There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
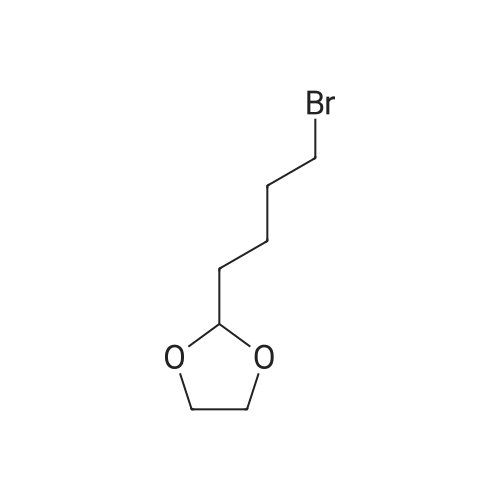
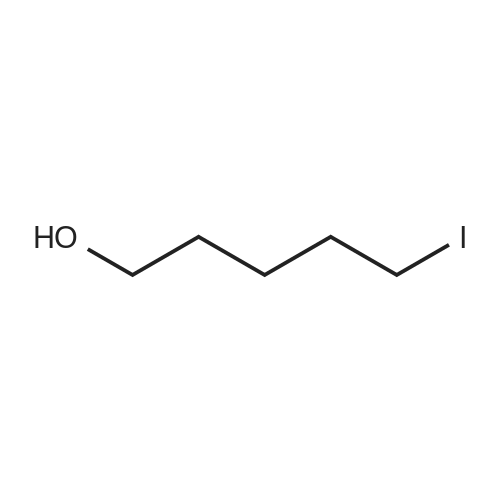
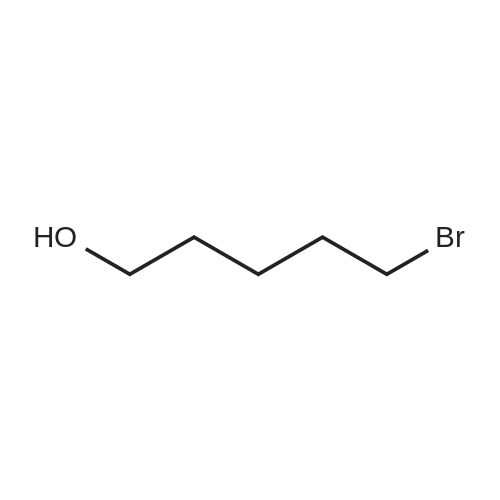
Structure of 34626-51-2 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Chemistry and Biology, 2010, vol. 17, # 1, p. 18 - 27
4
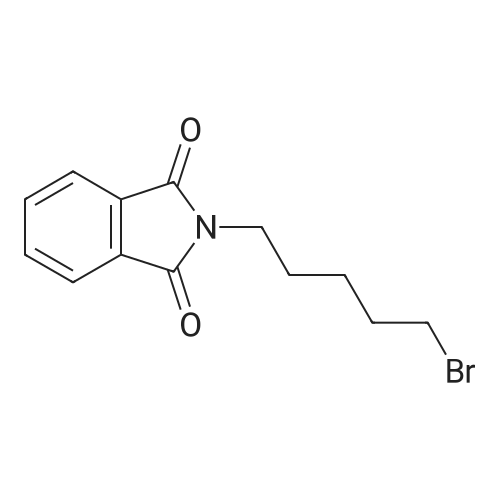
[ 111-29-5 ]
[ 34626-51-2 ]
Yield
Reaction Conditions
Operation in experiment
89.1%
With hydrogen bromide In benzene at 70 - 80℃; for 15 h;
In 250mL three-necked flask add 1a (14 g, 0.135 mol), 40percent hydrobromic acid (28 mL, 0.2 mol) and 60 mL of benzene, 70 ~ 80 ° C oil bath water for 15h, TLC detection, raw material points disappear. Washed successively with 5percent sodium hydroxide solution, 10percent hydrochloric acid and saturated brine, and dried over anhydrous sodium sulfate. The crude product was separated by silica gel column chromatography (petroleum ether: ethyl acetate, V / V = 8: 1) and concentrated to give colorless to pale yellow liquid 2a (20.1g, yield 89.1percent).
80%
With hydrogen bromide In water; toluene for 15 h; Dean-Stark; Reflux
A round bottom flask, equipped with a Dean-Stark trap, wascharged with 1,5-pentanediol (3.71 g, 35.6 mmol), concentratedhydrobromic acid (5 mL), and toluene (75 mL). The mixture wasstirred under refluxed for 15 h. After cooling, the reaction mixturewas extracted with 6 M sodium hydroxide (2 15 mL), 5percent HCl(2 15 mL), water (2 15 mL) and brine (20 mL). The organicswere dried over sodium sulfate and concentrated under reducedpressure to yield the title compound as an orange/brown oil(4.75 g, 2.84 mmol, 801H NMR (500 MHz, CDCl3) d 8.16(s, 1H), 7.65 (s, 1H), 7.46 (m, 6H), 7.25 (m 6H), 7.17 (m, 3H), 6.98(m, 3H), 6.48 (m, 4H), 3.59 (br, 2H), 3.11 (br, 4H), 2.65 (q, 12H,J = 7 Hz), 2.11 (br, 2H), 1.61 (m, 2H), 1.52 (m, 4H), 1.45 (m, 2H),1.41 (s, 9H), 1.31 (m, 2H), 1.01 (t, 18H, J = 7 Hz); 13C NMR(125 MHz, CDCl3) d 180.65, 171.21, 156.78, 155.79, 155.64, 146.13,140.35, 138.09, 130.98, 129.19, 128.54, 127.72, 126.14, 124.68,123.33, 120.15, 113.95, 103.13, 79.37, 79.15, 70.77, 47.23, 46.91,45.71, 44.64, 43.29, 29.65, 28.48, 28.14, 27.81, 26.53, 26.10,24.21, 9.07; HRMS (C54H56N4O7S): calculated 904.3870, observed904.3866.
Reference:
[1] Patent: CN105766907, 2016, A, . Location in patent: Paragraph 0026; 0041; 0042
[2] RSC Advances, 2015, vol. 5, # 37, p. 29114 - 29120
[3] Chemistry - A European Journal, 2015, vol. 21, # 30, p. 10721 - 10728
[4] Journal of Physical Chemistry, 1995, vol. 99, # 32, p. 12195 - 12203
[5] Organic Process Research and Development, 2003, vol. 7, # 3, p. 339 - 340
[6] Bioorganic and Medicinal Chemistry, 2014, vol. 22, # 17, p. 4602 - 4608
[7] Synthesis, 1985, # 12, p. 1161 - 1162
[8] Synthetic Communications, 2003, vol. 33, # 10, p. 1809 - 1814
[9] Tetrahedron Letters, 2002, vol. 43, # 2, p. 327 - 329
[10] Tetrahedron, 2005, vol. 61, # 5, p. 1127 - 1140
[11] Tetrahedron Letters, 1996, vol. 37, # 5, p. 625 - 628
[12] Canadian Journal of Chemistry, 1994, vol. 72, # 6, p. 1500 - 1511
[13] Journal of Agricultural and Food Chemistry, 2004, vol. 52, # 14, p. 4368 - 4374
[14] Journal of the American Chemical Society, 2013, vol. 135, # 32, p. 11692 - 11695
[15] Advanced Synthesis and Catalysis, 2000, vol. 342, # 8, p. 779 - 784
[16] Tetrahedron Letters, 1988, vol. 29, # 48, p. 6369 - 6372
[17] Tetrahedron, 1991, vol. 47, # 18, p. 3095 - 3128
[18] Synthetic Communications, 1989, vol. 19, # 7,8, p. 1369 - 1380
[19] Bioscience, Biotechnology, and Biochemistry, 1992, vol. 56, # 12, p. 1962 - 1965
[20] Agricultural and Biological Chemistry, 1985, vol. 49, # 1, p. 141 - 148
[21] Journal of Organic Chemistry, 1997, vol. 62, # 26, p. 9192 - 9202
[22] Bulletin de la Societe Chimique de France, 1994, vol. 131, # 6, p. 699 - 705
[23] Journal of Organic Chemistry, 1995, vol. 60, # 10, p. 2989 - 2999
[24] Synthetic Communications, 2007, vol. 37, # 15, p. 2491 - 2500
[25] Tetrahedron, 2010, vol. 66, # 6, p. 1267 - 1273
[26] Chemical Papers, 2015, vol. 69, # 2, p. 380 - 384
[27] Patent: CN105732654, 2016, A, . Location in patent: Paragraph 0067; 0068
[28] Patent: CN103102266, 2016, B, . Location in patent: Paragraph 0024
5
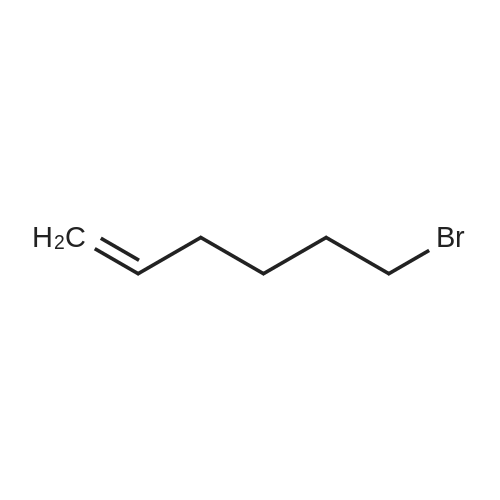
[ 142-68-7 ]
[ 34626-51-2 ]
Reference:
[1] Tetrahedron Letters, 1996, vol. 37, # 38, p. 6919 - 6922
[2] Journal of Organic Chemistry, 1987, vol. 52, # 9, p. 1680 - 1686
[3] Journal of Organic Chemistry, 2002, vol. 67, # 26, p. 9248 - 9256
[4] Organic Process Research and Development, 2003, vol. 7, # 3, p. 339 - 340
[5] Synthetic Communications, 1989, vol. 19, # 13-14, p. 2431 - 2440
[6] Heterocycles, 1982, vol. 18, p. 163 - 167
[7] Journal of Organic Chemistry, 1993, vol. 58, # 25, p. 7170 - 7179
[8] Synthesis, 1992, # 12, p. 1239 - 1241
[9] Justus Liebigs Annalen der Chemie, 1970, vol. 738, p. 170 - 173
[10] Journal of Organic Chemistry, 2016, vol. 81, # 16, p. 7288 - 7300
6
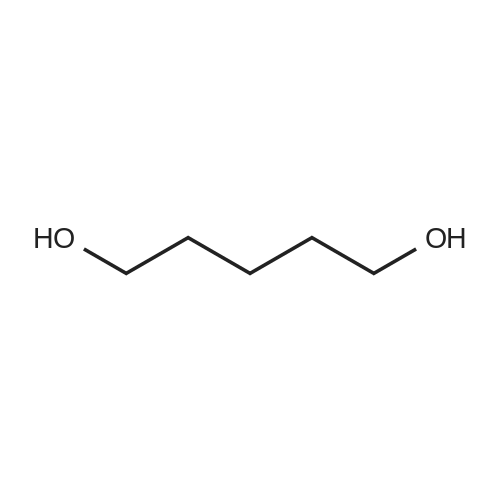
[ 15848-22-3 ]
[ 34626-51-2 ]
Reference:
[1] Journal of the Chemical Society - Perkin Transactions 1, 1999, # 19, p. 2771 - 2782
[2] Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry, 1989, vol. 28, # 1-11, p. 728 - 732
[3] Synthesis, 1992, # 12, p. 1239 - 1241
[4] Canadian Journal of Chemistry, 1992, vol. 70, # 5, p. 1427 - 1445
[5] Justus Liebigs Annalen der Chemie, 1970, vol. 738, p. 170 - 173
[6] Tetrahedron, 1971, vol. 27, p. 5979 - 5985
[7] Journal of the Chemical Society, 1963, p. 4363 - 4368
[8] Synthetic Communications, 1989, vol. 19, # 3and4, p. 645 - 658
[9] Bioscience, Biotechnology, and Biochemistry, 1992, vol. 56, # 12, p. 1962 - 1965
[10] Journal of Organic Chemistry, 1993, vol. 58, # 25, p. 7170 - 7179
[11] Patent: US5866560, 1999, A,
7
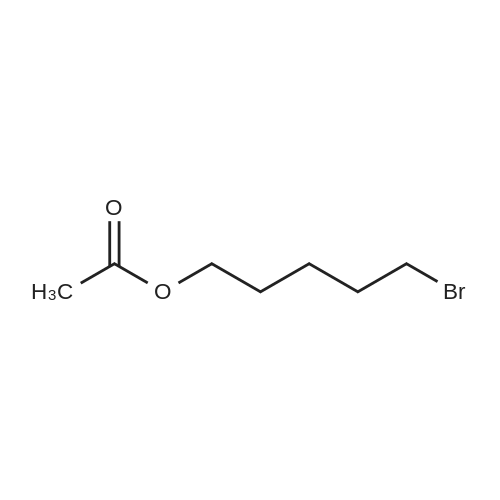
[ 2067-33-6 ]
[ 34626-51-2 ]
Reference:
[1] Journal of the American Chemical Society, 1996, vol. 118, # 50, p. 12541 - 12554
[2] Helvetica Chimica Acta, 1993, vol. 76, p. 1901 - 1915
[3] Farmaco, 1992, vol. 47, # 3, p. 379 - 385
[4] Bulletin of the Chemical Society of Japan, 2005, vol. 78, # 10, p. 1856 - 1861
8
[ 14660-52-7 ]
[ 34626-51-2 ]
Reference:
[1] Monatshefte fur Chemie, 1998, vol. 129, # 8-9, p. 931 - 936
[2] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1959, vol. 248, p. 2217
[3] Helvetica Chimica Acta, 1993, vol. 76, p. 1901 - 1915
a 5-Bromo-1-pentanol 50 ml of concentrated sulfuric acid is added in drops to a solution of 50 g of 5-<strong>[15848-22-3]bromopentyl acetate</strong> in 1.61 of methanol, and the mixture is stirred for 30 hours at room temperature. The methanol is drawn off in a vacuum, the residue is taken up in diethyl ether, washed neutral with saturated common salt solution, dried on sodium sulfate and concentrated by evaporation. 28 g of 5-bromo-1-pentanol is obtained as crude product.
With toluene-4-sulfonic acid; In dichloromethane; at 20℃;
General procedure: 3,4-Dihydropyran (DHP, 15 mmol) was added slowly to a stirred solution of bromohydrin (10 mmol) and p-toluenesulfonic acid (1 mmol) in dichloromethane (20 mL) on ice bath. After stirring at room temperature overnight, the reaction mixture was diluted with water and extracted with dichloromethane. The organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, and the residue was purified by flash column chromatography to afford 1-5 as a light yellow oil.
96.1%
With toluene-4-sulfonic acid; In dichloromethane; at 10 - 20℃; for 4h;
A mixture of 2a (8.40 g, 0.05 m 1) and a TsOH was dissolved in 10 mL of dichloromethane stirred at ice-salt bath, the freshly distilled 2,3-dihydropyran (5.04 g, 0.06 mol) was added dropwise to the above system, reaction exothermic, control the temperature of the mixture does not exceed 10 C, after the reaction at room temperature for 4 h. TLC detection 2a raw material point disappears, stop the reaction, add the same volume of ether dilution, and then 2M sodium hydroxide solution, washed with saturated brine, dried over anhydrous potassium carbonate. concentrated, and the crude product was chromatographed (petroleum ether: ethyl acetate, V / V = 8: l) to give colorless transparent liquid 3a (12.1 g) in 96.1% yield.
79%
With pyridinium p-toluenesulfonate; In tetrahydrofuran; dichloromethane; at 20℃;
The compound 41 5-bromo-1-pentanol (15.1 g, 91 mmol) and 37 pyridinium p-toluenesulfonate (25.1 g, 100 mmol) were dissolved in 100 mL of 18 DCM, added 8 THF (12.5 mL, 126 mmol). The reaction mixture was stirred at room temperature for overnight. Progress of reaction was monitored by TLC. Upon completion, most of the DCM was removed until about 30 mL left. Then, the residue was added 100 mL of 38 ethyl ether and stirred for 0.5 h. The solid was filtered off, and washed with ethyl ether. The filtrate was concentrated, and then, purified using column chromatography with PE:EA (v/v)=50/1 to give the compound 42 B9, 18 g, yield 79%.
33%
With pyridinium p-toluenesulfonate; In dichloromethane;
[00149] 5-bromo-l-pentanol (1), 3g, is treated with 1.5 eq of 3, 4-dihydro-2H-pyran and 0.1 eq of pyridinium para toluenesulfonate (PPTS) in 135mL of CH2CI2. Other starting materials, such as bromo C1.9 alcohols may be used to obtain other embodiments of compounds of formula VII. [00150] After work -up and purification, 1.45 g (33%) of product 2 is obtained. 1.0 eq of methyl malonitrile is deprotonated with 1 eq of NaH and 1.0 eq of bromide 2 is added along with catalytic amount of KI at 50C. A complete conversion can be observed after 10 hours and a 90% yield can be obtained. Deprotection of tetrahydo pyran (THP) can be done with PPTS in ethanol at 55C. After work-up, a quantitative yield of alcohol 4 is obtained and directly used for the mesylation reaction. With the mesylate 5 in hand, a kryptofix- mediated fluorination can be performed. Compound 6 is obtained in 68% yield. 12mL of DMF, 10 eq. of acetic acid, triethylamine and sodium azide each are added to compound 6. The resulting mixture is stirred at 140C for 19 hours. Water and IN HC1 are added. The solid is filtered off and washed with water. Compound 7 is obtained in a 60% yield. [00151] NMR [for example, Bruker Avance 400 (400 MHz, CDC13, TMS as internal standard] of the 5,5'(7-fluoroheptane-2,2-diyl)bis(lH-tetrazole) compound shows the following results: delta 1.29 (m, 4H, CH2), 1.49 (m, 2H, CH2), 1.77 (s, 3H, Me), 1.87 (t, 2H, CH2), 4.09 (m, 2H, CH2F).
With p-toluenesulfonic acid monohydrate; In dichloromethane; at 0 - 20℃; for 18h;Cooling with an ice bath;
5-bromopentanol (8 g) was dissolved in DICHLOROMETHANE (100 mL) and was chilled in an ice bath. Dihydropyran (9 g) was added dropwise followed by P-TOLUENE SULFONIC acid monohydrate (1 g). The mixture was allowed to slowly warm to room temperature and stirred for 18 HOURS. The mixture was diluted with 200 mL ether, washed with 10% INA (: H (100 mL) and dried over MgSO4 to give the title compound. DZ (CDC13, 62.9 MHz): 19.7, 25.0, 25.5, 28. 9,30. 7,32. 6,33. 7,62. 4,67. 2 and 89. 9.
With sodium chloride; trichlorophosphate; In hexane; ethyl acetate;
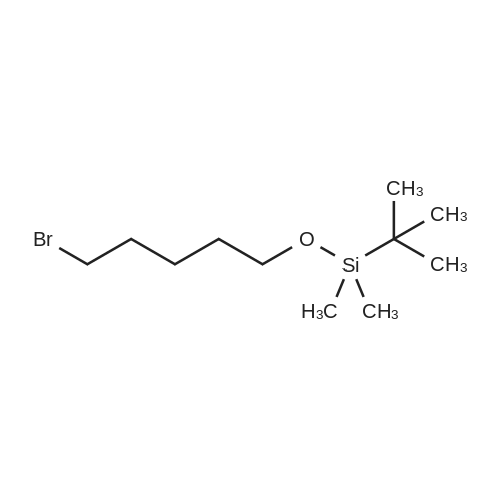
(1) 5-bromo-1-(tetrahydropyran-2-yloxy)pentane To 36 g of <strong>[34626-51-2]5-bromopentan-1-ol</strong> were added 24.72 ml of 2,3-dihydropyran and 3 drops of phosphorus oxychloride under cooling in an ice bath, and the mixture was stirred overnight at room temperature. The reaction mixture was diluted with diethyl ether, washed with 5% aqueous potassium hydroxide, water, a saturated aqueous solution of sodium chloride, dried over magnesium sulphate and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel using a mixture of ethyl acetate and hexane (1:19) as eluent to give 49.5 g of the subtitle compound having the following physical characteristics: TLC(benzene:ethyl acetate=4:1): Rf=0.79; IR: upsilon=2960, 2900, 1460, 1450, 1365, 1145, 1130, 1040, 1030, 980, 820, 780 cm-1. NMR:delta=4.5(1H,m), 4.1-3.1(6H,m), 2.5-1.1(12H,m). MS(%): m/e=250(5), 151(9.5), 149(10), 85(100), 69(37).
With toluene-4-sulfonic acid; In diethyl ether;
<strong>[34626-51-2]5-Bromo-1-pentanol</strong> (16.7 g, 100 mmol) was converted to the corresponding THP ether 15 (29.8 g) by treatment with DHP (9.0 g, 107 mmol) and TsOHxH2O (0.2 g) in Et2O (70 mL). A solution of n-BuLi (1.6 M in hexane, 52 mL, 82 mmol) was added dropwise to a stirred and cooled solution of 13 (9.0 g, 82 mmol) in dry THF (70 mL) and dry HMPA (7 mL) at -78 to -60 C under argon. The mixture was then warmed up to 0 C and again cooled to -78 C. A solution of 15 (29.8 g, 100 mmol) in dry THF (10 mL) was added through a syringe to the stirred and cooled mixture at -78 to -60 C. The mixture was left to stand overnight at room temperature, and then stirred and heated at 40-50 C for 1 h. After cooling, it was diluted with water, and extracted with hexane. The hexane extract was washed with water and brine, dried (MgSO4), and concentrated in vacuo to give crude 17 as an oil, numax (film): 2939 (s), 2860 (s), 1200 (m), 1136 (s), 1120 (s), 1077 (s), 1035 (s), 992 (m), 972 (m), 905 (m), 870 (m), 815 (m). TsOHxH2O (1.0 g) was added to a solution of 17 (31.9 g) in MeOH (170 mL), and the solution was left to stand overnight at room temperature. It was then stirred and heated under reflux for 1 h, and concentrated in vacuo. The residue was diluted with water, and extracted with hexane. The hexane extract was washed with water and brine, dried (MgSO4), and concentrated in vacuo. The residue was distilled to give 19 (5.73 g, 36% based on 13, two steps), bp 135-138 C/3 Torr; nD22=1.4648; numax (film): 3346 (br m), 2931 (s), 2859 (s), 1460 (m), 1073 (m), 1054 (m); deltaEta (CDCl3): 0.89 (3H, t, J 7.2), 1.22-1.40 (6H, m), 1.40-1.74 (9H, m), 2.10-2.24 (4H, m), 3.64 (2H, t, J 7); GC-MS (same conditions as those used for 12): tR 13.34 min (84.5%) [impurities at tR 14.27 (5.1%) and 18.04 min (7.3%)]; MS (70 eV, EI): m/z: 196 (<1) [M+], 178 (6), 149 (8), 135 (11), 121 (10), 109 (22), 93 (72), 79 (100), 67 (81), 55 (48), 41 (40). HRMS calcd for C13H24O: 196.1827, found: 196.1862.
With hydrogen bromide; In benzene; at 70 - 80℃; for 15h;
In 250mL three-necked flask add 1a (14 g, 0.135 mol), 40% hydrobromic acid (28 mL, 0.2 mol) and 60 mL of benzene, 70 ~ 80 C oil bath water for 15h, TLC detection, raw material points disappear. Washed successively with 5% sodium hydroxide solution, 10% hydrochloric acid and saturated brine, and dried over anhydrous sodium sulfate. The crude product was separated by silica gel column chromatography (petroleum ether: ethyl acetate, V / V = 8: 1) and concentrated to give colorless to pale yellow liquid 2a (20.1g, yield 89.1%).
80%
With hydrogen bromide; In water; toluene; for 15h;Dean-Stark; Reflux;
A round bottom flask, equipped with a Dean-Stark trap, wascharged with 1,5-pentanediol (3.71 g, 35.6 mmol), concentratedhydrobromic acid (5 mL), and toluene (75 mL). The mixture wasstirred under refluxed for 15 h. After cooling, the reaction mixturewas extracted with 6 M sodium hydroxide (2 15 mL), 5% HCl(2 15 mL), water (2 15 mL) and brine (20 mL). The organicswere dried over sodium sulfate and concentrated under reducedpressure to yield the title compound as an orange/brown oil(4.75 g, 2.84 mmol, 801H NMR (500 MHz, CDCl3) d 8.16(s, 1H), 7.65 (s, 1H), 7.46 (m, 6H), 7.25 (m 6H), 7.17 (m, 3H), 6.98(m, 3H), 6.48 (m, 4H), 3.59 (br, 2H), 3.11 (br, 4H), 2.65 (q, 12H,J = 7 Hz), 2.11 (br, 2H), 1.61 (m, 2H), 1.52 (m, 4H), 1.45 (m, 2H),1.41 (s, 9H), 1.31 (m, 2H), 1.01 (t, 18H, J = 7 Hz); 13C NMR(125 MHz, CDCl3) d 180.65, 171.21, 156.78, 155.79, 155.64, 146.13,140.35, 138.09, 130.98, 129.19, 128.54, 127.72, 126.14, 124.68,123.33, 120.15, 113.95, 103.13, 79.37, 79.15, 70.77, 47.23, 46.91,45.71, 44.64, 43.29, 29.65, 28.48, 28.14, 27.81, 26.53, 26.10,24.21, 9.07; HRMS (C54H56N4O7S): calculated 904.3870, observed904.3866.
With hydrogen bromide; In toluene; for 36h;Reflux;
1,5-pentadiol (300mg, 2mmol) dissolved in 10mL of toluene was added HBr (0.3mL, 2.4mmol), refluxed for 36h. The toluene layer was spin-dried to give a colorless oily liquid bodies.
With hydrogen bromide; In toluene; at 120 - 125℃; for 24h;
1,5_ pentanediol (20.8 g, 0.2 mol) was dissolved in 400 mlToluene, 48% hydrobromic acid (40.5 g, 0.24 mol) was added and the reaction was stopped after the reaction was refluxed at 120-125 C for 24 hours.Add appropriate amount of saturated sodium carbonate and, separated from the organic layer, and then extracted with toluene 2 times, dried over anhydrous sodium sulfate and evaporated solvent to get the crude product,5-bromo-pentanol 31.7 g, yield 95%, without further separation directly for the next
Sodium azide (2.33 g, 36.0mmol) was added to a solution of 5-bromo-1-pentanol (3.00 g, 18.0 mmol) inwater (50 mL). The reaction mixture was stirred at 80 C for 18 hrs. Aftercooling down to room temperature, the aqueous solution was extracted with CH2Cl2(3 x 20 mL). The combined organic layers were dried over anhydrous MgSO4,filtered and concentrated under reduced pressure to afford 7 as a colorless oil (2.10 g, yield 90%) that was used for the nextstep without any further purification. 1H NMR (250 MHz, CDCl3)delta 1.35-1.75 (m, 7 H), 3.27(t, 2 H, J = 6.6 Hz), 3.63 (t, 2 H, J = 6.6 Hz); 13C NMR (62.9MHz, CDCl3) delta 23.1, 28.7, 32.3, 51.5, 62.6; HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C5H11N3ONa+152.0794; found 152.0794.
With dimethylsulfide borane complex In tetrahydrofuran at 45℃; for 1h; Cooling with ice;
3.1.2. General Procedure for the Synthesis of Alcohols 4-6
General procedure: To an ice-cold solution of acid 1-3 (20 mmol) in THF (30 mL), a borane dimethylsulphide complex (2.7 mL, 28 mmol) was added dropwise. The solution was warmedat room temperature and stirred for 1 h at 45 C. Then, the reaction was cooled at 0 C,quenched with MeOH (60 mL) and concentrated under reduced pressure. The residuewas dissolved in EtOAc, washed with brine, dried over Na2SO4 and concentrated invacuo to obtain the projected alcohols 4-6, which were used in the next step withoutfurther purification.
96%
With dimethylsulfide borane complex
With lithium aluminium tetrahydride; sulfuric acid In tetrahydrofuran for 0.25h; T < 2 deg C; Yield given;
With dimethylsulfide; borane
63 % Spectr.
With diphenylsilane; triphenylphosphine In tetrahydrofuran at 20℃; for 48h;
With 1H-imidazole; In tetrahydrofuran; at 0 - 20℃; for 5h;Inert atmosphere;
To a solution of <strong>[34626-51-2]5-bromopentan-1-ol</strong> (0.43 mL, 3.00 mmol) in tetrahydrofuran (15 mL) at 0 C under an atmosphere of nitrogen was added imidazole (210 mg, 3.00 mmol) and t-butylchlorodiphenylsilane (0.78 mL, 3.00 mmol) at 0 C. After 5 h at room temperature, the reaction mixture was quenched by addition of water (5 mL) and the mixture was extracted with diethyl ether. The organic layer was washed with brine, dried over magnesium sulfate, filtered and evaporated in vacuo. The resulting crude residue was purified on a Biotage purification apparatus (silica gel, 0-10% ethyl acetate in hexanes gradient) to yield the title compound (17, 980 mg, 2.42 mmol, 81%) as a colorless oil. 1H NMR (400 MHz, CDCl3) delta 7.72-7.69 (m, 4H), 7.48-7.39 (m, 6H), 3.70 (t, J = 6.4 Hz, 1H), 3.42 (t, J = 6.8 Hz, 1H), 1.91-1.84 (m, 2H), 1.62-1.58 (m, 2H), 1.47-1.42 (m, 4H), 1.09 (s, 9H).
With 1H-imidazole; In dichloromethane; at 20℃; for 18h;
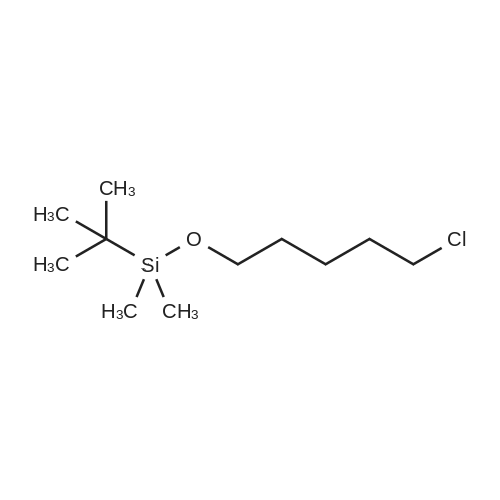
Step 1. Synthesis of [(5-bromopentyl)oxy](tert-butyl)dimethylsilane A solution of 5-bromo-1-pentanol (0.80 mL, 6.64 mmol), tert-butyldimethylsilyl chloride (1.20 g, 7.97 mmol) and imidazole (0.542 g, 7.97 mmol) in CH2Cl2 (30 mL) was stirred at room temperature for 18 h. The solution was washed with 1 N HCl(aq), dried over MgSO4(s), filtered, and concentrated to give [(5-bromopentyl)oxy](tert-butyl)dimethylsilane (1.62 g, 87%) as a colorless oil. 1H NMR (CDCl3, 400 MHz) delta 3.61 (t, 2H), 3.41 (t, 2H), 1.87 (quint, 2H), 1.57-1.48 (m, 4H), 0.88 (s, 9H), 0.05 (s, 6H).
With 2,6-dimethylpyridine; In dichloromethane; at -78℃; for 0.5h;Schlenk technique;
General procedure: According to a literature procedure,[1] a flame-dried Schlenk flask is charged with the correspondingprimary alcohol in dry CH2Cl2. At -78 C, 2,6-lutidine (1.6 equiv) is added followedby the slow addition of Tf2O (1.2 equiv). After completion of reaction (TLC monitoring), thereaction mixture is quenched with HCl (1M), washed with brine, and the aqueous phase isextracted with CH2Cl2. The combined organic layers are dried over Na2SO4. Evaporation ofthe solvents at room temperature (due to decomposition of the triflates at higher temperatures)affords the crude title compounds. Purification by vacuum filtration over silica gel usinga sintered funnel (grade 4) with the indicated eluent affords the analytically pure alkyltriflates.
[00490] A flame-dried 25 mL flask was charged with 2,6-lutidine (0.377 mL, 3.23 mmol, 1.08 equiv) and dichloromethane (4 mL). The solution was cooled to -20 C in a dry ice- acetone bath. Trifluoromethanesulfonic anhydride (0.526 mL, 3.11 mmol, 1.04 equiv) was added dropwise, generating a bright red mixture. After 10 min, 5-bromopentan-l-ol (0.361 mL, 2.99 mmol, 1 equiv) was added dropwise. The mixture partially clarifies and stirs more freely. The cooling bath was removed and the mixture allowed to warm slowly to 23 C. After 10 min, the mixture was diluted with 1: 1 dichloromethane-hexanes (20 mL) and filtered through a pad of silica (1 cm in a 30-mL sintered glass funnel), eluting with 1: 1 dichloromethane-hexanes (80 mL). The filtrate was concentrated, providing 5-bromopentyl trifluoromethanesulfonate as a colorless oil, which was used without further purification.
2-(N-Phenylsulfonylindol-3-yl)ethyl 3-Deoxy-2,4-di-O-benzyl-b-D-glucopyranoside[ No CAS ]
[ 38222-83-2 ]
[ 34626-51-2 ]
[ 170220-11-8 ]
2-(N-Phenylsulfonylindol-3-yl)-ethyl 2,4-Di-O-benzyl-3-deoxy-6-O-(5-azidopentyl)-β-D-glucopyranoside[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
With trifluoromethylsulfonic anhydride; In dichloromethane; dimethyl sulfoxide;
Z. 2-(N-Phenylsulfonylindol-3-yl)ethyl 2,4-Di-O-benzyl-3-deoxy-6-O-(5-azidopentyl)-beta-D-glucopyranoside (III-29a). A stirred solution of 5-bromo-1-pentanol (0.79 g, 4.7 mmol) in DMSO (15 ml) was treated with sodium azide (1.83 g, 28.2 mmol). The resultant mixture was stirred at room temperature for 2.5 h, diluted with water, and extracted with diethyl ether. The combined organic layers were washed with saturated aqueous sodium bicarbonate and brine, dried over magnesium sulfate, filtered, and concentrated in vacuo. The azide was used without purification in the next step. Crude 5-azido-1-pentanol (280 mg, equivalent to 2.17 mmol) and <strong>[38222-83-2]2,6-di-tert-butyl-4-methylpyridine</strong> (441 mg, 2.17 mmol) were dissolved in dichloromethane (9 ml) and triflic anhydride (0.36 ml, 2.17 mmol) was added dropwise. After 10 min the mixture was poured into brine (40 ml) and extracted with dichloromethane (2*40 ml). The combined organic layers were dried over magnesium sulfate, filtered, and concentrated in vacuo. The triflate was used without purification in the next step. Sodium hydride (16 mg, 0.40 mmol, 60percent dispersion in oil) was added to a solution of alcohol III-28 (120 mg, 0.198 mmol) and azido triflate (105 mg, equivalent to 0.40 mmol) in dichloromethane (3 ml) at room temperature. The mixture was stirred for 24 h, diluted with dichloromethane (40 ml) and poured into saturated ammonium chloride (40 ml). The aqueous phase was extracted with dichloromethane and the combined organic solutions were washed with brine, dried over magnesium sulfate, filtered, and concentrated in vacuo. Flash chromatography (15percent ethyl acetate/hexane) afforded III-29a (121 mg, 83percent yield) as a colorless oil: [alpha]D25 +4.0° (c 0.24, CHCl3); IR (CHCl3) 3022 (s), 2940 (s), 2880 (m), 2105 (s), 1455 (s), 1375 (s), 1270 (s), 1210 (m), 1180 (m), 1125 (m), 1090 (m), 725 (s), 599 (m) cm-1; 1 H NMR (500 MHz, CDCl3) delta7.96 (d, J=8.1 Hz, 1H), 7.82 (dd, J=8.2, 0.9 Hz, 2H), 7.50-7.43 (m, 3H), 7.29-7.19 (m, 14H), 4.65 (d, J=12.0 Hz, 1H), 4.58 (d, J=11.4 Hz, 1H), 4.52 (d, J=12.0 Hz, 1H). 4.42 (d, J=11.5 Hz, 1H), 4.18 (dt, J=9.5, 6.7 Hz, 1H), 3.81 (dt, J=9.5, 7.1 Hz, 1H), 3.71 (d, J=10.6 Hz, 1H), 3.57 (dd, J=10.8, 4.7 Hz, 1H), 3.51-3.38 (m, 4H), 3.31-3.21 (m, 1H), 3.16 (t, J=6.9 Hz, 2H), 3.00 (t, J=6.9 Hz, 2H), 2.50-2.46 (dt, J=12.1, 4.5 Hz, 1H),1.63-1.50 (m, 5H), 1.48-1.32 (m, 3H); 13 C NMR (62.5 MHz, CDCl3) delta138.52, 138.23, 137.00, 135.07, 133.59, 131.09, 129.14, 128.43, 128.31, 127.78, 127.68, 127.50, 126.70, 126.69, 124.70, 123.54, 123.09, 119.71, 119.48, 113.70, 105.26, 78.01, 74.92, 72.67, 72.25, 71.38, 71.24, 69.96, 68.41, 34.97, 29.62, 29.15, 28.66, 25.65, 23.39; high resolution mass spectrum (FAB, m-nitrobenzyl alcohol) m/z 761.2973 (M+; calcd for C41 H46 N4 O7 S: 761.2985).
With caesium carbonate In N,N-dimethyl-formamide at 20℃; for 66h;
60 2H-1,2,4-Benzothiadiazine-7-sulfonamide, 6-chloro-3,4-dihydro-2-(5-hydroxypentyl)-, 1,1-dioxide
To a stirred solution of hydrochlorothiazide (3.65 g, 12.3 mmol) in DMF (30 mL) were added cesium carbonate (2.00 g, 6.15 mmol), and 5-bromo-1-pentanol (2.05 g, 12.3 mmol). After being stirred for 66 hours at room temperature, the mixture was poured into water, and extracted with EtOAc. The organic layer was washed with aqueous NaCl, filtered through a pad of Na2SO4, and concentrated. Chromatography (silica gel; EtOAc, and then THF) and subsequent recrystallization from EtOAc:CH2Cl2:Hexane (1:1:1) gave the title compound (2.21 g, 47% yield) as a white solid: mp 177-179° C; 1H NMR (400 MHz, d4-MeOH) δ 8.17 (s, 1H), 6.96 (s, 1H), 4.91 (s, 2H), 3.54 (t, J=6.4 Hz, 2H), 2.98 (t, J=7.6 Hz, 2H), 1.65 (m, 2H), 1.53 (m, 2H), 1.42 (m, 2H); Mass spectrum (API-TIS) m/z 384.3 and 386.2 (MH+ for 35Cl and 37Cl respectively).
a 5-Bromo-1-pentanol 50 ml of concentrated sulphuric acid are added dropwise to a solution of 50 g of 5-<strong>[15848-22-3]bromopentyl acetate</strong> in 1.6 l or methanol and the mixture is stirred at room temperature for 30 hours. The methanol is stripped off in vacuo, the residue is taken up in diethyl ether, and the mixture is washed with saturated sodium chloride solution until neutral, dried over sodium sulphate and concentrated. 28 g of 5-bromo-1-pentanol are obtained as a crude product.
With potassium carbonate; potassium iodide; In N,N-dimethyl-formamide; at 110℃;
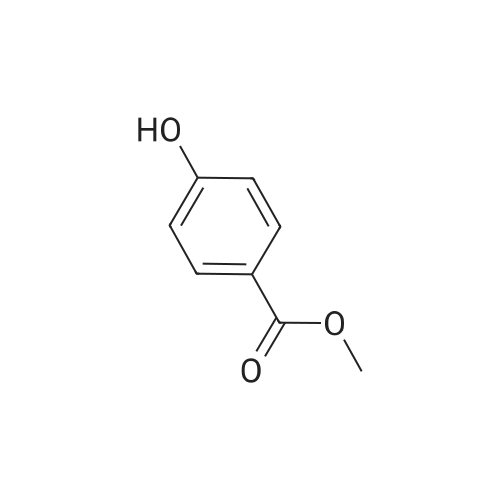
A mixture of methyl 4-hydroxybenzoate (3.0 g, 20 mmol), <strong>[34626-51-2]5-bromopentan-1-ol</strong> (3.3 g, 20 mmol), potassium carbonate (5.5 g, 40 mmol) and potassium iodide (0.3 g, 2 mmol) in N,N-dimethylformamide (20 mE) was heated at 1100 C. overnight. The reaction mixture was cooled to room temperature. Water (50 mE) was added. Extracted with ethyl acetate (50 mEx3) and combined organic layers were washed with water (30 mEx2) and brine (30 mEx2), dried over anhydrous sodium sulfate. The solvent was concentrated to give the residue, which was purified by column chromatography on silica gel (petroleum ether/ethyl acetate=10/1) to give methyl 4-(5-hydroxypentyloxy)benzo- ate (2.2 g, 46% yield) as a white solid. EC-MS (Agilent ECMS 1200-6120, Column:Waters X-l3ridge C18 (50 mmx4.6 mmx3.5 jim); Column Temperature: 40 C.; Flow Rate: 2.0 mE/mm; Mobile Phase:from 95% [water+10 mM NH4HCO3] and 5% [CH3CN] to 0% [water+10 mM NH4HCO3] and 100% [CH3CN] in 1.6 mm, then under this condition for 1.4 mm, finally changed to 95% [water+10 mM NH4HCO3] and 5% [CH3CN] in 0.1 mm and under this condition for 0.7 mi. Purity is 98.48%, Rt=1.637 mm; MS Calcd.: 238.1; MS Found: 239.2 [M+H].
38%
With potassium carbonate; In acetonitrile; for 24h;Reflux;
Condensation of a-bromo aldehyde with 2,4-diamino-4-oxo-pyrimidine 19 is the key step in the synthesis of compounds 9-11 as outlined in Scheme 1 and Scheme 2. Commercially available alcohol 14 was coupled to the corresponding phenyl esters and oxidized to the corresponding aldehydes 17a and 17b (Scheme 1).-? Alcohol 24 was coupled to the appropriate phenyl ester to afford the aldehyde 25 (Scheme 2). The aldehydes were reacted with Br2 in dioxane to give the desired a-bromoaldehydes?2 18a, 18b (Scheme 1) and 26 (Scheme 2) and immediately reacted with 2,4-diamino-6-hydroxypyrimidine 19 to cyclize to the 5-substituted pyrrolo[2,3-d]pyrimidines 20a, 20b (Scheme 1) and 27 (Scheme 2).? Hydrolysis of the esters provided the free acids 21 a, 2 lb and 28. Subsequent peptide coupling with diethyl L-glutamate using the activating agents N-methyl morpholine and 2,4-dimethoxy-6-chlorotriazine, afforded the diesters 22a, 22b and 29. Saponification of the diesters yielded the final compounds 9-11 of this invention.
21%
With potassium carbonate; In acetonitrile; at 80℃; for 24h;
5.5 g (36.0 mmol) of methyl 4-hydroxybenzoate, 6.0 g (36.0 mols) of 5-bromo-1-pentanol, 9.0 g (72 mmol) of potassium carbonate and 80 ml of acetonitrile were added to a 200 ml round bottom flask equipped with a condenser to provide a mixture, followed by reaction under stirring at 80C for 24 hours. After completion of the reaction, the reaction solution was filtered under reduced pressure and the solvent was distilled off under reduced pressure to obtain a yellow wet solid. This solid was purified by silica gel column chromatography (column: silica gel 60, 0.063-0.200 mm, made by Merck & Co., eluate: hexane/ethyl acetate = 1/1). The solvent was distilled off from the resulting solution to obtain 1.8 g of a white solid. This solid was subjected to measurement of NMR with the results shown below. From the results, it was confirmed that the white solid was made of an intermediate compound (P9) (yield: 21%). 1H-NMR(CDCl3) delta: 1.5-1,7(m, 4H), 2.85(m, 2H), 3.67(t, 2H), 3.88(s, 3H), 4.02(t, 2H), 6.90(d, 2H), 7.99(d, 2H).
21%
With potassium carbonate; In acetonitrile; at 80℃; for 24h;
In a 200 ml Nasar flask equipped with a cooling tube, 5.5 g (36.0 mmol) of methyl 4-hydroxybenzoate, 6.0 g (36.0 mmol) of 5-bromo-1-pentanol,9.0 g (72 mmol) of potassium carbonate,And 80 ml of acetonitrile were added to the mixture, The reaction was carried out with stirring at 80 for 24 hours. After completion of the reaction, The reaction solution was filtered under reduced pressure, the solvent was distilled off under reduced pressure,A yellow, wet solid was obtained. This solid was purified by silica gel column chromatography (column: silica gel 60, 0.063-0.200 mm, a solvent, eluent hexane / ethyl acetate = 1/1).The solvent was distilled off from the obtained solution,1.8 g of a white solid was obtained.The result of measurement of this solid by NMR is shown below.From this result, it was confirmed that the white solid was the intermediate compound (P2) (yield: 21%).
With 1H-imidazole; In dichloromethane; at 20℃; for 4h;
General procedure: To a solution of 100 mg of alcohol (4-bromo-1-butanol, 5-bromo-1-pentanol, or 6-bromo-1-hexanol) in 5 mL of CH2Cl2 were added 2 equiv. of imidazole and 1.0 equiv. of TIPSCl (triisopropylsilyl chloride). After being stirred at room temperature for 4 h,the reaction mixture was diluted with water, and the product was extracted with EtOAc. The extract was washed with brine, dried, and evaporated. The product was purified by chromatography (hexane/EtOAc 20:1) to give compounds 12, 13, and 14 as colorless oils. Compound 12: 85%; 1H NMR (400 MHz, CDCl3) 1.03-1.10 (m,21H), 1.68 (quin, J = 6.1 Hz, 2H), 1.98 (quin, J = 7.0 Hz, 2H), 3.46 (t,J = 6.8 Hz, 2H), 3.72 (t, J = 6.1 Hz, 2H); 13C NMR (100 MHz, CDCl3) 11.9, 18.0, 29.6, 31.5, 34.0, 62.4; ESI-HRMS (M+H)+m/z calcd. for C13H30BrOSi 309.1249, found 309.1248. Compound 13: 88%; 1H NMR (400 MHz, CDCl3) 1.05-1.11 (m,21H), 1.49-1.58 (m, 4H), 1.89 (t, J = 7.6 Hz, 2H), 3.41 (t, J = 6.9 Hz,2H), 3.69 (t, J = 6.1 Hz, 2H);13C NMR (100 MHz, CDCl3) 12.0, 18.0,24.6, 32.1, 32.7, 33.8; ESI-HRMS (M+H)+m/z calcd. for C14H32BrOSi 323.1406, found 323.1401.
With pyridine; In dichloromethane; at 0℃; for 2h;Inert atmosphere;
To a solution of 6 (3.00 g, 17.96 mmol) in dry CH2Cl2 (89.8 mL)were added TrCl (5.509 g, 19.76 mmol) and pyridine (1.88 mL,23.35 mmol) under Ar atmosphere at 0C. After being stirred for2 h at 0C, the reaction mixture was quenched with EtOH (ca.10 mL). The aqueous layer was extracted with EtOAc. The organiclayer was washed with 1 M HCl aq. and brine, dried over Na2SO4,filtered, and concentrated in vacuo. The residue was solidified from acetone and MeOH. After the filtration, the residue was washed byMeOH to give Tr protected 5-bromo-1-pentanol (5.70 g, 78%) as awhite solid.1H NMR (500 MHz, CDCl3) d 7.45-7.42 (m, 6 H, aromatic), 7.31-7.27 (m, 7 H, aromatic), 7.24-7.21 (m, 2H, aromatic), 3.89 (t,J = 6.9 Hz, 2H), 3.07 (t, J = 6.6 Hz, 2H), 1.83 (q, J = 6.9 Hz, 2H), 1.64(q, J = 6.6 Hz, 2H), 1.54-1.49 (m, 2H); 13C NMR (100 MHz, D2O) d101.7, 79.6, 76.8, 73.6, 71.9, 71.7, 71.0, 57.2, 16.0.; HR ESI-MS;m/z calcd for C24H25BrO [M+Na]+: 431.0981, found: 431.0987.
With tetra-(n-butyl)ammonium iodide; caesium carbonate; In N,N-dimethyl-formamide; at 0 - 20℃; for 4h;
Condensation of a-bromo aldehyde with 2,4-diamino-4-oxo-pyrimidine 19 is the key step in the synthesis of compounds 9-11 as outlined in Scheme 1 and Scheme 2. Commercially available alcohol 14 was coupled to the corresponding phenyl esters and oxidized to the corresponding aldehydes 17a and 17b (Scheme 1).-? Alcohol 24 was coupled to the appropriate phenyl ester to afford the aldehyde 25 (Scheme 2). The aldehydes were reacted with Br2 in dioxane to give the desired a-bromoaldehydes?2 18a, 18b (Scheme 1) and 26 (Scheme 2) and immediately reacted with 2,4-diamino-6-hydroxypyrimidine 19 to cyclize to the 5-substituted pyrrolo[2,3-d]pyrimidines 20a, 20b (Scheme 1) and 27 (Scheme 2).? Hydrolysis of the esters provided the free acids 21 a, 2 lb and 28. Subsequent peptide coupling with diethyl L-glutamate using the activating agents N-methyl morpholine and 2,4-dimethoxy-6-chlorotriazine, afforded the diesters 22a, 22b and 29. Saponification of the diesters yielded the final compounds 9-11 of this invention.
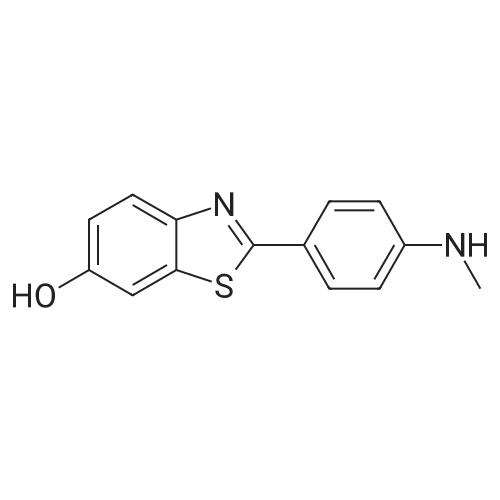
5-((2-(4-(methylamino)phenyl)benzo[d]thiazol-6-yl)oxy)pentan-1-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
68.4%
With potassium carbonate; In acetonitrile; at 90℃; for 4h;
General procedure: To a stirring solution of 1a - 1d (1 equiv.) in CH3CN was added3-bromo-1-propanol or 5-bromo-1-pentanol (5 equiv.) and K2CO3(3 equiv.) at 90 C for 4 h. Then, K2CO3 was filtered and CH3CN wasremoved under vacuum. The residue was suspended in petroleumether, and the productwas precipitated and filtered without furtherpurification unless otherwise purified by column chromatography
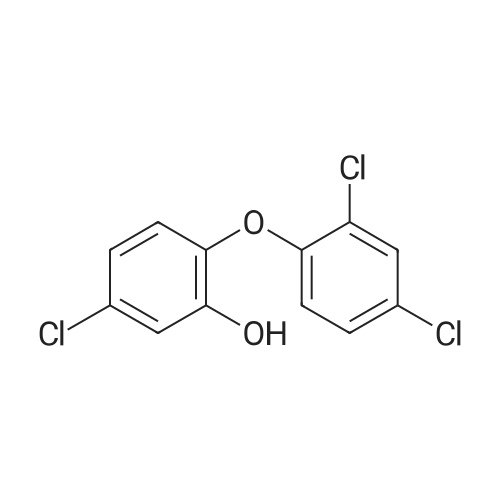
5-(5-chloro-2-(2,4-dichlorophenoxy)phenoxy)pentan-1-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
62%
General procedure: <strong>[3380-34-5]Triclosan</strong>, (3.1mmol, 900mg), potassium hydroxide (4.0mmol, 224mg) and acetonitrile (10mL), were placed into in a 50mL flat-bottomed flask equipped with a magnetic stirring bar. The mixture was stirred and heated to reflux under microwave irradiation for a period of 5min. Then, -bromoalkylalcohols (3.2mmol) were added and the reaction mixture was refluxed for 30min under microwave irradiation (200W). The crude reaction mixture was evaporated under reduced pressure and the residue was purified by column chromatography over silica gel eluting with Hexanes and a mixture of Hexanes-Ethyl acetate (9:1 ratio) to obtain the alkyl<strong>[3380-34-5]triclosan</strong>alcohols in yields ranging between 61% and 88%. Monitoring of the reaction progress and product purification was carried out by TLC.
3,4-dihydro-12-O-(5'-hydroxypentyl)-2,2-dimethyl-2H,6H-pyrano[3,2-b]xanthen-6-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85.6%
With sodium hydroxide; In N,N-dimethyl-formamide; at 20℃; for 24h;
General procedure: To a 50-ml flask, pyranoxanthone (0.296 g, 1.0 mmol) and NaOH (44 mg, 1.1 mmol) in 15 ml of dried DMF. 3-Bromopropanol (0.208 g, 1.5 mmol) in 5 ml dried DMF was then added dropwise with stirring. The reaction was lasted at r. t. for 24 h. After the finish of reaction, the mixture was pulled into 50 ml ice-water with violently stirring for 20 min. The precipitates were collected by filtrate, and were purified by flash column liquid chromatography led to slight yellow powder 0.318 g, yield 89.6%.
With dmap; sodium carbonate; In toluene; for 1h;Cooling with ice;
To a reaction flask, 33.4 g (200 mmol) of 5-bromo-1-pentanol, 25.4 g (240 mmol) of anhydrous sodium carbonate, 2.4 g (20 mmol) of N,N-dimethyl-4-aminopyridine and 200 mL of toluene were added. While the mixture was stirred under ice cooling, 22.5 g (220 mmol) of acetic anhydride was added dropwise thereto. After the completion of the dropwise addition, the mixture was stirred for 1 hour under ice cooling. The resultant reaction mixture was washed successively with water twice, with dilute hydrochloric acid and then with water. The toluene was distilled off under reduced pressure to obtain 41.8 g of 5-bromopentyl acetate. Yield: quantitative.
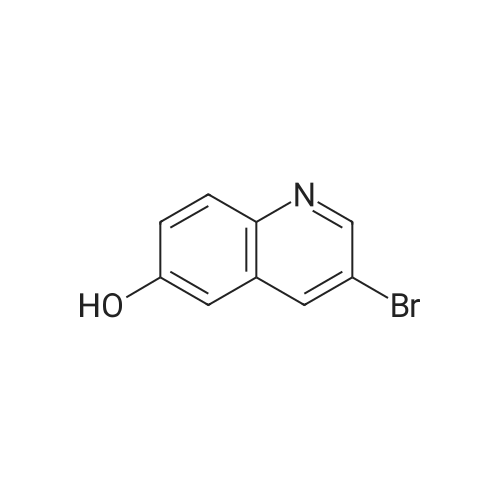
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃; for 6h;
A mixture of <strong>[13669-57-3]3-bromoquinolin-6-ol</strong> (700 mg, 3.1 mmol), 5-bromopentan-1-ol (518 mg, 3.1 mmol) and potassium carbonate (856 mg, 6.2 mmol) in N,N-dimethylformamide (5 mE) was heated at 80 C. for 6 hours. The reaction mixture was cooled to room temperature. Water (10 mE) was added and extracted with ethyl acetate (20 mEx3). The combined organic layers were washed with water (20 mEx2) and brine (20 mE), dried over anhydrous sodium sulfate. The solvent was concentrated to give the residue, which was purified by column chromatography on silica gel (petroleum ether/ethyl acetate=1/1) to give 5-(3-bromoquinolin-6- yloxy)pentan-1-ol (750 mg, 78% yield) as a yellow solid. EC-MS (Agilent ECMS 1200-6120, Column:Waters X-l3ridge C18 (50 mmx4.6 mmx3.5 jim); Column Temperature: 40 C.; Flow Rate: 2.0 mE/mm; Mobile Phase:from 95% [water+10 mM NH4HCO3] and 5% [CH3CN] to 0% [water+10 mM NH4HCO3] and 100% [CH3CN] in 1.6 mm, then under this condition for 1.4 mm, finally changed to 95% [water+10 mM NH4HCO3] and 5% [CH3CN] in 0.1 mm and under this condition for 0.7 mi. Purity is 91.43%, Rt=1.767 mm; MS Calcd.: 309.04; MS Found: 310.0 [M+H].
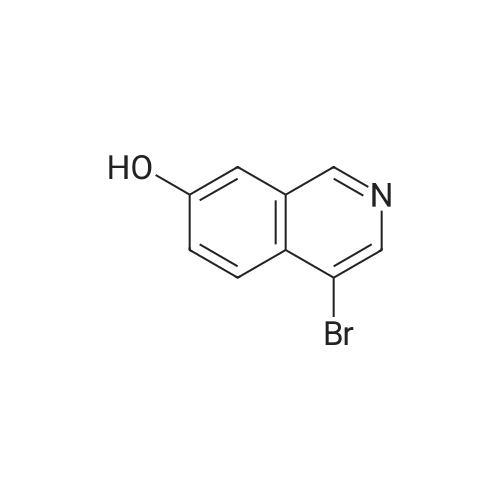
With potassium carbonate In N,N-dimethyl-formamide at 70℃; for 8h;
5 Step 5: Synthesis of5-(4-bromoisoquinolin-7-yloxy)pentan-1 -ol
To a solution of compound 4-bromoisoquinolin-7- ol (0.90 g, 4.02 mmol) in DMF (10 mE) was added 5-bro- mopentan-1 -ol (0.66 g, 4.02 mmol) and potassium carbonate (0.74 g, 8.04 mmol), then stirred at 70° C. for 8 hours. The reaction mixture was poured in cold water and extracted with dichioromethane/methanol (10 mEx3). The combined organic layer was dried over anhydrous sodium sulfate, filtered and concentrated in vacuo and purified by prep -TEC (dichloromethane/methanol=1 5:1) to give 5-(4-bromoiso- quinolin-7-yloxy)pentan-1 -ol (1.0 g, 81%) as a yellow solid. ‘H NMR (400 MHz, DMSO-d6) ö 1.49-1.5 1 (4H, m), 1.82 (2H, t, J6.8 Hz), 3.43-3.44 (2H, m), 4.16 (2H, t, J=6.4 Hz), 4.41 (1H, t, J=5.2 Hz), 7.58-7.64 (2H, m), 8.00 (1H, d, J=9.2 Hz), 8.59 (1H, s), 9.19 (1H, s). Chemical Formula: C,4H, 5BrNO2, Molecular Weight: 310.19. Total H count from HNMR data: 16.
With pyridine; In dichloromethane; at 0 - 20℃; for 3h;Inert atmosphere;
e added pyridine (7.1 g, 90 mmol) and benzoyl chloride (10.2 g, 72.5 mmol) at 0 oC. The resulting mixture was stirred for 3 h at room temperature under nitrogen atmosphere. The resulting mixture was concentrated under reduced pressure. The residue was purified by silica gel column chromatography, eluted with 1% ethyl acetate in petroleum ether to afford 5-bromopentyl benzoate (12.5 g, 77%) as a colorless oil. 1H NMR (400 MHz, CDCl3) d 8.07 (dd, J = 8.4, 1.5 Hz, 2H), 7.62-7.53 (m, 1H), 7.46 (dd, J = 8.4, 7.0 Hz, 2H), 4.36 (t, J = 6.5 Hz, 2H), 3.46 (t, J = 6.7 Hz, 2H), 1.96 (p, J = 7.0 Hz,2H), 1.82 (dt, J = 8.3, 6.5 Hz, 2H), 1.64 (dddd, J = 14.8, 9.5, 6.6, 3.5 Hz, 2H)
104 mg
With triethylamine; In diethyl ether; at 20℃; for 8h;Cooling with ice;
<strong>[34626-51-2]5-Bromo-1-pentanol</strong> (167 mg, 1.0 mmol) was dissolved in diethyl ether (5 mL).Add triethylamine (111 mg, 1.1 mmol),After cooling the reaction system with an ice water bath,Add benzoyl chloride (141 mg, 1.0 mmol),Move to room temperature and stir for 8 h. The solvent was evaporated to dryness, and ethyl acetate, water and brine were evaporated.And evaporating the solvent,The intermediate benzoic acid 5-bromopentyl ester (104 mg, 0.38 mmol) was obtained by silica gel column chromatography.
Stage #1: 5-bromopentan-1-ol; 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl-(1->4)-2,3,6-tri-O-acetyl-β-D-glucopyranosyl acetate In dichloromethane at 0℃; for 0.166667h; Molecular sieve; Inert atmosphere;
Stage #2: With boron trifluoride diethyl etherate In dichloromethane for 0.5h; Molecular sieve; Inert atmosphere;
With potassium carbonate; In N,N-dimethyl-formamide; at 60℃; for 8h;
General procedure: Respectively mixing intermediate scopoletin, 20a, and 20b(0.5 mmol) with indicated halo alcohol (0.6 mmol) in DMF (10 ml),K2CO3 (0.6 mmol) was added. The mixture was stirred at 60 C for8 h. The mixture was cooled to room temperature, 10% NaOH aqueous solution was added into the mixture. Intermediate 21-35were obtained by suction filtering, water washing, recrystallization with ethanol, and drying. Compounds 21-35 (0.3 mmol) separately reacted with 15 (0.45 mmol) in CH2Cl2 (10 ml) using DBU (1,0.36 mmol) as a trigger. The mixture was stirred at 15 C for 4 h.Then, it was diluted with CH2Cl2 (20 ml), washed with 5% HCl,water, and dried over anhydrous Na2SO4, and then concentrated.The crude products were purified by silica gel column chromatography using CH2Cl2/MeOH (V/V, 150:1) to provide target compounds 36-50.
With sodium hydroxide In toluene at 50 - 60℃; for 1h;
2 Preparation method:
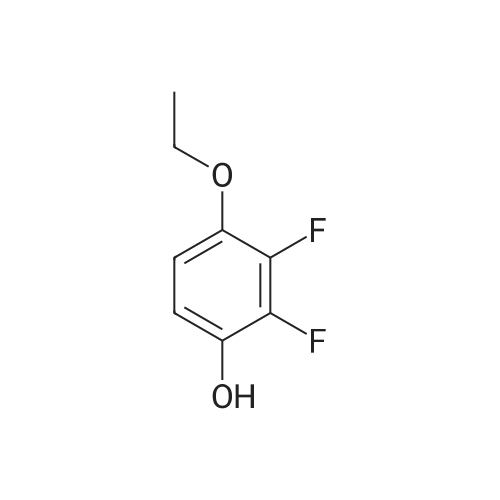
The compound 5-bromopentyl-1-ol 26.72g, NaOH 6g, toluene 40g,Put 25.7g of 4-ethoxy-2,3-difluorophenol into a three-necked flask,After the reaction was kept at 5060 for 1h, the reaction was stopped. The reaction liquid is extracted,After distillation, 28.3 g of compound (I) was obtained.
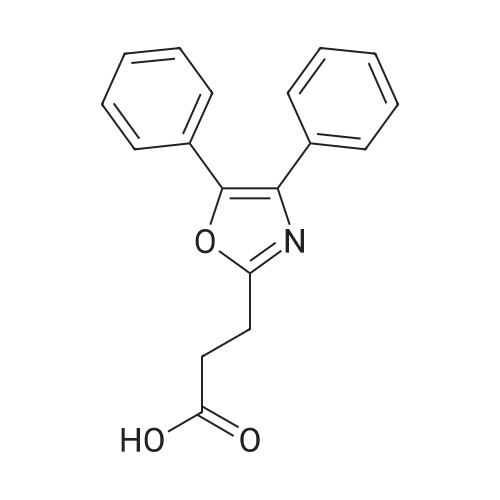
5-bromopentyl 3-(4,5-diphenyloxazol-2-yl)propanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
46%
Stage #1: 3-(4,5-Diphenyl-oxazol-2-yl)-propionic acid With diisopropyl-carbodiimide In dichloromethane at 0℃; for 0.166667h;
Stage #2: 5-bromopentan-1-ol With triethylamine In dichloromethane at 0 - 20℃;
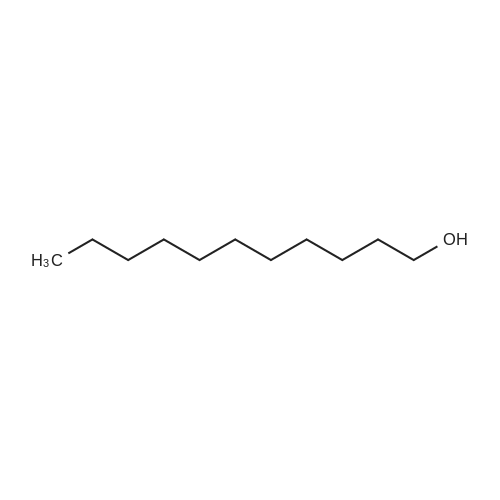
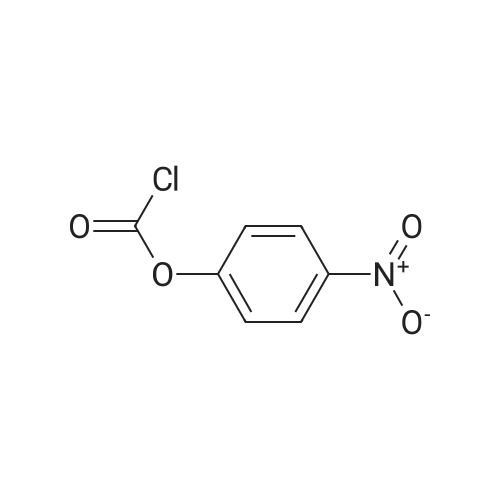
Stage #1: 5-bromopentyl alcohol; 4-Nitrophenyl chloroformate With 4-dimethylaminopyridine In dichloromethane at 20℃; for 3h;
Stage #2: undecyl alcohol In dichloromethane at 20℃;
2 Synthesis of 5-bromopentylundecyl carbonate (2c)
5-Bromopentanol (0.84 g, 5.0 mmol) was dissolved in 30 mL of dichloromethane, 4-dimethylaminopyridine (1.22 g, 10 mmol) was added, and phenyl p-nitrochloroformate (1.11 g) was added in batches , 5.5 mmol), the reaction was stirred at room temperature for 3 h, undecanol (0.97 g, 5.6 mmol) was added to the reaction solution, and the mixture was stirred at room temperature overnight. After TLC showed that the reaction was complete, 20 mL of dichloromethane was added to dilute, and then 30 mL of Washed with saturated brine, the organic phase was dried over anhydrous sodium sulfate, filtered and concentrated, and separated by column chromatography to obtain 5-bromopentylundecyl carbonate 2c (1.20 g, pale yellow oil) in a yield of 66%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping