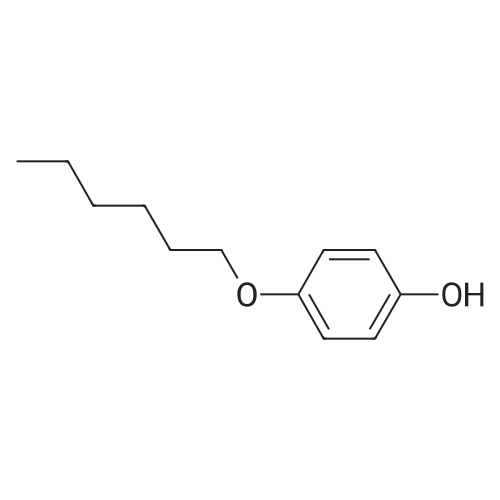
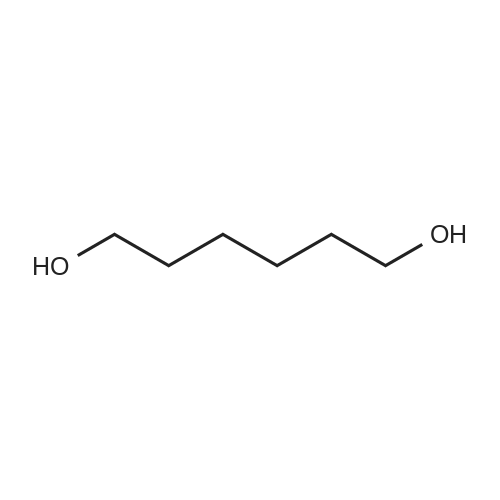
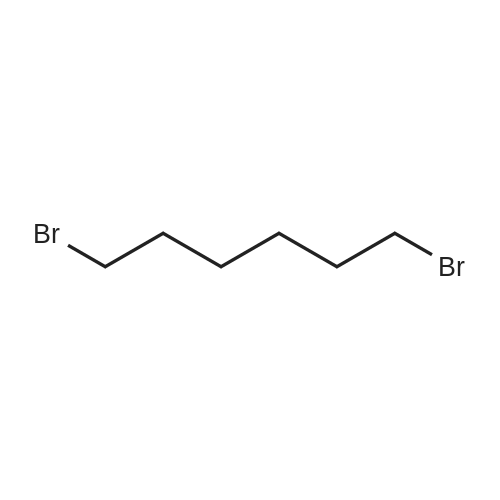
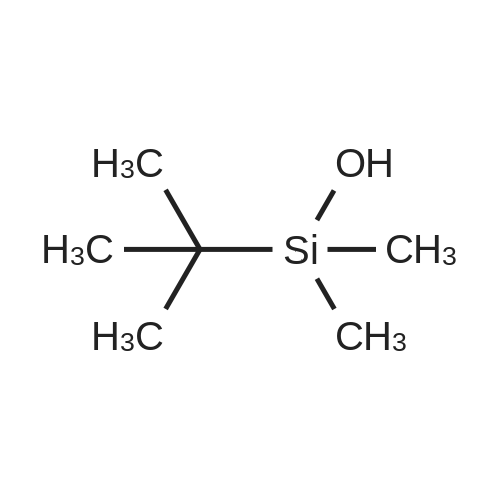
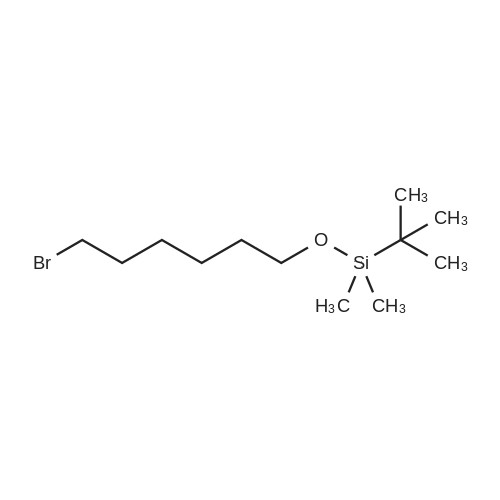
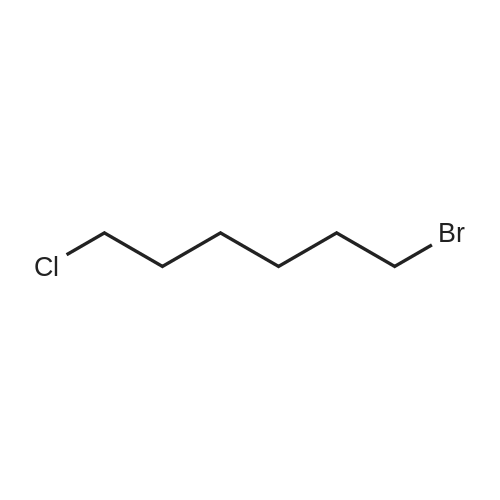
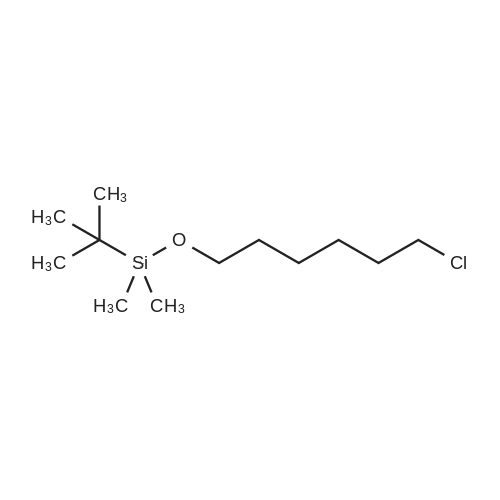
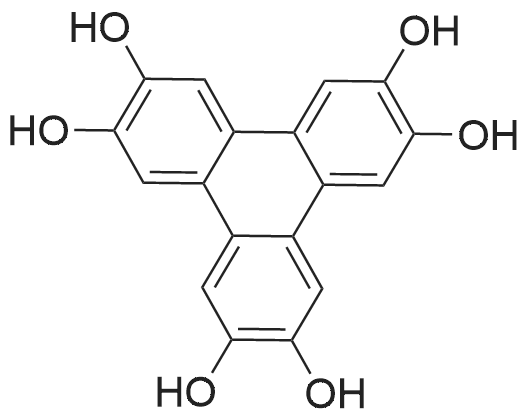
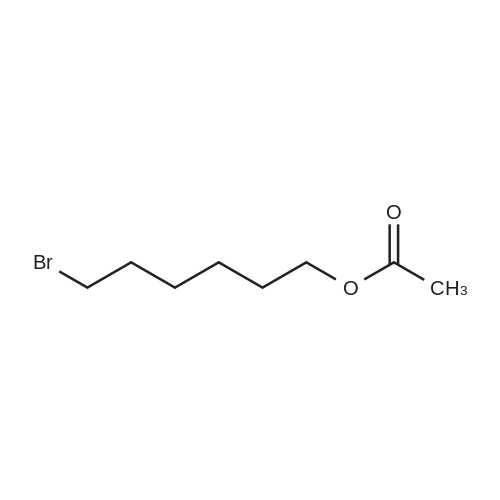
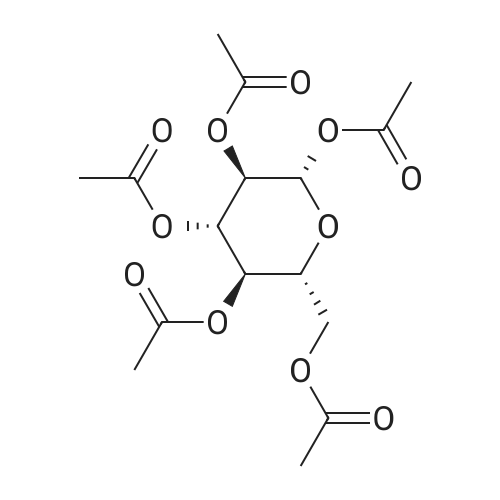
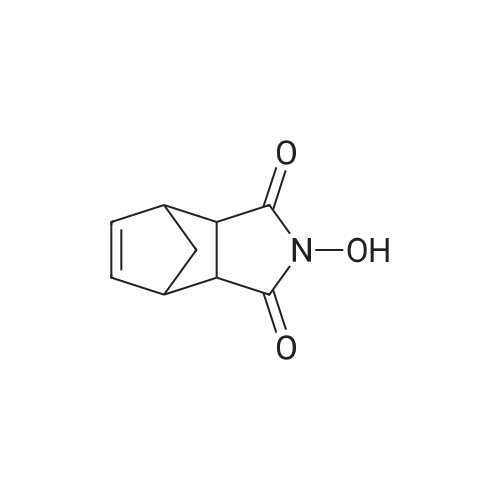
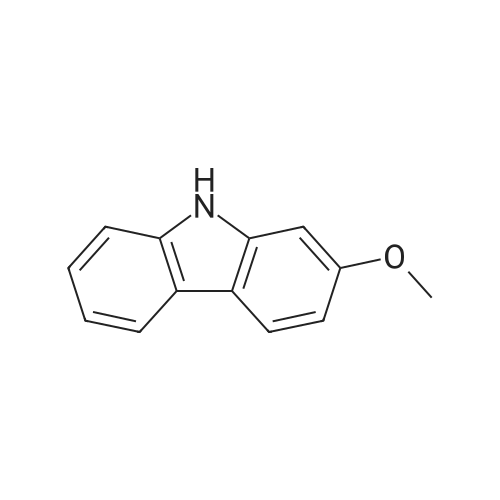
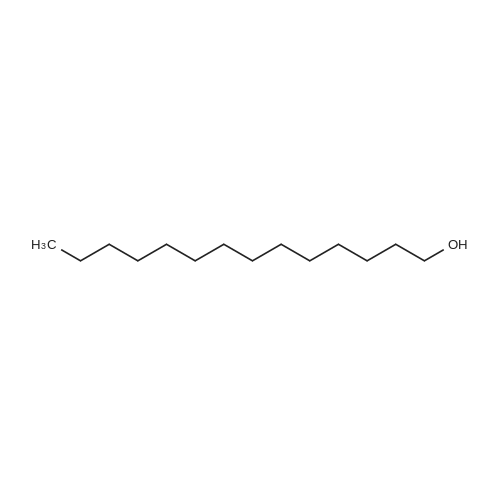
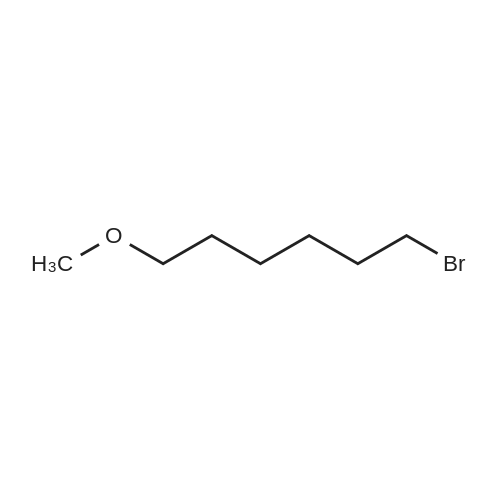
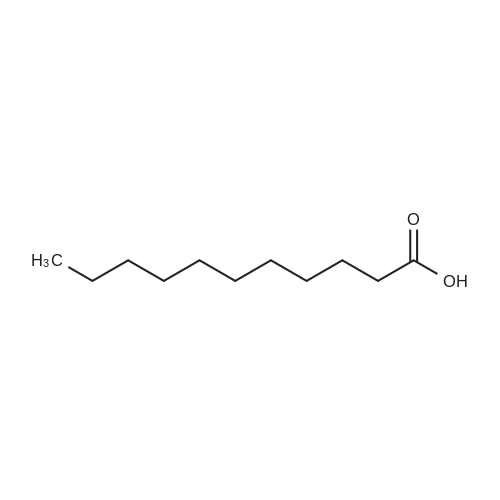
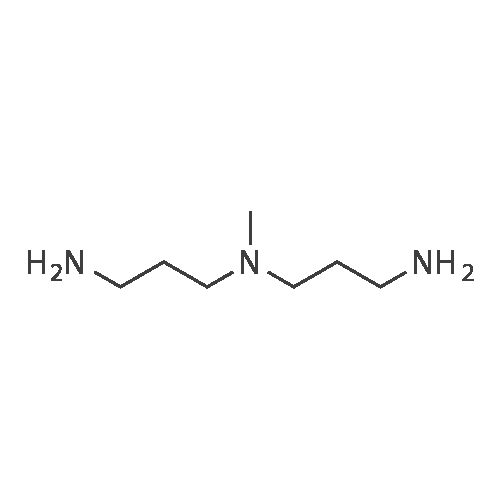
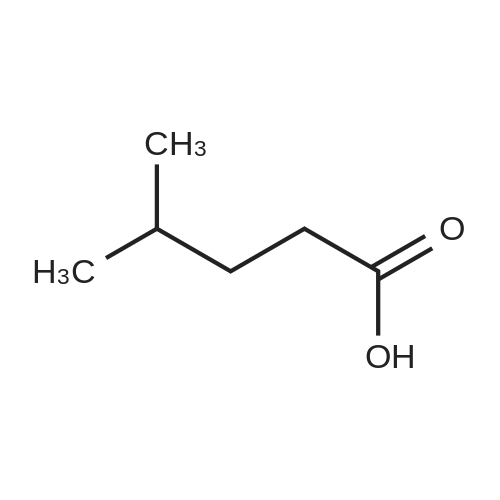
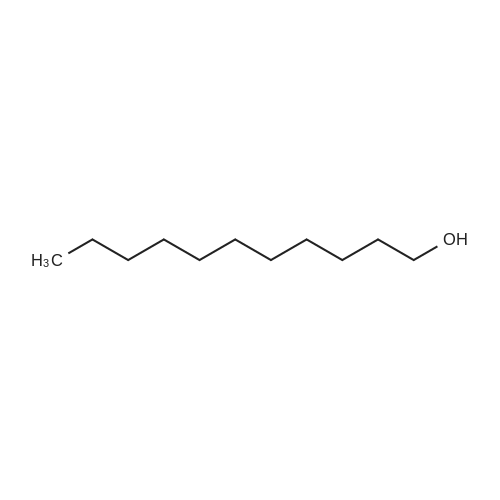
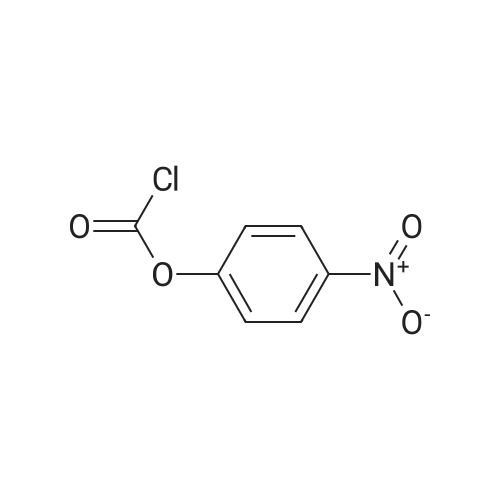
| 98% |
With pyridinium 4-toluenesulfonate In dichloromethane at 20℃; for 4h; Inert atmosphere; |
|
| 97% |
With pyridinium 4-toluenesulfonate In dichloromethane at 20℃; for 23h; |
|
| 96.4% |
With toluene-4-sulfonic acid In dichloromethane at 20℃; |
Synthesis of compounds 1-5
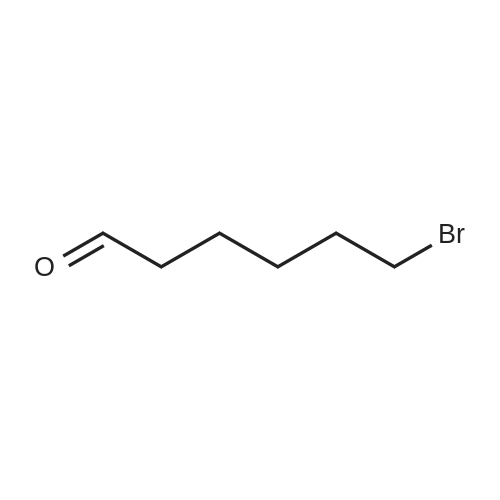
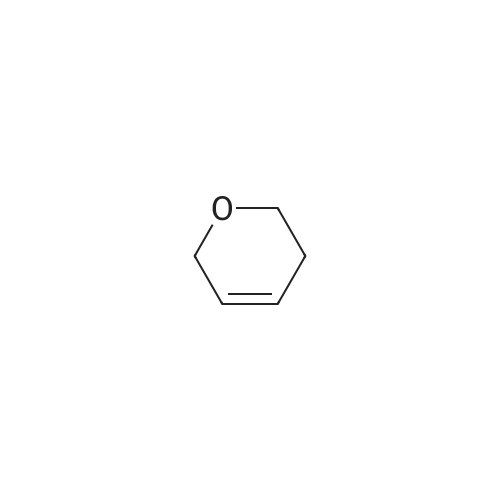
General procedure: 3,4-Dihydropyran (DHP, 15 mmol) was added slowly to a stirred solution of bromohydrin (10 mmol) and p-toluenesulfonic acid (1 mmol) in dichloromethane (20 mL) on ice bath. After stirring at room temperature overnight, the reaction mixture was diluted with water and extracted with dichloromethane. The organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the residue was purified by flash column chromatography to afford 1-5 as a light yellow oil. |
| 95% |
With toluene-4-sulfonic acid In diethyl ether for 12h; Ambient temperature; |
|
| 95% |
With pyridinium 4-toluenesulfonate In dichloromethane for 3h; Ambient temperature; |
|
| 95% |
With trifluoroacetic acid In dichloromethane |
|
| 95% |
With (1S)-10-camphorsulfonic acid In diethyl ether at 20℃; |
3.1.12. 6-(1-Heptyl-1H-1,2,3-triazol-4-yl)hexanoic acid (14) (Scheme 3)
General procedure: 3,4-Dihydro-2H-pyran (1.40g, 16.0mmol) was added to stirred solution of 6-bromo-1-hexanol (33a) (2.0g, 11.0mmol) and camphorsulfonic acid (116mg, 5mol%) in Et2O at room temperature overnight. The reaction was basified with satd K2CO3 and extracted into Et2O, washed with brine and dried over Na2SO4. Purification by flash column chromatography; neat CH2Cl2 gave 2-((6-bromohexyl)oxy)tetrahydro-2H-pyran (34a) (2.70g, 95%) as a colourless oil. 1H NMR [DMSO] δ 4.55-4.50 (m, 1H), 3.72 (ddd, J=11.2, 8.1, 3.1Hz, 1H), 3.61 (td, J=9.7, 6.7Hz, 1H), 3.52 (t, J=6.7Hz, 2H), 3.46-3.37 (m, 1H), 3.35-3.28 (m, 1H), 1.84-1.76 (m, 2H), 1.73-1.64 (m, 1H), 1.64-1.54 (m, 1H), 1.54-1.28 (m, 10H). HRMS (ESI+) Calcd for C11H21BrNaO2: 287.0617; (MNa+) 287.0613n-Butyllithium (2.07mL of a 2.0M solution in cyclohexanes, 4.1mmol) was added dropwise to a solution of trimethylsilylacetylene (0.60mL, as a solution in THF, 4.1mmol) in THF (8mL) at -60°C, then the reaction was allowed to warm to 0°C for 30min.12 The reaction was then cooled to -20°C, HMPA (2mL) was added dropwise followed by 34a (1.0g, 3.8mmol). The reaction was stirred for 5h at 0°C and room temperature for an additional 14h, then quenched with satd NH4Cl and extracted into EtOAc, washed with brine and dried over Na2SO4. The crude oil was filtered through a pad of silica with CH2Cl2 and concentrated to dryness to give (by 1H NMR) a 2:1 mixture of trimethyl-(8-((tetrahydro-2H-pyran-2-yl)oxy)oct-1-yn-1-yl)silane (35a) and 2-(oct-7-yn-1-yloxy)tetrahydro-2H-pyran (36a) (1.0g, 98%). This (1.0g, 3.8mmol) was taken up in THF (10mL) and stirred with TBAF (5.6mL as a 1molL-1 solution in THF, 5.6mmol) at room temperature for 48h. The reaction was diluted with EtOAc, and washed with satd NaHCO3, brine, dried over Na2SO4 and concentrated to dryness to give only 36a (740mg, 91%) as a colourless oil. |
| 95% |
With toluene-4-sulfonic acid In dichloromethane at 0 - 20℃; for 16h; Inert atmosphere; |
16 4.1.16 2-(6-Bromohexyl-1-oxy)tetrahydropyran 24
To a stirred solution of 6-bromohexyl-1-ol 23 (2.0 g, 9.61 mmol) in anhydrous CH2Cl2 (40 mL) at 0 °C, under N2, DHP (1.051 mL, 11.53 mmol) and PTSA (603 mg, 2.40 mmol) were added. The reaction mixture was allowed to stir for 16 h in the water bath, under N2 atmosphere, by which time the solution had turned slightly brown in color. After completion, the reaction mixture was neutralized with aqueous NaHCO3 (50 mL), the organic phase was collected and the aqueous phase re-extracted with CH2Cl2 (2 * 30 mL). Organic fractions were combined, washed with water (30 mL), brine (30 mL) over MgSO4, filtered and concentrated to a crude residue. This crude residue was purified by silica gel (60-120) flash column chromatography, eluting with hexane/EtOAc (9:0.5 v/v), to furnish 24 as a bright yellow oil (2.5 g, 95%): Rf 0.60 (hexane/EtOAc, 9:1 v/v). 1H NMR (400 MHz, CDCl3): δ 1.31-1.62 (m, 10H 5*CH2), 1.66-1.71 (m, 1H CH2), 1.88-2.0 (m, 3H), 3.37-3.48 (m, 3H), 3.50-3.54 (m, 1H), 3.72-3.75 (m, 1H, CHO), 3.85-3.87 (m, 1H, CHO), 4.57-4.59 (t, 1H, CHO); ESI+, m/z 264 [M+H]+. |
| 94% |
With 1-n-butyl-3-methylimidazolium hydrogen sulfate for 0.0333333h; microwave irradiation; |
|
| 93% |
With Amberlyst-15(Cat) In dichloromethane at 20℃; Inert atmosphere; |
Synthesis of Compound 2
Under argon protection, 6-bromo-1-hexanol (39.82 g, 0.22 mol) was dissolved in dry dichloromethane (250 mL), a catalytic amount of Amberlyst-15 ion exchange resin (0.6 g) was added, and at room temperature 3,4-dihydropyrane (18.7 g, 0.22 mol) was slowly added drop wise , and detected by TLC, after the reaction was completed, it was filtered, concentrated, and the crude product was subjected to column chromatography to give 54.1 g of a colorless transparent liquid, yield was 93%. |
| 92% |
With toluene-4-sulfonic acid In dichloromethane for 16h; Ambient temperature; |
|
| 92% |
With pyridinium 4-toluenesulfonate In dichloromethane at 20℃; for 5h; |
|
| 92% |
With toluene-4-sulfonic acid In dichloromethane at 20℃; |
|
| 90.9% |
|
W.1.1 Step 1:
Step 1: Synthesis of 6-bromo-1-(tetrahydropyranyloxy)hexane 197.8 g (1.09 mol) 6-bromo-1-hexanol were given into a 500 ml reactor vessel and cooled to 5 ØC or less. 102.1 g (1.21 mol) dihydropyran were added dropwise to this at a temperature of 10ØC or less. After dropping was finished, the mixture was returned to room temperature and stirred for one hour. The precipitate obtained in the reaction was purified in a silica gel column with hexane / IPE (diisopropyl ether) = 5 / 1, resulting in 263.4 g of 6-bromo- 1 -(tetrahydropyranyloxy)hexane. The yield was 90.9%. |
| 90% |
With o-toluenesulfonic acid In tert-butyl methyl ether at 0 - 3℃; |
2-(6-Bromo-hexyloxy)-tetrahydro-pyran (XX)Bromohexanol (54.67 g) is dissolved in TBME (250 ml.) and dried over Na2SC>4. After filtration, TsOH (0.6 mmol) is added and the solution is cooled in an ice bath. 3,4-Dihydropyran (394 mmol) is added dropwise, while maintaining the temperature around 2-30C. After complete addition the reaction mixture is allowed to reach ambient temperature overnight. The mixture is ished with sat. NaHCC>3 (2 x 200 ml_). The aqueous layer is extracted with TBME (200 ml.) and the combined organic layers are ished with brine (200 ml_). After drying over Na2SC>4 (containing some K2CO3), the solvent is evapo- rated to yield (XX) as a colourless oil (90 %), which is stored at 4 0C over K2CO3 and is used without further purification. |
| 90% |
With toluene-4-sulfonic acid In tert-butyl methyl ether at 0 - 20℃; |
2-(6-Bromo-hexyloxy)-tetrahydro-pyran (XX) Bromohexanol (54.67 g) is dissolved in TBME (250 mL) and dried over Na2SO4. After filtration, TsOH (0.6 mmol) is added and the solution is cooled in an ice bath. 3,4-Dihydropyran (394 mmol) is added dropwise, while maintaining the temperature around 2-3° C. After complete addition the reaction mixture is allowed to reach ambient temperature overnight. The mixture is ished with sat. NaHCO3 (2*200 mL). The aqueous layer is extracted with TBME (200 mL) and the combined organic layers are ished with brine (200 mL). After drying over Na2SO4 (containing some K2CO3), the solvent is evaporated to yield (XX) as a colourless oil (90%), which is stored at 4° C. over K2CO3 and is used without further purification. |
| 90% |
With pyridinium 4-toluenesulfonate In dichloromethane at 20℃; for 18h; |
|
| 89% |
With trichlorophosphate In diethyl ether 1.) 0 deg C, 30 min, 2.) RT, 2 h; |
|
| 89% |
With toluene-4-sulfonic acid In diethyl ether for 0.5h; |
|
| 89% |
With hydrogenchloride In diethyl ether at 0 - 20℃; for 14h; |
|
| 87% |
With pyridinium 4-toluenesulfonate In dichloromethane 1.) 0-5 deg C, 0.5 h, 2.) RT, 4 h; |
|
| 87% |
With iron(III) perchlorate In diethyl ether at 20℃; for 1.5h; |
|
| 86.9% |
With pyridinium 4-toluenesulfonate In dichloromethane at 20℃; for 3h; |
16.1 Step (1)
To a solution of 6-bromohexan-l-ol (10 g, 55.2 mmol) and 3,4-dihydro-2H-pyran (4.78 g,56.9 mmol) in DCM (150 mL) was added PPTS (1.61 g,6.41 mmol) then stirred at room temperature for 3 h. TLC (EA/PE 9/1, SM Rf: 0.2; product, Rf: 0.7) indicated that all the starting materials was consumed. The solvent was concentrated and purified by flash chromatography column (0-10% EA in PE (5%) to give 2-(6-bromohexoxy)tetrahydropyran (12.72 g, 48 mmol, 86.9% yield) as colorless oil. 1H NMR (500 MHz, CDCI3) d 4.60 - 4.53 (m, 1H), 3.90 - 3.83 (m, 1H), 3.77 - 3.71 (m, 1H), 3.53 - 3.47 (m, 1H), 3.44 - 3.35 (m, 3H), 1.93 - 1.79 (m, 3H), 1.76 - 1.67 (m, 1H), 1.65 - 1.36 (m, 10H). |
| 86.9% |
With pyridinium 4-toluenesulfonate In dichloromethane at 20℃; for 3h; |
16.1 Step (1)
To a solution of 6-bromohexan-l-ol (10 g, 55.2 mmol) and 3,4-dihydro-2H-pyran (4.78 g,56.9 mmol) in DCM (150 mL) was added PPTS (1.61 g,6.41 mmol) then stirred at room temperature for 3 h. TLC (EA/PE 9/1, SM Rf: 0.2; product, Rf: 0.7) indicated that all the starting materials was consumed. The solvent was concentrated and purified by flash chromatography column (0-10% EA in PE (5%) to give 2-(6-bromohexoxy)tetrahydropyran (12.72 g, 48 mmol, 86.9% yield) as colorless oil. 1H NMR (500 MHz, CDCI3) d 4.60 - 4.53 (m, 1H), 3.90 - 3.83 (m, 1H), 3.77 - 3.71 (m, 1H), 3.53 - 3.47 (m, 1H), 3.44 - 3.35 (m, 3H), 1.93 - 1.79 (m, 3H), 1.76 - 1.67 (m, 1H), 1.65 - 1.36 (m, 10H). |
| 85% |
With trifluoroacetic acid at 20℃; for 12h; |
|
| 85% |
With iodine In tetrahydrofuran for 0.05h; microwave irradiation; |
|
| 85% |
With pyridinium 4-toluenesulfonate In dichloromethane |
|
| 84% |
With hydrogen cation In diethyl ether |
|
| 83% |
With toluene-4-sulfonic acid In dichloromethane at 0 - 20℃; Inert atmosphere; |
|
| 82% |
With pyridinium 4-toluenesulfonate In dichloromethane at 20℃; for 1h; |
2
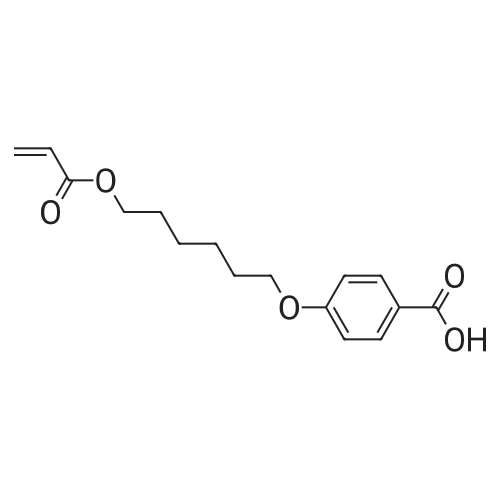
EXAMPLE 2 First, 2-(6-bromohexyloxy)-tetrahydro-2H-pyran (compound (1 B-1)) was synthesized according to the following equation. [Show Image] Into a 500 ml eggplant-shaped flask, 25 g (0.139 mol) of 6-bromohexanol, 12.83 g (0.152 mol) of dihydropyran, 1.74 g (0.007 mol) of pyridinium paratoluenesulfonate and 300 ml of dichloromethane were put, and such a mixture was stirred for 1 hour at room temperature. After reaction was completed, a reaction solvent was removed by distillation by a rotary evaporator, and 30 g of a transparent liquid of 2-(6-bromohexyloxy)-tetrahydro-2H-pyran (1 B-1) was obtained by distillation. The yield was 82%. Next, using the compound (1 B-1), trans-4-(4-(6-(tetrahydro-2H-pyran-2-yloxy)hexyloxy)phenyl)cyclohexanol (1 B-2) was synthesized according to the following equation. [Show Image] A reflux tube was attached to a 300 ml eggplant-shaped flask, 3 g (0.015 mol) of trans-(4-hydroxycyclohexyl)phenol, 10.8 g (0.078 mol) of potassium carbonate, 0.26 g (0.0016 mol) of potassium iodide, 50 ml of acetone and 4.97 g (0.016 mol) of compound (1B-1) were put into the flask, and such a mixture was stirred for 24 hours in a state that it was refluxed with heating. After reaction was completed, a reaction solvent from which solid state content was removed by filtering was distilled by a rotary evaporator, and the reacted product was purified by a silica gel column chromatography (development solvent:hexane/ethyl acetate=3:1). As a result, 5.6 g of compound (1B-2) was obtained as a white solid material. The yield was 95%. Next, using the compound (1B-1) and compound (1B-2), trans-2-(6-(4-(4-(6-(tetrahydro-2H-pyran-2-yloxy)hexyloxy)phenyl)cyclohexyloxy)hexyloxy)tetrahydro-2H-pyran (1B-3) was synthesized according to the following equation. [Show Image] A reflux tube was attached to a 100 ml eggplant-shaped flask, and in a nitrogen atmosphere, 2 g (0.0053 mol) of compound (1B-2), 20 ml of tetrahydrofuran and 2 ml of hexamethyl phosphoric acid triamide were put into the flask. Further, 0.85 g (0.021 mol) of sodium hydroxide (60% assay) was gently added, and such a mixture was stirred for 30 minutes in a state that it was refluxed with heating. Thereafter, 5.07 g (0.019 mol) of compound (1 B-1) was added, and they were further stirred for 24 hours in a state that they were refluxed with heating. After reaction was completed, 10 ml of a saturated ammonium chloride aqueous solution was added to the reacted mixture, and 50 ml of a saturated saline solution was added, and an extraction was carried out with 200 ml of ethyl acetate. An organic layer obtained was dried by using sodium sulfate anhydride, a solvent was removed by distillation by a rotary evaporator, and the reminder was purified by a silica gel column chromatography (development solvent:hexane/ethyl acetate/dichloromethane=4:1:0.5). As a result, 2.31 g of compound (1 B-3) was obtained with an yield of 78%, Next, using the compound (1 B-3), trans-6-(4-(4-(6-hydroxyhexyloxy)phenyl)cyclohexyloxy)hexan-1-ol (1B-4) was synthesized according to the following equation. [Show Image] In a 100 ml eggplant-shaped flask, 2.31 g (0.0041 mol) of compound (1 B-3), 20 ml of methanol and 0.08 g (0.0004 mol) of paratoluene sulfonic acid monohydrate were added, and such a mixture was stirred for 2 hours at room temperature. After reaction was completed, a few drops of saturated sodium hydrogencarbonate aqueous solution was dropped, reaction solvent was removed by distillation by a rotary evaporator, and the reminder was purified by a silica gel column chromatography (development solvent:hexane/ethyl acetate=1:1). As a result, 1.19 g of compound (1B-4) was obtained with an yield of 74%. Next, using the compound B4, a compound (1 B) was synthesized according to the following equation. [Show Image] 1.19 g (0.00303 mol) of compound (1B-4), 0.04 g (0.0003 mol) of 4-dimethylaminopyridine, 20 ml of dichloromethane and 0.67 g (0.0067 mol) of triethylamine were added, 0.6 g (0.0067 mol) of acrylic acid chloride was slowly dropped at 0°C, and they were stirred for 1 hour at room temperature. To such a stirred mixture, 50 ml of a saturated saline solution was added, and extraction was carried out with 100 ml of dichloromethane. An organic layer obtained was dried with sodium sulfate anhydride, the solvent was removed by distillation by a rotary evaporator, and the reminder was purified by a silica gel column chromatography (development solvent:hexane/ethyl acetate=4:1). As a result, 1.3 g of a transparent oil-like compound (1 B) was obtained. The yield was 86%. The compound (1 B) was identified by 1 H-NMR. 1H-NMR (400 MHz, CDCl3):δ=1.19-1.82(m,20H,-CH2-), 1.90(m,2H,-CH2-), 2.13(m,2H,-CH2-), 2.45(m,1H,Ar-CH), 3.25(m,1H,O-CH-)3.49(t,2H,-O-CH2-), 3.93(t,2H,-O-CH2-), 4.10-4.20(m,4H,-O-CH2-), 5.79-5.82(d,2H,acryl), 6.08-6.16(dd,2H,acryl), 6.37-6.44(d,2H,acryl), 6.82(d,2H, aromatic H), 7.10(d,2H, aromatic H) |
| 82% |
With toluene-4-sulfonic acid In dichloromethane at 0 - 20℃; for 1.5h; Inert atmosphere; |
|
| 82% |
With toluene-4-sulfonic acid In dichloromethane at 0 - 20℃; for 1.5h; |
8 Preparation of 2-(6-bromohexyloxy)tetrahydro-2H-pyran (23)
6-bromohexan-1-ol (22) (7.25 ml, 55.2 mmol) was brought up in DCM (55 mL) and cooled to 0° C. To this solution was added tosylic acid (0.095 g, 0.55 mmol), then dihydropyran (6.31 ml, 69.0 mmol) was added drop wise. Upon completion of addition, the reaction was stirred for 1.5 hours allowing it to warm to ambient temperature. TLC showed complete conversion to protected alcohol. The mixture was diluted with ether (300 mL), and washed with sat NaHCO3. The organic layer was dried over MgSO4 and concentrated in vacuo. The crude product was purified by column chromatography (100% DCM) to give the product as a clear colorless oil (12.05 g, 82%). Rf=0.74 (2:1 hexanes to EtOAc); 1H NMR [400 MHz, CDCl3] δ 4.55 (dd, 1H, J=4.3 Hz), 3.84 (m, 1H), 3.71 (dt, 1H, J=9.4, 7.0 Hz), 3.47 (m, 1H), 3.38 (t, 2H, J=7.1 Hz), 3.36 (dt, 1H, J=9.4, 7.0 Hz), 1.85 (q, 2H, J=7.2 Hz), 1.80-1.30 (m, 12H); 13C NMR [100 MHz, CDCl3] δ 99.2, 99.0, 67.6, 62.6, 34.1, 32.9, 31.0, 29.8, 28.2, 25.7, 19.9 ppm; HRMS [ESI] m/z 271.00278 (calc'd for C11H21BrO2+Li: 271.08795); Elem. Anal. C, 50.17; H, 8.00; Br, 29.80 (calc'd for C11H21BrO2: C, 49.82; H, 7.98; Br, 30.13). |
| 81% |
With DL-10-camphorsulphonic acid In dichloromethane for 0.5h; |
|
| 81% |
With toluene-4-sulfonic acid In dichloromethane at 0 - 20℃; Inert atmosphere; |
|
| 81% |
With toluene-4-sulfonic acid In dichloromethane at 0 - 20℃; for 12h; Inert atmosphere; |
1
2-(6-Bromohexyloxy)tetrahydro-2H-pyran 11, was first synthesized according to the following reaction scheme. In particular, a cooled, flame-dried round bottom flask equipped with a stir bar and septum was charged, under argon and at 0° C., with 6-bromo-1-hexanol 10 (7.6551 g, 42.28 mmol, 1 eq), dry DCM (10 ml), dihydropyran (4.25 ml, 46.51 mmol, 1.1 eq), and p-toluenesulfonic acid (0.4030 g, 2.12 mmol, 5 mol %). The reaction was allowed to stir at room temperature overnight, and was quenched by diluting with water (50 ml) and DCM (50 ml) in a separatory funnel. The organic layer was washed three times with brine (3×50 ml), dried (MgSO4), filtered, and evaporated to dryness under reduced pressure. Flash chromatography (SiO2: 15:1 hexanes to ethyl acetate) gave 11 (9.0902 g, 81% yield) as a clear oil. 1H NMR (500 MHz, CDCl3): δ 84.56 (t, J=2.75 Hz, 1H), 3.85 (m, 1H), 3.72 (m, 1H), 3.51 (m, 1H), 3.40 (m, 3H), 1.95-1.36 (br m, 14H). 13C NMR (126 MHz, CDCl3): δ 99.05, 67.56, 62.54, 34.02, 32.92, 30.94, 29.73, 28.18, 25.67, 25.65, 19.87. HRMS-FAB (m/z): [M+H] calcd for C11H22O2Br, 265.0803; found 265.0804. |
| 75% |
With hydrogenchloride for 5h; Ambient temperature; |
|
| 71% |
With toluene-4-sulfonic acid In toluene at 100℃; Inert atmosphere; |
|
| 70% |
With hydrogen cation |
|
| 66% |
In dichloromethane |
|
| 61% |
With pyridinium 4-toluenesulfonate In dichloromethane Cooling with ice; |
5 5.3.5. 2-((6-Bromohexyl)oxy)tetrahydro-2H-pyran (7)
1-Bromohexanol (7.0 g, 1 eq.) was dissolved in CH2Cl2 and cooled with an ice bath, then dihydropyran (5.3 ml, 1.5 eq.) and pyridinium p-toluenesulfonate (50 mg, 0.05 eq.) were added. After stirring overnight, the mixture was washed with H2O (×2), once with brine, dried with Na2SO4, filtered, and the solvent was removed under reduced pressure. The resulting solid was dissolved in hexane:Et2O (5 mL, 20:1), loaded onto a silica column, and eluted using hexane:Et2O (20:1). Product containing fractions were combined and the solvent removed to afford compound 7 (6.442 g, 61% yield). |
| 60% |
With trichlorophosphate |
|
|
With DL-10-camphorsulphonic acid In dichloromethane |
|
|
With toluene-4-sulfonic acid |
|
|
With hydrogen cation |
|
|
With pyridinium 4-toluenesulfonate In dichloromethane for 5h; |
|
|
With toluene-4-sulfonic acid In chloroform |
|
|
With trichlorophosphate In tetrahydrofuran |
|
|
With hydrogenchloride at 20℃; for 16h; |
|
|
With pyridinium 4-toluenesulfonate In dichloromethane at 20℃; for 5h; Cooling with ice bath; |
|
|
With pyridinium 4-toluenesulfonate In dichloromethane at 20℃; |
|
|
Stage #1: 1-bromo-6-hexanol With toluene-4-sulfonic acid In dichloromethane at 0℃; for 0.166667h;
Stage #2: 3,4-dihydro-2<i>H</i>-pyran In dichloromethane at 0℃; for 3h; |
21 THP-protected 6-bromo-1-hexanol:
[0317j THP-protected 6-bromo-1-hexanol: Into an oven-dried 50 mL round-bottomflask, 6-Bromo-1-hexanol (1 mmol) and p-toluenesulfonic acid monohydrate (catalyticamount) are dissolved in 20 mL of dichloromethane. The mixture was allowed to mix at 0°C for 10 mm. With stirring, 3,4-Dihydro-2H-pyran (3 mmol) dissolved in 10 mL of dichloromethane was then dropwise added to the reaction mixture at 0°C. The progress of the reaction was monitored by TLC and the reaction was observed to be completed within 3hours. The reaction mixture was concentrated under reduced pressure and the resulting residue was purified by silica gel chromatography. Fractions contain the desired product were pulled and the solvent was removed under reduced pressure to afford the desired product. |
|
With pyridinium 4-toluenesulfonate In dichloromethane at 20℃; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping