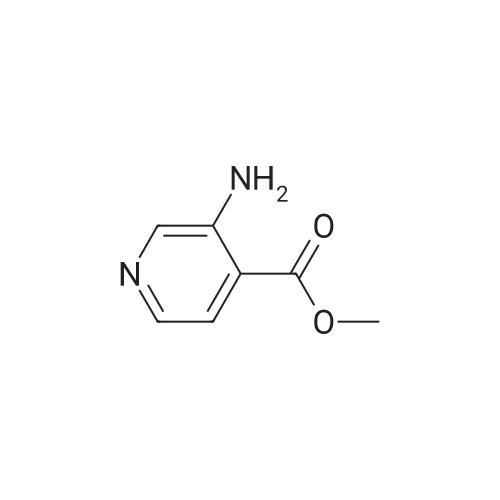
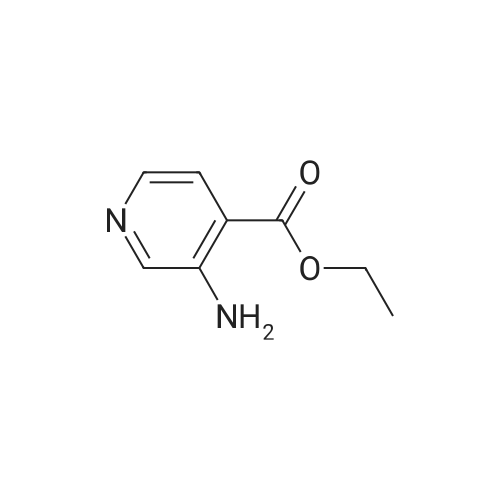
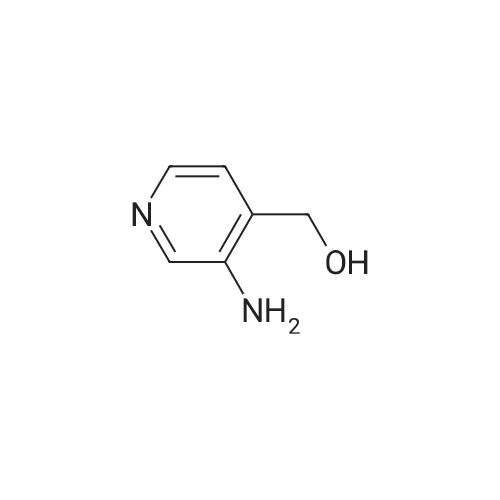
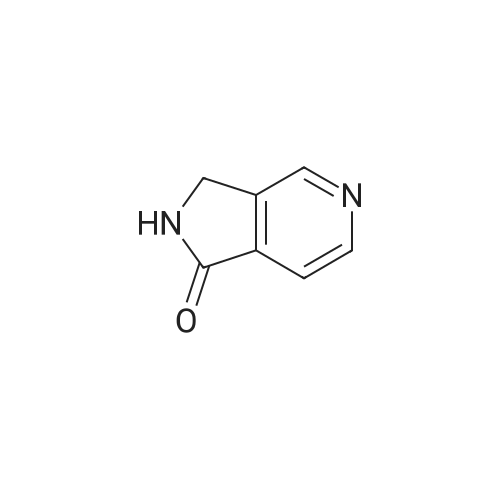
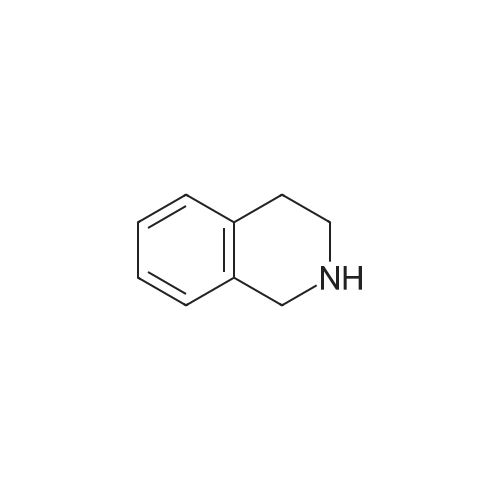
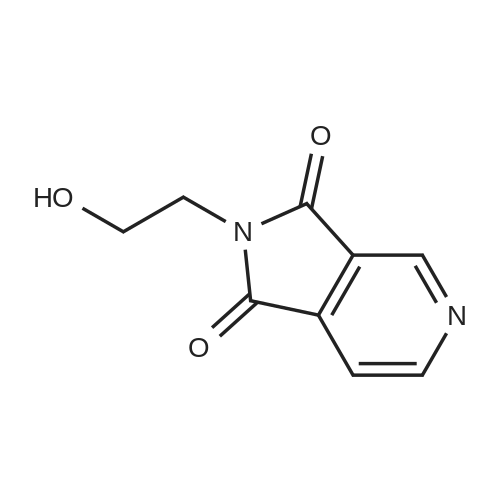
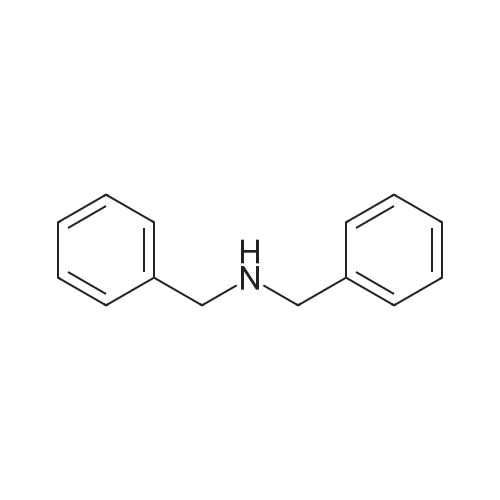
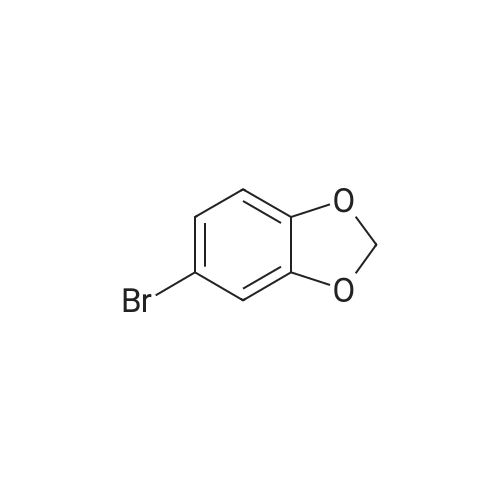
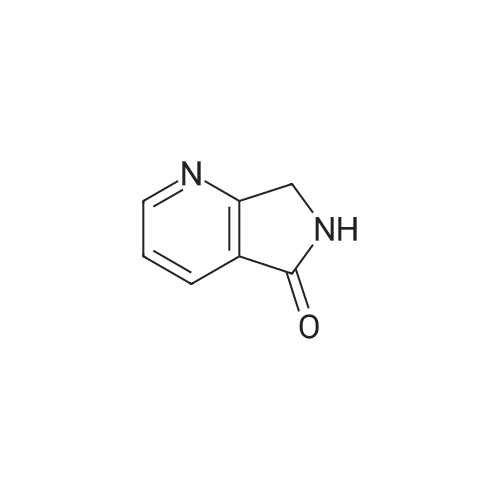
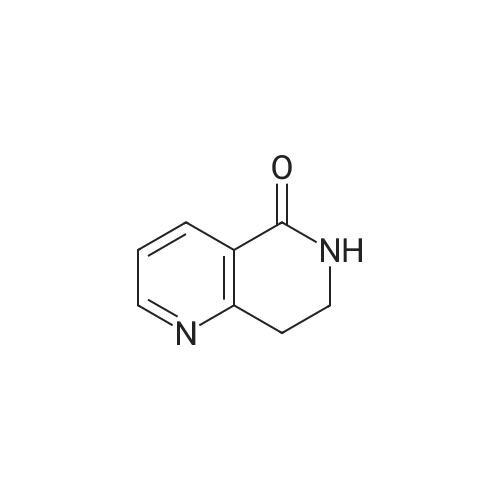
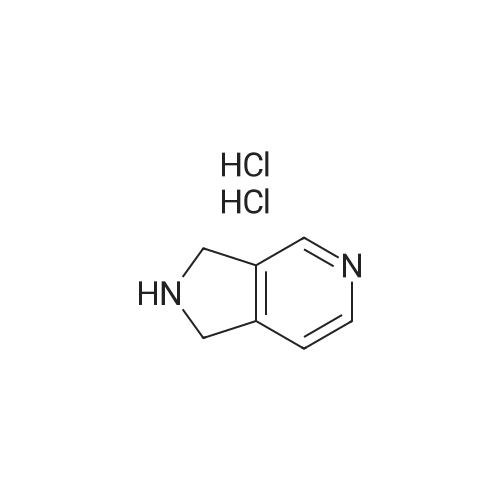
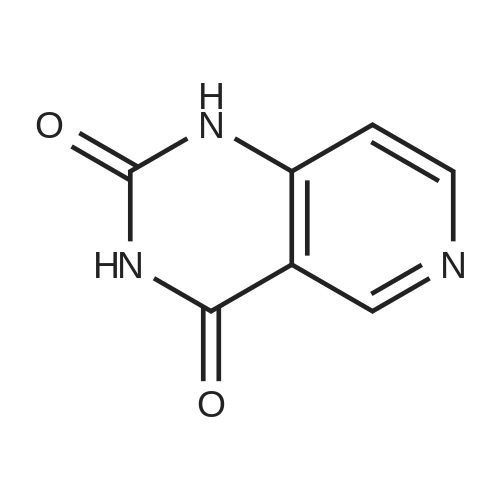
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: for 24 h; Reflux Stage #2: With calcium chloride In dichloromethane
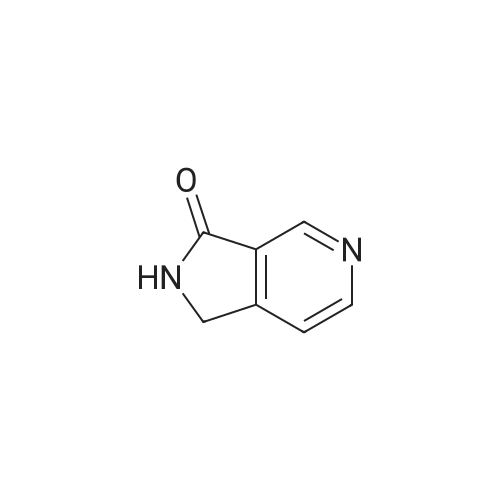
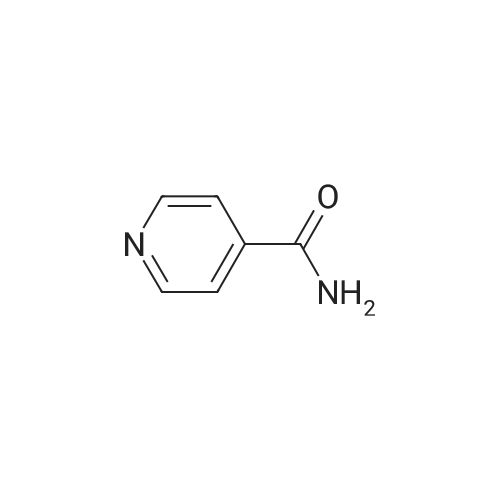
To a solution of 3,4-pyridindicarboxamide (0.40 g, 2.7 mmol, 1 eq) in acetic acid (12 mL), zinc (0.78 g, 11.9 mmol, 4.4 eq) was added and the suspension was heated at reflux for 24 h. After cooling at room temperature, the mixture was filtered off through a Celite pad. The filtrate was evaporated and the residue was taken up with CH2Cl2. To this solution CaCl2 was added and the suspension was filtered. The filtrate was evaporated and the residue was purified by column chromatography using EtOAc/MeOH 95/5 as eluant to give 10 as yellowish solid after crystallization with EtOH (0.061 g, 17percent). Mp. 190-193 °C dec. 1H NMR (300 MHz, DMSO-d6) δ 8.84 (s, 1-H), 8.70 (br d, 2-H), 7.62 (d, J = 5.5 Hz, 1-H), 4.42 (s, 2-H); 13C NMR (75 MHz, DMSO-d6) δ 169.1, 153.3, 151.6, 145.3, 129.0, 119.9, 45.2; GC-MS m/z 134 (M)+; IR (KBr) 2856, 1728, 1692, 1455, 1206, 1026 cm-1. Anal. Calcd for C7H6N2O: C, 62.68; H, 4.51; N, 20.88. Found: C, 62.75; H, 4.50; N, 20.63.
Reference:
[1] European Journal of Medicinal Chemistry, 2012, vol. 55, p. 58 - 66,9
[2] Patent: WO2005/103003, 2005, A2, . Location in patent: Page/Page column 106
2
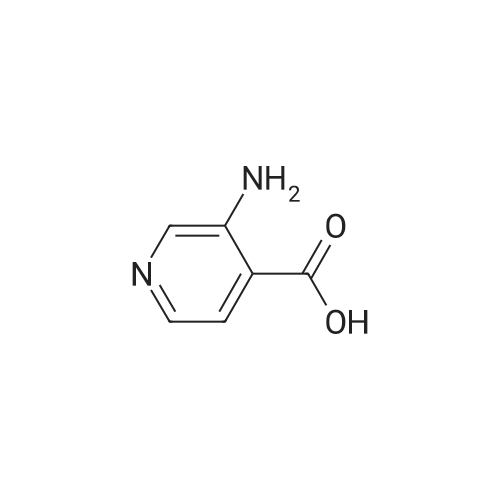
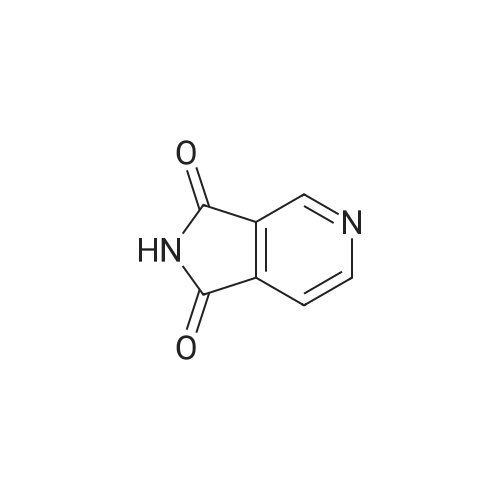
[ 490-11-9 ]
[ 4664-01-1 ]
Yield
Reaction Conditions
Operation in experiment
95.1%
Stage #1: at 140 - 150℃; Stage #2: at 140℃; for 3 h;
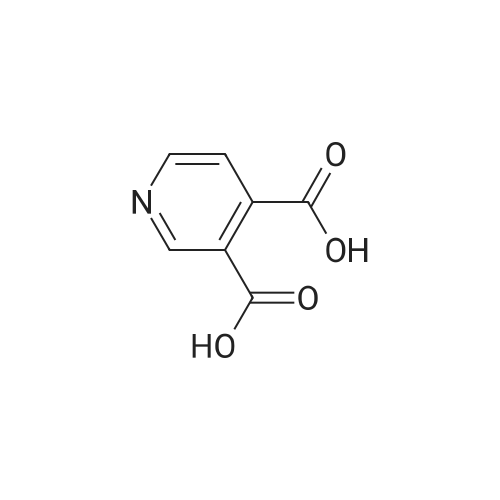
A suspension of cinchomeronic acid (30)(50 g, 300 mmol) in acetic anhydride (123.5 g, 1200 mmol) was heated to reflux (140-150° C.) until all solid material dissolved and the mixture was homogeneous. The mixture was then cooled and concentrated in vacuo. Acetamide (50 g, 846 mmol) was then added and the mixture heated to 140° C. for 3 hours whereupon it was then cooled to room temperature. The solid residue that formed upon cooling was pulverized and triturated with water (100 ml), filtered and washed with more water and dried in a desiccator to give the title compound (42.26 g, 95.1percent) in suitably pure form to be used without any further purification. m/z (LC-MS, ESP): 149 [M+H]+, R/T=0.44 mins.
Reference:
[1] Acta Chimica Hungarica, 1983, vol. 112, # 4, p. 487 - 500
[2] Tetrahedron Letters, 1993, vol. 34, # 39, p. 6225 - 6228
[3] Farmaco, Edizione Scientifica, 1980, vol. 35, # 11, p. 940 - 944
[4] Acta Poloniae Pharmaceutica, 1995, vol. 52, # 3, p. 245 - 248
4
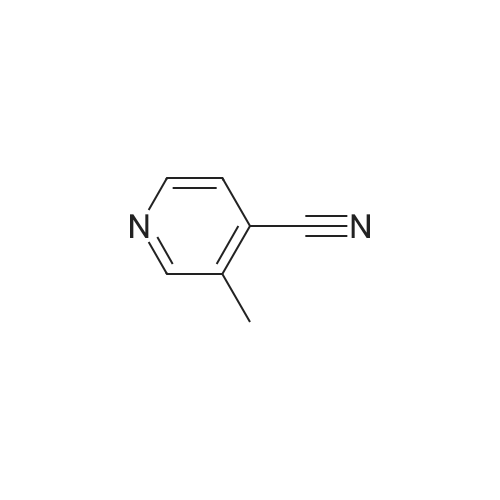
[ 1678-52-0 ]
[ 102-06-7 ]
[ 4664-01-1 ]
[ 65252-12-2 ]
[ 76240-10-3 ]
[ 76240-09-0 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1980, p. 1667 - 1670
5
[ 60-35-5 ]
[ 4664-08-8 ]
[ 4664-01-1 ]
Reference:
[1] Journal of Organic Chemistry, 1949, vol. 14, p. 97,101
6
[ 583-58-4 ]
[ 7584-05-6 ]
[ 4664-01-1 ]
Reference:
[1] Russian Journal of General Chemistry, 2012, vol. 82, # 12, p. 1987 - 1993[2] Zh. Obshch. Khim., 2012, vol. 82, # 12, p. 2033 - 2039,7
7
[ 4664-01-1 ]
[ 7579-20-6 ]
Yield
Reaction Conditions
Operation in experiment
100%
With bromine; sodium hydroxide In water at 90℃; for 0.666667 h; Cooling with ice
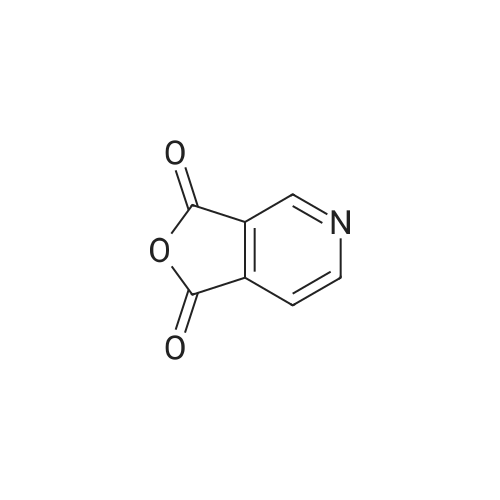
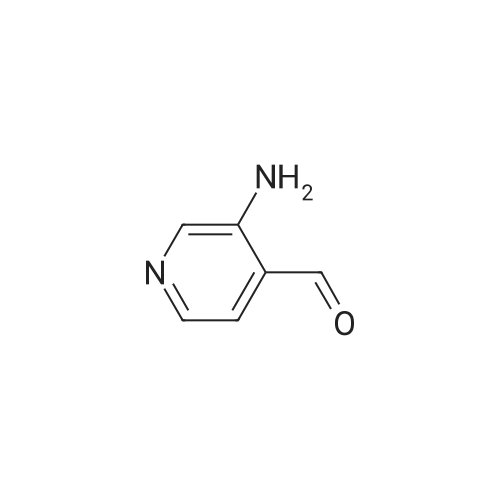
Step 1 To 10percent aqueous sodium hydroxide solution (100 mL) was added bromine (1.93 mL, 38.6 mmol) under ice-cooling, and 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-dione (5.20 g, 35.1 mmol) was subsequently added. 10percent-Aqueous sodium hydroxide solution (60 mL) was added to the reaction mixture, and the mixture was stirred with heating at 90° C. for 40 min. After allowing to cool to room temperature, 50percent sulfuric acid was added to adjust the reaction mixture to pH 3. The precipitated insoluble material was collected by filtration, and the solid was washed with water to give 3-aminoisonicotinic acid as a pale-yellow powder (5.00 g, 100percent). 1H-NMR (200 MHz, DMSO-d6) δ: 7.46 (1H, d, J=5.1 Hz), 7.73 (1H, d, J=5.1 Hz), 8.21 (1H, s).
68.33%
Stage #1: With sodium hydroxide; bromine In water at 7 - 80℃; for 0.5 h; Stage #2: With acetic acid In water at 37℃;
NaOH (10percent aqueous, 640 ml) was cooled to 7° C. and bromine (15 ml, 286.82 mmol) added dropwise. Pyrrolo[3,4-c]pyridine-1,3-dione (41.711 g, 281.6 mmol) was then added to the reaction mixture before it was heated to 80° C. for 30 minutes. After this time the reaction was allowed to warm to 37° C. and the pH modified to 5.5 by the addition of acetic acid (70 ml). A suspension formed that was removed by filtration and washed with 20 ml of ice cold methanol to give the title compound (26.58 g, 68.33percent) in a suitably clean form to be used without any further purification. m/z (LC-MS, ESP): 139 [M+H]+, R/T=0.72 mins.
67%
Stage #1: With sodium hydroxide; bromine In water at 0 - 80℃; for 0.75 h; Stage #2: With acetic acid In water
Bromine (3. 5 ml, 69.0 mmol) was added to a solution of 10 percent aqueous sodium hydroxide (120 ml) at 0 C to give a pale yellow solution. To this solution was added 3,4-pyridinedicarboximide (10 g, 67. 5 mmol) and the reaction was heated at 80 C for 45 min. The reaction was cooled in a water bath and acidified by the addition of acetic acid (12. 5 ml) causing precipitation. The solid was collected, rinsed with water(50 ml), thenMeOH(50 ml) and dried to give the title compound as a beige solid (6.28 g, 67 percent).1H NMR (360 MHz, DMSO)5 8. 06 (1H, s), 7.60(1H, d,J5. 1), 7.34(1H, d,J5. 1), 3.19 (2H, brs). M/z (ES+) 139 (M+H+).
64%
Stage #1: With sodium hydroxide; bromine In water at 80℃; for 1.1667 h; Stage #2: With acetic acid In water at 20℃; for 0.166667 h;
Following the procedures of Zhou et al (2001) Bioorg. Med. Chem. Lett.9(8):2061-2071, bromine (1.22 ml, 23.9 mmol) was slowly added to a cooled (50C) 2.5N solution of NaOH (60 ml, 150 mmol) and after stirring for 5 minutes pyrrolo[3,4-c]pyridine- 1,3-dione (3.5 g, 23.6 mmol) was added. The temperature was raised to 8O0C and the mixture was stirred for 1 hour before being cooled to ambient temperature. Acetic acid (5.9 ml, 98.3 mmol) was cautiously added (N.B.: gas evolution) and the solution stirred for 10 minutes whereby a precipitate formed that was collected by filtration. The solid was washed with water (20 ml) and MeOH (20 ml) then dried to afford the title compound as a yellow solid (2.1 g, 64percent).
62%
With sodium hydroxide; bromine In water at 0 - 80℃; for 0.75 h;
A solution of10percent aqueousNaOH (416 mL) was cooled to0 C and treated with bromine (28.2 g 176 mmol) portionwise via pipette while keeping the temperature below 5 C. To this mixture was added 3,4-pyridinedicarboximide (25.78 g, 174mmol), and the cooling bath was removed. The reaction was heated to80 C for 45 min, then allowed to cool in an ice bath. When the temperature fell to 60 C, the dropwise addition of HOAc (50 mL) was started. Cooling was continued until the temperature reached 15 C. A yellow precipitate formed. The solid was filtered, rinsed with water, then dried under vacuum to give 14.9 g of product (108 mmol,62percent). 1NMR IS-MS, m/e 139(m+1)
57%
With bromine In sodium hydroxide
Preparation 18 3-Aminopyridine-4-carboxylic acid To an ice cold mixture of 3,4-pyridinedicarboximide (5.2 g, 35.11 mmol) in 10percent sodium hydroxide (85 mL) was added bromine (1.84 mL, 35.8 mmol), dropwise. The resulting solution was heated to 80° C. for 1 hour, cooled on ice, and the acidity was carefully adjusted to pH 5.5 with acetic acid. The precipitate was collected, washed well with water and air dried to afford 3-aminopyridine-4-carboxylic acid (2.74 g, 57percent). NMR (DMSO d6) δ8.20 (s, 1H), 7.72 (d, J=5 Hz, 1H), 7.45 (d, J=5 Hz, 1H). The material was used without purification.
57%
Stage #1: With sodium hydroxide; bromine In water at 7 - 85℃; for 1.5 h; Stage #2: With acetic acid In water at 20 - 30℃;
Step A: 3-Aminoisonicotinic acid: 2H-Pyrrolo[3,4-c]pyridine-1,3-dione (204.16 g, 1378.4 mmol) was dissolved in 10percent NaOH (3.3 L) and the solution was cooled to an internal temperature of 7° C. (ice/salt bath). Bromine (73.424 mL, 1433.5 mmol) was added dropwise while maintaining the internal temperature below 10° C. After completion of the addition, the reaction was heated to an internal temperature of 80-85° C. for 90 minutes. The reaction mixture was cooled to 20-30° C. in an ice bath then acetic acid (323.21 mL, 5651.2 mmol) was added dropwise. The reaction was stirred and cooled to 5° C. The solids were collected by vacuum filtration, washed with cold water then air-dried to provide the product (108.86 g, 57percent).
57%
With bromine In sodium hydroxide
Preparation 18 3-Aminopyridine-4-carboxylic acid To an ice cold mixture of 3,4-pyridinedicarboximide (5.2 g, 35.11 mmol) in 10percent sodium hydroxide (85 mL) was added bromine (1.84 mL, 35.8 mmol), dropwise. The resulting solution was heated to 80° C. for 1 hour, cooled on ice, and the acidity was carefully adjusted to pH 5.5 with acetic acid. The precipitate was collected, washed well with water and air dried to afford 3-aminopyridine-4-carboxylic acid (2.74 g, 57percent). 1H NMR (DMSO d6) δ8.20 (s,1 H), 7.72 (d, J=5 Hz, 1 H), 7.45 (d, J=5 Hz, 1 H). The material was used without purification.
Reference:
[1] Patent: US2015/329556, 2015, A1, . Location in patent: Paragraph 1612-1614
[2] Bioorganic and Medicinal Chemistry, 2001, vol. 9, # 8, p. 2061 - 2071
[3] Australian Journal of Chemistry, 1993, vol. 46, # 7, p. 987 - 993
[4] Tetrahedron Letters, 1993, vol. 34, # 39, p. 6225 - 6228
[5] Patent: US2006/199804, 2006, A1, . Location in patent: Page/Page column 29
[6] Patent: WO2005/49613, 2005, A1, . Location in patent: Page/Page column 38
[7] Patent: WO2008/24725, 2008, A1, . Location in patent: Page/Page column 67
[8] Patent: WO2005/49604, 2005, A2, . Location in patent: Page/Page column 69
[9] Journal of Medicinal Chemistry, 2017, vol. 60, # 16, p. 6942 - 6990
[10] Patent: US5962457, 1999, A,
[11] Patent: US2007/49603, 2007, A1, . Location in patent: Page/Page column 78; 30
[12] Patent: US6323208, 2001, B1,
[13] Patent: WO2004/831, 2003, A1, . Location in patent: Page 22
[14] Patent: WO2008/108957, 2008, A2, . Location in patent: Page/Page column 95
8
[ 4664-01-1 ]
[ 7579-20-6 ]
Yield
Reaction Conditions
Operation in experiment
57%
With bromine In sodium hydroxide
Preparation 18 3-Aminopyridine-4-carboxvlic acid To an ice cold mixture of 3,4-pyridinedicarboximide (5.2 g, 35.11 mmol) in 10percent sodium hydroxide (85 mL) was added bromine (1.84 mL, 35.8 mmol), dropwise. The resulting solution was heated to 80°C for 1 hour, cooled on ice, and the acidity was carefully adjusted to pH 5.5 with acetic acid. The precipitate was collected, washed well with water and air dried to afford 3-aminopyridine4-carboxylic acid (2.74 g, 57percent). NMR (DMSO d6) δ 8.20 (s, 1 H), 7.72 (d, J = 5 Hz, 1 H), 7.45 (d, J = 5 Hz, 1 H). The material was used without purification.
Reference:
[1] Patent: EP807633, 1997, A2,
9
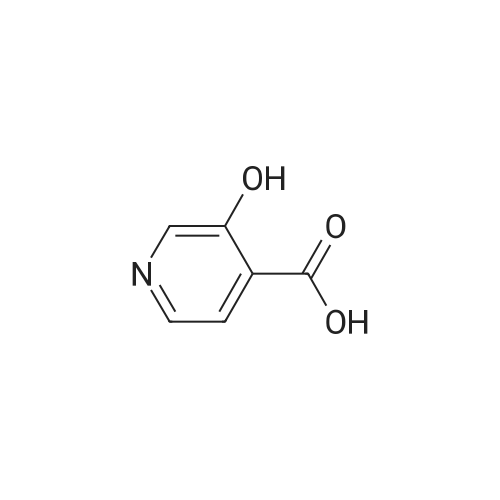
[ 4664-01-1 ]
[ 10128-71-9 ]
Reference:
[1] Journal of Medicinal Chemistry, 2017, vol. 60, # 16, p. 6942 - 6990
Reference:
[1] Bioorganic and Medicinal Chemistry, 2001, vol. 9, # 8, p. 2061 - 2071
[2] Australian Journal of Chemistry, 1993, vol. 46, # 7, p. 987 - 993
12
[ 4664-01-1 ]
[ 152398-05-5 ]
Reference:
[1] Patent: US2003/45519, 2003, A1,
13
[ 4664-01-1 ]
[ 152398-05-5 ]
Reference:
[1] Australian Journal of Chemistry, 1993, vol. 46, # 7, p. 987 - 993
[2] Patent: WO2008/108957, 2008, A2,
14
[ 4664-01-1 ]
[ 5655-00-5 ]
Yield
Reaction Conditions
Operation in experiment
62%
Stage #1: at 100℃; for 4 h;
Step lTo a solution of compound [137] (3 g, 20.2 mmol, leq) in glacial acetic acid (30 ml), Zn dust (5.26 g, 81.0 mmol, 4 eq.) was added and the reaction mixture was refluxed at 100 °C for 4 h. The reaction mixture was cooled to room temperature and the solvent was evaporated. The reaction mass was made basic with aqueous NaHC03 to pH 8 and extracted with chloroform. The aqueous layer was concentrated and further extracted with chloroform. The combined organic layer was dried over Na2S04 and evaporated to give [138] as a peach white solid (1.7 g, 62percent).ESIMS: 135 (M+ + 1)
With bromine; sodium hydroxide; In water; at 90℃; for 0.666667h;Cooling with ice;
Step 1 To 10percent aqueous sodium hydroxide solution (100 mL) was added bromine (1.93 mL, 38.6 mmol) under ice-cooling, and 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-dione (5.20 g, 35.1 mmol) was subsequently added. 10percent-Aqueous sodium hydroxide solution (60 mL) was added to the reaction mixture, and the mixture was stirred with heating at 90° C. for 40 min. After allowing to cool to room temperature, 50percent sulfuric acid was added to adjust the reaction mixture to pH 3. The precipitated insoluble material was collected by filtration, and the solid was washed with water to give 3-aminoisonicotinic acid as a pale-yellow powder (5.00 g, 100percent). 1H-NMR (200 MHz, DMSO-d6) delta: 7.46 (1H, d, J=5.1 Hz), 7.73 (1H, d, J=5.1 Hz), 8.21 (1H, s).
68.33%
NaOH (10percent aqueous, 640 ml) was cooled to 7° C. and bromine (15 ml, 286.82 mmol) added dropwise. Pyrrolo[3,4-c]pyridine-1,3-dione (41.711 g, 281.6 mmol) was then added to the reaction mixture before it was heated to 80° C. for 30 minutes. After this time the reaction was allowed to warm to 37° C. and the pH modified to 5.5 by the addition of acetic acid (70 ml). A suspension formed that was removed by filtration and washed with 20 ml of ice cold methanol to give the title compound (26.58 g, 68.33percent) in a suitably clean form to be used without any further purification. m/z (LC-MS, ESP): 139 [M+H]+, R/T=0.72 mins.
67%
Bromine (3. 5 ml, 69.0 mmol) was added to a solution of 10 percent aqueous sodium hydroxide (120 ml) at 0 C to give a pale yellow solution. To this solution was added 3,4-pyridinedicarboximide (10 g, 67. 5 mmol) and the reaction was heated at 80 C for 45 min. The reaction was cooled in a water bath and acidified by the addition of acetic acid (12. 5 ml) causing precipitation. The solid was collected, rinsed with water(50 ml), thenMeOH(50 ml) and dried to give the title compound as a beige solid (6.28 g, 67 percent).1H NMR (360 MHz, DMSO)5 8. 06 (1H, s), 7.60(1H, d,J5. 1), 7.34(1H, d,J5. 1), 3.19 (2H, brs). M/z (ES+) 139 (M+H+).
64%
Following the procedures of Zhou et al (2001) Bioorg. Med. Chem. Lett.9(8):2061-2071, bromine (1.22 ml, 23.9 mmol) was slowly added to a cooled (50C) 2.5N solution of NaOH (60 ml, 150 mmol) and after stirring for 5 minutes pyrrolo[3,4-c]pyridine- 1,3-dione (3.5 g, 23.6 mmol) was added. The temperature was raised to 8O0C and the mixture was stirred for 1 hour before being cooled to ambient temperature. Acetic acid (5.9 ml, 98.3 mmol) was cautiously added (N.B.: gas evolution) and the solution stirred for 10 minutes whereby a precipitate formed that was collected by filtration. The solid was washed with water (20 ml) and MeOH (20 ml) then dried to afford the title compound as a yellow solid (2.1 g, 64percent).
62%
With sodium hydroxide; bromine; In water; at 0 - 80℃; for 0.75h;
A solution of10percent aqueousNaOH (416 mL) was cooled to0 C and treated with bromine (28.2 g 176 mmol) portionwise via pipette while keeping the temperature below 5 C. To this mixture was added 3,4-pyridinedicarboximide (25.78 g, 174mmol), and the cooling bath was removed. The reaction was heated to80 C for 45 min, then allowed to cool in an ice bath. When the temperature fell to 60 C, the dropwise addition of HOAc (50 mL) was started. Cooling was continued until the temperature reached 15 C. A yellow precipitate formed. The solid was filtered, rinsed with water, then dried under vacuum to give 14.9 g of product (108 mmol,62percent). 1NMR IS-MS, m/e 139(m+1)
57%
With bromine; In sodium hydroxide;
Preparation 18 3-Aminopyridine-4-carboxylic acid To an ice cold mixture of 3,4-pyridinedicarboximide (5.2 g, 35.11 mmol) in 10percent sodium hydroxide (85 mL) was added bromine (1.84 mL, 35.8 mmol), dropwise. The resulting solution was heated to 80° C. for 1 hour, cooled on ice, and the acidity was carefully adjusted to pH 5.5 with acetic acid. The precipitate was collected, washed well with water and air dried to afford 3-aminopyridine-4-carboxylic acid (2.74 g, 57percent). NMR (DMSO d6) delta8.20 (s, 1H), 7.72 (d, J=5 Hz, 1H), 7.45 (d, J=5 Hz, 1H). The material was used without purification.
57%
Step A: 3-Aminoisonicotinic acid: 2H-Pyrrolo[3,4-c]pyridine-1,3-dione (204.16 g, 1378.4 mmol) was dissolved in 10percent NaOH (3.3 L) and the solution was cooled to an internal temperature of 7° C. (ice/salt bath). Bromine (73.424 mL, 1433.5 mmol) was added dropwise while maintaining the internal temperature below 10° C. After completion of the addition, the reaction was heated to an internal temperature of 80-85° C. for 90 minutes. The reaction mixture was cooled to 20-30° C. in an ice bath then acetic acid (323.21 mL, 5651.2 mmol) was added dropwise. The reaction was stirred and cooled to 5° C. The solids were collected by vacuum filtration, washed with cold water then air-dried to provide the product (108.86 g, 57percent).
57%
With bromine; In sodium hydroxide;
Preparation 18 3-Aminopyridine-4-carboxylic acid To an ice cold mixture of 3,4-pyridinedicarboximide (5.2 g, 35.11 mmol) in 10percent sodium hydroxide (85 mL) was added bromine (1.84 mL, 35.8 mmol), dropwise. The resulting solution was heated to 80° C. for 1 hour, cooled on ice, and the acidity was carefully adjusted to pH 5.5 with acetic acid. The precipitate was collected, washed well with water and air dried to afford 3-aminopyridine-4-carboxylic acid (2.74 g, 57percent). 1H NMR (DMSO d6) delta8.20 (s,1 H), 7.72 (d, J=5 Hz, 1 H), 7.45 (d, J=5 Hz, 1 H). The material was used without purification.
Preparation 5 Step 1: 3,4 Pyridine-dicarboximide (10.0 g, 67.5 [MMOL)] was dissolved in 10percent aqueous [NAOH] (162 g) and the solution was cooled to an internal temperature [OF 7 OC] in an ice-salt bath. Bromine (3.6 ml ; 70 [MMOL)] was added dropwise. After the addition, the solution was heated for 45 min at a bath temperature of 80-85 [°C.] The yellow solution was then cooled to an internal temperature of 37 [°C,] and [GLACIAL ACOH] (17 ml) were added dropwise to a pH of 5.5. The resulting mixture was refrigerated overnight. The solid formed was filtered and washed with water (5 ml) and CH30H (5 [ML).] The reaction yielded 6.35 g. of product, m. p. 280-285 [°C] (decomp. ).
With sodium hydroxide; bromine; In water; at 0 - 85℃; for 0.75h;
3,4 Pyridine-dicarboximide 288 (10.0 g; 67.5 mmoles) was dissolved in 162 g. of 10percent aqueous NaOH and the solution was cooled to an internal temperature of 70C in an ice-salt bath. Bromine (3.6 ml; 70 mmoles) was added dropwise. After the addition, the solution was heated for 45 minutes at a bath temperature of 80-85 0C. The yellow solution was then cooled to an internal temperature of 37 0C, then 17 ml of glacial acetic acid were added dropwise to a pH of 5.5. The resulting mixture was saved overnight in a refrigerator. The solid formed was filtered and washed with 5 ml of water and 5 ml of methanol. The reaction yielded 6.35 g. of product 289 melting at 280-285 0C (decomp.).
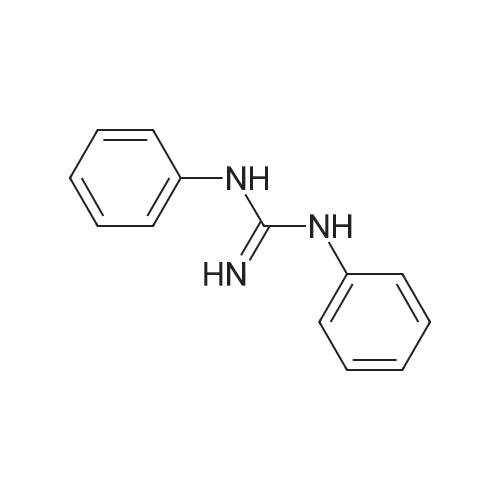
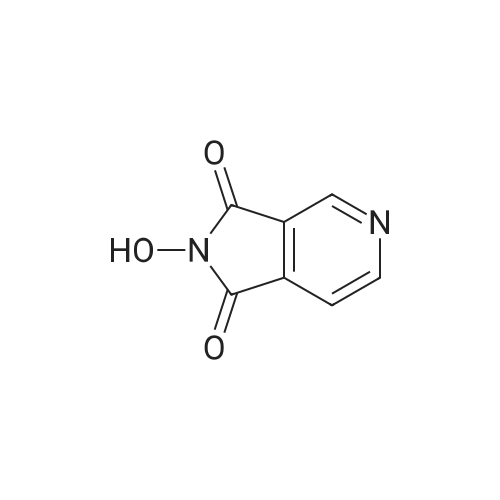
5-[2-(ethyloxy)-2-oxoethyl]-1-[1-(2-(ethyloxy)-2-oxoethyl)pyridinium-4-yl]-1-hydroxy-3-oxo-2,3-dihydro-1H-pyrrolo[3,4-c]pyridin-5-ium dibromide[ No CAS ]
Acetic acid (2S,3S,4S,5S)-4-acetoxy-5-acetoxymethyl-2-((1R,7aR)-1-hydroxy-3-oxo-1-phenyl-1,2,3,7a-tetrahydro-pyrrolo[3,4-c]pyridin-5-yl)-tetrahydro-furan-3-yl ester[ No CAS ]
5-(3,4-diacetoxy-5-acetoxymethyl-tetrahydro-furan-2-yl)-1-hydroxy-3-oxo-1-phenyl-2,3-dihydro-1<i>H</i>-pyrrolo[3,4-<i>c</i>]pyridin-5-ium; bromide[ No CAS ]
Stage #1: 3,4-pyridinecarboxylic acid With acetic anhydride at 140 - 150℃;
Stage #2: With acetamide at 140℃; for 3h;
4.i
A suspension of cinchomeronic acid (30)(50 g, 300 mmol) in acetic anhydride (123.5 g, 1200 mmol) was heated to reflux (140-150° C.) until all solid material dissolved and the mixture was homogeneous. The mixture was then cooled and concentrated in vacuo. Acetamide (50 g, 846 mmol) was then added and the mixture heated to 140° C. for 3 hours whereupon it was then cooled to room temperature. The solid residue that formed upon cooling was pulverized and triturated with water (100 ml), filtered and washed with more water and dried in a desiccator to give the title compound (42.26 g, 95.1%) in suitably pure form to be used without any further purification. m/z (LC-MS, ESP): 149 [M+H]+, R/T=0.44 mins.
68%
Stage #1: 3,4-pyridinecarboxylic acid With acetic anhydride at 140℃; for 16h;
Stage #2: With acetamide at 140℃; for 8h;
16 Intermediate 16A: lH-pyrrolo[3,4-c]pyridine-l,3(2H)-dione
A stirred solution of pyridine-3, 4-dicarboxylic acid (100 g, 600 mmol) in AC2O (0391) (160 mL, 170 mmol) was heated at 140 °C for 16 h and cooled to room temperature. The solvent was evaporated under reduced pressure to yield a crude mixture. Acetamide (53.0 g, 900 mmol) was added and the resulting mixture was heated at 140 °C for 8 h. The mixture was cooled to room temperature, diluted with hot water, and stirred for 30 min. The precipitated solid was filtered and dried to provide lH-pyrrolo[3,4-c]pyridine- l,3(2H)-dione (60 g, 68% yield) as a pale yellow solid. LC-MS: m/z 149.1 (M+H).
Multi-step reaction with 2 steps
1: acetic anhydride / 0.08 h / Heating
2: urea / 0.33 h / 150 - 160 °C
To a solution of 3,4-pyridindicarboxamide (0.40 g, 2.7 mmol, 1 eq) in acetic acid (12 mL), zinc (0.78 g, 11.9 mmol, 4.4 eq) was added and the suspension was heated at reflux for 24 h. After cooling at room temperature, the mixture was filtered off through a Celite pad. The filtrate was evaporated and the residue was taken up with CH2Cl2. To this solution CaCl2 was added and the suspension was filtered. The filtrate was evaporated and the residue was purified by column chromatography using EtOAc/MeOH 95/5 as eluant to give 10 as yellowish solid after crystallization with EtOH (0.061 g, 17%). Mp. 190-193 C dec. 1H NMR (300 MHz, DMSO-d6) delta 8.84 (s, 1-H), 8.70 (br d, 2-H), 7.62 (d, J = 5.5 Hz, 1-H), 4.42 (s, 2-H); 13C NMR (75 MHz, DMSO-d6) delta 169.1, 153.3, 151.6, 145.3, 129.0, 119.9, 45.2; GC-MS m/z 134 (M)+; IR (KBr) 2856, 1728, 1692, 1455, 1206, 1026 cm-1. Anal. Calcd for C7H6N2O: C, 62.68; H, 4.51; N, 20.88. Found: C, 62.75; H, 4.50; N, 20.63.
With zinc; In ethanol; at 20 - 100℃;
1,2-dihydro-3l(at)pyrrolo[3,4-c]pyridin-3-one. Pyridinedicarboximide (60 g, 0.405 mol) and zinc (160.2 g, 1.626 mol) were added to glacial acidic acid (1800 mL) at ambient temperature. The mixture was stirred and heated at 100 C for 4 h and left over night to cool to ambient temperature. The mixture was analyzed by TLC (20% MeOH / 80% EtOAc, Rf SM = 0.86, Rf P = 0.24). An additional 5 g of zinc was added and the reaction was stirred for 3 h more. No changes in the TLC were observed. The acidic acid was evaporated. The pH of the residue was adjusted to pH 8 by addition of aq. NaHC03. The mixture was extracted with CHCI3 (8x 1 L). The water of the aq. phase was evaporated and the residue extracted with chloroform (2x 2L). The solvent of the organic phases was evaporated and the combined residue recrystallized from isopropanol. Yield: 23.1 g. ¹H NMR (300 MHz, d6-DMSO) No. = 8.87 (s, 1 H), 8.71 (s, 2H), 7.64 (br s,1 H), 4.44 (s, 2H). LCMS: 9.38 min, M+H=134.03 u/e.
With bromine; In tetrahydrofuran; methanol; sodium hydroxide; water; acetic acid;
EXAMPLE 29 3,4 Pyridine-dicarboximide 288 (10.0 g; 67.5 mmoles) was dissolved in 162 g. of 10% aqueous NaOH and the solution was cooled to an internal temperature of 7 C. in an ice-salt bath. Bromine (3.6 ml; 70 mmoles) was added dropwise. After the addition, the solution was heated for 45 minutes at a bath temperature of 80-85 C. The yellow solution was then cooled to an internal temperature of 37 C., then 17 ml of glacial acetic acid were added dropwise to a pH of 5.5. The resulting mixture was saved overnight in a refrigerator. The solid formed was filtered and washed with 5 ml of water and 5 ml of methanol. The reaction yielded 6.35 g. of product 289 melting at 280-285 C. (decomp.). Solid Compound 289 (9.5 gr.; 69 mmoles) was carefully added in three aliquots to a slurry of lithium aluminum hydride (9.5 gr.; 250 mmoles) in 200 ml of dry tetrahydrofuran. The resulting hot mixture was stirred at room temperature for two days. After cooling in an ice bath, the reaction was quenched with very careful sequential dropwise addition of 10 ml of water, followed by 10 ml of 15% aqueous NaOH, then by 30 ml of water. The resulting solid was filtered through a pad of Celite and washed several times with THF. The oil obtained after evaporation of the solvent, solidified on standing. The reaction mixture was purified by flash chromatography on silica gel using 5%MeOH(NH3)/EtOAc as eluent yielding 6.21 (72%) of Compound 290. LC-MS: m/z=125 (M+1).
3-[2-(cis-6-Methoxy-2,3,3a,4,5,9b-hexahydro-[1H]-benz[e]isoindol-1-yl)ethyl]-pyrido[3,4-d]pyrimidine-2,4(1H,3H)-dione dihydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
0.68 g (71%)
In toluene;
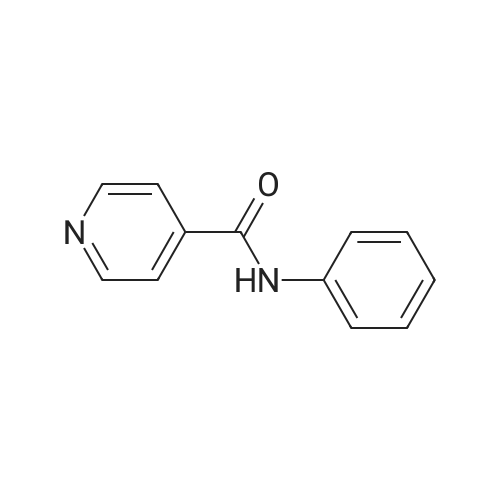
EXAMPLE 24 3-[2-(cis-6-Methoxy-2,3,3a,4,5,9b-hexahydro-[1H]-benz[e]isoindol-1-yl)ethyl]-pyrido[3,4-d]pyrimidine-2,4(1H,3H)-dione dihydrochloride Following the procedure described for Example 21, <strong>[14208-83-4]3-amino-4-ethoxycarbonylpyridine</strong> (0.58 g, 3,5 mmol), prepared by substituting 3,4-pyridinedicarboximide for quinolinimide and the procedure described in J. Chem. Soc., 1045 (1956), Et3 N (1.5 mL, 10.5 mmol), phosgene (1.8 mL of a 1.93M solution in toluene, 3.5 mmol), and the compound resulting from Example 1B (0.60 g, 2.4 mmol) provided 0.68 g (71%) of the desired product which was converted to its HCl salt. m.p. 228-230 C. 1 H NMR (300 MHz, CD3 OD) of the free base delta 1.45-1.49 (m, 1H), 1.66-1.78 (m, 1H), 2.22 (t, 1H), 2.33 (dt, 1H), 2.50-2.68 (m, 3H), 2.77-2.86 (m, 2H), 3.24-3.51 (m, 3H), 3.77 (s, 3H), 4.20 (t, 2 H), 6.71 (dd, 2H), 7.07 (t, 1H), 7.91 (d, 1H), 8.39 (d, 1H), 8.55 (s, 1H). MS (DCI/NH3) m/e 393 (M+H)+. Anal calcd for C22 H24 N4 O3 ·2 HCl: C, 56.78; H, 5.63; N, 12.04. Found: C, 56.3 1; H, 5.63; N, 11.82.
Preparation 18 3-Aminopyridine-4-carboxvlic acid To an ice cold mixture of 3,4-pyridinedicarboximide (5.2 g, 35.11 mmol) in 10percent sodium hydroxide (85 mL) was added bromine (1.84 mL, 35.8 mmol), dropwise. The resulting solution was heated to 80°C for 1 hour, cooled on ice, and the acidity was carefully adjusted to pH 5.5 with acetic acid. The precipitate was collected, washed well with water and air dried to afford 3-aminopyridine4-carboxylic acid (2.74 g, 57percent). NMR (DMSO d6) delta 8.20 (s, 1 H), 7.72 (d, J = 5 Hz, 1 H), 7.45 (d, J = 5 Hz, 1 H). The material was used without purification.
Step lTo a solution of compound [137] (3 g, 20.2 mmol, leq) in glacial acetic acid (30 ml), Zn dust (5.26 g, 81.0 mmol, 4 eq.) was added and the reaction mixture was refluxed at 100 C for 4 h. The reaction mixture was cooled to room temperature and the solvent was evaporated. The reaction mass was made basic with aqueous NaHC03 to pH 8 and extracted with chloroform. The aqueous layer was concentrated and further extracted with chloroform. The combined organic layer was dried over Na2S04 and evaporated to give [138] as a peach white solid (1.7 g, 62%).ESIMS: 135 (M+ + 1)
With potassium carbonate; In acetonitrile; at 80℃; for 16h;
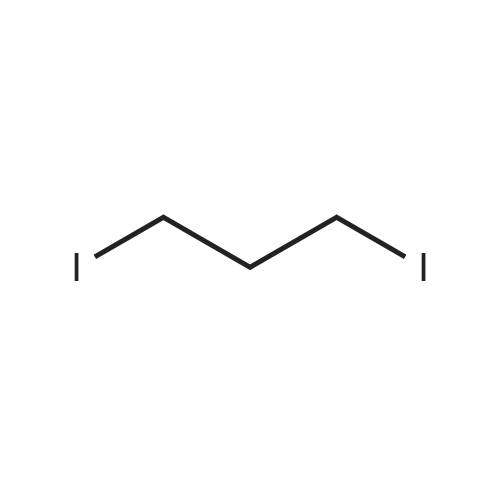
1,3-dibromopropane (1.0 g, 5 mmol) and NaI (3.0 g, 20 mmol) are added into acetonitrile (35 mL), the reaction is heated to 80 C. under stirring for 3 hours to obtain the crude product of 1,3-diiodopropane. This crude product can be used directly for the next step reaction without any purification. 3,4-pyridine-dicarboximide (740 mg, 5 mmol) and K2CO3 (828 mg, 6 mmol) are added into acetonitrile (20 mL), followed by 1,3-diiodopropane (5 mmol) After heated to 80 C., the reaction proceeds for 16 hours. After completion of the reaction, the reaction solution is filtered, then the filtrate is dried by rotary evaporation to obtain a crude product which is purified by column chromatography to get a yellow solid product, N-(3-iodopropyl)-3,4-pyridine-dicarboximide (200 mg, 13%).
With N-ethyl-N,N-diisopropylamine In [(2)H6]acetone for 5h; Reflux;
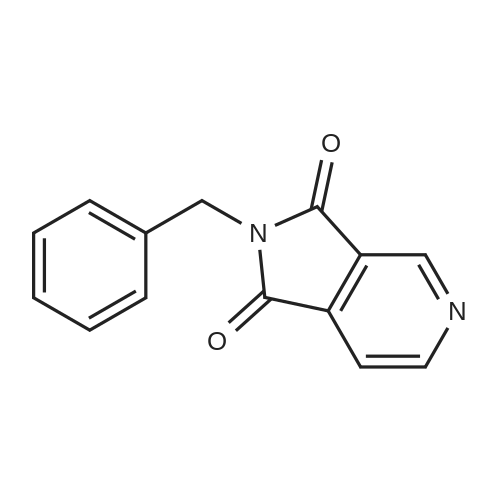
30.1 2-benzyl-1H-pyrrolo[3,4-c]pyridine-1,3(2H)-dione
To a solution of 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-dione (5.00 g) and N,N-diisopropylethylamine (20.00 mL) in acetone was added benzyl bromide (5.0 mL). The reaction mixture was heated to reflux for 5 h, then cooled to rt and filtered. The filtrate was poured into water and extracted with CH2Cl2 (50 mL×3). The combined organic phases were dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated in vacuo and the residue was chromatographed with a silica gel column (eluting agent: 2:1 (v/v) PE/EA) to give the title compound as a brown solid (4.00 g, 50.00%), HPLC: 92.00%. The compound was characterized by the following spectroscopic data: MS (ESI, pos. ion) m/z: 239.2 (M+1); 1H NMR (400 MHz, d6-DMSO) δ: 4.92 (s, 2H), 7.26-7.34 (m, 3H), 7.45 (m, 2H), 7.60 (m, 1H), 8.14 (d, J=8.4 Hz, 1H), 8.96 (d, J=10.4 Hz, 1H) ppm.
With N-ethyl-N,N-diisopropylamine In acetone for 5h; Reflux;
30.1 2-benzyl-l H-pyrrolor3,4-clpyridine-1.3(2HVdione
To a solution of l H-pyrrolo[3,4-c]pyridine-l ,3(2H)-dione (5.00 g) and N,N-diisopropylethylamine (20.00 mL) in acetone was added benzyl bromide (5.0 mL). The reaction mixture was heated to reflux for 5 h, then cooled to rt and filtered. The filtrate was poured into water and extracted with CH2C12 (50 mLx3). The combined organic phases were dried over anhydrous Na2S04 and filtered. The filtrate was concentrated in vacuo and the residue was chromatographed with a silica gel column (eluting agent: 2: 1 (v/v) PE/EA) to give the title compound as a brown solid (4.00 g, 50.00 %), HPLC: 92.00 %.The compound was characterized by the following spectroscopic data: MS (ESI, pos. ion) m/z: 239.2 (M+l ); ? NM (400 MHz, d6-DMSO) ?: 4.92 (s, 2H), 7.26-7.34 (m, 3H ), 7.45 (m, 2H), 7.60 (m, 1 H), 8.14 (d, J = 8.4 Hz, 1 H), 8.96 (d, J = 10.4 Hz, l H) ppm.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping