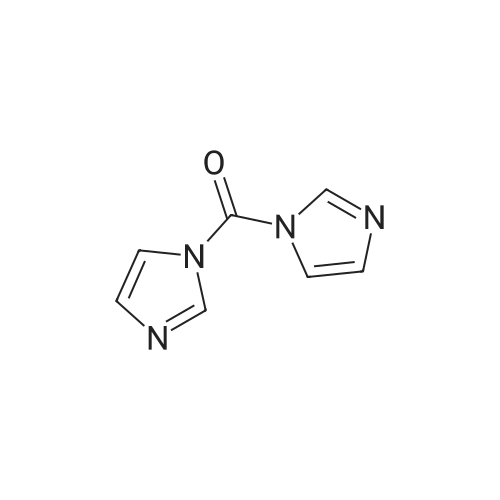
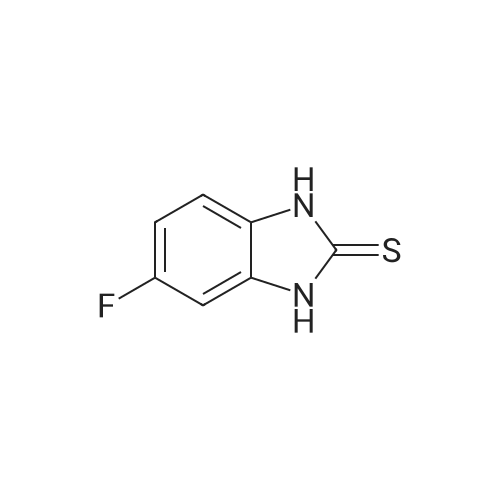
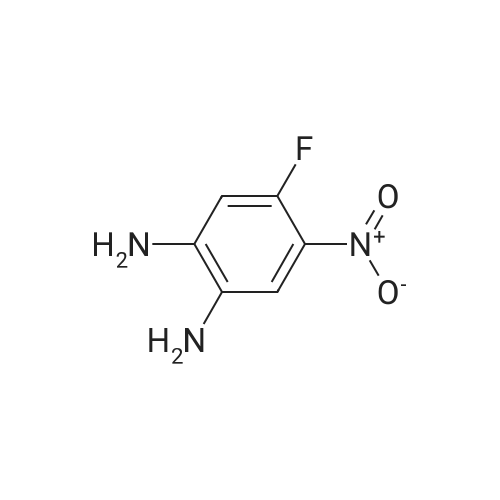
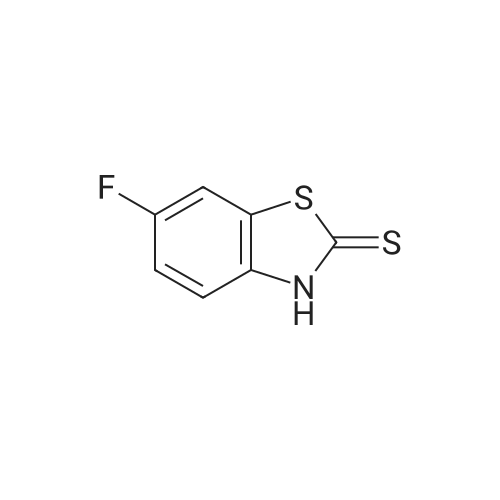
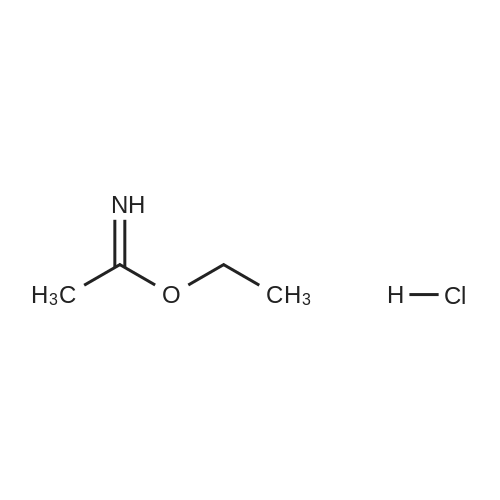
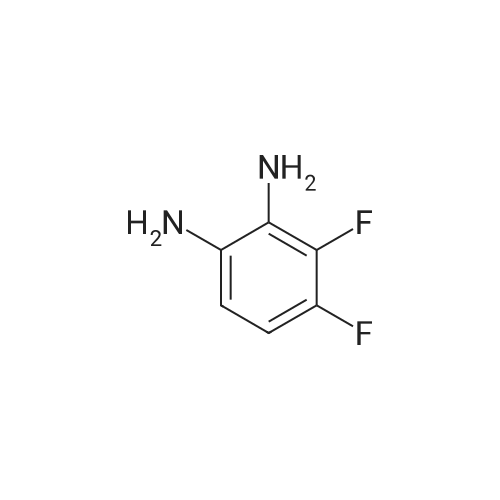
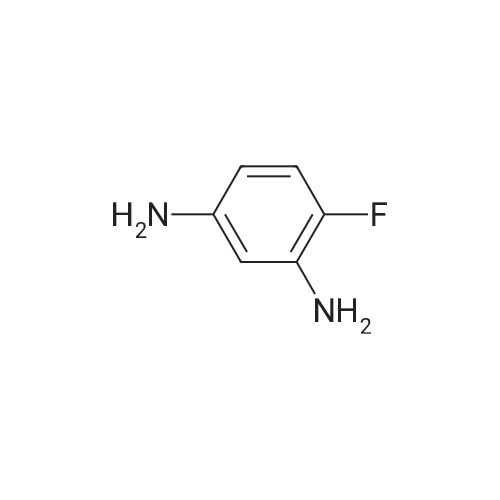
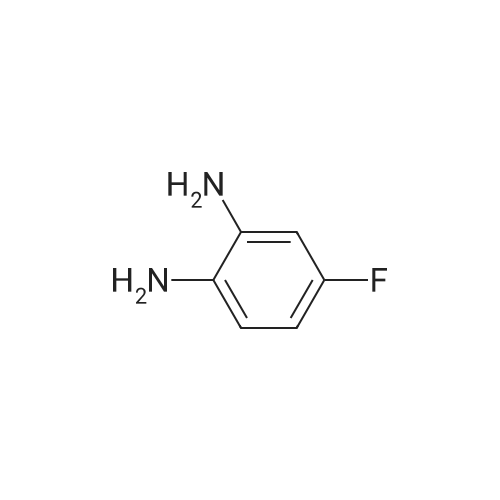
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With hydrogenchloride In water for 1.5 h; Cooling with ice; Reflux
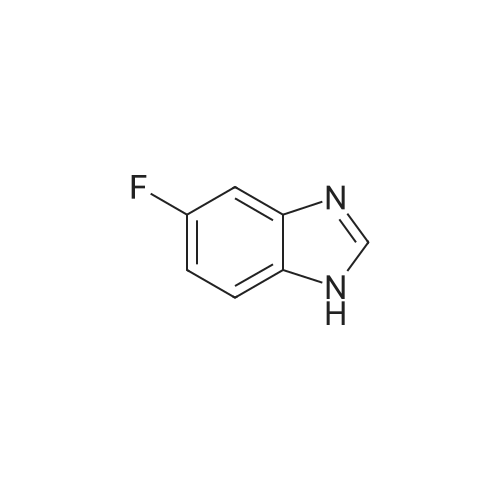
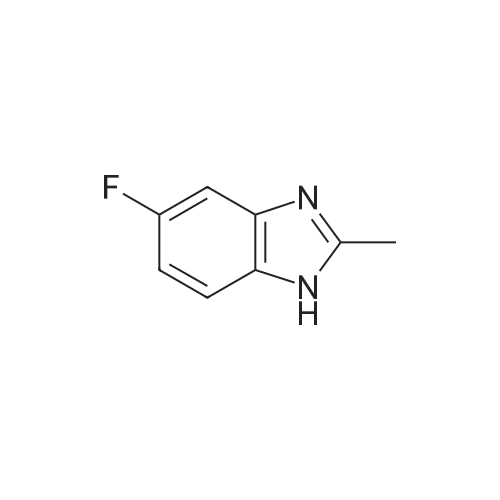
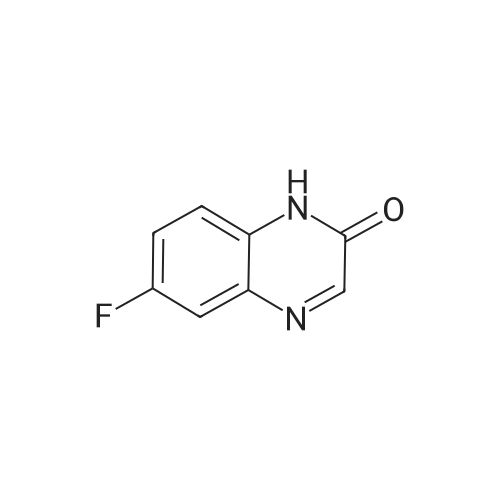
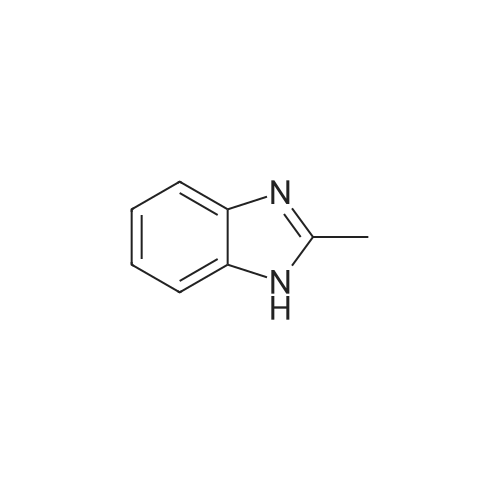
Reference Example 1Regioisomer mixture of 1-dimethoxymethyl-5-fluoro-1H-benzimidazole[0298]Under ice cooling, 4 mol/L hydrochloric acid (200 mL) and formic acid (38.3 g) were sequentially added to 4-fluoro-1,2-phenylenediamine (21.0 g), and the mixture was heated to reflux for 90 minutes while being stirred. Under ice cooling, the reaction mixture was basified with a 10percent aqueous solution of sodium hydroxide and extracted with ethyl acetate. The organic layer was washed with saturated brine and then dried over anhydrous sodium sulfate. After distilling off the solvent under reduced pressure, the residue was washed with diisopropyl ether, and 5-fluorobenzimidazole (19.7 g) was obtained. 5-Fluorobenzimidazole (19.7 g) thus obtained was dissolved in toluene (500 mL), and methyl orthoformate (38.7 g) and benzenesulfonic acid monohydrate (1.0 g) were sequentially added thereto. The mixture was heated to reflux for 40 hours while being stirred. After distilling off the solvent and methyl orthoformate under reduced pressure, the residue was diluted with toluene. Diisopropylamine (1 mL) and a saturated aqueous solution of sodium hydrogen carbonate were sequentially added thereto under ice cooling, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and then dried over anhydrous sodium sulfate. After distilling off the solvent under reduced pressure, the residue was purified by basic silica gel chromatography (20percent to 100percent ethyl acetate/hexane), and thus the title compound (25.2 g) was obtained.
Reference:
[1] Molecules, 2015, vol. 20, # 8, p. 15206 - 15223
[2] Synlett, 2001, # 11, p. 1755 - 1758
[3] Yakugaku Zasshi, 1958, vol. 78, p. 108[4] Chem.Abstr., 1958, p. 11013
[5] Journal of the American Chemical Society, 1953, vol. 75, p. 1292
[6] Patent: US2013/65896, 2013, A1, . Location in patent: Paragraph 0298
2
[ 367-31-7 ]
[ 144-62-7 ]
[ 1977-72-6 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2018, vol. 16, # 8, p. 1337 - 1342
Reference:
[1] European Journal of Organic Chemistry, 2011, # 15, p. 2827 - 2835
5
[ 124-38-9 ]
[ 367-31-7 ]
[ 1977-72-6 ]
Reference:
[1] Green Chemistry, 2013, vol. 15, # 1, p. 95 - 99
[2] New Journal of Chemistry, 2018, vol. 42, # 19, p. 15847 - 15851
6
[ 367-31-7 ]
[ 68-12-2 ]
[ 1977-72-6 ]
Reference:
[1] New Journal of Chemistry, 2016, vol. 40, # 10, p. 8282 - 8287
7
[ 57-55-6 ]
[ 367-31-7 ]
[ 118469-15-1 ]
[ 1977-72-6 ]
Reference:
[1] Bulletin of the Chemical Society of Japan, 2010, vol. 83, # 7, p. 831 - 837
8
[ 367-31-7 ]
[ 107-21-1 ]
[ 51-17-2 ]
[ 1977-72-6 ]
Reference:
[1] Bulletin of the Chemical Society of Japan, 2010, vol. 83, # 7, p. 831 - 837
9
[ 124-38-9 ]
[ 367-31-7 ]
[ 1544-75-8 ]
Yield
Reaction Conditions
Operation in experiment
87 %Chromat.
With tetrabutylammonium tungstate In 1-methyl-pyrrolidin-2-one at 140℃; for 24 h; Schlenk technique
General procedure: A typical procedure for the I-catalyzed reaction of aryl diamines with 1 atm CO2 was as follows: diamine (1 mmol), I (0.15 mmol), and N-methylpyrrolidone (NMP) (1 mL) were charged in a Schlenk tube with a magnetic stir bar. CO2 (1 atm) was introduced by a balloon, and the reaction mixture was stirred at 140 °C for 24 h. The reaction solution was periodically analyzed by GC, LC, GC–MS, or NMR. A Teflon vessel placed in a stainless steel autoclave was used for reactions with CO2 at 20 atm.
Reference:
[1] Angewandte Chemie - International Edition, 2012, vol. 51, # 27, p. 6700 - 6703
[2] Chemistry - An Asian Journal, 2016, vol. 11, # 19, p. 2735 - 2740
[3] ACS Catalysis, 2013, vol. 3, # 9, p. 2076 - 2082
[4] Catalysis Today, 2014, vol. 226, p. 160 - 166
10
[ 367-31-7 ]
[ 530-62-1 ]
[ 1544-75-8 ]
Yield
Reaction Conditions
Operation in experiment
88%
at 20℃; Inert atmosphere; Cooling with ice
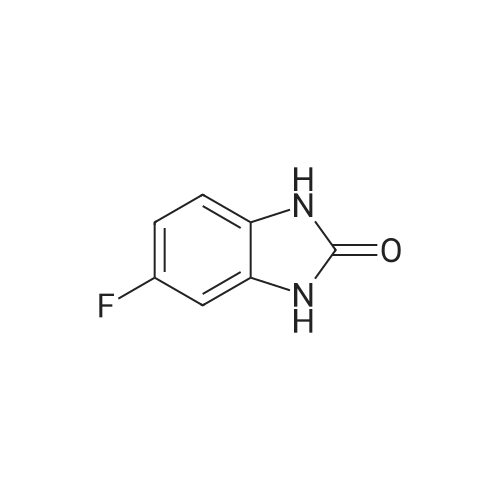
A stirred solution of 4-fluorobenzene-1 ,2-diamine (15.1 g, 120 mmol) in THF (120 mL) under nitrogen was cooled using an ice-bath and then was treated with di(1 -/-imidazol-1 - yl)methanone (23.4 g, 144 mmol) portion-wise over 15 min. The resulting mixture was slowly warmed to room temperature then was concentrated in vacuo after 2.5 h. The residue was suspended in a mixture of water and DCM (250 mL each) and filtered off. This residue was then washed with water (50 mL) and DCM (50 mL), before being dried at 40 °C under vacuum for 16 h to give the title compound (16.0 g, 105 mmol, 88percent) as a brown solid. LCMS (high pH): Rt 0.57 min; [M-H+]" = 151.1 δΗ NMR (400 MHz, DMSO-d6) ppm 10.73 (br s, 1 H), 10.61 (br s, 1 H), 6.91-6.84 (m, 1 H), 6.78-6.70 (m, 2H).
78%
at 150℃; for 0.333333 h; Microwave irradiation
Intermediate 15: 5-Fluoro-1 ,3-dihvdro-2H-benzimidazol-2-one; A mixture of 4-fluoro-1 ,2-diaminobenzene (commercially available, 1.0 g, 7.9 mmol), carbonyldiimidazole (1.4 g) and THF (4 ml) was heated to 150 0C in a microwave reactor and stirred for 10 minutes. The mixture was heated to 150 0C and stirred for a further 10 <n="38"/>minutes. The mixture was cooled to room temperature and concentrated under vacuum. The residue was suspended in dilute hydrochloric acid and filtered. The filter-cake was washed with water and cyclohexane then dried under vacuum to give the title compound as a dark grey solid (0.95 g, 78percent). 1 H-NMR (400 MHz, DMSO-d6): δ 10.73 (1 H, br s), 10.62 (1 H, br s), 6.87 (1 H, dd, J 8.5, 5 Hz), 6.78-6.70 (2H, m). UPLC-MS: 0.47 min, m/z 153 [M+H]+.
52%
at 20℃;
a) 4-Fluorobenzene-1,2-diamine (2 g, 15.86 mmol) was dissolved in THF (49.4 ml) and 1,1'-Carbonyldiimidazole (2.83 g, 17.44 mmol) was added at RT. The reaction mixture was stirred overnight at RT. To this was added concentrated ammonia solution (1.5 ml) and the mixture stirred for 30 minutes and then diluted with water (100 ml). The resultant solid was collected by filtration, washed with water, followed by Et2O and then dried in vacuo to afford 5-fluoro-1H-benzo[d]imidazol-2(3H)-one (1.250 g, 52percent); 1H NMR (400 MHz, DMSO-d6) 6.66-6.79 (2H, m), 6.81-6.94 (1H, m), 10.64 (1H, s), 10.76 (1H, s); m/z: (ES+) MH+, 151.19.
With triethylamine In dichloromethane at 0 - 20℃; for 2 h;
[0278] To a solution of 4-fluorobenzene-1,2-diamine lb (1.39 g, 11.0 mmol) and triethylamine (3.26 mE, 23.2 mmol) in methylene chloride (20 mE) was added dropwise a solution of diphosgene (0.69 mE, 5.72 mmol) in methylene chloride (5 mE) at 0 to 5° C. The resulting suspension was stirred for 2 hours at room temperature and filtered. The collected white solid was washed with water and dried to give compound 2b (1.56 g, 93percent). ‘H NMR (400 MHz, DMSO-d5) ö 10.73 (s, 1H), 10.61 (s, 1H), 6.97-6.81 (m, 1H), 6.74 (m, 2H); MS (ESI): mlz 153.1 [M+1]
General procedure: A mixture of 2-phenoxycarbonyl-4,5-dichloropyridazin-3(2H)-one (2a, 1.2 equiv), compounds 4 (1 equiv), and toluene (10 mL)was stirred at reflux conditions until amide intermediate wasconsumed (monitored by TLC). After cooling to r.t., the resultingprecipitate was filtered off and the solvent was evaporatedunder reduced pressure. The residue was transferred to anopen-bed silica gel column (3 × 7 cm). The column was elutedwith n-hexane–THF (1:2, v/v). Fractions containing compounds5 were combined and evaporated under reduced pressure togive compounds 5.
A solution of 5-fluoro-2-nitroaniline in MeOH (0.32 M) was added to Pd/C (5percentw/w, 10percentw). The reaction mixture was stirred at R.T.under H2 atmosphere (50 Psi) for 2 h. Then, the mixture was filtrated and the organic solution was concentrated to afford (90percent) the title compound as a brown oil. MS (ES+) C6H7FN2 required: 126, found: 127 (M+H)+.
76%
at 80℃; for 8 h;
General procedure: Nitro aromatic (1.0 mmol), B2(OH)4 (5.0 equiv, 5.0 mmol), and H2O (3.0 mL) were added in a10 mL tube. The reaction mixture was stirred at 80 °C for 8 h. When the reaction was completemonitored by TLC, the mixture was cooled to room temperature, extracted with ethyl acetate (3 ×20 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, andconcentrated under reduced pressure. The residue was purified by silica gel columnchromatography.
68%
With tetrahydroxydiboron; 5%-palladium/activated carbon; water In acetonitrile at 50℃; for 24 h;
General procedure: Nitrobenzene (0.6mmol), 5wtpercent Pd/C (0.5mmol percent, 0.003mmol), H2O (10 equiv, 6.0mmol), B2(OH)4 (3.3 equiv, 2.0mmol), and CH3CN (1.0mL) were added in a 10mL tube. The reaction mixture was stirred at 50°C for 24h. When the reaction was complete monitored by TLC, the mixture was cooled to room temperature. Water (5mL) was added, and extracted with EtOAc (3×5mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give aniline 2a (55mg, 99percent).
Reference:
[1] Macromolecules, 2005, vol. 38, # 2, p. 297 - 306
[2] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1994, # 19, p. 2751 - 2758
[3] Patent: WO2010/13037, 2010, A1, . Location in patent: Page/Page column 67
[4] Synlett, 2018, vol. 29, # 13, p. 1765 - 1768
[5] Tetrahedron, 2017, vol. 73, # 27-28, p. 3898 - 3904
[6] Patent: WO2008/75109, 2008, A1, . Location in patent: Page/Page column 102
[7] Bioorganic and Medicinal Chemistry Letters, 2012, vol. 22, # 2, p. 1156 - 1159
[8] Chemistry--A European Journal, 2012, vol. 18, # 36, p. 11433 - 11439,7
[9] Chemistry - A European Journal, 2012, vol. 18, # 36, p. 11433 - 11439
[10] Journal of Medicinal Chemistry, 2017, vol. 60, # 14, p. 6289 - 6304
19
[ 364-78-3 ]
[ 367-31-7 ]
Yield
Reaction Conditions
Operation in experiment
88%
With hydrogen In methanol at 20℃; for 4 h;
4-fluoro-2-nitroaniline (8.00 g, 51.22 mmol) was dissolved in methanol (100 mL), and 10percent palladium-carbon powder (0.80 g) was added thereto, followed by stirring for four hours at room temperature under hydrogen atmosphere. The reaction mixture was filtrated, and the filtrate was concentrated under reduced pressure. The residue was purified column chromatography (silica gel 150 g, hexane ethyl acetate=1:4), to thereby yield a brown oil (5.67 g, yield 88percent). The brown oil (5.64 g, 44.72 mmol) was dissolved in ethanol (150 mL), and potassium o-ethylxanthate (8.60 g, 53.65 mmol) was added thereto, followed by reflux for three hours. Potassium o-ethylxanthate (1.43 g, 8.92 mmol) was further added thereto, and the mixture was refluxed for two hours. The reaction mixture was concentrated under reduced pressure, and the residue was purified through column chromatography (silica gel 150 g, hexane:ethyl acetate=2:1), to thereby yield 5-fluoro-2-mercaptobenzimidazole (5.93 g, yield 79percent) as a brown powder.
70%
With hydrogenchloride; tin In water at 20℃; for 1 h;
When 5-fluoro-2-nitroaniline (4.496 g, 0.0288 mol) was dissolved in 150 ml of concentrated hydrochloric acid I turned it on. A small amount of tin (16.56 g, 0.140 mol) was added over 15 minutes at 0 . Room temperature Temperature), the mixture was stirred for 1 hour and then cooled overnight. Deionized water The mixture was poured into water and 4M sodium hydroxide (NaOH) was slowly added until pH 10 was reached. The organic layer was washed with Extracted with ethyl acetate (4 x 100 mL), washed with water (4 x 100 mL), dried over anhydrous magnesium sulfate 4). The solution was concentrated in vacuo to give 2.54 g of yellow
Reference:
[1] Macromolecules, 2005, vol. 38, # 2, p. 297 - 306
[2] Farmaco, Edizione Scientifica, 1986, vol. 41, # 12, p. 970 - 983
[3] Patent: US2005/20606, 2005, A1, . Location in patent: Page 10
[4] Heterocycles, 1987, vol. 26, # 9, p. 2433 - 2442
[5] Organic Preparations and Procedures International, 2000, vol. 32, # 5, p. 485 - 488
[6] Patent: KR2017/50088, 2017, A, . Location in patent: Paragraph 0192; 0193; 0194; 0195
[7] Journal of the Chemical Society, 1953, p. 3042
[8] Yakugaku Zasshi, 1958, vol. 78, p. 108[9] Chem.Abstr., 1958, p. 11013
[10] Journal of the American Chemical Society, 1953, vol. 75, p. 1292
[11] Journal of Fluorine Chemistry, 1990, vol. 46, # 2, p. 343 - 355
[12] Acta chemica Scandinavica. Series B: Organic chemistry and biochemistry, 1985, vol. 39, # 1, p. 31 - 34
[13] Journal de Chimie Physique et de Physico-Chimie Biologique, 1986, vol. 83, # 9, p. 577 - 588
[14] Journal of Medicinal Chemistry, 1995, vol. 38, # 25, p. 4906 - 4916
[15] Bioorganic and Medicinal Chemistry, 1998, vol. 6, # 2, p. 163 - 172
[16] Farmaco, 1998, vol. 53, # 7, p. 455 - 461
[17] Farmaco (Societa chimica italiana : 1989), 2001, vol. 56, # 11, p. 821 - 826
[18] European Journal of Medicinal Chemistry, 2006, vol. 41, # 1, p. 135 - 141
[19] Journal of the American Chemical Society, 2006, vol. 128, # 26, p. 8569 - 8574
[20] Molecules, 2006, vol. 11, # 12, p. 988 - 999
[21] Patent: US6632813, 2003, B1, . Location in patent: Page/Page column 37
[22] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 17, p. 4790 - 4793
[23] Journal of the Serbian Chemical Society, 2014, vol. 79, # 3, p. 277 - 282
[24] Patent: WO2014/114776, 2014, A1, . Location in patent: Page/Page column 24
[25] Molecules, 2014, vol. 19, # 7, p. 9736 - 9759
[26] Tetrahedron Letters, 2015, vol. 56, # 48, p. 6762 - 6767
20
[ 364-78-3 ]
[ 7732-18-5 ]
[ 367-31-7 ]
Reference:
[1] Patent: US5514680, 1996, A,
21
[ 351-83-7 ]
[ 367-31-7 ]
Reference:
[1] Journal of Medicinal Chemistry, 1995, vol. 38, # 25, p. 4906 - 4916
[2] Journal of Fluorine Chemistry, 1990, vol. 46, # 2, p. 343 - 355
22
[ 448-39-5 ]
[ 367-31-7 ]
Reference:
[1] Journal of Medicinal Chemistry, 1995, vol. 38, # 25, p. 4906 - 4916
[2] Journal of Fluorine Chemistry, 1990, vol. 46, # 2, p. 343 - 355
23
[ 371-40-4 ]
[ 367-31-7 ]
Reference:
[1] Journal of Medicinal Chemistry, 1995, vol. 38, # 25, p. 4906 - 4916
(52a) 5-fluoro-1H-benzimidazole-2-thiol A mixture of 3,4-diamino-1-fluorobenzene (10 g, 79.3 mmol), carbon disulfide (70 ml, 1164 mmol), and methanol (100 ml) was stirred at room temperature for 86 hours and 50 minutes. After the reaction mixture was concentrated, the residue was suspended in hexane. The resultant precipitate was collected by filtration and washed with hexane to obtain the title compound (13.1 g, yield: 98.2percent) as a brown solid. 1H NMR(400 MHz, DMSO-d6) δppm; 6.90-6.99(2H, m), 7.06-7.13(1H, m), 12.58(1H, s), 12.61(1H, s).
4-fluoro-2-nitroaniline (8.00 g, 51.22 mmol) was dissolved in methanol (100 mL), and 10percent palladium-carbon powder (0.80 g) was added thereto, followed by stirring for four hours at room temperature under hydrogen atmosphere. The reaction mixture was filtrated, and the filtrate was concentrated under reduced pressure. The residue was purified column chromatography (silica gel 150 g, hexane ethyl acetate=1:4), to thereby yield a brown oil (5.67 g, yield 88percent). The brown oil (5.64 g, 44.72 mmol) was dissolved in ethanol (150 mL), and potassium o-ethylxanthate (8.60 g, 53.65 mmol) was added thereto, followed by reflux for three hours. Potassium o-ethylxanthate (1.43 g, 8.92 mmol) was further added thereto, and the mixture was refluxed for two hours. The reaction mixture was concentrated under reduced pressure, and the residue was purified through column chromatography (silica gel 150 g, hexane:ethyl acetate=2:1), to thereby yield 5-fluoro-2-mercaptobenzimidazole (5.93 g, yield 79percent) as a brown powder.
Reference:
[1] Patent: US2005/20606, 2005, A1, . Location in patent: Page 10
[2] Chemical and Pharmaceutical Bulletin, 1989, vol. 37, # 6, p. 1517 - 1523
[3] Journal of Medicinal Chemistry, 1995, vol. 38, # 25, p. 4906 - 4916
Reference:
[1] Journal of the American Chemical Society, 2014, vol. 136, # 11, p. 4287 - 4299
[2] Patent: WO2017/222951, 2017, A1, . Location in patent: Page/Page column 84
31
[ 367-31-7 ]
[ 2208-07-3 ]
[ 118469-15-1 ]
Reference:
[1] Patent: US5180723, 1993, A,
32
[ 141-82-2 ]
[ 367-31-7 ]
[ 118469-15-1 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2018, vol. 16, # 8, p. 1337 - 1342
33
[ 57-55-6 ]
[ 367-31-7 ]
[ 118469-15-1 ]
[ 1977-72-6 ]
Reference:
[1] Bulletin of the Chemical Society of Japan, 2010, vol. 83, # 7, p. 831 - 837
34
[ 64-17-5 ]
[ 367-31-7 ]
[ 615-15-6 ]
[ 118469-15-1 ]
Reference:
[1] Bulletin of the Chemical Society of Japan, 2010, vol. 83, # 7, p. 831 - 837
35
[ 367-31-7 ]
[ 64-19-7 ]
[ 118469-15-1 ]
Reference:
[1] Journal of the American Chemical Society, 2017, vol. 139, # 24, p. 8267 - 8276
36
[ 24424-99-5 ]
[ 367-31-7 ]
[ 579474-47-8 ]
Yield
Reaction Conditions
Operation in experiment
69%
at 0 - 20℃; for 0.0833333 h;
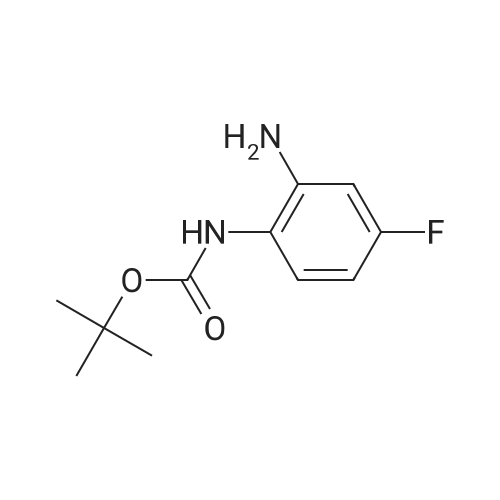
Step 1. To 4-fluorobenzene-1,2-diamine (13.88 g, 110.04 mmol) was added ditert-butyldicarbonate (50 mL) at 0°C. The resulting mixture was stirred at ambient temperature for 5 mm, then diluted with ice-water (500 mL) and extracted with 1:1 ethyl acetate-hexane (500 mL). The organic phase was washed with brine (300 mL), dried over Mg504, then filtered and the remaining liquid evaporated. The residual solid was washedwith hexane to afford tert-butyl (2-amino-4-fluorophenyl)carbamate (17.0 g, 69percent yield). 1H NMR (500 MHz, Acetone-d6) ö 7.48 (br. s, 1H), 7.05 - 7.26 (m, 1H), 6.51 - 6.59 (m, 1H), 6.27 - 6.38 (m, 1H), 4.80 (s, 2H), 1.47 (s, 9H).
44%
at 20℃; for 6 h;
Tert-butyl 2-amino-4-fluorophenylcarbamate (Compound 0113-60) A solution of 4-fluorobenzene-l,2-diamine (0.252 g, 2.0 mmol), (BoC)2O(0.436 g, 2.0 mmol) in dry THF (6 mL) was stirring at room temperature for 6 h. The reaction mixture was concentrated in vacuo, extracted with ethyl acetate. The organic phase was washed with saturated NaHCO3, dried over anhydrous Na2SO4, evaporated to afford crude product as oil. The product was further purified by flash column chromatography on silica gel (ethyl acetate in petroleum ether, 20percent v/v) to afford pure product 0113-60 (0.2 g, 44 percent) as a yellow solid. LCMS: 171 [M-56+l]+, 1H-NMR (400 MHz. DMSO-J6) "5 1.51 (s, 9H), 5.20 (s, 2H), 6.35 (m, IH), 6.52 (dd, J= 11.2, 2.8 Hz, IH), 7.17 (m, IH), 8.32 (s, IH).
With hydrogenchloride; In water; for 1.5h;Cooling with ice; Reflux;
Reference Example 1Regioisomer mixture of 1-dimethoxymethyl-5-fluoro-1H-benzimidazole[0298]Under ice cooling, 4 mol/L hydrochloric acid (200 mL) and formic acid (38.3 g) were sequentially added to 4-fluoro-1,2-phenylenediamine (21.0 g), and the mixture was heated to reflux for 90 minutes while being stirred. Under ice cooling, the reaction mixture was basified with a 10percent aqueous solution of sodium hydroxide and extracted with ethyl acetate. The organic layer was washed with saturated brine and then dried over anhydrous sodium sulfate. After distilling off the solvent under reduced pressure, the residue was washed with diisopropyl ether, and 5-fluorobenzimidazole (19.7 g) was obtained. 5-Fluorobenzimidazole (19.7 g) thus obtained was dissolved in toluene (500 mL), and methyl orthoformate (38.7 g) and benzenesulfonic acid monohydrate (1.0 g) were sequentially added thereto. The mixture was heated to reflux for 40 hours while being stirred. After distilling off the solvent and methyl orthoformate under reduced pressure, the residue was diluted with toluene. Diisopropylamine (1 mL) and a saturated aqueous solution of sodium hydrogen carbonate were sequentially added thereto under ice cooling, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and then dried over anhydrous sodium sulfate. After distilling off the solvent under reduced pressure, the residue was purified by basic silica gel chromatography (20percent to 100percent ethyl acetate/hexane), and thus the title compound (25.2 g) was obtained.
4-fluoro-2-nitroaniline (8.00 g, 51.22 mmol) was dissolved in methanol (100 mL), and 10% palladium-carbon powder (0.80 g) was added thereto, followed by stirring for four hours at room temperature under hydrogen atmosphere. The reaction mixture was filtrated, and the filtrate was concentrated under reduced pressure. The residue was purified column chromatography (silica gel 150 g, hexane ethyl acetate=1:4), to thereby yield a brown oil (5.67 g, yield 88%). The brown oil (5.64 g, 44.72 mmol) was dissolved in ethanol (150 mL), and potassium o-ethylxanthate (8.60 g, 53.65 mmol) was added thereto, followed by reflux for three hours. Potassium o-ethylxanthate (1.43 g, 8.92 mmol) was further added thereto, and the mixture was refluxed for two hours. The reaction mixture was concentrated under reduced pressure, and the residue was purified through column chromatography (silica gel 150 g, hexane:ethyl acetate=2:1), to thereby yield 5-fluoro-2-mercaptobenzimidazole (5.93 g, yield 79%) as a brown powder.
With hydrogenchloride In water at 95 - 105℃; for 21h; Large scale;
14A Example 14 A Production of Compound A
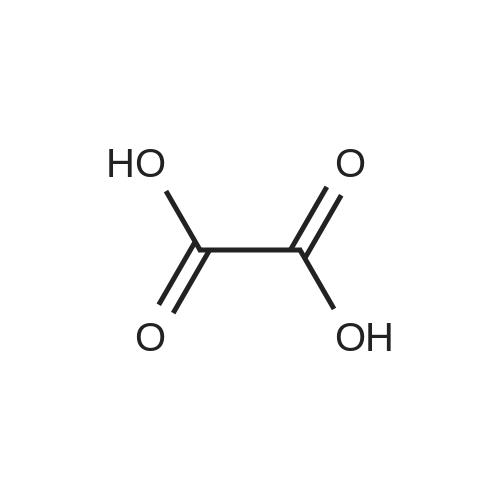
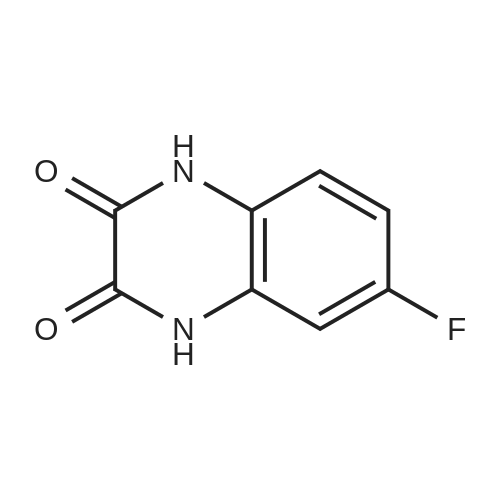
A 35 gallon reactor was charged with 1,2-diamino-4-fluorbenzene (7.50 kg, 59.46 mol, ChemiK), oxalic acid (5.35 kg, 59.42 mol) and 3 M HCl solution (95.8 kg water and 37.8 kg cone, hydrochloric acid). The dark mixture was heated at reflux (100±5 °C) for 21 h. An aliquot (1 mL) was taken while stirring and neutralized with saturated NaHCO3 solution (30 mL) to a pH of 8. HPLC analysis showed that the starting material was completely consumed. The mixture was removed from heating, allowed to cool to ambient temperature and then cold water (55 kg) was added. A dark solid precipitated and was collected by vacuum filtration. The filter cake was washed with cold water (50.0 kg) followed by Isopropyl Alcohol (IP A, 39.14 kg). The wet cake (11.43 kg) was then dried overnight in a vacuum oven (50±5 °C) to give 10.3 kg (96% molar yield) of 6-fluoro-1,4-dihydryoquinoxaline-2,3-dione (Compound A) as a blue-gray solid with an HPLC purity of >99%). The process was repeated on the identical scale to provide 9.95 kg >99%).
With ethanol; stannous chloride at 80℃; for 4h; Inert atmosphere;
2.3.1 Preparation of 5-Fluoro-1,2-diaminobenzene (1)
To a 50 mL round bottom flask was added 2-nitro-5-fluoroaniline (1.56 g, 10 mmol) and ethanol 20 mL. Anhydrous SnCl2 (9.47 g, 50 mmol, 5 equiv.) as a solid. The flask was equipped with a condenser and placed in an 80 °C sand bath under N2 and the mixture was refluxed for 4 h. The reaction was poured into a sep. funnel, diluted with 80 mL dichloromethane and then treated with 100 mL 2M NaOH. The resulting emulsion was filtered through Celite to remove all the tin, and the resulting biphasic mixture was separated in a sep. funnel. The aq. phase was washed with dichloromethane (50 mL) and the combined organic phases were dried with brine (30 mL), dried over MgSO4, filtered and concentrated to afford a yellow solid.1.21 g, 95%.1H NMR (300 MHz, Chloroform-d) δ 6.64 (dd, J = 8.4, 5.5 Hz, 1H), 6.46 (dd, J = 9.9, 2.8 Hz, 1H), 6.39 (td, J = 8.5, 2.8 Hz, 1H), 3.35 (s, 4H). Spectral data are in agreement with the literature. Chen et al., 2018.
95%
With ethanol; stannous chloride at 80℃; for 4h; Inert atmosphere;
2.3.1 Preparation of 5-Fluoro-1,2-diaminobenzene (1)
To a 50 mL round bottom flask was added 2-nitro-5-fluoroaniline (1.56 g, 10 mmol) and ethanol 20 mL. Anhydrous SnCl2 (9.47 g, 50 mmol, 5 equiv.) as a solid. The flask was equipped with a condenser and placed in an 80 °C sand bath under N2 and the mixture was refluxed for 4 h. The reaction was poured into a sep. funnel, diluted with 80 mL dichloromethane and then treated with 100 mL 2M NaOH. The resulting emulsion was filtered through Celite to remove all the tin, and the resulting biphasic mixture was separated in a sep. funnel. The aq. phase was washed with dichloromethane (50 mL) and the combined organic phases were dried with brine (30 mL), dried over MgSO4, filtered and concentrated to afford a yellow solid.1.21 g, 95%.1H NMR (300 MHz, Chloroform-d) δ 6.64 (dd, J = 8.4, 5.5 Hz, 1H), 6.46 (dd, J = 9.9, 2.8 Hz, 1H), 6.39 (td, J = 8.5, 2.8 Hz, 1H), 3.35 (s, 4H). Spectral data are in agreement with the literature. Chen et al., 2018.
93%
With hydrazine hydrate monohydrate In ethanol at 75℃; for 12h; Inert atmosphere;
90%
With hydrogen In methanol at 20℃; for 2h;
96.1
A solution of 5-fluoro-2-nitroaniline in MeOH (0.32 M) was added to Pd/C (5%w/w, 10%w). The reaction mixture was stirred at R.T.under H2 atmosphere (50 Psi) for 2 h. Then, the mixture was filtrated and the organic solution was concentrated to afford (90%) the title compound as a brown oil. MS (ES+) C6H7FN2 required: 126, found: 127 (M+H)+.
76%
With tetrahydroxydiborane; lithium hydroxide monohydrate at 80℃; for 8h;
2. General procedure for the reduction of nitroarenes
General procedure: Nitro aromatic (1.0 mmol), B2(OH)4 (5.0 equiv, 5.0 mmol), and H2O (3.0 mL) were added in a10 mL tube. The reaction mixture was stirred at 80 °C for 8 h. When the reaction was completemonitored by TLC, the mixture was cooled to room temperature, extracted with ethyl acetate (3 ×20 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, andconcentrated under reduced pressure. The residue was purified by silica gel columnchromatography.
68%
With tetrahydroxydiborane; 5%-palladium/activated carbon; lithium hydroxide monohydrate In acetonitrile at 50℃; for 24h; chemoselective reaction;
4.2 Typical procedure for reduction of nitrobenzene
General procedure: Nitrobenzene (0.6mmol), 5wt% Pd/C (0.5mmol %, 0.003mmol), H2O (10 equiv, 6.0mmol), B2(OH)4 (3.3 equiv, 2.0mmol), and CH3CN (1.0mL) were added in a 10mL tube. The reaction mixture was stirred at 50°C for 24h. When the reaction was complete monitored by TLC, the mixture was cooled to room temperature. Water (5mL) was added, and extracted with EtOAc (3×5mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give aniline 2a (55mg, 99%).
58%
With palladium 10% on activated carbon; hydrogen In methanol at 20℃; for 2h;
231.1 Step-1
To a solution of 5-fluoro-2-nitroaniline (300 mg, 1.92 mmol) in methanol (10.0 mL) was added 10% Palladium on Carbon (50% wet) (300 mg, 2.82 mmol) at room temperature and the reaction mixture stirred for 2h under hydrogen atmosphere. After completion, the reaction mixture passed through celite bed. The filtrate was concentrated to get 4-fluorobenzene-1,2-diamine (200 mg, 1.11 mmol) as green solid. Yield: 0.20 g, 58%
With hydrogen In methanol for 16h;
GP1
GENERAL PROCEDURE GPl - PREPARATION OF DIAMINOBENZENESDiaminobenzenes can be obtained commercially or prepared from nitroanilines using the reduction method set out below.A mixture of nitroarene (5.0 mmol) and 10% palladium on carbon (200 mg) in methanol (10 ml) was stirred for 16 hours under an atmosphere of hydrogen. The catalyst was removed by filtration and the solvent removed in vacuo to afford the desired 1,2- diaminobenzene which was used without further purification.The following compounds were prepared from the stated precursors according to the method described above:
With hydrogenchloride; iron(0) In ethanol; lithium hydroxide monohydrate Reflux;
With 5% Pd/C; hydrazine hydrate monohydrate In ethanol at -10 - 20℃; for 0.5h; Inert atmosphere;
With palladium 10% on activated carbon; hydrogen In methanol at 20℃; for 6h;
With palladium on activated charcoal; hydrogen In methanol at 20℃; Inert atmosphere;
General procedure: In a 100 mL flask with a magnetic stirring bar, N-benzyl-5-fluoro-2-nitroaniline (0.5 g, 2.03 mmol) and Pd/C (100 mg) weresuspended in 15 mL methanol. After sealing by rubber stopper, a2.0 L hydrogen balloon was connected with the reaction flask by asyringe needle. The reaction mixture was then stirred overnight atroom temperature. After removing the Pd/C by filtration, the solventwas removed by rotavapor. The residue was then purified bysilica gel column chromatography to afford 352 mg of N1-benzyl-5-fluorobenzene-1,2-diamine (80% yield).
To a solution of <strong>[2533-69-9]methyl 2,2,2-trichloroacetimidate</strong> (1.53 g) in acetic acid (10 ml) was added a solution of 4-fluorobenzene-1,2-diamine in acetic acid (10 ml) under ice-cooling, followed by stirring at room temperature for 3 hours. The reaction liquid was concentrated under reduced pressure and to the obtained residue was added water, followed by neutralization with a saturated aqueous sodium hydrogen carbonate solution and extraction with EtOAc. The organic layer was washed with saturated brine, then dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was recrystallized from iPr2O/EtOAc/MeOH to obtain 5-fluoro-2-(trichloromethyl)-benzimidazole (1.5 g) as a beige powder.
With acetic acid; at 20℃; for 2h;
To a solution of 4-fluorobenzene-1,2-diamine [Aldrich, 653586] (0.03 g, 0.2 mmol) in acetic acid (1 mL) was added <strong>[2533-69-9]methyl 2,2,2-trichloroethanimidoate</strong> [Acros, AC16093-0250] (0.030 mL, 0.24 mmol) dropwise at room temperature. The mixture was stirred at room temperature for 2 h and poured into water (25 mL). The precipitate was separated by filtration and air-dried, which was directly used in the next step without purification.
In tetrahydrofuran; at 20℃;Inert atmosphere; Cooling with ice;
A stirred solution of 4-fluorobenzene-1 ,2-diamine (15.1 g, 120 mmol) in THF (120 mL) under nitrogen was cooled using an ice-bath and then was treated with di(1 -/-imidazol-1 - yl)methanone (23.4 g, 144 mmol) portion-wise over 15 min. The resulting mixture was slowly warmed to room temperature then was concentrated in vacuo after 2.5 h. The residue was suspended in a mixture of water and DCM (250 mL each) and filtered off. This residue was then washed with water (50 mL) and DCM (50 mL), before being dried at 40 C under vacuum for 16 h to give the title compound (16.0 g, 105 mmol, 88%) as a brown solid. LCMS (high pH): Rt 0.57 min; [M-H+]" = 151.1 deltaEta NMR (400 MHz, DMSO-d6) ppm 10.73 (br s, 1 H), 10.61 (br s, 1 H), 6.91-6.84 (m, 1 H), 6.78-6.70 (m, 2H).
78%
In tetrahydrofuran; at 150℃; for 0.333333h;Microwave irradiation;
Intermediate 15: 5-Fluoro-1 ,3-dihvdro-2H-benzimidazol-2-one; A mixture of 4-fluoro-1 ,2-diaminobenzene (commercially available, 1.0 g, 7.9 mmol), carbonyldiimidazole (1.4 g) and THF (4 ml) was heated to 150 0C in a microwave reactor and stirred for 10 minutes. The mixture was heated to 150 0C and stirred for a further 10 <n="38"/>minutes. The mixture was cooled to room temperature and concentrated under vacuum. The residue was suspended in dilute hydrochloric acid and filtered. The filter-cake was washed with water and cyclohexane then dried under vacuum to give the title compound as a dark grey solid (0.95 g, 78%). 1 H-NMR (400 MHz, DMSO-d6): delta 10.73 (1 H, br s), 10.62 (1 H, br s), 6.87 (1 H, dd, J 8.5, 5 Hz), 6.78-6.70 (2H, m). UPLC-MS: 0.47 min, m/z 153 [M+H]+.
52%
In tetrahydrofuran; at 20℃;
a) 4-Fluorobenzene-1,2-diamine (2 g, 15.86 mmol) was dissolved in THF (49.4 ml) and 1,1'-Carbonyldiimidazole (2.83 g, 17.44 mmol) was added at RT. The reaction mixture was stirred overnight at RT. To this was added concentrated ammonia solution (1.5 ml) and the mixture stirred for 30 minutes and then diluted with water (100 ml). The resultant solid was collected by filtration, washed with water, followed by Et2O and then dried in vacuo to afford 5-fluoro-1H-benzo[d]imidazol-2(3H)-one (1.250 g, 52%); 1H NMR (400 MHz, DMSO-d6) 6.66-6.79 (2H, m), 6.81-6.94 (1H, m), 10.64 (1H, s), 10.76 (1H, s); m/z: (ES+) MH+, 151.19.
With 2-methylenesuccinic acid In water at 20℃; for 1h; Schlenk technique; Green chemistry;
In ethanol; water Heating;
0.49 g
With sodium bromate; sodium hydrogensulfite In water at 20℃;
General Synthetic Procedure for Quinoxaline Derivatives 1-10
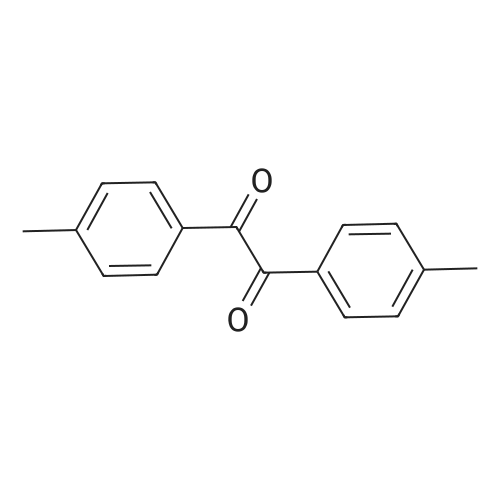
General procedure: Generally, to a stirring mixture of different substituted 1,2-phenylenedine (2 mmol) and substituted benzils (2mmol) in water, sodium bromate and sodium hydrogen sulfite(10%) were added and stirred for 2-3 h. The progress of reaction was monitored by TLC. After the completion of reaction, 20 mL ethyl acetate was added to reaction mixture,the organic layer was separated by a separating funnel. The quinoxaline derivatives 1-10 were obtained in high yields.Pure products were obtained by recrystallization from ethanol and in some cases through column chromatography (silicagel) using 2:8 ethyl acetate and n-hexane as eluent.The structures of compounds were confirmed by using various spectroscopic techniques, including 1H NMR, EI mass, and X-ray spectroscopy.
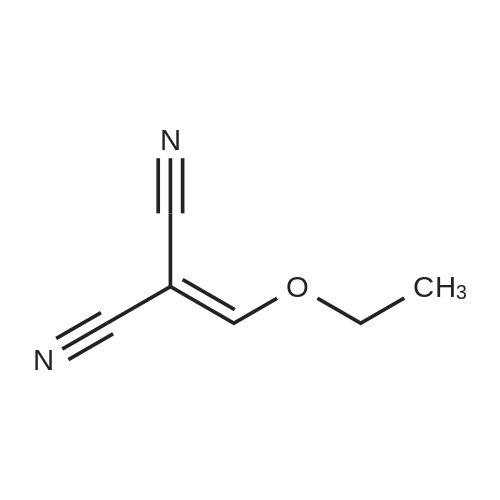
A solution of 4-fluoro-1,2-phenylenediamine (2.0 g) and ethoxymethylenemalononitrile (2.9 g) in isopropanol (80 mL) was heated to reflux overnight. The volatiles were removed in vacuo to yield 5-fluoro-1H-benzimidazole (1.8 g). ESI-MS (m/z) 137 [M+H]+.
In isopropyl alcohol; for 17.0h;Heating / reflux;
A solution OF 4-FLUORO-1, 2-phenylenediamine (2.0 g) and ETHOXYMETHYLENEMALONONITRILE (2. 9 g) in isopropanol (80 mL) was heated to reflux for 17 hours. The volatiles were removed in VCHO to yield 5-FLUORO-LH-BENZIMIDAZOLE (1.8 g). ESI-MS (MILZ) 137 [M+H] +.
(a) 2-Methyl-5-fluorobenzimidazole Ethyl acetimidate hydrochloride (37.1 g, 0.3 mol) was added to a stirred suspension of 4-fluoro-ortho-phenylenediamine (12.6 g, 0.1 mol) in ethanol (150 ml) at 0 C. The mixture was allowed to warm up to room temperature and stirred overnight. The solvent was removed under reduced pressure and the residue extracted into ethyl acetate (100 ml), washed with water (3*100 ml), dried over anhydrous magnesium sulphate, filtered and evaporated. Crystallisation from ethyl acetate gave 2-methyl-5-fluorobenzimidazole (7.7 g, 51%) as a brown crystalline solid. m.p. 177-178 C. deltaH 7.46 (1H, dd, J 8.8, 4.7 Hz), 7.22 (1H, dd, J 8.9, 2.4 Hz), 6.98 (1H, ddd, J 9.7, 8.9, 2.4 Hz), 2.65 (3H, s).
With hydrogenchloride; In water; for 20.0h;Heating / reflux;
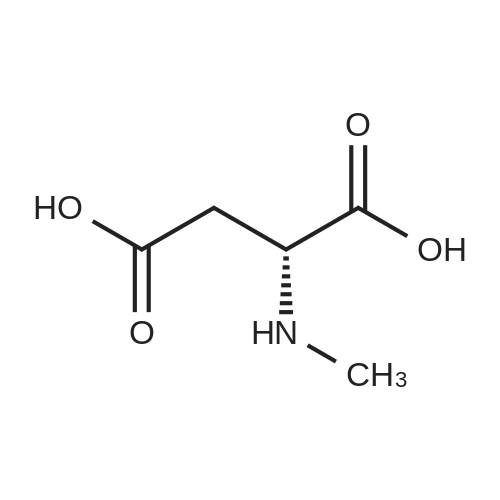
Compound 80a:; To an aqueous HCI solution (5.5 N, 5mL) was added 4-Fluorobenzene-1 ,2-diamine hydrochloride (0.572g, 4.53mmol) and 3-Aminopentanoic acid (1g, 6.8mmol). The mixture was heated at reflux for 2Oh. After being cooled down to 25C, the mixture was purified by preparative HPLC to afford 0.8g of the title compound (50% yield). LCMS (APCI positive): 238 (M+H+).
With hydrogenchloride; In water; for 72h;Heating / reflux;
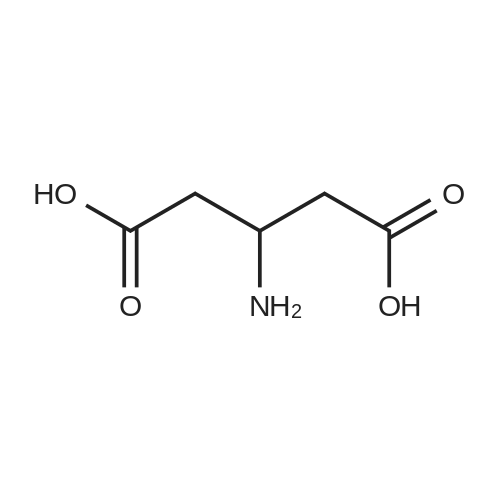
Compound 18a:; To an aqueous HCI solution (5.5 N, 87ml_) was added 4-Fluorobenzene-1 ,2-diamine (1 Og, 79.4mmol) and D-Aspartic acid (15.8g, 119mmol). The mixture was heated at reflux for 72h. The mixture was purified with preparative HPLC and recrystallization from EtOAc, affording the title compound (7.5g, 28% yield) as a pale tan solid. 1H NMR (CD3OD): delta 7.62 (1 H, dd, J=8.9, 4.8Hz), 7.36 (1 H, dd, J=8.9, 2.3Hz), 7.14 (1 H, td, J=9.6, 2.5 Hz), 4.53 (1 H, dd, J=7.4, 5.6Hz), 3.67 (1 H, dd, J=16.4, 5.8Hz), 3.54 (1 H, dd, J=16.2, 7.4Hz). LCMS (APCI): 224 (M+H) (positive).
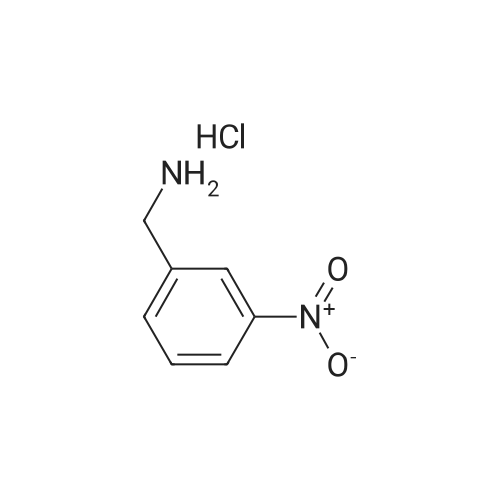
Preparation 124 To a mixture of 1,1'-thiocarbonyldiimidazole (1.87g), imidazole (143mg), acetonitrile (27ml) were added a suspension of <strong>[26177-43-5]3-nitrobenzylamine hydrochloride</strong> (1.32g) in acetonitrile (7ml) and imidazole (477mg) at 2~4C. After stirring for 3 hours at ambient temperature, to the reaction mixture was added 4-fluoro-1,2-benzenediamine (1,77g) under stirring. The reaction mixture was stirred at 50C for 3 hours, and at ambient temperature for 20 hours. The reaction mixture was evaporated under reduced pressure to give a residue which was purified by silica gel chromatography and eluted with ethyl acetate-n-hexane=2/3 to give N-(2-amino-4-fluorophenyl)-N'-(3-nitrobenzyl)thiourea (2.28g). MS:321(M+1) NMR(DMSO, delta):4.80(2H,d,J=5.9Hz), 5.17(2H,br s), 6.2-6.6(2H,m), 6.8-7.1(1H,m), 7.61(1H,t,J=7.9Hz), 7.78(1H,d,J=7.7Hz), 7.8-8.2(3H,m), 9.00(1H,br s)
(52a) 5-fluoro-1H-benzimidazole-2-thiol A mixture of 3,4-diamino-1-fluorobenzene (10 g, 79.3 mmol), carbon disulfide (70 ml, 1164 mmol), and methanol (100 ml) was stirred at room temperature for 86 hours and 50 minutes. After the reaction mixture was concentrated, the residue was suspended in hexane. The resultant precipitate was collected by filtration and washed with hexane to obtain the title compound (13.1 g, yield: 98.2%) as a brown solid. 1H NMR(400 MHz, DMSO-d6) deltappm; 6.90-6.99(2H, m), 7.06-7.13(1H, m), 12.58(1H, s), 12.61(1H, s).
A solution of 4-fluorobenzene-l ,2-diamine 5 (49 g, 320 mmol) in ethanol (500 ml) was stirred at 25C. The mixture was cooled to 0C. Ethyl 2-oxoacetate (24.48 g, 240 mmol) was added and stirred for 0.5 h at 0C. The mixture was stirred and refluxed for 3 h at 120C. The mixture was evaporated under vacuum. Dichloromethane (500 ml) was added and the mixture was stirred for 0.5 h at 25C. The precipitate was filtered off. The solid was washed with tetrahydrofuran (300 ml X 2) and washed with methanol (300 ml X 2). The filtrate was evaporated to dryness under vacuum. The residue was washed with methyl t-butyl ether (100 ml). 5.8 g of a mixture (50/50) of intermediates 6 and 7 was obtained as brown powder (Yield 1 1.24 %).
In ethanol;Reflux;
To a solution of 4-flourobenzene-1,2-diamine (5 g, 39.6 mmol) in ethanol (20 ml) was added ethyl 2-oxoacetate (4.45 g, 43.6 mmol)). The reaction mass was heated at reflux for overnight. The solvent was evaporated under reduced pressure and the residue was diluted with ethyl acetate and then evaporated to dryness to get the crude compound. The crude compound was washed with pet ether to get crude compound (4.6 g, 70% yield) as a mixture of regioisomers (black solid). This crude compound was taken to the next step without separation of isomers. MS: MS m/z 162.8 (M+-1).
In ethanol; toluene; at 20℃; for 2.0h;
Intermediate 143: 7-Fluoroquinoxalin-2(lH)-one andIntermediate 144: 6-Fluoroquinoxalin-2(lH)-oneA mixture of 4-fluorobenzene-l,2-diamine (5.0 g, 39.7 mmol) and ethyl oxoacetate (50 wt % in toluene, 17 mL, 79.4 mmol) in ethanol (100 mL) was stirred for two hours at room temperature. The precipitate was collected by filtration, washed with ethanol, and dried under vacuum giving 4.5 g of a solid. 1H NMR revealed a 1 : 1 mixture of Intermediates 143 and 144. This mixture was used for the next step.MS CBS): 165 (MH+) for C8H5FN2O
Under an N2 atmosphere, 4-fluorobenzene-l,2-diamine (5.0Og, 39.6 mmol) was suspended in EtOH (220 <n="84"/>mL) and 5M HCl (16OmL) was added. The reaction was warmed to 50C and 2,4- pentandione (8.14mL, 7.93mmol) was added and the reaction was heated to reflux for 30 min. Upon cooling to room temperature, the reaction was neutralized with a saturated solution of NaHCO3(aq) and extracted with dichloromethane. The layers were separated and the aqueous layer was washed with additional dichloromethane (2x). The organics were then combined, dried (Na2SO4), filtered and concentrated in vacuo. The resulting solid was triturated with dichloromethane to afford the title compound. 1H NMR (400MHz) (DMSO-d6) delta 7.43-7.19 (bm, 2H); 6.92 (bs, IH); 2.44 (s, 3H).
Stage #1: 3-aminopyrazinoic acid; 4-fluoro-1,2-phenylenediamine With N-ethyl-N,N-diisopropylamine; HATU In N,N-dimethyl-formamide at 20℃;
Stage #2: With acetic acid
12
The 5-bromo-3-(6-fluoro-lH-benzimidazol-2-yl)pyrazin-2- amine used as a reagent was prepared as follows:ηATU (4.2 g) was added to a mixture of 3-aminopyrazine-2-carboxylic acid (1.39 g), 1 ,2-diamine-4-fluoro-benzene ( 1.26 g) and Diisopropylethylamne (DIEA) ( 1.7 ml) in DMF (50 ml). The reaction mixture was stirred at ambient temperature overnight, and then poured into water. The precipitate was collected by filtration and recrystallised from acetic acid (10 ml). There was thus obtained 3-(6-fluoro-lH-benzimidazol-2-yl)pyrazin-2-amine (1.7 g); Mass Spectrum: M-H+ 228; NMR Spectrum: (DMSOd6) 8.15 (d, IH), 7.88 (d, IH), 7.45 (m, IH), 7.16 (m, 2H).
Cl3CC(NH)OMe (1 eq.) was added slowly to a solution of compound (Ql) in AcOH (0.8 M) at 00C, then the reaction mixture was stirred at RT for 2 h. The reaction mixture was diluted with EtOAc, and the resulting organic phase was washed The with sat. aq. NaHCO3 and dried(Na2SO4). Evaporation of the solvent under reduced pressure afforded (75%) the title compound.MS (ES+) C8H4Cl3FN2 required: 252, 254, found: 253, 255 (M+H)+.
With potassium hydrogencarbonate; In acetonitrile; at 0 - 30℃; for 4h;
A round-bottom flask charged with 4-fluorobenzene-1, 2-diamine 7 (126 mg, 1 mmol) and KHCO3 (200 mg, 2 mmol) wastaken up in CH3CN at 0 C. After 5 min, (Boc)2O (0.23 mL, 1 mmol)was added in portions and the resulting mixture was stirred overnight.Then, the mixture was concentrated and extracted withEtOAc. The combined organic extracts were washed with brine,dried over anhydrous Na2SO4 and concentrated in vacuo. Theproduct obtained by chromatography on a silica gel column. 90%isolated yield; white solid; Mp: 95-97 C. 1H NMR (300 MHz,DMSO-d6) delta: 8.18 (s, 1H), 7.22-7.07 (m, 1H), 6.45 (dd, J1 =2.85 Hz,J2 11.13 Hz, 1H), 6.28 (td, J1= 2.88 Hz, J2 = 8.55 Hz, 1H), 5.10 (s,2H), 1.44 (s, 9H); ESI-MS m/z: 227.1 [M+H]+.
69%
at 0 - 20℃; for 0.0833333h;
Step 1. To 4-fluorobenzene-1,2-diamine (13.88 g, 110.04 mmol) was added ditert-butyldicarbonate (50 mL) at 0C. The resulting mixture was stirred at ambient temperature for 5 mm, then diluted with ice-water (500 mL) and extracted with 1:1 ethyl acetate-hexane (500 mL). The organic phase was washed with brine (300 mL), dried over Mg504, then filtered and the remaining liquid evaporated. The residual solid was washedwith hexane to afford tert-butyl (2-amino-4-fluorophenyl)carbamate (17.0 g, 69% yield). 1H NMR (500 MHz, Acetone-d6) oe 7.48 (br. s, 1H), 7.05 - 7.26 (m, 1H), 6.51 - 6.59 (m, 1H), 6.27 - 6.38 (m, 1H), 4.80 (s, 2H), 1.47 (s, 9H).
44%
In tetrahydrofuran; at 20℃; for 6h;
Tert-butyl 2-amino-4-fluorophenylcarbamate (Compound 0113-60) A solution of 4-fluorobenzene-l,2-diamine (0.252 g, 2.0 mmol), (BoC)2O(0.436 g, 2.0 mmol) in dry THF (6 mL) was stirring at room temperature for 6 h. The reaction mixture was concentrated in vacuo, extracted with ethyl acetate. The organic phase was washed with saturated NaHCO3, dried over anhydrous Na2SO4, evaporated to afford crude product as oil. The product was further purified by flash column chromatography on silica gel (ethyl acetate in petroleum ether, 20% v/v) to afford pure product 0113-60 (0.2 g, 44 %) as a yellow solid. LCMS: 171 [M-56+l]+, 1H-NMR (400 MHz. DMSO-J6) «5 1.51 (s, 9H), 5.20 (s, 2H), 6.35 (m, IH), 6.52 (dd, J= 11.2, 2.8 Hz, IH), 7.17 (m, IH), 8.32 (s, IH).
With phosphoric acid In water at 20 - 165℃; for 8h; Inert atmosphere;
6.2. Standard procedure 1 for the preparation of 3-(benzimidazol-2′-yl)coumarins (3a-m)
General procedure: To a solution containing of 1,2-phenylenediamines 1 (1.0equiv) and 3-ethoxycarbonylcoumarins 2 (1.0equiv) were added o-phosphoric acid (85%, 15-16equiv). The reaction mixture was stirred at room temperature for 5.0min and at 165°C for another 8.0h. Then the solution was permitted to cool down to 100°C and poured in a large volume (500mL for 0.20mol of coumarin ester) of stirred water. The solution was neutralized with saturated aqueous NaHCO3 solution and extracted with EtOAc (3×15mL). The combined organic layers were washed with brine (15mL), dried over MgSO4 (s), filtered, and concentrated under reduced pressure to provide crude solids, which were then recrystallized in EtOH to give the pure coumarins 3a-m in 40-67% yields with purity >99.6%, as checked by GC.
65%
With phosphoric acid In water at 165℃; for 8h; Inert atmosphere;
With tetrabutylammonium tungstate; In 1-methyl-pyrrolidin-2-one; at 140℃; under 760.051 Torr; for 24h;Schlenk technique;Catalytic behavior;
General procedure: A typical procedure for the I-catalyzed reaction of aryl diamines with 1 atm CO2 was as follows: diamine (1 mmol), I (0.15 mmol), and N-methylpyrrolidone (NMP) (1 mL) were charged in a Schlenk tube with a magnetic stir bar. CO2 (1 atm) was introduced by a balloon, and the reaction mixture was stirred at 140 C for 24 h. The reaction solution was periodically analyzed by GC, LC, GC-MS, or NMR. A Teflon vessel placed in a stainless steel autoclave was used for reactions with CO2 at 20 atm.
With Oxone; In ISOPROPYLAMIDE; water; at 0 - 20℃;Inert atmosphere;
Intermediate 1 : 6-fluoro-2-(6-fluoropyridin-3-yl)-lH-benzimidazoleIn a 2L round-bottom flask equipped with magnetic stirring and nitrogen inlet, 6- fluoropyridine-3-carbaldehyde (25 g, 196 mmol) was dissolved in DMA (400 ml) and the solution was cooled to 0C. 4-Fluorobenzene-l,2-diamine (25.5 g, 196 mmol) was added (exotherm). Water (360 ml) was added followed by slow addition of potassiumperoxymonosulfate (78 g, 127 mmol). The dark brown slurry was allowed to age at room temperature. After 3h, the reaction mixture was diluted with water (2L) and the remaining slurry was allowed to age overnight at room tekperature. The reaction mixture was filtered (slow filtration) and the wet cake was washed with additional water. The wet cake was dried over nitrogen sweep and vacuum. The filter cake was later transferred to a round-bottom flask and triturated with MeCN. The mixture was filtered and the solid was dried over nitrogen sweep and under vacuum to afford solid product 6-fluoro-2-(6-fluoropyridin-3-yl)-lH-benzimidazole. LC- MS (ES, m/z) C12H7F2N3 : 231; Found: 232 [M+H]+.
(0618) A mixture of 1,1,1-trimethoxyethane (19.0 g, 159 mmol) and 4-fluorobenzene-1,2-diamine (2.00 g, 15.9 mmol) in MeOH (10 mL) was stirred at 60 C for 12 h. After cooling to ambient temperature, the mixture was concentrated. The residue was purified by flash silica gel chromatography (ISCO; 40 g SepaFlash Silica Flash Column, Eluent of 50100% EA/PE gradient (at) 40 mL/min) to give 5-fluoro-2-methyl-1H-benzo[d]imidazole as a solid.
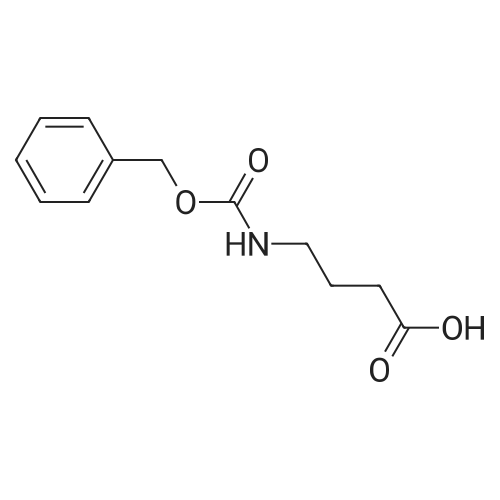
Stage #1: 4-(benzyloxycarbonylamino)butyric acid With triethylamine; isobutyl chloroformate In tetrahydrofuran at -15℃; Inert atmosphere;
Stage #2: 4-fluoro-1,2-phenylenediamine In tetrahydrofuran at -15 - 20℃; Inert atmosphere;
78%
Stage #1: 4-(benzyloxycarbonylamino)butyric acid With triethylamine; isobutyl chloroformate In tetrahydrofuran at -15℃; for 1h;
Stage #2: 4-fluoro-1,2-phenylenediamine In tetrahydrofuran at 20℃; for 2h;
27 Synthesis of benzyl (4 - ((2-amino-4-fluorophenyl) amino) -4-oxobutyl) carbamate
To the distilled tetrahydrofuran was added 4 - (((benzyloxy) carbonyl) amino) butanoic acid (799 mg, 3.36 mmol)Was added triethylamine (702 mL, 5.04 mmol) and isobutyl chloroformate (439 mL, 3.36 mmol) in tetrahydrofuranThe dissolved solution was slowly added in turn. After stirring at the same temperature for 1 hour, 4-fluoro-1,2-phenylenediamine(466 mg, 3.69 mmol) was dissolved in tetrahydrofuran and added. The reaction mixture was stirred for 2 hours at ambient temperatureAfter confirming that the reaction was terminated by TLC, the tetrahydrofuran was removed by decompression. Add ethyl acetate and distilled water.And washed with brine. The organic layer was dried using anhydrous magnesium sulfate and then reduced in pressure to remove the solvent. The mixture was purified by flash column chromatography (EtOAc: Hex = 4: 1, Rf = 0.3) to give the title compound 899Mg, 78%, brown solid)
General procedure: A solution of commercial nitrobenzaldehyde (1.0 equiv) and sodium metabisulfite (3.0 equiv) is warmed into refluxing DMF for 1 h. After cooling at room temperature, a solution of o-phenylenediamine (1.0 equiv) or 2-aminobenzenethiol (1.0 equiv) in DMF is carefully added. The mixture is stirred in refluxing DMF until the reaction is completed. After cooling at room temperature, the product is precipitated by addition of a frozen mixture of water and ethyl acetate (1:2). Crude material is filtrated and washed with cold ethyl acetate, then with water and finally with ether. Product is dried under vacuum and might be used for the next step without further purification.
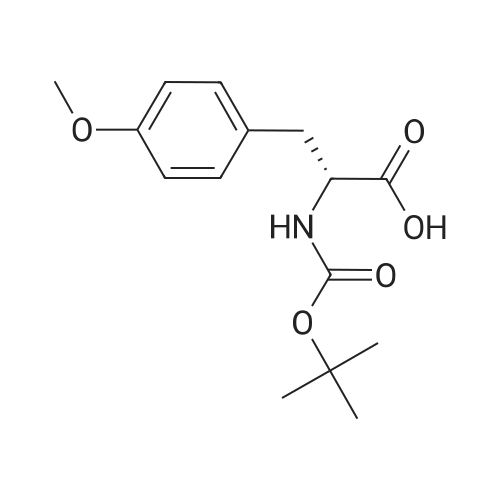
(R)-tert-butyl (1-(5-fluoro-1H-benzo[d]imidazol-2-yl)-2-(4-methoxyphenyl)ethyl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
(K)-iert-Butyl (1-(5-fluoro-1H-benzo[ ]imidazol-2-yl)-2-(4- methoxyphenyl)ethyl)carbamate: In a flame dried round-bottomed flask equipped with a magnetic stir bar and under inert atmosphere (N2), a solution of commercially available (ft)-2-((te/f-butoxycarbonyl)amino)- 3-(4-methoxyphenyl)propanoic acid (500 mg, 1.69 mmol) in AcCN (16.9 mL) was treated at 0 C with 4-ethylmorpholine (0.44 mL, 3.4 mmol), TBTU (544 mg, 1.69 mmol) and 4- fluorobenzene-1 ,2-diamine (220 mg, 1.69 mmol). The reaction mixture was stirred at 0 C for 1 h. The reaction mixture was diluted with EA and water. The org. phase was dried over MgS04, filtered, and the solvent was removed under reduced pressure. The residue was dissolved in glacial acetic acid (16.9 mL) and the reaction mixture was stirred at 60 C for 60 min. The mixture was cooled to rt and the solvent was removed under reduced pressure. The residue was partitioned between EA (30 mL) and sat. aq. NaHC03 (30 mL). The org. layer was washed with sat. aq. NaHC03 (twice 30 mL), dried over MgS04, filtered, and the solvent was removed under reduced pressure. Purification of the residue by combiflash (1 :1 hept-acetone) gave the title compound as yellow solid: TLC: rf (1 : 1 hept-acetone) = 0.46. LC-MS-conditions 07: tR = 0.72 min; [M+H]+ = 386.18.
With sodium metabisulfite; In N,N-dimethyl-formamide; at 120℃;
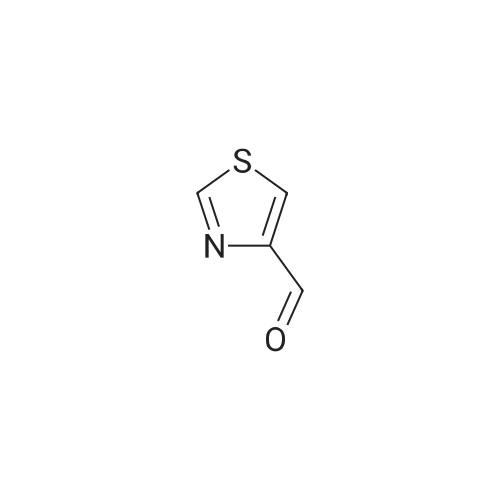
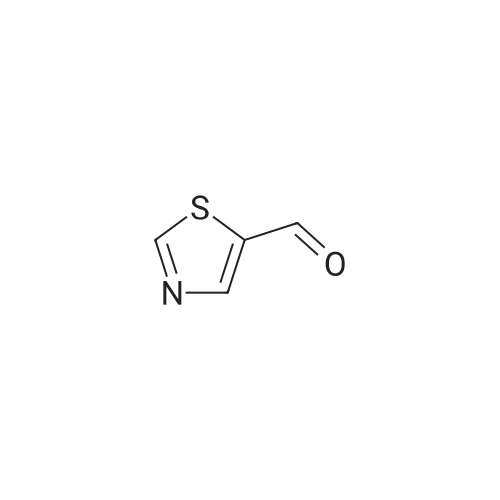
General procedure: A solution of substituted o-phenyldiamine (1.0 equiv), thiazole-4-aldehyde or pyridine-2-aldehyde (1.0 equiv) with sodium pyrosulfite in DMF was stirred at 120° C overnight. On completion of the reaction monitored by TLC, the solvent was evaporated and the residue was purified by silica gel chromatography by DCM/MeOH system to afford the final product. If necessary, the crudeproduct could be recrystallized in DCM or dichloroethane to afford pure sample.
General procedure: A mixture of 2-phenoxycarbonyl-4,5-dichloropyridazin-3(2H)-one (2a, 1.2 equiv), compounds 4 (1 equiv), and toluene (10 mL)was stirred at reflux conditions until amide intermediate wasconsumed (monitored by TLC). After cooling to r.t., the resultingprecipitate was filtered off and the solvent was evaporatedunder reduced pressure. The residue was transferred to anopen-bed silica gel column (3 × 7 cm). The column was elutedwith n-hexane-THF (1:2, v/v). Fractions containing compounds5 were combined and evaporated under reduced pressure togive compounds 5.
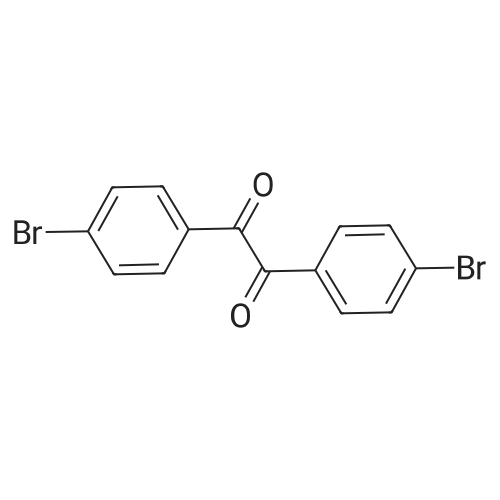
4-Fluoro-1,2-phenylenediamine (0.68 g, 5.40 mmol) and 4,4'-dibromobenzyl (2.00 g, 5.43 mmol) were dissolved in toluene-acetic acid consisting of 20 mL of toluene and 30 mL of acetic acid. In the solution, stir well and reflux for 8 hours. After the reaction is completed, cool to room temperature, then pour the reaction solution into water, first extract with dichloromethane, and then use dichloromethane/petroleum ether (v/v=2:3). Separation by silica gel column chromatography as a rinse agent,After rotary evaporation, it was dried to give Intermediate 1 (2.10 g, yield 84%).The product is a white powder.
3-(5-fluoro-1H-benzimidazol-2-yl)-2(1H)-quinolinone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
71.7%
In ethanol at 80℃; for 6h; Sealed tube;
3 Example 3: Preparation of 3-(5-fluoro-1H-benzimidazol-2-yl)-2(1H)-quinolinone (compound 1a3)
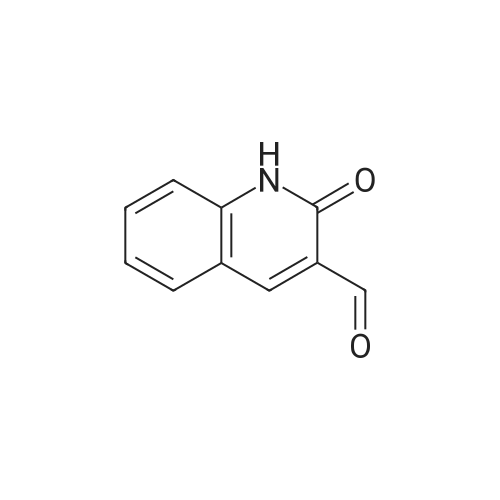
Referring to the implementation of the method of Example 2, compound 1a 0.1g (0.58mmol), 5-fluoro-1,2-phenylenediamine 0.08g (0.64mmol), and 6mL anhydrous ethanol were weighed and placed into pressure tubes (sealed tubes) at a temperature of 80°C with stirring until completion of reaction (TLC tracking tests at 6h). After cooling filtration, washed with anhydrous ethanol to give compound 1a3 0.116g, yield 71.7%.
General procedure: To a 10mL three-necked round bottle was charged with 1a (0.54 g, 5 mmol), imidazolium chloride (0.09 g, 0.5 mmol) and N,N-dimethylformamide 2 mL. The resulting solution was warmed to120 °C and stirred at this temperature for 6 h. When the reaction was completed, 25 mL water was added and the resulting mixture was extracted with 25 mL ethyl acetate twice. The combined organic layer was successively washed with H2O (50 mL) and then brine (50 mL), then dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel with petroleum ether and ethyl acetate or recrystallized from petroleum ether/EA to give the title products.
2-(2,5-dichlorophenyl)-6-fluoro-1H-benzo[d]imidazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
57%
In ethanol; at 80℃; for 8h;
General procedure: ZrO2-Al2O3 solid acid catalytic material was added to the stirred solution of O-phenylenediamines and different substituted aromatic aldehydes in a suitable solvent, and the resulting mixture was heated at a particular temperature and monitored by TLC. After the completion of the reaction, the reaction mixture was cooled and filtered and the residue was washed with ethanol to recover the solid acid catalyst. Filtrate was evaporated in vacuum to get the crude reaction product, which was then purified by silica-gel column chromatography using a suitable mobile phase to separate the desired product. The reaction products were characterized by melting point, 1H NMR and 13C NMR spectroscopy (Bruker, 400 MHz), LC-MS (Varian), and HPLC (Waters) techniques
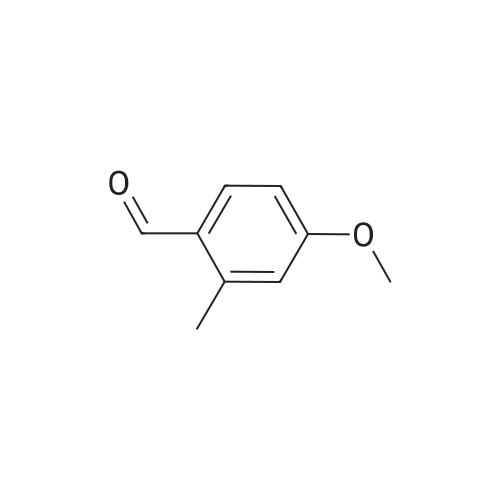
5-fluoro-2-(4-methoxy-2-methylphenyl)-1H-benzo[d]imidazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
72%
In ethanol at 80℃; for 8h;
Catalytic activity studies of solid acid catalytic materials in the synthesis of benzimidazoles
General procedure: ZrO2-Al2O3 solid acid catalytic material was added to the stirred solution of O-phenylenediamines and different substituted aromatic aldehydes in a suitable solvent, and the resulting mixture was heated at a particular temperature and monitored by TLC. After the completion of the reaction, the reaction mixture was cooled and filtered and the residue was washed with ethanol to recover the solid acid catalyst. Filtrate was evaporated in vacuum to get the crude reaction product, which was then purified by silica-gel column chromatography using a suitable mobile phase to separate the desired product. The reaction products were characterized by melting point, 1H NMR and 13C NMR spectroscopy (Bruker, 400 MHz), LC-MS (Varian), and HPLC (Waters) techniques
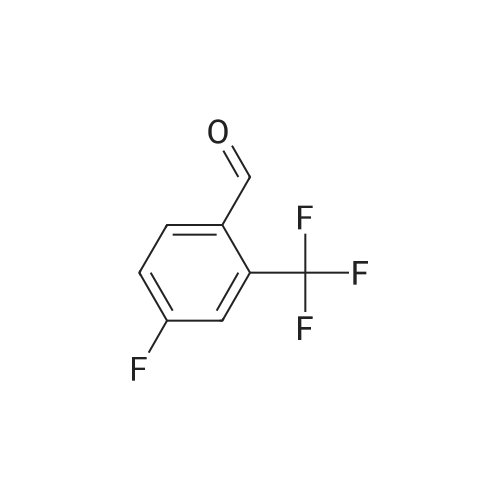
5-fluoro-2-(4-fluoro-2-(trifluoromethyl)phenyl)-1H-benzo[d]imidazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78%
In ethanol; at 80℃; for 4h;
General procedure: ZrO2-Al2O3 solid acid catalytic material was added to the stirred solution of O-phenylenediamines and different substituted aromatic aldehydes in a suitable solvent, and the resulting mixture was heated at a particular temperature and monitored by TLC. After the completion of the reaction, the reaction mixture was cooled and filtered and the residue was washed with ethanol to recover the solid acid catalyst. Filtrate was evaporated in vacuum to get the crude reaction product, which was then purified by silica-gel column chromatography using a suitable mobile phase to separate the desired product. The reaction products were characterized by melting point, 1H NMR and 13C NMR spectroscopy (Bruker, 400 MHz), LC-MS (Varian), and HPLC (Waters) techniques
(Z)-3-(2-(4-chlorophenyl)-2-oxoethylidene)-6-fluoro-3,4-dihydroquinoxalin-2(1H)-one[ No CAS ]
(Z)-3-(2-(4-chlorophenyl)-2-oxoethylidene)-7-fluoro-3,4-dihydroquinoxalin-2(1H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
39%
With Methyl p-tolyl sulfone; In N,N-dimethyl-formamide; at 20℃; for 72h;Inert atmosphere;
General procedure: A solution of <strong>[5814-38-0]ethyl 4-chlorobenzoylpyruvate</strong> 100 mg (0.39 mmol, 1.00 equiv) 12a, o-phenylenediamine (1.00 equiv) from 11a-f with or without an additive (1.00 equiv) (p-TsOH or DMAP) was stirred in 3.0 ml of DMF (abs) at rt under Ar for 72 h. A low soluble mixtureof ANTI/SYN regioisomers slowly precipitated within the reaction. The precipitate was collected by filtration or centrifugation, triturated by 3 ml of Et2O and crystallized from DMSO (if not otherwise stated) to yield the main solid regioisomer 16 or 17.
With triethylamine; In dichloromethane; at 0 - 20℃; for 2h;
[0278] To a solution of 4-fluorobenzene-1,2-diamine lb (1.39 g, 11.0 mmol) and triethylamine (3.26 mE, 23.2 mmol) in methylene chloride (20 mE) was added dropwise a solution of diphosgene (0.69 mE, 5.72 mmol) in methylene chloride (5 mE) at 0 to 5 C. The resulting suspension was stirred for 2 hours at room temperature and filtered. The collected white solid was washed with water and dried to give compound 2b (1.56 g, 93%). ?H NMR (400 MHz, DMSO-d5) oe 10.73 (s, 1H), 10.61 (s, 1H), 6.97-6.81 (m, 1H), 6.74 (m, 2H); MS (ESI): mlz 153.1 [M+1]
With C23H3BF16N2O; In toluene; at 25℃; for 3h;Green chemistry;
A 0.03 mol% Lewis acid-base bifunctional catalyst I (where Rf = CF3; R1, R2, R3, R4, R5, R6 = F) was added to a 100 mL single-mouth flask.0.2 mol <strong>[199287-52-0]4,5-difluorosalicylaldehyde</strong> (R7=4,5-2F-2-OH-C6H2), 0.1 mol p-fluoro o-phenylenediamine (R9=F; R8, R10, R11=H), 15 mL Toluene, the reaction was stirred at 25 C for 3 hours,TLC followed the reaction until the reaction was complete. The results showed that the yield of product II (R7=4,5-2F-2-OH-C6H2; R9=F; R8, R10, R11=H) was 93%; after catalyzing the catalyst system 10 times, it was catalyzed. No drop in performance.
With C23H3BF16N2O; In toluene; at 25℃; for 3.0h;Green chemistry;
A 0.03 mol% Lewis acid-base bifunctional catalyst I (where Rf = CF3; R1, R2, R3, R4, R5, R6 = F) was added to a 100 mL single-mouth flask.0.2 mol <strong>[77532-90-2]2-cyano-4-fluorobenzaldehyde</strong> (R7=4-F-2-CN-C6H3), 0.1 mol p-fluoro o-phenylenediamine (R9=F; R8, R10, R11=H), 15 mL of toluene The reaction was stirred at 25 C for 8 hours.TLC followed the reaction until the reaction was complete. The reaction results showed that the yield of product II (R7=4-F-2-CN-C6H3; R9=F; R8, R10, R11=H) was 99%; after 10 times of catalyst system was reused, its catalytic performance was not. See decline.
2-(2'-chloro-3'-methoxyphenyl)-5-fluoro-1H-benzo[d]imidazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
64.7%
With sodium metabisulfite; In N,N-dimethyl-formamide; for 4h;Reflux;
General procedure: o-Phenylene diamine derivative (1 mmol) and substitutedbenzaldehyde (1 mmol) were taken in N,N-dimethylformamide(10 mL) into a round-bottomed flask 100 mL. A pinch of sodiummetabisulfite (Na2S2O5) was added into it and refluxed solution for4 h. Progress of reactionwas checked by thin layer chromatography(TLC). After completion, reaction mixture was poured onto crushedice (100 mL). Precipitates were appeared immediately which werefiltered. The obtained solid crude products were crystallized fromethanol. Compounds were structurally characterized by variousspectroscopic techniques.
3'-bromo-2'-(5-fluoro-1H-benzo[d]imidazole-2-yl)-6'-methoxyphenol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65.0%
With sodium metabisulfite; In N,N-dimethyl-formamide; for 4h;Reflux;
General procedure: o-Phenylene diamine derivative (1 mmol) and substitutedbenzaldehyde (1 mmol) were taken in N,N-dimethylformamide(10 mL) into a round-bottomed flask 100 mL. A pinch of sodiummetabisulfite (Na2S2O5) was added into it and refluxed solution for4 h. Progress of reactionwas checked by thin layer chromatography(TLC). After completion, reaction mixture was poured onto crushedice (100 mL). Precipitates were appeared immediately which werefiltered. The obtained solid crude products were crystallized fromethanol. Compounds were structurally characterized by variousspectroscopic techniques.
2'-bromo-4'-chloro-6'-(5-fluoro-1H-benzo[d]imidazole-2-yl)phenol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
87.8%
With sodium metabisulfite; In N,N-dimethyl-formamide; for 4h;Reflux;
General procedure: o-Phenylene diamine derivative (1 mmol) and substitutedbenzaldehyde (1 mmol) were taken in N,N-dimethylformamide(10 mL) into a round-bottomed flask 100 mL. A pinch of sodiummetabisulfite (Na2S2O5) was added into it and refluxed solution for4 h. Progress of reactionwas checked by thin layer chromatography(TLC). After completion, reaction mixture was poured onto crushedice (100 mL). Precipitates were appeared immediately which werefiltered. The obtained solid crude products were crystallized fromethanol. Compounds were structurally characterized by variousspectroscopic techniques.
4'-(5-fluoro-1H-benzo[d]imidazole-2-yl)benzene-1',2',3'-triol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91%
With sodium metabisulfite In N,N-dimethyl-formamide for 4h; Reflux;
20 General procedure for the synthesis of 2-aryl benzimidazoles
General procedure: o-Phenylene diamine derivative (1 mmol) and substitutedbenzaldehyde (1 mmol) were taken in N,N-dimethylformamide(10 mL) into a round-bottomed flask 100 mL. A pinch of sodiummetabisulfite (Na2S2O5) was added into it and refluxed solution for4 h. Progress of reactionwas checked by thin layer chromatography(TLC). After completion, reaction mixture was poured onto crushedice (100 mL). Precipitates were appeared immediately which werefiltered. The obtained solid crude products were crystallized fromethanol. Compounds were structurally characterized by variousspectroscopic techniques.
2-(2'-chloro-3',4'-dimethoxyphenyl)-5-fluoro-1H-benzo[d]imidazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
62.3%
With sodium metabisulfite; In N,N-dimethyl-formamide; for 4h;Reflux;
General procedure: o-Phenylene diamine derivative (1 mmol) and substitutedbenzaldehyde (1 mmol) were taken in N,N-dimethylformamide(10 mL) into a round-bottomed flask 100 mL. A pinch of sodiummetabisulfite (Na2S2O5) was added into it and refluxed solution for4 h. Progress of reactionwas checked by thin layer chromatography(TLC). After completion, reaction mixture was poured onto crushedice (100 mL). Precipitates were appeared immediately which werefiltered. The obtained solid crude products were crystallized fromethanol. Compounds were structurally characterized by variousspectroscopic techniques.
4.1.1 General synthesis procedure for compounds 5(5a1-5d6)
General procedure: 2-Oxo-quinoline 3-carbaldehyde derivatives 4(4a-4d) (1mmol), o-phenylenediamine derivatives (1.0mmol) and 3mL methanol were mixed in a pressure tube and heated at 90°C for 4h. After the reaction, the mixture was cooled to room temperature and filtered to yield the product as yellow powder.
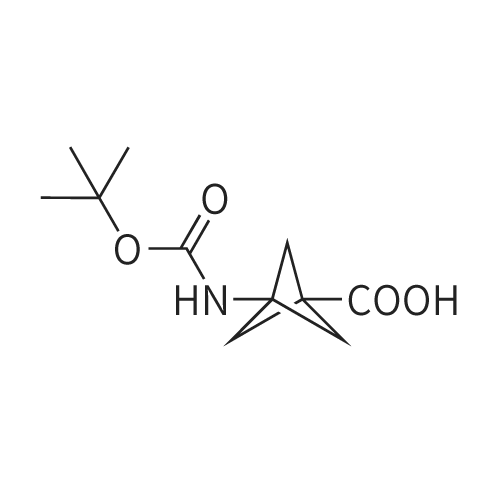
3-(5-fluoro-1H-benzo[d]imidazol-2-yl)bicyclo[1.1.1]pentan-1-amine hydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
45%
With hydrogenchloride; In water; at 160℃; for 0.833333h;Microwave irradiation;
[0275] A solution of 2-amino-4-fluoroaniline (0.278g, 2.20mmol) and 3-((tert- butoxycarbonyl)amino)bicyclo[l. l. l]pentane-l-carboxylic acid (0.500g, 2.20mmol) in a IN aqueous HCl (l l.OmL) was microwaved at 160C for 50 minutes. The dark mixture was filtered through a plug of Ci8 silica gel and washed with ACN. The filtrate was concentrated to provide the crude product which was further purified by flash chromatography (Ci8 Si02, H20/ACN buffered with 0.1% formic acid) to afford the desired product. The material was re-dissolved in EtOAc/Et20 and treated with 2N HCl in ether to generate the HCl salt. The resulting precipitate was collected via filtration and washed with Et20 to provide 0.250g (45%) of Compound 17 as an off-white solid. lU NMR (400 MHz, DMSO-J6) delta 9.17 (br s, NH, 3H), 7.76 (dd, =4.5, 8.9 Hz, 1H), 7.60 (dd, =2.3, 8.7 Hz, 1H), 7.36 (dt, =2.3, 9.4 Hz, 1H), 2.60 (s, 6H); LC/MS (APCI) mlz 218.1 [Ci2Hi2FN3+H]+.
5-fluoro-2-(3-(trifluoromethyl)phenyl)-1H-benzo[d]imidazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
With sodium hydrogensulfite In N,N-dimethyl-formamide at 80℃;
General procedure: Synthesis of 2-aryl benzimidazoles was done by the condensation reaction of o-phenylenediamine with the respective substituted aromatic aldehyde in the presence of NaHSO3. An equimolar concentration of 0.5mmol of ortho-phenylenediamine and the respective aryl-aldehyde 0.5mmol were systematically mixed in 2ml of DMF along with (0.15)mmol of sodium hydrogen sulphite. The reaction mixture was stirred at 80°C until completed and checked periodically with TLCs. By adding 20ml of distilled water (with constant stir) the mixture was cooled at room temperature. The product was formed and separated as free-floating solid. It was accumulated by suction filtration, washed with distilled water and finally dried. The gummy solid material was extracted with ethyl acetate and washed with water and brine solution. It was dried on sodium sulphate and the final residue was extracted with a column on silica gel by using hexane: ethyl acetate (2-6:1-3) as eluent [32]. The overall reaction for the synthesis of 2-Aryl-substituted benzimidazoles is given below.
N-ethyl-5-fluoro-1H-benzo[d]imidazol-2-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
In isopropyl alcohol; at 80℃; for 0.25h;Microwave irradiation;
General procedure: A mixture of o-phenylenediamine (1.0 mmol) and EDC.HCl (1.1 mmol) in 2-propanol (10 mL) was irradiated in microwave apparatus at 200 W (80 C) for 15 min. The reaction was monitored by TLC (eluent CH2Cl2 / MeOH 95:5). After completion, the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (2 x 25 mL). The combined organic layers were washed with brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure and the obtained crude product was purified by flash column chromatography using silica gel (100-200 mesh) with 1-5% MeOH/CH2Cl2 as eluent. The fractions containing product were collected and concentrated under reduced pressure to afford N-ethyl-1Hbenzo[d]imidazol-2-amine (1a, 90% yield) as an off white solid.
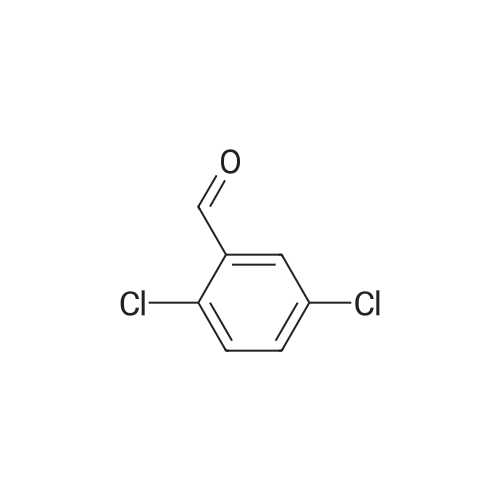
2-(2-bromo-5-fluorophenyl)-5-fluoro-1H-benzo[d]imidazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
68%
With polyphosphoric acid; at 140℃;
General procedure: The appropriate amount of polyphosphoric acid (PPA) was placed in a 100 mL round bottom flask and heated to 140 C. The substituted O-phenylenediamine (6 mmol) and substituted 2-bromobenzoic acid (6 mmol) were added and refluxed for 8 h with stirring. After the reaction was completed, it was cooled to 80-100 C, and an appropriate amount of ice-water mixture was poured into the flask, neutralized with a saturated aqueous sodium hydroxide solution, filtered, and the cake was taken and dried to give a solid powder. The crude product was purified by recrystallization from methanol to give the target compound (Z-1 to Z-6 and A-38).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping