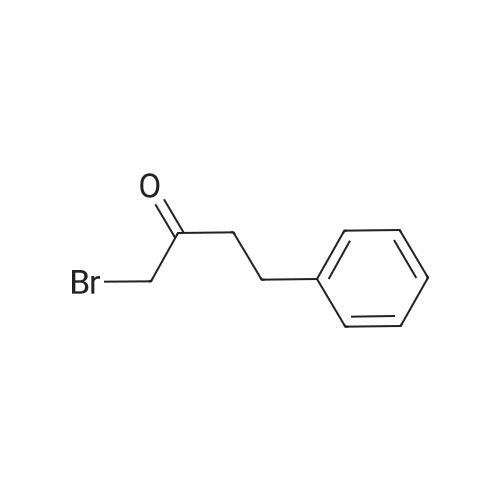
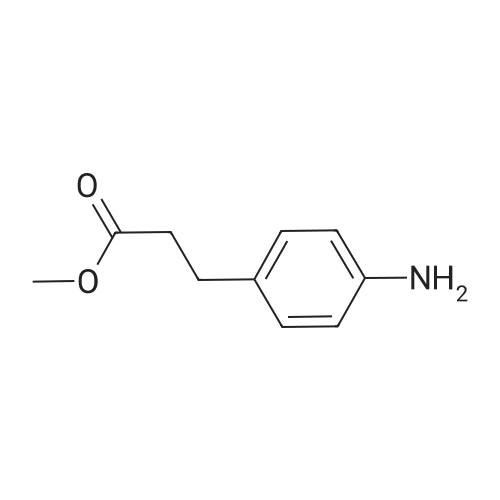
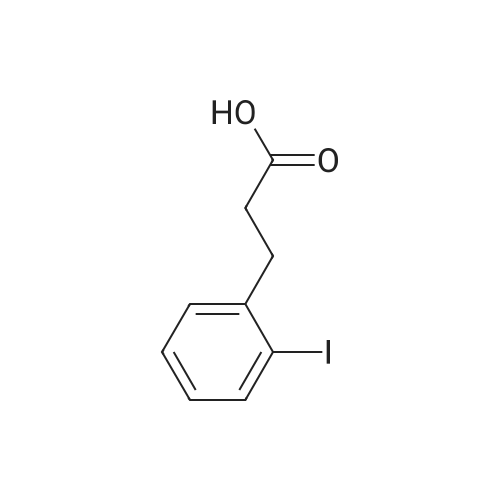
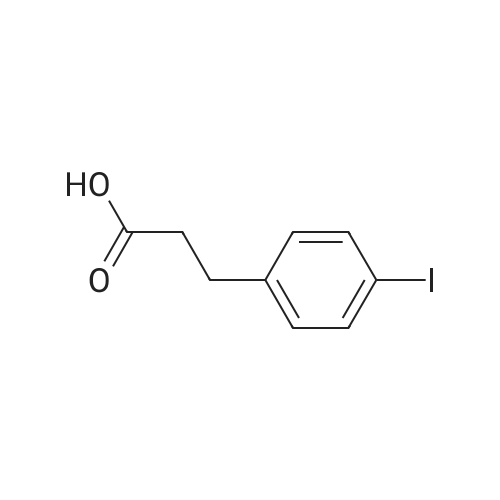
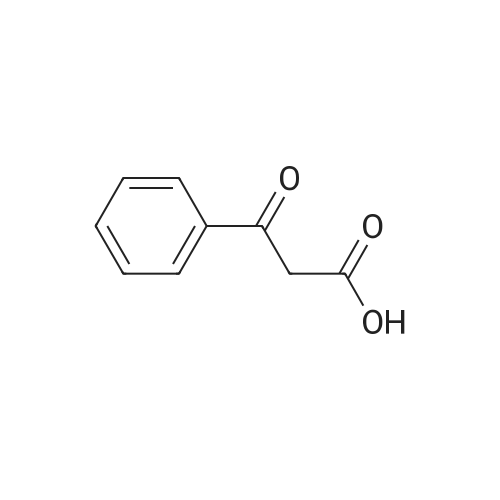
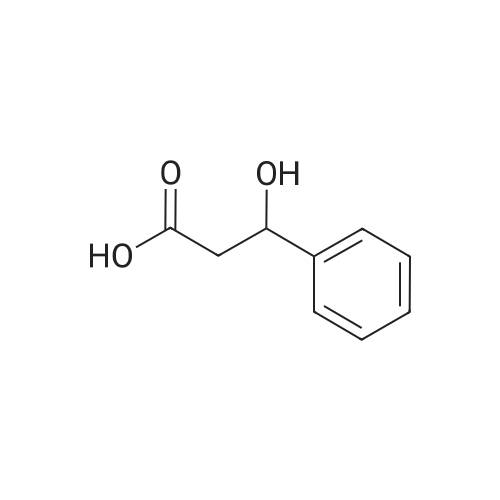
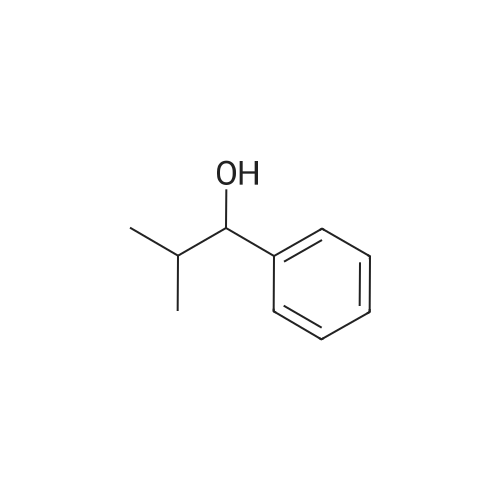
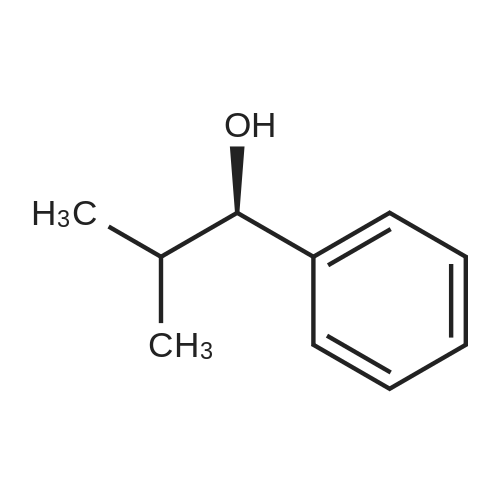
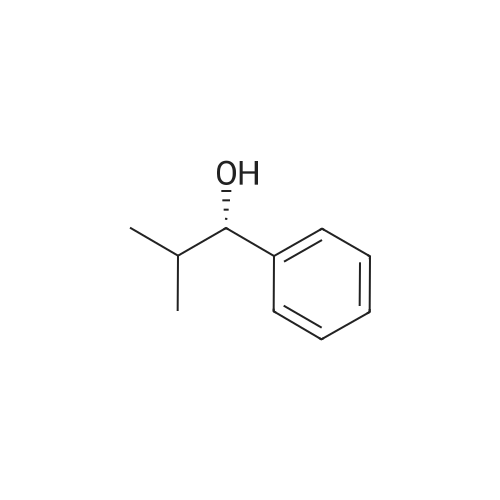
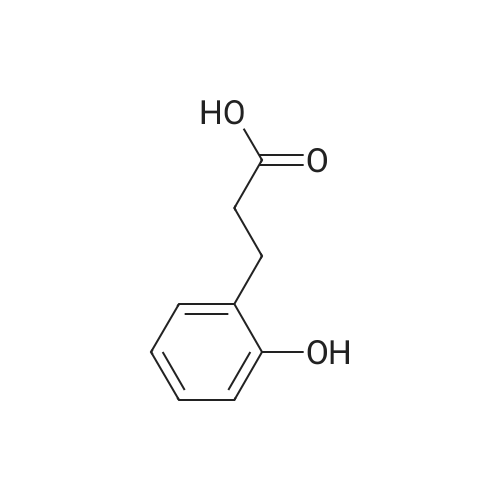
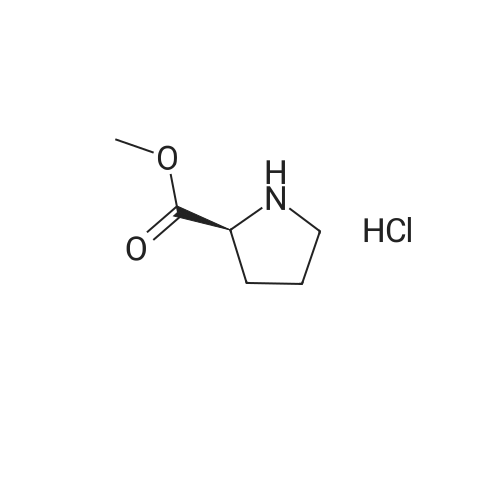
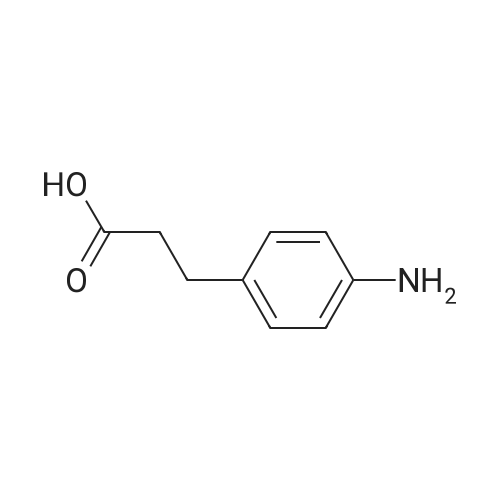
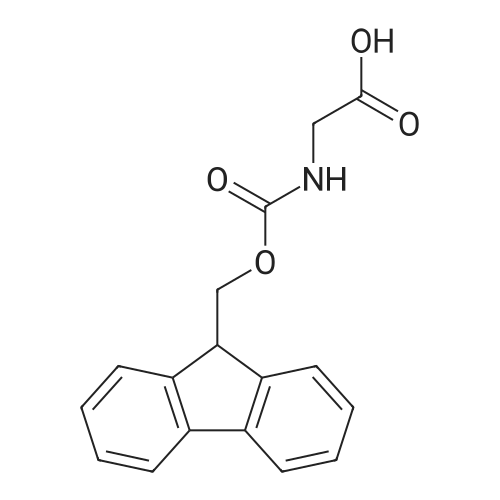
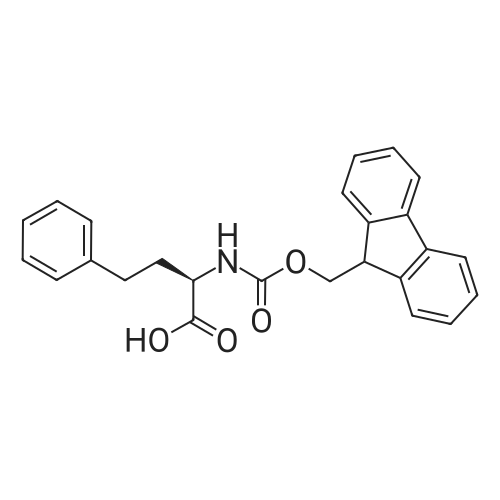
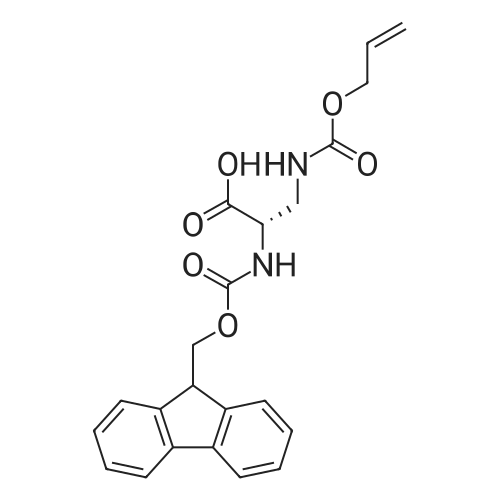
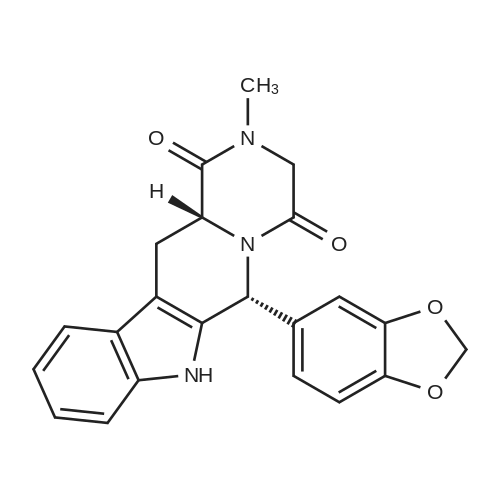
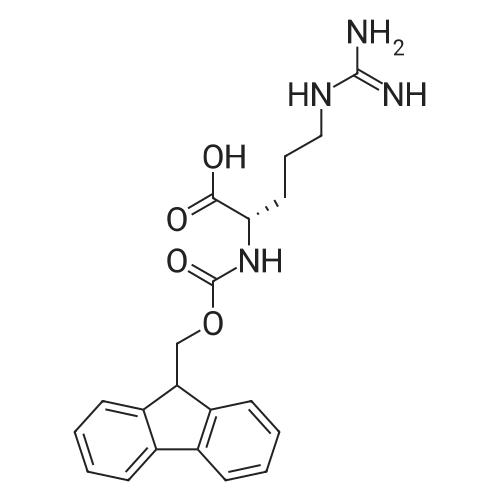
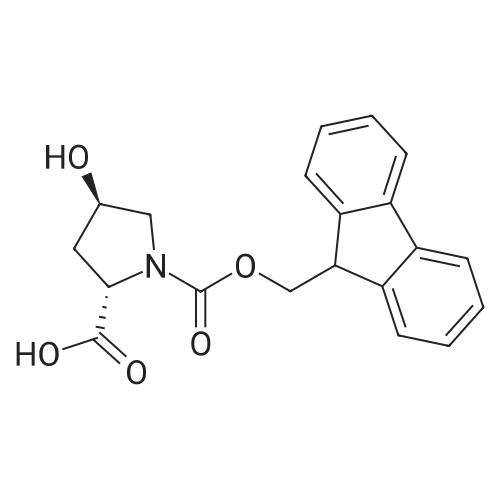
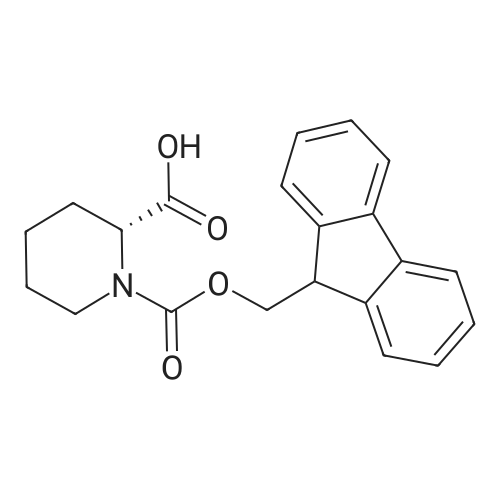
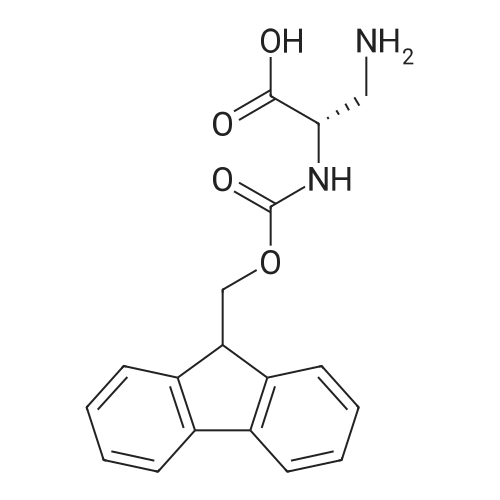
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Chinese Journal of Catalysis, 2015, vol. 36, # 7, p. 957 - 960
2
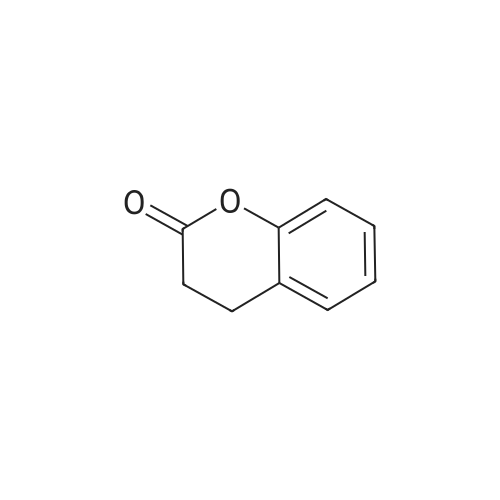
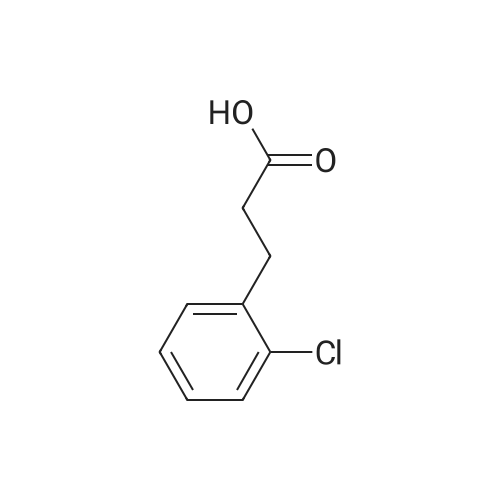
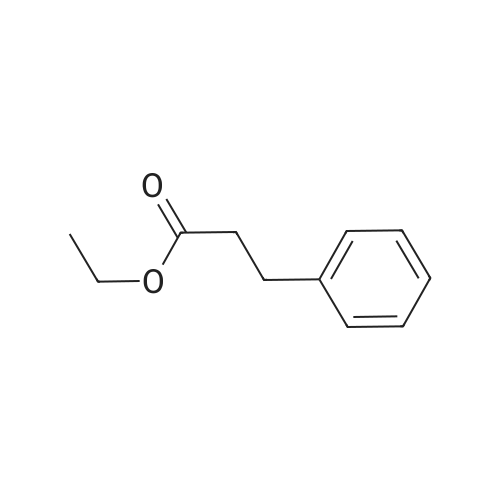
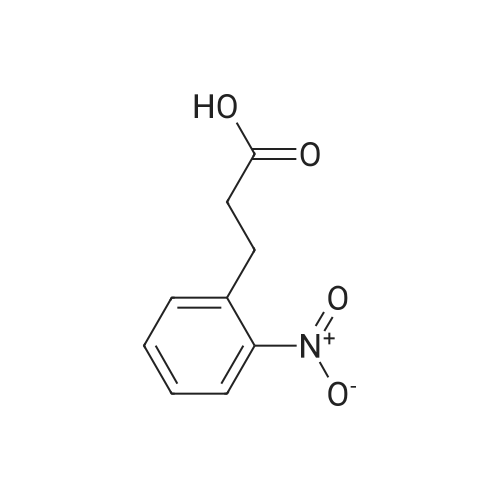
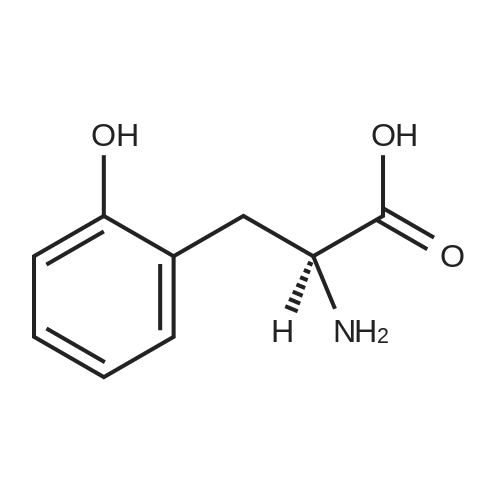
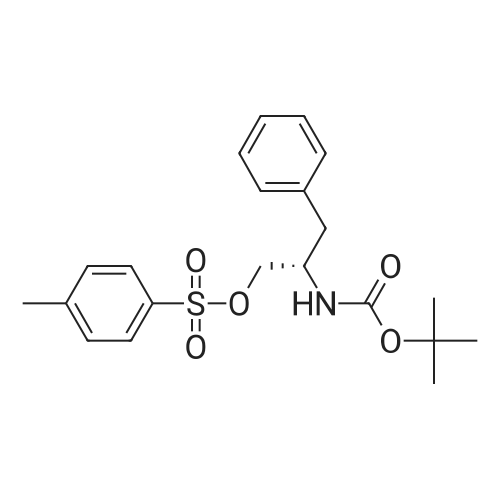
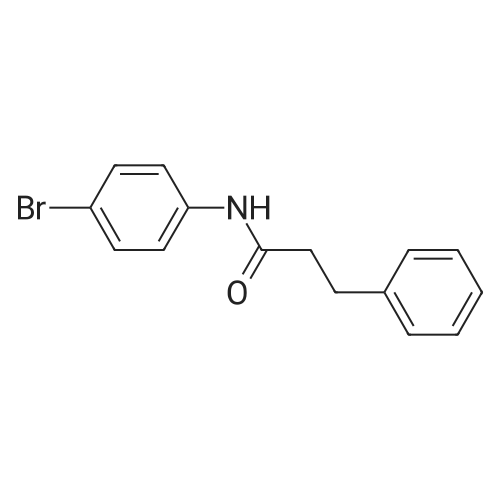
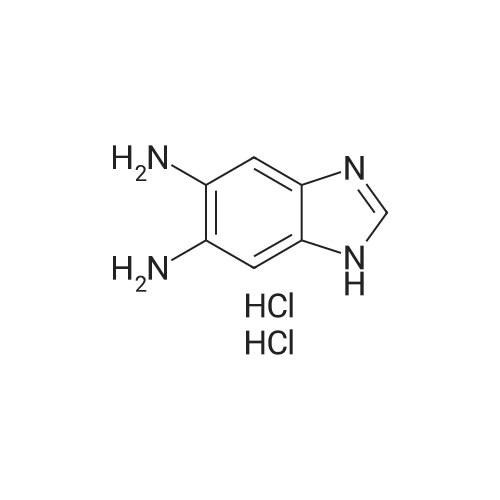
[ 91-64-5 ]
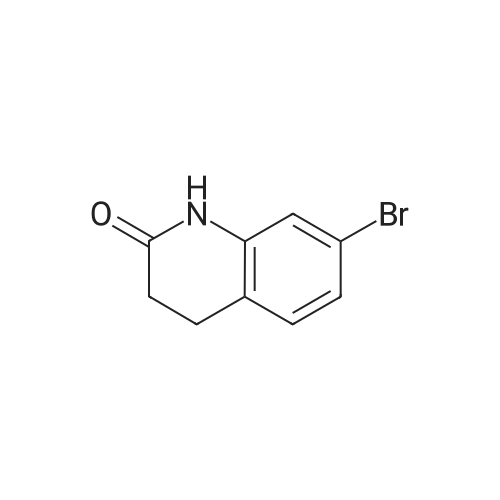
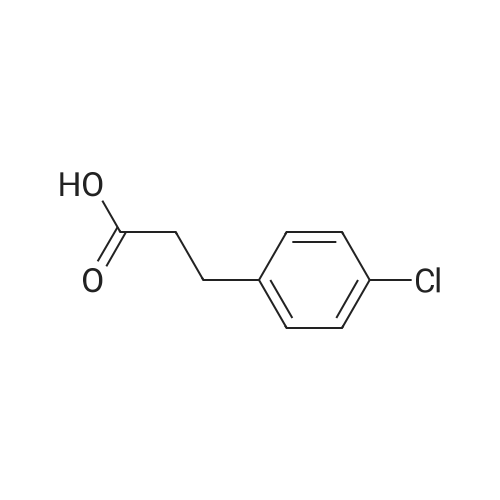
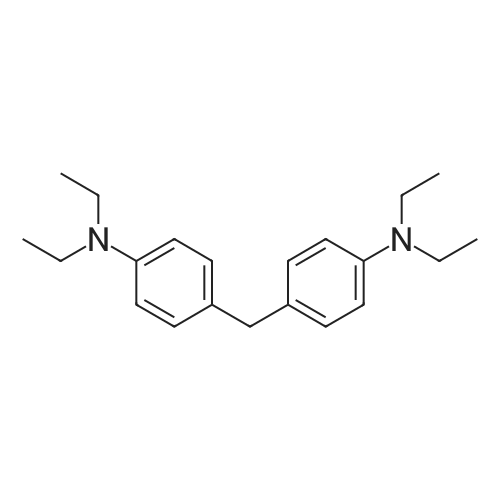
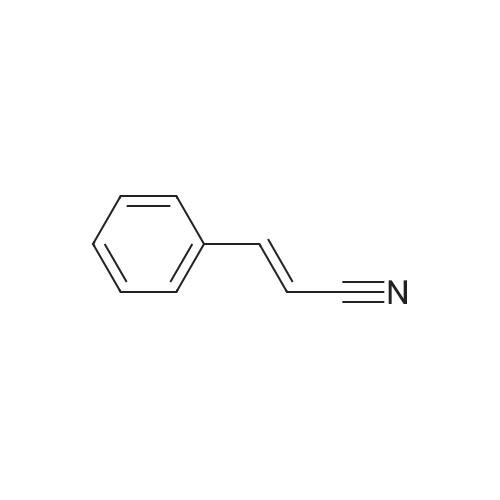
[ 4430-31-3 ]
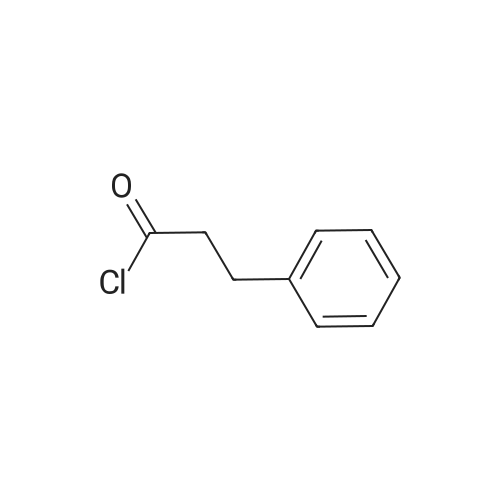
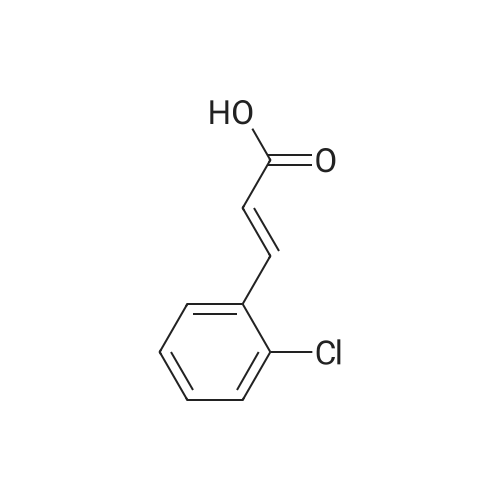
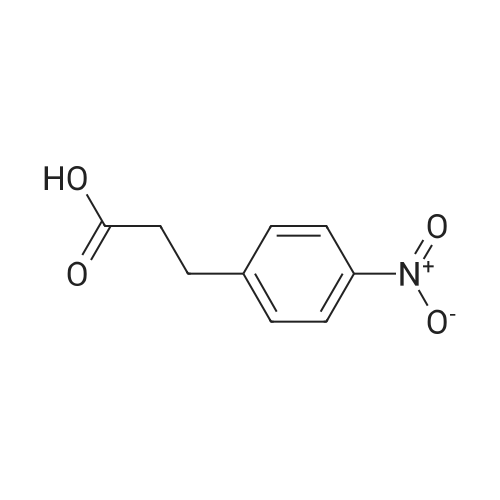
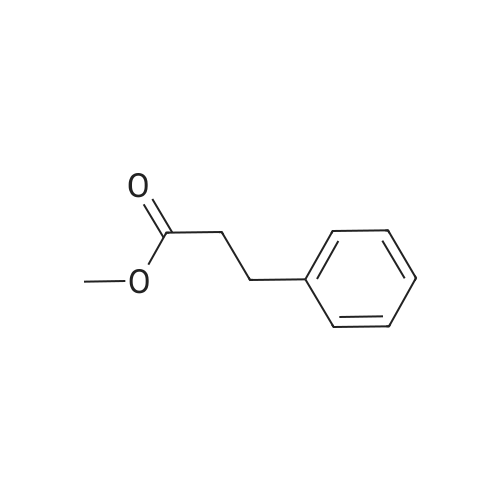
[ 701-97-3 ]
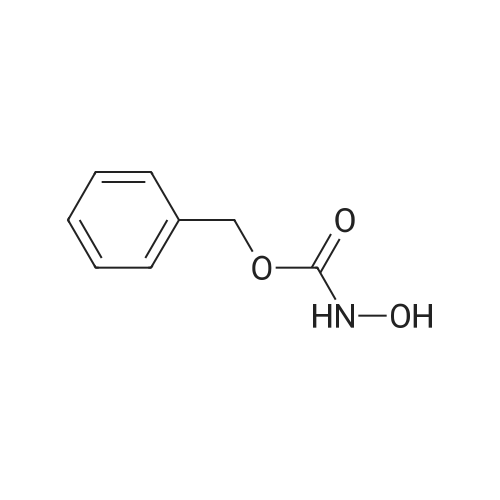
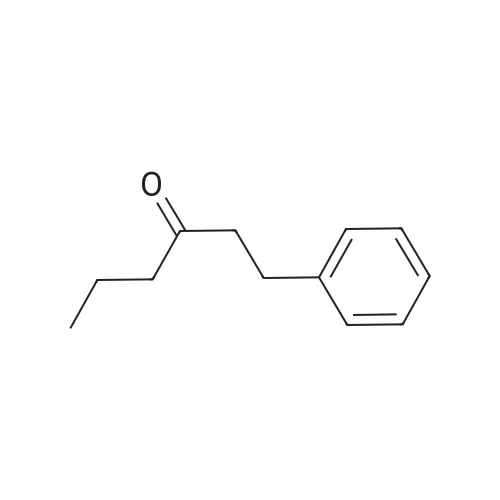
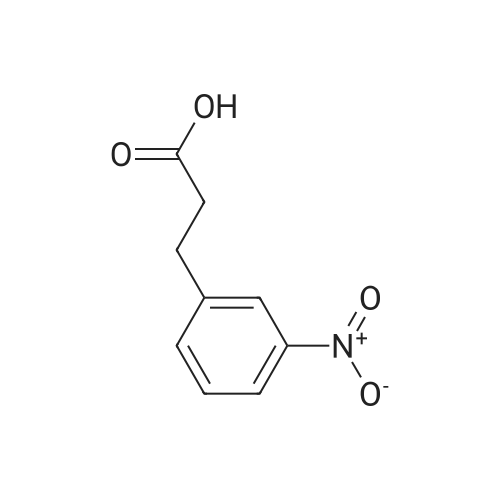
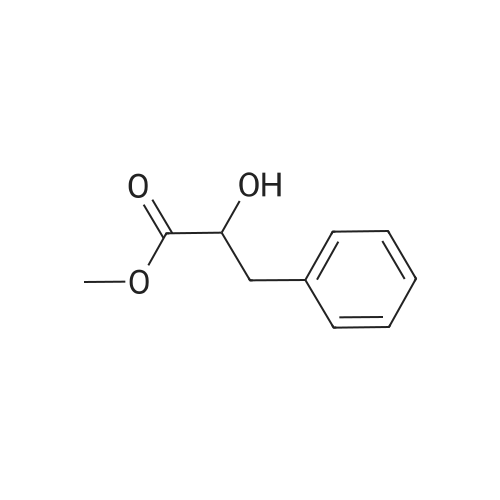
[ 20349-89-7 ]
[ 63714-95-4 ]
[ 119-84-6 ]
[ 501-52-0 ]
Reference:
[1] Chinese Journal of Catalysis, 2015, vol. 36, # 7, p. 957 - 960
3
[ 501-52-0 ]
[ 14548-51-7 ]
Reference:
[1] Chemische Berichte, 1880, vol. 13, p. 1680
4
[ 501-52-0 ]
[ 645-45-4 ]
Yield
Reaction Conditions
Operation in experiment
100%
With oxalyl dichloride In dichloromethane at 20℃;
3-Phenylpropanoyl chloride (74). A 200 mL flask fitted with a stir-bar and septum with an Ar inlet was charged with hydrocinnamic acid (5.00 g, 33.3 mmol), DMF (0.1 mL), and CH2CI2 (30 mL). To the resultant solution was added (COCI)2 (2M in CH2CI2, 20.0 mL, 40.0 mmol) over 30 min. The mixture stirred overnight at rt, and then was concentrated in vacuo to give 5.9 g of 74 as a yellow oil (100percent). HPLC analysis (15:10:75 H2O:A1 :MeOH) showed a purity of 99percent with a retention time of 5.3 min.
100%
With oxalyl dichloride In dichloromethane for 24 h; Inert atmosphere
To a stirred solution of 3-phenylpropanoic acid (3.0 g, 20 mmol, 1 eq.) in anhydrous CH2Cl2 (165 mL) under nitrogen was added oxalyl chloride (16.5 mL 190 mmol, 10 eq.) rapidly via syringe. The reaction mixture was stirred for 24 h, after which the solvent and unreacted oxalyl chloride were Removed by distillation under reduced pressure. The resulting yellow liquid was concentrated via azeotrope with benzene (3 x 50 mL) in vacuo and dried under a vacuum to yield 37 (3.4 g, ~100percent) as a yellow liquid, which was used directly in the next step.
95%
With thionyl chloride In benzene for 4 h; Reflux
Dihydrocinnamic acid (10b) (3.0g, 0.02mol) was dissolved in dry benzene (5mL, 0.06mol) at room temperature then SOCl2 (10mL, 0.14mol) was added in a dropwise manner. The reaction mixture was allowed to heat at reflux for 4h. The solvent was evaporated under reduced pressure to give (3.4g, 95percent) of 11b as yellow oil. The crude product was used in the next step without further purification or characterization.
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 10, p. 2899 - 2903
[2] Patent: WO2013/101911, 2013, A2, . Location in patent: Page/Page column 57
[3] Arkivoc, 2018, vol. 2018, # 5, p. 334 - 347
[4] Advanced Synthesis and Catalysis, 2015, vol. 357, # 18, p. 3825 - 3830
[5] Journal of the American Chemical Society, 1980, vol. 102, # 20, p. 6311 - 6314
[6] Tetrahedron Letters, 1995, vol. 36, # 19, p. 3397 - 3400
[7] Tetrahedron, 2010, vol. 66, # 29, p. 5349 - 5356
[8] Bioorganic and Medicinal Chemistry Letters, 2017, vol. 27, # 13, p. 2912 - 2919
[9] Organic and Biomolecular Chemistry, 2014, vol. 12, # 48, p. 9760 - 9763
[10] Journal of the Chemical Society - Perkin Transactions 1, 1998, # 24, p. 4077 - 4091
[11] Bioorganic and Medicinal Chemistry Letters, 1997, vol. 7, # 13, p. 1689 - 1694
[12] Journal of Medicinal Chemistry, 2001, vol. 44, # 10, p. 1491 - 1508
[13] Justus Liebigs Annalen der Chemie, 1902, vol. 323, p. 255
[14] Recueil des Travaux Chimiques des Pays-Bas, 1897, vol. 16, p. 35
[15] Journal fuer Praktische Chemie (Leipzig), 1905, vol. <2> 71, p. 330
[16] Justus Liebigs Annalen der Chemie, 1909, vol. 369, p. 319
[17] Journal fuer Praktische Chemie (Leipzig), 1905, vol. <2> 71, p. 330
[18] Journal of the American Chemical Society, 1980, vol. 102, # 11, p. 3917 - 3922
[19] Journal of the American Chemical Society, 1987, vol. 109, # 26, p. 8056 - 8066
[20] Bioorganic and Medicinal Chemistry Letters, 2005, vol. 15, # 22, p. 4973 - 4978
[21] Journal of the American Chemical Society, 1974, vol. 96, p. 1518 - 1522
[22] Bulletin de la Societe Chimique de France, 1970, # 3, p. 907 - 912
[23] Journal of Organic Chemistry, 1973, vol. 38, # 8, p. 1439 - 1444
[24] Journal of Organometallic Chemistry, 1968, vol. 13, p. 45 - 51
[25] Canadian Journal of Chemistry, 1991, vol. 69, # 12, p. 2053 - 2058
[26] Recueil des Travaux Chimiques des Pays-Bas, 1993, vol. 112, # 2, p. 143 - 150
[27] Monatshefte fuer Chemie, 1990, vol. 121, # 4, p. 267 - 274
[28] Journal of Heterocyclic Chemistry, 1980, vol. 17, # 5, p. 1081 - 1085
[29] Bulletin de la Societe Chimique de France, 1990, # 6, p. 824 - 829
[30] Tetrahedron, 1987, vol. 43, # 16, p. 3689 - 3694
[31] Tetrahedron, 1988, vol. 44, # 12, p. 3501 - 3512
[32] Journal of the American Chemical Society, 1980, vol. 102, # 17, p. 5530 - 5538
[33] Journal of Organic Chemistry, 1984, vol. 49, # 1, p. 207 - 209
[34] Canadian Journal of Chemistry, 1980, vol. 58, p. 458 - 462
[35] Steroids, 1983, vol. 41, # 3, p. 309 - 320
[36] European Journal of Medicinal Chemistry, 1987, vol. 22, p. 283 - 292
[37] Synthetic Communications, 1982, vol. 12, # 14, p. 1139 - 1146
[38] Journal of the American Chemical Society, 1984, vol. 106, # 11, p. 3344 - 3353
[39] Journal of Medicinal Chemistry, 1986, vol. 29, # 12, p. 2465 - 2472
[40] Journal of Medicinal Chemistry, 1984, vol. 27, # 3, p. 325 - 341
[41] Journal of Heterocyclic Chemistry, 1992, vol. 29, # 2, p. 335 - 341
[42] Tetrahedron, 1991, vol. 47, # 34, p. 7091 - 7108
[43] Recueil: Journal of the Royal Netherlands Chemical Society, 1981, vol. 100, # 1, p. 21 - 24
[44] Australian Journal of Chemistry, 1993, vol. 46, # 12, p. 1845 - 1860
[45] Tetrahedron, 1994, vol. 50, # 25, p. 7343 - 7366
[46] Chemical Communications, 1996, # 22, p. 2595 - 2596
[47] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 1996, vol. 35, # 6, p. 615 - 616
[48] Journal of the Chemical Society - Perkin Transactions 1, 1997, # 8, p. 1147 - 1156
[49] Revue Roumaine de Chimie, 1992, vol. 37, # 2, p. 289 - 298
[50] Journal of Medicinal Chemistry, 1999, vol. 42, # 21, p. 4446 - 4455
[51] Bioorganic and Medicinal Chemistry, 2001, vol. 9, # 3, p. 785 - 792
[52] Phytochemistry, 2002, vol. 59, # 1, p. 9 - 21
[53] Farmaco (Societa chimica italiana : 1989), 2002, vol. 57, # 3, p. 175 - 181
[54] Journal of Organometalic Chemistry, 2002, vol. 643-644, p. 404 - 408
[55] Journal of Medicinal Chemistry, 2003, vol. 46, # 13, p. 2589 - 2598
[56] Synthesis, 2003, # 15, p. 2415 - 2426
[57] Journal of Medicinal Chemistry, 2004, vol. 47, # 5, p. 1098 - 1109
[58] Bioorganic and Medicinal Chemistry Letters, 2004, vol. 14, # 5, p. 1239 - 1242
[59] Journal of Medicinal Chemistry, 2005, vol. 48, # 6, p. 1849 - 1856
[60] Bioorganic and Medicinal Chemistry Letters, 2005, vol. 15, # 13, p. 3257 - 3262
[61] Synthesis, 2005, # 15, p. 2521 - 2526
[62] Bioorganic and Medicinal Chemistry, 2006, vol. 14, # 7, p. 2209 - 2224
[63] Journal of Physical Organic Chemistry, 2006, vol. 19, # 11, p. 737 - 743
[64] Journal of Medicinal Chemistry, 2001, vol. 44, # 25, p. 4468 - 4474
[65] Journal of Organic Chemistry, 2000, vol. 65, # 13, p. 4179 - 4184
[66] Journal of the American Chemical Society, 2002, vol. 124, # 10, p. 2078 - 2079
[67] Journal of Organic Chemistry, 2000, vol. 65, # 5, p. 1442 - 1447
[68] Organic Letters, 2000, vol. 2, # 4, p. 539 - 541
[69] Russian Journal of Applied Chemistry, 2006, vol. 79, # 6, p. 1035 - 1037
[70] Organic and Biomolecular Chemistry, 2006, vol. 4, # 14, p. 2716 - 2723
[71] Tetrahedron, 1988, vol. 44, # 12, p. 3501 - 3512
[72] Patent: WO2006/84869, 2006, A1, . Location in patent: Page/Page column 80
[73] Patent: WO2007/149907, 2007, A2, . Location in patent: Page/Page column 52-53
[74] Patent: US2008/19930, 2008, A1, . Location in patent: Page/Page column 11
[75] Patent: US2008/19930, 2008, A1, . Location in patent: Page/Page column 12
[76] Patent: WO2008/64979, 2008, A2, . Location in patent: Page/Page column 50-51
[77] Patent: US2008/188533, 2008, A1, . Location in patent: Page/Page column 43
[78] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 10, p. 5514 - 5528
[79] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 17, p. 8054 - 8062
[80] Patent: US2004/9991, 2004, A1, . Location in patent: Page/Page column 50
[81] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 23, p. 9984 - 9990
[82] Australian Journal of Chemistry, 2008, vol. 61, # 11, p. 881 - 887
[83] Patent: EP2042494, 2009, A1, . Location in patent: Page/Page column 19
[84] Chemistry - A European Journal, 2008, vol. 14, # 30, p. 9240 - 9254
[85] Patent: US2009/298832, 2009, A1, . Location in patent: Page/Page column 13
[86] Patent: US2009/306214, 2009, A1, . Location in patent: Page/Page column 7
[87] Patent: WO2004/31129, 2004, A2, . Location in patent: Example 2
[88] Patent: EP1598353, 2005, A1, . Location in patent: Page/Page column 22
[89] Organic and Biomolecular Chemistry, 2009, vol. 7, # 17, p. 3561 - 3571
[90] Molecules, 2009, vol. 14, # 9, p. 3313 - 3338
[91] Bioorganic and Medicinal Chemistry, 2009, vol. 17, # 22, p. 7690 - 7697
[92] Patent: US2010/145060, 2010, A1, . Location in patent: Page/Page column 7-8
[93] Synthesis, 2010, # 11, p. 1845 - 1859
[94] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 17, p. 5123 - 5125
[95] Patent: WO2006/26430, 2006, A2, . Location in patent: Page/Page column 50; 67
[96] Organic Letters, 2010, vol. 12, # 18, p. 3990 - 3993
[97] Patent: WO2010/120854, 2010, A1, . Location in patent: Page/Page column 136
[98] Organic Letters, 2008, vol. 10, # 5, p. 885 - 887
[99] Langmuir, 2010, vol. 26, # 10, p. 7181 - 7187
[100] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 18, p. 5426 - 5430
[101] European Journal of Organic Chemistry, 2011, # 17, p. 3117 - 3121
[102] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 20, p. 6116 - 6121
[103] Angewandte Chemie - International Edition, 2011, vol. 50, # 41, p. 9708 - 9711
[104] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 23, p. 7089 - 7093
[105] Science China Chemistry, 2011, vol. 54, # 2, p. 375 - 379
[106] Angewandte Chemie - International Edition, 2012, vol. 51, # 1, p. 204 - 208
[107] Journal of the American Chemical Society, 2012, vol. 134, # 16, p. 6976 - 6979
[108] Chemistry - A European Journal, 2012, vol. 18, # 21, p. 6655 - 6662
[109] Russian Journal of Bioorganic Chemistry, 2012, vol. 38, # 2, p. 224 - 229
5
[ 79-37-8 ]
[ 501-52-0 ]
[ 645-45-4 ]
Reference:
[1] Journal of the American Chemical Society, 1920, vol. 42, p. 607
[2] Patent: US5618792, 1997, A,
[3] Patent: EP425134, 1991, A1,
6
[ 4885-02-3 ]
[ 501-52-0 ]
[ 107-31-3 ]
[ 645-45-4 ]
Reference:
[1] Chemische Berichte, 1959, vol. 92, p. 83,86,89
With sodium bromate; sulfuric acid; sodium bromide In water at 20℃; for 24 h;
General procedure: A total of 1.0 g of 1-octanol (7.69 mmol) was taken in a 50-mL round-bottomed flask, to it NaBr 0.523 g (0.66 eq.), NaBrO 3 0.383 g (0.33 eq.), and 10 mL of H 2 O [comprises the bromide and bromate in 2:1 molar ratio] were added[6f]. The reaction mixture was stirred vigorously to dissolve the contents completely. To the above reaction mixture, the aqueous H 2 SO 4 solution (0.5 eq.) was added slowly under stirring over a period of 2.5 h at room temperature (prepared by adding 0.21 mL of 98percent H 2 SO 4 to 1 mL of water). The reaction mixture was allowed to stir for up to 24 h. After the completion of reaction, the product was extracted with CH 2 Cl 2 (3 15 mL), the organic layer was dried with Na 2 SO 4 and removal of the solvent obtained octyloctanoate in 98percent yield (0.953 g) as colorless liquid. The product was confirmed by GC–MS as well as by NMR.
Reference:
[1] Organic and Biomolecular Chemistry, 2006, vol. 4, # 14, p. 2716 - 2723
[2] Organic and Biomolecular Chemistry, 2006, vol. 4, # 14, p. 2716 - 2723
10
[ 3752-25-8 ]
[ 1643-28-3 ]
[ 501-52-0 ]
Reference:
[1] Chemische Berichte, 1918, vol. 51, p. 582
11
[ 58-22-0 ]
[ 501-52-0 ]
[ 1255-49-8 ]
Reference:
[1] Patent: DE957030, 1953, ,
12
[ 107-92-6 ]
[ 501-52-0 ]
[ 29898-25-7 ]
Reference:
[1] Journal of the Chemical Society, 1925, vol. 127, p. 1097
[2] Annales de Chimie (Cachan, France), 1913, vol. <8> 28, p. 313
[3] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1911, vol. 152, p. 385[4] Bulletin de la Societe Chimique de France, 1911, vol. <4> 9, p. 952
13
[ 501-52-0 ]
[ 29898-25-7 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2005, vol. 15, # 13, p. 3257 - 3262
14
[ 501-52-0 ]
[ 31984-10-8 ]
Reference:
[1] Journal of Heterocyclic Chemistry, 1980, vol. 17, # 5, p. 1081 - 1085
[2] Bioorganic and Medicinal Chemistry, 2017, vol. 25, # 7, p. 2074 - 2083
15
[ 501-52-0 ]
[ 35418-07-6 ]
Reference:
[1] Journal of Labelled Compounds and Radiopharmaceuticals, 2001, vol. 44, p. S951 - S953
[2] Bioorganic and Medicinal Chemistry, 2017, vol. 25, # 8, p. 2445 - 2450
16
[ 501-52-0 ]
[ 96606-95-0 ]
[ 1643-29-4 ]
Reference:
[1] Journal of Medicinal Chemistry, 2008, vol. 51, # 22, p. 7061 - 7064
17
[ 501-52-0 ]
[ 1643-29-4 ]
Yield
Reaction Conditions
Operation in experiment
75%
With potassium iodate; sulfuric acid; iodine; acetic acid In water for 3 h; Reflux
lntermediate-1; 3-(4-lodophenyl)propanoic acid.; A mixture of H2SO4 (1.25 ml_), water (12.5 ml.) and AcOH (25 ml.) in a 250 ml. flask and 3-phenylpropanoic acid (3.00 g, 20.0 mmol), iodine (1.40 g, 5.5 mmol) and KIO3 (0.98 g, 4.6 mmol) was added. The reaction was heated to reflux and a solution of iodine (1.40 g, 5.5 mmol) in AcOH (25 ml.) was added in portions of 5 ml. as the colour of the reaction faded from purple to orange. After 3 hours when no further colour changes appeared the reaction was cooled to room temperature, before quenching with 1 M NaHSO3. The reaction was added water and extracted with EtOAc. The organic phases were combined, washed with brine, dried over MgSO4 and concentrated under vacuum. The product (4.15 g, 75percent), containing minor impurites of starting material and the orffro-iodinated product, was recrystallised from PE to provide 1.83 g (33percent) of the pure and white crystalline product. Rf: 0.10 (EtOAσhexanes, 1 :4); 1HNMR (CDCI3) δ 7.62-7.59 (m, 2H), 6.97-6.95 (m, 2H), 2.92-2.87 (t, 2H, J = 7.5 Hz), 2.68-2.63 (t, 2H, J = 7.5 Hz); 13CNMR (CDCI3) δ 178.5, 139.7, 137.6, 130.4, 91.6, 35.2, 30.0.
56%
With sulfuric acid; iodine; acetic acid; periodic acid In water at 67℃; for 17 h;
A mixture of 3-phenylpropanoic acid (6.00 g, 40.0 mmol), H5IO6 (2.00 g, 8.60 mmol), iodine (4.06 g, 16.0 mmol) and 98percent H2SO4 (1.2 mL) in water (8 mL) and acetic acid (40 mL) was heated at 67 °C for 17 h. The reaction mixture was cooled and quenched with water (100 mL). The crude product was then filtered, washed with water and hexane. The pure product (6.2 g, 56percent) was obtained by recrystallization from toluene. White solid.1H NMR (400 MHz, CDCl3) δ 7.62 (d, J =8.4 Hz, 2 H), 6.97 (d, J = 8.0 Hz, 2 H), 2.91 (t, J = 7.6 Hz, 2 H), 2.67 (t, J = 7.6 Hz, 2 H).
56%
With sulfuric acid; iodine; acetic acid; periodic acid In water at 67℃; for 17 h;
A mixture of 3-phenylpropanoic acid (6.00 g, 40.0 mmol), H5IO6 (2.00 g, 8.60 mmol), iodine (4.06 g, 16.0 mmol) and 98percent H2SO4 (1.2 mL) in water (8 mL) and acetic acid (40 mL) was heated at 67° C. for 17 h. The reaction mixture was cooled and quenched with water (100 mL). The crude product was then filtered, washed with water and hexane. The pure product (6.2 g, 56percent) was obtained by recrystallization from toluene. White solid. 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J=8.4 Hz, 2H), 6.97 (d, J=8.0 Hz, 2H), 2.91 (t, J=7.6 Hz, 2H), 2.67 (t, J=7.6 Hz, 2H).
Reference:
[1] Patent: WO2010/12650, 2010, A1, . Location in patent: Page/Page column 38
[2] Chemistry - A European Journal, 2009, vol. 15, # 10, p. 2278 - 2288
[3] Journal of the American Chemical Society, 2009, vol. 131, # 47, p. 17500 - 17521
[4] European Journal of Organic Chemistry, 2015, vol. 2015, # 27, p. 5919 - 5924
[5] Patent: WO2016/201125, 2016, A1, . Location in patent: Page/Page column 19
[6] Patent: US2018/66298, 2018, A1, . Location in patent: Paragraph 0162; 0163; 0164
[7] Tetrahedron Asymmetry, 2001, vol. 12, # 4, p. 585 - 596
[8] Journal of the American Chemical Society, 1943, vol. 65, p. 1273,1274
[9] Journal of the Chemical Society, 1930, p. 968,979
[10] Organic Letters, 2008, vol. 10, # 12, p. 2597 - 2600
[11] Patent: WO2004/63169, 2004, A1, . Location in patent: Page 52-53
[12] Patent: US2004/229889, 2004, A1, . Location in patent: Page 20
[13] Patent: US2006/111582, 2006, A1, . Location in patent: Page/Page column 16; 3/4
18
[ 501-52-0 ]
[ 96606-95-0 ]
[ 1643-29-4 ]
Reference:
[1] Journal of Medicinal Chemistry, 2008, vol. 51, # 22, p. 7061 - 7064
19
[ 91-66-7 ]
[ 501-52-0 ]
[ 2021-28-5 ]
[ 135-91-1 ]
[ 102100-44-7 ]
Reference:
[1] Chemische Berichte, 1930, vol. 63, p. 489,492
20
[ 501-52-0 ]
[ 16642-79-8 ]
[ 1664-57-9 ]
[ 2001-32-3 ]
Reference:
[1] Journal of Organic Chemistry, 1998, vol. 63, # 4, p. 952 - 958
21
[ 63-91-2 ]
[ 7423-92-9 ]
[ 587-33-7 ]
[ 60-18-4 ]
[ 501-52-0 ]
Reference:
[1] Applied Catalysis A: General, 2010, vol. 380, # 1-2, p. 142 - 148
[2] Applied Catalysis A: General, 2010, vol. 380, # 1-2, p. 142 - 148
22
[ 501-52-0 ]
[ 16642-79-8 ]
[ 2001-32-3 ]
Reference:
[1] Chemische Berichte, 1879, vol. 12, p. 600
[2] Zeitschrift fuer Chemie, 1869, p. 193
[3] Justus Liebigs Annalen der Chemie, 1872, vol. 163, p. 126
[4] Phosphorus, Sulfur and Silicon and the Related Elements, 2004, vol. 179, # 2, p. 221 - 226
[5] Tetrahedron Letters, 2005, vol. 46, # 48, p. 8307 - 8310
23
[ 501-52-0 ]
[ 16642-79-8 ]
[ 1664-57-9 ]
[ 2001-32-3 ]
Reference:
[1] Journal of Organic Chemistry, 1998, vol. 63, # 4, p. 952 - 958
24
[ 7697-37-2 ]
[ 501-52-0 ]
[ 16642-79-8 ]
[ 2001-32-3 ]
Reference:
[1] Chemische Berichte, 1879, vol. 12, p. 600
[2] Zeitschrift fuer Chemie, 1869, p. 193
[3] Justus Liebigs Annalen der Chemie, 1872, vol. 163, p. 126
General procedure: To a colorless solution of 75mg (0.50mmol) of 3-phenylpropanoic acid 1a in 10mL of THF were added at 0C 67muL (0.70mmol, 1.4equiv) of ClCO2Et and 209muL (1.5mmol, 3.0equiv) of Et3N. After stirring for 30min at 0C, 0.75 ml of a 1.0M aqueous solution of NH4Cl (0.75mmol, 1.5equiv) was added at 0C to the colorless suspension. The mixture was stirred for 30min at 0C and 5mL of H2O was added to the resulted mixture. The colorless clear solution was extracted with 30mL of EtOAc and the aqueous layer was extracted with 20mL of EtOAc. The organic layers were combined, washed with 5mL of brine, and dried over anhydrous MgSO4. The crude product was chromatographed on silica gel with EtOAc to afford 72mg (96% yield) of 3-phenylpropanamide 2a. .2.1 3-Phenylpropanamide 2a 72?mg (96%); colorless solid; mp: 92-95?C; 1H NMR (400?MHz, CDCl3): delta 2.54 (t, J?=?8.0?Hz, 2H, CH2C6H5), 2.98 (t, J?=?8.0?Hz, 2H, CH2CO), 5.29 (brs, 2H, NH2), 7.20-7.23, 7.26-7.32 (m, m, 3H, 2H, C6H5); 13C NMR (100?MHz, CDCl3): delta 31.4, 37.5, 126.3, 128.3, 128.6, 140.8, 174.6; IR (KBr, vmax/cm-1)?=?3394 (CONH), 3186 (CONH), 1646 (CON), 1628 (CON); HRMS (ESI-TOF): Calcd for C9H11NONa (M+Na)+: 172.0733, found: 172.0711.
With ammonia; at 165℃; for 0.5h;
General procedure: Into a 1L open reactor was added 500g of carboxylic acid feedstock (chemically pure), stirring was turned on (600 r/min), ammonia gas was continuously fed to the carboxylic acid feed from the bottom of the reactor (chemical purity, water content was 5.1wt%, Flow rate is 100g/min). After the reaction was allowed to proceed for TC hours at the reaction temperature TA, the ammonia gas flow was stopped. The contents of the reactor were sampled and nuclear magnetic proton spectra and elemental analysis were performed to characterize the amide intermediate. Specific reaction conditions and characterization results are shown in Table A-1, Table A-2, Table A-3, Table A-4, Table A-5 and Table A-6. These characterization results show that the obtained amide intermediate has an extremely high purity (above 99%).In this embodiment, the ammonia gas can be directly replaced with waste ammonia gas (from Yangzi Petrochemical Plant, containing approximately50wt% of ammonia gas, the rest were toluene, oxygen, nitrogen, steam, carbon monoxide, and carbon dioxide, and the flow rate of this waste ammonia was 130g/min).
With sulfuric acid; nitric acid; acetic acid; In water; at 5 - 25℃; for 5h;
Example 6; Preparation of 4-(4-methoxy-phenyl)-5-(4-nitro-benzyl)-thiazol-2-ylamine; [Show Image] Step 1: Preparation of 3-(4-nitro-phenyl)-propionic acid; To a 1000 ml three-necked flask equippeded with a thermometer were added 50.0 g (332.9 mmol) 3-phenylpropionic acid, 225 ml glacial acetic acid and 125 ml concentrated sulfuric acid, and added dropwise an acid mixture of 25.7 ml 65% nitric acid (366.2 mmol) and 35 ml concentrated sulfuric acid, premixed and cooled to about 5C. The mixture was placed in ice-bath to keep the inside temperature at 18-25C, maintained at 20-25C and stirred for 5 hours. The reaction was poured into 500 g ice, the precipitated solid was filtered, washed with water until neutrality, dried and recrystallized with ethanol to obtain a 35.8 g product as a white solid in a yield of 55.1%, 166-167 .1H-NMR(CDCl3, 400 MHz) delta: 2.62(2H, t, J=7.56Hz), 2.96(2H, t, J=7.56Hz), 7.53(2H, d, J=8.68Hz, ArH), 8.15(2H, d, J=8.68Hz, ArH), 12.22(1H, s, COOH); ESI-MS m/e (%): 218.0(M+Na, 27), 213.4(M+NH3, 3), 196.1(M+1, 100).
55.1%
With sulfuric acid; nitric acid; acetic acid; at 5 - 25℃;
Example 6; Preparation of 4-(4-methoxy-phenyl)-5-(4-nitro-benzyl)-thiazol-2-ylamine; Step 1: Preparation of 3-(4-nitro-phenyl)-propionic acid; To a 1000 ml three-necked flask equipped with a thermometer were added 50.0 g (332.9 mmol) 3-phenylpropionic acid, 225 ml glacial acetic acid and 125 ml concentrated sulfuric acid, and added dropwise an acid mixture of 25.7 ml 65% nitric acid (366.2 mmol) and 35 ml concentrated sulfuric acid, premixed and cooled to about 5 C. The mixture was placed in ice-bath to keep the inside temperature at 18-25 C., maintained at 20-25 C. and stirred for 5 hours. The reaction was poured into 500 g ice, the precipitated solid was filtered, washed with water until neutrality, dried and recrystallized with ethanol to obtain a 35.8 g product as a white solid in a yield of 55.1%, 166-167 . 1H-NMR (CDCl3, 400 MHz) delta: 2.62 (2H, t, J=7.56 Hz), 2.96 (2H, t, J=7.56 Hz), 7.53 (2H, d, J=8.68 Hz, ArH), 8.15 (2H, d, J=8.68 Hz, ArH), 12.22 (1H, s, COOH); ESI-MS m/e (%): 218.0 (M+Na, 27), 213.4 (M+NH3, 3), 196.1 (M+1, 100).
33.3%
With sulfuric acid; nitric acid; at 0℃; for 1h;
To a mixture of concentrated nitric acid (1.5 mL, 21.98 mmol) in concentrated sulfuric acid (20 mL) was added phenylpropionic acid (3 g, 19.98 mmol) in portions while maintaining the internal temperature below 0 C. After 1h, the resulting mixture was pour into 20 mL ice water and filtered, the solids were washed with water (5 ml × 2). The filter cake was dried in vacuum to afford product 2a (1.3 g, 33.3%) as a white solid. 1H NMR (300 MHz, DMSO-d6) delta: 12.53 (s, 1H, COOH), 8.14 (d, J = 8.61 Hz, 2H, ArH), 7.52 (d, J = 8.61 Hz, 2H, ArH), 2.95 (t, J = 7.45 Hz, 2H, COCH2), 2.83 (t, J = 7.45 Hz, 2H, ArCH2).
With sulfuric acid; nitric acid;
p-nitrohydrocinnamic acid STR21 In a 2-liter, 3-necked, round-bottomed flask equipped with mechanical stirrer, thermometer, and additional funnel was placed 361 g (2.41 mol) of hydrocinnamic acid which was warmed to 50 to maintain it in a liquid state. To the warm liquid was added slowly dropwise over a 4 hour period with good stirring a mixture of 241 g of nitric acid and 546 g of sulfuric acid. The reaction is very exothermic so that a water cooling bath was required to which ice was added occasionally to keep the temperature at 50 C. The very thick reaction mixture was stirred for an additional 2 hours and poured onto 3 liters of ice. The solid was collected by filtration and washed repeatedly with a total of 6 liters of water. After air drying, the entire sample was recrystallized from 800 ml of acetone, giving a first crop of 150.4 g mp. 162-164 C.
With potassium iodate; sulfuric acid; iodine; acetic acid; In water; for 3h;Reflux;
lntermediate-1; 3-(4-lodophenyl)propanoic acid.; A mixture of H2SO4 (1.25 ml_), water (12.5 ml.) and AcOH (25 ml.) in a 250 ml. flask and 3-phenylpropanoic acid (3.00 g, 20.0 mmol), iodine (1.40 g, 5.5 mmol) and KIO3 (0.98 g, 4.6 mmol) was added. The reaction was heated to reflux and a solution of iodine (1.40 g, 5.5 mmol) in AcOH (25 ml.) was added in portions of 5 ml. as the colour of the reaction faded from purple to orange. After 3 hours when no further colour changes appeared the reaction was cooled to room temperature, before quenching with 1 M NaHSO3. The reaction was added water and extracted with EtOAc. The organic phases were combined, washed with brine, dried over MgSO4 and concentrated under vacuum. The product (4.15 g, 75%), containing minor impurites of starting material and the orffro-iodinated product, was recrystallised from PE to provide 1.83 g (33%) of the pure and white crystalline product. Rf: 0.10 (EtOA?hexanes, 1 :4); 1HNMR (CDCI3) delta 7.62-7.59 (m, 2H), 6.97-6.95 (m, 2H), 2.92-2.87 (t, 2H, J = 7.5 Hz), 2.68-2.63 (t, 2H, J = 7.5 Hz); 13CNMR (CDCI3) delta 178.5, 139.7, 137.6, 130.4, 91.6, 35.2, 30.0.
56%
With sulfuric acid; iodine; acetic acid; periodic acid; In water; at 67℃; for 17h;
A mixture of 3-phenylpropanoic acid (6.00 g, 40.0 mmol), H5IO6 (2.00 g, 8.60 mmol), iodine (4.06 g, 16.0 mmol) and 98% H2SO4 (1.2 mL) in water (8 mL) and acetic acid (40 mL) was heated at 67 C for 17 h. The reaction mixture was cooled and quenched with water (100 mL). The crude product was then filtered, washed with water and hexane. The pure product (6.2 g, 56%) was obtained by recrystallization from toluene. White solid.1H NMR (400 MHz, CDCl3) delta 7.62 (d, J =8.4 Hz, 2 H), 6.97 (d, J = 8.0 Hz, 2 H), 2.91 (t, J = 7.6 Hz, 2 H), 2.67 (t, J = 7.6 Hz, 2 H).
56%
With sulfuric acid; iodine; acetic acid; periodic acid; In water; at 67℃; for 17h;
A mixture of 3-phenylpropanoic acid (6.00 g, 40.0 mmol), H5IO6 (2.00 g, 8.60 mmol), iodine (4.06 g, 16.0 mmol) and 98% H2SO4 (1.2 mL) in water (8 mL) and acetic acid (40 mL) was heated at 67 C. for 17 h. The reaction mixture was cooled and quenched with water (100 mL). The crude product was then filtered, washed with water and hexane. The pure product (6.2 g, 56%) was obtained by recrystallization from toluene. White solid. 1H NMR (400 MHz, CDCl3) delta 7.62 (d, J=8.4 Hz, 2H), 6.97 (d, J=8.0 Hz, 2H), 2.91 (t, J=7.6 Hz, 2H), 2.67 (t, J=7.6 Hz, 2H).
With sulfuric acid; iodine; periodic acid; In water; acetic acid; at 70℃; for 7h;
To a stirred solution of 3-phenylpropanoic acid (7.51 g) in acetic acid (70 mL) were added periodic acid (2.39 g), iodine (5.08 g), concentrated sulfuric acid (1.5 mL) and water (10 mL), and the mixture was stirred at 70C for 7 hours. The solvent was evaporated in vacuo, and the residue was diluted with water and extracted with ethyl acetate. The organic phase was washed with 10% sodium thiosulfate solution twice, then washed with brine, dried over magnesium sulfate and evaporated in vacuo. The precipitate was crystallized from ethyl acetate and hexane to give Compound (14) (5.80 g). LH-NMR (300 MHz, CDC13, 8) : 2. 66 (2H, t, J=7 Hz), 2. 90 (2H, t, J=7 Hz), 6.97 (2xlH, d, J=8.5 Hz), 7.61 (2xlH, d, J=8.5 Hz); MASS (ES-): m/e 275. Preparation 15 Compound (15) was obtained from Compound (14) according to a manner similar to Preparation 1 (9.50 g).
With sulfuric acid; iodine; periodic acid; In water; acetic acid; at 70℃; for 7h;
To a stirred solution of 3-phenylpropanoic acid (7.51 g) in acetic acid (70 mL) were added periodic acid (2.39 g), iodine (5.08 g), concentrated sulfuric acid (1.5 mL) and water (10 mL), and the mixture was stirred at 70 C. for 7 hours. The solvent was evaporated in vacuo, and the residue was diluted with water and extracted with ethyl acetate. The organic phase was washed with 10% sodium thiosulfate solution twice, then washed with brine, dried over magnesium sulfate and evaporated in vacuo. The precipitate was crystallized from ethyl acetate and hexane to give Compound (14) (5.80 g). 1H-NMR (300 MHz, CDCl3, delta): 2.66 (2H, t, J=7 Hz), 2.90 (2H, t, J=7 Hz), 6.97 (2×1H, d, J=8.5 Hz), 7.61 (2×1H, d, J=8.5 Hz); MASS (ES-): m/e 275.
With sulfuric acid; iodine; periodic acid; In water; acetic acid; at 105℃; for 6.5h;
Hydrocinnamic acid is iodinated to form 4-iodohydrocinnamic acid. The process is performed by mixing 201.2 g hydrocinnamic acid, 1 L acetic acid, 152 mL water, 52.8 g periodic acid, 76 mL sulfuric acid, and 144 g iodine. The mixture is heated at 105 C. for 6.5 hours. Next, 200 mL water is added, and the mixture is cooled to room temperature overnight. The solid product in the resultant mixture is filtered and washed with water, yielding 350 g unpurified 4-iodohydrocinnamic acid. Further purification is not necessary and is not conducted, and the product is used as is in the second step.
3-phenyl-propionic acid 4,6-dimethoxy-[1,3,5]triazin-2-yl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With 4-methyl-morpholine; In 1,4-dioxane; water; at 20℃; for 0.25h;
General procedure: To a solution of benzoic acid 1a (2.2 g, 18.0 mmol) and N-methylmorpholine (396 muL, 3.60 mmol) in 1,4-dioxane/H2O(100 mL:50 mL) was added DMT-MM (5.23 g, 18.9 mmol) at room temperature. After stirring for 15 min, a solution ofalanine 2a (1.76 g, 19.8 mmol) and aq 1 M NaOH (19.8 mL, 19.8 mmol) was added. After the reaction was completed(monitored by TLC), N-methylmorpholine (3.96 mL, 36.0 mmol) and DMT-MM (14.9 g, 54 mmol) were added in order.After stirring for 3 h, the reaction mixture was diluted in EtOAc (100 mL) and washed with aq 1 M HCl (40 mL), sat. aqNaHCO3 (40 mL), and brine (30 mL). The organic layer was dried over Na2SO4, filtered, and evaporated under reducedpressure. The residue was purified by column chromatography (EtOAc:hexane = 7:3) to give oxazole 3aa in 78% yield.5-((4,6-Dimethoxy-1,3,5-triazin-2-yl)oxy)-4-methyl-2-phenyloxazole (3aa) Yield: 78%. White solid.
3-Phenyl-propionic acid (3R,5S,8R,9S,10S,13R,14S,17R)-17-((R)-1,5-dimethyl-hexyl)-10,13-dimethyl-hexadecahydro-cyclopenta[a]phenanthren-3-yl ester[ No CAS ]
2-Methyl-6-nitro-benzoic acid (3R,5S,8R,9S,10S,13R,14S,17R)-17-((R)-1,5-dimethyl-hexyl)-10,13-dimethyl-hexadecahydro-cyclopenta[a]phenanthren-3-yl ester[ No CAS ]
With N-ethyl-N,N-diisopropylamine; HATU; In N,N-dimethyl-formamide; at 20℃;
General procedures:; Scheme 2; An acid (1 equiv), HATU (1 equiv) and DIEA (3 equiv) were added to a solution of 4-bromo-aniline (1 equiv) in DMF. The mixture was stirred at room temperature until the starting material disappeared (detection by LC-MS). The solution was diluted with EtOAc and washed with saturated aqueous NaHCO3. The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo to give the crude bromide 2-1. The bromide 2-1 (1 equiv) and the boronic acid ester (1.5 equiv) were dissolved in degassed dioxane/H2O (5:1 by volume) in a sealed tube. Pd(PPh3)4 (0.03 equiv) and 2M solution Of K2CO3 (3 equiv) were added sequentially. The mixture was heated at 95 0C for 2h. After cooling to room temperature, the mixture was diluted with water and extracted with ethyl acetate. The organic layers were combined, dried over sodium sulfate and concentrated in vacuo. The residue was then purified by preparative HPLC to give the product 2-2 as a solid (65percent-80percent yield in two steps).; Example 27. N-(4-(lH-pyrazol-4-yl)phenv0-3-phenylpropanamide; Procedures in Scheme 2 were utilized to synthesize this compound. LC/MS: Ci8HnN3O (M+l) 292. Single peak at both 215 nm and 254 nm in analytical HPLC traces.
With thionyl chloride;Reflux;
Case 1 Obtaining intermediate compound Va; [Show Image] To a boiling mixture of the 230 grams of the hydrocinnamic acid and 270 grams of the 4-bromoaniline in 1 liter of the toluol 160 ml of the thionyl chloride are added by drops; then 1 liter of water and 50 ml of the aqueous ammonia are added by drops. The sediment after cooling is filtered and dried. Yield: 405 grams of the intermediate compound Va.
4-(3-bromo-8-chloro-6,11-dihydro-5<i>H</i>-benzo[5,6]cyclohepta[1,2-<i>b</i>]pyridin-11-yl)-1-(3-phenyl-propionyl)-piperazine-2-carboxylic acid (pyridin-3-ylmethyl)-amide[ No CAS ]
With 4-methyl-morpholine; hydrogenchloride; In methanol; dichloromethane; water;
Example 68 Into a 100-ml egg plant-type flask were added 3.00 g (0.02 mols) of a 3-phenylpropionic acid as a carboxylic acid compound, 2.22 g (0.022 mols) of a 4-methylmorpholine as a tertiary amine compound and 50 ml of methanol as an alcohol compound, which were then stirred at room temperature for 10 minutes. Then, 6.35 g (0.02 mols) of the same <strong>[3945-69-5]4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride</strong> having a water content of 12.8% by weight as the one prepared in Example 4 was added thereto as a condensing agent to effect the reaction at room temperature for 4 hours. After the reaction, methanol was distilled off, 100 ml of water was added thereto, and the extraction operation was conducted twice with 30 ml of a methylene chloride. The separated methylene chloride solution was collected, and the organic layer was washed with 20 ml of a saturated sodium carbonate aqueous solution, 20 ml of a 1N hydrochloric acid and 20 ml of water. The obtained organic phase was dried on magnesium sulfate, the methylene chloride was distilled off, and the residue was isolated and refined by using a silica gel column chromatography to obtain 3.05 g of a methyl 3-phenylpropionate (yield, 93%).
With 4-methyl-morpholine; benzyl alcohol; In tetrahydrofuran; hydrogenchloride; dichloromethane; water;
Example 98 Into a 100-ml egg plant-type flask were added 3.00 g (0.02 mols) of a 3-phenylpropionic acid as a carboxylic acid compound, 6.06 g (0.06 mols) of a 4-methylmorpholine as a tertiary amine compound, 2.38 g (0.022 mols) of a benzyl alcohol as an alcohol compound and 50 ml of a tetrahydrofurane as a solvent, which were then stirred at room temperature for 10 minutes. Then, 6.85 g (0.02 mols) of the same <strong>[3945-69-5]4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride</strong> having a water content of 19.1% by weight as the one prepared in Example 5 was added thereto as a condensing agent to effect the reaction at room temperature for 24 hours. After the reaction, tetrahydrofurane was distilled off, 100 ml of water was added thereto, and the extraction operation was conducted twice with 30 ml of a methylene chloride. The separated methylene chloride solution was collected, and the organic layer was washed with 20 ml of a saturated sodium carbonate aqueous solution, 20 ml of a IN hydrochloric acid and 20 ml of water. The obtained organic phase was dried on magnesium sulfate, the methylene chloride was distilled off, and the residue was isolated and refined by using a silica gel column chromatography to obtain 4.51 g of a benzyl 3-phenylpropionate (yield, 94%).
WSCD.HCl (N-ethyl-N',N'-dimethylaminopropylcarbodiimide hydrochloride)[ No CAS ]
[ 2133-40-6 ]
[ 501-52-0 ]
[ 946438-16-0 ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In dichloromethane;
EXAMPLE 1 Synthesis of N-(3-phenylpropionyl)-proline methyl ester (Compound No. 4) STR7 Proline methyl ester hydrochloride (3.4 g), 3-phenylpropionic acid (3.1 g) and triethylamine (2.8 ml) were suspended in dry methylene chloride (30 ml). To the cooled suspension, WSCD.HCl (N-ethyl-N',N'-dimethylaminopropylcarbodiimide hydrochloride) (3.9 g) was added. Under cooling, the mixture was stirred for 1 hour, and after allowing the mixture to warm to room temperature, it was again stirred for 12 hours. The stirred mixture was washed successively with water, 1N HCl, water, saturated aqueous sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, the solvent was distilled off under vacuum. The resulting crude product was purified by medium-pressure liquid column chromatography on silica gel (solvent: carbon tetrachloride) to obtain the end compound as an oil (4.6 g). Instead of 3-phenylpropionic acid, a) 4-phenyl-n-butyric acid and b) 5-phenyl-n-valeric acid were used as starting compounds, and treated by the procedures described above to obtain the following end compounds as oil: STR8
A solution of <strong>[2393-17-1]3-(4-aminophenyl)propionic acid</strong> in methanol was cooled to 0 C. Thionyl chloride was added dropwise. Following addition, the reaction was heated to 40 C. overnight. The reaction was concentrated to provide the intermediate methyl ester which was used without further purification.The intermediate ester (0.115) was dissolved in anhydrous THF under nitrogen. EDC was added followed by DMAP and hydrocinnamic acid. The reaction was left at room temperature over night. The reaction was poured into water and extracted with EtOAc. The combined organic layers were washed once with water, dried over MgSO4, filtered and concentrated. The compound was purified by flash chromatography to give the intermediate amide (E208a).
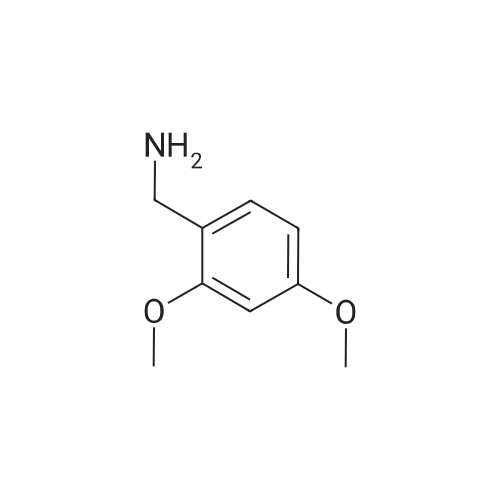
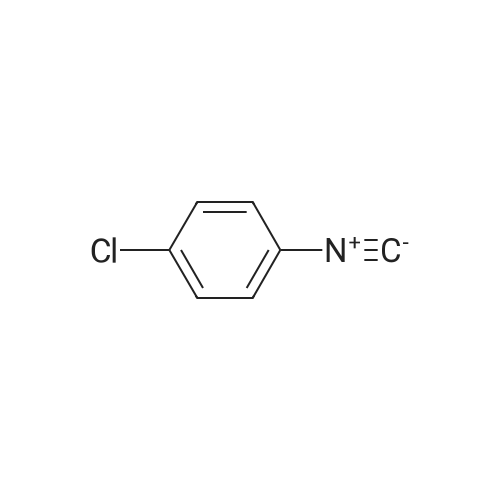
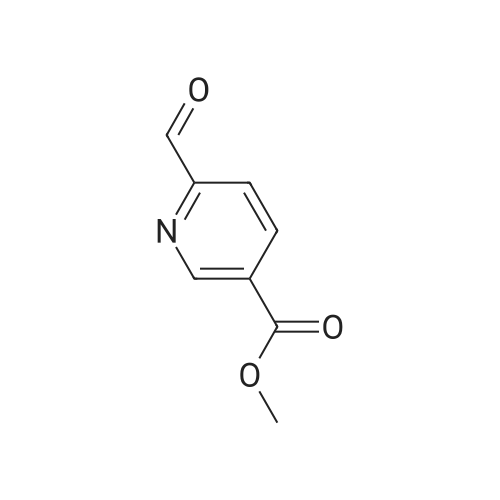
Step A: Methyl 6-{2-[(4-chlorophenyl)amino]-1-[(2,4-dimethoxybenzyl)(3-phenylpropanoyl)-amino]-2-oxoethyl}nicotinate Dihydrocinnamic acid (46 mg, 0.30 mmol), 4-chlorophenylisocyanide (42 mg, 0.30 mmol) and 2,4-dimethoxybenzylamine (61 mg, 0.36 mmol) were added to a solution of methyl 6-formylnicotinate (see Langlois, Y. et al, Tetrahedron. 1975, 31, 419-22) (50 mg, 0.30 mmol) in 400 muL of TFE. The solution was allowed to stir for 4 h at room temperature and then purified by flash chromatography (12-100percent ethyl acetate in hexanes to give the desired product. MS cal'd 602 (MH+), exp 602 (MH+).
176 mg of <strong>[71125-38-7]meloxicam</strong> was ground with 75 mg of hydrocinnamic acid and 400 muL of THF was added to the solid mixture. The solids gathered after grinding were stored in screw cap vials for subsequent analysis.
In acetonitrile;Product distribution / selectivity;
50 mg of tadaiafil was slurried with 288.75 mg of 3- phenylpropanoic acid and 1 mL of acetonitriie. The solids gathered after the slurry were dried and stored in a screw cap vial for subsequent analysis. [048] Example 6: Preparation of tadaiafii and malonic acid complex (1:1) [049] 110 mg of tadaiafil was ground with 30 mg of malonic acid and 40 muL of acetonitrile was added to the solid mixture. The solids gathered after grinding were stored in screw cap vials for subsequent analy
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 20℃; for 18h;
General procedure: A mixture 2-amino-4,5-dimethylthiophene-3-carboxamide (340 mg, 2 mmol), 2-(1-naphthalen-1-yl)acetic acid (372 mg, 2 mol), EDC (458 mg, 2.4 mmol), HOBt (324 mg, 2.4 mmol), DIEA ( 1.1 mL, 6 mmol) in DMF (10 mL) was stirred at room temperature for 18 h. The reaction mixture was poured into 100 mL of water then extracted with ethyl acetate (150 mL). The organic layer was washed with saturated NaHCO3 solution (3 x 50 mL), brine (3 x 50 mL), water (3 x 50 mL) respectively. The ethyl acetate layer was dried over MgSO4 and concentrated. The residue was chromatographed over silica gel (30 to 40% ethyl acetate in haxane) to afford compound 1 (277 mg, 41%).
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 20℃; for 24h;Reflux;
General procedure: To a stirred solution of 3a (692 mg, 2.4 mmol) in CH2Cl2 (20 mL) was added TFA (6.0 mL, 34.0 mmol), and the reaction mixture was refluxed for 48 h. After cooling, the reaction was quenched with 10% NaOH (aq), and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (10 mL 3). The organic layer and extracts were combined, dried over K2CO3, and evaporated to give amine, which was used directly in the next step. To a stirred solution of the amine obtained above in CH2Cl2 (15 mL) were added 2,6-difluorophenylacetic acid (409 mg, 2.4 mmol), EDC*HCl (910 mg, 2.62 mmol), and DMAP (29 mg, 0.24 mmol), and the resulting solution was stirred at room temperature for 24 h. The reaction was quenched with satd. NaHCO3 (aq), and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (10 mL x 3). The organic layer and extracts were combined, dried over K2CO3, and evaporated to give residue, which was chromatographed on silica gel (25 g, hexane:acetone = 8:1) to give 4a (925 mg, 84%) as a colorless solid.
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 0 - 20℃;Inert atmosphere;
General procedure: The solution of EDCI (3.14g, 16.38mmol) and DMAP (100mg, 0.82mmol) in 10mL DCM was added dropwise to a mixture of tert-butyl-3-aminobenzyl-carbamate (2.0g, 9.01mmol) and benzoic acid (1.0g, 8.19mmol) in 20mL DCM at 0C under nitrogen atmosphere, and the mixture was warmed to r.t. and stirred overnight. After quenched with water and extracted with DCM, the organic layer was washed with 1M NaOH (20mL), 1M HCl (20mL), and saturated NaHCO3 (10mL), dried over Na2SO4, and concentrated under reduced pressure. The Boc-protected 12a was dissolved in DCM (5mL), trifluoroacetic acid (5mL) was added. The reaction mixture was allowed to stir for 2hat r.t. and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel and further purified by recrystallization (ethyl ether) to obtain the title compound as a white solid (1.2g, yield 64.8%).
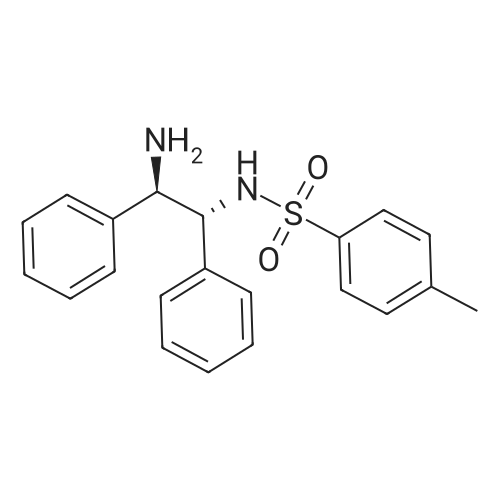
N-(1R,2R)-[1,2-diphenyl-2-(toluene-4-sulfonylamino)ethyl]-3-phenyl-propionamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
93%
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; N,N-dimethyl-formamide; at 0 - 20℃;
The preparation followed the method described for compound 10, using 3-phenyl-propanoic acid(1.00 g, 6.66 mmol), TsDPEN (2.68 g, 7.32 mmol), HOBt (0.99 g, 7.33 mmol) and EDC (1.23 g, 7.93mmol) in anhydrous DCM (20 mL) and anhydrous DMF (20 mL). The amide 23 was obtained as anoff-white solid (3.07g, 6.16 mmol, 93%).
With sodium bromate; sulfuric acid; sodium bromide; In water; at 20℃; for 24h;
General procedure: A total of 1.0 g of 1-octanol (7.69 mmol) was taken in a 50-mL round-bottomed flask, to it NaBr 0.523 g (0.66 eq.), NaBrO 3 0.383 g (0.33 eq.), and 10 mL of H 2 O [comprises the bromide and bromate in 2:1 molar ratio] were added[6f]. The reaction mixture was stirred vigorously to dissolve the contents completely. To the above reaction mixture, the aqueous H 2 SO 4 solution (0.5 eq.) was added slowly under stirring over a period of 2.5 h at room temperature (prepared by adding 0.21 mL of 98% H 2 SO 4 to 1 mL of water). The reaction mixture was allowed to stir for up to 24 h. After the completion of reaction, the product was extracted with CH 2 Cl 2 (3 15 mL), the organic layer was dried with Na 2 SO 4 and removal of the solvent obtained octyloctanoate in 98% yield (0.953 g) as colorless liquid. The product was confirmed by GC-MS as well as by NMR.
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl acetamide;
General procedure: To a solution of corresponding acids (RCOOH, 0.63 mmol), HOBt (170 mg, 1.26 mmol), EDCI (242 mg, 1.26 mmol), and DIPEA (0.208 mL, 1.26 mmol) were added to DMA (60 mL), and then 4 (186 mg, 1.26mmol) were added, the mixture was stirred overnight. The mixture was dissolved in water, then the aqueous solution was extracted with ethyl acetate (50 mL 3). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated to give the crude product, which was purified by column chromatography to give N-(4-nitrophenethyl)amide (5) in a good yield. And to a solution of 5 in MeOH was added 10% Pd/C under H2 at reflux overnight, then the mixture was dissolved in water, then the aqueous solution was extracted with ethyl acetate (50 mL 3). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated to give the crude product, which was purified by column chromatography to give N-(4-aminophenethyl)amide (6) in a good yield. Then the mixture of 3 and 6 was added NaCNBH3 in MeOH and the solution was stirred under reflux overnight. After reaction completed, the solvent was removed under reduced pressure and the residue was dissolved in water, the aqueous solution was extracted with ethyl acetate (50 mL 3). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated to give the crude product, which was purified by column chromatography to afford the corresponding compounds (7-26) in good yields.
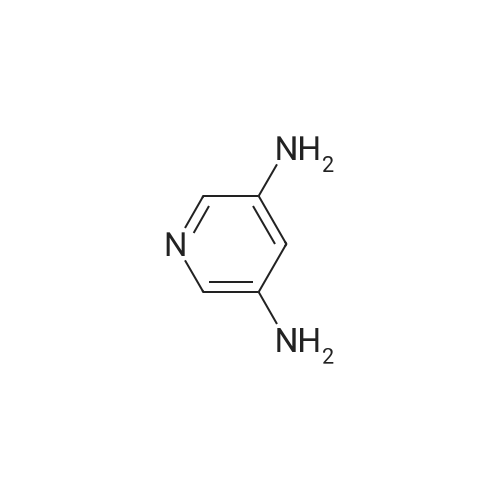
N-(5-aminopyridin-3-yl)-3-phenylpropanamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
59%
With pyridine; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; at 50℃; for 2.0h;
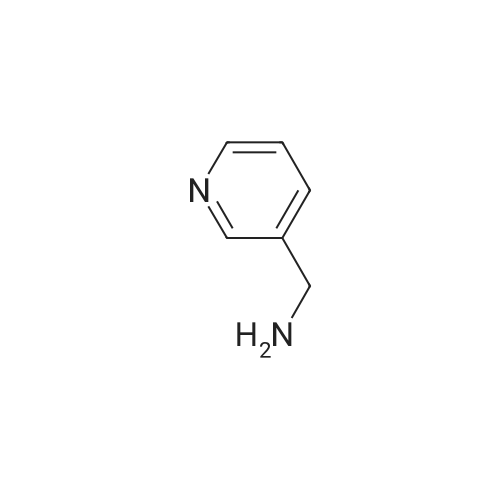
A mixture of 3-phenylpropanoic acid (413 mg, 2.75 mmol), pyridine-3, 5-diamine (300 mg, 2.75 mmol) and EDCI.HC1 (580 mg, 3.03 mmol) in pyridine (5 mL) was heated at 50 C for 2 h. A black solution was formed. The mixture was concentrated and the residue was poured into water (20 mL) and stirred for 2 minutes. The aqueous layer was extracted with ethyl acetate (20 mL x3). The combined organic layer was washed with water (20 mL x2) and brine (20 mL x2), dried over anhydrous Na2S04, filtered and concentrated. The residue was purified by Combi Flash (50% to 100% EtOAc in pentane) to give N-(5-aminopyridin-3- yl)-3-phenylpropanamide (400 mg, yield: 59%) as a light yellow solid. (1398) NMR (400 MHz DMSO-rie) d 2.63 (2H, t , J= 7.7 Hz), 2.91 (2H, t , J= 7.7 Hz), 5.34 (2H, brs), 7.15-7.22 (1H, m), 7.23-7.32 (4H, m), 7.38 (1H, t, J= 2.3 Hz), 7.63 (1H, d, J= 2.5 Hz), 7.86 (1H, d, .7= 2.0 Hz), 9.83 (1H, brs).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping