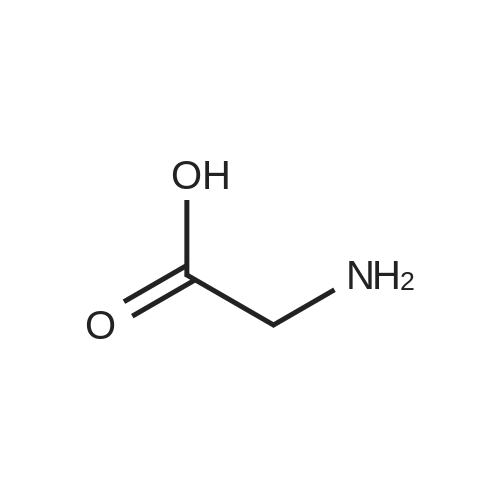
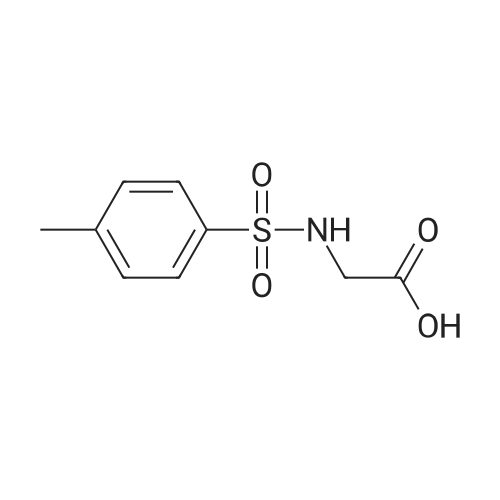
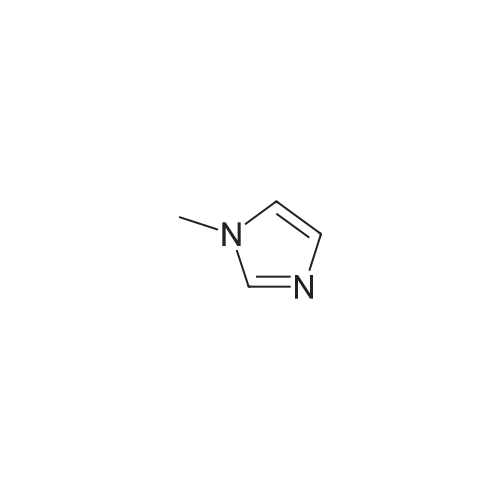
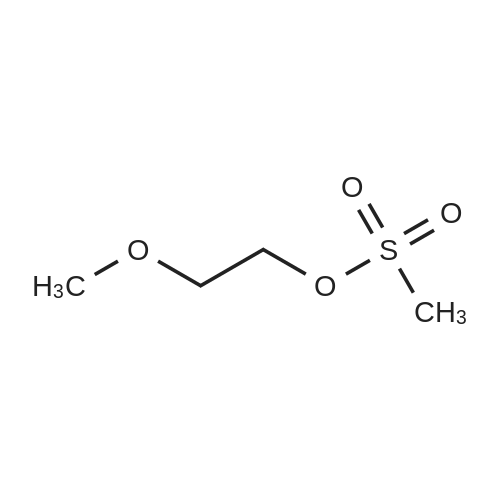
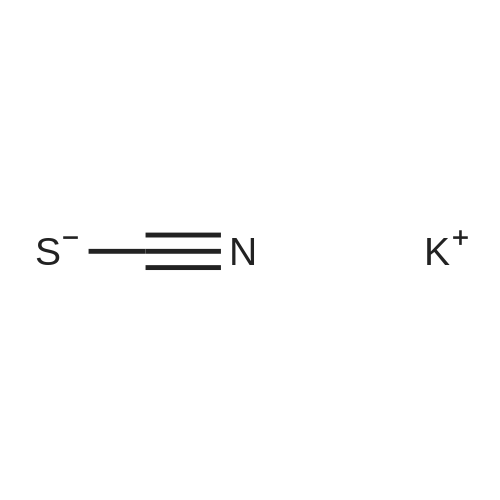|
With hydrogenchloride at 85 - 135℃; |
|
|
With hydrogenchloride In water at 135℃; |
|
|
With hydrogenchloride In toluene |
|
|
With hydrogenchloride In water |
|
|
With hydrogenchloride In water |
|
|
With hydrogenchloride In water for 5h; Cooling with ice; |
|
| > 99 %Spectr. |
With hydrogenchloride In water Cooling with ice; |
|
|
With hydrogenchloride In water Cooling with ice; |
|
|
With hydrogenchloride In water Inert atmosphere; |
|
|
With hydrogenchloride at 0℃; |
|
|
With hydrogenchloride at 60℃; |
|
|
With hydrogenchloride In water at 20℃; for 24h; Cooling with ice; |
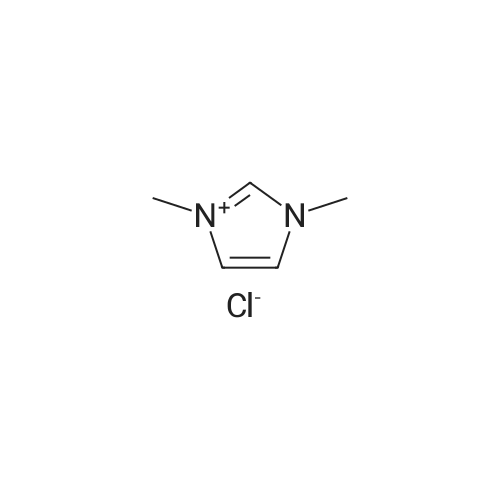
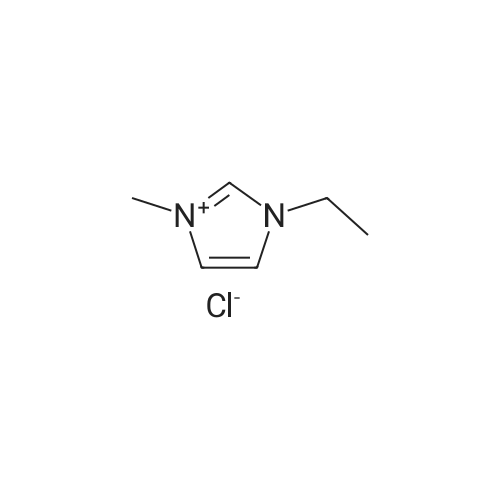
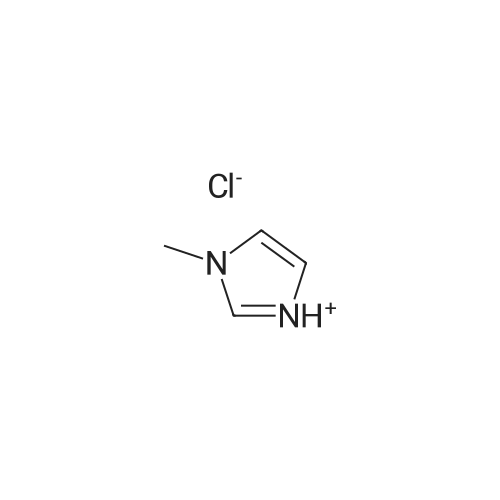
2.1 Synthesis of 1-H-3-methylimidazolium chloride ([HMIM]Cl)
General procedure: 1-Methyl imidazole (20 g, 0.244 mol) and hydrochloric acid (30 g, 0.296 mol) were mixed in a round-bottomed flask in ice-water bath and stirred for 24 h at room temperature. The resulting liquid was washed with ether (20 ml x3). Ether was then removed under rotary evaporation and the product was further dried under vacuum for 10 h to give a colorless liquid. The synthesis of ionic liquids [HMIM][HSO4], [HMIM][CH3COO] and [HMIM][H2PO4] followed a similar protocol as used for [HMIM]Cl. [HMIM]Cl: 1H NMR (400 MHz, DMSO-d6): 3.775 (s, 3H), 7.298 (s, 1H), 7.445 (s, 1H), 8.441(s, 1H). |
|
With hydrogenchloride In water at 0 - 5℃; for 2h; |
2.3. synthesis of bronsted acidic ionic liquids [Hmim]+L-
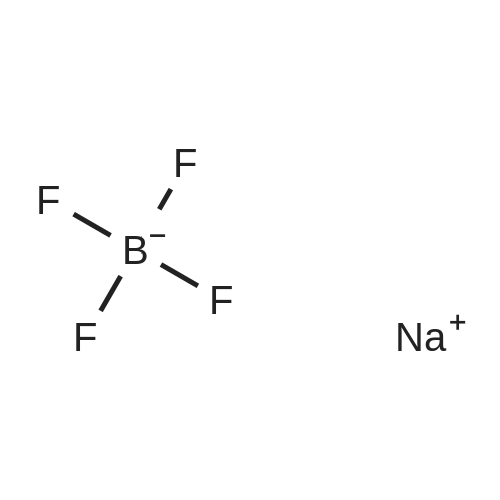
General procedure: The Bronsted acidic ionic liquids were synthesized as shown in Scheme 2. In a typical process [53,54], a 100 mL three necked,round-bottom flask was charged with 1-methylimidazole (6.15 g,75 mmol), which was allowed to cool to 0 °C in an ice bath with stirring. Then 40% aqueous tetrafluoroboric acid (75 mmol) was added at a rate sufficient to maintain the reaction temperature at 0-5 °C. After continuous stirring for another 2 h, water was evaporated under reduced pressure to afford the desired product 1-methylimidazolium tetrafluoroborate ([Hmim]+BF4-) as a colorless liquid. |
|
With hydrogenchloride In water at 50℃; for 5h; |
Synthesis of [mim][XCl] ILs
The [mim][XCl] ILs was prepared as follows: Firstly, N-methylimidazole was reacted with equal-mole hydrochloric acid at 50 C for 5 h to get [mim]Cl. After the reaction finished, the mixture was washed three times with ethyl acetate and dried under reduced pressure at 90 C for 4 h. Secondly, Lewis acidic ILs were synthesized through the reactions between the above products and different metal chlorides, including AlCl3, FeCl3, ZnCl2, TiCl4 and SnCl4, until these two materials completely mixed at 60 C under the protection of nitrogen. |
| 6.6g |
With hydrogenchloride In methanol; water at 25℃; for 1h; |
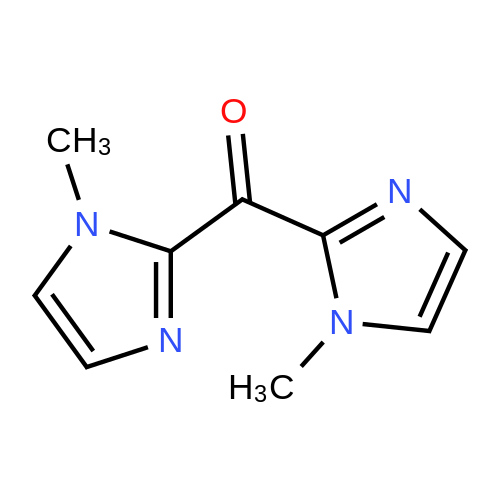
2 Preparation of intermediate B
Was dissolved in methanol 55 ml 1- methyl imidazole 4.5g (55mmol), 35% hydrochloric acid into a stirred 25 ° C.Aqueous solution of 5.8g (55mmol) was dropped for 1 hour reaction . Then , the solvent is removed in an evaporatorIntermediate Bs 6.6 g give ( compound represented by the following [ formula 10 ] ) of light brown solid dried under reduced pressure. |
|
With hydrogenchloride In water at 0℃; for 2.5h; |
Synthesis of ILs
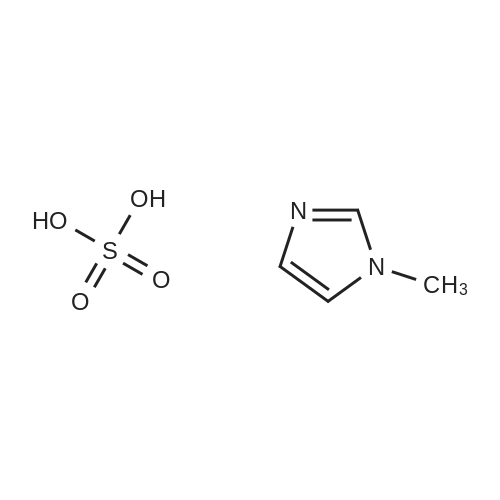
General procedure: Sulfuric acid (50% w/w) was added in a 1:1 M ratio with 1-methylimidazole over a period of 30 min with stirring and cooling to maintain the temperature at 0∘C. The reaction mixture was stirred for an additional period of 2 h.Then, water was removed under reduced pressure to yield the colorless liquid [Hmim][HSO4]. Thecrude productwaswashed several times with diethyl ether to remove non-ionic residue and then vacuum dried for 24 h under 80∘C. [Hmim][H2PO4], [Hmim][NO3], and [Hmim][Cl] were synthesized using similar procedures. |
|
With hydrogenchloride In acetonitrile at 60℃; for 24.5h; Cooling with ice; Inert atmosphere; Schlenk technique; |
|
|
With hydrogenchloride In water at 0℃; |
Synthesis of 1,3-dimethylimidazolium chloride (1) [35]
In a 250 mL round bottom flask immersed inan ice bath, 10 g of 1-methylimidazole was placed, and 10 mL of HCl (37% aqueous solution) was slowlyadded. After 20 min, the solvent was evaporated to yield the desired ionic liquid, 1-methylimidazoliumchloride, to be used in the following step without further purification. In a Q-tube reactor, 3.0 g of1-methylimidazolium chloride and 2.28 g of dimethyl carbonate (DMC; 0.025 mmol) were placed,and the mixture was heated at 170 °C for two hours to yield the desired product, 1,3-dimethylimidazolium chloride 1, in quantitative yield. 1H-NMR (200 MHz, DMSO-d6, ppm): δ = 9.46 (s, 1H),7.87 (s, 2H), 3.87 (m, 6H). |
|
Stage #1: 1-methyl-1H-imidazole With hydrogenchloride In water at 0 - 5℃; for 0.333333h;
Stage #2: In water at 50℃; for 12h; |
|
|
With hydrogenchloride In water at 20℃; for 24h; |
|
|
With sulfuric acid; sodium chloride at 20℃; |
[HMim]Cl was prepared by the reaction of 1-methylimidazole andHCl. HCl was liberated from the reaction of NaCl and H2SO4 in aseparated flask connected with the vessel containing 2 ml (25.1 mmol)1-methylimidazole. The reaction mixture was stirred at room temperatureovernight, then it was washed with 5 × 10 ml diethyl ether andwas dried in vacuum at 60 °C for 10 h. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping