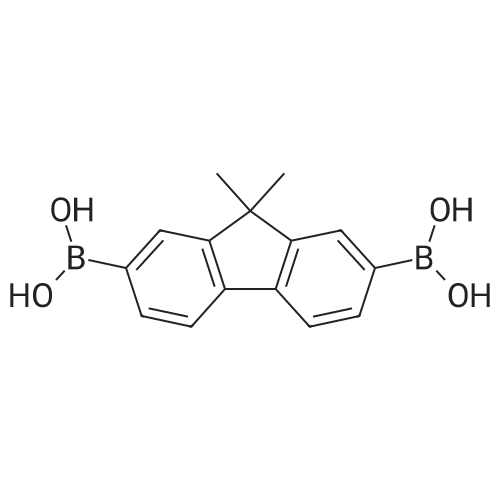
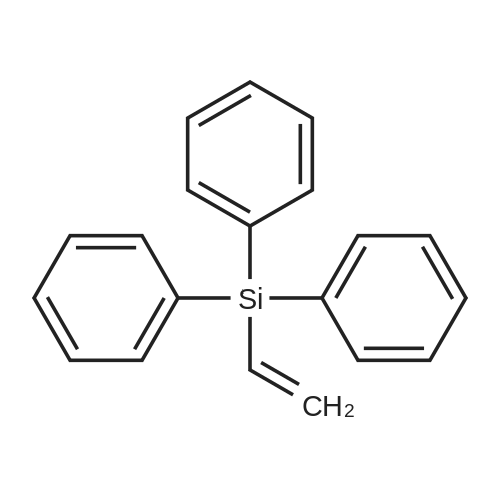
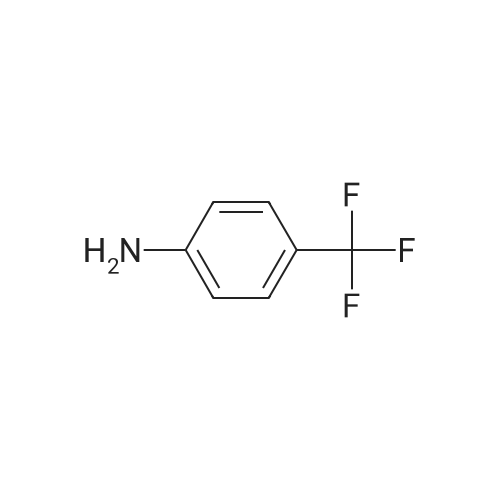
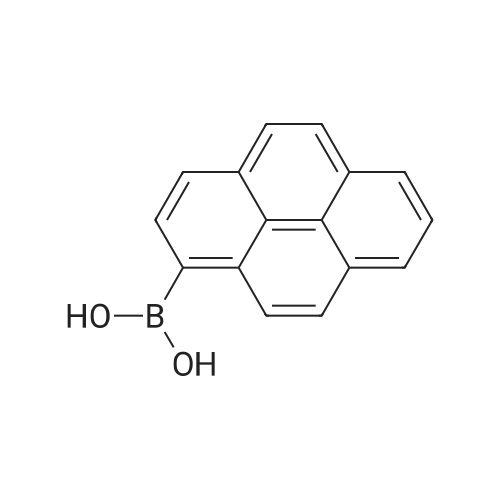
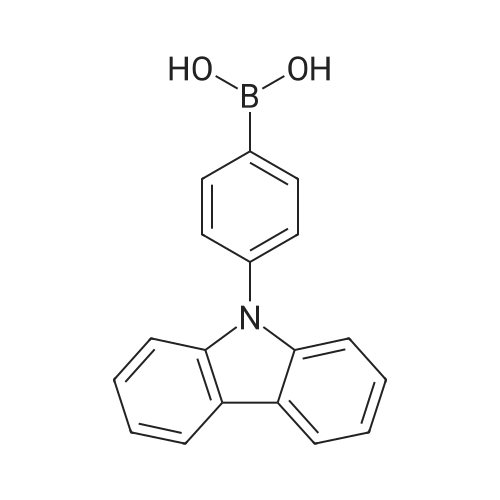
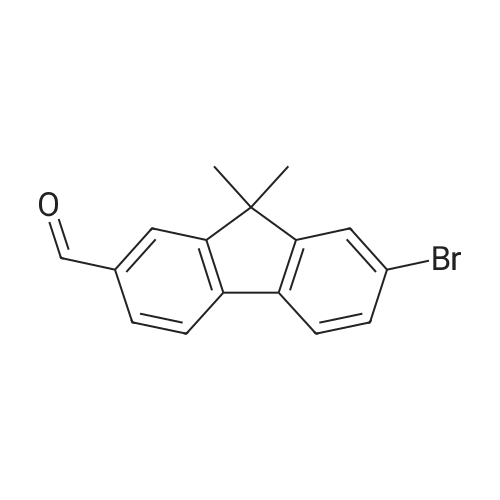
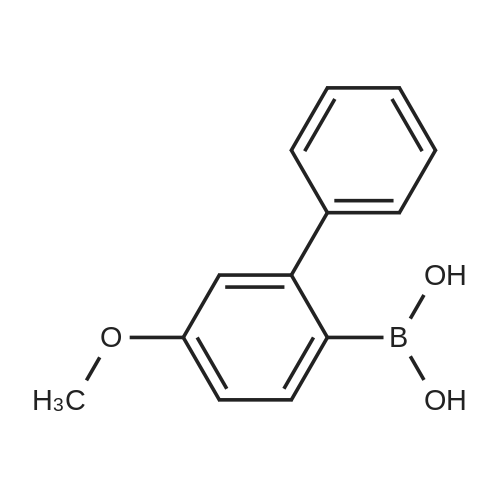
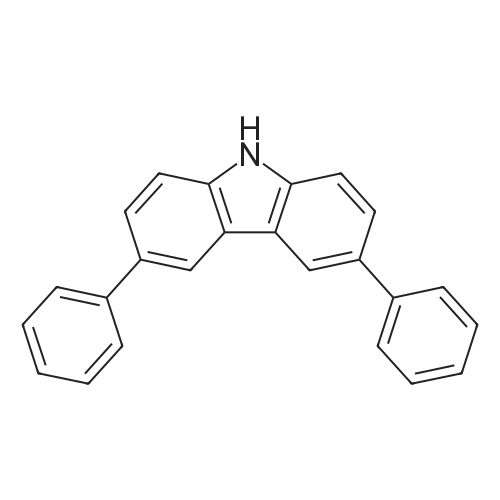
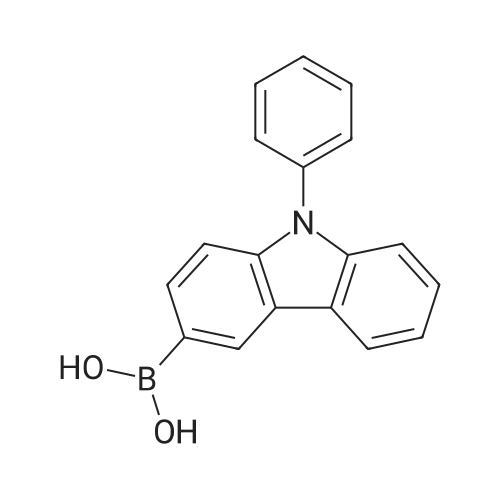
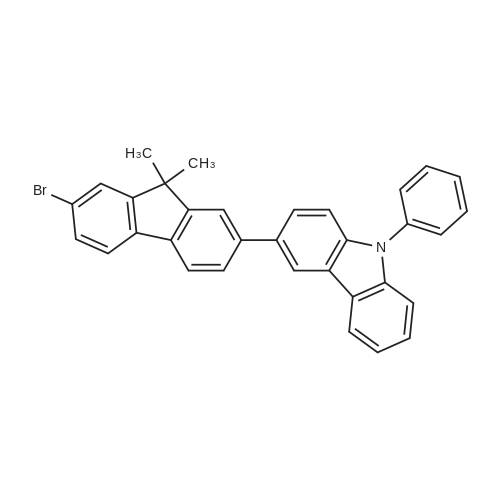
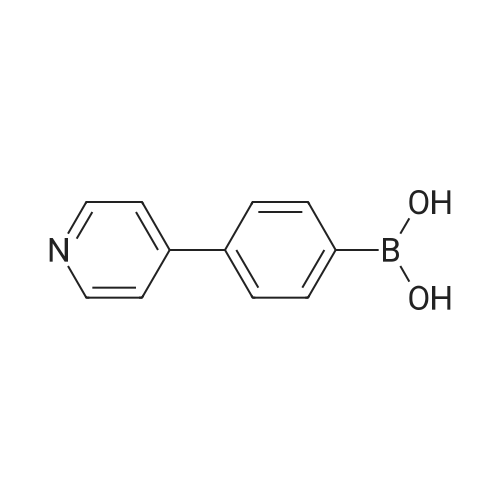
| 99% |
With tetrabutylammomium bromide; sodium hydroxide In dimethyl sulfoxide for 5h; Irradiation; |
1.1 Step1:
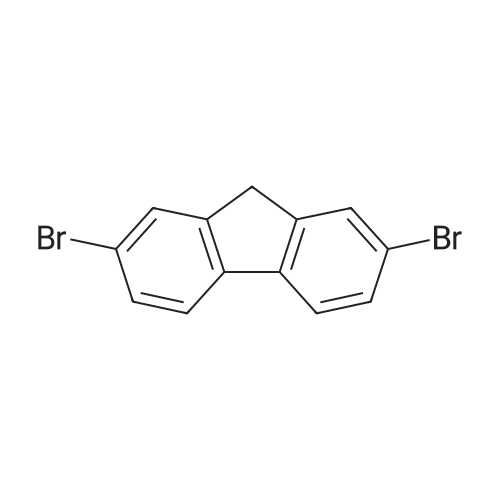
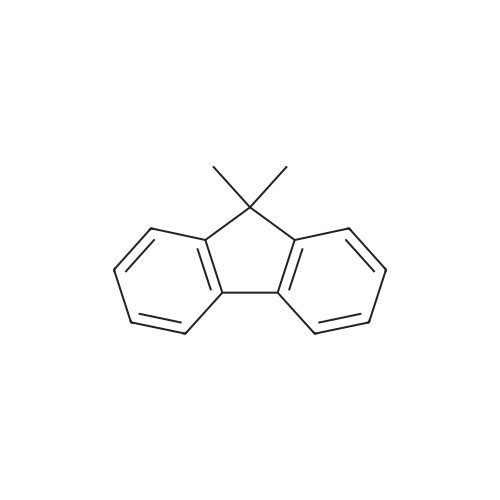
At room temperature, the reactor was added 2,7-dibromofluorene (9.72g, 30mmol),Tetra-n-butylammonium bromide (0.08g, 0.25mmol),Methyl iodide (12.77g, 90mmol),50% NaOH solution 10mL, DMSO 120mL,Ultrasonic reaction for 5 h, then add 500 mL of water to stop the reaction.Stirring, suction filtration, water washing and drying gave Intermediate 2-1 (10.46 g, 99%). |
| 98% |
With potassium <i>tert</i>-butylate In dimethyl sulfoxide at 5℃; |
2.1
In a stream of argon, 32 g (0.1 mol) of 2,7-dibromofluorene, 27 g (0.24 mol) of t-butoxypotassium, and 500 mL of DMSO were added to a 3-L three-necked flask, and the reaction system was cooled to 5°C. Subsequently, 34 g (0.24 mol) of methyl iodide were slowly dropped to the resultant, and then the whole was stirred overnight. After the completion of the reaction, water was added to the resultant, and an organic layer was extracted with ethyl acetate and washed with a saturated salt solution. After the washed product had been dried with magnesium sulfate, the solvent was removed by distillation with a rotary evaporator, whereby a coarse reaction product was obtained. The product waspurified by meansof column chromatography (silica gel (hexane solvent) : ethyl acetate = 95 : 5), whereby 34 g of Intermediate 6 (white crystal, 98% yield) as a target were obtained. |
| 98% |
With potassium <i>tert</i>-butylate In dimethyl sulfoxide at 5℃; Inert atmosphere; |
4-1
Synthesis Example 4; (4-1) Synthesis of Intermediate 7 Into a 3 liter three-necked flask, 32 g (0.1 mole) of 2,7-dibromofluorene, 27 g (0.24 moles) of t-butoxypotassium and 500 ml of DMSO were placed under the stream of argon, and the reaction system was cooled at 5° C. After 34 g (0.24 moles) of methyl iodide was slowly added dropwise, the resultant mixture was stirred for one night. After the reaction was completed, water was added, and the organic layer was separated by extraction with ethyl acetate, washed with a saturated aqueous solution of sodium chloride and dried with magnesium sulfate. The solvent was removed by distillation using a rotary evaporator, and a crude reaction product was obtained. The crude reaction product was purified in accordance with the column chromatography (silica gel; hexane:ethyl acetate=95:5), and 34 g of Intermediate 7 of the object compound was obtained (white crystals; the yield: 98%). |
| 97% |
With potassium iodide In dimethyl sulfoxide at 20℃; |
|
| 97% |
With potassium iodide; potassium hydroxide In dimethyl sulfoxide at 20℃; for 24h; |
|
| 97% |
With potassium iodide; potassium hydroxide In dimethyl sulfoxide at 20℃; for 24.5h; |
|
| 97.4% |
With tetrabutylammomium bromide; sodium hydroxide In dimethyl sulfoxide for 5h; Sonication; |
1.1 1. Preparation of ligand L:
At room temperature, 2,7-dibromofluorene(10.40 g, 32 mmol), tetrabutylammonium bromide (0.10 g, 0.3 mmol),10 mL of iodomethane (9.93 g, 90 mmol), 50% NaOH solution was added to 140 mL of DMSO,Ultrasonic reaction (100W) for 5 hours. After the reaction, pour into 500mL of water, stir for 20min and let stand for 1 hour.Suction filtration, the filter cake was washed with 1% NaCl aqueous solution, dried,This gave 10.97 g of a yellow solid with a yield of 97.40%. |
| 94% |
Stage #1: 2,7-dibromo-9H-fluorene In water; dimethyl sulfoxide
Stage #2: methyl iodide In water; dimethyl sulfoxide for 0.5h; |
4.1
2, 7-dibromofluorene (2.0 g, 6.2mmol), dimethyl sulfoxide (0.2 ml), benzyltriethylammonium chloride (0. 07 g, 0.3 mmol), and a 50 wt% aqueous solution of NaOH (2 g) were added to a flask. Next, iodomethane (2.2 g, 15 mmol) was dropped to the mixture, and the whole was stirred for 30 minutes. Water (100 mL) and toluene (100 mL) were added to the reaction liquid in such a manner that an organic layer would be separated. The organic layer was washed with a saturated brine, and was then dried with anhydrous magnesium sulfate. The resultant was concentrated under reduced pressure by using a rotary evaporator, and then the residue was purified by means of silica gel column chromatography, whereby Intermediate 4-1 was obtained (amount 2.0 g, yield 94 %). |
| 93.5% |
Stage #1: 2,7-dibromo-9H-fluorene With sodium t-butanolate In tetrahydrofuran at 0 - 20℃; Inert atmosphere;
Stage #2: methyl iodide In tetrahydrofuran for 2h; |
2 General procedure for the synthesis of the compounds (8,9)
These compounds were obtained following an essentially similar procedure. An illustrative example is provided for 8: 2-bromofluorene (4.90 g, 0.02 mol) was dissolved in anhydrous THF (100 mL) in a three-necked round-bottom flask fitted with a magnetic stirrer, a condenser and a N2 purge. t-BuONa (in THF, 8.64 g, 0.09 mol) was added dropwise at 0 °C and under N2 gas. The reaction mixture was stirred for 1.5 h at room temperature. Iodomethane (3.12 mL, 0.05 mol) was added dropwise and then the reaction was continued for another 2 h the reaction mixture was filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica gel chromatography with petroleum ether as the eluent to afford a white liquid (8) (5.21 g, 95.1%). |
| 92% |
Stage #1: 2,7-dibromo-9H-fluorene With potassium iodide; potassium hydroxide In dimethyl sulfoxide for 1h; Inert atmosphere; Schlenk technique; Cooling;
Stage #2: methyl iodide In dimethyl sulfoxide at 25℃; for 18h; Inert atmosphere; Schlenk technique; |
|
| 92% |
Stage #1: 2,7-dibromo-9H-fluorene With potassium iodide; potassium hydroxide In dimethyl sulfoxide for 1h; Inert atmosphere; Schlenk technique;
Stage #2: methyl iodide at 25℃; for 18h; Inert atmosphere; Schlenk technique; |
|
| 92% |
Stage #1: 2,7-dibromo-9H-fluorene With potassium iodide; potassium hydroxide In dimethyl sulfoxide for 1h; Inert atmosphere; Schlenk technique;
Stage #2: methyl iodide In dimethyl sulfoxide at 25℃; for 18h; Inert atmosphere; Schlenk technique; |
|
| 92% |
With potassium iodide; potassium hydroxide In dimethyl sulfoxide at 25℃; for 18h; Inert atmosphere; Schlenk technique; |
2.4.2 2,7-Dibromo-9,9-dimethyl-9H-fluorene (15)
A modified literature procedure was used.[24] 14 (16.2 g, 50.0 mmol, 1.00 eq) was suspended in DMSO(83 mL, 0.6 M) and KI (830 mg, 5.00 mmol, 0.10 eq) was added. To the water bath-cooled andvigorously stirred mixture were added KOH pellets (11.2 g, 200 mmol, 4.00 eq). The reaction was stirredfor one hour, while the solution turned intensive red. MeI (7.78 mL, 125 mmol, 2.50 eq) was added viasyringe pump (0.15 mL/min) through a rubber septum and stirring was continued at 25 °C for 18 hours.Excess of MeI was quenched by addition of NEt3 (13.9 mL, 100 mmol, 2.00 eq). The mixture was stirredfor 30 minutes, poured into water (500 mL) and extracted with CH2Cl2 (4 100 mL). The combinedorganic layers were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure ona rotary evaporator. Purification by flash column chromatography (SiO2, CH) afforded 15 (16.2 g,46.0 mmol, 92 %) as a colorless solid. If desired the product can be recrystallized from cyclohexane. |
| 90% |
Stage #1: 2,7-dibromo-9H-fluorene With potassium <i>tert</i>-butylate In tetrahydrofuran for 0.0833333h; Inert atmosphere;
Stage #2: methyl iodide In tetrahydrofuran for 12h; Inert atmosphere; |
|
| 86% |
With sodium methylate In N,N-dimethyl-formamide at 0 - 5℃; for 12h; Inert atmosphere; |
|
| 86.3% |
With potassium hydroxide In dimethyl sulfoxide at 35℃; for 12h; |
2.3. Synthesis of 2, 7-dibromo-9, 9-dimethyl-fluorene (3)
2, 7-Dibromo-9H-fluorene (13.2 g, 40 mmol) was poured into a250 mL three-necked flask. Then measuring 70 mL dimethyl sulfoxideand adding it to the three-necked flask. Stirring at roomtemperature until the solid is completely dissolved. Potassiumhydroxide (10.0 g, 179 mmol) was crushed in a mortar and added toa three-necked flask to turn the solution into purple black.Continuing to stirring and heating the oil bath to 35 °C.The mixture of methyl iodide (12.5 g, 88 mmol) and dimethyl sulfoxide (80 mL)was added by the constant pressure drop funnel for 8 h, and thenthe reactionwas continued for 12 h. Finally, the reactionwas cooledto room temperature. The reaction solution was poured into abeaker containing 250 mL water, and the purple solid was precipitated,then filtered and dried. The solid was added to 80-100 mL anhydrous ethanol and reflux dissolves it completely. Then thecrude product was filtered and recrystallized by the ethanol toobtain 12.38 g yellow needle-like crystal with the yield of 86.3%.2, 7-Dibromo-9, 9-dimethyl-fluorene: IR (KBr), ν/cm-1: 3028(=CH-stretching); 2966, 2923, 2866, (C-H stretching); 1595,1576, 1447 (Ph skeleton vibration); 1397 (C-H bending).1H NMR (500 MHz, CDCl3, δ/ppm): 7.-1.2 Hz, 2 H, Flu-H2 and Flu-H2’), 1.45 (s, 6 H, CH3). |
| 84% |
With potassium hydroxide In dimethyl sulfoxide at 20℃; for 32h; |
2,7-Dibromo-9,9-dimethylfluorene (2)
2,7-Dibromofluorene (13.2 g, 40 mmol), potassium hydroxide (10 g, 179 mmol) were stirred in dimethylsulfoxide (80 mL). Iodomethane (12.5 g, 88 mmol) in dimethylsulfoxide (80 mL) was added dropwise for 8 h. The reaction was stirred at room temperature for 24 h and then poured into 500 mL water. The product was extracted with dichloromethane and the combined organic layers were evaporated to dryness. The crude product was recrystallized in ethanol to give compound 2 as yellow solid (12.11 g, 84%). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.53 (d, J = 1.2 Hz, 2H), 7.51 (d, J =8.3 Hz, 2H), 7.45-7.43 (dd, J1= 8.3 Hz, J2=1.2 Hz, 2H), 1.45 (s, 6H). 13C NMR (125 MHz, CDCl3): δ (ppm) 155.22, 137.13, 130.33, 126.21, 121.43, 121.32, 47.36, 26.82. Anal. Calcd for C15H12Br2: C, 51.01; H, 3.68. Found: C, 51.17; H,3.44. |
| 81% |
With potassium hydroxide In dimethyl sulfoxide |
|
| 78% |
With potassium iodide; potassium hydroxide In dimethyl sulfoxide at 20℃; |
4 Example 4
The difference from Example 1 is that the raw material product bromoalkane in the reaction of step 1 can also be replaced with methyl iodide (chemical formula CH3I). The remaining steps are the same. The residual solid is purified by a silica gel column. The eluent is petroleum ether and ethyl acetate. The volume ratio is preferably 50:1 to obtain 2,7-dibromo-9,9-dimethylfluorene as a white solid with a yield of 78%. |
| 76% |
With triethylamine hydrochloride; sodium hydroxide In dimethyl sulfoxide at 20℃; |
|
| 72% |
Stage #1: 2,7-dibromo-9H-fluorene With potassium <i>tert</i>-butylate In tetrahydrofuran at 0℃; for 0.166667h; Inert atmosphere;
Stage #2: methyl iodide In tetrahydrofuran at 0 - 20℃; Inert atmosphere; |
|
| 72% |
With potassium iodide; potassium hydroxide In dimethyl sulfoxide |
|
| 61% |
Stage #1: 2,7-dibromo-9H-fluorene With potassium <i>tert</i>-butylate In tetrahydrofuran at 0 - 20℃; for 3h; Inert atmosphere;
Stage #2: methyl iodide In tetrahydrofuran at 0 - 20℃; for 18h; Inert atmosphere; |
|
|
With sodium hydroxide; N-benzyl-N,N,N-triethylammonium chloride In dimethyl sulfoxide |
|
|
Stage #1: 2,7-dibromo-9H-fluorene With potassium hydroxide In water; dimethyl sulfoxide at 0℃; Inert atmosphere;
Stage #2: methyl iodide In water; dimethyl sulfoxide at 0 - 20℃; Inert atmosphere; |
1
2,7-Dibromof luorene (50 g, 154.26 mmol) and potassium hydroxide (KOH) (69.2 g, 1234 mmol) were dissolved in DMSO (700 mL) under nitrogen atmosphere. After cooling to 0°C, followed by slowly adding distilled water (113 mL) dropwise, the mixture was stirred for an hour. Then, after slowly adding iodomethane (CH3I) (38.49 mL, 617.04 mmol) dropwise, followed by slowly heating to room temperature, the mixture was stirred for 15 hours. Excess water was added to terminate the reaction and extraction was carried out using dichloromethane. The resultant organic layer was dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Compound E (53.0 g, 150.50 mmol) was obtained by column separation. |
|
Stage #1: 2,7-dibromo-9H-fluorene With potassium hydroxide In water; dimethyl sulfoxide at 10℃; for 1h;
Stage #2: methyl iodide In water; dimethyl sulfoxide at 0 - 20℃; |
10
Compound (111) (2,7-dibromofluorene) (20 g, 61.7 mmol) and potassium hydroxide (27.7 g, 370 mmol) were dissolved in N,N-dimethylsulfoxide (250 mL) at 10° C. and distilled water (45 mL) was added thereto. After stirring for 1 hour, iodomethane (35.0 g, 144.6 mmol) was slowly added thereto. The mixture was stirred at 0° C. for 20 minutes and then at ambient temperature for 10 hours, and neutralized by using 2M HCl. The solid was filtered under reduced pressure and dissolved in dichloromethane (500 mL). Methanol (500 mL) was added to form crystals, which was then filtered to obtain Compound (133) (19.6 g, 55.6 mmol). |
|
With potassium hydroxide In dimethyl sulfoxide at 10 - 30℃; |
9
Preparation Example 9; Preparation of Compound (109); Preparation of Compound (231); In dimethylsulfoxide (200 mL), dissolved were 2,7-dibromofluorene (10.0 g, 30.9 mmol) and potassium hydroxide (KOH) (10.4 g, 185.4 mmol). Iodomethane (7.7 mL, 123.6 mmol) was added thereto at 10° C., and the temperature was raised to 30° C. After stirring for 12 hours, the reaction mixture was poured into distilled water, and the mixture was stirred for 30 minutes. The solid produced was filtered under reduced pressure and washed with hexane to obtain the objective compound (231) (8.6 g, 24.4 mmol). |
|
Stage #1: 2,7-dibromo-9H-fluorene With sodium t-butanolate In tetrahydrofuran at 0℃; for 2h; Inert atmosphere;
Stage #2: methyl iodide In tetrahydrofuran at 0℃; Inert atmosphere; |
4.2.2. 2,7-Dibromo-9,9-dimethyl-9H-fluorene (3FR).
All glasswareswere flame-dried under vacuum before any reaction procedurewasconducted. Anhydrous THF 40 ml was added to the mixture of 2,7-dibromofluorene (2) (4.0 g, 12.3 mmol) and sodium tert-butoxide(5.3 g, 55.5 mmol) under argon atmosphere and stirred for 2 h at0 C. Subsequently, methyl iodide (4.6 ml, 74.0 mmol) was addeddropwise and the reaction mixture was further stirred for 2 h atsame temperature. After confirmation of completion of the reaction,the reaction mixture was extracted with chloroform, andwashed with water and brine. Then, the combined organic layerwas dried over magnesium sulfate. The solvent was removed invacuo, and the residue was purified by recrystallization fromchloroform/methanol/ethanol mixed solvent to afford crude 3FR(2.81 g) as a colorless solid. Yield: 65%; 1H NMR (300 MHz, CDCl3):d 7.54 (d, 3J7.9 Hz, 2H), 7.54 (d, 4J1.8 Hz, 2H), 7.46 (dd, 4J1.7 Hz,3J8.2 Hz, 2H), 1.46 (s, 6H) ppm (Fig. S20). |
|
With potassium <i>tert</i>-butylate In dimethylsulfoxide-d6 |
|
|
With N-benzyl-N,N,N-triethylammonium chloride; sodium hydroxide In water; dimethyl sulfoxide at 20℃; |
|
| 79 % |
With potassium iodide; potassium hydroxide In dimethyl sulfoxide Inert atmosphere; |
|
| 80 % |
With potassium <i>tert</i>-butylate In tetrahydrofuran at 20℃; Inert atmosphere; |
3.2 Step 2 (Preparation Example 3):
Example 1 (1g, 3.09mmol) was weighed into a round bottom bottle, tetrahydrofuran (10mL) was added under nitrogen protection and stirred, then potassium tert-butoxide (1.51g, 12.36mmol) and iodine were added sequentially under an ice bath Methane (1.1g, 7.72mmol), after feeding, the reaction was returned to room temperature and stirred for two hours. After the reaction was completed, it was extracted with water and n-heptane. The organic layer was dewatered and filtered, concentrated and then precipitated with isopropanol. Compound Example 3 (0.87 g, yield: 80%) was obtained as a white solid after filtration. |
| 87 % |
With tetraethylammonium chloride; sodium hydroxide In water; dimethyl sulfoxide at 20℃; |
1-7 Preparation Example 1-7.Synthesis of Intermediate 34-7
2,7-dibromo-9H-fluorene (10.0 g, 30.8 mmol), iodomethane (9.64 g, 67.9 mmol), tetraethylammonium chloride (0.26 g, 1.5 mmol) were mixed with dimethyl sulfoxide (70 ml). After dissolving in ), 50% sodium hydroxide aqueous solution (15 ml) was added and stirred at room temperature for 5 hours.After completion of the reaction, it is cooled, distilled water is added to precipitate, filtered, and a product is obtained through column chromatography.(Yield: 87%) |
| 87 % |
With tetraethylammonium chloride; sodium hydroxide In water; dimethyl sulfoxide at 20℃; |
1-7 Preparation Example 1-7.Synthesis of Intermediate 34-7
2,7-dibromo-9H-fluorene (10.0 g, 30.8 mmol), iodomethane (9.64 g, 67.9 mmol), tetraethylammonium chloride (0.26 g, 1.5 mmol) were mixed with dimethyl sulfoxide (70 ml). After dissolving in ), 50% sodium hydroxide aqueous solution (15 ml) was added and stirred at room temperature for 5 hours.After completion of the reaction, it is cooled, distilled water is added to precipitate, filtered, and a product is obtained through column chromatography.(Yield: 87%) |
| 80 % |
With potassium <i>tert</i>-butylate In tetrahydrofuran at 20℃; Inert atmosphere; Cooling with ice; |
3.2 Step 2 (Preparation Example 3):
Example 1 (1g, 3.09mmol) was weighed into a round bottom bottle, tetrahydrofuran (10mL) was added under nitrogen protection and stirred, then potassium tert-butoxide (1.51g, 12.36mmol) and iodine were added sequentially under an ice bath Methane (1.1g, 7.72mmol), after feeding, the reaction was returned to room temperature and stirred for two hours. After the reaction was completed, it was extracted with water and n-heptane. The organic layer was dewatered and filtered, concentrated and then precipitated with isopropanol. Compound Example 3 (0.87 g, yield: 80%) was obtained as a white solid after filtration. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping