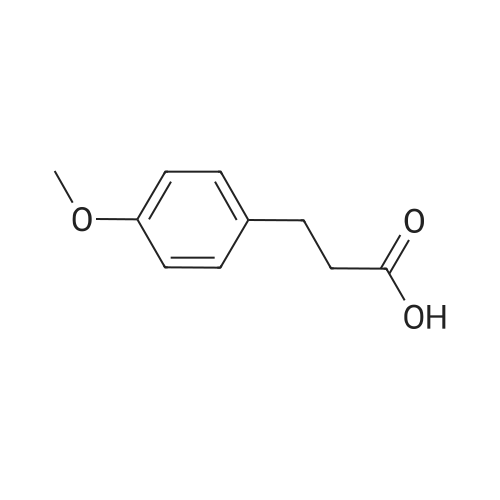
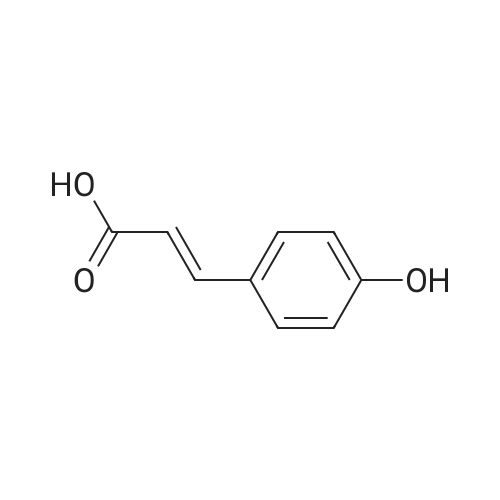
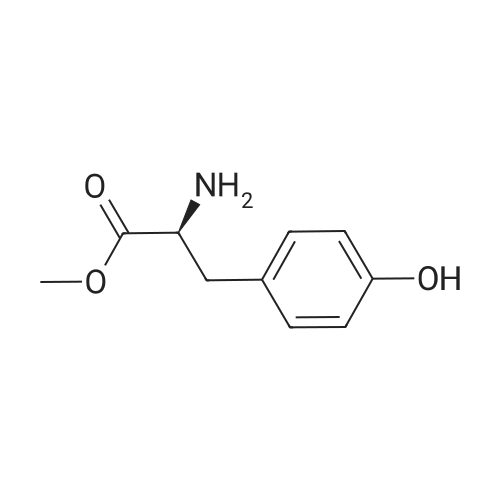
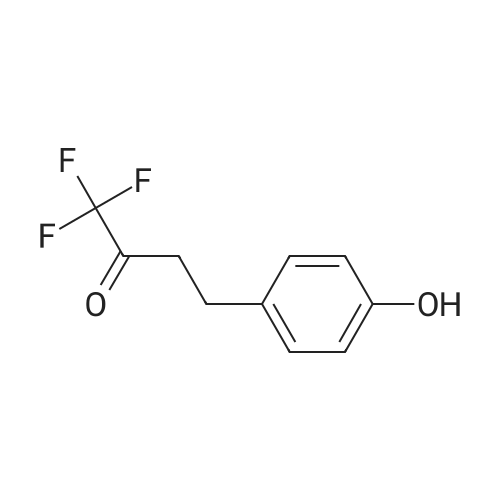
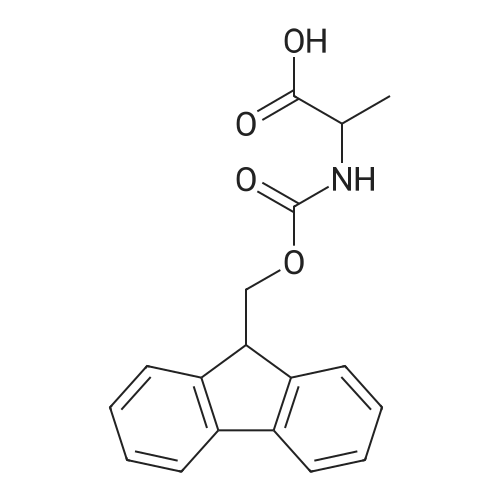
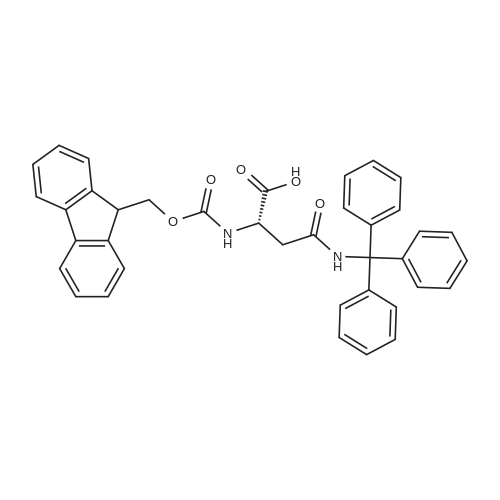
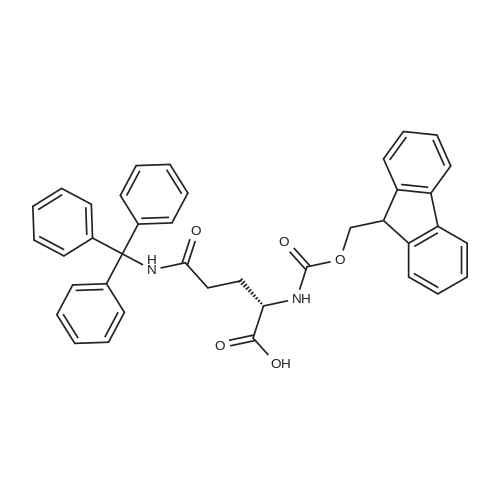
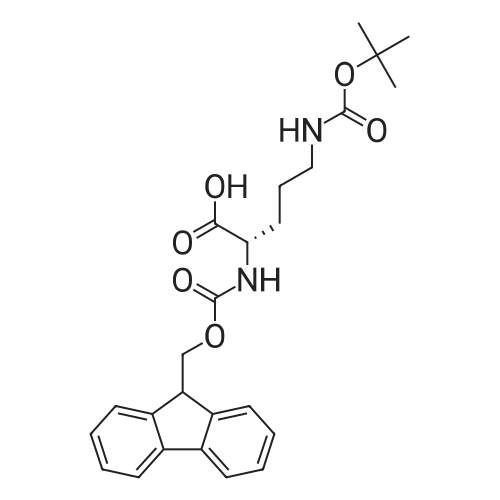
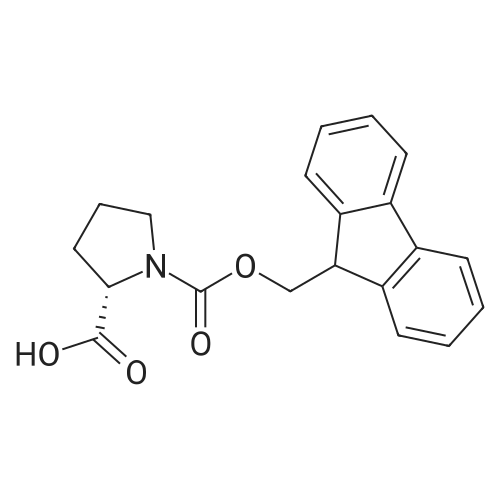
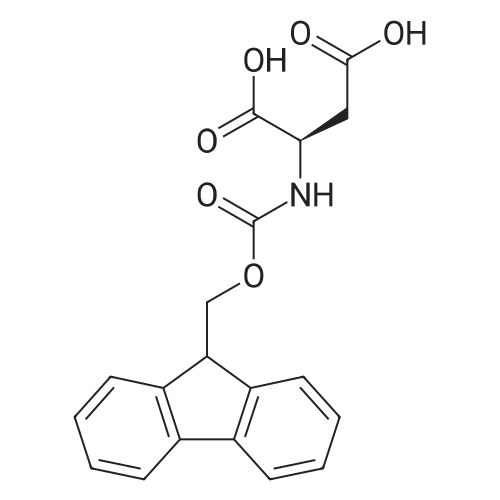
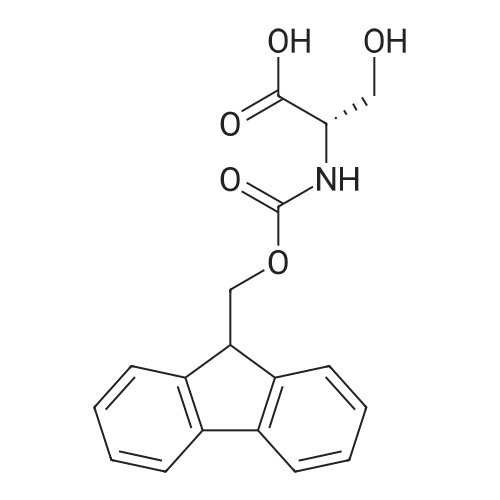
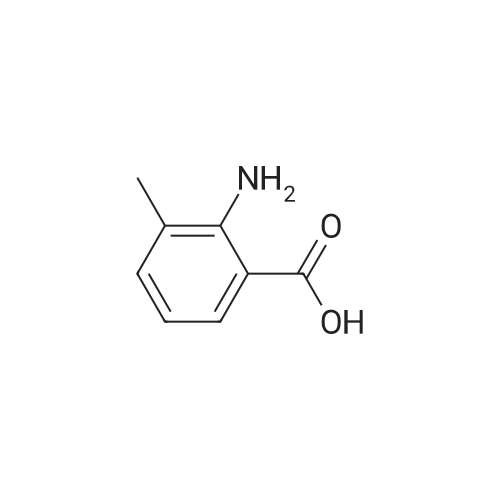
| 100% |
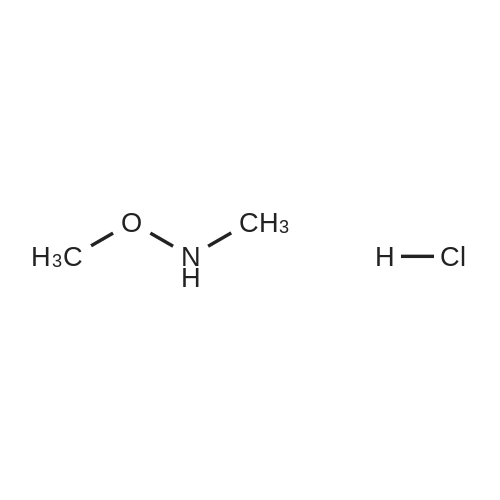
With chloro-trimethyl-silane at 25℃; for 24h; |
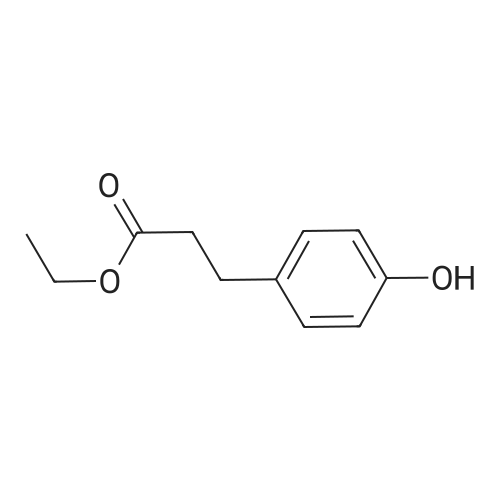
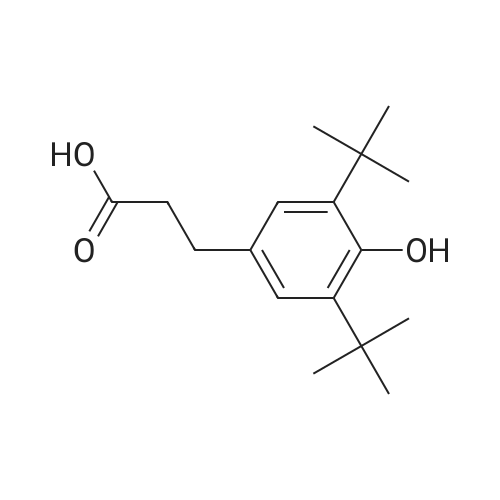
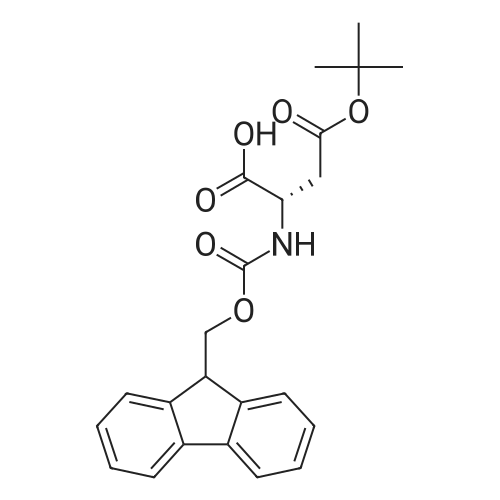
SI 4. General procedure for the preparation of esters 1a-d
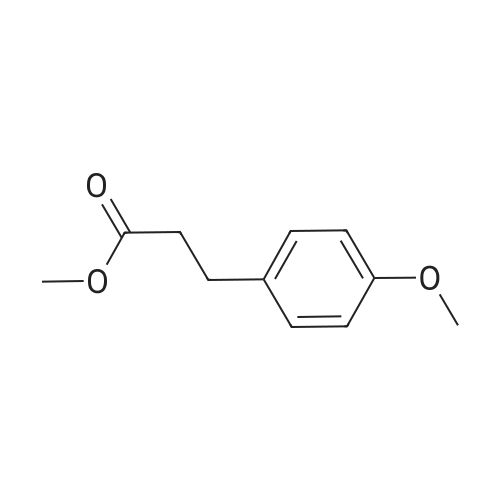
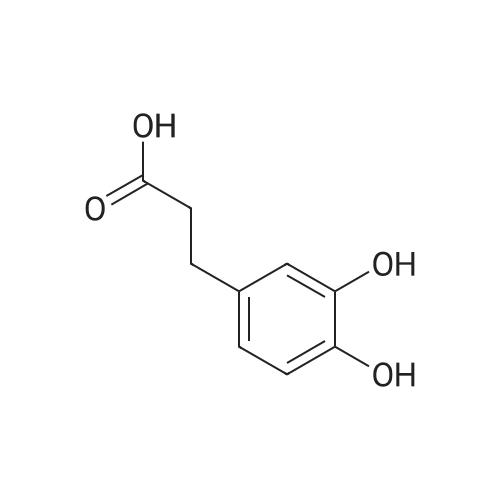
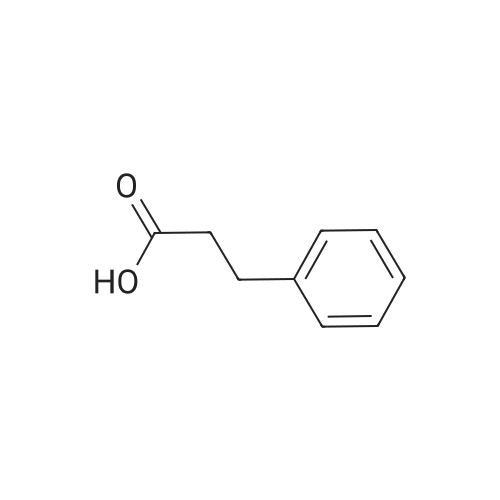
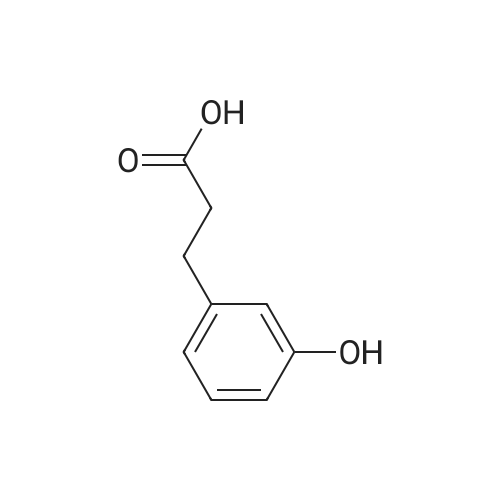
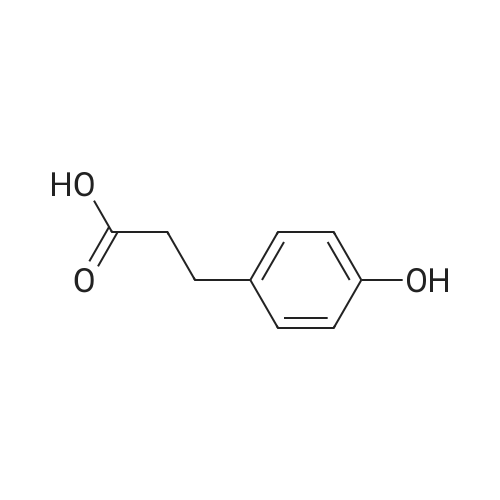
General procedure: To a suspension of 3(4-hydroxyphenyl)propanoic acid 1 (0.05 mmol) in TMCS (0.1 mmol), the appropriate alcohol (1.0 mL) was added and the reaction mixture was stirred at 25°C. After 24 h, the reaction was stopped and evaporated under reduced pressure to afford 1a-d in quantitative yield without the need of purification: 3-(4-Hydroxyphenyl)propionic acid methyl ester(1a) Oil; 1HNMR (400 MHz, CDCl3): δ H (ppm) = 2.66 (2H, m, CH2),3.05 (2H, m, CH2),3.65 (3H, m, OCH3), 6.73 (2H, m, Ph-H), 7.04(2H, m, Ph-H); 13C NMR (50 MHz, CDCl3): δ C (ppm) = 30.19 (CH2),35.23 (CH2), 50.99 (CH3),115.57 (2 CH), 129.54 (2 CH),131.59 (C), 155.71 (C), 172.74 (CO); MS (EI): m/z 252; Elemental analysis: calcd C, 66.65; H, 6.71; O, 26.64, found C, 66.64; H, 6.71;O, 26.63. |
| 100% |
With sulfuric acid for 2h; Reflux; |
|
| 100% |
With acetyl chloride In methanol at 0℃; for 1.5h; |
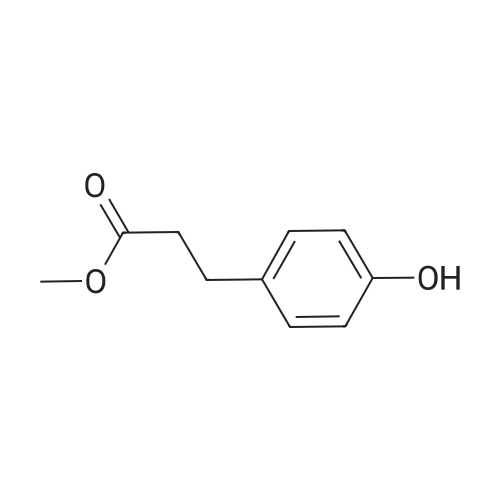
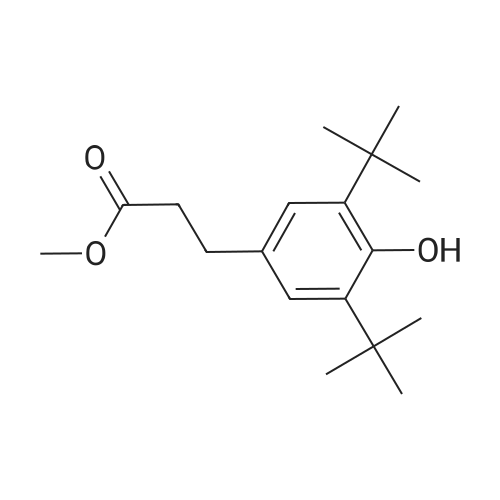
Methyl 3-(4-hydroxyphenyl)propanoate (38)
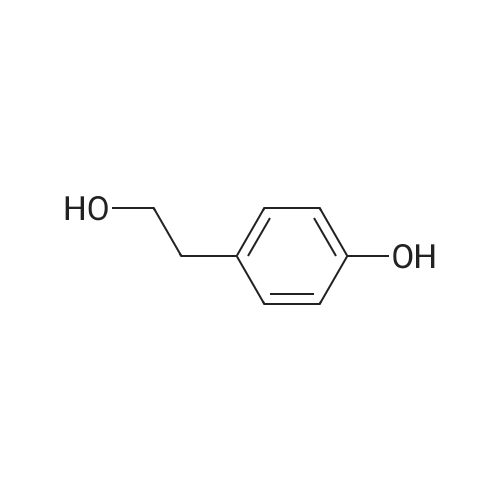
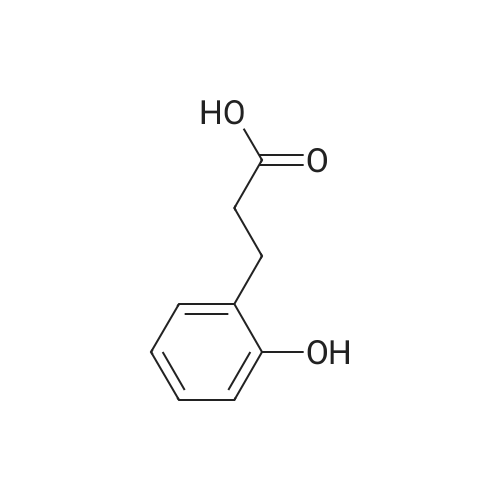
To a solution of3-(4-hydroxyphenyl)propanoic acid (2.50g, 15.04 mmol) in methanol(40 mL) were added dropwise 5.35 mL acetyl chloride (5.0eq,75.20 mmol) at 0 °C. The reaction mixture was stirred over 1.5 h at0 °C. After completion of the reaction, the mixture was concentratedin vacuo and ethyl acetate was added. The organic layer waswashed with NaHCO3 (3 x 50 mL) and concentrated in vacuo to givemethyl 3-(4-hydroxyphenyl)propanoate 38 (2.70 g, 100%) as acolorless oil. UPLC-MS: 1.62 min, 100%. 1H NMR (300 MHz, CD3OD) δ (ppm) 7.08-6.94 (m, 2H), 6.80-6.64 (m, 2H), 3.69-3.49 (m, 3H),2.87-2.70 (m, 2H), 2.60-2.45 (m, 2H). 13C NMR (75 MHz, CD3OD) δ (ppm) 174.4, 155.8, 131.8, 129.3, 115.3, 51.1, 36.1, 30.2. MS (ES) m/z181.7 (M + H)+. HRMS calculated for C10H12O3: 203.0679; found:203.0679 (M + Na)+. |
| 100% |
With sulfuric acid for 2h; Reflux; |
|
| 100% |
With sulfuric acid for 6h; Reflux; |
|
| 99% |
With hydrogen cation esterification; |
|
| 99% |
With hydrogenchloride In 1,4-dioxane at 20℃; for 3h; |
4 [355] Preparation Example 4: 3-(4-hydroxy-phenyl)-propionic acid methyl ester
[355] Preparation Example 4: 3-(4-hydroxy-phenyl)-propionic acid methyl ester[356] After 3-(4-hydroxy-phenyl)propionic acid (3 g, 18 mmol) was added with MeOH(6 mL), 4M HCl/dioxane solution (18 mL, 72 mmol) was added thereto, and themixture was stirred at room temperature for 3 hours. The reactant wasconcentrated, added with EtOAc, and then washed with NaCl aqueous solution. Theorganic layer was separated, dried with MgSO4, and filtered toobtain the title compound (3.26 g, 99%). [357] 1H-NMR (CDCl3) δ 7.06(2H, m), 6.75(2H, m),4.79(1H, brs, OH), 3.66(3H, s), 2.88(2H, t), 2.59(2H, t) |
| 99% |
With sulfuric acid In dichloromethane for 41h; Reflux; |
|
| 99.6% |
Stage #1: methanol With thionyl chloride at 5℃; for 0.5h;
Stage #2: 4-hydroxyphenylpropionic acid at 35℃; |
1.1
Add 70.00g methanol into the reaction flask, stir and cool to 5°C, add 15.00g (0.126mol) thionyl chloride dropwise at 5°C, and finish the dropwise addition at 0.5h, and react at 5°C for 0.5h.Add 20.00g (0.120mol) p-hydroxyphenylpropionic acid (II), raise the temperature to 35, control the temperature at 35 to react for 0.5-1.0h, TLC will detect the reaction is complete, control the temperature at 30-40 and distill under reduced pressure to obtain a light yellow oil 21.62g, purity 99.9%, 21.60g converted to methyl p-hydroxyphenylpropionate (III), molar yield 99.6% |
| 98% |
With sulfuric acid at 70℃; for 3h; |
4.61 g p-hydroxyphenpropionic acid, 50 ml methanol and 0.5 ml concentrated H2SO4 were refluxed at 70°C for 3 hours and cooled. Excessive acid was neutralized with saturated NaHCO3 solution. The neutralized solution was extracted with ethyl acetate for three times. The extract was dried, filtrated, evaporated and purified by column chromatograph (petroleum ether: ethyl acetate=2:1) to obtain 4.9 g white solid, yield 98%. 1H-NMR (400MHz, DMSO-d6, δ ppm) δ 2.5(2H, m), 2.7(2H, t), 3.567(3H, s), 6.6(2H, d, J=6.4Hz), 7.0(2H, d, J=8.4Hz), 9.0(1H, s). |
| 98% |
With sulfuric acid at 70℃; for 3h; |
Intermediate 10: Methyl p-hydroxyphenpropionate 4.61 g p-hydroxyphenpropionic acid, 50 ml methanol and 0.5 ml concentrated H2SO4 were refluxed at 70° C. for 3 hours and cooled. Excessive acid was neutralized with saturated NaHCO3 solution. The neutralized solution was extracted with ethyl acetate for three times. The extract was dried, filtrated, evaporated and purified by column chromatograph (petroleum ether:ethyl acetate=2:1) to obtain 4.9 g white solid, yield 98%. 1H-NMR (400 MHz, DMSO-d6, δ ppm) δ 2.5 (2H, m), 2.7 (2H, t), 3.567 (3H, s), 6.6 (2H, d, J=6.4 Hz), 7.0 (2H, d, J=8.4 Hz), 9.0 (1H, s). |
| 98% |
With sulfuric acid for 16h; Reflux; |
Typical procedure for synthesis of methyl esters
General procedure: According to a literature protocol.[1] Propanoic acid derivative (10 g) was dissolved inmethanol (80 ml) and H2SO4 (700μl, 6 mmol) added. The resulting solution was refluxed for16 h and then allowed to cool to room temperature. The reaction mixture was reduced todryness and re-dissolved in ethyl acetate (20 ml). The organic phase was washed withsaturated aqueous sodium hydrogen carbonate (10 ml), brine (10 ml) and dried over MgSO4.Removal of the solvent gave the desired methyl ester. (Note: column chromatography is oftennot required for the methyl ester starting materials). Methyl 3-(4-hydroxyphenyl)propanoate (1a) Synthesized according to general procedure A (98%); 1H NMR (500 MHz, d4-MeOH): δ 6.98(d, J = 8.4 Hz, 2H), 6.66 (d, J = 8.4 Hz, 2H), 3.60 (s, 3H), 2.78 (t, J = 7.6 Hz, 2H), 2.53 (t, J= 7.6 Hz, 2H); 13C NMR (125 MHz, d4-MeOH): 172.8, 155.6, 130.6, 129.1, 115.1, 51.2, 35.4,29.5; HRMS (EI) calculated for C10H12O3 [M]+: 180.078643; found: 180.078803. |
| 98.2% |
With sulfuric acid for 10h; Reflux; |
2.5 5) Preparation of methyl 3-(4-hydroxyphenyl)propanoate:
20 g of p-hydroxyphenylpropionic acid was added to 300 ml of anhydrous methanol and2 ml of concentrated sulfuric acid, heated to reflux for 10 h, the solvent was evaporated under reduced pressure, then dissolved with ethyl acetate and washed sequentially with sodium hydrogen carbonate and water.Polyester, organic layer dried over anhydrous magnesium sulfate,The solvent was evaporated to give 21.2 g (yield: 98.2%). |
| 97% |
With sulfuric acid Reflux; |
|
| 97% |
With sulfuric acid |
|
| 96% |
With thionyl chloride at 20℃; for 24h; |
|
| 95% |
With hydrogenchloride at 20℃; for 3h; |
|
| 95% |
With thionyl chloride at -40 - 20℃; for 12h; Cooling with acetonitrile-dry ice; |
|
| 95% |
With sulfuric acid at 60℃; for 6h; |
|
| 95% |
With sulfuric acid at 35℃; for 4h; |
|
| 95.3% |
With sulfuric acid for 6h; Reflux; |
1.1 1. Preparation of methylparaben:
150g of p-hydroxyphenylacetic acid, 400g of anhydrous methanol, 0.2ml of concentrated sulfuric acid, heated to reflux for 6 hours, after the reaction was completed, 3.0g of sodium carbonate was added to neutralize the sulfuric acid, then the methanol was evaporated under reduced pressure, 400g of toluene was added, and 500g of purified water was extracted and layered Then, an organic phase was obtained, and the organic phase was concentrated under reduced pressure to obtain 154.4 g of methylparaben, the purity detected by HPLC was 99.0%, and the yield was 95.3%. |
| 94% |
With sulfuric acid for 3h; Heating; |
|
| 93% |
With sulfuric acid In dichloromethane for 5h; Heating; |
|
| 92.7% |
With hydrogenchloride Heating; |
|
| 92% |
at 20℃; for 12h; |
|
| 92% |
With sulfuric acid at 20℃; for 16h; |
4.b
15 g (0.09 mol, 1 eq) of 3-(4-hydroxyphenyl)propanoic acid are dissolved in 50 ml of methanol and 4 drops of sulfuric acid are added. The reaction mixture is stirred for 16 hours at room temperature. The reaction is stopped by addition of 50 mL of saturated sodium hydrogen carbonate solution and then extracted with ethyl acetate. The organic phases are combined and dried over sodium sulfate. The solvents are evaporated off and the residue is then chromatographed on silica gel (70/30 heptane/ethyl acetate). 14.9 g of methyl 3-(4-hydroxyphenyl)propanoate are obtained. Yield = 92 % |
| 92% |
With sulfuric acid for 24h; Reflux; |
|
| 89% |
With acetyl chloride Heating; |
|
| 88% |
With sulfuric acid at 65℃; for 16h; Sealed tube; |
|
| 86% |
With toluene-4-sulfonic acid for 14h; Reflux; |
|
| 86% |
With thionyl chloride at 20℃; for 2h; |
Methyl 3-(4-hydroxyphenyl)propanoate (2).
3-(4-hydroxyphenyl)propanoic acid (1, 3.0 g, 18.1 mmol) was dissolved in methanol (50 mL), and SOCl2 (1.5 mL) was added. The solution was stirred at room temperature for 2 h. Then, the reaction mixture was concentrated in vacuo to afford 3.2 g (17.8 mmol) of crude product 2 as oil. The obtained crude product 2 was purified by silica gel column chromatography (dichloromethane). The process was monitored by TLC on silica gel plates (dichloromethane:ethyl acetate,4:1, Rf 0,79) with compound detection accomplished in an iodine chamber. The eluate was evaporated on a rotary evaporator to give compound 2 (2.8 g, 15.5 mmol, 86%) as an oil and with purity >98% (HPLC, method A, UV 220 nm). |
| 85% |
With sulfuric acid for 24h; Reflux; |
|
| 84% |
With boron trifluoride diethyl etherate at 20℃; for 8h; |
1 Methyl 3-(4-hydroxyphenyl)propanoate (A).
To a solution of 3-(4-hydroxyphenyl)propanoic acid (3 g, 18.1 mmol) in 50 mL MeOH was added ΒΡ3·Ε20 (0.3 mL). The mixture was stirred at rt for 8 h. The mixture was concentrated, and the residue was purified by flash chromatography (FC) (ethyl acetate (EtOAc) /hexane = 2/8) to give 2.72 g white solid A (yield: 84%): 1HNMR(400 MHz, CDC13) δ: 7.07(d, 2H, J= 8.4 Hz), 6.76(d, 2H, J= 8.4 Hz), 4.72(s, 1H), 3.68(s, 3H), 2.89(t, 2H, J= 7.6 Hz), 2.60(t, 2H, J= 7.6 Hz). HRMS (ESI) calculated for C10H13O3 (M+H+), 181.0865; found, 181.0889 |
| 84% |
With boron trifluoride diethyl etherate at 20℃; for 6h; |
1.1 Synthesis of compound 3 (methyl 3-(4-hydroxyphenyl)propionate):
Adding p-hydroxyphenylpropionic acid to a round bottom flask(3g, 18.1mmol) and 50mL of methanol,Subsequently, boron trifluoride diethyl ether (0.3 mL) was added dropwise to the mixed solution.After stirring at room temperature for 6 hours,The organic phase in the filtrate was removed under reduced pressure using a rotary evaporator using n-hexane/ethyl acetate (v/v, 2/8)Through the silica gel column separation,Collect target components,The solvent was removed under reduced pressure.2.72 g of a white solid were obtained (yield: 84%); |
| 83.5% |
With boron trifluoride diethyl etherate at 20℃; for 6h; |
|
| 70% |
Acidic conditions; Reflux; |
|
| 68% |
With hydrogenchloride for 12h; Heating / reflux; |
1.AV 32.A EXAMPLES EXAMPLE 1: Synthesis of compounds AV 32
AV 32 [ CNHISNOG] Mol. Wt.: 209.24 A. 0.8 gr 4-hydroxy cinnamic acid in 40 [ML METHANOL] and 10 drops HCI were [REFLUXED] for 12 hours. Workup as above gave 0.6 gr oil, 68% yield. NMR CDCl3 7.02, 6.75 (4H, Abq, J=8.6 Hz), 3.66 (3H, s), 2.86 (2H, t, J=7.4 [HZ),] 2.60 (2H, t, J=7. 4 Hz). B. 0.6 gr, 3.3 mM, ester from step A and 0.26 gr, 4.2 mM, ethanol amine were heated at 100 for 3 hours in an open vessel. Chromatography gave 0.3 gr recovered ester followed by amide. The viscous oil was triturated with acetone-methylene chloride and filtered to give 160 mg white solid, 23% yield, mp-102. NMR acetone d6 8.10 (1H, s, OH), 7. [03,] 6.74 (4H, Abq, J=8. [8 HZ),] 3.90 (1H, t, J=5.2 Hz, NH), 3.54 (2H, q, J=7.1 Hz), 3.28 (2H, t, J=7.1 [HZ),] 2.80 (2H, t, J=8.2 Hz), 2.41 (2H, t, J=8.2 Hz). |
| 68% |
With hydrogenchloride for 12h; Heating / reflux; |
1.A
0. 8g 4-hydroxy cinnamic acid in 40 ml methanol and 10 drops HCl were refluxed for 12 hours. Workup as above gave 0.6g oil, 68% yield. NMR CDC13 7.02, 6.75 (4H, Abq, J=8.6 Hz), 3.66 (3H, s), 2.86 (2H, t, J=7.4 Hz), 2.60 (2H, t, J=7. 4 Hz). |
| 65% |
With hydrogen cation |
|
| 62% |
With sulfuric acid for 24h; Reflux; |
General procedure for the synthesis of compounds 9-12 and 21-29
General procedure: After dissolving the carboxylic acid in the alcohol, three drops of H2SO4 95% were added to the solution and the mixture was refluxed for 24 h. The solvent was evaporated under reduced pressure and water was added to the crude mixture. The pH of the aqueous layer was adjusted to 7 adding drops of a saturated solution of NaHCO3 and brine was added in the mixture. The aqueous layer was extracted three times with ethyl acetate; the organic layer was dried over Na2SO4 and the solvent was evaporated under reduced pressure yielding the final compound. Further purification step was made when it was necessary. |
|
With sulfuric acid at 80℃; unter Druck; |
|
|
With sulfuric acid In 1,2-dichloro-ethane Heating; |
|
|
With hydrogenchloride Heating; |
|
|
With 3 A molecular sieve; sulfuric acid for 72h; Heating; |
|
|
With boron trifluoride for 0.0333333h; Heating; |
|
|
With Amberlyst 15 for 8h; Heating; |
|
|
With sulfuric acid Yield given; |
|
|
With thionyl chloride at 0 - 20℃; for 2h; |
A-1
[Example A-1]; Synthesis of methyl 3- (4-HYDROXYPHENYL) propionate (Intermediate 1); A solution obtained beforehand by adding thionyl chloride (18.3 ml, WAKO) dropwise to methanol (250 ml) and mixing the mixture under ice cooling was added dropwise with a solution of 3- (4-HYDROXYPHENYL) propionic acid (16. 6g, TCI) in methanol (50 ml) under ice cooling, stirred for 30 minutes, warmed to room temperature, and further stirred for 1.5 hours. The reaction mixture was concentrated under reduced pressure, and then extracted with diethyl ether (200 ml). The organic layer was washed successively with saturated aqueous sodium hydrogencarbonate, saturated aqueous ammonium chloride and saturated brine. The organic layer was dried, and then the solvent was evaporated under reduced pressure to obtain the title compound (Intermediate 1,17. 95 g). |
|
With hydrogenchloride In 1,4-dioxane |
B.67.1 Step 1: 3- (4-Hydroxyphenyl) propionic acid methyl ester.
A solution of HCI in dioxane (4.0 M, 7.4 mL) was added to a solution of 4- hydroxyphenylpropionic acid (15.0 g, 90.3 mmol) in MeOH (500 mL). The reaction mixture was stirred overnight and then evaporated. The residue was evaporated from benzene (2 x 50 mL) to provide the product as an oil, which was used without further purification. |
|
With hydrogenchloride In 1,4-dioxane |
B.67.1
A solution of HCl in dioxane (4.0 M, 7.4 mL) was added to a solution of 4-hydroxyphenylpropionic acid (15.0 g, 90.3 mmol) in MeOH (500 mL). The reaction mixture was stirred overnight and then evaporated. The residue was evaporated from benzene (2×50 mL) to provide the product as an oil, which was used without further purification. |
|
With sulfuric acid at 25℃; |
|
|
With thionyl chloride at 0 - 20℃; for 2h; |
1.c Reference Example 1; Synthesis of methyl 3-(4-hydroxyphenyl)Propionate (Intermediate 1) (Step c)
Reference Example 1 Synthesis of methyl 3-(4-hydroxyphenyl)Propionate (Intermediate 1) (Step c) A solution obtained beforehand by adding thionyl chloride (18.3 ml, WAKO) dropwise to methanol (250 ml) under ice cooling was added dropwise with a solution of 3-(4-hydroxyphenyl)propionic acid (16.6 g, TCI) in methanol (50 ml) under ice cooling, stirred for 30 minutes, then warmed to room temperature and further stirred for 1.5 hours.The reaction mixture was concentrated under reduced pressure and then extracted with diethyl ether (200 ml).The organic layer was washed successively with saturated aqueous sodium hydrogencarbonate, saturated aqueous ammonium chloride and saturated brine.The organic layer was dried, and then the solvent was evaporated under reduced pressure to obtain the title compound (Intermediate 1, 17.95 g). |
|
With hydrogenchloride In 1,4-dioxane |
1.1
A solution of HCl in dioxane (4.0 M, 7.4 mL) was added to a solution of 4- hydroxyphenylpropionic acid (15.0 g, 90.3 mmol) in MeOH (500 mL). The reaction mixture was stirred overnight and then evaporated. The residue was evaporated from benzene (2 x 50 mL) to provide the product as an oil, which was used without further purification. |
|
With hydrogenchloride |
|
|
With sulfuric acid |
|
|
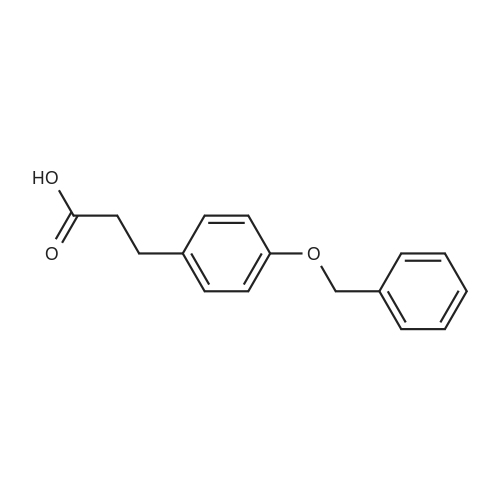
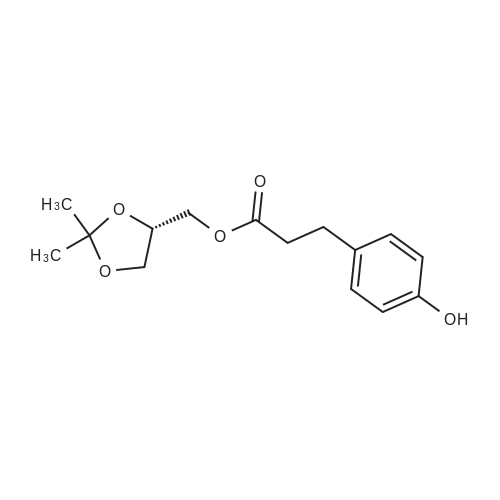
With chloro-trimethyl-silane at 25℃; for 24h; |
4.6 General procedure for the preparation of esters 2a-l
General procedure: To a suspension of acidic phenols 1a-d (0.05mmol) in TMCS (0.1mmol), the alcohol (1.0mL) was added and the reaction mixture was stirred at 25°C. After 24h, the reaction was stopped and evaporated under reduced pressure to afford 2a-l in quantitative yield without the need of purification: |
| 21.2 g |
With thionyl chloride at 0℃; Inert atmosphere; |
2
To a solution of 3-(4-hydroxyphenyl)propionic acid (20 mg, 120 mmol) in MeOH (300 mL) was added SOCl2 (28.6 g, 240 mmol) at 0° C. under nitrogen. The resulting mixture was stirred overnight then concentrated in vacuo. The residue was dissolved in EtOAc (50 mL) and washed with saturated aqueous NaHCO3 (2*50 mL). The organic layer was dried over Na2SO4 and concentrated in vacuo to yield Compound 1 as a red oil (21.2 g). A solution of Compound 1 (19 g, 106 mmol) in dry THF (200 mL) was slowly added to a suspension of LAH (6 g, 158 mmol) in dry THF (200 mL) at 0° C. under nitrogen. The mixture was allowed to reach room temperature and was then stirred for 2 hours. Then water (6 mL) and 15% aqueous NaOH (24 mL) were added. Additional water (6 mL) was added to quench the reaction. The solids were filtered and the filter cake was washed with EtOAc several times. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography (hexanes:EtOAc 8:1˜5:1˜2:1) to yield Compound 2 as a yellow oil (10 g). |
|
With sulfuric acid for 10h; Reflux; Inert atmosphere; |
Concentrated H2SO4 (40 μl, 0.83 mmol)was added to a solution of 3-(4-hydroxyphenyl)propionic acid (11) (2.76 g, 16.6 mmol) in MeOH (30mL), the resulting solution was heated to reflux and stirred for 10 h. The mixture was cooled to roomtemperature by removal of the oil bath, and the residual solvent was removed under reduced pressure.The crude residue was dissolved in EtOAc (40 mL), washed with saturated aqueous NaHCO3 (1 x 40mL) and the layers were separated. The organic layer was dried (MgSO4) and concentrated underreduced pressure to provide a clear, colorless oil that was dissolved in CH2Cl2 (55 mL) and cooled to 0°C. Imidazole (1.70 g, 24.9 mmol) was added, the solution stirred for 10 min, followed by a portion-wiseaddition of TBSCl (2.76 g, 18.3 mmol). The cooling bath was removed and the resulting heterogeneousmixture was stirred for 10 h. The mixture was diluted with H2O (50 mL), the layers were separated, andthe aqueous phase extracted with CH2Cl2 (3 x 50 mL). The combined organic extracts were washed withsaturated aqueous NaCl (50 mL), dried (MgSO4), and concentrated under reduced pressure to provide4.39 g of methyl 3-(4-((tert-butyldimethylsilyl)oxy)phenyl)propanoate as a clear, colorless crude oil.This crude residue was dissolved in CH2Cl2 ( 75 mL), cooled to -90 °C, and a solution of DIBAL-H (1.0M in hexanes, 16.6 mmol, 15 mL) was then added dropwise while ensuring that the internal reactiontemperature remained below -80 °C. The mixture was stirred for 1 h, then excess DIBAL-H wasquenched by the slow addition of MeOH (15 mL), while again being careful to ensure that the internaltemperature of the solution did not rise above -80 °C. The mixture was allowed to warm to roomtemperature by removal of the cooling bath, diluted with saturated aqueous Rochelle’s salt (70 mL), andstirred vigorously for 10 h. The layers were separated and the aqueous phase was extracted with CH2Cl2(3 x 50 mL). The combined organic extracts were washed with saturated aqueous NaCl (50 mL), dried(MgSO4), and concentrated under reduced pressure. The crude residue was purified by flash columnchromatography eluting with hexanes/EtOAc (7:1) to provide 12 in 76% yield (16.6 mmol scale, 3.36mg) over three steps as a clear colorless oil. 1H and 13C NMR data for 12 was consistent with literature reported values. |
| 365.8 g |
With sulfuric acid for 7h; Reflux; |
1.1 4-hydroxy-phenylpropionic acid methyl ester preparation
2L three-mouth bottle, is provided with a stirring, thermometer and a condenser. Adding 340.0g the hydroxy phenylpropanoic acid, 3.4g concentrated sulfuric acid and 1500 ml methanol, heating reflux for 7 hours. Cooling to room temperature, using NaHCO 3 solution to neutral, rotary evaporimeter desolventizing, by adding 600 ml of water and 600 ml toluene, liquid separation, the organic phase washed to neutral, and to obtain the product solvent 365.8g, GC purity: 98.4%. |
|
With sulfuric acid at 35℃; for 4h; |
1.1 Preparation of methyl 3-(4-hydroxyphenyl)propanoate
3-(4-Hydroxyphenyl)propionic acid (8 g, 48.14 mmol) was dissolved in methanol (40 mL),catalyzed by thedropwise addition of concentrated sulfuric acid (0.4 mL), reacted at 35° C. for 4 h, and concentrated under reduced pressure to remove some of the methanol. was added 10mL of water, adjusted to pH 6 with saturated sodium bicarbonate,with 20mL ethyl acetate three times, washed three times with 20mL saturated brine, dried over anhydrous sodium sulfate under reduced pressure to remove acetic acidethyl ester to give 3- (4- Hydroxyphenyl)propionic acid methyl ester. |
| 11 g |
With sulfuric acid for 16h; Reflux; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping