* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
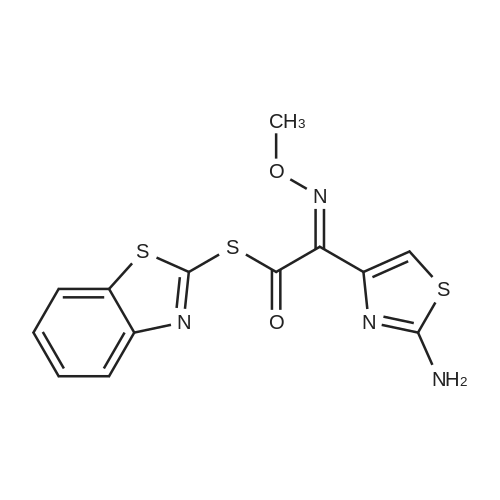
Stage #1: With dmap; dicyclohexyl-carbodiimide In ethanol at 20℃; for 0.5 h; Stage #2: With 1-hydroxy-pyrrolidine-2,5-dione In ethanol at 20℃; for 0.5 h;
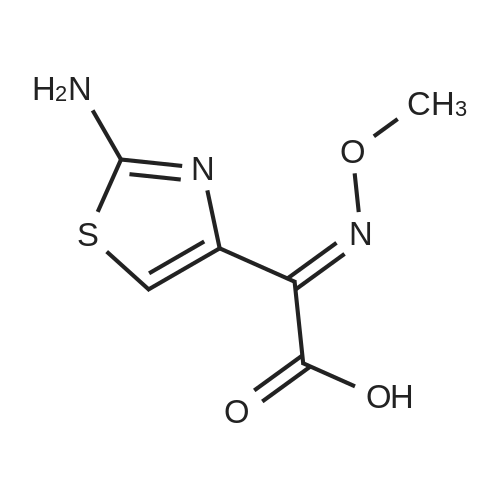
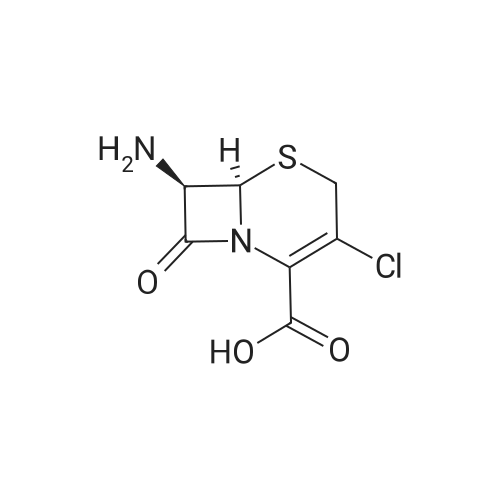
The cefotaxime sodium was prepared according to the preparation method described in Chinese Patent CN 102807573 A.2.05 g of 2- (2-amino-4-thiazolyl) -2-methoxyiminoacetic acid was dissolved in 20 ml of ethanol,Adding 1.85 g of 4-dimethylaminopyridine and 3.05 g of dicyclohexylcarbodiimide, and the mixture was stirred at room temperature for 30 min,Adding 1.20 g of N-hydroxysuccinimide and stirring at room temperature for 30 min,40 ml of saturated ammonium chloride solution was added, and the mixture was extracted with 40 ml of ethyl acetate. The ethyl acetate layer was separated,The organic phase was washed with saturated brine,Dried over anhydrous sodium sulfate, the solvent was evaporated, and the column chromatography (ethyl acetate / methanol = 10: 1)The active ester was 2.75 g. 2.75 g of the active ester was dissolved in 20 ml of dichloromethane,Join7-amino-none-3-Cephalosporin ring-4-carboxylic acid1.92 g and triethylamine (10 ml). After stirring for 8 h at room temperature,The pH was adjusted to 4.5 with 1 mol / L hydrochloric acid solution and reacted at -5 ° C for 2 h. Dichloromethane extraction, activated carbon decolorization, drying and then concentrated 3.04g cefotaxime. 3.04 g of ceftizole was dissolved in 10 ml of ethyl acetate,A 20 ml of ethyl acetate in which 5 g of sodium bicarbonate was dissolved was added dropwise,25 stirring crystallization, 30 under reduced pressure drying white powder 2.59g.
Reference:
[1] Patent: CN106279208, 2017, A, . Location in patent: Paragraph 0051; 0052; 0053
With trimethylsilyl bromide; In dichloromethane; at 20 - 25℃; for 5.5h;
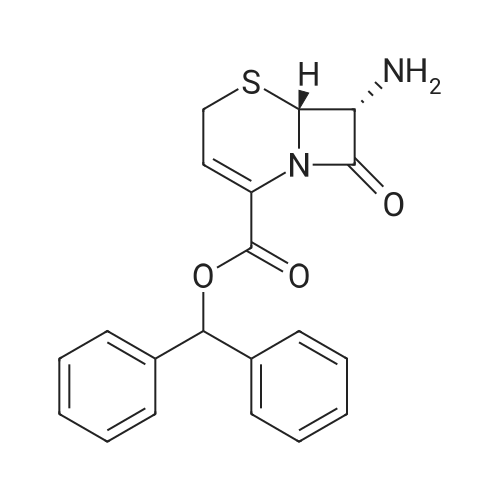
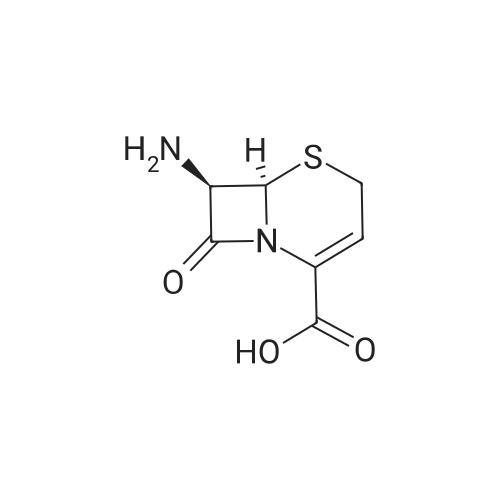
1000mL four-necked flask into 500mL methylene chloride, 37.18g (0.10mol) Compound B, temperature 20 ~ 25 , solution of 32.15g (0.21mol) of trimethylsilyl bromide in 30 minutes, dropping was completed, the temperature 20 ~ 25 , stirring reaction 5h, HPLC detection of compound B <1%, the reaction completed. Cooling to -5 ~ 0 C, add pre-cooled 300mL 10% hydrochloric acid solution (0 ~ 5 C), temperature 0 ~ 5 C, stirring 30 minutes, separated from the water phase, with 100mL dichloromethane stirring washing 10 minutes, Phase, add 1.00g activated carbon decolorization for 30 minutes, filter, filter cake with 100mL 10% hydrochloric acid leaching, combined filtrate and lotion. Temperature control 0 ~ 5 , 28% drop of ammonia to adjust the pH value between 3.5 to 4.0. After the pH value was adjusted, the temperature was controlled at 0 ~ 5 and stirred for 1h. The filter cake was rinsed with 100mL of purified water. The filter cake was vacuum dried at 40 ~ 50 for 6h to obtain 17.82g 7-ANCA. The yield of the powder was 89.0% and the purity was 99.1%.
The cefotaxime sodium was prepared according to the preparation method described in Chinese Patent CN 102807573 A.2.05 g of 2- (2-amino-4-thiazolyl) -2-methoxyiminoacetic acid was dissolved in 20 ml of ethanol,Adding 1.85 g of 4-dimethylaminopyridine and 3.05 g of dicyclohexylcarbodiimide, and the mixture was stirred at room temperature for 30 min,Adding 1.20 g of N-hydroxysuccinimide and stirring at room temperature for 30 min,40 ml of saturated ammonium chloride solution was added, and the mixture was extracted with 40 ml of ethyl acetate. The ethyl acetate layer was separated,The organic phase was washed with saturated brine,Dried over anhydrous sodium sulfate, the solvent was evaporated, and the column chromatography (ethyl acetate / methanol = 10: 1)The active ester was 2.75 g. 2.75 g of the active ester was dissolved in 20 ml of dichloromethane,Join7-amino-none-3-Cephalosporin ring-4-carboxylic acid1.92 g and triethylamine (10 ml). After stirring for 8 h at room temperature,The pH was adjusted to 4.5 with 1 mol / L hydrochloric acid solution and reacted at -5 C for 2 h. Dichloromethane extraction, activated carbon decolorization, drying and then concentrated 3.04g cefotaxime. 3.04 g of ceftizole was dissolved in 10 ml of ethyl acetate,A 20 ml of ethyl acetate in which 5 g of sodium bicarbonate was dissolved was added dropwise,25 stirring crystallization, 30 under reduced pressure drying white powder 2.59g.
(1) Add 50g of <strong>[36923-17-8]7-ANCA</strong> to 1L of chloroform, stir for 10min, slowly add 100ml of triethylamine, stir for20min, then add 96.5g of AE-active ester at 35 C for 4.5h, the reaction is completed, reduce the temperature to 15 C, 2L of water extraction,separation of the aqueous phase, 10g of activated carbon adsorption was stirred 20min, filtered, washed and the combined filtrate with 2mol / L hydrochloric acid solution was adjustedsection 2.2,15 PH value to crystallize with stirring 2h, filtered, washed with chloroform , dried at 40 C for 20 min under vacuum to obtain ceftizoxime acid 87.0 g;
<strong>[36923-17-8]7-ANCA</strong> 20.02 g (0.1 mol), dichloromethane 150 ml, and 4-dimethylaminopyridine 0.2 mol were sequentially added to the reactor to be stirred and dissolved, and the AE active ester 38.52 g was added dropwise at -5 to 0 C ( 0.11 mol), after 15 minutes of addition, add 4-dimethylaminopyridine 0.05 mol,Keep the temperature conditions unchanged, stir the reaction for 3-4h, after the reaction is over,Water is added at room temperature for stratification, the organic phase is separated, and the aqueous phase is retained.Adding dichloromethane to wash, then adding activated carbon to decolorize, adding hydrochloric acid to crystallize,Centrifugation and drying gave 37.13 g of ceftizozoate, the yield was 97%, and the purity was 99.8%.The maximum single impurity is 0.05%, and the total impurity is 0.18%.
With potassium hydroxide; In methanol; water; at 25 - 30℃; for 5h;pH 8 - 8.5;
Phase 500mL four-necked flask was charged with methanol (196.86g), water (65.62g) and compound B (32.81g, 0.10mol),Stir, control temperature 25 30 , add 10% KOH aqueous solution dropwise, control pH value 8.0 8.5,After the pH value was stable, the reaction was incubated for 5 hours. Compound B <1% was detected by HPLC.The temperature is controlled at 15 25 , and the pH value of the solution after the reaction is adjusted to 3.0 3.5 with 10% hydrochloric acid aqueous solution.After the pH value is in place, continue to stir for 1 h, suction filter, and dry the resulting filter cake at 50 C under vacuum for 6 h7-ANCA (18.19 g) was obtained with a molar yield of 90.85% and a HPLC purity of 99.86%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping