* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: With trifluorormethanesulfonic acid; [bis(pyridine)iodine]+ tetrafluoroborate In dichloromethane at 0 - 20℃; for 21 h; Stage #2: With water; sodium thiosulfate In dichloromethane
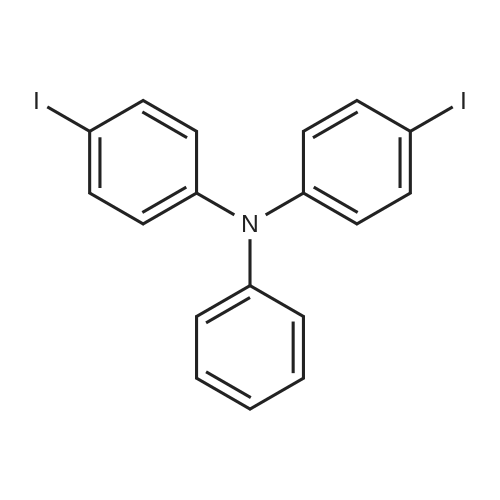
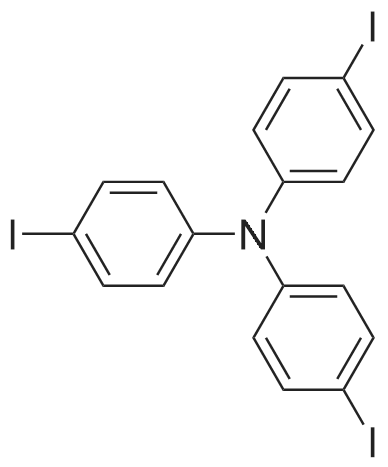
Synthesis Example 4Tris(4-iodophenyl)amine was prepared as follows. Firstly, a mixture of IPy2BF4 (5.3 g, 14.3 mmol) and triphenylamine (1 g, 4.1 mmol) was added with dist. CH2Cl2 (60 mL) under a nitrogen atmosphere, and then was added dropwise at 0° C. with trifluoromethanesulfonic acid (TfOH: 900 μL, 4.1 mmol). Thereafter, the resultant mixture was stirred under a nitrogen atmosphere at room temperature for 21 hours to obtain a reddish-brown reaction mixture. Subsequently, the obtained reaction mixture was added with sat. Na2S2O3, and an aqueous layer wad extracted with CH2Cl2. After that, an organic layer thus collected was washed with sat. NaCl, and dried with Na2SO4. Subsequently, the dried organic layer was filtered and concentrated to obtain a crude product. Then, the obtained crude product was separated and purified by silica gel column chromatography (hexane/EtOAc=5/1) to obtain tris(4-iodophenyl) amine (2.507 g, 99percent yield).The obtained compound was subjected to 1H NMR and 13C NMR measurements. The obtained results are shown below.1H NMR (CDCl3) δ7.54 (d, J=8.9 Hz, 6H), 6.81 (d, J=8.9 Hz, 6H).13C NMR (CDCl3) δ146.5, 138.4, 126.0, 86.6.From the NMR measurement results, it was confirmed that the obtained compound was tris(4-iodophenyl)amine.
99%
Stage #1: With trifluorormethanesulfonic acid; [bis(pyridine)iodine]+ tetrafluoroborate In dichloromethane at 0 - 20℃; for 21 h; Stage #2: With sodium thiosulfate In dichloromethane; water
A mixture of 5.3 g (14.3 mmol, 3.5 eq.) of bis(pyridine)iodonium tetrafluoroborate (IPy2BF4) and 1 g (4.1 mmol) of triphenylamine was added with 60 ml of dichloromethane (dist.CH2Cl2) under a nitrogen atmosphere to obtain a mixed solution. Then, the mixed solution thus obtained was cooled to 0° C., and added dropwise with 900 μl (4.1 mmol, 1 eq.) of trifluoromethanesulfonic acid (TfOH). The resultant mixed solution was stirred under a nitrogen atmosphere at room temperature for 21 hours to obtain a reaction mixture. Subsequently, the reaction mixture thus obtained was added with a saturated sodium thiosulfate (Na2S2O3) aqueous solution to suppress the reaction. Thereafter, the aqueous phase in the reaction solution was extracted with dichloromethane. Thereby, the organic phase containing the reddish-brown reaction mixture was obtained. After that, the organic phase thus obtained was washed with a saturated NaCl solution, dried with Na2SO4, filtered, and concentrated to obtain a crude product (2.9714 g). Then, the crude product thus obtained was separated and purified by silica gel column chromatography (hexane:ethyl acetate=5:1). Thereby, tris(4-iodophenyl)amine was obtained (a yield of 2.507 g and 99percent).The tris(4-iodophenyl)amine thus obtained was subjected to 13C NMR and 1H NMR measurements. Note that, the NMR spectra were measured with a JOEL JNM EX270 spectrometer (270 MHz for 1H). Moreover, TMS was used as a reference for the chemical shifts in 1H NMR, and CDCl3 was used as a reference for the chemical shifts in 13C NMR. The measurement results are shown below.1H NMR (CDCl3) δ7.54 (d, J=8.9 Hz, 6H), 6.81 (d, J=8.9 Hz, 6H);13C NMR (CDCl3) δ146.5, 138.4, 126.0, 86.6.In addition, the following reaction formula (H) shows an outline of the synthesis method for the tris(4-iodophenyl)amine.
90%
With iodine; mercury(II) oxide In ethanol at 20℃;
A mixture of triphenylamine (1 g, 0.004 mmol), HgO (4.06 g, 0.019 mmol) and I2 (5.08 g,0.020 mmol) in EtOH (50 mL) was stirred overnight at room temperature. The solvent was removed, and the product was separated from mercuric salts with boiling toluene. The solution was filtered through the short column of Al2O3, and the product was precipitated from hot toluene with MeOH to afford the title compound 4as white solid (2.33 g, 90percent). Mp: 170C. 1H NMR (300 MHz, CDCl3)d(ppm): 7.53 (d, J8.8 Hz, 6H), 6.81 (d,J8.8 Hz, 6H); 13C NMR (75 MHz, CDCl3) d (ppm): 86.76, 126.12, 138.53, 146.59.23
90%
Stage #1: With mercury(II) oxide In ethanol at 20℃; for 1 h; Stage #2: With iodine In ethanol at 20℃; for 12 h;
At room temperature, 24.5 g of triphenylamine, 300 mL of anhydrous ethanol, and 70 g of mercury oxide were added to the reaction system.Stir at room temperature for 1 h. Then 80 g of iodine was added and the reaction was maintained at room temperature for 12 h.The reaction mixture was distilled under reduced pressure and ethanol was recovered. 300 mL of benzene was added to the solid phase for recrystallization.A white crystalline product, tris-(4-iodophenyl)amine, was obtained, yield 56 g, yield 90percent.
88%
With N-iodo-succinimide; acetic acid In chloroform
Tris(4-iodophenyl)amine (1). To a stirred mixture of triphenylamine (7.36 g, 30.0 mmol) and N-iodosuccinimide (NIS, 21.60 g, 96.0 mmol) in chloroform (180 mL) was added acetic acid (120 mL) at room temperature under exclusion of light. The solution was stirred overnight at room temperature. The reaction mixture was poured into water, washed with sodium thiosulfate, and extracted with methylene chloride. The combined methylene chloride layers were washed with water, dried with Na2SO4, and concentrated. The crude product was purified over a silica gel column with hexane/methylene chloride (7:1) as eluent to afford a slightly brown solid (16.54 g, 88percent). 1H NMR (300 MHz, CDCl3): δ 7.56 (d, 6H, J=8.7 Hz), 6.83 (d, 6H, J=9.0 Hz).
Reference:
[1] Patent: US2008/227939, 2008, A1, . Location in patent: Page/Page column 17
[2] Patent: US2009/54649, 2009, A1, . Location in patent: Page/Page column 43-44
[3] Journal of Organic Chemistry, 2009, vol. 74, # 16, p. 6287 - 6290
[4] European Journal of Organic Chemistry, 2013, # 13, p. 2608 - 2620
[5] Tetrahedron, 2014, vol. 70, # 15, p. 2546 - 2555
[6] Patent: CN107827836, 2018, A, . Location in patent: Paragraph 0024
[7] Organic Letters, 2004, vol. 6, # 1, p. 47 - 50
[8] Patent: US2009/278445, 2009, A1,
[9] Journal of Materials Chemistry C, 2015, vol. 3, # 28, p. 7345 - 7355
[10] RSC Advances, 2016, vol. 6, # 16, p. 12819 - 12828
[11] New Journal of Chemistry, 2017, vol. 41, # 4, p. 1459 - 1472
[12] Journal of Polymer Science, Part A: Polymer Chemistry, 2011, vol. 49, # 4, p. 832 - 841
[13] Journal of the American Chemical Society, 2002, vol. 124, # 8, p. 1736 - 1743
[14] Chemistry - A European Journal, 2011, vol. 17, # 3, p. 969 - 975
[15] Tetrahedron Letters, 2010, vol. 51, # 3, p. 521 - 524
[16] Chinese Journal of Chemistry, 2013, vol. 31, # 4, p. 456 - 464
[17] Organic Letters, 2009, vol. 11, # 13, p. 2768 - 2771
[18] European Journal of Organic Chemistry, 2011, # 5, p. 912 - 921
[19] Journal of Organic Chemistry, 2011, vol. 76, # 21, p. 8726 - 8736
[20] Journal of Polymer Science, Part A: Polymer Chemistry, 2011, vol. 49, # 8, p. 1830 - 1839
[21] Journal of the Chemical Society. Perkin Transactions 1, 2001, # 20, p. 2548 - 2552
[22] Chemistry Letters, 1989, p. 1145 - 1148
[23] Journal of the American Chemical Society, 2006, vol. 128, # 36, p. 11840 - 11849
[24] Tetrahedron Letters, 2009, vol. 50, # 49, p. 6901 - 6905
[25] Chemical Communications, 2011, vol. 47, # 6, p. 1818 - 1820
[26] Crystal Growth and Design, 2010, vol. 10, # 9, p. 3964 - 3976
[27] CrystEngComm, 2011, vol. 13, # 10, p. 3402 - 3407
[28] Tetrahedron Letters, 2012, vol. 53, # 2, p. 196 - 199
[29] Dalton Transactions, 2012, vol. 41, # 17, p. 5280 - 5293
[30] RSC Advances, 2013, vol. 3, # 39, p. 17914 - 17917
[31] European Journal of Organic Chemistry, 2013, # 34, p. 7785 - 7799
[32] Zeitschrift fur Anorganische und Allgemeine Chemie, 2014, vol. 640, # 10, p. 2030 - 2034
[33] Journal of Materials Chemistry C, 2014, vol. 2, # 35, p. 7201 - 7215
[34] RSC Advances, 2015, vol. 5, # 87, p. 71046 - 71051
[35] Tetrahedron Letters, 2014, vol. 55, # 51, p. 7102 - 7105
[36] Advanced Functional Materials, 2014, vol. 24, # 48, p. 7645 - 7654
[37] Angewandte Chemie - International Edition, 2016, vol. 55, # 20, p. 5956 - 5960[38] Angew. Chem., 2016, vol. 128, # 20, p. 6060 - 6064,5
[39] Molecular Crystals and Liquid Crystals, 2017, vol. 645, # 1, p. 1 - 9
[40] Chemical Communications, 2017, vol. 53, # 62, p. 8778 - 8781
[41] RSC Advances, 2017, vol. 7, # 75, p. 47681 - 47688
2
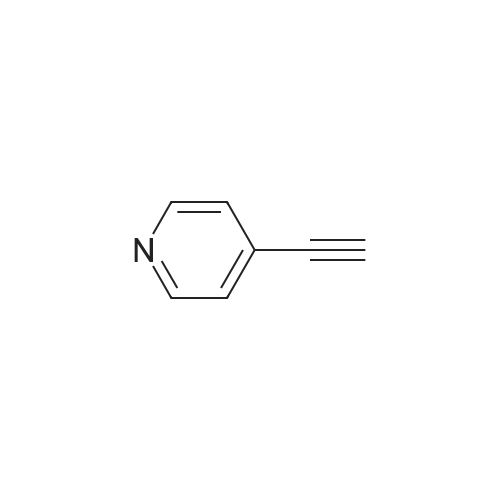
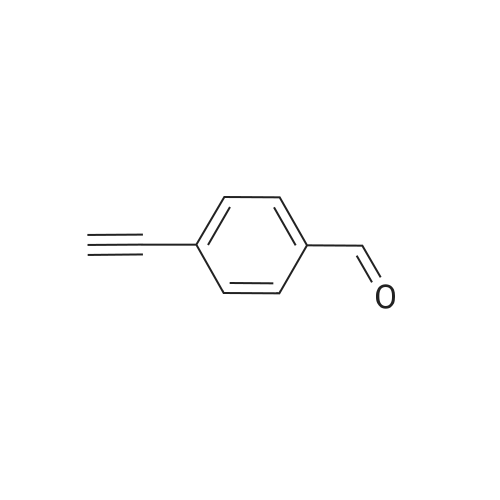
[ 603-34-9 ]
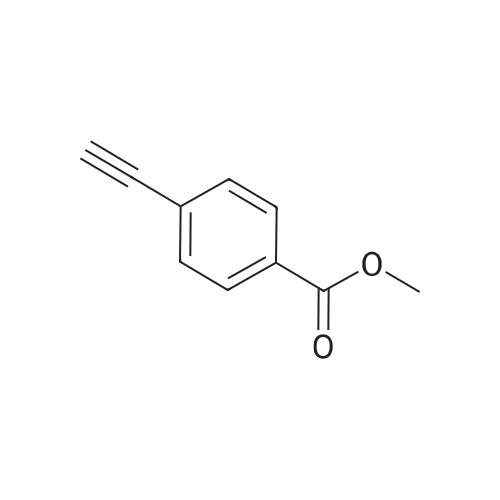
[ 218909-60-5 ]
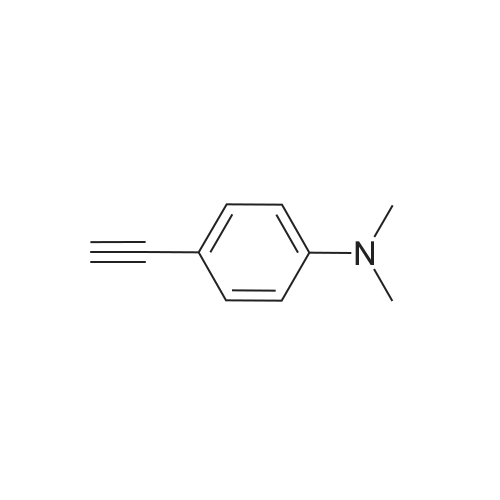
[ 4181-20-8 ]
Reference:
[1] Dyes and Pigments, 2015, vol. 113, p. 227 - 238
Synthesis Example 4Tris(4-iodophenyl)amine was prepared as follows. Firstly, a mixture of IPy2BF4 (5.3 g, 14.3 mmol) and triphenylamine (1 g, 4.1 mmol) was added with dist. CH2Cl2 (60 mL) under a nitrogen atmosphere, and then was added dropwise at 0 C. with trifluoromethanesulfonic acid (TfOH: 900 muL, 4.1 mmol). Thereafter, the resultant mixture was stirred under a nitrogen atmosphere at room temperature for 21 hours to obtain a reddish-brown reaction mixture. Subsequently, the obtained reaction mixture was added with sat. Na2S2O3, and an aqueous layer wad extracted with CH2Cl2. After that, an organic layer thus collected was washed with sat. NaCl, and dried with Na2SO4. Subsequently, the dried organic layer was filtered and concentrated to obtain a crude product. Then, the obtained crude product was separated and purified by silica gel column chromatography (hexane/EtOAc=5/1) to obtain tris(4-iodophenyl) amine (2.507 g, 99% yield).The obtained compound was subjected to 1H NMR and 13C NMR measurements. The obtained results are shown below.1H NMR (CDCl3) delta7.54 (d, J=8.9 Hz, 6H), 6.81 (d, J=8.9 Hz, 6H).13C NMR (CDCl3) delta146.5, 138.4, 126.0, 86.6.From the NMR measurement results, it was confirmed that the obtained compound was tris(4-iodophenyl)amine.
99%
A mixture of 5.3 g (14.3 mmol, 3.5 eq.) of bis(pyridine)iodonium tetrafluoroborate (IPy2BF4) and 1 g (4.1 mmol) of triphenylamine was added with 60 ml of dichloromethane (dist.CH2Cl2) under a nitrogen atmosphere to obtain a mixed solution. Then, the mixed solution thus obtained was cooled to 0 C., and added dropwise with 900 mul (4.1 mmol, 1 eq.) of trifluoromethanesulfonic acid (TfOH). The resultant mixed solution was stirred under a nitrogen atmosphere at room temperature for 21 hours to obtain a reaction mixture. Subsequently, the reaction mixture thus obtained was added with a saturated sodium thiosulfate (Na2S2O3) aqueous solution to suppress the reaction. Thereafter, the aqueous phase in the reaction solution was extracted with dichloromethane. Thereby, the organic phase containing the reddish-brown reaction mixture was obtained. After that, the organic phase thus obtained was washed with a saturated NaCl solution, dried with Na2SO4, filtered, and concentrated to obtain a crude product (2.9714 g). Then, the crude product thus obtained was separated and purified by silica gel column chromatography (hexane:ethyl acetate=5:1). Thereby, tris(4-iodophenyl)amine was obtained (a yield of 2.507 g and 99%).The tris(4-iodophenyl)amine thus obtained was subjected to 13C NMR and 1H NMR measurements. Note that, the NMR spectra were measured with a JOEL JNM EX270 spectrometer (270 MHz for 1H). Moreover, TMS was used as a reference for the chemical shifts in 1H NMR, and CDCl3 was used as a reference for the chemical shifts in 13C NMR. The measurement results are shown below.1H NMR (CDCl3) delta7.54 (d, J=8.9 Hz, 6H), 6.81 (d, J=8.9 Hz, 6H);13C NMR (CDCl3) delta146.5, 138.4, 126.0, 86.6.In addition, the following reaction formula (H) shows an outline of the synthesis method for the tris(4-iodophenyl)amine.
94.5%
With potassium iodate; acetic acid; potassium iodide; for 4h;Reflux;
In a 500 mL three-necked flask equipped with a spherical condenser and a stirrer, 4.90 g of triphenylamine was sequentially added.7.30 g of potassium iodide and 200 mL of acetic acid were heated to reflux, and then 6.40 g of potassium iodate was added to the reaction bottle in batches, and the reaction was performed at a constant temperature for 4 hours.The reaction solution changed from colorless to brown and finally colorless. Stop heating and cool to room temperature.Spin off the acetic acid, add saturated sodium thiosulfate solution and dichloromethane for extraction, and dry the organic phase over anhydrous sodium sulfate.Concentration gave 12.50 g of a pale yellow solid, which was recrystallized from ethyl acetate.11.77 g of light yellow platelets were obtained with a yield of 94.50%.
90%
With iodine; mercury(II) oxide; In ethanol; at 20℃;
A mixture of triphenylamine (1 g, 0.004 mmol), HgO (4.06 g, 0.019 mmol) and I2 (5.08 g,0.020 mmol) in EtOH (50 mL) was stirred overnight at room temperature. The solvent was removed, and the product was separated from mercuric salts with boiling toluene. The solution was filtered through the short column of Al2O3, and the product was precipitated from hot toluene with MeOH to afford the title compound 4as white solid (2.33 g, 90%). Mp: 170C. 1H NMR (300 MHz, CDCl3)d(ppm): 7.53 (d, J8.8 Hz, 6H), 6.81 (d,J8.8 Hz, 6H); 13C NMR (75 MHz, CDCl3) d (ppm): 86.76, 126.12, 138.53, 146.59.23
90%
At room temperature, 24.5 g of triphenylamine, 300 mL of anhydrous ethanol, and 70 g of mercury oxide were added to the reaction system.Stir at room temperature for 1 h. Then 80 g of iodine was added and the reaction was maintained at room temperature for 12 h.The reaction mixture was distilled under reduced pressure and ethanol was recovered. 300 mL of benzene was added to the solid phase for recrystallization.A white crystalline product, tris-(4-iodophenyl)amine, was obtained, yield 56 g, yield 90%.
88%
With N-iodo-succinimide; acetic acid; In chloroform;
Tris(4-iodophenyl)amine (1). To a stirred mixture of triphenylamine (7.36 g, 30.0 mmol) and N-iodosuccinimide (NIS, 21.60 g, 96.0 mmol) in chloroform (180 mL) was added acetic acid (120 mL) at room temperature under exclusion of light. The solution was stirred overnight at room temperature. The reaction mixture was poured into water, washed with sodium thiosulfate, and extracted with methylene chloride. The combined methylene chloride layers were washed with water, dried with Na2SO4, and concentrated. The crude product was purified over a silica gel column with hexane/methylene chloride (7:1) as eluent to afford a slightly brown solid (16.54 g, 88%). 1H NMR (300 MHz, CDCl3): delta 7.56 (d, 6H, J=8.7 Hz), 6.83 (d, 6H, J=9.0 Hz).
With potassium iodate; acetic acid; potassium iodide; at 120℃; for 4h;
Triarylamine(10 g, 40 mmol) was added to a three-necked flask, potassium iodide (14.36 g, 88.81 mmol), 150 ml of glacial acetic acid was added, and the reaction was refluxed at 120 C.Potassium iodate (9.52 g, 44.41 mmol) was added portionwise to the above reaction for 4 h.The treatment method after the end of the reaction is in agreement with the 2I-Cz treatment method.A pale yellow solid powder 3I-TPA was obtained with a yield of about 71%.
With copper; potassium carbonate; In decalin; at 220℃; for 24h;
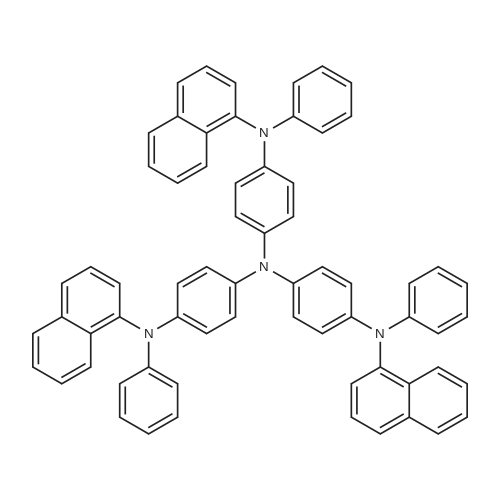
Compound 5; 4,4',4-tris[(1-naphtyl)phenylamino]tripnenylamine; 28.7 g (0.046 moles) of 4,4',4-triiodotriphenylamine, 50.4 g (0.23 moles) of N-(1-naphtyl)aniline, 44.2 g (0.32 moles) of anhydrous potassium carbonate, 4.32 g (0.068 moles) of copper powders and 50 ml of decalin were added to a reaction container of 200 ml, and the mixture was heated in an oil bath at 220° C. for 24 hours in an argon atmosphere. After completion of the reaction, 200 ml of toluene was added to the reaction product, followed by filtering to remove the insoluble. The filtrate was washed with water, and dried on sodium sulfate. After drying, the solvent was evaporated from the filtrate, and the residue was purified four times on a column chromatography using silica gel as a carrier and a mixture of n-hexane and toluene as a developing solvent. The recrystallization process was repeated using a mixture of n-hexane and toluene and ethyl acetate, followed by vacuum drying. 24.7 g (yield: 60.0percent) of 4,4',4-tris[(1-naphtyl)phenylamino]tripnenylamine was obtained. The resulting product was further purified by sublimation to obtain a high purity 4,4',4-tris[(1-naphtyl)phenylamino]tripnenylamine (yield of sublimation purification: 70.0percent).
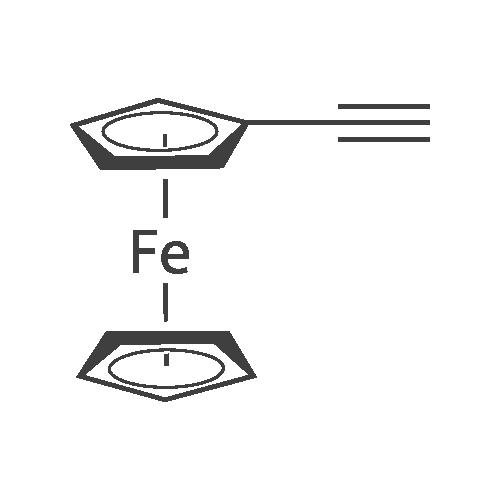
4,4',4''-tris(ferrocenylethynyl)triphenylamine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
With copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); triethylamine; In tetrahydrofuran; at 50℃; for 5h;Inert atmosphere;
To asolution of 1 (946 mg, 4.50mmol), <strong>[4181-20-8]tris(4-iodophenyl)amine</strong>(632 mg, 1.00mmol), and CuI (57 mg, 0.30mmol) in triethylamine(10 mL) and THF (10 mL) was added tetrakis(triphenylphosphine)palladium(0) (90 mg, 0.08mmol). The resultingmixture was stirred at 50 °C for 5 h under an Ar atmosphere.The reaction mixture was poured into a 10percent NH4Cl solutionand extracted with CH2Cl2. The organic layer was washed withbrine, dried with Na2SO4, and concentrated under reducedpressure. The residue was purified by column chromatographyon silica gel with CH2Cl2 to give 5 (828 mg, 95percent) as orangecrystals. Mp 158.0-161.0°C, decomp. (CH2Cl2); Melting point is not shown in the literature;12 1HNMR (500 MHz, CDCl3): deltaH 7.37 (d, 6H, J = 8.5 Hz, 3,3,3-H of C6H4), 7.03 (d, 6H, J =8.5 Hz, 2,2,2-H of C6H4), 4.48 (dd, 6H, J = 2.0, 2.0 Hz, Fc),4.24 (s, 15H, Cp), 4.23 (dd, 6H, J = 2.0, 2.0 Hz, Fc).
With tetra-(n-butyl)ammonium iodide; triethylamine;bis(acetonitrile)(1,5-cyclooctadiene)rhodium(I) tetrafluoroborate; In N,N-dimethyl-formamide; at 80℃; for 1h;
Synthesis Example 5Tris(4-triethoxysilylphenyl)amine was prepared as follows. Firstly, dist. DMF (4 mL), dist. Et3N (201 muL, 1.45 mmol) and (EtO)3SiH (178 muL, 0.96 mmol) were added dropwise to a mixture of <strong>[4181-20-8]tris(4-iodophenyl)amine</strong> (100 mg, 0.16 mmol) obtained in Synthesis example 4, [Rh(CH3CN)2(cod)]BF4 (5.4 mg, 0.014 mmol) and PPh3MeI (195 mg, 0.48 mmol). The resultant mixture was stirred under a nitrogen atmosphere at 80° C. for 1 hour to obtain a reaction mixture. Then, the solvent in the reaction mixture was distilled away with a vacuum pump, and a residue thus obtained was extracted with ether. A salt thus formed was removed through a celite filtration process, and then the solvent was distilled away from an organic layer with an evaporator to obtain a crude product. Subsequently, the obtained crude product was purified with an activated carbon (7 mm in a Kiriyama funnel (small) (available from Kiriyama Glass Co., Ltd.), ether: 15 mL) to obtain almost pure tris(4-triethoxysilylphenyl)amine (118.4 mg, 100percent yield).The obtained compound was subjected to 1H NMR and 13C NMR measurements. The obtained results are shown below.1H NMR (CDCl3) delta7.54 (d, J=8.6 Hz, 6H), 7.09 (d, J=8.6 Hz, 6H), 3.89 (q, J=7.0 Hz, 18H), 1.26 (t, J=7.0 Hz, 27H).13C NMR (CDCl3) delta148.9, 135.8, 124.7, 123.5, 58.7, 18.2.From the NMR measurement results, it was confirmed that the obtained compound was tris(4-triethoxysilylphenyl)amine.
100%
With methyl-triphenylphosphonium iodide; triethylamine;bis(acetonitrile)(1,5-cyclooctadiene)rhodium(I) tetrafluoroborate; In N,N-dimethyl-formamide; at 80℃; for 1h;
A mixture of 100 mg (0.16 mmol) of the <strong>[4181-20-8]tris(4-iodophenyl)amine</strong> obtained as described above, 5.4 mg (0.014 mmol, 9 mol percent) of a [Rh(CH3CN)2(cod)]BF4 complex, and 195 mg (0.48 mmol, 3 eq.) of PPh3MeI was added dropwise with 4 ml of DMF, 201 mul (1.45 mmol, 9 eq.) of triethylamine, and 178 mul (0.96 mmol, 6 eq.) of triethoxysilane (EtO)3SiH). Then, the resultant mixture was stirred under a nitrogen atmosphere at 80° C. for 1 hour to obtain a reaction mixture. Subsequently, a solvent in the reaction mixture thus obtained was removed by distillation with a vacuum pump, and a residue was extracted with ether. Thereafter, a salt thus formed was removed by filtering with celite. After that, the solvent was removed by distillation from the organic phase with an evaporator to obtain a crude product (128.4 mg). Then, the crude product thus obtained was dissolved in 15 ml of ether, and purified by filtering the resultant through activated carbon (Kiriyama funnel, thickness: 7 mm). Thereby, a triphenylamine-silane compound was obtained (118.4 mg, 100percent).The obtained triphenylamine-silane compound was subjected to 13C NMR and 1H NMR measurements. Note that, the NMR spectra were measured with a JOEL JNM EX270 spectrometer (270 MHz for 1H). Moreover, TMS was used as a reference for the chemical shifts in 1H NMR, and CDCl3 was used as a reference for the chemical shifts in 13C NMR. The measurement results are shown below.1H NMR (CDCl3) delta7.54 (d, J=8.6 Hz, 6H), 7.09 (d, J=8.6 Hz, 6H), 3.89 (q, J=7.0 Hz, 18H), 1.26 (t, J=7.0 Hz, 27H);13C NMR (CDCl3) delta148.9, 135.8, 124.7, 123.5, 58.7, 18.2.Based on the NMR measurement results, it was confirmed that the triphenylamine-silane compound obtained in Example 31 was tris(4-triethoxysilylphenyl)amine.In addition, the following reaction formula (1) shows an outline of the synthesis method for the tris(4-triethoxysilylphenyl)amine.
With copper(l) iodide; triethylamine;bis-triphenylphosphine-palladium(II) chloride; In toluene; at 40℃; for 3h;Inert atmosphere;
In reference to FIG. 3, a fourth compound according to the present invention, arbitrarily called CL108 and having a dendrimer core was also synthesized, according to diagram 3 below.More specifically, the compound N,N-bis(4-iodophenyl)-4-[(trimethylsilyl)ethynyl]benzenamine (6b) was first synthesized.The air is purged from a solution of compound 6a (1.00 g, 1.605 mmol) in 8 ml of toluene/Et3N (5/1) by argon bubbling for 20 min. Then, CuI (12 mg, 0.063 mmol), Pd(PPh3)2Cl2 (22.5 mg, 0.032 mmol) and trimethylsilyacetylene (0.227 mL, 1.605 mmol) are added, and the mixture is agitated at 40° C. for 3 h. After evaporation of the solvent under reduced pressure, the raw product is purified by chromatography in a silica column (heptane) to produce 352 mg (37percent) of compound 6b.The dendron 8a was then produced.The air is purged from a solution of compound 6b (200 mg, 0.337 mmol), compound 7 (341 mg, 0.843 mmol) and tri-o-furylphosphine (23 mg, 0.099 mmol) in 15 mL of toluene/Et3N (5/1) by argon bubbling for 30 min. Then, Pd2 dba3 (12.3 mg, 0.013 mmol) is added, and the mixture is agitated at 20° C. for 15 h. After evaporation of the solvent under reduced pressure, the raw product is purified by chromatography on a silica column (heptane/CH2Cl2 30:70) to produce 280 mg (73percent) of the dendron 8a.The dendron 8b was then synthesized.To do this, an aqueous solution of NaOH (1 M, 50 mL) is added to a solution of compound 8a (200 mg, 0.174 mmol) in THF (75 mL), and the mixture is agitated vigorously at 20° C. for 24 h. Dichloromethane is added, then the organic phase is separated, washed with water and dried (Na2SO4). The residue obtained after removal of the solvent is purified by chromatography in a silica column (heptane/CH2Cl2 25:75) to produce 98 mg (52percent) of the dendron 8b.Finally, the synthesis of compound CL108 is carried out according to the present invention.To this end, the air was purged from a solution of compound 6a (11.1 mg, 17.8 mumol) and dendron 8b (67 mg, 62.4 mumol) in 1.8 mL of toluene/Et3N (5/1) by argon bubbling for 25 min. Then, Pd(PPh3)2Cl2 (0.50 mg, 0.71 mumol) and CuI (0.27 mg, 1.42 mumol) are added, and the mixture is agitated at 40° C. for 16 h. After evaporation of the solvent under reduced pressure, the raw product is purified by chromatography on a silica column (heptane/CH2Cl2 20:80) to produce 15 mg (25percent) of CL108.
With copper(l) iodide; triethylamine;bis-triphenylphosphine-palladium(II) chloride; In toluene; at 40℃; for 16h;Inert atmosphere;
In reference to FIG. 3, a fourth compound according to the present invention, arbitrarily called CL108 and having a dendrimer core was also synthesized, according to diagram 3 below.More specifically, the compound N,N-bis(4-iodophenyl)-4-[(trimethylsilyl)ethynyl]benzenamine (6b) was first synthesized.The air is purged from a solution of compound 6a (1.00 g, 1.605 mmol) in 8 ml of toluene/Et3N (5/1) by argon bubbling for 20 min. Then, CuI (12 mg, 0.063 mmol), Pd(PPh3)2Cl2 (22.5 mg, 0.032 mmol) and trimethylsilyacetylene (0.227 mL, 1.605 mmol) are added, and the mixture is agitated at 40° C. for 3 h. After evaporation of the solvent under reduced pressure, the raw product is purified by chromatography in a silica column (heptane) to produce 352 mg (37percent) of compound 6b.The dendron 8a was then produced.The air is purged from a solution of compound 6b (200 mg, 0.337 mmol), compound 7 (341 mg, 0.843 mmol) and tri-o-furylphosphine (23 mg, 0.099 mmol) in 15 mL of toluene/Et3N (5/1) by argon bubbling for 30 min. Then, Pd2 dba3 (12.3 mg, 0.013 mmol) is added, and the mixture is agitated at 20° C. for 15 h. After evaporation of the solvent under reduced pressure, the raw product is purified by chromatography on a silica column (heptane/CH2Cl2 30:70) to produce 280 mg (73percent) of the dendron 8a.The dendron 8b was then synthesized.To do this, an aqueous solution of NaOH (1 M, 50 mL) is added to a solution of compound 8a (200 mg, 0.174 mmol) in THF (75 mL), and the mixture is agitated vigorously at 20° C. for 24 h. Dichloromethane is added, then the organic phase is separated, washed with water and dried (Na2SO4). The residue obtained after removal of the solvent is purified by chromatography in a silica column (heptane/CH2Cl2 25:75) to produce 98 mg (52percent) of the dendron 8b.Finally, the synthesis of compound CL108 is carried out according to the present invention.To this end, the air was purged from a solution of compound 6a (11.1 mg, 17.8 mumol) and dendron 8b (67 mg, 62.4 mumol) in 1.8 mL of toluene/Et3N (5/1) by argon bubbling for 25 min. Then, Pd(PPh3)2Cl2 (0.50 mg, 0.71 mumol) and CuI (0.27 mg, 1.42 mumol) are added, and the mixture is agitated at 40° C. for 16 h. After evaporation of the solvent under reduced pressure, the raw product is purified by chromatography on a silica column (heptane/CH2Cl2 20:80) to produce 15 mg (25percent) of CL108.
With copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); triethylamine; In tetrahydrofuran; at 70℃; under 7.50075e-05 Torr; for 10h;Inert atmosphere;
In this embodiment, a white light emitting material is synthesized, and the chemical structure formula of the white light emitting material is as follows:The preparation method includes the following steps:(1) 5.0 mmol of tris (4-iodophenyl) amine, 21.0 mmol of methyl 4-ethynylbenzoate, 30.0 mL of tetrahydrofuran,7.0mL of triethylamine was placed in a 100mL double-necked flask equipped with a stir bar and connected to a reflux tube;(2) Vacuum the reaction mixture with a vacuum pump so that the pressure reaches 1x10-2Pa,Pass in nitrogen, repeat pumping and nitrogen three times to maintain the nitrogen atmosphere of the reaction system;(3) adding 0.125 mmol of tetra (triphenylphosphine) palladium and 0.8 mmol of cuprous iodide to the above reaction solution under a nitrogen environment;(4) The above reaction mixture was heated to 70 C. under the protection of nitrogen and kept at reflux for 10 hours.The solvent was then evaporated to dryness on a rotary evaporator and washed with a small amount of ethyl acetate to give a yellow powder.Purified by column chromatography (silica gel column model: Yantai Jiangyou F-254 200-300 mesh,The mobile phase was a mixed solvent of methylene chloride and n-hexane, and the volume ratio of methylene chloride to n-hexane was 1:30) to obtain a white solid powder, that is, the white light-emitting material.The obtained white solid powder was characterized by hydrogen spectrum and carbon spectrum.
With tetraethylammonium hydroxide;(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In toluene; for 96h;Inert atmosphere; Reflux;
About 690 mg (about 3 mmol) of ferrocene boronic acid, about 310 mg (about 0.5 mmol) of <strong>[4181-20-8]tris(4-iodophenyl)amine</strong> and about 40 mg (about 10 mol percent) of Pd(dppf)Cl2 were placed in a flask. After a reflux condenser was connected to the flask, about 6 ml of toluene as a solvent and about 5 ml (about 1.33 M) of tetraethylammonium hydroxide as a base were injected into the flask using a syringe under a nitrogen atmosphere. The solution was degassed with nitrogen gas, and refluxed in an oil bath. The reaction was allowed to proceed for about 4 days. The reaction solution was diluted with about 20 ml of methylene chloride and neutralized with a saturated aqueous solution of ammonium chloride. The neutralized solution was transferred to a separatory funnel, followed by phase separation. The obtained organic layer was dried over anhydrous magnesium sulfate and passed through a glass filter to obtain a transparent polymer solution. The polymer solution was evaporated under reduced pressure to remove the solvents. The residue was purified by column chromatography using toluene/hexane (1/2), yielding the metallocenyl dendrimer (about 130 mg) of Formula 3 as an orange solid. The 1H-NMR spectrum of the metallocenyl dendrimer is shown in FIG. 4.
With tributyl-amine;tri-tert-butyl phosphine; palladium diacetate; In 1,4-dioxane; for 96h;Inert atmosphere; Reflux;
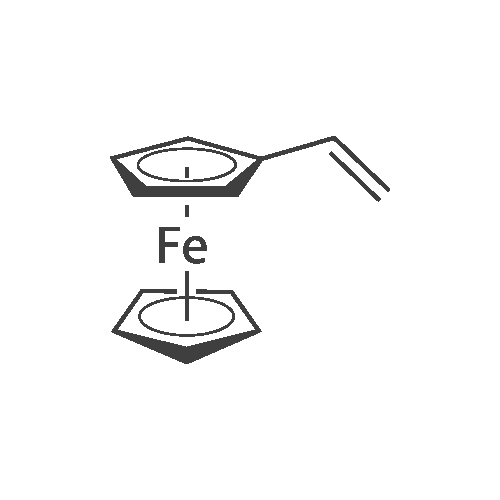
About 424 mg (about 2 mmol) of vinylferrocene, about 312 mg (about 0.5 mmol) of <strong>[4181-20-8]tris(4-iodophenyl)amine</strong> and about 7 mg (about 6 mol percent) of palladium acetate were placed in a flask. After a reflux condenser was connected to the flask, about 3 ml of 1,4-dioxene as a solvent, about 480 mul (about 2 mmol) of tri-n-butylamine as a base and about 11 mul (about 9 mol percent) of tri-t-butylphosphine were injected into the flask using a syringe under a nitrogen atmosphere. The solution was degassed with nitrogen gas, and refluxed in an oil bath. The reaction was allowed to proceed for about 4 days. The reaction solution was diluted with about 10 ml of methylene chloride and neutralized with a saturated aqueous solution of ammonium chloride. The neutralized solution was transferred to a separatory funnel, followed by phase separation. The obtained organic layer was dried over anhydrous magnesium sulfate and passed through a glass filter to obtain a transparent polymer solution. The polymer solution was evaporated under reduced pressure to remove the solvents. The residue was purified by column chromatography using toluene/hexane (1/2), yielding the metallocenyl dendrimer (about 301 mg) of Formula 2 as an orange solid. The 1H-NMR spectrum of the metallocenyl dendrimer is shown in FIG. 3.
With N,N,N,N,N,N-hexamethylphosphoric triamide; at 160℃; for 12h;Inert atmosphere;
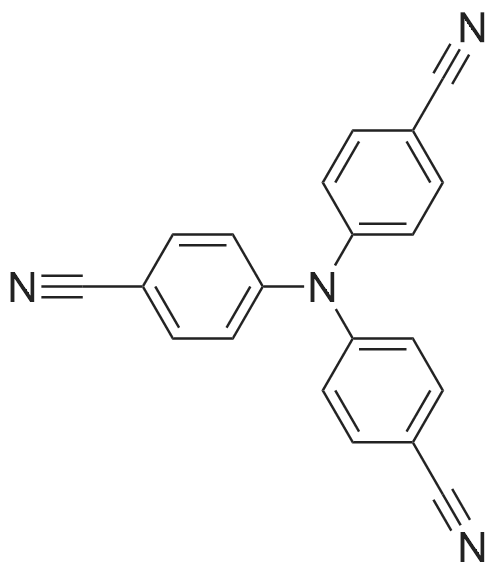
Under nitrogen atmosphere, 6.23 g of tri-(4-iodophenyl)amine and 4.5 g of CuCN were placed in a 250 ml four-necked flask. Then 100 ml of hexamethylphosphoric triamide was added. The reaction was heated at 160°C for 12 hours.After 12 hours, hexamethylphosphoric triamide was distilled off under reduced pressure with oil. Then cool naturally.To this was added a saturated aqueous solution of 50 mL of Na 2 CO 3 and 100 mL of CH 2 Cl 2 for extraction. Then separate the liquid.The organic phase is obtained and washed again. Atmospheric distillation, recovery of dichloromethane. The crude product was digested with dichloromethane Flash column chromatography gave a pale yellow solid, tris-(4-cyanophenyl)amine,Yield 2.57 g, yield 84percent.
tris(4-(4-(2,6-diphenylpyridin-4-yl)styryl)phenyl)amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
75%
With palladium diacetate; triethylamine; triphenylphosphine; In N,N-dimethyl-formamide; at 90℃; for 16h;Inert atmosphere;
A mixture of tris-(4-iodophenyl)amine (6) (200 mg, 0.32 mmol), PPh3 (4 mg, 0.016 mmol) and 2,6-diphenyl-4-(4-vinylphenyl)pyridine (1) (373 mg, 1.12 mmol) were dissolved in dry DMF (6 mL) and stirred for 15 min in argon atmosphere. Catalytic amount of Pd(OAc)2 (10 mg) and dry Et3N (2 mL) were added to the reaction mixture and heated for 16 h at 90 °C. The mixture was cooled to room temperature and extracted with dichloromethane (2.x.15 mL). The organic layer was washed with water (3.x.10 mL), brine (5 mL), and dried over anhydrous Na2SO4. Solvent was removed in a rotary evaporator and the crude product was purified using column chromatography (silica gel/ethyl acetate:petroleum ether 25:75) to yield the dendrimer TPA-TPP1 (297 mg, 75percent) as yellow solids. Mp: 91-92 °C; IR (KBr, cm-1): 3035, 2918, 1598, 1505; 1H NMR (500 MHz, CDCl3): delta 8.22 (d, 12H, J = 6.5 Hz), 7.91 (s, 6H), 7.76 (d, 6H, J = 7.0 Hz), 7.66 (d, 6H, J = 7.5 Hz), 7.61-7.42 (m, 24H), 7.24-7.07 (m, 12H); 13C NMR (100 MHz, CDCl3): delta 157.5, 149.5, 146.8, 139.5, 138.3, 137.6, 132.0, 129.6, 129.0, 128.7, 127.6, 127.4, 127.1, 127.0, 126.5, 124.3, 120.6, 116.6, 115.2; MALDI-TOF MS: m/e 1239.3 (MH+, 100percent).
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; In tetrahydrofuran; at 20℃; for 24h;Inert atmosphere;
To a mixture of tris-(4-iodophenyl)amine (6) (200 mg, 0.320 mmol), Pd(PPh3)2Cl2 (14 mg, 0.02 mmol), and copper iodide (2 mg, 0.01 mmol) was added THF (8 mL) under argon atmosphere and the suspension was stirred at room temperature for 15 min. Triethylamine (2 mL) was added to the reaction mixture and stirred for 0.5 h at room temperature. A solution of 4-(4-ethynylphenyl)-2,6-diphenylpyridine (2) (371 mg, 1.12 mmol) in THF (3 mL) was added to the reaction mixture and stirred for 24 h at room temperature. Solvent was removed in a rotary evaporator and the crude product was purified using column chromatography (silica gel/ethyl acetate:petroleum ether 25:75) to yield the dendrimer TPA-TPE2 (339 mg, 86percent) as yellow solids. Mp: 174-175 °C; IR (KBr, cm-1): 3023, 2369, 1598, 1517, 1388; 1H NMR (300 MHz, CDCl3): delta 8.21 (d, 12H, J = 7.2 Hz), 7.90 (s, 6H), 7.76 (d, 6H, J = 8.4 Hz), 7.68 (d, 6H, J = 8.1 Hz), 7.57-7.43 (m, 24H), 7.13 (d, 6H, J = 8.7 Hz); 13C NMR (125 MHz, CDCl3): delta 157.7, 157.6, 149.2, 146.8, 139.4, 139.3, 138.5, 133.2, 132.9, 132.2, 129.2, 129.1, 128.7, 127.3, 127.1, 124.2, 123.7, 117.8, 117.5, 116.8, 90.9, 89.0; MALDI-TOF MS: m/e 1233.4 (MH+).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping