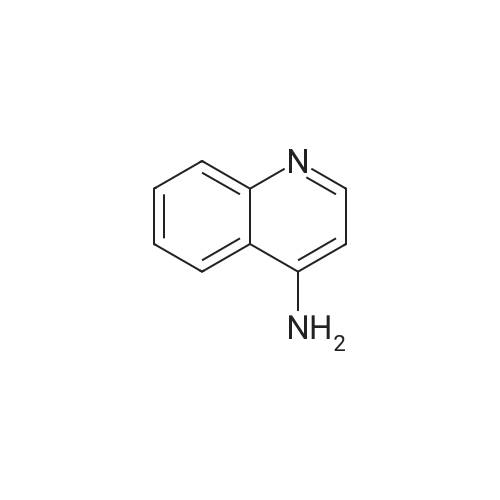
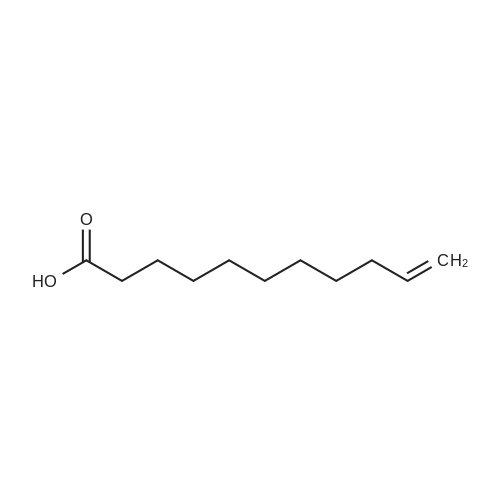
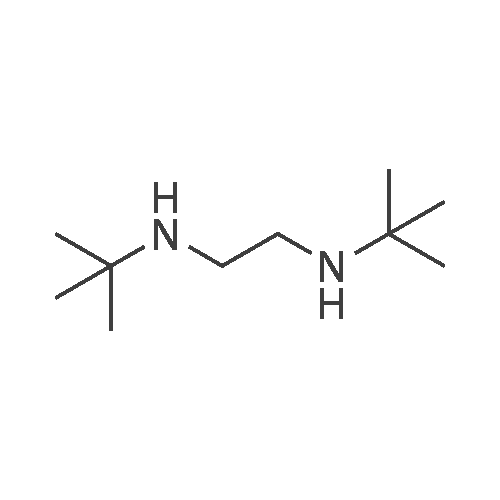
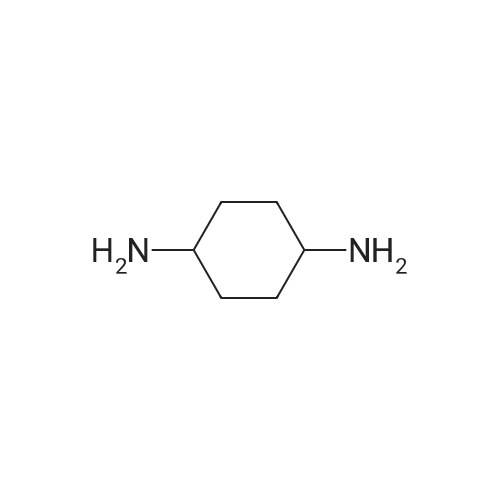
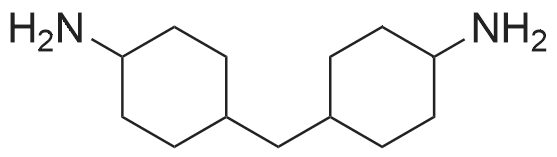
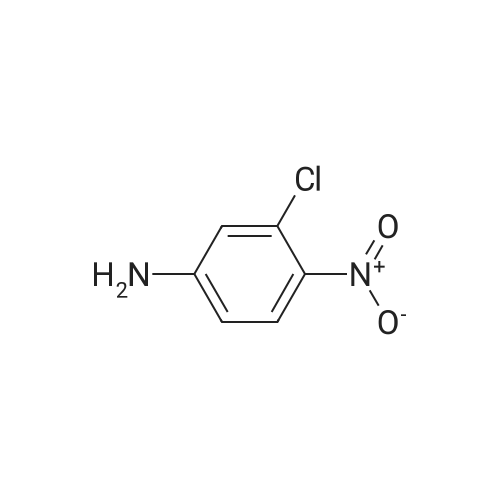
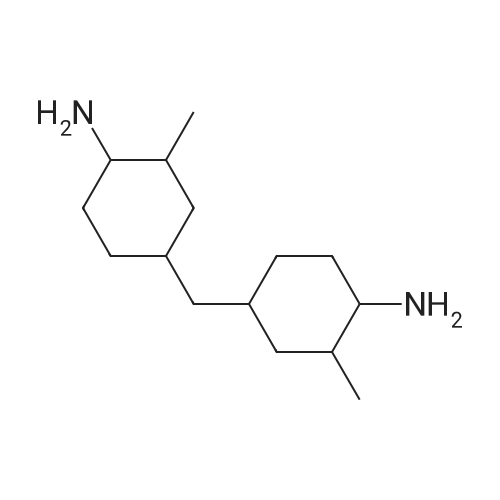
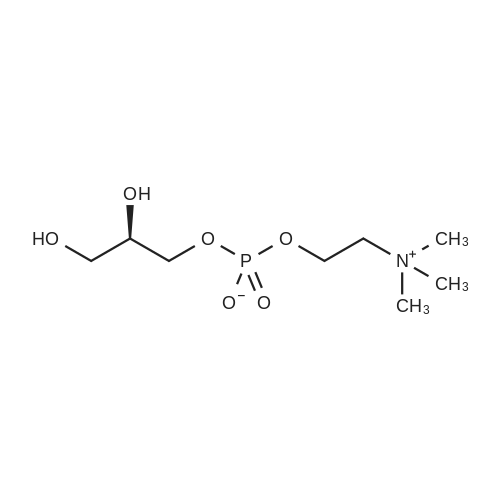
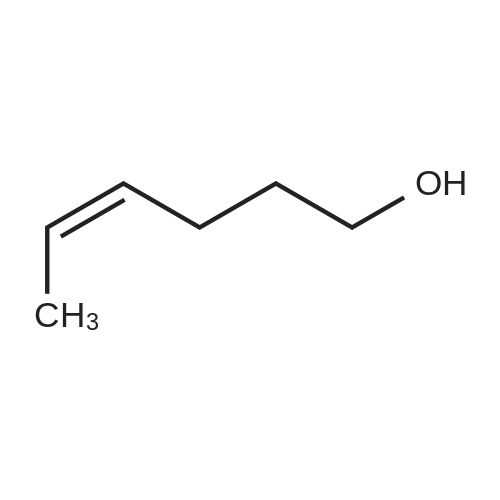
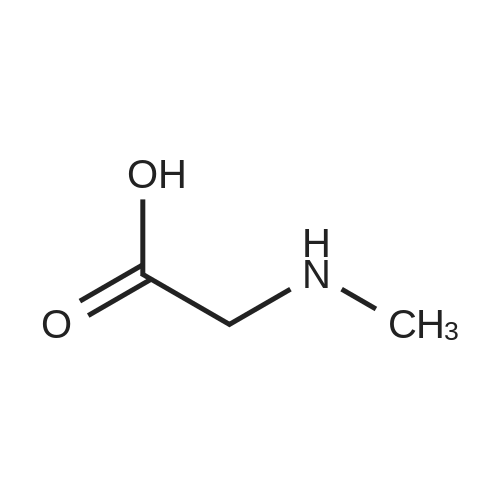
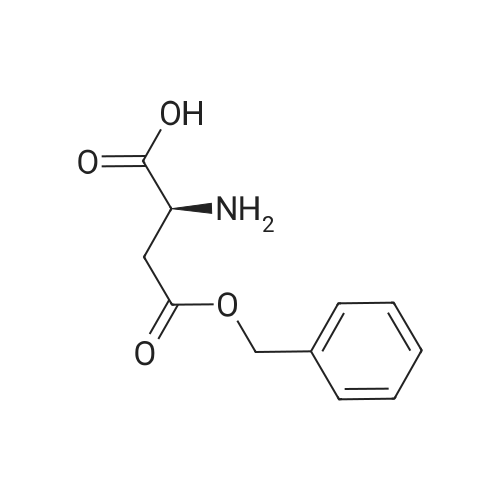
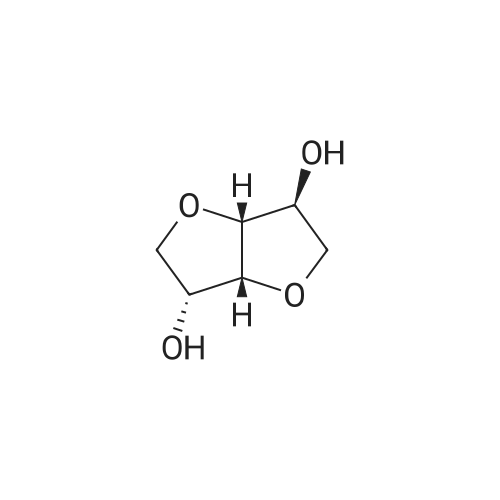
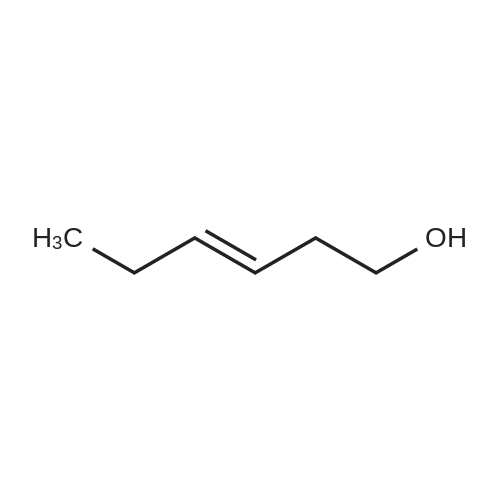
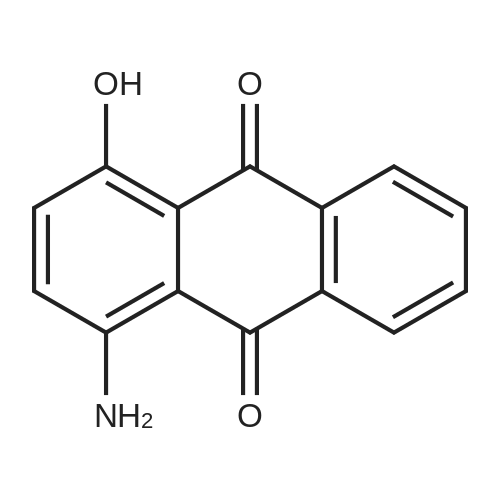
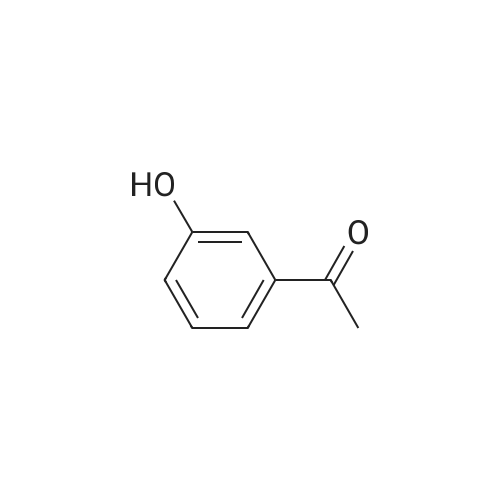
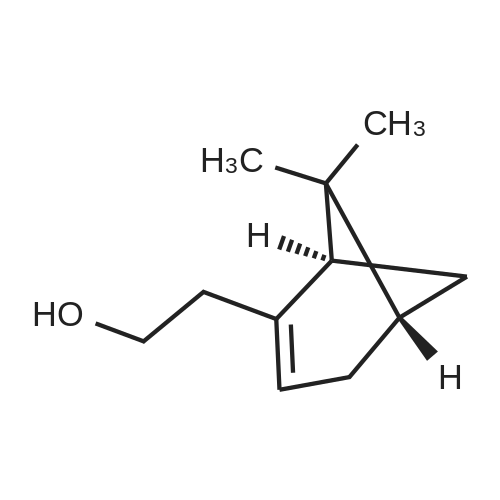
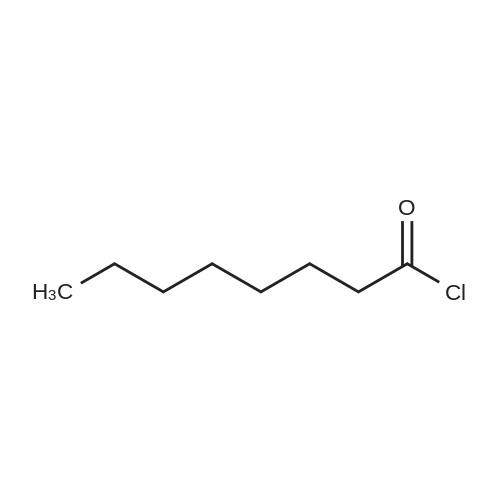
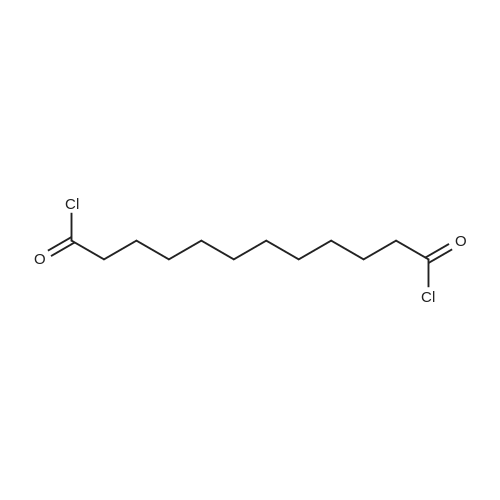
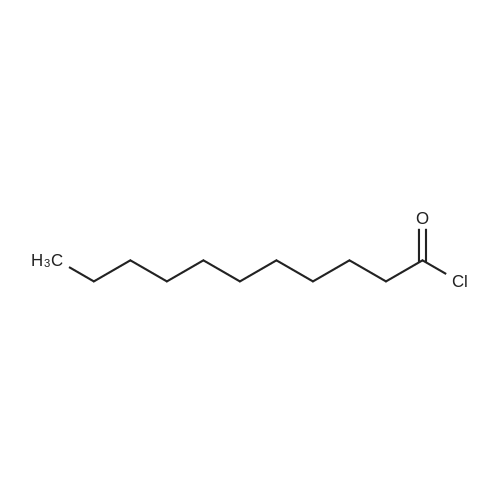
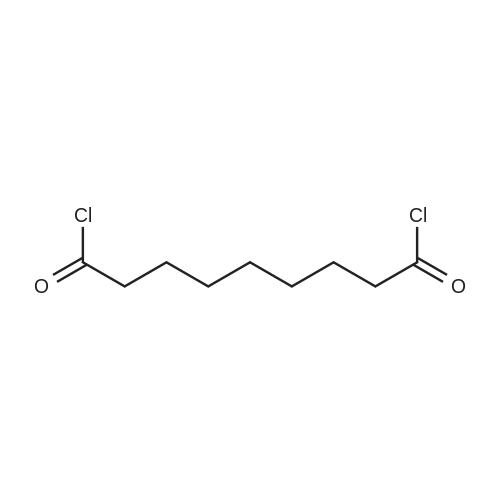
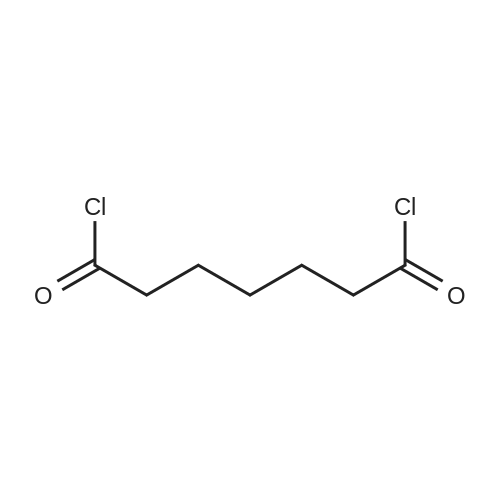
| 88% |
With thionyl chloride for 2h; Heating; |
|
| 84% |
With thionyl chloride In dichloromethane for 4h; Reflux; |
|
| 83% |
With thionyl chloride; N,N-dimethyl-formamide at 80℃; for 10h; Inert atmosphere; Sealed tube; |
|
| 83% |
With thionyl chloride; N,N-dimethyl-formamide at 80℃; for 10h; Inert atmosphere; Schlenk technique; |
|
| 61% |
With thionyl chloride at 20 - 60℃; for 5h; |
|
| 55% |
With thionyl chloride; hydroquinone at 65℃; for 6h; |
3.2.2.1 (2.1) Preparation of intermediate undecenoyloxybenzoyl chloride:
Dissolve 18.43g (0.10mol) of undecylenic acid and 16mL (0.20mol) of thionyl chloride in a 100mL three-necked flask, add a small amount of hydroquinone as a polymerization inhibitor, heat to reflux at 65°C for 6h, after the reaction is complete At 65°C, vacuum distillation was performed to remove excess thionyl chloride. The temperature was continued to increase, and the fractions at 139.0157.0°C/1.30KPa were collected to obtain the product undecenoyl chloride with a yield of 55%. In a 250mL three-necked flask, dissolve 16.57g (0.12mol) of p-hydroxybenzoic acid in 150mL of THF and 9.5mL of pyridine, stir until clear, and slowly add 0.10mol of undecenoyl chloride in THF solution at room temperature. Reaction for 10h, heating and refluxing for 10h. After the reaction is completed, pour the reaction solution into 1000 mL of water, add 37% concentrated hydrochloric acid to acidify the system, make the system appear strongly acidic, let stand overnight, filter with suction, wash the filter cake to neutral, then wash with hot water, and wait for the filter cake After drying, it was recrystallized with ethanol to obtain powder undecenoyloxy benzoic acid with a yield of 80%. In a 100mL round bottom flask, dissolve 9.13g (0.03mol) of undecenoyloxybenzoic acid in 20mL of thionyl chloride, and heat to reflux for 6h at 65°C in an oil bath. After the reaction is completed, vacuum distillation is carried out. A light yellow transparent liquid undecenoyloxy benzoyl chloride was obtained with a yield of 68%. |
|
With thionyl chloride |
|
|
With phosphorus pentachloride |
|
|
With thionyl chloride In benzene |
|
|
With oxalyl dichloride |
|
|
With thionyl chloride; N,N-dimethyl-formamide at 40℃; for 2h; |
|
|
With phosphorus pentachloride In benzene Heating; |
|
|
With oxalyl dichloride for 3h; |
|
|
With thionyl chloride for 12h; Heating; |
|
|
With oxalyl dichloride In toluene |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane |
|
|
With thionyl chloride for 4h; Ambient temperature; |
|
|
With oxalyl dichloride In dichloromethane at 20℃; for 2h; |
|
|
With thionyl chloride In dichloromethane for 10h; Heating; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane for 1h; |
|
|
With oxalyl dichloride In dichloromethane at 20℃; for 1h; |
|
|
With oxalyl dichloride In dichloromethane at 20℃; for 2h; |
|
|
With thionyl chloride at 80℃; for 2h; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0 - 25℃; Inert atmosphere; |
|
|
With phosphorus trichloride at 70℃; for 2h; |
|
|
With thionyl chloride |
|
|
With phosphorus trichloride at 70℃; for 3h; |
4.1.8. General procedure for the synthesis of aurone fatty acid esters
General procedure: A solution of fatty acid (3.75 mmol) and PCl3 (1.5 mmol) was stirred at 70 °C for 3 h, and a yellow solution containing fatty acid chloride could be gotten.A solution of aurone (1.86 mmol) and anhydrous pyridine (1 mL) in CH2Cl2 (10 mL) was stirred at boiling temperature, and the above reaction mixture was added slowly. Then the reaction continued for 1-2 h at boiling temperature. Washing with dilute hydrochloric acid (10% v/v aqueous solution), saturated NaHCO3 solution, drying (Na2SO4) and removal of solvent under reduced pressure gives the crude product. The product was further purified either by flash column chromatography or recrystallization. |
|
With oxalyl dichloride In tetrahydrofuran at 0℃; for 2h; Inert atmosphere; |
|
|
With thionyl chloride In toluene for 1.5h; Reflux; |
23
A mixture of toluene (12 mL) and SOCl2 (7.75 g, 65 mmol) and undec-10-enoic acid (1.00 g, 5.43 mmol) were refluxed for 1.5 h. The solvent and remaining SOCl2 were distilled off and the product (X) was further used in the thioester synthesis. |
|
With oxalyl dichloride; N,N-dimethyl-formamide In tetrahydrofuran at 0 - 23℃; for 1.08333h; Inert atmosphere; |
|
|
With thionyl chloride at 55℃; |
|
|
With thionyl chloride for 1.5h; Reflux; |
|
|
With thionyl chloride for 1.5h; Reflux; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0℃; for 3h; Inert atmosphere; |
General procedure for the synthesis of fatty acid chlorides (10a-h): General Procedure:
General procedure: To a stirred solution of fatty acid in DCM, catalytic amount of DMF was added. The contents were stirred at 0 °C. Oxalyl chloride was added under nitrogen atmosphere. The reaction mixture was further stirred at 0 °C for 3 h. Then reaction mixture was concentrated under reduced pressure to remove dichloromethane and excess of oxalyl chloride traces. Later crude acid chloride dissolved in DCM was used for the next step directly under nitrogen atmosphere. |
|
With thionyl chloride for 3h; Heating; |
9 Reference Production Example 9 (Trans-10-undecenoic acid methyl ester, compound 22)
Trans-10-undecenoic acid was dissolved in thionyl chloride (10 ml) and treated in water bath for 3 hours to distill off excess thionyl chloride. Methanol (30 ml) was added to chloride of trans-10-undecenic acid and refluxed in water bath for 2 hours. After cooling, the reaction mixture was added to 1N HCl (80 ml) to be acidic and distributed with EtOAc. The distributed liquid was purified with column chromatography (developing solvent: C6H14-EtOAc (3:1)) after concentration, and trans-10-undecenoic acid methyl ester shown in the following was isolated. Colorless oily substance, C12H22O2 MW 198, EIMS m/z (%): 198 (M+, 0), 166 (M+ - CH3OH, 22), 149 (13), 124 (36), 87 (53), 74 (100) |
|
With thionyl chloride; N,N-dimethyl-formamide; sodium hydroxide In dichloromethane; water for 1.5h; Reflux; |
|
|
With thionyl chloride for 1.5h; Reflux; |
|
|
With thionyl chloride for 2h; Reflux; |
1
10-undecenoic acid (3.7 g, 0.02 mol), the thionyl chloride and (8 mL) was added, and refluxed in water bath for 2 hours.Then, the excess of thionyl chloride was distilled off under reduced pressure to give the 10-undecenoic acid chloride |
|
With oxalyl dichloride In N,N-dimethyl-formamide at 20℃; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0℃; for 3h; Inert atmosphere; |
General Procedure for the Synthesis of Fatty Acid Chlorides (2a-2j)
To a stirred solution of fatty acid in dichloromethane (DCM), catalytic amount of dimethyl formamide (DMF) was added and the contents were stirred at 0 °C. Oxalyl chloride was added under nitrogen atmosphere and the reaction mixture was further stirred at 0 °C for 3 h. Then reaction mixture was concentrated under reduced pressure to remove DCM and excess oxalyl chloride. Later crude acid chloride dissolved in DCM was used for the next step directly under nitrogen atmosphere. |
|
With thionyl chloride In N,N-dimethyl-formamide at 80℃; for 4h; Inert atmosphere; |
|
|
With thionyl chloride; N,N-dimethyl-formamide In toluene at 70℃; for 3h; |
|
|
With oxalyl dichloride at 20 - 35℃; for 3.5h; Inert atmosphere; Green chemistry; |
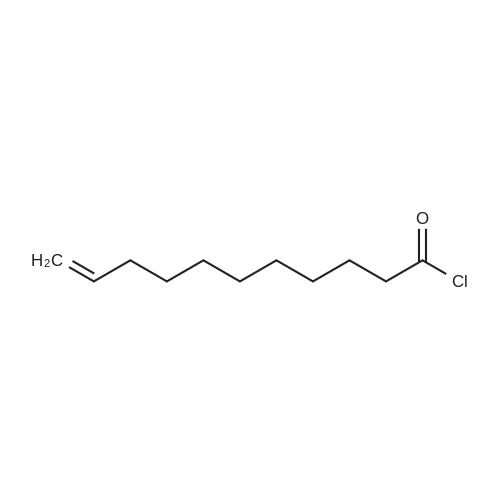
21 Synthesis of undecylenoyl chloride by chlorination of undecylenic acid Using Catalyst of Example 9
To a stirred mixture of undecylenic acid (37 g,200.8 mmol) and the polymeric catalyst of Example 9 (0.75 g) at room temperature under nitrogen atmosphere, oxalyl chloride (28 g, 220.5 mmol) is added over 30 minutes. The reaction is continued at 30-35° C. and the progress of the reaction is monitored by estimating the unconverted fatty acid. The reaction was stopped after 3 hours when the free undecylenic acid was found to be less than 1.0% by weight. |
|
With oxalyl dichloride In dichloromethane; N,N-dimethyl-formamide at 20 - 22℃; for 1h; |
|
|
With thionyl chloride |
|
|
With thionyl chloride In dichloromethane at 10℃; |
2.1 Step one amidation
At room temperature, the reaction vessel was added 1250ml of dichloromethane and 50g 10-undecenoic acid, while stirring 129.1g of thionyl chloride was added dropwise, the reaction was completed at 10 ° C, the reaction was completed, concentration, to obtain A; |
|
With thionyl chloride In tetrahydrofuran for 2h; Reflux; |
|
|
With oxalyl dichloride In dichloromethane at 0 - 20℃; for 3.16667h; Inert atmosphere; |
|
|
With thionyl chloride |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0 - 20℃; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0 - 20℃; |
Acyl chlorides
General procedure: The acid (5 mmol) was dissolved in anhydrous CH2Cl2 (10 mL) and DMF (a few drops) added.Oxalyl chloride (6 mmol, 1.2 equiv.) was added dropwise to the solution, that was cooled in an icewater bath. The resulting mixture was allowed to stir at room temperature for an additional 4 h andthe solvent was evaporated to afford the crude acyl chloride, which was used directly in the nextstep. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping