| 98% |
|
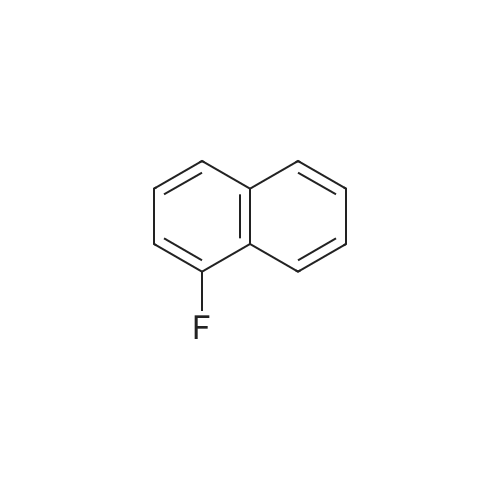
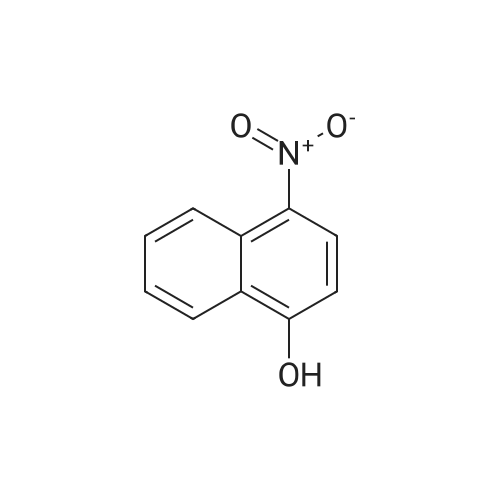
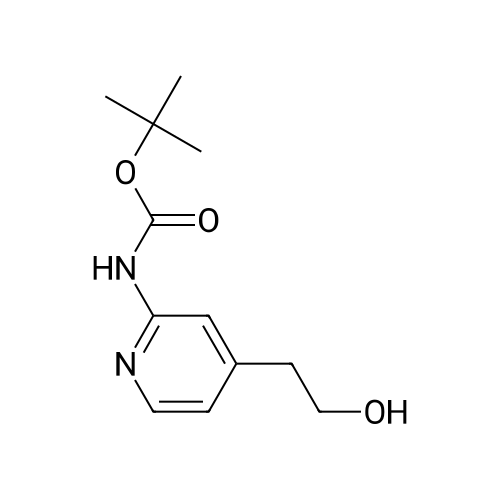
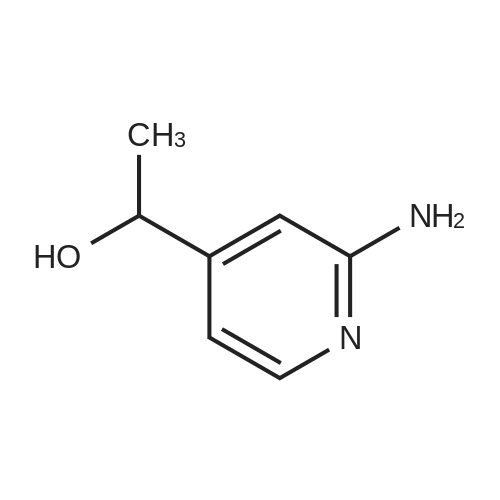
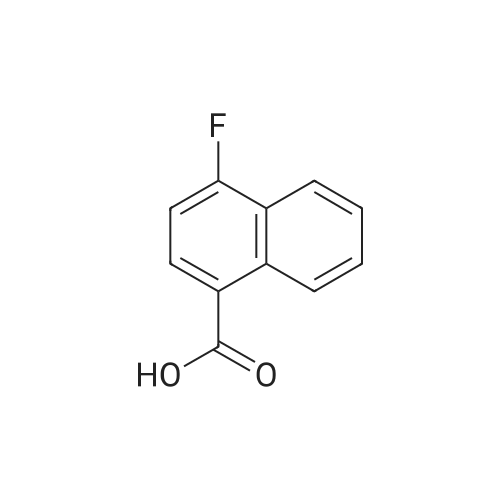
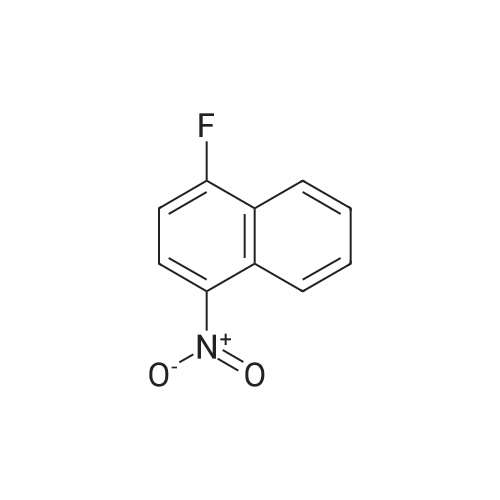
Intermediate BTo a solution of ethyl 2-(2-(fe/f-butoxycarbonylamino)pyridin-4-yl)acetate (WO 2007089512) (10.0 g, 35.7 mmol) under N2 in THF (100 mL), at -78C, was added a solution of DIBAL in THF (1 .0M, 71 .0 mL, 71 .0 mmol) over 1 hr. The reaction mixture was stirred at -78 to -60C for 40 min and was then warmed to -15C over 1 hr. The solution was re-cooled to -78C and was treated with a further aliquot of DIBAL solution (36.0 mL, 36.0 mmol) and was allowed to warm to -40C and stirred for 1 hr. The reaction was quenched by the cautious addition of water (10 mL), followed MgS04. The solids were removed by filtration and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (Si02, 330 g, EtOAc in hexanes, 65% v/v, isocratic elution) to give ferf-butyl 4-(2-hydroxyethyl)pyridin-2- ylcarbamate, (6.0 g, 64%) as a yellow solid: m/z 239 (M+H)+ (ES+).To a solution of ferf-butyl 4-(2-hydroxyethyl)pyridin-2-ylcarbamate (6.0 g, 25 mmol) in THF (70 mL) at 0C was added sodium hydride (2.52 g, 60% wt dispersion in mineral oil, 63.0 mmol) and the bright yellow suspension stirred for 20 min and then treated with 1 -fluoro-4- nitronaphthalene (4.81 g, 25.2 mmol) in a single portion. After stirring at RT for 2 hr the mixture was treated with water (100 mL) followed by EtOAc (100 mL) and the solid which formed at the interface was collected by filtration. The organic phase was separated and was washed with saturated aq. NaHC03 and brine and was then dried and evaporated in vacuo to furnish an orange solid. The two solids were combined and triturated with MeOH (50 mL) to provide ferf- butyl 4-(2-(4-nitronaphthalen-1 -yloxy)ethyl)pyridin-2-ylcarbamate, as a yellow solid (1 1 .0 g, 98%): m/z 410 (M+H)+ (ES+). To a suspension of ferf-butyl 4-(2-(4-nitronaphthalen-1 -yloxy)ethyl)pyridin-2-ylcarbamate (900 mg, 2.20 mmol) in DCM (10.0 mL) was added TFA (10.0 mL) and the reaction mixture was stirred at RT overnight. The resulting mixture was evaporated in vacuo and the residue subjected to SCX capture and release. The crude product so obtained was taken up into THF (8.0 mL) and DIPEA (660 muIota, 3.8 mmol) and then acetyl chloride (147 muIota, 2.06 mmol) were added. After stirring for 1 hr, the mixture was diluted with saturated aq. NaHC03 (10.0 mL) and was extracted with EtOAc (2 x 20 mL). The combined organic layers were washed with brine and then dried, and evaporated in vacuo. The residue was taken up in a mixture of acetonitrile and a solution of NH3 in MeOH (7M, 1 :1 v/v, 20 mL) and after 10 min was re-evaporated in vacuo. The residue was triturated with MeOH (10.0 mL) to afford W-(4-(2-(4-nitronaphthalen- 1 -yloxy)ethyl)pyridin-2-yl)acetamide, as a yellow solid (570 mg, 74%): m/z 352 (M+H)+ (ES+).A solution of /V-(4-(2-(4-nitronaphthalen-1 -yloxy)ethyl)pyridin-2-yl)acetamide (570 mg, 1.62 mmol) in a mixture of AcOH: MeOH (6:1 v/v, 54 mL) was subjected to hydrogenation by passage through a Thales H-cube (1 mLmin"1, 30 mm, 10% Pt/C Cat-Cart, full H2, 45C). The solvent was removed by evaporation in vacuo, and, the residue was subjected to SCX capture and release to furnish the title compound, Intermediate B, (550 mg, 100%): m/z 322 (M+H)+ (ES+). |
| 98% |
|
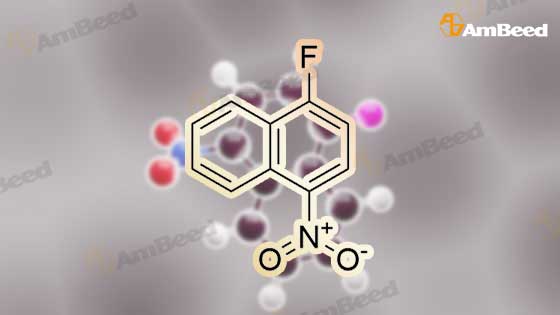
To a solution of tert-butyl 4-(2-hydroxyethyl)pyridin-2-ylcarbamate (20) (6.00 g, 25.2 mmol) in THF (70 mL) was added sodium hydride (2.52 g, 63.0 mmol, 60 wt %) at 0 C. The bright yellow suspension was stirred for 20 min at 0 C. and the <strong>[341-92-4]1-fluoro-4-nitronaphthalene</strong> (14) (4.81 g, 25.2 mmol) added in a single portion. After stirring at RT for 2 hr, water (100 mL) was added followed by EtOAc (100 mL). The solid formed between the layers was collected by filtration and the organic phase was washed with saturated aq NaHCO3 solution (100 mL), brine (100 mL) and dried. The volatiles were removed to give an orange solid. The solids were combined and triturated from MeOH (50 mL) to give tert-butyl 4-(2-(4-nitronaphthalen-1-yloxy)ethyl)pyridin-2-ylcarbamate (21) as a yellow solid (11.0 g, 98%): m/z 410 (M+H)+ (ES+). |
|
With sodium hydride; In tetrahydrofuran; methanol; water; ethyl acetate; |
To a solution of tert-butyl 4-(2-hydroxyethyl)pyridin-2-ylcarbamate (20) (6.00 g, 25.2 mmol) in THF (70 mL) was added sodium hydride (2.52 g, 63.0 mmol, 60 wt %) at 0 C. The bright yellow suspension was stirred for 20 min at 0 C. and the <strong>[341-92-4]1-fluoro-4-nitronaphthalene</strong> (14) (4.81 g, 25.2 mmol) added in a single portion. After stirring at RT for 2 hr, water (100 mL) was added followed by EtOAc (100 mL). The solid formed between the layers was collected by filtration and the organic phase was washed with saturated aq NaHCO3 solution (100 mL), brine (100 mL) and dried. The volatiles were removed to give an orange solid. The solids were combined and triturated from MeOH (50 mL) to give tert-butyl 4-(2-(4-nitronaphthalen-1-yloxy)ethyl)pyridin-2-ylcarbamate (21) as a yellow solid (11.0 g, 98%): m/z 410 (M+H)+ (ES+). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping