* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: With bis(acetylacetonate)nickel(II) In tetrahydrofuran at 20℃; for 0.0166667 h; Inert atmosphere Stage #2: With sodium triethylborohydride In tetrahydrofuran at 50℃; for 10 h; Inert atmosphere
A typical procedure (Table 2, entry 1) is as follows. To a stirredsolution of Ni(acac)2 (1a) (1.3 mg, 0.005 mmol) in THF (5 mL) wasadded 1-octene (112 mg, 1.0 mmol) and (EtO)3SiH (164 mg,1.0 mmol) at room temperature. After the mixture was stirred for1 min, NaBHEt3 (1.0Min THF, 5 mL, 0.005 mmol) was added and theresulting mixture was heated at 50 °C. The solution was stirred atthe same temperature, and the progress of the reaction wasmonitored by GLC. After completion of the reaction, mesitylene(60 mg, 0.50 mmol) was added as an internal standard to the reactionmixture. The GLC analysis of the resulting solution revealedthe formation of (EtO)3(nOct)Si (0.90 mmol, 90percent) and (EtO)4Si(0.05 mmol, 5percent). The solutionwas concentrated under vacuum, andthe residue was purified by gel permeation chromatography (GPC)using toluene as an eluent to give (EtO)3(nOct)Si (234 mg,0.85 mmol, 85percent). The 1H, 13C{1H} and 29Si{1H} NMR spectra of theisolated compound are consistent with the reported data. A similarprocedurewas employed for the hydrosilylation using other silanesand 1,3-diene/alkenes/alkynes. These reactions were carried out atroom temperature except for the reactions, Table 2, entries 2-3.The 1H/13C NMR spectroscopic data for the new compounds aregiven in the supplementary data.
Reference:
[1] Journal of Organometallic Chemistry, 2016, vol. 809, p. 57 - 62
2
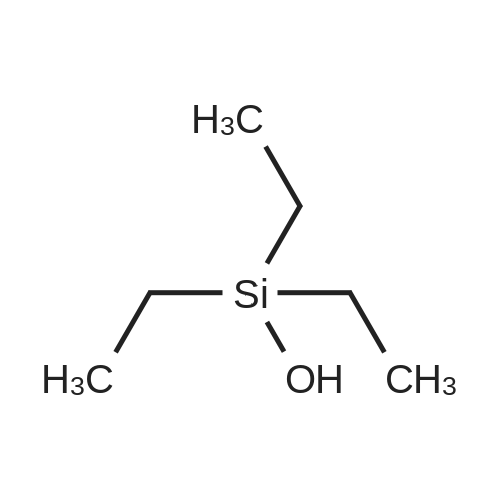
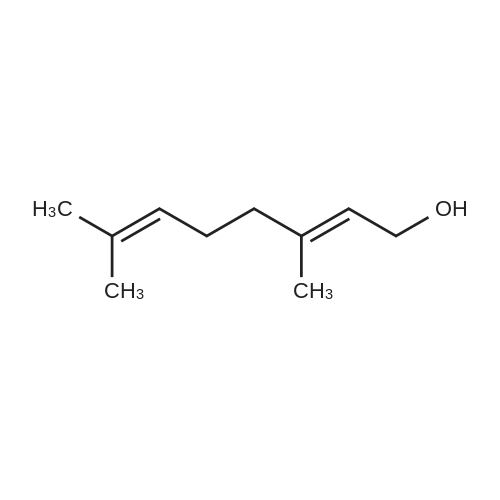
[ 111-66-0 ]
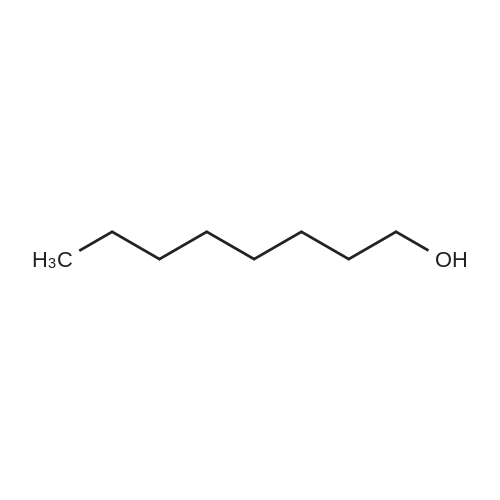
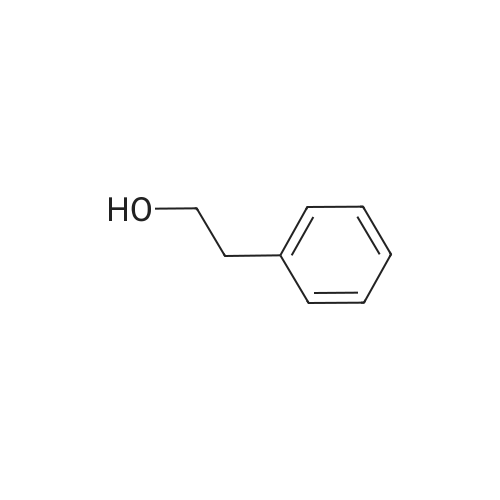
[ 998-30-1 ]
[ 78-10-4 ]
[ 2157-42-8 ]
[ 2943-75-1 ]
Yield
Reaction Conditions
Operation in experiment
54%
Stage #1: With bis(acetylacetonate)nickel(II); sodium triethylborohydride In tetrahydrofuran-d8 at 20℃; for 2 h; Inert atmosphere Stage #2: at 50℃; for 14 h; Inert atmosphere
To an NMR tube equipped with a Teflon valve were addedNi(acac)2 (13.0 mg, 0.05 mmol), 1-octene (11.3 mg, 0.1 mmol),(EtO)3SiH (16.0 mg, 0.1 mmol), and THF-d8 (0.5 mL). After theaddition of NaBHEt3 (1.0 M in THF, 50 mL, 0.05 mmol) to this solutionat room temperature, the reaction was followed by 1H NMR.After 2 h, the reaction did not proceed and only the signalsassignable to the starting materials [1-octene and (EtO)3SiH] weredetected by 1H NMR. The reaction was further followed at 50 °C for14 h, and formation of (EtO)3(nOct)Si (54percent) as well as (EtO)4Si (8percent)and (EtO)3SiOSi(OEt)3 (7percent) was observed. During the reaction, nohydride signals were detected in the high field region (δ 0 to -30 ppm).
Reference:
[1] Journal of Organometallic Chemistry, 2016, vol. 809, p. 57 - 62
3
[ 111-66-0 ]
[ 998-30-1 ]
[ 2943-75-1 ]
Yield
Reaction Conditions
Operation in experiment
100%
at 90℃; for 5 h; Inert atmosphere
In a 500-ml three-necked flask, octene (1.25 mol) was added, and Catalyst 1 (1.25 mmol) prepared in Example 1 was slowly heated to 90°C under a nitrogen atmosphere.While stirring, triethoxyhydrosilane (1.5 mol) was added dropwise through the dropping funnel. The dropwise addition time was 0.5 hours, the reaction temperature was maintained, and the stirring reaction was continued for 5 hours.After cooling to room temperature, the corresponding fractions were collected by distillation under reduced pressure. The conversion of octene was 94.7percent as determined by GC-MS.The yield of the β adduct 1-triethoxysilyloctane was 100percent.
100%
at 90℃; for 5 h; Inert atmosphere
In a 500-ml three-necked flask, octene (1.25 mol) was added, and Catalyst 1 (1.25 mmol) prepared in Example 1 was slowly heated to 90°C under a nitrogen atmosphere.While stirring, triethoxyhydrosilane (1.5 mol) was added dropwise through the dropping funnel. The dropwise addition time was 0.5 hours, the reaction temperature was maintained, and the stirring reaction was continued for 5 hours.After cooling to room temperature, the corresponding fractions were collected by distillation under reduced pressure. The conversion of octene was determined to be 94.3percent by GC-MS.The yield of the β adduct 1-triethoxysilyloctane was 100percent.
98%
at 20℃; for 1 h; Inert atmosphere; Sealed tube
o a scintillation vial equipped with a stir bar in a nitrogen filled glovebox was added (TFAPDI)Co(2-ethylhexanoate)2(5 mg, 0.009 mmol) followed by 1- octene (125 mg, 1.11 mmol) to give a heterogeneous mixture. Triethoxysilane (183 mg, 1.11 mmol) was then added resulting in formation of an olive green reaction mixture. The vial was sealed with a cap, removed from the box and stirred at room temperature for 1 hour. The cap was removed and any remaining volatiles were removed with a stream of air. The resulting residue was diluted with a solution of 5percent ethyl ether in pentane and passed through a small column of silica gel, eluting with additional 5percent ether in pentane solution (5 mL). The resulting clear, colorless eluent was concentrated to give the product as a clear, colorless oil (303 mg, 98percent). The product distribution was determined by analysis of the1H NMR spectrum of the isolated product.
96.5%
Stage #1: at 110℃; for 0.5 h; Stage #2: at 110℃; for 2.5 h;
Reactions were carried out in a three-necked flask with a magnetic stirrer and a reflux condenser with an attached drying system on the upper condenser. Olefin and platinum catalyst were stirred at the reaction temperature for 30 min before triethoxysilane which was added at a constant speed. The reaction mixture was heated at the reaction temperature within the stipulated time and then catalyst was separated from the raw product by decantation. After the removal of the raw product, a new portion of substrates was added and the reaction was repeated under the same conditions. Gas chromatography was employed to follow the course of the reaction by the appearance of product. All hydrosilylation products were characterized by 1H NMR and 13C NMR.
95.1%
Stage #1: for 0.0833333 h; Stage #2: at 90℃; for 5 h;
General procedure: All catalysis reaction operations were performed in a 10 mL flat-bottomed tube without protection from air. The alkene (4.0 mmol) and the requisite amount of catalyst were placed in a dried tube and the reaction mixture was stirred for 5 min. Thereafter, the silane (4.4 mmol) was added and the resulting mixture was heated and stirred for the requisite time and then cooled to room temperature. The product phase was separated by decantation and the conversion of the alkene and the selectivity were determined by GC–MS analysis on an Agilent 26890N/59731 apparatus equipped with a DB-5 column (30 m × 2.5 mm × 0.25 μm). 1H NMR (400 MHz) and 13C NMR (100.6 MHz) spectra were recorded on a Bruker Advance spectrometer using TMS as an internal standard. Methanol-d4 and DMSO-d6 were used as solvents.
95.8%
at 110℃; for 2 h;
General procedure: Hydrosilylation was carried out in a three-necked flask with amagnetic stirrer and a reflux condenser with an attached dryingsystem on the upper condenser. The olefin and platinumcomplex were stirred at the setting temperature for 30 min atfirst. Then the triethoxysilane was added at a constant speed.The reaction mixture was maintained at the reaction temperaturewithin the stipulated time. After the reaction, the catalystwas separated from the raw product by centrifugation. A newportion of substrates was added and the reaction was repeatedunder the same conditions. Gas chromatography was employedtofollowthe course of the reactionby the appearance of product.
86.9%
at 90℃; for 10 h;
General procedure: All the catalytic reactions were performed in a 10 mLat-bottomed tube without protection from air. The alkene(4.0 mmol) and the requisite amount of catalyst were placed ina dried tube and the reaction mixture was stirred for 5 min.Thereafter, the silane (4.4 mmol) was added and the resulting mixture was heated and stirred for the requisite time andthen cooled to room temperature. The product phase was separated by decantation and the alkene conversion and selectivity of the reaction were determined by GC-MS analysis on anAgilent 26890N/59731 apparatus equipped with a DB-5 column(30 m × 2.5 mm × 0.25 mm).
> 98 %Spectr.
With (terpy)Co(Ph2SiOC3H5) In neat (no solvent) at 20℃; for 1 h; Schlenk technique; Glovebox; Inert atmosphere; Sealed tube
A 50 mL Schlenk flask equipped with a stir bar was charged with 1-octene (112 mg, 1 mmol) and triethoxysilane (164 mg, 1 mmol) in the glove box. The flask was sealed with a glass stopper and transferred out of the box. (terpy)Co(Ph2SiOC3H5) (2 mg, 0.5 molpercent) was charged into a vial, brought out of the box and exposed to air for 10 minutes. The solid catalyst was added into the Schlenk flask under an Ar counterflow. The flask was sealed with a glass stopper and stirred for 1 hour. The reaction was quenched by exposure to air. The reaction mixture was analyzed by GC and 1H NMR spectroscopy which established >98percent yield of anti-Markovnikov hydrosilylation product and trace dehydrogenative silylation product.
90 %Chromat.
With nickel(II) bis(2,2,6,6-tetramethylheptane-3,5-dionate); sodium triethylborohydride In tetrahydrofuran at 50℃; for 12 h;
General procedure: Example 16: THF solution (5 mL) of Nickel complex compound (0.005mmol) shownin Table 2 was placed in vial, then for the nickel complex compound, 20-foldmolar amount 1-octene, hydrosilanes shown in Table 2, equimolar amount hydridereducing agent were added sequentially to initiate the reaction. After a lapseof time shown in Table 2, the reaction solution was exposed to air to terminatethe reaction (the color of the solution turned colorless.), the yield of thefollowing product was quantified by GC analysis. The results are shown in Table2.
Reference:
[1] Patent: CN107857776, 2018, A, . Location in patent: Paragraph 0048; 0049
[2] Patent: CN107857777, 2018, A, . Location in patent: Paragraph 0048; 0049
[3] ACS Catalysis, 2016, vol. 6, # 4, p. 2632 - 2636
[4] Journal of the American Chemical Society, 2017, vol. 139, # 5, p. 1798 - 1801
[5] Patent: WO2017/19473, 2017, A1, . Location in patent: Page/Page column 35
[6] ACS Catalysis, 2016, vol. 6, # 7, p. 4105 - 4109
[7] European Journal of Inorganic Chemistry, 2018, vol. 2018, # 45, p. 4867 - 4874
[8] Journal of Organometallic Chemistry, 2011, vol. 696, # 9, p. 1845 - 1849
[9] Journal of Organometallic Chemistry, 2014, vol. 765, p. 59 - 63
[10] Phosphorus, Sulfur and Silicon and the Related Elements, 2016, vol. 191, # 5, p. 728 - 733
[11] Journal of Organometallic Chemistry, 1981, vol. 208, # 3, p. 401 - 406
[12] Russian Chemical Bulletin, 1997, vol. 46, # 5, p. 1003 - 1006
[13] Phosphorus, Sulfur and Silicon and the Related Elements, 2017, vol. 192, # 12, p. 1271 - 1278
[14] Angewandte Chemie - International Edition, 2018, vol. 57, # 33, p. 10620 - 10624[15] Angew. Chem., 2018, vol. 130, p. 10780 - 10784,5
[16] Journal of the American Chemical Society, 2016, vol. 138, # 31, p. 9783 - 9786
[17] Chemistry Letters, 1981, p. 243 - 246
[18] Organometallics, 2018, vol. 37, # 16, p. 2760 - 2768
[19] Journal of Organometallic Chemistry, 1979, vol. 172, p. 153 - 163
[20] Journal of Organometallic Chemistry, 1977, vol. 137, p. 293 - 300
[21] Journal of Organometallic Chemistry, 1984, vol. 271, # 1-3, p. 153 - 168
[22] Journal of Organometallic Chemistry, 2010, vol. 695, # 3, p. 431 - 436
[23] Journal of Organometallic Chemistry, 2011, vol. 696, # 1, p. 263 - 268
[24] Patent: WO2011/6044, 2011, A2, . Location in patent: Page/Page column 25
[25] Journal of Organometallic Chemistry, 2011, vol. 696, # 10, p. 2116 - 2121
[26] New Journal of Chemistry, 2010, vol. 34, # 7, p. 1330 - 1334
[27] Science, 2012, vol. 335, # 6068, p. 567 - 570
[28] Dalton Transactions, 2013, vol. 42, # 1, p. 270 - 276
[29] Journal of Organometallic Chemistry, 2013, vol. 727, p. 28 - 36
[30] Patent: US2013/109772, 2013, A1, . Location in patent: Paragraph 0315; 0316; 0317; 0318; 0319; 0320; 0321; 0322;
[31] Patent: US2013/158281, 2013, A1, . Location in patent: Paragraph 0105
[32] Patent: WO2015/77304, 2015, A1, . Location in patent: Page/Page column 21; 22
[33] Journal of Organometallic Chemistry, 2015, vol. 794, p. 65 - 69
[34] Angewandte Chemie - International Edition, 2015, vol. 54, # 48, p. 14523 - 14526[35] Angew. Chem., 2015, vol. 127, # 48, p. 14731 - 14734,4
[36] Patent: JP2016/14005, 2016, A, . Location in patent: Paragraph 0033; 0034
[37] Patent: CN106380486, 2017, A, . Location in patent: Paragraph 0038; 0039
[38] Patent: CN106380488, 2017, A, . Location in patent: Paragraph 0051-0052; 0056
[39] Nature Chemistry, 2017, vol. 9, # 6, p. 595 - 600
[40] Journal of Organometallic Chemistry, 2018, vol. 855, p. 7 - 11
[41] Chemical Science, 2018, vol. 9, # 10, p. 2817 - 2825
[42] Applied Organometallic Chemistry, 2018, vol. 32, # 4,
[43] Journal of the American Chemical Society, 2018, vol. 140, # 24, p. 7407 - 7410
4
[ 111-66-0 ]
[ 998-30-1 ]
[ 2943-75-1 ]
[ 911808-73-6 ]
Yield
Reaction Conditions
Operation in experiment
90 %Spectr.
With (terpy)Co(Me2SiOC3H5) In neat (no solvent) at 20℃; for 1 h; Glovebox; Inert atmosphere
In a glove box, 1-octene (112 mg, 1 mmol) and triethoxysilane (164 mg, 1 mmol) were weighed into a vial equipped with a stir bar. Purple (terpy)Co(Me2SiOC3H5) (2 mg, 0.5 molpercent) was weighed into a separate vial, and was subsequently combined with the substrates. The contents of the vial were stirred at room temperature for 1 hour. The reaction was quenched by exposure to air. The product mixture was processed as described in the general procedure above. GC and 1H NMR spectroscopy which showed 90percent yield of anti-Markovnikov hydrosilylation product and 9percent dehydrogenative silylation product.
Reference:
[1] Journal of the American Chemical Society, 2013, vol. 135, # 51, p. 19154 - 19166
6
[ 111-66-0 ]
[ 998-30-1 ]
[ 111-65-9 ]
[ 2943-75-1 ]
Reference:
[1] Phosphorus, Sulfur and Silicon and the Related Elements, 2010, vol. 185, # 2, p. 484 - 490
[2] Journal of the American Chemical Society, 2017, vol. 139, # 5, p. 1798 - 1801
7
[ 64-17-5 ]
[ 111-66-0 ]
[ 2943-75-1 ]
Reference:
[1] Journal of the American Chemical Society, 1991, vol. 113, # 26, p. 9887 - 9888
[2] Journal of the American Chemical Society, 1991, vol. 113, # 26, p. 9887 - 9888
In a 500-ml three-necked flask, octene (1.25 mol) was added, and Catalyst 1 (1.25 mmol) prepared in Example 1 was slowly heated to 90C under a nitrogen atmosphere.While stirring, triethoxyhydrosilane (1.5 mol) was added dropwise through the dropping funnel. The dropwise addition time was 0.5 hours, the reaction temperature was maintained, and the stirring reaction was continued for 5 hours.After cooling to room temperature, the corresponding fractions were collected by distillation under reduced pressure. The conversion of octene was 94.7% as determined by GC-MS.The yield of the beta adduct 1-triethoxysilyloctane was 100%.
100%
at 90℃; for 5h;Inert atmosphere;
In a 500-ml three-necked flask, octene (1.25 mol) was added, and Catalyst 1 (1.25 mmol) prepared in Example 1 was slowly heated to 90C under a nitrogen atmosphere.While stirring, triethoxyhydrosilane (1.5 mol) was added dropwise through the dropping funnel. The dropwise addition time was 0.5 hours, the reaction temperature was maintained, and the stirring reaction was continued for 5 hours.After cooling to room temperature, the corresponding fractions were collected by distillation under reduced pressure. The conversion of octene was determined to be 94.3% by GC-MS.The yield of the beta adduct 1-triethoxysilyloctane was 100%.
98%
With (TFAPDI)Co(2-ethylhexanoate)2; at 20℃; for 1h;Inert atmosphere; Sealed tube;
o a scintillation vial equipped with a stir bar in a nitrogen filled glovebox was added (TFAPDI)Co(2-ethylhexanoate)2(5 mg, 0.009 mmol) followed by 1- octene (125 mg, 1.11 mmol) to give a heterogeneous mixture. Triethoxysilane (183 mg, 1.11 mmol) was then added resulting in formation of an olive green reaction mixture. The vial was sealed with a cap, removed from the box and stirred at room temperature for 1 hour. The cap was removed and any remaining volatiles were removed with a stream of air. The resulting residue was diluted with a solution of 5% ethyl ether in pentane and passed through a small column of silica gel, eluting with additional 5% ether in pentane solution (5 mL). The resulting clear, colorless eluent was concentrated to give the product as a clear, colorless oil (303 mg, 98%). The product distribution was determined by analysis of the1H NMR spectrum of the isolated product.
96.5%
Reactions were carried out in a three-necked flask with a magnetic stirrer and a reflux condenser with an attached drying system on the upper condenser. Olefin and platinum catalyst were stirred at the reaction temperature for 30 min before triethoxysilane which was added at a constant speed. The reaction mixture was heated at the reaction temperature within the stipulated time and then catalyst was separated from the raw product by decantation. After the removal of the raw product, a new portion of substrates was added and the reaction was repeated under the same conditions. Gas chromatography was employed to follow the course of the reaction by the appearance of product. All hydrosilylation products were characterized by 1H NMR and 13C NMR.
95.1%
General procedure: All catalysis reaction operations were performed in a 10 mL flat-bottomed tube without protection from air. The alkene (4.0 mmol) and the requisite amount of catalyst were placed in a dried tube and the reaction mixture was stirred for 5 min. Thereafter, the silane (4.4 mmol) was added and the resulting mixture was heated and stirred for the requisite time and then cooled to room temperature. The product phase was separated by decantation and the conversion of the alkene and the selectivity were determined by GC-MS analysis on an Agilent 26890N/59731 apparatus equipped with a DB-5 column (30 m × 2.5 mm × 0.25 mum). 1H NMR (400 MHz) and 13C NMR (100.6 MHz) spectra were recorded on a Bruker Advance spectrometer using TMS as an internal standard. Methanol-d4 and DMSO-d6 were used as solvents.
95.8%
With silica supported poly-3-(2-aminoethylamino)propylsiloxane platinum complex; at 110℃; for 2h;
General procedure: Hydrosilylation was carried out in a three-necked flask with amagnetic stirrer and a reflux condenser with an attached dryingsystem on the upper condenser. The olefin and platinumcomplex were stirred at the setting temperature for 30 min atfirst. Then the triethoxysilane was added at a constant speed.The reaction mixture was maintained at the reaction temperaturewithin the stipulated time. After the reaction, the catalystwas separated from the raw product by centrifugation. A newportion of substrates was added and the reaction was repeatedunder the same conditions. Gas chromatography was employedtofollowthe course of the reactionby the appearance of product.
86.9%
With N-heterocyclic carbene fuctionalized with methyl(trimethylsilane) and methoxypolyethylene glycol Pt(II) complex; at 90℃; for 10h;
General procedure: All the catalytic reactions were performed in a 10 mLat-bottomed tube without protection from air. The alkene(4.0 mmol) and the requisite amount of catalyst were placed ina dried tube and the reaction mixture was stirred for 5 min.Thereafter, the silane (4.4 mmol) was added and the resulting mixture was heated and stirred for the requisite time andthen cooled to room temperature. The product phase was separated by decantation and the alkene conversion and selectivity of the reaction were determined by GC-MS analysis on anAgilent 26890N/59731 apparatus equipped with a DB-5 column(30 m × 2.5 mm × 0.25 mm).
[[(1,1'-(pyridine-2,6-diyl)bis(N-(2,6-dimethylphenyl)ethan-1-imine))Fe(N2)]2(mu2-N2)]; In toluene; at 20℃; for 1h;Inert atmosphere;
Hydrosilylation of 1-Octene with Triethoxysilane[00127] In a nitrogen filled atmosphere, to a scintillation vial was added 150 mg (1.33 mmol) of 1-octene and 170 mg (1.03 mmol), of triethoxysilane. To a separate vial was added 5 mg of [(2'6~Me2PDI)Fe(N2)]2[mu-(N2)] and 200 mg of toluene. To the stirring solution of olefin and silane was added 19 mg of the catalyst solution (0.48 mg, 0.1 mole % to silane). The reaction was allowed to stir at room temperature in the drybox for about one hour. The resonance associated with the Si-H in the 1H NMR was observed to disappear during the course of the reaction, and a new resonance upfeld at 0.62 ppm assignable to methylene attached to silicon appeared, giving a spectrum consistent with that of the previously reported compound.
With chloro(eta4-1,5-cyclooctadiene)[(2,4,6-trimethylphenyl)-3-methylimidazole-2-ylidene]rhodium(I); 3-butyl-1-methyl-1H-imidazol-3-ium hexafluorophosphate; at 70℃; for 2h;
Typical hydrosilylation reaction procedures were as follows: Agiven amount of catalyst and ionic liquid were added to a 10 mLround bottomed flask equipped with a magnetic stirrer and thealkene and silane were then added. This mixture was heated to theappropriate temperature and the hydrosilylation reaction wasallowed to proceed with constant stirring for 5 h. At the end of thereaction, the product phase was separated from the catalyst bydecantation and the conversion of alkene and the selectivity weredetermined by GC. The catalyst was recharged with fresh alkeneand silane for the next catalytic run.
With Wilkinson's catalyst; 1-(2-carboxyethyl)-3-methylimidazolium inner salt; at 90℃; for 5h;
General procedure: Typical hydrosilylation reaction procedures were as follows: A given amount of catalyst and ionic liquid was added to a 10 mL round bottomed flask equipped with a magnetic stirrer and then the alkene and silane were added. After heating the mixture at the appropriate temperature, the hydrosilylation reaction was preceded with constant stirring for 5 h. At the end of the reaction, the product phase was separated from the catalyst by decantation and the conversion of alkene and the selectivity were determined by GC. The catalyst was recharged with fresh alkene and silane for the next catalytic run
With 5% platinum on aluminium oxide; acetic acid; at 110℃;
This Example illustrates the invention and was performed as a batch process.Reactants: Triethoxysilane (43.8 grams) and 1-octene (32.7 grams, in excess), 0.27 gram of acetic acid as catalyst promoterCatalyst: 5% Pt/Alumina from Johnson Matthey (0.035 gram of solid powder, 22.9 ppm Pt)Reaction Temp.: 110 C.Pressure: atmospheric pressureProcedure: Triethoxysilane and Pt/Alumina solid catalyst were introduced into the batch reactor first, then heated to 110 C. Then 1-Octene and acetic acid were added through an addition syringe after the temperature reached 110 C., with control of the exotherm of the reaction. After 1-octene/acetic acid addition, the reactor was held at 110 C. for an additional 2 hrs. to complete the reaction.Results: Final Product Mixture Compostion:1-Octene3.188%Octene Isomers3.486%Triethoxysilane0.461%tetraethoxysiane2.234%Octyltriethoxysilane Product85.348%Heavies3.813%
With (iPrPDI)Fe(N2)2; for 0.25h;Inert atmosphere;
Example 2 Hydrosilylation of 1-octene with triethoxysilane using (iPrPDI)Fe(N2)2 In a nitrogen-filled drybox, a 20 mL scintillation vial was charged with 245 mg (2.18 mmol) of 1-octene and 360 mg (2.19 mmol) of triethoxysilane. The solution was stirred and 1 mg (0.002 mmol, 2*103 ppm catalyst loading) of (iPrPDI)Fe(N2)2 was added. After 15 minutes, the reaction was quenched by exposure to air. GC analysis of the product showed 99% conversion of 1-octene (retention time=1.33 min) to the hydrosilylated product (retention time=5.97 min).
> 98%Spectr.
With (terpy)Co(Ph2SiOC3H5); In neat (no solvent); at 20℃; for 1h;Schlenk technique; Glovebox; Inert atmosphere; Sealed tube;
A 50 mL Schlenk flask equipped with a stir bar was charged with 1-octene (112 mg, 1 mmol) and triethoxysilane (164 mg, 1 mmol) in the glove box. The flask was sealed with a glass stopper and transferred out of the box. (terpy)Co(Ph2SiOC3H5) (2 mg, 0.5 mol%) was charged into a vial, brought out of the box and exposed to air for 10 minutes. The solid catalyst was added into the Schlenk flask under an Ar counterflow. The flask was sealed with a glass stopper and stirred for 1 hour. The reaction was quenched by exposure to air. The reaction mixture was analyzed by GC and 1H NMR spectroscopy which established >98% yield of anti-Markovnikov hydrosilylation product and trace dehydrogenative silylation product.
With Wilkinson's catalyst;Heating;Catalytic behavior;
General procedure: All catalytic reaction was performed in a 10 mL flat-bottomed tube without protection from air. The alkenes (4.0 mmol) and a requisite amount of catalyst and ionic liquid were placed in a dried tube and the reaction mixture stirred for 5 min. Thereafter, the silane (4.4 mmol) was added, the resulting reaction mixture heated with stirring for a requisite time and then allowed to cool to room temperature. The product phase was separated by decantation and the conversion of the alkenes and selectivity determined by GC-MS analysis using an Agilent 26890N/59731 apparatus equipped with a DB-5 column (30 m × 2.5 mm × 0.25 mum).
90%Chromat.
With nickel(II) bis(2,2,6,6-tetramethylheptane-3,5-dionate); sodium triethylborohydride; In tetrahydrofuran; at 50℃; for 12h;
General procedure: Example 16: THF solution (5 mL) of Nickel complex compound (0.005mmol) shownin Table 2 was placed in vial, then for the nickel complex compound, 20-foldmolar amount 1-octene, hydrosilanes shown in Table 2, equimolar amount hydridereducing agent were added sequentially to initiate the reaction. After a lapseof time shown in Table 2, the reaction solution was exposed to air to terminatethe reaction (the color of the solution turned colorless.), the yield of thefollowing product was quantified by GC analysis. The results are shown in Table2.
With silicon based polyether N-heterocyclic carbene Pt metal complex; at 70℃; for 5h;Sealed tube;
The silicon-based polyether chain N-heterocyclic carbene Pt metal complex 1 in Example 1 was used as a catalyst of 0.001 mmol,5.0 mmol octene,5.5 mmol of triethoxysilane was placed in a 25 mL reaction tube with a magnetic stirrer,seal,70 under the conditions of reaction 5h,The octene conversion was measured to be 99.9%Beta-adductThe selectivity was 99.6%.
With C40H66N4O3Pt2Si6; at 70℃; for 5h;
Take0.001 mmol of the complex 2 prepared in Example 2,5.0 mmol octene,5.5 mmol of triethoxysilane was placed in the belt A magnetic stirrer in a 25 mL reaction tube,Sealed at 70 C for 5 h,The octene conversion was measured at 98.9%The addition of beta-adduct was 99.5%. The upper product was separated and another portion of 5.0 mmol of octene was added,5.5 mmol triethoxysilane,Under the same conditions for 5h,Octene conversion rate of 99.2%The addition of beta-adduct was 99.8%.
With rhodium(III) chloride trihydrate; alpha-Oxo-phenylmethan-diphenylphosphin; at 70℃; for 10h;Inert atmosphere;
General procedure: A 10mL three-necked flask equipped with a magnetic stirrer was charged with RhCl3·3H2O (8.0×10-3mmol) and acylphosphines prepared (4.0×10-2mmol) under argon atmosphere. Then alkene (4mmol) and silane (4.4mmol) were added via syringe. The hydrosilylation reaction proceeded with constant stirring under an appropriate temperature for 5h. At the end of the reaction, the conversion of alkene and the selectivity of product were determined by GC (Scheme 3) .
Example 6 Reaction between octene-1 and triethoxysilane with platinum catalyst in the presence of trimethylsilyl ester of methanesulfonic acid. 325 mg Octene-1 and 475 mg triethoxysilane were introduced into a glass tube filled with argon gas, and 20 mg of a trimethylsilyl ester of methanesulfonic acid and 0.002 ml (1.7 mg) of a toluene solution of a 0-valent platinum complex of divinyltetramethyldisiloxane (0.04 wt % platinum content) were added. The tube was sealed with Teflon tape and a rubber septum, degassed, and then filled again with argon gas. The tube was placed in a 75 C. oil bath where it was heated for 1 hour. After cooling, the tube contents were analyzed by gas chromatography revealing an octyltriethoxysilane yield of 88%.
88%
Example 6. Reaction between octene-1 and triethoxysilane with platinum catalyst in the presence of trimethylsilyl ester of methanesulfonic acid. 325 mg Octene-1 and 475 mg triethoxysilane were introduced into a glass tube filled with argon gas, and 20 mg of a trimethylsilyl ester of methanesulfonic acid and 0.002 ml (1.7 mg) of a toluene solution of a 0-valent platinum complex of divinyltetramethyldisiloxane (0.04 wt% platinum content) were added. The tube was sealed with Teflon tape and a rubber septum, degassed, and then filled again with argon gas. The tube was placed in a 75C oil bath where it was heated for 1 hour. After cooling, the tube contents were analyzed by gas chromatography revealing an octyltriethoxysilane yield of 88%.
44%
With cobalt pivalate; 1-adamantyl isocyanide; at 80℃; for 24h;Inert atmosphere;
General procedure: [Example 22] Hydrosilylation Reaction of 1-Octene (0224) A screw-top vial was charged with 5 mg (0.03 mmol) of cobalt acetate (commercial product) as a catalyst, 5 mg (0.06 mmol) of t-butyl isocyanide as a ligand, 254 muL (1.3 mmol) of 1,1,3,3,3-pentamethyldisiloxane, and 157 muL (1.0 mmol) of 1-octene. The vial was closed, after which the contents were stirred at 80 C. for 24 hours. After cooling, analysis was made by 1H-NMR spectroscopy to determine the structure and yield of the product. As a result, it was confirmed that the signal assigned to the ethylene site of 1-octene as the reactant disappeared completely. Instead, a triplet at 0.51 ppm indicative of the signal assigned to proton on silicon-adjoining carbon in the desired product, 1,1,1,3,3-pentamethyl-3-octyldisiloxane was observed, from which a yield was computed. The results are shown in Table 6. (0225) 1H-NMR (396 MHz, CDCl3) delta: 0.03 (a, 6H), 0.06 (s, 9H), 0.50 (t, J=7.7 Hz, 2H), 0.88 (t, J=6.8 Hz, 3H), 1.19-1.34 (br, 12H)
With particular preference, the alkoxysilanes set out below are prepared by the process of the invention: ... isobutyltriethoxysilane, amyltrimethoxysilane, amyltriethoxysilane, octyltrimethoxysilane, octyltriethoxysilane, cyclohexyltrimethoxysilane, cyclohexylmethyldimethoxysilane, hexadecyltrimethoxysilane, ...
With C28H44FFeN2PSi2; at 20℃; for 20h;Inert atmosphere;
General procedure: 3a under inert gas (N2)(50-0.5 muL stock solution, 1.1-0.11 mumol) was added to silane (R3SiH) (10.7 mmol; R3 = PhH2: 1.32 mL, PhMeH: 1.47 mL, Ph2H: 1.97 mL, Ph3: 2.79 g, Et3: 1.71 mL, (EtO) 3: 2.10 mL), olefin (CH2 = CHR ') (10.7 mmol; R' = C6H13: 1.68 mL, nBu: 1.34 mL, tBu: 1. 0.38 mL, Ph: 1.23 mL, p-F-Ph: 1.28 mL, p-MeO-Ph: 1.44 mL, p-Br-Ph: 1.96 mL) were added (Table 4). The reaction mixture was stirred at room temperature for 20 hours, and the product was confirmed by 1H NMR spectrum and GC-MS (eq5). Table 5 shows the 1 H NMR spectrum measurement results. The iron complex 3a showed good catalytic activity also for eq10 and the hydrosilylation reaction of the olefins and silanes shown in Table 4, showing that the applicable range is wide.
18
magnesium(II) nitrate hexahydrate[ No CAS ]
aluminium(III) acetylacetonate[ No CAS ]
[ 2943-75-1 ]
[ 1310-73-2 ]
Mg0.5Na0.5[(CH3(CH2)7Si)3.4Al1.5Mg1.2O8.3(OH)2][ No CAS ]
Preparation was carried out by the same method as in Production Example 1, except that 0.01 mole of 3-mercaptopropyltriethoxysilane (manufactured by Gelest Inc.) and 0.04 mole of <strong>[2943-75-1]n-octyltriethoxysilane</strong> (manufactured by Tokyo Kasei Kogyo Co., Ltd.) were used in place of TESPT and that tin dibutyldioleate was used in place of tin dioleate.
20
[ 75-77-4 ]
N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed collagen[ No CAS ]
[ 78-62-6 ]
[ 2943-75-1 ]
N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed collagen-dimethyldiethoxysilane-octyltriethoxysilane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Production Example 2Production of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed collagen-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition [1:25:25 (molar ratio)] Into the reaction vessel made of glass shaped in cylinder with round bottom of its volume of 2 liters, 150 g of 10% aqueous solution of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed collagen (wherein the number average molecular weight of hydrolyzed collagen is about 500) and 7.6 g of 18% aqueous solution of hydrochloric acid were added to adjust pH of the mixture to 1.5 and said mixture was heated up to 60 C . While the mixture being stirred in 400 rpm, the mixture of 79.7 g of dimethyldiethoxysilane (a trade name of KBE-22, manufactured by Shin-Etsu Silicone Co. , Ltd. ) and 148.6 g of octyltriethoxysilane (a trade name of A-137, Nippon Unicar Co., Ltd.) was dropped into said mixture for 5 hours, and then, after said dropping being completed, the solution obtained was further stirred for 15 hours at 60 C. Then, while the solution being stirred, 22.9 g of 5% aqueous solution of sodium hydroxide was gradually dropped into said solution to adjust the solution pH to 6, and the solution was further stirred for one hour at 60 C. While the reaction solution obtained being stirred in 400 rpm at 60 C, 9.3 g of trimethylchlorosilane (a trade name of KA-31, manufactured by Shin-Etsu Silicon Co., Ltd.) was added, and then said reaction solution was stirred for one hour at 60 C. Then, after 68.5 g of 5% aqueous solution of sodium hydroxide being dropped into the reaction solution and pH of said solution being adjusted to 6, the solution was stirred for one hour at 60 C, and then the solution was heated up to 80 C and further stirred for one hour. Then, the reaction solution was concentrated to the solid content of 70% by vacuum concentration with the rotary-evaporator. Thus, 211.9 g of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed collagen-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition was obtained. The viscosity of 70% aqueous solution at 20 C of obtained N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed collagen-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition was measured according to the same condition applied in Production Example 1, thus the viscosity resulted in 1,116 mPa · s.
N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed silk[ No CAS ]
[ 78-62-6 ]
[ 2943-75-1 ]
N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed silk-dimethyldiethoxysilane-octyltriethoxysilane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Production Example 4Production of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed silk-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition [1:25:25 (molar ratio)] Into the reaction vessel made of glass shaped in cylinder with round bottom of its volume of 2 liters, 407.5 g of 5% aqueous solution of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed silk (wherein the number average molecular weight of hydrolyzed silk is about 600) and 13.0 g of 18% aqueous solution of hydrochloric acid were added to adjust pH of the mixture to 1.2 and said mixture was heated up to 60 C. While the mixture being stirred in 400 rpm, the mixture of 88.0 g of dimethyldiethoxysilane (a trade name of KBE-22, manufactured by Shin-Etsu Silicon Co., Ltd.) and 164.0 g of octyltriethoxysilane (a trade name of A-137, Nippon Unicar Co., Ltd. ) was dropped into said mixture for 5 and half hours, and then, after said dropping being completed, the solution obtained was further stirred for 15 hours at 60 C. Then, while the solution being stirred, 36.7 g of 5% aqueous solution of sodium hydroxide was gradually dropped into said solution to adjust the solution pH to 6, and the solution was further stirred for one hour at 60 C. While the reaction solution obtained being stirred in 400 rpm at 60 C, 10.3 g of trimethylchlorosilane (a trade name of KA-31, manufactured by Shin-Etsu Silicon Co., Ltd.) was added, and then said reaction solution was stirred for one hour at 60 C. Then, after 72.4 g of 5% aqueous solution of sodium hydroxide being dropped into the reaction solution and pH of said solution being adjusted to 6, the solution was stirred for one hour at 60 C, and then the solution was heated up to 80 C and further stirred for one hour. Then, the reaction solution was concentrated to the solid content of 70% by vacuum concentration with the rotary-evaporator. Thus, 220 g of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed silk-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition was obtained. The viscosity of 70% aqueous solution at 20 C of obtained N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed silk-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition was measured by the same condition applied in Production Example 1, thus the viscosity resulted in 8,050 mPa · s.
N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed silk[ No CAS ]
[ 78-62-6 ]
[ 2943-75-1 ]
-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed silk-dimethyldiethoxysilane-octyltriethoxysilane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Production Example 3Production ofN-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed silk-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition [1:40:40 (molar ratio)] Into the reaction vessel made of glass shaped in cylinder with round bottom of its volume of 2 liters, 127.3 g of 10% aqueous solution of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed silk (wherein the number average molecular weight of hydrolyzed silk is about 600) and 4.8 g of 18% aqueous solution of hydrochloric acid were added to adjust pH of the mixture to 1.5 and said mixture was heated up to 60 C. While the mixture being stirred in 400 rpm, the mixture of 88.0 g of dimethyldiethoxysilane (a trade name of KBE-22, manufactured by Shin-Etsu Silicon Co., Ltd.) and 164.0 g of octyltriethoxysilane (a trade name of A-137, Nippon Unicar Co. , Ltd.) was dropped into said mixture for 5 and half hours, and then, after said dropping being completed, the solution obtained was further stirred for 15 hours at 60 C. Then, while the solution being stirred, 17.1 g of 5% aqueous solution of sodium hydroxide was gradually dropped into said solution to adjust the solution pH to 6, and the solution was further stirred for one hour at 60 C. While the reaction solution obtained being stirred in 400 rpm at 60 C, 6.4 g of trimethylchlorosilane (a trade name of KA-31, manufactured by Shin-Etsu Silicon Co., Ltd.) was added, and then said reaction solution was stirred for one hour at 60 C. Then, after 45.7 g of 5% aqueous solution of sodium hydroxide being dropped into the reaction solution and pH of said solution being adjusted to 6, the solution was stirred for one hour at 60 C, and then the solution was heated up to 80 C and further stirred for one hour. Then, the reaction solution was concentrated to the solid content of 70% by vacuum concentration with the rotary-evaporator. Thus, 209 g of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed silk-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition was obtained. The viscosity of 70% aqueous solution at 20 C of obtained N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed silk-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition was measured by the same condition applied in Production Example 1, thus the viscosity resulted in 640 mPa · s.
N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed wheat protein[ No CAS ]
[ 78-62-6 ]
[ 2943-75-1 ]
N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed wheat protein-dimethyldiethoxysilane-octyltriethoxysilane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Production Example 6Production of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed wheat protein-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition [1:15:15 (molar ratio)] Into the reaction vessel made of glass shaped in cylinder with round bottom of its volume of 2 liters, 345.3 g of 10% aqueous solution of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed wheat protein (wherein the number average molecular weight of hydrolyzed soybean protein is about 600) and 26.8 g of 18% aqueous solution of hydrochloric acid were added to adjust pH of the mixture to 1.5 and said mixture was heated up to 60 C. While the mixture being stirred in 400 rpm, the mixture of 81 g of dimethyldiethoxysilane (a trade name of KBE-22, manufactured by Shin-Etsu Silicon Co. , Ltd. ) and 151 g of octyltriethoxysilane (a trade name of A-137, Nippon Unicar Co., Ltd.) was dropped into said mixture for 5 hours, and then, after said dropping being completed, the solution obtained was further stirred for 15 hours at 60 C. Then, while the solution being stirred, 73.2 g of 5% aqueous solution of sodium hydroxide was gradually dropped into said solution to adjust the solution pH to 6, and the solution was further stirred for one hour at 60 C. While the reaction solution obtained being stirred in 400 rpm at 60 C, 11.9 g of trimethylchlorosilane (a trade name of KA-31, manufactured by Shin-Etsu Silicon Co., Ltd.) was added, and then said reaction solution was stirred for one hour at 60 C. Then, after 87.4 g of 5% aqueous solution of sodium hydroxide being dropped into the reaction solution and pH of said solution being adjusted to 6, the solution was stirred for one hour at 60 C, and then the solution was heated up to 80 C and further stirred for one hour. Then, the reaction solution was concentrated to the solid content of 70% by vacuum concentration with the rotary-evaporator. Thus, 233 g of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed wheat protein-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition was obtained. The viscosity of 70% aqueous solution at 20 C of obtained N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed wheat protein-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition was measured by the same condition applied in Production Example 1, thus the viscosity resulted in 14,340 mPa · s.
N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed soybean protein[ No CAS ]
[ 78-62-6 ]
[ 2943-75-1 ]
N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed soybean protein-dimethyldiethoxysilane-octyltriethoxysilane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Production Example 5Production of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed soybean protein-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition [1:25:25 (molar ratio)] Into the reaction vessel made of glass shaped in cylinder with round bottom of its volume of 2 liters, 196 g of 10% aqueous solution of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed soybean protein ( wherein the number average molecular weight of hydrolyzed soybean protein is about 600) and 17.7 g of 18% aqueous solution of hydrochloric acid were added to adjust pH of the mixture to 1.5 and said mixture was heated up to 60 C. While the mixture being stirred in 400 rpm, the mixture of 86.2 g of dimethyldiethoxysilane (a trade name of KBE-22, manufactured by Shin-Etsu Silicon Co., Ltd.) and 160.7 g of octyltriethoxysilane (a trade name of A-137, Nippon Unicar Co., Ltd.) was dropped into said mixture for 8 hours, and then, after said dropping being completed, the solution obtained was further stirred for 15 hours at 60 C. Then, while the solution being stirred, 54.3 g of 5% aqueous solution of sodium hydroxide was gradually dropped into said solution to adjust the solution pH to 6, and the solution was further stirred for one hour at 60 C. While the reaction solution obtained being stirred in 400 rpm at 60 C, 10.1 g of trimethylchlorosilane (a trade name of KA-31, manufactured by Shin-Etsu Silicon Co., Ltd.) was added, and then said reaction solution was stirred for one hour at 60 C. Then, after 73.7 g of 5% aqueous solution of sodium hydroxide being dropped into the reaction solution and pH of said solution being adjusted to 6, the solution was stirred for one hour at 60 C, and then the solution was heated up to 80 C and further stirred for one hour. Then, the reaction solution was concentrated to the solid content of 70% by vacuum concentration with the rotary-evaporator. Thus, 222.7 g of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed soybean protein-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition was obtained. The viscosity of 70% aqueous solution at 20C of obtained N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed soybean protein-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition was measured by the same condition applied in Production Example 1, thus the viscosity resulted in 3,560 mPa ? s.
N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed sericin[ No CAS ]
[ 78-62-6 ]
[ 2943-75-1 ]
N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed sericin-dimethyldiethoxysilane-octyltriethoxysilane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Production Example 1Production of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed sericin-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition [1:25:25 (molar ratio)] Into the reaction vessel made of glass shaped in cylinder with round bottom of its inner diameter of 12 cm and its volume of 2 liters, 200 g of 10% aqueous solution of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed sericin (wherein the number average molecular weight of said hydrolyzed sericin is about 500) and 11.5 g of 18% aqueous solution of hydrochloric acid were added to adjust pH of a mixture to 1.5 and said mixture was heated up to 60 C. While the mixture being stirred in 400 rpm, the mixture of 99.1 g of dimethyldiethoxysilane (a trade name of KBE-22, manufactured by Shin-Etsu Silicone Co. , Ltd. ) and 184.7 g of octyltriethoxysilane (a trade name of A-137, Nippon Unicar Co., Ltd. ) was dropped into said mixture for 5 hours, and then, after said dropping being completed, the solution obtained was further stirred for 15 hours at 60 C. Then, while the solution being stirred, 40.8 g of 5% aqueous solution of sodium hydroxide was gradually dropped into said solution to adjust the solution pH to 6, and the solution was further stirred for one hour at 60 C. While the reaction solution obtained being stirred in 400 rpm at 60 C, 11.6 g of trimethylchlorosilane (a trade name of KA-31, manufactured by Shin-Etsu Silicone Co., Ltd.) was added, and then said reaction solution was stirred for one hour at 60 C. Then, after 78.8 g of 5% aqueous solution of sodium hydroxide being dropped into the reaction solution and pH of said solution being adjusted to 6, the solution was stirred for one hour at 60 C, and then the solution was heated up to 80 C and further stirred for one hour. Then, the reaction solution was concentrated to the solid content of 70% by vacuum concentration with the rotary-evaporator. Thus, 260 g of N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed sericin-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition was obtained. The viscosity of 70% aqueous solution at 20 C of obtained N-[2-hydroxy-3-(3'-methyldihydroxysilyl)propoxy]propyl hydrolyzed sericin-dimethyldiethoxysilane-octyltriethoxysilane copolymer composition was measured by B-type viscometer with the rotor 3 and number of revolution 30, thus the viscosity resulted in 2120 mPa · s.
With bis(cyclopentadienyl)titanium dichloride; samarium; at 100℃; for 12h;Inert atmosphere;
General procedure: Cp2TiCl2 and metal powder were placed into a 10mLround-bottomed flask equipped with a magnetic stir bar andflushed with nitrogen, and then olefin (4.0 mmol) andhydrosilane (4.4 mmol) were charged into the flask, and theresulting mixture was stirred at desired temperature fordesired time. At the end of the reaction, the products weredetermined by GC/MS, and the conversion of alkene as wellas the selectivity of products was determined by GC.
With titanium(IV) tetraethanolate; at 90℃; for 12h;Inert atmosphere;
General procedure: The calculated amount of catalyst was charged into a 10mLround-bottomed flask equipped with magnetic stirring under nitrogen, and then olefin (4.0 mmol) and hydrosilane(4.4 mmol) were added into the flask. The resulting mixturewas stirred at the desired reaction temperature for thedesired time. At the end of the reaction, the reaction mixturewas analyzed by GC/MS, and the conversion of alkene aswell as the selectivity of products were determined by GC
600-900 g of each of finely powdered titanium dioxide, talc, mica and EPO <DP n="16"/>sericite was introduced into a fluidized reactor, and then the coating mixture 4 was introduced into a feed cylinder. Then, the coating mixture 4 was vaporized by heating it to about 120-2000C depending on the mixing ratio between the components of the coating mixture 4. Said temperature is a temperature higher than the boiling temperature of the coating mixture 4. The coating mixture 4 was passed through the heating device of a nozzle unit using air as a carrier gas, and then vaporized and introduced into the fluidized reactor. Subsequent steps were carried out in the same manner as in Example 1. About 12O g of each of the first- surface-modified powders prepared in Example 4 above was introduced into a main reactor and then subjected to vapor phase reaction with 1-tetradecene introduced into a feed cylinder.Herein, organic metal catalyst H2PtCl6 was used and the reaction temperature was maintained at about 6O0C
Yield
Reaction Conditions
Operation in experiment
600-900 g of each of finely powdered titanium dioxide, talc, mica and sericite was introduced into a fluidized reactor, and then the coating mixture 3 was introduced into a feed cylinder. Then, the coating mixture 3 was vaporized by heating it to about 120-2000C depending on the mixing ratio between the components of the coating mixture 3. Said temperature is a temperature higher than the boiling temperature of the coating mixture 3.The coating mixture 3 was passed through the heating device of a nozzle unit using air as a carrier gas, and then vaporized and introduced into the fluidized reactor. Subsequent steps were carried out in the same manner as inExample 1. About 12O g of each of the first-surface-modified powders prepared inExample 3 above was introduced into a main reactor and then subjected to vapor phase reaction with 1-tetradecene introduced into a feed cylinder. Herein, organic metal catalyst H2PtCl6 was used and the reaction temperature was maintained at about 6O0C
Surface modification using triethoxy carpryryl silane (TCS) 350 g of each of titanium dioxide, talc and mica was introduced into a main reactor, and then triethoxy carpryryl silane (TCS) was introduced into a feed cylinder and vaporized by heating it to a temperature of about 86-9O0C, which is higher than the boiling point of TCS. The vaporized TCS was introduced into the main reactor using inert nitrogen gas as carrier gas. Herein, the vaporized TCS was introduced into the main reactor at a rate of about 100-150 m/sec through a nozzle. The residence time of the vaporized EPO <DP n="18"/>TCS in the main reactor was 2-3 hours, and it was allowed to react with the pigment particles in the main reactor to form an S-H group on the surface of the particles. About 120 g of each of the first-surface-modified powders prepared inComparative Example 1 above was introduced into a main reactor and then subjected to vapor phase reaction with 1-tetradecene introduced into a feed cylinder. Herein, organic metal catalyst H2PtCl6 was used and the reaction temperature was maintained at about 6O0C
Yield
Reaction Conditions
Operation in experiment
600-900 g of each of finely powdered titanium dioxide, talc, mica and EPO <DP n="16"/>sericite was introduced into a fluidized reactor, and then the coating mixture 4 was introduced into a feed cylinder. Then, the coating mixture 4 was vaporized by heating it to about 120-2000C depending on the mixing ratio between the components of the coating mixture 4. Said temperature is a temperature higher than the boiling temperature of the coating mixture 4. The coating mixture 4 was passed through the heating device of a nozzle unit using air as a carrier gas, and then vaporized and introduced into the fluidized reactor. Subsequent steps were carried out in the same manner as in Example 1. Second surface modification (wet coating) of powder subjected to first surface modification using coating mixture 3 15 g of the first-surface-modified powder prepared in Example 4 was introduced into a three-neck round bottom, to which about 45 ml of ethanol was then added. The solution was uniformly stirred. Then, 0.002 ml of organic metal catalyst H2PtCl6 was added thereto. Then, 0.1-0.4 mol of 1- dodecene or vinyl-terminated polydimethylsiloxane as a second coating compound was added thereto, and then the second surface modification of the powder was performed by stirring the mixture for about 2-3 hours while maintaining the reaction temperature at 7O0C
Yield
Reaction Conditions
Operation in experiment
600-900 g of each of finely powdered titanium dioxide, talc, mica and sericite was introduced into a fluidized reactor, and then the coating mixture 3 was introduced into a feed cylinder. Then, the coating mixture 3 was vaporized by heating it to about 120-2000C depending on the mixing ratio between the components of the coating mixture 3. Said temperature is a temperature higher than the boiling temperature of the coating mixture 3.The coating mixture 3 was passed through the heating device of a nozzle unit using air as a carrier gas, and then vaporized and introduced into the fluidized reactor. Subsequent steps were carried out in the same manner as inExample 1. 15 g of the first-surface-modified powder prepared in Example 3 was EPO <DP n="17"/>introduced into a three-neck round bottom, to which about 45 ml of ethanol was then added. The solution was uniformly stirred. Then, 0.002 ml of organic metal catalyst H2PtCl6 was added thereto. Then, 0.1-0.4 mol of 1- dodecene or vinyl-terminated polydimethylsiloxane as a second coating compound was added thereto, and then the second surface modification of the powder was performed by stirring the mixture for about 2-3 hours while maintaining the reaction temperature at 700C
Surface modification using triethoxy carpryryl silane (TCS) 350 g of each of titanium dioxide, talc and mica was introduced into a main reactor, and then triethoxy carpryryl silane (TCS) was introduced into a feed cylinder and vaporized by heating it to a temperature of about 86-9O0C, which is higher than the boiling point of TCS. The vaporized TCS was introduced into the main reactor using inert nitrogen gas as carrier gas. Herein, the vaporized TCS was introduced into the main reactor at a rate of about 100-150 m/sec through a nozzle. The residence time of the vaporized EPO <DP n="18"/>TCS in the main reactor was 2-3 hours, and it was allowed to react with the pigment particles in the main reactor to form an S-H group on the surface of the particles. 15 g of the powder, which has been surface-modified with TCS inComparative Example 1 , was introduced into a three-neck round bottom, to which about 45 ml of ethanol was then added. The solution was uniformly stirred. Then, 0.002 ml of organic metal catalyst H2PtCl6 was added thereto.Then, 0.1-0.4 mol of 1-dodecene or vinyl-terminated polydimethylsiloxane as a second coating compound was added thereto, and then the second surface modification of the powder was performed by stirring the mixture for about 2- 3 hours while maintaining the reaction temperature at 7O0C
Yield
Reaction Conditions
Operation in experiment
600-900 g of each of finely powdered titanium dioxide, talc, mica and EPO <DP n="16"/>sericite was introduced into a fluidized reactor, and then the coating mixture 4 was introduced into a feed cylinder. Then, the coating mixture 4 was vaporized by heating it to about 120-2000C depending on the mixing ratio between the components of the coating mixture 4. Said temperature is a temperature higher than the boiling temperature of the coating mixture 4. The coating mixture 4 was passed through the heating device of a nozzle unit using air as a carrier gas, and then vaporized and introduced into the fluidized reactor. Subsequent steps were carried out in the same manner as in Example 1. Second surface modification (wet coating) of powder subjected to first surface modification using coating mixture 3 15 g of the first-surface-modified powder prepared in Example 4 was introduced into a three-neck round bottom, to which about 45 ml of ethanol was then added. The solution was uniformly stirred. Then, 0.002 ml of organic metal catalyst H2PtCl6 was added thereto. Then, 0.1-0.4 mol of 1- dodecene or vinyl-terminated polydimethylsiloxane as a second coating compound was added thereto, and then the second surface modification of the powder was performed by stirring the mixture for about 2-3 hours while maintaining the reaction temperature at 7O0C
Yield
Reaction Conditions
Operation in experiment
600-900 g of each of finely powdered titanium dioxide, talc, mica and sericite was introduced into a fluidized reactor, and then the coating mixture 3 was introduced into a feed cylinder. Then, the coating mixture 3 was vaporized by heating it to about 120-2000C depending on the mixing ratio between the components of the coating mixture 3. Said temperature is a temperature higher than the boiling temperature of the coating mixture 3.The coating mixture 3 was passed through the heating device of a nozzle unit using air as a carrier gas, and then vaporized and introduced into the fluidized reactor. Subsequent steps were carried out in the same manner as inExample 1. 15 g of the first-surface-modified powder prepared in Example 3 was EPO <DP n="17"/>introduced into a three-neck round bottom, to which about 45 ml of ethanol was then added. The solution was uniformly stirred. Then, 0.002 ml of organic metal catalyst H2PtCl6 was added thereto. Then, 0.1-0.4 mol of 1- dodecene or vinyl-terminated polydimethylsiloxane as a second coating compound was added thereto, and then the second surface modification of the powder was performed by stirring the mixture for about 2-3 hours while maintaining the reaction temperature at 700C
Surface modification using triethoxy carpryryl silane (TCS) 350 g of each of titanium dioxide, talc and mica was introduced into a main reactor, and then triethoxy carpryryl silane (TCS) was introduced into a feed cylinder and vaporized by heating it to a temperature of about 86-9O0C, which is higher than the boiling point of TCS. The vaporized TCS was introduced into the main reactor using inert nitrogen gas as carrier gas. Herein, the vaporized TCS was introduced into the main reactor at a rate of about 100-150 m/sec through a nozzle. The residence time of the vaporized EPO <DP n="18"/>TCS in the main reactor was 2-3 hours, and it was allowed to react with the pigment particles in the main reactor to form an S-H group on the surface of the particles. 15 g of the powder, which has been surface-modified with TCS inComparative Example 1 , was introduced into a three-neck round bottom, to which about 45 ml of ethanol was then added. The solution was uniformly stirred. Then, 0.002 ml of organic metal catalyst H2PtCl6 was added thereto.Then, 0.1-0.4 mol of 1-dodecene or vinyl-terminated polydimethylsiloxane as a second coating compound was added thereto, and then the second surface modification of the powder was performed by stirring the mixture for about 2- 3 hours while maintaining the reaction temperature at 7O0C
34
[ 107-46-0 ]
[ 2370-88-9 ]
[ 556-67-2 ]
[ 2943-75-1 ]
poly(octamethylcyclotetrasiloxane-co-tetramethylcyclotetrasiloxane-co-hexamethyldisiloxane-co-octyltriethoxysilane)[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sulfuric acid; water; at 20℃; for 10h;
In a reactor, 32.4 parts by weight of hexamethyldisiloxane, 55.2 parts by weight of octyltriethoxysilane, 222 parts by weight of octamethylcyclotetrasiloxane and 60 parts by weight of tetramethylcyclotetrasiloxane were placed, to which 19.3 parts by weight of concentrated sulfuric acid and 5.4 parts by weight of water was added. A mixture obtained was stirred at room temperature for 10 hours. A reaction mixture obtained was washed with water and then unreacted silane and siloxane were distilled off. An organohydrogenpolysiloxane of the following average compositional formula (6) was obtained. [(CH3)3SiO1/2]3[(CH3)2SiO]30[(CH3)HSiO]10[C8H17SiO3/2]2 (6) This reaction product was colorless and transparent liquid having a viscosity of 42 mm2/s at 25 C.
poly(octamethylcyclotetrasiloxane-co-tetramethylcyclotetrasiloxane-co-hexamethyldisiloxane-co-octyltriethoxysilane)[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sulfuric acid; water; at 20℃; for 10h;
In a reactor, 48.6 parts by weight of hexamethyldisiloxane, 55.2 parts by weight of octyltriethoxysilane, 60 parts by weight of tetramethylcyclotetrasiloxane and 222 parts by weight of octamethylcyclotetrasiloxane were placed, to which 19 parts by weight of concentrated sulfuric acid and 5.4 parts by weight of water was added. A mixture obtained was stirred at room temperature for 10 hours. A reaction mixture obtained was washed with water and then unreacted silane and siloxane were distilled off. An organohydrogenpolysiloxane of the following average compositional formula (9) was obtained. [(CH3)3SiO1/2]6[(CH3)2SiO]30[(CH3)HSiO]10[C8H17SiO]30[SiO2] (9) This reaction product was colorless and transparent liquid having a viscosity of 58mm2/s at 25 C.
poly(tetramethylcyclotetrasiloxane-co-hexamethyldisiloxane-co-octyltriethoxysilane-co-tris(trifluoropropyl)trimethylcyclotrisiloxane)[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sulfuric acid; water; at 20℃; for 10h;
In a reactor, 32.4 parts by weight of hexamethyldisiloxane, 55.2 parts by weight of octyltriethoxysilane, 180 parts by weight of tetramethylcyclotetrasiloxane and 37.8 parts by weight of tris(trifluoropropyl)trimethylcyclotrisiloxane were placed, to which 15 parts by weight of concentrated sulfuric acid and 5.4 parts by weight of water was added. A mixture obtained was stirred at room temperature for 10 hours. A reaction mixture obtained was washed with water and then unreacted silane and siloxane were distilled off. An organohydrogenpolysiloxane of the following average compositional formula (7) was obtained. [(CH3)3SiO1/2]3[(CH3)(CF3C2H4)SiO]3[(CH3)HSiO]30[C8H17SiO3/2]2 (7) This reaction product was colorless and transparent liquid having a viscosity of 34 mm2/s at 25 C.
With sodium methylate; In methanol; at 110 - 119℃; under 60.006 Torr; for 5.5h;Inert atmosphere;
In a 300 mL four-neck flask, under nitrogen flow, 83.01 g of octyltriethoxysilane (0.30 mol), 127.76 g of geraniol (0.83 mol), and 0.857 mL of 2.8% sodium methoxide in methanol were stirred for 2.5 hours at 110 to 115 C. with distilling off ethanol. After 2.5 hours, an inner pressure of the flask was gradually reduced to 8 kPa, and the mixture was stirred for additional three hours at 110 to 119 C. with distilling off ethanol. After three hours, the mixture was cooled and the reduced pressure was released. Then, the mixture was filtered to give 173.61 g of yellow oil containing octylsilicic acid trigeranyl ester. The resultant oil was analyzed by GC. The result is shown in Table 4. From the result, contents of octylsilicic acid trigeranyl ester of the compound (1) and octylsilicic acid digeranyl ester of the compound (2) were 72.2% and 23.0%, respectively.
octylsilicic acid bis(2-phenylethyl) ethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium methylate; In methanol; at 115 - 121℃; under 60.006 Torr; for 5h;Inert atmosphere;
In a 200 mL four-neck flask, under nitrogen flow, 55.30 g of octyltriethoxysilane (0.20 ml), 65.99 g of 2-phenylethanol (0.54 mol), and 0.958 mL of 2.8% sodium methoxide in methanol were stirred for two hours at 115 C. with distilling off ethanol. After two hours, an inner pressure of the flask was gradually reduced to 8 kPa, and the mixture was stirred for additional three hours at 115 to 121 C. with distilling off ethanol. After three hours, the mixture was cooled and the reduced pressure was released. Then, the mixture was filtered to give 94.84 g of yellow oil containing octylsilicic acid tris (2-phenylethyl)ester. The resultant oil was analyzed by gas chromatography (hereinafter, referred to as GC). The result is shown in Table 1. From the result, contents of octylsilicic acid tris(2-phenylethyl)ester of the compound (1) and octylsilicic acid bis(2-phenylethyl)ethyl ester of the compound (2) were 68.8% and 24.7%, respectively.
0045] Diethyl H-phosphonate: (Table 2, entry 1). Aqueous H3PO2 (50 wt.-%, 25 mmol) was concentrated in vacuo for 30 min at r.t. Octyltriethoxysilane (1 equiv, 25 mmol) and toluene (reagent grade, 50 mL) were added, and the reaction was heated to reflux for 3 h. After completion, the solution was cooled to r.t. and Ni/SiO2 (64 wt.-%, 5 mol-%) and anhydrous ethanol (3 equiv, 75 mmol) were added. The reaction was heated to reflux and allowed to react under nitrogen for 40 h. After cooling down, the reaction was filtered through Celite and was rinsed with ethyl acetate (20 mL). The organic layer was concentrated and subsequently dissolved in CH3CN(HPLC grade, 50 mL). The CH3CN layer was partitioned with hexanes (4×15 mL), and concentrated in vacuo to obtain a discolored liquid in 2.59 g (75% isolated yield). 1H NMR (400 MHz, CDCl3) delta 6.82 (d, 1JHP=692.8 Hz, 1H, H-P); 4.16 (p, 3J=7.20 Hz, 4H, CH2); 1.37 (t, 6.80 Hz, 6H, CH3) 31P NMR (161.97 MHz, CDCl3) delta 7.77 (dm, 1JPH=691.61 Hz).
With (terpy)Co(Me2SiOC3H5); In neat (no solvent); at 20℃; for 1h;Glovebox; Inert atmosphere;
In a glove box, 1-octene (112 mg, 1 mmol) and triethoxysilane (164 mg, 1 mmol) were weighed into a vial equipped with a stir bar. Purple (terpy)Co(Me2SiOC3H5) (2 mg, 0.5 mol%) was weighed into a separate vial, and was subsequently combined with the substrates. The contents of the vial were stirred at room temperature for 1 hour. The reaction was quenched by exposure to air. The product mixture was processed as described in the general procedure above. GC and 1H NMR spectroscopy which showed 90% yield of anti-Markovnikov hydrosilylation product and 9% dehydrogenative silylation product.
With (MeAPDI)Co(OAc)2; at 80℃; for 12h;Inert atmosphere; Sealed tube;
To a scintillation vial equipped with a stir bar in a nitrogen filled glovebox was added (MeAPDI)Co(OAc)2(10 mg, 0.027 mmol) followed by 1-octene (75 mg, 0.67 mmol) to give a heterogeneous mixture. Triethylsilane (78 mg, 0.67 mmol) was then added and the vial was sealed with a cap, removed from the box and heated to 80oC in an oil bath with stirring for 12 hours. After cooling to room temperature, the cap was removed and any remaining volatiles were removed with a stream of air. The resulting residue was diluted with a solution of 5% ethyl ether in pentane and passed through a small column of silica gel, eluting with additional 5% ether in pentane solution (5 mL). The resulting clear, colorless eluent was concentrated to give the product as a clear, colorless oil (55 mg, 36%). The product distribution was determined by analysis of the1H NMR spectrum of the isolated product.
1-octyl-3,7,10-trimethyl-2,8,9-trioxa-5-aza-1-silabicyclo[3.3.3]undecane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
84.5%
With potassium hydroxide; at 90℃; under 100 - 250 Torr; for 4h;
Trisoprpanolamine (250.0 g), octyltriethoxysilane (368.6 g, from Aldrich) and potassium hydroxide (3.7 g) were mixed in a 1-liter 3-neck round bottle flask equipped with distillation apparatus. The mixture was then heated to 90 C. by heating mantle, and ethanol produced from the reaction was removed under reduced pressure of 250 mmHg. After 2 hour of reaction the pressure was reduced to 150 mmHg for 1 hour, then 100 mmHg for another hour. Total of 228 mL of ethanol was recovered from the distillation. The oily crude product was then distilled out under the pressure of 3 mmHg and the temperature of 170 C. Total of 364 g (84.5% yield) of clear oily liquid, 1-octyl-3,7,10-trimethylsilatrane (OSTI), was obtained. 1HNMR and 13CNMR spectroscopy analysis has shown larger than 95% purity of the desired product.
With methanesulfonic acid; In tetrahydrofuran; at 20℃; for 24h;
4.65 g (5 mmol) of Trisi-lanolphenyl POSS were dissolved in dry THF (40 ml) followed by theaddition of n-Octyltriethoxysilane (1.38 g, 5 mmol) and 10 drops ofmethanesulfonic acid under stirring. The solution was maintainedat room temperature for 24 h and then heated at 50-60C for 4days. The reactant mixture was cooled and solvent was reducedunder vacuum; methanol was added and the white solid obtainedwas crystallized from THF/MeOH (3.60 g, 59.7% yield). Yields andelemental analysis data of the obtained compounds are reported inTable 4.
With tetramethyl ammoniumhydroxide; In methanol; ethanol; at 0 - 5℃; for 24h;
4.37 g (5 mmol) of trisilanol-cyclopentyl POSS were dissolved in 100 mL of anhydrous ethanol,and 5.5 mL (1.37 g, 15 mmol of a 25% methanol solution) of tetram-ethylammonium hydroxide were added under stirring. The clearsolution was cooled at 0-5C in an ice bath, and 1.38 g (5 mmol)of n-Octyltriethoxysilane were added. The solution was stirredand temperature was maintained at 0-5C for 24 h. The resultingsolid was filtered and washed with two portions (10 mL) of anhy-drous ethanol. After drying under reduced pressure, the white solidobtained was crystallized from THF/MeCN mixture to give 4.18 g ofdesired compound (72.8% yield).
With tetramethyl ammoniumhydroxide; In methanol; ethanol; at 0 - 5℃; for 24h;
General procedure: 4.37 g (5 mmol) of trisilanol-cyclopentyl POSS were dissolved in 100 mL of anhydrous ethanol,and 5.5 mL (1.37 g, 15 mmol of a 25% methanol solution) of tetram-ethylammonium hydroxide were added under stirring. The clearsolution was cooled at 0-5C in an ice bath, and 1.38 g (5 mmol)of n-Octyltriethoxysilane were added. The solution was stirredand temperature was maintained at 0-5C for 24 h. The resultingsolid was filtered and washed with two portions (10 mL) of anhy-drous ethanol. After drying under reduced pressure, the white solidobtained was crystallized from THF/MeCN mixture to give 4.18 g ofdesired compound (72.8% yield).
20 g (0.072 mol) octyltriethoxysilane and 7.388 g (0.036 mol) of bis-(hydroxymethyl)-o-carborane were charged with stirring into a round-bottomed flask under argon flow. As the temperature rises up to 110 C (30 min) the bis-(hydroxymethyl)-o-carborane was dissolved and distillation of ethanol began. After 3 h, when the alcohol removal stopped, the reaction mass was cooled with stirring to room temperature and 22.6 g (92%) of a liquid product of a yellow color was obtained. 1H NMR (CDCl3, ppm): 0.55-0.68 (4H, m, SiCH2(CH2)5CH3), 0.82-0.84 (6H, m, Si(CH2)7CH3), 1.15-1.18 (12H, m, OCH2CH3), 1.23 (8H, m, Si(CH2)6CH2CH3 Si(CH2)4CH2CH2CH3), 1.31 (12H, m, Si(CH2)3CH2(CH2)3CH3, Si(CH2)4CH2(CH2)2CH3 Si(CH2)5CH2CH2CH3), 1.35 (8H, m, Si(CH2)6CH2CH3 SiCH2CH2(CH2)5CH3), 3.73-3.82 (8H, m, SiOCH2CH3), 4.08-4.57 (4H, m, OCH2CB10H10CCH2).
A typical procedure (Table 2, entry 1) is as follows. To a stirredsolution of Ni(acac)2 (1a) (1.3 mg, 0.005 mmol) in THF (5 mL) wasadded 1-octene (112 mg, 1.0 mmol) and (EtO)3SiH (164 mg,1.0 mmol) at room temperature. After the mixture was stirred for1 min, NaBHEt3 (1.0Min THF, 5 mL, 0.005 mmol) was added and theresulting mixture was heated at 50 C. The solution was stirred atthe same temperature, and the progress of the reaction wasmonitored by GLC. After completion of the reaction, mesitylene(60 mg, 0.50 mmol) was added as an internal standard to the reactionmixture. The GLC analysis of the resulting solution revealedthe formation of (EtO)3(nOct)Si (0.90 mmol, 90%) and (EtO)4Si(0.05 mmol, 5%). The solutionwas concentrated under vacuum, andthe residue was purified by gel permeation chromatography (GPC)using toluene as an eluent to give (EtO)3(nOct)Si (234 mg,0.85 mmol, 85%). The 1H, 13C{1H} and 29Si{1H} NMR spectra of theisolated compound are consistent with the reported data. A similarprocedurewas employed for the hydrosilylation using other silanesand 1,3-diene/alkenes/alkynes. These reactions were carried out atroom temperature except for the reactions, Table 2, entries 2-3.The 1H/13C NMR spectroscopic data for the new compounds aregiven in the supplementary data.
To an NMR tube equipped with a Teflon valve were addedNi(acac)2 (13.0 mg, 0.05 mmol), 1-octene (11.3 mg, 0.1 mmol),(EtO)3SiH (16.0 mg, 0.1 mmol), and THF-d8 (0.5 mL). After theaddition of NaBHEt3 (1.0 M in THF, 50 mL, 0.05 mmol) to this solutionat room temperature, the reaction was followed by 1H NMR.After 2 h, the reaction did not proceed and only the signalsassignable to the starting materials [1-octene and (EtO)3SiH] weredetected by 1H NMR. The reaction was further followed at 50 C for14 h, and formation of (EtO)3(nOct)Si (54%) as well as (EtO)4Si (8%)and (EtO)3SiOSi(OEt)3 (7%) was observed. During the reaction, nohydride signals were detected in the high field region (delta 0 to -30 ppm).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping