|
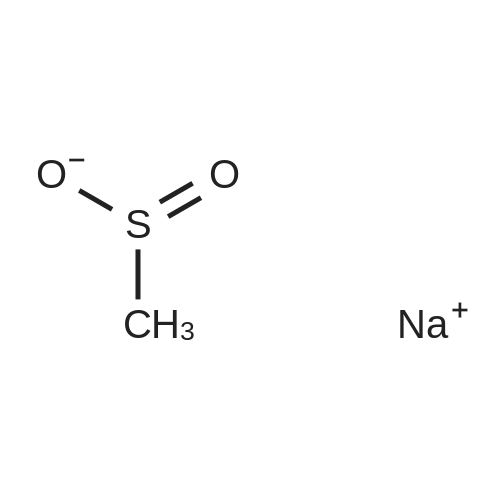
In tetrahydrofuran at 0℃; Molecular sieve; Inert atmosphere; |
4
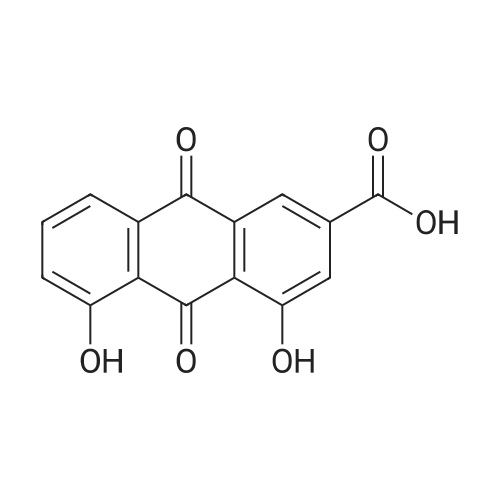
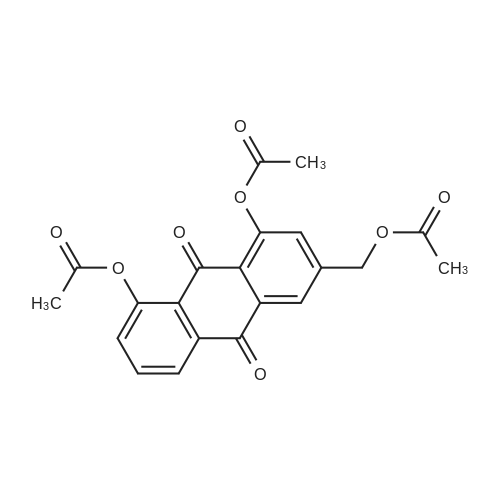
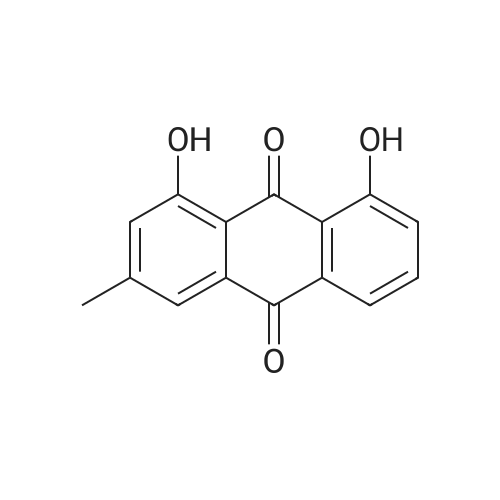
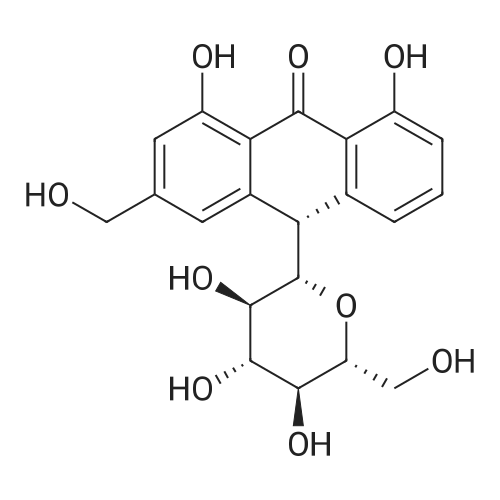
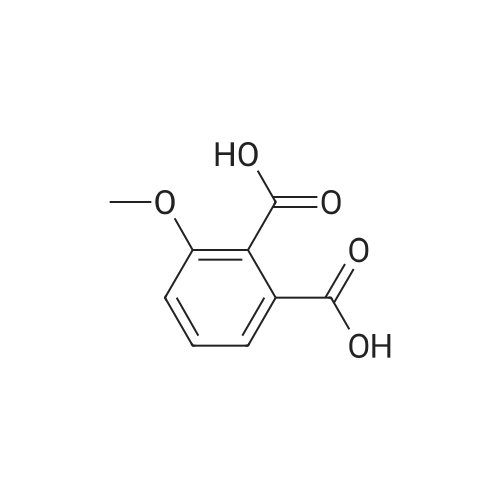
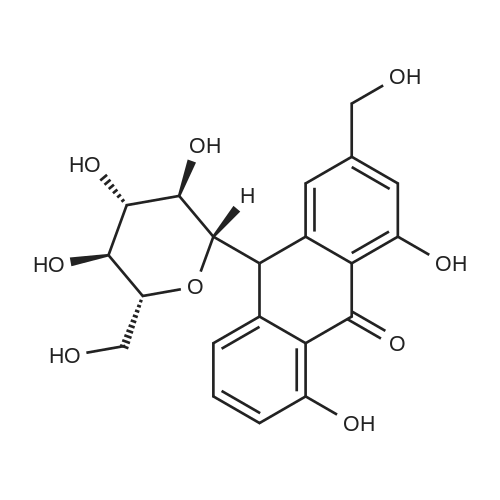
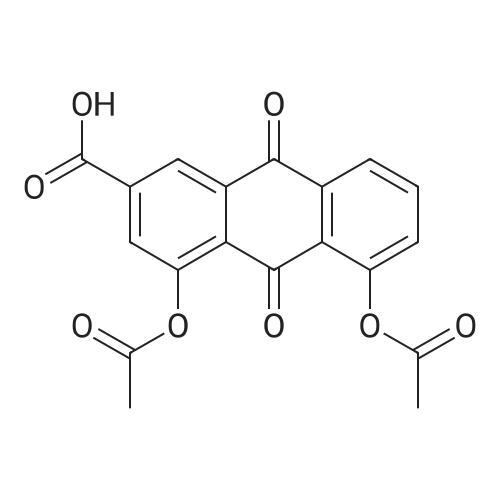
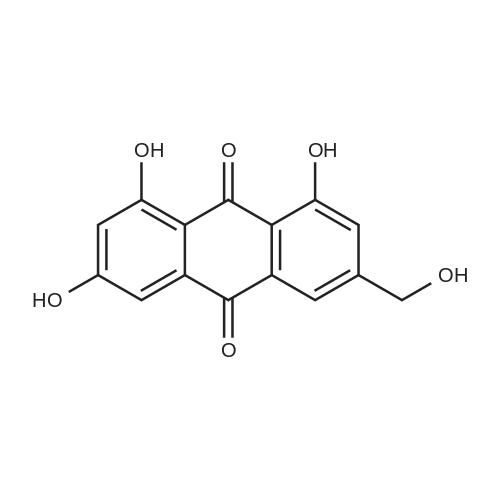
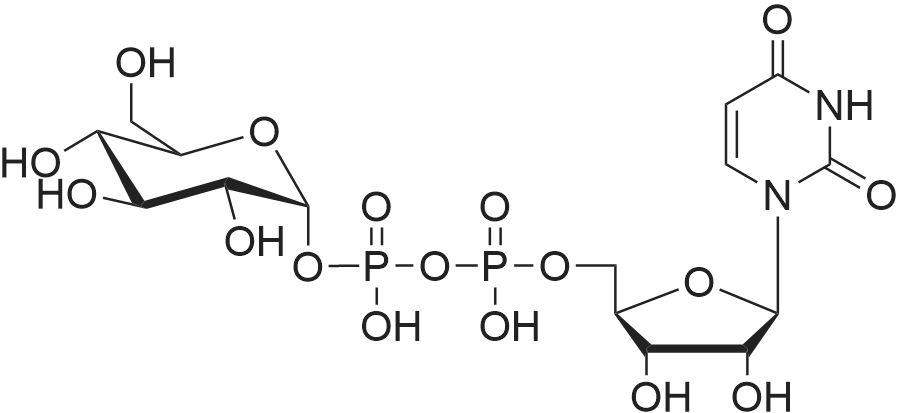
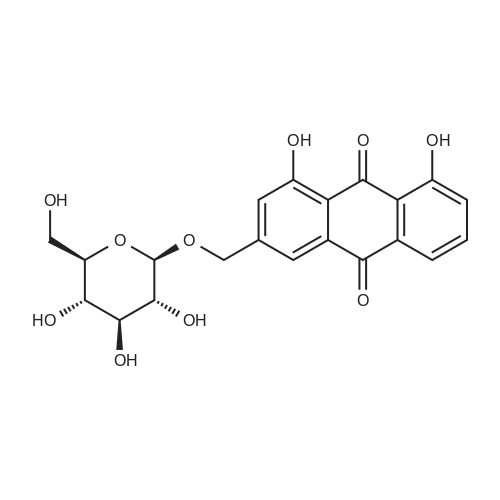
AEGs 15a and 16a: Ristosamine glycosyl donor D-2 (295.0 mg, 1.15 mmol) and AE (283.4 mg, 1.05 mmol) in dry THF (6.0 ml) were added flame dried molecular sieves (4 A, 400 mg) and stirred under argon atmosphere at ambient temperature for 20 min. The reaction mixture was then cooled to 0°C, added trimethylsilyl trifluoromethanesulfonate (60 L, 0.33mmol). Reaction progress was monitored by TLC (70% petroleum ether, 30% ethyl acetate) and indicated the formation of an anomeric mixture of products (a-anomer 15a Rf=0.51, β-anomer 16a Rf=0.69). Upon completion (18h at 0°C) the reaction was quenched by trimethylamine (60 / L) and the crude was filtered through a small plug of celite. The products were isolated by reverse-phase HPLC using a Phenomenex Luna axia 5 μτα C-18 (250 mm x 21.20 mm) column at a flow rate of 20.0 mL/min. The HPLC solvents were A: H20 (0.1% TFA) and B: ACN (0.1% TFA). The elution gradient was 80%B for 2 min followed by 80-100%B over 20 min and product elution was monitored at 256 nm by a UV detector. The product retention times were 8.1 minutes for the a-anomer 15a and 9.4 minutes for the β-anomer 16a. Fractions containing the pure product were concentrated under reduced pressure to yield the pure cc- anomer 15a (113.4mg, 0.24mmol), the pure β-anomer 16a (182.7mg, 0.39mmol) and a mixture of both anomers (373. lmg, 0.80mmol). The total isolated yield of the reaction was 69 % with an α:β ratio of 1 :1 as indicated HPLC. NMR (500 MHz, CDC13) for AEG 15a δ: 12.02(s, IH, OH), 12.01(s, IH, OH) 7.78(d, J=7.4 Hz, IH, H-5'), 7.74(s, IH, H-4'), 7.62(t, J=8.0 Hz, IH, H-6'), 7.32(s, IH, H-2'), 7.24(d, J=8.4 Hz, IH, H-7'), 4.85(d, J=4.0 Hz, IH, H-1) , 4.77(d, J=14.0 Hz, IH, H-15'), 4.62(dd, J.=3.5, J2=9.6 Hz, IH, H-4), 4.53(d, J=14.0 Hz,IH, H-15'), 4.17(dq, J,=6.3, J2=9.5, IH, H-5), 4.12(ddd, J,= J2= J3=3.5 Hz, IH, H-3), 2.20(bdd, Ji=3.0, J2=14.9 Hz, IH, H-2eq), 2.07(s, 3H, OCH3), 2.05(ddd, J,= J2=4.2, J3=14.7 Hz, IH, H-2ax), 1.1 l(d, J=6.3 Hz, 3H, H-6). 13C NMR (100.6 MHz, CDC13) for AEG 15a δ: 193.4(C-9'), 182.4(C-10'), 170.9 (COCH3), 163.6, 163.3, 149.5(C-3'), 137.9(C-6'), 134.4 (C- i r, C-14'), 125.4, 122.9, 120.8, 119.2, 1 16.6, 1 15.7, 95.9(C-1), 74.5, 68.7, 63.0, 56.3, 30.4(C-2) , 21.5(COCH5), 18.0(C-6). Positive HRESIMS, m/z calcd 490.1226 for C23H2iN308Na, found 490.1226 [M+Na]+.'H NMR (500 MHz, CDC13) for AEG 16a δ: 12.01(s, IH, OH),I I .99(s, 1Η, OH) 7.77(d, J=7.5 Hz, IH, H-5'), 7.70(s, IH, H-4'), 7.63(t, J=8.0 Hz, IH, H-6') 7.23(d, J=7.9 Hz, IH, H-7'), 7.19(s, IH, H-2'), 4.89(d, J-13.6 Hz, IH, H-15'), 4.77(dd, J,=2.0, J2=8.9 Hz, IH, H-1), 4.64(dd, Ji=3.4, J2=9.2 Hz, IH, H-4), 4.58(d, J=13.6 Hz, IH, H-15'), 4.15(ddd, J J2= J3=3.5 Hz, IH, H-3), 3.93(dq, Ji=6.3, J2=9.2 Hz, IH, H-5), 2.08(ddd, J,=2.2, J2=4.0, J3=12.0 Hz, IH, H-2eq), 2.07(s, 3H, OCH3), 1.87(ddd, J,=3.4, J2=9.0, J3=12.4 Hz, IH, H-2ax), 1.19(d, J=6.4 Hz, 3H, H-6) 13C NMR (125.7 MHz, CDC13) for AEG 16a δ: 192.2(C- 9'), 181.2(C-10'), 169.6 (COCH3), 162.3, 162.0, 148.0(C-3'), 136.7(C-6'), 133.1(C-1 1', C-14'), 124.2, 121.8, 1 19.6, 1 18.0, 115.4, 114.5, 96.7(C-1), 73.8, 68.7, 67.8, 57.1 , 34.8(C-2), 20.2(COCH3), 17.3(C-6). Positive HRESIMS, m/z calcd 490.1226 for C23H2iN308Na, found 490.1225 [M+Na]+. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping