* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With sodium tetrahydroborate In methanol at 0 - 20℃;
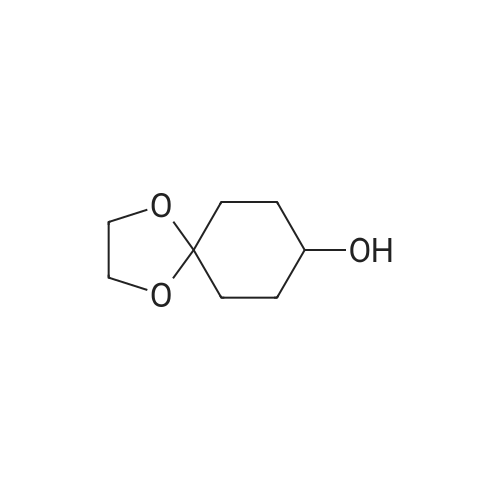
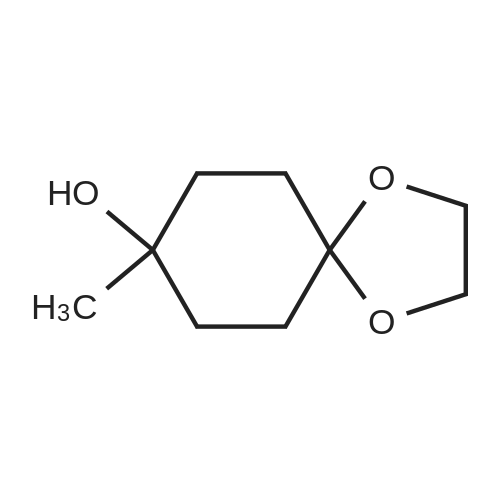
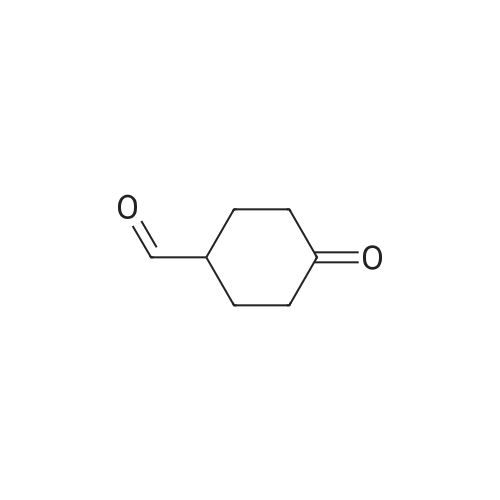
Step A: Preparation of 4-dioxaspiro[4.5]decan-8-ol A solution of 4-dioxaspiro[4.5]decan-8-on (5.0134 g, 32.10 mmol) in methanol (100 mL) was cooled in an ice bath. Sodium borohydride (3.64 g, 96.30 mmol) was added in portions over 20 minutes to the solution. The mixture was stirred at 0° C. for 30 minutes. The reaction mixture was warmed to room temperature and stirred at room temperature for 1 hour. After the standard work up, the title compound (5.56 g, 109percent) was obtained as yellow oil, which was used in the subsequent step without further purification. 1H NMR (400 MHz, CHLOROFORM-D) δ ppm 1.47-1.70 (m, 5 H), 1.72-1.92 (m, 4 H), 3.70-3.83 (m,1H), 3.85-3.96 (m, 4 H).
100%
With sodium tetrahydroborate; water In methanol at 0℃;
EXAMPLE 35 Preparation of 6-(((S)-l -amino- l-oxopropan-2-yl)amino)-2-(4-(4- (trifluoromethyl) phenoxy)cyclohex-l-en-l-yl)pyrimidine-4-carboxamide (Cpd No. 103) Scheme 77 Synthesis of l,4-dioxaspiro[4.5]decan-8-ol (compound 35-1) NaBH4 (370 mg, 10 mmol) in 5 mL H20 was slowly added to 10 mL of MeOH solution of l ,4-dioxaspiro[4.5]decan-8-one (1.56 g, 10 mmol) at 0°C. After the addition, the methanol was removed and the residue was extracted with EtOAc (2 x 20 mL). The EtOAc layer was dried over MgS04, filtered, and evaporated to give l,4-dioxaspiro[4.5]decan-8-ol (compound 35-1), which was used in next step without further purification (1.56 g, yield 100percent).
100%
With sodium tetrahydroborate In methanol; water at 0℃; for 0.166667 h;
NaBH4 (370 mg, 10 mmol) in 5 mL H2O was slowly added to 10 mL of MeOH solution of 1,4-dioxaspiro[4.5]decan-8-one (1.56 g, 10 mmol) at 0°C. After the addition, the methanol was removed and the residue was extracted with EtOAc (2 x 20 mL). The EtOAc layer was dried over MgSO4, filtered, and evaporated to give 1,4-dioxaspiro[4.5]decan-8-ol (compound 35-1), which was used in next step without further purification (1.56 g, yield 100percent).
100%
Stage #1: With sodium tetrahydroborate In methanol at 0 - 18℃; for 1 h; Stage #2: With water In dichloromethane
V) Synthesis of CP30347-8; 4,4-Ethylenedioxycyclohexan-l-ol (41); Following protocols reported by Kitano et al.,s a magnetically stirred solution of 1,4- cyclohexanedione monoethylene acetal 40 (5.00 g, 32.0 mmol) in MeOH (30 mL), maintained at 0 0C, was treated with sodium borohydride (1.57 g, 41.5 mmol). After 0.5 h, the reaction mixture was warmed to 18 °C and stirred at this temperature for an additional 0.5 h. The solvent was then removed under reduced pressure and the ensuing residue partitioned between H2O (30 mL) and CH2Cl2 (30 mL). The separated aqueous phase was extracted with CH2Cl2 (1 x 20 mL) and the combined organic fractions were then dried (MgSO4), filtered and concentrated under reduced pressure to afford the title compound 41 (5.06 g, quant.) as a colourless oil. <n="84"/>1H NMR (300 MHz) δ 3.96-3.88 (complex m, 4H), 3.77 (m, IH), 1.90-1.75 (complex m, 5H), 1.68-1.50 (complex m, 4H).
100%
at 0 - 20℃; for 3 h;
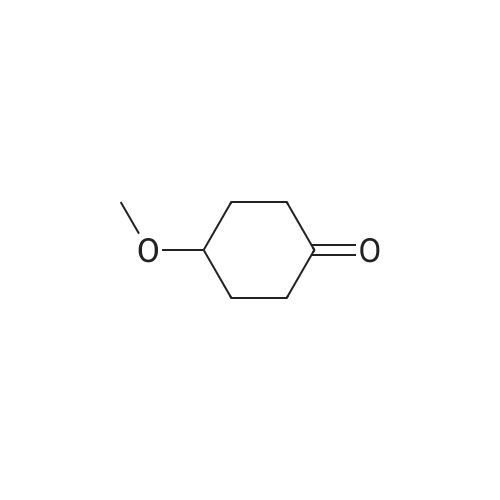
The reduction of the free keto group in 3 followed by the methylation of the resulting alcohol affords methoxy derivative 5.
99%
With methanol; sodium tetrahydroborate In tetrahydrofuran at 0℃; for 3 h; Inert atmosphere
Into a 2-L 3-necked round-bottom flunder nitrogen, was placed 153.1 (100 g, 640.29 mmol, 1.00 equiv), THF (0.8 L), MeOH (0.16 L) and NaBH4 (12.18 g, 321.97 mmol, 0.50 equiv). The reaction was stirred for 3 h at 0 °C in a water/ice bath. The reaction was then quenched by the addition of 1 L of H4C1 (aq). The resulting mixture was concentrated under vacuum, and then extracted with 3 x 2 L of EtOAc . Organic layers were combined and washed with 3 x 500 mL of Brine. The crude was purified by column chromatography to provide 100 g (99.0 percent) of 153.2 as light yellow oil.
96%
With lithium aluminium tetrahydride In diethyl ether at 0 - 20℃; Inert atmosphere
Synthesis of compound 7 (1,4-dioxaspiro[4.5]denan-8-ol)In a dry 250 ml flask 2.5 g (68.5 mmole, 1 eq.) LiAlH4 was suspended in 20 ml dry diethyl ether. The flask was placed under inert N2-atmosphere and cooled to 0° C. 10.4 g (66.6 mmole, 1 eq.) 1,4-dioxaspiro[4.5]decan-8-one 6 was dissolved in 100 ml dry diethyl ether and slowly added to the suspension. The reaction mixture was stirred for 30 minutes at room temperature. Water, diluted with THF, was added in order to remove the excess LiAlH4. The reaction mixture was filtrated over MgSO4 and the volatile components were removed by evaporation.
96%
at 20℃; for 3 h;
l,4-Dioxaspiro[4.5]decan-8-ol (14-1): Sodium borohydride (0.8 g, 21 mmol, 1.05 equiv) was added at 0 to 10 °C in portions to a solution ofl ,4-dioxaspiro[4.5]decan-8-one (SM14) (3.1 g, 20 mmol, 1.0 equiv) was dissolved in methanol (50 mL). The mixture was stirred at room temperature for 3 h and concentrated under reduced pressure to remove most of solvent. Water (50 mL) was added and the mixture and stirred for 30 minutes. The mixture was extracted with ethyl acetate (3 x 50 mL)and the combined organic layers were washed with sequentially with IN HC1 (30 mL), water and saturated brine . The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure to give 14-1 (3.0 g, 96percent yield) as a colorless oil which was used subsequently .
96%
Stage #1: With sodium tetrahydroborate In ethanol at 0 - 20℃; for 1 h; Stage #2: With sodium hydroxide; water In ethanol
Description 1; . 1,4-dioxaspiro[4.5]decan-8-ol (D1) <n="52"/>1 ,4-Dioxaspiro[4.5]decan-8-one (256 mmol, 40 g) was dissolved in EtOH (500 ml) and treated with NaBH4 (1.2eq., 307.2 mmol, 11.6 g), at O 0C and the mixture was stirred at room temperature for 1 hour. Reaction was quenched with NaOH (200 ml, 2N aqueous solution). The aqueous solution was extracted with dichloromethane. The organic extracts were combined, dried over Na2SO4, filtered and the solvent was evaporated to afford the title compound, (38.7 g, 96percent) as colourless oil. 1H NMR δ(d6-DMSO, 400 MHz) 1.446 (4H, m), 1.661 (4H, m), 3.537 (1 H, broad), 3.828 (4H, m), 4.529 (1 H, d).
95%
With sodium tetrahydroborate In methanol for 0.5 h;
1 ,4-Dioxaspiro[4.5]decan-8-one (60 g, 384 mmol) was dissolved in methanol (600 mL) under Argon, then sodium borohydride (15.99 g, 423 mmol) was added portionwise (the addition was exothermic and a huge gas evolution was observed). The resulting mixture was stirred for 30 min. The reaction was quenched with water (200 mL) and stirred for 10 min. Solvent was removed under reduced pressure and the residue taken-up with DCM (600 mL) and water (300 mL). Phases were separated then the aqueous phase extracted with DCM (1 x 600 mL). Combined organic phases were dried on Na2SO4 and concentrated under vacuum to obtain title material (58 g; 95percent) as colourless oil.1H NMR δ (CDCI3, 400 MHz): 1.64 (4H, m), 1.87 (4H, m), 3.83 (1 H, m), 3.97 (4H, dt).
95.2%
Stage #1: With sodium tetrahydroborate In methanol at 0℃; for 1.5 h; Stage #2: With hydrogenchloride In methanol; water
To a solution of 1,4-cyclohexanedione monoethylene acetal (3.41 g, 21.8 mmol) in 50 ml methanol cooled to 0° C. was added sodium borohydride (0.826 g, 21.8 mmol) in portions. The reaction was stirred for an additional 1.5 hr before being brought to pH 7 by the addition of 1 N HCl. The mixture was partitioned between ethyl acetate and brine. The aqueous layer was concentrated to the point that a precipitate began to form and this layer was extracted twice with ethyl acetate. The combined organic layers were dried over sodium sulfate, filtered and concentrated. This crude alcohol (3.28 g, 95.2percent) was used without further purification. 1H NMR (CDCl3) δ: 1.54-1.87 (8H, m, 4.x.-CH2-), 3.77 (1H, m, -CH-), 3.91 (4H, t, 2.x.O-CH2-).
94%
With sodium tetrahydroborate In methanol at 0℃; for 3 h;
NaBH4 (2.42 g, 64 mmol) was added portionwise to a cold (0 0C) solution of 1 ,4- dioxaspiro[4.5]decan-8-one (5.00 g, 32 mmol) in MeOH (30 ml_). The solution was stirred at 0 0C for 3 hours, concentrated under reduced pressure and 5percent aqueous NaOH was added. The solution was extracted with 2-propanol/CHCI3 (1 :4, 2x100 ml_). The combined organic extracts were dried (MgSO4), filtered and concentrated under reduced pressure to afford the title compound as an oil (4.74 g, 94 percent yield).1H NMR (300 MHz, CDCI3) 4.02-3.92 (4H, m), 3.85-3.77 (1 H, m), 1.94-1.80 (4H, m), 1.73- 1.56 (4H, m).
94%
at 20℃; for 2 h;
Commercially available 1,4-cyclohexadione monoethylene acetal (5 g, 32 mmol) was dissolved in methanol (65 mL) and the mixture was placed in a cold-water bath. Sodium borohydride (1.9 g, 50 mmol) was added in small portions (exotherm). After the addition was completed, the mixture was stirred at room temperature for 2 h. The solvent was removed and the residue was dissolved in ethyl acetate (150 mL), water (40 mL) and 1 M NaOH (10 mL). The organic phase was separated and the aqueous phase was extracted with ethyl acetate (4.x.75 mL). The combined organic phase was dried over Na2SO4, filtered and the solvent was removed to afford the title compound as a colorless liquid (4.8 g, 94percent).1H-NMR (400 MHz, CDCl3): δ=1.53-1.70 (m, 4H), 1.78-1.95 (m, 4H), 3.77-3.83 (m, 1H), 3.94-3.97 (m, 4H)
94%
at 20℃; for 1 h; Inert atmosphere; Cooling with ice
In a 250 ml three-mouth bottle by adding 15.8g (0.1mol) of 1,4-cyclohexanedione monoethylene ketal, 100 ml of anhydrous methanol, under nitrogen protection and ice bath stirring condition for 10 min, then added 1g (1.1mol) of NaBH4, this reaction stirred under room temperature for 60 min, raw material by thin layer chromatography (TLC) to determine a basic after the reaction is complete, add 30 ml of water to stop the reaction, with 30 ml dichloromethane sub-2 time extraction, from the separatory funnel for methylene chloride level, with 20 ml water 2 times of cleaning, the resulting organic phase is dried with anhydrous sodium sulfate sleepovers, pulls out worry, desolution of the oily liquid to obtain yellow product 14.8g, the yield is 94percent.
93%
With sodium hydroxide In methanol; sodium tetrahydroborate; isopropyl alcohol
1,4-dioxaspiro[4.5]decan-8-ol (Intermediate M) 1,4-dioxaspiro[4.5]decan-8-one (150 g, 0.96 mol) was stirred with MeOH (1200 mL) under N2 until dissolution occurred. The reaction mixture was cooled to -5° C. in a drykold/acetone bath and treated portionwise with NaBH4 (72.6g, 1.82 mol) over 2 hrs. (T<10° C.). On complete addition, the mixture was cooled to -10° C. and then left to warm to room temperature and stirred overnight at room temperature. The resulting mixture was evaporated and treated with ice-cold 5N NaOH (400 mL) and extracted with CH2Cl2 (2*500 mL) followed by extraction with 4:1 dichloromethane:isopropanol (2*250 mL). The combined extracts were washed with brine (2*200 mL), dried overnight (Na2SO4) and evaporated to give a colourless oil. This was further dried in vacuo (to remove residual isopropanol) to give 1,4-dioxaspiro[4.5]decan-8-ol 141.8 g, 93percent yield. 1H NMR: CDCl3 (250 MHz): 3.91 (4H, m), 3.81 (1H, m), 1.21-1.88 (8H, m, aliphatic H's).
93%
With sodium hydroxide In methanol; sodium tetrahydroborate
1,4-dioxaspiro[4.5]decan-8-ol (Intermediate M) 1,4-dioxaspiro[4.5]decan-8-one (150 g, 0.96mol) was stirred with MeOH (1200 mL) under N2 until dissolution occurred. Cooled to -5° C. in a drykold/acetone bath and treated portionwise with NaBH4 (72.6 g, 1.82 mol) over 2hrs. (T<10° C.). On complete addition, the mixture was cooled to -10° C. and then left to warm to room temperature. Stirred overnight at room temperature. The resulting mixture was evaporated and treated with ice-cold 5N NaOH (400 mL) and extracted with CH2Cl2 (2*500 mL) followed by extraction with 4:1 dichloromethane:isopropanol (2*250 mL). The combined extracts were washed with brine (2*200 mL), dried overnight (Na2SO4) and evaporated to give a colourless oil. This was further dried in vacuo to give 1,4-dioxaspiro[4.5]decan-8-ol (141.8 g, 93percent yield.)1H NMR: CDCl3 (250 MHz) 3.91 (4H, m), 3.81 (1H, m), 1.21-1.88 (8H, m, aliphatic H's).
93%
With sodium hydroxide In methanol; sodium tetrahydroborate; isopropyl alcohol
1,4-dioxaspiro[4.5]decan-8-ol (Intermediate M) 1,4-dioxaspiro[4.5]decan-8-one (150 g, 0.96 mol) was stirred with MeOH (1200 mL) under N2 until dissolution occurred. The reaction mixture was cooled to -5° C. in a drykold/acetone bath and treated portionwise with NaBH4 (72.6 g, 1.82 mol) over 2 hrs. (T<10° C.). On complete addition, the mixture was cooled to -10° C. and then left to warm to room temperature and stirred overnight at room temperature. The resulting mixture was evaporated and treated with ice-cold 5N NaOH (400 mL) and extracted with CH2Cl2 (2*500 mL) followed by extraction with 4:1 dichloromethane:isopropanol (2*250 mL). The combined extracts were washed with brine (2*200 mL), dried overnight (Na2SO4) and evaporated to give a colourless oil. This was further dried in vacuo (to remove residual isopropanol) to give 1,4-dioxaspiro[4.5]decan-8-ol 141.8 g, 93percent yield. 1H NMR: CDCl3 (250 MHz): 3.91 (4H, m), 3.81 (1H, m), 1.21-1.88 (8H, m, aliphatic H's).
93%
With sodium hydroxide In methanol; sodium tetrahydroborate
1,4-dioxaspiro[4.5]decan-8-ol (Intermediate M) 1,4-dioxaspiro[4.5]decan-8-one (150 g, 0.96 mol) was stirred with MeOH (1200 mL) under N2 until dissolution occurred. Cooled to -5° C. in a drykold/acetone bath and treated portionwise with NaBH4 (72.6 g, 1.82 mol) over 2hrs. (T<10° C.). On complete addition, the mixture was cooled to -10° C. and then left to warm to room temperature. Stirred overnight at room temperature. The resulting mixture was evaporated and treated with ice-cold 5N NaOH (400 mL) and extracted with CH2Cl2 (2*500 mL) followed by extraction with 4:1 dichloromethane:isopropanol (2*250 mL). The combined extracts were washed with brine (2*200 mL), dried overnight (Na2SO4) and evaporated to give a colourless oil. This was further dried in vacuo to give 1,4-dioxaspiro[4.5]decan-8-ol (141.8 g, 93percent yield.)1H NMR: CDCl3 (250 MHz) 3.91 (4H, m), 3.81 (1H, m), 1.21-1.88 (8H, m, aliphatic H's).
93%
at 0 - 20℃; for 3 h;
Step 1. Preparation of 1,4-dioxaspiro[4.5]decan-8-ol. To a solution of 1,4-dioxaspiro[4.5]decan-8-one (15.0 g, 96 mmol) in MeOH (240 mL) was added NaBH4 (4.00 g, 106 mmol) in small portions at 0° C. After being stirred for 3 hours at room temperature, the reaction mixture was concentrated in vacuo and brine was added. The mixture was extracted with EtOAc, dried over anhydrous Na2SO4, filtered and passed through a short pad of silica gel and concentrated in vacuo to give the desired product (14.2 g, 93percent) as a colorless oil. 1H-NMR (400 MHz, CDCl3) δ 1.54-1.67 (4H, m), 1.79-1.91 (5H, m), 3.77-3.83 (1H, m), 3.91-3.98 (4H, m).
92%
With sodium tetrahydroborate In methanol at 0 - 20℃; for 1 h;
To a solution of 1,4-dioxa-spiro [4.5] decan-8-one (16) (Aldrich, 10.0 g, 64.0 mmol) in anhydrous methanol (250 mL) at 0 C was added solid sodium borohydride (4.6 g, 121 mmol). The reaction mixture was allowed to warm to rt over 1 h, whereupon TLC analysis indicated complete reaction. Water (60 mL) was added, and the methanol was removed under reduced pressure. The aqueous residue was partitioned between ethyl acetate (200 mL) and saturated aqueous brine (50 mL). The layers were separated, and the aqueous extracted with addition ethyl acetate (200 mL). The combined organic layers were dried (Mg04), filtered and concentrated under reduced pressure to afford the crude alcohol 17 (9.3 g, 92percent) : Rf = 0.2 (CH2CI2) ; H NMR (300 MHz, CDCI3) (5 3. 95 (s, 4H), 3.85-3. 75 (m, 1 H), 2.00-1. 75 (m, 4H), 1.75-1. 50 (m, 4H).
92%
With sodium tetrahydroborate In methanol at 0 - 20℃; for 1 h;
To a solution of 1,4-dioxa-spiro [4.5] decan-8-one (Aldrich, 10.0 g, 64.0 mmol) in anhydrous methanol (250 mL) at 0 °C was added solid sodium borohydride (4.6 g, 121 mmol). The reaction mixture was then allowed to warm to room temperature over 1 h, whereupon TLC analysis indicated complete reaction. Water (60 mL) was added, and the methanol was removed under reduced pressure. The aqueous residue was partitioned between ethyl acetate (200 mL) and saturated aqueous brine (50 mL). The layers were separated, and the aqueous extracted with addition ethyl acetate (200 mL). The combined organic layers were dried (magnesium sulfate), filtered and concentrated under reduced pressure to afford the crude alcohol 2 (9.3 g, 92percent): Rf = 0.2 (CH2CI2) ; 1H NMR (300 MHz, CDC13) 6 3.95 (s, 4H), 3.85-3. 75 (m, 1 H), 2.00-1. 75 (m, 4H), 1.75-1. 50 (m, 4H).
92%
With sodium tetrahydroborate In methanol at 0 - 20℃; for 1 h;
To a solution of 1,4-dioxa-spiro [4.5] decan-8-one (Aldrich, 10.0 g, 64.0 mmol) in anhydrous methanol (250 mL) at 0°C was added solid sodium borohydride (4.6 g, 121 mmol). The reaction mixture was then allowed to warm to room temperature over 1 h, whereupon TLC analysis indicated complete reaction. Water (60 mL) was added, and the methanol was removed under reduced pressure. The aqueous residue was partitioned between ethyl acetate (200 mL) and saturated aqueous brine (50 mL). The layers were separated, and the aqueous extracted with addition ethyl acetate (200 mL). The combined organic layers were dried (magnesium sulfate), filtered and concentrated under reduced pressure yielding the crude alcohol 2 (9.3 g, 92percent): Rf = 0.2 (CH2CI2) ; H NMR (300 MHz, CDCI3) ; 6 3.95 (s, 4H), 3.85-3. 75 (m, 1 H), 2.00-1. 75 (m, 4H), 1.75-1. 50 (m, 4H).
92%
With sodium tetrahydroborate In methanol at 0 - 20℃; for 1 h;
1,4-dioxa-spiro [4. 5] decan-8-ol (66) from 1,4-dioxa-spiro [4. 5] decan-8-one (65) To a solution of 1,4-dioxa-spiro [4.5] decan-8-one (65) (Aldrich, 10.0 g, 64.0 mmol) in anhydrous methanol (250 mL) at 0 C was added solid sodium borohydride (4.6 g, 121 mmol). The reaction mixture was allowed to warm to rt over 1 h, whereupon TLC analysis indicated complete reaction. Water (60 mL) was added, and the methanol was removed under reduced pressure. The aqueous residue was partitioned between ethyl acetate (200 mL) and saturated aqueous brine (50 mL). The layers were separated, and the aqueous extracted with addition ethyl acetate (200 mL). The combined organic layers were dried (MgS04), filtered and concentrated under reduced pressure to afford the crude alcohol 66 (9.3 g, 92percent): Rf = 0.2 (CH2CI2) ; H NMR (300 MHz, CDCI3) 63. 95 (s, 4H), 3.85-3. 75 (m, 1 H), 2.00-1. 75 (m, 4H), 1.75-1. 50 (m, 4H).
90%
at 0 - 20℃; for 2.25 h;
Sodium borohydride (370 mg, 9.6 mmol) was added portion wise to a stirred solution of 1,4- dioxa-spiro[4.5]decan-8-one (1.0 g, 6.4 mmol) in methanol (10 mL) over a period of 15 mm at 0 °C. Reaction mass was warmed to room temperature and stirred for 2h. Methanol was evaporated and the residue diluted with water. Extracted with ethyl acetate, the organic layerdried over anhydrous sodium sulfate and evaporated to afford 0.9 g (90percent) of 1, 4-dioxa- spiro[4.5]decan-8-ol as a brown oil. [TLC system: 3:7 Ethyl acetate Pet ether; Rrvalue: 0.15]
87%
at 10 - 20℃; for 0.5 h;
To a solution of l,4-dioxaspiro[4.5]decan-8-one (244 g, 1.56 mol, 1.0 eq.) in MeOH (5 L) was slowly added NaBH4 (59 g, 1.56 mol, 1.0 eq.) keeping the internal temperature <10 °C using an ice bath. The ice bath was removed and the mixture was stirred for 30 min at room temperature. The solvent was then removed under reduced pressure and the resulting solid was dissolved in 50percent diethyl ether in EtOAc (5 L), washed with saturated aqueous NH4CI (800 mL X 3), brine (800 mL), dried over Na2S04, filtered, and evaporated to give 215 g (87percent yield) of l,4-dioxaspiro[4.5]decan-8-ol. GC-MS: 159 (M+l)..
84%
With sodium tetrahydroborate In methanol at 0 - 20℃; for 3 h;
1.28 g of Sodium borohydride (33.9 mmol, 1.06 eq) were added to a 0 °C cold solution of 5.00 g of 1,4-dioxaspiro[4.5]decan-8-one, S16, (32.0 mmol, 1.0 eq) in 75 mL of abs. methanol. The mixture was stirred for 3 hours at room temperature. After neutralization with 1 M hydrochlorid acid the solution was extracted with dichoromethane (3 x 100 mL). The combined organic phases were dried over Na2SO4, filtered and the solvent was removed under reduced pressure giving 4.25 g (84percent) of a colorless oil.
84%
With sodium borohydrid In ethanol; water
1. 1,4-Dioxaspiro[4.5]decan-8-ol A stirred, cooled (5° C.) solution of 1,4-dioxaspiro[4.5]decan-8-one (20 g, 0.128 mol) in ethanol (250 ml) was treated with sodium borohydride (7.3 g, 0.192 mol) in portions over 20 minutes. The reaction mixture was stirred at 5° C. for 1 hour then at room temperature for 1 hour. Water (20 ml) was added and the mixture was stirred vigorously for 10 minutes then concentrated in vacuo. The residue was partitioned between water (100 ml) and ethyl acetate (100 ml). The organic layer was separated and the aqueous re-extracted with ethyl acetate (100 ml). The combined organics were dried (sodium sulphate) then evaporated to give the title alcohol as a colourless oil (17.1 g, 84percent). MS ES+, m/z=159 for (M+H)+; δ(360 MHz, CDCl3) 1.52-1.71 (4H, m), 1.78-1.92 (4H, m), 3.76-3.83 (1H, m), 3.91-3.94 (4H, m).
83%
With sodium tetrahydroborate In methanol at 20℃; for 16 h; Inert atmosphere
Into a 500 ml 3-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed 1,4-dioxaspiro[4.5]decan-8-one (20 g, 128.06 mmol, 1.00 equiv.), methanol (200 ml). Then NaBH4 (3.9 g, 105.91 mmol, 0.83 equiv.) was added at 0 °C. The resulting solution was stuffed at room temperature for 16 h. The reaction was then quenched by the addition of 100 ml of NH4C1 (sat. aq.). The resulting mixture was concentrated under vacuum. The resulting solution was diluted with 100 ml of H20. The resulting solution was extracted with 3x200 ml of dichloromethane and the organic layers combined and dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1:15). This resulted in 16.84 g of the title compound as a colorless oil (83percent). ‘H NMR (300 MHz, CDC13) & 4.0 1-3.89 (m, 4H), 3.88-3.76 (m, 1H), 1.95-1.74 (m, 4H), 1.74-1.52 (m, 4H).
82%
With sodium tetrahydroborate In ethanol at 0 - 20℃; for 1 h;
1 ,4-Dioxaspiro[4.5]decan-8-one (64 mmol, 10 g) was dissolved in ethanol (125 ml_) and treated with NaBH4 (1.2 eq., 76.8 mmol, 2.9 g), at 0 0C portionwise and the mixture was stirred at room temperature for 1 hour. Reaction was quenched with NaOH (25 ml_, 2N aqueous solution). The aqueous solution was extracted with dichloromethane (2 x). The organics were combined, dried over Na2SO4, filtered and the solvent was evaporated to afford the title compound, 8.3 g, 82percent, as a colourless oil.1H NMR δ ( DMSO-d6, 400 MHz) 1.44 (4H, m), 1.64 (4H, m), 3.54 (1 H, d broad), 3.82 (4H, m), 4.48 (1 H, d).
82%
Stage #1: With sodium tetrahydroborate In ethanol at 0 - 20℃; for 1 h; Stage #2: With sodium hydroxide In ethanol; water
1 ,4-Dioxaspiro[4.5]decan-8-one (64 mmol, 10 g) was dissolved in ethanol (125 ml) and treated with NaBH4 (1.2eq., 76.8 mmol, 2.9 g), at O0C portionwise and the mixture was stirred at room temperature for 1 hour. Reaction was quenched with NaOH (25 ml, 2N aqueous solution). The aqueous solution was extracted with dichloromethane (2x). The organics were combined, dried over Na2SO4, filtered and the solvent was evaporated to afford the title compound, 8.3 g, 82percent, as a colourless oil.1H NMR δ (d6DMSO, 400 MHz) 1.44 (4H, m), 1.64 (4H, m), 3.54 (1 H, d broad), 3.82 (4H, m), 4.48 (1 H, d).
82%
Stage #1: With sodium tetrahydroborate In ethanol at 0 - 20℃; for 1 h; Stage #2: With sodium hydroxide; water In ethanol
Description s. 1 ,4-Dioxaspiro[4.5]decan-8-ol (D8); .1 ,4-Dioxaspiro[4.5]decan-8-one (64 mmol, 10 g) was dissolved in ethanol (125 ml) and treated portionwise with NaBH4 (1.2eq., 76.8 mmol, 2.9 g), at O0C and the mixture was stirred at room temperature for 1 hour. Reaction was quenched with NaOH (25 ml, 2N aqueous solution). The aqueous solution was extracted with dichloromethane. The organic extracts were combined, dried over Na2SC>4, filtered and the solvent was evaporated to afford the title compound, 8.3 g, 82percent, as a colourless oil. 1H NMR δ (d6DMSO, 400 MHz) 1.44 (4H, m), 1.64 (4H, m), 3.54 (1 H, d broad), 3.82 (4H, m), 4.48 (1 H, d).
82%
Stage #1: With sodium tetrahydroborate In ethanol at 0 - 20℃; for 1 h; Stage #2: With sodium hydroxide; water In ethanol
Description 3. 1,4-Dioxaspiro[4.5]decan-8-ol (D3); 1 ,4-Dioxaspiro[4.5]decan-8-one (64 mmol, 10 g) was dissolved in ethanol (125 ml) and treated with NaBH4 (1.2eq., 76.8 mmol, 2.9 g), at O0C and the mixture was stirred at room temperature for 1 hour. Reaction was quenched with NaOH (25 ml, 2N aqueous solution). The aqueous solution was extracted with dichloromethane (2x). The organics were combined, dried over Na2SO4, filtered and the solvent was evaporated to afford the title compound, 8.3 g, 82percent, as a colourless oil.1HNMR q (d6DMSO, 400 MHz) 1.443 (4H, m), 1.645 (4H, m), 3.549 (1 H, d broad), 3.827 (4H, m), 4.480 (1 H, d).
82%
Stage #1: With sodium tetrahydroborate In ethanol at 0 - 20℃; for 1 h; Stage #2: With sodium hydroxide; water In ethanol
Description 4. 1,4-Dioxaspiro[4.5]decan-8-ol (D4); 1 ,4-Dioxaspiro[4.5]decan-8-one (64 mmol, 10 g) was dissolved in ethanol (125 ml) and treated with NaBH4 (1.2eq., 76.8 mmol, 2.9 g), at O0C and the mixture was stirred at room temperature for 1 hour. Reaction was quenched with NaOH (25 ml, 2N aqueous solution). The aqueous solution was extracted with dichloromethane (2x). The organics were combined, dried over Na2SO4, filtered and the solvent was evaporated to afford the title compound, 8.3 g, 82percent, as a colourless oil. 1HNMR D (d6DMSO, 400 MHz): 1.44 (4H, m), 1.65 (4H, m), 3.55 (1 H, d broad), 3.83 (4H, m), 4.48 (1 H, d).
63%
With lithium aluminium tetrahydride In tetrahydrofuran at 0℃; for 2 h;
At RT, l,4-dioxaspiro[4.5]decan-8-one (5.00 g, 32.01 mmol, 1.00 equiv) was dissolved in THF (50 mL). This was followed by the addition of LiAlH4 (1.22 g, 32.14 mmol, 1.00 equiv) in portions at 0 °C over 5 min period. The mixture was then stirred for 2 h at 0 °C. The reaction was then quenched by the addition of 10 mL water. The pH value of this mixture was adjusted to 1.0 with HC1 solution (2 M), which was extracted with 3 x 100 mL ethyl acetate. The organic phases were combined and washed with 2 x 50 mL brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure to yield l,4-dioxaspiro[4.5]decan-8-ol as colorless oil (3.2 g, 63percent).
61%
Stage #1: With sodium tetrahydroborate In methanol at 20℃; for 0.0833333 h; Stage #2: With hydrogenchloride In methanol; water at 0℃;
Example 117 cis-2-methyl-3-(4-[4-(1-pyrrolidinyl)cyclohexyl]oxy}phenyl)-4(3H)-quinazolinone and trans-2-methyl-3-(4-[4-(1-pyrrolidinyl)cyclohexyl]oxy}phenyl)-4(3H)-quinazolinone (1) Manufacture of 1,4-dioxaspiro[4.5]decan-8-ol 1,4-dioxaspiro[4.5]decan-8-one (1.0 g, 6.40 mmol) was dissolved in methanol (10 mL), sodium borohydride (242 mg, 6.40 mmol) was slowly added, and stirred at room temperature for 5 minutes. The mixture was cooled on an ice bath, 10percent hydrochloric acid aqueous solution and sodium chloride were added, and the mixture extracted with ethyl acetate. The organic phase was washed with saturated brine, and dried by anhydrous sodium sulfate. The sodium sulfate was filtered off, and the product concentrated under reduced pressure to obtain the target compound (614 mg, 61percent) as a light yellow oily substance.
59%
With sodium tetrahydroborate In methanol at 0 - 20℃; for 2 h;
To a stirred solution of 1,4-dioxaspiro[4.5]decan-8-one (5 g, 32.0 mmol, Lobochem) in dry MeOH (50 mL) was added Sodium borohydride (1 .8 g, 48.0 mmol) portion wise at 0 00. The reaction mixture was stirred at RT for 2h. Reaction completion was monitored by TLC. Reaction mixture was quenched with ice. The reaction mixture was concentrated completely and extracted with dichloromethane, washed with water (2 x 30 mL) andbrine solution (2 x 20 mL). Combined organic layers were dried over anhydrous Na2504, filtered, concentrated and the crude mass obtained as brown liquid was used as such for next step without further purification (3 g, 59percent). 1H NMR (400 MHz, DMSO-d6: 6 4.47 (d, J = 4.0 Hz, 1 H), 3.85-3.80 (m, 4H), 3.55 (s, 1 H), 1.66-1.64 (m, 4H), 1.48-1.42 (m, 4H).
750 mg
With sodium tetrahydroborate In methanol at 30℃; for 1 h;
To a solution of 1 ,4-dioxaspiro[4.5]decan-8-one (1 .0 g) in methanol (3 mL) was added NaBH4 (0.2 g). The mixture was stirred at 30°C for 1 hour, and then poured into H20 (20 mL). The mixture was extracted with EA (10 mL). The organic layer was washed with brine, driedover Na2SO4, and concentrated to afford 1 ,4-dioxaspiro[4.5]decan-8-ol (750 mg).
57 g
at 0 - 25℃; Inert atmosphere
To a mixture of l,4-dioxaspiro[4.5]decan-8-one (60 g, 384 mmol) in MeOH (750 mL) was added NaBH4 (43.6 g, 1.15 mol) slowly at about 0° C under N2 atmosphere. The reaction was stirred at about 25 °C for about 2. The reaction mixture was cooled to about 0° C and water was added to quench the reaction. The reaction was concentrated to remove the MeOH and the residue was extracted with EtOAc (3 x200 mL). The organics were combined, dried over MgS04 filtered and concentrated to give the intermediate, 1,4- dioxaspiro[4.5]decan-8-ol (57 g).
Reference:
[1] Journal of Medicinal Chemistry, 1992, vol. 35, # 12, p. 2243 - 2247
[2] Journal of the American Chemical Society, [3] Journal of the American Chemical Society, 2009, vol. 131, p. 251 - 262
[4] Patent: US2009/275574, 2009, A1, . Location in patent: Page/Page column 16
[5] Patent: WO2013/30665, 2013, A1, . Location in patent: Page/Page column 175; 176
[6] Patent: WO2014/135955, 2014, A1, . Location in patent: Paragraph 0516
[7] Patent: WO2008/124878, 2008, A1, . Location in patent: Page/Page column 82-83
[8] Patent: WO2008/128058, 2008, A1, . Location in patent: Page/Page column 11
[9] Patent: WO2017/75056, 2017, A1, . Location in patent: Paragraph 0848
[10] Canadian Journal of Chemistry, 1994, vol. 72, # 7, p. 1699 - 1704
[11] Journal of the Chemical Society. Perkin Transactions 2, 1997, # 6, p. 1221 - 1234
[12] Journal of Organic Chemistry, 2006, vol. 71, # 22, p. 8424 - 8430
[13] ACS Medicinal Chemistry Letters, 2014, vol. 5, # 8, p. 851 - 856
[14] Bioorganic and Medicinal Chemistry Letters, 2015, vol. 25, # 3, p. 695 - 700
[15] Patent: US2009/275616, 2009, A1, . Location in patent: Page/Page column 7
[16] Patent: WO2012/151343, 2012, A1, . Location in patent: Page/Page column 35-36
[17] Angewandte Chemie - International Edition, 2016, vol. 55, # 5, p. 1816 - 1819[18] Angew. Chem., 2016, vol. 128, # 5, p. 1848 - 1851,4
[19] Patent: WO2008/119713, 2008, A1, . Location in patent: Page/Page column 50-51
[20] Patent: WO2009/37294, 2009, A1, . Location in patent: Page/Page column 98
[21] Patent: US2006/292073, 2006, A1, . Location in patent: Page/Page column 11-12
[22] Chemical and Pharmaceutical Bulletin, 1993, vol. 41, # 11, p. 1971 - 1986
[23] Patent: WO2008/53136, 2008, A1, . Location in patent: Page/Page column 41
[24] Patent: US2011/280808, 2011, A1, . Location in patent: Page/Page column 59
[25] Journal of Polymer Science, Part A: Polymer Chemistry, 2011, vol. 49, # 2, p. 369 - 380
[26] Patent: CN104151209, 2016, B, . Location in patent: Paragraph 0023-0025
[27] Patent: US2002/156081, 2002, A1,
[28] Patent: US2002/156081, 2002, A1,
[29] Patent: US6921763, 2005, B2,
[30] Patent: US6921763, 2005, B2,
[31] Patent: US2012/53180, 2012, A1, . Location in patent: Page/Page column 6
[32] Tetrahedron Letters, 2001, vol. 42, # 38, p. 6633 - 6636
[33] Patent: WO2005/87751, 2005, A2, . Location in patent: Page/Page column 166
[34] Patent: WO2005/70407, 2005, A1, . Location in patent: Page/Page column 243-244
[35] Patent: WO2005/87215, 2005, A1, . Location in patent: Page/Page column 277-278
[36] Patent: WO2005/87752, 2005, A2, . Location in patent: Page/Page column 110-111
[37] Patent: WO2017/9651, 2017, A1, . Location in patent: Page/Page column 49
[38] Patent: WO2011/143495, 2011, A1, . Location in patent: Page/Page column 92
[39] European Journal of Medicinal Chemistry, 2014, vol. 89, p. 503 - 523
[40] Patent: US6211219, 2001, B1,
[41] Patent: WO2016/44626, 2016, A1, . Location in patent: Paragraph 00518
[42] Patent: WO2009/37294, 2009, A1, . Location in patent: Page/Page column 97-98
[43] Patent: WO2007/107565, 2007, A1, . Location in patent: Page/Page column 48-49
[44] Patent: WO2008/119721, 2008, A1, . Location in patent: Page/Page column 53
[45] Patent: WO2007/107566, 2007, A1, . Location in patent: Page/Page column 46
[46] Patent: WO2007/107567, 2007, A1, . Location in patent: Page/Page column 45
[47] Patent: WO2015/17502, 2015, A1, . Location in patent: Paragraph 00312
[48] Patent: US2005/182045, 2005, A1, . Location in patent: Page/Page column 50
[49] Patent: WO2014/198808, 2014, A1, . Location in patent: Page/Page column 138
[50] Organic Mass Spectrometry, 1982, vol. 17, # 2, p. 102 - 106
[51] Journal of Materials Chemistry, 1997, vol. 7, # 3, p. 391 - 401
[52] Synlett, 1998, # 7, p. 723 - 724
[53] Journal of Mass Spectrometry, 1998, vol. 33, # 3, p. 229 - 241
[54] Journal of Organic Chemistry, 1998, vol. 63, # 23, p. 8320 - 8330
[55] Tetrahedron, 2002, vol. 58, # 1, p. 129 - 133
[56] Canadian Journal of Chemistry, 2003, vol. 81, # 1, p. 81 - 108
[57] Journal of Medicinal Chemistry, 2000, vol. 43, # 17, p. 3322 - 3334
[58] Journal of the Chemical Society. Perkin Transactions 1, 2002, # 20, p. 2251 - 2255
[59] Heterocycles, 2007, vol. 71, # 2, p. 419 - 428
[60] Patent: US2002/156081, 2002, A1,
[61] Patent: US6071954, 2000, A,
[62] Patent: US5536725, 1996, A,
[63] Patent: US5595992, 1997, A,
[64] Patent: US5691351, 1997, A,
[65] Patent: EP970067, 2003, B1,
[66] Patent: US6921763, 2005, B2,
[67] Journal of Medicinal Chemistry, 2008, vol. 51, # 23, p. 7523 - 7531
[68] Patent: WO2010/6086, 2010, A2, . Location in patent: Page/Page column 87-88
[69] European Journal of Organic Chemistry, 2010, # 6, p. 1017 - 1020
[70] Patent: WO2010/57121, 2010, A1, . Location in patent: Page/Page column 161
[71] Patent: US2007/197526, 2007, A1, . Location in patent: Page/Page column 24; 25
[72] Patent: WO2011/7146, 2011, A1, . Location in patent: Page/Page column 60
[73] Journal of Organic Chemistry, 2011, vol. 76, # 19, p. 7706 - 7719
[74] Organic Letters, 2012, vol. 14, # 1, p. 62 - 65
[75] Patent: US2012/71461, 2012, A1, . Location in patent: Page/Page column 143
[76] Organic and Biomolecular Chemistry, 2012, vol. 10, # 36, p. 7321 - 7326
[77] European Journal of Organic Chemistry, 2013, # 18, p. 3741 - 3750
[78] RSC Advances, 2014, vol. 4, # 5, p. 2226 - 2234
[79] Patent: WO2014/48065, 2014, A1, . Location in patent: Page/Page column 91
[80] Synthesis (Germany), 2014, vol. 46, # 11, p. 1455 - 1462
[81] Patent: WO2014/139325, 2014, A1, . Location in patent: Page/Page column 254
[82] Patent: EP2813505, 2014, A1, . Location in patent: Paragraph 0212
[83] Chemistry - A European Journal, 2015, vol. 21, # 7, p. 2785 - 2788
[84] Journal of the American Chemical Society, 2015, vol. 137, # 25, p. 7998 - 8001
[85] Patent: WO2015/138220, 2015, A1, . Location in patent: Page/Page column 88
[86] Patent: US2015/329529, 2015, A1, . Location in patent: Paragraph 0507-0508
[87] Patent: WO2015/181186, 2015, A1, . Location in patent: Page/Page column 126; 127
[88] Small, 2016, vol. 12, # 10, p. 1378 - 1390
[89] Patent: WO2016/168641, 2016, A1, . Location in patent: Page/Page column 196
[90] Patent: WO2017/46604, 2017, A1, . Location in patent: Paragraph 00286; 00287; 00288
[91] Patent: WO2017/4500, 2017, A1, . Location in patent: Page/Page column 162
[92] Patent: US2017/247323, 2017, A1, . Location in patent: Paragraph 0479; 0480
[93] Patent: WO2018/5860, 2018, A1, . Location in patent: Page/Page column 172; 209; 210
[94] Dalton Transactions, 2018, vol. 47, # 26, p. 8738 - 8745
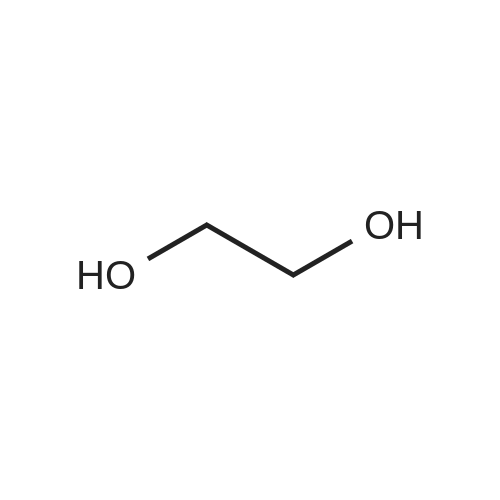
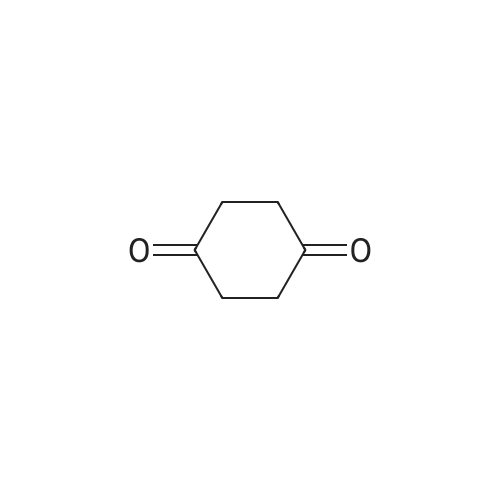
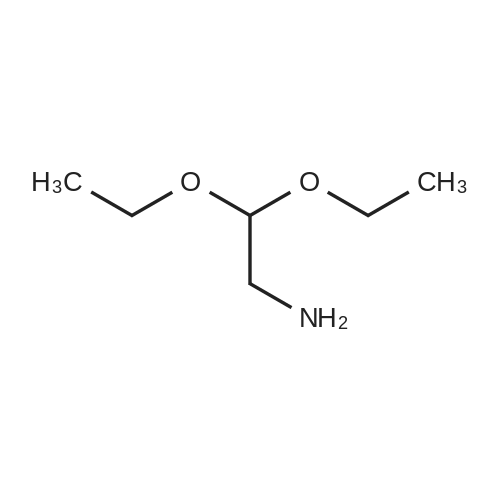
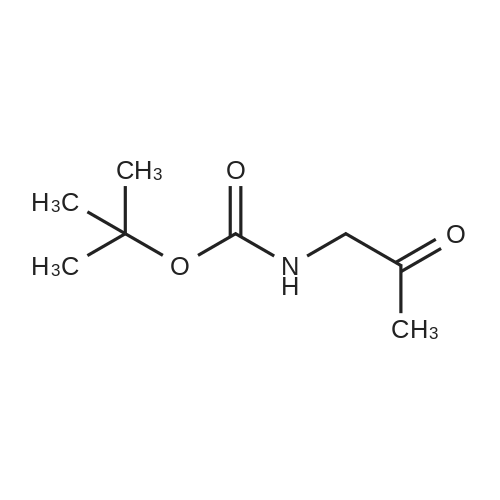
2
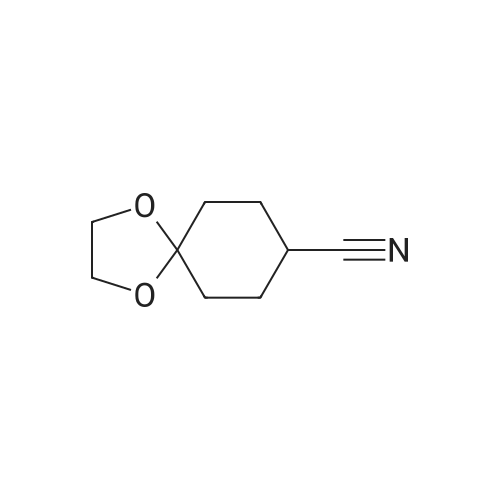
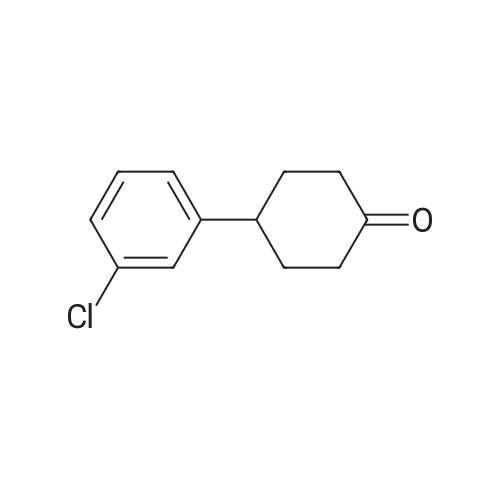
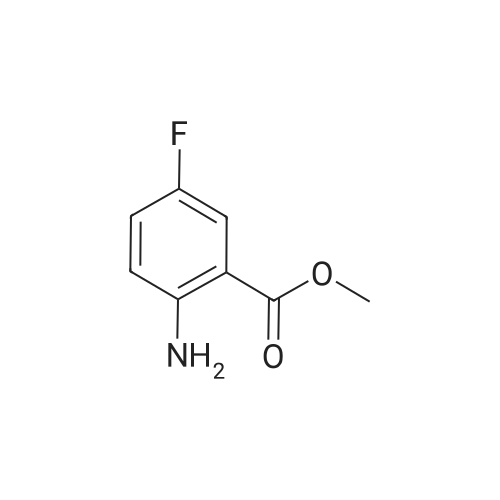
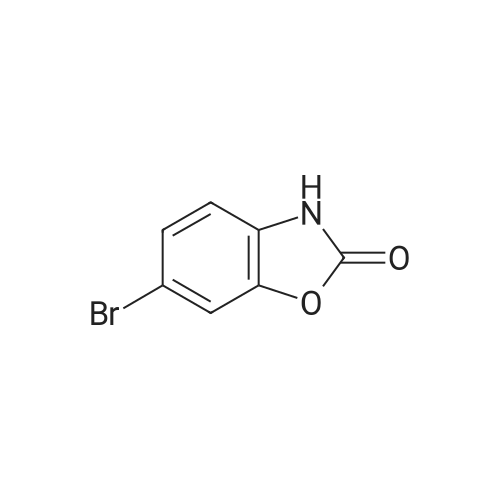
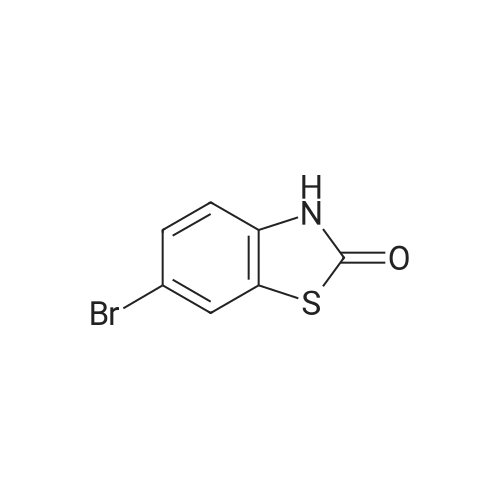
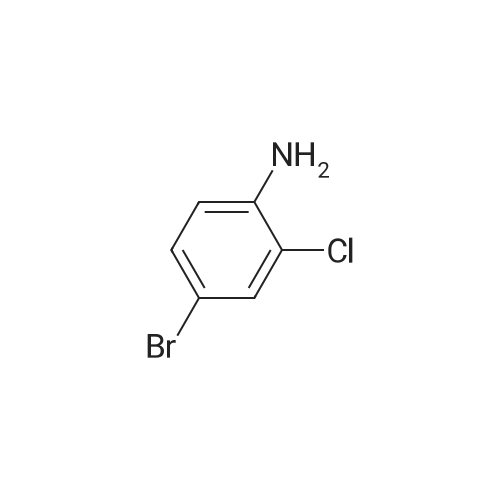
[ 78881-14-8 ]
[ 16940-66-2 ]
[ 4746-97-8 ]
[ 22428-87-1 ]
Reference:
[1] Patent: US5808146, 1998, A,
[2] Patent: US5817776, 1998, A,
3
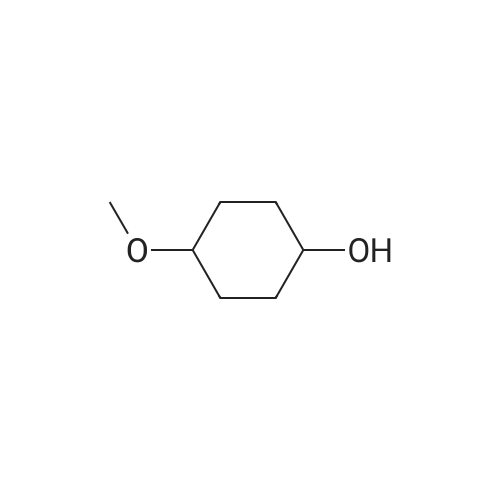
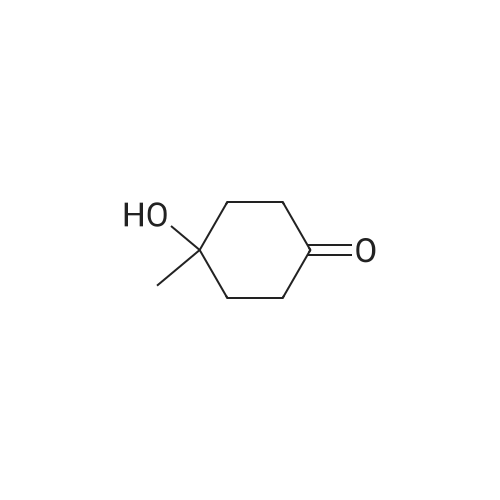
[ 4746-97-8 ]
[ 67-63-0 ]
[ 22428-87-1 ]
Reference:
[1] Patent: US2003/153752, 2003, A1,
4
[ 4746-97-8 ]
[ 621-84-1 ]
[ 22428-87-1 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2013, vol. 11, # 26, p. 4379 - 4382
5
[ 4746-97-8 ]
[ 506-59-2 ]
[ 40594-34-1 ]
Yield
Reaction Conditions
Operation in experiment
17%
With hydrogenchloride; triethylamine In water; 1,2-dichloro-ethane
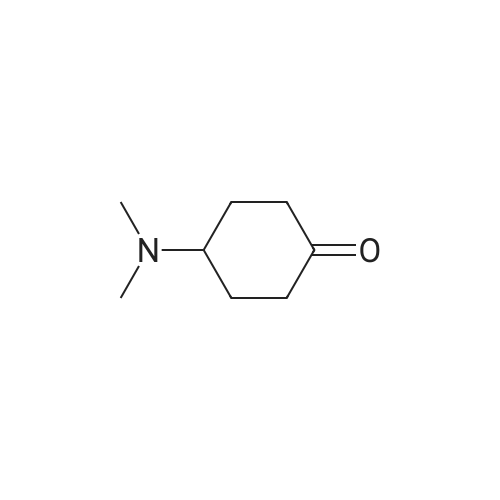
Reference Example 8 To a suspension of 1,4-cyclohexanedione monoethylene-ketal (3.82 g, 24.6 mmol) and dimethylamine hydrochloride (2.00 g, 24.6 mmol) in 1,2-dichloroethane (50 ml) were dropwise added triethylamine (4.2 ml, 29.6 mmol) and DBU (1,8-diazabicyclo-[5.4.0]-7-undecene) (4.4 ml), and the mixture was stirred for 10 minutes. To the mixture was added triacetoxyborohydride (7.68 g, 34.4 mmol), and the mixture was stirred for 4.5 hours. Precipitate was filtered off, and the filtrate was concentrated to give crude product (6.34 g), which was dissolved in water (10 ml). To the mixture was dropwise added concentrated hydrochloric acid (6 ml), and the mixture was stirred for 48 hours. The reaction mixture was diluted with water and washed twice with ether. The aqueous layer was made basic with sodium hydroxide and extracted with ether twice. The extract was washed with saturated sodium chloride solution, dried with potassium carbonate and purified by evaporation to give 4-dimethylaminocyclohexanone (0.59 g, 17percent). b.p.142-145° C. 1 H-NMR (CDCl3) δ: 1.69-2.13 (4H, m), 2.32 (6H, s), 2.20-2.41 (2H, m), 2.44-2.64 (3H, m).
Reference:
[1] Patent: US6096780, 2000, A,
6
[ 4746-97-8 ]
[ 506-59-2 ]
[ 40594-34-1 ]
Yield
Reaction Conditions
Operation in experiment
17%
With triethylamine In water; 1,2-dichloro-ethane
REFERENCE EXAMPLE 174 To a suspension of 1,4-cyclohexanedione monoethyleneketal (3.82 g, 24.6 mmol) and dimethylamine hydrochloride (2.00 g, 24.6 mmol) in 1,2-dichloroethane (50 ml) were dropwise added triethylamine (4.2 ml, 29.6 mmol) and DBU (1,8-diazabicyclo-[5.4.0]-7-undecene) (4.4 ml), and the mixture was stirred for 10 minutes. To the mixture was added triacetoxyborohydride (7.68 g, 34.4 mmol), and the mixture was stirred for 4.5 hours. Precipitate was filtered off, and the filtrate was concentrated to give crude product (6.34 g), which was dissolved in water (10 ml). To the mixture was dropwise added concentrated hydro-chloric acid (6 ml), and the mixture was stirred for 48 hours. The reaction mixture was diluted with water and washed twice with ether. The aqueous layer was made basic with sodium hydroxide and extracted with ether twice. The extract was washed with saturated sodium chloride solution, dried with potassium carbonate and purified by evaporation to give 4-dimethylaminocyclohexanone (0.59 g, 17percent). b.p.142-5° C.; 1 H-NMR (CDCl3) δ:1.69-2.13 (4H, m), 2.32 (6H, s), 2.20-2.41 (2H, m), 2.44-2.64 (3H, m).
Reference:
[1] Patent: US6166006, 2000, A,
7
[ 4746-97-8 ]
[ 40594-34-1 ]
Reference:
[1] Journal of Organic Chemistry, 2003, vol. 68, # 3, p. 770 - 778
[2] Patent: US5821240, 1998, A,
8
[ 4746-97-8 ]
[ 917-54-4 ]
[ 66336-42-3 ]
Yield
Reaction Conditions
Operation in experiment
80%
at -78 - -55℃; for 4 h; Inert atmosphere
Step 1: synthesis of 8-methyl-1,4-dioxaspiro[4.5]decan-8-ol:To a cooled solution of 1,4-dioxaspiro[4.5]decan-8-one (7.8 g, 50.0 mmol) in dry THF (75 mL)was added methyllithium solution (1.5M in ether, 43.3 mL, 65.0 mmol) at -78°C under argonwhile keeping inner temperature below -55 °C. After addition, the mixture was stirred at -55 °Cfor additional 4 hours. The reaction was warmed to room temperature and then quenched bysaturated NH4C1 solution. The separated organic layer was concentrated in vacuo and the residue was purified by column chromatography on silica gel (EA : PE =1:9 to 1:3) to afford the product as a white solid (6.8 g, yield: 80percent).
80%
at -78 - -55℃; for 4 h; Inert atmosphere
Step 1: synthesis of 8-methyl-1,4-dioxaspiro[4.5]decan-8-ol To a cooled solution of 1,4-dioxaspiro[4.5]decan-8-one (7.8 g, 50.0 mmol) in dry THF (75 mL) was added methyllithium solution (1.5M in ether, 43.3 mL, 65.0 mmol) at -78° C. under argon while keeping inner temperature below -55° C. After addition, the mixture was stirred at -55° C. for additional 4 hours. The reaction was warmed to room temperature and then quenched by saturated NH4Cl solution. The separated organic layer was concentrated in vacuo and the residue was purified by column chromatography on silica gel (EA:PE=1:9 to 1:3) to afford the product as a white solid (6.8 g, yield: 80percent).
80%
at -78 - -55℃; for 4 h;
To a cooled solution of 1,4-dioxaspiro[4.5]decan-8-one (7.8 g, 50.0 mmol) in dry THF (75 mL) was added methyllithium solution (1.5M in ether, 43.3 mL, 65.0 mmol) at −78° C. under argon while keeping inner temperature below −55° C. After addition, the mixture was stirred at −55° C. for additional 4 hours. The reaction was warmed to room temperature and then quenched by saturated NH4Cl solution. The separated organic layer was concentrated in vacuo and the residue was purified by column chromatography on silica gel (EA:PE=1:9 to 1:3) to afford the product as a white solid (6.8 g, yield: 80percent).
58%
Stage #1: at -12℃; for 2.16667 h; Stage #2: With water; ammonium chloride In tetrahydrofuran; diethyl ether
A solution of 1 ,4-dioxaspiro[4.5]decan-8-one (8.5g, 55mmole) in tetrahydrofuran (75ml) at - 120C under argon was treated dropwise over 10 minutes with a 1.6M solution of methyllithium in ether (45ml, 72mmole). The mixture was stirred at -120C under argon for 2 hours then quenched by addition of 1 M NH4CI solution (100ml) and the resulting mixture washed with water and extracted with ether (3 x 75ml). The combined extract was dried (Na2SO4) and concentrated under vacuum to leave a white solid. This was purified by chromatography on silica gel eluting with 10-40percent ethyl acetate/hexane to afford the title compound (5.5g, 58percent).1H NMR δ (CDCI3, 400MHz): 1.27 (3H, s), 1.55-1.75 (7H, m), 1.83-1.95 (2H, m), 3.90-4..00 (4H, m).
Reference:
[1] Canadian Journal of Chemistry, 1997, vol. 75, # 6, p. 656 - 664
[2] Patent: WO2014/9302, 2014, A1, . Location in patent: Page/Page column 89
[3] Patent: US2015/158879, 2015, A1, . Location in patent: Paragraph 0437; 0438
[4] Patent: US2016/200741, 2016, A1, . Location in patent: Paragraph 0438-0439
[5] Patent: WO2008/119716, 2008, A1, . Location in patent: Page/Page column 49
[6] Tetrahedron Letters, 1995, vol. 36, # 7, p. 1043 - 1046
[7] Bioorganic and Medicinal Chemistry, 1996, vol. 4, # 3, p. 413 - 418
[8] Synthesis, 2001, # 3, p. 437 - 443
9
[ 4746-97-8 ]
[ 75-16-1 ]
[ 66336-42-3 ]
Yield
Reaction Conditions
Operation in experiment
92%
at -78 - 0℃; for 1.33333 h; Inert atmosphere
To a cooled (−78° C.) solution of 1,4-dioxaspiro[4.5]decan-8-one (1 equiv) in THF (0.64 M) was added a solution of methylmagnesium bromide (1.8 equiv, 3 mol/L in ether) dropwise under nitrogen atmosphere. The resulting mixture was stirred at −78° C. for 20 minutes, then warmed to −30° C. for 30 minutes and then stirred at 0° C. for 30 minutes. After the reaction was complete, the resulting mixture was quenched with saturated aqueous ammonia chloride solution and extracted with ethyl acetate. The organic layers were combined, washed with brine, dried over sodium sulfate, filtered and concentrated to afford the title compound as a white solid (92percent yield), which was used in the next step without further purification. 1H NMR (300 MHz, CHLOROFORM-d1) δ ppm 4.00-3.91 (m, 4H), 1.95-1.80 (m, 3H), 1.77-1.67 (m, 4H), 1.59-1.58 (m, 1H), 1.27 (s, 3H), 1.17 (s, 1H).
92%
at -70 - 20℃;
A stirred solution of 1,4-dioxaspiro[4.5]decan-8-one (5 g, 32.0 mmol) in dry THF (70 mL) was cooled to -70° C. and methylmagnesium bromide (23.48 mL, 70.4 mmol) in ether was added dropwise over 10 min. The cooling bath was allowed to warm to room temperature and the mixture was stirred overnight. The mixture was quenched with sat. aq. NH4Cl (75 mL) and extracted with diethyl ether (2*300 mL). The combined ether extracts were washed with brine (50 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford 1A (yellow liquid, 5.1 g, 29.6 mmol, 92percent yield) which was used in next step without further purification. 1H NMR (400 MHz, CDCl3) δ 3.96-3.90 (m, 4H), 1.91-1.84 (m, 2H), 1.71-1.65 (m, 3H), 1.63-1.57 (m, 3H), 1.22 (s, 3H).
88%
at 0 - 5℃; for 6 h;
PREPARATION OF AUnder argon, 1000 ml of toluene and 1000 ml of 1 molar methylmagnesium bromide solution in THF are initially charged in a 4000 ml three-necked flask. At from 0 to 5 C., 156.2 g of 1,4-cyclohexanedione monoethylene glycol ketal in 130 ml of THF are added dropwise over 2 hours. The mixture is stirred at from 0 to 5 C. for 4 hours, and 200 ml of NH4Cl solution are then added. The phases are separated, the aqueous phase is extracted with CH2Cl2 and the organic phases are dried with MgSO4.The solvent is distilled off at atmospheric pressure and the residue is distilled at 1 mbar/110-115 C. using a 10 cm Vigreux column.This gives 152.3 g (=88percent of theory).
88%
at 0 - 5℃; for 6 h; Inert atmosphere
Preparation of Aj0599]A10600] Under argon, 1000 ml of toluene and 1000 ml of 1 molar methylmagnesium bromide solution in THF are initially charged in a 4000 ml three-necked flask. At from 0 to 5° C., 156.2 g of 1,4-cyclohexanedione monoethylene glycol ketal in 130 ml of THF are added dropwise over 2 hours. The mixture is stirred at from 0 to 5° C. for 4 hours, and 200 ml of NH4C1 solution are then added. The phases are separated, the aqueous phase is extracted with CH2C12 and the organic phases are dried with Mg504.10601] The solvent is distilled off at atmospheric pressure and the residue is distilled at 1 mbar/i 10-i 15° C. using a 10 cm Vigreux column.10602] This gives 152.3 g (=88percent of theory).
Stage #1: at 0 - 20℃; for 3 h; Stage #2: With water; ammonium chloride In tetrahydrofuran
Example 227: Synthesis of N -cyclopropyl- N -((1s,4s)-4-hydroxy-4-methylcyclohexyl)-6-(piperidin-1-yl)picolinamide and N -cyclopropyl- N -((1r,4r)-4-hydroxy-4-methylcyclohexyl)-6-(piperidin-1-yl)picolinamide [563] [564] Step 1: Synthesis of 8-methyl-1,4-dioxaspiro[4.5]decan-8-ol [565] 1,4-cyclohexandione mono-ethylene ketal (1.0 g, 6.4 mmol) was dissolved in THF (30 ml), followed by addition of MeMgCl (3.0M solution in THF, 2.6 ml, 7.7 mmol) at 0oC, and then the resulting mixture was stirred at room temperature under nitrogen stream for 3 hours. A saturated aqueous ammonium chloride solution was added to the resulting reaction liquid, followed by extraction with MC (50 ml x 2). The organic layer was dried over anhydrous sodium sulfate, followed by filtration and concentration, and then the residue thus obtained was subjected to MPLC (50percent EtOAc/Hexanes), to obtain 654 mg of white solid (59percent).[566]
Reference:
[1] Patent: WO2011/139107, 2011, A2, . Location in patent: Page/Page column 90
[2] Bioorganic and Medicinal Chemistry Letters, 2015, vol. 25, # 3, p. 695 - 700
11
[ 4746-97-8 ]
[ 75-16-1 ]
[ 917-54-4 ]
[ 66336-42-3 ]
Yield
Reaction Conditions
Operation in experiment
99%
Stage #1: at -78℃; for 1 h; Inert atmosphere Stage #2: at -78℃; for 1.5 h; Inert atmosphere
Step 1. Preparation of 8-methyl-l,4-dioxaspiro[4.5]decan-8-ol To THF (100 mL) in a round bottom flask at -78 °C under 2 was added methyllithium (48.0 mL, 77 mmol) and methylmagnesium bromide (12.81 mL, 38.4 mmol) via syringe. After stirring at -78 °C for 1 h, l,4-dioxaspiro[4.5]decan-8-one (5.00 g, 32.0 mmol) in THF (50 mL) was added via cannula. The reaction mixture was stirred at -78 °C for 1.5 h. The reaction was quenched by the addition of saturated aqueous NH4C1 solution (100 mL). The mixture was transferred to a separatory funnel containing water (50 mL) and the aqueous layer was extracted with ethyl acetate (3 x 150 mL). The combined organic layers were washed with brine (100 mL), dried over MgS04, filtered, and concentrated to afford 8-methyl-l,4-dioxaspiro[4.5]decan-8-ol (5.47 g, 99percent yield). The product was used in the next step without further purification. XH NMR (400MHz, CHLOROFORM- d) δ 4.03 - 3.91 (m, 4H), 1.97 - 1.84 (m, 2H), 1.79 - 1.56 (m, 6H), 1.28 (s, 3H), 1.17 (s, 1H); 13C NMR (100MHz, CHLOROFORM-d) δ 108.27, 68.52, 63.87, 63.81, 36.33, 30.47, 29.39.
To a solution of 1 ,4-cyclohexanedione monoethylene acetal (6.00 g, 38.4 mmol) in THF (48 mL) at 0 °C was added bromo(methyl)magnesium in diethyl ether (3 M, 19.2 mL, 42.26 mmol) under a nitrogen atmosphere. The reaction mixture was allowed to stir at room temperature for 2 h and then quenched with a saturated NH4CI solution. Water was added and the mixture was extracted with DCM. The combined organic extracts were dried over Na2S04, filtered and concentrated under reduced pressure to give 8-methyl-1 ,4-dioxaspiro[4.5]decan-8-ol (5.82 g, 27.7 mmol, 72percent yield), which was used without further purification. NMR (400 MHz, CDCI3, δ): 4.01 -3.93 (m, 4H), 1 .93-1 .86 (m, 4H), 1 .73-1 .68 (m 4H), 1 .29 (s, 3H), 1 .15 (s, 1 H).
Stage #1: With lithium hexamethyldisilazane In tetrahydrofuran at -78℃; for 0.85 h; Stage #2: at -78 - 20℃; for 2.33333 h;
TBS ether 11. To a cooled (-78 °C) solution of LHMDS (1.0 M in THF, 89.5 mL, 1.0 equiv) in THF (250 mL) was added a solution of ketone 5 (13.90 g, 89.00 mmol) in THF (40 mL) slowly over 15 min and the resulting mixture was stirred at -78 °C for 34 min. Iodomethane (6.5 mL, 1.2 equiv) was added to the reaction mixture and the resulting solution was stirred at -78 °C for 20 min and at room temperature for 2 h. The reaction mixture was quenched by addition of saturated NH4CI (250 mL) and extracted with ether (3 X 250 mL). Combined extracts were dried (MgSC^), filtered, and concentreated in vacuo. Flash chromatography (pentane-ether 4:1 -> 3:1 -> 2:1) afforded a-methylcyclohexanone (11.16 g, 74percent yield). The spectroscopic data of the product were in accord with the published data (Pfau, M.; Jabin, I.; Revial, G. J. Chem. Soc, Perkin Trans. 1 1993, 1935).
74.5%
Stage #1: With lithium hexamethyldisilazane In tetrahydrofuran; N,N-dimethyl-formamide at -60℃; for 1.5 h; Stage #2: at -60 - 20℃; for 20 h;
The reaction was set in two batches in parallel and work-up was combined. (1321) To a stirred solution of 1 M of Lithium hexamethyldisilazide in THF (1.056 L, 1.056 mol, 1.03 eq) at -60 °C was added compound 51-A (160 g, 1.026 mol, 1 eq) in DMF (600 mL) slowly using an addition funnel and keeping the temperature below -60 °C. The reaction mixture was allowed to stir below -60 °C for 1.5 h, and then treated with Methyl iodide (142 g, 1.00 mol, 0.99 eq) dropwise. Warmed to room temperature and stirred for 20 h. TLC (PE/EA = 3: 1) showed a little compound 51-A remained. The reaction mixture was quenched by half- saturated ammonium chloride solution (1.5 L) below 20 °C. Extracted with MTBE (1 L x 6). The combined organic phase was concentrated under reduced pressure to about 2 L, washed with brine (500 mL x 3). The brine layer was extracted with MTBE (500 mL x 3). The combined organic layer was dried over anhydrous Na2S04, filtered and concentrated. The residue was azeotroped with heptane 4 times (300 mL x 4). The resultant oil was slurried with heptanes (~ 800 mL) until it dissolved at room temperature, and then cooled to -10 °C with vigorous stirring. The reaction oiled out but eventually crystallized. The pale-yellow solid was filtered and rinsed with heptanes (300 mL x 3) to supply compound 51-B (260 g, 74.5percent yield) as pale-yellow solid. (1322) 1H-NMR: (400 MHz, CDC13) δ: 4.14 - 3.92 (m, 4H), 2.81 - 2.58 (m, 2H), 2.44 - 2.31 (m, 1H), 2.16 - 1.89 (m, 3H), 1.78 - 1.67 (m, 1H), 1.03 (d, 7 = 6.7 Hz, 3H)
67%
Stage #1: With lithium hexamethyldisilazane In tetrahydrofuran at -78℃; for 0.666667 h; Inert atmosphere Stage #2: With manganese(II) bromide; 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone In tetrahydrofuran at 20℃; for 0.666667 h; Stage #3: for 4 h;
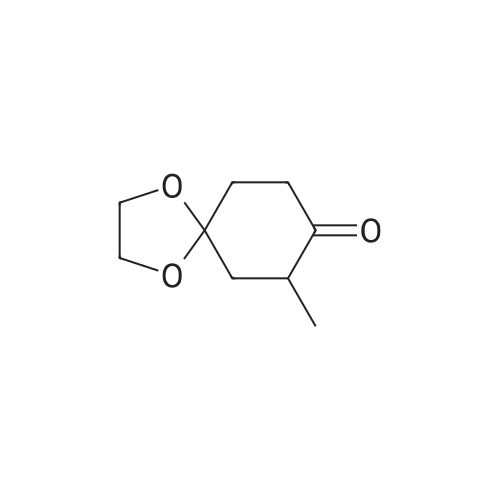
(a) α-Methylation of a ketone 1 (SCHEME I). 7-Methyl-1,4-dioxa-spiro[4.5]decan-8-one (2). A solution of 1,4-cyclohexanedione monoethylene ketal (1, 5.12 g, 32.96 mmol) in dry THF (20 mL) was added to a solution of LiHMDS (1.0 M in THF, 33.0 mL, 33.0 mmol) under argon at −78° C. and the mixture was stirred for 40 min. After warming up to room temperature DMPU (13.3 mL) was added. Stirring was continued for additional 10 min, and the enolate solution was cannulated to the flask containing anhydrous MnBr2 (7.83 g, 36.46 mmol) and the mixture was stirred until clear reddish-brown solution was obtained (approximately 30 min). The methyl iodide (2.5 mL, 40.0 mmol) was then added, and after 4 h the reaction was quenched by the addition of saturated NH4Cl and EDTA. Materials were extracted with diethyl ether, dried over MgSO4, and concentrated. Purification by column chromatography on silica (3→5percent ethyl acetate/hexane gradient) gave an oily α-methyl ketone 2 (3.72 g, 67percent).[0043]2: 1H NMR (200 MHz, CDCl3) δ 1.02 (3H, d, J=6.6 Hz, CH3), 1.72 (1H, br t, J=13.2 Hz), 2.04 (3H, br m), 2.35 (1H, ddd, J=14.4, 4.9, 2.9 Hz), 2.69 (2H, m), 4.02 (4H, m, O—CH2CH2—O); 13C NMR (50 MHz, CDCl3) δ 14.48, 34.82, 38.17, 41.44, 42.92, 64.78, 64.90, 107.55, 212.08; HRMS (ESI) exact mass calcd for C9H14O3Na (M++Na) 193.0841, measured 193.0836.
67%
Stage #1: With lithium hexamethyldisilazane In tetrahydrofuran at -78℃; for 0.666667 h; Inert atmosphere Stage #2: With 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone In tetrahydrofuran for 0.166667 h;
A solution of 1,4-cyclohexanedione monoethylene ketal (1, 5.12 g, 32.96 mmol) in dry THF (20 mL) was added to a solution of LiHMDS (1.0 M in THF, 33.0 mL, 33.0 mmol) under argon at -78° C. and the mixture was stirred for 40 min. After warming up to room temperature DMPU (13.3 mL) was added. Stirring was continued for additional 10 min, and the enolate solution was cannulated to the flask containing anhydrous MnBr2 (7.83 g, 36.46 mmol) and the mixture was stirred until clear reddish-brown solution was obtained (approximately 30 min). The methyl iodide (2.5 mL, 40.0 mmol) was then added, and after 4 h the reaction was quenched by the addition of saturated NH4Cl and EDTA. Materials were extracted with diethyl ether, dried over MgSO4, and concentrated. Purification by column chromatography on silica (3→5percent ethyl acetate/hexane gradient) gave an oily α-methyl ketone 2 (3.72 g, 67percent). 2: 1H NMR (200 MHz, CDCl3) δ 1.02 (3H, d, J=6.6 Hz, CH3), 1.72 (1H, br t, J=13.2 Hz), 2.04 (3H, br m), 2.35 (1H, ddd, J=14.4, 4.9, 2.9 Hz), 2.69 (2H, m), 4.02 (4H, m, O—CH2CH2—O); 13C NMR (50 MHz, CDCl3) δ 14.48, 34.82, 38.17, 41.44, 42.92, 64.78, 64.90, 107.55, 212.08; HRMS (ESI) exact mass calcd for C9H14O3Na (M++Na) 193.0841, measured 193.0836.
62%
Stage #1: With lithium hexamethyldisilazane In tetrahydrofuran at -78℃; for 1 h; Inert atmosphere Stage #2: at -78 - 20℃; for 2.5 h;
Under a nitrogen atmosphere, to a solution of lithium bis(trimethylsilyl)amide (1M, 100 ml, 100 mmol) in tetrahydrofuran (200 ml) was added dropwise a solution of 1,4-dioxaspiro[4.5]decan-8-one (15.6 g, 100 mmol) in tetrahydrofuran (50 ml) at -78° C. over about 30 min, and the mixture was stirred for 30 min. Then, methyl iodide (2.5 ml, 120 mmol) was added dropwise over 5 min, and the mixture was stirred at -78° C. for 30 min, and at room temperature for 2 hr. To the reaction mixture was added saturated aqueous ammonium chloride solution, and the mixture was extracted three times with diethyl ether. The combined organic layers were dried over magnesium sulfate. Magnesium sulfate was removed by filtration, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography to give the title compound (10.6 g, yield 62percent).1H-NMR (400 MHz, DMSO-d6) δ: 0.90 (3H, d, J=6.62 Hz), 1.65 (1H, t, J=13.01 Hz), 1.84-2.04 (3H, m), 2.19 (1H, ddd, J=14.50, 5.13, 3.03 Hz), 2.49-2.69 (2H, m), 3.85-4.06 (4H, m).
57%
Stage #1: With lithium hexamethyldisilazane In tetrahydrofuran at -78℃; for 2 h; Inert atmosphere Stage #2: at -78 - 28℃; for 15 h;
To a solution of Compound 1 (1 g, 6.4 mmol) in THF (30 mL) was added LiHMDS (12.8 mL, 12.8 mmol) at −78° C. under N2. The reaction mixture was stirred at −78° C. for 2 h. MeI (2.7 g, 19.2 mmol) was added to the mixture at −78° C., then warmed to 28° C. slowly and stirred for 15 h. The resulting mixture was quenched with H2O and extracted with EA (50 mL). The organic layer was dried over Na2SO4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography to give Compound 2 (0.62 g, 57percent). 1H NMR (400 MHz, CHLOROFORM-d) δ 3.92-4.17 (m, 4H), 2.55-2.83 (m, 2H), 2.31-2.43 (m, 1H), 1.87-2.13 (m, 3H), 1.69-1.81 (m, 1H), 1.04 (d, J=6.65 Hz, 3H). LCMS: 171.0 [M+1].
57%
Stage #1: With lithium hexamethyldisilazane In tetrahydrofuran at -78℃; for 2 h; Inert atmosphere Stage #2: at -78 - 28℃; for 15 h; Inert atmosphere
To a solution of Compound 1 (1 g, 6.4 mmol) in THF (30 mL) was added LiHMDS (12.8 mL, 12.8 mmol) at −78° C. under N2. The reaction mixture was stirred at −78° C. for 2 h. MeI (2.7 g, 19.2 mmol) was added to the mixture at −78° C., then warmed to 28° C. slowly and stirred for 15 h. The resulting mixture was quenched with H2O and extracted with EA (50 mL). The organic layer was dried over Na2SO4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography to give Compound 2 (0.62 g, 57percent). 1H NMR (400 MHz, CHLOROFORM-d) δ 3.92-4.17 (m, 4H), 2.55-2.83 (m, 2H), 2.31-2.43 (m, 1H), 1.87-2.13 (m, 3H), 1.69-1.81 (m, 1H), 1.04 (d, J=6.65 Hz, 3H). LCMS: 171.0 [M+1].
39%
Stage #1: With lithium hexamethyldisilazane In tetrahydrofuran-d8 at -78℃; for 1 h; Inert atmosphere Stage #2: at -78 - 20℃; Inert atmosphere
To a solution of 1,4-dioxaspiro[4.5]decan-8-one (1.04 g, 6.66 mmol, available from commercial suppliers such as Apollo Scientific) in THF (67 mL) under N2 at -78 °C was added 1M LiHMDS in THF (7.32 mL, 7.32 mmol). The reaction was stirred for 1 h and MeI (0.547 mL, 8.66 mmol) was added dropwise. The reaction was stirred for 3 h at -78 °C, and then left to warm up to rt overnight. It was then quenched with a sat. NH4Cl (aq) solution and extracted with EtOAc. The combined organics were filtered through a hydrophobic frit and concentrated in vacuo to a brown oil. This oil was purified by silica gel column chromatography eluting with a gradient of 0 to 32percent EtOAc : cyclohexane to give (+/-)-7-methyl-1,4-dioxaspiro[4.5]decan-8-one (443 mg, 2.60 mmol, 39 percent yield) as a white solid. (0636) 1H NMR (400 MHz, MeOH-d4) δ ppm 3.95 - 4.16 (m, 4 H) 2.58 - 2.88 (m, 2 H) 2.24 - 2.41 (m, 1 H) 2.04 - 2.17 (m, 2 H) 1.90 - 2.01 (m, 1 H) 1.71 (t, J=13.1 Hz, 1 H) 0.94 - 1.07 (m, 3 H)
Reference:
[1] Angewandte Chemie - International Edition, 2015, vol. 54, # 6, p. 1960 - 1964[2] Angew. Chem., 2015, vol. 127, # 6, p. 1983 - 1987,5
[3] Journal of the American Chemical Society, 2004, vol. 126, # 44, p. 14358 - 14359
[4] Journal of the American Chemical Society, 2006, vol. 128, # 3, p. 1016 - 1022
[5] Patent: WO2006/19673, 2006, A2, . Location in patent: Page/Page column 34; 60
[6] Patent: WO2018/125880, 2018, A1, . Location in patent: Page/Page column 125; 126
[7] Organic Letters, 2017, vol. 19, # 4, p. 878 - 881
[8] Tetrahedron Asymmetry, 2001, vol. 12, # 12, p. 1683 - 1688
[9] Synlett, 2007, # 15, p. 2379 - 2382
[10] Patent: US2013/102567, 2013, A1, . Location in patent: Paragraph 0042; 0043
[11] Patent: US2013/102573, 2013, A1, . Location in patent: Paragraph 0041; 0042; 0094
[12] Patent: US2011/306599, 2011, A1, . Location in patent: Page/Page column 36
[13] Patent: US2015/197493, 2015, A1, . Location in patent: Paragraph 0495; 0496; 0497
[14] Patent: US2015/225355, 2015, A1, . Location in patent: Paragraph 0488
[15] Patent: WO2018/158212, 2018, A1, . Location in patent: Page/Page column 81
[16] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1993, # 17, p. 1935 - 1936
[17] Patent: WO2012/170845, 2012, A2, . Location in patent: Page/Page column 108
17
[ 4746-97-8 ]
[ 54621-18-0 ]
Reference:
[1] Journal of Agricultural and Food Chemistry, 2012, vol. 60, # 19, p. 4779 - 4787
[2] Patent: WO2012/119941, 2012, A1,
18
[ 4746-97-8 ]
[ 51656-91-8 ]
Yield
Reaction Conditions
Operation in experiment
91%
Stage #1: With sodium hexamethyldisilazane In tetrahydrofuran at 0 - 20℃; for 0.5 h; Stage #2: at 0 - 20℃; for 18 h;
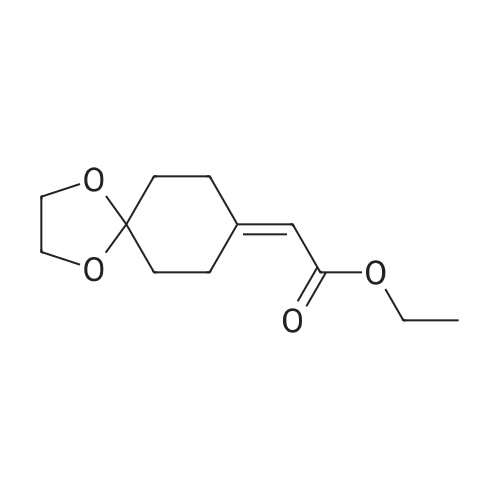
A solution of triethyl phosphonate (44.8 g, 200 mmol) in THF (30 ml) at 0° C. was treated with a 1M solution (200 ml) of sodium bis(trimethylsilylamide) in THF. The resulting mixture was stirred at room temperature for 0.5 hour, and then cooled to 0° C. A solution of 1,4-cyclohexanedione mono ethylene ketal (15.6 g, 200 mmol) in THF (50 ml) was added dropwise, and the resulting solution was stirred at room temperature for 18 hours. The reaction mixture was then cooled to 0° C., treated with cold aqueous citric acid, and the mixture was extracted with EtOAc. The extract was washed with satd. aqueous NaHCO3, brine, dried over Na2SO4, filtered, and the filtrate was concentrated. The residue was chromatographed on silica gel, eluting with a gradient of CH2Cl2/EtOAc to afford 223b (21 g, 91percent).
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at 0℃; for 1 h; Stage #2: at -20 - 20℃; for 3 h;
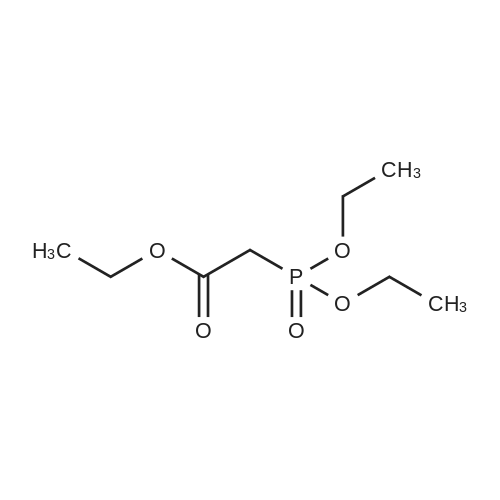
To a solution of NaH (60percent mineral oil suspension, 33.3 g, 832.38 mmol) in anhydrous THF (1 L) was added dropwise a solution of l,4-dioxaspiro[4.5]decan-8-one (100 g, 640.29 mmol) in anhydrous THF (500 mL) at 0 °C for 1 hour. The reaction was stirred at 0 °C for 1 hour, then triethyl phosphonoacetate (172.26 g, 768.35 mmol) was added dropwise at -20 °C for 1 hour. The reaction was allowed to warm to room temperature and stirred for 2 hours. The mixture was diluted with H2O (1 L) and extracted with EtOAc (1 L x 3). The combined organic phases were washed with brine (1 L), dried over anhydrous Na2SO i, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (PE/EtAOc (v/v) = 10/1) to give the title compound as light yellow oil (145 g, 100 percent). 1H M (600 MHz, CDCI3): δ (ppm) 5.62 (s, 1H), 4.10 (qd, / = 7.1, 2.9 Hz, 2H), 3.94 (d, J = 13.8 Hz, 4H), 2.99-2.89 (m, 2H), 2.37-2.27 (m, 2H), 1.77-1.66 (m, 4H), 1.23 (td, J = 7.1, 2.7 Hz, 3H).
100%
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at 0℃; for 2 h; Stage #2: at -20 - 20℃; for 3 h;
Step 1) ethyl 2-(l,4-dioxaspiro[4.51decan-8-ylidene)acetate [0342] To a suspension of NaH (60percent mineral oil suspension, 33.3 g, 832.38 mmol) in anhydrous THF (1 L) was added a solution of l,4-dioxaspiro[4.5]decan-8-one (100 g, 640.29 mmol) in anhydrous THF (500 mL) dropwise at 0 °C for 1 h and continued to stir for 1 h. Then triethyl phosphonoacetate (203.23 g, 832.38 mmol) was added to the above suspension dropwise at -20 °C in 1 h. The resulting mixture was allowed to warm to rt, stirred for 2 h, quenched with H20 (1 L) and extracted with EtOAC (1 L x 3). The combined organic phases were washed with brine (1 L), dried over anhydrous Na2S04, then filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (EtOAc/PE (v/v) = 1/10) to give the title compound as pale yellow oil (157 g, 100 percent). FontWeight="Bold" FontSize="10" H NMR (600 MHz, CDCI3): δ (ppm) 5.64 (s, 1H), 4.12 (q, J = 7.1 Hz, 2H), 3.95 (s, 4H), 2.97 (m, 2H), 2.36 (m, 2H), 1.74 (m, 4H), 1.25 (t, J= 7.2 Hz, 4H).
100%
Stage #1: With potassium <i>tert</i>-butylate In N,N-dimethyl-formamide at 20℃; for 0.166667 h; Stage #2: at 20℃; for 1 h;
Potassium tert-butylate (10.7 g, 95.6 mmol) was added to a solution of phosphonoacetic acid triethyl ester (21.4 g, 19 ml, 95.6 mmol) in anhydrous N,N-dimethylformamide (90 ml) underargon and the mixture was stirred for 10 mm at room temperature. A solution of 1,4- dioxaspiro[4.5]decan-8-one (10.0 g, 64 mmol) in anhydrous N,N-dimethylformamide (160 ml) was then added to the mixture and the mixture was stirred for 1 h at room temperature and then poured into ice-water (240 g). The aqueous suspension was extracted with diethyl ether (4 x 100 ml). The combined organic extracts were dried with sodium sulfate andconcentrated i. vac.Yield: 14.4 g (100 percent), yellowish oil.1H-NMR (CDCI3): 1.27 (3 H, t, J = 7.1 Hz): 1.73—1.80 (4 H, m); 2.35—2.40 (2 H, m); 2.92—3.02(2 H, m): 3.97 (4 H, s): 4.15 (2 H, q, J = 7.1 Hz): 5.66(1 H, s).
100%
Stage #1: With sodium hydride In tetrahydrofuran; water; mineral oil at -20 - 0℃; Stage #2: at 20℃; for 3 h;
The NaH (60percent mineral oil suspension, 33.3g, 832 . 38mmol) THF suspended water-free (1L) in, in 0 °C lower, added to the 1,4-dioxaspiro [4.5] decane-8-one (100g, 640.29mmol) anhydrous THF (500 ml) solution, 1-hour internal dropping end. Furthermore, at -20 °C lower, the phosphoryl acetic acid triethyl ester (203.23g, 832 . 38mmol) is dripped into the in the above-mentioned suspension system, 1-hour internal dropping end. The resulting system is moved to the room temperature, is continuously stirred for 2 hours, then water (1L) quenching the reaction, and using ethyl acetate (1Lx3) extraction. Combined with the phase, saturated salt water for (1L) washing, anhydrous Na2SO4drying, concentrated filtrate under reduced pressure, the resulting residue by a silica gel column chromatography (PE/EtOAc = 10/1 (v/v)) purification, to obtain the title compound as of bombycinous (157g, 100percent).
100%
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at 0℃; for 2 h; Stage #2: at -20 - 20℃; for 3 h;
Step 1) ethyl 2-(l ,4-dioxaspiror4.51decan-8-ylidene)acetate [0350] To a suspension of NaH (60percent mineral oil suspension, 33.3 g, 832.38 mmol) in anhydrous THF (1 L) was added a solution of l ,4-dioxaspiro[4.5]decan-8-one (100 g, 640.29 mmol) in anhydrous THF (500 mL) dropwise at 0 °C for 1 h and the reaction mixture was stirred for another 1 h. Then, triethyl phosphonoacetate was added dropwise to the above suspension at -20 °C in 1 h. The resulting mixture was allowed to warm to rt, and stirred for another 2 h, then quenched with 0 (1 L) and extracted with EtOAc (1 L x 3). The combined organic phases were washed with brine (1 L), then dried over anhydrous Na2S04, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (EtOAc/PE (v/v) = 1/10) to give the title compound as pale yellow oil (157 g, 100 percent). NMPv (600 MHz, CDCb): δ (ppm) 5.64 (s, 1H), 4.12 (q, J = 7.1 Hz, 2H), 3.95 (s, 4H), 2.97 (m, 2H), 2.36 (m, 2H), 1.74 (m, 4H), 1.25 (t, J= 7.2 Hz, 4H).
100%
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at 0℃; for 0.5 h; Inert atmosphere Stage #2: at 20℃; for 2 h;
To a suspension of NaH (60percent suspension in oil) (1.42 g, 35.45 mmol) in THF (190mL) at 0 °C, under N2, ethyl 2-(diethoxyphosphoryl)acetate (7 mL, 35.45 mmol) was addeddrop-wise. The mixture was stirred for 30’, then 1,4-dioxaspiro[4.5]decan-8-one (5g, 32 mmol)in THF (20 mL) was added drop-wise. The resulting mixture was stirred at RT for 2 hrs and thenconcentrated under vacuum. The residue was taken up with Et20, washed with water and Brine, dried over Na2SO4 and concentrated to obtain 7.58 g of title compound (p121, y= quant) as colourless oil. MS (m/z): 227.2 [IVIH]t
100%
With sodium hydride In tetrahydrofuran; mineral oil at -20 - 20℃; for 2 h;
NaH (60percent suspended in mineral oil, 33.3 g, 832.38 mmol) was suspended in dry THF (1 L), and then the suspension was placed at 0 ° C, 1,4-dioxaspiro[4.5]decan-8-one (100 g, 640.29 mmol) in dry THF (500 mL) dropwise over 1 hour to give a suspension. Then, triethyl phosphonoacetate (203.23 g, 832.38 mmol) was added dropwise to the above suspension at -20 ° C, and the mixture was dropwise added in 1 hour to obtain a reaction system. The resulting reaction was moved to room temperature and stirring continued for 2 hours, then the reaction was quenched with water (1 L) and extracted with ethyl acetate (1 L x 3). The combined organic phases were washed with brine (1 L), then dried over anhydrous Na2SO4, filtered and the filtrate was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (PE / EtOAc (v / v) = 10/1) to give the title compound as a pale yellow oil (157 g, 100percent).
100%
With sodium hydride In tetrahydrofuran; kerosene at -20 - 20℃; for 4 h;
At 0 ° C, NaH (60percent [w / w], 33.3 g, 832.38 mmol) suspended in kerosene was added to dry tetrahydrofuran (500 mL) and 1,4-dioxaspiro[4.5]decan-8-one (100 g, 640.29 mmol) was added dropwise over 1 hour. Then triethyl phosphonoacetate (172.26 g, 768.35 mmol) was added dropwise at -20 ° C for 1 hour. The reaction was warmed to room temperature and stirred for 2 hours before it was diluted with water (1 L), extracted with ethyl acetate (1 L × 3), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (petroleum ether / ethyl acetate (v / v) = 10/1) to give the title compound as a pale yellow oil (145 g, 100percent).
96%
Stage #1: With sodium hydride In tetrahydrofuran at 0 - 20℃; for 0.5 h; Stage #2: at 0 - 20℃; for 0.5 h;
Triethyl phosphonoacetate (21.79 ml, 109 mmol) was added to a suspension of sodium hydride (3.84 g, 96 mmol) in THF (64.0 ml) and 0 °C. Reaction was stirred at room temperature for 30 minutes. After 30 minutes, the reaction was recooled to 0 °C and a solution of 1,4-dioxaspiro[4.5]decan-8-one (10 g, 64.0 mmol) in 5 mL THF wasadded. The reaction was then stirred at room temperature for 30 minutes prior to quenching with water. The mixture was extracted with DCM three times. Combined organic extracts were dried with sodium sulfate, filtered, and concentrated in vacuo. Crude residue was purified via silica gel chromatography to give Intermediate 71A (13.88 g, 61.3 mmol, 96percent yield). TLC: product stains as purple spot in anisaldehyde (Rf= 0.75in 1:1 Hex/EtOAc). ‘H NMR (400 MHz, chloroform-d) ö: 5.65 (s, 1H), 4.13 (q, J=7.2 Hz,2H), 3.92-3.99 (m, 4H), 2.94-3.02 (m, 2H), 2.3 1-2.40 (m, 2H), 1.71-1.79 (m, 4H), 1.26 (t, J=7.2 Hz, 3H).
96%
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at 0 - 20℃; for 0.5 h; Stage #2: at 0 - 20℃; for 0.5 h;
Triethyl phosphonoacetate (21.79 ml, 109 mmol) was added to a suspension of sodium hydride (3.84 g, 96 mmol) in THF (64.0 ml) and 0 °C. Reaction was stirred at room temperature for 30 minutes. After 30 minutes, the reaction was recooled to 0 °C and a solution of l,4-dioxaspiro[4.5]decan-8-one (10 g, 64.0 mmol) in 5 mL THF was added. The reaction was then stirred at room temperature for 30 minutes prior to quenching with water. The mixture was extracted with DCM three times. Combined organic extracts were dried with sodium sulfate, filtered, and concentrated in vacuo. Crude residue was purified via silica gel chromatography to give Intermediate 83A (13.88 g, 61.3 mmol, 96percent> yield). TLC: product stains as purple spot in anisaldehyde (Rf = 0.75 in 1 : 1 Hex/EtOAc). 1H NMR (400 MHz, chloroform-d) δ: 5.65 (s, 1H), 4.13 (q, J=7.2 Hz, 2H), 3.92-3.99 (m, 4H), 2.94-3.02 (m, 2H), 2.31-2.40 (m, 2H), 1.71-1.79 (m, 4H), 1.26 (t, J=7.2 Hz, 3H)
96%
Stage #1: With sodium hydride In tetrahydrofuran at 0 - 20℃; for 1 h; Stage #2: at 0 - 20℃; for 0.5 h;
Triethyl phosphonoacetate (21.79 ml, 109 mmol) was added to a suspension ofsodium hydride (3.84 g, 96 mmol) in THF (64.0 ml) and 0 °C. Reaction was stirred atroom temperature for 30 minutes. After 30 minutes, the reaction was recooled to 0 °C and a solution of 1,4-dioxaspiro[4.5]decan-8-one (10 g, 64.0 mmol) in 5 mL THF was added. The reaction was then stirred at room temperature for 30 minutes prior to quenching with water. The mixture was extracted with DCM three times. Combinedorganic extracts were dried with sodium sulfate, filtered, and concentrated in vacuo. Crude residue was purified via silica gel chromatography to give Intermediate 305A (13.88 g, 61.3 mmol, 96percent yield). TLC: product stains as purple spot in anisaldehyde (Rf = 0.75 in 1:1 Hex/EtOAc). ‘H NMR (400 MHz, chloroform-d) ö: 5.65 (s, 1H), 4.13 (q, J7.2 Hz, 2H), 3.92-3.99 (m, 4H), 2.94-3.02 (m, 2H), 2.3 1-2.40 (m, 2H), 1.7 1-1.79 (m,4H), 1.26 (t, J7.2 Hz, 3H).
96%
Stage #1: With sodium hydride In tetrahydrofuran at 0 - 20℃; for 0.5 h; Stage #2: at 0 - 20℃; for 0.5 h;
[00187] Tri ethyl phosphonoacetate (21.79 ml, 109 mmol) was added to a suspension of sodium hydride (3.84 g, 96 mmol) in THF (64.0 ml) and 0 °C. Reaction was stirred at room temperature for 30 minutes. After 30 minutes, the reaction was recooled to 0 °C and a soution of l,4-dioxaspiro[4.5]decan-8-one (10 g, 64.0 mmol) in 5 mL THF was added. The reaction was then stirred at room temperature for 30 minutes prior to quenching with water. The mixture was extracted with DCM three times. Combined organic extracts were dried with sodium sulfate, filtered, and concentrated in vacuo. Crude residue was purified via silica gel chromatography to give intermeduate 3A (13.88 g, 61.3 mmol, 96 percent yield). TLC: product stains as purple spot in anisaldehyde (Rf = 0.75 in 1 : 1 Hex/EtOAc). NMR (400 MHz, CHLOROFORM-d) δ: 5.65 (s, 1H), 4.13 (q, J=7.2 Hz, 2H), 3.92-3.99 (m, 4H), 2.94-3.02 (m, 2H), 2.31 -2.40 (m, 2H), 1.71 -1.79 (m, 4H), 1.26 (t, J=7.2 Hz, 3H)
96%
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at 0 - 20℃; for 0.5 h; Stage #2: at 0 - 20℃; for 0.5 h;
Triethyl phosphonoacetate (21.79 ml. 109 mmoi) was added to a suspension of sodium hydride (3.84 g, 96 mrnol) in TI-iF (64.0 ml) and 0 °C. Reaction was stirred at room temperature for 30 minutes. After 30 minutes, the reaction was recooled to 0 C and a soution of 1,4-dioxaspiro[4.5Idecan-8-one (10 g, 64.0 mmol) in 5 mL THF was added. The reaction was then stirred at room temperature for 30 minutes prior to quenching with water, The mixture was extracted with DCM three times. Combined organic extracts were dried with sodium sulfate, filtered, and concentrated in vacuo. Crude residue was purified via silica gel chromatography to give intermediate 13A (13.88 g, 61.3 mmoi, 96 percent yield). TLC: product stains as purple spot in anisaldeliyde (RI 0 7 in 11 He [tO’\.c) ‘HMR(400 MHz (HLOROH)RNI-d) 5 565 (1H), 4.13 (q, J=7.2 Hz, 2Ff), 3.92-3.99 (m, 4H), 2.94-3.02 (in, 2H), 2.31-2.40 (m, 2H). 1.71-1.79 (in, 4H), 1.26 (t, J=7.2 Hz, 3H)
94%
Stage #1: With sodium hydride In tetrahydrofuran at 0℃; for 1 h; Stage #2: at -20 - 20℃;
Step-1: Ethyl 2-(1,4-dioxaspiro[4.5]decan-8-ylidene)acetate A solution of compound 1 (10.05 g, 64.04 mmol, 1.0 eq.) in THF (30 ml) was added dropwise to a suspension NaH (1.84 g, 76.85 mmol, 1.2 eq.) in THF (90 ml) at 0° C. and the mixture was stirred at same temperature for 1 h. Triethyl phosphonoacetate (16.5 ml, 83.25 mmol, 1.3 eq.) was added to the reaction mixture at -20° C. and the reaction mixture was allowed to warm to RT and stir for 2 h. The reaction mixture was diluted with ethyl acetate (100 ml), washed with water (2*100 ml) and the organic layer dried over sodium sulfate. The solvent was evaporated under reduced pressure to obtain the crude product which was purified by column chromatography (silica gel; 5percent ethyl acetate/hexanes) to yield compound 2. Yield: 94percent (17.28 g, 60.2 mmol).
93%
Stage #1: With lithium hydride In tetrahydrofuran at 20℃; for 1 h; Stage #2: at 65℃; for 16 h;
To a solution of THF (18 mL) under argon was added 0.38 g (47.8 mmol, 5 equiv) of LiH, followed by slow addition of 8.78 G (47.8 mmol, 5 equiv) of triethyl phosphonoacetate. The solution was stirred at rt for 1 h and 1.49 g (9.6 mmol, 1 equiv) of 1, 4-cyclohexanedione mono-ethylene ketal was added and the solution was heated at 65 °C for 16 h. Upon cooling the solution was treated with MeOH (10 mL) and water (5 mL) and concentrated in vacuo. The resulting yellow oil was purified by silica gel chromatography eluting with 4: 1 Hex/EtOAc to yield 1.89 g (93percent) of a clear oil.'H-NMR (CDCI3-D) 8 5.67 (s, 1H), 4.16 (t, 2H), 3.99 (m, 4H), 3.02 (m, 2H), 2.39 (m, 2H), 1.78 (m, 4H), 1.29 (t, 3H); LCMS RT = 2.56 min; [M+H] + = 226.9.
93%
Stage #1: With lithium hydride In tetrahydrofuran at 20℃; for 1 h; Stage #2: at 65℃; for 16 h;
Example 1; Preparation of ethyl [4-({3-chloro-4-[(3-fluorobenzyl)oxy] phenyl )amino)[1]benzothieno[2,3-d]pyrimidin-7-yl]acetate; Step 1. Preparation of ethyl 1,4-dioxaspiro[4.5]dec-8-ylideneacetate; To a solution of THF (18 mL) under argon was added 0.38 g (47.8 mmol, 5 equiv) of LiH, followed by slow addition of 8.78 g (47.8 mmol, 5 equiv) of triethyl phosphonoacetate. The solution was stirred at rt for 1 h and 1.49 g (9.6 mmol, 1 equiv) of l,4-dioxa-spiro[4.5]decan-8-one was added and the solution was heated at 65°C for 16 h. Upon cooling the solution was treated with MeOH (10 mL) and water (5 mL) and concentrated in vacuo. The resulting yellow oil was purified by silica gel chromatography eluting with 4: 1 Hex/EtOAc to yield 1.89 g (93percent) of a clear oil. 1H- NMR (CDCl3-d) δ 5.67 (s, 1H), 4.16 (t, 2H), 3.99 (m, 4H), 3.02 (m, 2H), 2.39 (m, 2H), 1.78 (m, 4H), 1.29 (t, 3H); LCMS RT = 2.56 min, [M+H]+ = 226.9.
93%
Stage #1: With sodium hydride In tetrahydrofuran at 0℃; for 0.5 h; Inert atmosphere Stage #2: at 0 - 25℃; for 3 h; Inert atmosphere
Triethyl phosphonoacetate (12.2g, 54.4mmol) was dissolved in tetrahydrofuran (100mL), at 0 °C was added sodium hydride (1.92g, 48.0mmol), the reaction mixture was stirred under nitrogen atmosphere for 30 minutes.Then at 0 °C dissolved in tetrahydrofuran (15mL) 1,4-cyclohexanedione monoethylene ketal (5.00g, 32.0mmol) was added dropwise to the reaction mixture, the reaction solution was stirred at 25 °C for 3 hours. Water was added (25mL) to quench the reaction and extracted with dichloromethane (20mLx3). The combined organic phase was washed with saturated brine (20 mL), dried over anhydrousOver sodium sulfate, and concentrated under reduced pressure, the residue was residue was purified by silica gel column chromatography (5: 1 petroleum ether / acetic acidEthyl ester, Rf = 0.3), give ethyl 2-(1,4-dioxa-spiro[4.5]decane-8-ylidene)acetate (6.30g, Colorless oil). Yield: 93percent.
90%
Stage #1: With sodium hydride In tetrahydrofuran at 0℃; for 0.5 h; Stage #2: at 0℃; for 16 h;
A solution of triethyl phosphonoacetate (11 mmol) in THF (50 ml) was added slowly to a suspension, cooled to 0° C., of NaH (10 mmol) in THF (50 ml), and the reaction mixture was stirred for 30 min. 1,4-Dioxa-spiro[4.5]decan-8-one (10 mmol) in THF (50 ml) was then added dropwise at 0° C., and stirring was carried out for 16 h. After addition of ice and aqueous saturated NaCl solution, the aqueous phase was washed with ethyl acetate and the organic phase with water and aqueous saturated NaCl solution. The combined organic phases were dried over Na2SO4 and, after filtration, the solvent was removed in vacuo. The product was purified by column chromatography (20percent ethyl acetate/hexane). Yield: 90percent.
83%
With sodium hydride In tetrahydrofuran at 0℃;
Into a 250-mL round-bottom flask, was placed ethyl 2-(diethoxyphosphoryl)acetate (14.4 g, 64.23 mmol, 1 equiv), tetrahydrofuran (150 mL), sodium hydride (5.12 g, 213.33 mmol, 3.33 equiv), l,4-dioxaspiro[4.5]decan-8-one (10 g, 64.03 mmol, 1 equiv). The resulting solution was stirred overnight at 0 °C. The reaction was then quenched by the addition of 50 mL of water. The resulting solution was extracted with 3x50 mL of ethyl acetate and the organic layers combined. The resulting mixture was washed with 3x50 mL of H2O. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 :5). This resulted in 12 g (83percent) of as a yellow liquid. Analytical Data: 1H NMR (300 MHz, Chloroform-d) δ 5.67 (p, J= 1.1 Hz, 1H), 4.15 (q, J= 7.1 Hz, 2H), 3.98 (s, 4H), 3.00 (ddd, J= 7.8, 5.1, 1.2 Hz, 2H), 2.44 - 2.32 (m, 2H), 1.84 - 1.70 (m, 4H), 1.28 (t, J= 7.1 Hz, 3H).
69%
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at 0℃; for 1 h; Stage #2: at 0 - 20℃;
To a solution of commercially available ethyl 2-diethoxyphosphorylacetate(9.5 g, 42.3 mmol) in THE (20 mL) was added NaH (1 .7 g, 42.3 mmol) at 0°C. The mixture solution was stirred at 0°C for 1 h. Then a solution ofcommercially available 1 ,4-dioxaspiro[4.5]decan-8-one (6 g, 38.5 mmol) in THE (5 mL) was added at 000. The solution was stirred at r.t overnight. The mixture was quenched with aqueous NH4CI and extracted with EtOAc, the organic layer was washed with brine, dried over anhydrous Na2SO4, concentrated to give the crude product which was purified by column to givereagent KR-46 (6.6 g, 69 percent yield) as a white solid. ESI-MS (Mi-i): 227.2; calc. for C12H1804: 226.1.
Reference:
[1] Tetrahedron, 1994, vol. 50, # 4, p. 1093 - 1104
[2] Organic Letters, 2005, vol. 7, # 19, p. 4185 - 4188
[3] Patent: WO2015/94803, 2015, A1, . Location in patent: Paragraph 340
[4] Patent: WO2015/148868, 2015, A1, . Location in patent: Paragraph 0342
[5] Patent: WO2016/8582, 2016, A1, . Location in patent: Page/Page column 125
[6] Patent: CN105367555, 2016, A, . Location in patent: Paragraph 0488; 0489; 0490
[7] Patent: WO2016/190847, 2016, A1, . Location in patent: Paragraph 0350; 0374
[8] Patent: WO2017/21920, 2017, A1, . Location in patent: Paragraph 1102; 1103
[9] Patent: CN104974163, 2017, B, . Location in patent: Paragraph 0551; 0552; 0553; 0644; 0645; 0646
[10] Patent: CN104672250, 2017, B, . Location in patent: Paragraph 0520; 0521; 0522
[11] Synthesis (Germany), 2012, vol. 44, # 23, p. 3623 - 3632
[12] Tetrahedron Asymmetry, 2008, vol. 19, # 2, p. 176 - 185
[13] Patent: WO2016/73774, 2016, A2, . Location in patent: Paragraph 0341
[14] Patent: WO2016/73770, 2016, A1, . Location in patent: Paragraph 0334
[15] Patent: WO2016/73738, 2016, A2, . Location in patent: Paragraph 0567
[16] Patent: WO2017/192840, 2017, A1, . Location in patent: Paragraph 00187
[17] Patent: WO2018/39512, 2018, A1, . Location in patent: Paragraph 00175; 00188
[18] Tetrahedron, 1999, vol. 55, # 36, p. 11095 - 11108
[19] Bioorganic and Medicinal Chemistry, 2005, vol. 13, # 23, p. 6309 - 6323
[20] Patent: US2010/249095, 2010, A1, . Location in patent: Page/Page column 128-129
[21] Patent: WO2005/10008, 2005, A1, . Location in patent: Page/Page column 104-105
[22] Patent: WO2006/44524, 2006, A1, . Location in patent: Page/Page column 45
[23] Patent: CN105566324, 2016, A, . Location in patent: Paragraph 0303; 0304; 0305; 0306
[24] Patent: US2008/153843, 2008, A1, . Location in patent: Page/Page column 47-48
[25] Patent: WO2017/181177, 2017, A1, . Location in patent: Paragraph 0568-0571
[26] Journal of Medicinal Chemistry, 2016, vol. 59, # 19, p. 8967 - 9004
[27] Patent: WO2014/131855, 2014, A1, . Location in patent: Page/Page column 65; 66
[28] Tetrahedron Letters, 1992, vol. 33, # 32, p. 4581 - 4584
[29] Journal of the Chemical Society, Chemical Communications, 1988, # 6, p. 425 - 426
[30] Patent: US2010/222324, 2010, A1, . Location in patent: Page/Page column 58
[31] Patent: WO2010/103429, 2010, A1, . Location in patent: Page/Page column 33
[32] Organic Letters, 2011, vol. 13, # 7, p. 1698 - 1701
[33] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 6, p. 1880 - 1886
[34] Patent: WO2011/146371, 2011, A1, . Location in patent: Page/Page column 24-25
[35] Patent: WO2014/89365, 2014, A1, . Location in patent: Paragraph 00395
[36] Patent: WO2018/93716, 2018, A1, . Location in patent: Page/Page column 20; 22; 23
20
[ 4746-97-8 ]
[ 311-46-6 ]
[ 51656-91-8 ]
Yield
Reaction Conditions
Operation in experiment
86%
Stage #1: With potassium <i>tert</i>-butylate In N,N-dimethyl-formamide at 0 - 20℃; for 1 h; Stage #2: at 0 - 20℃; for 16 h;
Triethylphosphono acetate (6lmL, 0.3Omol) was added to a suspention KOBu-t(33g, 0.3Omol) in DMF (200mL) at 0 00, stirred for lh at RT. A solution of 1,4- dioxaspiro[4.5]decan-8-one (40g, 0.25mo1) in DMF (200mL) was added at 0 C and the whole then stirred for 16h at RT. The reaction mixture was quenched with sat NH4CI solutionand extracted with ethyl acetate (2X500mL). The combined organic layer was washed with water, brine,dried over Na2SO4 and distilled under reduced pressure to afford crude, which was purified by column chromatography (silica gel; 60-l20mesh); the product eluted with 10- 15percentethyl acetate in hexane to yield 50.Og (86percent) of Ethyl-2-(1,4-dioxaspiro-[4.5]-decan-8- ylidene)-acetate as liquid.
Reference:
[1] Patent: WO2016/8582, 2016, A1, . Location in patent: Page/Page column 65
[2] Journal of the American Chemical Society, 1991, vol. 113, # 21, p. 8016 - 8024
[3] Tetrahedron Letters, 1993, vol. 34, # 22, p. 3505 - 3508
21
[ 4746-97-8 ]
[ 1099-45-2 ]
[ 51656-91-8 ]
Yield
Reaction Conditions
Operation in experiment
90%
for 24 h; Heating / reflux
Preparation of (l,4-Dioxa-spiro[4.5]dec-8-ylidene)-acetic acid ethyl ester 22; A solution of 1,4-cycloliexanedionemonoetliylketal 20 (6g, 40mmol) and ethyl- (triphenylphosphoranylidene)acetate 22 (15g, 44mmol) in dry benzene (80ml) were refluxed under argon for 24hours. The solvent was removed under vacuum and product purified by flash chromatography to give the product in 90percent.
Reference:
[1] Angewandte Chemie - International Edition, 2007, vol. 46, # 33, p. 6278 - 6283
[2] Patent: US2007/265243, 2007, A1, . Location in patent: Page/Page column 3
[3] Organic and Biomolecular Chemistry, 2006, vol. 4, # 24, p. 4431 - 4436
[4] Patent: WO2008/38030, 2008, A2, . Location in patent: Page/Page column 51
[5] Tetrahedron, 1995, vol. 51, # 37, p. 10259 - 10280
Reference:
[1] Journal of the American Chemical Society, 2012, vol. 134, # 41, p. 17023 - 17026,4
[2] Journal of the American Chemical Society, 2012, vol. 134, # 41, p. 17023 - 17026
[3] Journal of the American Chemical Society, 2012, vol. 134, # 49, p. 20208 - 20208
[4] Patent: WO2014/140279, 2014, A1,
[5] Patent: CN107573212, 2018, A,
[6] Patent: WO2018/54365, 2018, A1,
35
[ 4746-97-8 ]
[ 18068-06-9 ]
Reference:
[1] Journal of the Chemical Society. Perkin Transactions 2, 1997, # 6, p. 1221 - 1234
Reference:
[1] Patent: EP2471777, 2012, A1,
[2] Angewandte Chemie - International Edition, 2017, vol. 56, # 38, p. 11599 - 11603[3] Angew. Chem., 2017, vol. 129, p. 11757 - 11761,5
39
[ 4746-97-8 ]
[ 96184-81-5 ]
Reference:
[1] Molecular Crystals and Liquid Crystals (1969-1991), 1985, vol. 131, p. 327 - 342
40
[ 4746-97-8 ]
[ 120686-00-2 ]
Reference:
[1] Journal of the American Chemical Society, 1989, vol. 111, # 11, p. 4116 - 4117
41
[ 4746-97-8 ]
[ 74-88-4 ]
[ 134409-06-6 ]
Yield
Reaction Conditions
Operation in experiment
64%
Stage #1: With sodium amide In toluene at 20℃; for 0.5 h; Inert atmosphere Stage #2: at 0 - 20℃; for 4 h; Inert atmosphere
To a solution of sodium amide (10.0 g, 256.0 mmol) in dry toluene (50 mL) was added cyclohexane-1,3-dione 6 (10.0 g, 64.0 mmol). The reaction mixture was stirred under Ar for 30 min at rt. Then iodomethane (10.0mL, 160.7 mmol) was added dropwise to the above reaction mixture at 0°C and the resulting solution was stirred at rt for 4 h. The reaction mixture was quenched by additionof saturated NH4Cl (350 mL) aqueous solution and extracted with ether (3 ×250 mL). Combined extracts were dried (MgSO4), filtered, and concentrated invacuo. Silica gel flash chromatography afforded compound 7 (7.6 g, 64percent yield).
41%
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at 0 - 20℃; for 1 h; Inert atmosphere Stage #2: at 10 - 20℃; for 1.5 h; Inert atmosphere
To a solution of 1,4-dioxaspiro[4.5]decan-8-one (5.45 g, 34.9 mmol, commercially available from, for example, Sigma Aldrich) in dry THF (20 mL) was added sodium hydride (60percent in mineral oil, 2.79 g, 69.8 mmol) at 0 °C under nitrogen. The resulting reaction mixture was stirred at rt for 1 h followed by the addition of methyl iodide (5.45 mL, 87 mmol). The reaction mixture was stirred at 10 °C to rt over 1.5 h. The reaction mixture was quenched with saturated ammonium chloride solution (50 mL) and the aqueous layer was extracted with ethyl acetate (2x 80 mL). The combined organic layers were washed with brine and dried through a hydrophobic frit. The organic layer was concentrated in vacuo to give ~6.8 g of crude yellow residue. This was purified by chromatography on SiO2 (Biotage SNAP Ultra 100 g cartridge, eluting with 5-65percent diethyl ether/cyclohexane) to give 7,7-dimethyl-1,4- dioxaspiro[4.5]decan-8-one (3.07 g, 14.16 mmol, 41 percent yield) as a colourless oil. (0589) LCMS (2 min Formic): Rt = 0.78 min, [MH]+ = 185.0. (0590) 1H NMR (400 MHz, CDCl3) δ ppm 3.95 - 4.06 (m, 4 H) 2.56 - 2.64 (m, 2 H) 2.02 (d, J=1.26 Hz, 2 H) 1.87 - 1.94 (m, 2 H) 1.19 (s, 6 H)
40%
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at 0 - 20℃; for 1 h; Inert atmosphere Stage #2: at 10℃;
A. 7,7-Dimethyl-1,4-dioxaspiro[4.5]decan-8-one To a solution of 1,4-dioxaspiro[4.5]decan-8-one (1.0 equiv.) in anhydrous THF (0.6 M) was added sodium hydride (2.0 equiv, 60percent in mineral oil) at 0° C. under nitrogen. After the resulting reaction mixture was stirred at room temperature for 60 min, iodomethane (2.5 equiv.) was added. The reaction was stirred at 10° C. overnight. TLC (petroleum ether: ethyl acetate=10:1) showed that the reaction was completed. The reaction was quenched with saturated aqueous ammonium chloride solution and the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under vacuum, and the residue was purified by silica gel column chromatography (10percent ethyl acetate in petroleum ether) to afford the 7,7-dimethyl-1,4-dioxaspiro[4.5]decan-8-one (40percent yield) as colorless oil. 1H NMR (400 MHz, CDCl3) δ (ppm) 3.99 (s, 4H), 2.55-2.58 (m, 2H), 1.97-2.00 (m, 2H), 1.87 (s, 2H), 1.16 (s, 6H).
21%
With potassium <i>tert</i>-butylate In tetrahydrofuran at 0 - 20℃; for 16 h;
Stage (i): 7,7-Dimethyl-1,4-dioxaspiro[4.5]decan-8-one A solution of ButOK (39.0 g, 352.157 mmol, 2.2 eq) in THF (250 ml) was added dropwise at 0° C. to a solution of 1,4-dioxaspiro[4.5]decan-8-one (25 g, 160.7 mmol, 1 eq) in THF (500 ml). Then methyl iodide (60 ml, 960.43 mmol, 6 eq) was added dropwise at 0° C. and stirring was carried out for 16 h at RT. The reaction mixture was filtered off over Celite, and the filtrate was concentrated under reduced pressure and then purified by column chromatography (silica gel, 3percent ethyl acetate in hexane). The desired product was obtained in the form of a light-yellow oil. Yield: 21percent (6.18 g, 33.58 mmol).
Reference:
[1] Journal of Organic Chemistry, 2011, vol. 76, # 19, p. 7706 - 7719
43
[ 4746-97-8 ]
[ 38622-91-2 ]
[ 69947-09-7 ]
Yield
Reaction Conditions
Operation in experiment
85%
With potassium <i>tert</i>-butylate In 1,2-dimethoxyethane at 0 - 20℃;
The present invention will now be described in detail with reference to the foregoing,As shown above,In the present technical solution,The p-toluenesulfonyl methyl isocyanide of formula 1, and(1,4-dioxaspiro [4,5] decan-8-one)The contacting of the compounds shown is carried out in ethylene glycol dimethyl ether,And the molar ratio of p-toluenesulfonylmethyl isonitrile to the compound of formula 1 is 1: 1,The reaction temperature of the reaction system is 0-5 degrees Celsius,P-Methylphenylsulfonylmethylisonitrile (320 g, 1 mole)And compound 1 (156 g, 1 mole) were dissolved in ethylene glycol dimethyl ether (2000 ml)And the reaction system is cooled to 0-5 degrees;Potassium t-butoxide (224 g, 2 mol) was added in portions,Keep the system temperature does not exceed 5 degrees,After the addition was complete the system was slowly raised to room temperature with stirring,To the raw material reaction is complete;Saturated sodium chloride solution (3000 ml) was added,Extraction with methyl tert-butyl ether (500 ml * 2)Combine organic phase,dry,Spin dry,Distilled (100 ° C, 5 mm Hg) to give compound 2 (142 g, 85percent) as a colorless oil,
71%
With potassium <i>tert</i>-butylate In 1,2-dimethoxyethane; ethanol at -13 - 5℃; for 0.666667 h;
To a solution of Compound 58 (10.0 g, 64.0 mmol) in a mixture of 1,2-dimethoxyethane (218 ml)-ethanol (6.5 mL) was added p-toluenesulfonyl methyl isocyanide (16.3 g, 83.0 mmol), and the obtained reaction mixture was cooled to −13° C. Potassium t-butoxide (17.2 g, 154 mmol) was added thereto at 5° C. or less over 40 minutes. The reaction liquid was stirred under ice-cooling for one hour, and further stirred at room temperature for one hour, and the solvent was removed by distillation under reduced pressure. The residue was diluted with ethyl acetate, and then washed with water, and the solvent was removed by distillation under reduced pressure, followed by purification by silica gel chromatography to obtain Compound 67 (7.63 g, 45.6 mmol, 71percent) as a colorless oily substance. Compound 67: 1H-NMR (CDCl3) δ: 1.57-1.67 (2H, m), 1.79-2.03 (5H, m), 2.66 (1H, s), 3.95 (4H, s).
69%
Stage #1: With potassium <i>tert</i>-butylate In 1,2-dimethoxyethane; <i>tert</i>-butyl alcohol at 0 - 20℃; for 2 h; Stage #2: With water In 1,2-dimethoxyethane; <i>tert</i>-butyl alcohol
l,4-Dioxaspiro[4.5]decane-8-carbonitrileA solution of t-BuOK (22.8 g, 0.203 mol) in a 1: 1 mixture of t-BuOH and 1,2- dimethoxyethane (200 niL) was added to a solution of 1,4-cyclohexanedione monoethylene ketal (15.5 g, 0.099 mol) and tosylmethyl isocyanide (20.3 g, 0.104 mol) in dimethoxyethane (200 mL) at 00C. The reaction mixture was stirred for one hour at 00C, allowed to warm to ambient temperature and stirred for one extra hour. The reaction mixture was poured in water (500 mL). The product was extracted with hexane (3 x 200 mL) and ether (3 x 200 mL). The combined organics were dried with anhydrous Na2SO4 and the solvent was concentrated. The product was purified by flash chromatography on silica gel using EtOAc 40percent in hexane as eluent to afford the title compound as a colorless liquid. Yield: 11.5 g (69percent). 1H NMR (400 MHz, METHANOL-D4): δ 1.55 - 1.69 (m, 2 H), 1.70 - 1.80 (m, 2 H), 1.78 - 1.90 (m, 2 H), 1.90 - 2.05 (m, 2 H), 2.71 - 2.89 (m, 1 H), 3.86 - 3.98 (m, 4 H).
Reference:
[1] Patent: CN106467477, 2017, A, . Location in patent: Paragraph 0025; 0026; 0027
[2] Synthesis, 1992, # 11, p. 1080 - 1082
[3] Tetrahedron Letters, 2000, vol. 41, # 29, p. 5589 - 5592
[4] Tetrahedron, 2002, vol. 58, # 8, p. 1557 - 1563
[5] Patent: US9567330, 2017, B2, . Location in patent: Page/Page column 67
[6] Patent: WO2007/13848, 2007, A1, . Location in patent: Page/Page column 8
44
[ 4746-97-8 ]
[ 19158-51-1 ]
[ 69947-09-7 ]
Yield
Reaction Conditions
Operation in experiment
50%
With potassium <i>tert</i>-butylate In 1,2-dimethoxyethane at 0℃; for 2 h;
Step 1: t-BuOK (18.5 g, 165.2 mmol) was added into a solution of compound 116-1 (10 g, 64 mmol) and TosMIC(17.6 g, 90.27 mmol) in DME (10 mL) at 0 °C. The reaction mixture was stirred at 0°C for 2h. After the reaction wascomplete, the reaction mixture was quenched with saturated NH4Cl solution and extracted with EtOAc. The combinedextraction liquid was washed with brines, dried over sodium sulfate and concentrated to dry, finally purified by columnchromatography (PE:EtOAc = 1:1) to deliver 2 (6.7 g, yield 50percent) as colorless oil.
Reference:
[1] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[2] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[3] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[4] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[5] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[6] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[7] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[8] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[9] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[10] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[11] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[12] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[13] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[14] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[15] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[16] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[17] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[18] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[19] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
[20] Journal of Organic Chemistry, 2012, vol. 77, # 10, p. 4842 - 4848
52
[ 4746-97-8 ]
[ 84892-43-3 ]
Reference:
[1] Journal of Organic Chemistry, 1994, vol. 59, # 7, p. 1855 - 1862
53
[ 4746-97-8 ]
[ 105-36-2 ]
[ 189509-22-6 ]
Reference:
[1] Journal of the Chinese Chemical Society, 2003, vol. 50, # 3 A, p. 449 - 456
[2] Tetrahedron, 1997, vol. 53, # 13, p. 4693 - 4702
[3] Chemical and Pharmaceutical Bulletin, 1998, vol. 46, # 4, p. 581 - 586
[4] Organic and Biomolecular Chemistry, 2015, vol. 13, # 24, p. 6737 - 6741
Stage #1: With morpholinosulfur trifluoride In dichloromethane at 0 - 20℃; for 72 h; Inert atmosphere Stage #2: With potassium permanganate In dichloromethane; water for 24 h;
Method B A solution of ketone 6 (4.40 g, 28.2 mmol) in dry CH2Cl2 (100 mL) was cooled to 0 °C under argon atmosphere. Morph-DAST (11.85 g, 67.7 mmol, 2.4 equiv) was added dropwise upon stirring. The reaction mixture was allowed to warm to a room temperature and was stirred for 72 h. A saturated solution of NaHCO3 in water (50 mL) was added and the reaction mixture was stirred for 10 min to quench the unreacted fluorinating agent. The organic layer was separated and the water phase was extracted with CH2Cl2 (2*50 mL). Solution of KMnO4 (5.7 g, 36 mmol, 10 equiv) in water (100 mL) was added to the combined organic phase and the formed suspension was vigorously stirred for 24 h. The organic layer was separated and the water phase was extracted with CH2Cl2 (2*100 mL). The combined organic phases were dried over Na2SO4, and evaporated under vacuum at 30 °C to provide pure compound 7 (3.65 g, 20.5 mmol, 73percent yield) as a colourless oil, which crystallized upon storage.
60%
With (diethylamino)sulfur trifluoride In dichloromethane; water
EXAMPLE 46 8,8-Difluoro-1,4-dioxa-spiro[4.5]decane 1,4-Dioxa-spiro[4.5]decan-8-one (9.0 g, 56 mmol) and (diethylamino)sulfur trifluoride (19 g, 112 mmol) are reacted in dichloromethane (180 ml) for 2 h at room temperature. The mixture is poured in water (300 ml), the layers are separated and the aqueous phase back-extracted twice with dichloromethane (50 ml). The combined organic phases are dried with magnesium sulfate and evaporated. Distillation under reduced pressure over a vigreux-column afforded the title compound as colorless liquid (6.0 g, 60percent), bp 65-72° C. at 13-14 mbar, MS: m/e=186 (M+), contaminated with ~30percent 8-Fluoro-1,4-dioxa-spiro[4.5]dec-7-ene, MS: m/e=158 (M+).
45%
With N-methyl-N-phenylaminodifluorosulfiniumtetrafluoroborate; triethylamine tris(hydrogen fluoride) In dichloromethane at 20℃; for 3 h; Inert atmosphere
General procedure: To a solution of triethylamine trihydrofluoride (1equiv.) in dichloromethane (2mL) at room temperature was added the N,N-disubstituted aminodifluorosulfinium tetrafluoroborate (1.5equiv.) followed by 1,4-dioxaspiro[4.5]decan-8-one (1.0mmol, 1equiv.). After 3h of stirring under nitrogen, the reaction mixture was quenched at room temperature with a 5percent aqueous sodium bicarbonate solution, stirred for 15min, and the resulting mixture was extracted twice using dichloromethane. The organic phases were combined, dried over sodium sulfate and solvents were evaporated under low-vacuum at 40°C. NMR yield was calculated by integration of 19F NMR and/or 1H NMR signals of the resulting crude material+internal standard (2-fluoro-4-nitrotoluene).
Reference:
[1] Journal of Organic Chemistry, 2010, vol. 75, # 10, p. 3401 - 3411
[2] Organic Letters, 2013, vol. 15, # 5, p. 1088 - 1091
[3] Tetrahedron, 2013, vol. 69, # 20, p. 4066 - 4075
[4] Patent: US2003/149036, 2003, A1,
[5] Journal of Fluorine Chemistry, 2013, vol. 153, p. 57 - 60
[6] Patent: US2006/293392, 2006, A1, . Location in patent: Page/Page column 51
[7] Patent: WO2008/7930, 2008, A1, . Location in patent: Page/Page column 28
[8] Patent: WO2009/48547, 2009, A1, . Location in patent: Page/Page column 40
[9] Patent: US2010/120783, 2010, A1, . Location in patent: Page/Page column 10
[10] Patent: WO2010/56022, 2010, A2, . Location in patent: Page/Page column 23
[11] Patent: EP2786986, 2014, A2, . Location in patent: Paragraph 0122-0124
[12] Patent: CN105985355, 2016, A, . Location in patent: Paragraph 0670; 0671; 0672; 0673
56
[ 4746-97-8 ]
[ 176251-49-3 ]
[ 142273-44-7 ]
Yield
Reaction Conditions
Operation in experiment
60%
With pyrrolidinodifluorosulfiniumtetrafluoroborate; triethylamine tris(hydrogen fluoride) In dichloromethane at 20℃; for 3 h; Inert atmosphere
General procedure: To a solution of triethylamine trihydrofluoride (1equiv.) in dichloromethane (2mL) at room temperature was added the N,N-disubstituted aminodifluorosulfinium tetrafluoroborate (1.5equiv.) followed by 1,4-dioxaspiro[4.5]decan-8-one (1.0mmol, 1equiv.). After 3h of stirring under nitrogen, the reaction mixture was quenched at room temperature with a 5percent aqueous sodium bicarbonate solution, stirred for 15min, and the resulting mixture was extracted twice using dichloromethane. The organic phases were combined, dried over sodium sulfate and solvents were evaporated under low-vacuum at 40°C. NMR yield was calculated by integration of 19F NMR and/or 1H NMR signals of the resulting crude material+internal standard (2-fluoro-4-nitrotoluene).
Reference:
[1] Journal of Fluorine Chemistry, 2013, vol. 153, p. 57 - 60
[2] Organic Letters, 2009, vol. 11, # 21, p. 5050 - 5053
[3] Journal of Organic Chemistry, 2010, vol. 75, # 10, p. 3401 - 3411
[4] Tetrahedron, 2013, vol. 69, # 20, p. 4066 - 4075
[5] Patent: CN107827721, 2018, A, . Location in patent: Paragraph 0100-0105
Reference:
[1] Tetrahedron Letters, 2011, vol. 52, # 17, p. 2228 - 2231
[2] Journal of the American Chemical Society, 2012, vol. 134, # 41, p. 17023 - 17026,4
[3] Journal of the American Chemical Society, 2012, vol. 134, # 41, p. 17023 - 17026
[4] Journal of the American Chemical Society, 2012, vol. 134, # 49, p. 20208 - 20208
[5] Angewandte Chemie - International Edition, 2015, vol. 54, # 49, p. 14748 - 14752[6] Angew. Chem., 2015, vol. 127, # 49, p. 14961 - 14965,5
With n-butyllithium; In tetrahydrofuran; (2S)-N-methyl-1-phenylpropan-2-amine hydrate; hexane;
a 4-(4-Dimethylaminomethylphenyl)-4-hydroxycyclohexanone-ethylene ketal To a solution of 36.4 g (0.17 mol) of 4-bromo-N,N-dimethylbenzylamine in 250 ml of dry tetrahydrofuran, cooled to -70 C., are added dropwise, under a nitrogen atmosphere and with stirring, 112 ml (0.179 mol) of a 1.6 molar solution of n-butyllithium in hexane in such a way that the temperature does not exceed -65 C. The orange solution is stirred for a further 15 minutes at -70 C. and then within 10 minutes a solution of 27.6 g (0.172 mol) of 1,4-cyclohexanedione-monoethylene ketal in 110 ml of tetrahydrofuran is added, whilst the temperature must not exceed -65 C. The reaction mixture is stirred first for 30 minutes at -70 C. and then without external cooling until a temperature of +20 C. is reached, then poured into 600 ml of ice water and extracted with 200 ml of ethyl acetate. The organic phase is separated off and the aqueous phase is extracted several times with ethyl acetate. The combined organic extracts are dried with sodium sulphate, evaporated down in vacuo and the residue remaining is recrystallized from diisopropylether. 41.9 g (85% of theory) of 4-(4-dimethylaminomethylphenyl)-4-hydroxycyclohexanone-ethylene ketal are obtained, m.p. 84-86 C.
With n-butyllithium; In tetrahydrofuran; (2S)-N-methyl-1-phenylpropan-2-amine hydrate; hexane;
a) 4-(4-Dimethylaminomethylphenyl)-4-hydroxycyclohexanone-ethylene ketal To a solution of 36.4 g (0.17 mol) of 4-bromo-N,N-dimethylbenzylamine in 250 ml of dry tetrahydrofuran, cooled to -70 C., are added dropwise, under a nitrogen atmosphere and with stirring, 112 ml (0.179 mol) of a 1.6 molar solution of n-butyllithium in hexane in such a way that the temperature does not exceed -65 C. The orange solution is stirred for a further 15 minutes at -70 C. and then within 10 minutes a solution of 27.6 g (0.172 mol) of 1,4-cyclohexanedione-monoethylene ketal in 110 ml of tetrahydrofuran is added, whilst the temperature must not exceed -65 C. The reaction mixture is stirred first for 30 minutes at -70 C. and then without external cooling until a temperature of +20 C. is reached, then poured into 600 ml of ice water and extracted with 200 ml of ethyl acetate. The organic phase is separated off and the aqueous phase is extracted several times with ethyl acetate. The combined organic extracts are dried with sodium sulphate, evaporated down in vacuo and the residue remaining is recrystallized from diisopropylether. 41.9 g (85% of theory) of 4-(4-dimethylaminomethylphenyl)-4-hydroxycyclohexanone-ethylene ketal are obtained, m.p. 84-86 C.
With potassium tert-butylate; In 1,2-dimethoxyethane; at 0 - 20℃;
The present invention will now be described in detail with reference to the foregoing,As shown above,In the present technical solution,The p-toluenesulfonyl methyl isocyanide of formula 1, and(1,4-dioxaspiro [4,5] decan-8-one)The contacting of the compounds shown is carried out in ethylene glycol dimethyl ether,And the molar ratio of p-toluenesulfonylmethyl isonitrile to the compound of formula 1 is 1: 1,The reaction temperature of the reaction system is 0-5 degrees Celsius,P-Methylphenylsulfonylmethylisonitrile (320 g, 1 mole)And compound 1 (156 g, 1 mole) were dissolved in ethylene glycol dimethyl ether (2000 ml)And the reaction system is cooled to 0-5 degrees;Potassium t-butoxide (224 g, 2 mol) was added in portions,Keep the system temperature does not exceed 5 degrees,After the addition was complete the system was slowly raised to room temperature with stirring,To the raw material reaction is complete;Saturated sodium chloride solution (3000 ml) was added,Extraction with methyl tert-butyl ether (500 ml * 2)Combine organic phase,dry,Spin dry,Distilled (100 C, 5 mm Hg) to give compound 2 (142 g, 85%) as a colorless oil,
71%
With potassium tert-butylate; In 1,2-dimethoxyethane; ethanol; at -13 - 5℃; for 0.666667h;
To a solution of Compound 58 (10.0 g, 64.0 mmol) in a mixture of 1,2-dimethoxyethane (218 ml)-ethanol (6.5 mL) was added p-toluenesulfonyl methyl isocyanide (16.3 g, 83.0 mmol), and the obtained reaction mixture was cooled to -13 C. Potassium t-butoxide (17.2 g, 154 mmol) was added thereto at 5 C. or less over 40 minutes. The reaction liquid was stirred under ice-cooling for one hour, and further stirred at room temperature for one hour, and the solvent was removed by distillation under reduced pressure. The residue was diluted with ethyl acetate, and then washed with water, and the solvent was removed by distillation under reduced pressure, followed by purification by silica gel chromatography to obtain Compound 67 (7.63 g, 45.6 mmol, 71%) as a colorless oily substance. Compound 67: 1H-NMR (CDCl3) delta: 1.57-1.67 (2H, m), 1.79-2.03 (5H, m), 2.66 (1H, s), 3.95 (4H, s).
69%
l,4-Dioxaspiro[4.5]decane-8-carbonitrileA solution of t-BuOK (22.8 g, 0.203 mol) in a 1: 1 mixture of t-BuOH and 1,2- dimethoxyethane (200 niL) was added to a solution of 1,4-cyclohexanedione monoethylene ketal (15.5 g, 0.099 mol) and tosylmethyl isocyanide (20.3 g, 0.104 mol) in dimethoxyethane (200 mL) at 00C. The reaction mixture was stirred for one hour at 00C, allowed to warm to ambient temperature and stirred for one extra hour. The reaction mixture was poured in water (500 mL). The product was extracted with hexane (3 x 200 mL) and ether (3 x 200 mL). The combined organics were dried with anhydrous Na2SO4 and the solvent was concentrated. The product was purified by flash chromatography on silica gel using EtOAc 40% in hexane as eluent to afford the title compound as a colorless liquid. Yield: 11.5 g (69%). 1H NMR (400 MHz, METHANOL-D4): delta 1.55 - 1.69 (m, 2 H), 1.70 - 1.80 (m, 2 H), 1.78 - 1.90 (m, 2 H), 1.90 - 2.05 (m, 2 H), 2.71 - 2.89 (m, 1 H), 3.86 - 3.98 (m, 4 H).
With potassium tert-butylate; In 1,2-dimethoxyethane; tert-butyl alcohol; at 0 - 20℃; for 13h;
A solution of t-BuOK (147 g, 1.31 mol) in t-BuOH and DME (2.0 L, 1:1 v/v) was added dropwise to a 0-5 C. solution of 1,4-dioxaspiro[4.5]decan-8-one (100 g, 640 mmol) and TosMIC (131 g, 672 mmol) in DME (2.0 L), and the resulting mixture was stirred at 0-5 C. for 1 h before it was allowed to warm to rt over 12 h. After this time, the mixture was poured into water and then extracted three timed with MTBE. The organic layers were combined, washed with brine, dried with anhydrous Na2SO4, filtered, and then concentrated to afford the title compound, which was used in the next step without further purification.
With potassium tert-butylate; In 1,2-dimethoxyethane; tert-butyl alcohol; at 0 - 20℃; for 13h;
A solution of t-BuOK (147 g, 1.31 mol) in t-BuOH and DME (2.0 L, 1:1 v/v) was added dropwise to a 0-5 C. solution of 1,4-dioxaspiro[4.5]decan-8-one (100 g, 640 mmol) and TosMIC (131 g, 672 mmol) in DME (2.0 L), and the resulting mixture was stirred at 0-5 C. for 1 h before it was allowed to warm to rt over 12 h. After this time, the mixture was poured into water and then extracted three timed with MTBE. The organic layers were combined, washed with brine, dried with anhydrous Na2SO4, filtered, and then concentrated to afford the title compound, which was used in the next step without further purification.
With triethylmethylammonium chloride; at 50℃; for 1h;
Take 19.7 g of methyltriethylammonium chloride,Add 62g of ethylene glycol,Heat to 50 C,A colorless, transparent liquid is obtained. 11.2 g of 1,4-cyclohexanedione was weighed and added to the colorless transparent liquid, and reacted at 50 C for 1 h. After the reaction was completed, the temperature was lowered to room temperature, and 56 g of diethyl ether was added thereto to extract, the lower layer solution was taken, and the solvent was evaporated to give a white solid (15.6 g).The content of 1,4-cyclohexanedione monoethylene glycol ketal was 96.1%, and the yield was 96.0%.
toluene-4-sulfonic acid; In toluene;Reflux;
The synthesis of 2-(4-ammo-l-(34-hydroxycyclohexyl)-lH-pyrazolo [3, 4-d]py?midm-3- yl)-lH-mdol-5-ol (Compound 14-9) is described m Scheme 14. 1, 4 cyclohexyldione is selectively monoketahzed, then the remaining ketone is reduced with sodium borohydride to yield compound 14-3. The hydroxyl is converted to the tosylate (compound 14-4), which is then reacted with pyrazolopy?midme iodide 1-3, to produce compound 14-5. The ketone of the cyclohexyl moiety is unmasked with acid treatment and reduced to hydroxy compound 14-7. Palladium catalyzed coupling with mdolyl boronic acid and subsequent deprotection affords compound 14-9.
In tetrahydrofuran; diethyl ether; at -78 - -55℃; for 4h;Inert atmosphere;
Step 1: synthesis of 8-methyl-1,4-dioxaspiro[4.5]decan-8-ol:To a cooled solution of 1,4-dioxaspiro[4.5]decan-8-one (7.8 g, 50.0 mmol) in dry THF (75 mL)was added methyllithium solution (1.5M in ether, 43.3 mL, 65.0 mmol) at -78C under argonwhile keeping inner temperature below -55 C. After addition, the mixture was stirred at -55 Cfor additional 4 hours. The reaction was warmed to room temperature and then quenched bysaturated NH4C1 solution. The separated organic layer was concentrated in vacuo and the residue was purified by column chromatography on silica gel (EA : PE =1:9 to 1:3) to afford the product as a white solid (6.8 g, yield: 80%).
80%
In tetrahydrofuran; diethyl ether; at -78 - -55℃; for 4h;Inert atmosphere;
Step 1: synthesis of 8-methyl-1,4-dioxaspiro[4.5]decan-8-ol To a cooled solution of 1,4-dioxaspiro[4.5]decan-8-one (7.8 g, 50.0 mmol) in dry THF (75 mL) was added methyllithium solution (1.5M in ether, 43.3 mL, 65.0 mmol) at -78 C. under argon while keeping inner temperature below -55 C. After addition, the mixture was stirred at -55 C. for additional 4 hours. The reaction was warmed to room temperature and then quenched by saturated NH4Cl solution. The separated organic layer was concentrated in vacuo and the residue was purified by column chromatography on silica gel (EA:PE=1:9 to 1:3) to afford the product as a white solid (6.8 g, yield: 80%).
80%
In tetrahydrofuran; at -78 - -55℃; for 4h;
To a cooled solution of 1,4-dioxaspiro[4.5]decan-8-one (7.8 g, 50.0 mmol) in dry THF (75 mL) was added methyllithium solution (1.5M in ether, 43.3 mL, 65.0 mmol) at -78 C. under argon while keeping inner temperature below -55 C. After addition, the mixture was stirred at -55 C. for additional 4 hours. The reaction was warmed to room temperature and then quenched by saturated NH4Cl solution. The separated organic layer was concentrated in vacuo and the residue was purified by column chromatography on silica gel (EA:PE=1:9 to 1:3) to afford the product as a white solid (6.8 g, yield: 80%).
58%
A solution of 1 ,4-dioxaspiro[4.5]decan-8-one (8.5g, 55mmole) in tetrahydrofuran (75ml) at - 120C under argon was treated dropwise over 10 minutes with a 1.6M solution of methyllithium in ether (45ml, 72mmole). The mixture was stirred at -120C under argon for 2 hours then quenched by addition of 1 M NH4CI solution (100ml) and the resulting mixture washed with water and extracted with ether (3 x 75ml). The combined extract was dried (Na2SO4) and concentrated under vacuum to leave a white solid. This was purified by chromatography on silica gel eluting with 10-40% ethyl acetate/hexane to afford the title compound (5.5g, 58%).1H NMR delta (CDCI3, 400MHz): 1.27 (3H, s), 1.55-1.75 (7H, m), 1.83-1.95 (2H, m), 3.90-4..00 (4H, m).
With sodium tris(acetoxy)borohydride; In dichloromethane;Inert atmosphere;
2,2-diethoxyethan-1-amine 8 (3.63 g; 34.15 mmol) was dissolved in dry DCM (100 ml) and cooled to 0 C, then1,4-cyclohexanedione monoethylene acetal 7 (5.0 g; 31.05 mmol) was added dropwise. After10 min NaBH(OAc)3 (7.89 g; 37.26 mmol) was added in one portion and stirred for 2h atroom temp. The reaction mixture was diluted with DCM and saturated aq. NaHCO3 wasadded slowly, and then stirred for additional 15 min at rt. The layers were separated, theDCM layer was washed couple of times with saturated aq. NaHCO3, water, brine solutionand dried over anhydrous Na2SO4. After concentration the crude residue was filtered throughsilica column to afford pure compound 9a 7.2 g (85% yield).
In tetrahydrofuran; at -78 - 0℃; for 1.33333h;Inert atmosphere;
To a cooled (-78 C.) solution of 1,4-dioxaspiro[4.5]decan-8-one (1 equiv) in THF (0.64 M) was added a solution of methylmagnesium bromide (1.8 equiv, 3 mol/L in ether) dropwise under nitrogen atmosphere. The resulting mixture was stirred at -78 C. for 20 minutes, then warmed to -30 C. for 30 minutes and then stirred at 0 C. for 30 minutes. After the reaction was complete, the resulting mixture was quenched with saturated aqueous ammonia chloride solution and extracted with ethyl acetate. The organic layers were combined, washed with brine, dried over sodium sulfate, filtered and concentrated to afford the title compound as a white solid (92% yield), which was used in the next step without further purification. 1H NMR (300 MHz, CHLOROFORM-d1) delta ppm 4.00-3.91 (m, 4H), 1.95-1.80 (m, 3H), 1.77-1.67 (m, 4H), 1.59-1.58 (m, 1H), 1.27 (s, 3H), 1.17 (s, 1H).
92%
In tetrahydrofuran; diethyl ether; at -70 - 20℃;
A stirred solution of 1,4-dioxaspiro[4.5]decan-8-one (5 g, 32.0 mmol) in dry THF (70 mL) was cooled to -70 C. and methylmagnesium bromide (23.48 mL, 70.4 mmol) in ether was added dropwise over 10 min. The cooling bath was allowed to warm to room temperature and the mixture was stirred overnight. The mixture was quenched with sat. aq. NH4Cl (75 mL) and extracted with diethyl ether (2*300 mL). The combined ether extracts were washed with brine (50 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford 1A (yellow liquid, 5.1 g, 29.6 mmol, 92% yield) which was used in next step without further purification. 1H NMR (400 MHz, CDCl3) delta 3.96-3.90 (m, 4H), 1.91-1.84 (m, 2H), 1.71-1.65 (m, 3H), 1.63-1.57 (m, 3H), 1.22 (s, 3H).
88%
In tetrahydrofuran; toluene; at 0 - 5℃; for 6h;
PREPARATION OF AUnder argon, 1000 ml of toluene and 1000 ml of 1 molar methylmagnesium bromide solution in THF are initially charged in a 4000 ml three-necked flask. At from 0 to 5 C., 156.2 g of 1,4-cyclohexanedione monoethylene glycol ketal in 130 ml of THF are added dropwise over 2 hours. The mixture is stirred at from 0 to 5 C. for 4 hours, and 200 ml of NH4Cl solution are then added. The phases are separated, the aqueous phase is extracted with CH2Cl2 and the organic phases are dried with MgSO4.The solvent is distilled off at atmospheric pressure and the residue is distilled at 1 mbar/110-115 C. using a 10 cm Vigreux column.This gives 152.3 g (=88% of theory).
88%
In tetrahydrofuran; toluene; at 0 - 5℃; for 6h;Inert atmosphere;
Preparation of Aj0599]A10600] Under argon, 1000 ml of toluene and 1000 ml of 1 molar methylmagnesium bromide solution in THF are initially charged in a 4000 ml three-necked flask. At from 0 to 5 C., 156.2 g of 1,4-cyclohexanedione monoethylene glycol ketal in 130 ml of THF are added dropwise over 2 hours. The mixture is stirred at from 0 to 5 C. for 4 hours, and 200 ml of NH4C1 solution are then added. The phases are separated, the aqueous phase is extracted with CH2C12 and the organic phases are dried with Mg504.10601] The solvent is distilled off at atmospheric pressure and the residue is distilled at 1 mbar/i 10-i 15 C. using a 10 cm Vigreux column.10602] This gives 152.3 g (=88% of theory).
In diethyl ether; at 20℃; for 4.5h;
A solution of methylmagnesium bromide in diethyl ether (3 molar, 32 ml, 96 mmol) was added to a solution of 1 ,4-cyclohexanedione-monoethyleneacetal (10 g, 64.0 mmol) in diethyl ether (330 ml) at room temperature with cooling. After stirring for 4.5 hours at room temperature water was added, the layers separated and the aqueous layer extracted a further 2X with diethyl ether, the combined organic layers dried over magnesium sulphate and evaporated to give teh tile compound as a white solid.
In tetrahydrofuran; at 0℃; for 4h;
Example G8. General experimental for Grignard addition to a ketoneGeneral Scheme:mt R99cR"b R99cMgBr, THF HO.p99a - ^ R9p_99b KRepresentative Scheme:To a solution of the ketone starting material (e.g., l,4-dioxaspiro[4.5]decan-8-one, 1 equiv) in dry THF was added CHsMgBr (1.1 equiv) at 0 C until the reaction was complete (approximately 4 h). The mixture was quenched with NH4C1 aqueous and extracted with ethyl acetate, dried over Na2S04 and concentrated under reduced pressure to give the desired alcohol product (e.g., 8-methyl-l,4-dioxaspiro[4.5]decan-8-ol).
In tetrahydrofuran; diethyl ether; at 0 - 20℃; for 2h;
A solution of methylmagnesium bromide (2.2 M in diethylether, 2.82 mmol) was added to a solution of 1,4-cyclohexanedione monoethylene acetal (2.56 mmol) in THF(5 mL), cooled to 0 C. The reaction mixture was stirred atRT for 2 h, and then quenched with a saturated solution of aqueous ammonium chloride. The layers were partitioned between DCM (20 mL) and water (20 mL). The aqueous layer was extracted with DCM (x3). The combined organic extracts were filtered over a phase separator and concentrated under reduced pressure to give crude the titled compound (2.50 mmol,) as a clear oil. 1H NMR (400 MHz, CDCI3, ): 4.02-3.91 (m, 4H), 1.96-1.84 (m, 2H), 1.78-1.65 (m, 4H), 1.65-1.56 (m, 2H), 1.29 (5, 3H), 1.15 (5, 1H)
In diethyl ether; toluene; at 5 - 10℃; for 2h;
To a stirred solution of CH 3MgBr (344.0 ml, 1.032 mol, 3 M in Et 2O) in dried toluene (2 L) was added 1, 4-dioxaspiro [4.5] decan-8-one (70.0 g, 0.449 mol) solution in 350 ml dried toluene dropwise. The resulting mixture was stirred at 510 C for 2 hours. The mixture was poured into saturated aq. NH 4Cl solution (3L) and extracted with EtOAc (3 1L). The combined organic phase was washed with brine (1.5L), dried over Na 2SO 4 and concentrated to afford the 8-methyl-1,4-dioxaspiro [4.5] decan-8-ol (70.0 g, crude) as a white solid.
Example 20 Intermediate 20-trifluoro-methanesulfonic acid 1,4-dioxa-spiro[4,5]dec-7-en-8yl ester To a cold solution of LDA (2.35 g) in THF (20 ml) at -78 C., a solution of 1,4 cyclohexanedione mono ethylene ketal (3.0 g) in THF (20 ml) was added drop wise with cooling. After 90 min at -78 C., a solution of N-phenyl trifluoromethane sulfonimide (7.8 g) in THF (20 ml) was added drop wise with cooling and the reaction temperature was left to rise to room temperature. The THF is removed under vacuum, and the residue was filtered through silica gel (500 ml) and 2% ethyl acetate/hexane to give 2.0 g of the desired product.
Zu einer AUF-78C gekuehlten Loesung von Lithium-bis- (trimethylsilyl)-amid (1M in Tetrahydrofuran, 1. 1 mmol) in trockenem Tetrahydrofuran werden 1, 4-Dioxa-spiro [4. 5] decan- 8-on (1 mmol), geloest in Tetrahydrofuran (2 ml) gegeben. Man ruehrt 1. 5 Stunden BEI-78C weiter und tropft dann eine Loesung von N-Phenyl-trifluormethansulfonimid (1. 07 mmol) in Tetrahydrofuran (2 ml) zu. Anschliessend wird ueber Nacht bei Raumtemperatur geruehrt und das Loesungsmittel anschliessend im Vakuum entfernt. Nach Trocknen des Rueckstandes im Vakuum wird 1, 4-Dioxa-spiro [4. 5] dec-7-en-8- yl-trifluormethan-sulfonsaeureester erhalten, welcher ohne zusaetzliche Aufreinigung sofort weiter umgesetzt wird (Tetrahedron 1999, 55, 14479-14490) : 1H NMR (PPM, CDC13) : 5. 65 (1H) ; 4 (4H) ; 2. 55 (2H) ; 2. 4 (2H) ; 1. 9 (2H).
To a cold solution of LDA (2.35g) in THF (20 ml) AT-78 C, a solution of 1,4 cyclohexanedione mono ethylene ketal (3. 0g) in THF (20 ml) was added drop wise with cooling. After 90 min AT-78 C, a solution of N-phenyl trifluoromethane sulfonimide (7. 8 g) in THF (20 ml) was added drop wise with cooling and the reaction temperature was left to rise to room temperature. The THF is removed under vacuum, and the residue was filtered through silica gel (500 ml) and 2% ethyl acetate/hexane to give 2.0 g of the desired product.
PREPARATION C; 8-(Pinacolboronyl)-1,4-dioxaspiro[4.5]dec-7-ene; Step A: 8-(Trifluoromethyl)sulphonyloxy-1,4-dioxaspiro[4.5]dec-7-ene; Under an anhydrous atmosphere, a 2M solution of 6.4 mmol of LDA in a mixture of THF/heptane is diluted with 8 ml of THF. The temperature is lowered to -78 C. and then 6.4 mmol of 1,4-dioxaspiro[4.5]decan-8-one dissolved in 8 ml of THF are slowly added. The reaction mixture is stirred for 2 hours at that temperature and 9.6 mmol of N-phenyltrifluoromethanesulphonimide dissolved in 8 ml of THF are added. After stirring for 15 minutes at -78 C. and then returning to ambient temperature for a night, the mixture is concentrated. After purification on neutral alumina gel (petroleum ether/ethyl acetate: 95/5), the expected product is isolated. IR (NaCl film): vC=C=1692 cm-1; vSO2=1418 cm-1.
A solution of 2N lithium diisopropylamide (LDA) (48.25 mL, 96.5 mmol) was cooled to -78° C. and diluted with 25 mL of tetrahydrofuran (THF). To this was added dropwise, a solution of 2-(4-Benzyloxy-phenyl)-N,N-dimethyl-acetamide (20 g, 74.3 mmol) in 250 mL of THF. The mixture was warmed to 0° C., then cooled back to -78° C. A solution of 1,4-cyclohexanedione mono-ethylene ketal (14.1 g) in 350 mL of THF was added. The solution was allowed to warm to -20° C. Highthroughput liquid chromatography (HPLC) assay still showed starting material. Another 1 g of ketal was added and the solution was warmed to 0° C. for 2 hour. The reaction was quenched with a mixture of 25 g NH4Cl in 200 mL of water. EtOAc was added and the layers separated. The organic layer was dried with MgSO4, filtered and concentrated. Column chromatography (50percent EtOAc/hexanes) gave 30.3 g, 96percent yield of a solid
A suspension of <strong>[107818-55-3]3-amino-5-bromo-thiophene-2-carboxylic acid methyl ester</strong> (17.0 g, 72.0 mmol) in dry THF (21 ml) was treated with 1 ,4-cyclohexanedione monoethylene ketal (11.3 mg, 72.0 mmol), followed by dibutyltin dichloride (1.098 gr, 3.6 mmol). After 5 min, phenyl silane (9.74 ml, 79.2 mmol) was added and the reaction mixture was stirred overnight at room temperature. After concentration, the residue was dissolved in EtOAc washed with NaHCO3 then brine. The organic layer was separated, dried on Na2SO4, filtered and concentrated. The crude material was diluted in hexane (500 ml). After filtration, the mother liquor was evaporated to dryness to give 5- Bromo-3-(1 ^-dioxa-spiro^.SJdec-delta-ylaminoKhiophene^-carboxylic acid methyl ester (24.79 g, 92% yield). 1H NMR (CDCl3, 400 MHz): 6.90 (br s, 1 H), 6.65 (s, 1H), 3.95 (s, 4H), 3.78 (s, 3H), 3.35 (m, 1H), 2.00 (m, 2H), 1.80 (m, 2H), 1.65 (m, 4H).
92%
A suspension of S-amino-S-bromo-thiophene^-carboxylic acid methyl ester (17.0 g, 72.0 mmol) in dry THF (21 mL) is treated with 1 ,4-cyclohexanedione monoethylene ketal (11.3 mg, 72.0 mmol), followed by dibutyltin dichloride (1.098 g, 3.6 mmol). After 5 min, phenyl silane (9.74 mL, 79.2 mmol) is added and the reaction mixture is stirred over- night at room temperature. After concentration, the residue is dissolved in EtOAc washed with NaHCO3 then brine. The organic layer is separated, dried on Na2SO4, filtered and concentrated. The crude material is diluted in hexane (500 mL). After filtration, the mother liquor is evaporated to dryness to give 5-bromo-3-(1 ,4-dioxa-spiro[4.5]dec-8-ylamino)-thiophene-2-carboxylic acid methyl ester (24.79 g, 92% yield). Ref: WO2004/052885
92%
Example 1C: Preparation of Compound 1: 3-[(trans-4-Hydroxy- cyclohexyl)-(trans-4-methylcyclohexanecarbonyl)-amino]-5-d5-phenyl-thiophene-2- carboxylic acidCompound 1 can be prepared as described in Scheme C below:Step IA suspension of <strong>[107818-55-3]3-amino-5-bromo-thiophene-2-carboxylic acid methyl ester</strong> (17.0 g, 72.0 mmol) in dry THF (21 mL) is treated with 1 ,4-cyclohexanedione monoethylene ketal (11.3 mg, 72.0 mmol), followed by dibutyltin dichloride (1.098 g, 3.6 mmol). After 5 min, phenyl silane (9.74 mL, 79.2 mmol) is added and the reaction mixture is stirred overnight at room temperature. After concentration, the residue is dissolved in EtOAc washed with NaHCC>3 then brine. The organic layer is separated, dried on Na2S04, filtered and concentrated. The crude material is diluted with hexane (500 mL). After filtration, the mother liquor is evaporated to dryness to give 5-bromo-3-(l ,4-dioxa- spiro[4.5]dec-8-ylamino)-thiophene-2-carboxylic acid methyl ester (24.79 g, 92% yield). JH NMR (CDC13, 400 MHz): 6.90 (br s, IH), 6.65 (s, IH), 3.95 (s, 4H), 3.78 (s, 3H), 3.35 (m, IH), 2.00 (m, 2H), 1.80 (m, 2H), 1.65 (m, 4H).
92%
A suspension of <strong>[107818-55-3]3-amino-5-bromo-thiophene-2-carboxylic acid methyl ester</strong> (17.0 g, 72.0 mmol) in dry THF (21 mL) is treated with 1 ,4-cyclohexanedione monoethylene ketal (11.3 mg, 72.0 mmol), followed by dibutyltin dichloride (1.098 g, 3.6 mmol). After 5 min, phenyl silane (9.74 mL, 79.2 mmol) is added and the reaction mixture is stirred over- night at room temperature. After concentration, the residue is dissolved in EtOAc washed with NaHC03 then brine. The organic layer is separated, dried on Na2S04, filtered and concentrated. The crude material is diluted in hexane (500 mL). After filtration, the mother liquor is evaporated to dryness to give 5- bromo-3-(l,4-dioxa-spiro[4.5]dec-8-ylamino)-thiophene-2-carboxylic acid methyl ester (24.79 g, 92% yield). Ref: WO2004/052885
68%
A suspension of 3-Amino-5-bromo-thiophene-2-carboxylic acid methyl ester (1.03 g, 4.38 mmol) in dry THF (1.1 ml) was treated with 1,4-cyclohexanedione monoethylene ketal (684 mg, 4.38 mmol), followed by dibutyltin dichloride (133 mg, 0.44 mmol). After 5 min, phenyl silane (877 L, 4.8 mmol) was added and the reaction mixture was stirred at room temperature for 2 days when a clear solution resulted. The solution was then concentrated and the residue purified by silica gel column chromatography using EtOAc: hexanes as eluent to furnished 5-BROMO-3- (1, 4-dioxa-spiro [4.5] dec-8- ylamino) -thiophene-2-carboxylic acid methyl ester (1.11 g, 68% YIELD). 1H NMR (CDC13, 400 MHz): 6.90 (br s, 1H), 6.65 (s, 1H), 3.95 (s, 4H), 3.78 (s, 3H), 3.35 (m, 1H), 2.00 (M, 2H), 1.80 (m, 2H), 1.65 (m, 4H).
With potassium tert-butylate; In 1,2-dimethoxyethane; ethanol; at 0 - 20℃;
P-toluenesulfonylacetonitrile (6.25 g, 32.01 mmol) and 11 (5 g, 32.01 mmol) were dissolved in DME (160 mL) and EtOH (2 mL) and cooled to 0 C. Potassium t-butoxide (7.18 g, 64.03 mmol) was added. The reaction mixture stirred at 0 C. for thirty minutes and was warmed to room temperature. Upon completion, the reaction mixture was concentrated in vacuo. Water (40 mL) and NaCl were added to the residue, and a precipitate formed. After filtering through celite, the filtrate was extracted with Ether (5×50 mL). The combined organic phases were washed with brine, dried over sodium sulfate, filtered, and concentrated. Short-path distillation (105 to 115 C.) gave 1,4-dioxaspiro[4.5]decane-8-carbonitrile.
3H,4'H-spiro[7-aza-2-benzofuran-1,1'-cyclohexane]-3,4'-dione[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With hydrogenchloride; sodium hydroxide; n-butyllithium; In tetrahydrofuran; acetone;
(3) Preparation of spiro[7-azaisobenzofuran-1(3H),1'-cyclohexane]-3,4'-dione <strong>[20577-26-8]2-bromo-3-cyanopyridine</strong> (2.96 g) and 1,4-cyclohexanedione monoethyleneketal (3.47 g) were dissolved in anhydrous tetrahydrofuran (38 mL). After being cooled to -78 C., n-butyl lithium (1.5 M hexane solution, 12.64 mL) was added and the mixture was stirred at -78 C. for 30 minutes. The reaction temperature was allowed to rise up to the room temperature. The reaction mixture was poured into water (40 mL) and extracted with ethyl acetate (100 mL*3). The organic layer was dried over anhydrous MgSO4. The concentration of the solvent gave a residue, which was recrystallized from ethyl ether-hexane to give iminoether compound (2.93 g). This compound was dissolved in acetone (5 mL) and 2N hydrochloric acid (30 mL). The solution was refluxed for 2hours. After cooling, 2N sodium hydroxide aqueous solution was added to the reaction mixture to adjust pH to 4. The whole was extracted with ethyl acetate (100 mL*3). The organic layer was dried over anhydrous MgSO4. The concentration of the solvent gave a residue, which was recrystallized from ether-hexane to give the subject compound (1.07 g).
With hydrogenchloride; sodium hydroxide; n-butyllithium; In tetrahydrofuran; acetone;
(3) Preparation of spiro[7-azaisobenzofuran-1(3H),1'-cyclohexane]-3,4'-dione <strong>[20577-26-8]2-bromo-3-cyanopyridine</strong> (2.96 g) and 1,4-cyclohexanedione monoethyleneketal (3.47 g) were dissolved in anhydrous tetrahydrofuran (38 mL). After being cooled to -78 C., n-butyl lithium (1.5 M hexane solution, 12.64 mL) was added and the mixture was stirred at -78 C. for 30 minutes. The reaction temperature was allowed to rise up to the room temperature. The reaction mixture was poured into water (40 mL) and extracted with ethyl acetate (100 mL*3). The organic layer was dried over anhydrous MgSO4. The concentration of the solvent gave a residue, which was recrystallized from ethyl ether-hexane to give iminoether compound (2.93 g). This compound was dissolved in acetone (5 mL) and 2N hydrochloric acid (30 mL). The solution was refluxed for 2 hours. After cooling, 2N sodium hydroxide aqueous solution was added to the reaction mixture to adjust pH to 4. The whole was extracted with ethyl acetate (100 mL *3). The organic layer was dried over anhydrous MgSO4. The concentration of the solvent gave a residue, which was recrystallized from ether-hexane to give the subject compound (1.07 g).
With hydrogenchloride; sodium hydroxide; n-butyllithium; In tetrahydrofuran; acetone;
(3) Preparation of spiro[7-azaisobenzofuran-1(3H),1'-cyclohexane]-3,4'-dione <strong>[20577-26-8]2-bromo-3-cyanopyridine</strong> (2.96 g) and 1,4-cyclohexanedione monoethyleneketal (3.47 g) were dissolved in anhydrous tetrahydrofuran (38 mL). After being cooled to -78 C., n-butyl lithium (1.5 M hexane solution, 12.64 mL) was added and the mixture was stirred at -78 C. for 30 minutes. The reaction temperature was allowed to rise up to the room temperature. The reaction mixture was poured into water (40 mL) and extracted with ethyl acetate (100 mL*3). The organic layer was dried over anhydrous MgSO4. The concentration of the solvent gave a residue, which was recrystallized from ethyl ether-hexane to give iminoether compound (2.93 g). This compound was dissolved in acetone (5 mL) and 2N hydrochloric acid (30 mL). The solution was refluxed for 2hours. After cooling, 2N sodium hydroxide aqueous solution was added to the reaction mixture to adjust pH to 4. The whole was extracted with ethyl acetate (100 mL*3). The organic layer was dried over anhydrous MgSO4. The concentration of the solvent gave a residue, which was recrystallized from ether-hexane to give the subject compound (1.07 g).
8-(4-propylphenyl)-1,4-dioxaspiro[4.5]decan-8-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With ammonium chloride; In tetrahydrofuran;
(1) Metal magnesium in an amount of 12 g was suspended in 50 ml of THF under nitrogen gas stream, and a solution prepared by dissolving 100 g of 4-n-propylbromobenzene in 300 ml of THF was added dropwise and stirred at room temperature for 1 hour. A solution prepared by dissolving 78 g of 1,4-cyclohexanedione-monoethylene ketal in 300 ml of THF was added dropwise thereto and stirred at room temperature for 1 hour. After finishing of the reaction, 500 ml of a saturated aqueous solution of ammonium chloride was added and extracted with 500 ml of ethyl acetate. Organic layer was washed with water and dried over anhydrous magnesium sulfate, and the solvent was distilled off under a reduced pressure to obtain 98 g of 1,1-ethylenedioxy-4-hydroxy-4-(4-n-propylphenyl)cyclohexane.
19.8 g (0.1 mol) of 3-propylbromobenzene (reactant) was added dropwise1/10 to 150 ml containing 2.9 g (0.12 mol) of magnesium scrap (reactants)Tetrahydrofuran (solvent) solution,After initiation, the remaining 3-propylbromobenzene (reactants)Prepared into 50 ml of tetrahydrofuran (solvent)The solution was slowly added dropwise to the reaction solution, and the mixture was heated under reflux for 60 minutes,A solution of 15.6 g (0.1 mol) of 1,4-cyclohexanedione mono-diol ketal (reactant) in 50 ml of toluene (solvent) was added dropwise and the mixture was refluxed for 120 minutes and allowed to stand at room temperature for hydrolysis.The reaction solution was slowly poured into a beaker containing 5 ml of concentrated hydrochloric acid and 100 g of ice, and the mixture was separated and extracted with 80 ml of toluene,Mixed with water to neutral, 20g anhydrous sodium sulfate for 10 minutes, under reduced pressure distillation of tetrahydrofuran solvent,150 ml of a pale yellow toluene solution (I-6-a).
DBU (1,8-diazabicyclo-[5.4.0]-7-undecene)[ No CAS ]
[ 506-59-2 ]
[ 40594-34-1 ]
Yield
Reaction Conditions
Operation in experiment
17%
With hydrogenchloride; triethylamine; In water; 1,2-dichloro-ethane;
Reference Example 8 To a suspension of 1,4-cyclohexanedione monoethylene-ketal (3.82 g, 24.6 mmol) and dimethylamine hydrochloride (2.00 g, 24.6 mmol) in 1,2-dichloroethane (50 ml) were dropwise added triethylamine (4.2 ml, 29.6 mmol) and DBU (1,8-diazabicyclo-[5.4.0]-7-undecene) (4.4 ml), and the mixture was stirred for 10 minutes. To the mixture was added triacetoxyborohydride (7.68 g, 34.4 mmol), and the mixture was stirred for 4.5 hours. Precipitate was filtered off, and the filtrate was concentrated to give crude product (6.34 g), which was dissolved in water (10 ml). To the mixture was dropwise added concentrated hydrochloric acid (6 ml), and the mixture was stirred for 48 hours. The reaction mixture was diluted with water and washed twice with ether. The aqueous layer was made basic with sodium hydroxide and extracted with ether twice. The extract was washed with saturated sodium chloride solution, dried with potassium carbonate and purified by evaporation to give 4-dimethylaminocyclohexanone (0.59 g, 17%). b.p.142-145 C. 1 H-NMR (CDCl3) delta: 1.69-2.13 (4H, m), 2.32 (6H, s), 2.20-2.41 (2H, m), 2.44-2.64 (3H, m).
DBU (1,8-diazabicyclo-[5.4.0]-7-undecene)[ No CAS ]
[ 506-59-2 ]
[ 40594-34-1 ]
Yield
Reaction Conditions
Operation in experiment
17%
With triethylamine; In water; 1,2-dichloro-ethane;
REFERENCE EXAMPLE 174 To a suspension of 1,4-cyclohexanedione monoethyleneketal (3.82 g, 24.6 mmol) and dimethylamine hydrochloride (2.00 g, 24.6 mmol) in 1,2-dichloroethane (50 ml) were dropwise added triethylamine (4.2 ml, 29.6 mmol) and DBU (1,8-diazabicyclo-[5.4.0]-7-undecene) (4.4 ml), and the mixture was stirred for 10 minutes. To the mixture was added triacetoxyborohydride (7.68 g, 34.4 mmol), and the mixture was stirred for 4.5 hours. Precipitate was filtered off, and the filtrate was concentrated to give crude product (6.34 g), which was dissolved in water (10 ml). To the mixture was dropwise added concentrated hydro-chloric acid (6 ml), and the mixture was stirred for 48 hours. The reaction mixture was diluted with water and washed twice with ether. The aqueous layer was made basic with sodium hydroxide and extracted with ether twice. The extract was washed with saturated sodium chloride solution, dried with potassium carbonate and purified by evaporation to give 4-dimethylaminocyclohexanone (0.59 g, 17%). b.p.142-5 C.; 1 H-NMR (CDCl3) delta:1.69-2.13 (4H, m), 2.32 (6H, s), 2.20-2.41 (2H, m), 2.44-2.64 (3H, m).
In ammonia-saturated methanol; aluminum nickel; dimethyl sulfoxide;
Preparation of the starting material 4-isobutylidenecyclohexylamine 26.7 g (0.18 mol) of 4-isobutylidenecyclohexanone (prepared with <strong>[22884-29-3]isobutyltriphenylphosphonium bromide</strong>/sodium hydride and cyclohexane-1,4-dione monoethylene ketal in DMSO, followed by hydrolysis of the ketal) in 200 ml of ammonia-saturated methanol was subjected to reductive amination at 100 C. at a hydrogen pressure of 100 bar in the presence of 6 g of Raney nickel. After the catalyst had been removed by filtration and the methanol had been stripped off, the crude product was distilled on a thin-layer evaporator (135 C./0.6 bar). 16.5 g (61% of theory) of a pale yellow oil were obtained.
diethyl N-(1,4-dioxaspiro[4.5]decen-8-yl)aminomethylenemalonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In toluene, p-toluenesulfonic acid monohydrate; ethyl acetate; toluene;
1,4-Dioxaspiro[4.5]decan-8-one (40 g) and <strong>[6296-99-7]diethyl aminomethylenemalonate</strong> (47.94 g) are dissolved in 500 ml of toluene. p-Toluenesulfonic acid monohydrate (2.44 g) and 100 ml of toluene are then added and the mixture is refluxed for 3 days collecting water in a Dean-Stark trap. The resulting mixture is decanted and solvent is removed to leave on oil. This oil is flash chromatographed on 750 g silica gel using 9:1 CH2 Cl2, EtOAc as eluant to yield diethyl N-(1,4-dioxaspiro[4.5]decen-8-yl)aminomethylenemalonate as an oil.
To a DMF (50 mL) solution of <strong>[5927-18-4]trimethyl phosphonoacetate</strong> (26 mL), sodium hydride (purity: 55% or higher, 7.03 g) was added in small portions at 0C. The reaction mixture was warmed to room temperature and stirred for 30 minutes. A DMF (50 mL) solution of 1,4-cyclohexanedione monoethylene ketal (25.2 g) was added thereto in small portions at room temperature. This suspension was stirred for 19 hours and diluted with a saturated aqueous solution of ammonium chloride, followed by two extractions with ethyl acetate. The organic layer was washed with saturated brine, then dried over sodium sulfate, and then concentrated. The residue was purified by chromatography (hexane/ethyl acetate=5:1) to obtain the title compound (29.7 g, 87%) as a colorless oil. 1H NMR (400 MHz, CDCl3): delta (ppm) = 5.75-5.65 (1H, m), 3.99 (4H, s), 3.70 (3H, s), 3.01 (2H, t, J = 6.7 Hz), 2.39 (2H, t, J = 6.7 Hz), 1.81-1.75 (4H, m);. MS (EI) m/z: 212 (M)+.
87%
(1a) (1,4-dioxa-spiro[4.5]dec-8-ylidene)-acetic acid methyl ester To a DMF (50 mL) solution of <strong>[5927-18-4]trimethyl phosphonoacetate</strong> (26 mL), sodium hydride (purity: 55% or higher, 7.03 g) was added in small portions at 0C. The reaction mixture was warmed to room temperature and stirred for 30 minutes. A DMF (50 mL) solution of 1,4-cyclohexanedione monoethylene ketal (25.2 g) was added thereto in small portions at room temperature. This suspension was stirred for 19 hours and diluted with a saturated aqueous solution of ammonium chloride, followed by two extractions with ethyl acetate. The organic layer was washed with saturated brine, then dried over sodium sulfate, and then concentrated. The residue was purified by chromatography (hexane/ethyl acetate=5:1) to obtain the title compound (29.7 g, 87%) as a colorless oil. 1H NMR (400 MHz, CDCl3): delta (ppm) = 5.75-5.65 (1H, m), 3.99 (4H, s), 3.70 (3H, s), 3.01 (2H, t, J = 6.7 Hz), 2.39 (2H, t, J = 6.7 Hz), 1.81-1.75 (4H, m) MS (EI) m/z: 212 (M)+.
With sodium hydride; In tetrahydrofuran; 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone; at 0 - 20℃; for 12.0833h;
a). To a 0 C. solution of <strong>[5927-18-4]trimethyl phosphonoacetate</strong> (4.8 mL, 34 mmol) in DMPU (12 mL) and THF (30 mL) was added a suspension of NaH (0.74 g, 31 mmol) in THF (45 mL). To the resulting mixture was added a solution 1,4-dioxaspiro[4.5]decane-8-one (4.7 g, 30 mL) in THF (30 mL). After stirring at 0 C. for 5 min the suspension was allowed to reach room temperature and stirred for 12 h. The resulting solution was poured into water (900 mL) and extracted with Et2O (2×200 mL). The combined organics were washed with brine and dried over MgSO4, and organics were removed in vacuo. Silica gel chromatography (gradient of 5 to 7% ethyl acetate in hexanes) afforded the product.
NaH (9.2 g, 384 mmol, 1.5 eq) was suspended in THF (850 ml). This was cooled to C. and trimethylphosphonoacetate (40 ml, 277 mmol, 1.1 eq) was added dropwise keeping T<10 C. Mixture was then recooled to 0 C. and stirred for 15 min. Then compound 1 (40 g, 256 mmol, 1.0 eq) in THF (300 ml) was added dropwise. The reaction mixture was stirred over night allowing it to warm to room temperature. The mixture was poured into water (500 ml) and EtOAc (300 ml) were added, the layers were separated. The aqueous layer was extracted with EtOAc (400 ml). The combined organic layers were dried (Na2SO4) and evaporated. Yield: 51.6 g of methyl 2-(4-(1,3-dioxalane)cyclohexylidene)acetate; 1H NMR (CDCl3, 300 MHz) delta 1.78 (q, 4H), 2.37 (t, 2H), 2.99 (t, 2H), 3.68 (s, 3H), 3.99 (s, 4H), 5.65 (s, 1H)
methyl 2-(1,4-dioxaspiro[4.5]dec-8-yl-amino)-5-fluorobenzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
EXAMPLE 22 ;2-([4-({(1R)-1-(4-chlorobenzyl)-2-[4-cyclohexyl-4-(1H-1,2,4-triazol-1-ylmethyl)-piperidin-1-yl]-2-oxoethyl}amino)cyclohexyl]amino}-5-fluoro-N,N-dimethylbenzamide hydrochloride (compound No. 182); 22.1: methyl 2-(1,4-dioxaspiro[4.5]dec-8-yl-amino)-5-fluorobenzoate; 4.1 g of 1,4-dioxaspiro[4.5]decan-8-one are placed in 88 ml of acetic acid. 3.0 g of <strong>[319-24-4]2-amino-5-fluorobenzoic acid methyl ester</strong>, 11.3 g of sodium triacetoxyborohydride and 12.6 g of sodium sulphate are added. The reaction medium is stirred at ambient temperature for 18 h. After the addition of 1.5 g of 1,4-dioxaspiro[4.5]decan-8-one and stirring for a further 24 h, the acetic acid is evaporated off. The residue is treated with a 3N aqueous sodium hydroxide solution. Extraction is carried out with ethyl acetate until the aqueous phase is completely depleted. After drying over MgSO4 and concentration to dryness, the crude obtained is chromatographed on silica gel, elution being carried out with a gradient of methanol in dichloromethane ranging from 0% to 5%. 0.67 g of methyl 2-(1,4-dioxaspiro[4.5]dec-8-ylamino)-5-fluorobenzoate is obtained.
6-(8-hydroxy-1,4-dioxaspiro[4.5]dec-8-yl)-3H-benzoxazol-2-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Step 2: Bromide 1 (12.8 g, 59.6 mmol) was dissolved in anhydrous THF (220 mL), and the solution was cooled to ?78 C. Solutions of MeMgBr (21.9 mL of a 3.0 M solution in Et2O, 65.6 mmol), sec-BuLi (50.4 mL of a 1.3 M solution in cyclohexane, 65.6 mmol), and 1,4-Cyclohexanedione mono-ethylene ketal (11.2 g, 71.5 mmol) in anhydrous THF (10 mL) were added sequentially at 30-minute intervals. After the final addition, the reaction mixture was allowed to warm to room temperature. The reaction was quenched by the addition of 1N HCl (25 mL). The reaction mixture was diluted with EtOAc (500 mL), washed with saturated NaCl (250 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure to provide a mixture of 2 and 3, as a brown oil.
Step 1: Bromide 21 (2.88 g, 12.52 mmol) was dissolved in anhydrous THF (50 mL), and the solution was cooled to ?78 C. Solutions of MeMgBr (4.59 mL of a 3.0 M solution in Et2O, 13.77 mmol), sec-BuLi (10.59 mL of a 1.3 M solution in cyclohexane, 13.77 mmol), and 1,4-cyclohexanedione mono-ethylene ketal (1.95 g, 12.52 mmol) in anhydrous THF (10 mL) were added sequentially at 30-minute intervals. After the final addition, the reaction mixture was allowed to warm to room temperature. The reaction was quenched by the addition of 1N HCl (25 mL). The reaction mixture was diluted with EtOAc (300 mL), washed with saturated NaCl (250 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by silica gel chromatography (eluent 1:1 Hexanes:EtOAc) gave alcohol 22 (1.81 g, 47%), as a white solid: 1H NMR (500 MHz, CDCl3): ?8.48 (br s, 1H), 7.59 (s, 1H), 7.31 (d, J=5 Hz, 1H), 7.04 (d, J=5 Hz, 1H), 2.17-2.04 (m, 4H), 1.81 (d, J=8 Hz, 2H), 1.77 (d, J=8 Hz, 2H).
With sodium triacetoxyborohydride; acetic acid; In 1,2-dichloro-ethane; at 20℃; for 48h;
A mixture of <strong>[38762-41-3]4-bromo-2-chloro-phenylamine</strong> (6 g, 29.06 mmol), 1,4-cyclohexanedione monoethyleneketal (6.908 g, 43.59 mmol), and sodium triacetoxy-borohydride (9.239 g, 43.59 mmol) in DCE (100 ml) and acetic acid (0.2 ml) was stirred at r.t. for 2 days.The mixture was then filtered through a pad of diatomaceous earth and washed with dichloromethane. The filtrate was washed with NaHCO3 (aqueous sat. solution), sodium chloride (aqueous sat. solution), dried (MgSO4) and evaporated in vacuo. The crude product thus obtained was purified by column chromatography (silica gel;DCM/EtOAc 4:1 as eluent). The desired fractions were collected and evaporated in vacuo to yield intermediate compound D33 (8.57 g, 85%).
85%
With sodium tris(acetoxy)borohydride; acetic acid; In 1,2-dichloro-ethane; at 20℃; for 48h;
A mixture of <strong>[38762-41-3]4-bromo-2-chloro-phenylamine</strong> (6 g, 29.06 mmol), [CAS 38762-41-3], 1,4-cyclohexanedione monoethyleneketal [CAS 4746-97-8], (6.908 g, 43.59 mmol), and sodium triacetoxy-borohydride (9.239 g, 43.59 mmol) in DCE (100 ml) and acetic acid (0.2 ml) was stirred at r.t. for 2 days. The mixture was then filtered through a pad of diatomaceous earth and washed with DCM. The filtrate was washed with NaHCO3 (aqueous sat. solution), sodium chloride (aqueous sat. solution), dried (MgSO4) and concentrated in vacuo. The crude product thus obtained was purified by column chromatography (silica gel; DCM/ AcOEt 4:1 as eluent). The desired fractions were collected and concentrated in vacuo to yield intermediate compound D38 (8.57 g,OJ.
A solution of 3-bromoisopropylbenzene (25 mmol) in 20 mL of dry THF is added dropwise over 20 min to 1.22 g (50 mmol) of magnesium turnings in 10 mL of refluxing THF under nitrogen and the mixture is refluxed for an additional 25 min to form the Grignard reagent. The Grignard solution is cooled and added by cannula to a suspension of CuBr-dimethylsulfide complex (0.52 g, 2.5 mmol) in dry THF at-25 C. The suspension is stirred at-25 C for 20 min, and then a solution of 1,4 cyclohexanedione, monoethylene ketal (3.9 g, 25 mmol) in 15 mL of THF is added dropwise over 5 min. The mixture is allowed to gradually warm to ambient temperature. After chromatography over silica gel, eluting with 20% to 30% ethyl acetate in heptane, alcohol 62 (5.6 g, 20 mmol, 80%) as a colorless oil which crystallizes to a white solid on cooling : H NMR (CDCl3) No. 7.39 (s, 1 H), 7.33 (m, 1 H), 7.28 (t, J=7. 5Hz, 1 H), 7.13 (d, J=7. 5Hz, 1 H), 4.0 (m, 4 H), 2.91 (hept, J = 7 Hz, 1 H), 2.15 (m, 4H), 1.82 (brd, J=11. 5Hz, 2H), 1.70 (brd, J=11. 5Hz, 2H), 1.25 (d, J = 7 Hz, 6 H); MS (CI) m/z 259.2
80%
A solution of 3-bromoisopropylbenzene (25 mmol) in 20 ml_ of dry THF was added dropwise over 20 min to 1.22 g (50 mmol) of magnesium turnings in 10 mL of refluxing THF under nitrogen and the mixture was refluxed for an additional 25 min to form the Grignard reagent. The Grignard solution was cooled and added by cannula to a suspension of CuBr-dimethylsulfide complex (0.52 g, 2.5 mmol) in dry THF at -25 C. The suspension was stirred at -25 C for 20 min, and then a solution of 1,4 cyclohexanedione, monoethylene ketal (3.9 g, 25 mmol) in 15 ml of THF was added dropwise over 5 min. The mixture was allowed to gradually warm to ambient temperature. After chromatography over silica gel, eluting with 20% to 30% ethyl acetate in heptane, alcohol 1 (5.6 g, 20 mmol, 80%) as a colorless oil which crystallized to a white solid on cooling: 1H NMR (CDCIs) 8 7.39 (s, 1 H), 7.33 (m, 1 H), 7.28 (t, J= 7.5 Hz, 1 H), 7.13 (d, J= 7.5 Hz, 1 H), 4.0 (m, 4 H), 2.91 (hept, J=7Hz, 1 H), 2.15 (m, 4 H), 1.82 (br d, J= 11.5 Hz, 2 H), 1.70(br d, J = 11.5 Hz, 2 H), 1.25 (d, J = 7 Hz, 6 H); MS (Cl) m/z 259.2 (M-OH).
Cyclopropyltriphenylphosphonium bromide (10.8 g, 28 mmol) was suspended in dry THF (30 mL) and cooled to -60C. N-Butyllithium (1.6M in THF, 17 mL, 27 mmol) was added dropwise at -40 to -60C. The temperature was raised to 20C and after 3 hours at this temperature, the reaction mixture was cooled to -60C. 1,4-Cyclohexane dione mono ethylene ketal (4.0 g, 26 mmol) dissolved in THF (30 mL) was added at -60C whereafter the temperature was raised to 20C. After 16 hours the reaction mixture was diluted with THF (75 mL) filtered through hyflo and concentrated in vacuo. The residue was purified by flash chromatography (4 x 15 cm column of silica) using heptane and ethyl acetate (4:1) as eluent to give 2.5 g of 8-cyclopropylidene-1,4-dioxa-spiro[4.5]decane (cf Synthetic Communications 21(20), 2015-2023, 1991).
26.18 g Sodium triacetoxyborohydride are added at 5° C. to a solution of 10.0 g 4-methyl-3-oxo-piperazine, 13.51 g 1,4-dioxa-spiro[4,5]decan-8-one, 5.65 ml acetic acid and 200 ml dichloromethane. After 23 hours stirring at ambient temperature 100 ml dichloromethane and 100 ml 4N sodium hydroxide solution are added. The phases are separated and the organic phase is evaporated down. The residue is purified by chromatography.Mass spectrum (ESI+): m/z=255 [M+H]+
Example 40 ; n-BuLi (2.5 N in hexanes, 19 mL, 46 mmol) was slowly dropped into a solution of <strong>[78839-75-5](2-bromo-phenyl)-carbamic acid tert-butyl ester</strong> (Aldrich, 5.00 g, 18.5 mmol) in THF (25 mL) at -78 C. After addition, the reaction was stirred for another 30 min. A solution of 1,4-dioxa-spiro[4.5]decan-8-one (Aldrich, 2.89 g, 18.5 mmol) in THF (10 mL) was added into the reaction mixture at -78 C. The reaction was stirred for an additional 2 hours and warmed to room temperature. Solid t-BuOK (Aldrich, 4 g, 37 mmol) was added, and the reaction was stirred overnight. The reaction was then quenched with saturated NH4Cl solution. The solvent was removed in vacuo and the residue was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated to give a yellow solid, which was then purified by silica gel column on a CombiFlash system using hexanes and ethyl acetate (from 10% ethyl acetate to 100% ethyl acetate) to afford the product compound as a white solid.1H NMR (400 MHz, CDCl3) delta 9.45 (s, br, 1H), 7.25 (dt, J=7.0, 3.2 Hz, 1H), 7.20 (d, J=6.5 Hz, 1H), 7.05 (dt, J=7.0, 3.0 Hz, 1H), 6.90 (d, J=6.6 Hz, 1H), 3.98 (m 4H), 2.20 (m, 6H), 1.67 (d, J=6.5 Hz, 2H).
To a solution of 2-chloro-4-fluoro-l-iodobenzene (10.0 g, 39.0 mmol) in dry THF (150 niL) was added /-PrMgCl (29.3 niL, 2.0 N, 58.5 mmol) dropwise at 0 C . After stirring for 1 hr at 0 C , the reaction was warmed to room temperature and stirred for 2 h. 1,4-cyclohexanedione monoethylene acetal (4.87 g, 31.2 mmol) was added and the reaction was stirred at room temperature for 12 h. Water (100 mL) was added and the mixture was extracted with ethyl acetate (3 x 100 mL). The combined organic phases were washed with brine (100 mL), then dried over Na2SO4. After concentration in vacuo, the crude material was purified by column chromatography (petroleum ethe?ethyl acetate = 5:1) on silica gel to give the desired product as a white solid (5.38 g, 48%). 1H NMR (400 MHz, CDCl3): delta 7.61-7.57 (m, IH), 7.14-7.11 (m, IH), 6.99- 6.94 (m, IH), 4.00-3.95 (m, AzU), 2.63-2.57 (m, IH), 2.42-2.36 (m, 2H), 2.17-2.10 (m, 2H), 2.05-2.01 (m, 2H), 1.72-1.69 (m, 2H).
First Step Magnesium (dried; 6.1 g) and THF (20 ml) were put in a reaction vessel under a nitrogen atmosphere, and heated to 40 C. 1-Bromo-4-ethoxy-2,3-difluorobenzene (12) (59.2 g) dissolved in THF (300 ml) was slowly added dropwise thereto in the temperature range of 40 C to 60 C, and the stirring was continued for another 60 minutes. Then, 1,4-dioxaspyro[4.5]decane-8-one (13) (30.0 g) dissolved in THF (150 ml) was slowly added dropwise in the temperature range of 50 C to 60 C, and the stirring was continued for another 60 minutes. The obtained reaction mixture was cooled to 30 C, poured into to a vessel containing an aqueous solution of ammonium chloride (3%; 900 ml) and toluene (500 ml) which were cooled to 0 C, and mixed. The mixture obtained was allowed to stand until it had separated into organic and aqueous phases, and the extraction into an organic phase was carried out. The organic phase obtained was washed sequentially with water, a saturated aqueous solution of sodium hydrogencarbonate and water, and dried over anhydrous magnesium sulfate. Then, the solvent was distilled off under reduced pressure, giving 86.2 g of 8-(4-ethoxy-2,3-difluorophenyl)-1,4-dioxaspyro[4.5]decan-8-ol (14).
Compound (T-2) (142.2 g) obtained in the above step was dissolved in dry tetrahydrofuran (hereinafter, abbreviated as DryTHF) (500 ml), and the resultant solution was cooled to -70C, n-BuLi (364 ml) was added dropwise in a nitrogen atmosphere, and agitation was carried out at -70C for 2 hours. Then, a DryTHF solution of cyclohexanedione monoethylene ketal (T-3) (93.6 g) was slowly added dropwise, the resultant solution was heated to room temperature and agitated for 16 hours. After completion of the reaction, 2N-HCl (200 ml) was added, extraction was carried out with toluene, an organic layer was washed with water and a saturated aqueous solution of sodium chloride, and then the resultant solution was dried over anhydrous magnesium sulfate and concentrated under reduced pressure, and thus a light brown solid was obtained. The resultant material was dissolved in toluene (400 ml), p-TsOH (4.74 g) was added, and heating reflux was carried out for 4 hours while performing dehydration. After completion of the reaction, extraction was carried out with toluene, an organic layer was washed with water and a saturated aqueous solution of sodium chloride, and then the resultant solution was dried over anhydrous magnesium sulfate and concentrated under reduced pressure, and thus a light brown solid was obtained. The resultant material was subjected to silica gel column chromatography (heptane:ethyl acetate = 20:1 in a volume ratio) and recrystallization (Solmix), and thus (T-4) was obtained as a colorless crystal (126.1 g, yield: 71%).
Example 227: Synthesis of N -cyclopropyl- N -((1s,4s)-4-hydroxy-4-methylcyclohexyl)-6-(piperidin-1-yl)picolinamide and N -cyclopropyl- N -((1r,4r)-4-hydroxy-4-methylcyclohexyl)-6-(piperidin-1-yl)picolinamide [563] [564] Step 1: Synthesis of 8-methyl-1,4-dioxaspiro[4.5]decan-8-ol [565] 1,4-cyclohexandione mono-ethylene ketal (1.0 g, 6.4 mmol) was dissolved in THF (30 ml), followed by addition of MeMgCl (3.0M solution in THF, 2.6 ml, 7.7 mmol) at 0oC, and then the resulting mixture was stirred at room temperature under nitrogen stream for 3 hours. A saturated aqueous ammonium chloride solution was added to the resulting reaction liquid, followed by extraction with MC (50 ml x 2). The organic layer was dried over anhydrous sodium sulfate, followed by filtration and concentration, and then the residue thus obtained was subjected to MPLC (50% EtOAc/Hexanes), to obtain 654 mg of white solid (59%).[566]
8-(4-propylphenyl)-1,4-dioxaspiro[4.5]decane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
52%
Well-dried magnesium (14.5 g) and THF (100 ml) were placed in a reaction vessel under an atmosphere of nitrogen, and heated to 40°C. 1-Bromo-4-propylbenzene (s14) (100.0 g) dissolved in THF (300 ml) was added dropwise in the temperature range of 40° C. to 60° C, and the stirring was continued for another 60 minutes. 1,4-Dioxaspiro[4.5]decan-8-one (s7) (78.4 g) dissolved in THF (150 ml)was slowly added dropwise in the temperature range of -5° C. to 0° C, and the stirring was continued for another 60 minutes. After the reaction mixture was allowed to come to room temperature, it was treated with an aqueous solution of ammonium chloride (3percent), and the aqueous layer was extracted with toluene. The combined organic layers were washed with water, a saturated aqueous solution of sodium hydrogencarbonate and water, and then dried over anhydrous magnesium 55 sulfate. The solvent was distilled off under reduced pressure. The residue was mixed with p-toluenesulfonic acid (3.9 g) and toluene (300 ml), and the mixture was heatedto reflux for 2 hours while distilled water was removed. After the reactionmixture had been cooled to 30° C, it was treated with water, 60 and the aqueous layer wasextracted with toluene. The combined organic layers were washed with asaturated aqueous solution of sodium hydrogencarbonate and water, and thendried over anhydrous magnesium sulfate. The solution was concentrated underreduced pressure, and the residue was 65 purified by column chromatography with toluene as aneluent and silica gel as a stationary phase powder. The purified product was dissolved in a mixed solvent of toluene (250 ml) and Solmix A-11 (250 ml),to which Pd/C (5.0 g) was added. The mixture was stirred at room temperatureunder a hydrogen atmosphere until hydrogen absorption had ceased. After thereaction had been completed, Pd/C was removed, and then solvent was distilledoff. The residue was purified by column chromatography with toluene as an eluent and silica gel as a stationary phase powder, and then by recrystallization from heptane to give 8-(4-propylphenyl)-1,4-dioxaspiro[4.5]de-cane (si5) (68.0 g). The yield based on the compound (s14) was 52.0percent.
With ammonium chloride; sodium hydrogencarbonate; toluene-4-sulfonic acid; magnesium;palladium-carbon; In tetrahydrofuran; water; toluene;
Third StepWell-dried magnesium (14.5 g) and THF (100 ml) were placed in a reaction vessel under an atmosphere of nitrogen, and heated to 40° C. 1-Bromo-4-propylbenzene (s14) (100.0 g) dissolved in THF (300 ml) was added dropwise in the temperature range of 40° C. to 60° C., and the stirring was continued for another 60 minutes. 1,4-Dioxaspiro[4.5]decan-8-one (s7) (78.4 g) dissolved in THF (150 ml) was slowly added dropwise in the temperature range of -5° C. to 0° C., and the stirring was continued for another 60 minutes.After the reaction mixture was allowed to come to room temperature, it was treated with an aqueous solution of ammonium chloride (3percent), and the aqueous layer was extracted with toluene.The combined organic layers were washed with water, a saturated aqueous solution of sodium hydrogencarbonate and water, and then dried over anhydrous magnesium sulfate.The solvent was distilled off under reduced pressure.The residue was mixed with p-toluenesulfonic acid (3.9 g) and toluene (300 ml), and the mixture was heated to reflux for 2 hours while distilled water was removed.After the reaction mixture had been cooled to 30° C., it was treated with water, and the aqueous layer was extracted with toluene.The combined organic layers were washed with a saturated aqueous solution of sodium hydrogencarbonate and water, and then dried over anhydrous magnesium sulfate.The solution was concentrated under reduced pressure, and the residue was purified by column chromatography with toluene as an eluent and silica gel as a stationary phase powder.The purified product was dissolved in a mixed solvent of toluene (250 ml) and Solmix A-11 (250 ml), to which Pd/C (5.0 g) was added.The mixture was stirred at room temperature under a hydrogen atmosphere until hydrogen absorption had ceased.After the reaction had been completed, Pd/C was removed, and then solvent was distilled off.The residue was purified by column chromatography with toluene as an eluent and silica gel as a stationary phase powder, and then by recrystallization from heptane to give 8-(4-propylphenyl)-1,4-dioxaspiro[4.5]decane (s15) (68.0 g).The yield based on the compound (s14) was 52.0percent.
Then <strong>[38573-88-5]2,3-difluorobromobenzene</strong> (T-10) (115.2 g) was dissolved in DryTHF (600 ml), and the resultant solution was cooled to -70°C. In a nitrogen atmosphere, n-BuLi (364 ml) was added dropwise, and agitation was carried out at -70°C for 2 hours. Then, a DryTHF solution of cyclohexanedione monoethylene ketal (T-3) (93.6 g) was slowly added dropwise at -70°C, and the resultant solution was heated to room temperature and agitated for 16 hours. After completion of the reaction, 2N-HCl (200 ml) was added, extraction was carried out with toluene, an organic layer was washed with water and a saturated aqueous solution of sodium chloride, and then the resultant solution was dried over anhydrous magnesium sulfate and concentrated under reduced pressure, and thus a light brown solid was obtained. The resultant material was dissolved in toluene (400 ml), p-TsOH (4.74 g) was added, and heating reflux was carried out for 4 hours while performing dehydration. After completion of the reaction, extraction was carried out with toluene, an organic layer was washed with water and a saturated aqueous solution of sodium chloride, and then the resultant solution was dried over anhydrous magnesium sulfate and concentrated under reduced pressure, and thus a light brown solid was obtained. The resultant material was subjected to silica gel column chromatography (heptane:ethyl acetate = 20:1 in a volume ratio) and recrystallization (Solmix), and thus (T-11) was obtained as a colorless crystal (121.0 g, yield: 80percent).
8-(4-bromopyridin-2-yl)-1,4-dioxaspiro[4.5]decan-8-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Step 1 : <strong>[58530-53-3]2,4-Dibromopyridine</strong> (250 mg, 1.06 mmol) in toluene (8 mL) was cooled to -78 C, and w-buthyl lithium (0.791 ml, 1.266 mmol) was added dropwise. The reaction mixture was stirred for 1 hour at -78 C. 1,4-Cyclohanedione mono-ethylene ketal (165 mg, 1.055 mmol) in toluene (2 mL) was added to the reaction mixture and allowed to warm to room temperature. The reaction mixture was quenched with saturated aqueous ammonium chloride, extracted with CH2CI2 (3x), dried over a2S04 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate/hexanes) to afford 8-(4- bromopyridin-2-yl)-l,4-dioxaspiro[4.5]decan-8-ol as a pale yellow oil. MS ESI calc'd for Ci3H17BrN03 [M + H]+ 314 and 316, found 314 and 316
The title compound was prepared according to the procedure of Takada (J. Med. Chem. 1996, 39, 2844-2851). A 350 mL pressure vessel, equipped with a magnetic stir bar, was charged with <strong>[14150-94-8]1-methyl-3,5-dinitropyridin-2(1H)-one</strong> (5.0 g, 25.1 mmol) and 1,4-dioxaspiro[4.5]decan-8-one (4.7 g, 30.1 mmol). Ammonia in MeOH (1M, 200 mL) was added, the vessel was sealed with a screw cap, and the mixture was heated to 60 C. and stirred for 17 hours. The reaction mixture was concentrated and the residue was partitioned with ethyl acetate (200 mL) and water (200 mL). The layers were separated and the aqueous portion was extracted with ethyl acetate (2×100 mL). The organic portions were combined, dried (Na2SO4), filtered, and concentrated. The residue was purified by silica gel chromatography (7/3, hexanes/ethyl acetate) to afford the title compound (4.85 g, 82%).
Step 1. Preparation of 8-methyl-l,4-dioxaspiro[4.5]decan-8-ol To THF (100 mL) in a round bottom flask at -78 C under 2 was added methyllithium (48.0 mL, 77 mmol) and methylmagnesium bromide (12.81 mL, 38.4 mmol) via syringe. After stirring at -78 C for 1 h, l,4-dioxaspiro[4.5]decan-8-one (5.00 g, 32.0 mmol) in THF (50 mL) was added via cannula. The reaction mixture was stirred at -78 C for 1.5 h. The reaction was quenched by the addition of saturated aqueous NH4C1 solution (100 mL). The mixture was transferred to a separatory funnel containing water (50 mL) and the aqueous layer was extracted with ethyl acetate (3 x 150 mL). The combined organic layers were washed with brine (100 mL), dried over MgS04, filtered, and concentrated to afford 8-methyl-l,4-dioxaspiro[4.5]decan-8-ol (5.47 g, 99% yield). The product was used in the next step without further purification. XH NMR (400MHz, CHLOROFORM- d) delta 4.03 - 3.91 (m, 4H), 1.97 - 1.84 (m, 2H), 1.79 - 1.56 (m, 6H), 1.28 (s, 3H), 1.17 (s, 1H); 13C NMR (100MHz, CHLOROFORM-d) delta 108.27, 68.52, 63.87, 63.81, 36.33, 30.47, 29.39.
1-((isocyanomethyl)sulfonyl)-2-methylbenzene[ No CAS ]
[ 69947-09-7 ]
Yield
Reaction Conditions
Operation in experiment
With potassium tert-butylate; In 1,2-dimethoxyethane; ethanol; water; at -10 - 20℃; for 16h;
1,4-Dioxaspiro[4.5]decane-8-carbonitrile To a -10 C. solution of 1,4 dioxaspiro[4.5]decan-8-one (1 equiv.), ethanol (1.78 equiv.) and toluenesulfonylmethyl isocyanide (1.3 equiv.) in DME (0.3 M) was added potassium 2-methylpropan-2-olate (2.3 equiv.) portion wise. The reaction was stirred at -10 C. for 1 h, and 15 h at room temperature. The reaction mixture was concentrated to a beige solid, dissolved in water and extracted with ether. The combined extracts were washed with brine and dried. Concentration under reduced pressure gave an orange oil. This material was purified by distillation [(103 C. (oil bath 150 C.) at about 2-3 mbar] to give 1,4-dioxaspiro[4.5]decan-8-one as a colorless oil (87% yield). 1H NMR (400 MHz, CDCl3) delta ppm: 3.88-4.02 (m, 4H), 2.56-2.74 (m, 1H), 1.79-2.05 (m, 6H), 1.50-1.71 (m, 2H).
With potassium tert-butylate; In 1,2-dimethoxyethane; at 0℃; for 2h;
Step 1: t-BuOK (18.5 g, 165.2 mmol) was added into a solution of compound 116-1 (10 g, 64 mmol) and TosMIC(17.6 g, 90.27 mmol) in DME (10 mL) at 0 C. The reaction mixture was stirred at 0C for 2h. After the reaction wascomplete, the reaction mixture was quenched with saturated NH4Cl solution and extracted with EtOAc. The combinedextraction liquid was washed with brines, dried over sodium sulfate and concentrated to dry, finally purified by columnchromatography (PE:EtOAc = 1:1) to deliver 2 (6.7 g, yield 50%) as colorless oil.
In tetrahydrofuran; diethyl ether; at 0 - 20℃; for 2h;Inert atmosphere;
To a solution of 1 ,4-cyclohexanedione monoethylene acetal (6.00 g, 38.4 mmol) in THF (48 mL) at 0 C was added bromo(methyl)magnesium in diethyl ether (3 M, 19.2 mL, 42.26 mmol) under a nitrogen atmosphere. The reaction mixture was allowed to stir at room temperature for 2 h and then quenched with a saturated NH4CI solution. Water was added and the mixture was extracted with DCM. The combined organic extracts were dried over Na2S04, filtered and concentrated under reduced pressure to give 8-methyl-1 ,4-dioxaspiro[4.5]decan-8-ol (5.82 g, 27.7 mmol, 72% yield), which was used without further purification. NMR (400 MHz, CDCI3, delta): 4.01 -3.93 (m, 4H), 1 .93-1 .86 (m, 4H), 1 .73-1 .68 (m 4H), 1 .29 (s, 3H), 1 .15 (s, 1 H).
8-(3-chloro-4-methoxyphenyl)-1,4-dioxaspiro[4.5]decan-8-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
61%
To a 3-necked round bottom flask was added <strong>[50638-47-6]4-bromo-2-chloro-1-methoxy-benzene</strong>(45.00 g, 203.18 mmol) and THF (450 mL), n-Butyllithium (2.5 M in hexanes, 90.21 mL, 1.11 eq) was added at -78 C. The mixture was stirred for 2 h at -78 C. A solution of 1,4- dioxaspiro[4.5]decan-8-one (34.91 g, 223.50 mmol) in THF (90 mL) was added dropwise to the reaction mixture. The resulting mixture was stirred for 3 h at -78 C. The reaction was quenched with aqueous NH4C1 (lOOmL) and extracted with EtOAc (500 mL). The organic layer was dried (Na2504), filtered and concentrated. The residue was washed with hexanes (350 mL), filtered and dried under high vacuum. The solid was triturated with hexanes (15 mL), filtered and dried under high vacuum to give 8-(3-chloro-4-methoxy-phenyl)-1,4- dioxaspiro[4.5]decan-8-ol (37 g, 61%) as a white solid. ?H NMR (400 IVIHz, CDC13): 7.31 (d, 1H), 7.29 (dd, 1H), 7.10 (d, 1H), 3.90-3.92 (m, 4H), 3.89 (s, 3H), 1.99-2.02 (m, 4H), 1.70- 1.73 (m, 4H); LCMS: 281.2 [M-OH].
To a solution of 1,4-cyclohexanedione monoethylene acetal (500 mg, 3.2 mmol) in THF (10 ml) at 0 C was added methyl magnesium bromide (1.2 ml, 3.4M solution in 2-methyltetrahydrofuran, 4.16 mmol) and the reaction mixture was stirred at room temperature for 1hr. After reaction progress was confirmed by TLC, saturated ammonium chloride solution (15 ml) was added carefully prior to addition of ethyl acetate (20 ml). Workup was done with ethyl acetate (3 x 15 ml), organic portion was washed with water and brine. Sodium sulphate (2 g) was added to remove the remaining traces of water followed by removal of solvent under reduced pressure. Crude product obtained was re-dissolved in THF-H2O (2:1 v/v, 15 ml), cooled to 0 C and 5% HCl (10 ml) was added and reaction mixture was stirred for 4hr at room temperature. After completion, the reaction mixture was carefully neutralized with saturated sodium bicarbonate (20 ml), extracted with ethyl acetate (3 x 15 ml ) and combined organic layer was washed with brine (40 ml), dried over sodium sulphate and solvent removed under reduced pressure. Crude product was purified with column chromatography using ethyl acetate/hexanes as eluent (40:60). The product was obtained as a colorless liquid in 88% yield (360 mg, over 2-steps): IR (neat) 3426, 2966, 1705, 1420, 1135, 10, 773 cm-1; 1H NMR (CDCl3, 500 MHz): delta 1.29-1.30 (m, 3H), 1.74-1.80 (m, 2H), 1.91-1.94 (m, 2H), 2.14-2.17 (m, 2H), 2.62-2.69 (m, 2H); 13C NMR (CDCl3, 125 MHz): delta 29.7, 37.2, 38.7, 68.4, 212.9; HRMS calcd. for C7H12NaO2[M+Na]+: 151.0735. Found 151.0734.
1-(cyclopropylmethyl)-4-[1,4-dioxaspiro [4.5]decan-8-yl]piperazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
70.4%
With sodium tris(acetoxy)borohydride; acetic acid; In dichloromethane; at 0 - 20℃;
Into a 250-mL round-bottom flask, was placed 1, 4- dioxaspiro [4.5] decan-8- one (2.75 g, 0.018 mmol, 1.20 equiv), l-(cyclopropylmethyl)piperazine (2.06 g, 14.690 mmol, 1 equiv), HO Ac (0.88 g, 0.015 mmol, 1.00 equiv), DCM (100 mL). This was followed by the addition of NaBH(OAc)3 (3.74 g, 0.018 mmol, 1.20 equiv) at 0 degrees C. The resulting solution was stirred for overnight at room temperature. The pH value of the solution was adjusted to 8 with NaHC03 (2mol/L). The resulting solution was extracted with 3x100 mL of dichloromethane concentrated. The residue was applied onto a silica gel column with dichloromethane/methanol (10:1). The collected fractions were combined and concentrated. This resulted in 2.9 g (70.40%) of l-(cyclopropylmethyl)-4-[l, 4- dioxaspiro [4.5] decan-8- yl] piperazine as colorless oil. LC-MS (ES, m/z ): 28l[M+l] +






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






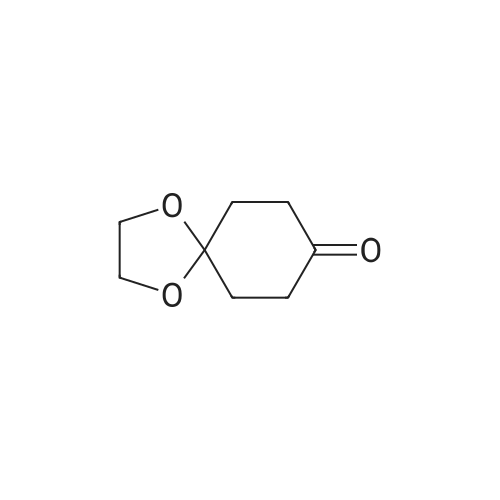

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping