* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With hydrogenchloride In water at 20 - 25℃; for 24 h;
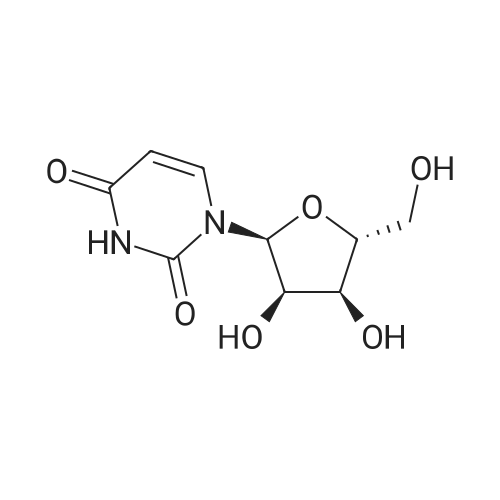
98 g of Compound D was dissolved in 500 ml of 0.1 N hydrochloric acid, and the reaction was stirred at 20-25 ° C;After 24 hours of reaction, the sample was monitored by HPLC.The remaining material is 0.21percent,Product A was 99.74percent and almost no other side reactions were produced.After the reaction is completed, the pH of the system is adjusted to 7-8 with concentrated ammonia water.Then concentrated under reduced pressure at 50 ± 5 ° C until no drop;The system was concentrated under reduced pressure at 50 ± 5 ° C until after dripping.Add 500 ml of anhydrous acetonitrile to dissolve the residue at 70 ± 5 ° C.Filter out insoluble impurities,The filtrate was stirred and crystallized at 0 ± 5 ° C for 12 hours.filter,Obtaining a white solid compound A,The purity of the sampled HPLC analysis was 99.92percent.The solid was blast dried at 45 ° C for 24 hours to obtain 71.8 g of the product.
Reference:
[1] Patent: CN108424431, 2018, A, . Location in patent: Page/Page column 10-20
3-Benzoyl-1-((2S,3R,3aS,9aR)-3-hydroxy-5,5,7,7-tetraisopropyl-tetrahydro-1,4,6,8-tetraoxa-5,7-disila-cyclopentacycloocten-2-yl)-1H-pyrimidine-2,4-dione[ No CAS ]
3-Benzoyl-1-((2S,3R,3aR,9aR)-5,5,7,7-tetraisopropyl-3-methoxy-tetrahydro-1,4,6,8-tetraoxa-5,7-disila-cyclopentacycloocten-2-yl)-1H-pyrimidine-2,4-dione[ No CAS ]
With hydrogenchloride; In water; at 20 - 25℃; for 24.0h;
98 g of Compound D was dissolved in 500 ml of 0.1 N hydrochloric acid, and the reaction was stirred at 20-25 C;After 24 hours of reaction, the sample was monitored by HPLC.The remaining material is 0.21%,Product A was 99.74% and almost no other side reactions were produced.After the reaction is completed, the pH of the system is adjusted to 7-8 with concentrated ammonia water.Then concentrated under reduced pressure at 50 ± 5 C until no drop;The system was concentrated under reduced pressure at 50 ± 5 C until after dripping.Add 500 ml of anhydrous acetonitrile to dissolve the residue at 70 ± 5 C.Filter out insoluble impurities,The filtrate was stirred and crystallized at 0 ± 5 C for 12 hours.filter,Obtaining a white solid compound A,The purity of the sampled HPLC analysis was 99.92%.The solid was blast dried at 45 C for 24 hours to obtain 71.8 g of the product.
With cation resin; In water; acetonitrile; at 50℃;
101 g of the crude compound III-1 obtained in the previous step, acetonitrile (432.3 ml) of water (21.8 ml) and a cation resin (10.9 g) were added to the reaction flask; after the addition was completed, the reaction was stirred in an oil bath at 50 C.Detection: Samples were taken every 2 hours after 2 hours of reaction. HPLC detection; if HPLC detected <1% of the raw materials, the reaction was completed and the heating was terminated; otherwise, the reaction was continued.Filtration: After the reaction is completed to room temperature, the cationic resin is filtered off and rinsed twice with 45 ml of water; neutralized: 1 N NaOH is added dropwise to adjust the pH of the reaction solution to 7.1-7.3, and the temperature is controlled below 30 degrees;Concentration: The filtrate was concentrated to dryness under reduced pressure at 45 ± 2 C.The crude product was subjected to silica gel column chromatography to obtain 8.1 g of alpha-uridine, and the total separation yield of the three steps was 10.1% (mol), 8.1% (w).The purity is 99.09%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping