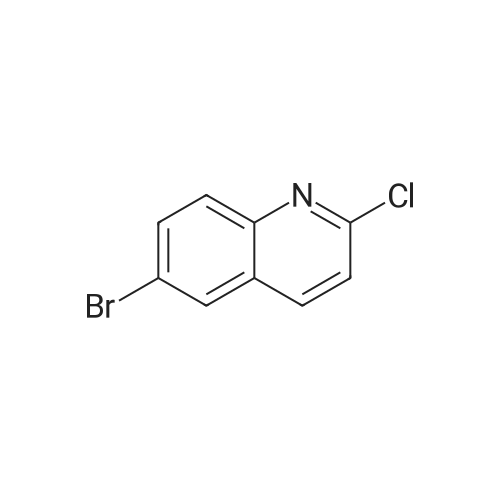
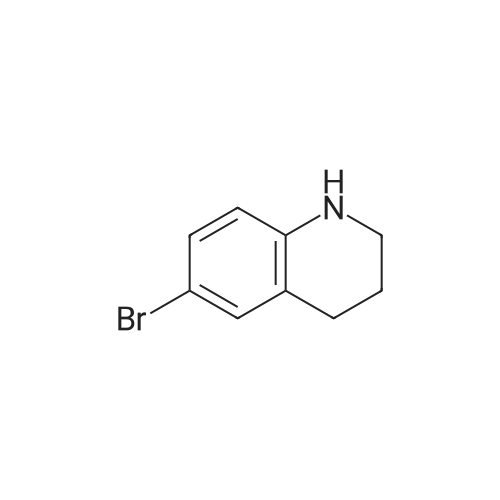
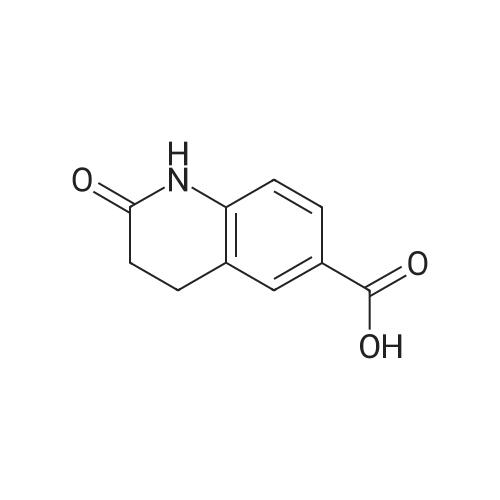
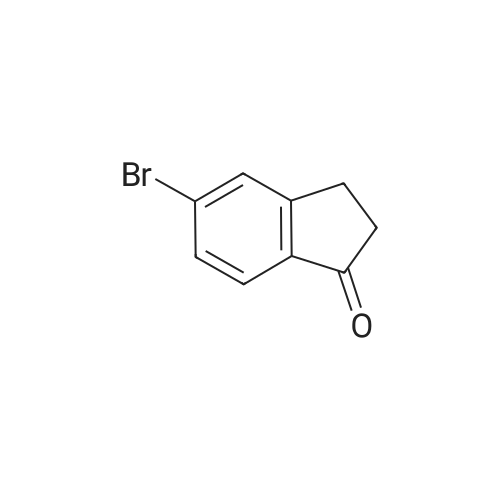
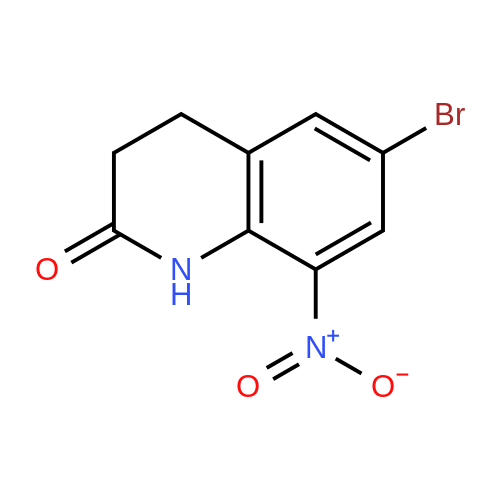
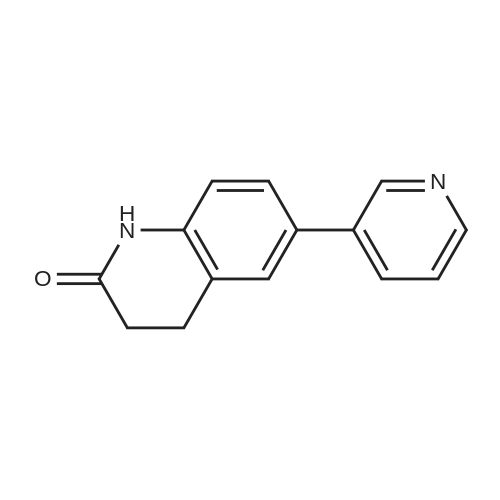
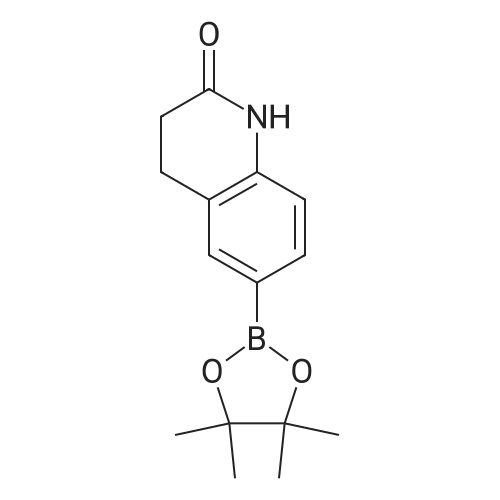
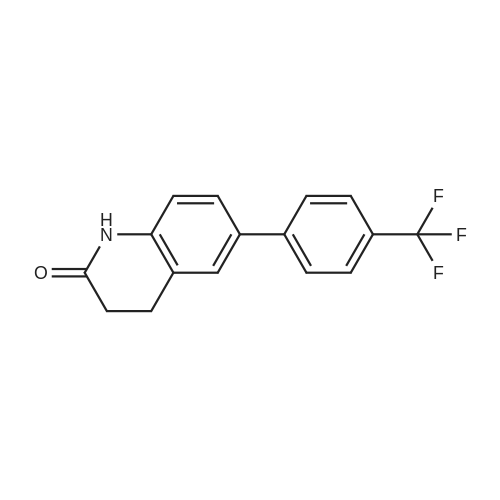
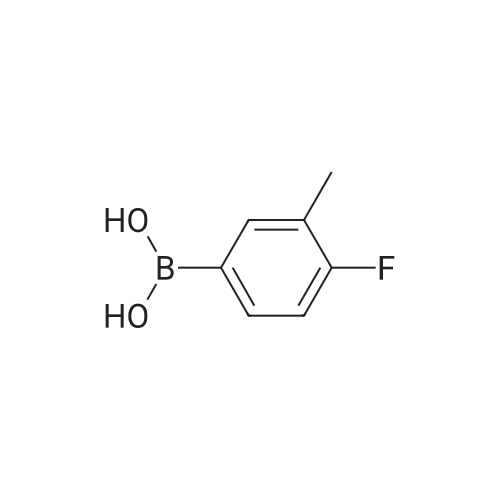
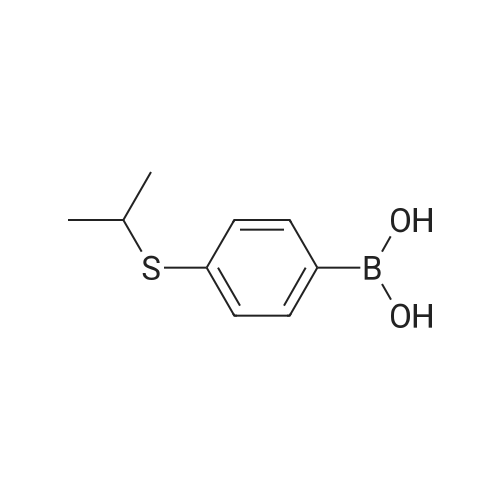
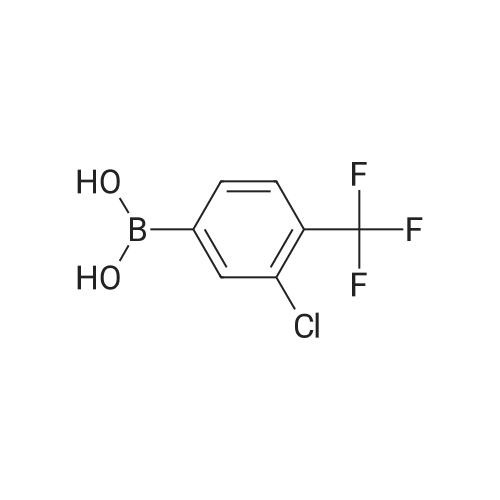
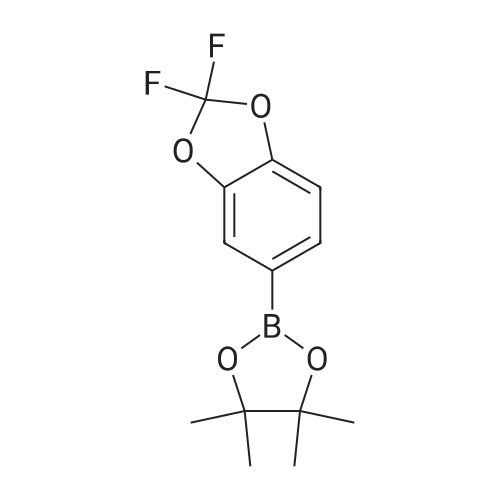
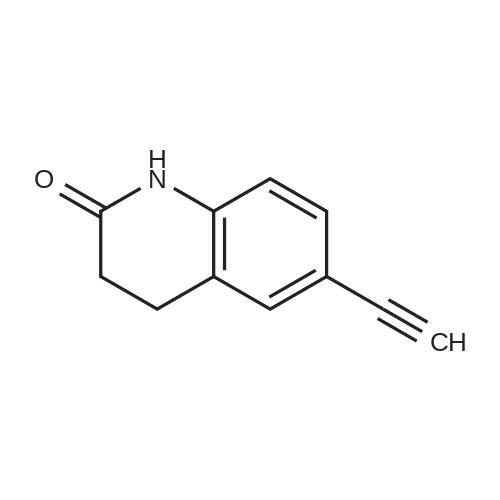
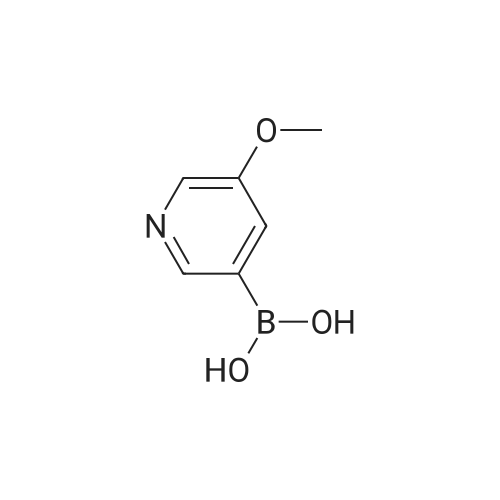
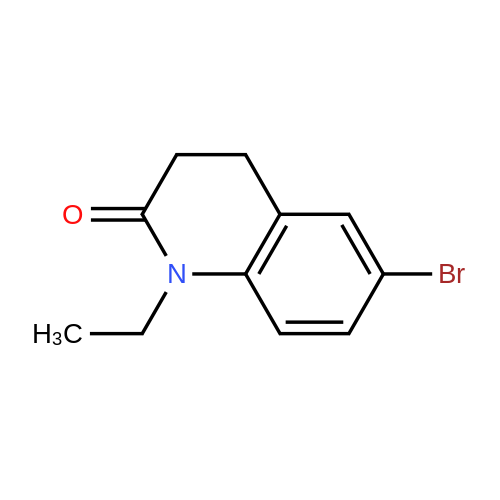
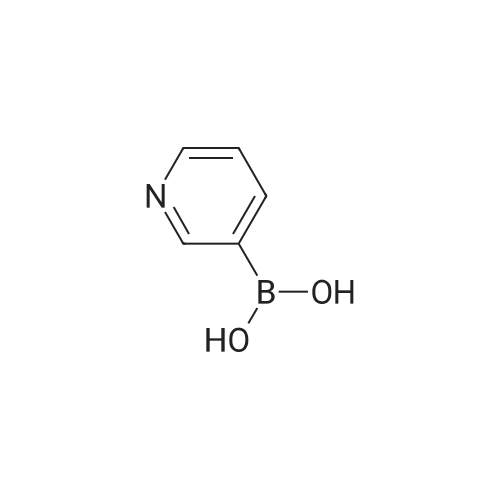
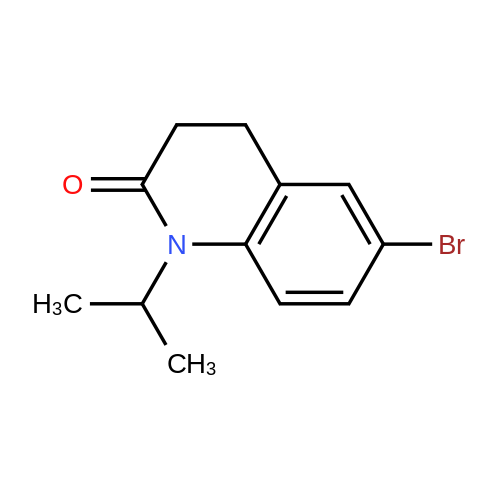
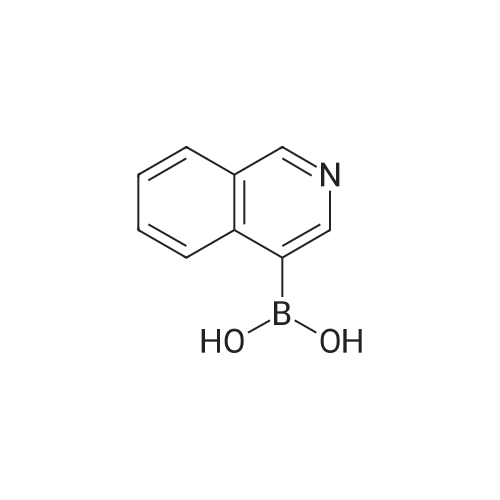
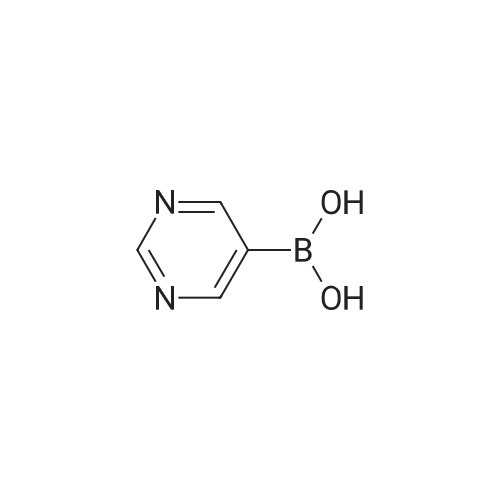
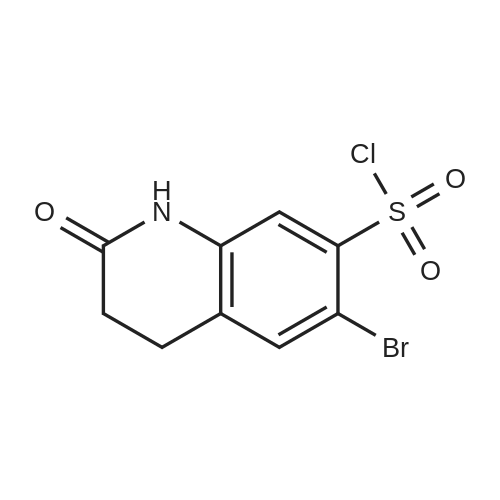
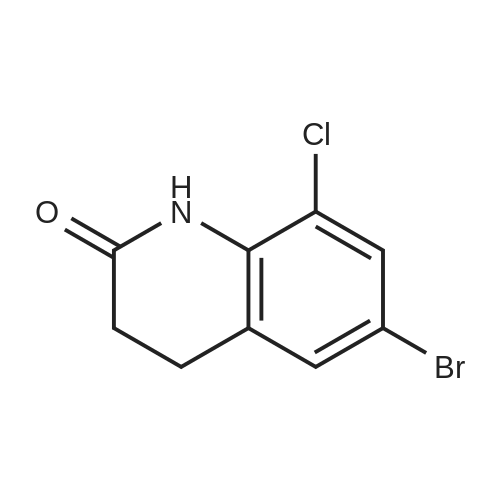
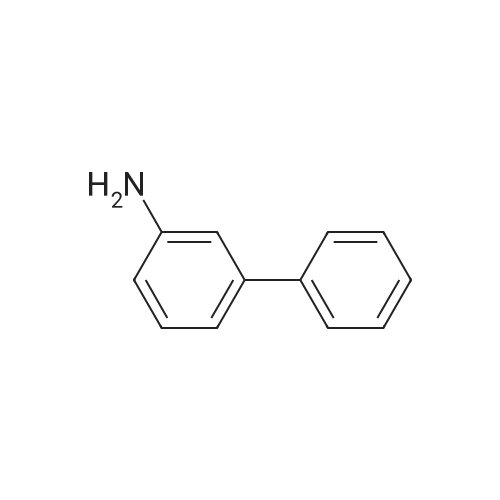
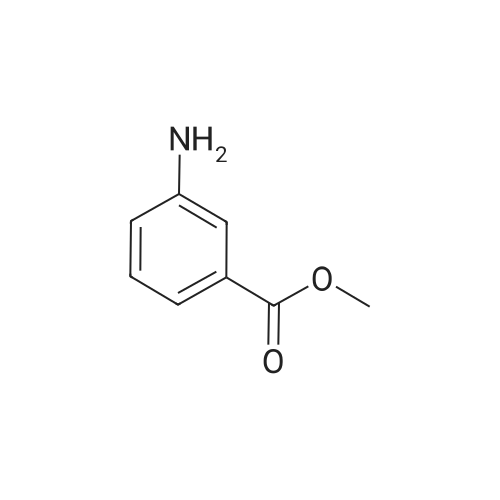
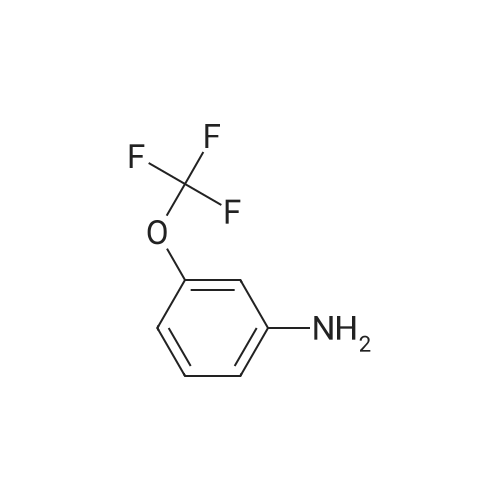
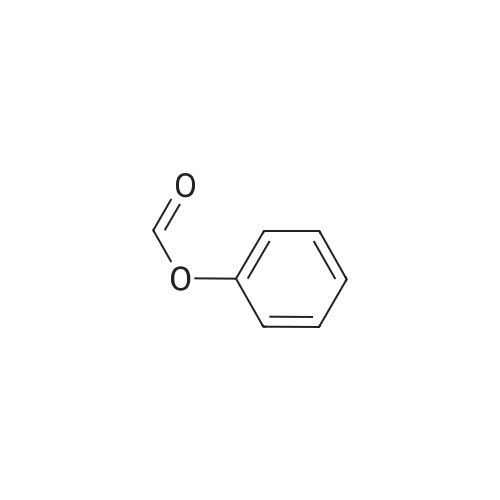
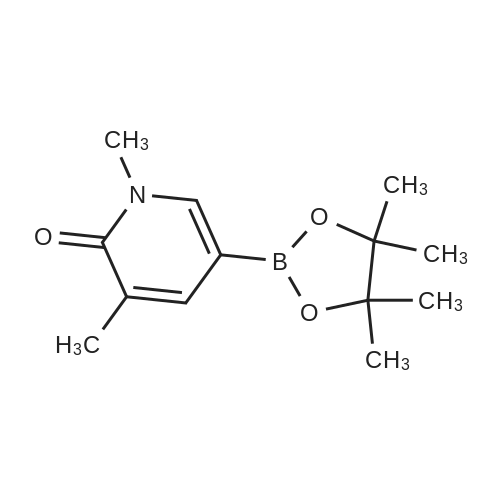
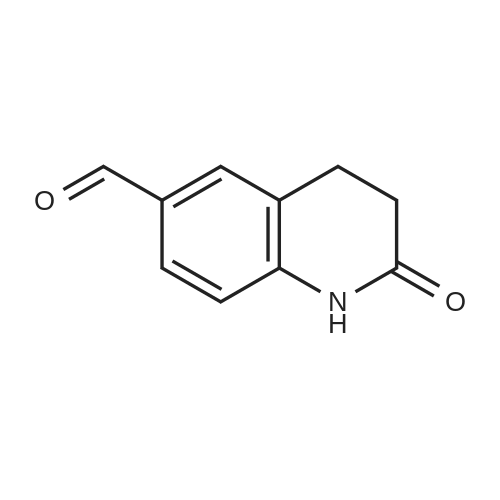
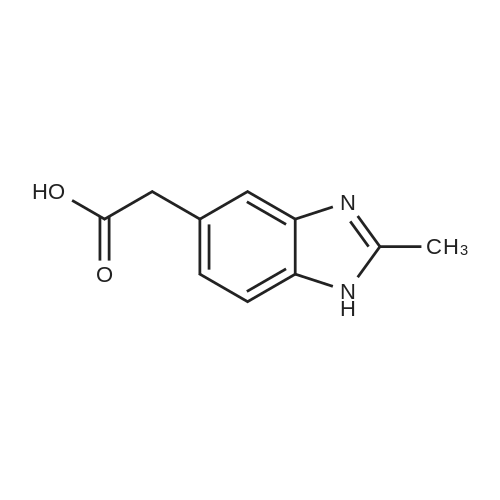
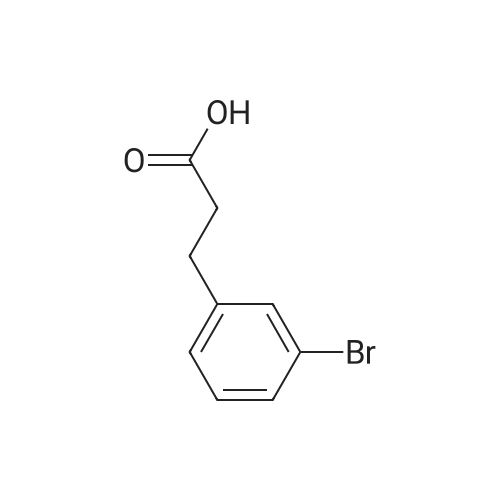
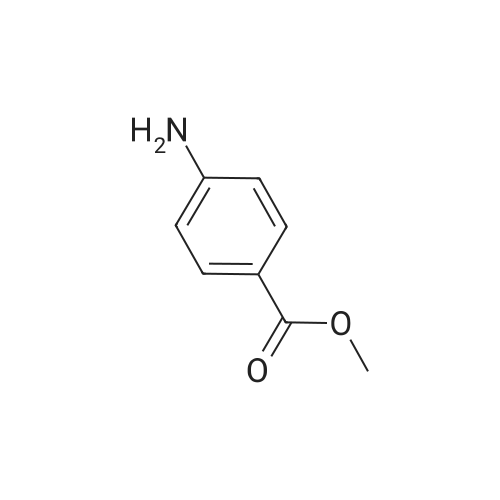
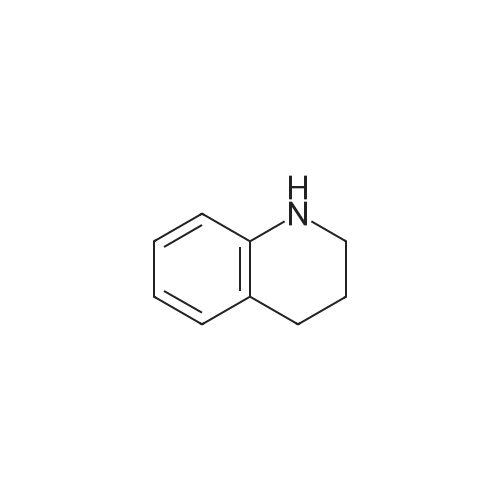
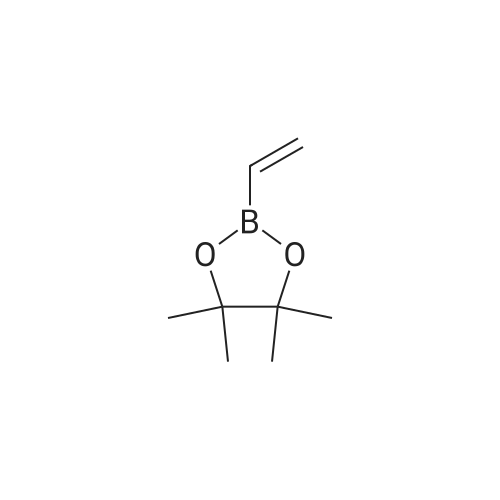
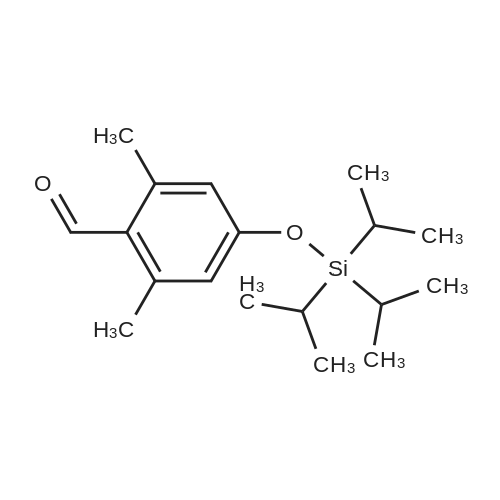
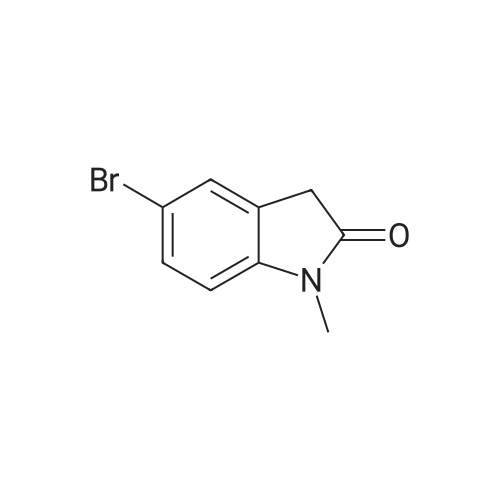
| 92% |
With N-Bromosuccinimide In N,N-dimethyl-formamide at 0℃; for 2h; |
|
| 90% |
With N-Bromosuccinimide In N,N-dimethyl-formamide at 0℃; for 6h; |
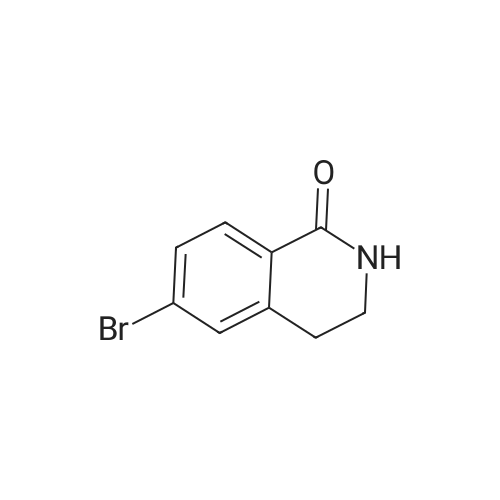
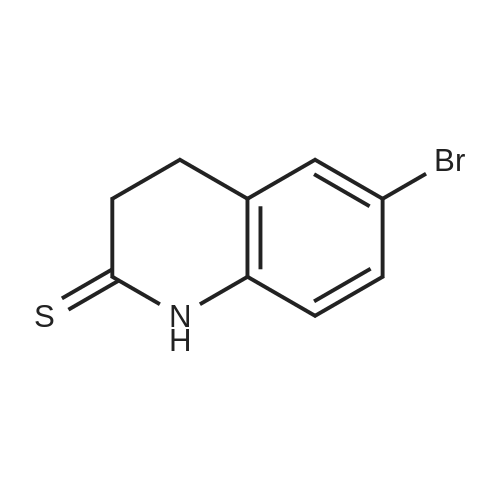
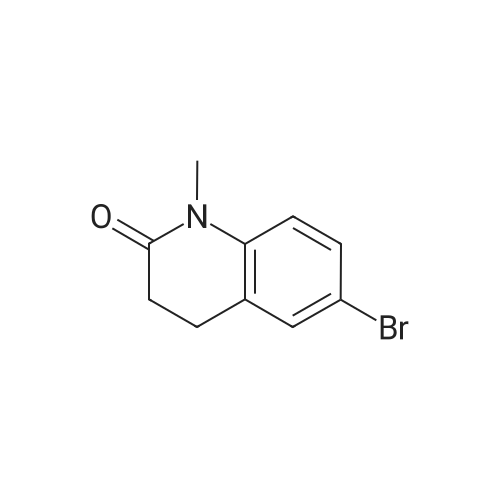
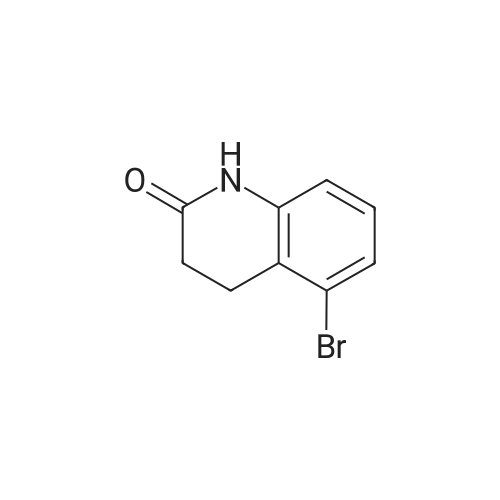
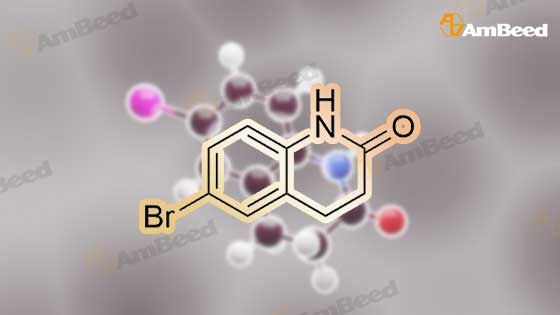
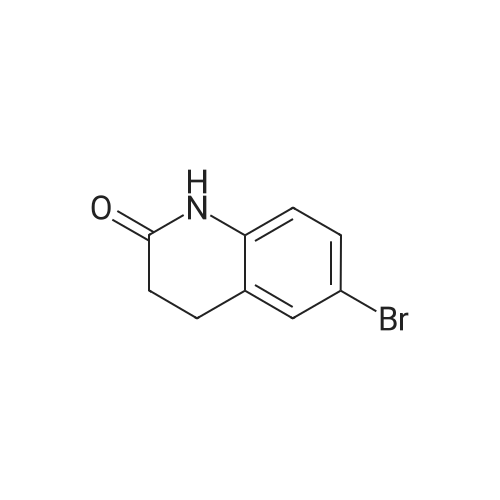
1.1 Step 1: 6-Bromo-3,4-dihydroquinolin-2(1H)-one
The compound 3,4-dihydro-2 (1H) -quinolinone (10.0 g, 67.95 mmol) was dissolved in N, N-dimethylformamide (100 mL).Cooled to 0 ° C,N-bromosuccinimide (12.7 g, 71.34 mmol) was added portionwise,The reaction was stirred for 6 hours under a slow temperature rise.The reaction solution was concentrated to dryness, and ethyl acetate was added thereto, followed by washing with sodium bicarbonate solution and brine. The organic phase was dried, filtered and concentrated to give 6-Bromo-3,4-dihydroquinolin-2(1H)-one (14 g) in 90% yield. |
| 89% |
With N-Bromosuccinimide In N,N-dimethyl-formamide at 0℃; for 2h; |
1
To a solution of 3,4-dihydro-1H-quinolin-2-one (10.0 g, 67.9 mmol) in 100 ml dry DMF was added dropwise a solution of N-bromosuccinimide (12.7 g, 71.3 mmol) in 150 ml dry DMF at 0° C. The mixture was stirred at 0° C. for 2 h, then 400 ml water was added and the solution was extracted with ethyl acetate (3×150 ml). The organic phase was washed with water (2×200 ml), then dried over MgSO4 and evaporated, affording a yellow solid which was purified by washing with cold ether providing pure 6-bromo-3,4-dihydro-1H-quinolin-2-one (13.6 g, 60.3 mmol, 89%) as colorless needles. |
| 89% |
With N-Bromosuccinimide In N,N-dimethyl-formamide at 0℃; Inert atmosphere; |
1
To a solution of 3,4-dihydro-1 H-quinolin-2-one (10.0 g, 67.9 mmol) in 100 ml dry DMF was added dropwise a solution of λ/-bromosuccinimide (12.7 g, 71.3 mmol) in 150 ml dry DMF at 0 °C. The mixture was stirred at 0 0C for 2 h, then 400 ml water was added and the solution was extracted with ethyl acetate (3 x 150 ml). The organic phase was washed with water (2 x 200 ml), then dried over MgSO4 and evaporated, affording a yellow solid which was purified by washing with cold ether providing pure 6-bromo-3,4-dihydro-1 H-quinolin-2-one (13.6 g, 60.3 mmol, 89 %) as colorless needles. |
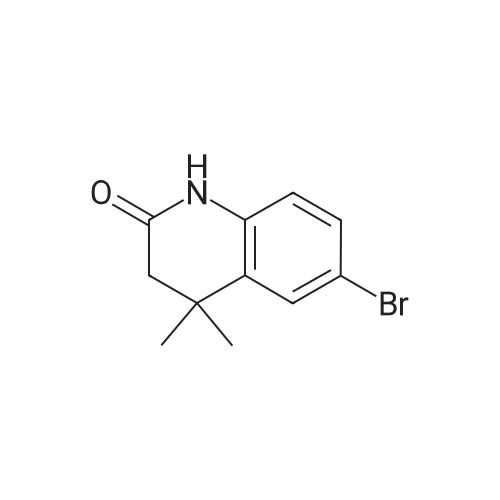
| 87% |
With N-Bromosuccinimide In N,N-dimethyl-formamide at 0℃; for 2h; |
|
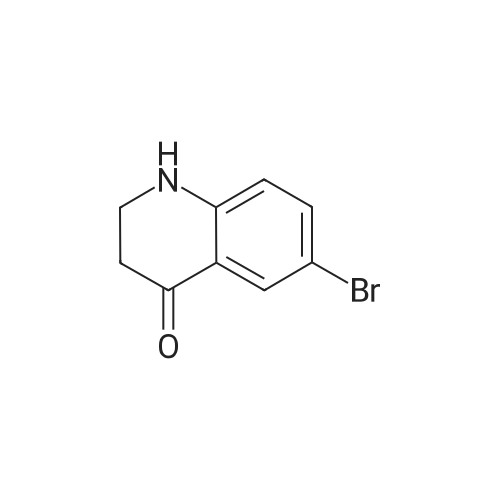
| 71% |
With bis-[(trifluoroacetoxy)iodo]benzene; sodium bromide In ethanol at 25℃; for 0.166667h; Green chemistry; regioselective reaction; |
|
| 69% |
With N-Bromosuccinimide In N,N-dimethyl-formamide at 100℃; for 0.25h; |
|
| 62% |
With N-Bromosuccinimide In DMF (N,N-dimethyl-formamide) at 20℃; |
5 Preparation 5; Preparation of 6-Bromo-3,4-dihydro-1H-quinolin-2-one.
Preparation 5 Preparation of 6-Bromo-3,4-dihydro-1H-quinolin-2-one. To a stirred solution of 3,4-dihydro-1H-quinolin-2-one (0.735 g, 5.0 mmol) in dry DMF (20.0 ML) under N2 at ambient temperature was added N-bromosuccimide (0.93 g, 5.2 mmol) portionwise.The solution was stirred under N2 overnight then the orange mixture was poured into H2O (200 ML) and the precipitated solid was extracted into ether(100 ML).The ether was separated extracted with of H2O, (4*150 ML) washed with brine and dried(MgSO4).Filtration and evaporation in vacuo gave the title compound (0.70 g, 62%) as a light tan powder. 1H NMR(CDCl3) δ 2.60 (2H, m), 2.90 (2H, m), 6.65 (1H, d), 7.2-7.3 (2H, m), 8.9(1H, br s). |
|
With bromine; acetic acid Edukt 2: 1 Mol.; |
|
|
With N-Bromosuccinimide In N,N-dimethyl-formamide |
56.a a)
a) 6-Bromo-3,4-dihydro-2(1H)-quinolinone To a solution of 3,4-dihydro-2(1H)-quinolinone (30 g, 204 mmol) in dried DMF (250 mL) at 0° C. was slowly added N-bromosuccinimide (38 g, 1.05 eq) via a dropping funnel. The reaction mixture was stirred at room temperature overnight, then poured into cold water (3.5L) and the precipitate formed filtered off and dried in vacuo at 45° C. to give the title compound as a white solid. |
|
With N-Bromosuccinimide In N,N-dimethyl-formamide at 0℃; |
|
|
With N-Bromosuccinimide In N,N-dimethyl-formamide at 0 - 20℃; for 12h; |
74.C
To a stirred solution of 3,4-dihydroquinolin-2(l H)-one (74-2; 15.0 g, 0.11 mol) in N,N-dimethylformamide (150 mL) was added N-bromosuccinimide (18.4 g, 0.11 mol) portion wise at 0 °C. Reaction mass was allowed to stir at room temperature for 12 h. The reaction mixture was concentrated and diluted with ice cold water (300 mL) with constant stirring and the solid residue was filtered and dried to obtain the title compound (1-3). 1H MR (400 MHz, DMSO- |
|
With N-Bromosuccinimide In N,N-dimethyl-formamide at 0℃; for 1h; |
3.1 Preparation of 6-bromo-3 ,4-dihydroquinolin-2( 1 H)-one, 13
3,4-Dihydroquinolin-2(1H)-one, 12 (25.0 g, 170 mmol) in DMF (50 mL) was added to a stirred solution of NBS (33.2 g, 186 mmol) in DMF (250 mL) at 0 °C and the mixture was stirred for 1 h. The progress of the reaction was monitored by TLC (TLC system: 50 % EtOAc/Pet ether, Rf value: 0.35). After completion of the reaction, the reaction mixture was quenched with water and then solid formed. The solid was filtered and dried under vacuum to afford the crude product. The crude product was purified over silica gel (100-200 mesh) column chromatography eluting with 3 % EtOAc/Pet ether to give 6-bromo-3,4-dihydroquinolin-2(1H)-one, 13 as a white solid. 1H NMR (400 MHz, CDC13) ö: 8.73 (s, 1 H), 7.28 (dd, 2 H, Jj = 10.8, J2 = 2.4 Hz), 6.69 (d, 1 H, J= 8.0 Hz), 2.95 (t, 2 H, J= 7.2 Hz), 2.65-2.61 (m, 2 H); LC-MS: 96.7 % at 215 nm (mlz 418 [M+H]). |
|
With N-Bromosuccinimide In N,N-dimethyl-formamide at 0℃; for 1h; |
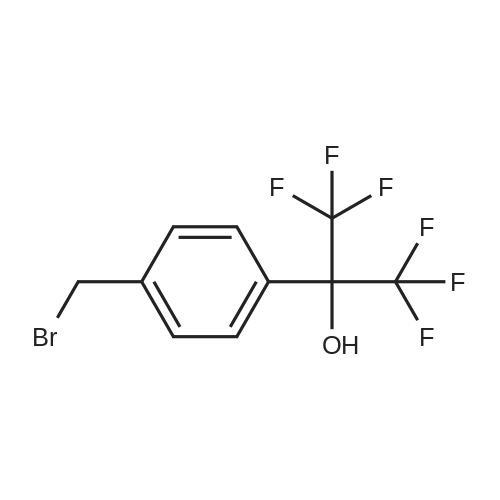
5.1 Example 5. Preparation of l-(4-(l,l,l,3,3,3-hexafluoro-2-hydroxypropan-2-yl)benzyl)-2-oxo- l,2,3,4-tetrahydroquinoline-6-carboxamide [00101] Reaction step 1. Preparation of 6-bromo-3,4-dihydroquinolin-2(lH)-one, 13
[00101] Reaction step 1. Preparation of 6-bromo-3,4-dihydroquinolin-2(lH)-one, 13 [00102] 3,4-Dihydroquinolin-2(lH)-one, 12 (25.0 g, 169 mmol) in DMF (50 mL) was added to a stirred solution of NBS (33.2 g, 186 mmol) in DMF (250 mL) at 0 °C and the mixture was stirred for 1 h. The progress of the reaction was monitored by TLC (TLC system: 50 % EtO Ac/Pet ether, Rf value: 0.35). [00103] After completion of the reaction, the reaction mixture was quenched with water and then a solid formed. The solid was filtered and dried under vacuum to afford the crude product. The crude product was purified over silica gel (100-200 mesh) column chromatography eluting with 3 % EtOAc/Pet ether to give 6-bromo-3,4-dihydroquinolin-2(lH)-one, 13 as a white solid. LC/MS: calc M+H 227, obs 227; lH NMR (400 MHz, CDC13) δ: 8.73 (s, 1 H), 7.28 (dd, 2 H, J, = 10.8, J2 = 2.4 Hz), 6.69 (d, 1 H, J= 8.0 Hz), 2.95 (t, 2 H, J= 7.2 Hz), 2.65-2.61 (m, 2 H). |
| 385.5 mg |
With N-Bromosuccinimide In N,N-dimethyl-formamide at 0 - 20℃; for 9h; |
4.2.7.1 6-Bromo-3,4-dihydroquinolin-2(1H)-one (89)
To a solution of 3,4-dihydro-2(1H)-quinolinone (454.8mg, 3.090mmol) in DMF (5mL) was added N-bromosuccinimide (587.1mg, 3.299mmol) in DMF (5mL) at 0°C. The mixture was then allowed to warm to ambient temperature, stirred for 9h, and diluted with ethyl acetate. The organic layer was washed with water and brine, dried, and concentrated. The residue was recrystallized from ethanol/water to yield the title compound (385.5mg, 1.705mmol) as an off-white solid. 1H NMR (CDCl3) δ: 8.56 (1H, br s), 7.30-7.26 (2H, m), 6.68 (1H, d, J=8.0Hz), 2.96 (2H, t, J=7.5Hz), 2.63 (2H, t, J=8.0Hz). 13C NMR (CDCl3) δ: 171.37, 136.34, 130.85, 130.39, 125.71, 116.84, 115.45, 30.32, 25.14. |
|
With N-Bromosuccinimide In acetonitrile at 0℃; for 2h; |
Step A: 6-bromo-3.4-dihydroiuinolin-2(1H)-one
Step A: 6-bromo-3.4-dihydroiuinolin-2(1H)-oneTo a solution of 3,4-dihydro-1H-quinolin-2-one (5.00 g, 31.0 mmol) in 40 mL of CH3CN was added NBS (6.80 g, 38.0 mmol) in portions at 0 °C. The mixture was stirred at 0 °C for 2 h, then 30 mL of water was added and the solution was extracted with ethyl acetate three times. The combined organic phase was dried over Na2504, filtered and then concentrated to afford the title compound. LC/MS[M+1] = 226. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping