* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: for 0.25 h; Green chemistry Stage #2: With tert.-butylhydroperoxide In decane for 48 h; Green chemistry
General procedure: B(C6F5)3 (1 mol percent) was added to a stirringsolution of aldehyde (1 mmol) in MeOH (6 mL). After 15 min., 5.5 M TBHP indecane (3 mmol) was added slowly and reaction mixture was refluxed untilthe complete conversion of starting material (monitored by TLC). Aftercompletion of reaction, the methanol was evaporated in vacuo. Later, thereaction mixture was diluted with water (20 mL) and extracted with ethylacetate (3 15 mL). The organic layer was washed with cold saturated sodiumbicarbonate solution (2 20 mL) followed by brine. The organic layer wasdried over MgSO4 and concentrated under reduced pressure and products werepurified over silica gel column chromatography in ethyl acetate/hexane. Allcompounds were characterized and confirmed by comparison of their spectraldata and physical properties with reported literature.
Reference:
[1] Advanced Synthesis and Catalysis, 2013, vol. 355, # 2-3, p. 499 - 507
3
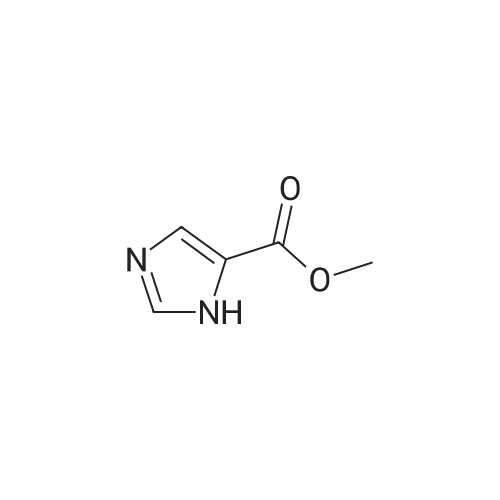
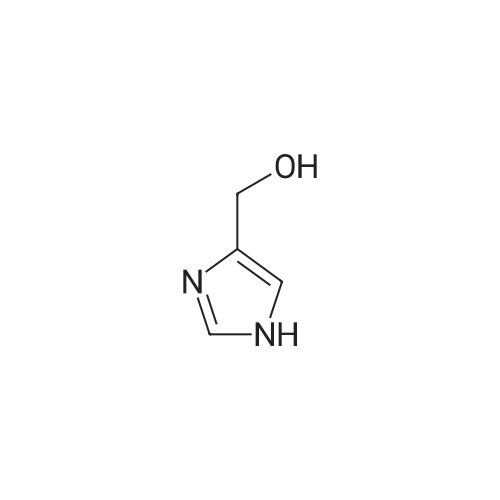
[ 2302-25-2 ]
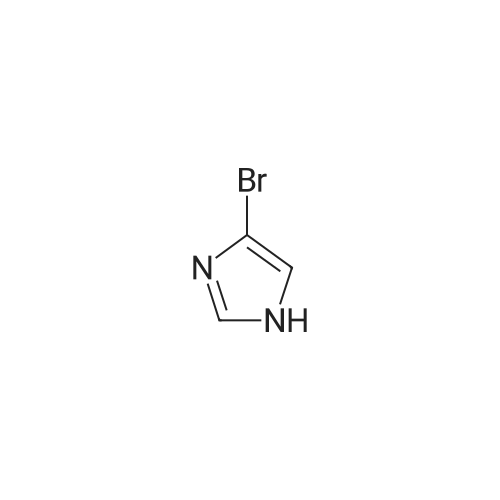
[ 68-12-2 ]
[ 3034-50-2 ]
Yield
Reaction Conditions
Operation in experiment
85%
Stage #1: With isopropylmagnesium chloride In tetrahydrofuran at 0℃; for 0.166667 h; Stage #2: With n-butyllithium In tetrahydrofuran; hexane at -20℃; for 0.5 h; Stage #3: at -20℃; for 0.5 h;
To a solution of 4-bromo-1H-imidazole (0.65 g, 4.4 mmol, 1.0 equiv.) indry THF (20 mL) at 0 C was added a 2 M solution of i-PrMgCl in THF (2.2 mL, 4.4 mmol, 1.0 equiv.)during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a2.5 M solution of n-BuLi in hexanes (3.5 mL, 8.8 mmol, 2.0 equiv.) was added dropwise during5 min, while maintaining the temperature below 20 C. The resulting mixture was stirred at thattemperature for 0.5 h, dry DMF (0.32 g, 4.4 mmol, 1.0 equiv.) was added to 20 C. The resultingmixture was warmed to 20 C in 0.5 h and quenched with water (6 mL). After stirring the mixturebelow 20 C for 10 min, the phases were separated and the water phase was extracted one additionaltime with ethyl acetate. The resulting suspension was allowed to reach room temperature and ®teredthrough a 0.5 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The ®ltrate was concentratedand the residue was puri®ed by ash chromatography on silica gel (eluent:petroleum ether/ethylacetate = 10:1) to afford product 3l as off-white solid, 0.36 g (yield: 85percent), m.p.: 175–177 C. 1H-NMR(600 MHz, DMSO) 9.74 (s, 1H), 7.99 (s, 1H), 7.94 (s, 1H). 13C-NMR (151 MHz, DMSO) 184.46, 139.44,134.9, 129.5.
Reference:
[1] Molecules, 2017, vol. 22, # 11,
4
[ 2644-71-5 ]
[ 3034-50-2 ]
Yield
Reaction Conditions
Operation in experiment
54%
With sodium periodate In water at 27 - 30℃; for 2 h;
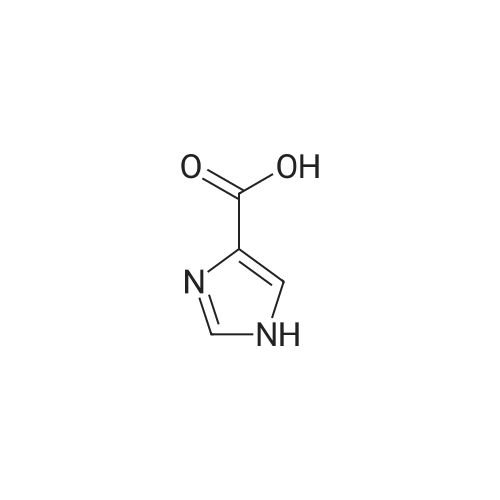
Reference Example 10Under nitrogen stream, to ( 1R, 2S, 3R) -1- (2-sulfanyl-lH- imidazol-4-yl) butane-1, 2, 3, 4-tetraol (10 g, 45.4 mmol) was added water (40 mL) , and to the obtained suspension was added dropwise an aqueous diluted solution- of 30percent aqueous hydrogen peroxide (15.4 g, 136 mmol, 3.0 equivalents) in water (40 mL) over 10 min at 17 to 43°C (the compound was gradually dissolved to give an uniform pale-yellow solution) . The reaction mixture was stirred at 24 to 36°C for 4 hr, and barium carbonate (27 g, 136 mmol, 3.0 equivalents) was added over 5 min at 24 to 26°C (neutralized to pH 7), and the mixture was stirred at 25 to26°C for 1 hr and 20 min. The insoluble material was filtered off, and washed with water (40 mL) . To the filtrate and washing was added sodium sulfite (11.4 g, 90.8 mmol, 2.0 equivalents) over 5 min at 20 to 32°C. The obtained aqueous solution was stirred at 26 to 32°C for 1 hr and 30 min to give an aqueous solution of (1R, 2S, 3R) -1- (lH-imidazol-4-yl) butane- 1 , 2 , 3 , 4-tetraol . To this aqueous solution was added sodium periodate (29.1 g, 136 mmol, 3.0 equivalents) over 10 min at 12 to 30°C, and the mixture was stirred at 27 to 30°C for 1 hr and 30 min. To the reaction mixture was added sodium periodate(2.91 g, 13.6 mmol, 0.3 equivalents) at 27 to 30°C, and the mixture was stirred at 27 to 30°C for 2 hr. The insoluble material was filtered off, and washed four times with water (10 mL) . To the filtrate and washing was added methanol (500 mL) , and the inorganic salt was filtered off, and washed twice with methanol (50 mL) . To the filtrate and washing was added activated carbon (3 g, SHIRASAGI A, trade name), and the mixture was stirred at room temperature for 1 hr. Theinsoluble material was filtered off, and washed with methanol. The filtrate and washing were concentrated under reduced pressure to give a crude compound (9.37 g) . To the crude compound were added water (3 mL) and seed crystals forcrystallization, and the mixture was stirred at roomtemperature for 24 hr, and then for 2 hr under ice-cooling. The crystals were collected by filtration-, washed with cooled water (1 mL) , and vacuum-dried (50°C) to a constant amount to give (5) -formylimidazole (2.35 g) . yield 54percent.
Stage #1: at 8 - 30℃; for 2 h; Stage #2: at 80 - 110℃; for 3.83333 h;
Example 72An alternate method'for the synthesis of the imidazole intermediate is described below:4-Cyano-l-(2-trimethylsilanyl-ethoxymethyl)-lH-imidazole-2-carboxylic acid potassium salt a) lH-Imidazole-4-carbonitrile; '-NHA 22-L, four-neck, round-bottom flask equipped with a mechanical stirrer, a temperature probe, a condenser, and an addition funnel with a nitrogen inlet was charged with lH-imidazole-4-carboxaldehyde (Aldrich, 1.10 kg, 11.5 mol) and pyridine (3.0 L, 3.0 mol). The reaction flask was cooled to 8 0C with an ice bath and hydroxylamine hydrochloride (871 g, 12.5 mol) was added slowly in portions to maintain the internal temperature below 30 °C. The reaction was allowed to cool to ambient temperature and stirred for 2 h at ambient temperature. The resulting thick yellow solution was heated to 80 0C with a heating mantle and acetic anhydride (2.04 L, 21.6 mol) was added dropwise EPO <DP n="148"/>over 200 min to maintain the temperature below 110 °C during the addition. The reaction mixture was heated at 100 0C for 30 min, after which time it was allowed to cool to ambient temperature and then further cooled in an ice bath. The pH was adjusted to 8.0 (pH meter) by the addition of 25 wt percent NaOH (5.5 L) at such a rate that the internal temperature was maintained below 30 °C. The reaction mixture was then transferred into a 22-L separatory funnel and extracted with ethyl acetate (6.0 L). The combined organic layer was washed with brine (2 x 4.0 L), dried over MgSO4, filtered, and concentrated to dryness under reduced pressure at 35 °C to give the crude product as a yellow semisolid. The resulting semisolid was suspended in toluene (3.0 L) and stirred for 1 h, after which time it was filtered to give a light yellow solid, which was resuspended in toluene (3.0 L) and stirred for 1 h. The resulting slurry was filtered and the filter cake washed with toluene (2 x 500 mL) to give the title compound as a light yellow solid [870 g, 82percent). The 1H and 13C NMR spectra were consistent with the assigned structure.
82%
at 0 - 110℃; for 5.83333 h;
A 22-L, four-neck, round-bottom flask equipped with a mechanical stirrer, a temperature probe, a condenser, and an addition funnel with a nitrogen inlet was charged with 1H-imidazole-4-carboxaldehyde (Aldrich, 1.10 kg, 11.5 mol) and pyridine (3.0 L, 3.0 mol). The reaction flask was cooled to 8° C. with an ice bath and hydroxylamine hydrochloride (871 g, 12.5 mol) was added slowly in portions to maintain the internal temperature below 30° C. The reaction was allowed to cool to ambient temperature and stirred for 2 h at ambient temperature. The resulting thick yellow solution was heated to 80° C. with a heating mantle and acetic anhydride (2.04 L, 21.6 mol) was added dropwise over 200 min to maintain the temperature below 110° C. during the addition. The reaction mixture was heated at 100° C. for 30 min, after which time it was allowed to cool to ambient temperature and then further cooled in an ice bath. The pH was adjusted to 8.0 (pH meter) by the addition of 25 wt percent NaOH (5.5 L) at such a rate that the internal temperature was maintained below 30° C. The reaction mixture was then transferred into a 22-L separatory funnel and extracted with ethyl acetate (6.0 L). The combined organic layer was washed with brine (2.x.4.0 L), dried over MgSO4, filtered, and concentrated to dryness under reduced pressure at 35° C. to give the crude product as a yellow semisolid. The resulting semisolid was suspended in toluene (3.0 L) and stirred for 1 h, after which time it was filtered to give a light yellow solid, which was resuspended in toluene (3.0 L) and stirred for 1 h. The resulting slurry was filtered and the filter cake washed with toluene (2.x.500 mL) to give the title compound as a light yellow solid [870 g, 82percent). The 1H and 13C NMR spectra were consistent with the assigned structure.
82%
Stage #1: at 8 - 30℃; for 2 h; Stage #2: at 80 - 110℃; for 0.5 h;
a) 1H-Imidazole-4-carbonitrile A 22-L, four-neck, round-bottom flask equipped with a mechanical stirrer, a temperature probe, a condenser, and an addition funnel with a nitrogen inlet was charged with 1H-imidazole-4-carboxaldehyde (Aldrich, 1.10 kg, 11.5 mol) and pyridine (3.0 L, 3.0 mol). The reaction flask was cooled to 8° C. with an ice bath and hydroxylamine hydrochloride (871 g, 12.5 mol) was added slowly in portions to maintain the internal temperature below 30° C. The reaction was allowed to cool to ambient temperature and stirred for 2 h at ambient temperature. The resulting thick yellow solution was heated to 80° C. with a heating mantle and acetic anhydride (2.04 L, 21.6 mol) was added dropwise over 200 min to maintain the temperature below 110° C. during the addition. The reaction mixture was heated at 100° C. for 30 min, after which time it was allowed to cool to ambient temperature and then further cooled in an ice bath. The pH was adjusted to 8.0 (pH meter) by the addition of 25 wt percent NaOH (5.5 L) at such a rate that the internal temperature was maintained below 30° C. The reaction mixture was then transferred into a 22-L separatory funnel and extracted with ethyl acetate (6.0 L). The combined organic layer was washed with brine (2*4.0 L), dried over MgSO4, filtered, and concentrated to dryness under reduced pressure at 35° C. to give the crude product as a yellow semisolid. The resulting semisolid was suspended in toluene (3.0 L) and stirred for 1 h, after which time it was filtered to give a light yellow solid, which was resuspended in toluene (3.0 L) and stirred for 1 h. The resulting slurry was filtered and the filter cake washed with toluene (2*500 mL) to give the title compound as a light yellow solid [870 g, 82percent). The 1H and 13C NMR spectra were consistent with the assigned structure.
82%
Stage #1: With hydroxylamine hydrochloride In pyridine at 8 - 30℃; for 2 h; Stage #2: at 0 - 110℃; for 0.5 h;
A 22-L, four-neck, round-bottom flask equipped with a mechanical stirrer, a temperature probe, a condenser, and an addition funnel with a nitrogen inlet was charged with lH-imidazole-4-carboxaldehyde (Aldrich, 1.10 kg, 11.5 mol) and pyridine (3.0 L, 3.0 mol). The reaction flask was cooled to 8 0C with an ice bath and hydroxylamine hydrochloride (871 g, 12.5 mol) was added slowly in portions to maintain the internal temperature below 30 0C. The reaction was allowed to cool to ambient temperature and stirred for 2 h at ambient temperature. The resulting thick yellow solution was heated to 80 0C with a heating mantle and acetic anhydride (2.04 L5 21.6 mol) was added dropwise over 200 min to maintain the temperature below 110 0C during the addition. The reaction mixture was heated at 100 0C for 30 min, after which time it was allowed to cool to ambient temperature and then further <n="149"/>cooled in an ice bath. The pH was adjusted to 8.0 (pH meter) by the addition of 25 wt percent NaOH (5.5 L) at such a rate that the internal temperature was maintained below 30 °C. The reaction mixture was then transferred into a 22-L separatory funnel and extracted with ethyl acetate (6.0 L). The combined organic layer was washed with brine (2 x 4.0 L), dried over MgSO4, filtered, and concentrated to dryness under reduced pressure at 35 0C to give the crude product as a yellow semisolid. The resulting semisolid was suspended in toluene (3.0 L) and stirred for 1 h, after which time it was filtered to give a light yellow solid, which was resuspended in toluene (3.0 L) and stirred for 1 h. The resulting slurry was filtered and the filter cake washed with toluene (2 x 500 nxL) to give the title compound as a light yellow solid [870 g, 82percent). The 1H and 13C NMR spectra were consistent with the assigned structure.
With bromine; sodium acetate; acetic anhydride In acetic acid at 20℃; for 3.5 h;
Bromine (8.0 g, 50 mmol, 2.3 eq) in anhydrous Ac2O (40 mL) was added dropwise to a solution of 1B (2.08 g, 21.7 mmol) and NaOAc (18.7 g, 228 mmol, 10.5 eq) in anhydrous HOAc (200 mL) over a period of 1 h at RT. The resulting mixture was stirred at RT for 2.5 h and then concentrated. The residue was partitioned between Et2O (200 mL) and water (200 mL), the layers were separated and the aqueous layer was extracted with Et2O (200 mL). The combined organic phase was dried, concentrated and chromatographed (EtOAc) to afford 5-bromo-4-formyl imidazole 39A (1.00 g, 26percent) as white crystals.
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at -78 - 20℃; for 0.166667 h; Inert atmosphere Stage #2: at -78 - 20℃; for 18 h; Inert atmosphere
To a suspension of 10 (4.94 g, 51.4 mmol) in dry THF (30 mL) was added NaH (2.48 g, assey 60percent, 61.9mmol, 1.2 eq) at -78 °C under argon atmosphere, and stirred for 10 min at rt. The reaction mixture was cooledto -78 °C and added MeI (3.9 ml, 62.6 mmol, 1.2 eq) dropwise at -78 °C. The mixture was gradually warmed to rt and stirred for 18 h. The mixture was quenched with MeOH, then concentrated in vacuo. The residue was dissolved with CH2Cl2 and the solution was filtered through a pad of Celite to afford the crude product, which was purified by silica gel chromatography (20:1 CHCl3 / MeOH) to afford 15 (5.58 g, 99percent) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 3.79 (s, 3H), 7.53 (s, 1H), 7.60 (s, 1H), 9.88 (s, 1H)
53%
With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20℃; for 1.5 h;
To a cooled (0 °C) solution of NaH (60 percent oil dispersion, 832 mg, 20.8 mmol) and 4-formylimidazole (2.0 g, 20.8 mmol) in dry DMF (30 mL) was added dropwise iodomethane (1.5 mL, 23.0 mmol). The reaction mixture was stirred at rt for 1.5 hr. After quenching the reaction with water, the solvent was removed by evaporation. The residue was dissolved in water and extracted with CHCl3. The extracts were dried over MgSO4 and concentrated by evaporation. The residue was purified by flash column chromatography on SiO2 (CHCl3 → CHCl3 : MeOH = 100 : 1 → 50 : 1 → 10 : 1) to give 36 (1.2 g, 53 percent) as a pale yellow solid.
34%
With sodium hydride In N,N-dimethyl-formamide at 0 - 20℃; for 2 h;
To a stirring solution of NaH (1.25 g, 52.1 mmol) in DMF (65 mL) was added SM1 (5 g, 52.1 mmol) followed by iodomethane (8.13 g, 57.3 mmol) at 0 °C. Thereaction mixture was stirred at room temperature for 2 h. The reaction mixture was diluted with water and extracted with DCM. Combined organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure to obtain crude product, which was purified by silica gel column chromatography eluting with 5percent MeOH/ DCM to compound 1(1.95 g, 34percent) as a yellow solid. LC-MS: m/z = 111[(M+1)].
Reference:
[1] Synlett, 2016, vol. 27, # 19, p. 2734 - 2736
[2] Bulletin of the Chemical Society of Japan, 2015, vol. 88, # 6, p. 784 - 791
[3] Patent: WO2016/44777, 2016, A1, . Location in patent: Page/Page column 61
[4] Patent: WO2003/87088, 2003, A2, . Location in patent: Page/Page column 28
22
[ 3034-50-2 ]
[ 74-88-4 ]
[ 39021-62-0 ]
[ 17289-26-8 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2005, vol. 3, # 1, p. 118 - 122
[2] Chemistry - A European Journal, 2009, vol. 15, # 14, p. 3560 - 3566
23
[ 3034-50-2 ]
[ 74-88-4 ]
[ 17289-26-8 ]
Reference:
[1] Combinatorial Chemistry and High Throughput Screening, 2011, vol. 14, # 7, p. 570 - 582
24
[ 3034-50-2 ]
[ 74-88-4 ]
[ 39021-62-0 ]
[ 17289-26-8 ]
Reference:
[1] Journal of Heterocyclic Chemistry, 2007, vol. 44, # 6, p. 1445 - 1451
25
[ 76-83-5 ]
[ 3034-50-2 ]
[ 33016-47-6 ]
Yield
Reaction Conditions
Operation in experiment
100%
With triethylamine In N,N-dimethyl-formamide at 0 - 20℃;
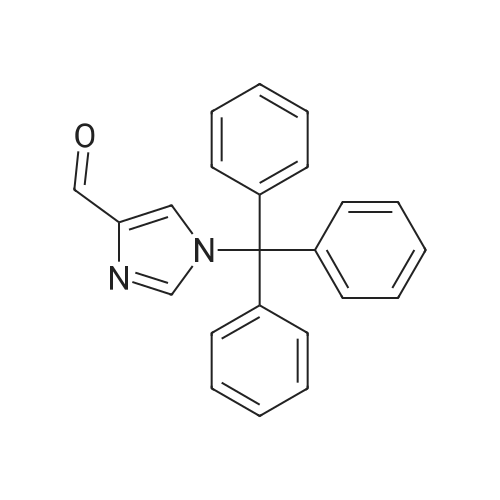
Example 33: 1 -trityl-1 H-imidazole-4-carbaldehydeTo a solution of 1W-imidazole-4-carbaldehyde {30.0 g, 0.30 mol) in DMF (200 mL) was added Et3N (70 mL, 0.375 mol) under ice bath, and then Trt-CI (105 g, 0.375 mol) was added in portions and the mixture stirred at RT overnight. The mixture was evaporated in vacuo and the residue was washed with anhydrous Et2O (4 x 50 mL), and the resulting precipitate was dried to yield the title compound (100 g, 100percent) as a yellow solid.
98%
With triethylamine In dichloromethane at 0 - 20℃;
[2372] To a stirred solution of 4(5)-imidazolecarboxaldehyde (20.0 g, 0.208 mmol) in CH2Cl2 (200 mL), was added Et3N (29.0 mL, 0.208 mmol). The solution was then cooled down at 0° C., followed by addition of triphenylmethylchloride (52.8 g, 0.18 mmol) at 0° C. The resulting solution was stirred at room temperature overnight and then washed it with brine, water and concentrated to dryness to give a white solid (63.0 g, 98percent yield, MH+=339.1)
96%
With triethylamine In acetonitrile
Dry triethylamine (12.6 mL, 90.0 mmol) was added drop wise over two hours to a slurry of (l,3)-H-imidazole-4-carbaldehyde (5.0 g, 52 mmol) and trityl chloride (16.0 g, 57.0 mmol) in acetonitrile (170 mL). After complete addition of the triethylamine, the resulting solution was stirred overnight and then hexane (16.6 mL) and water (170 mL) were added. After stirring for an additional 30 minutes, the resulting solid was collected and dried overnight under vacuum to provide the title compound as a white solid (16.8 g, 96percent). 1H NMR (400 MHz, CDCl3): δ 9.81 (s, IH), EPO <DP n="24"/>7.54 (s, IH), 7.46 (s, IH), 7.29 (m, 10H), 7.04 (m, 5H). 13C NMR (100 MHz, CDCl3): δ 186.9, 141.9, 141.2, 141.0, 130.0, 129.0, 128.9, 127.2, 76.7.
95%
With alkaline condition In acetonitrile at 20℃; for 2 h;
To a mixture of 10 (1.03 g, 10.7 mmol) and TrCl (3.29 g, 11.8 mmol, 1.1 eq) in MeCN (45 mL) was added Et3N (2.5 mL, 18.0 mmol, 1.7 eq) at rt. After stirring for 2 h, hexane (3 mL) and H2O (40 mL) were added and the mixture was filtered. The resultant cake was washed with water three times and dried in a house vacuumoven at 50 °C to afford 18 (3.42 g, 95percent) as a white powder.1H NMR (500 MHz, CDCl3) δ 7.10-7.13 (m, 5H), 7.35-7.38 (m, 10H), 7.53 (d, J = 1.1 Hz, 1H), 7.61 (d, J =1.7 Hz, 1H), 9.88 (s, 1H)
78%
With triethylamine; citric acid In <i>N</i>-methyl-acetamide; ethyl acetate
Synthesis of 1-Trityl-1H-imidazole-4-carbaldehyde STR49 To dimethylformamide (100 mL) was added imidazole-4-carboxaldehyde (10 g, 0.104 mol, Aldrich Chemical Corp.) and triethylamine (20.5 mL, 0.147 mol). A solution of trityl chloride (40 g, 0.143 mol) in dimethylformamide (300 mL) was added, followed by stirring at 25° C. for 6 hours. The solvent was removed in vacuo, and the residue was dissolved in ethyl acetate (1.5 L) and 1N citric acid (350 mL). The phases were separated, and the organic phase was washed with brine, dried over anhydrous magnesium sulfate, and filtered. Concentration of the solution in vacuo gave a white solid, 27.59 g, 78percent yield. NMR was consistent with structure. MS: APCI: M-1: 337.2 (M: 338.41).
67%
With triethylamine In N,N-dimethyl-formamide at 20℃; for 18 h;
Intermediate 18:l-trityl-lH-imidazole-4-carbaldehyde[00417 ] Triethylamine (3.05 ml, 21.86 mmol) was added to a solution of lH-imidazole-4- carbaldehyde (2 g, 20.82 mmol) and triphenylmethyl chloride (5.80 g, 20.82 mmol) in N5N- dimethylformamide (20 ml). The reaction mixture was stirred at room temperature for 18 hours. The crude mixture was concentrated in vacuo and partitioned between ethyl acetate and water. The organic phase was washed dried over sodium sulfate, filtered and concentrated in vacuo. The residue was recrystallised from dichloromethane and hexanes to afford the title compound as a cream solid (4.7 g, 67percent yield).[00418] 1H NMR (DMSO-d6, 400 MHz) δ 7.10-7.16 (6H, m), 7.36-7.57 (9H, m), 7.63 (IH, s), 7.64 (IH, s), 9.91 (IH, s).
65%
With triethylamine In ISOPROPYLAMIDE at 25℃; for 24 h;
Reference Example 11 To 4 (5) -formylimidazole (2 g, 20.8 mmol) were added D Ac (30 mL) and triethylamine (3.5 mL, 25.0 mmol, 1.2 equivalents), and then trityl chloride (4.06 g, 14.6 mmol, 0.7 equivalents) was added thereto at room temperature. The mixture was stirred at room temperature for 24 hr, and to the reaction mixture was added water (60 mL) at room temperature, and the mixture was stirred at room temperature for 2 hr. The crystals werecollected by filtration, washed with water, and vacuum-dried(50°C) to a constant amount to give a crude compound (4.6 g) . To the crude compound (0.2 g) was added methanol (1 mL) , and the. mixture was stirred at room temperature for 2 hr. The crystals were collected by filtration, was washed withmethanol (0.2 mL) , and vacuum-dried (50°C) to a constant amount to give l-trityl-4-formyl-lH-imidazole (0.14 g) . yield 65percent.
10 g
With triethylamine In N,N-dimethyl-formamide at 0 - 35℃;
A) 1-trityl-1H-imidazol-4-carbaldehyde A mixture of 1H-imidazol-4-carbaldehyde (3.00 g), triethylamine (7.00 g) and N,N-dimethylformamide (40 mL) was cooled to 0°C, trityl chloride (10.5 g) was added thereto. The reaction mixture was stirred at room temperature for 18 hr, water was added thereto, and the solid was collected by filtration. The obtained solid was washed with water and diethyl ether to give the title compound (10.0 g). 1H NMR (400 MHz, DMSO-d6) δ 7.11-7.14 (6H, m), 7.40-7.48 (9H, m), 7.67 (1H, d, J = 0.8 Hz), 7.80 (1H, d, J = 1.2 Hz), 9.23 (1H, s).
Reference:
[1] Bioorganic and Medicinal Chemistry, 2009, vol. 17, # 23, p. 7993 - 8002
[2] Patent: WO2008/4096, 2008, A1, . Location in patent: Page/Page column 79
[3] Journal of Medicinal Chemistry, 2002, vol. 45, # 1, p. 177 - 188
[4] European Journal of Organic Chemistry, 2015, vol. 2015, # 18, p. 3957 - 3962
[5] Patent: US2004/122018, 2004, A1, . Location in patent: Page 317
[6] Organic and Biomolecular Chemistry, 2011, vol. 9, # 7, p. 2274 - 2278
[7] Journal of Medicinal Chemistry, 2011, vol. 54, # 6, p. 1693 - 1703
[8] Angewandte Chemie - International Edition, 2005, vol. 44, # 31, p. 4903 - 4906
[9] Journal of Medicinal Chemistry, 2006, vol. 49, # 19, p. 5710 - 5727
[10] Patent: WO2006/102159, 2006, A2, . Location in patent: Page/Page column 21; 22
[11] Chemistry - A European Journal, 2006, vol. 12, # 16, p. 4321 - 4332
[12] Synlett, 2016, vol. 27, # 19, p. 2734 - 2736
[13] Chemical Communications, 2011, vol. 47, # 48, p. 12855 - 12857
[14] Patent: US6143766, 2000, A,
[15] Journal of Medicinal Chemistry, 2010, vol. 53, # 4, p. 1712 - 1725
[16] Patent: WO2008/94992, 2008, A2, . Location in patent: Page/Page column 188
[17] Tetrahedron Letters, 2006, vol. 47, # 8, p. 1253 - 1255
[18] Patent: WO2012/173280, 2012, A1, . Location in patent: Page/Page column 35-36
[19] Patent: US2003/199522, 2003, A1,
[20] Patent: US2002/82272, 2002, A1,
[21] Patent: WO2010/52256, 2010, A1, . Location in patent: Page/Page column 37
[22] Patent: WO2010/52255, 2010, A1, . Location in patent: Page/Page column 68
[23] Patent: WO2006/23720, 2006, A2, . Location in patent: Page/Page column 48
[24] Patent: US2007/49616, 2007, A1, . Location in patent: Page/Page column 29
[25] Patent: EP2848618, 2015, A1, . Location in patent: Paragraph 0816
[26] ChemMedChem, 2018, vol. 13, # 8, p. 842 - 851
26
[ 3034-50-2 ]
[ 76-83-5 ]
[ 33016-47-6 ]
Yield
Reaction Conditions
Operation in experiment
98%
at 0 - 20℃;
[1815] To a stirred solution of 4(5)-imidazolecarboxaldehyde (20.0 g, 0.208 mmol) in CH2Cl2 (200 mL), was added Et3N (29.0 mL, 0.208 mmol). The solution was then cooled down at 0° C., followed by addition of triphenylmethylchloride (52.8 g, 0.18 mmol) at 0° C. The resulting solution was stirred at room temperature overnight and then washed it with brine, water and concentrated to dryness to give a white solid (63.0 g, 98percent yield, MH+=339.1)
94%
With triethylamine In acetonitrile at 20℃; for 20.3333 h;
To a 1L three-necked round-bottomed flask with an addition funnel was added 3H-imidazole-4-carbaldehyde (4, 12 g, 0.125 mol) trityl chloride (38.3 g, 0.137 mol) and acetonitrile (400 mL). The mixture was stirred at rt to give a slurry. Triethylamine (30 mL, 0.215 mol) was added drop wise over 20 min. After the addition was complete, the reaction mixture was stirred at rt for 20 h. Hexane (40 mL) and water (400mL) were added. The slurry was stirred for 30 min and filtered. The cake was washed with water (3 × 100 mL) and dried in a vacuum oven at 50 °C for 20 h to give 5 as a white solid (39.8 g, 94percent).
Reference:
[1] Patent: US2003/229099, 2003, A1, . Location in patent: Page 269
[2] Journal of Medicinal Chemistry, 2010, vol. 53, # 10, p. 3887 - 3898
[3] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 23, p. 6492 - 6499
[4] Organic Process Research and Development, 2000, vol. 4, # 6, p. 613 - 614
[5] Journal of Medicinal Chemistry, 2007, vol. 50, # 19, p. 4585 - 4605
[6] Journal of Medicinal Chemistry, 2009, vol. 52, # 12, p. 3703 - 3715
27
[ 3034-50-2 ]
[ 66247-84-5 ]
Yield
Reaction Conditions
Operation in experiment
100%
Stage #1: With ammonia In methanol at 40℃; for 0.5 h; Stage #2: With hydrogen In methanol at 40 - 50℃; for 28 h; Stage #3: With hydrogenchloride In methanol; diethyl ether
12.0 g (124 mmol) 4-formyl-imidazole are placed together with 750 mg Raney nickel in 1000 ml of methanolic ammonia solution and shaken at 40° C. for 30 min. Then the mixture is hydrogenated in a Parr apparatus under a hydrogen atmosphere at 5 bars pressure at 40° C. for 14 h. Another 750 mg Raney nickel are then added and the mixture is again hydrogenated at 50° C. under a hydrogen atmosphere at 5 bars pressure for 14 h. The mixture is filtered, evaporated down i. vac., and in each case methanol, toluene and ethanol are added to the residue and it is again evaporated down completely i. vac. The residue is combined with ethereal hydrochloric acid in methanol and evaporated down completely i. vac. The residue is in each case combined with methanol and dichloromethane and evaporated down completely i. vac.Yield: 21.2 g (quant.)Rt value: 0.49 min (D)C4H7N3*2 HCl (170.04/97.12)Mass spectrum: (M+H)+=98
With manganese(IV) oxide In methanol at 40℃; for 6h;
3.1 (1) 1H-imidazole-4-formaldehyde preparation, steps are as follows
In the 500 ml four-mouth bottle in, adding 25.1 g (0.254 µM) 4 - hydroxy methyl imidazole and 125.5 g methanol, 4 - hydroxy methyl imidazole and methanol and the mass ratio of 1:5, stir, at room temperature by adding 123.7 g (1.422 µM) manganese dioxide, 4 - hydroxy methyl imidazole, manganese dioxide in a molar ratio of 1: 5.6, material feeding is finished, heating up to 40 °C reaction 6 hours, then the reaction liquid cooling to 25 °C, filtering [...], the filtrate is 1 H - imidazole -4 - formaldehyde reaction solution (0.229 µM), yield 92%, direct synthesis of the next step.
51%
With potassium osmate(VI) dihydrate; potassium carbonate; potassium hexacyanoferrate(III) In water; acetonitrile at 60℃; for 18h; chemoselective reaction;
1.73 g
With manganese(IV) oxide In 1,4-dioxane for 1h; Heating;
538 mg starting material (4 mmol) was dissolved in ~ 4 ml methanol and 500 mg NaHCO3 (6 mmol) was added. The tube was occasionally stirred for 60 min, alternatively at 4 C and at warm water temperature. CO2 was allowed to escape from the glass tube. The mix was divided across several Eppendorf tubes and subjected to Speedvac treatment for 1 hour. Residues were white solids with light-yellow sirupy liquids. Chloroform/methanol mix 1:1 was added to the tubes with subsequent gentle warming and stirring. NaHCO3 was separated by centrifugation of the combined fractions at 3500 rpm for 5 min. Clear supernatant was kept overnight at -20 C to allow the precipitation of additional NaHCO3. Then, the solution was cleared by filtration and evaporated to dryness with a Rotavapor device. The residue was taken up in 20 ml dioxane with magnetic stirring and 4.4 mg MnO2 (activated; for synthesis) was added in the same flask. The residue may not have been dissolved completely in first instantion. The mix was refluxed for 2 hours on a paraffin oil bath. The warm solution was filtered and MnO2 was washed once with warm dioxane. Dioxane was evaporated with the Rotavapor yielding a white and yellow fine cristalline solid. Crystallization was carried out in methanol repeated times. Small volumes of methanol were required, because the residue dissolved well in methanol
With triethylamine In N,N-dimethyl-formamide at 0 - 20℃;
33
Example 33: 1 -trityl-1 H-imidazole-4-carbaldehydeTo a solution of 1W-imidazole-4-carbaldehyde {30.0 g, 0.30 mol) in DMF (200 mL) was added Et3N (70 mL, 0.375 mol) under ice bath, and then Trt-CI (105 g, 0.375 mol) was added in portions and the mixture stirred at RT overnight. The mixture was evaporated in vacuo and the residue was washed with anhydrous Et2O (4 x 50 mL), and the resulting precipitate was dried to yield the title compound (100 g, 100%) as a yellow solid.
99%
With triethylamine In N,N-dimethyl-formamide at 20℃;
99%
With triethylamine In N,N-dimethyl-formamide at 20℃; for 4h;
98%
With triethylamine In dichloromethane at 0 - 20℃;
476.B Step B
[2372] To a stirred solution of 4(5)-imidazolecarboxaldehyde (20.0 g, 0.208 mmol) in CH2Cl2 (200 mL), was added Et3N (29.0 mL, 0.208 mmol). The solution was then cooled down at 0° C., followed by addition of triphenylmethylchloride (52.8 g, 0.18 mmol) at 0° C. The resulting solution was stirred at room temperature overnight and then washed it with brine, water and concentrated to dryness to give a white solid (63.0 g, 98% yield, MH+=339.1)
98%
With triethylamine In dichloromethane at 0 - 20℃; for 1.5h; Inert atmosphere;
98%
With triethylamine In N,N-dimethyl-formamide at 20℃; for 4h; Inert atmosphere;
96%
With triethylamine In acetonitrile
96%
With triethylamine In acetonitrile
96%
With triethylamine In acetonitrile
Dry triethylamine (12.6 mL, 90.0 mmol) was added drop wise over two hours to a slurry of (l,3)-H-imidazole-4-carbaldehyde (5.0 g, 52 mmol) and trityl chloride (16.0 g, 57.0 mmol) in acetonitrile (170 mL). After complete addition of the triethylamine, the resulting solution was stirred overnight and then hexane (16.6 mL) and water (170 mL) were added. After stirring for an additional 30 minutes, the resulting solid was collected and dried overnight under vacuum to provide the title compound as a white solid (16.8 g, 96%). 1H NMR (400 MHz, CDCl3): δ 9.81 (s, IH), EPO 7.54 (s, IH), 7.46 (s, IH), 7.29 (m, 10H), 7.04 (m, 5H). 13C NMR (100 MHz, CDCl3): δ 186.9, 141.9, 141.2, 141.0, 130.0, 129.0, 128.9, 127.2, 76.7.
95%
With triethylamine In dichloromethane at 20℃; for 14h;
95%
With alkaline condition In acetonitrile at 20℃; for 2h;
Synthesis of 1-trityl-1H-imidazole-4-carbaldehyde (18)
To a mixture of 10 (1.03 g, 10.7 mmol) and TrCl (3.29 g, 11.8 mmol, 1.1 eq) in MeCN (45 mL) was added Et3N (2.5 mL, 18.0 mmol, 1.7 eq) at rt. After stirring for 2 h, hexane (3 mL) and H2O (40 mL) were added and the mixture was filtered. The resultant cake was washed with water three times and dried in a house vacuumoven at 50 °C to afford 18 (3.42 g, 95%) as a white powder.1H NMR (500 MHz, CDCl3) δ 7.10-7.13 (m, 5H), 7.35-7.38 (m, 10H), 7.53 (d, J = 1.1 Hz, 1H), 7.61 (d, J =1.7 Hz, 1H), 9.88 (s, 1H)
89%
With triethylamine In acetonitrile at 20℃; for 12h;
84%
With triethylamine In acetonitrile at 20℃; for 18h;
82%
With triethylamine In N,N-dimethyl-formamide
78%
With triethylamine; citric acid In <i>N</i>-methyl-acetamide; ethyl acetate
4 Synthesis of 4-(3-Chloro-phenyl)-1-methyl-6-[(3-methyl-3H-imidazol-4-yl)-phenyl-methoxy]-1H-quinolin-2-one STR48 4a.
Synthesis of 1-Trityl-1H-imidazole-4-carbaldehyde STR49 To dimethylformamide (100 mL) was added imidazole-4-carboxaldehyde (10 g, 0.104 mol, Aldrich Chemical Corp.) and triethylamine (20.5 mL, 0.147 mol). A solution of trityl chloride (40 g, 0.143 mol) in dimethylformamide (300 mL) was added, followed by stirring at 25° C. for 6 hours. The solvent was removed in vacuo, and the residue was dissolved in ethyl acetate (1.5 L) and 1N citric acid (350 mL). The phases were separated, and the organic phase was washed with brine, dried over anhydrous magnesium sulfate, and filtered. Concentration of the solution in vacuo gave a white solid, 27.59 g, 78% yield. NMR was consistent with structure. MS: APCI: M-1: 337.2 (M: 338.41).
77%
With triethylamine In dichloromethane at 0℃;
69%
With triethylamine In chloroform; N,N-dimethyl-formamide at 20℃; Cooling with ice;
67%
With triethylamine In N,N-dimethyl-formamide at 20℃; for 18h;
18
Intermediate 18:l-trityl-lH-imidazole-4-carbaldehyde[00417 ] Triethylamine (3.05 ml, 21.86 mmol) was added to a solution of lH-imidazole-4- carbaldehyde (2 g, 20.82 mmol) and triphenylmethyl chloride (5.80 g, 20.82 mmol) in N5N- dimethylformamide (20 ml). The reaction mixture was stirred at room temperature for 18 hours. The crude mixture was concentrated in vacuo and partitioned between ethyl acetate and water. The organic phase was washed dried over sodium sulfate, filtered and concentrated in vacuo. The residue was recrystallised from dichloromethane and hexanes to afford the title compound as a cream solid (4.7 g, 67% yield).[00418] 1H NMR (DMSO-d6, 400 MHz) δ 7.10-7.16 (6H, m), 7.36-7.57 (9H, m), 7.63 (IH, s), 7.64 (IH, s), 9.91 (IH, s).
66.5%
With triethylamine In acetonitrile at 8 - 18℃; for 2.33h;
6b Example 6b: TRT protection reaction
1H-Imidazole-4-carbaldehyde (45.2g, 0.47M) and acetonitrile (0.8 liter) are introduced into a 2 liter flack and cooled to 8°C, then TRT-Cl (13 lOg, 0.47M) was added at 8°C and TEA (57.lg, 0.56M) was added dropwise during 20 mm. The reaction mixture was stirred at 8 to 18°C for 2 hrs.The reaction mixture was poured into a stirring mixture of water (0.72 liter) and MTBE (0.72 liter) and stirred for 10 minutes. The resulting solid was isolated by filtration on Buchner funnel and dissolved with THF (3 liter). The solution was dried over Na2504 and concentrated to remove most of the solvent.MTBE (400 ml) and PE (200m1) was added to the residue, the mixture stirred at 8°C for 16 hrs. The precipitated solid was isolated by filtration on Buchner filter and dried in air for 16 hrs at room temperature. Then the filter cake is dried by azeotropic drying with 2-Me-THF (2x500 ml) to give 129g of intermediate 2 as white solid with a yield of 66.5%.
65%
With TEA In N,N-dimethyl-formamide at 20℃; for 16h;
65%
With triethylamine In ISOPROPYLAMIDE at 25℃; for 24h;
11
Reference Example 11 To 4 (5) -formylimidazole (2 g, 20.8 mmol) were added D Ac (30 mL) and triethylamine (3.5 mL, 25.0 mmol, 1.2 equivalents), and then trityl chloride (4.06 g, 14.6 mmol, 0.7 equivalents) was added thereto at room temperature. The mixture was stirred at room temperature for 24 hr, and to the reaction mixture was added water (60 mL) at room temperature, and the mixture was stirred at room temperature for 2 hr. The crystals werecollected by filtration, washed with water, and vacuum-dried(50°C) to a constant amount to give a crude compound (4.6 g) . To the crude compound (0.2 g) was added methanol (1 mL) , and the. mixture was stirred at room temperature for 2 hr. The crystals were collected by filtration, was washed withmethanol (0.2 mL) , and vacuum-dried (50°C) to a constant amount to give l-trityl-4-formyl-lH-imidazole (0.14 g) . yield 65%.
32%
Stage #1: 1H-imidazole-4-carbaldehyde With triethylamine In dichloromethane at 0℃; for 0.166667h;
Stage #2: Chlorotriphenylmethane In dichloromethane at 0℃; for 16h;
Step-1: Synthesis of 1-trityl-1H-imidazole-4-carbaldehyde (2):
To a solution of 1H- imidazole-4-carbaldehyde (1, 10.0 g, 104 mmol) in DCM (100 mL), Et3N (28.9 mL, 110 mmol) was added to it. The reaction mixture was stirred for 10 min at 0 °C and added trityl chloride (34.7 g, 124.0 mmol) at same temperature. The resulting mixture was further stirred for 16 h. After completion, water was added and the aqueous layer was extracted with DCM (3 x 100 mL). The combined organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo to get crude material. The obtained material was triturated with hexane (200 mL) and hexane was decanted off. The resulting material was dried under vacuo to give 1-trityl-1H-imidazole-4-carbaldehyde (2, 11.2 g, 32%) as an off white solid.1H NMR (400 MHz; DMSO-d6): δ 7.06 - 7.18 (m, 6H), 7.37 - 7.50 (m, 9H), 7.65 (s, 1H), 7.79 (s, 1H), 9.72 (s, 10H).
32%
Stage #1: 1H-imidazole-4-carbaldehyde With triethylamine In dichloromethane at 0℃; for 0.166667h;
Stage #2: Chlorotriphenylmethane In dichloromethane at 0℃; for 16h;
Step-1: Synthesis of 1-trityl-1H-imidazole-4-carbaldehyde (2):
To a solution of 1H- imidazole-4-carbaldehyde (1, 10.0 g, 104 mmol) in DCM (100 mL), Et3N (28.9 mL, 110 mmol) was added to it. The reaction mixture was stirred for 10 min at 0 °C and added trityl chloride (34.7 g, 124.0 mmol) at same temperature. The resulting mixture was further stirred for 16 h. After completion, water was added and the aqueous layer was extracted with DCM (3 x 100 mL). The combined organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo to get crude material. The obtained material was triturated with hexane (200 mL) and hexane was decanted off. The resulting material was dried under vacuo to give 1-trityl-1H-imidazole-4-carbaldehyde (2, 11.2 g, 32%) as an off white solid.1H NMR (400 MHz; DMSO-d6): δ 7.06 - 7.18 (m, 6H), 7.37 - 7.50 (m, 9H), 7.65 (s, 1H), 7.79 (s, 1H), 9.72 (s, 10H).
With triethylamine In dichloromethane; water monomer; N,N-dimethyl-formamide
5 1-Trityl-1H-imidazole-4-carboxaldehyde
Preparation 5 1-Trityl-1H-imidazole-4-carboxaldehyde A solution of trityl chloride (9.5 g, 34.3 mmol) in N,N-dimethylformamide (50 ml) was added dropwise to an ice-cooled solution of imidazole-4-carboxaldehyde (3 g, 31.2 mmol) and triethylamine (17 ml, 125 mmol) in N,N-dimethylformamide (30 ml) and the solution was stirred for 2 hours. The reaction was then allowed to warm to room temperature, and was stirred for a further 18 hours. Water (200 ml) was added, and the resulting pink solid was collected and dried, then dissolved in dichloromethane (200 ml). The resulting solution was washed with water (2*100 ml), dried (MgSO4) and evaporated under reduced pressure. The product was recrystallized from ethanol to afford the title compound as a solid, 7.8 g. 1H-NMR (CDCl3, 400 MHz) δ: 7.06 (m, 6H), 7.32 (m, 9H), 7.48 (s, 1H), 7.58 (s, 1H), 9.82 (s, 1H). Microanalysis found: C, 81.54; H, 5.37; N, 8.24. C23H18N2O requires C, 81.63; H, 5.36; N, 8.28%.
With triethylamine In dichloromethane; water monomer
4.i (i)
(i) Preparation of 1-(triphenylmethyl)-1H-imidazol-4-carboxaldehyde (7): To a stirred suspension of 4-imidazole carboxaldehyde (from Maybridge Chemicals, United Kingdom) (35.0 g, 364 mmol) and triethylamine (55.8 mL; 400 mmol) in dichloromethane (2 L), was added a solution of triphenylmethyl chloride in dichloromethane (600 mL) while maintaining the reaction temperature at approximately 15° C. with a cooling bath. The resultant solution was warmed to room temperature and stirred for 19 h. Washed the reaction solution with a solution of saturated brine and water (1:3.5; 3*600 mL), followed by brine (1*800 mL). Dried over sodium sulfate; filtered to remove drying agent; and removed solvent under vacuum to obtain the desired tritylated product (7) as an off-white solid. MP 186.5-194° C. [Trituration of this product with ether yielded a cream-colored powder with mp 195-197° C.]
With triethylamine In acetonitrile at 20℃;
10.10-1-1
Example 10 : Ethyl 4-cyclohexyl-1-[(/?)-2-{3-[5-(1 H-imidazol-4-yl)pentyl]- ureido}-3-(4-methoxyphenyl)propionyl]piperidine-4-carboxylate10-1-1 1-Trityl- 1 H-imidazole-4-carbaldehyde:To a solution containing 1 g (10.4 mmol) of 1 H-imidazole-4-carbaldehyde and 3.18 g (11.4 mmol) of trityl chloride suspended in 28 ml of acetonitrile are added dropwise 2.5 ml (17.7 mmol) of triethylamine. After stirring for 2 hours at room temperature, 30 ml of water are added and the crude reaction product is filtered. 3.2 g in the form of a beige-coloured powder are obtained and used in the following step without further purification.
With triethylamine In acetonitrile at 20℃;
18.1.1
18-1-1 1-Trityl- 1 H-imidazole-4-carbaldehyde; To a solution containing 1 g (10.4 mmol) of 1 H-imidazole-4-carbaldehyde and 3.18 g (1 1 .4 mmol) of trityl chloride suspended in 28 ml. of acetonitrile are added dropwise 2.5 ml. (17.7 mmol) of triethylamine. After stirring for 2 hours at room temperature, 30 ml_ of water are added and the crude reaction product is filtered. 3.2 g in the form of a beige-coloured powder are obtained and used in the following step without further purification.
With triethylamine In N,N-dimethyl-formamide at 20℃; for 18h;
With triethylamine In N,N-dimethyl-formamide at 20℃; for 18h;
2.A
A. 1-Trityl-4-carboxaldehyde-1H-imidazole (cas No.33016-47-6) According to procedure outlined in J. Med. Chem. 2002, 45(1), 177, to imidazole-4-carboxaldehyde (15.0 g, 156.2 mmol) in DMF (300 mL) is added triethylamine (43.8 ml, 312 mmol) followed by trityl chloride (44.4 g, 159.0 mmol). The reaction mixture is stirred at ambient temperature for 18 h before the solvent is removed in vacuo. The resulting solid is dissolved in dichloromethane and washed with sodium bicarbonate and water. The organic phase is concentrated in vacuo to give the desired material as a solid.
10 g
With triethylamine In N,N-dimethyl-formamide at 0 - 35℃;
80.A A) 1-trityl-1H-imidazol-4-carbaldehyde
A) 1-trityl-1H-imidazol-4-carbaldehyde A mixture of 1H-imidazol-4-carbaldehyde (3.00 g), triethylamine (7.00 g) and N,N-dimethylformamide (40 mL) was cooled to 0°C, trityl chloride (10.5 g) was added thereto. The reaction mixture was stirred at room temperature for 18 hr, water was added thereto, and the solid was collected by filtration. The obtained solid was washed with water and diethyl ether to give the title compound (10.0 g). 1H NMR (400 MHz, DMSO-d6) δ 7.11-7.14 (6H, m), 7.40-7.48 (9H, m), 7.67 (1H, d, J = 0.8 Hz), 7.80 (1H, d, J = 1.2 Hz), 9.23 (1H, s).
With triethylamine In dichloromethane at 20℃; for 24h;
12.0 g (124 mmol) 4-formyl-imidazole are placed together with 750 mg Raney nickel in 1000 ml of methanolic ammonia solution and shaken at 40° C. for 30 min. Then the mixture is hydrogenated in a Parr apparatus under a hydrogen atmosphere at 5 bars pressure at 40° C. for 14 h. Another 750 mg Raney nickel are then added and the mixture is again hydrogenated at 50° C. under a hydrogen atmosphere at 5 bars pressure for 14 h. The mixture is filtered, evaporated down i. vac., and in each case methanol, toluene and ethanol are added to the residue and it is again evaporated down completely i. vac. The residue is combined with ethereal hydrochloric acid in methanol and evaporated down completely i. vac. The residue is in each case combined with methanol and dichloromethane and evaporated down completely i. vac.Yield: 21.2 g (quant.)Rt value: 0.49 min (D)C4H7N3*2 HCl (170.04/97.12)Mass spectrum: (M+H)+=98
B
[1815] To a stirred solution of 4(5)-imidazolecarboxaldehyde (20.0 g, 0.208 mmol) in CH2Cl2 (200 mL), was added Et3N (29.0 mL, 0.208 mmol). The solution was then cooled down at 0° C., followed by addition of triphenylmethylchloride (52.8 g, 0.18 mmol) at 0° C. The resulting solution was stirred at room temperature overnight and then washed it with brine, water and concentrated to dryness to give a white solid (63.0 g, 98% yield, MH+=339.1)
94%
With triethylamine In acetonitrile at 20℃;
94%
With triethylamine In acetonitrile at 20℃; for 20.3333h;
1-Triphenylmethyl-4-imidazole Carboxaldehyde (5)
To a 1L three-necked round-bottomed flask with an addition funnel was added 3H-imidazole-4-carbaldehyde (4, 12 g, 0.125 mol) trityl chloride (38.3 g, 0.137 mol) and acetonitrile (400 mL). The mixture was stirred at rt to give a slurry. Triethylamine (30 mL, 0.215 mol) was added drop wise over 20 min. After the addition was complete, the reaction mixture was stirred at rt for 20 h. Hexane (40 mL) and water (400mL) were added. The slurry was stirred for 30 min and filtered. The cake was washed with water (3 × 100 mL) and dried in a vacuum oven at 50 °C for 20 h to give 5 as a white solid (39.8 g, 94%).
94%
Stage #1: 5-imidazolecarboxaldehyde; trityl chloride In acetonitrile at 20℃; Inert atmosphere;
Stage #2: With triethylamine at 20℃; for 22h; regioselective reaction;
With triethylamine In N,N-dimethyl-formamide
With triethylamine In acetonitrile at 20℃; Inert atmosphere;
With dihydrogen peroxide;pH 7.2;aqueous phosphate buffer; UV-irradiation;Product distribution / selectivity;
Photooxidation; A 1-cm quartz cuvette, filled with 1.4 mL sample, was placed in the parallel beam of a filtered 1000 W xenon arc lamp (Oriel, Stratford, CT). The samples were magnetically stirred during irradiation. To minimize infrared (heat) and visible radiation, the beam was passed through a water filter (7 cm), reflected by a dichroic mirror and filtered through a 1-mm UG11 filter. Short-wave cut off was achieved by passing the beam through WG280, WG305 or WG335 filters with 3 mm thickness each (Schott-Jena, Mainz, Germany). Xenon lamp emission filtered through WG280 included W-C, UV-B and W-A; through WG305 W-B and W-A and through WG335 only W-A was included. Two narrow bands in the W-B and W-A spectral regions were selected to monitor the xenon-arc emission. The probe of a calibrated EGG 550 radiometer (Salem, MA, USA) was equipped with a neutral density filter and narrow band filter type UV-M-IL (Schott-Jena) with a transmission maximum of 21 percent at 303 nm and a half-width of 11.5 nm to monitor UV-B or with a type UV-PIL (Schott-Jena) with a transmission maximum of 46 percent at 363 nm and a half-width of 7.7 nm to monitor UV-A. Transmission spectra of the optical filters were checked on a Perkin Elmer Lambda 40 UV/VIS spectrometer (Norwalk, CT, USA). Additional irradiations were performed with fluorescent tubes TL12, used as a UV-B source, and TL10R, used as a UV-A source (Philips, Eindhoven, The Netherlands), on samples that were magnetically stirred in small Petri dishes. The UV-B output was measured with an IL 443 phototherapy radiometer, fitted with a SEE 1240 silicon detector probe and the UV-A output with an IL 442A phototherapy radiometer with a SEE 115 detector probe (International Light, Newburyport, MA, USA); UCA photo-oxidation on a preparative scale Concentrations of trans-UCA and hydrogen peroxide were largely increased, as was the UV exposure, to obtain larger amounts of UCA photo-oxidation products as collected fractions from the reversed phase column for further analysis. A typical chromatogram is shown in Fig. 4. Four fractions, designated as Rt 8, Rt 10, Rt 14, Rt 17, were finally selected for identification (peak A, 1-3 in Fig.4). Prior to analysis, tetrabutylammonium was removed by solid phase extraction on C18 silica.Identification Rt 8 was identified as imidazole-4-carboxaldehyde (ImCHO). Its UV-spectrum was identical to the synthesized (see below) reference compound with an absorption maximum of 257 nm. Co-injection of Rt 8 with synthesized imidazole-4-carboxaldehyde resulted in a single chromatographic peak with a retention time of 8.13 minutes. Further evidence is to be collected (peak A in Fig.4). The amount of ImCHO in the photooxidized UCA sample was gradually reduced upon storage at -20°C. Rt 10 was identified as imidazole-4-acetic acid. Its UV-spectrum was identical with an absorption maximum of 213 nm. Mass spectrum was obtained with electrospray technique and the dry sample was treated with methanol/HCl and n-butanol/HCl before analysis. A peak at mass 140 was obtained after methylation and at mass 183 after butylation. Consequently, the mass of the original compound was 126. Co-injection of Rt 10 with commercially available imidazole-4-acetic acid resulted in a single chromatographic peak with a retention time of 8.98 minutes (peak 1 in Fig.4). Rt 14 was identified as imidazole-4-carboxylic acid (ImCOOH). Its UV-spectrum was identical to the commercially obtained reference compound with an absorption maximum of 226 nm. Proton resonance (1H-NMR) analysis was done in D2O, showing imidazolic protons in a ratio 1:1 with shifts of 7.76 and 7.53 ppm. Mass spectrum was obtained with electrospray technique and the dry sample was treated with methanol/HCl and n-butanol/HCl before analysis. A peak at mass 126 was obtained after methylation and at mass 169 after butylation. Consequently, the mass of the original compound was 112. Co-injection of Rt 14 with commercially available ImCOOH resulted in a single chromatographic peak with a retention time of 14.73 minutes (peak 2 in Fig.4). The amount of ImCOOH in the photooxidized UCA sample was gradually increased upon storage at -20° C.
With dihydrogen peroxide;pH 7.2;aqueous phosphate buffer;Product distribution / selectivity;
Fenton oxidation UCA isomers (10 or 40 muM) were oxidized with a hydroxyl-radical- generating system that consisted of various concentrations of ferrous ions (10 - 500 muM) and a fixed hydrogen peroxide concentration of 500 muM (the Fenton reagent), either in a sodium phosphate (10 or 20 mM) medium of pH 7.2, or in ultrapure water. In addition, two hydroxyl-radical-generating systems with copper ions (Cu2+) were used, consisting of 50 muM Cu2+ with either 500 muM hydrogen peroxide or 5 mM ascorbic acid.
With dihydrogen peroxide; In water;pH 7.2;Product distribution / selectivity;
Fenton oxidation UCA isomers (10 or 40 muM) were oxidized with a hydroxyl-radical- generating system that consisted of various concentrations of ferrous ions (10 - 500 muM) and a fixed hydrogen peroxide concentration of 500 muM (the Fenton reagent), either in a sodium phosphate (10 or 20 mM) medium of pH 7.2, or in ultrapure water. In addition, two hydroxyl-radical-generating systems with copper ions (Cu2+) were used, consisting of 50 muM Cu2+ with either 500 muM hydrogen peroxide or 5 mM ascorbic acid.
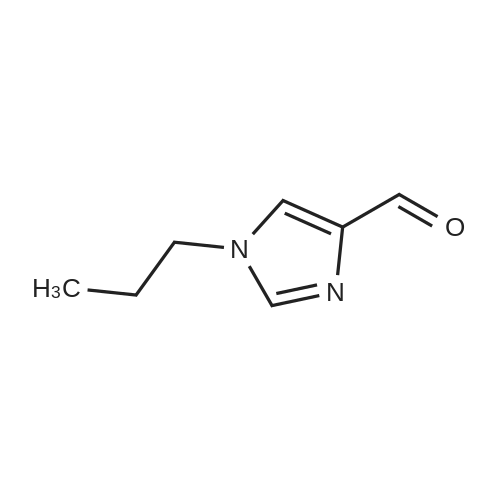
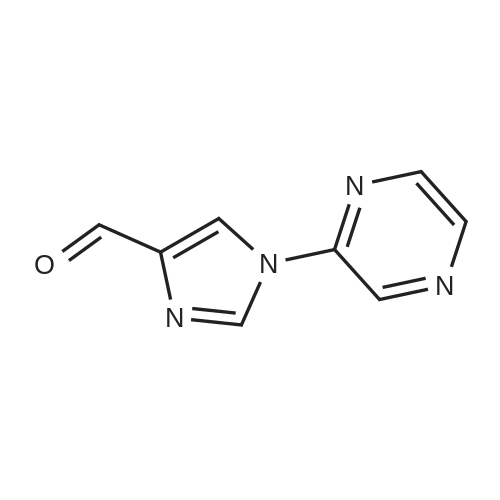
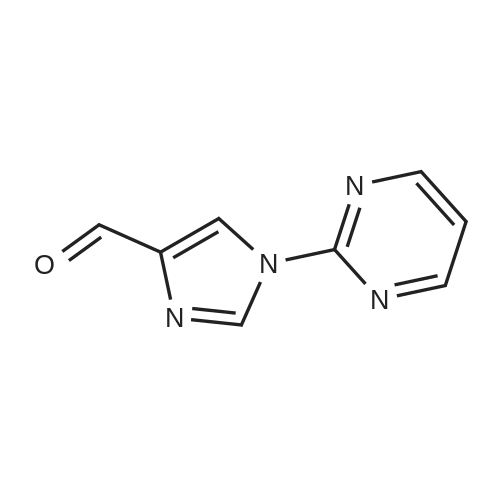
Step A 1-(2-pyrazinyl)-1H-imidazole-4-carboxaldehyde Sodium hydride (60% in oil, 720 mg, 18.0 mmol) was added to a 0 C. solution of 1H-imidazole-4-carboxaldehyde (2.20 g, 18.0 mmol) in DMF (8 mL). After stirring for 20 min at 0 C., the mixture was allowed to warm to RT and a solution of 2-chloropyrazine (2.06 g, 17.9 mmol) in DMF (4 mL) was added. The resulting mixture was heated to 100 C. for 18 h. The solvent was evaporated, the residue was dissolved in ethyl acetate, washed with water and brine, dried (MgSO4), and concentrated to provide 3.10 g (86%) of the title compound. MS 175 (M+H)+
With potassium carbonate In N,N-dimethyl-formamide at 100℃; for 2h; Inert atmosphere;
6.20 g (86%)
In ethyl acetate; N,N-dimethyl-formamide
R.22 1-(2-Pyrimidinyl)-1H-imidazole-4-carboxaldehyde
REFERENCE EXAMPLE 22 1-(2-Pyrimidinyl)-1H-imidazole-4-carboxaldehyde Sodium hydride (60% in oil, 1.44 g, 36.00 mmol) was added to a 0° C. solution of 1H-imidazole-4-carboxaldehyde (4.40 g, 36.03 mmol) in DMF (16 mL). After stirring for 20 min at 0° C., the mixture was allowed to warm to RT and a solution of 2-chloropyrimidine (4.12 g, 35.97 mmol) in DMF (8 mL) was added. The resulting mixture was heated to 100° C. for 18 h. The solvent was evaporated, the residue was dissolved in ethyl acetate, washed with water and brine, dried (MgSO4), and concentrated to provide 6.20 g (86%) of the title compound. MS 175 (M+H)+.
With sodium hydroxide; In methanol; 1,1-dichloroethane; dichloromethane;
N2-(4-{4-amino-1-[1-(1H-4-imidazolylmethyl)tetrahydro-1H-3-pyrrolyl]-1H-pyrazolo[3,4-d]pyrimidin-3-yl}-2-methoxyphenyl)-1-methyl-1H-2-indolecarboxamide A suspension of N2-[4-(4-amino-1-tetrahydro-1H-3-pyrrolyl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl]-1-methyl-1H-2-indolecarboxamide (0.200 g, 0.415 mmol) in dichloroethane (5 mL) was treated with 4-formylimidazole (0.08 g, 0.83 mmol) and sodium triacetoxy borohydride (0.176 g, 0.83 mmol). The reaction mixture was stirred at room temperature for 24 h under a nitrogen atmosphere. Sodium hydroxide (1N, 15 mL) was added to the reaction mixture and was stirred for 1 h. The organic layer was removed under reduced pressure and dichloromethane was added. The layers were partitioned and the aqueous layer was extracted with dichloromethane (200 mL). The combined organic layers were dried over magnesium sulfate, filtered and evaporated under reduced pressure. The crude material was purified by flash chromatography on silica gel using 10% methanol in dichloromethane (20 min), 15% methanol in dichloromethane (10 min), 20% methanol in dichloromethane (10 min) and 50% methanol in dichloromethane (8 min) as the eluent. The column afforded 0.074 g (25%) of pure N2-(4-{4-amino-1-[1-(1H-4-imidazolylmethyl)tetrahydro-1H-3-pyrrolyl]-1H-pyrazolo[3,4-d]pyrimidin-3-yl}-2-methoxyphenyl)-1-methyl-1H-2-indolecarboxamide. 1H NMR (DMSO-d6, 400 MHz) delta 9.446 (s, 1H), 8.252 (s, 1H), 8.126-8.1082 (d, 1H, J=8.16 Hz), 7.7198-7.7 (d, 1H, J=7.92 Hz), 7.6-7.569 (m, 2H), 7.35-7.298 (m, 4H), 7.171-7.134 (m, 1H), 6.946 (s, 1H), 5.422-5.385 (m, 1H), 4.043 (s, 3H), 3.961 (s, 3H), 3.691 (s, 2H), 3.175-3.162 (m, 2H), 2.9-2.883 (m, 3H), 2.385-2.332 (m, 2H); LCMS (Thermoquest AQA single-quad MS, Genesis C18 column, 3pm particle size, 33*4.6 mm; 70% 50 mM ammonium acetate in water to 95% acetonitrile over 6 min, 0.8 to 0.5 mL/min) Rt 2.13 min (100%), MH+ 563.3.
0.074 g (25%)
With sodium hydroxide; In methanol; 1,1-dichloroethane; dichloromethane;
N2-(4-{4-amino-1-[1-(1H-4-imidazolylmethyl)tetrahydro-1H-3-pyrrolyl]-1H-pyrazolo[3,4-d]pyrimidin-3-yl}-2-methoxyphenyl)-1-methyl-1H-2-indolecarboxamide A suspension of N2-[4-(4-amino-1-tetrahydro-1H-3-pyrrolyl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-2-methoxyphenyl]-1-methyl-1H-2-indolecarboxamide (0.200 g, 0.415 mmol) in dichloroethane (5 mL) was treated with 4-formylimidazole (0.08 g, 0.83 mmol) and sodium triacetoxy borohydride (0.176 g, 0.83 mmol). The reaction mixture was stirred at room temperature for 24 h under a nitrogen atmosphere. Sodium hydroxide (1N, 15 mL) was added to the reaction mixture and was stirred for 1 h. The organic layer was removed under reduced pressure and dichloromethane was added. The layers were partitioned and the aqueous layer was extracted with dichloromethane (200 mL). The combined organic layers were dried over magnesium sulfate, filtered and evaporated under reduced pressure. The crude material was purified by flash chromatography on silica gel using 10% methanol in dichloromethane (20 min), 15% methanol in dichloromethane (10 min), 20% methanol in dichloromethane (10 min) and 50% methanol in dichloromethane (8 min) as the eluent. The column afforded 0.074 g (25%) of pure N2-(4-{4-amino-1-[1-(1H-4-imidazolylmethyl)tetrahydro-1H-3-pyrrolyl]-1H-pyrazolo[3,4-d]pyrimidin-3-yl}-2-methoxyphenyl)-1-methyl-1H-2-indolecarboxamide. 1H NMR (DMSO-d6, 400 MHz) delta 9.446 (s, 1H), 8.252 (s, 1H), 8.126-8.1082 (d, 1H, J=8.16 Hz), 7.7198-7.7 (d, 1H, J=7.92 Hz), 7.6-7.569 (m, 2H), 7.35-7.298 (m, 4H), 7.171-7.134 (m, 1H), 6.946 (s, 1H), 5.422-5.385 (m, 1H), 4.043 (s, 3H), 3.961 (s, 3H), 3.691 (s, 2H), 3.175-3.162 (m, 2H), 2.9-2.883 (m, 3H), 2.385-2.332 (m, 2H); LCMS (Thermoquest AQA single-quad MS, Genesis C18 column, 3 mum particle size, 33*4.6 mm; 70% 50 mM ammonium acetate in water to 95% acetonitrile over 6 min, 0.8 to 0.5 mL/min) Rt 2.13 min (100%), MH+ 563.3.
Example 72An alternate method'for the synthesis of the imidazole intermediate is described below:4-Cyano-l-(2-trimethylsilanyl-ethoxymethyl)-lH-imidazole-2-carboxylic acid potassium salt a) lH-Imidazole-4-carbonitrile; '-NHA 22-L, four-neck, round-bottom flask equipped with a mechanical stirrer, a temperature probe, a condenser, and an addition funnel with a nitrogen inlet was charged with lH-imidazole-4-carboxaldehyde (Aldrich, 1.10 kg, 11.5 mol) and pyridine (3.0 L, 3.0 mol). The reaction flask was cooled to 8 0C with an ice bath and hydroxylamine hydrochloride (871 g, 12.5 mol) was added slowly in portions to maintain the internal temperature below 30 °C. The reaction was allowed to cool to ambient temperature and stirred for 2 h at ambient temperature. The resulting thick yellow solution was heated to 80 0C with a heating mantle and acetic anhydride (2.04 L, 21.6 mol) was added dropwise EPO <DP n="148"/>over 200 min to maintain the temperature below 110 °C during the addition. The reaction mixture was heated at 100 0C for 30 min, after which time it was allowed to cool to ambient temperature and then further cooled in an ice bath. The pH was adjusted to 8.0 (pH meter) by the addition of 25 wt percent NaOH (5.5 L) at such a rate that the internal temperature was maintained below 30 °C. The reaction mixture was then transferred into a 22-L separatory funnel and extracted with ethyl acetate (6.0 L). The combined organic layer was washed with brine (2 x 4.0 L), dried over MgSO4, filtered, and concentrated to dryness under reduced pressure at 35 °C to give the crude product as a yellow semisolid. The resulting semisolid was suspended in toluene (3.0 L) and stirred for 1 h, after which time it was filtered to give a light yellow solid, which was resuspended in toluene (3.0 L) and stirred for 1 h. The resulting slurry was filtered and the filter cake washed with toluene (2 x 500 mL) to give the title compound as a light yellow solid [870 g, 82percent). The 1H and 13C NMR spectra were consistent with the assigned structure.
82%
With pyridine; hydroxylamine hydrochloride; acetic anhydride; at 0 - 110℃; for 5.83333h;
A 22-L, four-neck, round-bottom flask equipped with a mechanical stirrer, a temperature probe, a condenser, and an addition funnel with a nitrogen inlet was charged with 1H-imidazole-4-carboxaldehyde (Aldrich, 1.10 kg, 11.5 mol) and pyridine (3.0 L, 3.0 mol). The reaction flask was cooled to 8° C. with an ice bath and hydroxylamine hydrochloride (871 g, 12.5 mol) was added slowly in portions to maintain the internal temperature below 30° C. The reaction was allowed to cool to ambient temperature and stirred for 2 h at ambient temperature. The resulting thick yellow solution was heated to 80° C. with a heating mantle and acetic anhydride (2.04 L, 21.6 mol) was added dropwise over 200 min to maintain the temperature below 110° C. during the addition. The reaction mixture was heated at 100° C. for 30 min, after which time it was allowed to cool to ambient temperature and then further cooled in an ice bath. The pH was adjusted to 8.0 (pH meter) by the addition of 25 wt percent NaOH (5.5 L) at such a rate that the internal temperature was maintained below 30° C. The reaction mixture was then transferred into a 22-L separatory funnel and extracted with ethyl acetate (6.0 L). The combined organic layer was washed with brine (2.x.4.0 L), dried over MgSO4, filtered, and concentrated to dryness under reduced pressure at 35° C. to give the crude product as a yellow semisolid. The resulting semisolid was suspended in toluene (3.0 L) and stirred for 1 h, after which time it was filtered to give a light yellow solid, which was resuspended in toluene (3.0 L) and stirred for 1 h. The resulting slurry was filtered and the filter cake washed with toluene (2.x.500 mL) to give the title compound as a light yellow solid [870 g, 82percent). The 1H and 13C NMR spectra were consistent with the assigned structure.
82%
a) 1H-Imidazole-4-carbonitrile A 22-L, four-neck, round-bottom flask equipped with a mechanical stirrer, a temperature probe, a condenser, and an addition funnel with a nitrogen inlet was charged with 1H-imidazole-4-carboxaldehyde (Aldrich, 1.10 kg, 11.5 mol) and pyridine (3.0 L, 3.0 mol). The reaction flask was cooled to 8° C. with an ice bath and hydroxylamine hydrochloride (871 g, 12.5 mol) was added slowly in portions to maintain the internal temperature below 30° C. The reaction was allowed to cool to ambient temperature and stirred for 2 h at ambient temperature. The resulting thick yellow solution was heated to 80° C. with a heating mantle and acetic anhydride (2.04 L, 21.6 mol) was added dropwise over 200 min to maintain the temperature below 110° C. during the addition. The reaction mixture was heated at 100° C. for 30 min, after which time it was allowed to cool to ambient temperature and then further cooled in an ice bath. The pH was adjusted to 8.0 (pH meter) by the addition of 25 wt percent NaOH (5.5 L) at such a rate that the internal temperature was maintained below 30° C. The reaction mixture was then transferred into a 22-L separatory funnel and extracted with ethyl acetate (6.0 L). The combined organic layer was washed with brine (2*4.0 L), dried over MgSO4, filtered, and concentrated to dryness under reduced pressure at 35° C. to give the crude product as a yellow semisolid. The resulting semisolid was suspended in toluene (3.0 L) and stirred for 1 h, after which time it was filtered to give a light yellow solid, which was resuspended in toluene (3.0 L) and stirred for 1 h. The resulting slurry was filtered and the filter cake washed with toluene (2*500 mL) to give the title compound as a light yellow solid [870 g, 82percent). The 1H and 13C NMR spectra were consistent with the assigned structure.
82%
A 22-L, four-neck, round-bottom flask equipped with a mechanical stirrer, a temperature probe, a condenser, and an addition funnel with a nitrogen inlet was charged with lH-imidazole-4-carboxaldehyde (Aldrich, 1.10 kg, 11.5 mol) and pyridine (3.0 L, 3.0 mol). The reaction flask was cooled to 8 0C with an ice bath and hydroxylamine hydrochloride (871 g, 12.5 mol) was added slowly in portions to maintain the internal temperature below 30 0C. The reaction was allowed to cool to ambient temperature and stirred for 2 h at ambient temperature. The resulting thick yellow solution was heated to 80 0C with a heating mantle and acetic anhydride (2.04 L5 21.6 mol) was added dropwise over 200 min to maintain the temperature below 110 0C during the addition. The reaction mixture was heated at 100 0C for 30 min, after which time it was allowed to cool to ambient temperature and then further <n="149"/>cooled in an ice bath. The pH was adjusted to 8.0 (pH meter) by the addition of 25 wt percent NaOH (5.5 L) at such a rate that the internal temperature was maintained below 30 °C. The reaction mixture was then transferred into a 22-L separatory funnel and extracted with ethyl acetate (6.0 L). The combined organic layer was washed with brine (2 x 4.0 L), dried over MgSO4, filtered, and concentrated to dryness under reduced pressure at 35 0C to give the crude product as a yellow semisolid. The resulting semisolid was suspended in toluene (3.0 L) and stirred for 1 h, after which time it was filtered to give a light yellow solid, which was resuspended in toluene (3.0 L) and stirred for 1 h. The resulting slurry was filtered and the filter cake washed with toluene (2 x 500 nxL) to give the title compound as a light yellow solid [870 g, 82percent). The 1H and 13C NMR spectra were consistent with the assigned structure.
1.B B.
B. 1H-Imidazole-4-carboxaldehyde A solution of Part A imidazole (7.21 g, 74.3 mmol) in 360 mL of dioxane with 86.9 g of activated MnO2 (220.5 mmol) was refluxed under argon for 40 minutes. After cooling to room temperature the suspension was filtered through a pad of Celite and rinsed with several portions of hot dioxane (55° C.); total rinse volume was 300 mL. Dioxane was removed in vacuo to yield 2.68 g of a white solid. The filter cake was rinsed with 300 mL each of boiling dioxane. Evaporation of the combined filtrates yielded an off-white solid which was combined with the above solid. The crude product was suspended in 50 mL of ethyl acetate, collected by filtration, rinsed with 25 mL of ethyl acetate then diethyl ether. Yield: 5.81 g of a white solid: mp 169°-170° C. dec.;
With bromine; sodium acetate; acetic anhydride; In acetic acid; at 20.0℃; for 3.5h;
Bromine (8.0 g, 50 mmol, 2.3 eq) in anhydrous Ac2O (40 mL) was added dropwise to a solution of 1B (2.08 g, 21.7 mmol) and NaOAc (18.7 g, 228 mmol, 10.5 eq) in anhydrous HOAc (200 mL) over a period of 1 h at RT. The resulting mixture was stirred at RT for 2.5 h and then concentrated. The residue was partitioned between Et2O (200 mL) and water (200 mL), the layers were separated and the aqueous layer was extracted with Et2O (200 mL). The combined organic phase was dried, concentrated and chromatographed (EtOAc) to afford 5-bromo-4-formyl imidazole 39A (1.00 g, 26%) as white crystals.
To a stirred solution of 4A (5 g, 30 mmol) in 1 ,2-dichloroethane (100 ml_) was added 4-imidazolecarboxaldehyde (2.9 g, 30 mmol) and HOAc (3 ml_). The mixture was stirred for 1 h, and NaBH(OAc)3 (13 g, 61 mmol) was added. The reaction was stirred overnight at RT, and washed with NaHCO3 and brine. The organic layer was dried (MgSO4), filtered and concentrated (7 g, 94%).
With HCl In methanol tris(2-aminoethyl)amine (1 mmol) was added to a soln. of 4-formylimidazole (3 mmol) in MeOH; the mixt. was stirred at 50°C for 10 min; solns. of Co-contg. compd. (1 mmol) in MeOH and NaClO4 (3 mmol) in MeOH were added; the mixed soln. was filtered, and the filtrate was allowed to stand overnight; the ppt. was collected by suction filtration, washed with a smallvol. of methanol, and dried in vac.; recrystn. from aq. soln. at pH 3; elem. anal.;
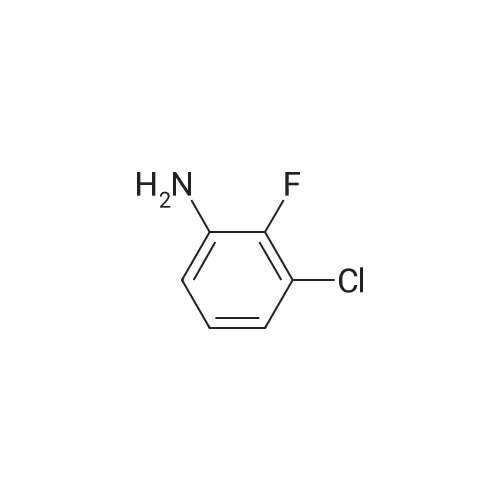
Stage #1: 3-chloro-2-fluoroaniline; 4(5)formylimidazole In methanol at 60℃;
Stage #2: With sodium tetrahydroborate In methanol at 20℃; for 4h;
Stage #3: With water In methanol
81.a
a) (3-Chloro-2-fluoro-phenyl)-(3H-imidazol-4-ylmethyl) -amine; To a solution of 3-chloro-2-fluoroaniline (0.44 g, 3 mmol) in methanol (5 ml) was added 4-formylimidazole (0.29 g, 3 mmol). After stirring the mixture overnight at 600C the solution was cooled and sodium borohydride (0.57 g, 15 mmol) was added. The reaction mixture was stirred at room temperature for 4 hours. Then water was added and the mixture was extracted with ethyl acetate. The organic layer was separated, washed with water, dried over magnesium sulfate and evaporated. The residue was purified by column filtration (heptane/ethyl acetate=l:l) to yield a light yellow liquid (0.55 mg, 81%); MS (ISP): 226.3 ((M+H)+').
With sodium tris(acetoxy)borohydride In tetrahydrofuran at 20℃; for 18.25h;
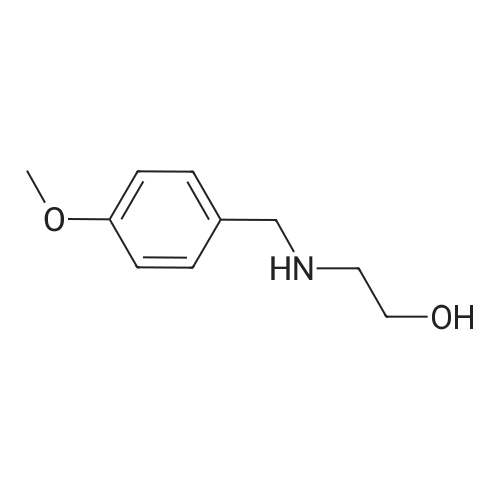
1.B 2-(((1H-imidazol-4-yl)methyl)(4-methoxybenzyl)amino)ethanol
To a heterogeneous mixture of 1H-imidazole-4-carbaldehyde (8.0 g, 83.33 mmol) and 2-(4-methoxybenzylamino)ethanol (Step A, 18.1 g, 99.9 mmol) in anhydrous THF (100 mL) was added sodium triacetoxyborohydride (21.2 g, 99.9 mmol) over a 15 minute period at room temperature. The reaction mixture was stirred at room temperature for 18 hours, concentrated under reduced pressure and partitioned between saturated aqueous sodium bicarbonate (200 mL) and dichloromethane (2×200 mL). The dichloromethane layer was dried over sodium sulfate, concentrated under reduced pressure and purified by silica gel flash chromatography using dichloromethane and methanol (9:1, 800 mL) followed by dichloromethane:methanol:2.0M ammonia in methanol (850:100:50 mL) to afford the title compound b as an oil (14.5 g), [M+H]+ 262.21.
To a suspension of 10 (4.94 g, 51.4 mmol) in dry THF (30 mL) was added NaH (2.48 g, assey 60%, 61.9mmol, 1.2 eq) at -78 C under argon atmosphere, and stirred for 10 min at rt. The reaction mixture was cooledto -78 C and added MeI (3.9 ml, 62.6 mmol, 1.2 eq) dropwise at -78 C. The mixture was gradually warmed to rt and stirred for 18 h. The mixture was quenched with MeOH, then concentrated in vacuo. The residue was dissolved with CH2Cl2 and the solution was filtered through a pad of Celite to afford the crude product, which was purified by silica gel chromatography (20:1 CHCl3 / MeOH) to afford 15 (5.58 g, 99%) as a yellow oil. 1H NMR (500 MHz, CDCl3) delta 3.79 (s, 3H), 7.53 (s, 1H), 7.60 (s, 1H), 9.88 (s, 1H)
53%
With sodium hydride; In N,N-dimethyl-formamide; mineral oil; at 20℃; for 1.5h;
To a cooled (0 C) solution of NaH (60 % oil dispersion, 832 mg, 20.8 mmol) and 4-formylimidazole (2.0 g, 20.8 mmol) in dry DMF (30 mL) was added dropwise iodomethane (1.5 mL, 23.0 mmol). The reaction mixture was stirred at rt for 1.5 hr. After quenching the reaction with water, the solvent was removed by evaporation. The residue was dissolved in water and extracted with CHCl3. The extracts were dried over MgSO4 and concentrated by evaporation. The residue was purified by flash column chromatography on SiO2 (CHCl3 ? CHCl3 : MeOH = 100 : 1 ? 50 : 1 ? 10 : 1) to give 36 (1.2 g, 53 %) as a pale yellow solid.
34%
With sodium hydride; In N,N-dimethyl-formamide; at 0 - 20℃; for 2h;
To a stirring solution of NaH (1.25 g, 52.1 mmol) in DMF (65 mL) was added SM1 (5 g, 52.1 mmol) followed by iodomethane (8.13 g, 57.3 mmol) at 0 C. Thereaction mixture was stirred at room temperature for 2 h. The reaction mixture was diluted with water and extracted with DCM. Combined organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure to obtain crude product, which was purified by silica gel column chromatography eluting with 5% MeOH/ DCM to compound 1(1.95 g, 34%) as a yellow solid. LC-MS: m/z = 111[(M+1)].
To a stirred suspension of imidazole-4-carboxaldehyde (l. Og) in dry tetrahydrofuran (60ml) under nitrogen at room temperature was added sodium hydride (60% suspension in oil, 0.437g) in small portions. The suspension was stirred until effervescence had ceased. A solution of methyl iodide (0. 65ml) in THF (lOml) was added dropwise and the resulting reaction stirred at 21C for 4h. Methanol was added and the mixture evaporated under reduced pressure to give a yellow oil which was chromatographed on silica gel using cyclohexane: ethyl acetate, ethyl acetate, to methanol: dichloromethane as eluents. This gave the desired product (0. 807g).'H-NMR (CDC13) 8 3.78 (3H, s), 7.58 (1H, s), 7.62 (1H, s), 9.85 (1H, s).
Stage #1: N-(4-tert-butylphenyl)-piperazine; 4(5)formylimidazole With triethylamine In 1,4-dioxane; methanol at 20℃;
Stage #2: With sodium tetrahydroborate In methanol at 20℃;
Y(III)(tris[[2-[(4-formylimidazole)methylidene]amino]ethyl]amine)(acetate)(ClO4)2*H2O[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
75%
In methanol MeOH soln. of tris(2-aminoethyl)amine and 4-formylimidazole mixed, stirred at 50°C for 1 h, cooled to room temp., added to MeOH soln. of Y(CH3CO2)3*4H2O, stirred for 30 min at room temp., MeOH soln. of NaClO4 added, stirred for 30 min; filtered, filtrate left to stand for several d; elem. anal.;
With 1,5,7-triazabicyclo[4.4.0]dec-5-ene supported polystyrene In N,N-dimethyl-formamide at 20℃; Combinatorial reaction / High throughput screening (HTS);
With 1,5,7-triazabicyclo[4.4.0]dec-5-ene supported polystyrene In N,N-dimethyl-formamide at 20℃; Combinatorial reaction / High throughput screening (HTS);
To a mixture of 4-Imidazolecarboxaldehyde (6 g, 62.44 mmol, 1.0 eq) in THF (60 mL) was added NaH (3 g, 74.9 mmol, 1.2 eq) at room temperature. After 10 min, the mixture was cooled to -78 C. and MeI (10.5 g, 74.9 mmol, 1.2 eq) was added. Then the mixture was gradually warmed to room temperature and stirred for 18 h. The reaction was quenched with methanol (10 mL) and concentrated under reduced pressure. The residue was purified by silica gel chromatography to afford OI-14a (4 g, 58%).
With triethylamine In dichloromethane at 20℃; for 18h;
91%
Stage #1: 1H-imidazole-4-carbaldehyde With sodium hydride In acetonitrile; mineral oil at 20℃; for 1.5h; Cooling with ice;
Stage #2: N,N-dimethylaminosulfonyl chloride In acetonitrile; mineral oil at 20℃;
87%
With triethylamine In tetrahydrofuran at 20℃; for 24h; Inert atmosphere; Schlenk technique;
86%
With 1,4-diaza-bicyclo[2.2.2]octane In acetonitrile at 0 - 20℃; for 18h;
A19
l,4-Diazabicyclo[2.2.2]octane (21 g, 187.33 mmol) and dimethyl sulfamoyl chloride (18.4 mL, 171.72 mmol) were added to a stirred suspension of lH-imidazole-4- carbaldehyde (15 g, 156.11 mmol) in acetonitrile (300 mL) at 0 °C. The mixture was allowed to warm to room temperature and stirred for 18 hours. The mixture was concentrated in vacuo and the residue was diluted with water and extracted withEtOAc. The organic layer was separated, dried (MgS04), filtered and the solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica; EtOAc in DCM 0/100 to 60/40). The desired fractions were collected and concentrated in vacuo to yield 4-formyl-N,N-dimethyl-lH-imidazole-l -sulfonamide (27.2 g, 86 % yield) as a cream solid.
79%
With triethylamine In tetrahydrofuran at 20℃; for 24h; Inert atmosphere; Schlenk technique;
68%
With triethylamine In N,N-dimethyl-formamide at 0 - 20℃; for 5.5h;
Synthesis of 33
To a cooled (0 °C) solution of 4-formylimidazole (2.5 g, 26.0 mmol) in NEt3 (7.8 mL, 52.0 mmol) was added dropwise a solution of dimethylsulfamoyl chloride (4.49 g, 31.2 mmol) in dry DMF (15 mL) toward 10 min. The reaction mixture was stirred at rt for 5.5 hr. After dilution with ice-cold water, the mixture was extracted with AcOEt. The extracts were washed with sat. NaHCO3, brine, dried over MgSO4, and then concentrated by evaporation. The residue was washed with diisopropyl ether-hexane (1 : 1) to give 33 (3.6 g, 68 %) as pale yellow powder.
62%
With triethylamine In dichloromethane at 20℃; for 16h; Inert atmosphere;
48%
With triethylamine In toluene at 70℃; for 20h;
1
Reference Example 1To 4-formylimidazole (30.0 g, 0.30 mol) were added toluene (300 mL) and triethylamine (35.0 g, 0.34 mol), and then N, -dimethylaminosulfonyl chloride (50.0 g, 0.34 mol) was added thereto at room temperature. The mixture was stirred at70°C for 20 hr, and the insoluble material was collected by filtration, and washed with toluene (300 mL) to give wet crystals. To the obtained wet crystals were added water (100 mL) and ethyl acetate (300 mL) , and the crystals were dissolved with stirring at room temperature. The organic layer and the aqueous layer were separated. The obtained aqueous layer was extracted with ethyl acetate (200 mL) , and the organic layer and the aqueous layer were separated. The obtained organic layer and the previously obtained organic layer were combined. These operations were repeated twice, and the organic layer was completely concentrated under reduced pressure to give crude crystals (32.0 g) . To the crudecrystals was added ethyl acetate (90 mL) , and crystals were dissolved with heating to about 60°C. The solution was slowly .cooled to 30°C for recrystallization, hexane (180 mL) was added thereto, and the mixture was stirred at room temperature for 2 hr to give crystals. The obtained crystals were collected by filtration, and washed with a mixed solvent (45 mL) of ethyl acetate/hexane (1:2, volume ratio). The obtained wet crystals were dried under reduced pressure to give 1-N,N- dimethylaminosulfonyl-4-formyl-lH-imidazole (29.4 g, 0.14 mmol) . yield 48%XH NMR (500 MHz, DMSO-d6) δ 2.93 (s, 6H) , 7.90 (d, J = 1.3 Hz, 1H) , 7.96 (d, J = 1.3 Hz, 1H) , 9.95 (s, 1H) ) ; HRMS (ESI) m/z Calcd for a C6Hi0N3O3S [m+H] +: 204.0365, Found: 204.0438.
161 g
With triethylamine In chloroform at 20℃;
36-1 Reference Example 36-1: 4-formyl-N,N-dimethyl-1H-imidazole-1-sulfonamide
Dimethylsulfamoyl chloride (91 mL, 859 mmol) was added dropwise to a chloroform solution (750 ml) of 1Himidazole-4-carbaldehyde (75g, 78 mmol) and triethylamine (163 mL, 1.17 mol) over 50 minutes at room temperature.The reaction solution was stirred for 3 days and then water (900 mL) was added, and the mixture was extracted withchloroform (500 mL, 3 times). The organic phase was dried over sodium sulfate, filtered, and concentrated to obtain thetitle compound (161 g) as a white solid with a brownish tinge. 1H-NMR (CDCl3) δ: 9.88 (1H, br s), 7.91 (1H, t, J = 7.3 Hz), 7.84 (1H, dd, J = 8.5, 1.2 Hz), 2.87 (6H, dd, J =9.8, 5.5 Hz) .
2-(((1H-imidazol-4-yl)methyl)(benzyl)amino)ethanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
With sodium tris(acetoxy)borohydride In tetrahydrofuran
85%
Stage #1: 4(5)formylimidazole; N-Benzylethanolamine In tetrahydrofuran at 20℃; for 1h;
Stage #2: With sodium tris(acetoxy)borohydride In tetrahydrofuran at 20℃;
Intermediate 6 2-(((lH-Imidazol-4-yl)methyl)(benzyl)amino)ethanol.
A flask was charged with lH-imidazole-4-carbaldehyde (5.0 g, 52 mmol), tetrahydrofuran (75 mL), and 2- (benzylamino)ethanol (9.44 g, 62.4 mmol). The reaction was stirred at room temperature for 1 h. To this was added sodium triacetoxyborohydride (13.2 g, 62.4 mmol) in one portion. A significant exotherm was noted. After 10 min, the solution had become a thick suspension which required occasional agitation by hand. The reaction was stirred at room temperature overnight. The reaction was concentrated, dissolved in water (100 mL), made basic with an aqueous solution of potassium carbonate, and extracted into dichloromethane (3X). The organics were dried over potassium carbonate and concentrated. Column chromatography (5% MeOH/DCM 10% MeOH/DCM -> 90: 10: 1 DCM/MeOH/2M ammonia in methanol) gave 10.2 g (85%>) as a viscous oil. Mass spec: 232.11 (MH)+.
26.9 g
Stage #1: 4(5)formylimidazole; N-Benzylethanolamine In ethanol for 3h;
Stage #2: With sodium tris(acetoxy)borohydride In ethanol at 20℃; Cooling with ice;
B.1 B.1. 2-(((1 H-Imidazol-4-yI)methyl)(benzyl)amino)ethanol
A 1 litre flask was charged with 1 H-imidazole-4-carbaldehyde (14 g, 146 mmol) and absolute ethanol (240 mL). Then 2-(benzylamino)ethanol (20.8 mL, 146 mmol) wasadded and the white suspension turned slowly into a yellow solution after 3 h The mixture was then cooled in an ice-bath and sodium triacetoxyborohydride (93 g, 437 mmol) was added portionwise. The mixture was then stirred overnight at room temperature. Water was added and the mixture was partially concentrated. After neutralising to pH -7 with 1 M aqueous NaOH, the aqueous phase was rinsed twicewith ethyl acetate, then it was basified further to pH 14. The aqueous layer was then extracted with thrice with ethyl acetate. The combined organic layers were dried over sodium sulfate, filtered and evaporated to dryness to give the title compound (26.9 g), which was directly used in the next step.1H NMR (ODd3) 67.52 (s, 1H), 7.37-7.18 (m, 6H), 6.87 (s, 1H), 3.69 (s, 2H), 3.68- 3.58 (m, 5H), 2.70 (t, J = 5.1 Hz, 2H).LC/MS: m/z 232 [M+H].
With sodium sulfate; In methanol; at 20℃;Sealed tube;
General procedure: To a solution of imidazole carbaldehyde (1a-d) (100 mg, 1 equiv) in methanol (3 mL) were added successively Na2SO4 (0.2 g), amine 2a-d (1.2 equiv), alkynoic acid 3a-c (1.2 equiv) and isonitrile 4a-c (1.2 equiv) in a screw capped vial equipped with a magnetic stir bar. The reaction mixture was stirred at room temperature for 24-48 h in closed vial. After completion of the reaction, the mixture was diluted with dichloromethane (100 mL) and was extracted with water (50 mL). Organic layer was washed with brine (50 mL), dried over magnesium sulfate and evaporated under reduced pressure to obtained residue which was subjected to silica gel column chromatography (1-5 % methanol in dichloromethane) to afford the desired product 5a-r as solid.
With sodium sulfate; In methanol; at 20℃;Sealed tube;
General procedure: To a solution of imidazole carbaldehyde (1a-d) (100 mg, 1 equiv) in methanol (3 mL) were added successively Na2SO4 (0.2 g), amine 2a-d (1.2 equiv), alkynoic acid 3a-c (1.2 equiv) and isonitrile 4a-c (1.2 equiv) in a screw capped vial equipped with a magnetic stir bar. The reaction mixture was stirred at room temperature for 24-48 h in closed vial. After completion of the reaction, the mixture was diluted with dichloromethane (100 mL) and was extracted with water (50 mL). Organic layer was washed with brine (50 mL), dried over magnesium sulfate and evaporated under reduced pressure to obtained residue which was subjected to silica gel column chromatography (1-5 % methanol in dichloromethane) to afford the desired product 5a-r as solid.
With sodium sulfate In methanol at 20℃; Sealed tube;
General procedure for synthesis of Ugi products
General procedure: To a solution of imidazole carbaldehyde (1a-d) (100 mg, 1 equiv) in methanol (3 mL) were added successively Na2SO4 (0.2 g), amine 2a-d (1.2 equiv), alkynoic acid 3a-c (1.2 equiv) and isonitrile 4a-c (1.2 equiv) in a screw capped vial equipped with a magnetic stir bar. The reaction mixture was stirred at room temperature for 24-48 h in closed vial. After completion of the reaction, the mixture was diluted with dichloromethane (100 mL) and was extracted with water (50 mL). Organic layer was washed with brine (50 mL), dried over magnesium sulfate and evaporated under reduced pressure to obtained residue which was subjected to silica gel column chromatography (1-5 % methanol in dichloromethane) to afford the desired product 5a-r as solid.
A solution consisting in 0.1242 g (0.50 mmol) 1,3-bis(3 aminopropyl)tetramethyldisiloxane in 10 mL methanol was added to a solution of4-imidazolecarboxaldehyde 0.0961 g (1 mmol) in 2.5 mL methanol,in a 50mL one-necked round-bottom flask. The reactionmixturewasstirred for 4 h at 60 °C, after which was allowed to stay in a closedflask at room temperature. Formation of colorless crystals was observedafter about 5 days. These were separated by filtration washedwith diethyl ether, dried and analyzed further. Crystalline compoundwas labeled as APCO: C11H27N2O3Si2
1-(4-chloro-2-fluorophenyl)-1H-imidazole-4-carbaldehyde[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
100 mg
With (R,R)-N,N'-dimethyl-1,2-diaminocyclohexane; caesium carbonate In N,N-dimethyl-formamide at 110℃; for 18h;
476.1
To 1H-imidazole-4-carbaldehyde (2.56 g, 26.6 mmol), 4-chloro-2-fluoro-1-iodobenzene (0.739 g, 2.88 mmol) and Cs2003 (1.564 g, 4.80 mmol) in DMF (5OmL) was added copper(l) iodide (0.023 g, 0.120 mmol) and (1 R,2R)-N 1, N2-dimethylcyclohexane-1 ,2-diamine (0.068 g, 0.480mmol). The reaction was heated to 110 00 for 18 hours. The reaction mixture was then cooled to room temperature and filtered, and washed with EtOAc (lOOmL). The organic was washed with water (2x3OmL) and brine (3OmL), dried (Na2SO4) and concentrated. The residue was purified by silica gel chromotography to give 1-(4-chloro-2-fluorophenyl)-1H-imidazole-4- carbaldehyde (451 mg), to which was added (S)-2-methylpropane-2-sulfinamide (0.29 g, 2.4mmol), CuSO4 (0.638 g, 4 mmol) and DOE (lOmL). The reaction was heated to 65 00 for 18 hours. The reaction was cooled to rt, filtered and concentrated to give (S,E)-N-((1-(4-chloro-2- fluorophenyl)- 1 H-imidazol-4-yl)methylene)-2-methylpropane-2-sulfinamide, to which was added DCM (2OmL). The reaction mixture was cooled to -70 00 and methylmagnesium bromide (1.33 ml, 4 mmol) was added dropwise to the solution. The reaction was stirred fortwo hours and the cold bath was then removed, and the reaction was allowed to warm to rt and stir for 30 minutes. The reaction was cooled to 0 00 and HCI (1M) was added cautiously to quench until aqueous pH=8. The phases were separated and the aqueous layer was extracted with DCM (2x3OmL). Combined organic was dried (Na2SO4) and concentrated to give (S)- N-((S)- 1 -(1 -(4-chloro-2-fl uorophenyl)- 1 H-imidazol-4-yl)ethyl)-2-methylpropane-2-sulfinamide (130mg), which was then dissolved in MeOH (5mL) and HCI (0.5 mL, 4M) was added. The reaction was stirred for one hour and concentrated to give product (100mg). LCMS m/z 240.1 (M + H) Rt-0.50 mm.
1-(2,4-difluorophenyl)-1H-imidazole-4-carbaldehyde[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
651 mg
With copper(l) iodide; (R,R)-N,N'-dimethyl-1,2-diaminocyclohexane; caesium carbonate; In N,N-dimethyl-formamide; at 110℃; for 18h;
To 1 H-imidazole-4-carbaldehyde (3.01 g, 31.3 mmol),<strong>[2265-93-2]2,4-difluoro-1-iodobenzene</strong> (1.008 g, 4.20 mmol), Cs2003 (2.281 g, 7.00 mmol) in DMF (5OmL) was added copper(l) iodide (0.033g, 0.175 mmol) and (1R,2R)-N1,N2-dimethylcyclohexane-1,2-diamine (0.100 g, 0.700 mmol). The reaction was heated to 110 00 for 18 hours. The reaction mixture was then cooled to room temperature and filtered, and washed with EtOAc (lOOmL). The organic was washed with water (2x3OmL) and brine (3OmL), dried (Na2SO4) and concentrated. The residue was purified by silica gel chromotography to give 1-(2,4-difluorophenyl)-1H-imidazole-4-carbaldehyde (651 mg), to which was added (S)-2-methylpropane-2-sulfinamide (0.424 g, 3.5 mmol), CuSO4 (0.798 g, 5 mmol) and DOE (lOmL). The reaction was heated to 65 00 for 18 hours. The reaction was cooled to rt, filtered and concentrated to give (S,E)-N-((1-(2,4- difluorophenyl)-1 H-imidazol-4-yl)methylene)-2-methylpropane-2-sulfinamide, to which was added DCM (2OmL). The reaction mixture was cooled to -70 00 and methylmagnesiumbromide (1.667 ml, 5 mmol) was added dropwise to the solution. The reaction was stirred for two hours and the cold bath was then removed, and the reaction was allowed to warm to rt and stir for 30 minutes. The reaction was cooled to 0 00 and HCI (1M) was added cautiously to quench until aqueous pH=8. The phases were separated and the aqueous layer was extracted with DCM (2x3OmL). Combined organic was dried (Na2SO4) and concentrated togive (S)-N-((S)-1-(1-(2,4-difluorophenyl)-1 H-imidazol-4-yl)ethyl)-2-methylpropane-2-sulfinamide (230mg), which was then dissolved in MeOH (5mL) and HCI (0.5 mL, 4M) was added. The reaction was stirred for one hour and concentrated to give product (212mg). LCMS m/z 224.1 (M + H) Rt-0.39 mm.
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In toluene; at 115℃; for 1.5h;
To a stirred solution of 3H-Imidazole-4-carbaldehyde (1) (1 g, 0.010 mole) in toluene (20 mL) was added Intermediate Wittig salt (A) (3.9 g, 0.01 1 mole) at room temperature. To this resulted suspension was added DBU (1.9 g, 0.013 mole) drop wise at room temperature and heated to reflux at 115C for 1.5 h. After completion of reaction, toluene was distilled off completely under vacuum. Resulted crude oily mass was purified by silica gel column chromatography (100-200 mesh). Pure compound was eluted at 100% DCM. Evaporation of solvent afforded compound 2 (1.0 g, 81%) as off white solid.
(E)-N'-((1H-imidazol-5-yl)methylene)cyclohexanecarbohydrazide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
46%
In ethanol at 20℃; for 2h;
General Procedure for Preparation of Cyclohexylacylhydrazones 10-26
General procedure: The corresponding aromatic or heteroaromatic aldehyde (2.11 mmol) was added to a solution of cyclohexanecarbohydrazide (28, 0.3 g; 2.11 mmol) in absolute ethanol (21 mL). The mixture was stirred for 2 h at room temperature. At the end of the reaction, the volume of ethanol was partially concentrated at reduced pressure and the resulting mixture was poured into cold water. The precipitate was filtered out, dried under vacuum and then the solid was washed with n-hexane and/or recrystallized from ethanol to give the desired cyclohexyl-N-acylhydrazones 10-26.
(E)-3-[(1H-imidazol-4-yl)methylene]chroman-4-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78%
With pyrrolidine In methanol at 20℃; for 3h;
13 4.1.2. General procedure for the synthesis of (E)-3-heteroarylidenechroman-4-ones (1a-r)
General procedure: METHOD A: Pyrrolidine (10 mmol) was added to a mixture of the appropriate chroman-4-one (6.7 mmol) and heteroaryl aldehyde (10 mmol) in dry MeOH (15 mL for 1a, 1b, 1e, 1h, 1k-r and 30mL for 1c, 1d, 1i, 1j). The reaction mixture was stirred at room temperature for 1-24 h (1h for 1f, 3h for 1l-q, 24h for 1a-c, 1h-j, 1n, and 1o). The mixture was diluted with ice-water, and the precipitate was filtered off and washed with water. The crude solid was purified by column chromatography on silica gel, eluting with a mixture of AcOEt and CHCl3 (3:1) (1a-g and 1i-r) or CHCl3 (1h) and crystallized from n-hexane. METHOD B: (synthesis of 1f and 1g) A mixture of chroman-4-one (6.7 mmol), the appropriate pyridinecarbaldehyde (10 mmol) and pyrrolidine (10 mmol) was heated at 120 °C under stirring for 1 h. After cooling, the mixture was diluted with ice-water, and the precipitate was filtered off and washed with water. The crude solid was purified by column chromatography on silica gel, eluting with a mixture of AcOEt and CHCl3 (3:1), and crystallized from n-hexane. 4.1.2.13 (E)-3-[(1H-Imidazol-4-yl)methylene]chroman-4-one (1m) Yield: 78%, mp = 197-198 °C. IR (KBr): 3385, 1667 cm-1. 1H NMR (DMSO-d6, 400 MHz): δ (ppm) 12.49 (bs, 1H, NH), 7.93 (s, 1H, =CH), 7.88 (dd, 1H, H5, J5-6 = 8.0 Hz, J5-7 = 1.6 Hz), 7.80 (s, 1H, H3'), 7.58-7.54 (m, 2H, H7, H5'), 7.11 (t, 1H, H6, J5-6 = J7-6 = 8.0 Hz), 7.06 (d, 1H, H8, J7-8 = 8.4 Hz), 5.85 (d, 2H, H2, Jall = 0.8 Hz). 13C NMR (DMSO-d6, 100 MHz): δ (ppm) 181.18, 160.99, 137.86, 136.23, 135.48, 127.33, 127.10, 125.31, 125.06, 121.87, 121.49, 117.76, 67.73. Anal. Calcd. for C13H10N2O2: C, 69.02; H, 4.46; N, 12.38. Found: C, 69.21; H, 4.52; N, 12.33.
With copper(l) iodide; caesium carbonate; rac-diaminocyclohexane; In N,N-dimethyl-formamide; at 110℃; for 16h;
Synthesis of 1-phenyl-1H-imidazole-4-carbaldehyde To a stirred solution of 1H-imidazole-5-carbaldehyde (10 g, 104 mmol) in DMF (135 mL), iodobenzene (31.8 g, 156 mmol), cyclohexane-1,2-diamine (2.377 g, 20.8 mmol), cesium carbonate (67.8 g, 208 mmol), and copper(I) iodide (0.991 g, 5.2 mmol) were added. The reaction mixture was stirred at 110 C. for about 16 hours. It was precipitated in water and filtered. The residue was column chromatographed with THF:hexane (1:4). 1-Phenyl-1H-imidazole-4-carbaldehyde (3.5 g, 20.3 mmol, 19.5% yield) was isolated as white solid.
1-(6-aminopyrimidin-4-yl)-1H-imidazole-4-formaldehyde[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
46%
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 12h;
The 0.96 g (10 mmol) of 1H-imidazole-4-carbaldehyde was dissolved in 20 ml of DMF, and 1.30 g (10 mmol) of <strong>[5305-59-9]4-amino-6-chloropyrimidine</strong>, 2.07 g (15 mm1) of potassium carbonate was added successively 100 C, stirring reaction 12h. The appropriate amount of water was added and extracted with ethyl acetate, washed with brine, dried over anhydrous sodium sulfate and then filtered to remove the solvent under reduced pressure. The residue was purified by silica gel column The eluent was methanol: dichloromethane = 1: 30 to give 0.87 g of a white solid (compound represented by the formula Pialpha) in a yield of 46%
46%
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 12h;
General procedure: A mixture of imidazole derivatives (4a-c, 10mmol), <strong>[5305-59-9]4-amino-6-chloropyrimidine</strong> (1.30g, 10mmol), and K2CO3 (2.07g, 15mmol, for 5a) or Cs2CO3 (3.91g, 12mmol, for 5b-c) was stirred in DMF (20mL) at 100C (for 5a) or at 140C (for 5b-c) for 12h. Water (100mL) was then added. The mixture was extracted with EtOAc (3×50mL). The organic layer was separated and dried over Na2SO4. Removal of the solvents produced a residue which was purified using column chromatography and eluted with a mixture of MeOH:CH2Cl2 (1:30, v:v) to afford 5a-c. 4.1.2.1 1-(6-Aminopyrimidin-4-yl)-1H-imidazole-4-carbaldehyde (5a) White solid, 46% yield. 1H NMR (400MHz, DMSO-d6) delta 9.83 (s, 1H), 8.70 (s, 1H), 8.64 (s, 1H), 8.37 (s, 1H), 7.35 (s, 2H), 6.72 (s, 1H).
1-(2-bromo-4-chlorophenyl)-1H-imidazole-4-carbaldehyde[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
43%
With potassium carbonate; In N,N-dimethyl-formamide; at 130℃; for 3h;Microwave irradiation;
440 p1 (3.4 mmol) of <strong>[1996-30-1]2-bromo-4-chloro-1-fluorobenzene</strong>, 337 mg (3.44 mmol) of 1H-imidazole-4-carbaldehyde, 1.43 g (10.3 mmol) of potassium carbonate and 17 ml DMF were stirred in the microwave at 130C for 3 hours, After cooling, methyl tert-butyl ether was added and the organic phase was washed three times with a saturated aqueous sodium chloride solution. The organic phase was dried over sodium sulphate and concentrated under reduced pressure. Yield: 430 mg (43% of theory).LC/MS [Method 10]: R = L44 mm; MS (ESIpos): m/z = 287 (M+H),?H-NMR (400 MHz, DM50-cl6): oe [ppm] = 9.83 (s, 1H), 8.36 (d, 1H), 8.13 (d, 1H), 8.08 (d, 1H),7.72-7.66 (m, 2H).
4-(1H-imidazol-4-yl)-3-phenyl-2,4-dihydroimidazo[1,5-a]pyrazolo[3,4-e][1,4]diazepine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
37%
With acetic acid In ethanol for 72h; Reflux;
General procedure for [1,4]-diazepine synthesis
General procedure: The corresponding amine (10 mmol) and aldehyde (20 mmol) were dissolved in absolute ethanol (40 mL) and a catalytic amount of acetic acid added. The mixture was refluxed for 72 h. The crude product was isolated from the reaction solution by precipitation with diethyl ether and recrystallization from hot methanol.
To a solution of <strong>[2302-25-2]4-bromo-1H-imidazole</strong> (0.65 g, 4.4 mmol, 1.0 equiv.) indry THF (20 mL) at 0 C was added a 2 M solution of i-PrMgCl in THF (2.2 mL, 4.4 mmol, 1.0 equiv.)during 5 min. The clear solution was stirred at that temperature for an additional 5 min, and a2.5 M solution of n-BuLi in hexanes (3.5 mL, 8.8 mmol, 2.0 equiv.) was added dropwise during5 min, while maintaining the temperature below 20 C. The resulting mixture was stirred at thattemperature for 0.5 h, dry DMF (0.32 g, 4.4 mmol, 1.0 equiv.) was added to 20 C. The resultingmixture was warmed to 20 C in 0.5 h and quenched with water (6 mL). After stirring the mixturebelow 20 C for 10 min, the phases were separated and the water phase was extracted one additionaltime with ethyl acetate. The resulting suspension was allowed to reach room temperature and teredthrough a 0.5 1 cm pad of silica gel eluted with 10 mL of ethyl acetate. The ltrate was concentratedand the residue was puried by ash chromatography on silica gel (eluent:petroleum ether/ethylacetate = 10:1) to afford product 3l as off-white solid, 0.36 g (yield: 85%), m.p.: 175-177 C. 1H-NMR(600 MHz, DMSO) 9.74 (s, 1H), 7.99 (s, 1H), 7.94 (s, 1H). 13C-NMR (151 MHz, DMSO) 184.46, 139.44,134.9, 129.5.
ethyl 4-((4-formyl-1H-imidazol-1-yl)methyl)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
(1235) Intermediate A-10 was synthesized starting from ethyl 4-(bromomethyl)benzoate and lH-imidazole-4-carbaldehyde following protocol given for A-8. LC-MS m/z calcd for Ci4Hi4N203, 258.1, found 259.1 [M+H]+.
2-(1-Ethylimidazol-4-yl)-4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazole-3-oxide-1-oxyl (L4Et) and 2-(1-ethylimidazol-5-yl)-4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazole-3-oxide-1-oxyl (L5Et)
Diethyl sulfate (2.4 g, 0.016 mol) was added to a suspension of imidazole-4-carbaldehyde (1.5 g, 0.016 mol) in anhydrous acetone (15 mL). The reaction mixture was refluxed for 18 h with stirring without contact with air moisture. Then the mixture was neutralized with an excess of moist NaHCO3 and the precipitate that formed was filtered and washed with EtOH. The filtrate was dried with Na2SO4 and concentrated using a rotary evaporator. The residue was filtered through a layer of Al2O3 (1.5×10 cm), the eluent was AcOEt. The filtrate was concentrated in vacuo and a light yellow oil (1.0 g) was obtained,which contained a mixture of isomers of 1-ethyl-1H-imidazole-4-carbaldehyde and 1-ethyl-1H-imidazole-5-carbaldehyde.
With potassium permanganate; In dichloromethane; at 40℃; for 4h;
(2) Mixing the obtained 1H-imidazole-4-carboxaldehyde with dichloromethane,Heat to 40C,Add KMnO4 and mix well.Stir the reaction for 4h,After filtration, distillation under reduced pressure, recrystallization, 1H-imidazole-4-carboxylic acid was obtained.The yield of 1H-imidazole-4-carboxylic acid produced was 94.3% and the purity was 99.3%.
6-(4-formyl-1H-imidazol-1-yl)-4-methoxynicotinonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
25%
With potassium carbonate; In N,N-dimethyl-formamide; at 90℃; for 1.0h;Inert atmosphere;
To a solution of iH4rnidazo1e-4carbaIdehyde (0.50 g, 5.20 mmol) and 6-chloro-4-- methoxynicotinonitrile (1.05 g, 6.24 mrno]) in DMF (10 mL) was added K2C03 (1.08g. 7.81rnmol) at ambient temperature under a nitrogen atmosphere. The resulting reaction mixture was heated at 90C for 1 h. The reaction mixture was cooled to ambient temperature and diluted with ice water (30 mL). The resulting precipitate was filtered and was washed with ethanol (2. rnL) to obtain Intermediate 18(0.30g. 25.00%). ?FINMR (400 MHz, DMSO-d6) oe ppm 413 (s.,3 Fl), 7.81 (s, I H), 8.83 (s, 2 H), 8.95 (d, J:::: 1.19 Hz, 1 H), 9.87 (s, I H). LCMS (Method-I);retention time 0.75 mm, FM-f-Hi 229.1.
25.00%
With potassium carbonate; In N,N-dimethyl-formamide; at 90℃; for 1.0h;Inert atmosphere;
To a solution of 1Himidazole4carbaldehyde (0.50 g, 5.20 mmol) and 6chloro4methoxynicotinonitrile (1.05 g, 6.24 mmol) in DMF (10 mL) was added K2CO3 (1.08 g, 7.81 mmol) at ambient temperature under a nitrogen atmosphere. The resulting reaction mixture was heated at 90C for 1 h. The reaction mixture was cooled to ambient temperature and diluted with ice water (30 mL). The resulting precipitate was filtered and was washed with ethanol (2 mL) to obtained Intermediate 11 (0.30 g, 25.00%).1H NMR (400 MHz, DMSOd6) G ppm 4.13 (s, 3 H), 7.81 (s, 1 H), 8.83 (s, 2 H), 8.95 (d, J = 1.19 Hz, 1 H), 9.87 (s, 1 H). LCMS (MethodL): retention time 0.75 min, [M+H] 229.1.
2-(1H-imidazol-4-yl)-1H-naphthalene[2,3-d]imidazole-4,9-dione[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
75%
With sodium metabisulfite In 1,2-dimethoxyethane at 120℃;
29 General procedure for the preparation of compounds T1-T43
General procedure: A solution of 2,3-diamino-1,4-naphthoquinone (1.0 mmol),appropriate aromatic aldehyde (1.0 mmol) with sodium pyrosulfitein DMF was stirred at 120 C overnight. On completion of the reactionmonitored by TLC, the solvent was evaporated and the residuewas purified by silica gel chromatography by DCM/MeOHsystem to afford the final product. If necessary, the crude productcould be recrystallized in methanol or DMSO to afford pure sample.4.1.5.1. 2-(2-Chlorophenyl)-1H-naphtho[2,3-d]imidazole-4,9-dione(T1). Pale yellow solid; yield 80%;
ethyl (Z)-2-(5-((1H-imidazol-4-yl)methylene)-2,4-dioxothiazolidin-3-yl)acetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
39%
With piperidine; acetic acid In toluene for 18h; Reflux;
32.1 Step 1 - Synthesis of Ethyl (Z)-2-(2,4-dioxo-5-(thiophen-3-yl methylene)thiazolidi n-3- yl)acetate
To a solution of 3-thiophenecarboxaldehyde (0.110 g, 0.984 mmol) and 1(0.200 g,0.984 mmol) in toluene (5 mL), three drops piperidine and two drops acetic acid were added.The reaction was heated under ref lux for 18 hours then concentrated in vacuo. The crude solid was washed with small amounts of methanol and collected via vacuum filtration to afford the desired compound (0.182 g, 62%). OH (400 MHz, 0D013) 7.94 (5, 1H, OH), 7.65 (5, 1H, ArH), 7.46 (d, J8.0, 1H, ArH), 7.31 (d, J8.0, 1H, ArH), 4.47 (s, 2H, OH2), 4.25 (q, J 12.0, 8.0, 2H,OH2), 1.30 (t, J8.0, 3H, OH3).
39%
With piperidine; acetic acid In toluene for 18h; Reflux;
4.3. General procedure B: Knoevenagel condensation
General procedure: Knoevenagel condensation was undertaken according to the methodsdescribed by Bhat et al.29 To a solution of thiazolidinedione 3 (0.200 g,0.984 mmol) and aryl aldehyde (0.984 mmol) in toluene (5 mL), threedrops piperidine and two drops acetic acid were added. The reaction washeated under reflux for 18 h then concentrated in vacuo. The desired product was afforded, sufficiently pure, after washes with small amountsof methanol and collection via vacuum filtration unless otherwisespecified.
4-(1H-imidazol-5-ylmethylene-amino)-4H-1,2,4-triazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91.89%
With sulfuric acid; In ethanol; at 80℃; for 10h;
The ligand 4-(1H-imidazol-5-ylmethylene-amino)-4H-1,2,4-triazole (5-imztrz) (Fig. 1) was prepared by the condensation reaction of a modified literature procedure [21]. The dissolved <strong>[584-13-4]4-amino-1,2,4-triazole</strong> (1.68 g, 0.02 mol, 1.0 eq.) in ethanol was heated to boiling (80 C). Then, the second reactant 1H-imidazole-4-carbaldehyde (2.11 g, 0.022 mol, 1.1 eq.) dissolved in ethanol was added dropwise though a dropping funnel. After the complete addition few drops of catalytic H2SO4 were added. The reaction mixture was kept for refluxing for another 10 h. After cooling down to r.t., the excess of solvent was removed by rotary evaporator at 35 C. The obtained product was washed with small amounts of cold ethanol, diethyl ether and then dried in the desiccator with phosphorus pentoxide as dry agent. A light-yellow powder was obtained. Yield: 2.98 g (91.89%). m.p. 253.5-265.7 C. IR (KBr, cm1): 3109 (s),2838 (w), 1625 (vs), 1504 (vs), 1434 (s), 1386 (s), 1299 (s), 1214 (w),1175 (vs), 1060 (vs), 995 (s), 945 (w), 853 (s), 691 (m), 630 (vs), 510(m). 1H NMR (Fig. S1) (d6-DMSO): 9.07 (s, 2H, triazole), 8.92 (s, 1H,HACN), 7.91 (s, 1H, imidazole ring), 7.79 (s, 1H, imidazole ring).
4-(4-formyl-1H-imidazol-1-yl)-2-methoxybenzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
33.80%
With copper(l) iodide; dimethylaminoacetic acid; caesium carbonate; In 1,4-dioxane; at 110℃; for 16h;Sealed tube;
To a stirred solution of 1H-imidazole-4-carbaldehyde (1.00 g, 10.41 mmol) in dioxane (5 mL) was added <strong>[330793-38-9]4-bromo-2-methoxybenzonitrile</strong> (2.20 g, 10.41 mmol), N,N- Dimethylglycine (1.073 g, 10.41 mmol) and Cs2CO3 (3.39 g, 10.41 mmol) followed by copper(I)iodide (1.98 g, 10.41 mmol). The resulting reaction mixture was heated at 110 °C for 16 h in a sealed tube. The reaction mixture was cooled to ambient temperature, concentrated to dryness under vacuum, diluted with water (40 mL) and extracted with DCM (2 x 100 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain Intermediate 48 (0.80 g, 33.80percent) as brown solid.1H NMR (400 MHz, DMSO-d6) G ppm 4.04 (s, 3 H), 7.56 (dd, J = 8.35, 1.95 Hz, 1 H), 7.66 (d, J = 1.44 Hz, 1 H), 7.97 (d, J = 8.41 Hz, 1 H), 8.67 - 8.72 (m, 1 H), 8.85 - 8.89 (m, 1 H), 9.84 - 9.87 (m, 1 H). LCMS (Method-O): retention time 0.76 min, [M+H] 228.1
1-(2-methoxypyridin-4-yl)-1H-imidazole-4-carbaldehyde[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
19.00%
With potassium carbonate; In N,N-dimethyl-formamide; at 90℃; for 1h;Inert atmosphere;
General procedure: To a solution of 1Himidazole4carbaldehyde (0.50 g, 5.20 mmol) and 6chloro4methoxynicotinonitrile (1.05 g, 6.24 mmol) in DMF (10 mL) was added K2CO3 (1.08 g, 7.81 mmol) at ambient temperature under a nitrogen atmosphere. The resulting reaction mixture was heated at 90C for 1 h. The reaction mixture was cooled to ambient temperature and diluted with ice water (30 mL). The resulting precipitate was filtered and was washed with ethanol (2 mL) to obtained Intermediate 11 (0.30 g, 25.00%).1H NMR (400 MHz, DMSOd6) G ppm 4.13 (s, 3 H), 7.81 (s, 1 H), 8.83 (s, 2 H), 8.95 (d, J = 1.19 Hz, 1 H), 9.87 (s, 1 H). LCMS (MethodL): retention time 0.75 min, [M+H] 229.1.
In N,N-dimethyl-formamide; at 100℃; for 2h;Inert atmosphere;
General procedure: To a flask containing lH-imidazole-4-carbaldehyde (20 mmol, 1.0 equiv.) and 1, 4/1,5- dichloropyrimidine (22 mmol, 1.1 equiv.) was added 40 mL DMF (0.5 M). This solution was then heated to l00C and stirred for approximately 2 hours while being monitored by TLC.Upon reaction completion, the reaction mixture was diluted with dichloromethane and washed with water and brine. The organic layers were then combined, dried over Na2S04 and concentrated via rotary-evaporation. The crude mixture was then dissolved in a minimal amount of dichloromethane and hexanes was added to the solution to precipitate the final product. The solid was then filtered and dried under vacuum and submitted to the next reduction reaction without any further purification. Reaction yields for the 1, 5-pyrimidine were 85% while the yield for the 1, 4-pyrimidine was around 40% due to an undesired regioisomer being formed.
2-(((1H-imidazol-5-yl)methyl)(4-methoxybenzyl)amino)ethanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
77%
Stage #1: 2-[(4-methoxybenzyl)amino]ethanol; 4(5)formylimidazole With acetic acid In methanol; acetonitrile at 20℃; for 1h;
Stage #2: With sodium cyanoborohydride In methanol; acetonitrile at 0 - 20℃; for 3h;
16 2-(((1H-Imidazol-5-yl)methyl)(4-methoxybenzyl)amino)ethanol
To a solution of 1H-imidazole-4-carbaldehyde (14.85 g, 154.5 mmol) and 2-(4-methoxybenzylamino)ethanol (20.00 g, 110.4 mmol) in MeCN (220 mL) and MeOH (22 mL) was added AcOH (10 mL). The mixture was stirred at RT for 1 h, cooled to 0° C., then treated with NaBH3CN (8.32 g, 132 mmol) portionwise. The resulting mixture was stirred at RT for 3 h, then concentrated under reduced pressure. The residue was partitioned between CH2Cl2 (200 mL) and sat. aq. NaHCO3 (200 mL). The aqueous layer was treated with 1 M aq. NaOH to adjust the pH to 14, then extracted with CH2Cl2 (75 mL*4). The combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure to give the title compound as a colorless oil (22.30 g, 77%). MS (ES+): C14H19N3O2 requires: 261, found: 262 [M+H]+.
1-Phenyl-1H-imidazole-4-carbaldehyde, IV-Ph-4. CuI (93.0 mg, 488 mumol), 1,10-phenanthroline(88.0 mg, 488 mumol), tripotassium phosphate (1.56 g, mmol, 7.35 mmol), and 4-imidazolecarboxaldehyde(330 mg, 3.43 mmol) were placed under inert conditions in a flask with 5 mL of dry DMF. After stirringthe mixture at room temperature for 10 min, bromobenzene (386 mg, 258 muL, 2.46 mmol) was added. Thereaction mixture was heated to 120 C over 65 h. After cooling to room temperature, the solution wasdiluted with ethyl acetate (20 mL) and filtered through silica gel. The organic phase was washed withbrine and then dried over MgSO4. Following to concentration in vacuo, the raw product wasrecrystallized from DCM and pentane, which afforded the pure product as a white solid (112 mg, 650mumol; 26% yield). The obtained spectroscopic data were in accordance with literature [66]. 1H-NMR (300MHz, CDCl3, rt): delta [ppm] = 9.94 (s, 1H), 7.92 (dd, J1 = 14.0 Hz, J2 = 1.3 Hz, 2H), 7.69-7.29 (m, 5H).
(Z)-1-acetyl-3-((1H-imidazol-4-yl)methylene-d)piperazine-2,5-dione[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
41%
With caesium carbonate; In N,N-dimethyl-formamide; at 20℃; for 20h;Inert atmosphere;
General procedure: Under nitrogen, a mixture of compound 10a (4.88g, 32.11mmol), 1, 4-diacetylpiperazine-2, 5-dione (12.73g, 64.21mmol), and Cs2CO3 (15.69g, 48.16mmol) in DMF (50ml) was stirred for 20h at room temperature. The solution was poured into cool water to precipitate a solid. The mixture was filtered to produce 4.43g of orange solid with a yield of 47%. 1H NMR (500MHz, DMSO-d6) delta 12.36 (s, 1H), 12.01 (s, 1H), 7.85 (s, 1H), 7.04 (s, 1H), 4.30 (s, 2H), 2.49 (s, 3H), 1.39 (s, 9H). MS (ESI) m/z: [M+H]+ Calcd for C14H19N4O3: 291.15, Found: 290.79.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping