There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
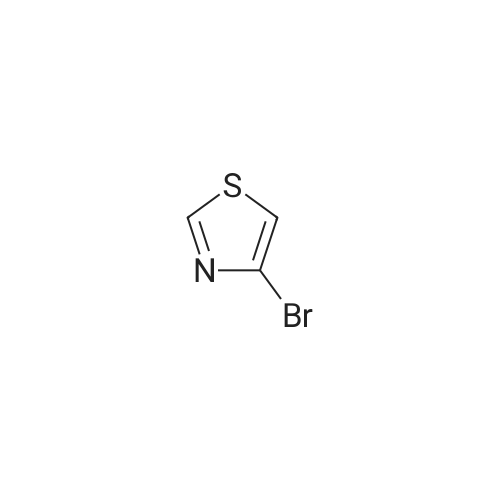
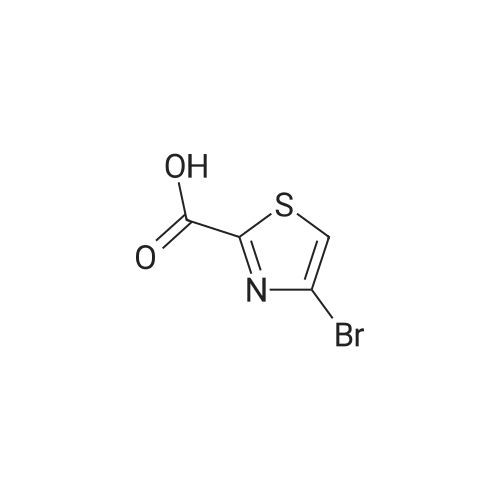
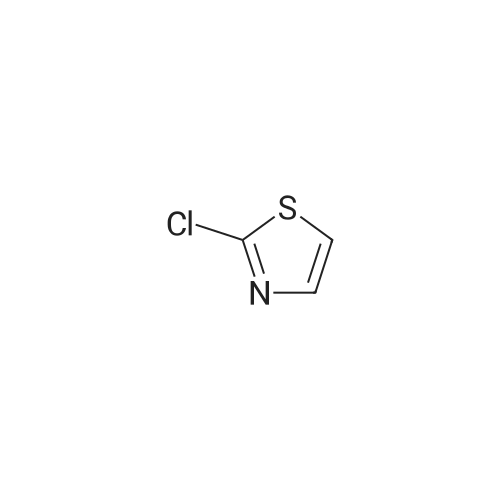
Structure of 4175-77-3 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: With n-butyllithium In diethyl ether; hexane at -78℃; for 0.5 h; Stage #2: With methanol In diethyl ether; hexane at -78 - 20℃; for 16 h;
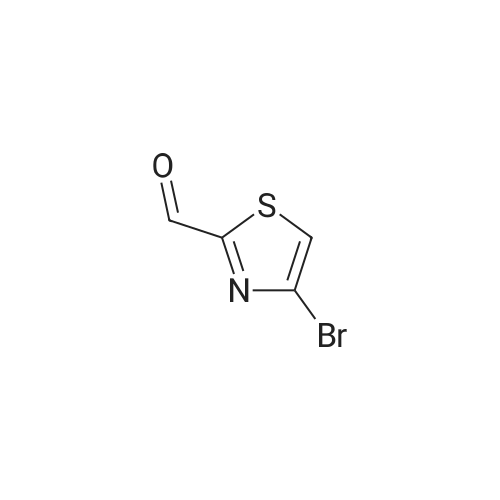
a) Synthesis of 4-bromothiazol A solution of 10.0 g (41.2 mmol) 2,4-dibromothiazol in ether (210 ml) was cooled to -78° C. and 28.3 ml (45.3 mmol, 15percent in hexane) n-butyllithium was added in drops at this temperature. After 30 min of stirring, 3.3 ml (82.3 mmol) methanol was added at -78° C. to the reaction mixture. Heating was subsequently performed to RT over a period of 16 h. The reaction mixture was filtered over silica gel and washed with a n-hexane/AE mixture (2:1). The filtrate was concentrated in a vacuum, whereby 6.7 g (40.9 mmol, 99percent) 4-bromothiazol was obtained.
Reference:
[1] Patent: US2008/261996, 2008, A1, . Location in patent: Page/Page column 44
[2] Journal of Materials Chemistry C, 2017, vol. 5, # 45, p. 11927 - 11936
[3] Journal of Organic Chemistry, 2006, vol. 71, # 10, p. 3754 - 3761
[4] Tetrahedron Letters, 1995, vol. 36, # 51, p. 9293 - 9296
[5] Patent: US4990520, 1991, A,
[6] Patent: WO2008/94737, 2008, A2, . Location in patent: Page/Page column 41-42
[7] Journal of Organic Chemistry, 2017, vol. 82, # 11, p. 5947 - 5951
2
[ 57314-13-3 ]
[ 34259-99-9 ]
[ 4175-77-3 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1992, # 2, p. 215 - 219
[2] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1992, # 2, p. 215 - 219
3
[ 2295-31-0 ]
[ 4175-77-3 ]
Yield
Reaction Conditions
Operation in experiment
71%
Stage #1: at 130℃; for 0.5 h; Stage #2: With sodium carbonate In water at 0℃;
EXAMPLE 41; Preparation of 2-Amino-3-methyl-5-(3-pyrimidin-5-ylphenyl)-5-(1,3-thiazol-4-yl)-3,5-dihydro-4H-imidazol-4-one; Step a); Preparation of Compound 2; A mixture of 2,4-thiazolidinedione 1 (2.28 g, 19.5 mmol) and phosphorous oxybromide (25.0 g, 87.0 mmol) was heated at 130° C. for 30 min, then cooled to room temperature. The reaction was diluted with ice water (300 mL) and carefully neutralized by the addition of solid sodium carbonate in small portions. Once neutral, the mixture was extracted with dichloromethane (3.x.150 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated. Purification of the residue by flash chromatography (silica, 0:100 to 5:95 ethyl acetate/hexanes) afforded 2 (3.34 g, 71percent) as light yellow crystals: 1H NMR (500 MHz, CDCl3) δ 7.21 (s, 1H).
50%
at 80 - 125℃; for 2 h;
2-4-thiazolidinione (10 g, 0.085 mol) was added portionwise to stirred phosphorus oxybromide (160 g) at 80° C. The mixture was then warmed slowly to 125° C. with stirring (vigorous evolution of HBr occurred at approximately 118° C.) and held at 125° C. for 2 hrs. After cooling, the resultant solid was added portionwise to ice/sodium bicarbonate and the brown solid was collected by filtration. The reaction was repeated as above on 10 g and then on 20 g scale, and all three batches of the brown solids were combined. The crude combined material was purified via short path silica gel column chromatograph. The purified product was isolated as a pale yellow solid (41.23 g, 50percent).
47%
at 110℃; for 3 h;
A mixture of 21-1 (5.0 g, 38.42 mmol) and POBr3 (55.07 g, 192.09 mmol) was stirred at 110 °C for 3 h, then cooled to 55 °C and poured onto ice (300 g). Solid Na2CO3 (40g) was added portionwise and the mixture was extracted with EA (150 ml x 3). The combined organic layers were washed with brine (80 ml), dried over Na2SO4, filtered, concentrated and purified by silica gel column chromatography (pure PE to PE : EA = 50 : 1) to afford 21-2 as a white solid (4.86 g, yield 47percent).
47%
at 110℃; for 3 h;
Compound 22-2 (0434) A mixture of 22-1 (5.0 g, 38.42 mmol) and POBr3 (55.07 g, 192.09 mmol) was stirred at 110° C. for 3 h, then cooled to 55° C. and poured onto ice (300 g). Solid Na2CO3 (40 g) was added portionwise and the mixture was extracted with EA (150 mL×3). The combined organic layers were washed with brine (80 mL), dried over Na2SO4, filtered, concentrated and purified by silica gel column chromatography (pure PE to PE:EA=50:1) to afford 22-2 as a white solid (4.86 g, yield 47percent).
Reference:
[1] Journal of Organic Chemistry, 2006, vol. 71, # 10, p. 3754 - 3761
[2] Synthesis, 2012, vol. 44, # 7, p. 1026 - 1029
[3] Journal of Materials Chemistry C, 2017, vol. 5, # 45, p. 11927 - 11936
[4] Patent: US2007/4786, 2007, A1, . Location in patent: Page/Page column 21
[5] Tetrahedron, 2006, vol. 62, # 38, p. 9017 - 9037
[6] Patent: US2004/254236, 2004, A1,
[7] Patent: US2008/234267, 2008, A1, . Location in patent: Page/Page column 17
[8] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 19, p. 5849 - 5853
[9] Patent: US9138427, 2015, B2, . Location in patent: Page/Page column 298-299
[10] Journal of Chemical Crystallography, 2015, vol. 45, # 10-12, p. 461 - 465
[11] Journal of Organic Chemistry, 2017, vol. 82, # 11, p. 5947 - 5951
[12] Journal of Organic Chemistry, 2018, vol. 83, # 2, p. 664 - 671
4
[ 2682-49-7 ]
[ 4175-77-3 ]
Yield
Reaction Conditions
Operation in experiment
57%
at 110℃; for 3 h;
Thiazolidinone (3.43 g, 29.32 mmol) and POBr3 (25 g, 87.96 mmol, 3 equiv.) are mixed under nitrogen as solids. The reaction mixture is then heated to HO0C under stirring for 3 h causing the formation of a black syrup. The reaction mixture is then allowed to cool down to room temperature and a mixture of water/ice (200 mL) is added very cautiously. The resulting grey suspension is extracted with diethyl ether (3 x 50 mL), the organic layers are combined, filtered through a silica plug and evaporated to afford the title compound as an orange oil (4 g, 57percent) which is used in the next step without further purification.
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1992, # 2, p. 215 - 219
[2] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1992, # 2, p. 215 - 219
7
[ 124-38-9 ]
[ 4175-77-3 ]
[ 88982-82-5 ]
Yield
Reaction Conditions
Operation in experiment
82%
Stage #1: With n-butyllithium In diethyl ether at -78℃; for 1 h; Inert atmosphere Stage #2: at -78 - 20℃; Inert atmosphere
To the solution of 2,4-dibromothiazole (50 g, 207 mmol, 1.0 eq.) in Et2O (1000 ml) was added n-BuLi (90 ml, 2.5 M, 1.1 eq.) at −78° C. dropwise and it was stirred for one hour. The reaction solution was poured into dry CO2 at −78° C. and the reaction mixture was warmed to the room temperature. TLC and LCMS showed the reaction was complete. It was quenched with water (100 ml). The Et2O phase was removed. The aqueous phase was adjusted to pH to 2-3 and extracted with ethyl acetate. The organic phase was dried, filtered and concentrated to obtain 35 g (82percent y) of the 4-bromothiazole-2-carboxylic acid. 1HNMR (400 MHz, DMSO): δ8.23 (1H, s)
Reference:
[1] Patent: US2014/179663, 2014, A1, . Location in patent: Paragraph 0174
[2] Journal of Fluorine Chemistry, 1991, vol. 55, # 2, p. 173 - 178
[3] Patent: WO2008/57336, 2008, A2, . Location in patent: Page/Page column 36
[4] Patent: US2015/175597, 2015, A1, . Location in patent: Paragraph 0179; 0180
8
[ 4175-77-3 ]
[ 68-12-2 ]
[ 139669-95-7 ]
Yield
Reaction Conditions
Operation in experiment
35%
Stage #1: With lithium diisopropyl amide In tetrahydrofuran at -78℃; for 0.666667 h; Inert atmosphere Stage #2: at -78℃; for 1 h;
Step 1: 2,4-Dibromothiazole-5-carbaldehyde (25a)To a solution of LDA (1M in THE, 183 mL, 183 mmol) was added a solution of 2,4- dibromothiazole (37 g, 154 mmol) in dry THF (500 mL) at —78°C under N2 and the solution was stirred under this condition for 40 mm. Then DMF (13 g, 178 mmol) was added slowly at this temperature and the solution was stirred for another 1 h, warmed to rt, quenched with sat.NH4CI and extracted with EA twice. The combined organic layers were washed with water and brine, dried over Na2SO4, filtered, concentrated and purified by CC (PE/EA = 15/1) to give compound 25a (14.5 g, 35percent) as a yellow solid.
Reference:
[1] Patent: WO2013/178362, 2013, A1, . Location in patent: Page/Page column 156
[2] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1992, # 2, p. 215 - 219
9
[ 124-38-9 ]
[ 4175-77-3 ]
[ 139669-96-8 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1992, # 2, p. 215 - 219
10
[ 4175-77-3 ]
[ 98-80-6 ]
[ 141305-40-0 ]
Yield
Reaction Conditions
Operation in experiment
84%
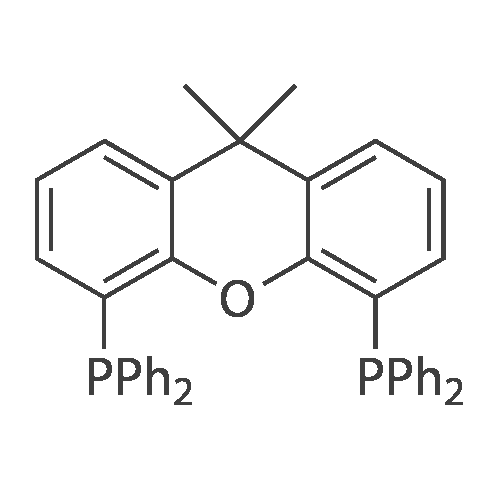
With potassium phosphate; palladium diacetate; [5-(diphenylphosphanyl)-9,9-dimethyl-9H-xanthen-4-yl]diphenylphosphane In tetrahydrofuran at 60℃; for 18 h; Inert atmosphere
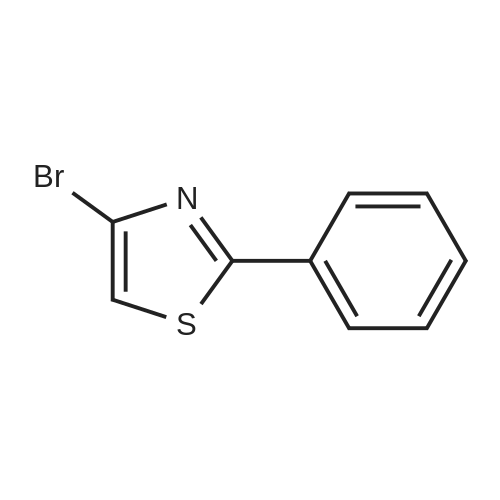
[00287] Under N2 atmosphere Pd(OAc)2 (0.084 g, 0.38 mmol) was combined with xantphos (0.220 g, 0.38 mmol) in 25 mL THF. After stirring for 5 min., the resulting solution was transferred to a round bottom flask containing 2,4 dibromothiazole (3.64 g, 15 mmol), phenylboronic acid (1.96 g, 16 mmol) and Κ3ΡO4 (9.55 g, 45 mmol). The resulting reaction mixture was heated at 60 °C for 18 h and then cooled to room temperature and filtered and washed with dichloromethane. Removal of the solvents under vacuum afforded the crude product which was purified by silica gel column chromatography using hexanes: ethyl acetate to afford 4- bromo-2-phenylthiazole as a white solid (3 g, 84percent). 1HNMR (400 MHz, CDC13): δ 7.95 (dd, J = 3.4 Hz, 8.2 Hz, 2H), 7.47-7.46 (m, 3H), 7.23 (s, 1H). LRMS (ESI) calcd. for C9H6BrNS [M+H]+: 241.12. Found: 241.00.
66%
With potassium phosphate; [5-(diphenylphosphanyl)-9,9-dimethyl-9H-xanthen-4-yl]diphenylphosphane In tetrahydrofuran at 60℃; for 18 h; Inert atmosphere
Frame-dried 200 mL four-necked flask was purged with nitrogen and Xantphos (Xantphos, 220 mg, 0.38 mmol),Tris (dibenzylideneacetone) dipalladium (0) (Pd 2 (dba) 3,840 mg, 0.38 mmol) and dry tetrahydrofuran (dry THF, 75 mL) were added and stirred for 5 minutes, Thereby preparing a THF solution.Another flame-dried 200 mL four-necked flask was purged with nitrogen, 2,4-dibromothiazole (Compound 101 in the following formula (A), 3.6 g, 15 mmol),Phenylboronic acid (1.9 g, 16 mmol),Triopotassium phosphate (9.6 g, 45 mmol),The above THF solution was added, And the mixture was heated under reflux at 60 ° C. for 18 hours.After returning to room temperature,Celite filtration was carried out using dichloromethane. Removal of the solvent and purification by column chromatography gave the compound of the following formula (A), And 2.4 g of a white solid compound represented by "102" (yield: 66percent).
0.71 g
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In ethanol; water; toluene for 6 h; Inert atmosphere; Reflux
A flask containing 238 mg of tetrakis(triphenylphosphine)palladium, 0.55 g of phenylboronic acid, and 1.00 g of 2,4-dibromothiazole was purged with nitrogen and then charged with 30 ml of toluene, 6.1 ml of ethanol, and 9.1 ml of a 2 M aqueous solution of sodium carbonate, and the mixture was stirred under reflux for 6 hours. After cooling to room temperature, 50 ml of water was added to the reaction mixture and two extractions were conducted with 50 ml of ethyl acetate. After being washed with 30 ml of saturated brine, the organic layer was dried over magnesium sulfate. After filtering the magnesium sulfate, the organic layer was concentrated and the residue was purified by column chromatography (Wakogel C-200; hexane:ethyl acetate=14:1) to give 0.71 g of 2-phenyl-4-bromothiazole.
Reference:
[1] Patent: WO2015/191630, 2015, A1, . Location in patent: Paragraph 00287
[2] Journal of Organic Chemistry, 2010, vol. 75, # 5, p. 1733 - 1739
[3] Patent: JP2018/140974, 2018, A, . Location in patent: Paragraph 0067; 0068
[4] Patent: EP2455371, 2012, A1, . Location in patent: Page/Page column 22
[5] Patent: US2013/296271, 2013, A1, . Location in patent: Paragraph 0175
11
[ 591-51-5 ]
[ 4175-77-3 ]
[ 141305-40-0 ]
Reference:
[1] Journal of Organic Chemistry, 2002, vol. 67, # 16, p. 5789 - 5795
[2] Synthesis, 2011, # 2, p. 199 - 206
12
[ 4175-77-3 ]
[ 161265-03-8 ]
[ 141305-40-0 ]
Reference:
[1] Chemistry - A European Journal, 2015, vol. 21, # 23, p. 8471 - 8482
13
[ 4175-77-3 ]
[ 68-12-2 ]
[ 167366-05-4 ]
Reference:
[1] Angewandte Chemie - International Edition, 1998, vol. 37, # 1-2, p. 84 - 87
[2] Journal of Medicinal Chemistry, 2007, vol. 50, # 14, p. 3256 - 3266
[3] Bioorganic and Medicinal Chemistry, 1999, vol. 7, # 5, p. 665 - 697
[4] Patent: WO2009/77990, 2009, A1, . Location in patent: Page/Page column 71
[5] Patent: US2010/298388, 2010, A1, . Location in patent: Page/Page column 14
[6] Patent: WO2010/143116, 2010, A1, . Location in patent: Page/Page column 46
[7] Patent: EP2264017, 2010, A1, . Location in patent: Page/Page column 119
14
[ 4394-85-8 ]
[ 4175-77-3 ]
[ 167366-05-4 ]
Yield
Reaction Conditions
Operation in experiment
61%
Stage #1: With TurboGrignard In tetrahydrofuran at -78℃; for 0.25 h; Inert atmosphere Stage #2: at -78 - 0℃; for 1.66667 h; Inert atmosphere
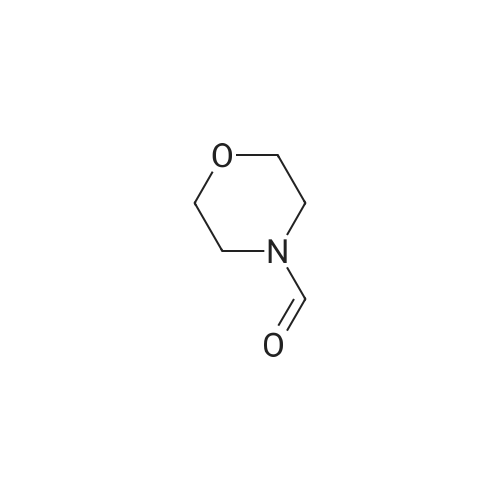
General procedure: The reaction was carried out in a sealed tube under N2 using THF (4 mL) as the solvent. To the solution of tert-butyl 2-bromothiazole-4-carboxylate 1a (0.397 mmol), iPrMgCl*LiCl (1.3 M, 0.52 mmol) was added dropwise at -78 °C. After 15 min the corresponding aldehyde (0.79-0.99 mmol) or N-formyl-morpholine (in the case of 2-formylthiazoles 3a-b) was added dropwise under stirring and the reaction mixture was stirred at -78 °C for 10 min, then 1.5 h at 0 °C. Saturated aqueous NH4Cl was added and the aqueous layer was extracted with Et2O (3 .x. 10 mL). The combined organic layers were washed with 5percent aqueous HCl (10 mL), brine (10 mL), dried over MgSO4 and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel using a mixture of EtOAc:P (petroleum ether) as eluent (see below) to give tert-butyl 2-(1-hydroxymethyl)thiazole-4-carboxylate derivatives rac-2a-h.
Reference:
[1] Russian Journal of Organic Chemistry, 2010, vol. 46, # 11, p. 1702 - 1708
[2] European Journal of Organic Chemistry, 2003, # 6, p. 1042 - 1049
[3] Organic and Biomolecular Chemistry, 2018, vol. 16, # 9, p. 1465 - 1479
[4] MedChemComm, 2013, vol. 4, # 3, p. 520 - 526
16
[ 67-56-1 ]
[ 201230-82-2 ]
[ 4175-77-3 ]
[ 1025468-06-7 ]
Yield
Reaction Conditions
Operation in experiment
13%
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; triethylamine In N,N-dimethyl-formamide at 70℃; for 3 h; Sealed tube
General procedure: To a nitrogen-purged solution of aryl iodide in TEA (3rriLlmmol), DMF (3mL/mmol) and MeOH (3mL/mmol) was added Palladium (II)Acetate (0.O3eq) and Xantphos (0.O6eq). The reaction mixture was flushed with Carbon Monoxide gas for several minutes and then sealed with CO balloon attached and heated to 60°C for 3 hours. Upon completion, the reaction was cooled to room temperature and the crude product was triterated via addition of water and collected by filtration. The crude interemediate was taken into the next step w/o further purification.Similar to the procedure as described in General Procedure O, 2,4-dibromo-l,3- thiazole was reacted with carbon monoxide to give the title compound (1.2 g, 13percent) as a Hi yellow solid. LC-MS (ES, m/z): 222 [M+H]+.
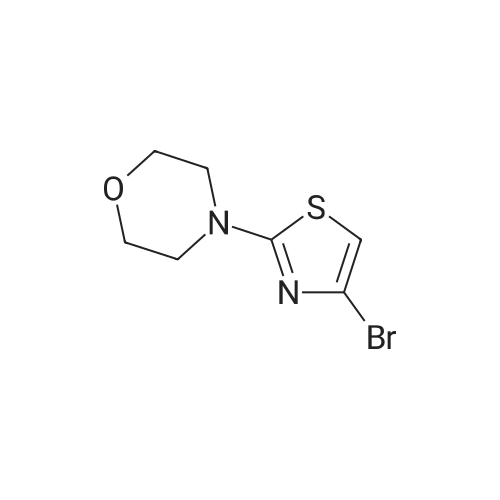
Example 89: 2,2,2-Trifluoro-l-(2-morpholin-4-yl-thiazol-4-yl)ethanone; A solution of 2,4-dibromothiazole (500 mg, 2.06 mmol) in morpholine (4.0 mL, 22.3 mmol) was warmed at 500C in a sealed tube. After 16 hours, the mixture was cooled to room temperature, diluted with 20 mL of water and extracted with three 30 mL portions of diethyl ether. The combined organic layers was washed with five 30 mL portions of water, 30 mL of brine, dried over magnesium sulfate, filtered, and concentrated in vacuo. The residue was chromatographed over SiO2 using ethyl acetate in hexanes (0-30 percent gradient) to afford 458 mg (89percent) of4-(4-bromothiazol-2-yl)morpholine.
Reference:
[1] Patent: WO2008/70507, 2008, A2, . Location in patent: Page/Page column 179
[2] Patent: WO2016/206101, 2016, A1, . Location in patent: Page/Page column 91; 92
[3] Chemical Biology and Drug Design, 2018, vol. 91, # 1, p. 172 - 180
18
[ 4175-77-3 ]
[ 74-88-4 ]
[ 1206708-88-4 ]
Yield
Reaction Conditions
Operation in experiment
56%
Stage #1: With n-butyllithium; diisopropylamine In tetrahydrofuran; hexanes at -78 - -70℃; Stage #2: at -78 - 20℃;
Diisopropylamine (1.375 g, 1.904 mL, 13.58 mmol) in THF (50 mL) was cooled to -78 0C before BuLi (5.432 mL, 2.5 M in hexanes, 13.58 mmol) was added dropwise, maintaining temperature below -70 0C. On complete addition, mixture allowed to 0 0C by removing cardice bath and putting RBF into ice-water bath. Mixture then cooled back down to -78 0C before 2,4- dibromothiazole (3 g, 12.35 mmol) in THF (10 mL) was added dropwise at -78 0C (temperature did not exceed -70 0C). On complete addition, stirred at -78 0C for a further 30 mins. iodomethane (1.928 g, 845.6 L, 13.58 mmol) added dropwise, mixture stirred at -78 0C for 30 mins and then allowed to warm to room temperature over 18 h. Quenched by addition of water, diluted with EtOAc and organic layer collected. Organics then washed with brine, dried (MgS?4), filtered and concentrated in vacuo. Crude product purified twice by flash chromatography (80g SiO2, 0 to 10percent EtOAc/petrol) then (80 g SiO2, 0 to 2.5percent EtOAC/petrol) to leave impure product as a colourless oil which turned to colourless solid on standing (2.7 g, 56percent based on 66percent purity); 1H NMR DMSO-d6 ? 2.35 (s, 3H); ES+ 257.80.
To the solution of <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (50 g, 207 mmol, 1.0 eq.) in Et2O (1000 ml) was added n-BuLi (90 ml, 2.5 M, 1.1 eq.) at ?78° C. dropwise and it was stirred for one hour. The reaction solution was poured into dry CO2 at ?78° C. and the reaction mixture was warmed to the room temperature. TLC and LCMS showed the reaction was complete. It was quenched with water (100 ml). The Et2O phase was removed. The aqueous phase was adjusted to pH to 2-3 and extracted with ethyl acetate. The organic phase was dried, filtered and concentrated to obtain 35 g (82percent y) of the 4-bromothiazole-2-carboxylic acid. 1HNMR (400 MHz, DMSO): delta8.23 (1H, s)
A solution of <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (5g, 20.6 mmol) in anhydrous Et2O (30ml) was added in a dropwise manner to a solution of butyl lithium (9.9ml of a 2.5M solution in hexanes, 24.7mmol) in anhydrous Et2O (70ml) cooled at -780C, at such a rate that the temperature did not15 rise above at -730C. After addition was complete the mixture was stirred at at -780C for 1 hour. Carbon dioxide gas was bubbled through the mixture for 5 mins than a pellet (~5g) of solid carbon dioxide added and the mixture allowed to warm to room temperature. Water (100 ml) added and the aqueous layer extracted further with Et2O. The aqueous layer was acidified with cone. HCl and extracted with Et2O (3 x 100 ml), combined Et2O layers dried over Na2SO4,20 filtered and evaporated. The residue was crystallized from Et2O/Hexanes to give of the title compound. 1HNMR (500 MHz, DMSO-d6) delta: 8.20 (s, IH).
Step 1: Preparation of 4-bromothiazole-2-carboxylic acid To the solution of <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (50 g, 207 mmol, 1.0 eq.) in Et2O (1000 mL) was added n-BuLi (90 mL, 2.5 M, 1.1 eq.) at -78° C. dropwise and it was stirred for one hour. The reaction solution was poured into dry CO2 at -78° C. and the reaction mixture was warmed to the room temperature. TLC and LCMS showed the reaction was complete. It was quenched with water (100 ml). The Et2O phase was removed. The aqueous phase was adjusted to pH to 2-3 and extracted with ethyl acetate. The organic phase was dried, filtered and concentrated to obtain 4-bromothiazole-2-carboxylic acid. 1HNMR (400 MHz, DMSO): delta 8.23 (1H, s).
Step 1: 2,4-Dibromothiazole-5-carbaldehyde (25a)To a solution of LDA (1M in THE, 183 mL, 183 mmol) was added a solution of 2,4- dibromothiazole (37 g, 154 mmol) in dry THF (500 mL) at -78C under N2 and the solution was stirred under this condition for 40 mm. Then DMF (13 g, 178 mmol) was added slowly at this temperature and the solution was stirred for another 1 h, warmed to rt, quenched with sat.NH4CI and extracted with EA twice. The combined organic layers were washed with water and brine, dried over Na2SO4, filtered, concentrated and purified by CC (PE/EA = 15/1) to give compound 25a (14.5 g, 35%) as a yellow solid.
a) Synthesis of 4-bromothiazol A solution of 10.0 g (41.2 mmol) 2,4-dibromothiazol in ether (210 ml) was cooled to -78° C. and 28.3 ml (45.3 mmol, 15percent in hexane) n-butyllithium was added in drops at this temperature. After 30 min of stirring, 3.3 ml (82.3 mmol) methanol was added at -78° C. to the reaction mixture. Heating was subsequently performed to RT over a period of 16 h. The reaction mixture was filtered over silica gel and washed with a n-hexane/AE mixture (2:1). The filtrate was concentrated in a vacuum, whereby 6.7 g (40.9 mmol, 99percent) 4-bromothiazol was obtained.
With sodium hydroxide; In dichloromethane; acetic acid;
Reference Example 2 4-Bromothiazole Following the procedure of M. Robba and R. C. Moreau, Annales pharm. franc. 22, #3, 201-210 (1965); 10 g of 2,4-dibromothiazole and 5 g of powdered zinc in 40 ml of glacial acetic acid is stirred at 60°-65° C. for 45 minutes. The mixture is cooled in an ice bath, as 40 ml of 10 N sodium hydroxide is added in portions. Stirring is continued for 30 minutes. Ten milliliters of 10 N sodium hydroxide is added and the reaction is extracted with diethyl ether followed by methylene chloride. The combined organic layers are dried over sodium sulfate, filtered and concentrated in vacuo to give 5.4 g of the product as a yellow oil.
2,4-Dibromothiazole (5.00 g, 20.7 mmol) is placed in a flask which has been back filled with Argon three times. Anhydrous ether (82 mL) is added and the solution is cooled to -78°C. n-Butyllithium (2.5 M in cyclohexane, 10.0 mL) is added and the reaction mixture is stirred for 90 minutes at -78°C before quenching with HCl/ether solution (2.0 m x 15 mL). The reaction mixture is warmed to room temperature. The mixture is washed with NaHCpsi3 (saturated aqueous solution, 60 mL) and the organic phase is dried with Na2SO4. After evaporation, 4-bromothiazole is obtained as a crude product.
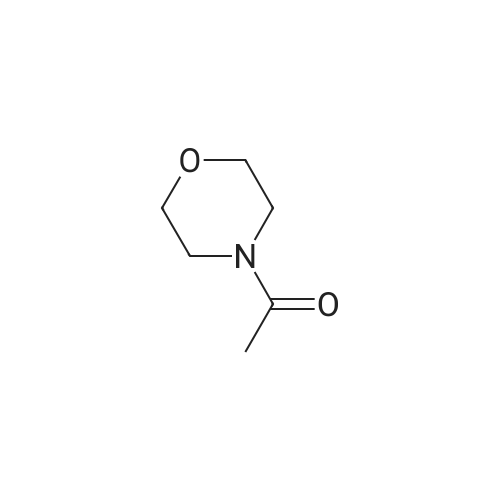
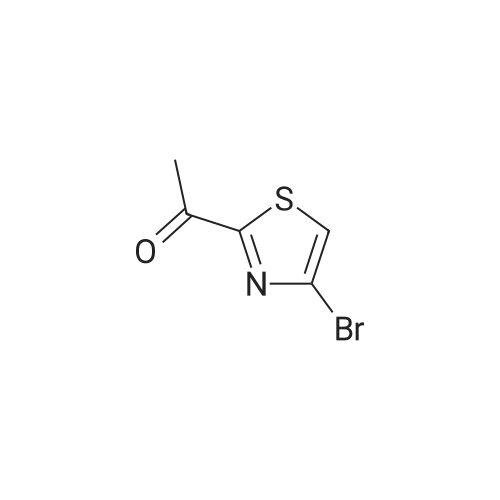
A solution of 2,4-dibromothiazole B.31 (5.0128 g, 20.63 mmol) in ether (52 mL) was cooled to -78 0C. To the cooled solution was added n-BuLi (1.6 M sol. in hexane, 14.2 mL, 22.72 mmol) and the mixture was stirred at -78 0C for 30 minutes. To the cooled mixture was then added dropwise N-acetylmorpholine (3.1 mL, 26.83 mmol). The mixture was stirred at -78 0C for 1.5 hours and then room temperature for 18 hours. The mixture was diluted with ether (200 mL), washed with saturated aqueous NaHCO3 (100 mL x 1), dried over MgSO4, filtered, and concentrated under reduced pressure. The product was purified by silica gel column chromatography using 0% to 50% gradient of ethyl acetate in hexane as eluent to give 1 -(4- bromothiazol-2-yl)ethanone B.32 (3.383 g, 79.5% yield): 1H NMR (500 MHz, CHLOROFORM-d) 5 ppm 7.59 (1 H, s), 2.73 (3 H, s); Mass Spectrum (ESI) m/e = 205.9 [M+l (79Br)] and 207.9 [M+l (81Br)].
50%
To a solution of compound 21-2 (3.0 g, 12.35 mmol) in THF (25 ml) was added dropwise n-BuLi (2.5 M in hexane, 2.5 ml) at -78 C. After addition, the reaction mixture was stirred at -78 C for 30 min. N-acetyl morpholine (1.9 ml, 16.06 mmol) was added dropwise during 15 min at -78 C. After addition, the reaction mixture was stirred at -78 C for 4 h, quenched with saturated NaHCO3 (15 ml) and extracted with ethyl acetate (25 ml x 4). The combined organic layers were washed with brine (30 ml), dried over Na2SO4, filtered, concentrated and purified by silica gel column chromatography (PE : EA = 50 : 1 to 10 : 1) to afford 21-3 as a white solid (1.28 g, yield 50%).
50%
Compound 22-3 (0435) To a solution of compound 22-2 (3.0 g, 12.35 mmol) in THF (25 mL) was added dropwise n-BuLi (2.5 M in hexane, 2.5 mL) at -78 C. After addition, the reaction mixture was stirred at -78 C. for 30 min. N-acetyl morpholine (1.9 mL, 16.06 mmol) was added dropwise during 15 min at -78 C. After addition, the reaction mixture was stirred at -78 C. for 4 h, then quenched with sat. NaHCO3 (15 mL) and extracted with ethyl acetate (25 mL×4). The combined organic layers were washed with brine (30 mL), dried over Na2SO4, filtered, concentrated and purified by silica gel column chromatography (PE:EA=50:1 to 10:1) to afford 22-3-3 as a white solid (1.28 g, yield 50%).
33%
To a stirring solution of 2, 4-dibromothiazole 84 (50 g, 205.82 mmol) in anhydrous THF (500 mL) under inert atmosphere was added n-butyllithium (193 mL, 308.74 mmol) dropwise for 30 min at -40 oC and stirred for 1 h at the same temperature. To this was added 1- morpholinoethan-1-one (32 g, 248 mmol) in anhydrous THF (100 mL) dropwise for 20 min at -40 oC and stirred for 3 h. The reaction was monitored by TLC; after completion of the reaction, the reaction mixture was quenched with saturated ammonium chloride solution and extracted using EtOAc. The combined organic extracts were dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to obtain the crude. The crude was purified through silica gel column chromatography using 1-2% EtOAc/ hexanes to afford compound 85 (14 g, 33%) as an off-white solid. TLC: 10% EtOAc/ hexanes (Rf: 0.8); 1H NMR (DMSO-d6, 400 MHz): delta 8.33 (s, 1H), 2.62 (s, 3H); LCMS Calculated for C5H4BrNOS: 204.92; LCMS observed: 208.0 (M+2)+.
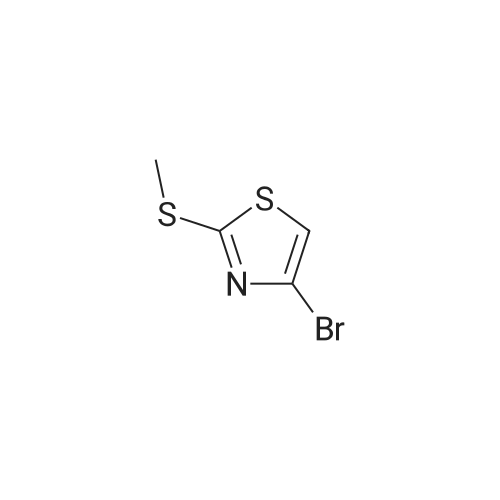
Methylthio thiazole 33: Methylthio thiazole 33 was prepared from commercially available 2,4- dibromothiazole (32) as previously described (Nicolaou, et al , 1998). The physical and spectral data are consistent with those reported (Nicolaou,
88%
In ethanol; at 0 - 25℃; for 6h;
To a stirred solution of <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> SI (1.97 g, 8.11 mmol, 1.0 equiv) in ethanol (10 mL) at 0 °C was added sodium thiomethoxide (1.70 g, 24.3 mmol, 3.0 equiv), and the reaction mixture was allowed to slowly warm to 25 °C. After 6 h, the reaction mixture was quenched with water (30 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were dried with anhydrous sodium sulfate and concentrated under reduced pressure. The obtained residue was purified by flash column chromatography (silica gel, 2? > 8percent ethyl acetate in hexanes) to afford pure methylthiothiazole S2 (Nicolaou, et al., 1998; Nicolaou, et al., 1999; and Nicolaou, et al. , 2002) (1.50 g, 7.14 mmol, 88percent yield) as a white amorphous solid. S2: = 0.43 (silica gel, 10percent ethyl ether in hexanes); FT-IR (film) vmax 3006, 2990, 1275, 1260, 764, 750 cm"1; NMR (600 MHz, CDC13) 5 = 7.07 (s, 1 H), 2.70 (s, 3 H) ppm; 13C NMR (151 MHz, CDCI3) 5 = 168.1, 124.5, 115.7, 16.8 ppm.
In a flame dried round-bottomed flask equipped with a magnetic stir bar and under inert atmosphere (N2), a solution of commercially available 2,4-dibromo-thiazole (3.50 g, 14.41 mmol) in dry Et2O (120 ml.) was treated with n-BuLi (5.9 ml. of a 2.5M solution in hexanes, 14.72 mmol) at -78 0C. The reaction mixture was stirred at this temperature for 30 min. lambda/,lambda/-Dimethylformamide (1.35 ml_, 14.47 mmol) was then added and the mixture allowed to warm to rt over a period of 1 h. The reaction was quenched by the addition of sat. aq. NH4CI (50 ml_). The layers were separated and the aq. layer extracted with Et2O (3 x 50 ml_). The combined org. extracts dried over Na2SO4, filtered, and the solvents were removed under reduced pressure. Purification of the residue by FC (10:1 -> 3:1 hept-EA) gave the title compound as a pale yellow solid. TLC: rf (1 :1 hept-EA) = 0.21. LC-MS-conditions 02: tR = 0.81 min.
With n-butyllithium; In diethyl ether; at -78 - 20℃; for 1.25h;
To a solution of 2,4-dibromothiazole (2.50 g, 10.28 mmol) in diethyl ether (50 ml) cooled to -78 C. was added n-butyl lithium (1.40 M, 8.80 ml, 12.38 mmol) and the resulting reaction mixture was stirred for 15 min at the same temperature followed by addition of DMF (5.0 ml, 64.30 mmol). The reaction mass was then allowed to come to room temperature and stirred for 1 h. After the completion of the reaction (TLC monitoring), the reaction mass was cooled to 0 C. and quenched with saturated NH4Cl solution (aqueous). Water was then added to the reaction mass and extracted with diethyl ether (3×100 ml). The combined organics was then dried over anhydrous Na2SO4, filtered and concentrated to get the desired product (2.10 g, quantitative crude yield) that was carried forward to the next step without further purification.
4-Bromo-thiazole-2-carbaldehyde:; In a flame dried round-bottomed flask equipped with a magnetic stir bar and under inert atmosphere (N2), a solution of commercially available 2,4-dibromo-thiazole (3.50 g, 14.41 mmol) in dry Et2O (120 mL) was treated with n-BuLi (5.9 mL of a 2.5M solution in hexanes, 14.72 mmol) at -78 0C. The reaction mixture was stirred at this temperature for 30 min. Lambda/,Lambda/-Dimethylformamide (1.35 mL, 14.47 mmol) was then added and the mixture allowed to warm to rt over a period of 1 h. The reaction was quenched by the addition of sat. aq. NH4CI (50 mL). The layers were separated and the aq. layer extracted with Et2O (3 x 50 mL). The combined org. extracts dried over Na2SO4, filtered, and the solvents were removed under reduced pressure. Purification of the residue by FC (10:1 ->; 3:1 hept-EA) gave the title compound as a pale yellow solid. TLC: rf (1 :1 hept-EA) = 0.21. LC-MS-conditions 02: tR = 0.81 min.
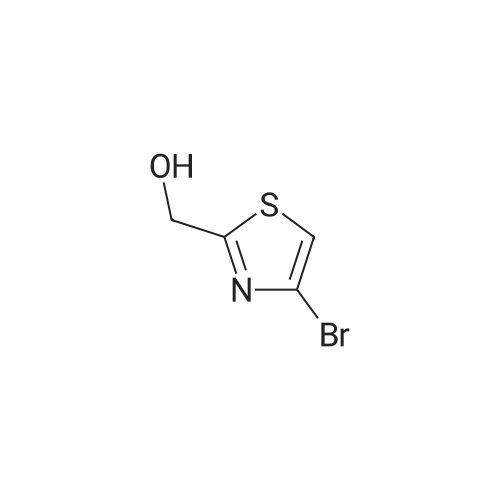
(Example 30a) (4-bromo-1,3-thiazol-2-yl)methanol [Show Image] Under a nitrogen atmosphere at -78C, to a solution (20 mL) of 2,4-dibromothiazole (5 g, 20.6 mmol) in diethyl ether was added n-butyllithium (1.6 M hexane solution, 15.4 mL, 24.7 mmol), and the mixture was stirred at the same temperature for 30 min. N,N-Dimethylformamide (2.3 g, 30.9 mmol) was added to the reaction mixture at -78C, and the mixture was gradually warmed to room temperature. After confirmation of the termination of the reaction by TLC, hexane was added. The resulting salt was filtered and the solvent was evaporated under reduced pressure to give 4-bromo-1,3-thiazole-2-carbaldehyde as a crude product. 1H NMR (400 MHz, CHLOROFORM-d) delta ppm 7.68 (1 H, d, J=1.0 Hz), 9.95 (1 H, d, J=1.2 Hz)
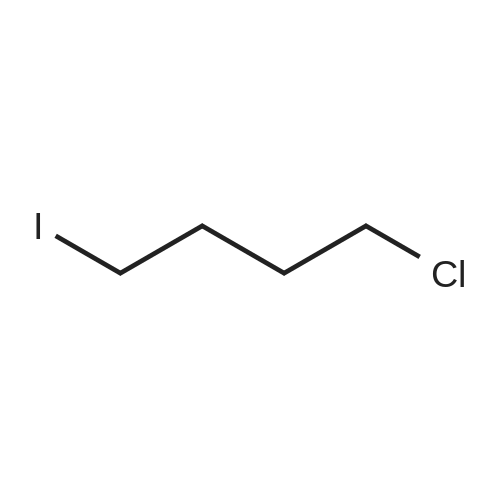
Stage #1: 1-chloro 4-iodobutane With tert.-butyl lithium In diethyl ether; pentane at -78℃; for 1h;
Stage #2: With zinc(II) chloride In tetrahydrofuran; diethyl ether; pentane at 20℃; for 0.5h;
Stage #3: 2,4-dibromo-1,3-thiazole With 1,1'-bis-(diphenylphosphino)ferrocene In tetrahydrofuran; diethyl ether; pentane at 20℃; for 16h; Further stages.;
80%
Stage #1: 1-chloro 4-iodobutane With tert.-butyl lithium In diethyl ether; pentane at -78℃; for 1h;
Stage #2: With zinc(II) chloride In tetrahydrofuran; diethyl ether; pentane at -78 - 20℃;
Stage #3: 2,4-dibromo-1,3-thiazole With 1,1'-bis-(diphenylphosphino)ferrocene; tris-(dibenzylideneacetone)dipalladium(0) In tetrahydrofuran; diethyl ether; pentane at 20℃; for 16h; regioselective reaction;
Stage #1: 2,4-dibromo-1,3-thiazole With tert.-butyl lithium In diethyl ether; hexane at -78℃; for 0.5h;
Stage #2: acetone In diethyl ether; hexane at -78℃; for 1h;
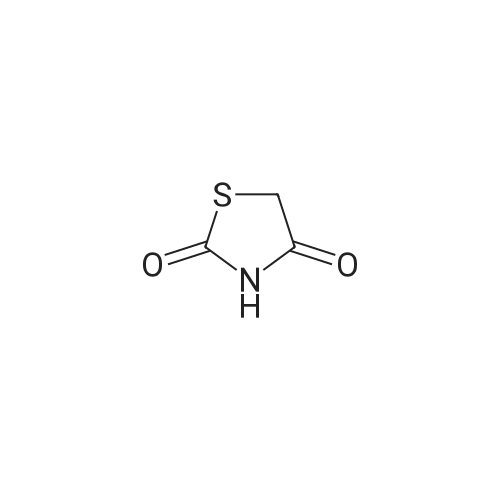
EXAMPLE 41; Preparation of 2-Amino-3-methyl-5-(3-pyrimidin-5-ylphenyl)-5-(1,3-thiazol-4-yl)-3,5-dihydro-4H-imidazol-4-one; Step a); Preparation of Compound 2; A mixture of 2,4-thiazolidinedione 1 (2.28 g, 19.5 mmol) and phosphorous oxybromide (25.0 g, 87.0 mmol) was heated at 130 C. for 30 min, then cooled to room temperature. The reaction was diluted with ice water (300 mL) and carefully neutralized by the addition of solid sodium carbonate in small portions. Once neutral, the mixture was extracted with dichloromethane (3×150 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated. Purification of the residue by flash chromatography (silica, 0:100 to 5:95 ethyl acetate/hexanes) afforded 2 (3.34 g, 71%) as light yellow crystals: 1H NMR (500 MHz, CDCl3) delta 7.21 (s, 1H).
55%
In methanol; phosphoryl(III) bromide; dichloromethane; ethyl acetate; acetone;
A. 2,4-Dibromo-thiazole A stirred solution of 2,4-thiazolidinedione (5 g, 42.7 mmol) in phosphorus oxybromide (25 g, 87.2 mmol) was heated to 110 C. for 3 hours. After cooling to room temperature, 250 g of crushed ice was added (CAUTION). The precipitate that is formed was filtered and dissolved in a mixture of acetone/methylene chloride/MeOH. The remaining aqueous phase was extracted with ether and methylene chloride. The combined organic phases were dried over MgSO4 and the volatiles removed in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc 20% to 100% in hexanes) to provide 2,4-dibromo-thiazole 26A as a yellow solid (5.36 g, 55% yield).
50%
With phosphorus(V) oxybromide; at 80 - 125℃; for 2h;
2-4-thiazolidinione (10 g, 0.085 mol) was added portionwise to stirred phosphorus oxybromide (160 g) at 80 C. The mixture was then warmed slowly to 125 C. with stirring (vigorous evolution of HBr occurred at approximately 118 C.) and held at 125 C. for 2 hrs. After cooling, the resultant solid was added portionwise to ice/sodium bicarbonate and the brown solid was collected by filtration. The reaction was repeated as above on 10 g and then on 20 g scale, and all three batches of the brown solids were combined. The crude combined material was purified via short path silica gel column chromatograph. The purified product was isolated as a pale yellow solid (41.23 g, 50%).
47%
With phosphorus(V) oxybromide; at 110℃; for 3h;
A mixture of 21-1 (5.0 g, 38.42 mmol) and POBr3 (55.07 g, 192.09 mmol) was stirred at 110 C for 3 h, then cooled to 55 C and poured onto ice (300 g). Solid Na2CO3 (40g) was added portionwise and the mixture was extracted with EA (150 ml x 3). The combined organic layers were washed with brine (80 ml), dried over Na2SO4, filtered, concentrated and purified by silica gel column chromatography (pure PE to PE : EA = 50 : 1) to afford 21-2 as a white solid (4.86 g, yield 47%).
47%
With phosphorus(V) oxybromide; at 110℃; for 3h;
Compound 22-2 (0434) A mixture of 22-1 (5.0 g, 38.42 mmol) and POBr3 (55.07 g, 192.09 mmol) was stirred at 110 C. for 3 h, then cooled to 55 C. and poured onto ice (300 g). Solid Na2CO3 (40 g) was added portionwise and the mixture was extracted with EA (150 mL×3). The combined organic layers were washed with brine (80 mL), dried over Na2SO4, filtered, concentrated and purified by silica gel column chromatography (pure PE to PE:EA=50:1) to afford 22-2 as a white solid (4.86 g, yield 47%).
With phosphorus pentoxide; tetrabutylammomium bromide; In toluene;Reflux;
This compound was prepared by reaction of commercially available thiazolidine-2,4-dione 5 with P2O5 and Bu4NBr in boiling toluene [3]. It was obtained directly as colourless crystals suitable for X-ray diffraction which gave the following data: mp 81-82 C (81.1-82.4 C [3]; 1H NMR(CDCl3) d 7.21 (1H, s); 13C NMR d 120.8 (CH), 124.3 (C-4) and 136.3 (C-2).
Thiazolidinone (3.43 g, 29.32 mmol) and POBr3 (25 g, 87.96 mmol, 3 equiv.) are mixed under nitrogen as solids. The reaction mixture is then heated to HO0C under stirring for 3 h causing the formation of a black syrup. The reaction mixture is then allowed to cool down to room temperature and a mixture of water/ice (200 mL) is added very cautiously. The resulting grey suspension is extracted with diethyl ether (3 x 50 mL), the organic layers are combined, filtered through a silica plug and evaporated to afford the title compound as an orange oil (4 g, 57%) which is used in the next step without further purification.
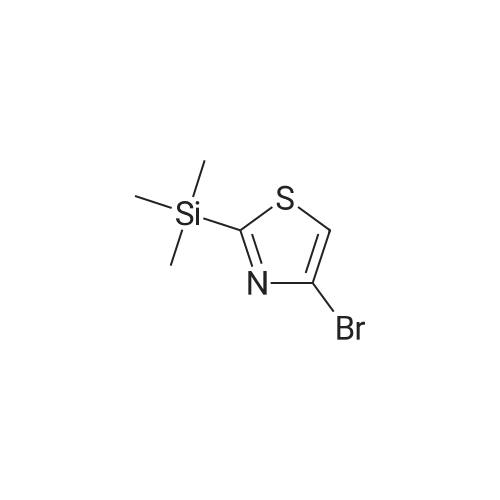
Example 1: (Referring to compounds as numbered in Schemes I and II) Synthesis of 7a: A) 4-Bromo-2-(trimethyl-silanyl)-thiazole (2). To a stirred solution of n-BuLi (2M in pentane; 12.35 ML, 0.0247 mol) in 70 mL of dry ether AT-78°C was added dropwise <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> in 30 mL of ether over a period of 30 minutes. The mixture was stirred for Ih, then TMSC1 (2. 87 mL, 0.0226 mol) was added dropwise over 10 minutes. After lh of stirring at-78°C the reaction mixture was washed with sat'd NAHCO3 and extracted with ether. The organic layer was dried with NA2S04 and concentrated in vacuo to give 4. 0g (82percent) of 2 as an oil that was used directly for the next step.
With triethylamine In dimethyl sulfoxide at 70℃; for 8h;
1 Step-i: Synthesis of 4-bromo-iV-cyclopropylthiazol-2-amine (3):
To a stirred solution of 2,4-dibromothiazole 1 (1.0 g, 4.12 mmol) in DMSO (10 mL) was added Et3N (2.30 mL, 16.46 mmol) followed by cyclopropanamine 2 (0.70 g, 12.28 mmol) and the reaction mixture was heated at 70 °C for 8 h. After completion of the reaction (monitored by TLC), the reaction mixture was diluted with water and extracted with ethyl acetate. Combined organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure. Crude compound was purified by silica gel column chromatography eluting with 10-15% ethyl acetate in n- hexane to afford 0.60 g (67% yield) of 3 as a brown solid. LCMS-Condition-i: (0228) [M+2+H]+ = 220.85; Rt = 1-86 min.
at 50℃; for 8h;
186.186F; F.1
186F. Commercially available 2,4-dibromothiazole (2 mmol) is dissolved in cyclopropylamine (3 mL) and the reaction mixture is heated to 50° C. for 8 hour. The cooled mixture is then poured into water and extracted 2 times with ether. After drying the combined organic fractions (MgSO4), evaporation of solvents, and purification by flash column chromatography (silica gel, 15% EtOAc/Hexane) furnished 2-cyclopropylamine-4-bromothiazole which is further converted to the corresponding stannane 186f. A solution of 2-cyclopropylamine-4-bromothiazole in degassed DME is treated with hexamethylditin and Pd(PPh3)4 and heated at 80° C. for 18 hour. The cooled mixture is concentrated under vacuum and the residue is purified by column chromatography eluting with 20% EtOAc/Hexane/2% Et3N to give Stannane 186f. [M+H]+=304.1.
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In water; toluene; at 90℃; for 18h;
Scheme Dl; [00314] sec-BuLi (4.3 mL, 6.0 mmol, 1.4M solution in cyclohexane) was added dropwise to a solution of 9-bromoanthracene (1.29 g, 5.0 mmol) in Et2O (20 mL) at O°C under N2. The reaction was held at O°C for 15 minutes then warmed to rt and stirred an additional 45 minutes. Triisopropylborate (1.5 mL, 6.5 mmol) was added and the reaction was stirred at rt for 18h. Concentrated HCl (1.0 mL) and MeOH (1.0 mL) was added and the reaction was stirred for 30 minutes. The layers were separated and the aqueous phase was extracted three times with CH2Cl2. The combined organic layers EPO <DP n="79"/>were washed with H2O, sat. NaHCO3 (aq.), brine, and then dried over Na2SO4. Filtration and concentration gave 1.05 g of 9-anthraceneboronic acid.[00315] A reaction flask was charged, under N2, with 9-anthraceneboronic acid(220 mg, 1.0 mmol), Pd(PPh3)4 (29 mg, 5.0 molpercent), <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (122 mg, 0.5 mmol), Na2CO3 (0.5 mL, 2.0 M solution in H2O), and toluene (4.0 mL). The reaction was heated at 9O°C for 18h. After cooling to rt, water was added and the layers were separated. The aqueous phase was extracted three times with CH2Cl2. The combined organic layers were washed with H2O, brine, and then dried over Na2SO4. The crude product mixture was purified by silica gel chromatography eiuting with 1percent Et2O in hexanes. Isolated 93 mg, 50percent yield, of 2-anthracenyl-4-bromothiazole.
With pyridine; potassium acetate; copper; copper(l) chloride; at 100℃; for 2h;
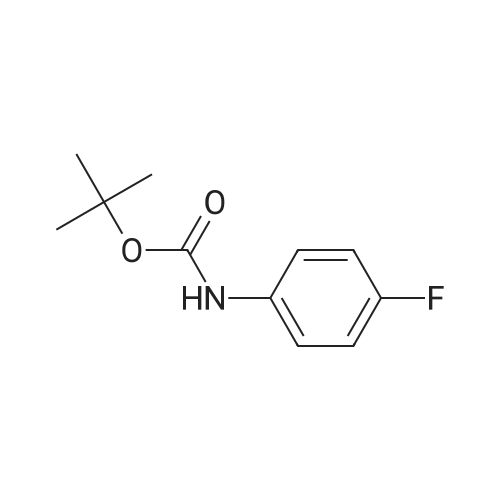
(4-Bromo-thiazol-2-yl)-<strong>[60144-53-8](4-fluoro-phenyl)-carbamic acid tert-butyl ester</strong>; A mixture of <strong>[60144-53-8](4-fluoro-phenyl)-carbamic acid tert-butyl ester</strong> (260 mg, 1.23 mmol), 2,4-dibromothiazole (100 mg, 0.41 mmol), copper powder (26 mg, 0.41 mmol), CuCl (41 mg, 0.41 mmol), and KOAc (40 mg, 0.41 mmol) in pyridine (4 ml) is heated at 1000C for 2 hours. The cooled mixture is diluted with EtOAc (30 ml) and washed with H2O (30 ml). The organic layer is dried over Na2SO4, filtered and concentrated in vacuum. The residue is purified by HPLC (C]8 column, eluted with CH3CN/H2O with 0.035% TFA) to give the title compound as solid: 1H NMR (DMSO-d6) delta 1.36 (s, 9H), 7.29 (t, 2H, J = 8.8 Hz), 7.37-7.43 (m, 3H); m/z [M+^t-Bu] 316.9.
Example 89: 2,2,2-Trifluoro-l-(2-morpholin-4-yl-thiazol-4-yl)ethanone; A solution of 2,4-dibromothiazole (500 mg, 2.06 mmol) in morpholine (4.0 mL, 22.3 mmol) was warmed at 500C in a sealed tube. After 16 hours, the mixture was cooled to room temperature, diluted with 20 mL of water and extracted with three 30 mL portions of diethyl ether. The combined organic layers was washed with five 30 mL portions of water, 30 mL of brine, dried over magnesium sulfate, filtered, and concentrated in vacuo. The residue was chromatographed over SiO2 using ethyl acetate in hexanes (0-30 % gradient) to afford 458 mg (89%) of4-(4-bromothiazol-2-yl)morpholine.
78%
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 20℃; for 12h;Inert atmosphere;
To a degassed mixture of 2,4-dibromothiazole 1 (0.5 g, 2.08 mmol) and morpholine 2 (0.54 g, 6.17 mmol) in DMF (15 mL) was added DIPEA (0.53 g, 4.16 mmol) and the reaction mixture was stirred at room temperature for 12 h. After completion of the reaction (monitored by TLC), solvent was removed under reduced pressure. Crude solid obtained was dissolved in ethyl acetate (50 mL) and washed with water (2 c 25 mL). Organic layer was separated, dried over anhydrous Na2S04 and concentrated under reduced pressure. Crude compound was purified by silica gel column chromatography eluting with 5-7% ethyl acetate in n- hexane to afford 0.40 g (78% yield) of 3 as a brown solid. LCMS-Condition-i: [M+H]+ = 248.9; Rt = 1.80 min.
With triethylamine; In N,N-dimethyl-formamide; at 80℃; for 1h;
Into a 2000-mL 4-necked round-bottom flask, was placed a solution of 2,4- dibromothiazole (150 g, 617.54 mmol, 1.00 equiv) inN,N-dimethylformamide (1000 mL), morpholine (60 g, 688.86 mmol, 1.00 equiv), and triethylamine (187 g, 1.85 mol, 3.00 equiv). The resulting solution was stirred for 1 hr at 80C in an oil bath. The mixture was cooled toroom temperature. The resulting solution was diluted with 3000 mL of H20. The solids were collected by filtration. The filter cake was washed with 3x1000 mL of water. The solid was dried in an oven to give the title compound as a solid.
A solution of Bu3SnCl (1.6M, 54 mL, 0.084 mol, 1.05 eq) was added dropwise via a dropping funnel to a cooled (-78° C.) solution of Intermediate 14 (20 g, 0.0823 moles) in dry ether (180 ml) under nitrogen. The resulting yellow-brown solution was stirred at -80° C. for 30 min before tributytin chloride (22.8 ml, 0.084 moles, 1.05 eg) was added using a dropping funnel at -80° C. The cooling bath was removed, allowing the reaction mixture to warm to -5° C. over 1 hr. The crude reaction was concentrated in vacuo followed by the addition of hexanes (200 ml), and the resultant precipitate was removed by filtration. The hexanes were removed from the filtrate in vacuo to provide the crude product (brown oil) which was purified by distillation (220° C., 0.56 mBar, Kugulrhor apparatus) in 2 batches, Batch 1 (8.81 g, 24percent) and Batch 2 (23.07 g, 62percent).
Stage #1: 2,4-dibromo-1,3-thiazole With bis(2,2,6,6-tetramethylpiperidinyl)zinc lithium chloride magnesium chloride complex In tetrahydrofuran at 25℃; for 0.75h; Inert atmosphere;
Stage #2: With copper(I) chloride bis(lithium chloride) complex In tetrahydrofuran at -50℃; for 0.5h; Inert atmosphere;
Stage #3: lithium hexamethyldisilazane Further stages;
80%
Stage #1: 2,4-dibromo-1,3-thiazole With bis(2,2,6,6-tetramethylpiperidinyl)zinc, lithium chloride, magnesium chloride complex In tetrahydrofuran at 25℃; for 0.75h; Inert atmosphere;
Stage #2: With copper(I) chloride bis(lithium chloride) complex In tetrahydrofuran at -50℃; Inert atmosphere;
Stage #3: lithium hexamethyldisilazane Further stages;
80%
Stage #1: 2,4-dibromo-1,3-thiazole With bis(2,2,6,6-tetramethylpiperidinyl)zinc, lithium chloride, magnesium chloride complex In tetrahydrofuran at 25℃; for 0.75h; Inert atmosphere;
Stage #2: With copper(I) chloride bis(lithium chloride) complex In tetrahydrofuran at -50℃; for 0.5h; Inert atmosphere;
Stage #3: lithium hexamethyldisilazane Further stages;
With triethylamine; In N,N-dimethyl-formamide; at 50 - 60℃; for 45h;
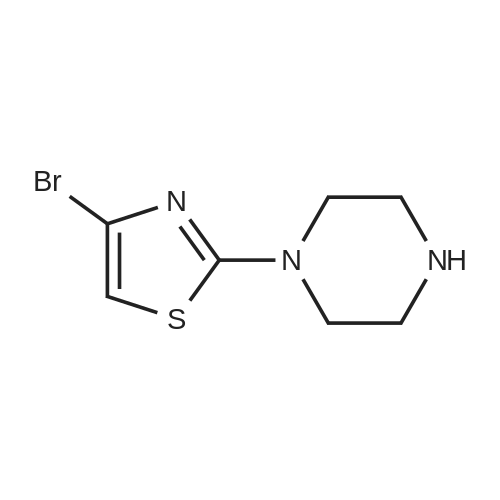
Production Example 95 2-{4-[2-(2-piperazin-l-yl-l,3-thiazol-4- yl) ethyl] phenyl }acetohydrazide dihydrochloride step 1[0300] [0301]To a solution of tert-butyl piperazine-1-carboxylate (767 mg, 4.12 mmol) , 2, 4-dibromo-l, 3-thiazole (1.0 g, 4.12 mmol) in N,N-dimethylformamide (9 ml) was added triethylamine (0..86 ml, 6.18 mmol) . The reaction mixture was stirred at 50°C for 6 hrs and at 6O0C for 39 hrs. After cooling to room temperature, the reaction mixture was poured into water, and the mixture was extracted with ethyl acetate. The combined organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and concentrated. The residue was purified by silica gel column chromatography (dichloromethane :methanol = 100:0 --> 98:2) to give tert-butyl 4- (4-bromo-l, 3-thiazol-2- yl) piperazine-1-carboxylate (1.09 g, yield 76percent)' as a pale- white brown solid.
23%
With triethylamine; In N,N-dimethyl-formamide; at 120℃; for 18h;
Step 1. tert-butyl 4-(4-bromothiazol-2-yl)piperazine-l-carboxylate [0660] A 100-mL round-bottom flask was charged with <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (1.00 g, 4.12 mmol), tert-butyl piperazine-l-carboxylate (0.918 g, 4.93 mmol), triethylamine (1.72 mL, 12.35 mmol) and N,N-dimethylformamide (20 mL). The resulting solution stirred for 18 h at 120 °C. The reaction mixture was cooled to room temperature, poured into water (50 mL), and extracted with ethyl acetate (3 x 20 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was purified via column chromatography on silica gel (eluting with 10: 1, dichloromethane/methanol) to afford tert-butyl 4-(4-bromothiazol-2- yl)piperazine-l-carboxylate (0.350 g, 23percent) as brown oil. MS (ESI, pos. ion) m/z 348, 350[M+H]+.
Hydroxymethyl thiazole 37: Hydroxymethyl thiazole 37 was prepared from commercially available <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (32) as previously described (Nicolaou, et al, 1998). The physical and spectral data are consistent with those reported (Nicolaou, et al, 1998).
34%
To a solution containing 0.5 mL (1.1 MMOL) of 2.3M n-butyllithium in hexane and 2 mL of THF AT-78°C was added a solution containing 243 mg (1.0 MMOL) of 2,4-dibromo-1, 3-thiazole and 5 mL of THF slowly. The bright yellow reaction mixture was allowed to stir for 15 min and 0. 15, UL of DMF was added. The orange reaction mixture was allowed to stir for a further 30 min and quenched by the addition of methanol. To this crude reaction mixture was added 63 UL of acetic acid and 34 mg (1.0 MMOL) of sodium borohydride. The mixture was allowed to warm slowly to room temperature, then quenched by the addition of water and extracted with ethyl acetate. The organic layers were washed with water and brine, then dried over MGS04. The solvents were removed under reduced pressure and the residue was subjected to silica gel chromatography to give the title compound (66mg, 34 H NMR (300 MHz, CDCI3) 6 4.00 (brs, 1H), 4.93 (s, 3H), and 7.20 (s, 1H).
With triethylamine; In N,N-dimethyl-formamide; at 80℃;
Intermediate 31 : Ethyl 1-(4-bromo-1 ,3-thiazol-2-yl)-4-piperidinecarboxylate; A mixture of the commercially available 2,4-dibromo-1 ,3-thiazole (7.3 g, 30 mmol), ethyl 4- piperidinecarboxylate (6.9 ml_, 45 mmol, 1.5 equiv.) and triethylamine (9.2 ml_, 66 mmol, 2.2 equiv.) in DMF (70 ml.) was stirred at 8O0C overnight before being concentrated to dryness. The crude product was taken up in EtOAc and washed successively with a saturated NH4CI solution and brine. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (cyclohexane/EtOAc: 100/0 to 80/20) to give the title compound (8.2 g, 86percent yield) as a colorless oil. 1H NMR (DMSO d6) J6.84 (s, 1 H), 4.08 (q, J = 7.1 Hz, 2H), 3.77 (m, 2H), 3.09 (m, 2H), 2.59 (m, 1 H), 1.91 (m, 1 H), 1.59 (m, 2H), 1.18 (t, J = 7.0 Hz, 3H). LCMS: (M+H)+= 319, 321 , Rt= 3.24 min.
79%
With triethylamine; In 1,4-dioxane; at 100℃; for 1h;Microwave irradiation;
EXAMPLE 4. ETHYL 1- 4-BROMOTHIAZOL-2-YL)PIPERIDINE-4-CARBOXYLATE (XJBOl-034)[0111] A mixture of <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (500 mg, 2.06 mmol), ethyl piperidine-4- carboxylate (380 mg, 2.42 mmol) and TEA (2.50 mL) in 1,4-dioxane (5.00 mL) was heated in mu\\.yen. at 100 °C for 1 hour. The reaction mixture was cooled to room temperature, filtered, and concentrated as light brown solid. The crude mixture was purified by Biotage with 0- 50percent EtOAc in hexanes to give 510 mg (79percent) product as a light yellow oil: 1H NMR (400 MHz, CHLOROFORM-d) delta ppm 6.41 (s, 1 H), 4.17 (q, 7=7.0 Hz, 2 H), 3.92 (ddd, 7=13.5, 4.3, 4.1 Hz, 2 H), 3.12 (ddd, 7=13.2, 11.2, 3.3 Hz, 2 H), 2.53 (tt, 7=11.0, 3.9 Hz, 1 H), 1.94 - 2.12 (m, 2 H), 1.70 - 1.90 (m, 2 H), 1.28 (t, 7=7.0 Hz, 3 H); LCMS RT = 6.110 min, m/z 319.0 [M+H+] ; HRMS (ESI) m/z calcd for CiiHi679BrN202S [M+H+] 319.0116.
Diisopropylamine (1.375 g, 1.904 mL, 13.58 mmol) in THF (50 mL) was cooled to -78 0C before BuLi (5.432 mL, 2.5 M in hexanes, 13.58 mmol) was added dropwise, maintaining temperature below -70 0C. On complete addition, mixture allowed to 0 0C by removing cardice bath and putting RBF into ice-water bath. Mixture then cooled back down to -78 0C before 2,4- dibromothiazole (3 g, 12.35 mmol) in THF (10 mL) was added dropwise at -78 0C (temperature did not exceed -70 0C). On complete addition, stirred at -78 0C for a further 30 mins. iodomethane (1.928 g, 845.6 L, 13.58 mmol) added dropwise, mixture stirred at -78 0C for 30 mins and then allowed to warm to room temperature over 18 h. Quenched by addition of water, diluted with EtOAc and organic layer collected. Organics then washed with brine, dried (MgS?4), filtered and concentrated in vacuo. Crude product purified twice by flash chromatography (80g SiO2, 0 to 10% EtOAc/petrol) then (80 g SiO2, 0 to 2.5% EtOAC/petrol) to leave impure product as a colourless oil which turned to colourless solid on standing (2.7 g, 56% based on 66% purity); 1H NMR DMSO-d6 ? 2.35 (s, 3H); ES+ 257.80.
With potassium phosphate; palladium diacetate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In tetrahydrofuran; at 80℃; for 0.15h;Inert atmosphere;
he mixture of compound 433a (5 g, 20.5 mmol), cyclopropyl boronic acid (2.12 g, 24.7 mmol), K3P04 (13.1 g, 61.7 mmol), Xantphos (0.6 g, 1.04 mmol) and Pd(OAc)2 (0.23 g, 1.04 mmol) in THF (140 mL) was stirred at 80 °C under N2 for about 15 hours. The reaction was complete detected by TLC (Petroleum Ether/EtOAc = 30: 1) and LCMS. The mixture was extracted with EtOAc (100 mL), water (50 mL). The combined organics was dried over Na2S04, purified with silica gel (Petroleum Ether /EtOAc = 50: 1) to provide compound 433b as oil (3.48 g, yield: 83.3percent)
83.3%
With potassium phosphate; palladium diacetate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In tetrahydrofuran; at 80℃; for 15h;Inert atmosphere;
The mixture of compound 433a (5 g, 20.5 mmol), cyclopropyl boronic acid (2.12 g, 24.7 mmol), K3P04 (13.1 g, 61.7 mmol), Xantphos (0.6 g, 1.04 mmol) and Pd(OAc)2 (0.23 g, 1.04 mmol) in THF (140 mL) was stirred at 80 °C under N2 for about 15 hours. The reaction was complete detected by TLC (Petroleum Ether/EtOAc = 30: 1) and LCMS. The mixture was extracted with EtOAc (100 mL), water (50 mL). The combined organics was dried over Na2S04, purified with silica gel (Petroleum Ether /EtOAc = 50: 1) to providecompound 433b as oil (3.48 g, yield: 83.3percent).
With potassium phosphate; palladium diacetate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In tetrahydrofuran; at 60℃; for 18h;Inert atmosphere;
[00287] Under N2 atmosphere Pd(OAc)2 (0.084 g, 0.38 mmol) was combined with xantphos (0.220 g, 0.38 mmol) in 25 mL THF. After stirring for 5 min., the resulting solution was transferred to a round bottom flask containing 2,4 dibromothiazole (3.64 g, 15 mmol), phenylboronic acid (1.96 g, 16 mmol) and Kappa3RhoO4 (9.55 g, 45 mmol). The resulting reaction mixture was heated at 60 C for 18 h and then cooled to room temperature and filtered and washed with dichloromethane. Removal of the solvents under vacuum afforded the crude product which was purified by silica gel column chromatography using hexanes: ethyl acetate to afford 4- bromo-2-phenylthiazole as a white solid (3 g, 84%). 1HNMR (400 MHz, CDC13): delta 7.95 (dd, J = 3.4 Hz, 8.2 Hz, 2H), 7.47-7.46 (m, 3H), 7.23 (s, 1H). LRMS (ESI) calcd. for C9H6BrNS [M+H]+: 241.12. Found: 241.00.
83%
With potassium phosphate; palladium diacetate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In tetrahydrofuran; at 60℃;Inert atmosphere; Schlenk technique;
According to the literature procedure,23 Pd(OAc)2(49.8 mg, 2.5 mol%) and Xantphos (129 mg, 2.5mol%) weredissolved in THF (10 mL) under an N2 atmosphere. After stirringfor 5min at room temperature, this solution was transferredto a two-neck round-bottom flask equipped with an N2balloon and a rubber cap containing 2,4-dibromothiazole(2.16 g, 8.88mmol), phenylboronic acid (1.16 g, 9.5mmol),K3PO4 (5.66 g, 26.6mmol), and THF (20 mL). The mixture washeated at 60 C overnight. The resulting suspension was dilutedwith H2O and was extracted with EtOAc three times. The combinedorganic layer was dried over Na2SO4 and concentratedin vacuo. The crude material was subjected to silica gel chromatography(eluent: hexane/EtOAc = 10/1) to give 4-bromo-2-phenylthiazole as white solid in 83% yield (1.76 g, 7.33mmol). NMR data were identical with those reported in theliterature.23 1HNMR (400 MHz, CDCl3) delta 7.22 (s, 1H), 7.427.47(m, 3H), 7.919.97(m, 2H).
66%
With potassium phosphate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In tetrahydrofuran; at 60℃; for 18h;Inert atmosphere;
Frame-dried 200 mL four-necked flask was purged with nitrogen and Xantphos (Xantphos, 220 mg, 0.38 mmol),Tris (dibenzylideneacetone) dipalladium (0) (Pd 2 (dba) 3,840 mg, 0.38 mmol) and dry tetrahydrofuran (dry THF, 75 mL) were added and stirred for 5 minutes, Thereby preparing a THF solution.Another flame-dried 200 mL four-necked flask was purged with nitrogen, 2,4-dibromothiazole (Compound 101 in the following formula (A), 3.6 g, 15 mmol),Phenylboronic acid (1.9 g, 16 mmol),Triopotassium phosphate (9.6 g, 45 mmol),The above THF solution was added, And the mixture was heated under reflux at 60 C. for 18 hours.After returning to room temperature,Celite filtration was carried out using dichloromethane. Removal of the solvent and purification by column chromatography gave the compound of the following formula (A), And 2.4 g of a white solid compound represented by "102" (yield: 66%).
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In ethanol; water; toluene; for 6h;Inert atmosphere; Reflux;
(7) In the general formula (I), R1 = 2-phenyl-4-thiazolyl, R2 = H, and R3-R4 = thiophene (Synthesis of compound No. 76 in Table 1)Stage A: 4-Bromo-2-phenylthiazole A flask containing 238 mg of tetrakis(triphenylphosphine)palladium, 0.55 g of phenyl boronic acid and 1.00 g of 2,4-dibromothiazole was subjected to nitrogen substitution. To this flask, 30 ml of toluene, 6.1 ml of ethanol, and 9.1 ml of a 2 M sodium carbonate aqueous solution were added, and the obtained mixture was then stirred under reflux for 6 hours. Thereafter, the reaction solution was cooled to a room temperature, and 50 ml of water was then added thereto, followed by extraction with 50 ml of ethyl acetate twice. The organic layer was washed with 30 ml of a saturated brine, and it was then dried over magnesium sulfate. The magnesium sulfate was filtrated, and the organic layer was then concentrated. The residue was then purified by column chromatography (Wako Gel C-200; hexane : ethyl acetate = 14 : 1), so as to obtain 0.71 g of 2-phenyl-4-bromothiazole.
0.71 g
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; for 6h;Inert atmosphere; Reflux;
A flask containing 238 mg of tetrakis(triphenylphosphine)palladium, 0.55 g of phenylboronic acid, and 1.00 g of 2,4-dibromothiazole was purged with nitrogen and then charged with 30 ml of toluene, 6.1 ml of ethanol, and 9.1 ml of a 2 M aqueous solution of sodium carbonate, and the mixture was stirred under reflux for 6 hours. After cooling to room temperature, 50 ml of water was added to the reaction mixture and two extractions were conducted with 50 ml of ethyl acetate. After being washed with 30 ml of saturated brine, the organic layer was dried over magnesium sulfate. After filtering the magnesium sulfate, the organic layer was concentrated and the residue was purified by column chromatography (Wakogel C-200; hexane:ethyl acetate=14:1) to give 0.71 g of 2-phenyl-4-bromothiazole.
Scheme C2; [00304] n-BuLi (2.4 niL, 3.84 mmol, 1.6M in cyclohexane) was added dropwise to a solution of methyl-protected BBl (1.Og, 3.34 mmol) in Et2O (10 mL) at -78°C under N2. After 30 min. at -78°C, triisopropylborate (0.88 mL, 3.84 mmol) was added and the reaction was warmed to rt. After Ih at rt, the reaction was quenched with MeOH (2 mL) then concentrated by rotary evaporation. The reaction flask was then charged, under N2, with <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (486 mg, 2.0 mmol), Pd(PPh3)4 (116 mg, 5 molpercent), Na2CO3 (1.1 mL, 2.0M solution in H2O), and toluene (10 mL). The reaction was heated overnight at 9O°C. After cooling to rt, H2O was added and the layers were separated. The aqueous phase was extracted several times with CH2Cl2. The combined organic layers were washed with H2O, brine, and then dried over Na2SO4. The crude reaction mixture was dissolved in CH2Cl2 (5.0 mL) and BBr3 (3.0 mL, 1.0M in CH2Cl2) was added dropwise at rt. After Ih, the reaction was quenched with several drops of MeOH, washed with H2O, sat. NH4Cl (aq.), brine, and then dried over Na2SO4. The crude product was purified by silica gel chromatography eluting with 1-2percent Et2O in hexanes. Isolated 715 mg, 97percent yield, of ligand C26 as a white solid.
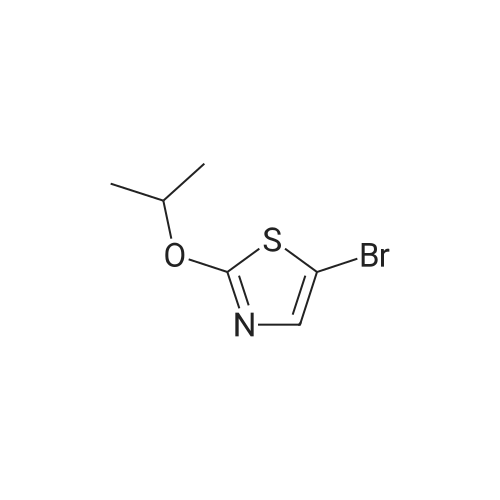
NaOH (3.4 g, 85.6 mmol, 13.0 eq.) was added portionwise to a solution of 2, 4- dibromothiazole (1.6 g, 6.6 mmol, 1.0 eq.) in isopropanol (50 ml) and the mixture was stirred at ambient temperature for overnight. The reaction mixture was diluted with diethyl ether (100 ml) and washed with water. The water layer was again extracted with ether (100 ml X 2). The combined organic layer was washed with water and brine, and was dried over sodium sulfate to afford 5-bromo-2-isopropoxythiazole.
With potassium carbonate; In N,N-dimethyl-formamide; at 70℃;
Step 1: K2CO3 (5.7 g, 41 mmol) was added into a solution of compound 179-1 (5.0 g, 20.5 mmol) and (2S,6R)-2, 6-dimethylmorpholine (3.55 g, 30.9 mmol) in DMF (60 mL). The mixture was stirred at 70°C overnight. The reactionmixture was cooled to room temperature, then poured into H2O, extracted with EtOAc, dried over sodium sulfate andconcentrated. The residue was purified by column chromatography to deliver compound 179-2 (5.56 g, yield 99percent) asyellow oil. MS ESI calcd for C9H13BrN2OS [M+H]+ 277, found 277.
77%
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 70℃;Inert atmosphere;
4-(4-Bromo-thiazol-2-yl)-cis-2,6-dimethyl-morpholine (Intermediate 1)(Intermediate 1)A I L flask equipped with a mechanical stirrer and a thermometer was charged with <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (60 g, 0.247 mol) and DMF (480 mL). After dissolution, cis-2,6-dimethylmorpholine (33.65 mL, 0.272 mol) and DIPEA (61.23 mL, 0.37 mol) were added under a stream of nitrogen. The reaction mixture was heated to 70°C under nitrogen and stirred overnight. The reaction mixture was cooled to 10°C in an ice/water bath. Then water (1 L) was added dropwise in a temperature range of 10-20°C. A slightly exothermic reaction was observed, with formation of an off-white precipitate. After the addition, the reaction mixture was stirred for 30 min. The precipitate was then filtered and washed with water (50 ml). The collected solid was dried in a vacuum oven at 50°C, yielding 52.8g (77 percent) of Intermediate 1 as an off-white solid.
77%
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 70℃;Inert atmosphere;
A 1 L flask equipped with a mechanical stirrer and a thermometer was charged with <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (60 g, 0.247 mol) and DMF (480 mL). After dissolution, cis-2,6-dimethylmorpholine (33.65 mL, 0.272 mol) and DIPEA (61.23 mL, 0.37 mol) were added under a stream of nitrogen. The reaction mixture was heated to 70° C. under nitrogen and stirred overnight. The reaction mixture was cooled to 10° C. in an ice/water bath. Then water (1 L) was added dropwise in a temperature range of 10-20° C. A slightly exothermic reaction was observed, with formation of an off-white precipitate. After the addition, the reaction mixture was stirred for 30 min. The precipitate was then filtered and washed with water (50 ml). The collected solid was dried in a vacuum oven at 50° C., yielding 52.8 g (77percent) of Intermediate 1 as an off-white solid.
Step 1. tert-butyl 4-(4-bromothiazol-2-yl)-4-hydroxypiperidine-l-carboxylate [0584] -Butyllithium (1.6 M in hexanes, 1.48 mL, 2.37 mmol) was added dropwise to a -78 °C solution of <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (0.524 g, 2.16 mmol) in dichloromethane (11 mL). The mixture stirred at -78 °C for 20 minutes, and then tert-butyl 4-oxopiperidine- 1 -carboxylate (0.516 g, 2.59 mmol) was added in 8 portions. The reaction mixture stirred at -78 °C for 10 minutes. The cooling bath was removed, and the reaction was allowed to warm to rt over 1 h. The reaction mixture was cooled to -30 °C and was then quenched by the addition of saturated aqueous ammonium chloride solution. The aqueous phase was separated and extracted with dichloromethane, and the combined organic phases were dried over anhydrous sodium sulfate, filtered, and concentrated. The residue was purified via column chromatography on silica gel (Biotage 50 g column, gradient elution with 5- 60percent ethyl acetate-hexane) to afford tert-butyl 4-(4-bromothiazol-2-yl)-4-hydroxypiperidine- 1 - carboxylate (0.744 g, 95percent). MS (ESI, pos. ion) m/z 363, 365 [M+H]+.
Preparation of Compounds of the Formula (I)2-[3,5-Bis(difluoromethyl)-1H-pyrazol-1-yl]-1-{4-fluoro-4-[4-(5-phenyl-4,5-dihydro-1,2-oxazol-3-yl)-1,3-thiazol-2-yl]piperidin-1-yl}ethanone (I-5)Process Atert-Butyl 4-(4-bromo-1,3-thiazol-2-yl)-4-hydroxypiperidine-1-carboxylate (IV-1)To a solution of <strong>[4175-77-3]2,4-dibromo-1,3-thiazole</strong> (8.8 g) in dichloromethane (180 ml) was added dropwise, at -78° C. under argon, n-butyllithium (1.6 M in tetrahydrofuran, 25 ml). The reaction mixture was stirred at -78° C. for 20 minutes and then tert-butyl 4-oxopiperidine-1-carboxylate was added. The mixture was stirred at room temperature for 30 minutes. The reaction mixture was subsequently admixed with saturated ammonium chloride solution at -30° C. and the aqueous phase was removed. After the aqueous phase had been extracted with dichloromethane, the combined organic phases were dried over sodium sulphate and concentrated under reduced pressure. The residue was purified by chromatography. This gave tert-butyl 4-(4-bromo-1,3-thiazol-2-yl)-4-hydroxypiperidine-1-carboxylate (15.3 g).1H NMR (DMSO-d6): deltappm: 1.43 (s, 9H), 1.70 (d, 2H), 1.88 (ddd, 2H), 3.11 (bs, 2H), 3.83 (d, 2H), 6.31 (s, 1H), 7.72 (s, 1H)log P (HCOOH): 2.74MS (ESI): 363 and 365 ([M+H]+)
Process Atert-Butyl 4-(4-bromo-1,3-thiazol-2-yl)-4-hydroxypiperidine-1-carboxylate (IV-1)To a solution of <strong>[4175-77-3]2,4-dibromo-1,3-thiazole</strong> (8.8 g) in dichloromethane (180 ml) was added dropwise, at -78° C. under an argon atmosphere, n-butyllithium (1.6 M in tetrahydrofuran, 25 ml). The reaction mixture was stirred at -78° C. for 20 minutes and then tert-butyl 4-oxopiperidine-1-carboxylate was added. The mixture was stirred at room temperature for 30 minutes. The reaction mixture was subsequently admixed with saturated ammonium chloride solution at -30° C. and the aqueous phase was removed. After the aqueous phase had been extracted with dichloromethane, the combined organic phases were dried over sodium sulphate and concentrated under reduced pressure. The residue was purified chromatographically. This gave tert-butyl 4-(4-bromo-1,3-thiazol-2-yl)-4-hydroxypiperidine-1-carboxylate (15.3 g).1H NMR (DMSO-d6): deltappm: 1.43 (s, 9H), 1.70 (d, 2H), 1.88 (ddd, 2H), 3.11 (bs, 2H), 3.83 (d, 2H), 6.31 (s, 1H), 7.72 (s, 1H)log P (HCOOH): 2.74MS (ESI): 363 and 365 ([M+H]+)
To a solution of <strong>[4175-77-3]2,4-dibromo-1,3-thiazole</strong> (8.8 g) in dichloromethane (180 ml) was added dropwise, at -78° C. under argon, n-butyllithium (1.6 M in tetrahydrofuran, 25 ml).The reaction mixture was stirred at -78° C. for 20 minutes and then tert-butyl 4-oxopiperidine-1-carboxylate was added.The mixture was stirred at room temperature for 30 minutes.The reaction mixture was subsequently admixed with saturated ammonium chloride solution at -30° C. and the aqueous phase was removed.After the aqueous phase had been extracted with dichloromethane, the combined organic phases were dried over sodium sulphate and concentrated under reduced pressure.The residue was purified by chromatography.This gave tert-butyl 4-(4-bromo-1,3-thiazol-2-yl)-4-hydroxypiperidine-1-carboxylate (15.3 g).
With triethylamine In 1,4-dioxane at 100℃; for 1h; Microwave irradiation;
57%
With triethylamine In 1,4-dioxane at 100℃; for 1h; Microwave irradiation;
7
EXAMPLE 7. 4-BROMO-2-(PIPERAZIN- 1 - YL)THIAZOLE (XJB02-056)[0114] A mixture of 2,4-dibromothiazole (1.00 g, 4.12 mmol), piperazine (426 mg, 4.94 mmol) in 1,4-dioxane (5.00 mL) and TEA (2.50 mL) was heated in μ¥ at 100 °C for 1 hour. The reaction mixture was cooled to room temperature, diluted with water and extracted with EtOAc and dichloromethane. The organic layer was separated, dried Na2S04, and concentrated as light yellow solid. The crude product was purified with Biotage using 0-10% MeOH in CH2C12 to give 581 mg (57%) product as a white solid.: 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 6.43 (s, 1 H), 3.39 - 3.58 (m, 4 H), 2.83 - 3.06 (m, 4 H), 1.80 (br. s., 1 H); LCMS RT = 3.038 min, m/z 247.9 [M+H+]; HRMS (ESI) m/z calcd forC7Hn79BrN3S [M+H+] 247.9857.
With triethylamine; In ethanol; at 100℃; for 1h;Microwave irradiation;
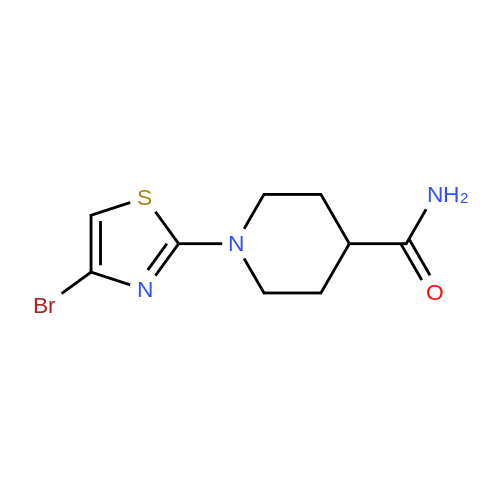
EXAMPLE 1 1-(4-BROMOTHIAZOL-2-YL)PIPERIDINE-4-CARBOXAMIDE (XJBOl-018) [0108] A mixture of <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (2.06 g, 8.48 mmol), piperidine-4- carboxamide (1.30 g, 10.1 mmol) and TEA (2.50 mL) in ethanol (5.00 mL) was heated in mu\\.yen. at 100 °C for 1 hour. The reaction mixture was cooled to room temperature, diluted with water and extracted with methanol and dichloromethane. The organic layer was separated, dried with Na2S04, and concentrated as light brown solid. The crude mixture was purified by Biotage with 0-10percent MeOH in CH2C12 with 1percent TEA to give 2.08 g (85percent) product as a white solid: 1H NMR (400 MHz, DMSO-d6) delta ppm 7.31 (br. s., 1 H), 6.85 (s, 1 H), 6.82 (br. s., 1 H), 3.77 - 3.90 (m, 2 H), 3.03 (td, 7=12.5, 2.8 Hz, 2 H), 2.29 - 2.39 (m, 1 H), 1.78 (dd, 7=13.5, 3.1 Hz, 2 H), 1.50 - 1.63 (m, 2 H); LCMS RT = 4.134 min, m/z 289.9 [M+H+] .
With triethylamine; In 1,4-dioxane; at 100 - 120℃; for 4h;Microwave irradiation;
EXAMPLE 6. TERT-BUTYL 1-(4-BROMOTHIAZOL-2-YL)PIPERIDIN-4-YLCARBAMATE (XJB02- 032)[0113] A mixture of <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (500 mg, 2.06 mmol), tert-butyl piperidin-4- ylcarbamate (495 mg, 2.47 mmol) in 1,4-dioxane (5.00 mL) and TEA (2.50 mL) was heated in mu\\.yen. at 100 °C for 1 h and at 120 °C for another 3 h. The reaction mixture was cooled to room temperature, diluted with water and extracted with EtOAc and dichloromethane. The organic layer was separated, dried Na2S04, and concentrated as light yellow solid. The crude mixture was purified with Biotage with 0-10percent MeOH in CH2C12 to give 654 mg (88percent) product as a white solid: 1H NMR (400 MHz, CHLOROFORM-d) delta ppm 6.41 (s, 1 H), 4.46 (br. s., 1 H), 3.92 (dt, 7=13.4, 3.3 Hz, 2 H), 3.70 (br. s., 1 H), 3.13 (ddd, 7=13.4, 11.6, 3.1 Hz, 2 H), 1.88 - 2.19 (m, 2 H), 1.40 - 1.54 (m, 11 H); LCMS RT = 6.137 min, m/z 362.0 [M+H+]; HRMS (ESI) m/z calcd for Ci3H2i79BrN302S [M+H+] 362.0538.
2-(1,3-benzodioxol-5-ylethynyl)-4-bromo-1,3-thiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
73%
With copper(l) iodide; tetrakis(triphenylphosphine) palladium(0); diisopropylamine; In tetrahydrofuran; at 20℃; for 2h;
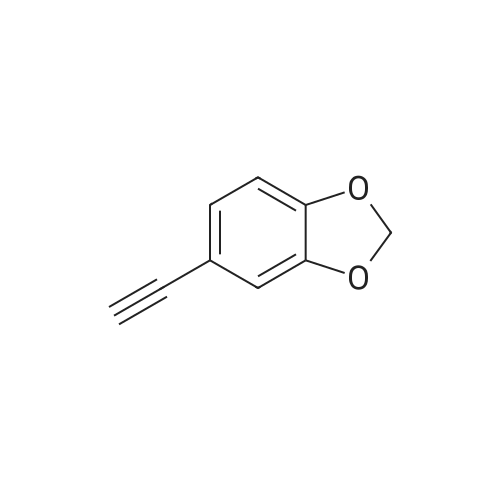
To a solution of 1 (975 mg, 6.68 mmol) and 2d (1.08 g, 4.45 mmol) in THF (12 mL) were added Pd(PPh3)4 (257 mg, 0.222 mmol), CuI (85 mg, 0.45 mmol), and NH(i-Pr)2 (675 mg, 6.67 mmol). The reaction mixture was stirred at room temperature for 2 h. Water (60 mL) was added to the reaction mixture and the reaction mixture was extracted with EtOAc (20 mL, 2×50 mL). The combined organic layer was washed with brine (60 mL), dried over MgSO4, and concentrated. The crude product was purified by column chromatography on silica gel eluting with 7% EtOAc/n-Hexane and then 50% CHCl3/n-Hexane to afford 3d as a light yellow powder (1.00 g, 73%): 1H NMR (300 MHz, CDCl3, delta): 6.02 (s, 2 H), 6.82 (d, J = 8.1 Hz, 1 H), 7.00 (d, J = 1.6 Hz, 1 H), 7.13 (dd, J = 8.1, 1.6 Hz, 1 H), 7.23 (s, 1 H); MS (ESI) m/z 308 [M+H]+, 10%, 310 [M+2+H]+, 10%, 330 [M+Na]+, 20%, 332 [M+2+Na]+, 20%.
With triethylamine; In N,N-dimethyl-formamide; at 100℃; for 2h;
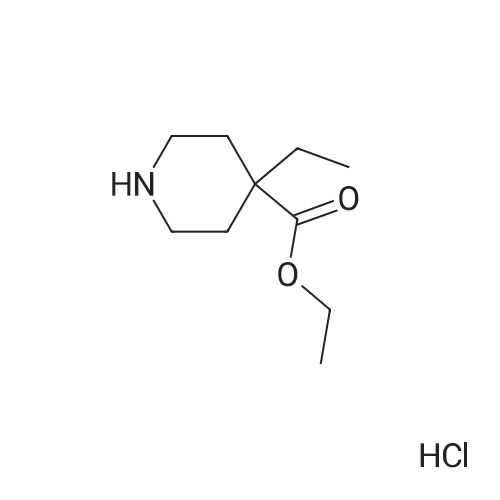
To a solution of ethyl 4-ethylpiperidine-4-carboxylate hydrochloride (140 mg, 0.62 mmol) and <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (100 mg, 0.41 mmol) in DMF (0.3 mL) was added triethylamine (0.26 mL, 1.9 mmol) at rt and the mixture heated to 100° C. and stirred for 2 h. The mixture was cooled to rt then diluted with EtOAc (2 mL) and MeOH (2 mL) then evaporated to dryness under reduced pressure. The crude residue was purified over silica-gel using EtOAc: cyclohexane (10percent to 60percent) to obtain the desired product as viscous oil that solidified upon standing (127 mg, 89percent yield). MS: 347.03, 349.01 [M+H]+.
With potassium carbonate In N,N-dimethyl-formamide at 140℃; for 6h;
1.9.A
(9) In the general formula (I), R1 = 2-phenoxy-4-thiazolyl, R2 = H, and R3-R4 = thiophene (Synthesis of compound No. 83 in Table 1)Stage A: 4-Bromo-2-phenoxythiazole 3.00 g of 2,4-dibromothiazole, 1.74 g of phenol, and 3.44 g of potassium carbonate were added to 75 ml of N,N-dimethylformamide, and the obtained mixture was then stirred at 140°C for 6 hours. Thereafter, the reaction solution was cooled to a room temperature, and 100 ml of water was then added thereto, followed by extraction with 100 ml of ethyl acetate twice. The organic layer was washed with 100 ml of water twice and then with 50 ml of a saturated brine once. The resultant was dried over magnesium sulfate. The magnesium sulfate was filtrated, and the organic layer was then concentrated. The residue was then purified by column chromatography (Wako Gel C-200; hexane : ethyl acetate = 9 : 1), so as to obtain 2.86 g of an ether compound.
2.86 g
With potassium carbonate In N,N-dimethyl-formamide at 140℃; for 6h;
9A A: 4-Bromo-2-phenoxythiazole
To 75 ml of N,N-dimethylformamide were added 3.00 g of 2,4-dibromothiazole, 1.74 g of phenol, and 3.44 g of potassium carbonate, and the mixture was stirred at 140° C. for 6 hours. After cooling to room temperature, 100 ml of water was added to the reaction mixture and two extractions were conducted with 100 ml of ethyl acetate. After being washed twice with 100 ml of water and once with 50 ml of saturated brine, the organic layer was dried over magnesium sulfate. After filtering the magnesium sulfate, the organic layer was concentrated and the residue was purified by column chromatography (Wakogel C-200; hexane:ethyl acetate=9:1) to give 2.86 g of an ether compound.
General procedure: The reaction was carried out in a sealed tube under N2 using THF (4 mL) as the solvent. To the solution of tert-butyl 2-bromothiazole-4-carboxylate 1a (0.397 mmol), iPrMgCl·LiCl (1.3 M, 0.52 mmol) was added dropwise at -78 C. After 15 min the corresponding aldehyde (0.79-0.99 mmol) or N-formyl-morpholine (in the case of 2-formylthiazoles 3a-b) was added dropwise under stirring and the reaction mixture was stirred at -78 C for 10 min, then 1.5 h at 0 C. Saturated aqueous NH4Cl was added and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organic layers were washed with 5% aqueous HCl (10 mL), brine (10 mL), dried over MgSO4 and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel using a mixture of EtOAc:P (petroleum ether) as eluent (see below) to give tert-butyl 2-(1-hydroxymethyl)thiazole-4-carboxylate derivatives rac-2a-h.
General procedure: The reaction was carried out in a sealed tube under N2 using THF (4 mL) as the solvent. To the solution of tert-butyl 2-bromothiazole-4-carboxylate 1a (0.397 mmol), iPrMgCl*LiCl (1.3 M, 0.52 mmol) was added dropwise at -78 °C. After 15 min the corresponding aldehyde (0.79-0.99 mmol) or N-formyl-morpholine (in the case of 2-formylthiazoles 3a-b) was added dropwise under stirring and the reaction mixture was stirred at -78 °C for 10 min, then 1.5 h at 0 °C. Saturated aqueous NH4Cl was added and the aqueous layer was extracted with Et2O (3 .x. 10 mL). The combined organic layers were washed with 5percent aqueous HCl (10 mL), brine (10 mL), dried over MgSO4 and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel using a mixture of EtOAc:P (petroleum ether) as eluent (see below) to give tert-butyl 2-(1-hydroxymethyl)thiazole-4-carboxylate derivatives rac-2a-h.
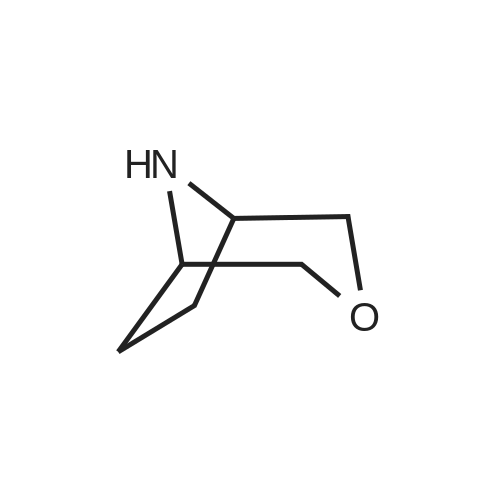
8-(4-bromothiazol-2-yl)-3-oxa-8-azabicyclo[3.2.1]octane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
46.7%
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 80℃; for 18h;Inert atmosphere;
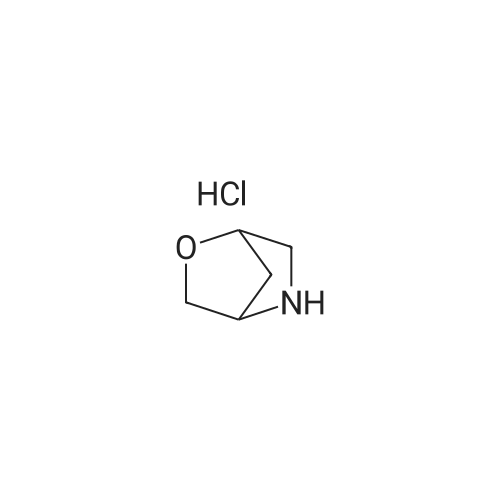
Step 1: 8-(4-bromothiazol-2-yl)-<strong>[280-07-9]3-oxa-8-azabicyclo[3.2.1]octane</strong> To a stirred solution of 2,4-dibromothiazole (0.585 g, 2.41 mmol) in DMF ( 10 ml) were added <strong>[280-07-9]3-oxa-8-azabicyclo[3.2.1]octane</strong> (prepared according to the procedure given in WO 2010/ 120854, 0.3 g, 2.65 mmol) and DIPEA (0.47 gm, 0.63 ml, 3.62 mmol) under a stream of nitrogen. The reaction mixture was then stirred at 80C for 18 hrs and the progress of reaction was monitored by TLC. The reaction mixture was cooled to room temperature; quenched with cold water (20 ml) and extracted with ethyl acetate (3 x 30 ml). The combined organic layer was washed with water (2 x 10 ml), dried over sodium sulphate and concentrated to give crude compound; which was purified by flash column chromatography using 20% ethyl acetate in hexanes as an eluent to obtain the title compound (0.31 g, 46.7%). MS: m/z 276 (M+ l). iHNMR (CDC13, 400 MHz): delta 6.47 (s, 1H), 4.17-4.12 (m, 2H), 3.90 (d, J = 10.8 Hz, 2H), 3.60 (d, J = 10.8 Hz, 2H), 2.13-2.02 (m, 4H).
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 120℃; for 1.5h;Microwave irradiation;
Example 77: 2-(1-(4-(4-((trans-4-(tert-Butyl)cyclohexyl)oxy)phenyl)thiazol-2-yl)azetidin- 3-yl)acetic acidStep 1 Methyl 2 -(1 -(4-bromothiazol-2 -yl)azetidin-3 -yl)acet ate [00225] To a mixture of 2,4-dibromo-thiazole (190 mg, 0.78 mmol) and azetidin-3-yl- acetic acid methyl ester HC1 salt (130 mg, 0.78 mmol) in acetonitrile (1.0 mL) was added N, N-diisopropylethylamine (273 jiL, 1.57 mmol). The reaction mixture was heated with microwave irritation at 120C for 30 mm. More azetidin-3-yl-acetic acid methyl ester HC1 salt (170 mg) and N, N-diisopropylethylamine (300 pL) were added, and the mixture was heated with microwave irritation at 120C for 1 hr. The reaction mixture was partitioned between EtOAc and saturated aqueous NH4C1. The organic phase was dried over MgSO4, filtered, and concentrated. The residue was purified by silica gel column to give the desired product as a tan colored solid (189 mg, 83% yield). ?H NMR (400 MHz, DMSO-d6) 6.85 (s, 1H), 4.11 (t, J= 8.16 Hz, 2H), 3.71 (dd, J= 6.02, 8.03 Hz, 2H), 3.60 (s, 3H), 2.99 - 3.15 (m, 1H), 2.76 (d, J = 7.78 Hz, 2H); LCMS mlz 290.9; 293.0 [M+Hf?.
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; triphenylphosphine; In toluene; at 140℃; for 48h;Inert atmosphere;
A mixture of commercially available <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> 5 (3.000 g, 12.34 mmol, 1 equiv), triphenylphosphine (486 mg, 1.851 mmol, 5 mol ), CuI (120 mg, 0.617 mmol, 5 mol ), and Pd(PPh3)2Cl2 (120 mg, 0.173 mmol, 1.4 mol ) was placed in a three-neck roundbottom flask under nitrogen. Anhydrous toluene (42 mL) was added followed by anhydrous Et3N (2.2 mL, 15.04 mmol, 1.3 equiv) and trimethylsilylacetylene (2.6 mL, 18.51 mmol, 1.5 equiv). The reaction mixture was refluxed at 140 C for 2 d. It was poured into water (50 mL) and extracted with EtOAc (3 x 20 mL). The combined organic extract was dried over anhydrous sodium sulfate and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel in CH2Cl2-hexanes to obtain 6 (2.625 g, 81%) as a brownish/red solid: TLC Rf= 0.72 (10 % EtOAc-hexanes); mp 37 C; IR: 3300, 2900, 2250 cm-1; 1H NMR (400 MHz, CDCl3): delta 7.18 (s, 1H), 0.24 (s, 9H); 13C NMR (CDCl3, 100 MHz): delta 149.54, 125.87, 118.85, 103.03, 95.52, -0.41 ; HRMS (m/z): [M + H]+ calculated for C8H11NSSiBr, 259.9565; found, 259.9565.
65.3%
With copper(l) iodide; trans-bis(triphenylphosphine)palladium dichloride; triethylamine; In acetonitrile; at 50℃; for 20h;Inert atmosphere;
To a solution of <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (500 mg, 2.07 mmol) in CH3CN (10 mL) was added Cul (20 mg, 0.10 mmol), TEA (1.4 mL), Pd(PPli3)2Cl2(73 mg, 0.10 mmol), ethynyl(trimethyl)silane (304 mg, 3.10 mmol). The mixture was heated under N2 at 50 C for 20 h. Removal of solvent gave a residue which was purified by column chromatography (RE/EA=10/1) to obtain the title compound (350 mg, 65.3%). m/z calcd for [C8Hl0BrNSSi] [M]: 258.9; found: 260.0 [M+H]
14%
With bis-triphenylphosphine-palladium(II) chloride; copper(l) iodide; triethylamine; In acetonitrile; at 50℃; for 20h;Inert atmosphere;
To a solution of <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (2.00g, 8.23mmol) in CH3CN (30 mL) was added Copper(I)Iodide (78.4mg, 0.412mmol), TEA (5.74mL), PdCl2(PPh3)2 (289mg, 0.412mmol), ethynyl(trimethyl)silane (1.21 g, 12.3mmol). The mixture was heated under N2 at 50? for 20 h. Removal of solvent gave a residue which was purified by column chromatography (PE/EA=10/1) to obtain 300mg(14%) of the title compound. ESI-MS m/z calcd for [C8H10BrNSSi]+ [M+H]": 259; found: 259
To a solution of 2-fluoroethanol (0.158 g, 2.470 mmol) in THF (5 mL) was added at 0°C NaH (0.053 g, 1.976 mmol). After 30 min at room temperature, <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (0.4 g, 1.647 mmol) was added and the suspension was degased with argon for 2 min. Then, the reaction mixture was heated under microwave irradiation at 50°C for 30 min. Water was added, whichwas then extracted several times with EtOAc. The combined organics were dried over Na2S04, filtered and concentrated under reduced pressure. The residue was purified on HP-Sil SNAP cartridges using a Biotage Isolera One purification system employing an EtOAc/n-heptane gradient (2/98 -> 5/95) to afford the title compound as a yellow solid (0.287 g, 77 percent). 1 H NMR (400 MHz, DMSO-d6) delta = 7.20 (s, 0.5H), 4.84 - 4.77 (m, 1 H), 4.72 - 4.63 (m, 2H), 4.61 - 4.55 (m, 1 H)
77%
Preparative Example 22 (0774) (0775) Step A (0776) To a solution of 2-fluoroethanol (0.158 g, 2.470 mmol) in THF (5 mL) was added at 0° C. NaH (0.053 g, 1.976 mmol). After 30 min at room temperature, <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (0.4 g, 1.647 mmol) was added and the suspension was degased with argon for 2 min. Then, the reaction mixture was heated under microwave irradiation at 150° C. for 30 min. Water was added, which was then extracted several times with EtOAc. The combined organics were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified on HP-Sil SNAP cartridges using a Biotage Isolera One purification system employing an EtOAc/n-heptane gradient (2/98->5/95) to afford the title compound as a yellow solid (0.287 g, 77percent). (0777) 1H NMR (400 MHz, DMSO-d6) delta=7.20 (s, 0.5H), 4.84-4.77 (m, 1H), 4.72-4.63 (m, 2H), 4.61-4.55 (m, 1H).
Stage #1: 2,4-dibromo-1,3-thiazole With isopropylmagnesium chloride In tetrahydrofuran at 0℃; for 0.166667h;
Stage #2: ethyl cyanoformate In tetrahydrofuran at 0 - 20℃; for 0.25h;
Ethyl 4-bromothiazole-2-carboxylate (10a)2
To asolution of 2,4-dibromothiazole (640 mg, 2.63mmol) in THF (10.0 mL) at 0 °C was added iPrMgCl(2.0 M in THF, 1.5 mL) dropwise. The reaction mixture was stirred at 0 °C for 10 minutes, and ethyl cyanoformate (0.65 mL, 6.60 mmol) was added. The resulting solution was stirred for additional 15 minutes at room temperature and quenched with NH4Cl solution. The mixture was extracted with H2O/Ethylacetate. The combined organic fraction was washed with brine and dried overMgSO4, concentrated under reduced pressure and purified by column chromatography on silica gel (hexane/ethyl acetate = 10:1) to afford the titlecompound 10a (349 mg, 56 %) as white solid: 1H-NMR (400 MHz, CDCl3) δ 7.52 (s, 1H), 4.42 (q, J = 7.12 Hz, 2H), 1.37 (t, J = 7.12 Hz, 3H). 13C-NMR (100MHz, CDCl3) δ 159.0, 158.9, 127.5, 123.7, 63.1, 14.2.
Stage #1: 2,4-dibromo-1,3-thiazole With magnesium chloride In tetrahydrofuran at 0℃; for 0.5h; Inert atmosphere;
Stage #2: ethyl cyanoformate In tetrahydrofuran at 15℃; for 1h;
046.1 Step 1. Synthesis of Compound WX046-3
WX046-1 (10.00 g, 41.17 mmol, 1.00 eq) was added to anhydrous tetrahydrofuran (100.00 mL), then isopropyl magnesium chloride (2M, 22.64 mL, 1.10 eq) was added dropwise at 0° C. under the protection of nitrogen atmosphere. The mixture was stirred at 0° C. for 0.5 hour, and WX046-2 (10.20 g, 102.92 mmol, 10.10 mL, 2.50 eq) was added. The mixture was stirred at 15° C. for 1 hour. The reaction solution was quenched with saturated aqueous ammonium chloride (150 mL), and extracted with dichloromethane (150 mL*2). The organic phase was dried over anhydrous sodium sulfate and filtered. The filtrate was dried on a rotary evaporator under reduced pressure. The crude product was purified by column (ethyl acetate/petroleum ether=05%10%20%) to obtain WX046-3.
(3aR,5S,7aR)-7a-(4-bromo-1,3-thiazol-2-yl)-5-methylhexahydro-1H-pyrano[3,4-c][1,2]oxazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
2,4-Dibromo-1 ,3-thiazole (44.7 g, 184 mmol) was dissolved in a mixture of toluene and tetrahydrofuran (10: 1 , 900 mL) and cooled to -78 C. To this solution was added boron trifluoride diethyl etherate (21.9 mL, 177 mmol), followed by drop-wise addition of n-butyllithium (2.5 M solution in hexanes, 68.0 mL, 170 mmol), and the reaction mixture was stirred for 30 minutes. A solution of P1 (20 g, 140 mmol) in a mixture of toluene and tetrahydrofuran (10:1 , 22 mL) was then added drop-wise; the reaction temperature was maintained below -72 C during the course of both additions. Stirring was continued for 1 hour at -78 C, whereupon the reaction was quenched via addition of saturated aqueous ammonium chloride solution. The aqueous layer was extracted three times with ethyl acetate, and the combined organic layers were washed with water and with saturated aqueous sodium chloride solution, dried over sodium sulfate, filtered, and concentrated in vacuo. Silica gel chromatography (Gradient: 0% to 100% ethyl acetate in heptane) afforded the product as a tacky amber oil. Yield: 36.34 g, 1 19.1 mmol, 85%. LCMS m/z 305.0, 307.0 [M+H]+. 1H N MR (400 MHz, CDCI3) delta 7.22 (s, 1 H), 3.97 (AB quartet, upfield doublet is broadened, JAB=12.6 HZ, AVAB=13.4 HZ, 2H), 3.67-3.76 (m, 3H), 3.38 (br ddd, J=1 1 .8, 6.9, 4.6 Hz, 1 H), 1 .90 (ddd, J=1 .1 , 6.9, 2.1 Hz, 1 H), 1 .42 (ddd, J=14.1 , 1 1.7, 1 1 .7 Hz, 1 H), 1 .27 (d, J=6.2 Hz, 3H).
85%
2,4-Dibromo-1,3-thiazole (44.7 g, 184 mmcl) was dissolved in a mixture of toluene and tetrahydrofuran (10:1, 900 mL) and cooled to -78 00 To this solution was added boron trifluoride diethyl etherate (21.9 mL, 177 mmcl), followed by drop-wise addition of n-butyllithium (2.5 M solution in hexanes, 68.0 mL, 170 mmcl), and the reaction mixture was stirred for 30 minutes. A solution of 03 (20 g, 140 mmol) in amixture of toluene and tetrahydrofuran (10:1, 22 mL) was then added drop-wise; the reaction temperature was maintained below -72 00 during the course of both additions. Stirring was continued for 1 hour at -78 0, whereupon the reaction was quenched via addition of saturated aqueous ammonium chloride solution. The aqueous layer was extracted three times with ethyl acetate, and the combined organic layers were washedwith water and with saturated aqueous sodium chloride solution, dried over sodium sulfate, filtered, and concentrated in vacuo. Silica gel chromatography (Gradient: 0% to100% ethyl acetate in heptane) afforded the product as a tacky amber oil. Yield: 36.34 g, 119.1 mmol, 85%. LCMS m/z 305.0, 307.0 [M+H]. 1H NMR (400 MHz, ODd3) oe 7.22 (s, 1H), 3.97 (AB quartet, upfield doublet is broadened, JAB=12.6 Hz, IVAB=l3.4 Hz, 2H), 3.67-3.76 (m, 3H), 3.38 (brddd, J=11.8, 6.9, 4.6 Hz, 1H), 1.90 (ddd, J=14.1, 6.9,2.1 Hz, 1H), 1.42 (ddd, J=14.1, 11.7, 11.7 Hz, 1H), 1.27 (d, J=6.2 Hz, 3H).
N-(2-methyl-5-nitrophenyl)-4-bromothiazol-2-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
2.2 g
With tris-(dibenzylideneacetone)dipalladium(0); potassium carbonate; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene In toluene at 100℃; Inert atmosphere;
1.1 Step (1): Preparation of N - (2-methyl-5-nitrophenyl) -4-bromo-thiazol-2-amine
2,4-dibromo-thiazole (20.6 mmol), 2-methyl _5_ nitroaniline (22.6 mmol), potassium carbonate (61.8 mmol), tris (dibenzylidene acetone) palladium (0.618 mmol) and 4,5-bis (diphenylphosphino) _9,9- dimethyl xanthene (1.9 mmol) were mixed in toluene (100 ml).Was replaced with nitrogen three times, and stirred overnight at 100 ° C under.Cooled to room temperature, 50 ml of water, and extracted three times with dichloromethane.The extract was dried over anhydrous sodium sulfate, and column chromatography (petroleum ether: ethyl acetate = I: I) was isolated the title compound 2.2 g.
2.2 g
With tris-(dibenzylideneacetone)dipalladium(0); potassium carbonate; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene In toluene at 100℃; Inert atmosphere;
5.A N-(2-methyl-5-nitro-phenyl)- 4-bromo-thiazol-2-amine preparation
The2,4-dibromo-thiazole (20.6 mmol), 2-methyl-5-nitroaniline (22.6 mmol),potassium carbonate (61.8 mmol), three (dibenzalacetone) palladium II (0.618mmol) and 4,5-bis (diphenyl phosphine) - 9,9-dimethyl-xanthene (1.9 mmol) oftoluene are mixed in (100 ml). Replacement by the nitrogen 3 times, in the 100°C lower stirring overnight. Cooling to room temperature, add 50 ml of water,dichloromethane is used for extracting 3 times. Extraction liquid drying byanhydrous sodium sulfate, column chromatography (petroleum ether: ethyl acetate= 1 the [...] 1) separation to obtain the title compound 2.2 g. 1H-NMR (CDCl 3) δ 7.55 (1H, dd, J=2.4Hz & J=2.4Hz), 7.50 (1H, d,J=2Hz), 7.17 (1H, d, J=8Hz), 3.89 (2H, brs), 2.24 (3H, s).
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; triethylamine; In N,N-dimethyl-formamide; at 70.0℃; for 3.0h;Sealed tube;
General procedure: To a nitrogen-purged solution of aryl iodide in TEA (3rriLlmmol), DMF (3mL/mmol) and MeOH (3mL/mmol) was added Palladium (II)Acetate (0.O3eq) and Xantphos (0.O6eq). The reaction mixture was flushed with Carbon Monoxide gas for several minutes and then sealed with CO balloon attached and heated to 60C for 3 hours. Upon completion, the reaction was cooled to room temperature and the crude product was triterated via addition of water and collected by filtration. The crude interemediate was taken into the next step w/o further purification.Similar to the procedure as described in General Procedure O, 2,4-dibromo-l,3- thiazole was reacted with carbon monoxide to give the title compound (1.2 g, 13%) as a Hi yellow solid. LC-MS (ES, m/z): 222 [M+H]+.
(3aR)-5,5-dimethyl-3,3a,4,5-tetrahydro-7H-pyrano[3,4-c][1,2]oxazole[ No CAS ]
[ 4175-77-3 ]
(3aR,7aR)-7a-(4-bromo-1,3-thiazol-2-yl)-5,5-dimethylhexahydro-1H-pyrano[3,4-c][1,2]oxazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
68%
Boron trifluoride diethyl etherate (3.00 mL, 23.7 mmol) was added to a ?65 00 slurry of 2,4-dibromo-1 ,3-thiazole (5.63 g, 23.2 mmcl) in toluene (50 mL). n-Butyllithium(2.5 M solution in hexanes; 10 mL, 25 mmol) was then slowly added, and the reactionmixture was allowed to stir at ?65 °C for 10 minutes. A solution of C920 (3.0 g, 19mmol) in tetrahydrofuran (5 mL) was added drop-wise, and stirring was continued for 30minutes at ?65 °C, whereupon the reaction was quenched via addition of saturatedaqueous ammonium chloride solution (150 mL) and the temperature warmed to ?10 °Cto 0 °C. The aqueous layer was extracted with ethyl acetate (2 x 100 mL), and the combined organic layers were washed with saturated aqueous sodium chloride solution (2 x 100 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Silica gel chromatography (Gradient: 0percent to 10percent ethyl acetate in petroleum ether) afforded theproduct as a yellow solid. Yield: 4.3 g, 13 mmol, 68percent. 1H NMR (400 MHz, CDCI3) 6 7.21 (s, 1H), 6.42 (s, 1H), 4.09 (d, J=13.0 Hz, 1H), 3.79-3.68 (m, 3H), 3.47-3.36 (m, 1H), 1.76 (dd, J=14.0, 6.6 Hz, 1H), 1.57 (brdd, J=13, 13Hz, 1H), 1.42 (5, 3H), 1.30 (5, 3H).
68%
Boron trifluoride diethyl etherate (3.00 mL, 23.7 mmol) was added to a ?65 °C slurry of 2,4-dibromo-1 3-thiazole (5.63 g, 23.2 mmol) in toluene (50 mL). n-Butyllithium(2.5 M solution in hexanes; 10 mL, 25 mmol) was then slowly added, and the reactionmixture was allowed to stir at ?65 °C for 10 minutes. A solution of C9 (3.0 g, 19 mmol)in tetrahydrofuran (5 mL) was added drop-wise, and stirring was continued for 30minutes at ?65 °C, whereupon the reaction was quenched via addition of saturatedaqueous ammonium chloride solution (150 mL) and the mixture was warmed to ?10 °Cto 0 °C. The aqueous layer was extracted with ethyl acetate (2 x 100 mL), and thecombined organic layers were washed with saturated aqueous sodium chloride solution (2 x 100 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Silica gel chromatography (Gradient: 0percent to 10percent ethyl acetate in petroleum ether) afforded the product as a yellow solid. Yield: 4.3 g, 13 mmol, 68percent. 1H NMR (400 MHz, CDCI3) oe 7.21(5, 1H), 6.42 (5, 1H), 4.09 (d, J=13.0 Hz, 1H), 3.79-3.68 (m, 3H), 3.47-3.36 (m, 1H), 1.76 (dd, J=14.0, 6.6 Hz, 1H), 1.57 (brdd, J=13, 13 Hz, 1H), 1.42 (5, 3H), 1.30 (5, 3H).
4-bromo-2-(pyrrolidin-l-yl)-1,3-thiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
With N-ethyl-N,N-diisopropylamine; In 1,4-dioxane; at 105℃; for 2h;
To a stirred solution of <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> 1 (1.0 g, 4.12 mmol) in 1,4 dioxane (30 mL) was added DIPEA (1.45 mL, 8.24 mmol) followed by pyrrolidine 2 (1.0 mL, 12.36 mmol) and the reaction mixture was heated at 105 C for 2 h. After completion of the reaction (monitored by TLC), the reaction mixture was concentrated under reduced pressure. Crude compound was purified by silica gel column chromatography eluting with 10-15% ethyl acetate in n- hexane to afford 0.80 g (83% yield) of 3 as a brown solid. LCMS-Condition-i: [M+H]+ = 232.8; Rt = 1.81 min.
With triethylamine; In N,N-dimethyl-formamide; at 30 - 100℃; for 2h;
Into a 2 L 4-necked round-bottom flask, was placed a solution of 2,4-dibromo- 1,3-thiazole (250 g, 1.03 mol, 1.00 equiv) inN,N-dimethylformamide (700 mL), pyrrolidine (73 g, 1.03 mol, 1.00 equiv), triethylamine (311.7 g, 3.08 mol, 3.00 equiv). The resulting solution was stirred for 2 hr at 100C and then cooled to 3 0C. The reaction mixture was then poured into 2 L of water/ice. The solid was collected by filtration. The filter cake was washed with 3x500 mL of water to give the title compound as a solid.
Hydroxyethyl thiazole 45: To a stirred solution of <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> 32 (10.2 g, 42.0 mmol, 1.0 equiv.) in diethyl ether (250 mL) at -78 °C was carefully added ra-butyllithium (2.5 M hexanes, 16.8 mL, 42.0 mmol, 1.0 equiv.). The reaction mixture was stirred for 20 min and then a solution of oxirane (2.5 M tetrahydrofuran, 16.8 mL, 42.0 mmol, 1.0 equiv.) was added, followed by dropwise addition of a solution of boron trifluoride diethyl etherate (5.18 mL, 42.0 mmol, 1.0 equiv.) in diethyl ether (30 mL). After 20 min, the reaction mixture was quenched with a saturated aqueous solution of ammonium chloride (50 mL) and allowed to warm to 25 °C. The two phases were separated, and the aqueous layer was extracted with ethyl acetate (3 chi 80 mL). The combined organic layers were dried with anhydrous sodium sulfate and concentrated in vacuo. The obtained residue was purified by flash column chromatography (silica gel, 30? 60percent ethyl acetate in hexanes) to afford pure thiazole 45 (5.42 g, 26.0 mmol, 62percent) as a colorless oil. 45: Rf = 0.24 (silica gel, 50percent ethyl acetate in hexanes); FT-1R (neat) v 3350, 3122, 2881, 1480, 1421, 1330, 1257, 1210, 1135, 1085, 1052, 938, 887, 857, 832, 733 cm 1; NMR (600 MHz, CDC13) delta = 7.12 (s, 1 H), 4.02 (td, J = 6.0, 6.0 Hz, 2 H), 3.22 (t, J = 6.0 Hz, 2 H), 2.67 (t, J = 6.0 Hz, 1 H) ppm;13C NMR (151 MHz, CDC13) delta = 169.7, 124.6, 116.5, 61.3, 36.2 ppm; HRMS (ESI) calcd for C5H7NOSBr [M+H]+ 207.9426, found 207.9421.
62%
To a stirred solution of <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> SI (10.2 g, 42.0 mmol, 1.0 equiv) in diethyl ether (250 mL) at -78 °C was added ra-butyllithium (2.5 M in hexanes, 16.8 mL, 42.0 mmol, 1.0 equiv) dropwise. After 20 min, ethylene oxide (2.5 M in tetrahydrofuran, 16.8 mL, 42.0 mmol, 1.0 equiv) was added, followed by dropwise addition of a solution of boron trifluoride diethyl etherate complex (5.18 mL, 42.0 mmol, 1.0 equiv) in diethyl ether (30 mL). After 20 min, the reaction mixture was quenched with saturated aqueous ammonium chloride solution (50 mL), and allowed to warm to 25 °C. The two phases were separated, and the aqueous layer was extracted with ethyl acetate (3 x 80 mL). The combined organic layers were dried with anhydrous sodium sulfate and concentrated under reduced pressure. The obtained residue was purified by flash column chromatography (silica gel, 30? > 60percent ethyl acetate in hexanes) to afford pure hydroxyethyl thiazole S16 (5.42 g, 26.0 mmol, 62percent yield) as a colorless oil. S16: =0.24 (silica gel, 50percent ethyl acetate in hexanes); FT-IR (neat) v 3350, 3122, 2881, 1480, 1421, 1330, 1257, 1210, 1135, 1085, 1052, 938, 887, 857, 832, 733 cm"1; NMR (600 MHz, CDC13) 5 = 7.12 (s, 1 H), 4.02 (td, 7=6.0, 6.0Hz, 2 H), 3.22 (t, /=6.0Hz, 2 H), 2.67 (t, /=6.0Hz, 1 H) ppm;13C NMR (151 MHz, CDC13) 5= 169.7, 124.6, 116.5, 61.3, 36.2 ppm; HRMS (ESI) calcd for C5H7BrNOS+ [M+H]+ 207.9426, found 207.9421.
tert-butyl (1-(4-bromothiazol-2-yl)-4-methylpiperidin-4-yl)-carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
87%
With triethylamine; In N,N-dimethyl-formamide; at 90℃; for 8h;
2,4-Dibromothiazole (121.5 mg, 0.5 mmol) and(4-methylpiperidin-4-yl) carbamic acidTert-butyl ester(160.7 mg, 0.75 mmol) were added to a 25 mL egg-shaped flask,5mL N, N-dimethylamide to dissolve,Triethylamine (0.3 mL, 1.5 mmol) was added dropwise,After the system was heated to 90 C for 8 hours.TLC tracking until the conversion of raw materials is completed, stop heating,After cooling to room temperature, add water to quench the reaction. Extraction with ethyl acetate,The organic phase was washed several times with anhydrous sodium sulfate.After concentration under reduced pressure, the crude product was purified by column chromatography (petroleum ether:Dichloromethane = 5: 1 ? petroleum ether: dichloromethane = 1:1) to give light yellow solid S9 (165 mg, yield 87%).
4-bromo-2-(1H-1,2,4-triazol-1-yl)thiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
86%
With potassium carbonate; In N,N-dimethyl-formamide; at 110℃; for 3h;
To a 50 mL two-neck flask were added <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (0.50 g, 2.1 mmol), 1H-1,2,4-triazole (0.22 g, 3.2 mmol) and anydrous DMF (9 mL), then potassium carbonate (0.57 g, 4.0 mmol) was added. The mixture was stirred at 110°C for 3 h. The reaction mixture was cooled to rt and quenched with water. The mixture was filtered by suction and the filtrate was concentrated in vacuo. The residue was purified by silica-gel column chromatography (DCM:EtOAc=25:1, V/V) to give a white solid (0.41 g, 86percent). MS(ESI, pos.ion)m/z: 232.1 (M+2);
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 80℃;Inert atmosphere;
General procedure: To a stuffed solution of 2,4-dibromothiazole (2) (4 mmol) in dimethylformamide (DMF) (15 mL), the coffesponding amine (1) (4 mmol) and diisopropylethylamine (DIPEA) (1.55 g, 2 mL, 12 mmol) were added under a stream of nitrogen.The reaction mixture was stirred at 80C for 4-10 h (monitored by Thin-layer chromatography (TLC)), then cooled to room temperature (rt), quenched with cold water (H20) (50 mL) and extracted with ethyl acetate (EtOAc) (3x30 mL). The combined organic layer was washed with H20 (2x10 mL), dried over sodium sulfate and concentrated to give the crude compound 3. The resulting product 3, the corresponding imidazole 4 (12 mmol),palladium(II)bis(triphenylphosphine) diacetate (Pd(OAc)2(PPh3)2) (0.1 mmol) and toluene (50 mL) were refluxed for 24 h. After cooling, the organic solvent was removed under vacuum. The resulting residue was purified by preparative high-performance liquid chromatography (HPLC) (EtOAc - Hexane (1:5) as eluent) to give the final product 5 as a solid. It was additionally purified by reverse phase HPLC (gradient acetonitrile (MeCN) - H20, 20 to 80% of MeCN aseluent). A compound of formula S are examples 1-15, 17-22, 24-40, 42-50 and 52-62.
(R)-3-oxohexahydroimidazo[1,5-a]pyrazine-7(1H)-carboxylic acid tert-butyl ester[ No CAS ]
[ 4175-77-3 ]
tert-butyl (S)-2-(4-bromothiazol-2-yl)-3-oxohexahydroimidazo[1,5-a]pyrazine-7(1H)-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
74%
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 90℃; for 12h;Inert atmosphere;
Under nitrogen protection, will(R)-3-oxo hexahydroimidazo[1,5-a]pyrazine-7(1H)-carboxylic acid tert-butyl ester(500 mg, 2.07 mmol), <strong>[4175-77-3]<strong>[4175-77-3]2,4-dibromothiazol</strong>e</strong> (503 mg, 2.07 mmol),Tris(dibenzylideneacetone)dipalladium (193 mg, 0.21 mmol),4,5-bisdiphenylphosphino-9,9-dimethyloxazepine(247mg, 0.41mmol)And cesium carbonate (1.35 g, 4.14 mmol) were dissolved in 1,4-dioxane (20 mL).The reaction solution is heated to 90 C for 12 h., then concentrated under reduced pressure, and the residue obtained was applied to silica gel column chromatography(PE/EtOAc (V/V) = 3/1) purificationThe title compound was obtained as a white solid (yield: 618 g, 74%).
3.1 Step 1: 4-Bromo-N, N-dimethylthiazol-2-amine
Add 2,4-dibromothiazole (2.6g, 11mmol) and N-methylmethanamine hydrochloride (1.7g, 21mmol) to a 100mL single-necked bottle, add DBU (4.8mL, 32mmol) at a temperature of 0 ), Warm up to room temperature and stir overnight. The reaction system was diluted with water (100 mL), and then extracted with EA (50 mL × 4). The organic phases were combined and washed with saturated brine (30 mL × 3). The combined organic phases were dried over anhydrous sodium sulfate, filtered, and depressurized. After concentration, the obtained residue was purified by thin layer chromatography (PE / EA (V / V) = 10/1) to obtain the title compound as a white solid (1.62 g, 7.82 mmol, 73%).
5-(4-bromothiazol-2-yl)-2-oxa-5-azabicyclo[2.2.1]heptane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
56%
With triethylamine; In dimethyl sulfoxide; at 0 - 60℃; for 6h;
To a stirred solution of 2,4-dibromothiazole 1 (0.5 g, 2.06 mmol) in DMSO (10 mL) was added Et3N (0.86 mL, 6.15 mmol) followed by 2-oxa-5-azabicyclo [2.2.1] heptane hydrochloride 2 (0.42 g, 3.09 mmol) at oC and the reaction mixture was heated at 60 C for 6 h. After completion of the reaction (monitored by TLC), the reaction mixture was diluted with ice cold water and extracted with ethyl acetate. Combined organic layer was washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure. Crude compound was purified by silica gel column chromatography eluting with 10% ethyl acetate in n- hexane to afford 0.30 g (56% yield) of 3 as an off-white solid. LCMS-Condition-i: [M+H]+ = 260.95; Rt = 1.93 min.
4-bromo-2-(6-(pyrrolidin-1-yl)pyridin-3-yl)thiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
84%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium carbonate In water; N,N-dimethyl-formamide at 100℃; for 1h; Inert atmosphere;
A71.1 Step 1 : 4-bromo-2-(6-(pyrrolidin- 1 -yl)pyridin-3-yl)thiazole
A mixture of 2,4-dibromothiazole (486 mg, 2.0 mmol, 1.0 eq.), 2-(pyrrolidin-l-yl)-5- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridine (603 mg, 2.2 mmol, 1.1 eq.), potassium carbonate (828 mg, 6.0 mmol, 3.0 eq.), and PdCh(dppf) (146 mg, 0.2 mmol, 0.1 eq.) in dioxane/water (10 ml 75 mL) was heated at 100 °C for 1 hour under nitrogen protection. The mixture was cooled to room temperature, poured into water, and extracted with ethyl acetate for three times. The organic extracts were combined, washed with brine, dried over anhydrous sodium sulfate, and concentrated. The residue was purified by silica gel column using hexane - EtOAc (0-100%)) to give 4-bromo-2-(6-(pyrrolidin-l-yl)pyridin-3-yl)thiazole (520 mg, 84% yield). LC-MS m/z [M+H]+ calc’d for Ci2Hi2BrN3S, 311; found, 311.
4-((4-bromothiazol-2-yloxy)methyl)-3-fluorobenzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
61%
Stage #1: 3-fluoro-4-(hydroxymethyl)benzonitrile With sodium hydride In tetrahydrofuran at 0℃; for 0.5h;
Stage #2: 2,4-dibromo-1,3-thiazole In tetrahydrofuran at 25℃; for 6h;
10.A Step A: The synthesis of 4- ( (4-bromothiazol-2-yloxy) methyl) -3-fluorobenzonitrile
To a solution of 3-fluoro-4- (hydroxymethyl) benzonitrile (1.51 g, 10 mmol) in dry THF (20 mL) were added to a solution of NaH (600 mg, 15mmol) in dry THF (15 mL) at 0. The mixture was stirred at 0 for 0.5h. 2, 4-dibromothiazole (2.42 g, 10 mmol) was added and the mixture was stirred at 25 for 6 h. The reaction was diluted with water (20 mL) and extracted with ethyl acetate (45 mL x 3) . The combined organic layers were dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by column chromatography to give 4- ( (4-bromothiazol-2-yloxy) methyl) -3-fluorobenzonitrile (1.9 g, yield: 61%) as white solid. MS Calcd.: 311.9; MS Found: 312.8 [M+H]+.
Stage #1: 2,4-dibromo-1,3-thiazole With isopropylmagnesium chloride lithium chloride In tetrahydrofuran at -10 - 0℃; for 1h;
Stage #2: trimethylsilan In tetrahydrofuran at -10 - 0℃; for 4h;
1.1 prepare 2-(trimethylsilyl)-4-bromothiazole:
Weigh 150g 2,4-dibromothiazole dissolved in 650mL tetrahydrofuran, temperature control -10 ~ 0 °C, dropwise add 264g of homemade 2.57mol / kg of isopropyl magnesium chloride tetrahydrofuran solution, after the end of the dropwise addition, the insulation reaction is 1h, the gas phase central control raw material reaction is complete, the temperature control -10 ~ 0 °C, Drop 73.8g trimethylsilane, after the end of the dropwise addition, the insulation reaction 4h, meteorological central control, intermediate conversion completely, temperature control -10 ~ 0 °C, drop into 50mL saturated brine quenching reaction, after the end of the drip stirring reaction for 10 minutes, filtration, filtrate concentrated to no dripping liquid, distillation and purification under reduced pressure, to give 2- (trimethylsilyl)-4-bromothiazole 89.4g, gas phase content of 97.8%, yield of 55%.
4-bromo-2-(7-tosyl-7H-pyrrolo[2,3-d]pyrimidin-5-yl)thiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65%
With palladium (II) [1,1'-bis(diphenylphosphanyl)ferrocene] dichloride; Cs2CO3 In 1,4-dioxane; lithium hydroxide monohydrate at 80℃; for 1h; Inert atmosphere; Microwave irradiation;
B; A Intermediate 7. 4-Bromo-2-(7-tosyl-7H-pyrrolo[2,3-b]pyrimidin-5-yl)thiazole.
To 5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-7-tosyl-7H-pyrrolo[2,3- djpyrimidine (900 mg, 2.25 mmol), 2,4-dibromothiazole (821 mg, 3.38 mmol), and CS2CO3 (1.84 g, 5.65 mmol) was added 1 ,4-dioxane (12.5 mL) and H2O (2.5 mL). The mixture was sparged with Ar for 5 minutes and then treated with Pd(dppf)Cl2 (165 mg, 0.226 mmol). The mixture was sparged with Ar for another 5 minutes and the resultant mixture was subjected to microwave irradiation at 80 °C for 1 h. The mixture was cooled to room temperature, diluted with water (50 mL) and extracted with ethyl acetate (50 mL x 3). The combined organic solvent extracts were dried over anhydrous Na2SO4, filtered, concentrated and purified by FCC (petroleum ether: ethyl acetate = 20:1 to 5:1 ) to yield 4-bromo-2-(7-tosyl-7H-pyrrolo[2,3-b]pyrimidin-5-yl)thiazole (900 mg, 65%) as a brown solid. LCMS (ESI): mass calcd. for C16H11BrN4O2S2, 433.95; m/z found, 434.9 [M+Hj+
4-bromo-2-(1H-pyrrolo[3,2-b]pyridin-1-yl)thiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
30%
With copper (I) iodide; Cs2CO3 In N,N-dimethyl-formamide at 120℃; for 16h; Inert atmosphere;
D Intermediate 170. 4-Bromo-2-(1H-pyrrolo[3,2-b]pyridin-1-yl)thiazole
1H-Pyrrolo[3,2-b]pyridine (1.0 g, 8.5 mmol) was added to a mixture of 2,4- dibromothiazole (2.5 g, 10 mmol), Cs2CO3 (6.9 g, 21 mmol), and DMF (25 ml). The resultant mixture was sparged with N2 for 5 minutes and then treated with Cul (1 .6 g, 8.4 mmol). The resultant mixture was sparged with N2 for another 5 minutes and then heated at 120 °C for 16 hours. After this time, the mixture was cooled to room temperature and filtered. The filtrate was diluted with saturated NH4CI (10 mL) and extracted with ethyl acetate (30 mL x 3). The combined organic extracts were washed with brine (10 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford the product, which was purified by FCC (eluent: petroleum ether: ethyl acetate = 1 :0 to 1 :1 ) to afford the title compound (900 mg, 30%) as a white solid. LCMS (ESI): mass calcd. for C10H6BrN3S 278.95 m/z, found 281.8 [M+1]+.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +
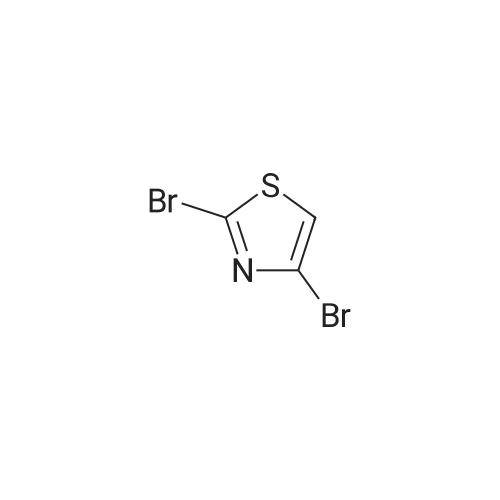

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping