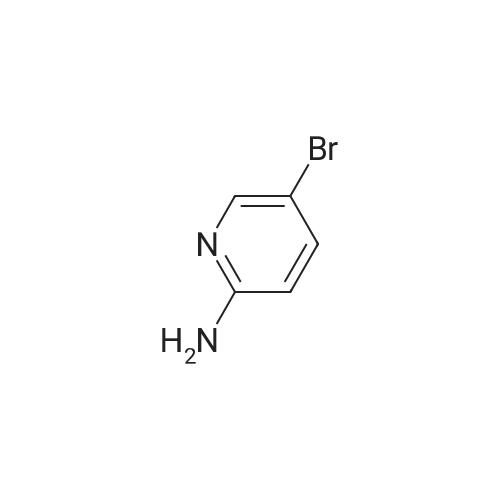
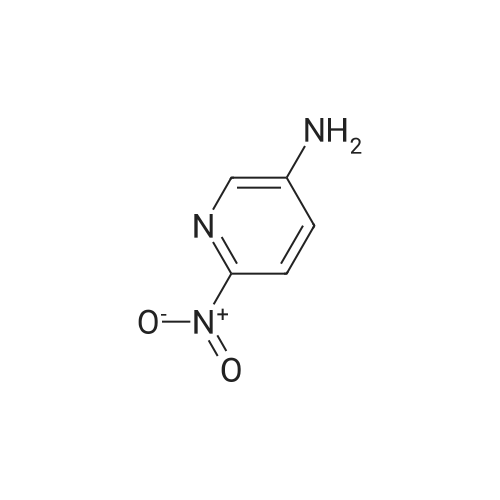
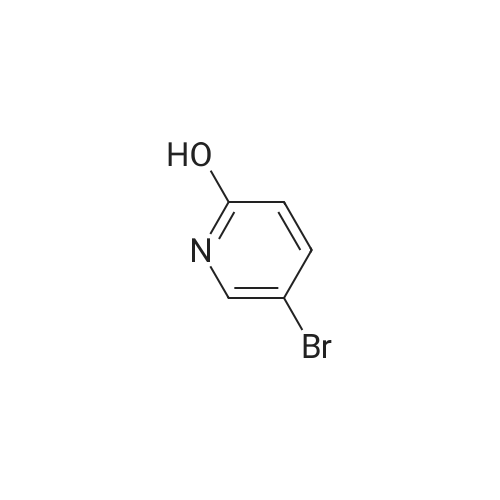
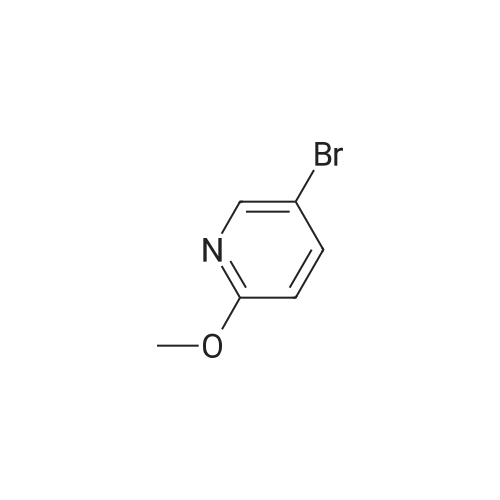
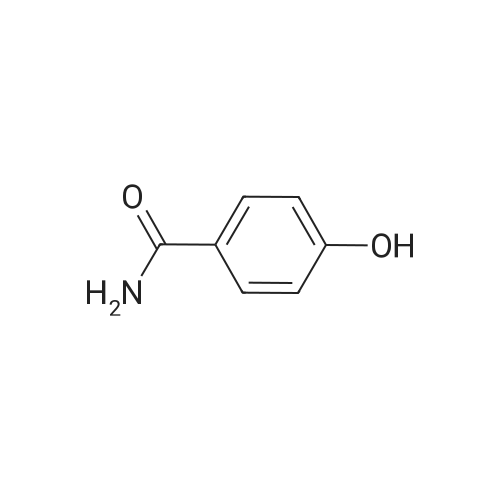
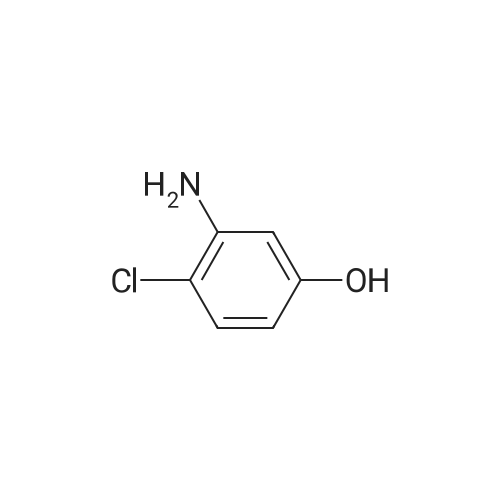
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
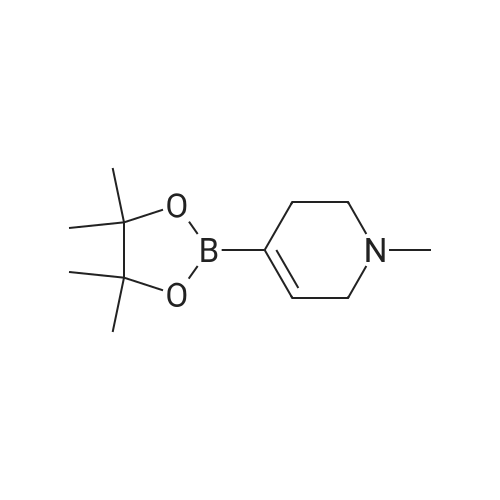
With tetrahydroxydiboron; 5%-palladium/activated carbon; water In acetonitrile at 50℃; for 24 h;
General procedure: Nitrobenzene (0.6mmol), 5wtpercent Pd/C (0.5mmol percent, 0.003mmol), H2O (10 equiv, 6.0mmol), B2(OH)4 (3.3 equiv, 2.0mmol), and CH3CN (1.0mL) were added in a 10mL tube. The reaction mixture was stirred at 50°C for 24h. When the reaction was complete monitored by TLC, the mixture was cooled to room temperature. Water (5mL) was added, and extracted with EtOAc (3×5mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give aniline 2a (55mg, 99percent).
Reference:
[1] Tetrahedron, 2017, vol. 73, # 27-28, p. 3898 - 3904
[2] Organic Process Research and Development, 2017, vol. 21, # 2, p. 247 - 252
[3] Organic Letters, 2016, vol. 18, # 11, p. 2774 - 2776
[4] ACS Catalysis, 2015, vol. 5, # 3, p. 1526 - 1529
[5] Yakugaku Zasshi, 1952, vol. 72, p. 381[6] Chem.Abstr., 1953, p. 6403
[7] Zhurnal Obshchei Khimii, 1940, vol. 10, p. 1105[8] Chem. Zentralbl., 1940, vol. 111, # II, p. 3474
[9] Chemical Communications, 2010, vol. 46, # 10, p. 1769 - 1771
[10] Chemical Communications, 2011, vol. 47, # 39, p. 10972 - 10974
[11] Green Chemistry, 2015, vol. 17, # 2, p. 898 - 902
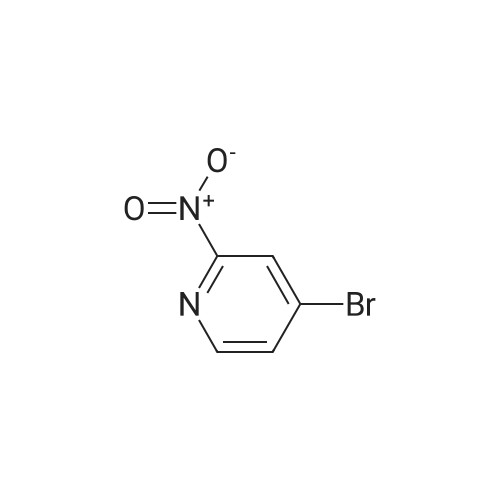
2
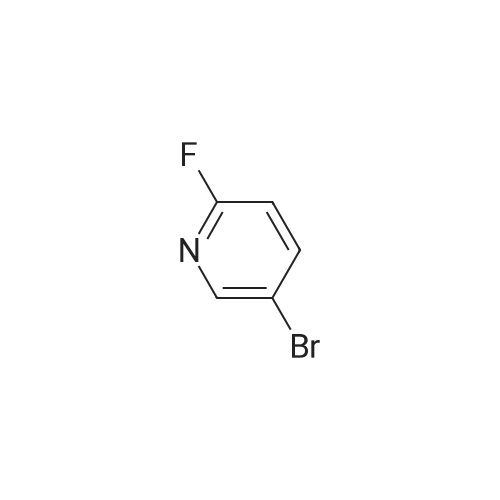
[ 39856-50-3 ]
[ 1072-97-5 ]
[ 14916-65-5 ]
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1953, vol. 72, p. 125,131
3
[ 39856-50-3 ]
[ 1072-97-5 ]
[ 14916-65-5 ]
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1953, vol. 72, p. 125,131
4
[ 39856-50-3 ]
[ 64-17-5 ]
[ 7664-41-7 ]
[ 1072-97-5 ]
[ 14916-65-5 ]
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1953, vol. 72, p. 125,131
A solution of 5-bromo-2-nitropyridine (5.0 g, 24.63 mmol), in methanol (100 mL) was added sodium methoxide (2.67 g, 49.44 mmol) and stirred at 75 00 for 2 h. The reaction mixture was diluted with water (150 mL) and extracted with ethyl acetate (2 x 200 mL). The combined extracts were washed with water (200 mL), brine (200 mL), dried over anhydrous Na2SO4 and concentrated. The crude compound was purified by column chromatography over silica gel (100-200 mesh) using a solvent gradient mixture of 5percent ethyl acetate in pet-ether as eluant to afford 2.5 g (54percent) of 5-bromo-2-methoxypyridine 122-1 as a colorless liquid. 1H NMR (400 MHz, CDCI3): c5 8.19 (d, J= 2.2 Hz, 1 H), 7.63 (dd, J= 1.8, 8.8 Hz, 1H), 6.66 (d, J= 8.7 Hz, 1H), 3.90 (5, 3H). ESI-LC/MS: m/z190.13 [(M+2)H+]; R = 3.13 mm [Agilent [C with Ion trap Detector; Waters Symmetry 018, 3.5 pm, 4.6 X 75 mm column; gradient of 50:50 H20 (0.1percent H000H): CH3CN (0.1percent H000H) to 10:90 H20 (0.1percent H000H): CH3CN (0.1percent H000H) in 4mm and hold for 3mm with flow rate of 1.0 mL/min].
Reference:
[1] Yakugaku Zasshi, 1952, vol. 72, p. 381[2] Chem.Abstr., 1953, p. 6403
9
[ 39856-50-3 ]
[ 141-52-6 ]
[ 55849-30-4 ]
Reference:
[1] Yakugaku Zasshi, 1952, vol. 72, p. 381[2] Chem.Abstr., 1953, p. 6403
[3] Recueil des Travaux Chimiques des Pays-Bas, 1949, vol. 68, p. 275,282
10
[ 7647-01-0 ]
[ 39856-50-3 ]
[ 142-08-5 ]
[ 13472-81-6 ]
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1951, vol. 70, p. 182,190
11
[ 504-29-0 ]
[ 35486-42-1 ]
[ 39856-50-3 ]
Reference:
[1] Organic Process Research and Development, 2017, vol. 21, # 3, p. 451 - 459
[2] Organic Process Research and Development, 2017, vol. 21, # 3, p. 451 - 459
12
[ 1072-97-5 ]
[ 35486-42-1 ]
[ 39856-50-3 ]
Reference:
[1] Organic Process Research and Development, 2017, vol. 21, # 3, p. 451 - 459
13
[ 39856-50-3 ]
[ 15206-26-5 ]
Yield
Reaction Conditions
Operation in experiment
21.8%
Stage #1: With potassium acetate; palladium diacetate; bis(pinacol)diborane In N,N-dimethyl-formamide at 100℃; for 16 h; Stage #2: With sodium perborate tetrahydrate In tetrahydrofuran; water at 20℃; for 16 h;
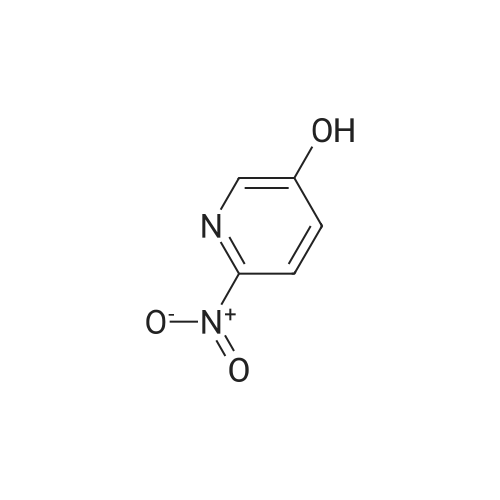
A suspension of 5-bromo-2-nitro-pyridine (10.0 g, 49 mmol), Boric acid ester (13.7 g, 54 mmol), Pd (OAc)2 (121 mg, 0.54 mmol) and KOAc (14.4 g, 147 mmol) in DMF (180 mL) was heated to 100 °C for 16 h. After the solvent was removed under vacuum, the remaining residue was mixed with EtOAc (600 mL). The EtOAc solution was washed with water (100 mL), brine (100 mL), and dried over Na2S04. It was concentrated to a residue that was mixed with NaBOs 4H20 (19.0g, 125 mmol), THF (180 mL) and H20 (180 mL). The resulting mixture was stirred at room temperature for 16 h. The aqueous phase was separated with the organic phase, and washed with EtOAc (100 mL x 2). The combined organic solution was then concentrated to give 6-nitro-pyridin-3-ol (1.5 g, 21.8 percent) as a yellow solid.
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1953, vol. 72, p. 125,131
15
[ 39856-50-3 ]
[ 1072-97-5 ]
[ 14916-65-5 ]
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1953, vol. 72, p. 125,131
16
[ 39856-50-3 ]
[ 64-17-5 ]
[ 7664-41-7 ]
[ 1072-97-5 ]
[ 14916-65-5 ]
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1953, vol. 72, p. 125,131
17
[ 1072-97-5 ]
[ 39856-50-3 ]
Yield
Reaction Conditions
Operation in experiment
86.1%
Stage #1: at 20℃; for 0.5 h; Autoclave; Large scale Stage #2: at 30 - 40℃; for 20 h; Large scale
500 L of glacial acetic acid and 150 L of acetic acid recovered in Example 1 were charged into a 1000 L autoclave, and the circulating water was stirred and stirred. After homogeneous mixing, 120 kg of 2-amino-5-bromopyridine was slowly added and the temperature was controlled at 20C. , Stirring 30min, slowly dropping 300kg peracetic acid, when dropping to about 50kg, the temperature began to slowly rise, stop dropping, when the temperature does not rise began to decline, and then dropping the remaining amount, until the peracetic acid plus Finished, the whole process temperature control at 30 . After completion of the dropwise addition, the temperature was controlled at 40 ° C and the reaction was carried out at constant temperature for 20 hours. When the reaction is complete, vacuum distillation, steaming about 600L acetic acid, to stop the distillation. The remaining liquid cooling to 25 , and then add 500L of water, with 40percent sodium hydroxide solution to adjust the pH value of the material to 8, cooled to -20 , filtration, drying, 125kg solid, crude The yield was 89.3percent. The recrystallized product was 120.5kg and the yield was 86.1percent.
73.4%
With sulfuric acid; dihydrogen peroxide In water at 5 - 15℃;
Add 1000ml of 30percent hydrogen peroxide solution to a 1000ml three-neck reaction flask, cool down to 15 °C with a low temperature reaction bath, and drop 320ml of 98percent concentrated sulfuric acid into the hydrogen peroxide water in the reaction bottle with a constant pressure dropping funnel. The temperature of the solution in the bottle is controlled at 5 In the range of -15 ° C, the solution B was added dropwise. At the same time, 160ml of 98percent concentrated sulfuric acid was added to another 1000ml three-neck reaction flask, and 60.00g of 2-amino-5-bromopyridine was added in batches by a low temperature reaction bath, and the temperature was controlled within the range of 5-15 °C.The solution A was obtained after the addition.The A and B solutions are all prepared, and the solution B is added dropwise to the solution A by a constant pressure dropping funnel, and the temperature of the reaction bottle is controlled by the low temperature reaction bath in the range of 0-10 ° C, and the optimum temperature is 5 ° C. The temperature change value was not more than 3 ° C. After the dropwise addition was completed, the reaction liquid was slowly heated to 50 ° C, the reaction was continued for 3 hours, and the sample was taken for TLC analysis to disappear.Add 5000 ml of water to a 5000 ml three-neck reaction flask, and cool to within 15 ° C. The above reaction solution is added to water with a constant pressure dropping funnel while stirring, and the temperature is controlled within a range of -15 ° C. After the dropwise addition is completed, Continue stirring for 18-20 minutes. At 5-15 ° C, 720 g of 30percent liquid alkali was added, stirring was continued for 0.5 hours, then filtered under reduced pressure, washed with 100 ml of water, and the filter cake was neutralized with a dilute alkali to pH = 7 and filtered again. The resulting solid was recrystallized from methanol and filtered hot.Dry to give a pale yellow solid of 51.13 g of product.Yield 73.4percent,The purity of the product liquid chromatography is greater than 99percent.
72%
With dihydrogen peroxide In sulfuric acid at 0 - 20℃;
Reference Example 11: 5-Bromo-2-nitro-pyridine. A solution of 2-amino-5-bromo-pyridine (5 g, 28.9 mmol) in cone, sulfuric acid (10 mL) was added dropwise to a cold (0 0C) mixture of hydrogen peroxide (10 mL, 38percent) and cone, sulfuric acid (10 mL). The mixture was warmed to r.t. and stirred overnight, then poured into ice cold water and filtered. The filtrate was basified with potassium hydroxide and extracted with ethyl acetate. The organic phase was dried over anhydrous sodium sulfate and concentrated to afford 5-bromo-2-nitro-pyridine (4.2 g, 72percent).
Reference:
[1] Patent: CN106187867, 2016, A, . Location in patent: Paragraph 0035; 0044; 0045
[2] Bioorganic and Medicinal Chemistry, 1999, vol. 7, # 3, p. 467 - 479
[3] Patent: CN108558745, 2018, A, . Location in patent: Paragraph 0023; 0024; 0025; 0026
[4] Patent: WO2008/62182, 2008, A1, . Location in patent: Page/Page column 113
[5] Journal of Organic Chemistry, 2008, vol. 73, # 23, p. 9326 - 9333
[6] Journal of the American Chemical Society, 1945, vol. 67, p. 668
[7] Yakugaku Zasshi, 1952, vol. 72, p. 381[8] Chem.Abstr., 1953, p. 6403
[9] Recueil des Travaux Chimiques des Pays-Bas, 1949, vol. 68, p. 275,282
[10] Zhurnal Obshchei Khimii, 1940, vol. 10, p. 1105[11] Chem. Zentralbl., 1940, vol. 111, # II, p. 3474
[12] Patent: US2002/52349, 2002, A1,
[13] Organic Process Research and Development, 2017, vol. 21, # 3, p. 451 - 459
18
[ 2402-97-3 ]
[ 39856-50-3 ]
[ 1678-49-5 ]
Reference:
[1] Asian Journal of Chemistry, 2013, vol. 25, # 8, p. 4632 - 4636
19
[ 504-29-0 ]
[ 39856-50-3 ]
Reference:
[1] Organic Process Research and Development, 2017, vol. 21, # 3, p. 451 - 459
[2] Organic Process Research and Development, 2017, vol. 21, # 3, p. 451 - 459
20
[ 504-29-0 ]
[ 35486-42-1 ]
[ 39856-50-3 ]
Reference:
[1] Organic Process Research and Development, 2017, vol. 21, # 3, p. 451 - 459
[2] Organic Process Research and Development, 2017, vol. 21, # 3, p. 451 - 459
21
[ 626-55-1 ]
[ 39856-50-3 ]
[ 1678-49-5 ]
Reference:
[1] Asian Journal of Chemistry, 2013, vol. 25, # 8, p. 4632 - 4636
22
[ 504-29-0 ]
[ 39856-50-3 ]
Reference:
[1] Organic Process Research and Development, 2017, vol. 21, # 3, p. 451 - 459
23
[ 1072-97-5 ]
[ 35486-42-1 ]
[ 39856-50-3 ]
Reference:
[1] Organic Process Research and Development, 2017, vol. 21, # 3, p. 451 - 459
24
[ 1072-97-5 ]
[ 39856-50-3 ]
Reference:
[1] Organic Process Research and Development, 2017, vol. 21, # 3, p. 451 - 459
25
[ 39856-50-3 ]
[ 683-60-3 ]
[ 870521-31-6 ]
Reference:
[1] Yakugaku Zasshi, 1952, vol. 72, p. 381[2] Chem.Abstr., 1953, p. 6403
26
[ 39856-50-3 ]
[ 100-51-6 ]
[ 83664-33-9 ]
Reference:
[1] Journal of Organic Chemistry, 2008, vol. 73, # 23, p. 9326 - 9333
With triethylamine; lithium chloride In dimethyl sulfoxide at 60 - 65℃; for 12 h;
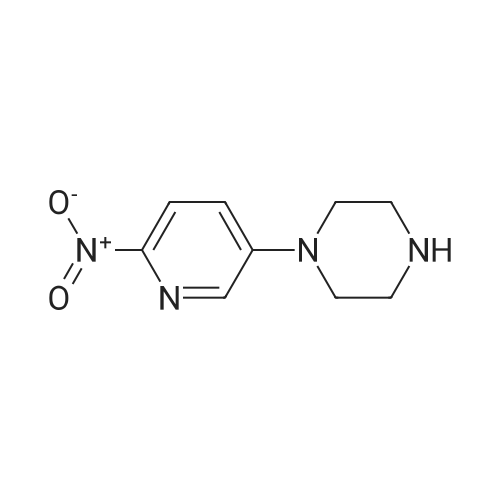
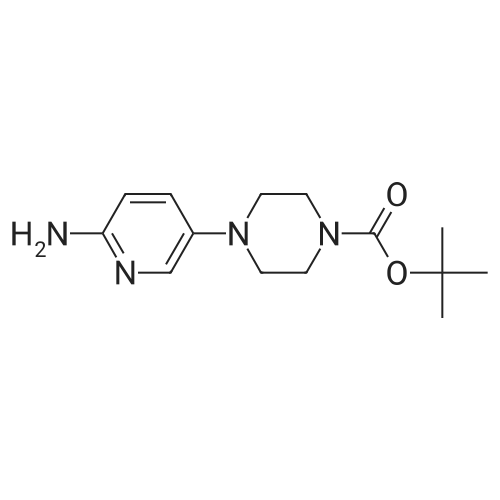
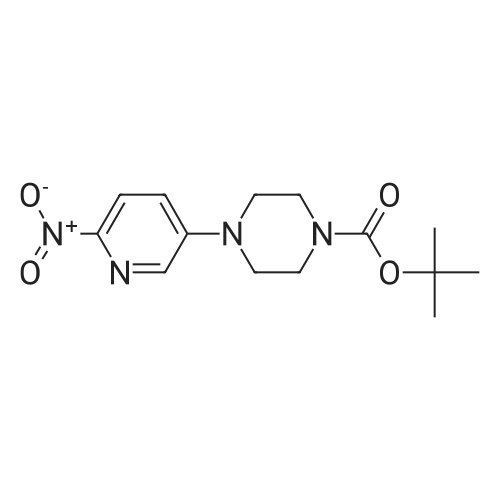
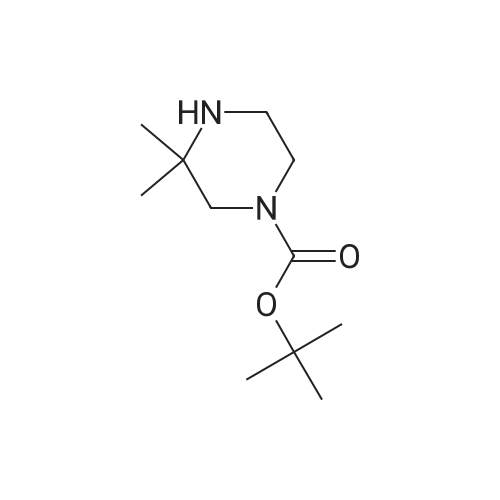
To a vessel was added 5-bromo-2-nitropyridine (10.0 g, 1.0 equiv.) along with DMSO (25 ml_, 2.5 vol). N-Boc piperazine (13.8 g, 1.5 equiv.) was added, followed by triethylamine (7.5 g, 1.5 equiv.) and LiCI (2.1 g, 1.0 equiv.). The mixture was warmed to 60-65°C for a minimum of 12 hours.Water (5 ml_, 0.5 vol) was added slowly to the vessel at 60-65°C. The mixture was kept at 60-65 °C for one hour, then cooled to room temperature. The slurry was kept at 20-25 °C for 1 hour and then filtered onto a 2 Whatman™ paper filter. The cake was rinsed with water (50 ml_, 5 vol.). The crude solids were collected and transferred back to a clean vessel. Water (100 mL, 10 vol.) was added to the vessel containing the solids and the mixture was warmed to 35-40°C for 2 hours, then filtered while warm onto a 2 Whatman paper™ filter. The solids were rinsed with water (40 mL, 4 vol.) and allowed to dry overnight in the vacuum oven at 50-55°C. The 4-(6-nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester was isolated as a yellow solid (14.1 g collected; -93percent yield).
90%
With triethylamine; lithium chloride In dimethyl sulfoxide at 60 - 65℃; for 12 h;
500 mL of the reaction flask was added to the starting material III20.3 g (0.1 mol) and IV27.9 g (0.15 mol) of starting material,200mL DMSO,15.2 g (0.15 mol) of triethylamine and 4.2 g (0.1 mol) of lithium chloride were added with stirring,System temperature to 60-65 incubation reaction 12h.65 ° C to the system slowly adding 200mL of water,Stir for 1 h. The system was cooled to room temperature and stirred for 1 h. The mixture was filtered and washed thoroughly with water at 50-55 ° C overnight to give 27.7 g of pure product as a pale yellow solid. Yield: 90.0percent
88.87%
With triethylamine In dimethyl sulfoxide at 60℃; for 18 h; Inert atmosphere
Step 1 tert-butyl 4-(6-nitropyridin-3-yl)piperazine-1-carboxylate To a solution of 5-bromo-2-nitropyridine (20.00 g, 98.53 mmol, 1.00 equivalent) in dimethylsulfoxide (52 mL) were added tert-butyl piperazine-1-carboxylate (24.00 g, 128.86 mmol, 1.31 equivalentalents) and triethylamine (20.00 g, 197.65 mmol. 2.01 equivalents). The solution was heated to 60°C and stirred for 18 hours. TLC (petroleum ether: ethyl acetate = 3: 1) showed completion of the reaction. The solution was diluted with water (200 mL), stirred for 30 minutes, and then filtered. The filter cake was washed with water and dried in vacuo to give a crude product. The crude product was purified by silica gel column (petroleum ether: ethyl acetate = 50: 1 to 20: 1) to give the title compound (27.00 g, 87.57 mmol, 88.87percent yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 9.03 Hz, 1H), 8.13 (d, J = 2.89 Hz, 1H), 7.21 (dd, J = 9.10, 2.95 Hz, 1H), 3.69-3.59 (m, 4H), 3.51-3.40 (m, 4H), 1.49 (s, 9H).
84.1%
With potassium carbonate In water; dimethyl sulfoxide at 70℃; for 28 h; Inert atmosphere
Step a): synthesis of 1-Boc-4-(6-nitro-3-pyridyl)piperazine (formula 3)Step b): A 1 -L RBF, was charged with 50 g of 5-Bromo-2-nitro-pyridine (0.246 mol, 1 .00 eq.), 175 ml DMSO and 35 ml water. To the resulting mixture was added 37.45 g potassium carbonate (0.271 mol, 1 .1 eq.) followed by 59.64 g boc-piperazine (0.320 mol, 1 .3 eq.). The mixture was heated to 70^ and stirred under argon until completion (approx. 28 h). The reaction was diluted with 315 ml of water and allowed to cool down to RT. After 2 h stirring, the solid was collected by filtration, washed with water (2 x 250 ml) and left in air at RT overnight to give crude wet 1 -Boc-4-(6-nitro-3- pyridyl)piperazine as yellow solid. HPLC (Method 1 ): 6.49 min (96.4percent) (230 nm)Crude product was suspended in 250 ml toluene, followed by concentrated under reduced pressure. The residue was dissolved in 200 ml toluene, under reflux. Resulting orange solution was allowed to cool down slowly to RT without stirring. After 3 h, precipitated solid was filtered, washed with toluene (50 ml), TBME (2 x 100 ml) and dried at 50<C/20 mbar for 7 h to yield 63.88 g of 1 -Boc-4-(6-nitro-3-pyridyl)piperazine as yellow solid (84.1 percent yield). HPLC (Method 1 ): 6.49 min (99.1 percent) (230 nm).
82%
With N-ethyl-N,N-diisopropylamine In dimethyl sulfoxide at 80℃;
To a solution of 5-bromo-2-nitropyridine (40.0 g,197 mmol) in DMSO (150 mL) were added 1-Bocpiperazine(47.4 g, 252 mmol) and DIPEA (38 mL,219 mmol) . The reaction mixture was heated at 80 °C for11 h. The reaction mixture was poured into ice-water andthen extracted with EtOAc. The combined extracts werewashed with water and brine. The organic layer was driedover Na2SO4, filtered, and concentrated under reducedpressure. Purification by column chromatography (1:9methanol/ dichloromethane) gave 1-Boc- 4-(6-nitro-pyridin-3-yl)-piperazine (49.9 g, 82percent) as yellow solid. 1H NMR(400 MHz, CDCl3): δ 8.11 (d, J = 9.1 Hz, 1H), 8.08 (d, J =2.7 Hz, 1H), 7.18 (dd, J = 9.1, 2.8 Hz, 1H), 3.65–3.56 (m,4H), 3.48–3.38 (m, 4H), 1.45 (s, 9H).
80%
With N-ethyl-N,N-diisopropylamine In acetonitrile for 72 h; Reflux
To a stirred solution of 5-bromo-2-nitropyridine (4.93 g, 24.3 mmol) and piperazine-1-carboxylic acid tert-bλxtyl ester (4.97 g, 26.7 mmol) in CH3CN (60 ml) is added DIPEA (4.65 mL, 26.7 mmol). The mixture is heated at reflux for 72 hours then cooled to room temperature and the precipitated product collected by filtration. The filtrate is concentrated and purified by flash column chromatography eluting with 30percent EtO Ac/petrol. The combined products are re-crystallized from EtO Ac/petrol to give 4-(6-nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-huty\\ ester, (4.50 g, 80percent yield). MS(ESI) m/z 308 (M+H)+
80%
at 120℃; for 3 h;
A solution of 1-Boc-piperazine (15.0 mmol) and 5-bromo-2-nitropyridine (5.00 mmol) in N-methyl pyrrolidone (15 mL) was stirred at 120 °C for 3 h. The reaction mixture was diluted with water and the precipitate was collected by filtration to give 2 (80percent) as a colorless powder. 1H NMR (400 MHz, DMSO-d6): δ 8.10–8.08 (d, J = 9.2 Hz, 1H, ArH), 7.96–7.95 (d, J = 3.0 Hz, 1H, ArH), 7.33–7.30 (dd, J = 3.4 Hz, 1H, ArH), 3.66–3.63 (m, 4H, piperazine-CH2), 3.43–3.41 (m, 4H, piperazine-CH2), 1.47 (s, 9H, t-butyl-CH3).
80%
With N-ethyl-N,N-diisopropylamine In acetonitrile for 72 h; Reflux
Nitrile analogues can be made by the following. To a stirred solution of 5-bromo-2-nitropyridine (4.93 g, 24.3 mmol) and piperazine-1-carboxylic acid tert-butyl ester (4.97 g, 26.7 mmol) in CH3CN (60 ml) is added DIPEA (4.65 mL, 26.7 mmol). The mixture is heated at reflux for 72 hours then cooled to room temperature and the precipitated product collected by filtration. The filtrate is concentrated and purified by flash column chromatography eluting with 30percent EtOAc/petrol. The combined products are re-crystallized from EtOAc/petrol to give 4-(6-nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester, (4.50 g, 80percent yield). MS(ESI) m/z 308 (M+H)+
79%
With triethylamine In dimethyl sulfoxide at 65 - 70℃; for 30 h;
EXAMPLE 2: Preparation of 4-(6-Nitro-pyridin-3yl)-piperazine-1-carboxylic acid tert-butyl ester; B Exampl e 2A: Preparation of 4-(6-Nitro-pyridin-3yl)-piperazine-1-carboxylic acid tert-butyl ester To 1.0 kg (5 mol) 5-bromo-2-nitropyridine was added 1.2 kg {6.4 mol) boc piperazine (tert-Butyl piperazine-1-carboxylate) in 2.6L DMSO and 0.5 kg triethylamine under nitrogen. The mixture was heated to 65-700C and held for 30~hours after which some solids precipitated. Water was added and the reaction cooled to 25°C over 2hrs. The resulting slurry was filtered, washed and dried at 45°C to give 1.2kg (79percent crude yield) of canary yellow solid intermediate (2A), which was used without further purification in the subsequent step.
79%
With tetra-(n-butyl)ammonium iodide; potassium carbonate In dimethyl sulfoxide at 120℃; for 16 h;
Synthesis of compound 107.1 To a solution of 78.4(1. Og, 4.92mmol, 1.0 eq) in DMSO (10 ml) was added Bu4NI (0.18g, 0.049 mmol, 0.1 eq), tert-butyl piperazine-1- carboxylate (1.37g, 7.38mmol, 1.5eq), and K2CO3 (1.36g, 9.85mmol, 2eq). Reaction mixture was heated at 120° C for 16 hours.Upon completion reaction mixture was poured into water and product was extracted with ethyl acetate. Organic layers were combined, dried over sodium sulphate and concentrated under reduced pressure. The crude was purified using column chromatography to provide 107.1 (1.2 g, 79percent). MS (ES): m/z 279.23 [M+H]+.
67%
for 72 h; Heating / reflux
Preparation XX <n="116"/>Synthesis of 4-r6-amino-pyridin-3-ylVpiperazine-l-carboxylic acid tert-butyl ester Step 1. Synthesis of 4-f6-nitro-pyridin-3-yl)-piperazine-l-carboxyric acid tert-butyl ester; A mixture of 5-bromo-2-nitropyridine (4.93g, 24.30 mmol) and tert-butyl piperazine-1-carboxylate (5.Og, 26.7 mmol) in acetonitrile (60ml) was heated at refluxed for 3 days. The solvent was evaporated and the solid residue purified by flash chromatography on silica eluting with EtO Ac/Petrol (1:3) and recrystallised from EtO Ac/Petrol to afford the title compound as an orange solid (5.Og, 67percent) (LCMS: Rt 2.8, [M+H]+ 309).
60%
With N-ethyl-N,N-diisopropylamine In acetonitrile at 110℃; for 72 h;
To the reaction flask was added 5-bromo-2-nitropyridine (4.93 g, 24.3 mmol), Piperazine-1-carboxylic acid tert-butyl ester (4.97 g, 26.7 mmol)Diisopropylethylamine (4.65 mL, 26.7 mmol) and acetonitrile (60 mL)The mixture was stirred at 110 ° C for 72 hours.Cooled to room temperature and concentrated under reduced pressure. The residue was purified by column chromatography (DCM / MeOH = 10/1)The resulting residue was purified to give the title compound (4.5 g, white solid) in 60percent yield.
60%
With potassium carbonate; potassium iodide In dimethyl sulfoxide at 120℃;
Compound 11a (13.7 g, 73.9 mmol), compound 11b (10 g, 49.3 mmol), potassium iodide (81.8 mg, 0.493 mmol)and potassium carbonate (13.6 g, 98.6 mmol) were added into DMSO (100 mL). The reaction solution was stirred at120 °C overnight and then cooled to room temperature, adjusted to pH 7 with hydrochloric acid (1 mol) and then extractedwith dichloromethane. The aqueous phase was alkalize by saturated solution of sodium carbonate, and then extractedwith dichloromethane again. The organic phase was combined and dried over anhydrous Na2SO4, concentrated andthen slurried by water to give compound 11c (9.2 g, 60percent). LCMS:309(M+H)+, RT=1.710min.
50%
With N-ethyl-N,N-diisopropylamine In acetonitrile for 2 h; Reflux
N,N-Diisopropylethylamine (4.77g, 36.94mmol) was added to a solution of 82 5-bromo-2-nitropyridine (5.00g, 24.63mmol) and 100 tert-butyl piperazine-1-carboxylate (5.10g, 27.09mol) in 101 acetonitrile ([ACN] 30mL). The mixture was refluxed for 2h, cooled to RT, concentrated under a vacuum, and purified by silica gel column chromatography (from 102 PE/86 EA=1:1 to 103 DCM/87 MeOH=20:1) to obtain 104 tert-butyl 4-(6-nitropyridin-3-yl)piperazine-1-carboxylate (3.80g; yield, 50percent) as a yellow solid. Pd/C (100.0mg) was added to a solution of tert-butyl 4-(6-nitropyridin-3-yl) piperazine-1-carboxylate (925.0mg, 3.0mmol) in EA/MeOH (10 mL/10mL). The mixture was degassed by flushing with H2, stirred at RT under a H2 atmosphere for 2h, and filtered and concentrated under a vacuum to obtain INT-3 (792.0mg; yield, 95percent) as an off-white solid. ESI-MS: m/z 279.2 [M+H]+.
45.2%
With triethylamine In dimethyl sulfoxide for 12 h; Heating
4.1.39 Tert-Butyl 4-(6-nitropyridin-3-yl)piperazine-1-carboxylate (10a) A mixture of 5-bromo-2-nitropyridine (3.4 g, 16.9 mmol), tert-butyl piperazine-1-carboxylate (3.2 g, 16.9 mmol) and Et3N (3.4 g, 33.8 mmol) in DMSO (20 mL) was stirred at 75 °C for 12 h. The reaction mixture was cooled down to room temperature and diluted with water (10 mL), and then the product was extracted three times with EtOAc (25 mL). The combined organic layer was dried over Na2SO4, and the solvent was removed in vacuo. The residue product was purified on a silica gel column using petroleum ether/EtOAc (4:1, v/v) as eluent to afford 10a (2.19 g, 45.2percent) as a white solid. MS (ESI) m/z 309.2 [M+H]+; 1H NMR (400 MHz, CDCl3): δ 8.19 (d, J = 9.10 Hz, 1H), 8.13 (d, J = 3.00 Hz, 1H), 7.22 (dd, J = 3.20 Hz, 9.20 Hz, 1H), 3.65 (t, J = 5.00 Hz, 4H), 3.46 (t, J = 5.00 Hz, 4H), 1.49 (s, 9H).
40.63%
With N-ethyl-N,N-diisopropylamine In N,N-dimethyl-formamide at 50℃;
Compound 38 (10.2 g, 49.52 mmol) was added to the reaction flask, respectively,Compound 36 (10.40 g, 54.47 mmol)Dissolved in DMF,DIPEA (7.78 g, 59.42 mmol) was added.50 ° C under the conditions of reaction,TLC tracking, to be completely complete,The reaction system is poured into ice water,There is a yellow solid precipitation,Filter solids, beat with EA,Dried to obtain 6.20 g of compound 39,Yield: 40.63percent.
37%
With potassium carbonate In dimethyl sulfoxide at 65℃;
Into a solution of 5-bromo-2-nitropyridine (30 g, 148 mmol) in DMSO (1 L) were added K2CO3 (40 g, 296 mmol) and teri-butyl piperazine-l-carboxylate (28g, 148 mmol). The mixture was stirred at 65 degree for overnight. After cooling down, it was poured into water (2 L). The solid precipitated was collected and dried under vacuum. It was then further purified by flash column eluting with 20:1 petroleum ether/ethyl acetate and then with methylene chloride to give 188a as a yellow solid (17 g, 37percent). MS: [M+H]+ 309.
37%
With potassium carbonate In dimethyl sulfoxide at 65℃;
To a solution of 5-bromo-2-nitropyridine (30 g, 148 mmol) in DMSO (1 L) was added K2CO3 (40 g, 296 mmol) and tert-butyl piperazine-1-carboxylate (28 g, 148 mmol). The mixture was stirred at 65° C. overnight. After cooling down, it was poured into water (2 L). The precipitated solid was collected and dried under vacuum. It was then further purified by flash column eluting with 20:1 petroleum ether/ethyl acetate and then with methylene chloride to give 115a as a yellow solid (17 g, 37percent). MS: [M+H]+309.
37%
With potassium carbonate In dimethyl sulfoxide at 65℃;
Example 101g tert-Butyl 4-(6-Nitropyridin-3-yl)piperazine-1-carboxylate 101g Into a solution of 5-bromo-2-nitropyridine (30 g, 148 mmol) in DMSO (1 L) were added K2CO3 (40 g, 296 mmol) and tert-butyl piperazine-1-carboxylate (28g, 148 mmol). The mixture was stirred at 65 °C overnight. After cooling down, it was poured into water (2 L). The solid precipitated was collected and dried under vacuum. It was then further purified by flash column eluting with 20:1 petroleum ether/ethyl acetate and then with methylene chloride to give 101g as a yellow solid (17 g, 37percent). MS: [M+H]+ 309.
33%
With potassium carbonate In N,N-dimethyl-formamide at 120℃; for 36 h;
4-(6-nitro-3-pyridinyl)-1-piperazinecarboxylic acid, 1,1-dimethylethyl ester A solution is prepared of 1 g (4.9 mM) of 2-nitro-5-bromopyridine and 917 mg (4.9 mM) of N-Boc-piperazine in 10 ml of dimethylformamide and 1.02 g (7.4 mM) of potassium carbonate are added. The reaction mixture is agitated at 120° C. for 36 hours and then cooled. 50 ml of water are then added and extraction is carried out with ethyl acetate. The organic phase obtained is washed with water, dried and concentrated under reduced pressure. The crude product is purified by silica gel chromatography in eluding with the aid of a toluene/isopropanol mixture (98/2; v/v). The product sought after is thus obtained as a yellow solid (yield=33percent). 1H NMR (250 MHz, DMSO) δ: 8.24 (d, 1H); 8.17 (d, 1H); 7.46 (dd, 1H); 3.50 (m, 8H); 1.42 (s, 9H).
400 mg
at 120℃; for 18 h;
Dissolved 5-bromo-2-nitropyridine (1.0 g, 4.92 mmol) and tert-butyl piperazine-1- carboxylate (1.1 g, 5.91 mmol) in N-methylpyrrolidine and stirred at 120 C for 18 h. Thereafter the reaction mixture was cooled to 30 C and diluted with water and extracted with ethyl acetate (2 x 200 mL). The combined organic extract was washed with brine (50 mL). The organic layer was dried over sodium sulfate and concentrated under vacuum to give crude product. The crude product was purified by column chromatography to give 400 mg of the desired product. LC-MS: m/z calcd for C14H20N4O4, 308.33, no ionization 1H NMR (500 MHz, CDC13): δΗ 1.4 (9H, s, 0(CH3)3), 3.38 (4H, t, J = 5 Hz, NCH2CH2N), 3.58 (4H, t, J = 5 Hz, NCH2CH2N), 7.14 (1H, dd, Jl = 5 Hz, J2 = 10 HZ, ArCH), 8.06 (1H, d, J = 5 Hz, ArCH) and 8.11 (1H, d, J = 10 Hz, ArCH).
Reference:
[1] Patent: WO2014/128588, 2014, A1, . Location in patent: Page/Page column 32; 33
[2] Patent: CN105153149, 2017, B, . Location in patent: Page/Page column 2; 7
[3] Patent: EP3269715, 2018, A1, . Location in patent: Paragraph 0061; 0121; 0122
[4] Patent: WO2016/30439, 2016, A1, . Location in patent: Page/Page column 29; 38
[5] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 6, p. 3125 - 3140
[6] Medicinal Chemistry Research, 2018, vol. 27, # 6, p. 1666 - 1678
[7] Patent: WO2010/20675, 2010, A1, . Location in patent: Page/Page column 50
[8] European Journal of Medicinal Chemistry, 2014, vol. 81, p. 341 - 349
[9] Patent: US2016/8367, 2016, A1, . Location in patent: Paragraph 0204; 0205
[10] Patent: WO2008/32157, 2008, A2, . Location in patent: Page/Page column 25
[11] Patent: WO2015/131080, 2015, A1, . Location in patent: Paragraph 00669; 00670
[12] Patent: WO2008/7123, 2008, A2, . Location in patent: Page/Page column 114-115
[13] Patent: CN106608879, 2017, A, . Location in patent: Paragraph 0154-0157
[14] Patent: EP3284746, 2018, A1, . Location in patent: Paragraph 0095
[15] European Journal of Medicinal Chemistry, 2018, vol. 144, p. 1 - 28
[16] Bioorganic and Medicinal Chemistry, 2015, vol. 23, # 2, p. 348 - 364
[17] Patent: CN106905245, 2017, A, . Location in patent: Paragraph 0304; 0305; 0306; 0307; 0308
[18] Journal of Medicinal Chemistry, 2018, vol. 61, # 6, p. 2227 - 2245
[19] Patent: WO2011/140488, 2011, A1, . Location in patent: Page/Page column 270
[20] Patent: US2013/116262, 2013, A1, . Location in patent: Paragraph 0313; 0314
[21] Patent: EP2773638, 2015, B1, . Location in patent: Paragraph 0168; 0169; 1404
[22] ACS Medicinal Chemistry Letters, 2017, vol. 8, # 6, p. 608 - 613
[23] Patent: US2006/178360, 2006, A1, . Location in patent: Page/Page column 27
[24] Patent: WO2010/101849, 2010, A1, . Location in patent: Page/Page column 61
[25] Patent: WO2013/90497, 2013, A1, . Location in patent: Page/Page column 50; 51
[26] Patent: WO2014/195274, 2014, A1, . Location in patent: Page/Page column 48-49
[27] Patent: CN106749259, 2017, A, . Location in patent: Paragraph 0011; 0046; 0110; 0122; 0134
[28] Patent: CN108586452, 2018, A, . Location in patent: Paragraph 0024-0035
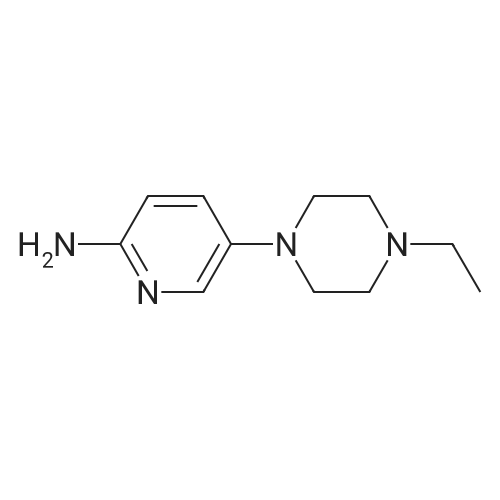
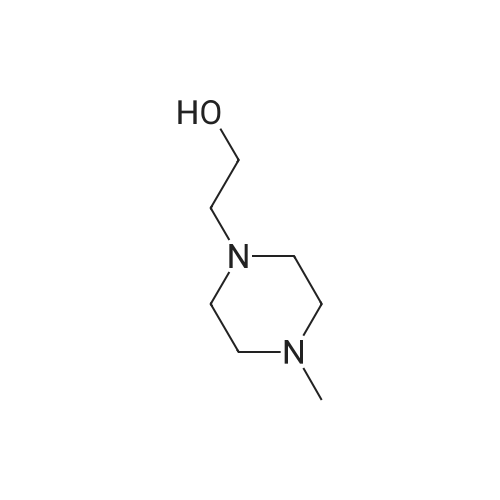
35
[ 39856-50-3 ]
[ 571189-16-7 ]
Reference:
[1] Journal of Medicinal Chemistry, 2005, vol. 48, # 7, p. 2388 - 2406
[2] Journal of Medicinal Chemistry, 2010, vol. 53, # 22, p. 7938 - 7957
[3] Journal of Medicinal Chemistry, 2017, vol. 60, # 5, p. 1892 - 1915
[4] Patent: EP3305785, 2018, A1,
[5] British Journal of Pharmacology, 2018, vol. 175, # 12, p. 2399 - 2413
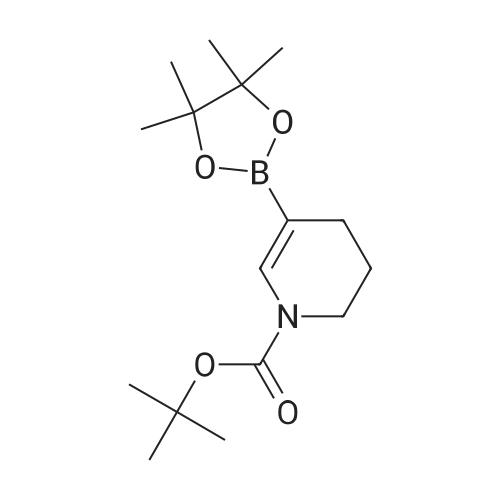
Stage #1: With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate In 1,4-dioxane at 80 - 90℃; Inert atmosphere Stage #2: With hydrogen In 1,4-dioxane at 20℃;
Under nitrogen protection, 550 mL of dioxane and 2-nitro-5-bromopyridine (20.3 g, After 0.10 mol) of pinacol borate (25.4 g, 0.10 mol) and potassium acetate (14.7 g, 0.15 mol), after stirring uniformly, the catalyst PdCl2dppf (0.74 g, 0.001 mol) was finally added, and the temperature was slowly raised to 80- 90 °C, Stir the reaction for 2-3 h. After the completion of the GC reaction, the reaction was stopped by cooling, and the reaction solution was filtered through celite to obtain a dark-black reaction solution. After charging at 1 atm of hydrogen, the mixture was reacted at room temperature overnight. After the reaction was completed, the activated carbon was decolorized, and the filtrate was distilled under reduced pressure until no liquid was poured. /Heptane mixed solvent was cooled and beaten for half an hour, filtered to obtain 18.5 g of light gray product, heated again with ethanol, and then cooled down, and the filter cake was rinsed with -20 ° C anhydrous ethanol, and dried to obtain 16.9 g, yield 77 percent HPLC purity 99.6percent
Reference:
[1] Patent: CN108047258, 2018, A, . Location in patent: Paragraph 0005; 0012; 0013
With tetrahydroxydiboron; 5%-palladium/activated carbon; water; In acetonitrile; at 50℃; for 24h;
General procedure: Nitrobenzene (0.6mmol), 5wt% Pd/C (0.5mmol %, 0.003mmol), H2O (10 equiv, 6.0mmol), B2(OH)4 (3.3 equiv, 2.0mmol), and CH3CN (1.0mL) were added in a 10mL tube. The reaction mixture was stirred at 50C for 24h. When the reaction was complete monitored by TLC, the mixture was cooled to room temperature. Water (5mL) was added, and extracted with EtOAc (3×5mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give aniline 2a (55mg, 99%).
With modified hectorite-loaded ionic liquid; In dimethyl sulfoxide; at 80℃; for 5h;Green chemistry;
1) 5-Bromo-2-nitropyridine (1 mol, 203 g) was dissolved in 1.5 L of dimethyl sulfoxide, and then an organically modified hectorite-supported ionic liquid material MHIL (40.6 g, 20 wt%) was added. Agitating and dispersing uniformly to obtain a first mixed liquid;2) Piperazine (129.2 g, 1.5 mol) was dissolved in 800 ml of dimethyl sulfoxide to form a piperazine solution, and then the piperazine solution was added dropwise to the first mixture to carry out a condensation reaction at 80 C;3) After 5 hours, the reaction solution was taken for HPLC detection (the percentage of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> in the reaction solution was 0.08%, and the target product 1-(6-nitropyridin-3-yl)piperazine was 99.68%. Double condensation by-product 0.17%, the balance is unknown impurities), stop the reaction, use an organic microporous membrane with a pore size of 0.5 mum to filter and remove the organic modified hectorite-loaded ionic liquid material to obtain a filtrate;4) The filtrate is warmed to 60-65 C, and then the aqueous solution of methylamine at a concentration of 3 V% is added dropwise. When the turbidity occurs in the system, the dropwise addition is stopped, the mixture is kept warm for 20-30 min, and then the aqueous solution of methylamine having a concentration of 3 V% is continuously added dropwise. The concentration of 1-(6-nitropyridin-3-yl)piperazine in the solution was no longer decreased, and the temperature was naturally lowered to room temperature, filtered, and dried under vacuum at 45 C to obtain 197.4 g of a solid. The yield was 94.8%. 99.92% (external standard method); LC-MS: m/z = 209.1 [M+H].The ionic liquid material supported by the organic modified hectorite filtered by filtration was air-dried by acetone and recovered, and the yield of 1-(6-nitropyridin-3-yl)piperazine was 94.2%, and the content was 99.89%. Compared with the catalytic effect of fresh preparation, the catalytic material prepared by the invention can be recycled and used to reduce the production cost of the condensation step.
82%
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 60℃;Inert atmosphere;
General procedure: A mixture of compound 7 (24.7 mmol), morpholine orsubstituted piperazine (29.2 mmol) and DIPEA (37.1 mmol) inacetonitrile (100 mL) was refluxed for 8 h. Progress of reaction wasmonitored by tlc and after complete conversion of starting materialreaction mixture was cooled to rt. The solvent was evaporated andthe residue was purified via column chromatography to affordcompounds 8c-8e.4.1.10.1. 1-(6-nitropyridin-3-yl)piperazine (8c). Yellow solid. Yield:82%; 1H NMR (DMSO-d6, 400 MHz) delta: 7.96 (d, J = 9.3 Hz, 1H, Pyr-H),7.88 (d, J = 2.9 Hz, 1H, Pyr-H), 7.26 (dd, J = 9.3, 3.0 Hz, 1H, Pyr-H),3.64 (t, 4H, Pyr-NCH2CH2N), 3.32 (t, 4H, Pyr-NCH2CH2N). 13C NMR(DMSO-d6, 100 MHz): delta 151.7, 146.2, 136.6, 125.9, 118.4, 51.3, 45.8;HRMS (ESI): calcd for C9H12N4O2, [(M+H)+], 209.1039, found209.1030.
With potassium carbonate;tetra-(n-butyl)ammonium iodide; In acetonitrile; for 16h;Heating / reflux;
Under a nitrogen purge, the solid 4-bromo-2-nitropyridine (10. Ig; 0.05mol), potassium carbonate (10.5g; 0.075mol; -325mesh), tetrabutylammonium iodide (1.25g; 5mol%), and piperazine (5.4g; 0.0625mol) were sequentially added to 80ml acetonitrile. The suspension was heated to reflux, and maintained for 16 h. The now-bright yellow suspension was filtered hot, and the filter cake washed with a few portions of hot acetonitrile, such that the filtrate flows only slightly yellow. The filtrate quickly deposited a yellow/orange solid. This was reheated to obtain a clear solution, which was placed in the refrigerator for 16 h. The yellow/orange solid was isolated by filtration and the filter cake was washed with small portion cold CH3CN, followed by a small portion of petroleum ether. Air drying the solid provided ca 10.2g of solid material, about 65% of theory. Another 2g of material was isolated by evaporating the acetonitrile filtrate down to a semi-solid and then recrystallizing the residue from a minimum amount of hot isopropanol (treated with activated charcoal). NMR: 1.63 (s, IH), 2.99 (m, 4H), 3.36 (m, 4H), 7.14 (m, IH), 8.08 (m, 2H), m/z 209.
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 80℃;
The title compound was prepared by the method described in J Med. Chem. 2005, 48 (7), 2388-2406. Thus, a mixture of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (5.03g; 24.8 mmol), potassium carbonate (3.8 g; 1.1 eqiuv.), piperazine (2.8 g; 1.3 equiv.) and tetra-n-butylammoniurn iodide (0.46 g; 0.05 equiv.) in 60 ml of DMSO was heated at 80 C overnight, then cooled and poured into 300 ml of water. The solid was collected by filtration washed with water (50 ml) and DCM (50 ml) and sucked dry to give 650 mg of yellow solid. The aqueous filtrate was extracted with CHCl3 (4 x 150 ml), the combined organics were washed with brine (100 ml), dried (MgSO4), filtered and evaporated to give 5.5g of yellow solid.The 2 batches of product were dissolved in THF / water (40 ml : 10 ml), treated with sodium hydrogen carbonate (3.1 g; 1.5 equiv.) and di-tert-butyl dicarbonate (6.5g; 1.2 equiv.), stirred at room temperature overnight and then evaporated. The residue was partitioned between DCM and brine (100 ml : 100 ml), and the DCM layer separated, dried (MgSO4), filtered and evaporated. The crude material was purified by flash column chromatography (eluting with 1 :2 to 2: 1 EtOAc / P.E.). Product-containing fractions were combined and evaporated to give 4.5 g of 4-(6- nitro-pyridin-3-yl)-piperazine-l-carboxylic acid tert-butyl ester.A mixture of 4-(6-nitro-pyridin-3-yl)-piperazine-l-carboxylic acid tert-butyl ester and 10% palladium on carbon in ethanol / ethyl acetate (100 ml / 100 ml) was hydrogenated at room temperature and pressure overnight, then filtered and the filtrate evaporated to give to give 4 g of 4-(6-amino-pyridin-3-yl)-piperazine-l- carboxylic acid tert-butyl ester as a brown solid. 1H NMR (d6-DMSO) 7.63 (IH, d), 7.18 (IH, dd), 6.40 (IH, d), 5.45 (2H, s), 3.45 (4H, m) 2.85 (4H, m), 1.43 (9H, s).
With N-ethyl-N,N-diisopropylamine; In acetonitrile; for 18h;Reflux;
A mixture of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (3.00 g, 14.8 mmol) and piperazine (12.7 g, 147 mmol) in acetonitrile (10 mL) was stirred at reflux for 18 h. After this time, the reaction was cooled to room temperature and concentrated under reduced pressure. The residue was diluted in ethyl acetate (50 mL), washed with water (2 chi 25 mL), then brine (25 mL) and dried over sodium sulfate. The drying agent was removed by filtration and the filtrate concentrated under reduced pressure. The resulting residue was purified by chromatography (silica, gradient, heptane to ethyl acetate) to afford l-(6-nitropyridin-3-yl)piperazine as a yellow solid: 1H NMR (400 MHz, DMSO-i¾d 8.23 (d, J= 2.8 Hz, 1H), 8.13 (d, J= 9.2 Hz, 1H), 7.88 (dd, J = 9.2, 2.8 Hz, 1H), 3.40 (t, J= 4.8 Hz, 4H), 2.82 (t, J= 4.8 Hz, 4H), NH (1H, not observed).
In acetonitrile; for 18h;Reflux;
Example 3 Preparation of 1-(6-nitropyridin-3-yl)piperazine A mixture of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (3.00 g, 14.8 mmol) and piperazine (12.7 g, 147 mmol) in acetonitrile (10 mL) was stirred at reflux for 18 h. After this time, the reaction was cooled to room temperature and concentrated under reduced pressure. The residue was diluted in ethyl acetate (50 mL), washed with water (2*25 mL), then brine (25 mL) and dried over sodium sulfate. The drying agent was removed by filtration and the filtrate concentrated under reduced pressure. The resulting residue was purified by chromatography (silica, gradient, heptane to ethyl acetate) to afford 1-(6-nitropyridin-3-yl)piperazine as a yellow solid: 1H NMR (400 MHz, DMSO-d6.) d 8.23 (d, J=2.8 Hz, 1H), 8.13 (d, J=9.2 Hz, 1H), 7.88 (dd, J=9.2, 2.8 Hz, 1H), 3.40 (t, J=4.8 Hz, 4H), 2.82 (t, J=4.8 Hz, 4H), NH (1H, not observed).
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 80℃;
A mixture of 5-Bromo-2-nitropyridine (203 g, 1.37 mol), piperazine (153 g, 1.77 mol), tetrabutylammonium iodide (25.2 g, 0.068 mol), and potassium carbonate (207 g, 1.50 mol) in dimethyl sulfoxide (2.6 L) was stirred at 80C overnight. The resultant reaction mixture was cooled to room temperature, and the mixture was poured into water (7 L). The resultant solid was collected by filtration, and the solid was washed with dichloromethane (1 L × 2) and dried. The filtrate was extracted with chloroform (2 L × 7). The resultant organic phase was washed with water (2 L) and then with saturated brine (2 L), and the organic phase was concentrated under reduced pressure to yield solid. The resultant solid products were combined together and used for the subsequent reaction without further purification. (0173) The solid product (490 g) was dissolved in THF (2 L) and water (500 mL), and sodium hydrogen carbonate (119 g, 1.42 mol) was added to the solution. To the resultant suspension was added di-tert-butyl dicarboxylate (262 g, 1.2 mol), and the mixture was stirred at room temperature for three hours. The reaction mixture was concentrated under reduced pressure, and the residue was diluted with water (1 L) and extracted with dichloromethane (1 L × 3). The resultant organic phases were combined together and then washed with water (1 L). The aqueous phase was extracted with dichloromethane (300 mL). The resultant organic phases were combined together and dried over anhydrous magnesium sulfate. The solid was separated by filtration, and the filtrate was concentrated under reduced pressure. The resultant solid was suspended in ethyl acetate (2 L) and heated to 60C, and the solid was separated by filtration at 60C. The solid was dried under reduced pressure to yield the title compound (191 g, 62%) APCI-MS (M+H)+ 309.1, C14H20N4O4=308.15 1H-NMR delta(400 MHz, CDCl3) : 8.16 (d, J=9 Hz, 1H), 8.11 (d, J=3 Hz, 1H), 7.19 (dd, J=9.3 Hz, 1H), 3.64-3.61 (m, 4H), 3.45-3.42 (m, 4H), 1.47 (s, 9H).
With potassium carbonate; In dimethyl sulfoxide; at 60 - 70℃;
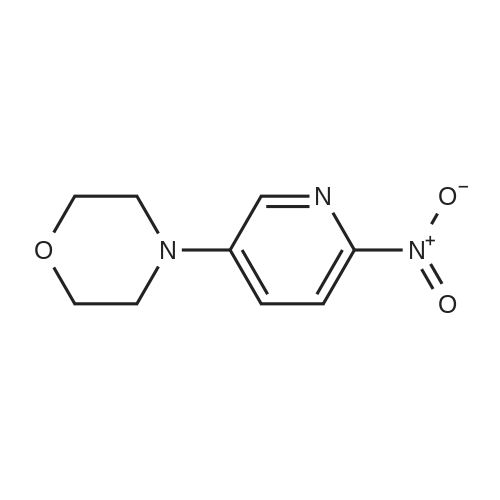
Example 2.3: Preparation of 3-Hydroxy-7-morpholin-4-yl-4-oxo-4H-pyrido[l,2- alpha]pyrimidine-2-carboxylic acid methyl ester; 5-Morpholin-4-yl-pyridin-2-ylamine was prepared by adapting he procedure described in J. Med. Chem., 2005, 48(7), 2388-2406. Briefly, <strong>[39856-50-3]5-bromo-2-nitro-pyridine</strong> was reacted with morpholine and potassium carbonate in DMSO at 60-70 C to afford 4-(6- nitro-pyridin-3-yl)-morpholine in 84% yield. Reduction with palladium on carbon under a hydrogen atmosphere provided 5-morpholin-4-yl-pyridin-2-ylamine in 70% yield. This was converted into 3-hydroxy-7-morpholin-4-yl-4-oxo-4H-pyrido[l,2- alpha]pyrimidine-2-carboxylic acid methyl ester in 25% yield by adapting the procedure described in Example 2, where glacial acetic acid was used instead of p- toluenesulphonic acid.1H NMR (300 MHz, D6-DMSO): delta 3.22 (4H, t, J=5.0 Hz, -NCH2CH2O-), 3.89 (4H, t, J=5.0 Hz, -NCH2CH2O-), 4.10 (3H, s, OCH3), 7.45 (IH, dd, J=9.9, 2.7 Hz, H8), 7.63 (IH, d, J=9.9 Hz, H9), 8.17 (IH, d, J=2.7 Hz, H6), 10.32 (IH, s, OH)MS (ESI+) m/z 305 (M+l)HPLCmethod 7 97.4%/l 1.7 min.
80%
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 60℃;Inert atmosphere;
General procedure: A mixture of compound 7 (24.7 mmol), morpholine orsubstituted piperazine (29.2 mmol) and DIPEA (37.1 mmol) inacetonitrile (100 mL) was refluxed for 8 h. Progress of reaction wasmonitored by tlc and after complete conversion of starting materialreaction mixture was cooled to rt. The solvent was evaporated andthe residue was purified via column chromatography to affordcompounds 8c-8e.
74%
With N-ethyl-N,N-diisopropylamine; In dimethyl sulfoxide; at 120℃; for 6h;Microwave irradiation;
To a solution of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (1.0 g, 4.93 mmol) in DMSO (4 mL) was added morpholine (0.43 mL, 4.9 mmol) followed by DIPEA (0.94 mL, 5.4 mmol). The reaction mixture was then subjected to microwave irradiation maintaining a reaction temperature of 120C for 3h. The mixture was cooled to room temperature, added more DIPEA (0.94 mL, 5.4 mmol), and then subjected to microwave irradiation maintaining a reaction temperature of 120C for 3h. The mixture was then cooled to room temperature, diluted with EtOAc (50 mL) and washed with water (50 mL). The aqueous phase was back extracted with EtOAc (2X50 mL) and the organic layers combined and washed with brine (1X50 mL). The organic phase was then dried (Na2SO4), filtered, and concentrated in vacuo. The crude residue was purified by FCC (SiO2, elution with 0-100% (0270) EtOAc/hexanes) to provide 0.76 g (74%) of 4-(6-nitropyridin-3-yl)morpholine. 1H NMR (400 MHz, CDCl3) d ppm 8.19 (d, 1H), 8.15 (d, 1H), 7.22 (dd, 1H), 3.90 (m, 4H), 3.42 (m, 4H); LCMS (Method B): tR= 0.92 min, m/z 210.2 (M+H)+.
68.6%
at 110℃; for 0.5h;Microwave irradiation;
A mixture of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (1.0 g, 1.0 eq) and morpholine (8 mL) was irradiated with microwave radiation (Biotage) at 110C for 30 min. After completion of starting material on TLC, the mixture was concentrated to obtain crude material which was purified by Combiflash chromatography (silica gel, 230-400) using 3% methanol in DCM as eluent to obtain 4-(6-nitropyridin-3-yl)morpholine (0.7 g, 68.6%) as yellow solid. LCMS calculated for (M) 209.08 and found (M+H) 210.08, LCMS showed 99.64% purity.
58.2%
at 120℃; for 3h;Microwave irradiation;
Synthesis of compound 196.3. Compound 78.4 (l .Og, 4.92 mmol, 1.0 eq) was dissolved in morpholine (5 mL). Reaction mixture was heated in microwave at 120 C for 3 h. After completion of reaction, mixture was poured in water, quenched with NH4C1 solution and product was extracted with EtOAc. Organic layers were combined,dried over Na2S04 and concentrated under reduced pressure to obtain crude which was purified by column chromatography to furnish 196.3 (0.6 g, 58.2 %). MS (ES): m/z 209.20 [M+H]+.
46%
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 80℃; for 20h;
Step 1 4-(6-Niotatro-pyriotadiotan-3-yl)-morphohne[00399] A mixture of 5-bromo-2-mtro-py?dme (5g, 24 75mmol), tetrabutyl ammonium iodide(0 46g, 1 24mmol), K2CO3 (3 76g, 27 22mmol) and morpholme (2 34g, 27 22mmol) in DMSO (5OmL) is stirred at 8O0C in a stem tube for 20 hours The reaction mixture is diluted with ethyl acetate and filtered The organic solution is washed with water, dried over MgSO4 and concentrated in vacuo The residue is triturated with dichloromethane and hexane to afford the title compound as a solid (2 39g,46%)
With N-ethyl-N,N-diisopropylamine; In acetonitrile; for 16h;Reflux;
A mixture of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (1.00 g, 4.93 mmol), morpholine (515 mg, 5.91 mmol) and N,N-diisopropylethylamine (1 .91 g, 14.8 mmol) in acetonitrile (12 mL) was stirred at reflux for 16 h. After this time, the reaction was cooled to room temperature and concentrated under reduced pressure to afford 4-(6-nitropyridin-3-yl)morpholine as a yellow solid: NMR (400 MHz, DMSO-i¾d 8.26 (d, J = 3.2 Hz, 1H), 8.17 (d, J = 9.2 Hz, 1H), 7.49 (dd, J = 9.2, 3.2 Hz, 1 H), 3.75 (t, J = 4.8 Hz, 4H), 3.46 (t, J = 4.8 Hz, 4H)
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 80℃; for 30h;
<strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (1.0 g, 4.93 mmol), morpholine (0.47 g, 5.42 mmol), BU4NI (0.09 g, 0.25 mmol), K2C03 (0.75 g, 5.42 mmol) were stirred in DMSO (10 mL) at 80 C for 30 h. Water was added and the solid separated by filtration was purified by column chromatography to give (4-(6-nitropyridin-3-yl)morpholine).
With N-ethyl-N,N-diisopropylamine; In acetonitrile; for 16h;Reflux;
Preparation of 4-(6-nitropyridin-3-yl)morpholine A mixture of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (1.00 g, 4.93 mmol), morpholine (515 ma, 5.91 mmol) and N,N-diisopropylethylamine (1.91 g, 14.8 mmol) in acetonitrile (12 mL) was stirred at reflux for 16 h. After this time, the reaction was cooled to room temperature and concentrated under reduced pressure to afford 4-(6-nitropyridin-3-yl)morpholine as a yellow solid: 1H NMR (400 MHz, DMSO-d.6d 8.26 (d, J=3.2 Hz, 1H), 8.17 (d, J=9.2 Hz, 1H), 7.49 (dd, J=9.2, 3.2 Hz, 1H), 3.75 (t, J=4.8 Hz, 4H), 3.46 (t, J=4.8 Hz, 4H)
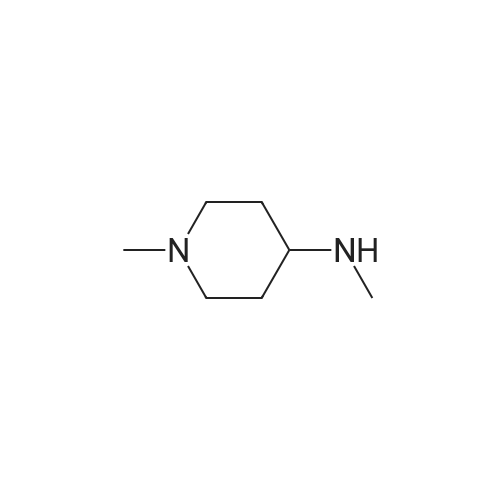
With palladium diacetate; caesium carbonate; 2,2'-bis-(diphenylphosphino)-1,1'-binaphthyl; In 1,4-dioxane; at 90℃; for 16h;Inert atmosphere;
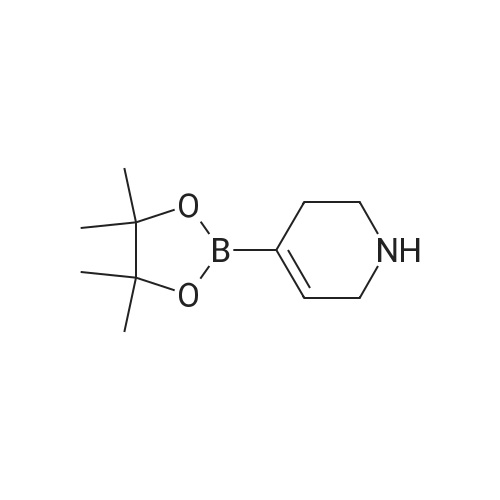
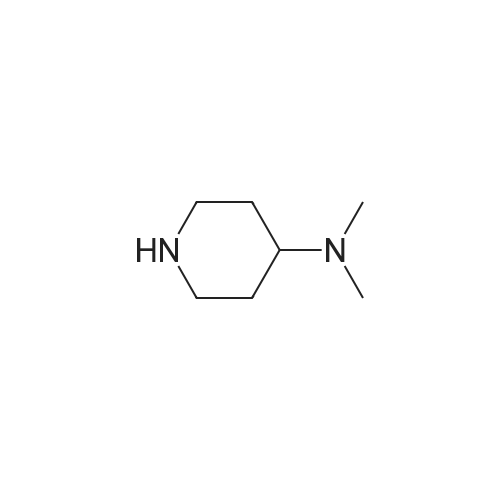
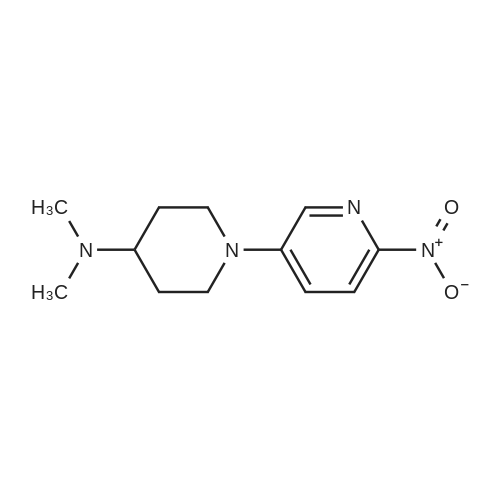
Step 1 N,N-dimethyl-1-(6-nitropyridin-3-yl)piperidine-4-amine In a nitrogen atmosphere, to a solution of N,N-dimethylpiperidine-4-amine (100.00 mg, 779.97 mumol, 1.00 equivalent) and 5-bromo-2-nitropyridine (158.33 mg, 779.97 mumol, 1.00 equivalent) in dioxane (5 mL) were added B1NAP (48.57 mg, 78.00 mumol. 0.10 equivalent), cesium carbonate (508.26 mg, 1.56 mmol, 2.00 equivalents) and Pd(OAc)2 (17.51 mg , 78.00 mumol, 0.10 equivalent). The reaction mixture was then heated to 90C and stirred for 16 hours. TLC showed completion of the reaction of the starting material. The reaction mixture was filtered and concentrated to give a crude product. The crude product was purified by preparative TLC (dichloromethane: methanol = 10: 1) to give the title compound (125.00 mg, 499.40 mumol, 64.03% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) delta 8.20-8.14 (m, 2H), 7.21 (dd, J = 9.2, 3.2 Hz, 1H), 3.99 (d, J = 13.2 Hz, 2H), 3.12-3.02 (m, 2H), 2.47-2.39 (m, 1H). 2.33 (s, 6H), 2.01 (d, J = 12.4 Hz, 2H), 1.70-1.64 (m, 2H). LC/MS (ESI) m/z: 251.1 (M+1).
38%
With N-ethyl-N,N-diisopropylamine; In acetonitrile; for 60h;Heating / reflux;
Preparation XXI; Synthesis of 4N.4N-dimethyl-3.4,5.6 tetrahvdro-2H-? ,3'lbvpyridmylV4.6'-diamine; <n="117"/>Step 1. Synthesis of dimethyl-r6'-nitro-3.4.5.6-tetrahvdro-2H-ri,3'1bvpyridinyl-4- ylVamine5-Bromo-2-nitropyridine; (3.7Og, 18.40 mmol) was dissolved in acetonitrile (40ml), 4-dimethylamino piperidine (2.5g, 19.5 mmol) and Hunig's base (3.4 ml, 19.5 mmol) were added and the mixture heated at reflux for 60 h. The reaction mixture was allowed to cool to room temperature and the desired product was collected by filtration as a yellow solid (1.73 g, 38 %) (LCMS: R12.29, [M+H]+251).
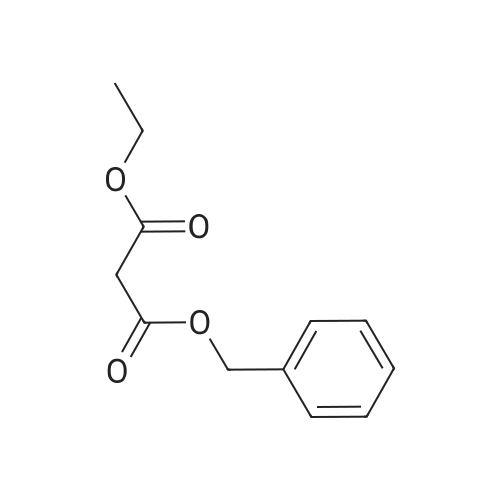
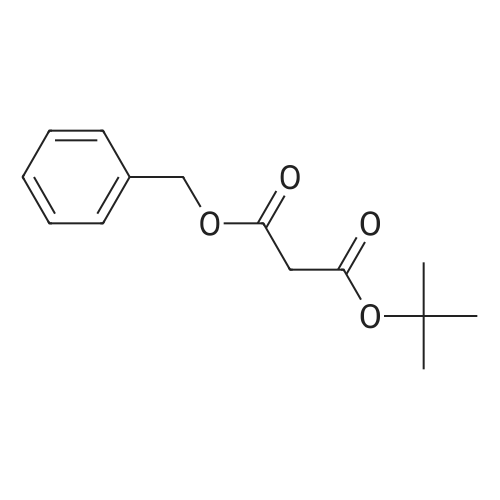
benzyl ethyl 2-(6-nitro-3-pyridinyl)malonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium carbonate In tetrahydrofuran; DMF (N,N-dimethyl-formamide) for 20h; Heating / reflux;
287.1 EXAMPLE 287; 2-{5-[6-Chloro-2-ethyl-5-(trifluorometyl)-1H-benzimidazol-1-yl]-3-pyridinyl}ethyl(4-methylphenyl)sufonylcarbamate; Step 1. Benzyl Ethyl 2-(6-Nitro-3-pyridinyl)malonate
To a mixture of 5-bromo-2-nitropyridine (8.66 g, 42.7 mmol) and benzyl ethyl malonate (9.50 g, 42.7 mmol) in tetrahydrofuran (160 ml) and dimethylformamide (40 ml) was added K2CO3 (5.90 g, 42.7 mmol) and stirred under reflux temperature for 20 h. The mixture was diluted with water (11) and extracted with ethyl acetate (3×200 ml). The organic layer was washed with brine, dried (MgSO4) and concentrated to give 5.26 g of title compound as orange oil. [2635] 1H-NMR (CDCl3) δ: 8.61 (1H, d, J=2.2 Hz), 8.26 (1H, d, J=8.4 Hz), 8.19 (1H, dd, J=2.1, 8.6 Hz), 7.29-7.38 (5H, m), 5.22 (2H, d, J=3.6 Hz), 4.84 (1H, s), 4.22 (2H, m), 1.23 (3H, t, J=7.1 Hz).
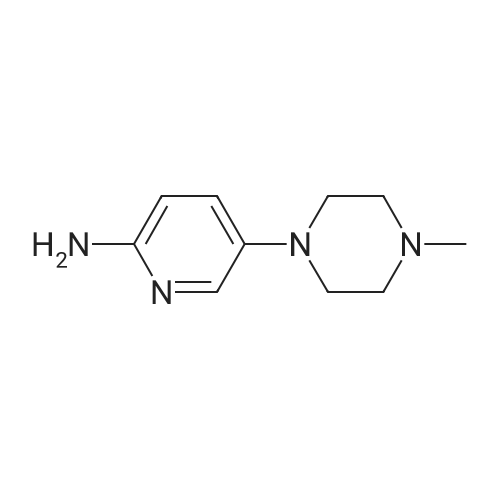
Into a solution of <strong>[39856-50-3]5'-bromo-2-nitropyridine</strong> (2.0 g, 9.85 mmol) in DMSO (10 mL) were added K2CO3 (2.72 g, 19.7 mmol), 1-methylpiperazine (1.64 mL, 14.8 mmol), and tetrabutylammonium iodide (36 mg). The mixture was stirred at 120 C. overnight, allowed to cool, and acidified with 1N HCl. The mixture was extracted with CH2Cl2, the aqueous layer was basified with sat aq Na2CO3, and further extracted with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated to give a wet brown solid, which was washed with small amount of water. The solid dried under vacuum to 2.16 g (99% yield) of yellow powder. 1H NMR (CDCl3): delta 8.16 (d, 1H, J=9.1 Hz), 8.14 (d, 1H, J=3.0 Hz), 7.20 (dd, 1H, J=3.0, 9.1 Hz), 3.49 (dd, 4H, J=4.9, 5.3 Hz), 2.61 (dd, 4H, J=4.9, 5.3 Hz), 2.39 (s, 3H).
99%
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 120℃;
Into a solution of [5'-BROMO-2-NITROPYRIDINE] (2.0 g, 9.85 mmol) in DMSO (10 mL) were added [K, C03] (2.72 g, 19.7 [MMOL),] 1-methylpiperazine (1.64 mL, 14. [8] mmol), and tetrabutylammonium iodide (36 mg). The mixture was stirred at [120C] overnight, allowed to cool, and acidified with 1N [HC1.] The mixture was extracted with [CH2C12,] the aqueous layer was basified with sat aq Na2CO3, and further extracted with [CHIC'2.] The combined organic layers were dried over [NA2SO4] and concentrated to give [A] wet brown solid, which was washed with small amount of water. The solid dried under vacuum to 2.16 g (99% yield) of yellow powder. 'H NMR (CDCl3) : [8] 8. 16 (d, 1H, J = 9.1 Hz), 8.14 (d, 1H, J = 3.0 Hz), 7.20 (dd, 1H, J = 3.0, 9.1 Hz), 3.49 (dd, 4H, J = 4.9, 5.3 Hz), 2.61 (dd, 4H, J = 4.9, 5.3 Hz), 2.39 (s, 3H).
99%
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 120℃;
According to US 2009/0318448, into a solution of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (2.0 g, 9.85 mmol) in DMSO (10 mL) was added K2C03 (2.72 g, 19.7 mmol) and 1- methylpiperazine (1.64 mL, 14. 8 mmol), and tetrabutylammonium iodide (36 mg). The mixture was stirred at 120 degree C for overnight. It was allowed to cool down and acidified with IN HC1. The mixture was extracted with DCM. The aqueous layer was basified with saturated Na2C03, and further extracted with DCM. The combined organic layers were dried over Na2S04 and concentrated to give a brown solid, which was washed with small amount of water. The solid was dried under vacuum to give 2.16 g of l-methyl-4-(6-nitropyridin-3- yl)piperazine (99% yield) as a yellow powder. LCMS: (M+H)+ 223
99.1%
With tetrabutylammomium bromide; potassium carbonate; In dimethyl sulfoxide; at 120℃; for 12h;
4.1.41 1-Methyl-4-(6-nitropyridin-3-yl)piperazine (10c) A suspension of 1-methylpiperazine (16.4 mL, 148 mmol), <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (20 g, 98.5 mmol), Bu4NBr (200 mg, 0.985 mmol) and K2CO3 (27.2 g, 197 mmol) in DMSO (100 mL) was stirred at 120 C for 12 h. The reaction mixture was poured into ice water, and the product was collected by filtration and washed by water for three times. The filter cake was dried under vacuum oven at 50C overnight to offord 10c (21.6g, 99.1%) as a yellow solid. MS (ESI) m/z 223.1 [M+H]+; 1H NMR (400MHz, DMSO-d6): delta 8.27 (d, J=4.00Hz, 1H), 8.15 (d, J=8.00Hz, 1H), 7.49 (dd, J=4.00Hz, 8.00Hz, 1H), 3.49 (t, J=4.00Hz, 4H), 2.44 (t, J=4.00Hz, 4H), 2.22 (s, 3H).
91%
With tetrabutyl ammonium fluoride; potassium carbonate; In dimethyl sulfoxide; at 80℃; for 16h;
To a solution of compound 5-6 (100 mg, 0.985 mmol) in DMSO (3 ml) was added compound 5-6-1 (148 mg, 1.48 mmol), K2CO3 (204.25 mg, 1.48 mmol) and tetrabutylammonium fluoride (TBAF) (17.5 mg, 0.049 mmol), and then the reaction mixture was reacted at 80 C. for 16 h. The reaction was monitored by TLC until the completion of reaction. The reaction solution was poured into ice water (100 mL) and filtered, and the resulting filter cake was washed with water (50 mL) to give compound 5-7 (100 mg, Yield 91%). 1HNMR (400 MHz, d6-DMSO): delta ppm 8.22-8.23 (m, 1H), 8.10-8.12 (m, 1H), 7.43-7.47 (m, 1H), 3.44-3.47 (m, 4H), 2.39-2.42 (m, 4H), 2.19 (s, 3H).
91%
With tetrabutylammomium bromide; potassium carbonate; In dimethyl sulfoxide; at 20 - 100℃; for 6h;
To a stirred solution of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (0.5 g, 2.46 mmol) in DMSO (5 ml) was added l-methylpiperazine (0.369 g, 3.69 mmol), K2C03 (0.679 g, 4.92 mmol) and TBAB (0.079 g, 0.0246 mmol) at room temperature. The reaction mixture was stirred at 100 C for 6 hours. After completion of the reaction (monitored by TLC), the reaction was quenched with N HC1 (15 ml) and extracted with ethyl acetate (2 x 15 ml). The aqueous layer was treated with IN NaOH solution and extracted with ethyl acetate (2 x 25 ml). The combined organic layers were dried over anhydrous Na2S04 and concentrated under reduced pressure to afford title compound (0.5 g, 91 %). 1H NMR (400 MHz, DMSO-d6): 2.39 (s, 3H), 2.61 (t, 7=5.2 Hz, 4H), 3.50 (t, 7=5.2 Hz, 4H), 7.22 (dd, 7=8.8 Hz, 2.8 Hz, 1H), 8.15-8.20 (m, 2H).
85%
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 60℃;Inert atmosphere;
General procedure: A mixture of compound 7 (24.7 mmol), morpholine orsubstituted piperazine (29.2 mmol) and DIPEA (37.1 mmol) inacetonitrile (100 mL) was refluxed for 8 h. Progress of reaction wasmonitored by tlc and after complete conversion of starting materialreaction mixture was cooled to rt. The solvent was evaporated andthe residue was purified via column chromatography to affordcompounds 8c-8e.
83%
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 120℃; for 16h;
To a solution of 5-bromo-2- nitropyridine (475 mg, 2.34 mmol) in DMSO (2.5 mL) was added potassium carbonate (651 mg, 4.71 mmol), 1-methylpiperazine (343 mg, 3.43 mmol) and TBAI (9 mg, 0.02 mmol) at room temperature. The resulting mixture was stirred for 16 h at 120 C. When the reaction was done, the reaction mixture was diluted with H20 (30 mL) and extracted with dichloromethane (50 mL x 3). The organic phases were combined, washed with brine and dried over Na2S04. The solvent was removed under reduced pressure and the residue was purified by flash chromatography eluting with MeOH in EtOAc (0 % to 50 % gradient) to yield l-methyl-4-(6- nitropyridin-3-yl)piperazine as a yellow solid (433 mg, 83 %). MS: m/z = 222.9 [M+H]+.
56%
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 80℃; for 16h;
1-methylpiperazine (3.00g, 29.6mmol), potassium carbonate ([K2CO3] 4.10g, 29.6mmol), and tetrabutylammonium iodide ([TBAI] 0.42g, 1.2mmol) were added to a solution of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (4.00g, 19.7mmol) in dimethyl sulfoxide ([DMSO] 50mL). The mixture was heated at 80C for 16h, and the reaction was poured into ice water and extracted with dichloromethane (DCM). The combined organic layers were washed with water, dried over anhydrous sodium sulfate (Na2SO4), concentrated under a vacuum, and purified by silica gel column chromatography (DCM/methanol [MeOH]=50:1-10:1) to obtain 1-methyl-4-(6-nitropyridin-3-yl)piperazine (2.45g; yield, 56%) as a yellow solid. Then palladium on carbon ([Pd/C], 0.10g) was added to the 1-methyl-4-(6-nitropyridin-3-yl)piperazine (1.00g, 4.5mmol) in ethanol (EA)/MeOH (10 mL/10mL) solution. The mixture was degassed by flushing with hydrogen (H2), stirred at room temperature (RT) under a H2 atmosphere for 2h, and filtered and concentrated under vacuum to obtain INT-1 (825mg; yield, 95%) as a white solid. ESI-MS: m/z 193.3 [M+H]+.
54.6%
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 90℃; for 20h;
10 g of compound 38 was weighed,Was added to a 250 mL reaction flask,To the flask was added 80 mL of DMF,9 mL DIPEA and 8.47 g of compound 19,The reaction was carried out in an oil bath at 90 C for 20 h,The reaction was quenched with 100 mL x 3 ethyl acetate,The organic phase was washed with saturated brine,Dried over anhydrous sodium sulfate,Concentrated, the residue was beaten with methyl t-butyl ether,To give 6 g of a yellow solid compound 42,Yield 54.6%.
With rac-2,2'-bis(phenylphosphino)-1,1'-binaphthyl; caesium carbonate;tris-(dibenzylideneacetone)dipalladium(0); In toluene; at 80℃; for 48h;
A mixture of 30 mmol of 9, 30 mmol of 7, 0.04 mmol of Pd2(dba)3, 0.08 mmol of rac-2,2'-bis(phenylphosphino)-I,I'-binaphthyl (BINAP), and 42 mmol of Cs2C03 in 100 mL of dry toluene was stirred at 80 C for two days under N2. The reaction mixture was diluted with 400 mL of ethyl acetate and the organic solution was washed with saturated NaCI, dried over MgS04, and concentrated under reduced pressure. The residue was crystallized in ethyl acetate to yield 15.8 mmol of 10. [0182] A solution or a suspension of 15 mmol of 10 and 0.5 g ofPd/C (10%) in 150 mL of methanol was stirred overnight under H2 (1 atm). After filtering through celite, the solution was concentrated under a reduced pressure to give 15 mmol of 8.
With potassium carbonate;tetra-(n-butyl)ammonium iodide; In dimethyl sulfoxide; at 120℃; for 18h;
To 5-Bromo-2-nitro-pyridine (2.0Og, 9.85mmol) in 10 mL dimethylsulfoxide was added potassium carbonate ( 2.72g, 19.7mmol), 1-methylpiperazine (1.64mL, 14.8mmol), and tetra- butylammonium iodide (36mg, 0.097mmol) and was heated at 1200C for 18 hours. The mixture was made acidic with IM aq. HCl and was partitioned between DCM and water. The aqueous layer was made basic with 2M aq. sodium carbonate and was extracted with DCM. The organic layer was dried over anhydrous magnesium sulfate, concentrated in vacuo, and was triturated with water to yield l-Methyl-4-(6-nitro-pyridin-3-yl)-piperazine (1.82g, 8.19mmol). MS (ESI) 223.1 (M+H)+.
With caesium carbonate;tris-(dibenzylideneacetone)dipalladium(0); 2,2'-bis-(diphenylphosphino)-1,1'-binaphthyl; In toluene; at 80℃; for 48h;
A mixture of 30 mmol of 9, 30 mmol of 7, 0.04 mmol of Pd2(dba)3, 0.08 mmol of nac-2,2'-bis(phenylphosphino)-1,1'-binaphthyl (BINAP), and 42 mmol of Cs2C03 in 100 mL of dry toluene was stirred at 80 C for two days under N2. The reaction mixture was diluted with 400 mL of ethyl acetate and the organic solution was washed with saturated NaCl, dried over MgS04, and concentrated under reduced pressure. The residue was crystallized in ethyl acetate to yield 15.8 mmol of 10. [0209] A solution or a suspension of 15 mmol of 10 and 0.5 g of Pd/C (10%) in 150 mL of methanol was stirred overnight under H2 (1 atm). After filtering through celite, the solution was concentrated under a reduced pressure to give 15 mmol of 8. ¹H NMR (300 MHz, DMSO-d6) No. 7.56 (d, J = 2.7 Hz, 1H), 7.13 (dd, J1 = 8.9 Hz, J2 = 2.9 Hz, 1H), 6.36 (d, J = 8.8 Hz, 1H), 5.36 (s, 2H), 2.89 (t, J = 5.0 Hz, 4H), 2.40 (t, J = 5.0 Hz, 4H), 2.18 (s, 3H) ; MS m/z: 193 (M + 1).
With rac-2,2'-bis(phenylphosphino)-1,1'-binaphthyl; caesium carbonate;tris-(dibenzylideneacetone)dipalladium(0); In toluene; at 80℃;
Example 5 Preparation of 8 5.1 Buchwald Cross-Coupling A mixture of 30 mmol of 9, 30 mmol of 7, 0.04 mmol of Pd2(dba)3, 0.08 mmol of rac-2,2'-bis(phenylphosphino)-1,1'-binaphthyl (BINAP), and 42 mmol of Cs2CO3 in 100 mL of dry toluene was stirred at 80 C. for two days under N2. The reaction mixture was diluted with 400 mL of ethyl acetate and the organic solution was washed with saturated NaCl, dried over MgSO4, and concentrated under reduced pressure. The residue was crystallized in ethyl acetate to yield 15.8 mmol of 10. A solution or a suspension of 15 mmol of 10 and 0.5 g of Pd/C (10%) in 150 mL of methanol was stirred overnight under H2 (1 atm). After filtering through celite, the solution was concentrated under a reduced pressure to give 15 mmol of 8.
at 80℃; for 2h;
Example 38 1-Methyl-4-(6-nitro-pyridin-3-yl)-piperazine A mixture of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (500 mg, 2.46 mmol) and 1-methylpiperazine (1 mL) is heated at 80 C. for 2 hour. Then water is added. The aqueous layer is extracted with EtOAc, and the organic extracts were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product is purified by column chromatography (SiO2, MeOH:CH2Cl2=0.7:99.3 to 6:93) to give 520 mg of the title compound as yellow solid. MS (ESI) m/z 223 (M+H)+
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 120℃; for 18h;
To 5-Bromo-2-nitro-pyridine (2.00 g, 9.85 mmol) in 10 mL dimethylsulfoxide was added potassium carbonate (2.72 g, 19.7 mmol), 1-methylpiperazine (1.64 mL, 14.8 mmol), and tetrabutylammonium iodide (36 mg, 0.097 mmol) and was heated at 120 C. for 18 hours. The mixture was made acidic with 1M aq. HCl and was partitioned between dichloromethane and water. The aqueous layer was made basic with 2M aq. sodium carbonate and was extracted with dichloromethane. The organic layer was dried over anhydrous magnesium sulfate, concentrated in vacuo, and was triturated with water to yield 1-Methyl-4-(6-nitro-pyridin-3-yl)-piperazine (1.82 g, 8.19 mmol). MS (ESI) 223.1 (M+H)+.
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 90℃; for 24h;
1-methyl-4-(6-nitro-3-pyridyl)piperazine To <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (4.93 g, 24.3 mmole) in DMF (20 mL) was added N-methylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 degrees for 24 hrs. After addition of ethyl acetate (200 mL) water 100 mL was added and the layers separated. Drying followed by concentration afforded the crude product which was columned using (0-10%) DCM/Methanol. 1HNMR (delta6-DMSO) 8.26 (s, 1H), 8.15 (1H, d, J=9.3 Hz), 7.49 (1H, d, J=9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
1.5 g
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 120℃; for 18h;
To 5-Bromo-2-nitro-pyridine (2 g, 9.85 mmol) in DMSO (10 mL) was added potassium carbonate (2.04 g, 15.0 mmol), 1-methylpiperazine (1.73 g, 17.0 mmol), and tetrabutylammonium iodide (70 mg, 0.197 mmol) and was heated at 120C for 18 h. The mixture was made acidic with 1M aq. HCl and was partitioned between DCM and water. The aqueous layer was made basic with 2M aq. sodium carbonate and was extracted with DCM. The organic layer was dried over anhydrous Na2S04, filtered and concentrated in vacuo, and was triturated with water to yield l-Methyl-4-(6-nitro-pyridin-3-yl)-piperazine (1.5 g) as a yellow solid. 1H NMR (400 MHz, CDC13) 5 8.15 (dd, J= 11.6, 6.1 Hz, 2H), 7.20 (dd, J= 9.2, 3.1 Hz, 1H), 3.53 - 3.45 (m, 4H), 2.60 (dd, J= 12.7, 7.5 Hz, 4H), 2.37 (d, J= 6.0 Hz, 3H).
2 g
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 80℃;
5-Bromo-2-nitro-pyridine (4.06 g, 20.0 mmol), 1-Methyl-piperazine (2.21 g, 22.1 mmol), K2C03 (3.1 g, 22.1 mmol), Bu4N+T(371 mg, 1.004 mmol) were mixed in DMSO (50 mL). The mixture was stirred at 80 C overnight. TLC showed the starting material was consumed completely; water was added and extracted with DCM. The organic layer was washed with water and brine, dried over anhydrous Na2S04. It was then concentrated to give the crude product l-methyl-4-(6-nitro-pyridin-3-yl)-piperazine (2.0 g, 45%) which was used in the next step directly.
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 90℃; for 24h;
Example 9 Synthesis of l-methyl-4-(6-nitro-3-pyridyl)piperazine, Compound 9 To <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (4.93 g, 24.3 mmole) in DMF (20 mL) was added N- methylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 degrees for 24 hrs. After addition of ethyl acetate (200 mL), water (100 mL) was added and the layers separated. Drying followed by concentration afforded the crude product which was purified by silica gel column chromatography using (0-10%) DCM/Methanol. 1HNMR (d6-DMSO) delta ppm 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 90℃; for 24h;
To <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (4.93 g, 24.3 mmole) in DMF (20 mL) was added N- methylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 degrees for 24 hrs. After addition of ethyl acetate (200 mL), water (100 mL) was added and the layers separated. Drying followed by concentration afforded the crude product which was purified by silica gel column chromatography using (0-10%) DCM/Methanol. 1HNMR (d6-DMSO) delta ppm 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 90℃; for 24h;
To <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (4.93 g, 24.3 mmole) in DMF (20 mL) was added Nmethylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole).The contents were heated at 90 degrees for 24 hrs. After addition of ethyl acetate (200 mL), water (100 mL) was added and the layers separated. Drying followed by concentration afforded the crude product which was purified by silica gel column chromatography using (0-10%) DCM/Methanol. ?HNMR (d6-DMSO) oe ppm 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 90℃; for 24h;
To <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (4.93 g, 24.3 mmole) in DMF (20 mL) was added Nmethylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 C for 24 hrs. After the addition of ethyl acetate (200 mL), water (100 mL) was added and the layers were separated. Drying followed by concentrationafforded the cmde product which was purified on a silica gel column using (0-10%) DCM/Methanol.?H NMR (DMSO- d6) 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 90℃; for 24h;
To <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (4.93 g, 24.3 mmole) in DMF (20 mL) was added Nmethylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 C for 24 hrs. After the addition of ethyl acetate (200 mL), water (100 mL) was added and the layers were separated. Drying followed by concentrationafforded the cmde product which was purified on a silica gel column using (0-10%) DCM/Methanol.?H NMR (DMSO- d6) 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 90℃; for 24h;
To <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (4.93 g, 24.3 mmole) in DMF (20 mL) was added N- methylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 C for 24 hrs. After the addition of ethyl acetate (200 mL), water (100 mL) was added and the layers were separated. Drying followed by concentration afforded the crude product which was purified on a silica gel column using (0-10%) DCM/Methanol. (0496) NMR (DMSO- de) delta 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
1.94 g
With potassium carbonate; In dimethyl sulfoxide; at 82℃; for 15h;
1-methylpiperazine (1.180 g) and K2CO3 (2.720 g) were added in sequence to <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (2.010 g)In DMSO (20 mL) solution. The reaction solution was stirred in an oil bath at 82 C for 15 hours. Add water (50 mL) and extract with DCM (20 mL×8). The combined organic phases were dried over anhydrous Na2SO4 .Concentration under reduced pressure and purification by column chromatography (DCM / MeOH = 10/1) to afford 1.940 g of 1-methyl-4-(6-nitropyridin-3-yl)piperazine.
With N-ethyl-N,N-diisopropylamine; In ethyl acetate; N,N-dimethyl-formamide; at 90℃; for 24h;
To <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (4.93 g, 24.3 mmole) in DMF (20 ml.) was added N- methylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 C for 24 hrs. After the addition of ethyl acetate (200 mL), water (100 mL) was added and the layers were separated. Drying followed by concentration afforded the crude product which was purified on a silica gel column using (0-10%) DCM/Methanol. 1H NMR (DMSO- de) d 8.26 (s, 11 1). 8.15 (11 1. d, J = 9.3 Hz), 7.49 . i l l. d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
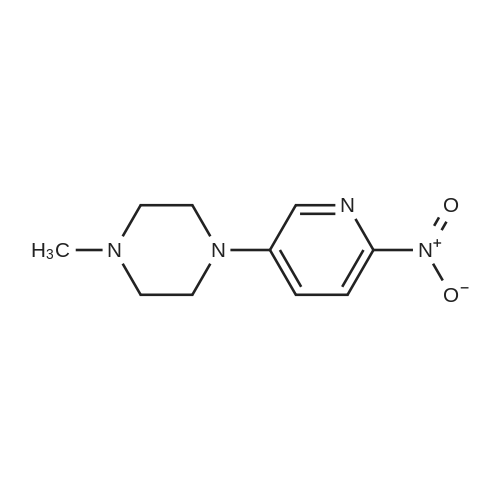
With caesium carbonate In DMF (N,N-dimethyl-formamide) at 50℃;
9.a
a) 4-[(6-n itro-3-pyrid invl) oxy] benzamide 4-hydroxybenzamide (150 mg, 1.1 mmol) and caesium carbonate (394 mg, 1.21 mmol) was dissolved in dimethylformamide (7ml). 5-bromo-2-nitropyridine (244 mg, 1.21 mmol) was then added. The mixture was left at 50 degrees until the starting material was consumed according to TLC (1 % methanol in ether). Purification by flash chromatography yielded 4-[(6-nitro-3-pyridinyl) oxy] benzamide (110 mg, 38 %) 1H NMR (CDCl3) : 8. 37 (d, 1H), 8. 29 (d, 1H), 7.94 (m, 2H), 7.51 (dd, 1H), 7.18 (m, 2H).
With potassium phosphate; copper(l) iodide; trans-1,2-cyclohexanediamine; In 1,4-dioxane; at 100℃; for 12.0h;
To a solution of 24.6 mmol of 9 and 27.3 mmol of 7 in 50 mL of 1,4-dioxane was added 4.92 mmol of copper (I) iodide followed by the addition of 49.2 mmol of K3P04 and 4.92 mmol of trans-cyclohexanediamine, then the resulting mixture was stirred at 100C for 12 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. The residue was diluted with CHC13, poured into water, and insoluble material was removed by celite filtration. The filtrate was extracted with CHC13, dried over MgS04 and concentrated in vacuo. The crude product was purified by column chromatography to give 7.87 mmol of nitro derivative. [0202] A solution of 7.66 mmol of nitro derivative and 0.5 g of Pd/C (10%) in 150 mL of methanol was stirred overnight under H2 (1 atm). After filtering through celite, the solution was concentrated under reduced pressure to give 4.75 mmol of 8.
With potassium phosphate;copper(l) iodide; trans-1,2-cyclohexanediamine; In 1,4-dioxane; at 100℃; for 12.0h;
To a solution of 24.6 mmol of 9 and 27.3 mmol of 7 in 50 mL of 1,4-dioxane was added 4.92 mmol of copper (I) iodide followed by the addition of 49.2 mmol ofK3P04 and 4.92 mmol of trans-cyclohexanediamine, then the resulting mixture was stirred at 100C for 12 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. The residue was diluted with CHC13, poured into water, and insoluble material was removed by celite filtration. The filtrate was extracted with CHCl3, dried over MgS04 and concentrated in vacuo. The crude product was purified by column chromatography to give 7.87 mmol of nitro derivative. [0233] A solution of 7.66 mmol of nitro derivative and 0.5 g of Pd/C (10%) in 150 mL of methanol was stirred overnight under H2 (1 atm). After filtering through celite, the solution was concentrated under reduced pressure to give 4.75 mmol of 8. ¹H NMR (400 MHz, DMSO-d6) 8 7.70 (d, J = 2.4 Hz, 1H), 7.17 (dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 1H), 7.30-7.36 (m, 5H), 6.40 (d, J = 8.8 Hz, 1H), 5.90 (br s, 2H), 3.66-3.72 (m, 2H), 3.59 (br s, 2H), 2.59-2.71 (m, 6H); MS m/z: 327 (M+I).
With potassium phosphate; copper(l) iodide; trans-1,2-cyclohexanediamine; In 1,4-dioxane; at 100℃; for 12.0h;
Example 6 Preparation of 8 6.1 Ullmann Cross-Coupling To a solution of 24.6 mmol of 9 and 27.3 mmol of 7 in 50 mL of 1,4-dioxane was added 4.92 mmol of copper (I) iodide followed by the addition of 49.2 mmol of K3PO4 and 4.92 mmol of trans-cyclohexanediamine, then the resulting mixture was stirred at 100 C. for 12 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. The residue was diluted with CHCl3, poured into water, and insoluble material was removed by celite filtration. The filtrate was extracted with CHCl3, dried over MgSO4 and concentrated in vacuo. The crude product was purified by column chromatography to give 7.87 mmol of nitro derivative. A solution of 7.66 mmol of nitro derivative and 0.5 g of Pd/C (10%) in 150 mL of methanol was stirred overnight under H2 (1 atm). After filtering through celite, the solution was concentrated under reduced pressure to give 4.75 mmol of 8.
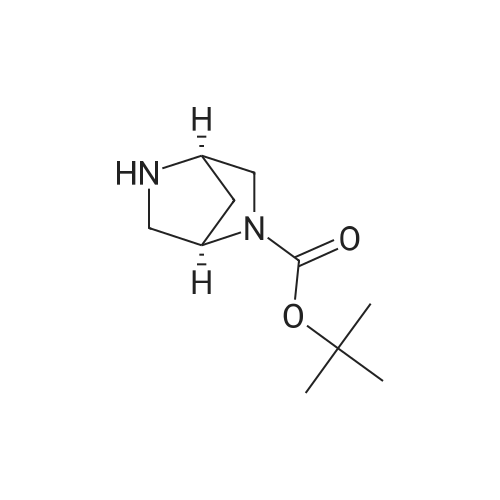
Example 10A tert-butyl 3-(6-nitro-3-pyridinyl)-3,8-diazabicyclo[3.2.1]octane-8-carboxylate tert-Butyl 3,8-diazabicyclo[3.2.1]octane-8-carboxylate (0.4 g; 1.9 mmol), prepared as described in (J. Med. Chem., (1998) 41, 674), 5-bromo-2-nitropyridine (0.43 g; 2.27 mmol), prepared as described in (J. Am. Chem. Soc., (1945) 67, 668), and triethylamine (0.23 g; 2.27 mmol) in toluene (10 mL) were heated at reflux for 14 hours. After evaporation of the solvent, additional triethylamine (0.23 g) was added and the mixture further heated at 140 C for 2 hours. The residue was purified on SiO2 (CH2Cl2:EtOAc 9:1) to provide the title compound.
With triethylamine; In dimethyl sulfoxide; at 110℃; for 16h;Inert atmosphere;
Step I tert-butyl 3-(6-nitro-3-pyridyl}-3,8-diazabicyclo[3.2.1]octane-8-carboxylate To a solution of <strong>[149771-44-8]tert-butyl 3,8-diazabicyclo[3.2.1]octane-8-carboxylate</strong> (1.00 g, 4.71 mmol, 1.00 equivalent) in dimethylsulfoxide (20.00 mL) were added triethylamine (1.43 g, 14.13 mmol, 3.00 equivalents) and 5-bromo-2-nitro-pyridine (1.15 g, 5.65 mmol, 1.20 equivalents). The mixture was stirred at 100C for 16 hours. LC/MS showed completion of the reaction. 40 mL of water was added to the mixture until the mixture was cooled to 20C. The aqueous phase was subjected to extraction using dichloromethane (50 mL*2), washed with saturated brine (50 mL*1), dried over anhydrous sodium sulfate, filtered and concentrated. The crude product was purified by being beaten in methanol (2 mL*3) to give the title compound (639.00 mg, crude product) as a yellow solid. 1H NMR (400MHz, CDCl3) delta 8.18 (d, J = 9.2 Hz, 1H), 8.09 (d, J = 3.0 Hz, 1H), 7.15 (dd, J = 9.2 Hz, 3.0 Hz, 1H), 4.47 (br. s., 2H), 3.58 (d, J = 10.0 Hz, 2H), 3.27 (br. s., 2H), 2.12-1.99 (m, 2H), 1.86-1.76 (m, 2H), 1.49 (s, 9H). LC/MS (ESI) m/z: 335.2 (M+1).
101 6'-Nitro-3,4,5,6-tetrahydro-2H-[1,3']bipyridinyl
EXAMPLE 101 6'-Nitro-3,4,5,6-tetrahydro-2H-[1,3']bipyridinyl 5-Bromo-2-nitropyridine (5.6 g, 27.6 mmol), tetra-n-butyl ammonium iodide (0.510 g, 1.38 mmol), piperidine (2.58 g, 30.3 mmol) and potassium carbonate (3.85 g, 30.3 mmol) were mixed in DMSO (50 mL). The reaction mixture was warmed to 80° C. for 4 hours. The reaction mixture was diluted with ethyl acetate and filtered. The volume was reduced to remove ethyl acetate, the remaining solution was diluted with water (50 mL). A precipitate immediately formed and was collected by filtration and washed on the funnel with water to provide 6'-nitro-3,4,5,6-tetrahydro-2H-[1,3']bipyridinyl as an orange-brown solid (4.90 g, 85.7%). 1H NMR δ (400 MHz, CDCl3) 7.76 (s, 1H), 7.15 (d, J=7.3 Hz, 1H), 6.49 (d, J=8.5 Hz, 1H), 3.84 (m, 5H), 3.00 (m, 4H), 2.60 (s, 1H).
With potassium carbonate; triethylamine; In dichloromethane; dimethyl sulfoxide;
EXAMPLE 85 2,2-Dimethyl-4-(6-nitro-pyridin-3-yl)-piperazine-1-carboxylic Acid Tert-butyl Ester 5-Bromo-2-nitropyridine (10.67 g, 52.6 mmol), tetra-n-butyl ammonium iodide (0.97 g, 02.63 mmol), <strong>[84477-72-5]2,2-dimethyl-piperazine</strong> (6.60 g, 57.8 mmol) and potassium carbonate (8.00 g, 57.8 mmol) were mixed in DMSO. (50 mL). The reaction mixture was warmed to 95 C. for 5 hours. The reaction mixture was poured onto ice chips (approximately 200 mL) then extracted with dichloromethane (6*75 mL). The combined organics were dried over MgSO4, the inorganic salts were removed by filtration and the remaining solvents were concentrated to provide an orange solid. This solid was dissolved in dichloromethane (100 mL) to which triethylamine (10.65 g, 14.7 mL, 105 mmol) and di-tert-butyl dicarbonate (13.8 g, 63.12 mmol) were added. After 16 hours, more di-tert-butyl dicarbonate (3.8 g, 17.41 mmol) was added and the mixture was brought to reflux for 3 hours. The reaction mixture was then cooled to room temperature and diluted with dichloromethane (100 mL) and washed with water (1*100 mL). The organic layer was then dried over MgSO4, filtered, and the solvent evaporated to yield 2,2-dimethyl-4-(6-nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester as an orange solid (14.91 g, 84.2%). 1H NMR delta (400 MHz, CDCl3) 8.17 (d, J=9.3 Hz, 1H), 7.97 (d, J=2.9 Hz, 1H), 7.01 (dd, J=2.9, 9.0 Hz, 1H), 3.91 (m, 2H), 3.54 (m, 4H), 1.48 (s, 9H), 1.43 (s, 6H).
With potassium carbonate; triethylamine; In tetrahydrofuran; dichloromethane; dimethyl sulfoxide;
EXAMPLE 91 4-(6-Amino-pyridin-3-yl)-2,6-dimethyl-piperazine-1-carboxylic Acid Tert-butyl Ester 5-Bromo-2-nitropyridine (10.81 g, 53.3 mmol), tetra-n-butyl ammonium iodide (0.98 g, 2.66 mmol), 2,6-dimethyl-piperazine (6.69 g, 58.6 mmol) and potassium carbonate (8.10 g, 58.6 mmol) were mixed in DMSO (50 mL). The reaction mixture was warmed to 80 C. for 4 hours by which time the reaction was complete by TLC analysis. The reaction mixture was diluted with dichloromethane and washed with water (3*75 mL). The combined organics were dried over MgSO4, the inorganic salts were removed by filtration and the remaining solvents were concentrated to provide an orange solid. This solid was dissolved in dichloromethane (150 mL) to which triethylamine (10.8 g, 14.8 mL, 108 mmol) and di-tert-butyl dicarbonate (13.95 g, 63.9 mmol) were added. The reaction mixture was heated to reflux for 3 hours then cooled to room temperature and diluted with dichloromethane (100 mL) and washed with water (1*100 mL). The organic layer was then dried over MgSO4, filtered and the solvent evaporated to yield an orange solid. The orange solid was dissolved in THF (500 mL) to which Raney Nickel (9.23 g) was added. The reaction mixture was shaken under a hydrogen atmosphere (50 psi) for 4 hours. The catalyst was removed by filtration, and the solvent evaporated to give a crude purple solid. This solid was purified by chromatography eluding with ethyl acetate to give 4-(6-amino-pyridin-3-yl)-2,6-dimethyl-piperazine-1-carboxylic acid tert-butyl ester as a purple solid (4.36 g, 26.7%). 1H NMR delta (400 MHz, CDCl3) 7.72 (d, J=2.4 Hz, 1H), 7.18 (dd, J=2.9, 8.8 Hz, 1H), 6.51 (d, J=8.8 Hz, 1H), 4.35 (s, 2H), 4.21 (m, 2H), 3.08 (dd, J=4.4, 11.7 Hz 2H), 1.48 (s, 9H), 1.35 (d, J=6.8 Hz, 6H).
With potassium carbonate; In N,N-dimethyl-formamide; at 120℃; for 12h;
) 3-Methyl-l-(6-nitropyridin-3-yl)pyrrolidin-3-ol; A mixture of 5-bromo-2-nitropyridine (2 g, 0.010 mol), HCl salt of pyrrolidinol derivative (1.62 g, 0.012 mol) and dry K2CO3 (4 g, 0.030 mol) in dry DMF (25 mL) was heated at 120 C for 12 h under nitrogen atm. The reaction mixture was brought to RT and filtered. The filtrate was concentrated, added with ethyl acetate and washed with water (3 x 10OmL) and brine, dried over Na2SO4 and concentrated. The crude product was purified by column chromatography using 50 % EtOAc in pet. ether. Yield =1.1 g (50 %). 1H-NMR (400 MHz, CDCi): delta 8.16 (d, IH), 7.79 (m, IH), 6.83 (m, IH), 3.70 (m, IH), 3.57 (m, IH), 3.46 (d, IH), 3.42 (d, IH), 2.05-2.20 (m, 2H), 1.57 (s, 3H).
With N-ethyl-N,N-diisopropylamine; In ethanol; at 90℃; for 96h;
Preparation of (2-methoxyethyl)-methy-(6-nitropyridin-3-yl)-amine A solution of 2-nitro-5-bromopyridine (500 mg, 3.15 mmol) in EtOH (15 mL) was treated with methoxyethyl-N-methylamine (1.12 g, 12.6 mmol) and diisopropylethylamine (2.2 mL, 12.6 mmol). The resulting mixture was then heated in a sealed tube at 90 C. for 4 days then cooled and partitioned between EtOAc and water. The organic layer was dried over Na2SO4, filtered and concentrated. The residue was chromatographed on a silica gel column with a 40-100% EtOAc in hexanes gradient to afford the product as a thick yellow oil that crystallized slowly upon standing (270 mg, 41% yield). HRMS m/z calcd for C9H13N3O3 [M+Na]+: 234.0849; Found: 234.0850.
b) Potassium carbonate (780 mg, 5.65 mmol) was added to a solution of 4-cyanophenylpiperazine (880 mg, 4.7 mmol) and 2-nitro-5-bromopyridine (860 mg, 4.24 mmol) in dimethylsulfoxide (6 ml) and heated at 95 C. for 20 hours. The reaction was cooled, poured into water (90 ml) and extracted into dichloromethane (3*50 ml). The organic solution was washed with water then dried and concentrated in vacuo. Purification by flash chromatography on alumina, eluding with dichloromethane, followed by recrystallisation from methanol/ethyl acetate yielded 1-(4-cyanophenyl)-4-(5-nitropyridin-2-yl)piperazine (160 mg, 12% yield) as a white solid: 1H-NMR (CDCl3): 9.05 (s, 1H), 8.25 (d, 1H), 7.55 (d, 2H), 6.85 (d, 2H), 6.60 (d, 1H), 3.97 (m, 4H), 3.50 (m, 4H): MS (+ve ESI): 310 (M+H)+.
A mixture of 5-bromo-2-nitropyridine (5 g, 24.63 mmol), tributyl(l-ethoxyvinyl)tin (9.785 g, 27.1 mmol) and Ph(PPh3)4 (0.284 g, 0.246 mmol) in DMF (100 ml) were stirred at 120 0C for 3 h. After cooling, a 1 N HCl sol. was added and the r.m. was stirred at r.t. for 18 h. The r.m. was neutralized with a sat. aq. NaHCO3 sol. and the product was extracted with DCM. The organic phase was dried (Na2SO4), filtered and the solvent was evaporated in vacuo. The residue was purified by flash chromatography over Silica gel (eluent: DCM/MeOH from 100/0 to 98/2). The product fractions were collected and the solvent was evaporated. Yield: 3.44 g of intermediate 29 (83 %).
triphenylphosphine; bis(dibenzylideneacetone)-palladium(0); In toluene; for 15h;Heating / reflux;
1-(6-Nitropyridin-3-yl)ethanone may be prepared in the following manner:1.14 g (1.99 mmol) of bis(dibenzylideneacetone)palladium and 10.1 g (49.75 mmol) of 5-bromo-2-nitropyridine are added at a temperature in the region of 20 C. under an argon atmosphere to a solution of 1.044 g (3.98 mmol) of triphenylphosphine in 10 cm3 of toluene. After stirring for 15 minutes at this temperature, a solution of 16.9 cm3 (49.75 mmol) of 1-ethoxy-vinyltributyltin in 60 cm3 of toluene is added. After heating under reflux for 15 hours, the reaction medium is cooled to 20 C. and then poured into 500 cm3 of a 1N hydrochloric acid solution and stirred for 16 hours at this temperature. The medium is then extracted with three times 150 cm3 of ethyl acetate. The organic phases are combined and then washed with 300 cm3 of water, dried over anhydrous magnesium sulphate, filtered and concentrated to dryness under reduced pressure (2.7 kPa) to give a residue which is purified by flash chromatography [eluent: pure dichloromethane]. After concentrating the fractions under reduced pressure, 3.4 g of 1-(6-nitropyridin-3-yl)ethanone are obtained. 1H NMR (400 MHz, (CD3)2SO, -delta in ppm) 2.71 (s, 3H); 8.42 (d, J=8.5 Hz, 1H); 8.66 (dd, J=2.5 and 8.5 Hz, 1H); 9.15 (d, J=2.5 Hz, 1H).
Reference Example 55; tert-butyl { 2-chloro-5- [ ( 6-nitropyridin-3-yl) oxy] phenyl }carbamate; 5-Bromo-2-nitropyridine (14.O g, 69.0 mmol) was added to a mixture of <strong>[16026-77-0]3-amino-4-chlorophenol</strong> (9.44 g, 65.7 mmol), cesium carbonate (32.2 g, 99.0 mmol) and N,N-dimethylformamide (70 mL) at 00C, and the mixture was stirred at room temperature for 17 hr. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate and filtrated. The filtrate was concentrated under reduced pressure, and the residue was dissolved in acetonitrile (100 mL) . di-tert- Butyl dicarbonate (24.0 g, 110 mmol), 4-dimethylaminopyridine (8.04 g, 65.8 mmol) and acetonitrile (150 mL) were added, and the <n="199"/>mixture was stirred at room temperature for 30 min. tert-Butyl alcohol (11.0 mL, 115 mmol) was added to the reaction mixture, and the mixture was stirred at 800C for 68 hr. The reaction solution was concentrated under reduced pressure, and ethyl acetate was added to the residue. The mixture was washed with saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous magnesium sulfate and filtrated. The filtrate was concentrated under reduced pressure, and the residue was purified by column chromatography (silica gel, hexane/ethyl acetate=100/0-»80/20) to give the title compound (13.0 g, 54%) as a pale-yellow oil.1H-NMR (CDCl3, 300 MHz) delta 1.52 (9H, s) , 6.73 (IH, dd, J = 8.8, 2.8 Hz), 7.38 - 7.47 (2H, m) , 8.10 (IH, d, J = 2.6 Hz), 8.18 (IH, d, J = 1.5 Hz), 8.25 (IH, d, J = 8.8 Hz), 8.32 (IH, d, J = 2.6 Hz) .
With caesium carbonate; In N,N-dimethyl-formamide; at 50℃; for 15h;
A mixture of 3 (7.76g, 55.8mmol), 5-bromo-2-nitropyridine (10.3g, 50.7mmol), cesium carbonate (24.8g, 76.1mmol), and DMF (150mL) was stirred at 50C for 15h. The mixture was diluted with water and extracted with AcOEt. The extract was washed with water and brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was purified by basic silica gel column chromatography (AcOEt/hexane) to give 4 (4.37g, 33%) as a yellow solid: 1H NMR (DMSO-d6) delta 7.73-7.85 (3H, m), 8.10 (1H, t, J=2.1Hz), 8.15-8.19 (1H, m), 8.38 (1H, dd, J=9.0, 0.6Hz), 8.52-8.54 (1H, m).
44%
With caesium carbonate; In N,N-dimethyl-formamide; at 20℃; for 15h;
Reference Example 33; tert-butyl {4-methyl-3- [ (6-nitropyridin-3-yl) oxy] phenyl} carbamate; A mixture of tert-butyl (3-hydroxy-4-methylphenyl) carbamate (3.14 g, 14.1 mmol), 5-bromo-2-nitropyridine (2.38 g, 11.7 mmol), <n="184"/>cesium carbonate (5.72 g, 17.6 mmol) and N,N-dimethylformamide (25 mL) was stirred at room temperature for 15 hr. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate and filtrated. The filtrate was concentrated under reduced pressure, and the residue was purified by column chromatography (NH silica gel, hexane/ethyl acetate=80/20-»0/100) to give the title compound (1.86 g, 44%) as a colorless oil. 1H-NMR (DMSOd6, 300 MHz) delta 1.45 (9H, s) , 2.06 (3H, s) , 7.24 - 7.34 (3H, m) , 7.45 (IH, dd, J = 9.2, 2.9 Hz), 8.32 (IH, d, J = 9.2 Hz), 8.38 (IH, d, J = 2.9 Hz), 9.50 (IH, s) .
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl acetamide; at 90℃;
The compound of formula 38 (10.0 g, 49.2 mmol) was added to the reaction flask at room temperature,100 mL of N, N-dimethylacetamide,1-acetylpiperazine 16 (8.2 g, 64.0 mmol)N, N-diisopropylethylamine (19.1 g, 147.8 mmol)90 reaction 7 ~ 8h,TLC monitoring reaction is complete,Down to room temperature,The reaction solution was poured into 300 mL of water,Extracted with ethyl acetate (80 mL x 3)Combined organic layer,Washed with saturated brine,Dried over anhydrous sodium sulfate,The compound of formula 41 (9.9 g) was obtained by steaming,As a beige solid (yield 80.6%).
With caesium carbonate; 2,2'-bis-(diphenylphosphino)-1,1'-binaphthyl;tris-(dibenzylideneacetone)dipalladium(0); In toluene; at 100℃; for 16h;
Example 39 1-[4-(6-Nitro-pyridin-3-yl)-piperazin-1-yl]-ethanone To a mixture of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (406 mg, 2 mmol) and 1-acetylpiperazine (256 mg, 2 mmol) in toluene (5 mL) is added Cs2CO3, then Pd2(dba)3 (74 mg, 0.08 mmol) and BINAP (100 mg, 0.16 mmol) are added. The mixture is degassed, and heated at 100 C. for 16 hours. Then the mixture is cooled down to room temperature, diluted with EtOAc, and filtered through celite. The filtrate is concentrated under reduced pressure. The crude product is purified by column chromatography (SiO2, MeOH:CH2Cl2=0.7:99.3 to 6:93) to give 270 mg of the title compound as yellow solid. MS (ESI) m/z 251 (M+H)+
With triethylamine; In dimethyl sulfoxide; at 120℃; for 16h;Sealed tube;
To a sealed tube was added <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> 111-1 (2.3 g, 11.4 mmol), l-(piperazin-l-yl)ethanone 111-2 (1.6 g, 12.8 mmol), triethylamine (4.8 mL, 34.2 mmol) and DMSO (5 mL). The reaction was heated to 120C and stirred for 16 hours. The reaction was cooled to room temperature. Triethylamine was removed by rotary evaporation. The residue was triturated in 15 mL ethyl acetate. The solid was collected by filtration and washed with small amount of ethyl acetate to give l-(4-(6-nitropyridin-3-yl)piperazin-l-yl)ethanone 111-3 as a light yellow solid. MS m/z 251.1 (M + 1).
With triethylamine; In dimethyl sulfoxide; at 120℃; for 16h;
To a solution of acetylpiperazine (5.00 g, 39.0 mmol) and <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (5.00 g, 24.6 mmol) in DMSO (10 mL) was added triethylamine (10.2 mL, 73.9 mmol). The reaction mixture was heated at 120 C for 16 h, cooled down to room temperature and triturated with EtOAc. The formed solid was filtered and washed with EtOAc (10 mL) and i12O (30 mL) to give the first portion of 1-(4-(6- nitropyridin-3-yl)piperazin-l-yl)ethan-1-one as a yellow solid. The filtrate and washing were combined and extracted with DCM (3 x lOOmL). The organic extracts were combined, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by chromatography (silica gel, DCM ramping to DCM:MeOH = 94:6) to give the second portion of l-(4-(6-nitropyridin-3-yl)piperazin-1- yl)ethan-i-one. ?HNMR (CDCI3) 2.l6 (s, 3H), 3.47 (t, 2H,J5.5), 3.52 (t, 2H,J5.5), 3.71 (t, 2H,J5.5), 3.83 (t, 2H, .1 5.5), 7.23 (dd, 1H,J9.5 & 3.0), 8.14 (d, IH,.J3.0), 8.20 (d, 1H,J9.0). HRIVIS (ESI)251.1130 (IM+Hr); calcd. for C,,H,5N403r ([M+Hj) 251.1139.
Example 121Exam le 121a ieri-Butyl 4-(6-Nitropyridin-3-yl)-3-oxopiperazine-l-carboxylate 121aA 250-mL single-neck round-bottomed flask equipped with a magnetic stirrer and reflux condenser was charged with 2-nitro-5-bromopyridine (1.00 g, 5.00 mmol), 2-oxo-4- (ieri-butoxycarbonyl)piperazine (1.01 g, 5.00 mmol), cesium carbonate (3.58 g, 11.0 mmol) and 1,4-dioxane (40 mL). After bubbling nitrogen through the result-ing solution for 30 min, Xantphos (246 mg, 0.425 mmol) and tris(dibenzylidene-acetone)dipalladium(0) (230 mg, CGIPHARM60WO0.250 mmol) were added, and the reaction mixture was heated at reflux for 6 h. Water (30 mL) and ethyl acetate (150 mL) were added after the reaction mixture was cooled to room temperature. The resulting mixture was filtered through a bed of Celite 521. The organic layer of the filtrate was separated and the aqueous layer was extracted with ethyl acetate (2 x 50 mL). The combined organic layers were washed with brine (30 mL), dried over sodium sulfate and purified by column chromatography to afford a 96% yield (1.55 g) of 121a as an amber oil: ]H NMR (300 MHz, CDC13) delta 8.67 (d, 1H, J = 2.4 Hz), 8.32 (d, 1H, J = 8.7 Hz), 8.15 (dd, 1H, / = 8.7, 2.4 Hz), 4.33 (s, 1H), 3.89 (m, 4H), 1.48 (s, 9H); MS (ESI+) mJz 323.1 (M+H).
77%
With caesium carbonate;palladium diacetate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 120℃; for 0.5h;Microwave irradiation;
A mixture of 5-bromo-2-nitropyridine (200 mg, 1 mmol), l-Boc-3-oxopiperazine ( 240 mg, 1.2 mmol), Xantphos (43 mg, 0.075 mmol), cesium carbonate (326 mg, 1 mmol), palladium(II) acetate (11 mg, 0.049 mmol) in dioxane (5.5 mL) is heated to 120 C in a Personal Chemistry microwave apparatus for 0.5 h. TLC and LCMS analysis indicates completion of the reaction. The reaction mixture is filtered through Celite, evaporated in vacuo, and the residue is partitioned between water and ethyl acetate. The organic layer is washed with brine, dried (Na2SO4) and evaporated in vacuo. Purification by flash chromatography on silica (ethyl acetate) provides 4-(6-Nitropyridin-3-yl)-3- oxopiperazine-1-carboxylic acid tert-butyl ester as a pale brown solid (248 mg, 77%). MS (ESI) m/z 323 [M+H+]
67%
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; for 2h;Inert atmosphere; Reflux;
In reference to the process disclosed in WO2012/031004, 2-nitro-5-bromopyridine (1.01 g, 5.0 mmol), tert-butyl 2-oxo-4-piperazinecarboxylate (1.00 g, 5.0 mmol, and cesium carbonate (3.26 g, 10.0 mmol) were suspended in 1,4-dioxane, and the suspension was bubbled with nitrogen gas for 30 minutes. To the suspension was added Xantphos (246 mg, 0.43 mmol) and tris(dibenzylideneacetone)dipalladium (229 mg, 0.25 mmol), and the mixture was stirred under reflux for two hours. The resultant reaction mixture was cooled to room temperature, and water and ethyl acetate were then added to the mixture, followed by filtration with Celite. The organic phase was separated from the filtrate, and the aqueous phase was extracted with ethyl acetate. The resultant organic phases were combined together and dried over anhydrous sodium sulfate, and the resultant solid was separated by filtration. The filtrate was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography to yield the title compound (1.08 g, 67%). 1H-NMR (CDCl3) delta: 8.67 (1H, d, J=2.4 Hz), 8.32 (1H, d, J=8.8 Hz), 8.15 (1H, dd, J=8.8, 2.4 Hz), 4.33 (2H, s), 3.93-3.83 (4H, m), 1.51 (9H, s)
67%
With reference to the process described in , 2-nitro-5-bromopyridine (1.01 g, 5.0 mmol), tert-butyl 2-oxo-4-piperazinecarboxylate (1.00 g, 5.0 mmol) and cesium carbonate (3.26 g, 10.0 mmol) were suspended in 1,4-dioxane. Nitrogen gas was bubbled into the suspension for 30 minutes. Xantphos (246 mg, 0.43 mmol) and tris(dibenzylideneacetone)dipalladium (229 mg, 0.25 mmol) were added to the suspension and the reaction mixture was stirred under reflux with heating for two hours. The reaction mixture was cooled to room temperature. Water and ethyl acetate were added to the mixture. The solution was filtered through a Celite pad. The organic phase was separated from the filtrate. The aqueous phase was extracted with ethyl acetate. The extracted organic phases were combined and dried over anhydrous sodium sulfate. The solid was filtered out, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give the title compound (1.08 g, yield: 67%). 1H-NMR(CDCl3) delta: 8.67 (1H,d,J=2.4 Hz), 8.32 (1H,d,J=8.8 Hz), 8.15 (1H,dd,J=8.8,2.4 Hz), 4.33 (2H,s), 3.93-3.83 (4H,m), 1.51 (9H,s).
47.86%
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 100℃; for 1h;Inert atmosphere;
Synthesis of compound 188.1. To a solution of 78.4 (l .Og, 4.92mmol, l .Oeq) in 1,4-dioxane (15ml) was added tert-butyl 3-oxopiperazine-l-carboxylate (1.18g, 5.91mmol, 1.2eq) and CS2CO3 (3.2 g, 9.85 mmol, 2.0eq). Reaction mixture was degassed for 10 minutes using argon, then Pd2(dba)3 (0.450g, 0.492mmol, 0.1 eq) and xantphos (0.560g, 0.985mmol, 0.2eq) were added, and again degassed for 5 minutes. The reaction was stirred at 100C in for lh. After completion of the reaction, mixture was poured into water and product was extracted with EtOAc. Organic layers were combined and dried over Na2S04 and concentrated under reduced pressure to obtain crude which was purified by column chromatography to furnish pure 188.1 (0.760g, 47.86%). MS (ES): m/z 323.6 [M+H]+.
To 4-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1 -carboxylic acid tert- butyl ester (8.81 g, 43.4 mmol) and <strong>[39856-50-3]5-bromo-2-nitro-pyridine</strong> (13.8 g, 44.7 mmol) in 1 ,4- dioxane/H20 (90 ml_/22.5 mL) is added K2C03 (18.0 g, 130 mmol) and degassed with argon for 5 min. Then PdCI2(PPh3)2 (152 mg, 0.22 mmol) is added. The reaction mixture is stirred at 90C for 3h. The reaction mixture is diluted with water (70 mL) and 1 ,4-dioxane is distilled off to 130 g. Subsequently it is charged with MTBE (22.5 mL) and heptane (90 mL) and stirred for 30 min. The precipitate is filtered. The cake is washed with water, 1 :4 MTBE/heptane and dried under vacuum. (0652) Yield: 13.2 g (quantitative) ESI-MS: m/z = 306 (M+H)+
95%
With bis-triphenylphosphine-palladium(II) chloride; potassium carbonate; In 1,4-dioxane; water; at 20 - 80℃;Inert atmosphere;
Method B A 12.0 L three neck flask equipped with mechanical stirrer, heating mantle, condenser, thermometer was charged with <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (322 g, 1.59 mol), potassium carbonate (658 g, 4.76 mol), tert-butyl 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-5,6- dihydropyridine-l(2H)-carboxylate (500 g, 1.62 mol), 1,4-dioxane (3.24 kg, 3.14 1) and water (314 g, 314 ml). The solution was degassed thrice by pulling vacuum then releasing with N2. Under a strong N2 flow, bis(triphenylphosphine)palladium(ii) dichloride (11.1 g, 15.9 mmol) was added. The reaction mixture was stirred at 80 C for 5 hours. The heat was turned off and the reaction mixture stirred at ambient temperature, overnight. The mixture was heated to 40 C, and then charcoal (200 g, 16.7 mol) was added. The mixture stirred at 40 C for 1 hour. The mixture was cooled to 30 C, then filtered through Celite and washed with plenty of dioxane. To the combined filtrate and wash was added hydrogen peroxide (50.0 g, 45.0 ml, 441 mmol) and the mixture was stirred at ambient temperature for 1 hour. The reddish solution was stored at room temperature for 48 hours then concentrated under vacuum at 50 C to a low volume solution (weight ca 3226 g). The solution was transferred into a 12L three neck flask and stored at room temperature under N overnight. The solution was heated to 40 C, then added slowly to water (3.38 kg, 3.38 1, 188 mol). A solid crystallized out. The heat was turned off and the mixture was allowed to cool to room temperature. The mixture was cooled to 8 C and stirred for 2 hours. The solids were filtered and washed with 1 :2 dioxane/water, followed by water. The solid was dried by vacuum for 30 minutes. The solid was transferred into a drying tray then dried in a vacuum oven at 50 C/26 inches Hg with a N2 bleed to a constant weight to afford 460 g (95%) of the desired product. *H NMR (300 MHz, DMSO- ) delta ppm 1.43 (s, 9 H) 2.55 (d, 7=1.89 Hz, 2 H) 3.47 - 3.73 (m, 2 H) 4.08 (d, 7=2.64 Hz, 2 H) 6.57 (br. s., 1 H) 8.20 - 8.26 (m, 1 H) 8.27 - 8.33 (m, 1 H) 8.77 (d, 7=1.89 Hz, 1 H).
90%
With bis-triphenylphosphine-palladium(II) chloride; caesium carbonate; In water; N,N-dimethyl-formamide; at 90℃; for 1h;Inert atmosphere;
Mixture of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (2.6 g, 12.8 mmol, 1.0 eq.), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydropyridine-1(2H)-carboxylic acid tert-butyl ester (tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate) (4.8 g, 15.5 mmol, 1.2 eq), bis(triphenylphosphine) palladium dichloride (0.9 g, 1.28 mmol, 0.1 eq.), cesium carbonate (8.4 g, 25.8 mmol, 2 eq.) was dissolved in 15 ml of water and 150 ml N,N- dimethylformamide. Under nitrogen atmosphere, the reaction system was placed in a preheated 90 C oil bath and heated for 1 hour. Completion of the reaction into the reaction system was cooled to room temperature and extracted with ethyl acetate, and the organic phase was washed with saturated brine and more concentrated once, then dried over anhydrous sodium sulfate, and reduced pressure. Finally, to give a yellow solid was purified by silica gel column, which is 4-(6-nitropyridin-3-yl)-3,6-dihydropyridin-1(2H)-carboxylic acid tert-butyl ester (3.53 g, yield: 90%). The above compound (3.53 g, 11.6 mmol, 1.0 eq.) was dissolved solution of 100 ml of methanol, followed by addition of palladium on carbon (0.353 g). Under the conditions of hydrogen, the reaction was stirred at room temperature overnight. After completion of the reaction, was filtered to remove palladium on carbon in the reaction mixture, the filter cake washed with methanol with several times, the filtrate was concentrated under reduced pressure. Finally, to give a yellow solid was purified by silica gel column, which is 4- (6-amino-pyridin-3-yl) piperidine-1-carboxylate (2.5 g, yield: 78%).
90%
With bis-triphenylphosphine-palladium(II) chloride; caesium carbonate; In water; N,N-dimethyl-formamide; at 90℃; for 1h;Inert atmosphere;
5-Bromo-2-nitropyridine (107A) (2.6 g, 12.8 mmol, 1.0 eq), 33 tert-butyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (107B) (4.8 g, 15.5 mmol, 1.2 eq), bis(triphenylphosphine)palladium(II) chloride (0.9 g, 1.28 mol, 0.1 eq) and cesium carbonate (8.4 g, 25.8 mmol, 2.0 eq) were dissolved in 15 mL of 9 water and 150 mL of N,N-dimethylformamide. Under nitrogen protection, the reaction system was placed in a pre-heated 90 C. oil bath and reacted for 1 hour. After the reaction was completed, the reaction system was cooled to room temperature and extracted with ethyl acetate. The organic phase was washed with saturated brine several times, dried over anhydrous sodium sulfate, concentrated in vacuo and finally purified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate=10:1 to 3:1) to obtain a yellow solid, which was tert-butyl 6-nitropyridine-3?,6?-dihydro-[3,4?-pyridine]-1?(2?H)-carboxylate (107C) (3.53 g, yield: 90%). LCMS (ESI): m/z 306 [M+1]+.
79%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate; In 1,4-dioxane; water; at 80℃; for 8h;Inert atmosphere;
N-Boc-1,2,5-6-tetrahydropyridine-4-boronic acid pinacol ester (18,3.1 g, 10 mmol), <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (19, 1.8 g, 9 mmol ) , cesium carbonate (6.5 g, 20 mmol) dissolved in 100 mL of dioxane / water (16: 1) in a mixed solvent, under nitrogen, [1,1'-bis(diphenylphosphine) D] palladium dichloride (Pd (dppf) Cl2, 0.7 g, 1mmol), heated to 80 deg.] C for 8 hours, cooled to room temperature, concentrated under reduced pressure, purification by silica gel column chromatography (petroleum ether: ether) to give 20 as a pale yellow solid ( 2.4g, 79%).
76%
With trans-bis(triphenylphosphine)palladium dichloride; caesium carbonate; In 1,4-dioxane; water; at 80℃;Inert atmosphere;
To a mixture of compound 23 (20 g, 98.5 mmol, 1 eq) and 24 (32 g, 103 mmol, 1.05eq) in dioxane (480 mL) was added Cs£03 (64 g, 196.4 mmol, 2 eq), H20 (20 mL) under nitrogen. The reaction mixture was evacuated and purged with nitrogen three times, thenPdCh(PPhJ)2 (7 g, 9.85 mmol, 0.1 eq) was added and purged with nitrogen again. The reactionmixture was stirred at 80 C overnight. After TLC and LCMS indicated completion; the mixturewas cooled to room temperature, H20 (480 ml) was added and extracted with EA (300 mL x3), the organic layer was washed with NaCl(salt) (200 mL x 3), dried over Na2S04, concentratedand purified by silica column to give the desired product as a yellow solid (15 g, 76%). LCMS:(M+Ht: 305.9.
74%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium phosphate tribasic trihydrate; sodium acetate; In water; acetonitrile; at 80℃; for 6h;Inert atmosphere;
Example 200a tert-Butyl 4-(6-Nitropyridin-3-yl)-5,6-dihydropyridine-1(2H)-carboxylate 200a A mixture of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (2.0 g, 9.7 mmol), tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-dihydropyridine-1(2H)-carboxylate (3.0 g, 9.7 mmol), Pd(dppf)Cl2 (792 mg, 0.97 mmol), K3PO4.3H2O (5.2 g, 19.4 mmol), and sodium acetate (1.59 g, 19.4 mmol) in acetonitrile (100 mL) and water (5 mL) was evacuated and then refilled with N2. The reaction mixture was heated at 80C for 6 h. It was then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was purified by silica-gel column chromatography eluting with 1:5 ethyl acetate/petroleum ether to afford 200a as a yellow solid (2.2 g, 74%).
71.2%
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; sodium carbonate; In 1,4-dioxane; water;Inert atmosphere; Heating;
4.1.40 Tert-Butyl 6-nitro-5',6'-dihydro-[3,4'-bipyridine]-1'(2'H)-carboxylate (10b) A solution of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (500 mg, 2.46 mmol) and tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-dihydropyridine-1(2H)-carboxylate (912 mg, 2.95 mmol) in 1,4-dioxane (15 mL) was degassed with nitrogen for 5 min, followed by addition of Na2CO3 (782 g, 2 M in water) under continuous flow of nitrogen. Pd(dppf)Cl2·CH2Cl2 (200 mg, 0.246 mmol) was added to the reaction mixture under a nitrogen atmosphere, and the reaction mixture was stirred at 80 C for 5 h. The mixture was diluted with H2O (10 mL) and extracted three times with EtOAc (15 mL). The combined organic layer was dried over Na2SO4 and the solvent was removed in vacuo. The residue product was purified on a silica gel column using petroleum ether/EtOAc (4:1, v/v) as eluent to afford 10b (540 mg, 71.2%) as a white solid. MS (ESI) m/z 306.2 [M+H]+; 1HNMR (400 MHz, CDCl3): delta 8.65 (s, 1H), 8.24 (d, J = 8.44 Hz, 1H), 7.95 (dd, J = 1.48 Hz, 8.48 Hz, 1H), 6.32 (s, 1H), 4.16 (d, J = 1.92 Hz, 2H), 3.78-3.65 (m, 2H), 2.57 (s, 2H), 1.51 (s, 9H).
69%
With caesium carbonate;bis-triphenylphosphine-palladium(II) chloride; In 1,4-dioxane; water; ethyl acetate; at 80℃; for 15h;Inert atmosphere;
Step 1 In a 50 mL round-bottomed flask, <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (3.28 g, 16.2 mmol, Eq: 1) and tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-dihydropyridine-1(2H)-carboxylate (5 g, 16.2 mmol, Eq: 1.00) were combined with dioxane (80.0 ml) to give a light yellow solution. Cs2CO3 (10.5 g, 32.3 mmol, Eq: 2) and 3 mL H2O were added. The solution was degassed with Ar before bis(triphenylphosphine)palladium(ii) dichloride (1.13 g, 1.62 mmol, Eq: 0.1) was added. The reaction mixture was heated to 80 C. and stirred for 15 h. The reaction mixture was poured into 300 mL H2O and extracted with EtOAc (3*100 mL). The combined organic extracts were washed with brine and dried over MgSO4. The crude reaction mixture was concentrated in vacuo. The crude material was purified by flash chromatography (silica gel, 220 g, 10% to 100% EtOAc/Hex gradient) to give a dark brown impure solid. The solid was triturated with Et2O to afford tert-butyl 4-(6-nitropyridin-3-yl)-5,6-dihydropyridine-1(2H)-carboxylate (685 mg, 69%) as a tan solid. LC/MS-ESI observed [M+H]+ 306.
64%
With bis-triphenylphosphine-palladium(II) chloride; caesium carbonate; In 1,4-dioxane; at 85℃;Inert atmosphere;
Step 1. Preparation of 6-nitro-3',6'-dihydro-2'H-[3,4']bipyridinyl-1'-carboxylic acid tert-butyl ester A solution of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylic acid tert-butyl ester (5.38 g, 17.40 mmol), <strong>[39856-50-3]5-bromo-2-nitro-pyridine</strong> (3.52 g, 17.40 mmol), Cs2CO3 (11.34 g, 34.8 mmol), and Pd(PPh3)2Cl2 (1.27 g, 1.74 mmol) in dioxane (50 mL) was stirred at 85 C. under N2 atmosphere overnight. TLC showed a complete reaction. The solution was poured onto water and extracted with ethyl acetate. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. Purification by column chromatography on silica gel (petroleum ether:ethyl acetate, 2:1 eluent) afforded the desired product as yellow solid (3.37 g, 64%). 1H NMR (300 MHz, CDCl3): delta 8.63 (d, J=2.4 Hz, 1H), 8.20 (d, J=8.4 Hz, 1H), 7.93 (dd, J=8.4, 2.4 Hz, 1H), 6.31 (s, 1H), 4.17-4.14 (m, 2H), 3.68 (t, J=5.7 Hz, 1H), 2.57-2.54 (m, 2H), 1.49 (s, 9H).
36%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate; In 1,4-dioxane; water; at 85℃; for 12h;Inert atmosphere;
Cs2CO3 (66.00g, 0.2mol), and Pd(dppf)Cl2 (7.33g, 0.01mol) were added to a solution of 82 <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (20.30g, 0.1mol) and 118 tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-dihydropyridine-1(2H)-carboxylate (31.00g, 0.1mol) in 111 dioxane/112 H2O (250 mL/30mL). The mixture was allowed to stir at 85C for 12h under N2, cooled to RT and concentrated under a vacuum, and purified by silica gel column chromatography (from 102 PE/86 EA=1:1 to 103 DCM/87 MeOH=20:1) to obtain 119 tert-butyl 6-nitro-5?,6?-dihydro-[3,4?-bipyridine]-1?(2?H)-carboxylate (11.00g; yield, 36%) as a yellow solid. Then 121 TFA (20mL) was added to the solution of 119 tert-butyl 6-nitro-5?,6?-dihydro-[3,4?-bipyridine]-1?(2?H)-carboxylate (5.00g, 24.4mmol) in 103 DCM (100mL). The mixture was stirred at RT for 1h, after which the reaction mixture was concentrated, washed with sodium carbonate (Na2CO3) (aq.), and extracted with DCM. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, concentrated under a vacuum, and purified by silica gel column chromatography (PE/EA=5:1) to obtain 122 6-nitro-1?,2?,3?,6?-tetrahydro-3,4?-bipyridine (2.61g; yield, 52%) as a yellow solid. 124 Sodium hydride (270.0mg, 11.2mmol) in 125 dimethyl fluoride ([DMF] 6mL) was added at 0C to a solution of 122 6-nitro-1?,2?,3?,6?-tetrahydro-3,4?-bipyridine (2.11g, 10.2mmol), and the reaction was stirred at 0C for 0.5h. Then 126 iodoethane (2.39g, 15.28mmol) was added and stirred for 2h. The mixture was poured into water and extracted with DCM. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, concentrated under a vacuum, and purified by silica gel column chromatography (PE/EA=5:1) to obtain 127 1?-ethyl-6-nitro-1?,2?,3?,6?-tetrahydro-3,4?-bipyridine (800.2mg; yield, 33.7%) as a yellow solid. Then Pd/C (0.10g) was added to a solution of 1?-ethyl-6-nitro-1?,2?,3?,6?-tetrahydro-3,4?-bipyridine (699.0mg, 3.0mmol) in EA/MeOH (10 mL/10mL). The mixture was degassed by flushing with H2, stirred at RT under a H2 atmosphere for 2h, and filtered and concentrated under a vacuum to obtain INT-5 (284.0mg; yield, 95%) as a white solid. ESI-MS: m/z 206.2 [M+H]+.
With sodium carbonate;bis-triphenylphosphine-palladium(II) chloride; In 1,4-dioxane; water; at 80℃;Inert atmosphere;
Preparation 146-nitro-3',6'-dihydro-2'H-[3,4]bipyridinyl-1'-carboxylic acid tert-butyl esterBubble nitrogen into a mixture of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylic acid tert-butyl ester (19.6 g), <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (12.87 g), sodium carbonate 2M in water (63.39 mL) and bis(triphenylphosphine)palladium(II) chloride (4.45 g) in 1,4-dioxane (316.94 mL) for 5 min and stir at 80 C. for 5 h. Dilute with DCM and wash with water. Dry over magnesium sulfate and remove the solvent under vacuum. Purify by silica gel chromatography eluting with DCM/EA (0-40%) to afford 8.72 g of the title compound. MS (ES+): m/z=306 (M+H)+.
7 g
With bis-triphenylphosphine-palladium(II) chloride; caesium carbonate; In 1,4-dioxane; water; at 80℃; for 15h;
In a 500 mL round-bottomed flask, <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (6.56 g, 32.3 mmol, Eq: 1) and tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-dihydropyridine-1(2H)-carboxylate (10 g, 32.3 mmol, Eq: 1.00) were combined with dioxane (160 ml) to give a light yellow solution. CS2CO3 (21.1 g, 64.7 mmol, Eq: 2) and water (6 ml) were added. The reaction mixture was degassed with argon before bis(triphenylphosphine)palladium(II) dichloride (2.27 g, 3.23 mmol, Eq: 0.1) was added. The reaction mixture was heated to 80 C and stirred for 15 h. The reaction mixture was poured into 500 mL H2O and extracted with EtOAc (3 x 200 mL). The combined extracts were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The crude material was purified by flash chromatography (silica gel, 220 g, 10% to 40% EtOAc in hexanes) to afford a pink solid. The resulting solid was triturated with ether to afford the desired product as a solid (4.8 g). The filtrate from the trituration and mixed fractions from the first chromatography were combined and purified by flash chromatography (silica gel, 220 g, 20% to 40% EtOAc in hexanes) affording additional product (2.2g). 1H NMR (400 MHz, CHLOROFORM-d) delta ppm 1.52 (s, 9 H) 2.59 (d, J=1.52 Hz, 2 H) 3.72 (t, J=5.56 Hz, 2 H) 4.19 (d, J=3.03 Hz, 2 H) 6.35 (br. s., 1 H) 7.97 (dd, J=8.46, 2.40 Hz, 1 H) 8.27 (d, J=8.34 Hz, 1 H) 8.67 (d, J=2.27 Hz, 1 H).
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; sodium carbonate; In 1,2-dimethoxyethane; water; at 130℃; for 1h;Microwave irradiation;
To a mixture of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (800 mg), tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-dihydropyridine-1(2H)-carboxylate (1.2 g) and sodium carbonate (840 mg) in 1,2-dimethoxyethane/water (v/v=5/1, 12 mL) was added 1,1?-bis(diphenylphosphino)ferrocene-palladium(II) dichloride-dichloromethane complex (160 mg), and the mixture was stirred in a microwave reactor at 130 C. for 1 hr. To the reaction mixture was added water, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, and dried over magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane/ethyl acetate) to give tert-butyl 6-nitro-3?,6?-dihydro-3,4?-bipyridine-1?(2?H)-carboxylate. (1710) To a solution of the obtained tert-butyl 6-nitro-3?,6?-dihydro-3,4?-bipyridine-1?(2?H)-carboxylate in methanol (10 mL) was added 4M hydrogen chloride ethyl acetate (10 mL) solution, and the mixture was stirred overnight at room temperature. The solvent was evaporated under reduced pressure to give the title compound (620 mg). (1711) MS(ESI+): [M+H]+ 206.6
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate; In 1,4-dioxane; water; at 85℃; for 12h;
Add to the reaction flask<strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (20.3 g, 0.1 mol)4- (4,4,5,5-tetramethyl-1,3,2-diboron-2-yl) -5,6-dihydropyridine-1 (2H) -carboxylate (30.9 g, 0.1 mol)Cesium carbonate (65 g, 0.2 mol),Pd (dppf) Cl2 (7.33 g, 0.01 mol) and dioxane / water (250 mL / 30 mL).The mixture was stirred at 85 C for 12 hours, cooled to room temperature, concentrated under reduced pressure,The resulting residue was purified by column chromatography (petroleum ether / ethyl acetate = 1: 1)To give the title compound (11 g, white solid) in 36% yield.
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium carbonate; In 1,4-dioxane; water; at 80℃; for 18h;Inert atmosphere;
Step 1 tert-butyl 6-nitro-5',6'-dihydro-[3,4'-bipyridyl]-1'(2'H)carboxylate In a nitrogn atmosphere, to a mixed solution of <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (10.00 g, 49.26 mmol, 1.02 equivalents) in dioxane (120 mL) and water (10 mL) were added tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dtoxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylate (15.00 g, 48.51 mmol, 1.00 equivalent), potassium carbonate (5.9 g, 72.77 mmol, 1.50 equivalents) and Pd(dppf)Cl2 (1.77 g, 2.43 mmol, 0.05 equivalent). The reaction mixture was heated to 80C and stirred for 18 hours. TLC (petroleum ether: ethyl acetate = 3: 1) showed completion of the reaction of the starting material. The reaction solution was filtered, dried over anhydrous sodium sulfate, filtered, and concentrated to give a crude product. The crude product was purified by column chromatography (petroleum ether: ethyl acetate = 3: 1) to give the title compound (18.70 g, crude product) as a light yellow solid. 1H NMR (400 MHz, CDCl3) delta 8.64 (d, J = 2.0 Hz, 1H) 8.24 (d, J = 8.4 Hz, 1H) 7.95 (dd, J = 8.5, 2.3 Hz, 1H) 6.33 (br s, 1H) 4.16 (d, J = 2.6 Hz, 2H) 3.69 (t, J = 5.6 Hz, 2H) 2.57 (br s, 2H) 1.50 (s, 9H).
With palladium catalyst;Heating;
3-Bromo-6-nitropyridine was reacted with tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-dihydropyridine-1(2H)-carboxylate under heating in the presence of a palladium catalyst by using the process described in J. Med. Chem. 2010, 53, p.7938-7957, to yield the title compound.
2.2 g
With bis-triphenylphosphine-palladium(II) chloride; caesium carbonate; In 1,4-dioxane; water; at 80℃; for 15h;Inert atmosphere;
5-Bromo-2-nitropyridine (6.56 g, 32.3 mmol, Eq=1) and4-(4,4,5,5-tetramethyl-1,3,2-dioxaborol-2-yl)-5,6-dihydropyridine-1(2-hydrogen)-carboxylic acid tert-butyl ester(10 g, 32.3 mmol, Eq=1) was added to a flask containing dioxane (160 mL).Form a yellow solution. caesium carbonate (21.1 g, 64.7 mmol, Eq=2) and water (6 mL) were added to the solution. After argon was replaced, bis(triphenylphosphine)palladium dichloride (2.27 g, 3.23 mmol, Eq=0.1) was added. ). The reaction mixture was heated to 80C and stirred for 15 hours. The reaction solution was poured into 500 mL of water and extracted with ethyl acetate, which was used as an organic layer. The solution was washed with brine, dried over magnesium sulfate, filtered, and spin-dried to give a pink solid after flash silica gel column purification. The solid is hit with ether The slurry was filtered to give 2.2 g of product.
2.2 g
With bis-triphenylphosphine-palladium(II) chloride; caesium carbonate; In 1,4-dioxane; at 80℃; for 15h;Inert atmosphere;
5-Bromo-2-nitropyridine (6.56 g, 32.3 mmol, Eq=1) and4-(4,4,5,5-tetramethyl-1,3,2-dioxaborol-2-yl)-Tert-butyl 5,6-dihydropyridine-1(2-hydrogen)-carboxylate(10g, 32.3 mmol, Eq=1)Add to a flask containing dioxane (160 mL),Form a yellow solution. Bismuth carbonate (21.1 g, 64.7 mmol, Eq=2) and water (6 mL) were added to the solution. After argon was replaced, bis(triphenylphosphine)palladium dichloride (2.27 g, 3.23 mmol, Eq=0.1) was added. ).The reaction mixture was heated to 80C and stirred for 15 hours. The reaction solution was poured into 500 mL of water and extracted with ethyl acetate. The organic layer was washed with brine, dried over magnesium sulfate, filtered, and spin-dried to give a pink solid after purification by flash silica gel column. The solid was slurried with ether and filtered to give 2.2 g of product.
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate; In 1,4-dioxane; water; at 85℃; for 12h;Inert atmosphere;
A reaction flask was charged with <strong>[39856-50-3]5-bromo-2-nitropyridine</strong> (20.3 g, 0.1 mol), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-dihydropyridine-1(2H)-carboxylic acid t-butyl ester (31 g, 0.1 mol), dioxane/water (250 mL/30 mL), cesium carbonate (66 g, 0.2 mol) and Pd(dppf)Cl2 (7.33 g, 0.01 mol), and protected by nitrogen gas. The mixture was heated to 85C for 12 h. The reaction product was cooled to room temperature, concentrated and separated by column chromatography (PE/EA = 1:1 to DCM/MeOH = 20:1) to give the title product (11 g, yellow solid). MS (ESI): mass calcd. for C15H19N3O4 305.1, m/z found 306.1 [M+H]+.
Step 1 :Preparation of 5-(6-Nitro-pyridin-3-yl)-2,5-diaza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester.2-Bromo-5-Nitropyridine (4.75 g, 23.4 mmol, 1.0 eq) and (R,R)-2,5-Diaza- bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester (5.04 g, 25.4 mmol, 1.1 eq) were combined in DMF (60mL) and heated to 100C. The reaction was cooled and diluted with Ethyl Acetate, washed with water several times followed by brine, the organic layer dried (Na2S04), filtered, and the organic concentrated. Crude was purified using chromatography eluting with MeOH/DCM mixtures giving the desired product (4.3g, 57%). MS m/z 278.4 (M-C4H9).
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 80℃; for 16h;
Add to the reaction flask5-bromo-2-nitropyridine (4.0 g, 19.7 mmol), 1-ethylpiperazine (3.4 g, 29.8 mmol)K2CO3 (4.1 g, 29.6 mmol), TBAI (0.42 g, 1.2 mmol) and dimethylsulfoxide (40 mL) were added and the mixture was heated with stirring at 80 C for 16 hours.The mixture was cooled to room temperature and the reaction solution was poured into ice water and extracted with dichloromethane (20 mL x 3).The combined organic layers were washed with saturated brine (20 mL), dried over anhydrous sodium sulfate,The filtrate was concentrated in vacuo. The residue was purified by column chromatography (DCM / MeOH = 10: 1)The resulting residue was purified to give the title compound (3.59 g, brown solid) in 77% yield.
65%
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 120℃; for 2h;
To a solution of compound 88 (2 g, 9.8 mmol, 1 eq) in DMSO (10 mL) was added K2CO3 (2.7 g, 19.7 mmol, 2 eq), 1-ethylpiperazine (compound 89, 1.7 g, 14.7 mmol, 1.5 eq) and TBAI (36 mg, 0.098 mmol, 0.01 eq). The reaction mixture was stirred at 120 C for 2 h. After cooling down to rt, the mixture was diluted with water (30 mL), thus formed solid was filtered, rinsed with water (5 mL x 3), and dried to afford the desired product (1.5 g, 65%) as a yellow solid which was used to the next step directly. NMR (300 MHz, CDC ): delta 8.19- 8.14 (m, 2 H), 7.23-7.19 (m, 1 H), 3.53-3.45 (m, 4 H), 2.79-2.65 (m, 4 H), 2.57-2.50 (m, 2 H), 1.19 (t, J= 7.2 Hz, 3 H).
56%
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 80℃; for 16h;
1-ethylpiperazine (3.40g, 29.8mmol), 80 K2CO3 (4.10g, 29.6mmol), and 93 TBAI (0.42g, 1.2mmol) were added to a solution of 82 5-bromo-2-nitropyridine (4.00g, 19.7mmol) in 83 DMSO (40mL). The mixture was heated at 80C for 16h, and the reaction was poured into ice water, and extracted with DCM. The combined organic layers were washed with water, dried over anhydrous Na2SO4, concentrated under a vacuum, and purified by silica gel column chromatography (DCM/MeOH=100:1-10:1) to obtain 94 1-ethyl-4-(6-nitropyridin-3-yl)piperazine (3.59g; yield, 56%) as a yellow solid. Pd/C (0.10g) was added to the 1-ethyl-4-(6-nitropyridin-3-yl)piperazine (0.90g, 3.8mmol) in EA/MeOH (9 mL/9mL) solution. The mixture was degassed by H2, stirred at RT under a H2 atmosphere for 2h, and filtered and concentrated under a vacuum to obtain INT-2 (720.0mg; yield, 92%) as an off-white solid. ESI-MS: m/z 207.2 [M+H]+.
56.1%
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 80℃; for 16h;
4.00 g (19.7 mmol) of 5-bromo-2-nitropyridine, 3.40 g (2.98 mmol) of 1-ethylpiperazine, 4.10 g (29.6 mmol) of potassium carbonate, 0.4 g (1.2 mmol) of tetrabutylammonium iodide, and 40mL of DMSO were added to a reaction flask, and reacted at 80C for 16 h. The reaction solution was then poured into ice-water and extracted three times with dichloromethane (20 ml for each time). The organic phases were combined, dried with anhydrous sodium sulfate, filtered, concentrated and separated by column chromatography (DCM/MeOH = 100:1-10:1) to give 3.59 g of yellow solid, yield 56.1%. MS (ESI): m/z 237.2 [M+H]+.
In 1-methyl-pyrrolidin-2-one; at 120℃; for 3h;
A mixture of 5-bromo-2-nitropyridine (1.02 g, 5.02 mmol) and 1 -ethylpiperazine (1 .71 g, 15.0 mmol) in N-methyl-2-pyrrolidinone (5 mL) was stirred at 120 C for 3 h. After this time, the reaction was cooled to room temperature, poured into water (100 mL) and extracted with methylene chloride (2 chi 100 mL). The combined organic layers were dried over sodium sulfate, filtered and the filtrate concentrated under reduced pressure. The resulting residue was purified by chromatography (silica, gradient, 1 :49 methanol/methylene chloride to 1 :9 methanol/methylene chloride) to afford l-ethyl-4-(6-nitropyridin-3- yl)piperazine as a yellow solid: 1H NMR (400 MHz, DMSO- )d 8.25 (d, J = 2.8 Hz, 1 H), 8.14 (d, J = 9.2 Hz, 1H), 7.48 (dd, J = 9.2, 2.8 Hz, 1H), 3.50-3.46 (m, 4H), 2.50-2.38 (m, 4H, merged with DMSO peak), 2.37 (q, J = 7.2 Hz, 2H), 1.02 (t, J= 7.2 Hz, 3H).
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 90℃;sealed tube;
To a sealed tube equipped with a stirring bar, 5-bromo-2-nitropyridine (3.00 g, 14.78 mmol), 1-ethylpiperazine (5.06 g, 44.34 mmol), tetrabutylammonium iodide (273 mmol, 0.739 mmol), K2C03 (6.128 g, 44.34 mmol), and DMSO (30 mL) were added. The tube was sealed and heated at 90 C overnight. Water (200 mL) was added and the precipitation was filtered to afford 138a as a yellow solid, 1.24 g.
In 1-methyl-pyrrolidin-2-one; at 120℃; for 3h;
Example 4 Preparation of 1-ethyl-4-(6-nitropyridin-3-yl)piperazine A mixture of 5-bromo-2-nitropyridine (1.02 g, 5.02 mmol) and 1-ethylpiperazine (1.71 g, 15.0 mmol) in N-methyl-2-pyrrolidinone (5 mL) was stirred at 120 C. for 3 h. After this time, the reaction was cooled to room temperature, poured into water (100 mL) and extracted with methylene chloride (2*100 mL). The combined organic layers were dried over sodium sulfate, filtered and the filtrate concentrated under reduced pressure. The resulting residue was purified by chromatography (silica, gradient, 1:49 methanol/methylene chloride to 1:9 methanol/methylene chloride) to afford 1-ethyl-4-(6-nitropyridin-3-yl)piperazine as a yellow solid: 1H NMR (400 MHz, DMSO-d6.) d 8.25 (d, J=2.8 Hz, 1H), 8.14 (d, J=9.2 Hz, 1H), 7.48 (dd, J=9.2, 2.8 Hz, 1H), 3.50-3.46 (m, 4H), 2.50-2.38 (m, 4H, merged with DMSO peak), 2.37 (q, J=7.2 Hz, 2H), 1.02 (t, J=7.2 Hz, 3H).
Step 1To 5-bromo-2-nitropyridine (1.0 g, 4.93 mmol, Eq: 1.00) in DMSO (10.0 ml) was added <strong>[4318-42-7]1-isopropylpiperazine</strong> (632 mg, 4.93 mmol, Eq: 1.00), and the resulting solution was heated at 70° C. for 18 hours.The solution was cooled to room temperature.The solution was diluted with 50 ml water.The resulting solid was filtered.The solid was washed with water and dried under vacuum.The crude material was purified by flash chromatography (silica gel, 80 g, 0percent to 3percent MeOH/DCM gradient) to give 1-isopropyl-4-(6-nitropyridin-3-yl)piperazine (788 mg, 64percent). LC/MS-ESI observed [M+H]+ 251.
33.66%
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 90℃; for 2h;Inert atmosphere;
Synthesis of compound 202.1. To a solution of 83.1 (0.20 g, 0.99 mmol, l .Oeq) in 1,4-dioxane (5ml) was added <strong>[4318-42-7]1-isopropylpiperazine</strong> (0.15 lg, 1.182mmol, 1.2eq) and cesium carbonate (0.94 g, 2.96 mmol, 3.0 eq.). The reaction mixture was degassed for 10 min. under argon atmosphere, then Pd2(dba)3 (0.09 g, 0.098 mmol, 0.1 eq) and Xantphos (0.077 g, 0.197 mmol, 0.2 eq) were added, and again degassed for 5 min. The reaction was then heated at 90°C for 2h. After completion of the reaction, mixture was poured in water and extracted with EtOAc. Organic layers were combined, dried over Na2S04 and concentrated under reduced pressure to obtain crude which was purified by column chromatography to get pure 202.1 (0.083g, 33.66percent). MS (ES): m/z 250.30 [M+H]+.
With potassium carbonate; In dimethyl sulfoxide; at 100℃; for 16h;
A mixture of 5-bromo-2-nitropyridine (18.0 g, 88.7 mmol), N-isopropylpiperazine (17.1 g, 133 mmol) and potassium carbonate (36.9 g, 267 mol) in dimethylsulfoxide (200 mL) was stirred at 100 °C for 16 h. After this time, the reaction was cooled to room temperature, poured into ice water (500 mL), stirred for 15 min, then extracted with ethyl acetate (2 x 500 mL). The combined organic layers were dried over sodium sulfate, filtered and the filtrate concentrated under reduced pressure. The resulting residue was dried under vacuum to a constant weight to afford l -isopropyl-4-(6-nitropyridin-3-yl)piperazine as a yellow solid: 1H NMR (400 MHz, CDC13) d 8.15 (d, J = 9.2 Hz, 1H), 8.12 (d, J = 2.8 Hz, 1 H), 7.18 (dd, J = 9.2, 2.8 Hz, 1 H), 3.46 (t, J = 4.8 Hz, 4H), 2.78-2.74 (m, 1 H), 2.69 (t, J = 5.2 Hz, 4H), 1.09 (d, J = 10.8 Hz, 6H).
With potassium carbonate; In dimethyl sulfoxide; at 100℃; for 16h;
Example 16 Preparation of 1-isopropyl-4-(6-nitropyridin-3-yl)piperazine A mixture of 5-bromo-2-nitropyridine (18.0 g, 88.7 mmol), N-isopropylpiperazine (17.1 g, 133 mmol) and potassium carbonate (36.9 g, 267 mol) in dimethylsulfoxide (200 mL) was stirred at 100° C. for 16 h. After this time, the reaction was cooled to room temperature, poured into ice water (500 mL), stirred for 15 min, then extracted with ethyl acetate (2*500 mL). The combined organic layers were dried over sodium sulfate, filtered and the filtrate concentrated under reduced pressure. The resulting residue was dried under vacuum to a constant weight to afford 1-isopropyl-4-(6-nitropyridin-3-yl)piperazine as a yellow solid: 1H NMR (400 MHz, CDCl3). d 8.15 (d, J=9.2 Hz, 1H), 8.12 (d, J=2.8 Hz, 1H), 7.18 (dd, J=9.2, 2.8 Hz, 1H), 3.46 (t, J=4.8 Hz, 4H), 2.78-2.74 (m, 1H), 2.69 (t, J=5.2 Hz, 4H), 1.09 (d, J=10.8 Hz, 6H).
Preparation of 5-(2-chloro-5-(trifluoromethyl)phenoxy)-2-nitropyridine (3).; Under an atmosphere of argon, sodium hydride (23.7 mg, 0.99 mmol, 60% dispersion in mineral oil) was added to a solution of phenol 2 (194 mg, 0.99 mmol) in DMF (2.5 ml) and stirred for 10 minutes. To this solution was added bromide 1 (100 mg, 0.49 mmol). The resulting mixture was heated under argon at 90 C. with stirring for 4 h. The reaction mixture was cooled to room temperature and quenched with water (10 mL), extracted with ethyl acetate (3×5 mL) and combined extracts dried over Na2SO4 and concentrated under reduced pressure. Flash chromatography (ISCO system, silica, 0-50% ethyl acetate in hexane) provided 3 (113 mg, 72%) as a solid.
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate; In 1,4-dioxane; water; at 85℃; for 12h;
Add to the reaction flask5-bromo-2-nitropyridine (20.3 g, 0.1 mol)1-methyl-4- (4,4,5,5-tetramethyl-1,3,2-dioxaboride-2-yl) -1,2,3,6-tetrahydropyridine (22.3 g , 0.1 mol), cesium carbonate (65 g, 0.2 mol), Pd (dppf) Cl2 (7.33 g,0.01 mol) and dioxane / water (250 mL / 30 mL).The mixture was heated and stirred at 85 ° C for 12 hours, cooled to room temperature, concentrated under reduced pressure,The resulting residue was purified by column chromatography (petroleum ether / ethyl acetate = 1: 1)To give the title compound (5.7 g, white solid) in 26percent yield.
26%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate; In 1,4-dioxane; water; at 85℃; for 12h;Inert atmosphere;
Cesium carbonate ([Cs2CO3] 66.00g, 0.2mol) and Pd(dppf)Cl2 (7.33g, 0.01mol) were added to a solution of 82 5-bromo-2-nitropyridine (20.30g, 0.1mol) and 110 <strong>[454482-11-2]1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,2,3,6-tetrahydropyridine</strong> (22.30g, 0.1mol) in 111 dioxane/112 H2O (250 mL/30mL). The mixture was allowed to stir at 85°C for 12h under nitrogen (N2). Then the solution was cooled to RT, concentrated under a vacuum, and purified by silica gel column chromatography (from 102 PE/86 EA=1:1 to 103 DCM/87 MeOH=20:1) to obtain 113 1?-methyl-6-nitro-1?,2?,3?,6?-tetrahydro-3,4?-bipyridine (5.70g; yield, 26percent) as a white solid. Next, Pd/C (0.10g) was added to the solution of 1?-methyl-6-nitro-1?,2?,3?,6?-tetrahydro-3,4?-bipyridine (657.0mg, 3.0mmol) in EA/MeOH (10 mL/10mL). The mixture was degassed by flushing with H2, stirred at RT under a H2 atmosphere for 2h, and then filtered and concentrated under a vacuum to obtain INT-4 (550.0mg; yield, 96percent) as a white solid. ESI-MS: m/z 192.2 [M+H]+.
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,4-dioxane; water; at 100℃; for 10h;
To a round-bottomed flask equipped with a stirring bar, l-methyl-4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)-l,2,3,6-tetrahydropyridine (1.50 g, 6.72 mmol), 5- bromo-2-nitropyridine (1.64 g, 8.07 mmol), Pd(PPh3)4 (388 mg, 0.336 mmol), Na2C03 aqueous solution (1.0 N, 20.2 mL, 20.2 mmol), dioxane (60.6 mL) were added. The reaction mixture was heated at 100 °C for 10 hrs. CH2CI2 (200 mL) was added to the resulting mixture was washed with water (30 mL X 3). CH2CI2 (200 mL) was added and the resulting mixture was washed with water (30 mL X 3), brine (30 mL X 1), dried over MgS04, filtered, and removed solvent in vacuo. Silica gel column chromatography (MeOH: DCM = 5: 95) gave 5- (l-methyl-l,2,3,6-tetrahydropyridin-4-yl)-2-nitropyridine (130a) as a yellow solid.
5.7 g
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate; In 1,4-dioxane; water; at 85℃; for 12h;Inert atmosphere;
A reaction flask was charged with 5-bromo-2-nitropyridine (20.3 g, 0.1 mol), <strong>[454482-11-2]1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,2,3,6-tetrahydropyridine</strong> (22.3 g, 0.1 mol), dioxane/water (250 mL/30 mL), cesium carbonate (66 g, 0.2 mol) and Pd(dppf)Cl2 (7.33 g, 0.01 mol). The mixture was stirred to react at 85°C under the protection of nitrogen gas for 12 h. The reaction product was cooled to room temperature, concentrated and separated by column chromatography (PE/EA = 1:1 to DCM/MeOH = 20:1) to give the titled product (5.7 g, white solid). MS (ESI): mass calcd. for C11H13N3O2 219.1, m/z found 220.1 [M+H]+.
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 120℃; for 0.5h;Microwave irradiation;
Synthesis of compound 80.2. To a solution of 78.4 (0.25g 1.23mmol, l .Oeq.) in dry DMSO (3.0mL) was added <strong>[3970-68-1]4-methylpiperidin-4-ol</strong>e (0.17g, 1.47mmol, 1.2eq.), potassium carbonate (0.68g, 4.92mmol, 4.0eq.) and tetrabutylammonium iodide (88mg, 0.24mmol, 0.2 eq.) at room temperature. Reaction mixture was heated at 120 C for 30 minutes in microwave. After completion of the reaction, mixture was poured into water and extracted using ethyl acetate. Organic layer was washed by brine (25 mL x3) solution, dried over sodium sulfate and concentrated under reduced pressure. Crude was purified by column chromatography to furnsh compound 80.2 (0.19g, 65%). MS (ES): m/z 238.2 [M+H]+.
tetra-(n-butyl)ammonium iodide; In dimethyl sulfoxide; at 120℃; for 18h;
Step 1. Preparation of 4-methyl-1-(6-nitropyridin-3-yl)piperidin-4-ol 4-Methylpiperidin-4-ol (2.72 g, 23.6 mmol), 5-bromo-2-nitropyridine (3.2 g, 15.8 mmol), and tetrabutylammonium iodide (72 mg, 195 umol) in DMSO (20 ml) was heated to 120 C. for 18 hr. After cooling to room temperature, the reaction mixture was then diluted with EtOAc and washed with water (3*20 mL). The organic phase was then concentrated and triturated with ether to give 4-methyl-1-(6-nitropyridin-3-yl)piperidin-4-ol as a yellow solid (2.55 g, 68.2%). (M+H)+=327.9 m/e.
With triethylamine; In dimethyl sulfoxide; at 90℃; for 40h;
Step 1. Preparation of 6-(6-Nitro-pyridin-3-yl)-2,6-diaza-spiro[3.3]heptane-2-carboxylic acid tert-butyl ester In a 25 mL pear-shaped flask, <strong>[1041026-70-3]tert-butyl 2,6-diazaspiro[3.3]heptane-2-carboxylate</strong> (2.81 g, 14.2 mmol), 5-bromo-2-nitropyridine (2.88 g, 14.2 mmol), and TEA (1.58 g, 2.17 ml, 15.6 mmol) were combined with DMSO (12 ml) to give a light yellow solution. The reaction mixture was heated to 90 C. and stirred for 40 h. Cooled to 25 C. and the reaction mixture was diluted with 50 mL H2O and extracted with EtOAc (3*50 mL). The organic layers were combined, washed with sat NaCl (1*50 mL), dried over Na2SO4 and concentrated in vacuo. The purple oil was used crude in the subsequent reduction. (M+H)+=321 m/e.
A suspension of <strong>[10010-93-2]5-methyl-3-(trifluoromethyl)pyrazole</strong> (300 mg, 2 mmol) and potassium carbonate (828 mg, 3 eq) in 4 ml DMSO was heated at 120 C. for 30 min before addition of 5-bromo-2-nitropyridine (406 mg, 2 mmol). The resulting suspension was heated at 120 C. for 4 more hours. After cooling down, the reaction mixture was worked up with EA/brine. Flash silica gel column followed by prep HPLC purification furnished nitropyridine intermediate 113 (127 mg, yield 23.3%, purity>95%) as a light yellow solid.
Example 57 Preparation of N-(2,6-difluorobenzyl)-5-(2-methyl-4-(trifluoromethyl)-1H-imidazol-1-yl)pyridin-2-amine (106) A suspension of <strong>[33468-67-6]2-methyl-4-(trifluoromethyl)imidazole</strong> (300 mg, 2 mmol) and potassium carbonate (828 mg, 3 eq) in 4 mL DMSO was heated at 120 C. for 30 min before addition of 5-bromo-2-nitropyridine (406 mg, 2 mmol). The resulting suspension was heated at 120 C. for 4 more hours. After cooling down, the reaction mixture was worked up with EA/brine. Flash silica gel column purification furnished nitropyridine intermediate 103 (125 mg, yield 23%, purity>90%) as a light yellow solid.
With potassium carbonate; In acetonitrile; at 80℃; for 10h;
D56 5-(4-chloro-3-(trifluoromethyl)phenoxy)- 2-nitropyridine To a solution of 5-bromo-2-nitropyridine (15 g, 73.9 mmol) and 4-chloro-3- (trifluoromethyl)phenol (14.52 g, 73.9 mmol) in acetonitrile (200 mL) was added potassium carbonate (18.38 g, 133 mmol) and the reaction mixture was stirred at 80 °C for 10 hr. Then, 200 mL of EtOAc and 200 mL of H20 were added. The organic layer was separated and concentrated under reduced pressure. The crude product was purified by a silica gel column chromatography eluting and was eluted with Hex/EtOAc (5: 1) to provide 5-(4-chloro-3-(trifluoromethyl)phenoxy)- 2-nitropyridine (8 g, 32.8percent),LC-MS (ESI): m/z 319 [M+l]+; 1.23 min (ret time).
32.8%
With potassium carbonate; In acetonitrile; at 80℃; for 10h;
D56 5-(4-chloro-3-(trifluoromethyl)phenoxy)-2-nitropyridine To a solution of 5-bromo-2-nitropyridine (15 g, 73.9 mmol) and <strong>[6294-93-5]4-chloro-3-(trifluoromethyl)phenol</strong> (14.52 g, 73.9 mmol) in acetonitrile (200 mL) was added potassium carbonate (18.38 g, 133 mmol) and the reaction mixture was stirred at 80° C. for 10 hr. Then, 200 mL of EtOAc and 200 mL of H2O were added. The organic layer was separated and concentrated under reduced pressure. The crude product was purified by a silica gel column chromatography eluting and was eluted with Hex/EtOAc (5:1) to provide 5-(4-chloro-3-(trifluoromethyl)phenoxy)-2-nitropyridine (8 g, 32.8percent), LC-MS (ESI): m/z 319 [M+1]+; 1.23 min (ret time).
32.8%
With potassium carbonate; In acetonitrile; at 80℃; for 10h;
To a solution of 5-bromo-2-nitropyridine (15 g, 73.9 mmol) and <strong>[6294-93-5]4-chloro-3-(trifluoromethyl)phenol</strong> (14.52 g, 73.9 mmol) in acetonitrile (200 mL) was added potassium carbonate (18.38 g, 133 mmol) and the reaction mixture was stirred at 80° C. for 10 hr. Then, 200 mL of EtOAc and 200 mL of H2O were added. The organic layer was separated and concentrated under reduced pressure. The crude product was purified by a silica gel column chromatography eluting and was eluted with Hex/EtOAc (5:1) to provide 5-(4-chloro-3-(trifluoromethyl)phenoxy)-2-nitropyridine (8 g, 32.8percent)
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 110℃;Inert atmosphere;
Example 114a (S)-tert-Butyl 3-Ethyl-4-(6-nitropyridin-3-yl)piperazine-1-carboxylate 114a A 250-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 1,4-dioxane (50 mL), 5-bromo-2-nitropyridine (2.02 g, 10 mmol), <strong>[928025-56-3](S)-tert-butyl 3-ethylpiperazine-1-carboxylate</strong> (2.14 g, 10.0 mmol), Pd2(dba)3 (458 mg, 0.50 mmol), XantPhos (576 mg, 1.0 mmol), and cesium carbonate (6.52 g, 20 mmol). After three cycles of vacuum/argon flush, the mixture was heated at 100 C. overnight. After this time the reaction was cooled to room temperature. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 3:1 petroleum ether/ethyl acetate to afford 114a (700 mg, 22%) as a yellow solid. MS: [M+H]+ 336
22%
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 100℃;Inert atmosphere;
Example 111a (S)-tert-Butyl 3-Ethyl-4-(6-nitropyridin-3-yl)piperazine-1-carboxylate 111a A 250-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 1,4-dioxane (50 mL), 5-bromo-2-nitropyridine (2.02 g, 10 mmol), <strong>[928025-56-3](S)-tert-butyl 3-ethylpiperazine-1-carboxylate</strong> (2.14 g, 10.0 mmol), Pd2(dba)3 (458 mg, 0.50 mmol), XantPhos (576 mg, 1.0 mmol), and cesium carbonate (6.52 g, 20 mmol). After three cycles of vacuum/argon flush, the mixture was heated at 100 C. overnight. After this time the reaction was cooled to room temperature. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 3:1 petroleum ether/ethyl acetate to afford 111a (700 mg, 22%) as a yellow solid. MS: [M+H]+ 336
22%
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 100℃;Inert atmosphere;
Example 161a (S)-tert-Butyl 3-Ethyl-4-(6-nitropyridin-3-yl)piperazine-1-carboxylate 161a A 250-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 1,4-dioxane (50 mL), 5-bromo-2-nitropyridine (2.02 g, 10 mmol), <strong>[928025-56-3](S)-tert-butyl 3-ethylpiperazine-1-carboxylate</strong> (2.14 g, 10.0 mmol), Pd2(dba)3 (458 mg, 0.50mmol), XantPhos (576 mg, 1.0 mmol), and cesium carbonate (6.52 g, 20 mmol). After three cycles of vacuum/argon flush, the mixture was heated at 100C overnight. After this time the reaction was cooled to room temperature. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 3:1 petroleum ether/ethyl acetate to afford 161a (700 mg, 22%) as a yellow solid. MS: [M+H]+ 336
With tris-(dibenzylideneacetone)dipalladium(0); 2,2'-bis-(diphenylphosphino)-1,1'-binaphthyl; In 1,4-dioxane; at 120℃; for 24h;Inert atmosphere;
Example 125a tert-Butyl 3,3-Dimethyl-4-(6-nitropyridin-3-yl)piperazine-1-carboxylate 125a A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 5-bromo-2-nitropyridine (5.6 g, 28.0 mmol), <strong>[259808-67-8]tert-butyl 3,3-dimethyl-piperazine-1-carboxylate</strong> (3.0 g, 14.0 mmol), cesium carbonate (9.1 g, 28 mmol), and 1,4-dioxane (50 mL). After bubbling nitrogen through the resulting solution for 30 min, Binap (870 mg, 1.4 mmol) and tris(dibenzylideneacetone)dipalladium(0) (1.2 g, 1.4 mmol) were added. The reaction mixture was subjected to three cycles of vacuum/argon flush and stirred at 120 C. for 24 h. After this time the reaction was cooled to room temperature it was filtered and the filtrate was partitioned between ethyl acetate (200 mL) and water (50 mL). The aqueous layer was separated and extracted with ethyl acetate (3*50 mL). The combined organic layer was washed with brine (50 mL) and dried over sodium sulfate. The drying agent was removed by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 5:1 petroleum ether/ethyl acetate to afford 125a (1.27 g, 27%). LCMS: [M+H]+ 337.2.
27%
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 2,2'-bis-(diphenylphosphino)-1,1'-binaphthyl; In 1,4-dioxane; at 120℃; for 24h;Inert atmosphere;
Example 190a tert-Butyl 3,3-Dimethyl-4-(6-nitropyridin-3-yl)piperazine-1-carboxylate 190a A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 5-bromo-2-nitropyridine (5.6 g, 28.0 mmol), tert-butyl 3,3-dimethyl-4-piperazine-1-carboxylate (3.0 g, 14.0 mmol), cesium carbonate (9.1 g, 28 mmol), and 1,4-dioxane (50 mL). After bubbling nitrogen through the resulting solution for 30 min, Binap (870 mg, 1.4 mmol) and tris(dibenzylideneacetone)-dipalladium(0) (1.2 g, 1.4 mmol) were added. The reaction mixture was subjected to three cycles of vacuum/argon flush and stirred at 120 C for 24 h. After this time the reaction was cooled to room temperature, filtered and the filtrate was partitioned between ethyl acetate (200 mL) and water (50 mL). The aqueous layer was separated and extracted with ethyl acetate (3 * 50 mL). The combined organic layers were washed with brine (50 mL) and dried over sodium sulfate. The drying agent was removed by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 5:1 petroleum ether/ethyl acetate to afford 190a (1.27 g, 27%). LCMS: [M+H]+ 337.2.
With triethylamine; In dimethyl sulfoxide; at 120℃; for 3h;Microwave irradiation;
Example 3 Synthesis of Substituted 2-Aminopyridines To 5-bromo-2-nitropyridine (1.2 g, 5.9 mmol) in DMSO (4 mL) was added 1-(4-piperidyl)piperidine (1.0 g, 5.9 mmole) and triethyl amine (0.99 mL, 7.1 mmole). The contents were heated to 120 degrees in a CEM Discovery microwave system for 3 hours. The crude reaction was then loaded over a silica gel column and eluted with DCM/methanol (0-20%) to afford 2-nitro-5-[4-(1-piperidyl)-1-piperidyl]pyridine as an oil (457 mg). 1H NMR (600 MHz, DMSO-d6) delta ppm 1.26-1.36 (m, 2H) 1.43 (m, 6H) 1.76 (m, 2H) 2.37 (m, 5H) 2.94 (t, J=12.74 Hz, 2H) 4.06 (d, J=13.47 Hz, 2H) 7.41 (dd, J=9.37, 2.64 Hz, 1H) 8.08 (d, J=9.37 Hz, 1H) 8.20 (d, J=2.64 Hz, 1H).
457 mg
With triethylamine; In dimethyl sulfoxide; at 120℃; for 3h;Microwave irradiation;
Example 24 Synthesis of 2-nitro-5-[4-(l-piperidyl)-l-piperidyl]pyridine, Compound 24 To 5-bromo-2-nitropyridine (1.2 g, 5.9 mmol) in DMSO (4 mL) was added l-(4- piperidyl)piperidine (1.0 g, 5.9 mmole ) and triethylamine (0.99 mL, 7.1 mmole). The contents were heated to 120 C in a CEM Discovery microwave system for 3 hours. The crude reaction was then purified by silica gel column chromatography with DCM/methanol (0-20% ) to afford 2-nitro-5-[4-(l-piperidyl)-l-piperidyl]pyridine as an oil (457 mg). 1HNMR (600 MHz, DMSO- d6) delta ppm 1.26 - 1.36 (m, 2 H) 1.43 (m, 6 H) 1.76 (m, 2 H) 2.37 (m, 5 H) 2.94 (t, J=12.74 Hz, 2 H) 4.06 (d, J=13.47 Hz, 2 H) 7.41 (dd, J=9.37, 2.64 Hz, 1 H) 8.08 (d, J=9.37 Hz, 1 H) 8.20 (d, J=2.64 Hz, 1 H).
457 mg
With triethylamine; In dimethyl sulfoxide; at 120℃; for 3h;Microwave irradiation;
To 5-bromo-2-nitropyridine (1.2 g, 5.9 mmol) in DMSO (4 mL) was added l-(4- piperidyl)piperidine (1.0 g, 5.9 mmole ) and triethylamine (0.99 mL, 7.1 mmole). The contents were heated to 120 C in a CEM Discovery microwave system for 3 hours. The crude reaction was then purified by silica gel column chromatography with DCM/methanol (0-20% ) to afford 2-nitro-5-[4-(l-piperidyl)-l-piperidyl]pyridine as an oil (457 mg). 1HNMR (600 MHz, DMSO- de) delta ppm 1.26 - 1.36 (m, 2 H) 1.43 (m, 6 H) 1.76 (m, 2 H) 2.37 (m, 5 H) 2.94 (t, J=12.74 Hz, 2 H) 4.06 (d, J=13.47 Hz, 2 H) 7.41 (dd, J=9.37, 2.64 Hz, 1 H) 8.08 (d, J=9.37 Hz, 1 H) 8.20 (d, J=2.64 Hz, 1 H).
457 mg
With triethylamine; In dimethyl sulfoxide; at 120℃; for 3h;Microwave irradiation;
To 5-bromo-2-nitropyridine (1.2 g, 5.9 mmol) in DMSO (4 mL) was added 1-(4- piperidyl)piperidine (1.0 g, 5.9 mmole ) and triethylamine (0.99 mL, 7.1 mmole). The contents were heated to 120 C in a CEM Discovery microwave system for 3 hours. The crude reactionwas then purified by silica gel column chromatography with DCM/methanol (0-20% ) to afford2-nitro-5-[4-(1-piperidyl)-1-piperidyl]pyridine as an oil (457 mg). ?HNMR (600 MHz, DMSOd 6) oe ppm 1.26 - 1.36 (m, 2 H) 1.43 (m, 6 H) 1.76 (m, 2 H) 2.37 (m, 5 H) 2.94 (t, J=12.74 Hz, 2 H) 4.06 (d, J=13.47 Hz, 2 H) 7.41 (dd, J=9.37, 2.64 Hz, 1 H) 8.08 (d, J9.37 Hz, 1 H) 8.20 (d, J2.64 Hz, 1 H).
457 mg
With triethylamine; In dimethyl sulfoxide; at 120℃; for 3h;Microwave irradiation;
To 5-bromo-2-nitropyridine (1.2 g, 5.9 mmole) in DMSO (4 mL) was added l-(4- piperidyl)piperidine (1.0 g, 5.9 mmole) and triethylamine (0.99 mL, 7.1 mmole). The contents were heated to 120 C in a CEM Discovery microwave system for 3 hours. The cmde reactionwas then loaded over a silica gel column and eluted with DCMlmethanol (0-20%) to afford 2-nitro-5- [4-( 1 -piperidyl)- 1 -piperidyll pyridine as an oil (457 mg).?H NMR (600 MHz, DMSO-d6) ppm 1.26 - 1.36 (m, 2 H) 1.43 (m, 6 H) 1.76 (m, 2 H) 2.37(m, 5 H) 2.94 (t, J=12.74 Hz, 2 H) 4.06 (d, J=13.47 Hz, 2 H) 7.41 (dd, J=9.37, 2.64 Hz, 1 H)8.08 (d, J9.37 Hz, 1 H) 8.20 (d, J=2.64 Hz, 1 H).
457 mg
With triethylamine; In dimethyl sulfoxide; at 120℃; for 3h;Microwave irradiation;
To 5-bromo-2-nitropyridine (1.2 g, 5.9 mmole) in DMSO (4 mL) was added l-(4- piperidyl)piperidine (1.0 g, 5.9 mmole) and triethylamine (0.99 mL, 7.1 mmole). The contents were heated to 120 C in a CEM Discovery microwave system for 3 hours. The cmde reaction was then loaded over a silica gel column and eluted with DCMlmethanol (0-20%) to afford 2-nitro-5- [4-( 1 -piperidyl)- 1 -piperidyll pyridine as an oil (457 mg).?H NMR (600 MHz, DMSO-d6) ppm 1.26 - 1.36 (m, 2 H) 1.43 (m, 6 H) 1.76 (m, 2 H) 2.37(m, 5 H) 2.94 (t, J=12.74 Hz, 2 H) 4.06 (d, J=13.47 Hz, 2 H) 7.41 (dd, J9.37, 2.64 Hz, 1 H)8.08 (d, J9.37 Hz, 1 H) 8.20 (d, J2.64 Hz, 1 H).
457 mg
With triethylamine; In dimethyl sulfoxide; at 120℃; for 3h;Microwave irradiation;
tert-Butyi 4-(6-ami -3-pyridyl)piperazine-l-carboxylate (0500) The compound was prepared as described in WO 2010/020675 Al . (0501) (0502) To 5-bromo-2-nitropyridine (1.2 g, 5.9 mmole) in DMSO (4 mL) was added l -(4- piperidyl)piperidine (1.0 g, 5.9 mmole) and triethylamine (0.99 mL, 7.1 mmole). The contents were heated to 120 C in a CEM Discovery microwave system for 3 hours. The crude reaction was then loaded over a silica gel column and eluted with DCM/methanol (0-20%) to afford 2- nitro-5-[4-(l-piperidyl)-l -piperidyl]pyridine as an oil (457 mg). NMR (600 MHz, DMSO-c e) delta ppm 1.26 - 1.36 (m, 2 H) 1.43 (m, 6 H) 1.76 (m, 2 H) 2.37 (m, 5 H) 2.94 (t, J=12.74 Hz, 2 H) 4.06 (d, J=13.47 Hz, 2 H) 7.41 (dd, J=9.37, 2.64 Hz, 1 H) 8.08 (d, J=9.37 Hz, 1 H) 8.20 (d, J=2.64 Hz, 1 H)
457 mg
With triethylamine; In dimethyl sulfoxide; at 120℃; for 3h;Microwave irradiation;
To 5-bromo-2-nitropyridine (1.2 g, 5.9 mmole) in DMSO (4 raL) was added 1~(4~ piperidyl)piperidme (1.0 g, 5.9 mmole) and triethylamine (0.99 mL, 7.1 mmole). The contents were heated to 120 °C m a CEM Discover ' microwave system for 3 horns. The crude reaction was then loaded over a silica gel column and eluted with DCM/methanol (0-20percent) to afford 2- nitro-5-[4-(1-piperidyl)-1-piperidyl]pyridme as an oil (457 mg). NMR (600 MHz, DMSO-rfe) d ppm 1.26 - 1.36 (m, 2 H) 1.43 (m, 6 H) 1.76 (m, 2 H) 2.37 (m, 5 H) 2 94 (t, J= 12.74 Hz, 2 1 1} 4.06 (d,./ 13.47 Hz, 2 H) 7.41 (dd,.7=9 37, 2.64 Hz, 1H) 8.08 (d,,7=9.37 Hz, 1H) 8.20 (d,./ 2.6-1Hz, 1H).
With potassium carbonate; In tetrahydrofuran; N,N-dimethyl-formamide; at 20℃;Inert atmosphere; Schlenk technique; Reflux;
To a solution of 5-bromo-2-nitropyridine (8.0 g, 39.41 mmol) and <strong>[72594-86-6]benzyl tert-butyl malonate</strong> (11.84 g, 47.29 mmol) in a 1:1 mixture of THF-DMF (200 ml) was added potassium carbonate (11.98 g, 86.69 mmol) at room temperature, under an inert atmosphere. The resultant mixture was refluxed overnight. The reaction mixture was cooled to room temperature and diluted with diethyl ether (300 ml) and washed with water (3 x 100 ml). The organic layer was dried over anhydrous sodium sulphate, filtered and evaporated to dryness. The residue obtained was purified by flash silica gel column chromatography to yield 6.5 g of product as brown viscous liquid.
With palladium diacetate; caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In toluene; at 90℃; for 12h;
To mixture of Scheme 41 compound 1 (2.00 g, 9.80 mmol), Scheme 41 compound 2 (1.41 g, 9.85 mmol), Cs2C03 (9.60 g, 29.55 mmol) and Xantphos (569 mg, 0.98 mmol) in dry toluene (20 mL) was added Pd(OAc)2 (221 mg, 0.98 mmol) and the reaction mixture was heated to 90°C for 12 h. After TLC showed the starting material was completely consumed, the reaction mixture was cooled to RT, passed through a pad of celite and washed with EtOAc. The filtrate was washed with water and brine, dried over Na2S04 and concentrated to give a residue which was purified by flash chromatography on silica gel (eluting with CH2Cl2/MeOH 100/0 gradually increasing to 95/5) to give Scheme 41 compound 3 (1.10 g, 33percent) as a pale yellow solid. MS [ESI, MH+] = 267.13.
With palladium diacetate; caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In toluene; at 100℃; for 4h;
To a mixture of Scheme 43 compound 1 (1.0 g, 4.90 mmol), Scheme 43 compound 2 (630 mg, 4.90 mmol), Cs2C03 (4.8 g, 14.80 mmol) and Xantphos (286 mg, 0.49 mmol) in dry toluene (10 mL) was added Pd(OAc)2 (111 mg, 0.49 mmol) and the reaction mixture was heated to 100 °C for 4 h. After TLC showed the starting material was completely consumed, the reaction mixture was cooled to RT, passed through a pad of celite and washed with EtOAc. The filtrate was washed with water and brine, dried over Na2S04 and concentrated to give a residue which was purified by flash chromatography on silica gel (eluting with CH2Cl2/MeOH 100/0 gradually increasing to 95/5) to give Scheme 43 compound 3 (1.0 g, 83percent) as a yellow solid. MS [ESI, MH+] = 251.15.
With potassium carbonate; In acetonitrile; at 20℃; for 2h;Inert atmosphere;
A mixture of 3,5- difluorobenzene- 1 -thiol (2 g, 13.68 mmol, 1.00 equiv), 5-bromo-2-nitropyridine (3.2 g, 15.76 mmol, 1.00 equiv), and potassium carbonate (4.8 g, 34.73 mmol, 2.00 equiv) in DMSO (30 mL) was stirred under nitrogen at rt for 2 h. The reaction mixture was diluted with 200 mL of EtOAc then washed with 3x50 mL of H2O. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated under vacuum to give 2 g of the title compound as a gray solid. The crude product was used in the next step without further purification. TLC: 1 : 1 EtO Ac/petroleum ether, Rf = 0.8.
With triethylamine; In dimethyl sulfoxide; at 70℃; for 5h;
5-Bromo-2-nitropyridine (10.0 g, 49.3 mmol) and tert-butyl pyrrolidin-3- ylcarbamate (11.9 g, 64.0 mmol) were dissolved in DMSO (30 mL), and triethylamine (8.93 mL, 64.0 mmol) was added. The mixture was stirred at 70 C for 5 h, then cooled to rt and poured into water. The mixture was extracted with ethyl acetate. The organic layer was dried over sodium sulphate, and the solvent was evaporated. The residue was triturated with ethyl acetate, and the precipitate was collected by suction filtration. The mother liquor was purified by preparative HPLC to yield a second batch. 10.5 g (69%) of the title compound were obtained. 1H-NMR (400MHz, DMSO-d6): delta [ppm]= 1.39 (s, 9H), 1.82 - 1.91 (m, 1 H), 2.07 - 2.16 (m, 1 H), 3.17 (dd, 1 H), 3.29 - 3.37 (m, 1 H), 3.42 - 3.50 (m, 1 H), 3.55 (dd, 1 H), 4.05 - 4.14 (m, 1 H), 6.42 (d, 1 H), 7.20 (d, 1 H), 7.62 (dd, 1 H), 8.11 (d, 1 H).
With tris-(dibenzylideneacetone)dipalladium(0); potassium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 105℃; for 4h;Inert atmosphere;
Synthesis of compound 78.5. To a solution of 78.4 (0.5 g, 2.46mmol, 1.0 equiv ) in dry Dioxane (5.0mL) was added 78.3 (404mg, 2.46mmol, 1.1 eq.) and potassium carbonate (l .Olg, 7.38mmol, 3.0eq.) at room temperature under argon purge for 15 minutes. To the above reaction mixture was added Pd2(dba)3 (22mg, 0.246mmol, 0.1 eq.) and Xantphos (27mg, 0.48mmol, 0.2eq.) under argon purge for 10 minutes. Reaction mixture was heated at 105C for 4 hours. After completion of the reaction, reaction mixture was poured into water and extracted using ethyl acetate. Organic layer was washed with by brine solution. Organic layers were combined, dried over sodium sulphate and concentrated under reduced pressure to obtain crude material which was purified using column chromatography to get pure 78.5 (0.3 g, 42.54%). MS (ES): m/z 287.2 [M+H]+.
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 120℃; for 1h;Microwave irradiation;
Synthesis of compound 173.1. To a solution of 78.4 (0.350g, 1.72mol, l .Oeq) in DMSO (5ml) was added TBAI (0.127g, 0.34mmol, 0.2eq), l-(tert-butyl) piperazine (0.293sg, 2.06mmol, 1.2eq), and K2CO3 (0.713g, 5.17 mmol, 3.0eq). Reaction mixture was heated in microwave at 120 C for lh. Reaction mixture was poured into water and product was extracted with EtOAC. Organic layers were combined, dried over sodium sulphate and concentrated under reduced pressure to obtain crude which was purified by column chromatography to furnish 173.1 (0.30 g, 65.8 %). MS (ES): m/z 264.2 [M+H]+.
5-(3,3-difluoroazetidin-1-yl)-2-nitropyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
94.35%
With tris-(dibenzylideneacetone)dipalladium(0)
A.2.1 5-(3,3-difluoroazetidin-1-yl)-2-nitropyridine A2b
Step 1 5-(3,3-difluoroazetidin-1-yl)-2-nitropyridine A2b A mixture of 5-bromo-2-nitropyridine A2a (1 g, 4.93 mmol), 3,3-difluoroazetidine HCl salt (957.20 mg, 7.39 mmol), Pd2(dba)3 (451.25 mg, 492.63 umol), Xantphos (570.46 mg, 985.26 umol) and Cs2CO3 (8.03 g, 24.63 mmol) in dioxane (25 mL) was stirred at 120° C. under Ar for 15 hours. The mixture was filtered and the filtrate was concentrated under reduced pressure. The crude product was purified by silica gel chromatography (PE:EA=4:1) to afford A2b (1 g, 4.65 mmol, 94.35% yield). LCMS: MS m/z (ESI): 215.9 [M+1]+
94.35%
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene In 1,4-dioxane at 120℃; for 15h; Inert atmosphere;
A2.1 Step 1 5-(3,3-difluoroazetidin-1-yl)-2-nitropyridine A2b
A mixture of 5-bromo-2-nitropyridine A2a (1 g, 4.93 mmol), 3,3-difluoroazetidine HCl salt (957.20 mg, 7.39 mmol), Pd2(dba)3 (451.25 mg, 492.63 umol), Xantphos (570.46 mg, 985.26 umol) and Cs2CO3 (8.03 g, 24.63 mmol) in dioxane (25 mL) was stirred at 120° C. under Ar for 15 hours. The mixture was filtered and the filtrate was concentrated under reduced pressure. The crude product was purified by silica gel chromatography (PE:EA=4:1) to afford A2b (1 g, 4.65 mmol, 94.35% yield). LCMS: MS m/z (ESI): 215.9 [M+1]+
94.35%
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene In 1,4-dioxane at 120℃; for 15h; Inert atmosphere;
A2.1 Step 1 5-(3,3-difluoroazetidin-1-yl)-2-nitropyridine A2b
A mixture of 5-bromo-2-nitropyridine A2a (1 g, 4.93 mmol), 3,3-difluoroazetidine HCl salt (957.20 mg, 7.39 mmol), Pd2(dba)3 (451.25 mg, 492.63 umol), Xantphos (570.46 mg, 985.26 umol) and Cs2CO3 (8.03 g, 24.63 mmol) in dioxane (25 mL) was stirred at 120° C. under Ar for 15 hours. The mixture was filtered and the filtrate was concentrated under reduced pressure. The crude product was purified by silica gel chromatography (PE:EA=4:1) to afford A2b (1 g, 4.65 mmol, 94.35% yield). LCMS: MS m/z (ESI): 215.9 [M+1]+
27.81%
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene In 1,4-dioxane at 100℃; for 3h; Inert atmosphere;
22.1 Step 1: 5-(3,3-difluoroazetidin-1-yl)-2-nitropyridine .
To a stirred solution of 5-bromo- 2-nitropyridine(500 mg, 2.340 mmol, 1 equiv, 95%), 3,3-difluoroazetidine hydrochloride (350.95 mg, 2.574 mmol, 1.1 equiv, 95%) and Cs2CO3 (4012.66 mg, 11.700 mmol, 5.00 equiv, 95%) in dioxane (37.50 mL, 425.622 mmol, 179.71 equiv, 95%) was added Pd2(dba)3 (112.78 mg, 0.117 mmol, 0.05 equiv, 95%) and XantPhos (142.52 mg, 0.234 mmol, 0.10 equiv, 95%) at room temperature under nitrogen atmosphere. The resulting mixture was stirred for 3 h at 100 degrees C under nitrogen atmosphere. The resulting mixture was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with PE/EtOAc (1:1) to afford 5-(3,3-difluoroazetidin-1-yl)-2-nitropyridine(200 mg,27.81%) as a light yellow solid.
20.44%
With tetra-(n-butyl)ammonium iodide; potassium carbonate In dimethyl sulfoxide at 110℃; for 2.5h;
175 Synthesis of compound 175.1.
Synthesis of compound 175.1. To a solution of 78.4 (0.300 g, 1.477 mmol, 1.Oeq) in DMSO (5ml) were added TBAI (0.109 g, 0.259 mmol, 0.2eq), 3, 3-difluoroazetidine hydrochloride (0.209 g, 1.625 mmol, l . leq), and K2C03 (0.61 lg, 4.433 mmol, 3.Oeq). Reaction mixture was heated at 110° C for 2.5 h. After completion of the reaction, mixture was poured in water to give solid product. The solid product was filtered, washed with water and dried and purified by column chromatography and prep HPLC to get pure 175.1 (0.065 g, 20.44%). MS (ES): m/z 215.1 [M+H]+.
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; DavePhos; In 1,4-dioxane; at 90℃; for 1h;Inert atmosphere;
Synthesis of compound 190.1. To a solution of 78.4 (0.2g, 0.985mmol, 1.Oeq) in 1,4-dioxane (3ml) was added <strong>[42185-06-8]3-methylmorpholine</strong> (0.119g, 1.182mmol, 1.2eq) and Cs2C03 (0.960g, 2.955mmol, 3. Oeq). The reaction mixture was degassed for 10 min. under argon atmosphere, then Pd2(dba)3 (0.090g, 0.098mmol, 0.1 eq) and 2-Dicylcohexylphosphino-2'-(N,N- dimethylamino)-biphenyl (0.113g, 0.197mmol, 0.2eq) were added, and again degassed for 5 min. The reaction was stirred at 90 C for 1 hour. After completion of the reaction, mixture was poured into water and extracted with EtOAc. Organic layers were combined,dried over Na2S04 and concentrated under reduced pressure to obtain crude which was purified by column chromatography to furnish 190.1 (0.127g, 57.74%). MS (ES): m/z 223.23 [M+H]+.
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene; In 1,4-dioxane; at 120℃; for 2h;Inert atmosphere;
Synthesis of compound 194.1. To a solution of 78.4 (0.6g, 2.955mmol, 1.Oeq) in 1,4-dioxane (5ml) was added <strong>[109-11-5]morpholin-3-one</strong> (0.298g, 2.955mmol, l .Oeq) and CS2CO3 (1.921g, 5.911mmol, 2. Oeq). Mixture was degassed for 10 min. under argon atmosphere, then Pd2(dba)3 (0.270g, 0.295mmol, 0.1 eq) and xantphos (0.34 lg, 0.591mmol, 0.2eq) were added, and again degassed for 5 min. The reaction was stirred at 120 C in for 2h. After completion of the reaction, mixture was poured into water and product was extracted with EtOAc. Organic layers were combined and dried over Na2S04 and concentrated under reduced pressure to obtain crude which was purified by column chromatography to yield 194.1 (0.550g, 83.37%). MS (ES): m/z 223.19 [M+H]+.
With tris-(dibenzylideneacetone)dipalladium(0); 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; sodium t-butanolate; In 1,4-dioxane; at 100℃; for 2h;Inert atmosphere;
Synthesis of compound 198.1. To a solution of 83.1 (0.250g, 1.23 lmmol, 1.Oeq) in 1,4-dioxane (2 mL) was added <strong>[38646-68-3]4,4-dimethylpiperidine hydrochloride</strong> (0.184g, 1.23 lmmol, l .Oeq) and sodium tert-butoxide (0.236g, 2.46 mmol, 2.0 eq.). Reaction mixture was degassed for 10 min under argon atmosphere, then Pd2(dba)3 (0.112g, 0.123mmol, 0.1 eq) and Xantphos (0.096 g, 0.246 mmol, 0.2 eq) were added, then again degassed for 5 min. The reaction was then heated at 100 C for 2 hours. After completion of the reaction, mixture was poured into water and extracted with EtOAc. Organic layers were combined, dried over Na2S04 and concentrated under reduced pressure to obtain crude which was purified by column chromatography to furnish 198.1 (0.160g, 55.22%). MS (ES): m/z 235.29 [M+H]+.
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 120℃; for 3h;
Synthesis of compound 212.1. To a solution of 83.1 (0.3g, 1.477mmol, l .Oeq) in DMSO (5ml) was added <strong>[373603-88-4]3,3-dimethylpiperidin-4-ol</strong> (0.190g, 1.477mmol, l .Oeq.), K2C03 (0.61 lg, 4.433mmol, 3.0eq.) and tetra n-butyl ammonium iodide (0.054g, 0.147mmol, O.leq.). Reaction was stirred at 120 C for 3h. After completion of the reaction, mixture was poured in water and product was extracted with EtOAc. Organic layers were combined, dried over sodium sulphate and concentrated under reduced pressure to obtain crude which was purified by flash chromatography to furnish 212.1 (0.220g, 59.24%), MS (ES): m/z 251.29 [M+H]+.
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 100℃; for 1h;Inert atmosphere;
Synthesis of compound 218.3. To a solution of 83.1 (0.3g, 1.477 mmol, l .Oeq) in 1,4-dioxane (5 mL) was added 218.2 (0.17, 1.48 mmol, 1.0 eq.) and Cs2C03 (1.445g, 4.433mmol, 3.0eq.). Reaction mixture was degassed for 10 min. under argon atmosphere, then Pd2(dba)3 (0.135g, 0.147mmol, O.leq) and Xantphos (0.170g, 0.295mmol, 0.2eq) were added, and again degassed for 5 min. The reaction was stirred at 100 C for lh. After completion of the reaction, mixture was poured into water and product was extracted with EtOAc. Organic layers were combined , dried over sodium sulphate and concentrated under reduced pressure to obtain crude which was purified by column chromatography to furnish 218.3 (0.200g, 57.53%). MS(ES): m/z 235.24 [M+H]+.
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 110℃;Inert atmosphere;
Example 152a tert-Butyl 8-(6-Nitropyridin-3-yl)-3,8-diazabicyclo[3.2.1]octane-3-carboxylate 152a A 250-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 1,4-dioxane (100 mL), 5-bromo-2-nitropyridine (2.5 g, 12.4 mmol), <strong>[201162-53-0]tert-butyl 3,8-diazabicyclo[3.2.1]octane-3-carboxylate</strong>(869 g, 4.1 mmol), Pd2(dba)3 (193 mg, 0.21 mmol), XantPhos (237 mg, 0.41 mmol), and cesium carbonate (2.7 g, 8.2 mmol). After three cycles of vacuum/argon flush, the mixture was stirred at 110C overnight. The reaction was cooled to room temperature. It was then filtered and the filtrate was evaporated in vacuo. The residue was purified by silica-gel column chromatography eluting with 3:1 petroleum ether/ethyl acetate to afford 152a (2.63 g, 66.8%) as a yellow solid. MS-ESI: [M+H]+ 335.2.
(6-nitropyridin-3-yl)(pyridin-1-ium-1-yl)amide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
With potassium carbonate; In acetonitrile; at 23℃; for 10h;
General procedure: Potassium carbonate (9.0 mmol) for 3a-t or triethylamine (9.0 mmol) for 3u was added to a solution of <strong>[6295-87-0]1-aminopyridinium iodide</strong> 1 (3.0 mmol) in acetonitrile (25 mL) for 3a-t or EtOH (25 mL) for 3u. The reaction mixtures were stirred vigorously for 10-20 min at room temperature to give a dark purple solution, to which electrophiles 2a-u (3.0 mmol) were added in one portion. The mixtures were stirred at rt for 3-24 h except for 3e, which was heated under reflux for 5h. Mixtures 3a-t were filtered to remove inorganic salts, which were washed with acetonitrile 3-5 times. The combined filtrates were concentrated in vacuo and the residues were purified either by gradient elution chromatography over silica gel and/or by recrystallization.
5-(4-methoxypiperidin-1-yl)-2-nitropyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1.6 mmol
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 80℃; for 15h;
Step a. A solution of 5-bromo-2-nitro-pyridine (2.0 mmol) and 4-methoxypiperidine HCl (4.0 mmol) in DMSO (10 ml) was stirred at rt for 5 min. K2C03 (4.0 mmol) and TBAI (0.2 mmol) were then added to the reaction mixture at rt. The reaction mixture was stirred at 80C for 15 h. The resulting reaction mixture was poured into water (200 ml) and extracted with EtOAc (2 x 150 ml). The combined organic phase was collected, dried over Na2SC>4, filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (15% EtOAc in hexane) yielding 5-(4-methoxypiperidin-l-yl)-2- nitropyridine (1.6 mmol). MS: ES+ 238.35; XH NMR (400 MHz, DMSO-d6) delta ppm 8.27 (d, J=3.05 Hz, 1 H), 8.14 (d, J=9.16 Hz, 1 H), 7.49 (dd, J=9.16, 3.05 Hz, 1 H), 3.75 - 3.84 (m, 2 H), 3.46 - 3.48 (m, 1 H), 3.30 - 3.35 (m, 2 H), 3.28 (s, 3 H), 1.90 - 1.95 (m, 2 H), 1.48 - 1.53 (m, 2 H).
tert-butyl 4-(1-(6-nitropyridin-3-yl)piperidin-4-yl)piperazine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In dimethyl sulfoxide; at 80℃; for 16h;
A mixture of tert-butyl 4- (piperidin-4-yl) piperazine-1-carboxylate (4b) (6.28 g, 23.3 mmol) , 5-bromo-2-nitropyridine (4.74 g, 23.3 mmol) , tetrabutylammonium iodide (0.52 g, 1.4 mmol) and K2CO3(4.84 g, 35.0 mmol) in DMSO (100 mL) was stirred at 80 for 16 h. The mixture was cooled dwon, poured into ice water (500 mL) and extracted with DCM (3 × 200 mL) . The extracts were dried over Na2SO4. Solvents were evaporated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with 1MeOH in DCM to give tert-butyl 4- (1- (6-nitropyridin-3-yl) piperidin-4-yl) piperazine-1-carboxylate (4c) . MS-ESI (m/z) : 392 [M + 1]+.
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate; In 1,4-dioxane; water; at 85℃; for 12h;Inert atmosphere;
Cs2CO3 (66.00g, 0.2mol), and Pd(dppf)Cl2 (7.33g, 0.01mol) were added to a solution of 82 5-bromo-2-nitropyridine (20.30g, 0.1mol) and 118 tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-dihydropyridine-1(2H)-carboxylate (30.90g, 0.1mol) in 111 dioxane/112 H2O (250 mL/30mL). The mixture was allowed to stir at 85C for 12h under N2, after which it was cooled to RT, concentrated under a vacuum, and purified by silica gel column chromatography (from 102 PE/86 EA=1:1 to 103 DCM/87 MeOH=20:1) to obtain 119 tert-butyl 6-nitro-5?,6?-dihydro-[3,4?-bipyridine]-1?(2?H)-carboxylate (11.01g, yield: 36%) as a yellow solid. Pd/C (100.0mg) was added to a solution of tert-butyl 6-nitro-5?,6?-dihydro-[3,4?-bipyridine]-1?(2?H)-carboxylate (915.0mg, 3.0mmol) in EA/MeOH (10 mL/10mL). The mixture was degassed by flushing with H2, stirred at RT under a H2 atmosphere for 2h, and filtered and concentrated under a vacuum to obtain INT-6 (790.0mg; yield, 95%) as an off-white solid. ESI-MS: m/z 278.2 [M+H]+.
1-methyl-4-(6-nitropyridin-3-yl)-1-4-homopiperazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85.94%
With potassium carbonate; In dimethyl sulfoxide; at 80℃; for 16h;Inert atmosphere;
Step 1 1-methyl-4-(6-nitropyridin-3-yl)-1-4-homopiperazine To a solution of 5-bromo-2-nitro-pyridine (2.00 g, 9.85 mmol, 1.00 equivalent) and 1-methyl-1,4-homopiperazine (1.69 g. 14.78 mmol, 1.50 equivalents) in dimethyl sulfoxide (20.00 mL) was added potassium carbonate (2.72 g. 19.70 mmol. 2.00 equivalents). The mixture was heated to 80C and stirred for 16 hours. LC/MS showed completion of the reaction of the starting material. The reaction mixture was cooled to 25C. and water (50 mL) was added. The mixture was then subjected to extraction using ethyl acetate (100 mL*3). The combined organic phases was washed with saturated brine (100 mL*3), dried over anhydrous sodium sulfate, filtered and concentrated to give a crude product. The crude product was purified by column chromatography (petroleum ether: ethyl acetate = 30: 1 to 1: 3) to give the title compound (2.00 g. 8.46 mmol, 85.94% yield) as a white solid. LC/MS (ESI) m/z: 237.1 (M+1).
Under nitrogen protection, 550 mL of dioxane and 2-nitro-5-bromopyridine (20.3 g, After 0.10 mol) of pinacol borate (25.4 g, 0.10 mol) and potassium acetate (14.7 g, 0.15 mol), after stirring uniformly, the catalyst PdCl2dppf (0.74 g, 0.001 mol) was finally added, and the temperature was slowly raised to 80- 90 C, Stir the reaction for 2-3 h. After the completion of the GC reaction, the reaction was stopped by cooling, and the reaction solution was filtered through celite to obtain a dark-black reaction solution. After charging at 1 atm of hydrogen, the mixture was reacted at room temperature overnight. After the reaction was completed, the activated carbon was decolorized, and the filtrate was distilled under reduced pressure until no liquid was poured. /Heptane mixed solvent was cooled and beaten for half an hour, filtered to obtain 18.5 g of light gray product, heated again with ethanol, and then cooled down, and the filter cake was rinsed with -20 C anhydrous ethanol, and dried to obtain 16.9 g, yield 77 % HPLC purity 99.6%
tert-butyl 6′-nitro-5,6-dihydro-[3,3′-bipyridine]-1(4H)-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
18.5%
With bis-triphenylphosphine-palladium(II) chloride; caesium carbonate; In water; N,N-dimethyl-formamide; at 85℃; for 1h;Inert atmosphere;
5-Bromo-2-nitropyridine (107A) (1.0 g, 4.98 mmol, 1.0 eq), 15 bis(triphenylphosphine)palladium(II) chloride (174 mg, 0.25 mmol, 0.05 eq) and anhydrous cesium carbonate (2.43 g, 7.47 mmol, 1.5 eq) were mixed in 5 mL of water and 50 mL of N,N-dimethylformamide. Under nitrogen protection, compound <strong>[1121057-77-9]tert-butyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,4-dihydropyridine-1(2H)-carboxylate</strong> (203A) (2.13 g, 6.89 mmol, 1.4 eq) was added and heated to 85 C. After 1 hour, the reaction was cooled to room temperature, extracted with ethyl acetate and washed with saturated brine twice. The organic phase was dried over anhydrous sodium sulfate and concentrated in vacuo to give the crude product which was purified by silica gel column chromatography (eluent: ethyl acetate/petroleum ether=10/100 to 50/100) to obtain a yellow solid, which was compound tert-butyl 6?-nitro-5,6-dihydro-[3,3?-bipyridine]-1(4H)-carboxylate (203B) (282 mg, yield: 18.5%). LCMS (ESI): m/z 306 [M+1]+.
With caesium carbonate In acetonitrile at 70℃; for 2h; Sealed tube;
160.1; 162.1; 164.1 Step 1 : Preparation of 5-(3-fluorophenoxy)-2-nitropyridine
To a sealed tube was added 3-fluorophenol (0.551 g, 4.92 mmol), 5-bromo-2-nitropyridine (1 .0 g, 4.92 mmol), and cesium carbonate (2.40 g, 7.37 mmol) and suspended in acetonitrile (10 ml_). Reaction was heated to 70 °C for 2 h. Reaction was cooled to room temperature and concentrated. The crude product was purified over silica gel (ISCO, 40 g, 0-15% ethyl acetate/hexanes, over 25 minutes) to give 5-(3-fluorophenoxy)-2-nitropyridine (644 mg, 2.74 mmol, 56%) as a yellow solid. NMR (300 MHz, Chloroform-d) d 8.37 (d, J = 2.8 Hz, 1 H), 8.29 (d, J = 8.9 Hz, 1 H), 7.53 - 7.35 (m, 2H), 7.03 (tdd, J = 8.3, 2.4, 0.9 Hz, 1 H), 6.97 - 6.78 (m, 2H); LCMS (ESI) m/z: 235.1 [M+H]+.
56%
With caesium carbonate In acetonitrile at 70℃; for 2h; Sealed tube;
160.1; 162.1; 164.1 Step 1 : Preparation of 5-(3-fluorophenoxy)-2-nitropyridine
To a sealed tube was added 3-fluorophenol (0.551 g, 4.92 mmol), 5-bromo-2-nitropyridine (1.0 g, 4.92 mmol), and cesium carbonate (2.40 g, 7.37 mmol) and suspended in acetonitrile (10 ml_). Reaction was heated to 70 °C for 2 h. Reaction was cooled to room temperature and concentrated. The crude product was purified over silica gel (ISCO, 40 g, 0-15% ethyl acetate/hexanes, over 25 minutes) to give 5-(3-fluorophenoxy)-2-nitropyridine (644 mg, 2.74 mmol, 56%) as a yellow solid. NMR (300 MHz, Chloroform-d) d 8.37 (d, J= 2.8 Hz, 1H), 8.29 (d, J= 8.9 Hz, 1H), 7.53 - 7.35 (m, 2H), 7.03 (tdd, J = 8.3, 2.4, 0.9 Hz, 1H), 6.97 - 6.78 (m, 2H); LCMS (ESI) m/z: 235.1 [M+H]+.
42%
With caesium carbonate In dimethyl sulfoxide at 50℃; for 4h; Sealed tube; Inert atmosphere;
1.1 Step 1
5-bromo-2-nitro-pyridine (750 mg, 3.69 mmol), cesium carbonate (2.4 g, 7.39 mmol) and 3-fluorophenol (335 uL, 3.69 mmol) were mixed in DMSO (7.5 mL), purged with nitrogen and stirred in a sealed vial at 50 °C for 4 hours. The reaction mixture was diluted with water and extracted twice with EtOAc. The combined organic extracts were dried over MgSO4, filtered, concentrated under reduced pressure and purified by column chromatography [Biotage SNAP cartridge KP-Sil 50 g; 0-50% EtOAc in heptane]. The mixed fractions were purified further by prep HPLC (Method F) to afford 5-(3-fluorophenoxy)-2-nitro-pyridine as an off-white solid (361 mg, 42% yield). 1H NMR (500 MHz, DMSO-d6) δ 8.46 (d,J= 2.8 Hz, 1H), 8.35 (d,J= 8.9 Hz, 1H), 7.73 (dd,J= 9.0, 2.9 Hz, 1H), 7.54 (td,J= 8.3, 6.8 Hz, 1H), 7.22 (dt,J= 10.0, 2.4 Hz, 1H), 7.17 (tdd,J= 8.6, 2.5, 0.7 Hz, 1H), 7.10 (dd,J= 8.2, 2.2 Hz, 1H)
5-(3-chloro-4-fluorophenoxy)-2-nitropyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
69.7%
With caesium carbonate; In acetonitrile; at 70℃; for 2h;Sealed tube;
To a sealed tube was added 4-chloro-3-fluorophenol (0.597 m, 4.08 mmol), 5-bromo-2-nitropyridine (0.830 g, 4.08 mmol), and cesium carbonate (1 .99 g, 6.12 mmol) and suspended in acetonitrile (10 ml_). Reaction was heated to 70 C for 2 h. Reaction was cooled to room temperature and concentrated. The crude product was purified over silica gel (ISCO, 40 g, 0-15% ethyl acetate/hexanes, over 25 minutes) to give 5-(4-chloro-3-fluorophenoxy)-2-nitropyridine (0.760 g, 2.82 mmol, 69.7%) as a yellow solid. NMR (300 MHz, Methanol-aL) d 8.44 - 8.18 (m, 2H), 7.47 (ddt, J = 8.9, 2.8, 1 .0 Hz, 1 H), 7.26 (s, 2H), 7.16 - 6.97 (m, 1 H); LCMS (ESI) m/z: 269.2 [M+H]+.
With dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II)*CH2Cl2; sodium carbonate; In 1,4-dioxane; at 90℃; for 3h;Inert atmosphere;
Compound 131 (10 g, 49.26 mmol) , Compound 132 (12 g, 54.19 mmol) , sodium carbonate (15.7 g, 147.79 mmol) , [1, 1'-bis (diphenylphosphine) ferrocene] palladium dichloride dichloromethane complex (4.0 g, 9.86 mmol) were sequentially added to a 500 mL round bottom flask containing 300 mL of dioxane and 60 mL water, protected with nitrogen, and stirred at 90 for 3 h. LCMS monitoring, after the reaction was completed, cooled to room temperature, concentrated under reduced pressure. Added 200 mL of water, extracted with ethyl acetate (300 mL ×2) , and the combined extracts were washed with saturated aqueous sodium chloride (200 mL) , dried with anhydrous Na 2SO 4, concentrated under reduced pressure, and then separated by column chromatography (eluent: methanol/dichloromethane, 1/100, v/v) , obtained 7.8 g of a brown soild, yield: 72.2%. 1H NMR (400 MHz, DMSO-d 6) : delta ppm 2.30 (s, 3H) , 2.57-2.60 (m, 4H) , 3.09 (br. s, 2H) , 6.59 (s, 1H) , 8.21-8.29 (m, 2H) , 8.77 (s, 3H) . LCMS: Rt =0.54 min, MS Calcd.: 219.1, MS Found: 220.0 [M+H] +
1-(6-nitropyridin-3-yl)-4-(trifluoromethyl)piperidin-4-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium carbonate In N,N-dimethyl-formamide at 100℃; for 10h; Inert atmosphere;
5.C Step C:
Step C: 1-(6-Nitropyridin-3-yl)-4-(trifluoromethyl)piperidin-4-ol Under a protection of nitrogen, after a mixture of Example 5B (1.67g, 5.90mmol), 5-bromo-2-nitropyridine (1.32g, 6.49mmol), potassium carbonate (4.08g, 29.49mmol) in DMF (50mL) was stirred at 100°C for 10 hours, it was diluted with water (50mL).The aqueous layer was extracted with ethyl acetate (50mL*3). After the combined organic layers were washed with brine (50mLx2), dried over sodium sulfate, filtered and evaporated. The residue was purified by column chromatography to give the title compound. 1H NMR (400MHz, DMSO-d6) δ=8.29 (d, J=3.0 Hz, 1H), 8.14 (d, J=9.3 Hz, 1H), 7.52 (dd, J=3.0, 9.3 Hz, 1H), 6.18 (s, 1H), 4.10-4.03 (m, 2H), 3.29-3.19 (m, 2H), 1.80-1.72 (m, 4H).
With potassium carbonate In dimethyl sulfoxide at 100℃;
52%
With potassium carbonate In dimethyl sulfoxide at 100℃;
1.1 Step 1 : Synthesis of 30b:
General procedure: (Ref: WO 2015038417): To a stirred mixture of compound 30a (2.0 g, 16.9 mmol) in DMF (50 ml.) at room temperature were added 26b (3.0 g, 18.9 mmol) and K2CO3 (13.1 g, 94.5 mmol). The resulting mixture was stirred at 1 10 °C for 16 h. After cooling down to room temperature, the reaction mixture was diluted with water (200 ml.) and extracted with EtOAc (3 x 150 ml_). The organic layer was washed with brine solution (50 ml_), dried (Na2SC>4), and concentrated to provide crude residue. The crude was triturated with n-hexane and filtered to give compound 30b as a yellow solid (3.8 g, 80% yield).
1-benzyl 3-tert-butyl 2-(6-nitropyridin-3-yl)malonate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
43%
A solution of <strong>[72594-86-6]benzyl tert-butyl malonate</strong> (2, 24.66 g, 98.53 mmol) in DMSO (200 mL) was treated with NaH (60 purity dispersion in mineral oil) (3.94 g, 10.28 mol) at 10 C and stirred at 20 C for 1 hour.5-bromo-2-nitropyridine (1, 10 g, 49.26 mmol) was added to the mixture and stirred at 60 C for 5 h. The reaction mixture was quenched with saturated NH4Cl (600 mL), extracted with ethyl acetate (3 x 300 mL). The organic phases were combined and washed with brine (300 mL), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by reversed phase flash chromatography (flow: 200 mL/min; gradient: from 30%-60% MeCN-Water(0.1% TFA) over 40 min; column: I.D.95 mm x H365 mm, Welch Ultimate XB_C18 20-40μm; 120 Å) to afford 1-benzyl 3-tert-butyl 2-(6-nitropyridin-3-yl)malonate (3, 8 g, 21.27 mmol, 43% yield) as a yellow oil. LCMS (ESI): m/z 373.2 [M + H]+






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






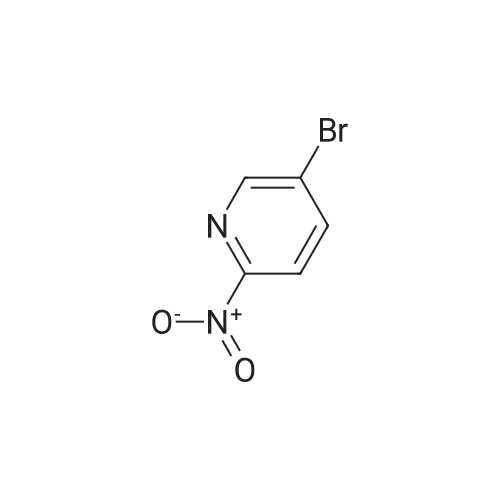

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping