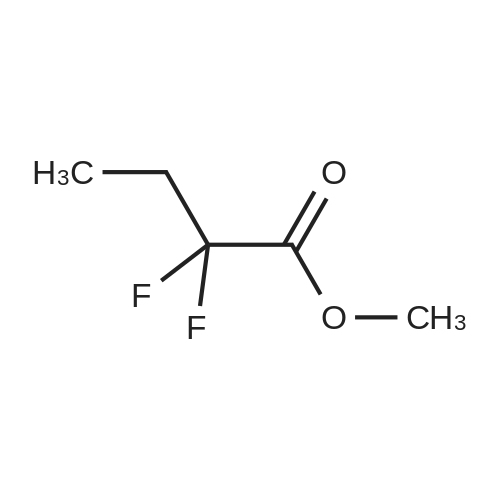
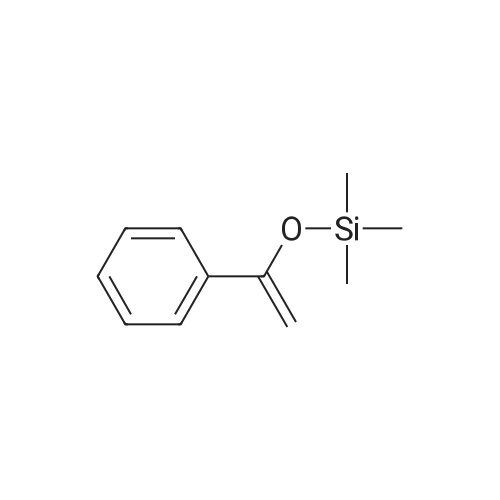
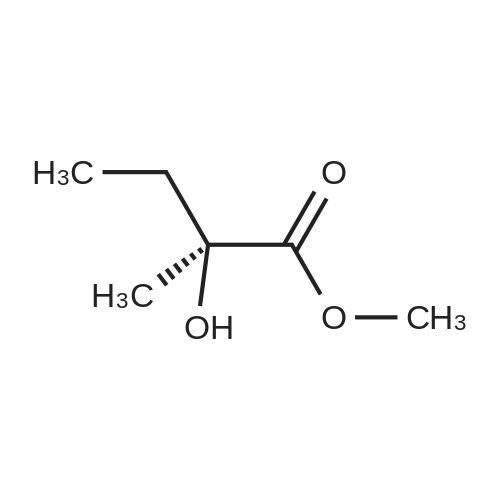
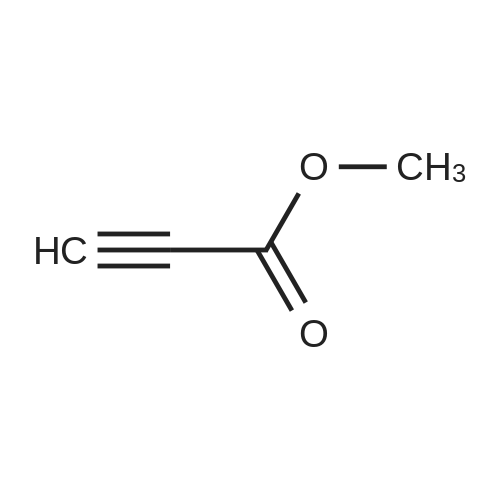
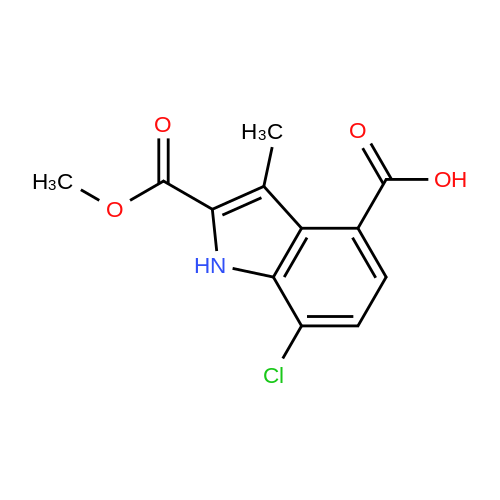
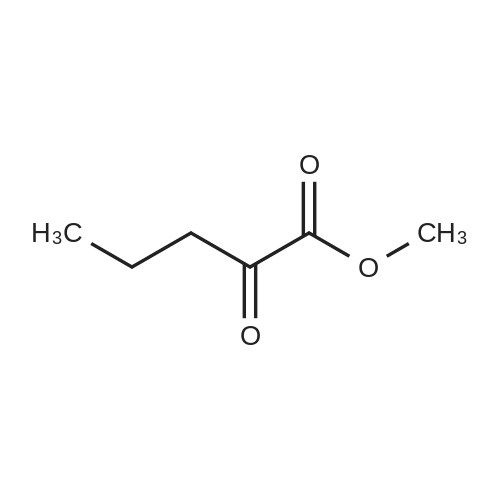
| 68% |
With sodium hydride; lithium chloride; In tetrahydrofuran; at 0 - 20℃; for 2.5h; |
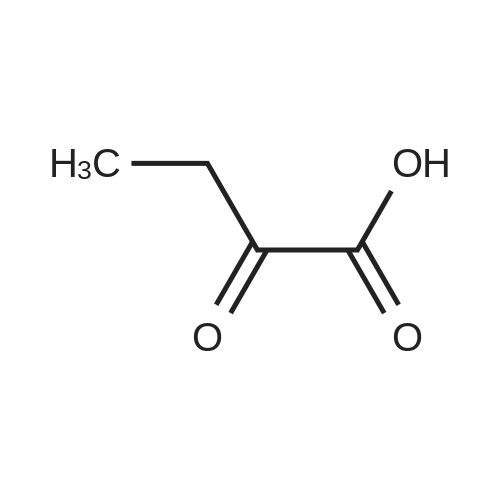
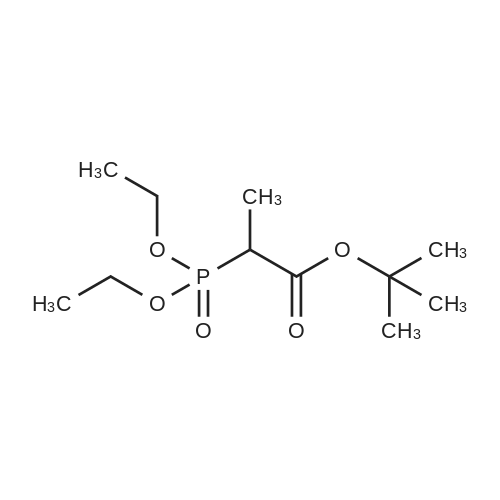
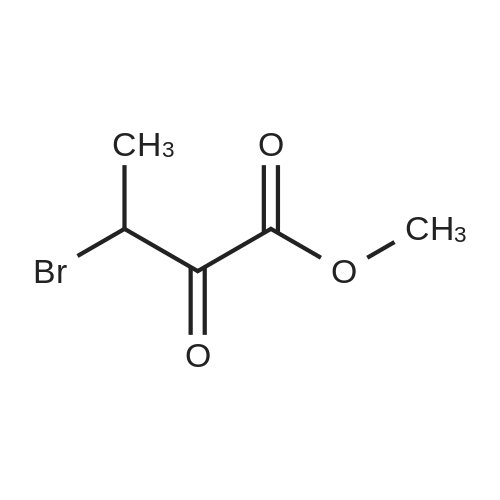
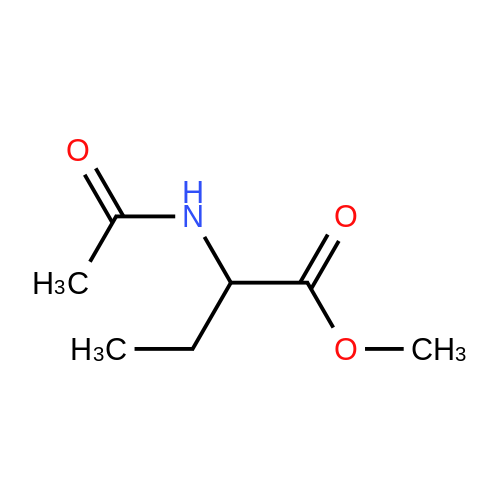
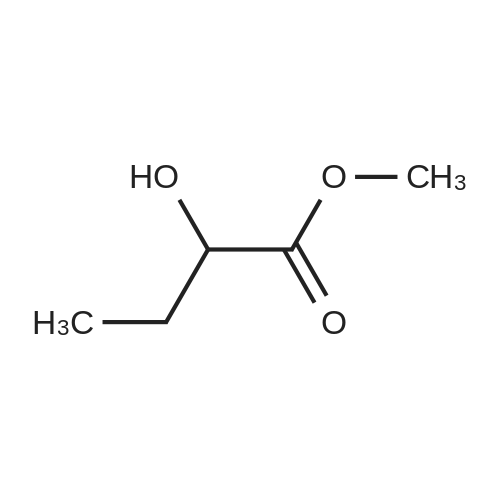
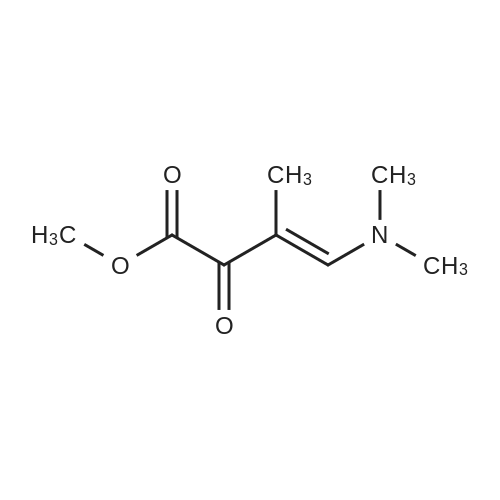
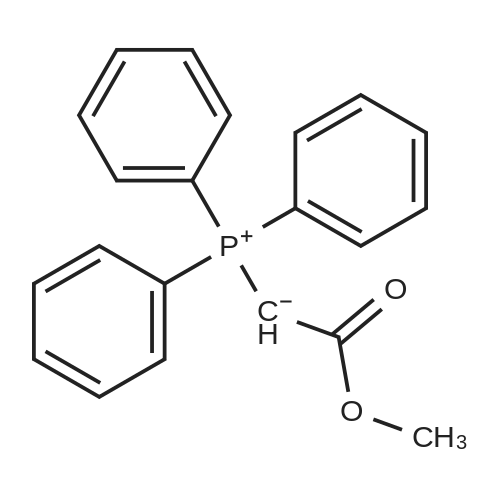
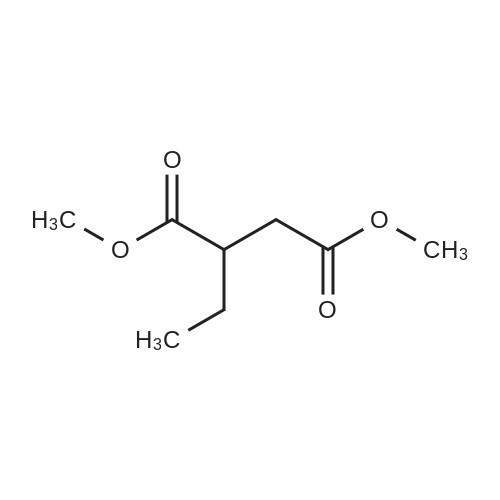
Preparation 31: M ethyl 2-ethyl-4- (1-isoquinolinyl)-4-oxobutanoate (VIi) [473] [474] At 0 C, anhydrous tetrahydrofuran (100 mi) was added to sodium hydride (60% dispersion in mineral oit, 649 mg, 1.1 eq, washed with anhydrous tetrahydrofuran in advance) under nitrogen atmosphere. The phosphonate compound prepared in Preparation 30 (4.12 g, 14.75 mmd) was dissolved in anhydrous tetrahydrofuran (20 mi) and was added to the mixture. The resulting mixture was stirred for about 10 minutes. Lithium chbride (1.25 g, 2.0 eq) was added thereto and the resuming mixture was further stirred at 0 C for about 10 minutes. Methyl 2-oxobutylate (2.4 g, 1.1 eq) dissolved in anhydrous tetrahydrofuran (20 mi) was added threrto at 0 C, and the mixture was stirred at room temperature for 2.5 hours. The reaction was completed by saturated ammonium chbride solution, and then extracted twice with ethyl acetate (100 moi). The filtrate was washed with water, saturated sodium bicarbonate solution (NaHCO, 50 mi x 2), and aqueous sodium chbride solution in turn, dried (anhydrous 3 Na2 SO4), and concentrated under reduced pressure. The residue was separated by column chromatography (20% ethyl acetate/hexane) to give the title compound (2.7 g, 68%) as yelow liquid in the form of cis. [475] [476] The obtained cis isomer (2.7 g, 10.03 mmol) was dissdved in ethyl acetate (50 mi ), and Pd/C (800 mg, 10%, Zurich) was added thereto. The resulting mixture was reacted by using Parr Hydrogenater under 40 psi of hydrogen atmospheric pressure for 2 hours. The reaction was filtered through Cette, and the filtrate was concentrated under reduced pressure. The residue was separated by cdumn chromatography (20% ethyl acetate/hexane) to give per-reduced (ketone was also reduced) methyl 2-ethyl-4-(1-isoquinolinyl)-4-hydroxybutanoate (1.95 g, 71%) as yelow liquid. [477] [478] NMR (400MHz, CDC1) 8 8.43 (d, 1H), 8.28 (d, 1H), 7.85 (d, 1H), 7.73-7. 59 (m, 3 2H), 7.59 (d, 1H), 5.40 (m, 1H), 3.83 (s, 3H), 3.05 (m, 1H), 2.37 (t, 1H), 1.69-1. 46 (m, 3H), 0.89 (t, 3H) [479] [480] Oxfam chbride (0.51 mi, 1.6 eq) was dissdved in dichbromethane (30 moi), DMSO (1.04 mi, 4.0 eq) was added thereto at-78 C, and the resulting mixture was stirred for about 10 minutes. The above compound (1.0 g, 3.66 mmd) was dissolved in dichbtomethane (30 moi), and then was added to the above mixture sbwly. The mixture was stirred at-78 C for 15 minutes. DIPEA (3.19 mi, 5.0 eq) was added to the reaction mixture and the mixture was stirred at-78 C for 10 minutes. The temperature was slowly raised to room temperature. Ethyl acetate/hexane (100 mi was added to the mixture, and organic layer was washed with 0.5N HCl and saturated sodium chbride solution, dried (anhydrous Na SO), and concentrated under reduced 2 4 pressure. The residue was separated by cdumn chromatography (25%-30% ethyl acetate/hexan) to give the title compound (900 mg, 91%) as yelow liquid. Each enantiomer was separated by Chiral OD Cdumn (Daicel Chemical Industries, 2. 00cm x 25cm, ODOOCJ-1C005, 3% i-PrOH in Hexane, 220nm). [481] [482] NMR (400MHz, CDCl 8 8.89 (d, 1H), 8.58 (d, 1H), 7.86 (d, 1H), 7.82 (d, 1H), 3 7.73-7. 65 (m, 2H), 3.74 (dd, 1H), 3.70 (s, 3H), 3.54 (dd, 1H), 3.05 (m, 1H), 1.77 (m, 2H), 0.98 (t, 3H) |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping