With tetrakis(triphenylphosphine) palladium(0); tetrabutylammomium bromide; potassium carbonate; In water; toluene; at 110℃; for 72h;Inert atmosphere;
(1) K2CO3 (14.8 g, 107 mmol) and 43 mL of water were added to a 1000 ml single-mouth round bottom with a magnetic stir bar.In the bottle, add a small amount of tetrabutylammonium bromide TBAB,The reactant 9,10-dibromoanthracene (6 g, 17.9 mmol),The reactant <strong>[85199-06-0]2,5-dimethylphenylboronic acid</strong> (6.7 g, 44.7 mmol). Further, 130 mL of a toluene solution (V toluene: V water = 3:1) was added, and a catalyst of tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) (0.6 g, 0.52 mmol) was added.Under the protection of oxygen-free inert gas,The reaction was carried out at 110 C for 72 h. The reaction mixture was a pale yellow solid solution. After the reaction was cooled to room temperature, the organic phase was extracted with methylene chloride and water. The crude product was separated and purified by column chromatography, and the eluent was petroleum ether. After spin-drying, 6.2 g of Compound 1 of the formula was obtained, and the yield of Compound 1 was 89%.
diethyl 4,4'-(anthracene-9,10-diyl)dibenzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65%
With palladium diacetate; caesium carbonate; triphenylphosphine; In N,N-dimethyl-formamide; at 53.84℃; for 24h;Inert atmosphere;
To anoven-dried 50 ml round-bottomed flask (RBF) were added9,10-dibromoanthracene (0.900 g, 2.68 mmol), 4-ethoxyphenylboronicacid (2.08 g, 10.7 mmol), palladium(II) acetate(60 mg, 0.27 mmol), triphenylphosphane (141 mg, 0.54 mmol),and caesium carbonate (3.49 g, 10.7 mmol). The flask waspurged with N2 three times, followed by the addition of DMF(10 ml). The reaction was purged another three cycles,followed by heating under reflux under positive N2 pressure at372 K for 24 h. Afterwards, the crude reaction mixture wasvacuum filtered over Celite while still hot, followed by washingthe filter with hot ethyl acetate (3 x 50 ml) until the washingwas only barely fluorescent. The filter was then washed withhot dichloromethane (6 x 25 ml) until the washing was onlybarely fluorescent. The dichloromethane layer was thenevaporated in vacuo to give the product as a beige solidconsistent with data reported in the literature (yield 826 mg,65%) (Young, 2015). 1HNMR (400 MHz, CDCl3): delta 8.30 (d, J=7.3 Hz, 4H), 7.63 (dd, J = 6.8, 3.2 Hz, 4H), 7.58 (d, J = 7.4 Hz,4H), 7.35 (dd, J = 6.9, 3.1 Hz, 4H), 4.49 (q, J = 7.1 Hz, 4H), 1.48(t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3): delta 1667, 144.0,136.5, 131.5, 130.0, 129.8, 129.6, 126.7, 125.6, 61.3, 14.6.
With potassium hydroxide semihydrate; phosphorus; water In dimethyl sulfoxide at 100℃; for 3h; Inert atmosphere;
General Procedure for the reduction of haloanthracenes 1a-c
General procedure: The mixture of haloanthracene 1a-c (2.0 mmol), red phosphorus (18.7 mg-atom), KOH·0.5H2O (29.0 mmol), H2O (11.0 mmol) in DMSO (48 mL) was stirred under Ar atmosphere in 3 h at 100oC. After the completion of the reaction the mixture was cooled, it was then filtered to get rid of any solid. DMSO was distilled in vacuum, the solid residue was treated in water (3×8 mL), the solid filtered off and dried on the air to get pure 9,10-dihydroanthracene (2a).
4-(2,4,6-tricyanophenyl)benzeneboronic acid[ No CAS ]
C53H30N4[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
53%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; sodium hydroxide; for 4h;Inert atmosphere; Reflux;
Put 9,10-dibromoanthracene in a three-necked bottle.6,9-diphenyl-9H-carbazolyl-3-boronic acid/4-(1,3,5-tricyanophenyl)-benzeneboronic acid, solvent 50 ml, equipped with a mechanical stir bar,Under nitrogen for 10 min, the catalyst PdCl2 (dppf) 0.25 mol% to 3 mol%, 2M alkali solution 0.018 mol was added under the protection of nitrogen, and the mixture was heated under reflux for 4 hours.After the reaction, it was suction filtered, washed with toluene and washed with ethanol.After recrystallization from xylene, a powder L1 having a purity of 99% or more was obtained, and the product yield was 53%
cyclohexyl (E)-3-(9,10-dibromoanthracen-2-yl)acrylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
53%
With copper(II) 2-ethylhexanoate; chlorobis(ethylene)rhodium(I) dimer; silver sulfate In cyclohexane at 140℃; for 24h; Inert atmosphere;
2-1. General procedure for Rh-catalyzed non-directed alkenylation (Scheme 1 and 2)
General procedure: A screw-top glass tube was charged with aromatic compound 1 (1.25 mmol, 5.0 eq.), alkene 2 (0.25 mmol, 1.0 eq.), [RhCl(C2H2)2]2 (2.5 mol %), Cu(eh)2 (0.5 mmol, 2.0 eq.), and Ag2SO4 (0.5mmol, 2.0 eq.). Cyclohexane (3.0 mL) was added via syringe, and the resulting mixture was stirred at 140 °C for 24 h. After cooling to room temperature, the reaction mixture was extracted with EtOAc several times. The combined organic layers was washed with H2O containing ethylenediamine (ca. 1.0 mL), dried over Na2SO4, and concentrated in vacuo. The residue was subjected to silica gel column chromatography to give the corresponding product 3.
With n-butyllithium; In tetrahydrofuran; at -78 - 20℃; for 3h;
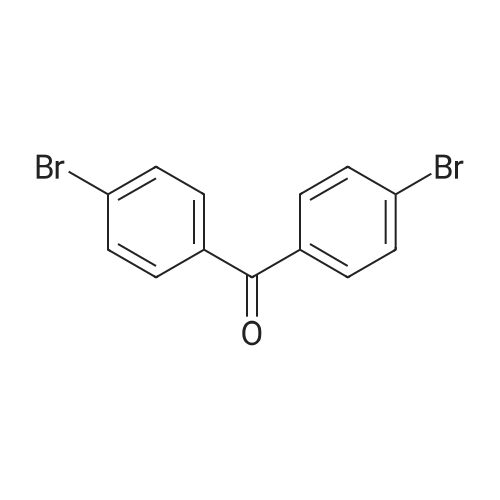
To the solution of compound S3 (3.34 g, 10.00 mmol) in 500 mL ofdegassed anhydrous THF was added n-BuLi solution (2.00 M, 12.00 mL, 2.40 eq) dropwiseat -78 oC, and the reaction mixture was allowed to stir in at this temperature for 2 hour,until a THF solution (100 mL) of 4,4?-dibromobenzophenone (compound S1, 4.05 g, 11.98mmol, 1.20 eq) was added. The reaction mixture was slowly warmed to room temperature,and stirred for an additional 1 hour at room temperature, until it was quenched by 60 ml 2.0 M HCl and keep stirring in air overnight. After extraction by ethyl acetate (EA) (3 ×60 ml), the organic phase was collected and dried over Na2SO4. The organic solvent was then removed by rotary evaporation and the residue was dried under vacuum and purifiedby column chromatography (hexanes/DCM = 4:1 to 1:1) to afford compound S4 (4.32 g,84.05%) as a white solid. 1H NMR (500 MHz, CD2Cl2): delta = 8.10 (d, J = 7.8 Hz, 4H), 7.43- 7.39 (m, 8H), 7.35 (m, 4H), 7.17 (d, J = 3.8 Hz, 8H), 7.11 - 7.08 (d, J = 3.9 Hz, 8H). 13CNMR (126 MHz, CD2Cl2): delta = 185.33, 143.02, 141.58, 138.90, 133.03, 131.83, 130.93,130.75, 129.10, 127.65, 126.41, 121.39 ppm. HR-MS (APCI): m/z = 514.9639 [M+H]+,calcd. for C27H17Br2O, 514.9639.
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In water; toluene; for 24h;Reflux; Inert atmosphere;
250mL round-bottomed flask, 9,10-dibromo-anthracene (3.33g, 10mmol), M2 ( 4.6g, 10mmol),tetrakis(triphenylphosphine)palladium (240mg, 0.21mmol), potassium carbonate (13.8 g, 100 mmol) was dissolved in 100mL toluene and 50mL aqueous solution under nitrogen atmosphere at reflux for 90 C for 24 hours.Liquid separation was extracted with dichloromethane, and concentrated to give the crude product purified by column chromatography (petroleum ether: dichloromethane = 1: 1) to give a white solid (2.70 g of, yield: 46%).
9,10-Bis{2-[4-(N,N-diphenylamino)phenyl]ethynyl}anthracene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1.45 g
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; triphenylphosphine; In N,N-dimethyl-formamide; at 80℃; for 0.5h;Inert atmosphere;
This embodiment discloses a conjugated alkynyl fluorene derivative, and the synthesis method thereof is as follows:Weigh 9,10-dibromoindole (1.01 g, 3 mmol),Cuprous iodide (28.5 mg, 0.15 mmol),Triphenylphosphine (39.3 mg, 0.15 mmol),Di(triphenylphosphine)palladium dichloride (21.1 mg, 0.01 mmol),Add to a 100 ml three-necked flask equipped with a magnet and a thermometer.20 ml of DMF and 15 ml of TEA were added to dissolve.4-Ethynyltriphenylamine (2.02 g, 7.5 mmol) was weighed.Dissolve in 10 ml DMF and transfer to a constant pressure dropping funnel.After vacuuming under nitrogen for three times, the temperature was raised to 80 C at 5 C / min.A solution of 4-ethynyltriphenylamine in DMF was added dropwise after stirring for half an hour.TCL monitors the 9,10-dibromofluorene raw material and stops heating after the reaction is completed.The reaction solution was cooled to room temperature and solid precipitated, and dried by filtration.The crude product was recrystallized from chloroform to give a yellow powder(1.45g, conversion rate 68%),
9,10-bis (4-bromo-5-methylthien-2-yl)anthracene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
63.3%
Dissolve 9,10-dibromoanthracene (1.0g) and Pd (PPh3) 4 (0.05g) in 150.0mL of THF and stir for 20min, then add <strong>[154566-69-5]2-methyl-3-bromo-5-boronic acid thiophene</strong> (1.3g ) And 50.0 mL of a Na2CO3 solution with a concentration of 2.0 mol / L, and refluxed at 80 C for 12 hours to perform a coupling reaction. After the reaction is completed, the resulting system is cooled to room temperature, separated, and extracted with dichloromethane. The magnesium was dried, filtered with suction, and the filtrate was subjected to rotary evaporation to remove the solvent. The column chromatography was performed with petroleum ether as the eluent to obtain9,10-bis (4-bromo5-methylthien-2-yl) anthracene (1.0 g), recorded as BTA, yield: 63.3%.
63%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In tetrahydrofuran; water; for 12h;Inert atmosphere; Reflux;
Compound 3 was synthesized from 9,10-dibromoanthracene (2, 1.0g, 3.0mmol) and 3-bromo-2-methyl-5-thienylboronic acid (1, 2.0g, 10.0mmol) in THF (170.0mL containing 10% water) under N2 atmosphere with Pd (PPh3)4 (0.05g, 0.043mmol) and Na2CO3 (2.0molL-1, 60.0mL). After 12h of refluxing, the product was extracted with dichloromethane. After the organic layer was dried with MgSO4, filtrated and evaporated, the crude product was purified through recrystallization in CH2Cl2. The obtained compound 3 (1.0g, 1.9mmol) was a yellow solid with a yield of 63%. Its m. p. was 275-276C with 1H NMR (400MHz, CDCl3): delta 7.94 (m, 4H), 7.46 (m, 4H), 7.02 (s, 2H), 2.56 (s, 6H); 13C NMR (100MHz, CDCl3): delta 14.99, 109.14, 126.10, 126.63, 129.67, 131.42, 131.96, 136.00, 136.26; ESI-MS (m/z): [M+H+]+ of (C24H17Br2S2+): 528.9 (calcd.), 529.1 (found).
9,10-bis{(E)-2-[4-(trimethylsilyl)phenyl]ethenyl}anthracene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
With potassium phosphate; palladium diacetate; In N,N-dimethyl acetamide; at 140℃; for 6h;Inert atmosphere;
A mixture of 0.6 g (1.8 mmol) of 9,10-dibromoanthracene 4, 0.76 g (4.3 mmol) of (4-vinylphenyl)trimethylsilane (2), 1.06 g (5.0 mmol) of K3PO4, and 8.0 mg (0.036 mmol) of Pd(OAc)2 in 6 mL of DMA was stirred for 6 h at 140C. The mixture was cooled to room temperature, diluted with2 0 mL of water, and extracted with toluene (3 × 10 mL). The combined extracts were washed with water (3 × 10 mL) and dried over Na2SO4, the solvent was distilled off under reduced pressure, and the residue was subjected to column chromatography using toluene-hexane (1 : 5) as eluent. The product was recrystallized from cyclohexane-hexane (1 : 5) and dried at 80C under reduced pressure.
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In tetrahydrofuran; water at 100℃; for 24h; Inert atmosphere;
2
Add 9mmol of 2,4,6-trifluorophenylboronic acid, 4mmol of 9,10-dibromoanthracene, 0.4mmol of Pd(PPh3)4, 16mmol of K2CO3 into a 250mL two-necked flask, add a stirring magnet, and proceed The operation of vacuuming and changing nitrogen was repeated three times, keeping the reaction flask in a nitrogen atmosphere, adding 120ml of mixed solvent tetrahydrofuran (THF)/pure water (V/V=2:1), and then refluxing at 100°C for 24h; after the reaction was completed, it was cooled to At room temperature, the reaction solution was evaporated to remove the solvent, and then extracted with dichloromethane three times. The organic layer was dried with anhydrous MgSO4, filtered, and the solvent was removed by rotating. Eluent, rotary evaporation to remove the solvent to collect the product H2, yield: 74%,






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






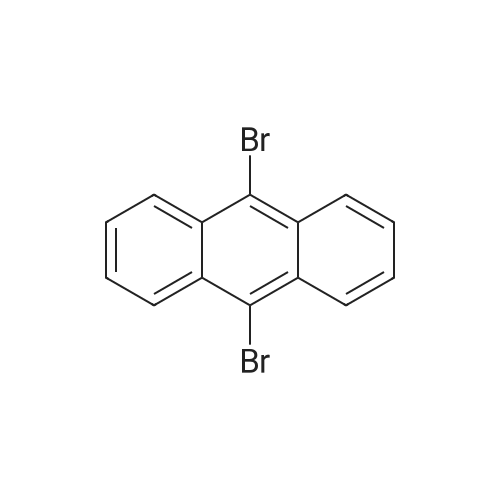

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping