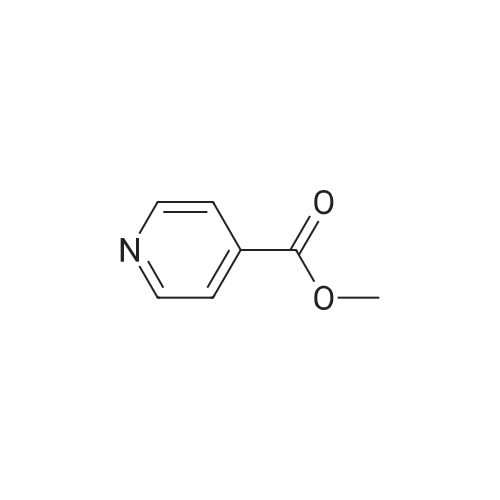
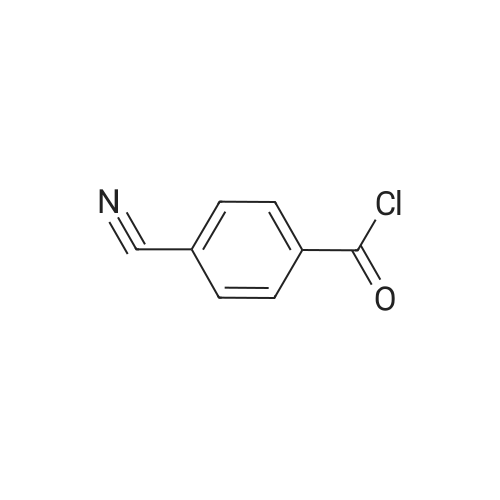
There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
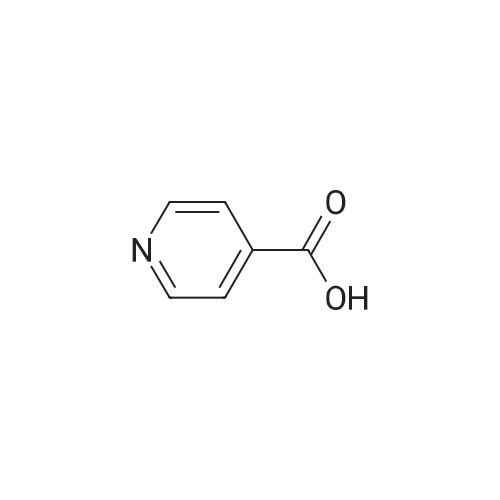
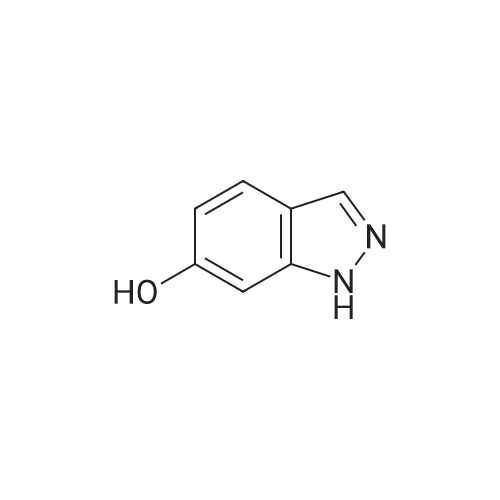
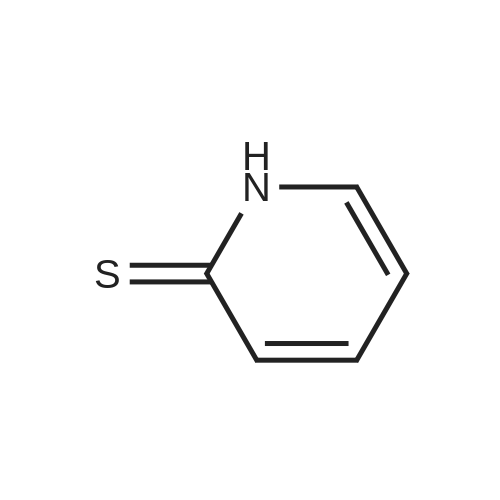
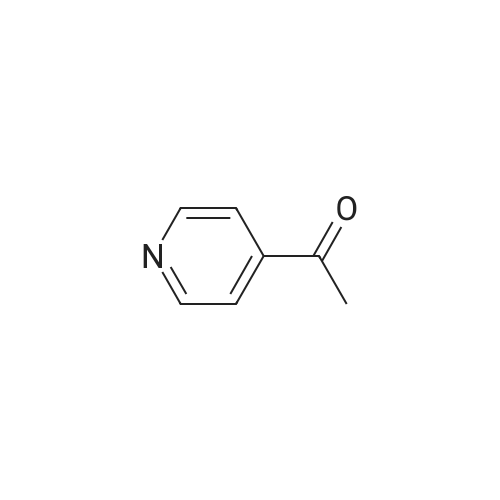
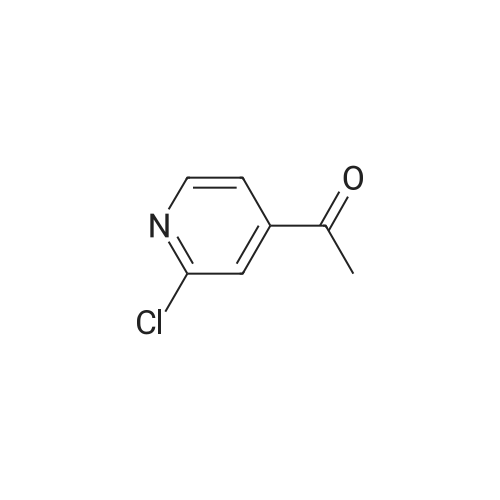
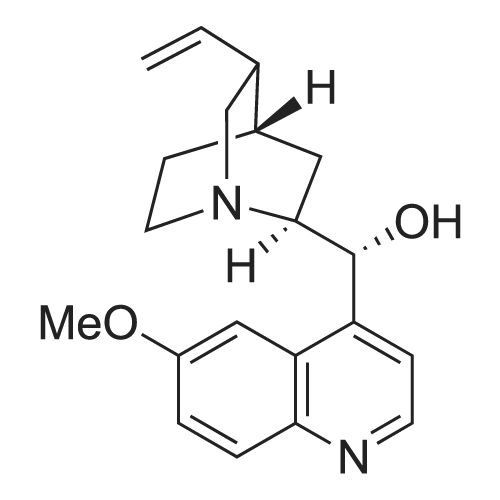
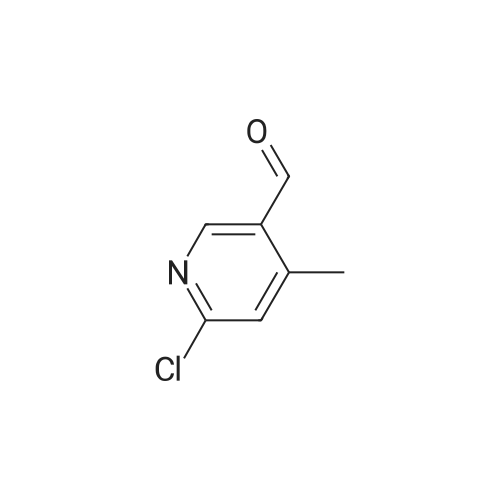
Structure of 39178-35-3 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With triethylamine In dichloromethane at 0℃; for 0.5 h;
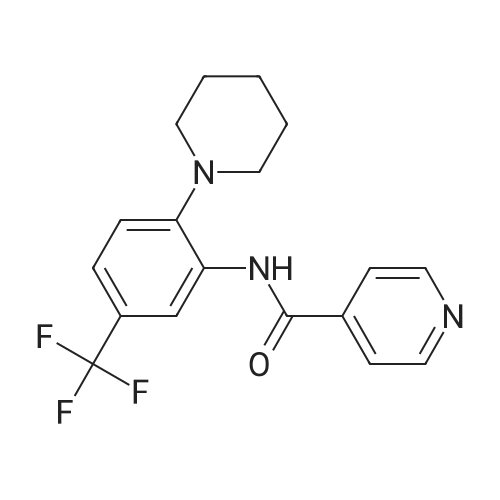
Isonicotinoyl chloride hydrochloride (6.48 g, 36.4 mmol, commercially available product) and triethylamine (5.57 ml, 54.6 mmol) were sequentially added at 0°C to a dichloromethane (10 ml) solution of 2-(1-piperidinyl)-5-(trifluoromethyl)aniline (4.45 g, 18.2 mmol) obtained as described in Referential Example 1-2B. The mixture was stirred for half an hour. Water was added to the mixture, and the resulting mixture was extracted three times with ethyl acetate. The obtained organic layer was washed with a saturated sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (200 g, hexane/ethyl acetate = 1/1) and recrystallization. Thus, N-[2-(1-piperidinyl)-5-(trifluoromethyl) phenyl]isonicotinamide (SRPIN-1, GIF-0340) (5.49 g, 86.3percent) was yielded as a colorless solid.
75%
With triethylamine In dichloromethane at 0 - 20℃; for 3 h;
A 25 mL round bottom flask initiallyplaced in an ice bath was charged with 0.629 g (3.389 mmol) ofisonicotinoyl chloride hydrochloride, 0.800 mL of triethylamine,8.00 mL of dichoromethane and 0.400 (1.64 mmol) of 2-(piperidin-1-yl)-5-(trifluoromethyl) aniline (8). The ice-bathwas removed andthe mixture was magnetically stirred at room temperature for 3 h.Then, 10.0 mL of distilled water was added, and the mixture wastransferred to a separatory funnel. The aqueous layer was extractedwith ethyl acetate (4 x 30.0 mL). The organic extracts were combinedand the resulting organic layer was washed with brine, driedover sodium sulphate, filtered, and concentrated under reducedpressure. The residue was purified by silica gel column chromatographyeluted with hexane-ethyl acetate (3:1 v/v). The solid wasfurther recrystallized with acetone. The compound SRPIN340 wasobtained as a white solid in 75percent yield (430 mg, 1.23 mmol).TLC Rf = 0.13 (hexane - ethyl acetate 3:1 v/v). mp 95.6-96.7 °C.IR (ATR, cm-1) νmax: 3347, 2945, 2917, 2811, 1679, 1611, 1587, 1556,1527, 1455, 1434, 1380, 1334, 1308, 1239, 1165, 1107, 1093, 1061,1022, 915, 895, 878, 839, 826, 751, 728, 681, 662, 644. 1H NMR(300 MHz, CDCl3) δ: 1.65-1.81 (m, 6H), 2.86 (t, 4H, J = 5.1 Hz), 7.28(d, 1H, J = 8.4 Hz), 7.37 (dd, 1H, J = 8.4 Hz and J = 1.8 Hz), 7.76 (dd,2H, J = 4.5 Hz and J = 1.5 Hz), 8.83-8.85 (m, 3H), 9.55 (s, 1H, NH).13C NMR (75 MHz, CDCl3) δ: 24.0, 27.1, 53.8, 116.6, 120.8, 121.1, 121.6(q, J C-F =3.7 Hz), 124.2 (q, J C-F = 270.5 Hz), 127.5 (q, J C-F = 32.3 Hz),133.4, 141.8, 145.9, 151.1, 163.0. HRMS (M + H+): Calculated forC18H19F3N3O, 350.1480; found: 350.1420.
33.9%
With dmap; triethylamine In dichloromethane at 0 - 20℃; for 19.5 h;
Isonicotinoyl chloride hydrochloride (151 mg, 0.850 mmol, commercially available product), triethylamine (450 μl, 3.23 mmol), and a catalytic amount of 4-(dimethylamino)pyridine were sequentially added at 0°C to a dichloromethane (5 ml) solution of 2-(1-piperidinyl)-5-(trifluoromethyl)aniline (173 mg, 0.708 mmol), obtained as described in Referential Example 1-2A. The resulting mixture was warmed to room temperature and stirred for 19.5 hours. Water was added to the mixture, and the resulting mixture was extracted three times with ethyl acetate. The obtained organic layer was washed with a saturated aqueous solution of sodium bicarbonate, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (10 g, hexane/ethyl acetate = 1.5/1) and recrystallization (hexane). Thus, N-[2-(1-piperidinyl)-5-(trifluoromethyl)phenyl]isonicotinamide (SRPIN-1, code name GIF-0340) (83.8 mg, 0.240 mmol, 33.9percent) was yielded as a colorless solid. The melting point, and results of TLC and 1H NMR (CDCl3, 400 MHz), are as follows: m.p. 96-98°C; TLC Rf 0.40 (hexane/ethyl acetate = 1/1); 1H NMR (CDCl3, 400 MHz) δ 1.67-1.68 (m, 2H, CH2), 1.78 (tt, 4H, J = 5.5, 5.5 Hz, 2CH2), 2.88 (t, 4H, J = 5.5 Hz, 2CH2), 7.29 (d, 1H, J = 8.2 Hz, aromatic), 7.40 (dd, 1H, J = 1.8, 8.2 Hz, aromatic), 7.76 (dd, 2H, J = 2.0, 4.4 Hz, aromatic), 8.86 (dd, 2H, J = 2.0, 4.4 Hz, aromatic), 8.87 (d, 1H, J = 1.8 Hz, aromatic), 9.53 (s, 1H, NH).
Reference:
[1] Patent: EP1712242, 2006, A1, . Location in patent: Page/Page column 18; 31
[2] European Journal of Medicinal Chemistry, 2017, vol. 134, p. 97 - 109
[3] Patent: EP1712242, 2006, A1, . Location in patent: Page/Page column 18; 30; 31
[4] Patent: EP2279750, 2011, A1,
With triethylamine; In 1,2-dichloro-ethane; at 0 - 20℃; for 2.41667h;Heating / reflux;
To a slurry comprising 500 ml of a 1,2-dichloroethane solutioncontaining 50 g of isonicotinic acid chloride hydrochloride andcooled to 0C were gradually added dropwise 31.4 g of aniline and50 ml of a 1,2-dichloroethane solution containing 60 g oftriethylamine over 25 minutes or longer. After stirring themixture at room temperature for 30 minutes, it was stirred underreflux for 1.5 hours. To the reaction mixture was added 100 ml ofwater and the mixture was gradually cooled to 0C. The formedprecipitates were collected by filtration, dried under reducedpressure, washed with diethyl ether, and dried under reduced 'pressure to give 45 g of N-phenylisonicotinic amide shown in Table55 below.
With triethylamine In tetrahydrofuran for 24h; Reflux;
82%
With triethylamine In tetrahydrofuran for 24h;
75%
With triethylamine In tetrahydrofuran
75%
Stage #1: isonicotinoyl chloride hydrochloride; 1,4-phenylenediamine In tetrahydrofuran at 20℃; for 0.5h;
Stage #2: With triethylamine In tetrahydrofuran
0.82%
With triethylamine In tetrahydrofuran for 24h; Reflux;
ethyl 2-(isonicotinamido)-4-methylthiazole-5-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
With triethylamine; In tetrahydrofuran; at 20℃; for 48h;
Synthesis of Ethyl 2-(lsonicotinamido)-4-methylthiazole~5~carboxylateTo a mixture of ethyl 2-amino-4-methylthiazole-5-carboxylate (5.70 g, 30.60 <n="93"/>mmol) and triethylamine (10.0 ml_, 71.80 mmol) in tetrahydrofuran (100 ml_) was added isonicotinoyl chloride hydrochloride (6.00 g, 32.00 mmol). The reaction mixture was stirred at ambient temperature for 2 days. The solvent was removed by evaporation and the resulting white solid was washed sequentially with water, 10% sodium bicarbonate solution, and water, then dried to afford the title compound in 90% yield (8.10 g); 1H NMR (DMSO-d6, 300 MHz) δ 8.77 (d, J = 6.0 Hz, 2H), 8.77 (d, J = 6.0 Hz, 2H), 4.23 (q, J = 7.2 Hz, 2H), 2.34 (s, 3H). 1.27 (t, J = 7.2 Hz, 3H); MS (ES+) m/z 292.0 (M + 1).
4-Isonicotinoyl chloride hydrochloride (6.43 g, 36.1 mmol) was suspended in 50 mL toluene in a 3-neck flask. Methyl-N-methyl-2-pyrroleacetate (4.3 mL, 29.9 mmol) was added to the reaction. The flask was equipped with a reflux condenser and a nitrogen bubbler. Nitrogen was gently bubbled through the reaction as it was heated at reflux for 14.5 hours. The reaction was then diluted with 200 mL chloroform and washed once with 200 mL 10% Na2CO3 solution. The organics were dried with MgS04 and treated with charcoal then filtered and the solvents removed in vacuo. The residue was triturated with 120 mL 5: 1 hexanes: EtOAc. The solid was then filtered off and air-dried to yield the pyrrolyl ester (6.170 g, 23.9 mmol) as a brown powder.
In toluene; for 14.5h;Heating / reflux;
A. 4-Isonicotinoyl chloride hydrochloride (6.43 g, 36.1 mmol) was suspended in 50 mL toluene in a 3-neck flask. Methyl-N-methyl-2-pyrroleacetate (4.3 mL, 29.9 mmol) was added to the reaction. The flask was equipped with a reflux condenser and a nitrogen bubbler. Nitrogen was gently bubbled through the reaction as it was heated at reflux for 14.5 hours. The reaction was then diluted with 200 mL chloroform and washed once with 200 mL 10% Na2CO3 solution. The organics were dried with MgSO4 and treated with charcoal then filtered and the solvents removed in vacuo. The residue was triturated with 120 mL 5:1 hexanes:EtOAc. The solid was then filtered off and air-dried to yield the pyrrolyl ester (6.170 g, 23.9 mmol) as a brown powder.
With sodium hydroxide; triethylamine In dichloromethane; water
4 S-2-Pyridyl isonicotinothioate (2c)
EXAMPLE 4 S-2-Pyridyl isonicotinothioate (2c) Following a general procedure (Rao et al., J. Org. Chem. 2000, 65: 1084), to a suspension of isonicotinoyl chloride hydrochloride (4.45 g, 25.0 mmol) in CH2Cl2 (50 mL) was added triethylamine (7 mL, 50 mmol). The mixture was stirred for 5 min under argon. To the resulting solution was added 2-mercaptopyridine (2.77 g, 25.0 mmol) in portions over 5 min with stirring. After 1 h, water was added and the layers were separated. The organic layer was washed (2 N NaOH, then water), dried (Na2SO4) and concentrated to dryness. The solid residue was recrystallized from ethyl acetate/hexanes to afford brown crystals (4.21 g, 77%): mp 115° C.; 1H NMR δ 7.34-7.38 (m, 1H), 7.69-7.38 (m, 4H), 8.69 (m, 1H), 8.83 (m, 2H); 13C NMR δ 120.5, 124.2, 130.8, 137.5, 142.8, 150.1, 150.8, 151.1; HRMS (FAB) obsd 217.0426, calcd 217.0436 (M++H, C11H9N2OS). Anal. Calcd for C11H8N2OS: C, 61.09; H, 3.73; N, 12.95. Found: C, 60.97; H, 3.72; N, 12.95.
59.93%
In tetrahydrofuran at 20℃; for 2.5h; Inert atmosphere;
45%
In tetrahydrofuran Inert atmosphere;
S-2-Pyridyl isonicotinothioate 8
An oven dried flask was charged with 2-mercaptopyridine (1.000 g, 8.99 mmol) and purged with argon. The solid was dissolved in anhydrous THF (20 mL) with stirring. The solution was treated with isonicotinoyl chloride hydrochloride (1.600 g, 8.99 mmol). The resulting slurry was stirred overnight. The reaction mixture was filtered. The filtrate was washed with hexanes. The resulting orange solid was added to a biphasic solution of diethyl ether and saturated sodium bicarbonate and stirred until it no longer bubbled. The organic layer was collected and the aqueous layer was washed with diethyl ether (3 ×50 mL). The organic layers were combined and dried with Na2SO4 and concentrated in vacuo to yield a yellow solid. Minimal THF was added to the solid to form a slurry which was filtered and washed with hexanes to yield 8 (0.8 g, 45%). 1H NMR (300 MHz, DMSO-d6): δ= 8.65 (m, 2H), 8.719 (m, 1H), 7.820-7.854 (m, 3H), 7.749 (d, J=7.8 Hz, 1H), 7.40 (m, 1H).
1.15.I I.
Thioester 2c was prepared by treating isonicotinoyl chloride hydrochloride with 2-mercaptopyridine in the presence of triethylamine (Scheme 2)(Scheme 2).
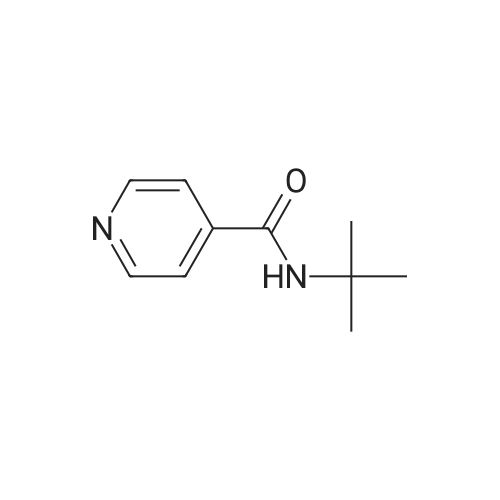
17 N-t-Butylisonicotinamide
Example 17 N-t-Butylisonicotinamide The title compound (0.3017 g; 84%) was prepared by the same method as that described in Example 1, using isonicotinoyl chloride hydrochloride (0.3606 g, 2.02 mmol). 1H-NMR(270 MHz, CDCl3) 8.72 (dd, 2H, J=4.3, 1.7 Hz), 7.56 (dd, 2H, J=4.3, 1.7 Hz), 5.95 (br, 1H), 1.48 (s, 9H)
2R-hydroxy-3-[(2-methylpropyl)(4-methoxyphenyl)sulfonyl] amino-1S-(phenylmethyl)propylamine[ No CAS ]
[ 39178-35-3 ]
[ 1453-82-3 ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine In dichloromethane; ethyl acetate; isopropyl alcohol
10.A Preparation of 4-Pyridinecarboxamide, N-[2R-hydroxy-3-[[(4-methoxyphenyl)sulfonyl](2-methylpropyl)amino]-1S-(phenylmethyl)propyl]
EXAMPLE 10A Preparation of 4-Pyridinecarboxamide, N-[2R-hydroxy-3-[[(4-methoxyphenyl)sulfonyl](2-methylpropyl)amino]-1S-(phenylmethyl)propyl] To a solution of 231 mg (0.57 mmol) of 2R-hydroxy-3-[(2-methylpropyl)(4-methoxyphenyl)sulfonyl]amino-1S-(phenylmethyl)propylamine in 3 mL of methylene chloride at 0 C., was added 288 mg (2.85 mmol) of triethylamine and then 112 mg (0.63 mmol) of isonicotinoyl chloride hydrochloride. After 19 hours at room temperature, the solvent was removed, ethyl acetate added, then washed with saturated sodium bicarbonate, brine, dried with magnesium sulfate, filtered and concentrated to afford 290 mg of crude product. This was chromatographed on silica gel using 3-5% isopropanol/methylene chloride as eluent to afford 190 mg of the desired compound; mass spectrum calc. for C27H34N3O5S (M+H) 512.2219; found 512.2280.
With triethylamine In dichloromethane; ethyl acetate; isopropyl alcohol
10.A Preparation of 4-Pyridinecarboxamide, N-[2R-hydroxy-3-[[(4-methoxyphenyl)sulfonyl](2-methylpropyl)amino]-1S-(phenylmethyl)propyl]
EXAMPLE 10A STR105 Preparation of 4-Pyridinecarboxamide, N-[2R-hydroxy-3-[[(4-methoxyphenyl)sulfonyl](2-methylpropyl)amino]-1S-(phenylmethyl)propyl] To a solution of 231 mg (0.57 mmol) of 2R-hydroxy-3-[(2-methylpropyl)(4-methoxyphenyl)sulfonyl] amino-1S-(phenylmethyl)propylamine in 3 mL of methylene chloride at O C, was added 288 mg(2.85 mmol) of triethylamine and then 112 mg(0.63 mmol) of isonicotinoyl chloride hydrochloride. After 19 hours at room temperature, the solvent was removed, ethyl acetate added, then washed with saturated sodium bicarbonate, brine, dried with magnesium sulfate, filtered and concentrated to afford 290 mg of crude product. This was chromatographed on silica gel using 3-5% isopropanol/methylene chloride as eluent to afford 190 mg of the desired compound; mass spectrum calc. for C27 H34 N3 O5 S (M+H) 512.2219; found 512.2280.
With triethylamine In dichloromethane; ethyl acetate; isopropyl alcohol
10.A Preparation of 4-Pyridinecarboxamide, N-[2R-hydroxy-3-[[(4-methoxyphenyl)sulfonyl](2-methylpropyl)amino]-1S-(phenylmethyl)propyl]
EXAMPLE 10A STR102 Preparation of 4-Pyridinecarboxamide, N-[2R-hydroxy-3-[[(4-methoxyphenyl)sulfonyl](2-methylpropyl)amino]-1S-(phenylmethyl)propyl] To a solution of 231 mg (0.57 mmol) of 2R-hydroxy-3-[(2-methylpropyl)(4-methoxyphenyl)sulfonyl]amino-1S-(phenylmethyl)propylamine in 3 mL of methylene chloride at 0 C., was added 288 mg(2.85 mmol) of triethylamine and then 112 mg(0.63 mmol) of isonicotinoyl chloride hydrochloride. After 19 hours at room temperature, the solvent was removed, ethyl acetate added, then washed with saturated sodium bicarbonate, brine, dried with magnesium sulfate, filtered and concentrated to afford 290 mg of crude product. This was chromatographed on silica gel using 3-5% isopropanol/methylene chloride as eluent to afford 190 mg of the desired compound; mass spectrum calc. for C27 H34 N3 O5 S (M+H) 512.2219; found 512.2280.
With triethylamine In dichloromethane; ethyl acetate; isopropyl alcohol
10.A Preparation of 4-Pyridinecarboxamide, N-[2R-hydroxy-3-[[(4-methoxyyhenyl)sulfonyl](2-methylpropyl)amino]-1S-(-phenylmethyl)propyl]
EXAMPLE 10A STR103 Preparation of 4-Pyridinecarboxamide, N-[2R-hydroxy-3-[[(4-methoxyyhenyl)sulfonyl](2-methylpropyl)amino]-1S-(-phenylmethyl)propyl] To a solution of 231 mg (0.57 mmol) of 2R-hydroxy-3-[(2-methylpropyl)(4-methoxyphenyl)sulfonyl] amino-1S-(phenylmethyl)propylamine in 3 mL of methylene chloride at 0 C, was added 288 mg(2.85 mmol) of triethylamine and then 112 mg(0.63 mmol) of isonicotinoyl chloride hydrochloride. After 19 hours at room temperature, the solvent was removed, ethyl acetate added, then washed with saturated sodium bicarbonate, brine, dried with magnesium sulfate, filtered and concentrated to afford 290 mg of crude product. This was chromatographed on silica gel using 3-5% isopropanol/methylene chloride as eluent to afford 190 mg of the desired compound; mass spectrum calc. for C27 H34 N3 O5 S (M+H) 512.2219; found 512.2280.
With triethylamine In dichloromethane; ethyl acetate; isopropyl alcohol
10.A Preparation of 4-Pyridinecarboxamide, N-[2R-hydroxy-3-[[(4-methoxyphenyl)sulfonyl](2-methylpropyl)amino]-1S-(phenylmethyl)propyl]
EXAMPLE 10A STR95 Preparation of 4-Pyridinecarboxamide, N-[2R-hydroxy-3-[[(4-methoxyphenyl)sulfonyl](2-methylpropyl)amino]-1S-(phenylmethyl)propyl] To a solution of 231 mg (0.57 mmol) of 2R-hydroxy-3-[(2-methylpropyl)(4-methoxyphenyl)sulfonyl]amino-1S-(phenylmethyl)propylamine in 3 mL of methylene chloride at 0° C., was added 288 mg (2.85 mmol) of triethylamine and then 112 mg (0.63 mmol) of isonicotinoyl chloride hydrochloride. After 19 hours at room is temperature, the solvent was removed, ethyl acetate added, then washed with saturated sodium bicarbonate, brine, dried with magnesium sulfate, filtered and concentrated to afford 290 mg of crude product. This was chromatographed on silica gel using 3-5% isopropanol/methylene chloride aseluent to afford 190 mg of the desired compound; mass spectrum calc. for C27 H34 N3 O5 S (M+H) 512.2219; found 512.2280.
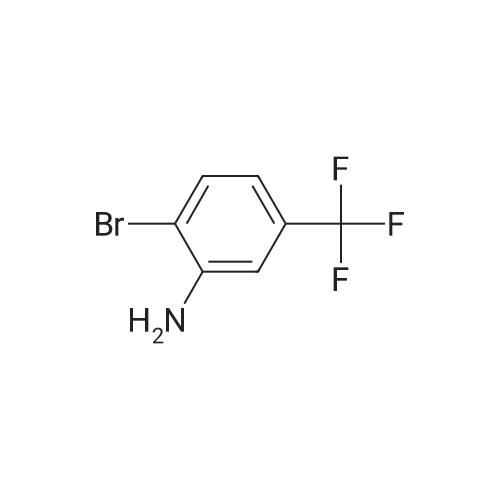
N-[2-bromo-5-(trifluoromethyl)phenyl]isonicotinamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
44.8%
With triethylamine In dichloromethane at 0 - 20℃; for 24h;
23
Isonicotinoyl chloride hydrochloride (427 mg, 2.39 mmol, commercially available product) and triethylamine (410 µl, 2.94 mmol) were sequentially added at 0°C to a dichloromethane (5 ml) solution of 2-bromo-5-(trifluoromethyl)aniline (480 mg, 2.00 mmol; commercially available product). The resulting mixture was warmed to room temperature and stirred for 24 hours. Water was added to the mixture, and the resulting mixture was extracted three times with ethyl acetate. The obtained organic layer was washed with a saturated sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by recrystallization (ethyl acetate). Thus, N-[2-bromo-5-(trifluoromethyl)phenyl]isonicotinamide (GIF-0612) (308 mg, 44.8%) was yielded as a colorless solid. TLC Rf 0.46 (hexane/ethyl acetate = 1/1).
With triethylamine; In dichloromethane; at 0℃; for 0.5h;Product distribution / selectivity;
Isonicotinoyl chloride hydrochloride (6.48 g, 36.4 mmol, commercially available product) and triethylamine (5.57 ml, 54.6 mmol) were sequentially added at 0C to a dichloromethane (10 ml) solution of <strong>[1496-40-8]2-(1-piperidinyl)-5-(trifluoromethyl)aniline</strong> (4.45 g, 18.2 mmol) obtained as described in Referential Example 1-2B. The mixture was stirred for half an hour. Water was added to the mixture, and the resulting mixture was extracted three times with ethyl acetate. The obtained organic layer was washed with a saturated sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (200 g, hexane/ethyl acetate = 1/1) and recrystallization. Thus, N-[2-(1-piperidinyl)-5-(trifluoromethyl) phenyl]isonicotinamide (SRPIN-1, GIF-0340) (5.49 g, 86.3%) was yielded as a colorless solid.
75%
With triethylamine; In dichloromethane; at 0 - 20℃; for 3h;
A 25 mL round bottom flask initiallyplaced in an ice bath was charged with 0.629 g (3.389 mmol) ofisonicotinoyl chloride hydrochloride, 0.800 mL of triethylamine,8.00 mL of dichoromethane and 0.400 (1.64 mmol) of 2-(piperidin-1-yl)-5-(trifluoromethyl) aniline (8). The ice-bathwas removed andthe mixture was magnetically stirred at room temperature for 3 h.Then, 10.0 mL of distilled water was added, and the mixture wastransferred to a separatory funnel. The aqueous layer was extractedwith ethyl acetate (4 x 30.0 mL). The organic extracts were combinedand the resulting organic layer was washed with brine, driedover sodium sulphate, filtered, and concentrated under reducedpressure. The residue was purified by silica gel column chromatographyeluted with hexane-ethyl acetate (3:1 v/v). The solid wasfurther recrystallized with acetone. The compound SRPIN340 wasobtained as a white solid in 75% yield (430 mg, 1.23 mmol).TLC Rf = 0.13 (hexane - ethyl acetate 3:1 v/v). mp 95.6-96.7 C.IR (ATR, cm-1) numax: 3347, 2945, 2917, 2811, 1679, 1611, 1587, 1556,1527, 1455, 1434, 1380, 1334, 1308, 1239, 1165, 1107, 1093, 1061,1022, 915, 895, 878, 839, 826, 751, 728, 681, 662, 644. 1H NMR(300 MHz, CDCl3) delta: 1.65-1.81 (m, 6H), 2.86 (t, 4H, J = 5.1 Hz), 7.28(d, 1H, J = 8.4 Hz), 7.37 (dd, 1H, J = 8.4 Hz and J = 1.8 Hz), 7.76 (dd,2H, J = 4.5 Hz and J = 1.5 Hz), 8.83-8.85 (m, 3H), 9.55 (s, 1H, NH).13C NMR (75 MHz, CDCl3) delta: 24.0, 27.1, 53.8, 116.6, 120.8, 121.1, 121.6(q, J C-F =3.7 Hz), 124.2 (q, J C-F = 270.5 Hz), 127.5 (q, J C-F = 32.3 Hz),133.4, 141.8, 145.9, 151.1, 163.0. HRMS (M + H+): Calculated forC18H19F3N3O, 350.1480; found: 350.1420.
33.9%
With dmap; triethylamine; In dichloromethane; at 0 - 20℃; for 19.5h;Product distribution / selectivity;
Isonicotinoyl chloride hydrochloride (151 mg, 0.850 mmol, commercially available product), triethylamine (450 mul, 3.23 mmol), and a catalytic amount of 4-(dimethylamino)pyridine were sequentially added at 0C to a dichloromethane (5 ml) solution of <strong>[1496-40-8]2-(1-piperidinyl)-5-(trifluoromethyl)aniline</strong> (173 mg, 0.708 mmol), obtained as described in Referential Example 1-2A. The resulting mixture was warmed to room temperature and stirred for 19.5 hours. Water was added to the mixture, and the resulting mixture was extracted three times with ethyl acetate. The obtained organic layer was washed with a saturated aqueous solution of sodium bicarbonate, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (10 g, hexane/ethyl acetate = 1.5/1) and recrystallization (hexane). Thus, N-[2-(1-piperidinyl)-5-(trifluoromethyl)phenyl]isonicotinamide (SRPIN-1, code name GIF-0340) (83.8 mg, 0.240 mmol, 33.9%) was yielded as a colorless solid. The melting point, and results of TLC and 1H NMR (CDCl3, 400 MHz), are as follows: m.p. 96-98C; TLC Rf 0.40 (hexane/ethyl acetate = 1/1); 1H NMR (CDCl3, 400 MHz) delta 1.67-1.68 (m, 2H, CH2), 1.78 (tt, 4H, J = 5.5, 5.5 Hz, 2CH2), 2.88 (t, 4H, J = 5.5 Hz, 2CH2), 7.29 (d, 1H, J = 8.2 Hz, aromatic), 7.40 (dd, 1H, J = 1.8, 8.2 Hz, aromatic), 7.76 (dd, 2H, J = 2.0, 4.4 Hz, aromatic), 8.86 (dd, 2H, J = 2.0, 4.4 Hz, aromatic), 8.87 (d, 1H, J = 1.8 Hz, aromatic), 9.53 (s, 1H, NH).
With triethylamine; In dichloromethane; water;
[Reference example 1-3B] Isonicotinoyl chloride hydrochloride (6.48 g, 36.4 mmol; commercial product) and triethylamine (5.57 mL, 54.6 mmol) were added sequentially at 0C to a dichloromethane (10 mL) solution of <strong>[1496-40-8]2-(1-piperidinyl)-5-(trifluoromethyl)aniline</strong> (4.45 g, 18.2 mmol) obtained in Reference example 1-2B. The mixture was stirred for 0.5 hours. Water was added to the mixture and then the mixture was subjected to extraction with ethyl acetate (x3). The thus obtained organic layer was washed with saturated sodium chloride solution, dried using anhydrous sodium sulfate, filtered, and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (200 g, hexane/ethyl acetate = 1/1) and recrystallization (hexane), so that N-[2-(1-piperidinyl)-5-(trifluoromethyl)phenyl]isonicotinamide (Compound 1) (5.49 g, 86.3%) was obtained as colorless solid.
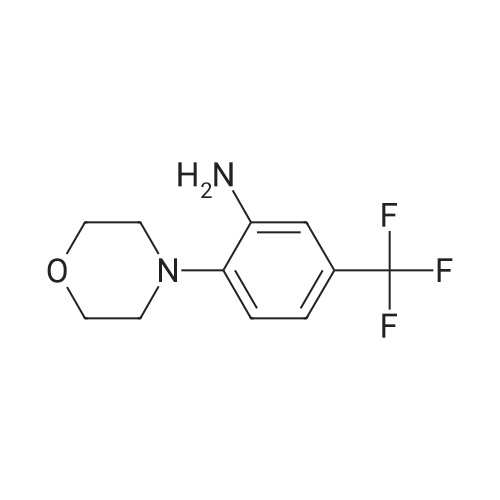
N-(2-morpholino-5-(trifluoromethyl)phenyl)isonicotinamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78%
With triethylamine In dichloromethane at 0 - 20℃; for 3h;
4.1.4.1. N-(2-(piperidin-1-yl)-5-(trifluoromethyl)phenyl)isonicotinamide(SRPIN340).
General procedure: A 25 mL round bottom flask initiallyplaced in an ice bath was charged with 0.629 g (3.389 mmol) ofisonicotinoyl chloride hydrochloride, 0.800 mL of triethylamine,8.00 mL of dichoromethane and 0.400 (1.64 mmol) of 2-(piperidin-1-yl)-5-(trifluoromethyl) aniline (8). The ice-bathwas removed andthe mixture was magnetically stirred at room temperature for 3 h.Then, 10.0 mL of distilled water was added, and the mixture wastransferred to a separatory funnel. The aqueous layer was extractedwith ethyl acetate (4 x 30.0 mL). The organic extracts were combinedand the resulting organic layer was washed with brine, driedover sodium sulphate, filtered, and concentrated under reducedpressure. The residue was purified by silica gel column chromatographyeluted with hexane-ethyl acetate (3:1 v/v). The solid wasfurther recrystallized with acetone. The compound SRPIN340 wasobtained as a white solid in 75% yield (430 mg, 1.23 mmol).
23.2%
With triethylamine In dichloromethane at 0 - 20℃; for 60h;
5-3
Isonicotinoyl chloride hydrochloride (320 mg, 1.80 mmol, commercially available product) and triethylamine (480 µl, 3.44 mmol) were sequentially added at 0°C to a dichloromethane (5 ml) solution of 4-[2-amino-4-(trifluoromethyl)phenyl]morpholine (196 mg, 0.796 mmol), obtained as described in Referential Example 5-2. The resulting mixture was warmed to room temperature and stirred for 60 hours. Water was added to the mixture, and the resulting mixture was extracted three times with dichloromethane. The obtained organic layer was washed with a saturated sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (30 g, hexane/ethyl acetate = 2/1). Thus, N-[2-(4-morpholinly)-5-(trifluoromethyl)phenyl]isonicotinamide (GIF-0346) (65.1 mg, 23.2%) was yielded as a colorless solid. The melting point, and results of TLC and 1H NMR (CDCl3, 400 MHz), are as follows: m.p. 172-173°C; TLC Rf 0.23 (hexane/ethyl acetate = 1/3); 1H NMR (CDCl3, 400 MHz) δ 2.96 (t, 4H, J = 4.4 Hz, 2CH2), 3.92 (t, 4H, J = 4.4 Hz, 2CH2), 7.34 (d, 1H, J = 8.4 Hz, aromatic), 7.44 (dd, 1H, J = 1.6, 8.4 Hz, aromatic), 7.75 (dd, 1H, J = 1.6, 4.4 Hz, aromatic), 8.87-8.88 (m, 3H , aromatic) 9.48 (s, 1H, NH).
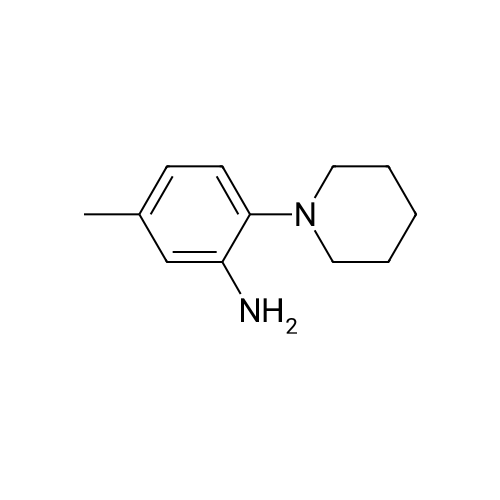
N-[5-methyl-2-(1-piperidinyl)phenyl]isonicotinamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
28.8%
With dmap; triethylamine In dichloromethane at 20℃; for 19h;
16-3
Isonicotinoyl chloride hydrochloride (172 mg, 0.966 mmol, commercially available product), triethylamine (340 µl, 2.44 mmol), and a catalytic amount of 4-(dimethylamino) pyridine were sequentially added at room temperature to a dichloromethane (5 ml) solution of 5-methyl-2-(1-piperidinyl)aniline (155 mg, 0.815 mmol), obtained as described in Referential Example 16-2. The resulting mixture was stirred for 19 hours. Water was added to the mixture, and the resulting mixture was extracted three times with ethyl acetate. The obtained organic layer was washed with a saturated sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (10 g, hexane/ethyl acetate = 1/1). Thus, N-[5-methyl-2-(1-piperidinyl)phenyl]isonicotinamide (GIF-0342) (69.5 mg, 28.8%) was yielded as a colorless solid. The melting point, and results of TLC and 1H NMR (CDCl3, 400 MHz), are as follows: m.p. 142-144°C; TLC Rf 0.35 (hexane/ethyl acetate = 1/1); 1H NMR (CDCl3, 400 MHz) δ 1.62-1.70 (m, 2H, CH2), 1.75 (tt, 4H, J = 4.9, 4.9 Hz, 2CH2), 2.37 (s, 3H, CH3), 2.82 (t, 4H, J = 4.9 Hz, 2CH2), 6.94 (dd, 1H, J = 1.6, 8.1 Hz, aromatic), 7.12 (d, 1H, J = 8.1 Hz, aromatic), 7.76 (dd, 2H, J = 1.3, 4.5 Hz, aromatic), 8.38 (d, 1H, J = 1.6 Hz, aromatic), 8.84 (dd, 2H, J = 1.3, 4.5 Hz, aromatic), 9.75 (s, 1H, NH).
EXAMPLE 4 Following a procedure similar to that of Example 3 but using 0.636 g. of 4-carbamoylimidazolium-5-olate and 1.068 g. of isonicotinoyl chloride hydrochloride there was obtained 1.16 g. of 5-carbamoyl-1H-imidazole-4-yl isonicotinate, m.p. 186 C. (charred). Recrystallized was crude material from dimethylsulfoxide and water. m.p.: 192.5 C. (charred).
5-carbamoyl-1H-imidazole-4-yl isonicotinate hydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In pyridine;
EXAMPLE 5 To a suspension of 0.636 g. of 4-carbamoylimidazolium-5-olate in 15 ml of dry pyridine was slowly added 1.068 g. of isonicotinoyl chloride hydrochloride at a temperature below 5 C. in N2 atmosphere. After being stirred for an hour at 41-43 C., the reaction mixture was cooled to room temperature. Then separated crystals was filtered off, washed with pyridine, chloroform, ethyl acetate and diethyl ether, and dried to give 1.194 g. of 5-carbamoyl-1H-imidazole-4-yl isonicotinate hydrochloride.
2 1,3-Dihydro-4-isonicotinoyl-5-methyl-2H-imidazole-2-one
EXAMPLE 2 1,3-Dihydro-4-isonicotinoyl-5-methyl-2H-imidazole-2-one In 80 ml tetrachloroethane are placed 3.87 g (39.5 mmole) 1,3-dihydro-4-methyl-2H-imidazole-2-one and 7 g (39.5 mmole) isonicotinoyl chloride hydrochloride. Aluminum chloride (26 g, 194 mmole) is added and the mixture is stirred at 85° C. for 3 hours. The tetrachloroethane is decanted from the reaction mixture and the residue is quenched with water and neutralized with sodium bicarbonate. The suspension is filtered and the filtrate evaporated to dryness. Chromatography over silica gel affords the title compound; m.p. 295°-96°. Anal. calcd. for C10 H9 N3 O2; C, 59.10; H, 4.46; N, 20.68. Found: C, 59.00; H, 4.45; N, 20.32.
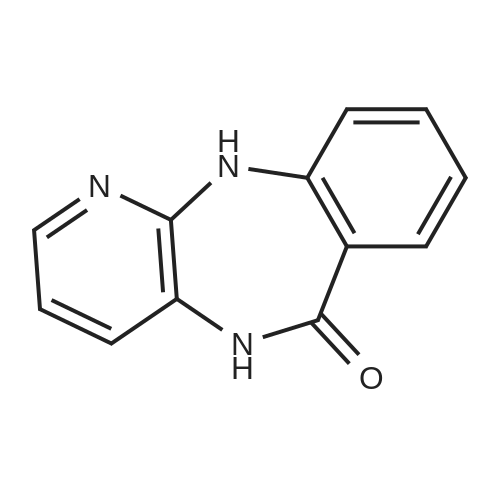
5,11-dihydro-11-[(4-pyridinyl)carbonyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With pyridine; In 1,4-dioxane; dichloromethane; water;
(a) 5,11-Dihydro-11-[(4-pyridinyl)carbonyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one A mixture of 40.4 gm (0.191 mol) of <strong>[885-70-1]5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one</strong>, 46.0 gm (0.258 mol) of isonicotinic acid chloride hydrochloride, 42 ml (0.52 mol) of pyridine, and 600 ml of anhydrous dioxane was refluxed for three hours. After cooling, the mixture was filtered, the filter residue was taken up in 300 ml of water, and the solution obtained was extracted twice, each time with 100 ml of dichloromethane. The aqueous phase was made alkaline with sodium hydroxide solution and exhaustively extracted with methylene chloride. The methylene chloride extracts thus obtained were washed with water and dried over sodium sulphate, and the solvent was eliminated in vacuo. The residue was recrystallized from hot methanol. After washing with ether, the resulting beige-colored crystals melted at 265-266 C. Yield: 21.0 gm (35% of theory).
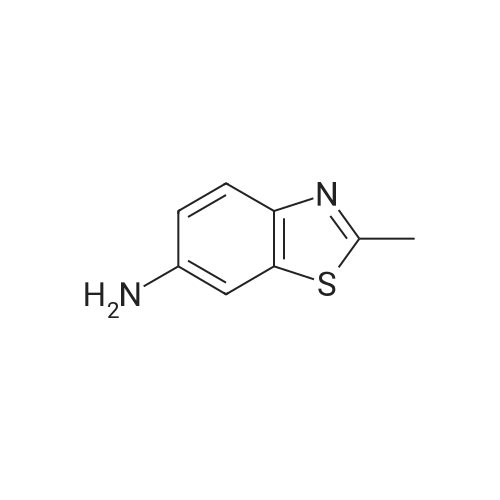
2-methyl-6-(pyridin-4-ylcarbonylamino)-benzthiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine In dichloromethane
8.a 2-Methyl-6-(4-pyridyl)-5H-imidazo[4,5-f]benzthiazole
(a) 13.2 g. (80.0 mMole) 6-amino-2-methylbenzthiazole and 22.2 ml. (160 mMole) triethylamine are dissolved in 120 ml. dichloromethane. 14.3 g. (80.0 mMole) 4-pyridinecarbonyl chloride hydrochloride are added thereto with ice cooling. The solvent is removed in a vacuum and the residue is digested with water. After crystallisation from ethanol, there are obtained 17.0 g. (79% of theory) of colourless crystals of 2-methyl-6-(4-pyridylcarbonylamino)-benzthiazole; m.p. 209°-210° C.
XI EXAMPLE XI
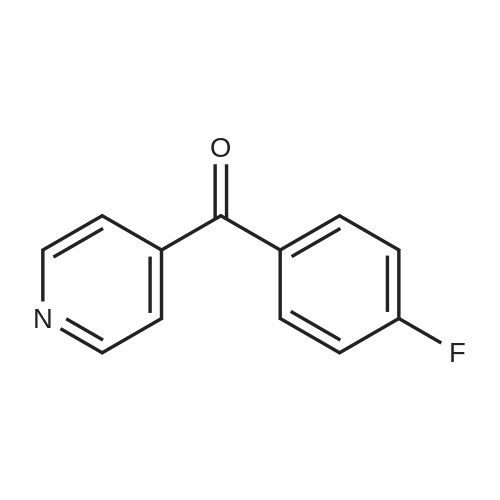
EXAMPLE XI To a stirred and cooled (ice-bath) solution of 141.5 parts of 4-pyridinecarbonyl chloride hydrochloride in 400 parts of fluorobenzene were added portionwise 399 parts of aluminum chloride. Upon completion, the whole was slowly heated to reflux and stirring at reflux was continued for 6 hours. The reaction mixture is cooled, poured onto crushed ice and acidified with 240 parts of a hydrochloric acid solution 10N. The layers were separated. The acid aqueous phase was washed twice with 180 parts of methylbenzene and strongly alkalized with a sodium hydroxide solution 60%. The product was extracted three times with dichloromethane. The combined extracts were dried, filtered and evaporated. The residue was dissolved in 900 parts of methylbenzene and the solution was treated with activated charcoal. The latter was filtered off and the filtrate was evaporated. The residue was crystallized from 2,2'-oxybispropane, yielding 152 parts (75.5%) of (4-fluorophenyl)(4-pyridinyl)methanone; mp. 85.5° C. (intermediate 25).
In fluorobenzene
XXXV EXAMPLE XXXV
EXAMPLE XXXV Following the procedure of Example XXXIV there is prepared (4-fluorophenyl)(4-pyridinyl)methanone; mp. 85.5° C., by the reaction of 4-pyridinecarbonyl chloride hydrochloride with fluorobenzene.
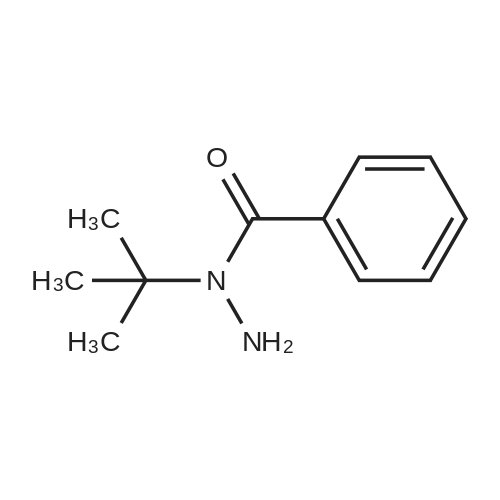
N-t-butyl-N-benzoyl-N-isonicotinoylhydrazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium hydroxide In methanol; dichloromethane; water; toluene
EXAMPLE 1 - Preparation of N--t-butyl-N-benzoyl-N--isonicotinoylhydrazine
EXAMPLE 1 - Preparation of N--t-butyl-N-benzoyl-N--isonicotinoylhydrazine N-- t -butyl-N-benzoylhydrazine (1.0 g, 0.0052 mol) was suspended in 20 ml of toluene. Isonicotinoyl chloride hydrochloride (0.93 g, 0.0052 mol) was added and then a solution of sodium hydroxide (1.25 g of 50% aqueous NaOH) in 5 ml of water was added dropwise. After stirring at 23°C for 2 hours, the solids were removed by filtration, washed with water and dried in air. The crude product was chromatographed on silica gel using 5% methanol/methylene chloride as eluant to afford pure N-- t -butyl-N-benzoyl-N--isonicotinoylhydrazine. m.p. >210°C.
With triethylamine; In dichloromethane; at 20℃; for 2 - 14h;Product distribution / selectivity;
In a solution of 2-methoxy-6-methylbenzoic acid ethyl ester (5 g, 25.77 mmol) and NaOH (6.18 g, 154.64 mmol) in EtOH (100 mL) and water (40 ml_) was stirred at reflux for 24 hours. EtOH was then removed using a rotary evaporator and the aqueous was acidified with HCI (1 N) to pH=4. Extract with CH2Cb (3x100 mL) followed by concentration using a rotary evaporator afforded 4.25 g of 2-methoxy-6-methylbenzoic acid (100%) as a white solid. To a solution of 2-methoxy-6-methylbenzoic acid (1.66 g, 10 mmol) in CH2CI2 (80 mL) at room temperature was added BBr3 in CH2CI2 (20 mL, 20 mmol). The reaction mixture was stirred at room temperature for 20 hour and then concentrated using a rotary evaporator. The resulting residue was re-dissolved in CH2CI2 (50 mL), diluted with HCI (0.5 N), extracted with CH2CI2 (3x100 mL) and concentrated using a rotary evaporator to afford 1.52 g of 2-hydroxy-6-methylbenzoic acid (100%). To a solution of 2-hydroxy-6-methylbenzoic acid (1.52 g, 10 mmol) in THF (50 mL) at room temperature was slowly added CH3Li in ether (22 mL, 35 mmol) and the suspension was stirred at 6O0C for 6 hour. The reaction was quenched with HCI EPO <DP n="110"/>(0.5 N) aqueous and extracted with CH2CI2 ( 3x100 mL). Concentration using a rotary evaporator afforded 1 g of <strong>[41085-27-2]2'-hydroxy-6'-methylacetophenone</strong> (67%) as a brown oil. A solution of 2'~hydroxy-6'-methylacetophenone (1.0 g, 6.67 mmol) in CH2CI2 (50 mL) at room temperature was mixed with isonicotinoyl chloride hydrochloride (2.136 g, 12 mmol) and triethylamine (3.9 mL, 28 mmol) sequentially. The resulting mixture was stirred at room temperature for 2 hours, quenched with water, and extracted with CH2CI2 ( 3x100 mL) The volume was reduced using a rotary evaporator to minimal and triturated with hexanes. The solid was collected by filtration to afford 1.2 g of the corresponding isonicotinic aryl ester (70%). A solution of the above isonicotinic aryl ester (1.2 g, 4.70 mmol) in THF (50 mL) was mixed with potassium tert-butoxide (2.24 g, 20 mmol) and stirred at 65C foji2 hours. The reaction was quenched with water and acidified with HCI (0.5 N) to pH = 6. Extract with CH2CI2 (3x100 mL) followed by concentration using a rotary evaporator afforded a yellow solid residue. It was purified by column (SiO2, hexane / EtOAc = 1 :1 ) to provide 0.72 g of the diketone (60%). A solution of the above diketone (0.7 g, 2.745 mmol) in HOAc (50 mL) was stirred at reflux for 2 hours. All the solvent was removed using a rotary evaporator to afford a solid residue. It was then diluted with water and neutralized with NaOH (0.5 N) to pH = 8. The solid was collected by filtration and washed with water and hexanes sequentially to afford 0.32 g of 5-methyl-2-(pyridin-4-yl)-4H- chromen-4-one as a light yellow solid (49%).; In a solution of 2-methoxy-6-methylbenzoic acid ethyl ester (5 g, 25.77 mmol) and NaOH (6.18 g, 154.64 mmol) in EtOH (100 mL) and water (40 mL) was stirred at reflux for 24 hours. EtOH was then removed using a rotary evaporator and the aqueous was acidified with HCI (1 N) to pH=4. Extract withCH2CI2 (3x100 mL) followed by concentration using a rotary evaporator afforded EPO <DP n="112"/>4.25 g of 2-methoxy-6-methylbenzoic acid (100%) as a white solid. To a solution of 2-methoxy-6-methylbenzoic acid (3.5 g, 21 mmol) in CH2CI2 (100 mL) at room temperature was added BBr3 in CH2CI2 (42 mL, 42 mmol). The reaction mixture was stirred at room temperature for 14 hrs and then concentrated using a rotary evaporator. The resulting residue was re-dissolved in CH2CI2 (50 mL), diluted with HCI (0.5 N), extracted with CH2CI2 (3x100 mL) and concentrated using a rotary evaporator to afford 3.3 g of 2-hydroxy-6-methylbenzoic acid (100%). To a solution of 2-hydroxy-6-methylbenzoic acid (3.3 g, 21.7 mmol) in THF (200 mL) at room temperature was slowly added CH3Li in ether (47 mL, 76 mmol) and the suspension was stirred at 600C for 6 hour. The reaction was quenched with HCI (0.5 N) aqueous and extracted with CH2CI2 (3x100 mL). Concentration using a rotary evaporator afforded an oily residue. It was purified by column (SiO2, hexane / EtOAc = 4:1 ) to provide 3 g of 2.-hydroxy-6'-methylacetophenone (92%) as a light yellow oil. A solution of <strong>[41085-27-2]2'-hydroxy-6'-methylacetophenone</strong> (1.5 g, 10 mmol) in CH2CI2 (50 mL) at room temperature was mixed with isonicotinoyl chloride hydrochloride (2.0 g, 11 mmol) and triethylamine (4.9 mL, 35 mmol) sequentially. The resulting mixture was stirred at room temperature for 14 hours, quenched with water, and extracted with CH2CI2 (3x100 mL). Concentration using a rotary evaporator afforded a solid residue. It was purified by column (SiO2, hexane / EtOAc = 3:1) to provide 1.5 g of the corresponding isonicotinic aryl ester (59%) as a light yellow solid. A solution of the above isonicotinic aryl ester (1.5 g, 5.88 mmol) in THF (100 mL) was mixed with potassium tert-butoxide (1.384 g, 12.35 mmol) and stirred at reflux for 2 hours. The reaction was quenched with water and...
With pyridine; In pyridine; at 100℃; for 0.0833333h;Microwave irradiation;
Example 21 N-(4-Methoxy-3-nitro-phenyl)-isonicotinamide A mixture of <strong>[577-72-0]4-methoxy-3-nitroaniline</strong> (168 mg, 1 mmol) and Isonicotinoyl chloride hydrogen chloride (267 mg, 0.2 M in 1.5 mmol) in pyridine (1 mL) is sealed in a microwave reactor and heated at 100 C. under microwave radiation for 5 mins. Then 1N NaOH aqueous solution is added to the reaction mixture. After stirring at room temperature for several minutes, the mixture is filtered. The solid is washed with H2O and air dried to give 263 mg of the title compound as a yellow solid. MS (ESI) m/z 274 (M+H)+
With triethylamine; In N,N-dimethyl-formamide; for 3.3h;Cooling with ice;
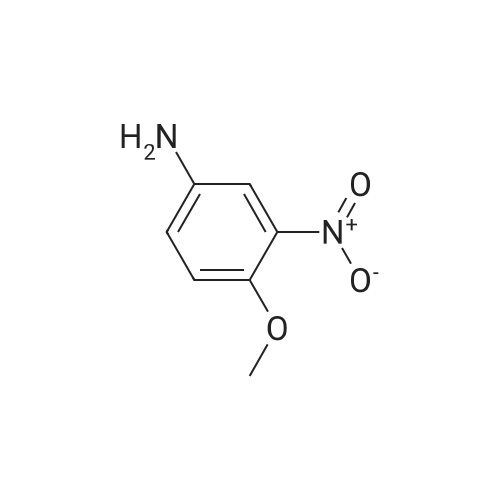
To a mixture of 2.5 g of <strong>[461699-34-3]2-amino-4-trifluoromethoxy phenol</strong>, 2.62 g of triethylamine and 15 ml of DMF, 2.31 g of 4-isonicotinic acid chloride hydrochloride was added while ice-cooling. The reaction mixture was stirred for 3.3 hours. The reaction mixture was poured into water and precipitated crystals were filtered and dried under reduced pressure to give 2.19 g of N-[5-(trifluoromethoxy)-2-hydroxyphenyl]isonicotinamide. [Show Image] 1H-NMR (DMSO-d6) delta: 8.78 (dd, J=4,4, 1.7 Hz, 2H), 7.86 (dd, J=4.4, 1.6 Hz, 2H), 7.80-7.77 (m, 1H), 7.10-7.05 (m, 1H), 6.99 (d, J=8.7 Hz, 1H)
With triethylamine; In N,N-dimethyl-formamide; for 3.3h;Cooling with ice;
Reference Production Example 28To a mixture of 4.0 g of 4-trifluoromethoxy phenol and 25 ml of acetic acid, a mixture of 2.02 g of 70% nitric acid and 10 ml of acetic acid was added dropwise with the temperature kept at 10-15 C. The reaction mixture was stirred for five hours. The reaction mixture was poured into ice water and extracted with ethyl acetate. The combined organic layers were washed with water, a saturated aqueous solution of sodium hydrogencarbonate and a saturated sodium chloride solution, dried over sodium sulfate, and then concentrated under reduced pressure to give 4.53 g of 4-trifluoromethoxy-2-nitrophenol.1H-NMR (CDCl3) delta: 10.50 (s, 1H), 8.02-7.99 (m, 1H), 7.50-7.45 (m, 1H), 7.22 (d, J=9.1 Hz, 1H)A mixture of 4.53 g of 4-trifluoromethoxy-2-nitrophenol, 35 ml of ethyl acetate and 1.0 g of 5% palladium on carbon was stirred under about one atmosphere of hydrogen at room temperature for 1.7 hours. The mixture was filtered through Celite. The filtrate was concentrated under reduced pressure to give 3.92 g of <strong>[461699-34-3]2-amino-4-trifluoromethoxy phenol</strong>.To a mixture of 2.5 g of <strong>[461699-34-3]2-amino-4-trifluoromethoxy phenol</strong>, 2.62 g of triethylamine and 15 ml of DMF, 2.31 g of 4-isonicotinic acid chloride hydrochloride was added while ice-cooling. The reaction mixture was stirred for 3.3 hours. The reaction mixture was poured into water and precipitated crystals were filtered and dried under reduced pressure to give 2.19 g of N-[5-(trifluoromethoxy)-2-hydroxyphenyl]isonicotinamide.1H-NMR (DMSO-d6) delta: 8.78 (dd, J=4.4, 1.7 Hz, 2H), 7.86 (dd, J=4.4, 1.6 Hz, 2H), 7.80-7.77 (m, 1H), 7.10-7.05 (in, 1H), 6.99 (d, J=8.7 Hz, 1H)
With triethylamine; In N,N-dimethyl-formamide; for 3.3h;Cooling with ice;
To a mixture of 2.5 g of <strong>[461699-34-3]2-amino-4-trifluoromethoxy phenol</strong>, 2.62 g of triethylamine and 15 ml of DMF, 2.31 g of 4-isonicotinic acid chloride hydrochloride was added while ice-cooling. The reaction mixture was stirred for 3.3 hours. The reaction mixture was poured into water and precipitated crystals were filtered and dried under reduced pressure to give 2.19 g of N-[5-(trifluoromethoxy)-2-hydroxyphenyl]isonicotinamide.1 H-NMR (DMSO-d6) delta: 8.78 (dd, J=4.4, 1.7 Hz, 2H), 7.86 (dd, J=4.4, 1.6 Hz, 2H), 7.80-7.77 (m, 1H), 7.10-7.05 (m, 1H), 6.99 (d, J=8.7 Hz, 1H)
With triethylamine; In N,N-dimethyl-formamide; for 3.3h;Cooling with ice;
To a mixture of 2.5 g of <strong>[461699-34-3]2-amino-4-trifluoromethoxy phenol</strong>, 2.62 g of triethylamine and 15 ml of DMF, 2.31 g of 4-isonicotinic acid chloride hydrochloride was added while ice-cooling. The reaction mixture was stirred for 3.3 hours. The reaction mixture was poured into water and precipitated crystals were filtered and dried under reduced pressure to give 2.19 g of N-[5-(trifluoromethoxy)-2-hydroxyphenyl]isonicotinamide.1H-NMR (DMSO-de) 6: 8.78 (dd, J=4.4, 1.7 Hz, 2H), 7.86 (dd, J=4.4, 1.6 Hz, 2H), 7.80-7.77 (m, 1H), 7.10-7.05 (m, 1H), 6.99 (d, J=8.7 Hz, 1H)
With triethylamine; In N,N-dimethyl-formamide; for 3.3h;Cooling with ice;
To a mixture of 2.5 g of <strong>[461699-34-3]2-amino-4-trifluoromethoxy phenol</strong>, 2.62 g of triethylamine and 15 ml of DMF, 2.31 g of 4-isonicotinic acid chloride hydrochloride was added while ice-cooling. The reaction mixture was stirred for 3.3 hours. The reaction mixture was poured into water and precipitated crystals were filtered and dried under reduced pressure to give 2.19 g of N-[5-(trifluoromethoxy)-2-hydroxyphenyl]isonicotinamide.1 H-NMR (DMSO-d6) delta: 8.78 (dd, 5=4.4, 1.7 Hz, 2H), 7.86 (dd, J=4.4, 1.6 Hz, 2H), 7.80-7.77 (m, 1H), 7.10-7.05 (m, 1H), 6.99 (d, J=8.7 Hz, 1H)
With triethylamine; In N,N-dimethyl-formamide; for 3.3h;Cooling with ice;
To a mixture of 2.5 g of <strong>[461699-34-3]2-amino-4-trifluoromethoxy phenol</strong>, 2.62 g of triethylamine and 15 ml of DMF, 2.31 g of 4-isonicotinic acid chloride hydrochloride was added while ice-cooling. The reaction mixture was stirred for 3.3 hours. The reaction mixture was poured into water and precipitated crystals were filtered and dried under reduced pressure to give 2.19 g of N-[5-(trifluoromethoxy)-2-hydroxyphenyl]isonicotinamide. 1 H-NMR (DMSO-d6) delta: 8.78 (dd, 3=4 A, 1.7 Hz, 2H), 7.86 (dd, J=4.4, 1.6 Hz, 2H), 7.80-7.77 (m, 1H), 7.10-7.05 (m, 1H), 6.99 (d, J=8.7 Hz, 1H)
With triethylamine; In N,N-dimethyl-formamide; for 3.3h;Cooling with ice;
To a mixture of 2.5 g of <strong>[461699-34-3]2-amino-4-trifluoromethoxy phenol</strong>, 2.62 g of triethylamine and 15 ml of DMF, 2.31 g of 4-isonicotinic acid chloride hydrochloride was added while ice-cooling. The reaction mixture was stirred for 3.3 hours. The reaction mixture was poured into water and precipitated crystals were filtered and dried under reduced pressure to give 2.19 g of N-[5-(trifluoromethoxy)-2-hydroxyphenyl]isonicotinamide.1 H-NMR (DMSO-d6) delta: 8.78 (dd, J=4.4, 1.7 Hz, 2H), 7.86 (dd, J=4.4, 1.6 Hz, 2H), 7.80-7.77 (m, 1H), 7.10-7.05 (m, 1H), 6.99 (d, J=8.7 Hz, 1H)
With triethylamine In N,N-dimethyl-formamide at 20 - 50℃; for 3.5h;
71
Reference Production Example 71To a mixture of 0.80 g of 3-amino-2-hydroxy-6-trifluoromethylpyridine, 1.14 g of triethylamine and 10 ml of DMF, 0.88 g of isonicotinic acid chloride hydrochloride was added while ice-cooling. The reaction mixture was stirred at room temperature for one hour and further stirred while heating at 50°C for one hour. To the reaction mixture, 0.88 g of isonicotinic acid chloride hydrochloride and 1.1 g of triethylamine were added, and the reaction mixture was stirred while heating at 50°C for further 1.5 hours. The reaction mixture was cooled to room temperature, and water was added to the reaction mixture.Precipitated crystals were collected by filtration. The resultant solid was dissolved in ethyl acetate, then washed with a saturated sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was subjected to silica gel column chromatography to give 0.91 g of N-[2-hydroxy-6-(trifluoromethyl)pyridin- 3-yl]-isonicotinamide.-NMR (DMSO-de) δ: 9.98 (br s, 1H), 8.79 (dd, J=4.4, 1.5 Hz, 2H), 8.39 (d, J=7.8 Hz, 1H), 7.85 (dd, J=4.5, 1.6 Hz, 2H), 7.40-7.19 (m, 1H)
Synthesis of 4-tert-butylcalix[8]arene 1,2,3,5,6,7-hexaisonicotinate (3a).
Triethylamine (1.29 mL, 9.24 mmol) was added to a stirred mixture of 4-tert-butylcalix[8]arene (0.65 g, 0.5 mmol) and isonicotinoyl chloride hydrochloride (0.75 g, 4.2 mmol) in chloroform (240 mL) at room temperature. The reaction mixture was refluxed for 2 days. The solution was filtered, and then the filtrate was washed with water several times. The chloroform layer was dried using magnesium sulfate and filtered. Evaporation of the chloroform gave a crude product. The crude product was finally recrystallized in a mixture of nitrobenzene and acetonitrile (1 : 3) to obtain colorless needle-like crystals, 3a, in a 44% (0.62 g). IR (KBr): 3450(m), 2960(s), 1747(s), 1483(s), 1270(s), 1116(s), 754 (m) cm-1. 1H NMR (CDCl3): δ 8.69 (s, Py, 12H), 7.60 ~ 6.89 (br, Py, ArH, 28H), 4.42 ~ 3.47 (br, ArCH2Ar, 16H), 1.27 (s, -CH3, 18H), 1.20 (s, -CH3, 54H). Anal. Calcd. for C140H146N10O18 (C124H130N6O142(C4H8O2)2(CH3CN)): C, 74.51; H, 6.52; N, 6.21. Found: C, 74.50; H, 6.60; N, 6.21. FAB-mass: m/z = 1928.7 [M - e-]+, 1928), 1912.7 ([M - CH3 - e-]+, 1913).
The <strong>[82379-38-2]4-hydroxymethyl-3-nitrobenzoic acid</strong> (B) was dissolved in pyridine and reacted with isonicotinoyl chloride hydrochloride at room temperature for 2 days. The resultant solution was poured onto ice, stirred overnight at room temperature and extracted with ethyl acetate. The organics were dried over magnesium sulfate and rotary evaporated to remove the solvents. Residual pyridine was removed by the addition of toluene followed by rotary evaporation. The resultant solid was slurried in ethanol at room temperature and the product, 4-carboxy-2-nitrobenzyl isonicotinate, was isolated by filtration and dried under vacuum.
L-valine-N-(4-pyridylcarbonyl) methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
With triethylamine; In acetonitrile; at 5 - 20℃; for 3.25h;
Valine ethyl ester HCI (19 g, 0.1 1 mol) is suspended in acetonitrile (164 mL) and cooled to 10 C. Isonicotinic acid chloride HCI (28.2 g, 0.16 mol) is added to the mixture. Triethylamine (42.4 g, 0.42 mol) is added drop wise at 5 - 10C during 2.5 h. The red-brown solution is stirred at room temperature for 45 min and treated with water (50 mL) after complete conversion. Acetonitrile is evaporated under vacuum (50 mbar). Methyl isobutyl ketone (95 mL) is added to the aqueous phase and the pH is adjusted to 8.5 by addition of NaOH solution (50% w/w%). Additional water (32 mL) and methyl isobutyl ketone (32 mL) are added, the aqueous phase is separated and extracted with methyl isobutyl ketone (32 mL). The combined organic layers are washed three times with water (3 x 32 mL). For the washings, the pH of the aqueous phase is adjusted to 10. The solvent of the organic phase is evaporated and the red- brown, crystalline solid is dried at 50 C under vacuum. Yield: 24.0 g (90%). 1H-NMR (600 MHz, DMSO): delta = 8.92 (d, J = 7.8 Hz, 1 H), 8.74 (d, J = 4.8 Hz, 2H), 7.78 (d, J = 4.2 Hz, 2H), 4.32 (t, J = 7.8 Hz, 1 H), 3.67 (s, 3H), 2.21-2.17 (m, 1 H), 0.98 (d, J = 6.6 Hz, 3H), 0.94 (d, J = 7.2 Hz, 3H) ppm.
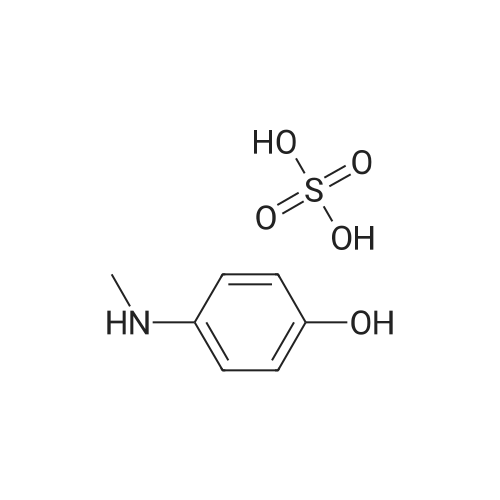
N-(4-hydroxyphenyl)-N-methylisonicotinamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
43%
With triethylamine; In tetrahydrofuran; at 0 - 50℃; for 23h;
To a suspension of <strong>[51-72-9]N-methyl-p-aminophenol sulfate</strong> (6.89 g,20.0 mmol) and isonicotinoyl chloride hydrochloride (7.12 g,40.0 mmol) in THF (120 mL) at 0 C, was added Et3N (12.14 g,16.74 mL, 120 mmol) dropwise over 5 min. The mixture was thenheated to 50 C for 23 h, after which it was cooled to RT, concentrated in vacuo and the crude residue purified by gradientcolumn chromatography (SiO2, flash, 0-100% (10% MeOH/5%Et3N/EtOAc)/EtOAc) to yield the title compound, N-(4-hydroxyphenyl)-N-methylisonicotinamide (10) after solvent removal, asa pale yellow solid (1.95 g, 8.54 mmol, 43%). 1H NMR (600 MHz,DMSO-d6) d 9.58 (s, 1H), 8.42 (d, J = 5.5 Hz, 2H), 7.15 (d,J = 5.5 Hz, 2H), 7.00 (d, J = 8.5 Hz, 2H), 6.60 (d, J = 8.5 Hz, 2H),3.30 (s, 3H). 13C NMR (151 MHz, DMSO-d6) d 167.56, 156.29,149.33, 144.34, 134.75, 128.72, 122.16, 115.69, 37.57. LC-MS(APPI+, H2O/MeCN/TFA, m/z) found 229 [M+H]+ calculated 229.1[M+H]+, rt (PDA) 0.21 min.
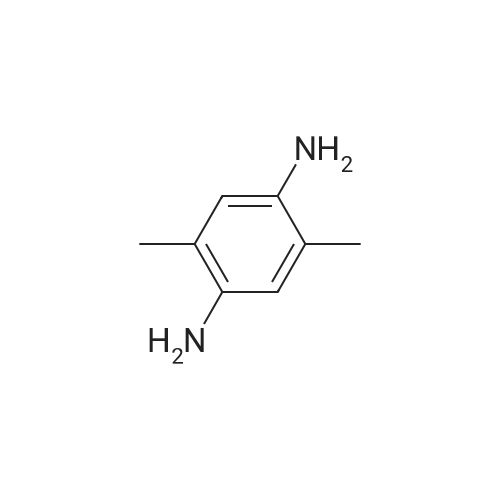
N,N'-(2,5-dimethyl-1,4-phenylene)diisonicotinamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76.77%
With pyridine; at 25℃; for 120h;
Isonicotinoyl chloride hydrochloride (2.94mmol) was reacted with <strong>[6393-01-7]2,5-dimethyl-1,4-phenylenediamine</strong> (1.47mmol) in 50ml of pyridine at 25C for five days. The colourless product so obtained was filtered, washed with water at least three times to remove the byproduct pyridine hydrochloride and dried in vacuo at 60C. Yield (76.77%). Anal. Calc. for C20H18N4O2: C, 69.35; H, 5.24; N, 16.17. Found: C, 69.29; H, 5.25; N, 16.18%. 1H NMR (DMSO-d6): delta 10.17 (s, 2H, NHCO), 8.80 (d, 4H, J=5.90Hz), 7.89 (d, 4H, J=5.60Hz), 7.28 (s, 2H), 2.22 (s, 6H) ppm. 13C NMR (DMSO-d6): delta 164.46, 150.82, 141.98, 134.29, 132.18, 128.91, 122.00, 17.85ppm. I.R (cm-1): nu(NH), 3287; nu(C=O), 1650. ESI-MS peak at m/z 347.14 corresponding to [M+H+].
76.77%
With pyridine; at 25℃; for 120h;
Isonicotinoyl chloride hydrochloride (2.94mmol) was reacted with <strong>[6393-01-7]2,5-dimethyl-1,4-phenylenediamine</strong> (1.47mmol) in 50mL of pyridine at 25C for five days. The colorless product so obtained was filtered, washed with water at least three times to remove byproduct pyridine hydrochloride and dried in vacuo at 60C. Yield (76.77%). Anal. Calc. for C20H18N4O2: C, 69.35; H, 5.24; N, 16.17. Found: C, 69.29; H, 5.25; N, 16.18. 1H NMR (DMSO-d6): delta 10.17 (s, 2H, NHCO), 8.80 (d, 4H, J=5.90Hz), 7.89 (d, 4H, J=5.60Hz), 7.28 (s, 2H), 2.22 (s, 6H) ppm. 13C NMR (DMSO-d6): delta 164.46, 150.82, 141.98, 134.29, 132.18, 128.91, 122.00, 17.85ppm. I.R (cm-1): nu(NH), 3287; nu(C=O), 1650. ESI-MS peak at m/z 347.14 corresponding to [M+H+].
Stage #1: methyl (4-pyridyl) ketone With lithium hexamethyldisilazane In toluene at 0℃; for 0.0166667h; Inert atmosphere;
Stage #2: isonicotinoyl chloride hydrochloride With acetic acid In tetrahydrofuran; ethanol; toluene Inert atmosphere;
General procedure for the synthesis of title compounds (7-27)
General procedure: Appropriate acetone (intermediates 1a-6a; 2 m mol) was dissolved in 5 ml of dry toluene in a 100-ml RBF with a septum, and the solution was cooled to 0 °C under nitrogen.LiHMDS (2.1 ml, 1.0 M in THF, 2.1 m mol) wasadded quickly via syringe with stirring. After 1 min.,substituted isonicotinoyl chloride hydrochloride (intermediates1b-6b; 1 m mol) was added in one portion withstirring. The flask was then removed from the ice bath andallowed to stand for 1 min, and then 2 ml of AcOH wasadded with stirring. EtOH (10 ml) and THF (5 ml) wereadded to form a homogenous mixture, and then hydrazinehydrate (2 ml, 1.1 g, and 34.43 m mol) was added. Themixture was allowed to auto reflux and was held at thattemperature for 5 min or until the reaction completes. Theresulting solution was added to 1.0 M NaOH solution andextracted with EtOAc. The organic fraction was thenwashed with brine, dried over Na2SO4, and evaporatedunder reduced pressure. The resulting residue was recrystallizedfrom 2-propanol/water to afford the product(compounds 7-27).
General procedure: Appropriate acetone (intermediates 1a-6a; 2 m mol) was dissolved in 5 ml of dry toluene in a 100-ml RBF with a septum, and the solution was cooled to 0 C under nitrogen.LiHMDS (2.1 ml, 1.0 M in THF, 2.1 m mol) wasadded quickly via syringe with stirring. After 1 min.,substituted isonicotinoyl chloride hydrochloride (intermediates1b-6b; 1 m mol) was added in one portion withstirring. The flask was then removed from the ice bath andallowed to stand for 1 min, and then 2 ml of AcOH wasadded with stirring. EtOH (10 ml) and THF (5 ml) wereadded to form a homogenous mixture, and then hydrazinehydrate (2 ml, 1.1 g, and 34.43 m mol) was added. Themixture was allowed to auto reflux and was held at thattemperature for 5 min or until the reaction completes. Theresulting solution was added to 1.0 M NaOH solution andextracted with EtOAc. The organic fraction was thenwashed with brine, dried over Na2SO4, and evaporatedunder reduced pressure. The resulting residue was recrystallizedfrom 2-propanol/water to afford the product(compounds 7-27).
General procedure: Appropriate acetone (intermediates 1a?6a; 2 m mol) was dissolved in 5 ml of dry toluene in a 100-ml RBF with a septum, and the solution was cooled to 0 °C under nitrogen.LiHMDS (2.1 ml, 1.0 M in THF, 2.1 m mol) wasadded quickly via syringe with stirring. After 1 min.,substituted isonicotinoyl chloride hydrochloride (intermediates1b?6b; 1 m mol) was added in one portion withstirring. The flask was then removed from the ice bath andallowed to stand for 1 min, and then 2 ml of AcOH wasadded with stirring. EtOH (10 ml) and THF (5 ml) wereadded to form a homogenous mixture, and then hydrazinehydrate (2 ml, 1.1 g, and 34.43 m mol) was added. Themixture was allowed to auto reflux and was held at thattemperature for 5 min or until the reaction completes. Theresulting solution was added to 1.0 M NaOH solution andextracted with EtOAc. The organic fraction was thenwashed with brine, dried over Na2SO4, and evaporatedunder reduced pressure. The resulting residue was recrystallizedfrom 2-propanol/water to afford the product(compounds 7?27).
(E)-1-(pyridin-4-yl)ethan-1-one O-isonicotinoyl oxime[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89%
With N-ethyl-N,N-diisopropylamine In tetrahydrofuran at 0 - 25℃; for 24h; Inert atmosphere;
3.2. Synthesis: General Procedure for the Synthesis of Oxime Ester Conjugates
General procedure: Parent amidoxime, ethanone oxime or aldoxime (2 mmol) was dissolved in THF or CHCl3(0.15 M) and Et3N or DIPEA (2.2 mmol) was added at 0 C under an Ar atmosphere, followed by the corresponding acid chloride (2.2 mmol). p-Pyridoyl chloride was commercially available as an HClsalt, therefore in those cases the amount of the amine used was doubled. The temperature was allowed to slowly rise to 25 C. The reaction was monitored by TLC and, upon completion, water (100 mL) was added and the mixture was extracted with dichloromethane (2 100 mL). The organic layers werefurther washed with water (100 mL), dried with Na2SO4, and the solvents were evaporated to dryness.The crude residue was then recrystallized, unless otherwise mentioned, to give the desired product,which was found to be sufficiently pure.
(E)-isonicotinaldehyde O-isonicotinoyl oxime[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
77%
With N-ethyl-N,N-diisopropylamine In tetrahydrofuran at 0 - 25℃; for 24h; Inert atmosphere;
3.2. Synthesis: General Procedure for the Synthesis of Oxime Ester Conjugates
General procedure: Parent amidoxime, ethanone oxime or aldoxime (2 mmol) was dissolved in THF or CHCl3(0.15 M) and Et3N or DIPEA (2.2 mmol) was added at 0 C under an Ar atmosphere, followed by the corresponding acid chloride (2.2 mmol). p-Pyridoyl chloride was commercially available as an HClsalt, therefore in those cases the amount of the amine used was doubled. The temperature was allowed to slowly rise to 25 C. The reaction was monitored by TLC and, upon completion, water (100 mL) was added and the mixture was extracted with dichloromethane (2 100 mL). The organic layers werefurther washed with water (100 mL), dried with Na2SO4, and the solvents were evaporated to dryness.The crude residue was then recrystallized, unless otherwise mentioned, to give the desired product,which was found to be sufficiently pure.
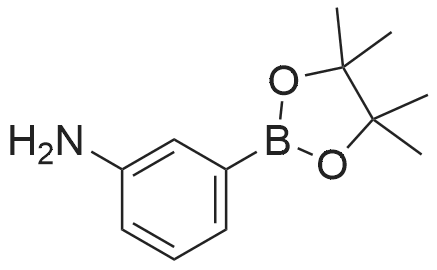
N-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)isonicotinamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
62%
With triethylamine In chloroform at 20℃;
174.174A Example 174A [0776] N-(3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)isonicotinamide
[0777] To a mixture of 3-aminophenylboronic acid pinacol ester (43 mg, 0.20 mmol) in a 1-dram vial was added dry CHCI3 (0.80 mL) followed by nicotinyl chloride hydrochloride (37 mg, 0.21 mmol) followed by TEA (64 mg, 0.088 mL, 0.63 mmol) dropwise at rt. The reaction mixture was stirred overnight at rt. LCMS showed one peak for the desired product mass. The reaction mixture was diluted with water and extracted with EtOAc. The aqueous layer was separated and extracted once with EtOAc. The organic layers were combined, dried over sodium sulfate, decanted from the drying agent, and concentrated in vacuo to give dark yellow oil. This oil was dissolved in DCM and subjected to column chromatography on silica gel to give 39.7 mg, 62%, of the title compound as a yellow oil. MS (ES+) m/e 325 (M+H)+.
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride In tetrahydrofuran at 23℃;
2 Synthesis of compound [MA6]
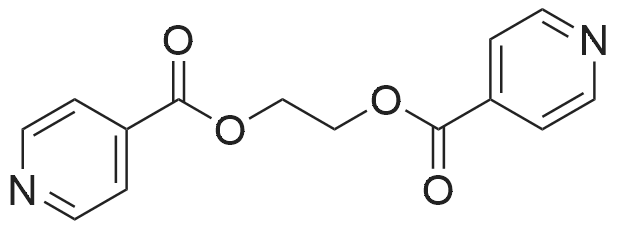
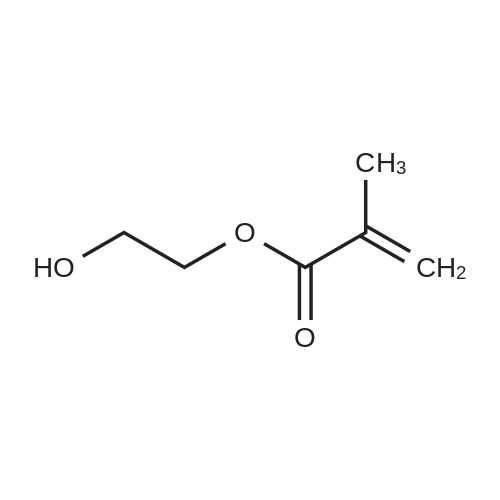
Add 2-hydroxyethyl methacrylate to a 1L four-neck flask[MA6-1] (63.42 g, 487 mmol),Isonicotinic acid salt [MA6-2] (50.00 g, 406 mmol),1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (hereinafter abbreviated as EDC) (93.43 g, 487 mmol), 4-dimethylaminopyridine (hereinafter abbreviated as DMAP) (4.96g, 40.6mmol),THF (500 g) was reacted at 23°C.Reaction tracing by HPLC,After confirming the end of the reaction,The reaction solution was poured into distilled water (3 L) and ethyl acetate (1 L) was added.The aqueous layer was removed by a liquid separation operation.After washing the organic layer with distilled water (1L) twice,The organic layer was dried over magnesium sulfate.after that,By filtering,Distill the solvent with an evaporator,86.3 g of compound [MA6] was obtained as an oily compound (yield 93%).
With pyridine In tetrahydrofuran at 20 - 65℃; for 26h;
2.3. Synthesis of the monomers
The synthetic route of LC monomers [1,1′-biphenyl]-4,4′-diyldiisonicotinate (BDI) and cyclohexane-1,4-diyl diisonicotinate (HDI) are shown in Scheme 1. The detailed synthesis and characterization ofBDI were reported in the previous work [33]. The experimental detailsand results of compound BDIwere consistentwith the report. The compoundHDIwas synthesized using cyclohexane-1,4-diol and isonicotinicacid. Isonicotinic acid (36.93 g, 0.3 moL) and thionyl chloride (70 mL)was added into a round flask and stirred at room temperature for 2 h,then heated at 65 °C for 25 h in awater bath. The excess thionyl chloridewas distilled out under reduced pressure to obtain 40.6 g isonicotinoylchloride hydrochloride. Cyclohexane-1,4-diol (8.13 g, 0.07 moL) and20 mL of pyridine were dissolved in 50 mL freshly distilled THF in athree-necked flask. Isonicotinoyl chloride hydrochloride (25 g,0.14 moL) was dissolved in 100 mL of THF and then added dropwiseinto the flask. Subsequently, stirred at room temperature for 2 h, thesemixed solutions reacted at reflux temperature for 24 h. The solventwas removed under reduced pressure after the reaction finished.Then, the mixture was poured into 1000 mL of sodium bicarbonate solution.The precipitates were filtrated and washed with water and recrystallizedin ethanol to obtain 20.5 g white crystals of compoundHDI. Yield: 90%. FTIR (KBr, cm-1): 2935, 2866 (CH2); 1730(C_O); 1594-1399 (C_C, C_N in pyridine); 1264 (CN in pyridine);1176 (COC). Anal. calc. for HDI: C, 66.21%; H, 5.50%; N, 8.52%. 1HNMR (300 MHz, DMSO): δ = 1.46-2.04 (m, 8H,CH2), 4.53-5.07(m, 2H,CHO), 7.89 (d, J = 6 Hz, 4H, pyridyl-H), 8.76 ppm (d, J= 9 Hz, 4H, pyridyl-H). 13C NMR (75 MHz, DMSO): δ = 26.72, 26.86(CH2); 72.45, 72.80 (CHO); 124.04, 124.74 (C_C inpyridyl);141.84, 142.07 (O_CC_C in pyridyl); 148.66, 149.19 (C_N in pyridyl);163.42, 163.59 ppm (O_C).
With triethylamine; In dichloromethane; at 20℃; for 2.25h;
To isoleucine tert-butyl ester HCl (H-Ile-OtBu HCl, 0.671 g, 3 mmol) in DCM (50 mL) was added Et3N (1.821 g, 18 mmol). Isonicotinoyl chloride hydrochloride (0.961 g, 5.4 mmol) was added in 4 portions over 15 min in a water bath (room temperature). The reaction was stirred for 2 hr. Subsequently, 5% of aq. NH4Cl (20 mL) was added to quench the reaction. The DCM layer was dried over Na2SO4. The product was purified by silica gel column chromatography (4-5% MeOH/DCM). 0.780 g of 7t was obtained as syrup. The yield was 90%.
With triethylamine In dichloromethane at 0 - 50℃; for 24h; Inert atmosphere;
2.3. Synthesis of Azobenzene-4-pyridinecarboxylic Acid methyl Ester (Compound, 1)
4-Hydroxyazobenzene (2.97 g, 15 mmol) was dissolved in CH2Cl2 (40 mL), and then 4.2 mL (3.03 g, 30 mmol) of dry Et3N was added under nitrogen. The reaction mixture was cooled to 0 C before isonicotinoyl chloride hydrochloride (3.20 g, 18 mmol) in 30 mL of CH2Cl2 was added dropwise. The reaction mixture was then warmed to room temperature and stirred at 50 C for further 24 h. After cooling to room temperature, the mixture was filtered, the filtrate was washed six times with water (6 × 60 mL), and the organic layer was dried over anhydrous MgSO4. Solvent was then removed under reduced pressure afforded 4.12 g of compound, 1 as a red-brown powder in 90.6% yield. 1 H NMR (CDCl3), δ (ppm): 8.93-8.87 (d, 2H, o-ArHNArH), 8.08-7.95 (m, 4H, oArN- -NArH + m-ArHNAr), 7.95-7.81, (m, 4H, m-OCOArHN- -NAr), 7.57-7.45 (t, 3H, p-ArN- -NArH + m-ArN- -NArH), 7.44-7.37, (d, 2H, oArOCOArH). EI/MS: Calcd. for C18H13N3O2: 303.10; found: 303.12. Anal. calcd for C: 71.28, H: 4.32, O: 10.55, N: 13.85; found C: 71.24, H: 4.32, O: 10.60, N: 13.84.
With triethylamine In dichloromethane at 20 - 23℃; for 16h;
3.4.1. General Procedure for the Synthesis of Compounds 2a-f
General procedure: Quinine (2.6 g, 0.008 mol) was dissolved in 100 mL of dry dichloromethane. Then,1.0 g (0.01 mol) of triethylamine and 0.009 mol of 1,2-azole-3- (2a-c) or adamantane- (2f)carbonyl chlorides were successively added to the resulting solution under stirring. Themixture was stirred for 1 h and left for 15 h at 20-23 C. The mixture was washed withwater (2 200 mL, 1 h stirring) and 5% sodium bicarbonate solution (2 200 mL, 1 hstirring). The organic layer was separated and dried over anhydrous Na2SO4. The solventwas removed, and the residue was crystallized from a mixture of ether and hexane (1:1).The procedure for the synthesis of quinine esters with a pyridine fragment (2d,e) issimilar to the previous one, except for the amount of the used triethylamine. Here, 2.6 g(0.008 mol) of quinine, 2.4 g (0.024 mol) of Et3N and 1.6 g (0.009) mol of hydrochloride ofnicotinic or isonicotinic carbonyl chlorides were taken into the reaction.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +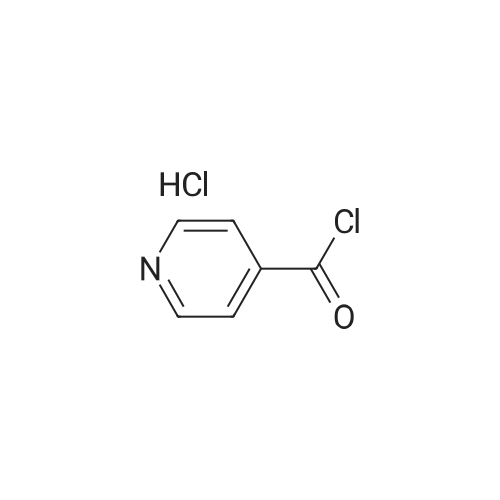

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping