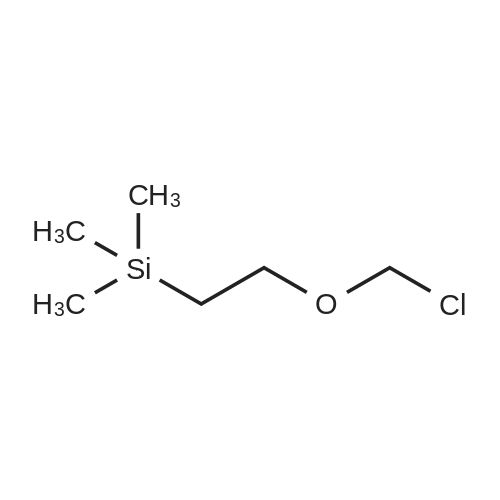
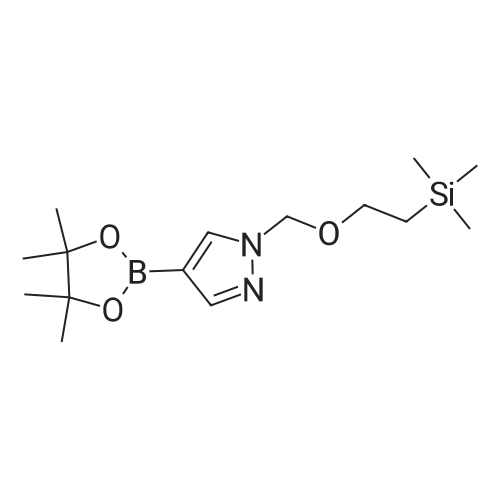
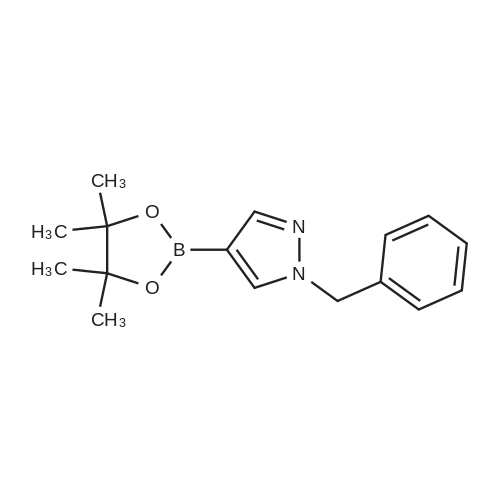
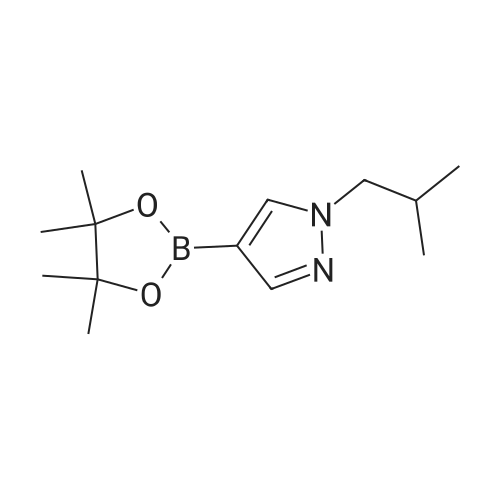
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With sodium hydride In tetrahydrofuran at 0 - 20℃; for 4 h;
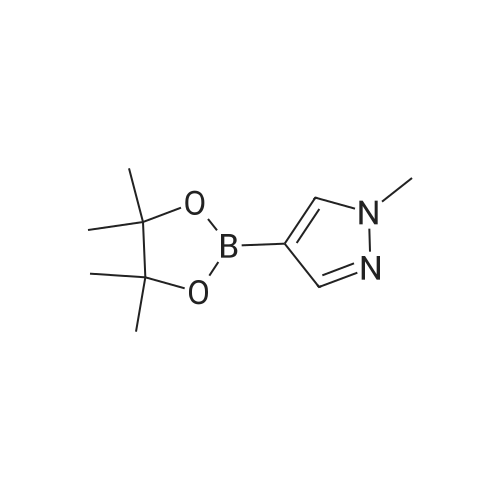
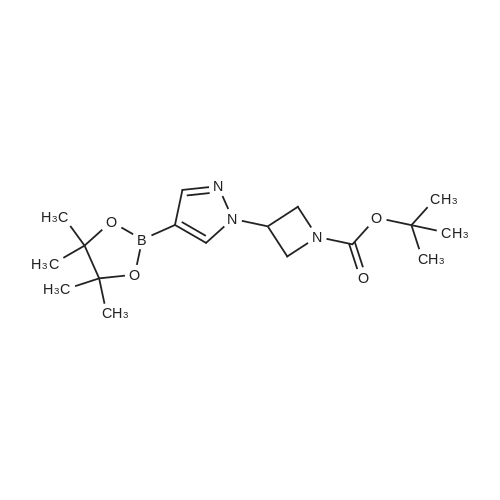
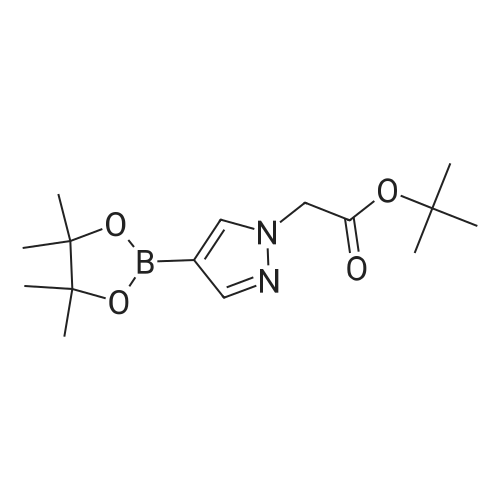
To the solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.40 g, 2.06 mmol) in THF (10 mL) was added NaH (0.1.00 g, 4.12 mmol) followed by methyl iodide (0.25 mL, 4.12 mmol) at 0° C and the reaction mixture was stirred at ambient temperature for 4 hr. The reaction was quenched with saturated NH4Cl solution (20 mL) and extracted with ethyl acetate (2 x 50 mL). The combined organic layers were washed with brine (20 mL), dried over sodium sulfate, and distilled under reduced pressure to obtain Intermediate 108 (0.40 g, 93.00percent) as yellow viscous liquid.1H NMR (400 MHz, CDCl3) δ ppm 1.31 (s, 12 H), 3.91 (s, 3 H), 7.65 (s, 1 H), 7.77 (s, 1 H). LCMS (Method-D): retention time 1.58 min, [M+1] 209.1.
90%
With sodium hydride In tetrahydrofuran at 20℃;
To the solution of 4-pinacolatoboron-lH-pyrazole (1.0 g, 5.0 mmol) in THF (30 mL) was added NaH (0.4 g, 10 mmol). After addition of NaH was completed, to the reaction mixture was added CH3I (1.42 g, 10 mmol) and stirred overnight at room temperature. The reaction was quenched <n="139"/>with MeOH (1 mL). The result mixture was concentrated to give residues, purification by chromatography (EA:PE=l : 10) to give compound l-methyl-4-(4,4,5,5-tetramethyl- 1,3,2- dioxaborolan-2-yl)-lH-pyrazole (0.9 g, 90percent) as light yellow solid. MS (m/z) (M++Η): 209
72%
With caesium carbonate In DMF (N,N-dimethyl-formamide) at 20℃; for 3 h;
To a solution of 4- (4, 4,5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl)-1H- pyrazole (0. 19g, 1.0 mmol) in 4 ml DMF was added Mel (0. 067 ml, 1.1 eq) and Cs2C03 (0.39g, 1.2 eq). The reaction mixture was stirred at RT for 3h. The solution was taken up into EtOAc, washed with water, brine, dried over Na2SO4 and concentrated. 150mg crude product was obtained (yield 72percent).
Reference:
[1] Journal of Medicinal Chemistry, 2009, vol. 52, # 24, p. 7934 - 7937
4
[ 269410-08-4 ]
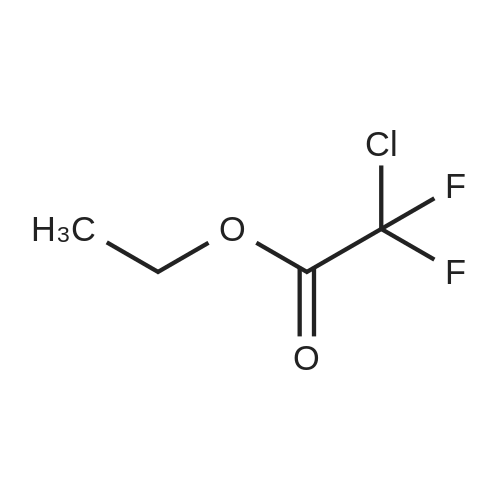
[ 75-03-6 ]
[ 847818-70-6 ]
Yield
Reaction Conditions
Operation in experiment
77%
With caesium carbonate In N,N-dimethyl-formamide at 20℃; for 12 h;
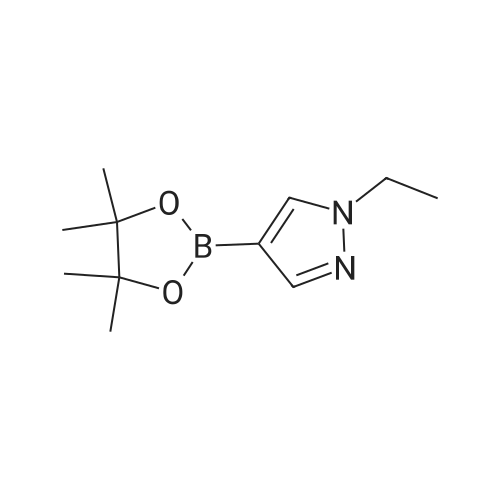
To a solution of 4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -1H-pyrazole (1.0 g, 5.2 mmol) in DMF (10 mL) were added cesium carbonate (2.5 g, 7.7 mmol) and iodoethane (1.2 mL, 15 mmol) . The mixture was stirred at rt for 12 h and diluted with water (30 mL) . The resulting mixture was extracted with DCM (30 mL × 3) . The combinded orgainc layers were dried over anhydrous Na2SO4and concentrated in vacuo. The residue was purified by silica gel column chromatography eluted with PE/EtOAc (v/v) 2/1 to give a colorless oily product (880 mg, 77) .[1505]MS (ESI, pos. ion) m/z: 223.25 [M+1]+
74%
Stage #1: With sodium hydride In N,N-dimethyl-formamide at 20℃; for 1 h; Stage #2: at 20℃; for 72 h;
(107a) 1-Ethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole Sodium hydride (55percent, oil, 0.54 g, 12 mmol) was suspended in N,N-dimethylformamide (20 mL), and a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (2.0 g, 10 mmol) in N,N-dimethylformamide (5 mL) was added thereto. The resulting mixture was stirred at room temperature for 1 hr, and then iodoethane (2.4 g, 16 mmol) was slowly added dropwise thereto. The resulting mixture was stirred at room temperature for 3 days. To this reaction solution, water was added. After extraction with diethyl ether, the organic layer was washed with water and brine, and then dried with anhydrous sodium sulfate. The solvent was evaporated under reduced pressure, and the resulting crude product was purified by silica gel column chromatography (Yamazen, eluding solvent: hexane and ethyl acetate) to obtain 1.7 g (yield: 74percent) of the title compound as a colorless oily material. 1H-NMR (400 MHz, CDCl3) δ ppm: 7.78 (1H, s), 7.70 (1H, s), 4.19 (2H, q, J = 7.3 Hz), 1.49 (3H, t, J = 7.3 Hz), 1.32 (12H, s).
38%
With potassium carbonate In N,N-dimethyl acetamide at 60℃;
Step 8 1-ethyl-4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole 4,4,5,5-Tetramethyl-2-(1H-pyrazol-4-yl)-1,3,2-dioxaborolane (5.0 g), N,N-dimethylacetamide (50 ml), potassium carbonate (5.3 g) and ethyl iodide (2.1 ml) were mixed, and the mixture was stirred at 60° C. overnight. The reaction mixture was cooled to room temperature, water (100 ml) and ethyl ether (100 ml) were added, and the mixture was partitioned in a separatory funnel. The aqueous layer was further extracted with ethyl ether (100 ml), and the organic layers were combined. The organic layer was washed 3 times with water (100 ml) and once with saturated brine (100 ml), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure, hexane (50 ml) was added to the obtained residue and the mixture was partitioned in a separatory funnel. The organic layer was washed 3 times with water (40 ml) and once with saturated brine (40 ml), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to give the title compound (2.1691 g, 38percent). 1H-NMR (CDCl3) δ: 7.79 (1H, s), 7.70 (1H, s), 4.19 (2H, q, J=7.3 Hz), 1.49 (3H, t, J=7.3 Hz), 1.32 (12H, s).
27%
Stage #1: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20℃; for 1 h; Stage #2: for 48 h;
NaH 60percent dispersion in mineral oil (50.0 mg, 1.24 mmol) was suspended in DMF (2 mL) followed by the addition of a solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)-1H-pyrazole (200 mg, 1.03 mmol) in DMF (550 pL). The resulting mixture was stirred at r.t. for ihour. lodomethane (132 pL, 1.6Smmol) was added dropwise and stirring was continued for 2 days. Water was added and the reaction mixture was extracted with EtOAc. The organic layer was washed with water and brine. Dried Mg504, filtered and concentrated in vacuo. The product was purified by flash chromatography (dry packing) on silica gel using a gradient 0 to 30percent EtOAc in hexanes and afforded the title compound (62.7 mg, 0.28 mmol, 27percent) as a yellow oil.
Reference:
[1] Patent: WO2016/615, 2016, A1, . Location in patent: Paragraph 00655
[2] Patent: EP1798229, 2007, A1, . Location in patent: Page/Page column 130
[3] Patent: US2010/240634, 2010, A1, . Location in patent: Page/Page column 67-68
[4] Patent: WO2018/102452, 2018, A2, . Location in patent: Paragraph 425; 426
[5] Bioorganic and Medicinal Chemistry Letters, 2008, vol. 18, # 19, p. 5299 - 5302
[6] Patent: WO2010/75270, 2010, A1, . Location in patent: Page/Page column 163
[7] Patent: WO2013/91096, 2013, A1, . Location in patent: Page/Page column 67
[8] Patent: WO2014/111871, 2014, A1, . Location in patent: Page/Page column 162; 163
[9] European Journal of Medicinal Chemistry, 2018, vol. 158, p. 270 - 285
Reference:
[1] European Journal of Medicinal Chemistry, 2018, vol. 158, p. 270 - 285
9
[ 269410-08-4 ]
[ 6482-24-2 ]
[ 847818-71-7 ]
Yield
Reaction Conditions
Operation in experiment
90%
With potassium carbonate In N,N-dimethyl-formamide at 160℃; for 2 h; Microwave irradiation
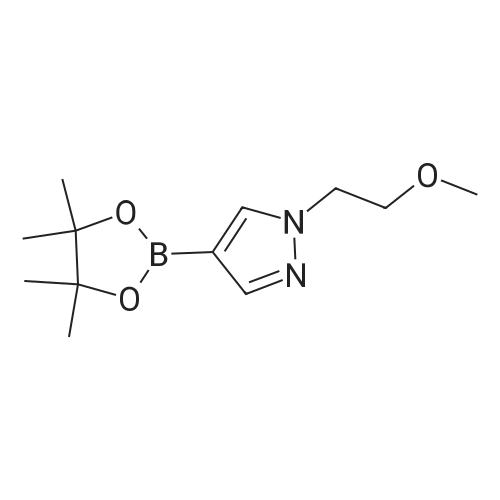
A mixture of 4-(4,4,5 ,5 -tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1H-pyrazole (1.94 g, 10 mmol), 2-bromoethyl methyl ether (1.68 g, 12 mmol) and K2C03 (2.76 g, 20 mmol) in DMF (16 mL) was stirred at 160 °C for 2 h in the microwave. The reaction mixture was concentrated and purified by silica gel chromatography (30percent EA:PE) to give 2.2 g (90percent) of the title compound as yellow oil. ‘H NMR (400 MHz, CDC13): ö 1.32 (12H, s), 3.32 (3H, s), 3.75 (2H, t, J= 5.2 Hz), 4.30 (2H, t, J= 5.2 Hz), 7.77 (1H, s), 7.79 (1H, s). [M+H] Calc’d for C,2H2,BN203, 253; Found, 253.
68%
With caesium carbonate In N,N-dimethyl-formamide at 150℃; for 0.5 h; Microwave irradiation
Example A68Preparation of intermediate 68: l-(2-Methoxy-ethyl)-4-(4,4,5,5-tetramethyl- [l,3,21dioxaborolan-2-yl)-7H-pyrazoleA mixture of 4-(4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-7H-pyrazole (1 g, 5.15 mmol), 2-bromoethyl methyl ether (0.63 ml, 6.7 mmol) and cesium carbonate (2.52 g, 7.73 mmol) in N,N-dimethylformamide (7 ml) was stirred at 150 °C for 30 min. under microwave irradiation. The mixture was partitioned between water and diethyl ether. The organic layer was separated, dried (Na2S04), filtered and the solvents evaporated in vacuo. The crude product was purified by flash column chromatography (silica; ethyl acetate in heptane 30/70). The desired fractions were collected and concentrated in vacuo to yield intermediate 68 (0.88 g, 68percent) as a pale yellow oil.
68%
With caesium carbonate In N,N-dimethyl-formamide at 150℃; for 0.5 h; microwave irradiation
Example A68 Preparation of intermediate 68: 1-(2-Methoxy-ethyl)-4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole A mixture of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (1 g, 5.15 mmol), 2-bromoethyl methyl ether (0.63 ml, 6.7 mmol) and cesium carbonate (2.52 g, 7.73 mmol) in N,N-dimethylformamide (7 ml) was stirred at 150° C. for 30 min. under microwave irradiation. The mixture was partitioned between water and diethyl ether. The organic layer was separated, dried (Na2SO4), filtered and the solvents evaporated in vacuo. The crude product was purified by flash column chromatography (silica; ethyl acetate in heptane 30/70). The desired fractions were collected and concentrated in vacuo to yield intermediate 68 (0.88 g, 68percent) as a pale yellow oil.
57%
With caesium carbonate In N,N-dimethyl-formamide at 0 - 20℃;
2-Bromoethyl methyl ether (0.93 g, 6.70 mmol, 0.64 mL) was added to a mixture of 4-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (1 .00 g, 5.15 mmol) and caesium carbonate (3.49 mg, 10.72 mmol) in dry N,N-dimethylformamide (20 mL) at 0°O. After stirring for 30 mm the ice-water bath was removed. The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with ethyl acetate (150 mL) and washed with brine(3x100 mL). The organic layer was dried with sodium sulfate and concentrated in vacuo. Purification by flash column chromatography (Method L7; 12 g; heptane, 10percent-30percent ethyl acetate) afforded 0.74 g (2.92 mmol; 57percent of theory) of the title compound.GO-MS (Method L9): R1 = 4.21 mm; m/z = 251 MH NMR (300 MHz, Ohloroform-d, Method M2) 6 7.79 (s, 1 H), 7.76 (s, 1 H), 4.29 (t, J = 5.3 Hz,2H), 3.75 (t, J = 5.3 Hz, 2H), 3.32 (s, 3H), 1.31 (s, 12H).
56%
Stage #1: With sodium hydride In N,N-dimethyl-formamide for 0.25 h; Stage #2: at 80℃; for 1 h; Microwave irradiation
General procedure: Preparation 122: 1 -(2-Methoxyethyl)-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- Method H NaH (60percent, 83 mg) was added to a solution of 4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 - -pyrazole (204 mg, 1 .05 mmol) in DMF (4 mL). After stirring for 15 minutes, 1 -bromo-2-methoxyethane (175 mg, 1 .26 mmol) in DMF (1 mL) was added. The resulting solution was stirred at 80°C under microwave irradiation for 60 minutes. The reaction mixture was diluted with brine and extracted with EtOAc. The combined organic layers were washed with water, dried with Na2S04j and concentrated in vacuo to afford the title compound as a yellow oil that was used directly in the next step (148 mg, 56percent). 1 H NMR (500 MHz, CDCI3): δ 7.80 (d, J = 0.7 Hz, 1 H), 7.77 (d, J = 0.7 Hz, 1 H), 4.31 (t, = 5.3 Hz, 2H), 3.76 (t, J = 5.3 Hz, 2H), 3.33 (s, 3H), 1.32 (s, 12H). LCMS (ESI) Rt = 2.17 minutes MS m/z 253 [M+H]+
26%
Stage #1: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20℃; for 1 h; Stage #2: at 0 - 20℃; for 1 h;
To a solution of 4-(tetramethyl-1 ,3, 2-dioxaborolan-2-yl)-1H-pyrazole (1 .00 g, 5.1 mmol) in DMF (5 mL) was added sodium hydride (245 mg of a 60percent dispersion in mineral oil, 6.1 mmol) at room temperature. The mixture was then stirred at room temperature for 60 minutes. The mixture was cooled to 0 ° C and 2-bromoethyl methyl ether (0.72 mL, 7.7 mmol) was added drop wise via syringe. After complete addition, the mixture was allowed to warm to room temperature and stirred for an hour prior to addition of ethyl acetate (70 mL) and water (30 mL). The organic layer was separated, washed with water (2 x 30 mL) and brine (30 mL), dried (Na2S04), filtered and concentrated at reduced pressure. The residue was purified by Biotage Isolera™ chromatography [Biotage SNAP Cartridge KP-Sil 50 g; using a gradient of eluents, 0-50percent EtOAc in heptane] to give the title compound (333 mg, 26percent yield) as a colourless oil. 1H NMR (250 MHz, DMSO-d6) δ [ppm] 7.88 (s, 1 H), 7.57 (s, 1 H), 4.32 - 4.20 (m, 2H), 3.73 - 3.60 (m, 2H), 3.21 (s, 3H), 1 .25 (s, 12H). LCMS (Analytical Method A): Rt = 1 .01 mins, MS (ESIPos): m/z = 253 (M+H)\
20%
With potassium hydroxide In ethanol at 50℃; for 76 h;
Intermediate 1171 -[2-(Methyloxy)ethyl]-4-(4,4,5,5-tetrameth l-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazolA solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (10 g, 51 .5 mmol) in ethanol (50 mL) was treated with KOH (3.47 g, 61 .8 mmol) and 1 -bromo-2- (methyloxy)ethane (5.81 mL, 61.8 mmol) at room temperature and the resulting mixture was stirred at 50°C under nitrogen for 16 h then cooled to room temeprature. 1 -Bromo-2- (methyloxy)ethane (2 ml_, 21 .3 mmol) was added and the resulting mixture was stirred at 50°C for 60 h then cooled to room temperature. The mixture was filtered through celite and the insoluble were washed with ethanol. The combined filtrate and washings were concentrated in vacuo. Purification of the residue on SP4 using a 100 G silica cartridge (gradient: 0 to 100percent AcOEt in Hexanes) gave 1 -[2-(methyloxy)ethyl]-4-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (2.72 g, 10.25 mmol, 20percent) as a yellow oil. LCMS (method G): Retention time 0.87 min, [M+H]+ = 252.9
20%
With potassium hydroxide In ethanol at 50℃; for 66 h; Inert atmosphere
A solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (10 g, 51.5 mmol) in ethanol (50 mL) was treated with KOH (3.47 g, 61.8 mmol) and 1-bromo-2-(methyloxy)ethane (5.81 mL, 61.8 mmol) at room temperature and the resulting mixture was stirred at 50° C. under nitrogen for 16 h then cooled to room temperature. 1-Bromo-2-(methyloxy)ethane (2 mL, 21.3 mmol) was added and the resulting mixture was stirred at 50° C. for 60 h then cooled to room temperature. The mixture was filtered through celite and the insoluble were washed with ethanol. The combined filtrate and washings were concentrated in vacuo. Purification of the residue on SP4 using a 100 G silica cartridge (gradient: 0 to 100percent AcOEt in Hexanes) gave 1-[2-(methyloxy)ethyl]-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (2.72 g, 10.25 mmol, 20percent) as a yellow oil. LCMS (method G): Retention time 0.87 min, [M+H]+=252.9
25.4 g
With caesium carbonate In N,N-dimethyl-formamide at 80℃;
4-(4,4,5,5-Tetramethyl-[1 ,3,2]clioxaborolan-2-yl)-1 H-pyrazole (19.4 g, 0.10 mol), 1- bromo-2-methoxy-ethane (14.18 ml, 0.15 mol), and caesium carbonate (32.58 g, 0.1 mol) are dissolved in DMF (200 ml). The suspension is stirred for 16 h at 80°C, filtered and the solvent is removed in vacuum. The residue is treated with tert-butyl methyl ether (200 ml), filtered over Celite and then the solvent is removed in vacuum; yield: 25.4 g 1-(2-methoxy-ethyl)-4-(4,4,5,5-tetramethyl-[1,3,2]dioxa- borolan-2-yl)-1H-pyrazole; HPLC/MS: 1.82 min, [M+H] = 253.
1.31 g
With caesium carbonate In acetonitrile at 50℃; Inert atmosphere
1H-pyrazole-4-boronic acid pinacol ester (1.0 g, 5.155 mmol),2-Bromoethyl methyl ether (0.788 g, 5.669 mmol)And cesium carbonate (5.04 g, 15.469 mmol) were dissolved in acetonitrile (20 mL),The mixture was stirred under nitrogen at 50 ° C overnight,filter,Concentrate to dryness to give 1- (2-methoxyethyl) -1H-pyrazole-4-boronic acid pinacol ester (1.310 g).
With caesium carbonate In N,N-dimethyl-formamide at 160℃; for 0.5 h; Microwave irradiation
4-( 4,4,5,5-Tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1H-pyrazole (1 00 mg, 0.52mmol), 1-chloro-2-methoxyethane (0.056 mL, 0.62 mmol) and cesium carbonate (252mg, 0.73 mmol) in DMF (1 mL) were heated in microwave reactor at 160 °C for 30 min.The reaction mixture was concentrated under reduced pressure and purified by silica gelchromatography (ISCO, hexanes/ethyl acetate 0-100percent over 15 min) to isolate CompoundB162a (130 mg, 100percent yield). HPLC: RT = 1.0 min (LCMS Method M). MS (ES): m/z= 253.0 [M+H( 1H NMR (500MHz, CHLOROFORM-d) 8 8.04 (s, 1H), 7.83 (d,J=18.4 Hz, 1H), 4.37 (t, J=5.2 Hz, 1H), 3.78 (t, J=5.2 Hz, 1H), 3.38-3.31 (m, 2H), 2.98(s, 3H), 2.90 (s, 3H), 1.34 (s, 6H), 1.26 (s, 3H).
72%
With caesium carbonate In N,N-dimethyl-formamide at 160℃; for 0.5 h; microwave irradiation
2-Chloroethyl methyl ether (0.050 ml, 0.63 mmol) was added to a stirred solution of 4- (4,4,5, 5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-lH-pyrazole (5.0 g, 25.77 mmol) and cesium carbonate (12.59 g, 38.65 mmol) in DMF (27 ml). The mixture was stirred at 160 °C for 30 min. under microwave irradiation and then the solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica; MeOH in DCM 2/98). The desired fractions were collected and evaporated in vacuo to yield intermediate 67 (4.6 g, 72percent) as a pale yellow oil.
72%
With caesium carbonate In N,N-dimethyl-formamide at 160℃; for 0.5 h; Microwave irradiation
Example A67 1-(2-Methoxy-ethyl)-4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole 2-Chloroethyl methyl ether (0.050 ml, 0.63 mmol) was added to a stirred solution of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (5.0 g, 25.77 mmol) and cesium carbonate (12.59 g, 38.65 mmol) in DMF (27 ml). The mixture was stirred at 160° C. for 30 min. under microwave irradiation and then the solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica; MeOH in DCM 2/98). The desired fractions were collected and evaporated in vacuo to yield intermediate 67 (4.6 g, 72percent) as a pale yellow oil.
Reference:
[1] Patent: WO2014/15088, 2014, A1, . Location in patent: Paragraph 00223
[2] Journal of Medicinal Chemistry, 2014, vol. 57, # 10, p. 4196 - 4212
[3] Patent: WO2011/110545, 2011, A1, . Location in patent: Page/Page column 95-96
[4] Patent: US2012/329792, 2012, A1, . Location in patent: Page/Page column 46
11
[ 269410-08-4 ]
[ 5407-04-5 ]
[ 847818-72-8 ]
Reference:
[1] Journal of Medicinal Chemistry, 2011, vol. 54, # 12, p. 4092 - 4108
12
[ 269410-08-4 ]
[ 109-54-6 ]
[ 847818-72-8 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2008, vol. 18, # 19, p. 5299 - 5302
13
[ 269410-08-4 ]
[ 76-83-5 ]
[ 863238-73-7 ]
Reference:
[1] Journal of the American Chemical Society, 2017, vol. 139, # 1, p. 211 - 217
14
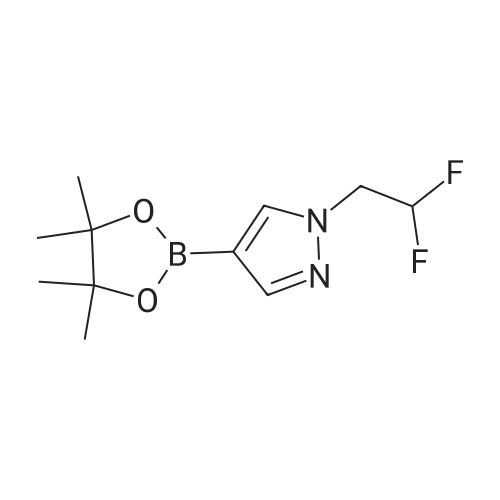
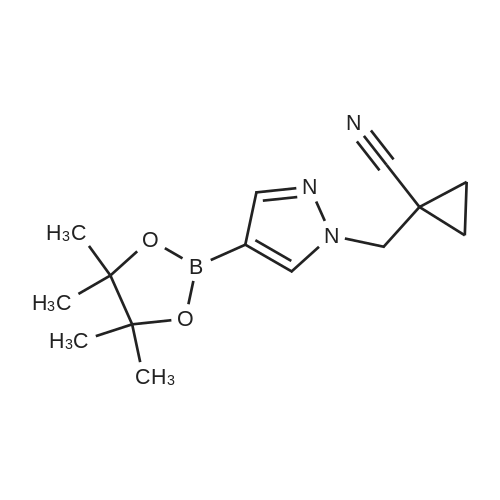
[ 1029716-44-6 ]
[ 269410-08-4 ]
Yield
Reaction Conditions
Operation in experiment
94%
With hydrogenchloride; 2,3-dimethyl-2,3-butane diol In water at 10 - 25℃; for 4.083 h;
1 -( 1 -Ethoxyethyl)-4-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)-l H-pyrazole (60.00 g, 224 mmol) is combined with CPME (120 mL) and 2,3-dimethylbutane-2,3-dtol (26.49 g, 224 mmol). The reaction is cooled to 5-10 °C then a solution of anhydrous HQ in CPME (3.1 M, 86.8 mL, 269 mmol) is added over 15 minutes, followed by additional CPME (15 mL). The reaction is stirred at 20-25 °C and monitored for completion. After 7 hours, additional HCI solution (3 mL, 9.3 mmol) is added to the reaction and stirring is continued for an additional 15 hours to give 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)-l H-pyrazole hydrochloride salt, which is not isolated.mL) is added over 10 minutes. The reaction mixture temperature increases to 20 °C following the addition. Additional CPME (10 mL) is added and the reaction mixture is stirred for 15 minutes then cooled in an ice bath. After 3 hours the reaction mixture is filtered and the solid (triethylamine hydrochloride) is washed with cold CPME (3 x 60 mL). The filtrate and washes are combined to give 426 g of solution containing 102,3 mg of 4- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazo]e/g solution (total 8.15 g, 94percent )yield of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyTazole) which is used directly in the next step.
77.3%
With hydrogenchloride; 2,3-dimethyl-2,3-butane diol In tert-butyl methyl ether; 1,2-dichloro-ethane at 0 - 20℃; Large scale
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1) [0166] To a mixture of 2,3-dimethylbutane-2,3-diol (25.0 kg, 211.6 mol) and 1-(1-ethoxyethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (24, 55.0 kg, 206.7 mol) in 1,2-dichloroethane (750 kg) was slowly added a solution of HCl in MTBE (25.0 kg, 20-30percent of HCl) at 0-5° C. The resulting reaction mixture was then stirred at 10-20° C. for 3-5 hours. After the selective deprotection reaction was complete as monitored by HPLC (1: below 1percent), the reaction mixture was degassed and refilled with nitrogen before being cooled to −15° C. The cooled reaction mixture was then added triethylamine (TEA, 30.0 kg, 296.5 mol) to adjust pH to 7-8. The mixture was then gradually warmed to ambient temperature before being treated with water (150 kg). The two phases were separated and the organic layer was washed with brine (60 kg) and dried over sodium sulfate (Na2SO4). The drying reagent, sodium sulfate (Na2SO4), was removed by filtration and the resulting solution was concentrated under reduced pressure at 40-50° C. to a thick oil. The residue was warmed to 60-70° C. and diluted with petroleum ether (100 kg) at the same temperature. The resulting mixture was then gradually cooled to ambient temperature and subsequently to −5° C. and stirred at the same temperature for 3 hours. The solids was collected by centrifugation and dried at 50-60° C. under vacuum to afford the crude desired product (1, 33.75 kg, 40.11 kg theoretical, 84.1percent). The crude desired product was then suspended in 1,2-dichloroethane (30 kg) and the resulting mixture was heated to reflux until a clear solution was formed. To the hot solution was then added petroleum ether (150 kg) at the same temperature. The resulting mixture was then gradually cooled to ambient temperature and subsequently to −5° C. and stirred and the same temperature for 3 hours. The solids were collected by centrifugation and dried under vacuum at 50-60° C. to afford 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1, 31.0 kg, 40.11 kg theoretical, 77.3percent) as an off-white solid, which is identical in every comparable aspect to the material synthesized by the synthetic method as described above in Example 5.
With n-Bu3MgLi In tetrahydrofuran at -20 - -10℃; Large scale
In the reaction kettle, adding tetrahydrofuran 4.5 kg and 1 - BOC - 4 - brompyrazole (2.01 kg, 9.2 µM), stirring 0.5 hours, cooling to -20 °C, maintain -10 °C -0 °C dropwise 3.2molBu3MgLi [preparation method: 1.0eq butyl magnesium chloride in -10 °C -0 °C lower drop by adding 2.0 equivalent butyl lithium in], TLC detection reaction, maintain the exchange completion -10 °C -0 °C dropwise dimethylamine fundamental frequency that mellow boron ester (1.61 kg, 9.4 µM) dissolved in 1 kg of the mixed solution in tetrahydrofuran, the completion of the dropping, thermal insulation 2 hours, natural heating stirring overnight. The control temperature of not more than 30 °C after adding glacial acetic acid, after detecting the protecting group removal, stop stirring filtration, the mother liquor recovered, solid add triethylamine (1.01 kg, 10 µM) and ethyl acetate 8 kg after, heating to reflux reaction, the proportion of internal standard detecting product is not increased when, after lowering the filtering, the filtrate obtained after the distillation is a kind of white solid, by adding heptane cooling to 0 °C beating, filtering, 50 - 60 °C vacuum drying to obtain white solid pyrazole -4 - boric acid frequency that alcohol ester 1.36 kg, GC: 99.1percent, HNMR consistent with the literature value, yield 76percent.
Reference:
[1] Patent: CN106188116, 2016, A, . Location in patent: Paragraph 0016; 0017
16
[ 121669-70-3 ]
[ 557087-01-1 ]
[ 269410-08-4 ]
Yield
Reaction Conditions
Operation in experiment
79%
With iPrBu2MgLi In 2-methyltetrahydrofuran at -20 - -10℃; Large scale
In the reaction kettle, add 2 - methyl tetrahydrofuran 4.5 kg and 1 - BOC - 4 - iodine pyrazole (2.65 kg, 9.0 µM), stirring 0.5 hours, cooling to -20 °C, maintain -10 °C -0 °C dropwise 2.95 µM Bu3MgLi [preparation method: 1.0eq isopropyl magnesium chloride in -10 °C -0 °C lower drop by adding 2.0 equivalent butyl lithium in], TLC detection reaction, maintain the exchange completion -10 °C -0 °C dropwise dimethylamine fundamental frequency that mellow boron ester (1.57 kg, 9.2 µM) dissolved in 1 kg of the mixed solution in tetrahydrofuran, the completion of the dropping, thermal insulation 2 hours, natural heating stirring overnight. The control temperature of not more than 20 °C hydrogen chloride gas, after detecting the protecting group removal, stop stirring filtration, the mother liquor recovered, solid add triethylamine (1.01 kg, 10 µM) and ethyl acetate 10 kg after, heating to reflux reaction, the proportion of internal standard detecting product is not increased when, after lowering the filtering, the filtrate obtained after the distillation yellow solid, adding hexane cooling to 0 °C beating, filtering, 50 - 60 °C vacuum drying to obtain white solid pyrazole -4 - boric acid frequency that alcohol ester 1.38 kg, GC: 99.5percent, HNMR consistent with the literature value, yield 79percent.
Reference:
[1] Patent: CN106188116, 2016, A, . Location in patent: Paragraph 0018; 0019
17
[ 61676-62-8 ]
[ 269410-08-4 ]
Yield
Reaction Conditions
Operation in experiment
54.8%
Stage #1: With isopropylmagnesium chloride In tetrahydrofuran at -6 - 0℃; Inert atmosphere Stage #2: at 0 - 20℃; Inert atmosphere
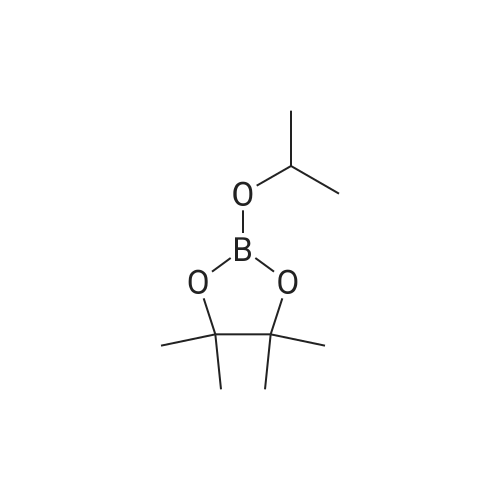
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (17).; A flask equipped with a mechanical stirrer, nitrogen inlet, addition funnel and thermowell was charged with 1-trimethylsilyl-4-iodopyrazole (15, 225.1 g, 0.85 mol) and THF (2200 mL). This mixture was cooled to -6° C. in an ice/salt/brine bath and isopropyl magnesium chloride (2 M in THF, 510 ml, 1.02 mol, 1.2 equiv) was added at a rate such that the temperature did not exceed 0° C. The extent of metal/halogen exchange was monitored by GC and was found complete after about 10 min. To the orange brown solution was added 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (isopropylpinacolborate, 16, 347 mL, 1.7 mol, 2.0 equiv) slowly at first keeping the temperature below 0° C. and then fairly rapidly after about 1/2 of the compound was added allowing the temperature to reach 5° C. (the reaction becomes quite thick and then thins out slowly). The reaction is then stirred at 0° C. for 10 min before being warmed to room temperature over 1 hr and stirred at room temperature for an additional 1 hr. The reaction was cooled to 6° C. and saturated aqueous ammonium chloride solution (2.2 L) was added with a temperature increase to 25° C. The mixture was stirred for 5 minutes before being diluted with toluene (10 L). The layers were separated (a large amount of solid is present in the aqueous layer) and the organic layer was sequentially washed with water (6.x.2.2 L), brine (2.x.2.2 L), dried over sodium sulfate, filtered, and concentrated under reduced pressure. Residual toluene was co-evaporated with heptane to afford 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (17, 90.3 g, 164.9 g theoretical, 54.8percent) as a white solid. For 17: 1H NMR (DMSO-d6, 400 MHz) δ ppm 13.08 (bs, 1H), 7.94 (s,1H), 7.62 (s,1H), 1.23 (s, 12H); C9H15BN2O2 (MW, 194.04), LCMS (EI) m/e 195 (M++H).
54.8%
Stage #1: With isopropylmagnesium chloride In tetrahydrofuran at -6 - 0℃; for 0.166667 h; Inert atmosphere Stage #2: at 0 - 20℃; for 2 h; Inert atmosphere Stage #3: With water; ammonium chloride In tetrahydrofuran at -6 - 0℃;
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1) [0161] A flask equipped with a mechanical stirrer, a nitrogen inlet, an addition funnel and a thermowell was charged with 1-trimethylsilyl-4-iodopyrazole (225.1 g, 0.85 mol) and THF (2200 mL) at ambient temperature. This mixture was cooled to approximately −6° C. in an ice/salt/brine bath before a solution of isopropyl magnesium chloride in THF (2 M solution in THF, 510 mL, 1.02 mol, 1.2 equiv) was added at a rate such that the internal temperature did not exceed 0° C. The extent of metal/halogen exchange was monitored by GC and was found complete after about 10 min. To the orange brown solution was then added 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (isopropylpinacolborate, 347 mL, 1.7 mol, 2.0 equiv) slowly at first keeping the temperature below 0° C. and then fairly rapidly after about half of the compound was added allowing the temperature to reach 5° C. (the reaction becomes quite thick and then thins out slowly). The reaction is then stirred at 0° C. for 10 min before being warmed to ambient temperature over 1 h and stirred at ambient temperature for an additional 1 h. The reaction mixture was cooled to approximately 6° C. and the saturated aqueous ammonium chloride solution (NH4Cl, 2.2 L) was added with a temperature increase to 25° C. The mixture was stirred for 5 minutes before being diluted with toluene (10 L). The layers were separated (a large amount of solid is present in the aqueous layer) and the organic layer was sequentially washed with water (6×2.2 L) and brine (2×2.2 L) before being dried over sodium sulfate (Na2SO4). The drying reagent, sodium sulfate (Na2SO4), was removed by filtration and the solution was concentrated under reduced pressure. Residual toluene was co-evaporated with n-heptane to afford 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1, 90.3 g, 164.9 g theoretical, 54.8percent) as a white solid. For 1: 1H NMR (400 MHz, DMSO-d6) δ 13.08 (bs, 1H), 7.94 (s, 1H), 7.62 (s, 1H), 1.23 (s, 12H) ppm; C9H15BN2O2 (MW, 194.04), LCMS (EI) m/e 195 (M++H).
With bis-triphenylphosphine-palladium(II) chloride; tetrabutylammomium bromide; potassium carbonate In 1,4-dioxane for 12 h; Inert atmosphere; Reflux
Under a nitrogen atmosphere, 7.35 g of 1-H-4-bromopyrazole, 14.0 g of diboronic acid pinacol ester, 6.0 g of potassium acetate, 2.15 g of tetrabutylammonium bromide, 0.1 g were placed in a 500 ml four-necked flask. Pd(PPh3)2Cl2, and 100 ml of 1,4-dioxane was added thereto, and the mixture was stirred under reflux with heating for 12 hours, and was subjected to HPLC. After completion of the reaction, the mixture was cooled to room temperature, and the reaction mixture was filtered under reduced pressure.Swirled white product 8.8g,Yield 94.0percent,HPLC 98.4percent.
Reference:
[1] Patent: CN108997309, 2018, A, . Location in patent: Paragraph 0016
[2] Patent: WO2017/107089, 2017, A1, . Location in patent: Page/Page column 43; 44
Reference:
[1] Angewandte Chemie - International Edition, 2013, vol. 52, # 49, p. 12915 - 12919[2] Angew. Chem., 2013, vol. 125, # 49, p. 13153 - 13157,5
23
[ 288-13-1 ]
[ 73183-34-3 ]
[ 269410-08-4 ]
Reference:
[1] Journal of the American Chemical Society, 2014, vol. 136, # 11, p. 4287 - 4299
24
[ 552846-17-0 ]
[ 269410-08-4 ]
Reference:
[1] Journal of Organic Chemistry, 2009, vol. 74, # 23, p. 9199 - 9201
25
[ 269410-08-4 ]
[ 24424-99-5 ]
[ 552846-17-0 ]
Yield
Reaction Conditions
Operation in experiment
85%
With dmap In dichloromethane at 20℃; for 4 h;
4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) 1H-pyrazole (1.16 g, 6.0mmol) in methylene chloride (40 mL) was added di-tert-butyl dicarbonate (1.58 g, 7.2 mmol), was added a catalytic amount of 4-dimethylaminopyridine (102 mg, 0.84 mmol), reaction at room temperature for 4 hours.Water was added to the system, extracted with ethyl acetate, washed with saturated aqueous sodium chloride, the organic Xiangde removed by rotary evaporation the crude product (1.5 g, 85percent yield).
76%
at 20℃; for 12 h;
Di-tert-butyl dicarbonate (7.2 molar equivalent), 4-(dimethylamino)pyridine (0.84 molar equivalent) were added to a solution of 4,4,5,5-tetramethyl-2-(1H-pyrazole-4-yl)-1,3,2-dioxaborolane (6 mmol) in 40 mL of DMF. The reaction mixture was stirred at room temperature for 12 h. Water was added to the reaction mixture to quench the reaction. EtOAc was then added to extract the aqueous solution. Dry EtOAc layer over Na2SO4. The Na2SO4 was filtered off and the filtrate was evaporated to give a brown yellow oil residue as compound 21-1 (1.32 g; 4.56 mmol; 76percent). 1H NMR (400 MHz, chloroform-D) δ ppm 1.32 (s, 12H) 1.63 (s, 9H) 7.91 (s, 1H) 8.37 (s, 1H). The residue was used for the next step reaction without further purification.
64%
With dmap In acetonitrile at 20℃; for 18 h;
To a mixture of 4-pyrazoleboronic acid pinacol ester (0.485 g, 2.5mmol) and DMAP (0.153 g, 1.25 mmol) in MeCN (12.5 ml) was added di-tert-butyldicarbonate (0.709 g, 3.25 mmol). The resulting mixture was stirred at rt for 18 h before itwas concentrated under reduced pressure. The obtained residue was purified by flashcolumn chromatography (silica gel, 0-10percent EtOAc in petroleum ether) to afford the titlecompound (0.473 g, 64percent yield) as a white solid. 1H NMR (400 MHz, CDCb) o 8.38 (s, 1 H),7.92 (s, 1H), 1.64 (s, 9H), 1.33 (s, 12H).
57%
With N-ethyl-N,N-diisopropylamine In dichloromethane at 0 - 20℃; for 48 h;
To a solution of 33a (l .Og, 5.15mmol) in CH2Cl2 (3OmL) was added DIEA (2.0g, 15.5mmol), followed by (Boc)2O (1.55g, 7.4mmol) drop-wise at O0C. The resulting mixture was stirred at room temperature for 2 days. After the reaction was complete detected by TLC, the mixture was evaporated and the residue was purified by silica column chromatography (PE:EA=4:1) to provide 35a (0.86g, 57percent yield).
With caesium carbonate In acetonitrile for 5 h; Reflux
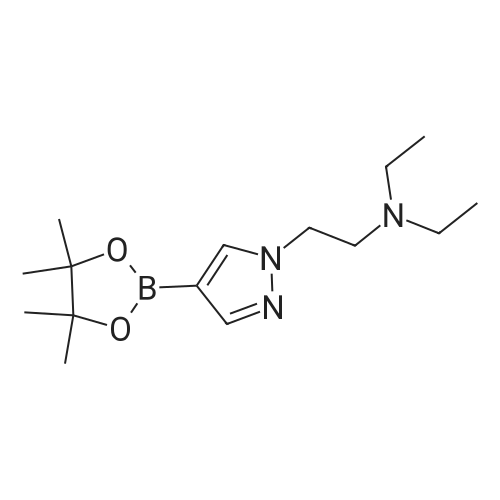
Intermediate 115yV,yV-Dimethyl-2-[4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazol-1 - yljethanamine4-(4,4,5,5-Tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (2 g, 10.31 mmol), cesium carbonate (6.72 g, 20.61 mmol) and 2-chloro-/V,/V-dimethylethanamine hydrochloride (2.227 g, 15.46 mmol) were suspended in acetonitrile (30 mL) and the mixture was heated at reflux for 5 h, then cooled to room temperature, diluted with Et20 and filtered. The filtrate was concentrated in vacuo to give N,N-dimethyl-2-[4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 H-pyrazol-1 -yl]ethanamine dimethyl{2-[4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 H-pyrazol-1 -yl]ethyl}amine (2.61 g, 9.84 mmol, 95 percent yield) as an amber oil which was used in the next step without further purification.
88%
With caesium carbonate In acetonitrile at 90℃;
A mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (60.0 mg, 0.309 mmol), 3-dimethylaminoethyl chloride hydrochloride (49.0 mg, 0.340 mmol) and cesium carbonate (0.302 g, 0.928 mmol) in acetonitrile (1 mL) was stirred at 90° C. overnight. The reaction mixture was cooled to ambient temperature, quenched with water, and extracted with methylene chloride. The combined organic layers were dried over Mg504, then filtered and concentrated under reduced pressure to give the desired product (60 mg, 88percent). LCMS calculated for C13H25BN3O2 (M+H)+: m/z=266.2. Found: 266.2.
66%
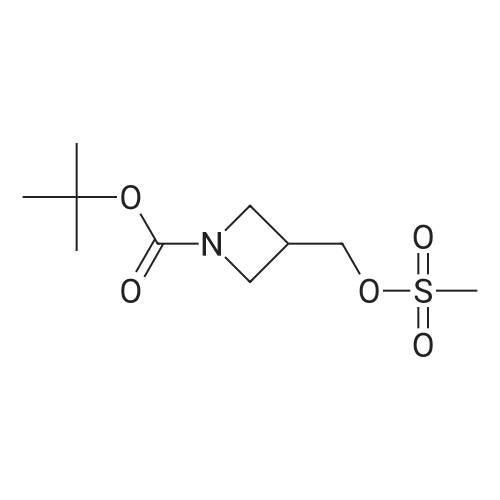
With caesium carbonate In N,N-dimethyl-formamide at 0 - 20℃; for 72.5 h;
2-Dimethylaminoethyl chloride hydrochloride (0.97 g, 6.70 mmol) was added to a mixture of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (1 .00 g, 5.15 mmol) and caesiumcarbonate (5.54 g, 17.01 mmol) in dry N,N-dimethylformamide (20 mL) at 0°C. After stirring for30 mm the ice-water bath was removed. The reaction mixture was stirred at room temperaturefor three days. The reaction mixture was diluted with ethyl acetate (150 mL) and washed with brine (3x100 mL). The organic layer was dried with sodium sulfate and concentrated in vacuo to afford 1 .00 g (3.41 mmol; 66percent of theory) of the title compound.GO-MS (Method L9): R1 = 4.48 mm; (no mass detected)1 H NMR (300 MHz, Chloroform-d, Method M2) 6 7.78 (s, 1 H), 7.74 (s, 1 H), 4.23 (t, J = 6.8 Hz,2H), 2.76 (t, J = 6.8 Hz, 2H), 2.26 (s, 6H), 1.31 (s, 12H).
56%
With caesium carbonate In acetonitrile at 90℃; for 72 h;
A mixture of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (100 mg, 0.515 mmol), 2-chloro-A/,A/-dimethylethanamine hydrochloride (82 mg, 0.567 mmol), and Cs2C03 (504 mg, 1.546 mmol) in acetonitrile (2 ml_) was heated at 90 °C for 3 d. After cooling to RT, the reaction mixture was partitioned between DCM and water. The aqueous layer was extracted with DCM and the combined extracts were washed (water), dried (MgS04), and concentrated in vacuo to give the title compound (76 mg, 56percent) as a colourless oil. LCMS (Method B): RT = 0.61 min, m/z = 266 [M+H]+.
With caesium carbonate In N,N-dimethyl-formamide at 70℃;
A mixture of 4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -1H-pyrazole (1.5 g, 7.7 mmol) , 2-bromo-N, N-dimethylethanamine hydrobromide (4 g, 20 mmol) and Cs2CO3(9 g, 17.6 mmol) in DMF (15 mL) was stirred at 70 overnight. The reaction mixture was concentrated to remove DMF. The residue was diluted with water (20 mL) . The resulting mixture was extracted with DCM (30 mL × 3) . The combined organic layers were dried over anhydrous Na2SO4and concentrated in vacuo. The residue was purified by silica gel column chromatography eluted with DCM/MeOH (v/v) 25/1 to give a white solid product (0.5 g, 20) .[1634]MS (ESI, pos. ion) m/z: 266.1 [M+1]+.
With potassium carbonate In N,N-dimethyl-formamide at 80℃; for 8 h;
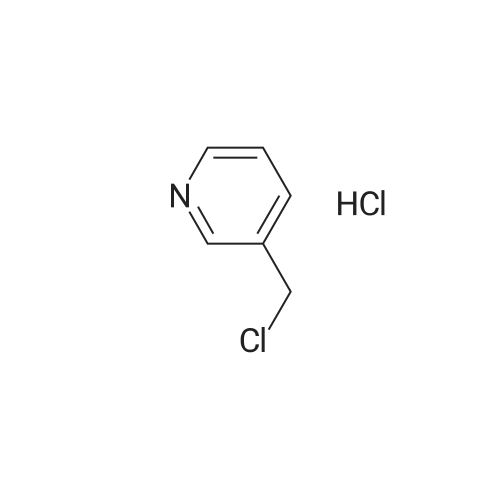
3- (Bromomethyl) pyridine (2.67 g, 15.5 mmol)And 4-pyrazole boronic acid pinacol ester(3.01 g, 15.5 mmol) was dissolved in DMF (20 mL)Potassium carbonate (2.2 g, 16 mmol) was added to the system, and the mixture was heated to 80 ° C. for 8 h.The reaction mixture was poured into water (50 mL) and extracted with ethyl acetate (50 mL × 3)Saturated brine (20 mL), dried over anhydrous sodium sulfate, and the solvent was removed.The residue was subjected to column chromatography (eluent: PE / EtOAc (v / v) = 1 / 1.5)This gave 280 mg of a pale yellow oil, yield: 6.33percent.
Reference:
[1] Patent: CN106749268, 2017, A, . Location in patent: Paragraph 0607; 0608
Reference:
[1] Journal of Medicinal Chemistry, 2011, vol. 54, # 18, p. 6342 - 6363
38
[ 269410-08-4 ]
[ 141699-59-4 ]
[ 877399-74-1 ]
Yield
Reaction Conditions
Operation in experiment
44%
With caesium carbonate In N,N-dimethyl-formamide at 90℃; Inert atmosphere
Step 2: Compound 96-2 (134 mg, 0.5 mmol) and cesium carbonate (245 mg, 0.75 mmol) was added to a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (97 mg, 0.5 mmol) in DMF, the reaction mixture was stirred at 90° C. for 12-16 h, and the reaction solution was cooled to room temperature, diluted with water, extracted with EA, and the combined organic phase was washed with brine, dried over anhydrous Na2SO4 and concentrated to give the crude product which was purified by Combi-flash column chromatography [PE:EA=100:0-50:50] to give the title compound 96-3 (2 g, yield 44percent), MS m/z (ESI): 378.2[M+1]+.
39%
With caesium carbonate In N,N-dimethyl-formamide at 100℃; for 24 h;
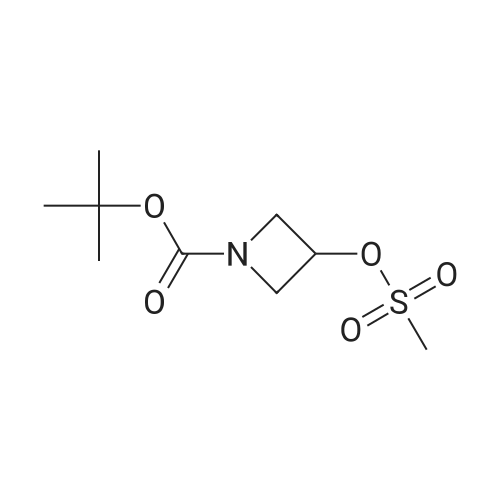
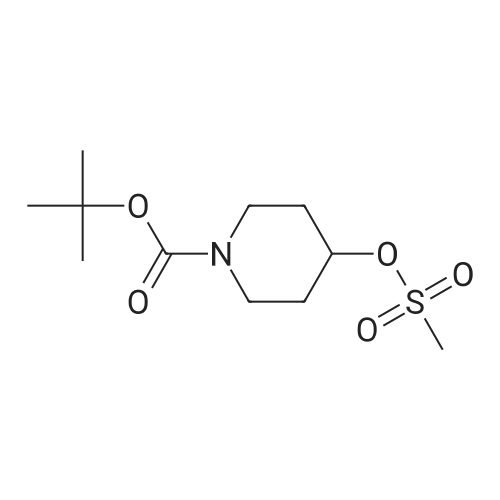
Step B: ferf-butyl 4-[4-(4l4l5l5-tetramethyl-1 l3l2-dioxaborolan-2-yl)-1 -/-pyrazol-1-yllpiperidine- 1-carboxylate (Title Compound)A mixture of terf-butyl 4-[(methylsulfonyl)oxy]piperidine-1-carboxylate (7.33 g, 26.2 mmol), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (5.09 g, 26.2 mmol), and Cs2C03 (12.8 g, 39.3 mmol) in DMF (50 mL) was heated at 100 °C for 24 h. The mixture was cooled to RT and diluted with water (100 mL) and extracted with EtOAc (3 x 60 mL). The combined organic phases were washed with water (3 x 50 mL), brine (50 mL), and dried over anhydrous sodium sulfate. The residue was purified by flash chromatography (20 to 40percent ethyl acetate:hexanes) to afford 3.84 g (39percent) of the title compound as a white solid. 1H NMR (400 MHz, CDCI3): 57.81 (s, 1H), 7.74 (s, 1H), 4.17-4.35 (m, 3H), 2.89 (m, ^-.-12 Hz, 2H), 2.14 (d, =14.65 Hz, 2H), 1.90 (qd, J=12.25, 4.42 Hz, 3H), 1.48 (s, 9H), 1.33 (s, 12H); MS (ESI): 379.15 [M+H]+; HPLC tR = 3.17 min.
35.2%
With caesium carbonate In N,N-dimethyl-formamide at 100℃; for 24 h;
Tert-butyl-1-carboxylate 4-methanesulfonyloxy piperidine (2.00 g, 7.16 mmol)Was added to a solution of 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1H-pyrazole (1.50 g, 7.70 mmol) andCesium carbonate (3.50 g, 11.00 mmol)In N, N-dimethylformamide (15 mL)The reaction solution was reacted at 100 ° C for 24 h,Cooled to room temperature,Extracted with water (100 mL) and ethyl acetate (100 mL)The aqueous phase was extracted with ethyl acetate (100 mL x 3), the organic phases were combined and washed with saturated brine (100 mL x 3)Dried over anhydrous sodium sulfate, concentrated under reduced pressure, and the concentrated solution was passed through a column (petroleum ether / ethyl acetate (v / v) = 2/1)To give 950 mg of a colorless solid in a yield of 35.2percent.
28.74%
Stage #1: With sodium hydride In N,N-dimethyl-formamide at 0 - 20℃; Stage #2: at 90℃;
Step 2: tert-butyl 4-[4-(4,4, 5, 5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl]piperidine-l- carboxvlate\\ S; To a solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (4.12 g, 21.2 mmol,Aldrich, Cat. 525057) in N,N-dimethylformamide (20 mL) was added sodium hydride (1.78 g, 44.5 mmol) at 0 0C. The resulting solution was stirred at r.t. for one hour, and then tert-butyl 4- [(methylsulfonyl)oxy]ρiρeridine-l-carboxylate (6.3 g, 22 mmol) in DMF (2 mL) was added. The reaction mixture was heated at 9O0C overnight. Then the reaction mixture was cooled to r.t., quenched with water, 0 and extracted with AcOEt. The organic layer was washed with NaHCO3 aqueous solution and brine successively, dried with MgSO4, and concentrated. The residue was purified by flash column chromatography on a silica gel column using 30percent ethyl acetate in hexane as eluent to afford the desired compound (2.30 g, 28.74percent). LCMS (M+H)+: m/z = 378.4.
884 mg
With caesium carbonate In N,N-dimethyl-formamide at 100℃;
To tert-butyl 4-hydroxypiperidine-1-carboxylate (402 mg, 2.0 mmol) in methylene chloride (20 mL) and triethylamine (303 mg, 3.0 mmol) at 4° C. was added methanesulfonyl chloride (274 mg, 2.4 mmol) drop-wise. The reaction was brought to ambient temperature and was stirred for 1 hour. The reaction mixture was concentrated in vacuo and diluted in diethyl ether (20 mL). The solution was washed with 1N hydrochloric acid (3 mL), water (3 mL), and saturated sodium bicarbonate (3 mL). The organics were dried (sodium sulfate) and concentrated in vacuo to afford tert-butyl4-(methylsulfonyloxy)piperidine-1-carboxylate in quantitative yield.quantitative yield. The product was used directly in the next step without thrther purification. A mixture of 4-(4,4,5,5- tetramethyl- 1 ,3,2-dioxaborolan-2-yl-1H-pyrazole (427 mg, 2.2 mmol), tert-butyl 4-(methylsulfonyloxy)piperidine-1-carboxylate (2.0 mmol), and cesium carbonate (847 mg, 2.6 mmol) in DMF (5 mL) was stirred at 100° C. overnight. The mixture was diluted with saturated aqueous NaHCO3 and extracted with EtOAc (3x). The combined organic layers were dried over Na2S04, filtered and concentrated to provide crude pale yellow oil 884 mg. MS (mlz): 378 (M+H).
Reference:
[1] Patent: US2017/8889, 2017, A1, . Location in patent: Paragraph 0309; 0311
[2] Patent: WO2011/100502, 2011, A1, . Location in patent: Page/Page column 37
[3] Patent: CN106336413, 2017, A, . Location in patent: Paragraph 0509; 0510; 0511; 0512
[4] Patent: WO2010/75270, 2010, A1, . Location in patent: Page/Page column 68-69
[5] Patent: WO2011/143646, 2011, A1, . Location in patent: Page/Page column 41-42
[6] Patent: US2012/165305, 2012, A1, . Location in patent: Page/Page column 29
[7] Journal of the American Chemical Society, 2014, vol. 136, # 11, p. 4287 - 4299
[8] Patent: US2014/121200, 2014, A1, . Location in patent: Paragraph 0333; 0334
[9] Patent: WO2010/59771, 2010, A1, . Location in patent: Page/Page column 30-31
39
[ 269410-08-4 ]
[ 76513-69-4 ]
[ 894807-98-8 ]
Yield
Reaction Conditions
Operation in experiment
86%
With potassium carbonate In 1-methyl-pyrrolidin-2-one at 20℃; for 16 h; Inert atmosphere
Synthesis of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2~yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1 t-15a)lnt-15aTo a solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)- H- pyrazole (8.2 g, 41 mmol) in NMP (60 mL) was added K2C03 (12 g, 82 mmoi) and 2-(trimethylsilyi)ethoxymethy. chloride (7.8 mL, 43 mmol) in sequence. The reaction mixture was stirred at r.t. under N2 for 16 h. Then, the reaction mixture was diluted and filtered, and then the filtrate was diluted with EtOAc (300 mL). The resulting solution was washed with sat. NaHC03 (aq) (3 x 200 mL), H20 (4 x 200 mL), brine (1 x 200 mL), dried over Na2S04, filtered, concentrated and dried in vacuo to yield intermediate .nt-15a (11.4 g, 86 percent) as a clear yellowish oil.
86%
Stage #1: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0 - 20℃; for 0.5 h; Inert atmosphere Stage #2: at 0 - 20℃;
[96] SEM-pyrazolo-4-boronic acid pinacol ester was prepared according the procedure from WO2011/130146, page 84. A solution of pyrazolboronic acid pinacolester (20 g, 103 mmol) in DMF (180 mL) was cooled to 0° C and treated with sodium hydride (60 percent dispersion in oil) (6.2 g, 150 mmol) in nitrogen athmosphere. [97] The reaction mixture was stirred at ambient temperature for 30 minutes. The reaction mixture was then cooled to 0° C and (2-(chloromethoxy)ethyl)trimethylsilane (23.65 ml, 134 mmol) was added. The reaction mixture was stirred at ambient temperature overnight. [98] The reaction mixture was poured into aqueous saturated ammonium chloride (200 mL) containing ice (approximately 200 mL) and stirred until the ice melted. The cold mixture was extracted with ethyl acetate twice. The combined organic extracts were washed with water, dried over Na2SO4, and concentrated under reduced pressure to afford SEM-pyrazolo-4-boronic acid pinacol ester (27.6 g, 86 percent yield).
72%
With sodium hydride In tetrahydrofuran at 20℃; Inert atmosphere
Compound 280.1. 4-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)-l-((2- (trimethylsilyl)ethoxy)methyl)-lH-pyrazole. Into a 250-mL three neck round-bottom flask, which was purged and maintained with an inert atmosphere of nitrogen, was placed a solution of 4-(tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (5.82 g, 30.0 mmol) in tetrahydrofuran (80 mL). This was followed by the addition of NaH (70percent) (2.05 g, 85.4 mmol) in portions at 0 °C. To this was added SEMC1 (6.4 mL, 36.1 mmol) dropwise. The reaction mixture was stirred overnight at room temperature, then quenched with 50 mL of NH4CI (sat). The aqueous phase was extracted with 2 x 100 mL of ethyl acetate and the combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. This resulted in 7 g (72percent) of the title compound as colorless oil.
65.94%
With caesium carbonate In tetrahydrofuran; acetonitrile at 20℃; for 2 h;
13.2 4-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-yl)-1-(2-trimethylsilanyl- ethoxymethyl)-1 H-p razole To a solution of 1H-pyrazole-4-boronic acid pinacol ester (0.5 g, 2.57 mmol), in tetrahydrofuran/acetonitrile (3:2, 20ml), 2-(chloromethoxylethyl)trimethyl- silane (0.51 g, 3.09 mmol) and cesium carbonate (1.67 g, 5.15 mmol) are added and stirred for 2 hours at room temperature. The reaction mixture is filtered through celite, and concentrated, the crude mass is taken in ethylacetate (30 ml), washed with water, brine solution, dried over anhydrous MgS04 and concentrated to get the product as brown oil (0.55 g, 65.94 percent); TLC: Pet ether/ethyl acetate(8/2) R - 0.5; 1H NMR: 400 MHz, DMSO-d6: δ [ppm] 8.08 (s, 1H), 7.64 (s, 1 H), 5.40 (s, 2H), 3.48-3.54 (m, 2H), 1.24 (s, 12H), 0.81-0.85 (m, 2H), -0.049(s, 9H);
61%
Stage #1: With sodium hydride In N,N-dimethyl-formamide at 60℃; for 0.333333 h; Stage #2: for 16.0833 h;
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (348 mg, 1.8 mmol) was dissolved in DMF (5 mL) and sodium hydride (60percent dispersion, 86 mg, 2.15 mmol) added and the mixture heated to 60° C. for 5 min. Upon cooling and stirring for an additional 15 min, trimethylsilylethoxymethyl chloride (358 mg, 2.15 mmol, 381 μL) was added dropwise over 5 min and mixture stirred for 16 h. The reaction mixture was diluted with ethyl acetate (25 mL), washed with 5percent lithium chloride (5.x.), dried over sodium sulfate and concentrated. The residue was purified by column chromatography (40 g ISCO column eluting with hexanes and ethyl acetate; gradient 100percent hexanes to 50percent hexanes over 30 min at 30 mL/min) to provide the SEM-protected pyrazole (360 mg, 61percent) as a colorless oil; 1H NMR (500 MHz, CDCl3) δ 7.84 (s, 1H), 7.80 (s, 1H), 5.42 (s, 2H), 3.56-3.53 (t, J=8.3 Hz, 2H), 1.31 (s, 12H), 0.91-0.87 (t, J=8.3 Hz, 2H), -0.03 (s, 9H).
56%
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 3 h;
To a solution of methyl 4-(tetramethyl-1 ,3, 2-dioxaborolan-2-yl)-1H-pyrazole (1 .238 g, 6.316 mmol) in DMF (20 mL) was added potassium carbonate (2.62 g, 18.95 mmol) and [2-(chloromethoxy)ethyl](trimethyl)silane (1 .68 mL, 9.48 mmol) at room temperature. The mixture was stirred for 3 hours and then partitioned between TBME (100 ml.) and water (50 ml_). The organic layer was separated, washed with water (2 x 30 mL) and brine (30 ml_), dried (Na2S04) and concentrated at reduced pressure. The residue was purified by Biotage Isolera™ chromatography [Biotage SNAP Cartridge KP-Sil 50 g; using a gradient of eluents, 0-50percent EtOAc in heptane]. The product containing fractions were combined, concentrated in vacuo to give the title compound (1 .20 g, 56percent yield) as colourless oil. 1H NMR (500 MHz, chloroform-d) δ [ppm] 7.88 (s, 1 H), 7.84 (s, 1 H), 5.46 (s, 2H), 3.61- 3.55 (m, 2H), 1 .35 (s, 12H), 0.96 - 0.90 (m, 2H), 0.00 (s, 9H). LCMS (Analytical Method A): Rt = 1 .34 mins; MS (ESIPos) m/z = 324.95 (M+H)\
46%
Stage #1: With sodium hydride In tetrahydrofuran at 0℃; for 0.0833333 h; Stage #2: at 20℃; for 2 h;
32-(a) 4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-1-(2-trimethylsilylethoxymethyl)-1H-pyrazole; Under argon atmosphere, to 20 ml of tetrahydrofuran solution containing 1.09 g (5.62 mmol) of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole was added 443 mg (11.1 mmol) of 60percent sodium hydride under ice-cooling, and the mixture was stirred for 5 minutes. Then, 3 ml (17.0 mmol) of (2-trimethylsilylethoxy)methyl chloride was added dropwise to the mixture, and the mixture was reacted at room temperature for 2 hours. After completion of the reaction, water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was separated, and the solutions were washed successively with water and then with a saturated aqueous solution of sodium chloride, dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The obtained residue was applied to silica gel column chromatography (Eluent; hexane:ethyl acetate=9:1 (V/V)), and the fractions containing the desired compound were concentrated under reduced pressure to obtain 832 mg of the title compound as a colorless oil. (46percent) Mass Spectrum (CI, m/z): 325 (M++1). 1H-NMR Spectrum (CDCl3, δ ppm): -0.03 (s, 9H), 0.86-0.94 (m, 2H), 1.32 (s, 12H), 3.51-3.59 (m, 2H), 5.43 (s, 2H), 7.81 (d, J=0.5 Hz, 1H), 7.86 (d, J=0.5 Hz, 1H).
0.9 g
Stage #1: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 10 - 35℃; for 1 h; Cooling with ice Stage #2: at 10 - 35℃; for 15 h;
A) 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole [1064] To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (770 mg) in DMF (10 mL) was added sodium hydride (60percent, 114 mg) under ice-cooling. The reaction mixture was stirred at room temperature for 1 hr. To the reaction mixture was added dropwise (2-(chloromethoxy)ethyl)(trimethyl)silane (990 mg) at room temperature, and the mixture was stirred for 15 hr. The reaction mixture was diluted with ethyl acetate, and the mixture was washed with 5percent aqueous lithium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give the title compound (0.90 g). MS(ESI+): [M+H]+ 325.2. MS(ESI+), found: 325.2.
With potassium carbonate In N,N-dimethyl-formamide at 20℃;
A mixture of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (3.0 g, 15.5 mmol), bromomethyl-benzene (3.2 g, 18.7 mmol) and K2CO3 (4.3 g, 31.2 mmol) in DMF (30 mL) was stirred at room temp overnight. After dilution with EtOAc (50 mL) and H2O (50 mL), the organic layer was separated and washed with brine (50 mL), dried over Na2SO4, filtered and concentrated in vacuum. The residue was purified by column chromatography on silica gel eluting with PE/EtOAc (5:1) to give the compound 1-benzyl-4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (3.5 g, 12.3 mmol) as a light yellow solid in 79percent yield. 1H NMR (400 MHz, CDCl3): δ 7.81 (s, 1H), 7.66 (s, 1H), 7.37-7.29 (m, 3H), 7.24-7.22 (m, 2H), 5.30 (s, 2H), 1.29 (s, 12H). LCMS (M+H)+ 285.
66%
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at 0℃; for 0.5 h; Stage #2: at 20℃;
Compound 244.1. l-Benzyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazole. _:Into a 100-mL three neck round-bottom flask, was placed a solution of 4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (3.60 g, 18.6 mmol) in THF (50 mL). Sodium hydride (742 mg, 18.6 mmol, 60percent dispersion in mineral oil) was carefully added in portions at 0 °C. The resulting mixture was stirred for 30 min at 0 °C, then benzyl bromide (2.21 mL, 18.6 mmol) was added. The resulting mixture was stirred overnight at room temperature, then carefully quenched with water (10 mL). The pH of the mixture was adjusted to 9-10 with aqueous HC1 (2 M) and the aqueous phase was extracted with EtOAc (300 mL). The combined organic layers were washed with brine (2 x 150 mL), dried (Na2S04), filtered and concentrated under vacuum. The residue was purified by silica gel chromatography with ethyl acetate/petroleum ether (1/2) as eluent to yield the title compound as a yellow oil (3.47 g, 66percent).
59%
Stage #1: With sodium hydride In tetrahydrofuran at 0℃; for 0.5 h; Stage #2: at 0 - 20℃; for 16 h;
1 -benzyl-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (i34): To a stirred solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (3.0 g, 15.46 mmol) in THF (50 ml_), NaH (0.408 g, 17.01 mmol) was added at 0°C and the reaction was stirred for 30 min. Benzyl bromide (2.9 g, 17.01 mmol) was then added at the same temperature and the reaction was stirred at room temperature for 16h. The progress of the reaction was monitored by TLC. After completion, the mixture was diluted with water and the pH adjusted to 7 using 2 M HCI. The aqueous layer was extracted with ethyl acetate. The organic layer was separated, dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by silica gel (100-200 mesh) column chromatography using 8percent ethyl acetate in n-hexanes as eluent to afford 1-benzyl-4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 H-pyrazole (i34) (2.6 g, Yield 59percent). 1H NMR (400 MHz, DMSO-d6) δ 1 .24 (s, 12H), 5.33 (s, 2H), 7.38-7.20 (m, 5H), 7.60 (s, 1 H), 8.03 (s, 1 H), MS (ESI) m/e (M+1 )+: 285.00
0.28 g
With caesium carbonate In N,N-dimethyl-formamide at 20℃; Inert atmosphere
0.25g 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole was dissolved in 5mL of DMF and 0.63g cesium carbonate was added. To this suspension was added 0.17mL of benzyl bromide and the reaction was stirred overnight at room temperature. The reaction was allowed to settle and the DMF was decanted into a flask. The remaining residue was washed and decanted twice with ethyl acetate and these washes were added to the flask with the DMF. Water was added to the ethyl acetate and DMF and the organic layer removed. The aqueous layer was extracted with ethyl acetate and the combined organic layers were dried over MgS04 and concentrated to give 0.28g of l -benzyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazole which was used without further purification.
With caesium carbonate In acetonitrile at 80℃; for 4 h;
A mixture of 4-(tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1 g, 5.15 mmol, 1.00 equiv), 1-bromo-2-methylpropane (1.05 g, 7.66 mmol, 1.49 equiv) and Cs2CO3 (3.36 g, 10.31 mmol, 2.00 equiv) in acetonitrile (60 mL) was stirred at 80° C. for 4 h. The reaction was cooled to room temperature and the solid material was removed by filtration. The filtrate was diluted with ethyl acetate (30 mL) and then washed with brine (40 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated under vacuum to give 1.14 g (88percent) of 1-iso-butyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole as a colorless oil. 1H-NMR (300 MHz, CDCl3): δ 7.78 (s, 1H), 7.65 (s, 1H), 3.91 (d, J=7.2 Hz, 2H), 2.24-2.19 (m, 1H), 1.32 (s, 12H), 0.90 (d, J=7.2 Hz, 6H) ppm. LCMS (method D, ESI): RT=1.51 min, m/z=251.0 [M+H]+.
31.8%
With caesium carbonate In N,N-dimethyl-formamide at 100℃; for 2 h;
To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H- pyrazole 1a (5.00 g, 25.77 mmol) in DMF(50 mL) was added Cs2CO3 (16.79 g, 51.54 mmol) and 1-bromo-2-methylpropane (7.06 g, 51.54 mmol). The mixturewas stirred at 100°C for 2h. It was cooled to room temperature, water (10 mL) was added and extracted with EA (100mL*3). The combined organic phases was washed with water (100 mL*2) and brine (100 mL*2), dried over Na2SO4,filtrated and evaporated. The residue was purified by silica gel chromatography to give 1-isobutyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole 1b (2.05 g, yield:31.8percent) as a yellow liquid.1H NMR (400 MHz, CDCl3) δ 7.71 (s, 1H), 7.58 (s, 1H), 3.84 (d, 1H), 4.08-4.00 (m, 1H), 2.05-2.20 (m, 1H), 1.25 (s, 12H),0.82 (d, 6H)
Reference:
[1] Patent: US2014/288105, 2014, A1, . Location in patent: Paragraph 0216-0217
[2] Patent: EP3042907, 2016, A1, . Location in patent: Paragraph 0277; 0278
[3] Journal of Medicinal Chemistry, 2009, vol. 52, # 24, p. 7934 - 7937
42
[ 75-30-9 ]
[ 269410-08-4 ]
[ 879487-10-2 ]
Yield
Reaction Conditions
Operation in experiment
57%
With caesium carbonate In N,N-dimethyl-formamide at 0 - 20℃;
2-lodopropane (1.14 g, 6.70 mmol, 0.67 mL)was added to a mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1.00 g, 5.15 mmol) and caesium carbonate (3.49 g,10.72 mmol) in dry N,N-dimethylformamide (20 mL) at 0°O. After stirring for 30 mm the ice- water bath was removed. The reaction mixture was stirred at room temperature overnight. Thereaction mixture was diluted with ethyl acetate (150 mL) and washed with brine (3x100 mL).The organic layer was dried with sodium sulfate and concentrated in vacuo. Purification byflash column chromatography (Method L7; 12 g; heptane, 10percent-30percent ethyl acetate) afforded0.69 g (2.32 mmol; 57percent of theory) of the title compound.GO-MS (Method L9): R1 = 3.86 mm; m/z = 236 M1 H NMR (300 MHz, Ohloroform-d, Method M2) 6 7.79 (s, 1 H), 7.74 (s, 1 H), 4.52 (p, J = 6.7 Hz,1H), 1.50 (d, J = 6.7 Hz, 6H), 1.32 (s, 12H).
Reference:
[1] Journal of Medicinal Chemistry, 2011, vol. 54, # 18, p. 6342 - 6363
[2] Patent: WO2017/178416, 2017, A1, . Location in patent: Page/Page column 114
[3] Patent: US2009/197862, 2009, A1, . Location in patent: Page/Page column 46-47
[4] Patent: US2011/152273, 2011, A1, . Location in patent: Page/Page column 73
[5] European Journal of Medicinal Chemistry, 2018, vol. 158, p. 270 - 285
[6] Patent: WO2009/154557, 2009, A1, . Location in patent: Page/Page column 116-117
[7] Patent: WO2010/59771, 2010, A1, . Location in patent: Page/Page column 36
43
[ 269410-08-4 ]
[ 75-26-3 ]
[ 879487-10-2 ]
Reference:
[1] Journal of Medicinal Chemistry, 2016, vol. 59, # 4, p. 1370 - 1387
[2] Journal of Medicinal Chemistry, 2014, vol. 57, # 10, p. 4196 - 4212
[3] Patent: WO2010/75270, 2010, A1, . Location in patent: Page/Page column 160-161
[4] Patent: WO2014/111871, 2014, A1, . Location in patent: Page/Page column 163; 164
With caesium carbonate In N,N-dimethyl-formamide at 90℃;
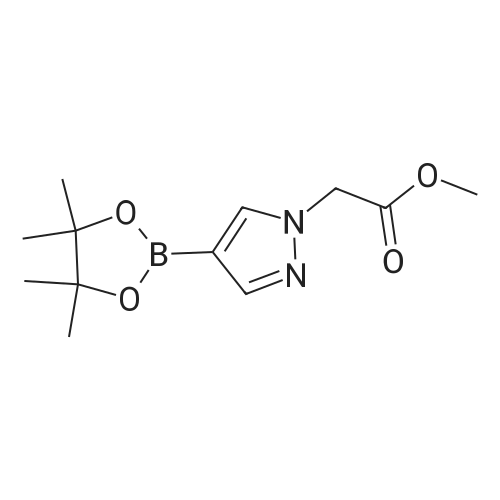
To a solution of 4-(4,4,5,5-Tetramethyl-[1 ,3,2]dioxaborolan-2-yl)-1 H-pyrazole (5.0 g, 25.8 mmol) in DMF (52 mL) was added Cs2CO3 (8.396 g, 25.8 mmol) and bromo-acetic acid methyl ester (2.52 mL, 25.8 mmol). The reaction mixture was heated at 9O0C under nitrogen for overnight. After cooling, the reaction mixture was diluted with water, and extracted with ethyl acetate. The combined extracts were washed with water for three times and brine, dried over Na2SO4., and concentrated to provide 4-(4,4,5,5- tetramethyl-[1 ,3,2]dioxaborolan-2-yl)-pyrazol-1-yl]-acetic acid methyl ester (4.27 g, 62percent yield).
47%
With caesium carbonate In N,N-dimethyl-formamide at 90℃; for 18 h; Inert atmosphere
Intermediate 10: Methyl [4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yI)-1 Hpyrazol-1 -yI]acetate Methyl [4-(4,4, 5, 5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)- 1 H-pyrazol- 1 -yl]acetate was prepared according to the synthetic route shown in Scheme 5. To a solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (560 mg, 2.9 mmol) in anhydrous DMF (10 mL) was added 052003 (1.03 g, 3.16 mmol) and methyl bromoacetate (0.30 mL, 3.16 mmol) and the reaction was stirred at 9000 under a nitrogen atmosphere for 18 hrs. The reaction was cooled, diluted with EtOAc (50 mL), washed with water (2 x 25 mL), brine (25 mL), dried over MgSO4, filtered and concentrated in vacuo. The crude material was purified by column chromatography (50percent EtOAc/heptane) to give the title compound as a colourless oil (360 mg, 47percent).LCMS: m/z 267 [M+H].
With caesium carbonate In N,N-dimethyl-formamide at 0 - 20℃;
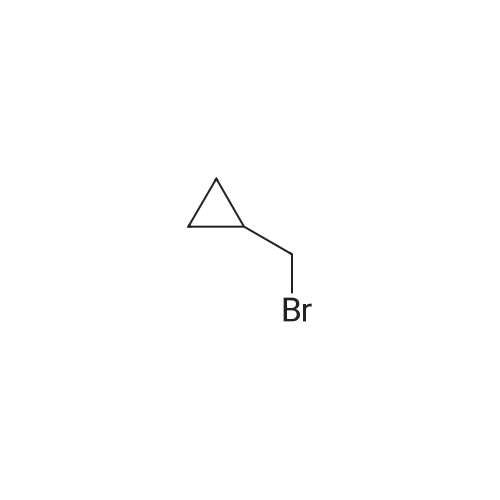
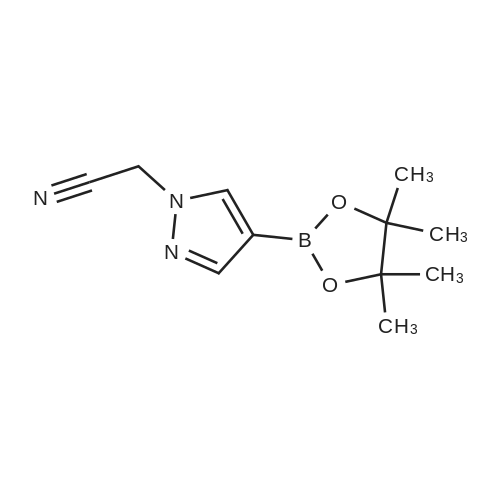
N-N(Bromomethyl)cyclopropane (0.95 mg, 6.70 mmol, 0.70 mL, 95 percent) was added to a mixture of4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (1 .00 g, 5.15 mmol) and caesium carbonate (3.49 mg, 10.72 mmol) in dry N,N-dimethylformamide (20 mL) at 0°O. After stirring for 30 mm the ice-water bath was removed. The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with ethyl acetate (150 mL) and washed with brine (3x100 mL). The organic layer was dried with sodium sulfate and concentrated in vacuo to afforded 1.30 g (4.38 mmol, 85percent of theory) of the title compound. GO-MS (Method L9): R1 = 4.35 mm; mlz = 247 M1 H NMR (300 MHz, Ohloroform-d, Method M2) 6 7.81 (s, 1 H), 7.79 (s, 1 H), 3.99 (d, J = 7.1 Hz,2H), 1.32 (s, 12H), 1.27 (m, 1 H), 0.71 - 0.58 (m, 2H), 0.41 - 0.33 (m, 2H).
49%
Stage #1: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20℃; for 0.333333 h; Inert atmosphere Stage #2: for 24 h; Inert atmosphere
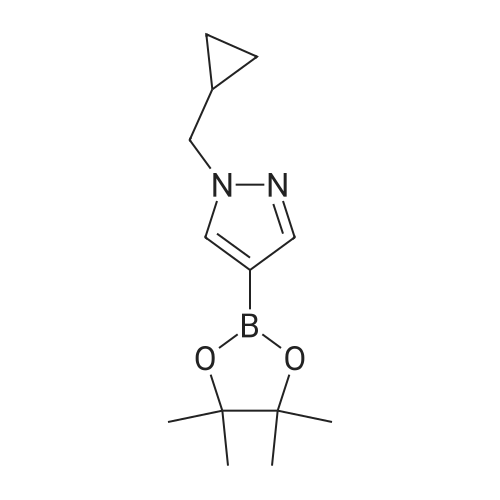
To a reaction vessel containing sodium hydride (60 wtpercent dispersion in mineral oil)(3.09 g, 77.32 mmol) was added NN-dimethylformamide (30 mL) under a stream of nitrogen followed by 4- (4,4,5, 5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (10 g, 51.55 mmol) and the reaction mixture was stirred for 20 min at room temperature. Cyclopropylmethylbromide (8.7 g, 61.86 mmol) was then added and the reaction mixture was stirred for 24 h under an atmosphere of nitrogen. The reaction mixture was slowly poured over saturated aqueous ammonium chloride and extracted with EtOAc. The organic layer was then washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified via flash chromatography on silica gel (solvent gradient: 0percent-100percent EtOAc in cyclohexane) to afford 1- (cyclopropylmethyl)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (6.29 g, 49percent). LCMS (ESI): [M+H]+ = 249.2; lH NMR (400 MHz, CDC13) δ 7.80 (d, J= 7.4 Hz, 2H), 3.99 (d, J = 7.0 Hz, 2H), 1.32 (s, 12H), 1.29 - 1.26 (m, 1H), 0.69 - 0.60 (m, 2H), 0.41 - 0.33 (m, 2H).
48%
With potassium carbonate In N,N-dimethyl-formamide at 60℃; for 14 h;
To a stirred solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (8.00 g,41.2 mmol) in DMF (130 mL) was added potassium carbonate (17.1 g, 124 mmol) and(bromomethyl)cyclopropane (6.0 mL, 62 mmol). The mixture was stirred at 600 C for 14 h. Water was added and the mixture was extracted with ethyl acetate. The organic phase was washed with half-saturated sodium chloride solution, dried (sodium sulfate) and the solvent was removed in vacuum. Silicagel chromatography gave 4.95 g (48 percent yield) of the titlecompound.LC-MS (Method 2): Rt = 1.12 mm; MS (ESIpos): mlz = 249 [M+H]1HNMR (400 MHz, DMSO-d6) O [ppm]: 0.314 (0.66), 0.318 (0.59), 0.325 (0.56), 0.330 (0.65),0.471 (0.53), 0.476 (0.61), 0.492 (0.62), 0.496 (0.57), 1.045 (0.78), 1.219 (1.70), 1.224 (16.00),3.295 (1.54), 3.927 (1.24), 3.946 (1.22), 7.542 (1.07), 7.544 (1.08), 7.932 (1.09).
48%
With potassium carbonate In N,N-dimethyl-formamide at 60℃; for 14 h;
To a stirred solution of 4-(4, 4,5, 5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (purchased from Acros, CAS 269410-08-4, 8.00 g, 41 .2 mmol) in DMF (130 mL) was added potassium carbonate (17.1 g, 124 mmol) and (bromomethyl)cyclopropane (6.0 mL, 62 mmol). The mixture was stirred at 60° C for 14 h. Water was added and the mixture was extracted with ethyl acetate. The organic phase was washed with half-saturated sodium chloride solution, dried (sodium sulfate) and the solvent was removed in vacuum. Silicagel chromatography gave 4.95 g (48 percent yield) of the title compound. LC-MS (Method 2): Rt = 1.12 min; MS (ESIpos): m/z = 249 [M+H]+ 1H-NMR (400 MHz, DMSO-d6) _ [ppm]: 0.314 (0.66), 0.318 (0.59), 0.325 (0.56), 0.330 (0.65), 0.471 (0.53), 0.476 (0.61 ), 0.492 (0.62), 0.496 (0.57), 1.045 (0.78), 1 .219 (1 .70), 1 .224 (16.00), 3.295 (1.54), 3.927 (1.24), 3.946 (1.22), 7.542 (1 .07), 7.544 (1 .08), 7.932 (1.09).
With caesium carbonate In N,N-dimethyl-formamide at 0 - 20℃;
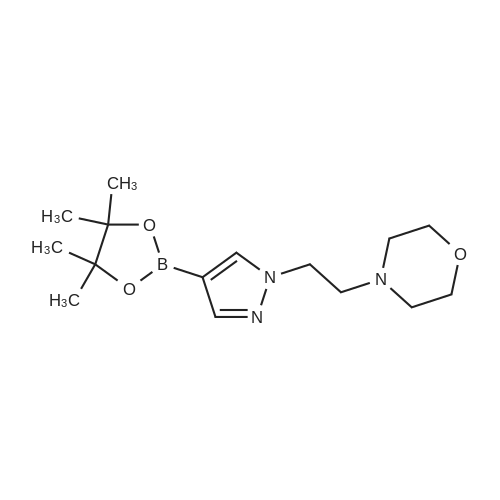
4-(2-Ohloroethyl)morpholine hydrochloride (1 .25 mg, 6.70 mmol) was added to a mixture of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (1 .00 g, 5.15 mmol) and caesium carbonate (5.54 g, 17.01 mmol) in dry N,N-dimethylformamide (20 mL) at 0°O. After stirring for 30 mm the ice-water bath was removed. The reaction mixture was stirred at room temperature for four days. The reaction mixture was diluted with ethyl acetate (150 mL) and washed withbrine (3x100 mL). The organic layer was dried with sodium sulfate and concentrated in vacuo.Purification by flash column chromatography (Method L7; 12 g; ethyl acetate) afforded 0.75 g(2.44 mmol; 47percent of theory) of the title compound.GO-MS (Method L9): R1 = 5.49 mm; (no mass detected)1 H NMR (300 MHz, Ohloroform-d, Method M2) 6 7.80 - 7.75 (m, 1 H), 7.73 (s, 1 H), 4.25 (t, J =6.8 Hz, 2H), 3.73 - 3.64 (m, 4H), 2.81 (t, J = 6.8 Hz, 2H), 2.50 - 2.42 (m, 4H), 1.32 (s, 12H).
Reference:
[1] Journal of Medicinal Chemistry, 2009, vol. 52, # 24, p. 7934 - 7937
49
[ 269410-08-4 ]
[ 5292-43-3 ]
[ 1006875-83-7 ]
Yield
Reaction Conditions
Operation in experiment
85%
With potassium carbonate In acetone at 65℃; for 13 h; Inert atmosphere
A mixture of 4-(4,4, 5, 5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole (2.0 g, 10.31 mmol), tert-butyl 2-bromoacetate (2.212 g, 11.34 mmol) and K2C03 (1.709 g, 12.37 mmol) in acetone (20 mL) was stirred at 65 °C for 13 hours under nitrogen. The reaction was poured into ice water (20 mL) and extracted with ethyl acetate (50 mL x 2). The combined organic phase was washed with brine, dried over magnesium sulfate, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 5/1) to give the title compound (3 g, 8.76 mmol, 85 percent yield) as an oil. LCMS (Method C): m/z 309.2 (M+H), retention time: 1.946 minutes; ‘H NIVIR (400 MHz, CDC13) ö 7.82 (s, 1H), 7.75 (s, 1H), 4.82 (s, 1H), 1.47 (s, 9H), 1.28 (s, 12H).
84%
With caesium carbonate In N,N-dimethyl-formamide at 90℃;
To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1.5 g, 0.0077 mol) in DMF (25 mL) was added 2-bromoacetic acid 1,1-dimethylethyl ester (1.2 mL, 0.0085 mol) and cesium carbonate (3.8 g, 0.012 mol). The suspension was stirred at 90° C. overnight. The reaction mixture was cooled to RT and partitioned with ethyl acetate and water. The organic layer was washed with water, brine, and dried over MgSO4. The solution was concentrated to afford the desired compound (2.0 g, 84percent). Analytical LC/MS: (M+H)+=309.4.
81%
Stage #1: With potassium <i>tert</i>-butylate In tetrahydrofuran; N,N-dimethyl-formamide at 0 - 20℃; for 0.0833333 h; Stage #2: at 0 - 20℃; for 2 h;
1.0 M Potassium tert-butoxide in THF (2.4 mL, 2.4 mmol) was added to a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.39 g, 2.0 mmol) in N,N-dimethylformamide (6.0 mL) at 0° C. The reaction mixture was stirred at room temperature for 5 min. After cooled to 0° C., to the mixture was added t-butyl bromoacetate (0.5 mL, 3 mmol). The reaction was stirred at room temperature for 2 h, then diluted with ethyl acetate, washed with sat. NaHCO3, water, brine, dried over Na2SO4, filtered and concentrated. The product (0.5 g, 81percent) was purified by chromatography eluting with hexanes/EtOAc (max. EtOAc 30percent). LCMS calculated for C15H26BN2O4 (M+H)+: m/z=309.2; Found: 309.1
81%
Stage #1: With potassium <i>tert</i>-butylate In tetrahydrofuran; N,N-dimethyl-formamide at 0 - 20℃; for 0.0833333 h; Stage #2: at 0 - 20℃; for 2 h;
Step 1. tert-Butyl [4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl]acetate 1.0M Potassium tert-butoxide in THF (2.4 mL, 2.4 mmol) was added to a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.39 g, 2.0 mmol) in N,N-dimethylformamide (6.0 mL) at 0° C. The reaction mixture was stirred at room temperature for 5 min. After cooled to 0° C., to the mixture was added t-butyl bromoacetate (0.5 mL, 3 mmol). The reaction was stirred at room temperature for 2 h, then diluted with ethyl acetate, washed with sat. NaHCO3, water, brine, dried over Na2SO4, filtered and concentrated. The product (0.5 g, 81percent) was purified by chromatography eluting with hexanes/EtOAc (max. EtOAc 30percent). LCMS calculated for C15H26BN2O4 (M+H)+: m/z=309.2. Found: 309.1
77%
With potassium carbonate In N,N-dimethyl acetamide at 20℃; for 4 h;
Example 2; Synthesis of (+)-2-[4-(2-fluoro-9-hydroxy-9-trifluoromethyl-9H-fluoren-4-yl)-pyrazol-1-yl]-propane-1,3-diol (compound No. 595); Step 1; [4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyrazol-1-yl]-acetic acid t-butyl ester; 4,4,5,5-Tetramethyl-2-(1H-pyrazol-4-yl)-1,3,2-dioxaborolane (10 g), N,N-dimethylacetamide (100 ml), potassium carbonate (17.8 g) and t-butyl bromoacetate (9.9 ml) were mixed, and the mixture was stirred at room temperature for 4 hr. The reaction mixture was filtered through celite. Water and ethyl ether were added to the filtrate, and the mixture was partitioned in a separatory funnel. The aqueous layer was extracted with ethyl ether, and the organic layers were combined. The organic layer was washed 3 times with water and once with saturated brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure, hexane (50 ml) was added to the obtained residue and the mixture was stirred. This slurry was filtered, and the obtained solid was washed with hexane, and dried under reduced pressure to give the title compound (12.23 g, 77percent).1H-NMR (DMSO-D6) δ: 7.92 (1H, d, J=0.7 Hz), 7.59 (1H, d, J=0.5 Hz), 4.95 (2H, s), 1.42 (9H, s), 1.25 (12H, s).
63%
With caesium carbonate In N,N-dimethyl-formamide at 20℃; for 8 h;
4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1H-pyrazole (0.5 g, 2.6 mmol) Cesium carbonate (1.3 g, 3.9 mmol) was dissolved in N, N-dimethylformamide (10 mL) Tert-butyl bromoacetate (0.6 mL, 3.9 mmol) was added, and the mixture was stirred at room temperature for 8 hours. After completion of the reaction, the reaction mixture was cooled to room temperature, distilled water (50 mL) was added thereto, and the mixture was extracted with ethyl acetate. The organic layer was washed with distilled water and saturated brine, dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel column chromatography (ethyl acetate: n-hexane = 1: 3) to give the title compound 56-a (1.1 g, 63percent) as a yellow solid.
12.23 g
With potassium carbonate In N,N-dimethyl-d6-formamide at 20℃; for 4 h;
Step 8 t-Butyl[4-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)-1H-pyrazol-1-yl]acetate 4,4,5,5-Tetramethyl-2-(1H-pyrazol-4-yl)[1,3,2]dioxaborolane (10 g), N,N-dimethylacetamide (100 ml), potassium carbonate (17.8 g) and t-butyl bromoacetate (9.9 mL) were mixed, and the mixture was stirred at room temperature for 4 hr. The reaction mixture was filtered through celite. Water and diethylether were added to the filtrate, and the mixture was poured into a separating funnel and partitioned. The aqueous layer was extracted again with diethyl ether, and combined with the organic layer. The obtained organic layer was washed three times with water, once with saturated brine, and dried over anhydrous sodium sulfate. The insoluble material was filtered off, and the filtrate was concentrated under reduced pressure. To the obtained residue was added hexane (50 ml) and the mixture was slurry washed (suspension stirred). The suspension was filtered, and the obtained solid was washed with hexane, and dried under reduced pressure to give the title compound (12.23 g). 1H-NMR (400 MHz, DMSO-D6) δ: 7.92 (1H, d, J=0.7 Hz), 7.59 (1H, d, J=0.5 Hz), 4.95 (2H, s), 1.42 (9H, s), 1.25 (12H, s).
Stage #1: With sodium hydride In N,N-dimethyl-formamide; mineral oil at 0 - 20℃; for 0.333333 h; Inert atmosphere Stage #2: at 20℃; for 2 h;
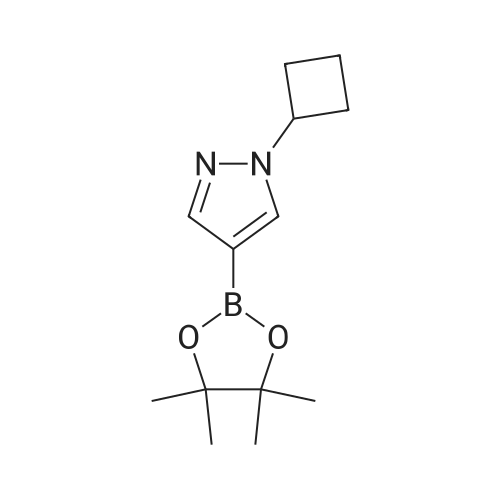
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (300 mg, 1.55 mmol) was dissolved in DMF (5 mL) at 0 °C under Ar. NaH (60percent in oil, 40.8 mg, 1.70 mmol) was added portionwise, and the reaction mixture was then stirred for 20 minutes at rt. A solution of bromocyclobutane (209 mg, 1.55 mmol) in 1 mL of DMF was added, and stirring was continued 2 h at rt. The reaction mixture was quenched with saturated aq. NH4Cl, and then extracted 3x with EtOAc, washed with brine, dried with Na2SO4, filtered and concentrated. The product was purified by silica gel chromatography to provide pyrazole boronate 20A (40 mg, 5.21 percent) . MS (ESI): m/z 249.0 (M+H).
With trifluoroacetic acid In toluene at 90℃; for 2 h;
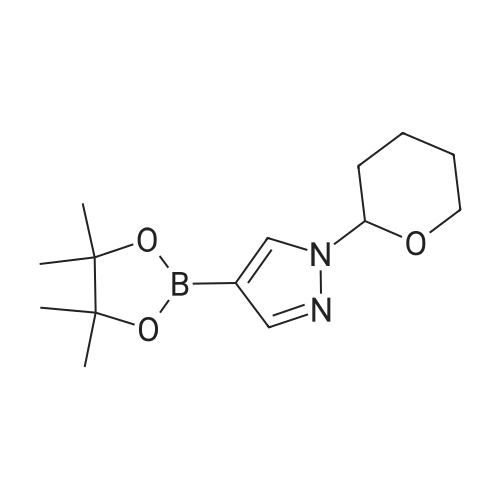
3,4-Dihydro-2H-pyran (5.6 g, 67 mmol) and trifluoroacetic acid (1.17 g, 10.3 mmol) were added to a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H- pyrazole (10.0 g, 51.5 mmol) in toluene (200 mL), and the reaction mixture was heated to 90 °C for 2 hours. After cooling to room temperature, the reaction mixture was partitioned between ethyl acetate (200 mL) and saturated aqueous sodium bicarbonatesolution (100 mL), and the aqueous layer was extracted with ethyl acetate (100 mL). The combined organic layers were washed with saturated aqueous sodium chloride solution (100 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Silica gel chromatography (Gradient: 10percent to 50percent ethyl acetate in petroleum ether) provided the product as a white solid. Yield: 13.4 g, 48.2 mmol, 94percent. 1H NMR (400 MHz, CDCl3) 7.94 (s, 1H), 7.83 (s, 1H), 5.41 (dd, J=9.5, 2.5 Hz, 1H), 4.01-4.08 (m, 1H), 3.65-3.74(m, 1H), 1.98-2.18 (m, 3H), 1.6-1.76 (m, 3H), 1.32 (s, 12H).
90%
With toluene-4-sulfonic acid In dichloromethane at 20℃; for 4 h;
To a solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (110.00 g, 566.89 mmol) in DCM (1000 mL) was added dihydropyran (95.37 g, 1.13 mol, 103.66 mL) and TsOH.H2O (53.92 g, 283.45 mmol). The mixture was stirred at 20 °C for 4 h. TLC (petroleum ether/EtOAc = 5/1) indicated starting material was consumed completely and one main new spot (Rf=0.4) formed. LCMS showed starting material was consumed completely and one main peak with desired mass was detected. The reaction mixture was concentrated under reduced pressure to remove volatiles. The residue was diluted with sat. NaHCCb (350 mL) and the mixture was extracted with EtOAc (2 x 500 mL). The combined organic layers were dried over Na2S04, filtered, and concentrated under reduced pressure to provide a residue. The residue was purified by column chromatography with petroleum ether/ethyl acetate (from 20/1 to 15/1) to afford the title compound (142.00 g, 90percent) as colorless oil. 1H NMR (400 MHz, DMSO-d6) 400 MHz δ 8.06 (s, 1H), 7.61 (s, 1H), 5.42 (dd, J= 10.0, 2.0 Hz, 1H), 3.94-3.86 (m, 1H), 3.66-3.54 (m, 1H), 2.17- 2.03 (m, 1H), 1.96- 1.82 (m, 2H), 1.77-1.37 (m, 3H), 1.33-1.18 (m, 12H). MS (ES+) m/e 279 (M+H).
73%
With toluene-4-sulfonic acid In dichloromethane at 30℃; for 4 h;
To a solution of 4-(4,4, 5, 5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole (10.00 g, 51.54 mmol) in CH2C12 (100.00 mL) was added DHP (8.67 g, 103.08 mmol, 9.42 mL) and TsOHH20 (4.90 g, 25.77 mmol). The mixture was stirred at 30 °C for 4 hour. LCMS showed 4-(4,4, 5,5 -tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazolewas consumed completely and one main peak with desired mass was detected. The reaction mixture was concentrated under reduced pressure to remove solvent. The residue was diluted with sat.NaHCO3 (35 mL) and the mixture was extracted with EtOAc (100 mL). The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (Si02, Petroleum ether/Ethyl acetate = 20/1 to 1/1) to afford the title compound (10.5 g, 73percent) as colorless oil. ‘H NIVIR (400 MHz, DMSO-d6) ö 8.06 (s, 1H), 7.62 (s, 1H), 5.44-5.41 (m, 1H), 3.94-3.89 (m, 1H), 3.76-3.58 (m, 2H), 3.46-3.41 (m, 1H), 2.15-2.06 (m, 1H), 1.95-1.84 (m, 2H), 1.76-1.40 (m, 9H), 1.26 (s, 12H).
73%
With toluene-4-sulfonic acid In dichloromethane at 30℃; for 4 h;
[0406] To a solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (10.00 g, 51.54 mmol) in CH2C12 (100.00 mL) was added DHP (8.67 g, 103.08 mmol, 9.42 mL) and TsOH H20 (4.90 g, 25.77 mmol). The mixture was stirred at 30 °C for 4 hour. LCMS showed 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazolewas consumed completely and one main peak with desired mass was detected. The reaction mixture was concentrated under reduced pressure to remove solvent. The residue was diluted with sat.NaHCC (35 mL) and the mixture was extracted with EtOAc (100 mL). The organic layer was dried over Na2S04, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (Si02, Petroleum ether/Ethyl acetate = 20/1 to 1/1) to afford the title compound (10.5 g, 73percent) as colorless oil. 1H NMR (400 MHz, DMSO-i/6) δ 8.06 (s, 1H), 7.62 (s, 1H), 5.44-5.41 (m, 1H), 3.94-3.89 (m, 1H), 3.76-3.58 (m, 2H), 3.46-3.41 (m, 1H), 2.15-2.06 (m, 1H), 1.95-1.84 (m, 2H), 1.76-1.40 (m, 9H), 1.26 (s, 12H).
5.04 g
With toluene-4-sulfonic acid In 1,2-dichloro-ethane at 50℃; for 2 h;
To a stirred solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (5.0 g, 25.76 mmol) in dry dichloroethane (128 ml), 3,4-dihydro-2H-pyran (4.71 ml, 51.52 mmol) was added followed by p-toluenesulfonic acid (490 mg, 2.576 mmol) and the reaction mixture was stirred at 50 °C for 2 h. The reaction mixture was quenched with aqueous saturated solution of sodium bicarbonate (75 ml) and extracted with ethyl acetate (2 x 150 ml). The combined organic layer was washed with brine (100 ml) and dried over sodium sulphate. The mixture was evaporated under reduced pressure and residue obtained was purified by column chromatography to yield 5.04 g of product as a white solid. 1H NMR (300 MHz, CDCI3): δ I .31 (s, 12H), 1.54-1.70 (m, 3H), 1.98-2.20 (m, 3H), 3.68 (t, / = 10.2 Hz, 1H), 4.04 (d, / = I I .7 Hz, 1H), 5.35-5.45 (m, 1H), 7.82 (s, 1H), 7.93 (s, 1H).
With sodium hydroxide In N,N-dimethyl-formamide at 140℃; for 16 h;
Intermediate 1182-[4-(4,4,5,5-tetramethyl-1 ,3,2-dioxa H-pyrazol-1 -yl]ethanolA solution of 1 ,3-dioxolan-2-one (2.496 g, 28.3 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 H-pyrazole (5 g, 25.8 mmol) and sodium hydroxide (0.103 g, 2.58 mmol) in N,N-Dimethylformamide (DMF) (18 mL) was stirred at 140°C for 16 h. then cooled to room temperature and treated with activated charcoal (200mg). The resulting mixture was stirred at room temperature for 1 h then filtered through celite (10 g). The insoluble were washed with EtOAc (50 mL) and EtOH (50ml_). The combined filtrate and washings were concentrated in vacuo. Purification of the residue by flash chromatography on silica gel using a 50 G silica cartyridge (gradient: 0 to 40percent MeOH in DCM) gave a residue which was further purified by SP4 using a 100 G silica cartridge (eluant: 0 to 20percent MeOH in DCM) to give 2-[4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazol-1 - yl]ethanol (5.58 g, 23.44 mmol, 91 percent yield) as a colourless oil which was used in the next step without further purification.LCMS (method A): Retention time 0.68 min, [M+H]+ = 239.13
85%
With sodium hydroxide In N,N-dimethyl-formamide for 2.5 h; Heating / reflux
INTERMEDIATE 34; 2-[4-f4.4.5.5-Tetramethyl-ri.3.21dioxaborolan-2-ylVρyrazol-l-yl1-ethanol; 4-Pyrazoleboronic acid pinacol ester (0.25 g, 1.29 mmol), ethylene carbonate(0.125 g, 1.42 mmol) and sodium hydroxide (5 mg, 0.13 mmol) were dissolved in DMF (1 mL) and the reaction mixture was heated to reflux for 2 Vi h. It was cooled to r.t. before addition of activated charcoal (25 mg). The resulting suspension was stirred at r.t. for Ih and then filtered through celite, washed with DMF (6 mL) and concentrated in vacuo to give the title compound (0.26g, 85percent) as a yellow oil. LCMS (ES+) 239.18 (M+H)+.
76%
With caesium carbonate In N,N-dimethyl-formamide at 140℃; for 0.5 h;
100 mg of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole and 91 mg of 1,3-dioxolan-2-one weredissloved in 2 ml of dimethylformamide. 336 mg of cesium carbonate was heated to 140 °C, stirred for 0.5 h and thencooled to room temperature and concentrated. The residue was purified by column chromatography (ethyl acetate:petroleum ether = 30: 70) to give 93 mg of 2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)ethanol aspale yellow oil. Yield: 76percent.1H NMR (300 MHz, DMSO-d6) δ (ppm): 1.25 (s, 12H), 3.71 (q, J= 5.4 Hz, 2H), 4.15 (t, J = 5.4 Hz, 2H), 4.87 (t, J = 5.4Hz, 1H), 7.57 (s, 1H), 7.88 (s, 1H).
7.53 g
With sodium hydroxide In N,N-dimethyl-formamide at 140℃;
A solution of I ,3-dioxolan-2-one (1 .902 mL, 28.5 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)-IH-pyrazole (5.0239 g, 25.9 mmol) and sodium hydroxide (0.0998 g, 2.495 mmol) in N,Ndimethylformamide (DMF) (20 mL) was heated to 140 °C overnight. The mixture was cooled down to rt and then activated charcoal (200 mg) was added and this was stirred for 4 h before filtering through celite cartridge (10 g). The mixture was washed with EtOAc (50 mL) and EtCH (50 mL andthe combined filtrate was concentrated in vacuo to afford 7.53 g of brown oil. This was used crude in further reactions. LCMS: Not recorded.
With caesium carbonate In N,N-dimethyl-formamide at 70℃;
4-(4,4,5,5-Tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (1 .00 g, 5.15 mmol) and cesium carbonate (5.04 g, 15.47 mmol) were suspended in 10 mL of dimethylformamide. 2-Bromoethanol (0.73 mL, 10.30 mmol) was added and the mixture was stirred at 70 °C for 3 hours. Additional amounts of cesium carbonate (5.04 g, 15.47 mmol) and 2-bromoethanol (0.73 mL, 10.30 mmol) were added and the mixture was left at 70 °C overnight. The solvent was evaporated under reduced pressure and the residue was partitioned between water and ethyl acetate. The organic layer was washed with water and brine, dried over magnesium sulphate, filtered and the solvent was evaporated to give 770 mg (63percent yield) of a yellowish oil that was used in the next step without further purification.LRMS (m/z): 239 (M+1 )+.
31.47%
With caesium carbonate In N,N-dimethyl-formamide at 100℃; for 48 h; Inert atmosphere; Sealed
Step A: Preparation of 2-(4-(4 A5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 H- pyrazol- 1 -yDethanol : To a flask charged with 4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2- yl)-lH-pyrazole (1.000 g, 5.154 mmol), Cs2C03 (2.687 g, 8.246 mmol), and 2-bromoethanol (0.5479 mL, 7.730 mmol) was added 10 mL of DMF and the flask was sealed under nitrogen and heated to 100 °C for 48 hours. The reaction was diluted with ethyl acetate (100 mL) and stirred for 30 minutes before it was passed through a glass microfiber filter and the cake was washed with ethyl acetate. The organic was concentrated under reduced pressure. The crude was then purified by silica gel chromatography eluting with a gradient of 75-100percent ethyl acetate in Hexanes to afford 2-(4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-lH-pyrazol- l-yl)ethanol (0.4290 g, 1.622 mmol, 31.47percent yield). MS (apci) m/z = 239.2 (M+H).
Stage #1: With hydrogenchloride In 1,4-dioxane; toluene at 35 - 40℃; Inert atmosphere Stage #2: With sodium hydrogencarbonate In 1,4-dioxane; toluene for 1 h;
1-(Ethoxyethyl)-4-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (19).; A 22 L 4-neck flask equipped with a mechanical stirrer, thermowell, addition funnel, condenser and N2 inlet was charged with 4-(4,4,5,5-tetra-methyl[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (17, 1.42 kg, 7.32 mol), toluene (9.5 L) and ethyl vinyl ether (18, 790.5 g, 1050 mL, 10.98 mol, 1.50 equiv). A 4 M HCl in dioxane (50 mL) was added via an addition funnel over 10 minutes and the resulting reaction mixture was heated at 35-40° C. for 7 hr to give a clear homogeneous solution. When the reaction was shown to be complete by GC, solid NaHCO3 (130 g) was added and the mixture was stirred for 1 hr before being filtered. The filtrate was concentrated under reduced pressure. Heptane (200 mL) was added to the residue to affect crystallization. The solid was collected by filtration and dried in a vacuum oven to afford 1-(ethoxyethyl)-4-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (19, 1.896 Kg, 1.948 Kg theoretical, 97.3percent) as a white to off-white solid. For 19: 1H NMR (DMSO-d6, 400 MHz) δ ppm 8.09 (s, 1H), 8.58 (s,1H), 7.62 (s,1H), 5.55 (q, 1H, J=6.1 Hz), 3.37 (dq, 1H, J=7.1, 9.6 Hz), 3.12 (dq, 1H, J=7.0, 9.7 Hz), 1.56 (d, 3H, J=6.0 Hz), 1.24 (s, 12H), 1.00 (t, 3H, J=7.0 Hz); C13H23BN2O3 (MW, 266.14), LCMS (EI) m/e 267 (M++H).
Reference:
[1] Patent: US2010/190981, 2010, A1, . Location in patent: Page/Page column 103-104
[2] Journal of Medicinal Chemistry, 2017, vol. 60, # 20, p. 8336 - 8357
[3] Bioorganic and Medicinal Chemistry Letters, 2018, vol. 28, # 8, p. 1357 - 1362
With potassium carbonate In 1,4-dioxane; water at 90℃; for 2 h; Inert atmosphere
4-(1H-Pyrazol-4-yl)-7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (5).; Method A.; To a flask equipped with a reflux condenser, a nitrogen inlet, mechanical stirrer, and a thermowell was added 4-chloro-7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (3a, 817 g, 2.88 mol) and dioxane (8 L). To this solution was added 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (4, 728 g, 3.75 mol, 1.30 equiv) followed by a solution of potassium carbonate (K2CO3, 1196 g, 8.67 mol, 3.0 equiv) in water (4 L). The solution was degassed by passing a stream of nitrogen through the solution for 15 minutes before being treated with tetrakis(triphenylphosphine)palladium(0) (167 g, 0.145 mol, 0.05 equiv) and the resulting reaction mixture was heated at reflux (about 90° C.) for 2 hours. When the reaction was deemed complete by TLC (1:1 heptane/ethyl acetate) and LCMS, the reaction mixture was cooled to room temperature, diluted with ethyl acetate (24 L) and water (4 L). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (4 L). The combined organic layers were washed with water (2.x.2 L), brine (2 L), dried over sodium sulfate (Na2SO4), and concentrated under reduced pressure. The residue was suspended in toluene (4 L) and the solvent was removed under reduced pressure. The residue was finally triturated with methyl tert-butyl ether (MTBE, 3 L) and the solids were collected by filtration and washed with MTBE (1 L) to afford 4-(1H-pyrazol-4-yl)-7-(2-trimethylsilanyl-ethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (5, 581.4 g, 908.5 g theoretical, 64percent yield) as white crystalline solids. For 5: 1H NMR (DMSO-d6, 400 MHz) δ ppm 13.41 (bs, 1H), 8.74 (s, 1H), 8.67 (bs, 1H), 8.35 (bs, 1H), 7.72 (d, 1H, J=3.7 Hz), 7.10 (d, 1H, J=3.7 Hz), 5.61 (s, 2H), 3.51 (t, 2H, J=8.2 Hz), 0.81 (t, 2H, J=8.2 Hz), 0.13 (s, 9H); C15H21N5OSi (MW, 315.45), LCMS (EI) m/e 316 (M++H).
27%
Stage #1: With potassium carbonate In 1,4-dioxane; water at 20℃; for 0.166667 h; Inert atmosphere Stage #2: With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride In 1,4-dioxane; waterInert atmosphere
At room temperature, 4-chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (12f) (800 mg, 2.82 mmol) and 4-pyrazoleboronic acid pinacol ester (842 mg, 4.34 mmol) were dissolved in dioxane (10 mL), water (2 mL) and potassium carbonate (857 mg, 6.2 mmol) were then added, nitrogen atmosphere protection was applied, and the reaction was stirred at room temperature for 10 min. Under protection of nitrogen, Pd(dppf)Cl2 (227 mg, 0.31 mmol) was added. The reaction was placed in an oil bath at 95°C, and stirred overnight. TLC indicated starting materials substantially disappeared. The reaction was quenched with water, extracted with EA, and the organic phase was dried over anhydrous sodium sulfate, and purified by preparative flash chromatography (PE:EA=2:3), to afford 4-(1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (12g) (240 mg, brown solid), yield: 27percent. MS (ESI, m/z): 316 [M+H]+.
166.4 g
Stage #1: With potassium carbonate In water at 25 - 30℃; Inert atmosphere Stage #2: at 78 - 80℃; for 2 h; Inert atmosphere
Nitrogen gas was purged through a mixture of 4-chloro-7- { [2- (0224) (trimemylsilyl)ethoxy]memyl}-7H-pyIτolo[2,3-
A mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (30.0 mg, 0.154 mmol), and tetrahydro-2H-pyran-4-yl methanesulfonate (0.150 g, 0.460 mmol) in acetonitrile (1.0 mL) was stirred at 90° C. for 2 hours. The reaction was quenched with water, extracted with ethyl acetate. The combined organic layer was dried over MgSO4, then filtered and concentrated under reduced pressure to afford the desired product (35 mg, 80percent), which was used directly in the next step without further purification. LCMS calculated for C14H24BN2O3 (M+H)+: m/z=279.2. Found: 279.2.
40%
Stage #1: With sodium hydride In N,N-dimethyl-formamide at 0 - 20℃; for 0.5 h; Stage #2: at 110℃;
To a solution of 4-(4,4,5 ,5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1H-pyrazole (1.0 g, 5.2 mmol) in DMF (10 mL) was added NaH (0.3 g, 7.5 mmol) at 0 °C and the mixture was stirred at rt for 30 mi Tetrahydro-2H-pyran-4-yl methanesulfonate (1.1 g, 6.1 mmol) was added and the mixture was stirred at 110 °C overnight. The reaction mixture was cooled to rt and filtered. The filtrate was concentrated and the residue was purified by silica gel chromatography (30percent EA:PE) to give 550 mg of the title compound (40percent). [M+H] Calc’d for C,4H23BN203, 279; Found, 279.
38%
With caesium carbonate In N,N-dimethyl-formamide at 100℃;
A mixture of tetrahydro-2H-pyran-4-yl methanesulfonate (2.8 g, 16 mmol) , 4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -1H-pyrazole (2.01 g, 10.4 mmol) and cesium carbonate (5.4 g, 17 mmol) in DMF (30 mL) was stirred at 100 overnight. The reaction mixture was concentrated in vacuo to remove DMF. The residue was diluted with water (20 mL) . The resulting mixture was extracted with DCM (30 mL × 3) . The combined organic layers were dried over anhydrous Na2SO4and concentrated in vacuo. The residue was purified by pre-HPLC to give a light yellow oily product (1.1 g, 38) .[1589]MS (ESI, pos. ion) m/z: 279.3 [M+1]+.
16.4%
With caesium carbonate In N,N-dimethyl-formamide at 100℃; for 1 h; Inert atmosphere; glass bomb
Step B: Preparation of 1 -(tetrahydro-2H-pyran-4-yl)-4-(4,4,5,5-tetramethyl-1 ,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole: In 8 mL of Ν,Ν-dimethylformamide were combined 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.75 g, 3.87 mmol), tetrahydro- 2H-pyran-4-yl methanesulfonate (1.04 g, 5.80 mmol), and CS2CO3 (2.01 g, 6.18 mmol) in a glass bomb and then sealed under nitrogen and heated to 100 °C. After one hour the reaction was cooled and water was added to dissolve all solids. The solution was diluted with water and then extracted with EtOAc (250 mL). The organic layer was then washed with water and brine. The combined aqueous layer was then extracted with EtOAc (200 mL). The organic layers were combined, dried over MgS04, filtered, and concentrated under reduced pressure. The crude material was dried overnight on high vacuum. The crude was purified by way of silica gel chromatography eluting with a gradient of 15-25percent EtOAc in DCM to afford 1- (tetrahydro-2H-pyran-4-yl)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.176 g, 0.633 mmol, 16.4percent yield). MS (apci) m/z = 279.2 (M+H).
With potassium carbonate; sodium iodide In acetonitrile at 70℃;
A mixture of 4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)- lH- pyrazole (0.50 g, 2.58 mmol), sodium iodide (39 mg, 0.26 mmol) bromoacetonitrile (1 .3 g, 10.8 mmol) and potassium carbonate (1.0 g, 7.8 mmol) in acetonitrile (10 mL) was heated at 70 °C overnight. Water was added and the solution was extracted with EtOAc (3x). The organic was dried over Na2S04, filtered and concentrated. The residue was purified by silica gel column chromatography ( 10percent to 100percent EtOAc/hexane) to obtain 2-(4-(4,4,5,5-tetramethyl-l ,3,2- dioxaborolan-2-yl)-lH-pyrazol-l-yl)acetonitrile (0.38g, 63percent yield). MS (ESI) m/z: 234.1 (M+H+).
39%
With potassium carbonate In N,N-dimethyl-formamide at 100℃; for 24 h;
A solution of 4-(4,4, 5,5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1H- pyrazole (5.11 g, 26.3 mmol) in DMF (50 mL) was treated with bromoacetonitrile (2.20 mL, 31.6 mmol) and K2CO3() (5.46 g, 39.5 mmol). The resulting suspension was stirred for 24 h at 100 °C. The reaction mixture was cooled to ambient temperature, diluted with water (100 mL) then extracted with EtOAc (3 x 250 mL). The combined organic extracts were washed with water (3 x 50 mL) and brine (50 mL) then dried over anhydrous Na2SO4(). Following filtration, the organic extracts were concentrated under vacuum then purified by silica chromatography (10-60percent Hexanes/EtOAc as the gradient eluent) to afford the title compound (2.42 g, 39percent yield). ‘H NIVIR (400 MHz, DMSO-d6) 8.04 (s, 1H), 7.71 (s, 1H), 5.49 (s, 2H), 1.25 (s, 12H).
With caesium carbonate In acetonitrile at 90℃; for 3 h;
Step 1: Preparation of 2-(4-(4,4,5 ,5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 Hpyrazol- 1 -vi) acetamide (Intermediate 24) A mixture of 4- (4,4,5 ,5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole(200 mg, 1.03 mmol), 2-bromoacetamide (213 mg, 1.54 mmol) and Cs2CO3 (1.27 g,3.92 mmol) in CH3CN (5.0 mL) was stirred at 90 °C for 3 hours. After being cooledto room temperature, the reaction mixture was diluted with water and extracted withEtOAc twice. The combined organic layers were dried over Na2SO4, filtered andconcentrated in vacuo to obtain the title compound (174 mg, 67percent) as a white solid, which was used for the next reaction without further purification.1H-NMR (400 MHz, CDC13): ö 7.89 (s, 1H), 7.76 (s, 1H), 6.21 and 5.41 (brs,2H), 4.83 (s, 2H), 1.32 (s, 12H).
70 mg
With caesium carbonate In N,N-dimethyl-formamide at 100℃; for 0.583333 h; Sealed tube; Microwave irradiation
To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole 1a (100 mg, 0.52 mmol) in DMF(3 mL) was added 2-bromoacetamide 1b (143.5 mg, 1.04 mmol) and Cs2CO3 (253.5 mg, 0.78 mmol) in a sealed tubeat RT. Reaction mixture was stirred at 100 °C for 35 min under microwave irradiation. After cooling at room temperature,the mixture was diluted with EA (10 mL) and water (10 mL), extracted with EA (10 mL*3), and the organic layers werecombined and washed with water (30 mL*3) and brine (50 mL), dried over Na2SO4, filtrated and evaporated to afford2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)acetamide 1c (70 mg) as a yellow oil and used in thenext step directly.MS m/z (ESI): 252.2 [M+1]1H NMR (100 MHz, CDCl3) δ 7.9 (d, 2 H), 2.05 (s, 2 H), 1.32 (s, 12 H)
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In 1,4-dioxane; water for 14 h; Heating
(4-Chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate (10 g; Formula V), water (50 mL), and potassium carbonate (15.5 g) were added into a reaction vessel at ambient temperature. 4-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazole (8.7 g; Formula VIII), 1,4-dioxane (100 mL), and tetrakis(triphenylphosphine)palladium(0) (0.08 g) were added to the reaction mixture. The reaction mixture was heated to a temperature of 80°C to 85°C, and then stirred at the same temperature for 14 hours. The progress of the reaction was monitored by thin layer chromatography. On completion, ethyl acetate (100 mL) was added to the reaction mixture. The contents were stirred for 1 hour,then filtered through a Hyflo, and then washed with ethyl acetate (40 mL). The organic layer was separated, and then concentrated under reduced pressure to obtain [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3- d]pyrimidin-7-yl]methyl pivalate. Yield: 82.27percent
With 18-crown-6 ether In acetonitrile for 18 h; Reflux
Step 1: l-(difluoromethyl)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole [00218] A 100 mL round bottom flask was charged with 4-pyrazoleboronic acid pinacol ester (1.0 g, 5.15 mmol), 18-crown-6 (0.27 g, 1.03 mmol) and anhydrous acetonitrile (25 mL). The reagents were stirred until a colorless solution formed then sodium chlorodifluoroacetate (0.94 g, 6.18 mmol) was added and the reaction mixture heated to reflux for 18 h. After this time the reaction mixture was cooled to r.t. and the precipitated solid removed by filtration through celite, washing with EtOAc (3 x 20 mL). Combined organics were filtered through a hydrophobic frit and condensed to give a pale yellow oil. The crude product was purified by flash silica column chromatography (gradient elution ο-hexane to 33percent EtOAc in /so-hexane) to give the title compound as a colorless solid (1.06 g, 84percent). MS (ES+) consistent with target (M+H)+.
84%
With 18-crown-6 ether In acetonitrile for 18 h; Reflux
Step 1 : 1 -(difluoromethyl)-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- pyrazole [00256] A 100 mL round bottom flask was charged with 4-pyrazoleboronic acid pinacol ester (1 .0 g, 5.15 mmol), 18-crown-6 (0.27 g, 1 .03 mmol) and anhydrous acetonitrile (25 mL). The reagents were stirred until a colorless solution formed then sodium chlorodifluoroacetate (0.94 g, 6.18 mmol) was added and the reaction mixture heated to reflux for 18 h. After this time the reaction mixture was cooled to r.t. and the precipitated solid removed by filtration through Celite, washing with EtOAc (3 x 20 mL). Combined organics were filtered through a hydrophobic frit and condensed to give a pale yellow oil. The crude product was purified by flash silica column chromatography (gradient elution, 0-33percent EtOAc in /so-hexane) to give the title compound as a colorless solid (1 .06 g, 84percent). MS (ES+) consistent with target (M+H)+.
80%
for 24 h; Reflux; Inert atmosphere
Step 1: To acetonitrile (100 ml) was added compound 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole 101-1 (4.0 g, 0.21 mol), sodium chlorodifluoroacetate (3.77 g 0.25 mol, 15-crown-5 (103.8 g, 0.65 mol), and the reaction mixture was heated to reflux under nitrogen and stirred for 24 h, cooled to room temperature, the reaction mixture was poured into water (50 ml) to quench the reaction, and extracted with EA (100 ml×3), washed with saturated brine (20 ml), dried over Na2SO4, and filtered, and the filtrate was evaporated to give compound 101-2 (4.1 g, yield 80percent) as a white solid. MS m/z (ESI): 245.1 [M+H]+.
47%
With 18-crown-6 ether In acetonitrile for 20 h; Reflux
Reference Example 1711-(Difluoromethyl)-1H-pyrazole-4-boronic acid pinacol ester A suspension of 1H-pyrazole-4-boronic acid pinacol ester (5.16 g, 26.6 mmol), CF2ClCO2Na (4.86 g, 31.9 mmol), and 18-crown-6 (1.41 g, 5.32 mmol) in CH3CN (100 mL) was refluxed for 20 h. After cooling to room temperature, the reaction mixture was poured into water and extracted with AcOEt. The extract was washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by basic silica gel column chromatography eluting with AcOEt to give the title compound (3.03 g, 47percent yield) as a yellow oil: 1H NMR (300 MHz, CDCl3): δ ppm 1.33 (12H, s), 7.22 (1H, t, J=60.7 Hz), 7.89 (1H, s), 8.13 (1H, s).
500 mg
With 18-crown-6 ether In acetonitrile for 20 h; Reflux
A suspension of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (516 mg, 2.66 mmol), CF2ClCO2Na (486 mg, 3.19 mmol), and 18-crown-6 (141 mg, 0.532 mmol) in CH3CN (150 mL) was heated to reflux for 20 hours. After cooled to room temperature, thereaction mixture was poured into water and extracted with EtOAc. The extracts were washed with brine, dried over MgSO4, and concentrated under reduced pressure to afford a crude 1-(difluoromethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (500 mg), which was used for the next step without further purification. MS (ESI) m/z: 245 [M+H]+.
1.54 g
With 18-crown-6 ether In acetonitrile at 90℃; for 16 h;
E) 1-(difluoromethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole To a mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1.50 g), sodium chlorodifluoroacetate (1.41 g) and acetonitrile (35 mL) was added 18-crown-6 (0.409 g). The reaction mixture was stirred at 90° C. for 16 hr, poured into water, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure to give the title compound (1.54 g). MS (API+): [M+H]+245.1.
With potassium carbonate In N,N-dimethyl-formamide at 80℃; for 6 h;
A mixture of 1- (2-chloroethyl) pyrrolidine hydrochloride (1.48 g, 8.70 mmol) , 4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -1H-pyrazole (1.7 g, 8.8 mmol) and potassium carbonate (3.7 g, 27.00 mmol) in DMF (15 mL) was stirred at 80 for 6 h. The reaction mixture was diluted with water (50 mL) . The resulting mixture was extracted with DCM (50 mL × 3) . The combined organic layers were washed with saturated aqueous NaCl (15 mL) , dried over anhydrous Na2SO4and concentrated in vacuo. The residue was purified by silica gel column chromatography eluted with DCM/EtOAc (v/v) 20/1 to give a light yellow liquid product (1.27 g, 50.10) .[1772]MS (ESI, pos. ion) m/z: 292.10 [M+1]+
Stage #1: With sodium hydride In tetrahydrofuran; mineral oil at 0 - 20℃; for 1.5 h; Stage #2: With potassium iodide In tetrahydrofuran at 50℃;
To a solution of 4,4,5,5-tetramethyl-2-(1H-pyrazol-4-yl)-1,3,2-dioxaborolane (250 mg, 1.29 mmol) and sodium hydride (61.8 mg, 2.58 mmol) in tetrahydrofuran at 0° C. was added 2-bromo-N,N-diethylethanamine (558 mg, 2.58 mmol). The reaction was allowed to warm up to room temperature and was monitored by LCMS. After 90 minutes there was still no reaction and potassium iodide (1.71 g, 10.3 mmol) was added and the reaction was heated at 50° C. overnight. The reaction mixture was diluted with a large volume of ethyl acetate and water and partitioned. The organic layer containing the product was washed with brine and concentrated in vacuo to give clear thick oil confirmed by LCMS to be 100percent pure N,N-diethyl-2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)ethanamine (340 mg, yield 90percent, M+1 294.2)
With potassium carbonate In acetonitrile at 35℃; for 3 h; Inert atmosphere
Intermediate 941 -[(Methyloxy)methyl]-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- pyrazole 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (10.00 g, 51.5 mmol) was dissolved in acetonitrile (40 mL) and stirred at 35°C for 5 min under nitrogen atmosphere then cooled to room temperature, lodomethyl methyl ether (21.83 mL, 258 mmol) and potassium carbonate (35.6 g, 258 mmol) were added and the resulting mixture was stirred at 35°C for 3 h then cooled to room temeprature. The insoluble material was filtered off and the filtrate was concentrated in vacuo. The residue was dissolved in EtOAc and the organic phase was washed with water, dried over MgS04 and concentrated in vacuo. Purification of the residue by SP4 using a 100 G silca cartridge (gradient: 0 to 50percent AcOEt in hexanes) gave 1 -[(methyloxy)methyl]-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)- 1 H-pyrazole (6.45 g, 50percent pure by LCMS, 26percent) as a yellow liquid which was used in the next step without further purification.LCMS (method G): Retention time 0.85 min, [M+H]+ = 238.8
260 mg
With potassium carbonate In acetonitrile at 35℃; for 4.5 h;
Intermediate 12: 1-[(Methyloxy)methyl]-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole[0399]4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (400 mg, 2.061 mmol) (Aldrich) was dissolved in acetonitrile (25 ml) and stirred for 5 min at 35° C. Iodomethyl methyl ether (0.873 ml, 10.31 mmol) and potassium carbonate (1424 mg, 10.31 mmol) were added and the mixture was allowed to stir at 35° C. for 3 h.[0401]The LCMS showed incomplete reaction. The mixture was left to stir at 35° C. for one additional hour. The LCMS showed no progression so 2 eq (349 μl) of the alkylating agent was added and the mixture was allowed to stir at 35° C. for 30 min. Ammonium chloride was then added. The mixture was partitioned between ethyl acetate and water. The aqueous layer was re-extracted with ethyl acetate and the combined organic phases were washed with water, dried using a hydrophobic frit and concentrated to an oil. The oil was purified on 50 g silica using an SP4 and eluted with a 0-100percent ethyl acetate/cyclohexane gradient. The product was found in the waste, which was concentrated to give the title compound as an oil (260 mg).[0402]LCMS (Method B): Rt=0.84 min, MH+=238.85
With caesium carbonate In acetonitrile at 90℃; for 4 h;
A mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.40 g, 2.1 mmol, Aldrich, Cat. 525057), ethyl 2-bromopropanoate (290 μL, 2.3 mmol), and cesium carbonate (1.5 g, 4.6 mmol) in acetonitrile (8 mL) was stirred at 90° C. for 4 h. After cooling, the reaction mixture was worked up with aqueous Na2CO3, extracted with ethyl acetate (3.x.20 mL), and washed with brine. The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on a silica gel column with ethyl acetate in hexanes (0-50percent) to afford the desired product (0.42 g, 61percent). LCMS (M+H)+: m/z=295.1.
21.6 g
With potassium carbonate In N,N-dimethyl-formamide at 80℃; for 14 h;
Step 9 Ethyl 2-[4-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)-1H-pyrazol-1-yl]propionate To a suspension of 4-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (21.3 g) and potassium carbonate (20.7 g) in dimethylformamide (100 ml) was added ethyl 2-bromopropionate (13 ml), and the mixture was stirred at 80° C. for 14 hr. The reaction mixture was cooled to 0° C., and toluene (100 ml) and water (150 ml) were successively added dropwise. The mixture was partitioned, and the aqueous layer was extracted with toluene (50 ml). The combined organic layer was successively washed once with 10percent aqueous potassium carbonate solution (50 ml), twice with water (50 ml) and once with saturated brine (50 ml). The obtained organic layer was dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to give the title compound (21.6 g). 1H-NMR (400 MHz, CDCl3) δ: 7.85 (1H, s), 7.81 (1H, s), 5.10 (1H, q, J=7.3 Hz), 4.19 (2H, q, J=7.1 Hz), 1.78 (3H, d, J=7.4 Hz), 1.32 (12H, s), 1.25 (3H, t, J=7.2 Hz).
With caesium carbonate; In N,N-dimethyl-formamide; at 90.0℃;
To a solution of 4-(4,4,5,5-Tetramethyl-[1 ,3,2]dioxaborolan-2-yl)-1 H-pyrazole (5.0 g, 25.8 mmol) in DMF (52 mL) was added Cs2CO3 (8.396 g, 25.8 mmol) and bromo-acetic acid methyl ester (2.52 mL, 25.8 mmol). The reaction mixture was heated at 9O0C under nitrogen for overnight. After cooling, the reaction mixture was diluted with water, and extracted with ethyl acetate. The combined extracts were washed with water for three times and brine, dried over Na2SO4., and concentrated to provide 4-(4,4,5,5- tetramethyl-[1 ,3,2]dioxaborolan-2-yl)-pyrazol-1-yl]-acetic acid methyl ester (4.27 g, 62% yield).
47%
With caesium carbonate; In N,N-dimethyl-formamide; at 90.0℃; for 18.0h;Inert atmosphere;
Intermediate 10: Methyl [4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yI)-1 Hpyrazol-1 -yI]acetate Methyl [4-(4,4, 5, 5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)- 1 H-pyrazol- 1 -yl]acetate was prepared according to the synthetic route shown in Scheme 5. To a solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (560 mg, 2.9 mmol) in anhydrous DMF (10 mL) was added 052003 (1.03 g, 3.16 mmol) and methyl bromoacetate (0.30 mL, 3.16 mmol) and the reaction was stirred at 9000 under a nitrogen atmosphere for 18 hrs. The reaction was cooled, diluted with EtOAc (50 mL), washed with water (2 x 25 mL), brine (25 mL), dried over MgSO4, filtered and concentrated in vacuo. The crude material was purified by column chromatography (50% EtOAc/heptane) to give the title compound as a colourless oil (360 mg, 47%).LCMS: m/z 267 [M+H].
With caesium carbonate; In acetonitrile; at 20.0℃; for 72.0h;
3.11 g (5.051 mmol) of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole and 1.808 g (5.550 mmol) of caesium carbonate are suspended in 20 ml of ACN. 468 mul (5.050 mmol) of methyl bromoacetate are added, and the mixture is stirred at RT for 72 hours. The precipitate is filtered off with suction and rinsed with ACN. The mother liquor is distilled off to dryness, ethyl acetate is added, and the mixture is washed rapidly 2× with water. The organic phase is immediately dried using sodium sulfate and distilled off to dryness: methyl [4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazol-1-yl]acetate as solid; EI-MS [M]+ 266.
With potassium carbonate; In acetone; at 65℃; for 13h;Inert atmosphere;
A mixture of 4-(4,4, 5, 5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole (2.0 g, 10.31 mmol), tert-butyl 2-bromoacetate (2.212 g, 11.34 mmol) and K2C03 (1.709 g, 12.37 mmol) in acetone (20 mL) was stirred at 65 C for 13 hours under nitrogen. The reaction was poured into ice water (20 mL) and extracted with ethyl acetate (50 mL x 2). The combined organic phase was washed with brine, dried over magnesium sulfate, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 5/1) to give the title compound (3 g, 8.76 mmol, 85 % yield) as an oil. LCMS (Method C): m/z 309.2 (M+H), retention time: 1.946 minutes; ?H NIVIR (400 MHz, CDC13) oe 7.82 (s, 1H), 7.75 (s, 1H), 4.82 (s, 1H), 1.47 (s, 9H), 1.28 (s, 12H).
84%
With caesium carbonate; In N,N-dimethyl-formamide; at 90℃;
To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1.5 g, 0.0077 mol) in DMF (25 mL) was added 2-bromoacetic acid 1,1-dimethylethyl ester (1.2 mL, 0.0085 mol) and cesium carbonate (3.8 g, 0.012 mol). The suspension was stirred at 90 C. overnight. The reaction mixture was cooled to RT and partitioned with ethyl acetate and water. The organic layer was washed with water, brine, and dried over MgSO4. The solution was concentrated to afford the desired compound (2.0 g, 84%). Analytical LC/MS: (M+H)+=309.4.
81%
1.0 M Potassium tert-butoxide in THF (2.4 mL, 2.4 mmol) was added to a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.39 g, 2.0 mmol) in N,N-dimethylformamide (6.0 mL) at 0 C. The reaction mixture was stirred at room temperature for 5 min. After cooled to 0 C., to the mixture was added t-butyl bromoacetate (0.5 mL, 3 mmol). The reaction was stirred at room temperature for 2 h, then diluted with ethyl acetate, washed with sat. NaHCO3, water, brine, dried over Na2SO4, filtered and concentrated. The product (0.5 g, 81%) was purified by chromatography eluting with hexanes/EtOAc (max. EtOAc 30%). LCMS calculated for C15H26BN2O4 (M+H)+: m/z=309.2; Found: 309.1
81%
Step 1. tert-Butyl [4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl]acetate 1.0M Potassium tert-butoxide in THF (2.4 mL, 2.4 mmol) was added to a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.39 g, 2.0 mmol) in N,N-dimethylformamide (6.0 mL) at 0 C. The reaction mixture was stirred at room temperature for 5 min. After cooled to 0 C., to the mixture was added t-butyl bromoacetate (0.5 mL, 3 mmol). The reaction was stirred at room temperature for 2 h, then diluted with ethyl acetate, washed with sat. NaHCO3, water, brine, dried over Na2SO4, filtered and concentrated. The product (0.5 g, 81%) was purified by chromatography eluting with hexanes/EtOAc (max. EtOAc 30%). LCMS calculated for C15H26BN2O4 (M+H)+: m/z=309.2. Found: 309.1
77%
With potassium carbonate; In N,N-dimethyl acetamide; at 20℃; for 4h;
Example 2; Synthesis of (+)-2-[4-(2-fluoro-9-hydroxy-9-trifluoromethyl-9H-fluoren-4-yl)-pyrazol-1-yl]-propane-1,3-diol (compound No. 595); Step 1; [4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pyrazol-1-yl]-acetic acid t-butyl ester; 4,4,5,5-Tetramethyl-2-(1H-pyrazol-4-yl)-1,3,2-dioxaborolane (10 g), N,N-dimethylacetamide (100 ml), potassium carbonate (17.8 g) and t-butyl bromoacetate (9.9 ml) were mixed, and the mixture was stirred at room temperature for 4 hr. The reaction mixture was filtered through celite. Water and ethyl ether were added to the filtrate, and the mixture was partitioned in a separatory funnel. The aqueous layer was extracted with ethyl ether, and the organic layers were combined. The organic layer was washed 3 times with water and once with saturated brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure, hexane (50 ml) was added to the obtained residue and the mixture was stirred. This slurry was filtered, and the obtained solid was washed with hexane, and dried under reduced pressure to give the title compound (12.23 g, 77%).1H-NMR (DMSO-D6) delta: 7.92 (1H, d, J=0.7 Hz), 7.59 (1H, d, J=0.5 Hz), 4.95 (2H, s), 1.42 (9H, s), 1.25 (12H, s).
63%
With caesium carbonate; In N,N-dimethyl-formamide; at 20℃; for 8h;
4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1H-pyrazole (0.5 g, 2.6 mmol) Cesium carbonate (1.3 g, 3.9 mmol) was dissolved in N, N-dimethylformamide (10 mL) Tert-butyl bromoacetate (0.6 mL, 3.9 mmol) was added, and the mixture was stirred at room temperature for 8 hours. After completion of the reaction, the reaction mixture was cooled to room temperature, distilled water (50 mL) was added thereto, and the mixture was extracted with ethyl acetate. The organic layer was washed with distilled water and saturated brine, dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel column chromatography (ethyl acetate: n-hexane = 1: 3) to give the title compound 56-a (1.1 g, 63%) as a yellow solid.
18%
With potassium carbonate; In acetone; at 65℃; for 16h;
To a stirred solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1.0 g, 5.15 mmol) in Acetone (10 mL) in a two neck RB were added K2C03 (0.85 g, 6.18 mmol ) and followed by tert-butyl bromoacetate (1.20 gm, 6.18 mmol) at RT and stirred for 16 h at 65 C. The reaction mixture was evaporated by vacuum to get residue which was quenched with water (30 mL) and extracted with EtOAc (2 X 50 mL). The combined organic layer was washed with water (50 mL), brine (50 mL), dried over anhydrous sodium sulphate and concentrated under reduced pressure to give the crude product which was purified by combi flash using 50 % ethyl acetate in hexane as eluent to afford the title compound (0.27 g, 18 %).1H NMR (400 MHz, DMSO-d6): d 7.91 (s, 1H), 7.58 (s, 1H), 4.95 (s, 2H), 1.42 (s, 9H), 1.25 (s, 12H).
With caesium carbonate; In N,N-dimethyl-formamide; at 20℃;
To a solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (1.5 g, 7.7 mmol) in N,N-dimethylformamide (25 mL) was added tert-butyl 2-bromoacetate (1.2 mL, 8.5 mmol) and cesium carbonate (3.8 g, 0.012 mol). The suspension was stirred overnight at RT and partitioned with EtOAc and water. The organic phase was washed with brine, dried with MgSO/t, filtered, and concentrated to afford the desired compound which was directly used in next step. LCMS: (M+H) =309.4.
With caesium carbonate; at 20℃;
Step I: tert-butyl [4-(4.4, 5,5-letramethyl-},3,2-dioxaborolan-2-yl)-lH-pyra?ol-I-yl]acetate; To a solution of 4-(4,4,5,5-tetramethyl-L3,2-dioxaborolan-2-yl)-l H-pyrazole (400 mg, 0.002 mol, AIdrich, Cat. No. 525057) in acetonitrile (10 mL,) was added tert-butyl 2-bromoacetate (0.36 mL, 0.0025 mol, AIdrich. Cat. No. 124230) and cesium carbonate (1.0 g, 0.0031 mol). The suspension was stirred at r.t. overnight. The reaction mixture was partitioned with AcOEt and water. The organic layer was separated, washed with water and brine, dried over MgSCV After filtration, the filtrate was concentrated to afford the desired compound which was directly used in the next step reaction without further purification. LCMS (M+H)+: m/z = 309.1.
12.23 g
With potassium carbonate; In N,N-dimethyl-d6-formamide; at 20℃; for 4h;
Step 8 t-Butyl[4-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)-1H-pyrazol-1-yl]acetate 4,4,5,5-Tetramethyl-2-(1H-pyrazol-4-yl)[1,3,2]dioxaborolane (10 g), N,N-dimethylacetamide (100 ml), potassium carbonate (17.8 g) and t-butyl bromoacetate (9.9 mL) were mixed, and the mixture was stirred at room temperature for 4 hr. The reaction mixture was filtered through celite. Water and diethylether were added to the filtrate, and the mixture was poured into a separating funnel and partitioned. The aqueous layer was extracted again with diethyl ether, and combined with the organic layer. The obtained organic layer was washed three times with water, once with saturated brine, and dried over anhydrous sodium sulfate. The insoluble material was filtered off, and the filtrate was concentrated under reduced pressure. To the obtained residue was added hexane (50 ml) and the mixture was slurry washed (suspension stirred). The suspension was filtered, and the obtained solid was washed with hexane, and dried under reduced pressure to give the title compound (12.23 g). 1H-NMR (400 MHz, DMSO-D6) delta: 7.92 (1H, d, J=0.7 Hz), 7.59 (1H, d, J=0.5 Hz), 4.95 (2H, s), 1.42 (9H, s), 1.25 (12H, s).
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In ethanol; water; toluene; at 100℃;
Example A41 4-Chloro-2-methylsulfanyl-pyrimidine (1.4 g, 8.8 mmol), 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (2.0 g, 10.3 mmol), Na2CO3 (2.8 g, 26.4) and Pd(PPh3)4 (500 mg, 0.43 mmol) were combined in a solvent comprised of toluene/EtOH/H2O (4/4/1, 20 mL). The mixture was degassed by applying a vacuum and backfilling the headspace with argon. The reaction mixture was heated overnight at 100° C. The insoluble portion was filtered and the filtrate was concentrated and purified by silica gel chromatography to provide 2-(methylthio)-4-(1H-pyrazol-4-yl)pyrimidine (1.2 g, 71percent yield). 1H NMR (400 MHz, CDCl3) delta 8.45 (d, J=6.4 Hz, 1H), 8.24 (s, 1H), 7.23 (s, 1H), 7.05 (d, J=6.4 Hz, 1H), 2.51 (s, 3H).
71%
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In ethanol; water; toluene; at 100℃;Inert atmosphere;
4-Chloro-2-methylsulfanyl-pyrimidine (1.4 g, 8.8 mmol), 4-(4,4,5,5- tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (2.0 g, 10.3 mmol), Na2CO3 (2.8 g, 26.4) and Pd(PPh3)4 (500 mg, 0.43 mmol) were combined in a solvent comprised of toluene/EtOH/H2psi (4/4/1, 20 mL). The mixture was degassed by applying a vacuum and backfilling the headspace with argon. The reaction mixture was heated overnight at 100 °C. The insoluble portion was filtered and the filtrate was concentrated and purified by silica gel chromatography to provide 2-(methylthio)-4-(1H-pyrazol-4- yl)pyrimidine (1.2 g, 71percent yield). 1H NMR (400 MHz, CDC13) delta 8.45 (d, J= 6.4 Hz, 1 H), 8.24 (s, 1 H), 7.23 (s, 1 H), 7.05 (d, J= 6.4 Hz, 1 H), 2.51 (s, 3 H).
71%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; at 100℃;Inert atmosphere;
4-Chloro-2-methylsulfanyl-pyrimidine (1.4 g, 8.8 mmol), 4- (4,4,5,5-tetramethyl-[l ,3,2]dioxaborolan-2-yl)-lH-pyrazole (2.0 g, 10.3 mmol), Na2C03 (2.8 g, 26.4) and Pd(PPh3)4 (500 mg, 0.43 mmol) were combined in a solvent comprised of toluene/EtOH/H20 (4/4/1 , 20 mL). The mixture was degassed by applying a vacuum and backfilling the headspace with argon. The reaction mixture was heated overnight at 100 °C. The insoluble portion was filtered and the filtrate was concentrated and purified by silica gel chromatography to provide 2-(methylthio)-4-(lH-pyrazol-4-yl)pyrimidine (1.2 g, 71percent yield). FontWeight="Bold" FontSize="10" H NMR (400 MHz, CDC13) delta 8.45 (d, J = 6.4 Hz, 1 H), 8.24 (s, 1 H), 7.23 (s, 1 H), 7.05 (d, J= 6.4 Hz, 1 H), 2.51 (s, 3 H).
With sodium hydroxide; In N,N-dimethyl-formamide; at 140℃; for 16h;
Intermediate 1182-[4-(4,4,5,5-tetramethyl-1 ,3,2-dioxa H-pyrazol-1 -yl]ethanolA solution of 1 ,3-dioxolan-2-one (2.496 g, 28.3 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 H-pyrazole (5 g, 25.8 mmol) and sodium hydroxide (0.103 g, 2.58 mmol) in N,N-Dimethylformamide (DMF) (18 mL) was stirred at 140C for 16 h. then cooled to room temperature and treated with activated charcoal (200mg). The resulting mixture was stirred at room temperature for 1 h then filtered through celite (10 g). The insoluble were washed with EtOAc (50 mL) and EtOH (50ml_). The combined filtrate and washings were concentrated in vacuo. Purification of the residue by flash chromatography on silica gel using a 50 G silica cartyridge (gradient: 0 to 40% MeOH in DCM) gave a residue which was further purified by SP4 using a 100 G silica cartridge (eluant: 0 to 20% MeOH in DCM) to give 2-[4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazol-1 - yl]ethanol (5.58 g, 23.44 mmol, 91 % yield) as a colourless oil which was used in the next step without further purification.LCMS (method A): Retention time 0.68 min, [M+H]+ = 239.13
85%
With sodium hydroxide; In N,N-dimethyl-formamide; for 2.5h;Heating / reflux;
INTERMEDIATE 34; 2-[4-f4.4.5.5-Tetramethyl-ri.3.21dioxaborolan-2-ylVrhoyrazol-l-yl1-ethanol; 4-Pyrazoleboronic acid pinacol ester (0.25 g, 1.29 mmol), ethylene carbonate(0.125 g, 1.42 mmol) and sodium hydroxide (5 mg, 0.13 mmol) were dissolved in DMF (1 mL) and the reaction mixture was heated to reflux for 2 Vi h. It was cooled to r.t. before addition of activated charcoal (25 mg). The resulting suspension was stirred at r.t. for Ih and then filtered through celite, washed with DMF (6 mL) and concentrated in vacuo to give the title compound (0.26g, 85%) as a yellow oil. LCMS (ES+) 239.18 (M+H)+.
76%
With caesium carbonate; In N,N-dimethyl-formamide; at 140℃; for 0.5h;
100 mg of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole and 91 mg of 1,3-dioxolan-2-one weredissloved in 2 ml of dimethylformamide. 336 mg of cesium carbonate was heated to 140 C, stirred for 0.5 h and thencooled to room temperature and concentrated. The residue was purified by column chromatography (ethyl acetate:petroleum ether = 30: 70) to give 93 mg of 2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)ethanol aspale yellow oil. Yield: 76%.1H NMR (300 MHz, DMSO-d6) delta (ppm): 1.25 (s, 12H), 3.71 (q, J= 5.4 Hz, 2H), 4.15 (t, J = 5.4 Hz, 2H), 4.87 (t, J = 5.4Hz, 1H), 7.57 (s, 1H), 7.88 (s, 1H).
With sodium hydroxide; In N,N-dimethyl-formamide; for 2h;Reflux;
2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl)ethanol was synthesized according to the procedure in PCT Publication No. WO 2008/44022, which is incorporated herein in its entirety. Specifically, 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)-l/f-pyrazole (5.0 g, 25.8 mmol), l,3-dioxolan-2-one (2.5 g, 28.3 mmol), and sodium hydroxide (pellets, 1.0 g, 25.8 mmol) were dissolved in DMF (206 mL). The reaction mixture was heated to reflux for 2 hours. Activated charcoal was added after reaction was cooled to ambient temperature and the reaction was stirred for 1 hr and then filtered through Celite. The filter cake was then rinsed with DMF (120 mL), and the filtrate was concentrated to provide 2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl)ethanol as a yellow oil (6 g). The resulting material was used without further purification. ESI-MS :m/z 239.3 (M+H)+.
With caesium carbonate; at 20 - 100℃;
4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (9.66 g, 49.8 mmol), 1,3-dioxolan-2-one (21 g, 238 mmol) and cesium carbonate (16 g, 49.1 mmol) were combined in a 100 mL round bottom flask. The reaction was warmed from room temperature to 100 C. in an oil bath, by which time the carbonate had melted and served as the solvent for the reaction, which remained a slurry. After heating for 3.5 hours, the reaction was cooled to room temperature and diluted with ethyl acetate, then filtered through Celite washing repeatedly with ethyl acetate. The filtrate was concentrated, then purified by chromatography on an Analogix Intelliflash purification system using a SF60-200 g column at a flow rate of 80 mL/min, eluding as follows: 5 minutes at 20% ethyl acetate/hexanes, then ramped from 40% to 90% ethyl acetate/hexanes over 35 minutes, then 100% ethyl acetate for another 20 minutes, to provide the title compound.
With caesium carbonate; at 100℃; for 0.583333h;oil bath;
Example 39A 2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)ethanol 4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (9.66 g, 49.8 mmol), 1,3-dioxolan-2-one (21 g, 238 mmol) and cesium carbonate (16 g, 49.1 mmol) were combined in a 100 mL round bottom flask. The reaction was warmed from room temperature to 100 C. in an oil bath, by which time the carbonate had melted and served as the solvent for the reaction, which remained a slurry. After heating for 3.5 hours, the reaction was cooled to room temperature and was diluted with ethyl acetate, then filtered through diatomaceous earth washing repeatedly with ethyl acetate. The filtrate was concentrated, then purified by chromatography on an Analogix(R) Intelliflash(TM) purification system using a SF60-200 g column at a flow rate of 80 mL/minute, eluting as follows: 5 minutes at 20% ethyl acetate/hexanes, then ramped from 40% to 90% ethyl acetate/hexanes over 35 minutes, then 100% ethyl acetate for another 20 minutes, to provide the title compound. MS ESI(+) m/z 239.0 (M+H)+.
With caesium carbonate; at 100℃; for 3.5h;
Example 10A2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)ethanol; 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (9.66 g, 49.8 mmol), 1,3-dioxolan-2-one (21 g, 238 mmol), and cesium carbonate (16 g, 49.1 mmol) were combined in a 100 mL round bottom flask At room temperature all reagents were solids. The reaction was warmed from room temperature to 100 C. in an oil bath, at which time the carbonate had melted and served as the solvent for the reaction, which then remained a slurry. After heating for 3.5 hours, the reaction was cooled to room temperature, diluted with ethyl acetate, and filtered through Celite (diatomaceous earth) washing repeatedly with ethyl acetate. The filtrate was concentrated, and the residue was purified by chromatography on an Analogix Intelliflash purification system using a SF60-200 g column at a flow rate of 80 mL/minute, eluting as follows: 5 minutes at 20% ethyl acetate/hexane, then ramped from 40% to 90% ethyl acetate/hexanes over 35 minutes, and then 100% ethyl acetate for another 20 minutes, to afford the title compound.
In N,N-dimethyl-formamide; at 140℃; for 16h;
A solution of 1,3-dioxolan-2-one (2.496 g, 28.3 mmol), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (5 g, 25.8 mmol) and sodium hydroxide (0.103 g, 2.58 mmol) in N,N-Dimethylformamide (DMF) (18 mL) was stirred at 140 C. for 16 h. then cooled to room temperature and treated with activated charcoal (200 mg). The resulting mixture was stirred at room temperature for 1 h then filtered through celite (10 g). The insoluble were washed with EtOAc (50 mL) and EtOH (50 mL). The combined filtrate and washings were concentrated in vacuo. Purification of the residue by flash chromatography on silica gel using a 50 G silica cartyridge (gradient: 0 to 40% MeOH in DCM) gave a residue which was further purified by SP4 using a 100 G silica cartridge (eluant: 0 to 20% MeOH in DCM) to give 2-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl]ethanol (5.58 g, 23.44 mmol, 91% yield) as a colourless oil which was used in the next step without further purification.LCMS (method A): Retention time 0.68 min, [M+H]+=239.13
7.53 g
With sodium hydroxide; In N,N-dimethyl-formamide; at 140℃;
A solution of I ,3-dioxolan-2-one (1 .902 mL, 28.5 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)-IH-pyrazole (5.0239 g, 25.9 mmol) and sodium hydroxide (0.0998 g, 2.495 mmol) in N,Ndimethylformamide (DMF) (20 mL) was heated to 140 C overnight. The mixture was cooled down to rt and then activated charcoal (200 mg) was added and this was stirred for 4 h before filtering through celite cartridge (10 g). The mixture was washed with EtOAc (50 mL) and EtCH (50 mL andthe combined filtrate was concentrated in vacuo to afford 7.53 g of brown oil. This was used crude in further reactions. LCMS: Not recorded.
In a 250 mL round-bottomed flask, 2-chloro-N,N-dimethylethanamine (998 mg, 9.28 mmol), 4- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (1.2 g, 6.18 mmol) and cesium carbonate (4.03 g, 12.4 mmol) were combined with acetonitrile (20 mL) to give a white suspension. The reaction mixture was heated at 100 C overnight. In the morning, the reaction mixture was cooled to room temperature, filtered, and the filtrate was concentrated to give title compound (1.26 g, 77 yield) as a colorless oil. The crude product was used in subsequent reactions without purification.
With potassium carbonate; In N,N-dimethyl-formamide; at 190℃; for 1h;Microwave irradiation;
Example 19; N,N-dimethyl-2-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl]ethanamine; 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.250 g, 1,29 mmol), dimethylamino ethyl chloride (0.37 g, 2.58 mmol) and potassium carbonate (0.534 g, 3.87 mmol) were dissolved in 3 mL of dry dimethylformamide. The reaction mixture was heated in a Biotage Initiator series microwave at 190 C. for 1 hour. The reaction mixture was poured into 300 mL of ethyl acetate and 50 of mL brine. The organic layer was separated, dried with magnesium sulfate, filtered, and concentrated to afford the title compound. 1H NMR (600 MHz, CDCl3) delta 7.76 (s, 1H); 7.72 (s, 1H); 4.22 (t, 2H); 2.94 (s, 3H); 2.86 (s, 3H); 2.74 (t, 2H); 1.29 (s, 12H).
With potassium carbonate; In 1-methyl-pyrrolidin-2-one; at 20℃; for 16h;Inert atmosphere;
Synthesis of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2~yl)-1-((2- (trimethylsilyl)ethoxy)methyl)-1 t-15a)lnt-15aTo a solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)- H- pyrazole (8.2 g, 41 mmol) in NMP (60 mL) was added K2C03 (12 g, 82 mmoi) and 2-(trimethylsilyi)ethoxymethy. chloride (7.8 mL, 43 mmol) in sequence. The reaction mixture was stirred at r.t. under N2 for 16 h. Then, the reaction mixture was diluted and filtered, and then the filtrate was diluted with EtOAc (300 mL). The resulting solution was washed with sat. NaHC03 (aq) (3 x 200 mL), H20 (4 x 200 mL), brine (1 x 200 mL), dried over Na2S04, filtered, concentrated and dried in vacuo to yield intermediate .nt-15a (11.4 g, 86 %) as a clear yellowish oil.
86%
[96] SEM-pyrazolo-4-boronic acid pinacol ester was prepared according the procedure from WO2011/130146, page 84. A solution of pyrazolboronic acid pinacolester (20 g, 103 mmol) in DMF (180 mL) was cooled to 0 C and treated with sodium hydride (60 % dispersion in oil) (6.2 g, 150 mmol) in nitrogen athmosphere. [97] The reaction mixture was stirred at ambient temperature for 30 minutes. The reaction mixture was then cooled to 0 C and (2-(chloromethoxy)ethyl)trimethylsilane (23.65 ml, 134 mmol) was added. The reaction mixture was stirred at ambient temperature overnight. [98] The reaction mixture was poured into aqueous saturated ammonium chloride (200 mL) containing ice (approximately 200 mL) and stirred until the ice melted. The cold mixture was extracted with ethyl acetate twice. The combined organic extracts were washed with water, dried over Na2SO4, and concentrated under reduced pressure to afford SEM-pyrazolo-4-boronic acid pinacol ester (27.6 g, 86 % yield).
72%
With sodium hydride; In tetrahydrofuran; at 20℃;Inert atmosphere;
Compound 280.1. 4-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)-l-((2- (trimethylsilyl)ethoxy)methyl)-lH-pyrazole. Into a 250-mL three neck round-bottom flask, which was purged and maintained with an inert atmosphere of nitrogen, was placed a solution of 4-(tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (5.82 g, 30.0 mmol) in tetrahydrofuran (80 mL). This was followed by the addition of NaH (70%) (2.05 g, 85.4 mmol) in portions at 0 C. To this was added SEMC1 (6.4 mL, 36.1 mmol) dropwise. The reaction mixture was stirred overnight at room temperature, then quenched with 50 mL of NH4CI (sat). The aqueous phase was extracted with 2 x 100 mL of ethyl acetate and the combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. This resulted in 7 g (72%) of the title compound as colorless oil.
65.94%
With caesium carbonate; In tetrahydrofuran; acetonitrile; at 20℃; for 2h;
13.2 4-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-yl)-1-(2-trimethylsilanyl- ethoxymethyl)-1 H-p razole To a solution of 1H-pyrazole-4-boronic acid pinacol ester (0.5 g, 2.57 mmol), in tetrahydrofuran/acetonitrile (3:2, 20ml), 2-(chloromethoxylethyl)trimethyl- silane (0.51 g, 3.09 mmol) and cesium carbonate (1.67 g, 5.15 mmol) are added and stirred for 2 hours at room temperature. The reaction mixture is filtered through celite, and concentrated, the crude mass is taken in ethylacetate (30 ml), washed with water, brine solution, dried over anhydrous MgS04 and concentrated to get the product as brown oil (0.55 g, 65.94 %); TLC: Pet ether/ethyl acetate(8/2) R - 0.5; 1H NMR: 400 MHz, DMSO-d6: delta [ppm] 8.08 (s, 1H), 7.64 (s, 1 H), 5.40 (s, 2H), 3.48-3.54 (m, 2H), 1.24 (s, 12H), 0.81-0.85 (m, 2H), -0.049(s, 9H);
61%
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (348 mg, 1.8 mmol) was dissolved in DMF (5 mL) and sodium hydride (60% dispersion, 86 mg, 2.15 mmol) added and the mixture heated to 60 C. for 5 min. Upon cooling and stirring for an additional 15 min, trimethylsilylethoxymethyl chloride (358 mg, 2.15 mmol, 381 muL) was added dropwise over 5 min and mixture stirred for 16 h. The reaction mixture was diluted with ethyl acetate (25 mL), washed with 5% lithium chloride (5×), dried over sodium sulfate and concentrated. The residue was purified by column chromatography (40 g ISCO column eluting with hexanes and ethyl acetate; gradient 100% hexanes to 50% hexanes over 30 min at 30 mL/min) to provide the SEM-protected pyrazole (360 mg, 61%) as a colorless oil; 1H NMR (500 MHz, CDCl3) delta 7.84 (s, 1H), 7.80 (s, 1H), 5.42 (s, 2H), 3.56-3.53 (t, J=8.3 Hz, 2H), 1.31 (s, 12H), 0.91-0.87 (t, J=8.3 Hz, 2H), -0.03 (s, 9H).
56%
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 3h;
To a solution of methyl 4-(tetramethyl-1 ,3, 2-dioxaborolan-2-yl)-1H-pyrazole (1 .238 g, 6.316 mmol) in DMF (20 mL) was added potassium carbonate (2.62 g, 18.95 mmol) and [2-(chloromethoxy)ethyl](trimethyl)silane (1 .68 mL, 9.48 mmol) at room temperature. The mixture was stirred for 3 hours and then partitioned between TBME (100 ml.) and water (50 ml_). The organic layer was separated, washed with water (2 x 30 mL) and brine (30 ml_), dried (Na2S04) and concentrated at reduced pressure. The residue was purified by Biotage Isolera chromatography [Biotage SNAP Cartridge KP-Sil 50 g; using a gradient of eluents, 0-50% EtOAc in heptane]. The product containing fractions were combined, concentrated in vacuo to give the title compound (1 .20 g, 56% yield) as colourless oil. 1H NMR (500 MHz, chloroform-d) delta [ppm] 7.88 (s, 1 H), 7.84 (s, 1 H), 5.46 (s, 2H), 3.61- 3.55 (m, 2H), 1 .35 (s, 12H), 0.96 - 0.90 (m, 2H), 0.00 (s, 9H). LCMS (Analytical Method A): Rt = 1 .34 mins; MS (ESIPos) m/z = 324.95 (M+H)
46%
32-(a) 4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-1-(2-trimethylsilylethoxymethyl)-1H-pyrazole; Under argon atmosphere, to 20 ml of tetrahydrofuran solution containing 1.09 g (5.62 mmol) of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole was added 443 mg (11.1 mmol) of 60% sodium hydride under ice-cooling, and the mixture was stirred for 5 minutes. Then, 3 ml (17.0 mmol) of (2-trimethylsilylethoxy)methyl chloride was added dropwise to the mixture, and the mixture was reacted at room temperature for 2 hours. After completion of the reaction, water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was separated, and the solutions were washed successively with water and then with a saturated aqueous solution of sodium chloride, dried over anhydrous magnesium sulfate and then concentrated under reduced pressure. The obtained residue was applied to silica gel column chromatography (Eluent; hexane:ethyl acetate=9:1 (V/V)), and the fractions containing the desired compound were concentrated under reduced pressure to obtain 832 mg of the title compound as a colorless oil. (46%) Mass Spectrum (CI, m/z): 325 (M++1). 1H-NMR Spectrum (CDCl3, delta ppm): -0.03 (s, 9H), 0.86-0.94 (m, 2H), 1.32 (s, 12H), 3.51-3.59 (m, 2H), 5.43 (s, 2H), 7.81 (d, J=0.5 Hz, 1H), 7.86 (d, J=0.5 Hz, 1H).
44%
With caesium carbonate; In N,N-dimethyl-formamide; at 20℃; for 3h;
To a stirred solution of 4-(4 , 4,5 , 5-tetra methyl- 1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (2 g, 10.3 mmol) in DMF (30 mL), (2-(chloromethoxy)ethyl)trimethylsilane (2 g, 12.3 mmol), and CS2CO3 (10 g, 30.9 mmol) were added. The resulting mixturen was stirred at rt for 3 h. Solvents were evaporated and the crude residue was diluted with ice cold water and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SC>4 and filtered. The filtered solution was concentrated under reduced pressure and the resulting crude compound was purified by flash column chromatography using 20-30% EtOAc/Pet ether to get the title compound (1.5 g, 44%) as pale yellow gummy.LC-MS (method 14): R, = 3.08 min; m/z = 325.2 (M+H?).
With potassium carbonate; In 1-methyl-pyrrolidin-2-one; at 25℃; for 0.75h;
Preparative Example 1; To a suspension of potassium carbonate (5.85 g, 1.5 equiv) and 4- (4,4,5,5-tetramethyM ,3,2-dioxaborolan-2-yl)-1 h-pyrazole (5.48 g, 1.0 equiv) in NMP (50 mL) at rt was added SEMCI (5.2 mL, 1.05 equiv) dropwise (mildly exothermic). The resulting mixture was allowed to stir an additional 45 min at rt. The reaction was diluted with ethyl acetate, rinsed with water (2x), brine and dried (sodium sulfate). Filtration and concentration afforded the title compound that was used without purification. MH+ = 325.
With potassium carbonate; In 1-methyl-pyrrolidin-2-one; for 1h;
A mixture of 4,4,5,5-tetramethyl-2-(1H-pyrazol-4-yl)-1,3,2-dioxaborolane (5.48 g), SEMCl (5.2 mL), and K2CO3 (5.85 g) in NMP (50 mL) was stirred under N2 for 1 hr. The reaction mixture was diluted with EtOAc, rinsed with H2O, brine, and dried over Na2SO4. The mixture was filtered, the solvents were evaporated and the residue was used directly in the next step.
With potassium carbonate; In 1-methyl-pyrrolidin-2-one; at 20℃; for 0.75h;
To a suspension of potassium carbonate (5.85 g, 1.5 equiv) and 1H-pyrazole-4-boronate (5.48 g, 1.0 equiv) in NMP (50 mL) at room temperature was added SEMCI (5.2 mL, 1.05 equiv) dropwise (mildly exothermic). The resulting mixture was allowed to stir for an additional 45 min at room temperature. The reaction was diluted with ethyl acetate, rinsed with water (×2), brine and dried (sodium sulfate). Filtration and concentration afforded the title compound (270) that used directly in the next step.
With potassium carbonate; In 1-methyl-pyrrolidin-2-one; at 20℃; for 0.75h;
Example 270; To a suspension of potassium carbonate (5.85 g, 1.5 equiv) and 1H-pyrazole-4-boronate (5.48 g, 1.0 equiv) in NMP (50 mL) at room temperature was added SEMCl (5.2 mL, 1.05 equiv) dropwise (mildly exothermic). The resulting mixture was allowed to stir for an additional 45 min at room temperature. The reaction was diluted with ethyl acetate, rinsed with water (×2), brine and dried (sodium sulfate). Filtration and concentration afforded the title compound (270) that used directly in the next step.
Preparation E4-(4A5 -tetramethyl-l ,3,2-dioxaborolan-2-yl)-l-((2-(trimethylsilyl)ethoxy)methyl)-lH- pyrazole [00624] A solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole(10.00 g, 51.54 mmol) in DMF (100 mL) was cooled to 0 C in an ice bath and treated with sodium hydride (60% dispersion in oil) (3.092 g, 77.30 mmol) in one portion. The reaction mixture was stirred at 0 C for 2 minutes, then at ambient temperature for 30 minutes. The reaction mixture was cooled to 0 C and (2-(chloromethoxy)ethyl)trimethylsilane (11.82 mL, 67.00 mmol) was added. The reaction mixture was warmed to ambient temperature and allowed to stir overnight. The reaction mixture was poured into aqueous saturated ammonium chloride (100 mL) containing ice (approximately 100 mL) and stirred until the ice melted. After the ice melted, the cold mixture was extracted with ethyl acetate. The combined organic extracts were washed with water, brine, dried over MgS04, and concentrated under reduced pressure to afford the title compound (14.45 g, 44.56 mmol, 86.46% yield). MS (apci) m/z = 325.0 (M+H).
With potassium carbonate; In 1-methyl-pyrrolidin-2-one; at 20℃; for 16h;Inert atmosphere;
To a solution of 4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-lH-pyrazole (8.2 g, 41 mmol) in NMP (60 mL) were added K2C03 (12 g, 82 mmol) and 2-(trimethylsilyl)ethoxymethyl chloride (7.8 mL, 43 mmol) in sequence. The reaction mixture was stirred at RT under N2 for 16 h. Then, the reaction mixture was diluted filtered and the filtrate was diluted with EtOAc (300 mL), The resulting solution was washed with sat NaHC03(aq) (3 x 200 mL), ¾0 (4 x 200 mL), brine (1 x 200 mL), dried over Na2S0 , filtered, concentrated and dried in vacuo to yield Int-37 as clear yellowish oil
0.9 g
A) 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole [1064] To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (770 mg) in DMF (10 mL) was added sodium hydride (60%, 114 mg) under ice-cooling. The reaction mixture was stirred at room temperature for 1 hr. To the reaction mixture was added dropwise (2-(chloromethoxy)ethyl)(trimethyl)silane (990 mg) at room temperature, and the mixture was stirred for 15 hr. The reaction mixture was diluted with ethyl acetate, and the mixture was washed with 5% aqueous lithium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give the title compound (0.90 g). MS(ESI+): [M+H]+ 325.2. MS(ESI+), found: 325.2.
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 16h;
To a solution of 4-(4,4,5,5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole (1.00 g, 5.15 mmol) in DMF (20 mL) was added K2C03 (1.07 g, 7.73 mmol) and (2-(chloromethoxy)ethyl)trimethylsilane (1.03 g, 6.18 mmol). The reaction was stirred at ambient temperature for 16 h and then diluted with water (20 mL). The mixture was extracted with EtOAc (2 x 30 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the crude 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy) methyl)-1H-pyrazole (1.00 g, 60% yield) as ayellow oil.A mixture of 2-(3-bromophenyl)- 1,1,1 -trifluorobut-3 -yn-2-ol (500 mg, 1.79 mmol), 4-(4,4,5,5- tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 -((2-(trimethylsilyl)ethoxy)methyl)- 1 H-pyrazole (872 mg, 2.69 mmol), Cs2CO3 (1.17 g, 3.58 mmol) and 1,1?-Bis(diphenylphosphion) ferrocene dichloride palladium(II) (66 mg, 0.09 mmol) in dioxane/H20 (5 mL/1 mL) was heated at 110 Cfor 0.5 h under microwave conditions. The solvent was removed and the crude residue was purified by column chromatography (petroleum ether: EtOAc = 4:1) to give the title compound (300 mg, 42% yield) as a colorless oil.
To a reaction mixture of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazole (210 mg, 1.08 mmol) in 2.0 mL of NMP was added cesium carbonate (672 mg, 2.06 mmol). The reaction mixture was stirred for 5 min and then l,l-difluoro-2-iodoethane (197 mg, 1.03 mmol) was added and stirred at room temperature for 40 hours. From the above crude reaction mixture, 0.8 mL (0.432 mol) was removed and used. (The remaining 1.2 mL was stored in freezer). To the 0.8 mL reaction mixture above was added (lR,2R)-2- (6-(5-bromopyridin-3-yloxy)benzo[d]thiazol-2-ylamino)cyclohexanol (15.0 mg, 0.0357 mmol, see Example 19 above), Pd(dppf)2Cl2 (8.8 mg, 0.0107 mmol) and 2 M Na2CO3 (0.108 mL, 0.216 mmol). The reaction solution was stirred at 105-110 C for 90 min or until done by LC. The crude reaction mixture was filtered, purified on preparative HPLC and lyophilized to give the title compound as TFA salt (3.3 mg). ES/MS m/z 472.1 (MH+), Rt = 2.03 min.
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; for 2h;
To a solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (5.00 g, 25.8 mmol) in acetonitrile (50 mL) was added acrylonitrile (3.4 mL, 52 mmol) followed by l,8-diazabicyclo[5.4.0]undec-7-ene (1.94 mL, 12.9 mmol). After about 2 hours, the reaction mixture was concentrated under reduced pressure. The crude material was then dissolved in a minimal amount of dichloromethane and purified via silica gel chromatography eluting with a gradient of 10-50% ethyl acetate in heptane to afford the title compound. 1H NMR (400 MHz, DMSO-d6) delta ppm 8.03 (s, IH), 7.64 (s, IH), 4.40 (t, J = 6.4 Hz, 2H), 3.06 (t, J = 6.4 Hz, 2H), 1.26 (s, 12H). MS (ESI+) m/z 247.3 (M+H)+.
With caesium carbonate; In acetonitrile; at 90.0℃; for 3.0h;
Step 1: Preparation of 2-(4-(4,4,5 ,5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 Hpyrazol- 1 -vi) acetamide (Intermediate 24) A mixture of 4- (4,4,5 ,5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole(200 mg, 1.03 mmol), 2-bromoacetamide (213 mg, 1.54 mmol) and Cs2CO3 (1.27 g,3.92 mmol) in CH3CN (5.0 mL) was stirred at 90 C for 3 hours. After being cooledto room temperature, the reaction mixture was diluted with water and extracted withEtOAc twice. The combined organic layers were dried over Na2SO4, filtered andconcentrated in vacuo to obtain the title compound (174 mg, 67%) as a white solid, which was used for the next reaction without further purification.1H-NMR (400 MHz, CDC13): oe 7.89 (s, 1H), 7.76 (s, 1H), 6.21 and 5.41 (brs,2H), 4.83 (s, 2H), 1.32 (s, 12H).
With caesium carbonate; In acetonitrile; at 90.0℃; for 2.0h;
Step 1 : 2-[4-(4, 4, 5, 5-tetramethyl-l, 3, 2~dioxaborolan-2~yl)-lH-pyrazol- -yl]acetamide; H2NO N^ O ^-A mixture of 4<4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (50 mg, 0.2 mmol), 2- Bromoacetamide (43 mg, 0.31 mmol, Aldrich, Cat. No. 301272), and cesium carbonate (250 mg, 0.77 mmol) in acetonitrile (1 mL) was stirred at 90 0C for 2 h. After cooling it was quenched with water, extracted with ethyl acetate. The extract was washed with water twice, brine once; dried over Na^SCv After filtration the filtrate was concentrated to yield 45 mg of the product which was directly used in the next step reaction without further purification. LCMS (M+H)+: m/z = 252.1
With caesium carbonate; In acetonitrile; at 90.0℃; for 3.0h;Sealed tube;
[0223] To a solution of compound I (5 g, 25.7 mmol; 1 eq) in acetonitrile in a reaction tube (150 mL) was added bromo acetamide (5.68 g, 41.2 mmol; 1.6 eq) and cesium carbonate (33.5 g, 102 mmol; 4 eq), and the reaction tube was sealed and heated at 90 C for 3 h. The mixture was cooled to room temperature and filtered through a Celite pad and the filtrate was concentrated in vacuo to obtain the title compound (4.2 g, 65%), as a crude product which was directly used in next step without further purification. 1H NMR (400 MHz, DMSO-d6) oe 7.67 (d, 1H, J = 2 Hz), 7.57 (m, 1H), 7.48 (brs, 1H), 7.42 (s, 1H), 4.76 (s, 2H), 0.89 (s, 12H)
70 mg
With caesium carbonate; In N,N-dimethyl-formamide; at 100.0℃; for 0.583333h;Sealed tube; Microwave irradiation;
To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole 1a (100 mg, 0.52 mmol) in DMF(3 mL) was added 2-bromoacetamide 1b (143.5 mg, 1.04 mmol) and Cs2CO3 (253.5 mg, 0.78 mmol) in a sealed tubeat RT. Reaction mixture was stirred at 100 C for 35 min under microwave irradiation. After cooling at room temperature,the mixture was diluted with EA (10 mL) and water (10 mL), extracted with EA (10 mL*3), and the organic layers werecombined and washed with water (30 mL*3) and brine (50 mL), dried over Na2SO4, filtrated and evaporated to afford2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)acetamide 1c (70 mg) as a yellow oil and used in thenext step directly.MS m/z (ESI): 252.2 [M+1]1H NMR (100 MHz, CDCl3) delta 7.9 (d, 2 H), 2.05 (s, 2 H), 1.32 (s, 12 H)
With potassium carbonate; In acetone; at 50.0℃; for 84.0h;
A suspension of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (2.00 g, 10.3 mmol), 2-bromoacetamide (2.14 g, 15.5 mmol), and potassium carbonate (2.14 g, 15.5 mmol) in acetone (60 mL) was heated at about 50 0C for about 3.5 days. The reaction mixture was then cooled to ambient temperature, filtered through diatomaceous earth, while washing with additional acetone, and then concentrated under reduced pressure. The crude material was then dissolved in a minimal amount of dichloromethane and purified via silica gel chromatography eluting with a gradient of 80-100% ethyl acetate in heptane to afford the title compound. 1H NMR (400 MHz, DMSO-d6) delta ppm 7.88 (s, IH), 7.57 (s, IH), 7.46 (s, IH), 7.24 (s, IH), 4.77 (s, 2H), 1.26 (s, 12H). MS (ESI+) m/z 252.2 (M+H)+
With caesium carbonate; In N,N-dimethyl-formamide; at 20℃; for 12h;
To a solution of 4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -1H-pyrazole (1.0 g, 5.2 mmol) in DMF (10 mL) were added cesium carbonate (2.5 g, 7.7 mmol) and iodoethane (1.2 mL, 15 mmol) . The mixture was stirred at rt for 12 h and diluted with water (30 mL) . The resulting mixture was extracted with DCM (30 mL × 3) . The combinded orgainc layers were dried over anhydrous Na2SO4and concentrated in vacuo. The residue was purified by silica gel column chromatography eluted with PE/EtOAc (v/v) 2/1 to give a colorless oily product (880 mg, 77) .[1505]MS (ESI, pos. ion) m/z: 223.25 [M+1]+
74%
(107a) 1-Ethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole Sodium hydride (55percent, oil, 0.54 g, 12 mmol) was suspended in N,N-dimethylformamide (20 mL), and a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (2.0 g, 10 mmol) in N,N-dimethylformamide (5 mL) was added thereto. The resulting mixture was stirred at room temperature for 1 hr, and then iodoethane (2.4 g, 16 mmol) was slowly added dropwise thereto. The resulting mixture was stirred at room temperature for 3 days. To this reaction solution, water was added. After extraction with diethyl ether, the organic layer was washed with water and brine, and then dried with anhydrous sodium sulfate. The solvent was evaporated under reduced pressure, and the resulting crude product was purified by silica gel column chromatography (Yamazen, eluding solvent: hexane and ethyl acetate) to obtain 1.7 g (yield: 74percent) of the title compound as a colorless oily material. 1H-NMR (400 MHz, CDCl3) delta ppm: 7.78 (1H, s), 7.70 (1H, s), 4.19 (2H, q, J = 7.3 Hz), 1.49 (3H, t, J = 7.3 Hz), 1.32 (12H, s).
38%
With potassium carbonate; In N,N-dimethyl acetamide; at 60℃;
Step 8 1-ethyl-4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole 4,4,5,5-Tetramethyl-2-(1H-pyrazol-4-yl)-1,3,2-dioxaborolane (5.0 g), N,N-dimethylacetamide (50 ml), potassium carbonate (5.3 g) and ethyl iodide (2.1 ml) were mixed, and the mixture was stirred at 60° C. overnight. The reaction mixture was cooled to room temperature, water (100 ml) and ethyl ether (100 ml) were added, and the mixture was partitioned in a separatory funnel. The aqueous layer was further extracted with ethyl ether (100 ml), and the organic layers were combined. The organic layer was washed 3 times with water (100 ml) and once with saturated brine (100 ml), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure, hexane (50 ml) was added to the obtained residue and the mixture was partitioned in a separatory funnel. The organic layer was washed 3 times with water (40 ml) and once with saturated brine (40 ml), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to give the title compound (2.1691 g, 38percent). 1H-NMR (CDCl3) delta: 7.79 (1H, s), 7.70 (1H, s), 4.19 (2H, q, J=7.3 Hz), 1.49 (3H, t, J=7.3 Hz), 1.32 (12H, s).
27%
NaH 60percent dispersion in mineral oil (50.0 mg, 1.24 mmol) was suspended in DMF (2 mL) followed by the addition of a solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)-1H-pyrazole (200 mg, 1.03 mmol) in DMF (550 pL). The resulting mixture was stirred at r.t. for ihour. lodomethane (132 pL, 1.6Smmol) was added dropwise and stirring was continued for 2 days. Water was added and the reaction mixture was extracted with EtOAc. The organic layer was washed with water and brine. Dried Mg504, filtered and concentrated in vacuo. The product was purified by flash chromatography (dry packing) on silica gel using a gradient 0 to 30percent EtOAc in hexanes and afforded the title compound (62.7 mg, 0.28 mmol, 27percent) as a yellow oil.
With caesium carbonate; In acetonitrile; at 90℃; for 2h;
Step 1: l-ethyI-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazoJe; A mixture of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.05 g, 0.2 mmol), iodoethane (31 uL, 0.39 mmol, Sigma-Aldrich, Cat. No. 17780), and cesium carbonate (250 mg, 0.77 mmol) in acetonitrile (1 mL) was stirred at 90 0C for 2 h. After cooling it was quenched with water. The product was extracted with ethyl acetate. The extract was washed with water twice, brine once; dried over Na2SO4. After filtration the filtrate was concentrated to yield 34 mg of the product which was directly used in the next step reaction without further purification.
To a solution of boronate ester 45e (Aldrich, 210 mg, 1.08 mmol) in NMP (2.0 mL) is added Cs2C03 (672 mg, 2.1 mmol). The mixture is stirred for 5 min at RT and then iodoethane (0.13 mL, 1.6 mmol) is added, followed by Nal (15 mg, 0.1 mmol). The mixture is stirred at RT for 16 h, filtered through a Waters Acrodisc filter, and concentrated to afford boronate ester 45f, which is used as such.
With caesium carbonate; In acetonitrile; at 80℃; for 16h;
To a solution of 4-(4,4,5,5-tetramemyl-l ,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.194 g, 1.000 mmol) in acetonitrile (10 mL) was added cesium carbonate (0.652 g, 2.0 mmol), followed by ethyl iodide (0.187 g, 1.2 mmol) at room temperature and stirred at 80°C for 16 h. After the completion of reaction, cooled to room temperature filtered through pad of celite and evaporated off the filtrate to give the titled compound (0.180 g) as a crude. LCMS: m/z 223.1 [M+H] +. lU NMR (600 MHz, Chloroform-d) delta 7.79 (d, / = 4.5 Hz, 1H), 7.71 (d, / = 4.6 Hz, 1H), 4.19 (q, / = 7.2 Hz, 2H), 1.71 (t, / = 7.2 Hz, 3H), 1.33 (d, / = 4.9 Hz, 12H).
With caesium carbonate; In acetone;Reflux;
General procedure: To a solution of 4-pyrazoleboronic acid pinacol esterdissolved in acetone, 2 equiv of Cs2CO3 and 2 equiv of the appropriatealkyl iodide were added. The reaction mixture was left to refluxovernight. The solvent was evaporated in vacuo and the productentered the following reaction without further purification.
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃;Inert atmosphere; Reflux;
Racemic 3-Cyclopentyl-3-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)propanenitrile (23).; To a 500 mL round bottom flask equipped with a stir bar, condenser and nitrogen inlet was charged <strong>[591769-05-0]3-cyclopentylacrylonitrile</strong> (8, a mixture of E and Z isomers, 8.46 g, 0.067 mol, 1.3 equiv), acetonitrile (242 mL, 4.64 mol), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (4, 10.0 g, 0.0515 mol), and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 16.2 ml, 0.108 mol, 2.1 equiv) at room temperature. The resulting solution was then warmed to reflux, and the reaction mixture was stirred at reflux for 18 hours. When the reaction was deemed complete, the reaction mixture was allowed to cool to room temperature followed by concentration under reduced pressure. The residue was purified directly by flash column chromatography (SiO2, 0percent to 30percent ethyl acetate/hexane gradient elution) to afford 3-cyclopentyl-3-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl]propanenitrile (23, 13.1 g, 16.2 g theoretical, 81percent) as off-white solids. This racemic mixture was directly used for subsequent chiral column separation with out further purification. For 23: 1H NMR (DMSO-d6, 400 MHz) delta ppm 8.07 (d, 1H, J=0.53 Hz), 7.65 (s, 1H), 4.42 (td, 1H, J=19.2, 4.5 Hz), 3.14 (dd, 1H, J=9.39, 17.2 Hz), 3.08 (dd, 1H, J=4.58, 17.2 Hz), 2.31 (m, 1H), 1.75 (m, 1H), 1.62-1.32 (m, 4H), 1.29-1.01 (m, 15H); C17H26BN3O2 (MW, 315.22) LCMS (EI) m/e 316 [M++H].
73%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 60℃;
To a solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (2.00 g, 0.0103 mol) in acetonitrile (30 mL) was added <strong>[591769-05-0]3-cyclopentylacrylonitrile</strong> (1.25 g, 0.0103 mol), followed by 1,8- diazabicyclo[5.4.0]undec-7-ene (1.54 mL, 0.0103 mol). The resulting mixture was stirred at 60 0C <n="54"/>overnight, then evaporated to dryness. The residue was purified on silica gel, eluting with 0 to 50percent EtOAc in hexanes, to give the desired product (2.36 g, 73percent). LCMS (M+H) 316.1.
27.5%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 60℃; for 18h;
To a solution of compound 3-b (1 g, 8.26 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (2.4 g, 12.39 mmol) in acetonitril (10 mL) was added 1,8-diazabicyclo[5.4.0]undec-7-ene (2.5 g, 16.52 mmol). The mixture was stirred at 60° C. for 18 hours. The mixture was concentrated in vacuum. To the residue was added water (50 mL), then the mixture was extracted with ethyl acetate (100 mL×3). The organic layers were combined, washed with water (60 mL×3) and saturated brine (60 mL) in sequence, dried over anhydrous sodium sulfate. The mixture was filtrated, the filtrate was concentrated in vacuum, the residue was purified by silica column chromatography (petroleum ether:ethyl acetate=3:1) to give yellow oil 3-a (715 mg, yield: 27.5percent). LC-MS (ESI): m/z=316 [M+H]+.
With trifluoroacetic acid; In toluene; at 90℃; for 2h;
3,4-Dihydro-2H-pyran (5.6 g, 67 mmol) and trifluoroacetic acid (1.17 g, 10.3 mmol) were added to a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H- pyrazole (10.0 g, 51.5 mmol) in toluene (200 mL), and the reaction mixture was heated to 90 C for 2 hours. After cooling to room temperature, the reaction mixture was partitioned between ethyl acetate (200 mL) and saturated aqueous sodium bicarbonatesolution (100 mL), and the aqueous layer was extracted with ethyl acetate (100 mL). The combined organic layers were washed with saturated aqueous sodium chloride solution (100 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Silica gel chromatography (Gradient: 10% to 50% ethyl acetate in petroleum ether) provided the product as a white solid. Yield: 13.4 g, 48.2 mmol, 94%. 1H NMR (400 MHz, CDCl3) 7.94 (s, 1H), 7.83 (s, 1H), 5.41 (dd, J=9.5, 2.5 Hz, 1H), 4.01-4.08 (m, 1H), 3.65-3.74(m, 1H), 1.98-2.18 (m, 3H), 1.6-1.76 (m, 3H), 1.32 (s, 12H).
90%
With toluene-4-sulfonic acid; In dichloromethane; at 20℃; for 4h;
To a solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (110.00 g, 566.89 mmol) in DCM (1000 mL) was added dihydropyran (95.37 g, 1.13 mol, 103.66 mL) and TsOH .H2O (53.92 g, 283.45 mmol). The mixture was stirred at 20 C for 4 h. TLC (petroleum ether/EtOAc = 5/1) indicated starting material was consumed completely and one main new spot (Rf=0.4) formed. LCMS showed starting material was consumed completely and one main peak with desired mass was detected. The reaction mixture was concentrated under reduced pressure to remove volatiles. The residue was diluted with sat. NaHCCb (350 mL) and the mixture was extracted with EtOAc (2 x 500 mL). The combined organic layers were dried over Na2S04, filtered, and concentrated under reduced pressure to provide a residue. The residue was purified by column chromatography with petroleum ether/ethyl acetate (from 20/1 to 15/1) to afford the title compound (142.00 g, 90%) as colorless oil. 1H NMR (400 MHz, DMSO-d6) 400 MHz delta 8.06 (s, 1H), 7.61 (s, 1H), 5.42 (dd, J= 10.0, 2.0 Hz, 1H), 3.94-3.86 (m, 1H), 3.66-3.54 (m, 1H), 2.17- 2.03 (m, 1H), 1.96- 1.82 (m, 2H), 1.77-1.37 (m, 3H), 1.33-1.18 (m, 12H). MS (ES+) m/e 279 (M+H).
73%
With toluene-4-sulfonic acid; In dichloromethane; at 30℃; for 4h;
To a solution of 4-(4,4, 5, 5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole (10.00 g, 51.54 mmol) in CH2C12 (100.00 mL) was added DHP (8.67 g, 103.08 mmol, 9.42 mL) and TsOHH20 (4.90 g, 25.77 mmol). The mixture was stirred at 30 C for 4 hour. LCMS showed 4-(4,4, 5,5 -tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazolewas consumed completely and one main peak with desired mass was detected. The reaction mixture was concentrated under reduced pressure to remove solvent. The residue was diluted with sat.NaHCO3 (35 mL) and the mixture was extracted with EtOAc (100 mL). The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (Si02, Petroleum ether/Ethyl acetate = 20/1 to 1/1) to afford the title compound (10.5 g, 73%) as colorless oil. ?H NIVIR (400 MHz, DMSO-d6) oe 8.06 (s, 1H), 7.62 (s, 1H), 5.44-5.41 (m, 1H), 3.94-3.89 (m, 1H), 3.76-3.58 (m, 2H), 3.46-3.41 (m, 1H), 2.15-2.06 (m, 1H), 1.95-1.84 (m, 2H), 1.76-1.40 (m, 9H), 1.26 (s, 12H).
73%
With toluene-4-sulfonic acid; In dichloromethane; at 30℃; for 4h;
[0406] To a solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (10.00 g, 51.54 mmol) in CH2C12 (100.00 mL) was added DHP (8.67 g, 103.08 mmol, 9.42 mL) and TsOH H20 (4.90 g, 25.77 mmol). The mixture was stirred at 30 C for 4 hour. LCMS showed 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazolewas consumed completely and one main peak with desired mass was detected. The reaction mixture was concentrated under reduced pressure to remove solvent. The residue was diluted with sat.NaHCC (35 mL) and the mixture was extracted with EtOAc (100 mL). The organic layer was dried over Na2S04, filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (Si02, Petroleum ether/Ethyl acetate = 20/1 to 1/1) to afford the title compound (10.5 g, 73%) as colorless oil. 1H NMR (400 MHz, DMSO-i/6) delta 8.06 (s, 1H), 7.62 (s, 1H), 5.44-5.41 (m, 1H), 3.94-3.89 (m, 1H), 3.76-3.58 (m, 2H), 3.46-3.41 (m, 1H), 2.15-2.06 (m, 1H), 1.95-1.84 (m, 2H), 1.76-1.40 (m, 9H), 1.26 (s, 12H).
Example 14: Synthesis of (2R,4S)-4-{(3,5-Bis-trifluoromethyl-benzyl)-[5-(1H-pyrazol-4- yl)-pyrimidin-2-yl]-amino}-2-ethyl-pyrrolidine-1-carboxylic acid isopropyl ester; A solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (5 mmol; 970 mg) and 3,4-dihydro-2H-pyran (10 mmol; 841 mg) and p-toluenesulfonic acid monohydrate (0.5 mmol; 95 mg) in 1 ,2-dichloroethane (25 ml_) is stirred at 40 0C for 2 hours. The reaction mixture is cooled to ambient temperature, and then quenched with saturated aqueous NaHCO3 solution. The product is extracted with dichloromethane. The organic layer is <n="82"/>washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure. The resulting material is used in the next step without further purification.
5.04 g
With toluene-4-sulfonic acid; In 1,2-dichloro-ethane; at 50℃; for 2h;
To a stirred solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (5.0 g, 25.76 mmol) in dry dichloroethane (128 ml), 3,4-dihydro-2H-pyran (4.71 ml, 51.52 mmol) was added followed by p-toluenesulfonic acid (490 mg, 2.576 mmol) and the reaction mixture was stirred at 50 C for 2 h. The reaction mixture was quenched with aqueous saturated solution of sodium bicarbonate (75 ml) and extracted with ethyl acetate (2 x 150 ml). The combined organic layer was washed with brine (100 ml) and dried over sodium sulphate. The mixture was evaporated under reduced pressure and residue obtained was purified by column chromatography to yield 5.04 g of product as a white solid. 1H NMR (300 MHz, CDCI3): delta I .31 (s, 12H), 1.54-1.70 (m, 3H), 1.98-2.20 (m, 3H), 3.68 (t, / = 10.2 Hz, 1H), 4.04 (d, / = I I .7 Hz, 1H), 5.35-5.45 (m, 1H), 7.82 (s, 1H), 7.93 (s, 1H).
With toluene-4-sulfonic acid; In dichloromethane; at 40℃; for 2h;Inert atmosphere;
step 1: DTSA (463 mg, 2.6 mmol) was added to a solution of compound 101-1 (5.0 g, 25.8 mmol) and 2,3-dihydropyran (4.33 g, 51.5 mmol) in dichloromethane (100 ml) with stirring, and the reaction mixture was stirred at 40 C. for 2 h. After completion of the reaction, the reaction solution was washed with water, extracted with EA, and concentrated under reduced pressure to give the crude product 102-2 (7.0 g, 60%) which was used for the next step without purification, MS m/z (ESI): 279 [M+H]+.
With toluene-4-sulfonic acid; In dichloromethane; at 60℃; for 1h;
A solution of 4-(4,4,5, 5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1H-pyrazole (10 g, 51.5 mmol), 3,4- dihydro-2H-pyran (9.40 ml, 103 mmol), and p-toluenesulfonic acid monohydrate (1.274 g, 6.70 mmol) in DCM (100 ml) was stirred at 60 C for 1 hour to see complete conversion of the startmaterial, by LCMS. It was quenched with saturated NaHCO3, and extracted with DCM. The organic phase was purified with a silica gel column, eluting with 30 % EtOAc in hexanes, to get the desired product as a white solid. MS (M+1): 279
5.49 g
With toluene-4-sulfonic acid; In chloroform; at 50℃; for 3.5h;
(1) 3,4-Dihydro-2H-pyran (4.00 mL) and p-toluenesulfonic acid monohydrate (421 mg) were added to a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (4.29 g) in chloroform (44.2 mL), and the mixture was stirred at 50C for 3.5 hours. The mixture was cooled to room temperature, diluted with chloroform, and then washed sequentially with an aqueous solution of saturated sodium hydrogen carbonate and brine. The organic layer was separated by a phase separator and concentrated under reduced pressure. The obtained residue was purified by column chromatography (n-hexane/ethyl acetate = 19:1 to ethyl acetate only) to give 1-(oxan-2-yl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (5.49 g) as a light brown oily substance.
With caesium carbonate; In N,N-dimethyl-formamide; at 0 - 20℃;
2-lodopropane (1.14 g, 6.70 mmol, 0.67 mL)was added to a mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1.00 g, 5.15 mmol) and caesium carbonate (3.49 g,10.72 mmol) in dry N,N-dimethylformamide (20 mL) at 0O. After stirring for 30 mm the ice- water bath was removed. The reaction mixture was stirred at room temperature overnight. Thereaction mixture was diluted with ethyl acetate (150 mL) and washed with brine (3x100 mL).The organic layer was dried with sodium sulfate and concentrated in vacuo. Purification byflash column chromatography (Method L7; 12 g; heptane, 10%-30% ethyl acetate) afforded0.69 g (2.32 mmol; 57% of theory) of the title compound.GO-MS (Method L9): R1 = 3.86 mm; m/z = 236 M1 H NMR (300 MHz, Ohloroform-d, Method M2) 6 7.79 (s, 1 H), 7.74 (s, 1 H), 4.52 (p, J = 6.7 Hz,1H), 1.50 (d, J = 6.7 Hz, 6H), 1.32 (s, 12H).
With caesium carbonate; In N,N-dimethyl-formamide; at 100℃; for 19h;
Method C: General Procedure for N-alkylation of Substituted Pyrazoles, Using Halogenated (Bromo- or Iodo-) and Mesylated SpeciesIn a sealed tube, to a suspension of 4-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (567 mg, 2.92 mmol, 1.0 eq) and Cs2CO3 (1.544 g, 4.739 mmol, 1.6 eq) in DMF (6 mL), the halide or mesylate (4.43 mmol, 1.5 eq) was added and the reaction was allowed to stir at 100 C. for 19 h. Water was added to dilute the reaction and dissolve all salts that had formed, after which EtOAc was added and the two layers were separated. The organic layer was washed with water (2×) and brine (1×). The combined aqueous layers were back extracted with EtOAc (1×), and the combined EtOAc extracts were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give the target material.; 1-Isopropyl-4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole; Method C was followed, using isopropyl iodide (753.3 mg, 4.431 mmol, 1.5 eq). The title compound was obtained as a yellow oil that was used without further purification. 1H NMR (400 MHz, CDCl3): delta=1.33 (s, 12H), 1.51 (d, J=6.8 Hz, 6H), 4.53 (septet, J=6.7 Hz, 1H), 7.75 (s, 1H), 7.80 (s, 1H). MS (AP+): m/z=235.98 (76) [MH+]. HPLC: tR=3.22 min (ZQ3, polar-5 min).
With caesium carbonate; In N,N-dimethyl-formamide; at 100℃;
Preparation 221-Isopropyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole In a microwave vial, a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1 g, 5.15 mmol) in DMF (10 mL) was added cesium carbonate (5.04 g, 15.46 mmol) and 2-iodopropane (2.58 mL, 25.8 mmol). The mixture was heated at 100 C. overnight. After cooling to RT, H2O (300 mL) was added and the aqueous layer was extracted with EtOAC (2×200 mL). The organic extracts were combined, dried over Na2SO4, filtered, and solvent was removed in vacuo. The resulting crude material was reconstituted in MeOH (1.0 mL) and purified by HPLC. The fractions were collected and stripped to dryness via rotary evaporation to yield the title compound. [M+H] calc'd for C12H21BN2O2, 237. found 237.
With caesium carbonate; In acetone;Reflux;
General procedure: To a solution of 4-pyrazoleboronic acid pinacol esterdissolved in acetone, 2 equiv of Cs2CO3 and 2 equiv of the appropriatealkyl iodide were added. The reaction mixture was left to refluxovernight. The solvent was evaporated in vacuo and the productentered the following reaction without further purification.
Example 20; (R)-N-(2-(2-(5-Hydroxy-3-oxo-3,4-dihydro-2H-benzo[b][l,4]oxazin-8- yl)ethylamino)ethyl)-3 -(3 -( 1 -isopropyl- 1 H-pyrazol-4-yl)phenethoxy)-N-(3 -methylbutan-2- yl)propanamide Trifluoroacetic Acid Salt Step i) 1 -Isopropyl-4-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole <n="118"/> Sodium hydride (0.412 g, 60% in oil) was washed with 2 ml of dry THF. DMF (6 mL) was added followed by portionwise addition of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)-lH-pyrazole (1 g). The mixture was stirred for 30 min. and 2-iodopropane (1.546 mL) was added. The mixture was stirred at 600C for 50 min. and at 200C for 16 h. The mixture was quenched with sat. ammonium chloride and ethyl acetate (70 ml). The mixture was washed with water (4 x). The organic was dried over magnesium sulfate, filtered and evaporated to afford crude product. The crude product was purified by flash silica chromatography, elution gradient 0 to 60% ethyl acetate in isohexane to afford the subtitled compound (840 mg) as a solid.MS [M+H]+ = 237 (MultiMode+)1H NMR (400 MHz, CDCl3) delta 7.79 (s, IH), 7.74 (s, IH), 4.52 (septet, J = 6.7 Hz, IH), 1.50 (d, J = 6.7 Hz, 6H), 1.32 (s, 12H)
With caesium carbonate; In N,N-dimethyl-formamide; at 100℃; for 19h;Sealed tube;
1-lsopropyl-4-(4,4,5,5-tetramethyl[1 ,3,2]dioxaborolan-2-yl)-1H-pyrazole [287] In a sealed tube, to a suspension of 4-(4,4,5,5-tetramethyl[1 ,3,2]dioxaborolan-2-yl)-1H- pyrazole (566.9 mg, 2.922 mmol) and Cs2CO3 (1.5442 g, 4.739 mmol) in DMF (6 ml_), isopropyl iodide (753.3 mg, 4.431 mmol) was added and the reaction was allowed to stir at 100 C for 19 h. Water was added to dilute the reaction and dissolve all salts that had formed, after which EtOAc was added and the two layers were separated. The organic layer was washed twice with water and once with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The combined aqueous layers were back extracted once with EtOAc, which was combined with the other organic batch. One obtained the title material as yellow oil. It was used without further purification in the next step. 1H NMR (400 MHz, CDCI3): delta = 1.33 (s, 12H), 1.51 (d, J = 6.8 Hz, 6H), 4.53 (spt, J = 6.7 Hz, 1 H), 7.75 (s, 1 H), 7.80 (s, 1 H). MS (AP+): m/z 235.98 (76) [MH+]. HPLC: tR = 3.22 min (ZQ3, polar_5min).
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In water; N,N-dimethyl-formamide; at 150℃; for 3h;Inert atmosphere; Microwave irradiation;
Example 5; Preparation of N-(5-(1H-pyrazol-4-yl)thiazolo[5,4-b]pyridin-2-yl)-4-cyclopropylbenzamide (5); Step A: To a microwave vial, <strong>[934266-82-7]5-bromothiazolo[5,4-b]pyridin-2-amine</strong> (1B, 100 mg, 1.0 equivalent), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (5A, 2.0 equivalent), Na2CO3 (4.0 equivalents), DMF (0. 15 M) and H2O (0.73 M) were added. While mixture was under N2, Pd(PPh3)4 was added and the mixture was then heated in a microwave for 3 h at 150 C. The crude mixture was filtered, and purified by preparatory LCMS. Intermediate 5-(1H-pyrazol-4-yl)thiazolo[5,4-b]pyridin-2-amine (5B) was obtained as a white solid. ESI-MS: m/z 218.0 (M+H)+.
With caesium carbonate; In N,N-dimethyl-formamide; at 0 - 20℃;
N-N(Bromomethyl)cyclopropane (0.95 mg, 6.70 mmol, 0.70 mL, 95 %) was added to a mixture of4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (1 .00 g, 5.15 mmol) and caesium carbonate (3.49 mg, 10.72 mmol) in dry N,N-dimethylformamide (20 mL) at 0O. After stirring for 30 mm the ice-water bath was removed. The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with ethyl acetate (150 mL) and washed with brine (3x100 mL). The organic layer was dried with sodium sulfate and concentrated in vacuo to afforded 1.30 g (4.38 mmol, 85% of theory) of the title compound. GO-MS (Method L9): R1 = 4.35 mm; mlz = 247 M1 H NMR (300 MHz, Ohloroform-d, Method M2) 6 7.81 (s, 1 H), 7.79 (s, 1 H), 3.99 (d, J = 7.1 Hz,2H), 1.32 (s, 12H), 1.27 (m, 1 H), 0.71 - 0.58 (m, 2H), 0.41 - 0.33 (m, 2H).
49%
To a reaction vessel containing sodium hydride (60 wt% dispersion in mineral oil)(3.09 g, 77.32 mmol) was added NN-dimethylformamide (30 mL) under a stream of nitrogen followed by 4- (4,4,5, 5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (10 g, 51.55 mmol) and the reaction mixture was stirred for 20 min at room temperature. Cyclopropylmethylbromide (8.7 g, 61.86 mmol) was then added and the reaction mixture was stirred for 24 h under an atmosphere of nitrogen. The reaction mixture was slowly poured over saturated aqueous ammonium chloride and extracted with EtOAc. The organic layer was then washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified via flash chromatography on silica gel (solvent gradient: 0%-100% EtOAc in cyclohexane) to afford 1- (cyclopropylmethyl)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (6.29 g, 49%). LCMS (ESI): [M+H]+ = 249.2; lH NMR (400 MHz, CDC13) delta 7.80 (d, J= 7.4 Hz, 2H), 3.99 (d, J = 7.0 Hz, 2H), 1.32 (s, 12H), 1.29 - 1.26 (m, 1H), 0.69 - 0.60 (m, 2H), 0.41 - 0.33 (m, 2H).
48%
With potassium carbonate; In N,N-dimethyl-formamide; at 60℃; for 14h;
To a stirred solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (8.00 g,41.2 mmol) in DMF (130 mL) was added potassium carbonate (17.1 g, 124 mmol) and(bromomethyl)cyclopropane (6.0 mL, 62 mmol). The mixture was stirred at 600 C for 14 h. Water was added and the mixture was extracted with ethyl acetate. The organic phase was washed with half-saturated sodium chloride solution, dried (sodium sulfate) and the solvent was removed in vacuum. Silicagel chromatography gave 4.95 g (48 % yield) of the titlecompound.LC-MS (Method 2): Rt = 1.12 mm; MS (ESIpos): mlz = 249 [M+H]1HNMR (400 MHz, DMSO-d6) O [ppm]: 0.314 (0.66), 0.318 (0.59), 0.325 (0.56), 0.330 (0.65),0.471 (0.53), 0.476 (0.61), 0.492 (0.62), 0.496 (0.57), 1.045 (0.78), 1.219 (1.70), 1.224 (16.00),3.295 (1.54), 3.927 (1.24), 3.946 (1.22), 7.542 (1.07), 7.544 (1.08), 7.932 (1.09).
48%
With potassium carbonate; In N,N-dimethyl-formamide; at 60℃; for 14h;
To a stirred solution of 4-(4, 4,5, 5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (purchased from Acros, CAS 269410-08-4, 8.00 g, 41 .2 mmol) in DMF (130 mL) was added potassium carbonate (17.1 g, 124 mmol) and (bromomethyl)cyclopropane (6.0 mL, 62 mmol). The mixture was stirred at 60 C for 14 h. Water was added and the mixture was extracted with ethyl acetate. The organic phase was washed with half-saturated sodium chloride solution, dried (sodium sulfate) and the solvent was removed in vacuum. Silicagel chromatography gave 4.95 g (48 % yield) of the title compound. LC-MS (Method 2): Rt = 1.12 min; MS (ESIpos): m/z = 249 [M+H]+ 1H-NMR (400 MHz, DMSO-d6) _ [ppm]: 0.314 (0.66), 0.318 (0.59), 0.325 (0.56), 0.330 (0.65), 0.471 (0.53), 0.476 (0.61 ), 0.492 (0.62), 0.496 (0.57), 1.045 (0.78), 1 .219 (1 .70), 1 .224 (16.00), 3.295 (1.54), 3.927 (1.24), 3.946 (1.22), 7.542 (1 .07), 7.544 (1 .08), 7.932 (1.09).
With caesium carbonate; In acetonitrile; at 90℃;
Step 1: J- (cyclopropylmethyl)-4- (4, 4, 5, 5-tetramethyl- 1, 3, 2-dioxaborolan-2-yl)-lH-pyrazole; A mixture of 4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.1 g, 0.5 mmol), cyclopropylmethyl bromide (60.0 uL, 0.62 mmol, Aldrich, Cat. No. 242403), and cesium carbonate (500.0 mg, 1.5 mmol) in acetonitrile (1 mL) was stirred at 90 0C overnight. After cooling it was diluted with ethyl acetate. Then the organic solution was washed with water, brine; dried over Na?SO-i. After filtration the filtrate was concentrated to yield 120 mg of the product which was directly used in the next step reaction without further purification.
Example 19; N-Cyclopentyl-3-(3-(l-(cyclopropylmethyl)-lH-pyrazol-4-yl)phenethoxy)-N-(2-(2-(5- hydroxy-3-oxo-3,4-dihydro-2H-benzo[b][l,4]oxazin-8-yl)ethylamino)ethyl)propanamide Trifluoroacetic Acid Salt Step i) 1 -(Cyclopropylmethyl)-4-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 H- pyrazole Sodium hydride (0.195 g) was washed with 2 ml of dry THF. DMF (4 mL) was added followed by portionwise addition of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazole (0.63 g). The mixture was stirred for 30 min and (bromomethyl)cyclopropane (0.313 mL) added. The mixture was stirred at 600C for 50 min and at rt for 16 h. The mixture was quenched with sat. ammonium chloride and ethyl acetate (70 mL). The mixture was washed with water (4x). The organic was dried over magnesium sulfate, filtered and evaporated to afford crude l-(cyclopropylmethyl)-4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-lH-pyrazole (717 mg) that was used in the next step without purification.MS [M+H]+ = 249 (MultiMode+) <n="115"/>1H NMR (400 MHz, CDCl3) delta 7.81 (s, IH), 7.79 (s, IH), 3.99 (d, J = 6.9 Hz, 2H), 1.32 (s, 12H), 1.31 - 1.23 (m, IH), 0.70 - 0.61 (m, 2H), 0.42 - 0.33 (m, 2H)
With caesium carbonate; In acetone; at 20℃;Reflux; Inert atmosphere;
To a solution of 4-(4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-lH-pyrazole (2.27 mmol; 1.0 equiv ) in acetone (10.0 mL) at room temperature were added under argon 3-chloromethyl-pyridine hydrochloride (3.18 mmol; 1.4 aquiv.) and cesium carbonate (2.8 equiv.). The reaction mixture was heated for 4 hours at reflux. The mixture was then cooled to room temperature, quenched by addition of a saturated aqueous solution of sodium hydrogen carbonate (100 mL) and extracted with dichloromethane (2 x 100 mL) The organic layer was dried over sodium sulfate, filtered and concentrated to dryness. The resulting residue was purified by flash chromatography over silica gel (dichloromethane/ethyl acetate) to afford the expected boronate as a white solid which was used in the next step without further purification.
With potassium carbonate; In N,N-dimethyl-formamide; at 60℃; for 2h;
This compound was prepared via Method T using l-Difluoromethyl-4-(4,4,5,5- tetramethyl-[l,3,2]dioxaborolan-2-yl)-lH-pyrazole prepared by the following Method:[00513] ClCHF2 was added to a solution of 4-(4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)- lH-pyrazole and K2CO3 in DMF. The reaction was heated at 6O0C for 2h. The reaction mixture was allowed to cool to room temperature. EtOAc and water were added to the reaction. The organic phase was separated, dried over MgSO4, evaporated under reduced pressure. The crude was used without further purification.
With potassium carbonate; In N,N-dimethyl-formamide; at 160℃; for 2h;Microwave irradiation;
A mixture of 4-(4,4,5 ,5 -tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1H-pyrazole (1.94 g, 10 mmol), 2-bromoethyl methyl ether (1.68 g, 12 mmol) and K2C03 (2.76 g, 20 mmol) in DMF (16 mL) was stirred at 160 °C for 2 h in the microwave. The reaction mixture was concentrated and purified by silica gel chromatography (30percent EA:PE) to give 2.2 g (90percent) of the title compound as yellow oil. ?H NMR (400 MHz, CDC13): oe 1.32 (12H, s), 3.32 (3H, s), 3.75 (2H, t, J= 5.2 Hz), 4.30 (2H, t, J= 5.2 Hz), 7.77 (1H, s), 7.79 (1H, s). [M+H] Calc?d for C,2H2,BN203, 253; Found, 253.
68%
With caesium carbonate; In N,N-dimethyl-formamide; at 150℃; for 0.5h;Microwave irradiation;
Example A68Preparation of intermediate 68: l-(2-Methoxy-ethyl)-4-(4,4,5,5-tetramethyl- [l,3,21dioxaborolan-2-yl)-7H-pyrazoleA mixture of 4-(4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-7H-pyrazole (1 g, 5.15 mmol), 2-bromoethyl methyl ether (0.63 ml, 6.7 mmol) and cesium carbonate (2.52 g, 7.73 mmol) in N,N-dimethylformamide (7 ml) was stirred at 150 °C for 30 min. under microwave irradiation. The mixture was partitioned between water and diethyl ether. The organic layer was separated, dried (Na2S04), filtered and the solvents evaporated in vacuo. The crude product was purified by flash column chromatography (silica; ethyl acetate in heptane 30/70). The desired fractions were collected and concentrated in vacuo to yield intermediate 68 (0.88 g, 68percent) as a pale yellow oil.
68%
With caesium carbonate; In N,N-dimethyl-formamide; at 150℃; for 0.5h;microwave irradiation;
Example A68Preparation of intermediate 68: 1-(2-Methoxy-ethyl)-4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazoleA mixture of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (1 g, 5.15 mmol), 2-bromoethyl methyl ether (0.63 ml, 6.7 mmol) and cesium carbonate (2.52 g, 7.73 mmol) in N,N-dimethylformamide (7 ml) was stirred at 150° C. for 30 min. under microwave irradiation.The mixture was partitioned between water and diethyl ether.The organic layer was separated, dried (Na2SO4), filtered and the solvents evaporated in vacuo.The crude product was purified by flash column chromatography (silica; ethyl acetate in heptane 30/70).The desired fractions were collected and concentrated in vacuo to yield intermediate 68 (0.88 g, 68percent) as a pale yellow oil.
57%
With caesium carbonate; In N,N-dimethyl-formamide; at 0 - 20℃;
2-Bromoethyl methyl ether (0.93 g, 6.70 mmol, 0.64 mL) was added to a mixture of 4-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (1 .00 g, 5.15 mmol) and caesium carbonate (3.49 mg, 10.72 mmol) in dry N,N-dimethylformamide (20 mL) at 0°O. After stirring for 30 mm the ice-water bath was removed. The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with ethyl acetate (150 mL) and washed with brine(3x100 mL). The organic layer was dried with sodium sulfate and concentrated in vacuo. Purification by flash column chromatography (Method L7; 12 g; heptane, 10percent-30percent ethyl acetate) afforded 0.74 g (2.92 mmol; 57percent of theory) of the title compound.GO-MS (Method L9): R1 = 4.21 mm; m/z = 251 MH NMR (300 MHz, Ohloroform-d, Method M2) 6 7.79 (s, 1 H), 7.76 (s, 1 H), 4.29 (t, J = 5.3 Hz,2H), 3.75 (t, J = 5.3 Hz, 2H), 3.32 (s, 3H), 1.31 (s, 12H).
56%
General procedure: Preparation 122: 1 -(2-Methoxyethyl)-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- Method H NaH (60percent, 83 mg) was added to a solution of 4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 - -pyrazole (204 mg, 1 .05 mmol) in DMF (4 mL). After stirring for 15 minutes, 1 -bromo-2-methoxyethane (175 mg, 1 .26 mmol) in DMF (1 mL) was added. The resulting solution was stirred at 80°C under microwave irradiation for 60 minutes. The reaction mixture was diluted with brine and extracted with EtOAc. The combined organic layers were washed with water, dried with Na2S04j and concentrated in vacuo to afford the title compound as a yellow oil that was used directly in the next step (148 mg, 56percent). 1 H NMR (500 MHz, CDCI3): delta 7.80 (d, J = 0.7 Hz, 1 H), 7.77 (d, J = 0.7 Hz, 1 H), 4.31 (t, = 5.3 Hz, 2H), 3.76 (t, J = 5.3 Hz, 2H), 3.33 (s, 3H), 1.32 (s, 12H). LCMS (ESI) Rt = 2.17 minutes MS m/z 253 [M+H]+
26%
To a solution of 4-(tetramethyl-1 ,3, 2-dioxaborolan-2-yl)-1H-pyrazole (1 .00 g, 5.1 mmol) in DMF (5 mL) was added sodium hydride (245 mg of a 60percent dispersion in mineral oil, 6.1 mmol) at room temperature. The mixture was then stirred at room temperature for 60 minutes. The mixture was cooled to 0 ° C and 2-bromoethyl methyl ether (0.72 mL, 7.7 mmol) was added drop wise via syringe. After complete addition, the mixture was allowed to warm to room temperature and stirred for an hour prior to addition of ethyl acetate (70 mL) and water (30 mL). The organic layer was separated, washed with water (2 x 30 mL) and brine (30 mL), dried (Na2S04), filtered and concentrated at reduced pressure. The residue was purified by Biotage Isolera? chromatography [Biotage SNAP Cartridge KP-Sil 50 g; using a gradient of eluents, 0-50percent EtOAc in heptane] to give the title compound (333 mg, 26percent yield) as a colourless oil. 1H NMR (250 MHz, DMSO-d6) delta [ppm] 7.88 (s, 1 H), 7.57 (s, 1 H), 4.32 - 4.20 (m, 2H), 3.73 - 3.60 (m, 2H), 3.21 (s, 3H), 1 .25 (s, 12H). LCMS (Analytical Method A): Rt = 1 .01 mins, MS (ESIPos): m/z = 253 (M+H)\
20%
With potassium hydroxide; In ethanol; at 50℃; for 76h;
Intermediate 1171 -[2-(Methyloxy)ethyl]-4-(4,4,5,5-tetrameth l-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazolA solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (10 g, 51 .5 mmol) in ethanol (50 mL) was treated with KOH (3.47 g, 61 .8 mmol) and 1 -bromo-2- (methyloxy)ethane (5.81 mL, 61.8 mmol) at room temperature and the resulting mixture was stirred at 50°C under nitrogen for 16 h then cooled to room temeprature. 1 -Bromo-2- (methyloxy)ethane (2 ml_, 21 .3 mmol) was added and the resulting mixture was stirred at 50°C for 60 h then cooled to room temperature. The mixture was filtered through celite and the insoluble were washed with ethanol. The combined filtrate and washings were concentrated in vacuo. Purification of the residue on SP4 using a 100 G silica cartridge (gradient: 0 to 100percent AcOEt in Hexanes) gave 1 -[2-(methyloxy)ethyl]-4-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (2.72 g, 10.25 mmol, 20percent) as a yellow oil. LCMS (method G): Retention time 0.87 min, [M+H]+ = 252.9
20%
With potassium hydroxide; In ethanol; at 50℃; for 66h;Inert atmosphere;
A solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (10 g, 51.5 mmol) in ethanol (50 mL) was treated with KOH (3.47 g, 61.8 mmol) and 1-bromo-2-(methyloxy)ethane (5.81 mL, 61.8 mmol) at room temperature and the resulting mixture was stirred at 50° C. under nitrogen for 16 h then cooled to room temperature. 1-Bromo-2-(methyloxy)ethane (2 mL, 21.3 mmol) was added and the resulting mixture was stirred at 50° C. for 60 h then cooled to room temperature. The mixture was filtered through celite and the insoluble were washed with ethanol. The combined filtrate and washings were concentrated in vacuo. Purification of the residue on SP4 using a 100 G silica cartridge (gradient: 0 to 100percent AcOEt in Hexanes) gave 1-[2-(methyloxy)ethyl]-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (2.72 g, 10.25 mmol, 20percent) as a yellow oil. LCMS (method G): Retention time 0.87 min, [M+H]+=252.9
With caesium carbonate; In N,N-dimethyl-formamide; at 90℃; for 2h;microwave irradiation;
A solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.8 g, 4.1 mmol), cesium carbonate (2.0 g, 6.2 mmol), and l-bromo-2-methoxyethane (0.41 rnL, 4.3 mmol) in DMF (14 rnL) was heated in a microwave at 900C for 1 hr. After the initial heating, additional l-bromo-2-methoxyethane (0.41 rnL) was added to the reaction. Heating was repeated for an additional 1 hr. The crude reaction mixtures were then diluted with water (250 mL) and extracted with ethyl acetate (3 x 50 mL). Product was purified by silica gel column using DCM/EtOAc/MeOH (8/1.5/0.5) as eluent to give l-(2-methoxyethyl)-4- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (1.2 g) as a light yellow oil. ESI- MS :m/z 253.2 (M+H)+.
With caesium carbonate; In acetonitrile; at 90℃;
Step I : l-(2-methoxyethyl)-4-(4, 4, 5, 5-tetramethyl-L 3, 2-dioxaborolan-2-yl)-W-pyrazoIe; A mixture of [A] 4-(4,4,5s5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l H-pyrazole ( 100 mg, 0.5 mmol), 2-bromoethyl methyl ether (58 uL, 0.62 mmol, Aldrich, Cat. No. 238155), and cesium carbonate (500.0 mg, 1.5 mmol) in acetonitrile (1 mL) was stirred at 90 0C overnight. After cooling it was quenched with water, extracted with ethyl acetate. The extract was washed with water twice, brine once; dried over Na2SO4. After filtration the filtrate was concentrated to yield 90 mg of the product. LCMS (M+H)+: m/z = 253.1
With sodium hydride; In tetrahydrofuran; mineral oil; for 24h;Reflux;
ntermediate 1 1 1 -(2-methoxyethyl)-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole[094] A mixture of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (200 mg, 1 .03 mmol),1 -bromo-2-methoxyethane (280 mg, 2.01 mmol), and NaH (200 mg, 4 mmol) in THF (15 mL) was stirred at reflux for 24 hours, cooled to the ambient temperature, and concentrated in vacuo. The residue was treated with HCI(aq) and extracted with EtOAc. The insoluble solid was removed by filtration, and the organic solution was concentrated to give the title compound. MS (m/z): 253 (M.bul.H)+.
25.4 g
With caesium carbonate; In N,N-dimethyl-formamide; at 80℃;
4-(4,4,5,5-Tetramethyl-[1 ,3,2]clioxaborolan-2-yl)-1 H-pyrazole (19.4 g, 0.10 mol), 1- bromo-2-methoxy-ethane (14.18 ml, 0.15 mol), and caesium carbonate (32.58 g, 0.1 mol) are dissolved in DMF (200 ml). The suspension is stirred for 16 h at 80°C, filtered and the solvent is removed in vacuum. The residue is treated with tert-butyl methyl ether (200 ml), filtered over Celite and then the solvent is removed in vacuum; yield: 25.4 g 1-(2-methoxy-ethyl)-4-(4,4,5,5-tetramethyl-[1,3,2]dioxa- borolan-2-yl)-1H-pyrazole; HPLC/MS: 1.82 min, [M+H] = 253.
With sodium hydride; In tetrahydrofuran; for 24h;Reflux;
A mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (200 mg, 1.03 mmol), 1-bromo-2-methoxyethane (280 mg, 2.01 mmol), and NaH (200 mg, 4 mmol) in THF (15 mL) was stirred at reflux for 24 hours, cooled to the ambient temperature, and concentrated in vacuo. The residue was treated with HCl(aq) and extracted with EtOAc. The insoluble solid was removed by filtration, and the organic solution was concentrated to give the title compound. MS (m/z): 253 (M+H)+.
1.31 g
With caesium carbonate; In acetonitrile; at 50℃;Inert atmosphere;
1H-pyrazole-4-boronic acid pinacol ester (1.0 g, 5.155 mmol),2-Bromoethyl methyl ether (0.788 g, 5.669 mmol)And cesium carbonate (5.04 g, 15.469 mmol) were dissolved in acetonitrile (20 mL),The mixture was stirred under nitrogen at 50 ° C overnight,filter,Concentrate to dryness to give 1- (2-methoxyethyl) -1H-pyrazole-4-boronic acid pinacol ester (1.310 g).
Step 1: l-Methyl-3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolcm-2-yl)-lH-pyrazole; N O Potassium tert-butoxide in THF (1.0 M, 0.309 mL, 0.309 mmol) was added to a solution of 3- (4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-lH-pyrazole (30.0 mg, 0.155 mmol, Alfa Aesar, Cat. No. H27619) in DMF (0.5 mL) and stirred at r.t. for 10 min, and then methyl iodide (28.9 uL, 0.464 mmol) was added and the stirred at r.t. for 1 h. LCMS showed the reaction was complete. The mixture was diluted with ethyl acetate, washed with water and brine, dried over Na2SCu, filtered, and concentrated to give the crude product which was directly used in the next reaction without further purification. LCMS (M+H-82)+: m/z = 127.2.
With caesium carbonate; In acetonitrile; at 90℃; for 2h;
Step 1: l-isopropyl-4-(4, 4,5, 5-tetramethyl-l, 3,2-dioxaborolan-2-yl)-lH-pyrazole; A mixture of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.05 g, 0.2 mmol), 2- Bromopropane (36 uL, 0.39 mmol, Aldrich, Cat. No. 239909), and cesium carbonate (250 mg, 0.77 mmol) in acetonitrile (1 mL) was stirred at 90 0C for 2 h. After cooling it was quenched with water. The product was extracted with ethyl acetate. The extract was washed with water twice, brine once; dried over Na2SO4. After filtration the filtrate was concentrated to yield 53 mg of the product which was directly used in the next step reaction without further purification.
With caesium carbonate; In acetonitrile; at 80℃; for 4h;
To a solution of 4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.194 g, 1.000 mmol) in acetonitrile (10 mL) was added cesium carbonate (0.652 g, 2.000 mmol), followed by 2-bromopropane (0.189 g, 1.538 mmol) at room temperature and stirred at 80C for 4 h. After the completion of reaction, cooled to room temperature filtered through pad of celite and evaporated off the filtrate to give the titled compound (0.200 g) as crude. LCMS: m/z 237.2 [M+H] +; H NMR (300 MHz, Chloroform-d) delta 7.89 (s, 2H), 4.61 - 4.44 (m, 1H), 1.50 (d, / = 6.6 Hz, 6H), 1.32 (d, / = 3.1 Hz, 12H).
Step 1 : S- [4- (4, 4, 5, 5-tetramethyl- 113, 2~dioxaborolan-2-y{)- 1 H-pyrazol- 1 -yljpropanenitrile; N A mixture of 4-(4,4,5,5-tetramethyl-L3,2-dioxaborolan-2-yl)-l H-pyrazole (50 mg, 0.2 mmol), 3- bromopropionitrile (23 uL, 0.28 mmol, Aldrich, Cat. No. 109231) and cesium carbonate (250 mg, 0.77 mmol) in acetonitrile ( 1 mL) was stirred at 90 0C overnight. After cooling, it was diluted with ethyl acetate, washed with water twice and brine once, dried over Na2SO4. After filtration the filtrate was concentrated to yield 55 mg of the desired product.
A mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (30.0 mg, 0.154 mmol), and <strong>[134419-59-3]tetrahydro-2H-pyran-4-yl methanesulfonate</strong> (0.150 g, 0.460 mmol) in acetonitrile (1.0 mL) was stirred at 90 C. for 2 hours. The reaction was quenched with water, extracted with ethyl acetate. The combined organic layer was dried over MgSO4, then filtered and concentrated under reduced pressure to afford the desired product (35 mg, 80%), which was used directly in the next step without further purification. LCMS calculated for C14H24BN2O3 (M+H)+: m/z=279.2. Found: 279.2.
40%
To a solution of 4-(4,4,5 ,5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1H-pyrazole (1.0 g, 5.2 mmol) in DMF (10 mL) was added NaH (0.3 g, 7.5 mmol) at 0 C and the mixture was stirred at rt for 30 mi Tetrahydro-2H-pyran-4-yl methanesulfonate (1.1 g, 6.1 mmol) was added and the mixture was stirred at 110 C overnight. The reaction mixture was cooled to rt and filtered. The filtrate was concentrated and the residue was purified by silica gel chromatography (30% EA:PE) to give 550 mg of the title compound (40%). [M+H] Calc?d for C,4H23BN203, 279; Found, 279.
38%
With caesium carbonate; In N,N-dimethyl-formamide; at 100℃;
A mixture of <strong>[134419-59-3]tetrahydro-2H-pyran-4-yl methanesulfonate</strong> (2.8 g, 16 mmol) , 4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -1H-pyrazole (2.01 g, 10.4 mmol) and cesium carbonate (5.4 g, 17 mmol) in DMF (30 mL) was stirred at 100 overnight. The reaction mixture was concentrated in vacuo to remove DMF. The residue was diluted with water (20 mL) . The resulting mixture was extracted with DCM (30 mL × 3) . The combined organic layers were dried over anhydrous Na2SO4and concentrated in vacuo. The residue was purified by pre-HPLC to give a light yellow oily product (1.1 g, 38) .[1589]MS (ESI, pos. ion) m/z: 279.3 [M+1]+.
16.4%
With caesium carbonate; In N,N-dimethyl-formamide; at 100℃; for 1h;Inert atmosphere; glass bomb;
Step B: Preparation of 1 -(tetrahydro-2H-pyran-4-yl)-4-(4,4,5,5-tetramethyl-1 ,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole: In 8 mL of Nu,Nu-dimethylformamide were combined 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.75 g, 3.87 mmol), tetrahydro- 2H-pyran-4-yl methanesulfonate (1.04 g, 5.80 mmol), and CS2CO3 (2.01 g, 6.18 mmol) in a glass bomb and then sealed under nitrogen and heated to 100 C. After one hour the reaction was cooled and water was added to dissolve all solids. The solution was diluted with water and then extracted with EtOAc (250 mL). The organic layer was then washed with water and brine. The combined aqueous layer was then extracted with EtOAc (200 mL). The organic layers were combined, dried over MgS04, filtered, and concentrated under reduced pressure. The crude material was dried overnight on high vacuum. The crude was purified by way of silica gel chromatography eluting with a gradient of 15-25% EtOAc in DCM to afford 1- (tetrahydro-2H-pyran-4-yl)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.176 g, 0.633 mmol, 16.4% yield). MS (apci) m/z = 279.2 (M+H).
With sodium hydride; In N,N-dimethyl-formamide; at 110℃; for 2h;
Step2: l-(tetrahydro-2H-pyran-4-yl)-4-(4, 4, 5, 5-tetramethyl- 1, 3, 2-dioxaborolan-2-yl)-lH-pyrazole; A mixture of 4-(4,4,5,5-tetramethyl-l,3>2-dioxaborolan-2-yl)-lH-pyrazole (0.1 g, 0.5 mmol; Aldrich, Cat. No. 525057), tetrahydro-2H-pyran-4-y. methanesulfonate (0.11 g, 0.62 mmol) and sodium hydride (31 mg, 0.77 mmol) in N,N-dimethylformamide (1 mL) was stirred at 1 10 0C for 2 h. After cooling, it was diluted with ethyl acetate, washed with water and brine, dried over Na2SO4. After filtration, the filtrate was concentrated to yield 0.15 g of the crude product which was directly used in the next step reaction without further purification.
The following Preparations were prepared according to Method H (Preparation 122) using 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole and the appropriate alkyl electrophile.
A mixture of bromocyclopentane (1.0 g, 6.71 mmol), 4-(4,4,5 ,5-tetramethyl- 1,3,2- dioxaborolan-2-yl)-1H-pyrazole (1.32 g, 6.84 mmol) and Cs2CO3 (6.54 g, 20.1 mmol) in CH3CN (50 mL) was refluxed overnight. The mixture was cooled, concentrated and purified by flashcolumn chromatography (PE:EA = 5:1) to give the title compound as a white solid (1.5 g, yield85.7 %). MS (ES+) C14H23BN202 requires: 262, found 263 [M+H].
With caesium carbonate; In acetonitrile; at 90℃; for 2h;
Step 1: l-cyclopentyl-4-(4,4,5,5'tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole; Lambda.O, >T~NB- // ? f O A mixture of 4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-l H-pyrazole (50 mg, 0.2 mmol), cyclopentyl bromide (46 mg, 0.31 mmol, Aldrich, Cat. No. Cl 15207), and cesium carbonate (250 mg, 0.77 mmol) in acetonitrile (1 mL) was stirred at 90 0C for 2 hours. After cooling it was quenched with water, extracted with ethyl acetate. The extract was washed with water, brine; dried over Na2SO4. After filtration the filtrate was concentrated to yield 40 mg of the product which was directly used in the next step reaction without further purification. LCMS (M+H)+: m/z = 263.3
Intermediate 7: 1-Cvclopentyl-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)- 1 H-pyrazole; 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (2g, 10.31 mmol) (Aldrich) and cesium carbonate (5.04g, 15.46mmol) were suspended in acetonitrile (30ml) and stirred at room temperature for 10min. Bromocyclopentane (1.658ml, 15.46mmol) was added and the reaction stirred at 60C for 4h. LCMS showed the reaction had not gone to completion. The reaction was stirred for 2h. The reaction was allowed to cool, diluted with ether and filtered. The filtrate was concentrated, re-dissolved in ether and filtered again; the filtrate was again concentrated and dried to give the title compound (2.2g).LCMS (Method B): Rt = 1.12min, MH+ = 262.89
2.2 g
Intermediate 7: 1-Cyclopentyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (2 g, 10.31 mmol) (Aldrich) and cesium carbonate (5.04 g, 15.46 mmol) were suspended in acetonitrile (30 ml) and stirred at room temperature for 10 min. Bromocyclopentane (1.658 ml, 15.46 mmol) was added and the reaction stirred at 60 C. for 4 h. LCMS showed the reaction had not gone to completion. The reaction was stirred for 2 h. The reaction was allowed to cool, diluted with ether and filtered. The filtrate was concentrated, re-dissolved in ether and filtered again; the filtrate was again concentrated and dried to give the title compound (2.2 g). LCMS (Method B): Rt=1.12 min, MH+=262.89
1-(Ethoxyethyl)-4-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (19).; A 22 L 4-neck flask equipped with a mechanical stirrer, thermowell, addition funnel, condenser and N2 inlet was charged with 4-(4,4,5,5-tetra-methyl[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (17, 1.42 kg, 7.32 mol), toluene (9.5 L) and ethyl vinyl ether (18, 790.5 g, 1050 mL, 10.98 mol, 1.50 equiv). A 4 M HCl in dioxane (50 mL) was added via an addition funnel over 10 minutes and the resulting reaction mixture was heated at 35-40 C. for 7 hr to give a clear homogeneous solution. When the reaction was shown to be complete by GC, solid NaHCO3 (130 g) was added and the mixture was stirred for 1 hr before being filtered. The filtrate was concentrated under reduced pressure. Heptane (200 mL) was added to the residue to affect crystallization. The solid was collected by filtration and dried in a vacuum oven to afford 1-(ethoxyethyl)-4-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (19, 1.896 Kg, 1.948 Kg theoretical, 97.3%) as a white to off-white solid. For 19: 1H NMR (DMSO-d6, 400 MHz) delta ppm 8.09 (s, 1H), 8.58 (s,1H), 7.62 (s,1H), 5.55 (q, 1H, J=6.1 Hz), 3.37 (dq, 1H, J=7.1, 9.6 Hz), 3.12 (dq, 1H, J=7.0, 9.7 Hz), 1.56 (d, 3H, J=6.0 Hz), 1.24 (s, 12H), 1.00 (t, 3H, J=7.0 Hz); C13H23BN2O3 (MW, 266.14), LCMS (EI) m/e 267 (M++H).
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,4-dioxane; water; at 90℃; for 2h;Inert atmosphere;
4-(1H-Pyrazol-4-yl)-7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (5).; Method A.; To a flask equipped with a reflux condenser, a nitrogen inlet, mechanical stirrer, and a thermowell was added 4-chloro-7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (3a, 817 g, 2.88 mol) and dioxane (8 L). To this solution was added 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (4, 728 g, 3.75 mol, 1.30 equiv) followed by a solution of potassium carbonate (K2CO3, 1196 g, 8.67 mol, 3.0 equiv) in water (4 L). The solution was degassed by passing a stream of nitrogen through the solution for 15 minutes before being treated with tetrakis(triphenylphosphine)palladium(0) (167 g, 0.145 mol, 0.05 equiv) and the resulting reaction mixture was heated at reflux (about 90 C.) for 2 hours. When the reaction was deemed complete by TLC (1:1 heptane/ethyl acetate) and LCMS, the reaction mixture was cooled to room temperature, diluted with ethyl acetate (24 L) and water (4 L). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (4 L). The combined organic layers were washed with water (2×2 L), brine (2 L), dried over sodium sulfate (Na2SO4), and concentrated under reduced pressure. The residue was suspended in toluene (4 L) and the solvent was removed under reduced pressure. The residue was finally triturated with methyl tert-butyl ether (MTBE, 3 L) and the solids were collected by filtration and washed with MTBE (1 L) to afford 4-(1H-pyrazol-4-yl)-7-(2-trimethylsilanyl-ethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (5, 581.4 g, 908.5 g theoretical, 64% yield) as white crystalline solids. For 5: 1H NMR (DMSO-d6, 400 MHz) delta ppm 13.41 (bs, 1H), 8.74 (s, 1H), 8.67 (bs, 1H), 8.35 (bs, 1H), 7.72 (d, 1H, J=3.7 Hz), 7.10 (d, 1H, J=3.7 Hz), 5.61 (s, 2H), 3.51 (t, 2H, J=8.2 Hz), 0.81 (t, 2H, J=8.2 Hz), 0.13 (s, 9H); C15H21N5OSi (MW, 315.45), LCMS (EI) m/e 316 (M++H).
27%
At room temperature, <strong>[941685-26-3]4-chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine</strong> (12f) (800 mg, 2.82 mmol) and 4-pyrazoleboronic acid pinacol ester (842 mg, 4.34 mmol) were dissolved in dioxane (10 mL), water (2 mL) and potassium carbonate (857 mg, 6.2 mmol) were then added, nitrogen atmosphere protection was applied, and the reaction was stirred at room temperature for 10 min. Under protection of nitrogen, Pd(dppf)Cl2 (227 mg, 0.31 mmol) was added. The reaction was placed in an oil bath at 95C, and stirred overnight. TLC indicated starting materials substantially disappeared. The reaction was quenched with water, extracted with EA, and the organic phase was dried over anhydrous sodium sulfate, and purified by preparative flash chromatography (PE:EA=2:3), to afford 4-(1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (12g) (240 mg, brown solid), yield: 27%. MS (ESI, m/z): 316 [M+H]+.
166.4 g
Nitrogen gas was purged through a mixture of 4-chloro-7- { [2- (0224) (trimemylsilyl)ethoxy]memyl}-7H-pyItauolo[2,3-
With 18-crown-6 ether; In acetonitrile; for 18h;Reflux;
Step 1: l-(difluoromethyl)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole [00218] A 100 mL round bottom flask was charged with 4-pyrazoleboronic acid pinacol ester (1.0 g, 5.15 mmol), 18-crown-6 (0.27 g, 1.03 mmol) and anhydrous acetonitrile (25 mL). The reagents were stirred until a colorless solution formed then sodium chlorodifluoroacetate (0.94 g, 6.18 mmol) was added and the reaction mixture heated to reflux for 18 h. After this time the reaction mixture was cooled to r.t. and the precipitated solid removed by filtration through celite, washing with EtOAc (3 x 20 mL). Combined organics were filtered through a hydrophobic frit and condensed to give a pale yellow oil. The crude product was purified by flash silica column chromatography (gradient elution omicron-hexane to 33% EtOAc in /so-hexane) to give the title compound as a colorless solid (1.06 g, 84%). MS (ES+) consistent with target (M+H)+.
84%
With 18-crown-6 ether; In acetonitrile; for 18h;Reflux;
Step 1 : 1 -(difluoromethyl)-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- pyrazole [00256] A 100 mL round bottom flask was charged with 4-pyrazoleboronic acid pinacol ester (1 .0 g, 5.15 mmol), 18-crown-6 (0.27 g, 1 .03 mmol) and anhydrous acetonitrile (25 mL). The reagents were stirred until a colorless solution formed then sodium chlorodifluoroacetate (0.94 g, 6.18 mmol) was added and the reaction mixture heated to reflux for 18 h. After this time the reaction mixture was cooled to r.t. and the precipitated solid removed by filtration through Celite, washing with EtOAc (3 x 20 mL). Combined organics were filtered through a hydrophobic frit and condensed to give a pale yellow oil. The crude product was purified by flash silica column chromatography (gradient elution, 0-33% EtOAc in /so-hexane) to give the title compound as a colorless solid (1 .06 g, 84%). MS (ES+) consistent with target (M+H)+.
80%
With 15-crown-5; for 24h;Reflux; Inert atmosphere;
Step 1: To acetonitrile (100 ml) was added compound 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole 101-1 (4.0 g, 0.21 mol), sodium chlorodifluoroacetate (3.77 g 0.25 mol, 15-crown-5 (103.8 g, 0.65 mol), and the reaction mixture was heated to reflux under nitrogen and stirred for 24 h, cooled to room temperature, the reaction mixture was poured into water (50 ml) to quench the reaction, and extracted with EA (100 ml×3), washed with saturated brine (20 ml), dried over Na2SO4, and filtered, and the filtrate was evaporated to give compound 101-2 (4.1 g, yield 80%) as a white solid. MS m/z (ESI): 245.1 [M+H]+.
47%
With 18-crown-6 ether; In acetonitrile; for 20h;Reflux;
Reference Example 1711-(Difluoromethyl)-1H-pyrazole-4-boronic acid pinacol ester A suspension of 1H-pyrazole-4-boronic acid pinacol ester (5.16 g, 26.6 mmol), CF2ClCO2Na (4.86 g, 31.9 mmol), and 18-crown-6 (1.41 g, 5.32 mmol) in CH3CN (100 mL) was refluxed for 20 h. After cooling to room temperature, the reaction mixture was poured into water and extracted with AcOEt. The extract was washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by basic silica gel column chromatography eluting with AcOEt to give the title compound (3.03 g, 47% yield) as a yellow oil: 1H NMR (300 MHz, CDCl3): delta ppm 1.33 (12H, s), 7.22 (1H, t, J=60.7 Hz), 7.89 (1H, s), 8.13 (1H, s).
500 mg
With 18-crown-6 ether; In acetonitrile; for 20h;Reflux;
A suspension of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (516 mg, 2.66 mmol), CF2ClCO2Na (486 mg, 3.19 mmol), and 18-crown-6 (141 mg, 0.532 mmol) in CH3CN (150 mL) was heated to reflux for 20 hours. After cooled to room temperature, thereaction mixture was poured into water and extracted with EtOAc. The extracts were washed with brine, dried over MgSO4, and concentrated under reduced pressure to afford a crude 1-(difluoromethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (500 mg), which was used for the next step without further purification. MS (ESI) m/z: 245 [M+H]+.
1.54 g
With 18-crown-6 ether; In acetonitrile; at 90℃; for 16h;
E) 1-(difluoromethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole To a mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1.50 g), sodium chlorodifluoroacetate (1.41 g) and acetonitrile (35 mL) was added 18-crown-6 (0.409 g). The reaction mixture was stirred at 90 C. for 16 hr, poured into water, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure to give the title compound (1.54 g). MS (API+): [M+H]+245.1.
With 18-crown-6 ether; In acetonitrile; for 16h;Reflux;
(A) 1-(Difluoromethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole Sodium 2-chloro-2,2-difluoroacetate (471 mg) was added to a mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (500 mg), 18-crown-6 (136 mg) and acetonitrile (10 mL), and the reaction mixture was heated at reflux for 16 hours. Water was added to the reaction mixture, followed by extraction with ethyl acetate. The organic layer was washed with brine and then dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure to obtain the title compound as a crude product (466 mg). This compound was used in the next step without being further purified. 1H NMR (300 MHz, CDCl3) delta 1.33 (12H, s), 7.01-7.43 (1H, m), 7.88 (1H, s), 8.12 (1H, s).
With 18-crown-6 ether; In acetonitrile; for 16h;Reflux;
A solution of sodium 2-chloro-2,2-difluoroacetate (471 mg)Add to4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1H-pyrazole (500 mg)18-crown-6 (18-crown-6) (136 mg) and acetonitrile (10 mL)The reaction mixture was heated under reflux for 16 hours.Water was added to the reaction mixture, followed by extraction with ethyl acetate.The organic layer was washed with brine and dried over anhydrous magnesium sulfate,And the solvent was removed by distillation under reduced pressure to obtain the crude product of the title compound (466 mg).This compound was used in the next step without further purification.
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,4-dioxane; water; at 95℃; for 2h;sealed tube;
General procedures:; Scheme 2; An acid (1 equiv), HATU (1 equiv) and DIEA (3 equiv) were added to a solution of 4-bromo-aniline (1 equiv) in DMF. The mixture was stirred at room temperature until the starting material disappeared (detection by LC-MS). The solution was diluted with EtOAc and washed with saturated aqueous NaHCO3. The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo to give the crude bromide 2-1. The bromide 2-1 (1 equiv) and the boronic acid ester (1.5 equiv) were dissolved in degassed dioxane/H2O (5:1 by volume) in a sealed tube. Pd(PPh3)4 (0.03 equiv) and 2M solution Of K2CO3 (3 equiv) were added sequentially. The mixture was heated at 95 0C for 2h. After cooling to room temperature, the mixture was diluted with water and extracted with ethyl acetate. The organic layers were combined, dried over sodium sulfate and concentrated in vacuo. The residue was then purified by preparative HPLC to give the product 2-2 as a solid (65percent-80percent yield in two steps).; Example 27. N-(4-(lH-pyrazol-4-yl)phenv0-3-phenylpropanamide; Procedures in Scheme 2 were utilized to synthesize this compound. LC/MS: Ci8HnN3O (M+l) 292. Single peak at both 215 nm and 254 nm in analytical HPLC traces.
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃; for 6.0h;
A mixture of 1- (2-chloroethyl) pyrrolidine hydrochloride (1.48 g, 8.70 mmol) , 4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -1H-pyrazole (1.7 g, 8.8 mmol) and potassium carbonate (3.7 g, 27.00 mmol) in DMF (15 mL) was stirred at 80 for 6 h. The reaction mixture was diluted with water (50 mL) . The resulting mixture was extracted with DCM (50 mL × 3) . The combined organic layers were washed with saturated aqueous NaCl (15 mL) , dried over anhydrous Na2SO4and concentrated in vacuo. The residue was purified by silica gel column chromatography eluted with DCM/EtOAc (v/v) 20/1 to give a light yellow liquid product (1.27 g, 50.10) .[1772]MS (ESI, pos. ion) m/z: 292.10 [M+1]+
With caesium carbonate; In acetonitrile; at 20℃; for 18.0h;
1.4 15.0 g (86.7 mmol) of N-(2-chloroethyl)pyrrolidine hydrochloride and 42.4 g (130 mmol) of caesium carbonate are added to a solution of 8.58 g (43.3 mmol) of pinacolyl pyrazole-4-boronate in 86 ml of acetonitrile, and the suspension formed is stirred at room temperature for 18 hours. The reaction mixture is filtered, and the residue is washed with acetonitrile. The filtrate is evaporated, and the residue is partitioned between saturated sodium chloride solution and ethyl acetate. The organic phase is dried over sodium sulfate and evaporated: 10.9 g of 1-(2-pyrrolidin-1-ylethyl)-4-(4,4,5,5-tetra-methyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole as yellow oil; ESI 292.
With caesium carbonate; In acetonitrile; at 20℃; for 18.0h;
A solution of 10.0 g (50.5 mmol) of pinacolyl pyrazole-4-boronate is dissolved in 100 ml of acetonitrile, and 17.5 g (101 mmol) of N-(2-chloroethyl)pyrrolidine hydrochloride and 49.4 g (152 mmol) of caesium carbonate are added. The suspension formed is stirred for 18 hours at room temperature. The reaction mixture is filtered off with suction and washed with acetonitrile The filtrate is evaporated and partitioned between ethyl acetate and saturated sodium chloride solution. The organic phase is dried over sodium sulfate and evaporated: 1-(2-pyrrolidin-1-ylethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole as pale-orange oil, which gradually crystallises;1H-NMR (d6-DMSO): delta [ppm]=1.25 (s, 12H), 1.65 (m, 4H), 2.44 (m, 4H), 2.79 (t, J=6.8 Hz, 2H), 4.21 (t, J=6.8 Hz, 2H), 7.56 (s, 1H), 7.93 (s, 1H).
With caesium carbonate; In acetonitrile; at 20℃; for 18.0h;
1.5 A solution of 10.0 g (50.5 mmol) of pinacolyl pyrazole-4-boronate is dissolved in 100 ml of acetonitrile, and 17.5 g (101 mmol) of N-(2-chloroethyl)pyrrolidine hydrochloride and 49.4 g (152 mmol) of caesium carbonate are added. The resultant suspension is stirred at room temperature for 18 hours. The reaction mixture is filtered off with suction and washed with acetonitrile. The filtrate is evaporated and partitioned between ethyl acetate and saturated sodium chloride solution. The organic phase is dried over sodium sulfate and evaporated: 1-(2-pyrrolidin-1-ylethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole as pale-orange oil;
With caesium carbonate; In acetonitrile; at 20℃; for 18.0h;
2.1 17.5 g (101 mmol) of N-(2-chloroethyl)pyrrolidine hydrochloride and 49.4 g (152 mmol) of caesium carbonate are added to a solution of 10.0 g (50.5 mmol) of pinacolyl pyrazole-4-boronate in 100 ml of acetonitrile. The resultant suspension is stirred at room temperature for 18 hours. The reaction mixture is filtered with suction and washed with acetonitrile. The filtrate is evaporated and partitioned between ethyl acetate and saturated sodium chloride solution. The organic phase is dried over sodium sulfate and evaporated: 1-(2-pyrrolidin-1-ylethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole as pale- orange oil, which gradually crystallises;1H-NMR (d6-DMSO): delta [ppm]=1.25 (s, 12H), 1.65 (m, 4H), 2.44 (m, 4H), 2.79 (t, J=6.8 Hz, 2H), 4.21 (t, J=6.8 Hz, 2H), 7.56 (s, 1H), 7.93 (s, 1H).
With caesium carbonate; In acetonitrile; at 20℃; for 18.0h;
EXAMPLE 10: Synthesis of 2-(5-Chloro-2-fluoro-phenyl)-4-{5-[1-(2-pyrrolidin-1-yl-ethyl)-1 H- pyrazol-4-yl]-pyridin-3-yl}-[1 ,8]naphthyridine (no. 21)DMETo a solution of 19.4 g (100 mmol) pyrazol-4-boronic acid pinacol-ester in 150 ml acetonitrile were added 32.5 g (191 mmol) N-(2-Chloroethyl)-pyrrolidine hydrochloride and 87.7 g (300 mmol) cesium carbonate. The resulting slurry was stirred for 18 hours at room temperature. The reaction mixture was filtered with suction and the residue was washed well with acetonitrile. The filtrate was evaporated and dissolved in ethyl acetate. This solution was extracted four times with water and finally washed with brine. The organic phase was dried over sodium sulfate and evaporated in vacuo yielding 1-(2-pyrrolidin-1-yl- ethyl)-4-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-yl)-1 H-pyrazol as light-brown oil.1H-NMR (d6-DMSO): delta = 1.25 (s, 12H), 1.65 (m, 4H), 2.44 (m, 4H), 2.79 (t, J = 6.8 Hz, 2H), 4.21 (t, J = 6.8 Hz, 2H), 7.56 (s, 1H), 7.93 (s, 1 H) ppm
With caesium carbonate; In acetonitrile; at 20℃; for 18.0h;
3.4 A solution of 10.0 g (50.5 mmol) of pinacolyl pyrazole-4-boronate is dissolved in 100 ml of acetonitrile, and 17.5 g (101 mmol) of N-(2-chloroethyl)pyrrolidine hydrochloride and 49.4 g (152 mmol) of caesium carbonate are added. The suspension formed is stirred at room temperature for 18 hours. The reaction mixture is filtered off with suction and washed with acetonitrile. The filtrate is evaporated and partitioned between ethyl acetate and saturated sodium chloride solution. The organic phase is dried over sodium sulfate and evaporated: 1-(2-pyrrolidin-1-ylethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole as pale-orange oil, which gradually crystallises;1H-NMR (d6-DMSO): delta [ppm] 1.25 (s, 12H), 1.65 (m, 4H), 2.44 (m, 4H), 2.79 (t, J=6.8 Hz, 2H), 4.21 (t, J=6.8 Hz, 2H), 7.56 (s, 1H), 7.93 (s, 1H).
With sodium hydride; In N,N-dimethyl-formamide; at 20 - 65℃; for 3.5h;
Example 20A 1-(2-(pyrrolidin-1-yl)ethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole To a suspension of sodium hydride (0.272 g, 11.34 mmol) in N,N-dimethylformamide (13 mL) was added 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1.0 g, 5.15 mmol) and 2-chloroethylpyrrolidine HCl salt (0.964 g, 5.67 mmol) as solutions in N,N-dimethylformamide (2 mL each). The reaction was stirred at room temperature for 0.5 hours and then heated to 65 C. for 3 hours. The reaction was cooled to room temperature and diluted with ethyl acetate (50 mL). Water (50 mL) was added, and the layers were separated. The aqueous layer was extracted with additional ethyl acetate (2*30 mL). The combined organics were washed with water, dried with anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give the title compound, which was used without further purification. MS ESI(+) m/z 292.2 [M+H]+;
With caesium carbonate; In acetonitrile;
EXAMPLE 4 The preparation of 6-phenoxy-3-(3-{5-[1-(2-pyrrolidin-1-ylethyl)-1H-pyrazol-4-yl]pyrimidin-2-yl}benzyl)-1,2,4-triazolo[4,3-b]pyridazine ("A16?) is carried out analogously to the following scheme
With caesium carbonate; In acetonitrile; at 20℃; for 18.0h;
To a solution of 19.4 g (100 mmol) pyrazol-4-boronic acid pinacol-ester in 150 ml acetonitrile were added 32.5 g (191 mmol) N-(2-Chloroethyl)-pyrrolidine hydrochloride and 87.7 g (300 mmol) cesium carbonate. The resulting slurry was stirred for 18 hours at room temperature. The reaction mixture was filtered with suction and the residue was washed well with acetonitrile. The filtrate was evaporated and dissolved in ethyl acetate. This solution was extracted four times with water and finally washed with brine. The organic phase was dried over sodium sulfate and evaporated in vacuo yielding 1-(2-pyrrolidin-1-yl-ethyl)-4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazol as light-brown oil. [0308] 1H-NMR (d6-DMSO): delta=1.25 (s, 12H), 1.65 (m, 4H), 2.44 (m, 4H), 2.79 (t, J=6.8 Hz, 2H), 4.21 (t, J=6.8 Hz, 2H), 7.56 (s, 1H), 7.93 (s, 1H) ppm.
With caesium carbonate; In acetonitrile; at 78℃;
4-pyrazoleboronic acid pinacol ester 9 (4.00 g, 20.62 mmol) was dissolved in 170 mL of acetonitrile.Stir well, add cesium carbonate (23.50g, 70.09mmol), stir for a while,1-(2-Chloroethyl)pyrrolidine hydrochloride 10a (5.30 g, 30.18 mmol) was added and stirred at 78 C overnight.The heating was stopped, suction filtered, and ethyl acetate (50 mL) was washed three times and evaporated to dryness to afford white solid 11a (6.50 g). Without purification, go directly to the next step.
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 0℃; for 48h;Inert atmosphere;
4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-IH-pyrazole (3.964 g, 20.43 mmol), DIAD (4.42 mL,22.47 mmol), triphenylphosphine (5.89 g, 22.47 mmol) and tert-butyl (2-hydroxyethyl)(methyl)carbamate (for a preparation see Intermediate 134, 3.58 g, 20.43 mmol) were dissolved in THF at 0 C under nitrogen for 48 h. The reaction mixture was concentrated and the orange oil triturated with diethyl ether. The precipitated solid was removed by filtration and washedwith more diethyl ether. The filtrate was concentrated to give 12.45 g of crude thick orange oil. This was purified by chromatography on silica (220 g cartridge, eluting with 0-100% ethyl acetate/cyclohexane over 13 CVs, collecting all fractions). Product fractions were combined to give the product (4.29 g, 12.21 mmol, 59.8%) as a yellow oil.LCMS (2 mm Formic): Rt = 1.07 mi [MH] = 352.
53.6%
With diisopropyl (E)-azodicarboxylate;triphenylphosphine; In tetrahydrofuran; at 0℃; for 4h;Inert atmosphere;
Intermediate 561 ,1 -Dimethylethyl methyl{2-[4-(4A5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazol-1 - yllethvDcarbamate o4-(4,4,5,5-Tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (5 g, 25.8 mmol), bis(1 - methylethyl) (£)-1 ,2-diazenedicarboxylate (5.73 g, 28.3 mmol), triphenylphosphine (7.43 g, 28.3 mmol) and 1 ,1 -dimethylethyl (2-hydroxyethyl)methylcarbamate (4.52 g, 25.8 mmol) were dissolved in THF at 0C under nitrogen and the resulting mixture was stirred at this temperature for 4 h, then evaporated in vacuo and the residue triturated with MTBE. The precipitated solid was removed by filtration and the filtrate evaporated in vacuo, then purified by chromatography on silica gel (gradient: 0-100% EtOAc in Hexanes) to give 1 ,1 -dimethylethyl methyl{2-[4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)-1 H-pyrazol-1 -yl]ethyl}carbamate (4.85g, 13.81 mmol, 53.6 % yield) as a colourless oil.Intermediate 57
With potassium carbonate; In acetonitrile; at 35℃; for 3h;Inert atmosphere;
Intermediate 941 -[(Methyloxy)methyl]-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- pyrazole 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (10.00 g, 51.5 mmol) was dissolved in acetonitrile (40 mL) and stirred at 35C for 5 min under nitrogen atmosphere then cooled to room temperature, lodomethyl methyl ether (21.83 mL, 258 mmol) and potassium carbonate (35.6 g, 258 mmol) were added and the resulting mixture was stirred at 35C for 3 h then cooled to room temeprature. The insoluble material was filtered off and the filtrate was concentrated in vacuo. The residue was dissolved in EtOAc and the organic phase was washed with water, dried over MgS04 and concentrated in vacuo. Purification of the residue by SP4 using a 100 G silca cartridge (gradient: 0 to 50% AcOEt in hexanes) gave 1 -[(methyloxy)methyl]-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)- 1 H-pyrazole (6.45 g, 50% pure by LCMS, 26%) as a yellow liquid which was used in the next step without further purification.LCMS (method G): Retention time 0.85 min, [M+H]+ = 238.8
With potassium carbonate; In acetonitrile; at 35℃; for 4.5h;
4-(4,4,5,5-Tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (400mg, 2.061 mmol) (Aldrich) was dissolved in acetonitrile (25ml) and stirred for 5min at 35C. lodomethyl methyl ether (0.873ml, 10.31 mmol) and potassium carbonate (1424mg, 10.31 mmol) were added and the mixture was allowed to stir at 35C for 3h.The LCMS showed incomplete reaction. The mixture was left to stir at 35C for one additional hour. The LCMS showed no progression so 2eq (349muIota) of the alkylating agent was added and the mixture was allowed to stir at 35C for 30min. Ammonium chloride was then added. The mixture was partitioned between ethyl acetate and water. The aqueous layer was re-extracted with ethyl acetate and the combined organic phases were washed with water, dried using a hydrophobic frit and concentrated to an oil. The oil was purified on 50g silica using an SP4 and eluted with a 0-100% ethyl acetate/cyclohexane gradient. The product was found in the waste, which was concentrated to give the title compound as an oil (260mg).LCMS (Method B): Rt = 0.84min, MH+ = 238.85
4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (10.00 g, 51.5 mmol) was dissolved in acetonitrile (40 mL) and stirred at 35 C. for 5 min under nitrogen atmosphere then cooled to room temperature. Iodomethyl methyl ether (21.83 mL, 258 mmol) and potassium carbonate (35.6 g, 258 mmol) were added and the resulting mixture was stirred at 35 C. for 3 h then cooled to room temperature. The insoluble material was filtered off and the filtrate was concentrated in vacuo. The residue was dissolved in EtOAc and the organic phase was washed with water, dried over MgSO4 and concentrated in vacuo. Purification of the residue by SP4 using a 100 G silica cartridge (gradient: 0 to 50% AcOEt in hexanes) gave 1-[(methyloxy)methyl]-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (6.45 g, 50% pure by LCMS, 26%) as a yellow liquid which was used in the next step without further purification.LCMS (method G): Retention time 0.85 min, [M+H]+=238.8
260 mg
With potassium carbonate; In acetonitrile; at 35℃; for 4.5h;
Intermediate 12: 1-[(Methyloxy)methyl]-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole[0399]4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (400 mg, 2.061 mmol) (Aldrich) was dissolved in acetonitrile (25 ml) and stirred for 5 min at 35 C. Iodomethyl methyl ether (0.873 ml, 10.31 mmol) and potassium carbonate (1424 mg, 10.31 mmol) were added and the mixture was allowed to stir at 35 C. for 3 h.[0401]The LCMS showed incomplete reaction. The mixture was left to stir at 35 C. for one additional hour. The LCMS showed no progression so 2 eq (349 mul) of the alkylating agent was added and the mixture was allowed to stir at 35 C. for 30 min. Ammonium chloride was then added. The mixture was partitioned between ethyl acetate and water. The aqueous layer was re-extracted with ethyl acetate and the combined organic phases were washed with water, dried using a hydrophobic frit and concentrated to an oil. The oil was purified on 50 g silica using an SP4 and eluted with a 0-100% ethyl acetate/cyclohexane gradient. The product was found in the waste, which was concentrated to give the title compound as an oil (260 mg).[0402]LCMS (Method B): Rt=0.84 min, MH+=238.85
With caesium carbonate; In acetonitrile; for 5h;Reflux;
Intermediate 115yV,yV-Dimethyl-2-[4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazol-1 - yljethanamine4-(4,4,5,5-Tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (2 g, 10.31 mmol), cesium carbonate (6.72 g, 20.61 mmol) and 2-chloro-/V,/V-dimethylethanamine hydrochloride (2.227 g, 15.46 mmol) were suspended in acetonitrile (30 mL) and the mixture was heated at reflux for 5 h, then cooled to room temperature, diluted with Et20 and filtered. The filtrate was concentrated in vacuo to give N,N-dimethyl-2-[4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 H-pyrazol-1 -yl]ethanamine dimethyl{2-[4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 H-pyrazol-1 -yl]ethyl}amine (2.61 g, 9.84 mmol, 95 % yield) as an amber oil which was used in the next step without further purification.
88%
With caesium carbonate; In acetonitrile; at 90℃;
A mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (60.0 mg, 0.309 mmol), 3-dimethylaminoethyl chloride hydrochloride (49.0 mg, 0.340 mmol) and cesium carbonate (0.302 g, 0.928 mmol) in acetonitrile (1 mL) was stirred at 90 C. overnight. The reaction mixture was cooled to ambient temperature, quenched with water, and extracted with methylene chloride. The combined organic layers were dried over Mg504, then filtered and concentrated under reduced pressure to give the desired product (60 mg, 88%). LCMS calculated for C13H25BN3O2 (M+H)+: m/z=266.2. Found: 266.2.
66%
With caesium carbonate; In N,N-dimethyl-formamide; at 0 - 20℃; for 72.5h;
2-Dimethylaminoethyl chloride hydrochloride (0.97 g, 6.70 mmol) was added to a mixture of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (1 .00 g, 5.15 mmol) and caesiumcarbonate (5.54 g, 17.01 mmol) in dry N,N-dimethylformamide (20 mL) at 0C. After stirring for30 mm the ice-water bath was removed. The reaction mixture was stirred at room temperaturefor three days. The reaction mixture was diluted with ethyl acetate (150 mL) and washed with brine (3x100 mL). The organic layer was dried with sodium sulfate and concentrated in vacuo to afford 1 .00 g (3.41 mmol; 66% of theory) of the title compound.GO-MS (Method L9): R1 = 4.48 mm; (no mass detected)1 H NMR (300 MHz, Chloroform-d, Method M2) 6 7.78 (s, 1 H), 7.74 (s, 1 H), 4.23 (t, J = 6.8 Hz,2H), 2.76 (t, J = 6.8 Hz, 2H), 2.26 (s, 6H), 1.31 (s, 12H).
56%
With caesium carbonate; In acetonitrile; at 90℃; for 72h;
A mixture of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (100 mg, 0.515 mmol), 2-chloro-A/,A/-dimethylethanamine hydrochloride (82 mg, 0.567 mmol), and Cs2C03 (504 mg, 1.546 mmol) in acetonitrile (2 ml_) was heated at 90 C for 3 d. After cooling to RT, the reaction mixture was partitioned between DCM and water. The aqueous layer was extracted with DCM and the combined extracts were washed (water), dried (MgS04), and concentrated in vacuo to give the title compound (76 mg, 56%) as a colourless oil. LCMS (Method B): RT = 0.61 min, m/z = 266 [M+H]+.
With caesium carbonate; In acetonitrile; for 5h;Reflux;
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (2 g, 10.31 mmol), cesium carbonate (6.72 g, 20.61 mmol) and 2-chloro-N,N-dimethylethanamine hydrochloride (2.227 g, 15.46 mmol) were suspended in acetonitrile (30 mL) and the mixture was heated at reflux for 5 h, then cooled to room temperature, diluted with Et2O and filtered. The filtrate was concentrated in vacuo to give N,N-dimethyl-2-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl]ethanamine dimethyl{2-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl]ethyl}amine (2.61 g, 9.84 mmol, 95% yield) as an amber oil which was used in the next step without further purification
With caesium carbonate; In acetonitrile; at 80℃; for 4h;
General procedure: To a solution of 4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.194 g, 1.000 mmol) in acetonitrile (10 mL) was added cesium carbonate (0.652 g, 2.000 mmol), followed by 2-bromopropane (0.189 g, 1.538 mmol) at room temperature and stirred at 80C for 4 h. After the completion of reaction, cooled to room temperature filtered through pad of celite and evaporated off the filtrate to give the titled compound (0.200 g) as crude. 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.200 g, 1.025 mmol) in acetonitrile (10 mL) was treated with cesium carbonate (0.668 g, 2.051 mmol) and 2- chloro-N,N-dimethylethanamine hydrochloride (0.221 g, 1.538 mmol) 80 C for 4h as described in the synthesis of step-1 of example- 139 to give the titled compound (0.200 g, crude). LCMS: m z 266.2 [M+H] +; lU NMR (300 MHz, Chloroform-d) delta 7.89 (d, / = 1.9 Hz, 2H), 4.23 (t, /= 6.9 Hz, 2H), 2.77 (t, / = 6.8 Hz, 2H), 1.40 - 1.20 (m, 18H).
With potassium carbonate; In N,N-dimethyl-formamide; at 60℃;
To a solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (V-1 ) (3 g, 15 mmol) in DMF (6 mL) were added bromoethane (3.24 g, 30 mmol) and K2C03 (4.26 g, 30 mmol). The reaction mixture was stirred at 60°C overnight, then diluted with EtOAc, washed with water and then brine. The organic layer was separated, then dried over Na2S04, and concentrated to afford the title compound (3.40 g). MS (m/z): 223 (M+1 )+.
With potassium carbonate; In N,N-dimethyl-formamide; at 60℃;
1-Ethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (V)To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (V-1) (3 g, 15 mmol) in DMF (6 mL) were added bromoethane (3.24 g, 30 mmol) and K2CO3 (4.26 g, 30 mmol).The reaction mixture was stirred at 60° C. overnight, then diluted with EtOAc, washed with water and then brine.The organic layer was separated, then dried over Na2SO4, and concentrated to afford the title compound (3.40 g). MS (m/z): 223 (M+1)+.
1.1 For the preparation of ?E2?, a suspension of 5.94 g (30 mmol) of pyrazole-4-boronic acid and ?E1? 19.55 g (60 mmol) of caesium carbonate in 60 ml of acetonitrile is stirred at room temperature for 15 minutes. 6.48 g (45 mmol) of 2-dimethylaminoethyl chloride hydrochloride (?E1a?) are added, and the mixture is stirred at room temperature for 2 days, filtered off with suction and rinsed with acetonitrile. The filtrate is evaporated, and a saturated NaCl solution is added, and the mixture is extracted three times with ethyl acetate. The organic phase is washed with water, dried over sodium sulfate and evaporated: 3.5 g of yellowish, gradually crystallising oil is obtained. According to NMR, it was essentially a mixture of product and pinacol in the ratio 1:1. This gives a content of about 64% (yield 21%)
With caesium carbonate; In N,N-dimethyl-formamide; at 160℃; for 0.5h;Microwave irradiation;
4-( 4,4,5,5-Tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1H-pyrazole (1 00 mg, 0.52mmol), 1-chloro-2-methoxyethane (0.056 mL, 0.62 mmol) and cesium carbonate (252mg, 0.73 mmol) in DMF (1 mL) were heated in microwave reactor at 160 °C for 30 min.The reaction mixture was concentrated under reduced pressure and purified by silica gelchromatography (ISCO, hexanes/ethyl acetate 0-100percent over 15 min) to isolate CompoundB162a (130 mg, 100percent yield). HPLC: RT = 1.0 min (LCMS Method M). MS (ES): m/z= 253.0 [M+H( 1H NMR (500MHz, CHLOROFORM-d) 8 8.04 (s, 1H), 7.83 (d,J=18.4 Hz, 1H), 4.37 (t, J=5.2 Hz, 1H), 3.78 (t, J=5.2 Hz, 1H), 3.38-3.31 (m, 2H), 2.98(s, 3H), 2.90 (s, 3H), 1.34 (s, 6H), 1.26 (s, 3H).
72%
With caesium carbonate; In N,N-dimethyl-formamide; at 160℃; for 0.5h;microwave irradiation;
2-Chloroethyl methyl ether (0.050 ml, 0.63 mmol) was added to a stirred solution of 4- (4,4,5, 5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-lH-pyrazole (5.0 g, 25.77 mmol) and cesium carbonate (12.59 g, 38.65 mmol) in DMF (27 ml). The mixture was stirred at 160 °C for 30 min. under microwave irradiation and then the solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica; MeOH in DCM 2/98). The desired fractions were collected and evaporated in vacuo to yield intermediate 67 (4.6 g, 72percent) as a pale yellow oil.
72%
With caesium carbonate; In N,N-dimethyl-formamide; at 160℃; for 0.5h;Microwave irradiation;
Example A671-(2-Methoxy-ethyl)-4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole2-Chloroethyl methyl ether (0.050 ml, 0.63 mmol) was added to a stirred solution of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (5.0 g, 25.77 mmol) and cesium carbonate (12.59 g, 38.65 mmol) in DMF (27 ml).The mixture was stirred at 160° C. for 30 min. under microwave irradiation and then the solvent was evaporated in vacuo.The crude product was purified by flash column chromatography (silica; MeOH in DCM 2/98).The desired fractions were collected and evaporated in vacuo to yield intermediate 67 (4.6 g, 72percent) as a pale yellow oil.
With caesium carbonate; In N,N-dimethyl-formamide; at 70℃;
4-(4,4,5,5-Tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (1 .00 g, 5.15 mmol) and cesium carbonate (5.04 g, 15.47 mmol) were suspended in 10 mL of dimethylformamide. 2-Bromoethanol (0.73 mL, 10.30 mmol) was added and the mixture was stirred at 70 C for 3 hours. Additional amounts of cesium carbonate (5.04 g, 15.47 mmol) and 2-bromoethanol (0.73 mL, 10.30 mmol) were added and the mixture was left at 70 C overnight. The solvent was evaporated under reduced pressure and the residue was partitioned between water and ethyl acetate. The organic layer was washed with water and brine, dried over magnesium sulphate, filtered and the solvent was evaporated to give 770 mg (63% yield) of a yellowish oil that was used in the next step without further purification.LRMS (m/z): 239 (M+1 )+.
31.47%
With caesium carbonate; In N,N-dimethyl-formamide; at 100℃; for 48h;Inert atmosphere; Sealed;
Step A: Preparation of 2-(4-(4 A5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 H- pyrazol- 1 -yDethanol : To a flask charged with 4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2- yl)-lH-pyrazole (1.000 g, 5.154 mmol), Cs2C03 (2.687 g, 8.246 mmol), and 2-bromoethanol (0.5479 mL, 7.730 mmol) was added 10 mL of DMF and the flask was sealed under nitrogen and heated to 100 C for 48 hours. The reaction was diluted with ethyl acetate (100 mL) and stirred for 30 minutes before it was passed through a glass microfiber filter and the cake was washed with ethyl acetate. The organic was concentrated under reduced pressure. The crude was then purified by silica gel chromatography eluting with a gradient of 75-100% ethyl acetate in Hexanes to afford 2-(4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-lH-pyrazol- l-yl)ethanol (0.4290 g, 1.622 mmol, 31.47% yield). MS (apci) m/z = 239.2 (M+H).
24%
With caesium carbonate; In N,N-dimethyl-formamide; at 85℃;
To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (500 mg, 2.58 mmol) in DMF (7 mL) was added 2-bromoethanol (645 mg, 5.16 mmol) and cesium carbonate (2.52 g, 7.74 mmol) and the mixture was heated at 85C for 3 h. More cesium carbonate (2.52 g, 7.74 mmol) and 2-bromoethanol (645 mg, 5.16 mmol) were added and the mixture was again heated at 85C overnight. The solvent was removed and the residue was diluted with water and extracted with EtOAc. The organic layer was washed with water and brine, dried over anhydrous sodium sulfate, filtered and the solvent was evaporated to give the desired product (150 mg, 24% yield) as a yellow oil. LCMS (ES-API): Rt2.0 min; m/z 239.1 [M+H]+.
With potassium carbonate; In acetonitrile; at 60℃;
General procedure: To a solution of 1-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)piperazine hydrochloride (500 mg, 1.54 mmol) and potassium carbonate (430 mg, 3.1 mmol) in acetonitrile (20 mL) was added 2-bromoethanol (388 mg, 3.1 mmol), then the mixture was heated at 60 C. overnight under an atmosphere of nitrogen. Then the mixture was filtered over celite and washed with DCM, concentrated to give product as a light brown solid. 2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)ethanol The title compound was prepared according to the procedures of Intermediate 52 using 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole. MS (m/z): 239 (M+H)+.
With potassium carbonate; sodium iodide; In acetonitrile; at 70℃;
A mixture of 4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)- lH- pyrazole (0.50 g, 2.58 mmol), sodium iodide (39 mg, 0.26 mmol) bromoacetonitrile (1 .3 g, 10.8 mmol) and potassium carbonate (1.0 g, 7.8 mmol) in acetonitrile (10 mL) was heated at 70 C overnight. Water was added and the solution was extracted with EtOAc (3x). The organic was dried over Na2S04, filtered and concentrated. The residue was purified by silica gel column chromatography ( 10% to 100% EtOAc/hexane) to obtain 2-(4-(4,4,5,5-tetramethyl-l ,3,2- dioxaborolan-2-yl)-lH-pyrazol-l-yl)acetonitrile (0.38g, 63% yield). MS (ESI) m/z: 234.1 (M+H+).
39%
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 24h;
A solution of 4-(4,4, 5,5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1H- pyrazole (5.11 g, 26.3 mmol) in DMF (50 mL) was treated with bromoacetonitrile (2.20 mL, 31.6 mmol) and K2CO3() (5.46 g, 39.5 mmol). The resulting suspension was stirred for 24 h at 100 C. The reaction mixture was cooled to ambient temperature, diluted with water (100 mL) then extracted with EtOAc (3 x 250 mL). The combined organic extracts were washed with water (3 x 50 mL) and brine (50 mL) then dried over anhydrous Na2SO4(). Following filtration, the organic extracts were concentrated under vacuum then purified by silica chromatography (10-60% Hexanes/EtOAc as the gradient eluent) to afford the title compound (2.42 g, 39% yield). ?H NIVIR (400 MHz, DMSO-d6) 8.04 (s, 1H), 7.71 (s, 1H), 5.49 (s, 2H), 1.25 (s, 12H).
With potassium carbonate; sodium iodide; In acetonitrile; at 70℃; for 24h;
[00769] Compound 48 was prepared in 2 steps according to the following procedures: 4-(4,4,5,5- tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole (2.61 mmol, 1.0 equiv), bromoacetonitrile (4.2 equiv), sodium iodide (0.1 equiv) and potassium carbonate (3.0 equiv) were suspended in acetonitrile (10 mL) and heated to 70 C for 24h after which the reaction was cooled, partioned between water and brine and extracted with ethyl acetate. The combined organics were concentrated onto silica gel (4g) and purified using flash silica gel chromatography (Silicycle Si-40g, gradient 10-100% ethyl acetate/hexanes) to provide pinacol ester 47.47
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃;
Compound 3F: 1 -[3-(Benzyloxy)propyl]-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- pyrazoleA mixture of 4-(4,4,5,5-tetramethyl[1 ,3,2]dioxaborolan-2-yl)-1 /-/-pyrazole (5.0 g, 26 mmol), potassium carbonate (4.27 g, 30.9 mmol) and 1-bromo-3-benzyloxypropane (6.20 g, 27.0 mmol) in DMF (20 mL) was stirred at 80 °C overnight, quenched with water (20 mL) and extracted with EtOAc (100 mL). The organic layer was washed with water (2 x 30 mL), brine (30 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (ISCO system: EtOAc/Heptane = 30- 70percent) to afford 7.67 g of the title compound as a light-yellow oil (87percent). 1H NMR (CDCI3, 400 MHz): delta = 7.80 (s, 1 H), 7.66 (s, 1 H), 7.30-7.36 (m, 5H), 4.48 (s, 2H), 4.28 (t, J = 6.8 Hz, 2H), 3.43 (t, J = 5.8 Hz, 2H), 2.17 (m, 2H), 1.33 (s, 12H). MS (ES+): m/z = 343.36 [MH+]. HPLC: iR = 4.20 min ( ZQ3: polar_5 min).
With caesium carbonate; In acetonitrile; at 90.0℃; for 4.0h;
A mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.40 g, 2.1 mmol, Aldrich, Cat. 525057), ethyl 2-bromopropanoate (290 muL, 2.3 mmol), and cesium carbonate (1.5 g, 4.6 mmol) in acetonitrile (8 mL) was stirred at 90 C. for 4 h. After cooling, the reaction mixture was worked up with aqueous Na2CO3, extracted with ethyl acetate (3×20 mL), and washed with brine. The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on a silica gel column with ethyl acetate in hexanes (0-50%) to afford the desired product (0.42 g, 61%). LCMS (M+H)+: m/z=295.1.
21.6 g
With potassium carbonate; In N,N-dimethyl-formamide; at 80.0℃; for 14.0h;
Step 9 Ethyl 2-[4-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)-1H-pyrazol-1-yl]propionate To a suspension of 4-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (21.3 g) and potassium carbonate (20.7 g) in dimethylformamide (100 ml) was added ethyl 2-bromopropionate (13 ml), and the mixture was stirred at 80 C. for 14 hr. The reaction mixture was cooled to 0 C., and toluene (100 ml) and water (150 ml) were successively added dropwise. The mixture was partitioned, and the aqueous layer was extracted with toluene (50 ml). The combined organic layer was successively washed once with 10% aqueous potassium carbonate solution (50 ml), twice with water (50 ml) and once with saturated brine (50 ml). The obtained organic layer was dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to give the title compound (21.6 g). 1H-NMR (400 MHz, CDCl3) delta: 7.85 (1H, s), 7.81 (1H, s), 5.10 (1H, q, J=7.3 Hz), 4.19 (2H, q, J=7.1 Hz), 1.78 (3H, d, J=7.4 Hz), 1.32 (12H, s), 1.25 (3H, t, J=7.2 Hz).
With caesium carbonate; In N,N-dimethyl-formamide; at 20 - 100℃; for 2h;
To a stirred solution of 4-(4,4,5 ,5 -tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 Hpyrazole (100 mg, 0.5 15 mmol) in DMF (1 ml) were added Cs2CO3 (252 mg, 0.773mmol) and 2,2,2-trifluoroethyl trifluoromethanesulfonate (0.144 ml, 1.031 mmol) at rt. After stirring at 100 C for 2 h, the reaction mixture was evaporated to dryness, partitioned between EtOAc and water, and the layers were separated. The organic layer was dried over Na2SO4, filtered, and concentrated to afford 4-(4,4,5,5-tetramethyl-i,3,2- dioxaborolan-2-yl)- 1 -(2,2,2-trifluoroethyl)- 1 H-pyrazole (0.111 g, 78% yield). MS(ESI)m/z: 277.4 (M+H).
76%
With caesium carbonate; In N,N-dimethyl-formamide; at 100℃; for 0.833333h;
To a solution of 4-(4, 4,5,5, -tetramethyl-1 ,3, 2-dioxaborolan-2-yl)-1 H-pyrazole (250 mg, 1.29 mmol) in N,N-dimethylformamide (4 mL) were added cesium carbonate (630 mg, 1.93 mmol) and 2,2,2-trifluoroethyl trifluoro methanesulfonate (0.37 mL, 2.58 mmol). The mixture was stirred at 100C for 50 minutes. The mixture was then poured in water (15 mL) and extracted with ethyl acetate (2x10 mL). The organic layer was washed with a saturated solution of sodium hydrogenocarbonate (10 mL), brine (10 mL), dried over sodium sulfate and concentrated in vacuo to provide 4-(4, 4,5,5,- tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 -(2,2,2-trifluoroethyl)-1 H-pyrazole (22a) (271 mg, 0.98 mmol, 76%) which was used without further purification. 1H NMR (300 MHz, CDCl3) delta 1.32 (s, 12H), 4.71 (q, J = 8.4 Hz, 2H), 7.80 (s, 1 H), 7.84 (s, 1 H).
64%
With caesium carbonate; In N,N-dimethyl-formamide; at 0 - 20℃; for 3.5h;
2,2,2-Trifluoroethyl trifluoromethanesulfonate (0.66 g, 2.86 mmol, 0.4 mL) was added to a mixture of 4-(4,4,5,5-tetramethyl-i ,3,2-dioxaborolan-2-yl)-i H-pyrazole (0.46 g, 2.37 mmol) and cesium carbonate (1.60 g, 4.92 mmol) in dry N,N-dimethylformamide (10.0 mL) at 0C. After stirring for 30 mm the reaction mixture was allowed to warm to room temperature. After stirring for 3 h the reaction mixture was poured out into water (200 mL) and the mixture was extracted with ethyl acetate (3x50 mL). The combined organic layers were washed with water (3x100 mL) and brine and were dried with sodium sulfate. Solvents were removed in vacuo to afford 0.42 g (1.51 mmol; 64% of theory) of the title compound.GO-MS (Method L9): R1 = 3.36 mm; m/z = 276 M
62%
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃; for 15h;
To a stirred solution of 4-(4,4,5,5-tetramethyl- I ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (2.00 g, 10.3 mmcl) in DMF (20 ml) was added potassium carbonate (4.27 g, 30.9 mmol) and 2,2,2-trifluoroethyl trifluoromethanesulfonate (2.2 ml, 15 mmcl). The mixture was stirred at 80C for14 h. Further 2,2,2-trifluoroethyl trifluoromethanesulfonate (420 p1, 2.9 mmcl) was added and the mixture was stirred at 80C for 1 h. Water was added and the mixture was extracted with ethyl acetate. The organic phase was washed with half-saturated sodium chloride solution, dried (sodium sulfate), filtered and the solvent was removed in vacuum. Silicagelchromatography gave 1.76 g (62 % yield) of the title compound. 1H-NMR (400 MHz, DMSO-d6) oe [ppm]: 1.066 (1.33), 1.256 (16.00), 5.144 (0.85), 5.167 (0.81),7.701 (1.31), 8.055 (1.26).
With caesium carbonate; In N,N-dimethyl-formamide; at 0 - 20℃; for 2.5h;Inert atmosphere;
Intermediate 14: 4-(4,4,5,5-Tetramethyl-1 , 3,2-dioxaborolan-2-yl)-1 -(2,2,2- trif luoroethyl)-1 H-pyrazole; 2,2,2-Trifluoroethyl trifluoromethanesulfonate (28.7g, 124mmol) (Apollo Scientific) was added to a mixture of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- pyrazole (20g, 103mmol) (Aldrich) and cesium carbonate (67.2g, 206mmol) in N,N- Dimethylformamide (DMF) (150ml) at 0C under nitrogen . The mixture was stirred for 30 min at 0C then allowed to warm to room temperature and stirred for a further 2h. The mixture was quenched with water (200ml) and extracted with EtOAc (200ml). The organic layer was washed with water (200ml), dried and evaporated to give a brown oil. This was dissolved in DCM (30ml), the fine precipitate was filtered off and the filtrate loaded onto a 330g silica column, then eluted with 0-50% EtOAc/cyclohexane. Appropriate fractions were combined and evaporated to give the title compound as a colourless oil (14.7g)1 H NMR (CDCI3) 7.86ppm (1 H, s, CH); 7.82ppm (1 H, s, CH); 4.73ppm ( 2H, q, CH2); 1.34ppm (12H, s, 4xCH3).
14.7 g
With caesium carbonate; In N,N-dimethyl-formamide; at 0 - 20℃;Inert atmosphere;
Intermediate 14: 4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2-trifluoroethyl)-1H-pyrazole 2,2,2-Trifluoroethyl trifluoromethanesulfonate (28.7 g, 124 mmol) (Apollo Scientific) was added to a mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (20 g, 103 mmol) (Aldrich) and cesium carbonate (67.2 g, 206 mmol) in N,N-Dimethylformamide (DMF) (150 ml) at 0 C. under nitrogen. The mixture was stirred for 30 min at 0 C. then allowed to warm to room temperature and stirred for a further 2 h. The mixture was quenched with water (200 ml) and extracted with EtOAc (200 ml). The organic layer was washed with water (200 ml), dried and evaporated to give a brown oil. This was dissolved in DCM (30 ml), the fine precipitate was filtered off and the filtrate loaded onto a 330 g silica column, then eluted with 0-50% EtOAc/cyclohexane. Appropriate fractions were combined and evaporated to give the title compound as a colourless oil (14.7 g) 1H NMR (CDCl3) 7.86 ppm (1H, s, CH); 7.82 ppm (1H, s, CH); 4.73 ppm (2H, q, CH2); 1.34 ppm (12H, s, 4*CH3).
The following Preparations were prepared according to Method H (Preparation 122) using 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole and the appropriate alkyl electrophile.
With sodium hydride; In N,N-dimethyl-formamide; at 0 - 20℃; for 12h;
. Sodium hydride (61.8 mg, 1.55 mmol) was added to a solution of 4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-lH-pyrazole (150 mg, 0.77 mmol) and 2,2,2-trifluoroethyl trifluoromethanesulfonate (269 mg, 1.16 mmol) in DMF (5 ml) at 0C and the mixture was stirred at 20C for 12 h. The mixture was quenched with aqueous saturated ammonium chloride (6 mL) and extracted with ethyl acetate (3 x 8 mL). The combined organic fractions were washed with brine, dried over sodium sulfate, filtered, and the solvent was evaporated under reduced pressure to give 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l-(2,2,2-trifluoroethyl)- lH-pyrazole. MS (ESI) calc'd for (C11H17BF3N2O2) [M+H]+, 277 ; found, 277
With caesium carbonate; In 1,4-dioxane; at 20℃; for 3.5h;
[00771] Compound 50 was prepared in 2 steps according to the following procedures: 4-(4,4,5,5- tetramethyl- 1 ,3,2-dioxaborolan-2-yl)- 1H-pyrazole (2.6 mmol, 1.0 equiv) and cesium carbonate (2.0 equiv) were suspended in dioxane (10 mL). Trifluoroethyltriflate (1.24 equiv) was added and the reaction was stirred at room temperature for 3 .5h after which the mixture partionned between water and brine and extracted with ethyl acetate. The combined organics were concentrated onto silica gel (4g) and purified using flash silica gel chromatography (Silicycle Si-40g, gradient 10-100% ethyl acetate/hexanes) to provide pinacol ester 49.
Step 1: 4-(4,4, 5,5-Tetramethyl- 1 ,3,2-dioxaborolan-2-yl)- 1 -(2 ,2,2-trifl uoroethyl)- 1 H-pyrazole Cesium carbonate (3.36 g, 10.31 mmol) was added to a stirred solution of 4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1.0 g, 5.15 mmol) in dry DMF (12 ml).After stirring at RT for 10 mm 2,2,2-trifluoroethyl trifluoromethanesulfonate (1.11 ml, 7.73 mmol) was added. The reaction was stirred for 2 days at RT then the solvent was removed and the residue was partitioned between diethyl ether and water. The organic extract was separated, dried over MgSO4 and the solvent removed to give an oil;LCMS: Rt 1.00 mins; MS MS mlz 277.4 [M+H]+; Method 2minLCvOO3
With caesium carbonate; In acetonitrile; at 60℃; for 2h;
Intermediate D1 4-(4,4,5,5-Tetramethyl-1,3,2-dioxoborolan-2-yl)-1-(2,2,2-trifluoroethyl)-1 H-pyrazoleA stirred mixture of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (0.56 g, 2.89 mmol), 2,2,2-trifluoroethyl trifluoromethanesulfonate (0.633 ml, 4.39 mmol) and cesium carbonate (2.81 g, 8.62 mmol) in acetonitrile (20 ml) was heated at 60C. After 2h the solvent was removed under reduced pressure and the residue was partitioned between EtOAc and water. The organic extract was dried over MgS04 and the solvent was removed under reduced pressure to afford the title compound as a gum;LCMS: Rt 1.00 mins; MS m/z 277.4 [M+H]+; Method 2minl_C_v0031H NMR (400MHz, CDCI3) delta 7.88 (1 H, s), 7.82 (1 H, s), 4.73 (2H, q), 1.35 (12H, s).
With caesium carbonate; In N,N-dimethyl-formamide; at 100℃; for 2h;
Intermediate 255A: 4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-l -(2,2,2- trifluoroethyl)- lH-pyrazole To a stirred solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazole (400 mg, 2.06 mmol) in DMF (2 mL) was added CS2CO3 (1.0 g mg, 3.1 mmol) and 2,2,2-trifluoroethyl triflate (0.58 mL, 4.1 mmol). The mixture was heated at 100 C for 2 hours. After cooling, the mixture was concentrated to dryness then partitioned between EtOAc and water. The layers were separated and the organic layer was dried over Na2S04, filtered, and concentrated to afford the crude product which was used directly in the next step. NMR (400 MHz, CHLOROFORM-^ delta 7.85 (s, 1 H) 7.80 (s; 1 H) 4.64-4.76 (m, 2 H), 1.32 (s, 12 H).
With caesium carbonate; In acetonitrile; at 20℃; for 3h;
Step A. tert-Butyl 3-((4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l- yl)methyl)phenylcarbamate [00239] To a solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.88 g, 4.54 mmol) in acetonitrile (20 mL) was added cesium carbonate (4.43 g, 13.61 mmol) and tert-butyl 3-(bromomethyl)phenylcarbamate (1.298 g, 4.54 mmol). The reaction was stirred at room temperature for 3 h. After this time, the reaction mixture was diluted with ethyl acetate and washed with water and brine. The aqueous phase was extracted with CHCI3. The combined organic layer was dried over anhydrous MgS04, filtered, and concentrated. The resulting residue was purified by (0-100% ethyl acetate:hexanes) to afford the title compound (1.06 g, 2.65 mmol, 59% yield) as a colorless oil. LCMS, [M+H]+ = 400.3
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; toluene; at 70℃; for 4h;Inert atmosphere;
Step 1: 2-(lH-Pyrazol-4-yl)thiazole-5-carbaldehyde To a solution of <strong>[464192-28-7]2-bromothiazole-5-carbaldehyde</strong> (500 mg, 2.6 mmol) in toluene (15 mL) and ethanol (7 mL) was added 4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-lH-pyrazole (606 mg, 3.1 mmol), 2M sodium carbonate solution (7.3 mL) and Pd(PPh3)4 (30 mg, 0.02 mmol). The reaction mixture was purged with argon for 15 min and the resulting mixture was heated at 70C for 4 h. The reaction mixture was concentrated, diluted with water (100 mL), and extracted with ethyl acetate (2 x 200 mL). The combined extracts were washed with brine solution (20 mL), dried over Na2S04, filtered and concentrated to under vacuum to obtain crude product. The crude product was purified by flash chromatography (60-120 mu; 50% ethyl acetate in hexane) to afford 2-(lH-pyrazol-4-yl)thiazole-5-carbaldehyde as yellow solid (250 mg, 54% yield). 1H NMR (400 MHz, DMSO-d6): delta 13.49 (br s, 1H), 10.00 (s, 1H), 8.61(s, 1H), 8.55(s, 1H), 8.09 (s, 1H); LC-MS m/z calculated for [M+H]+ 180.02, found 180.1.
The following Preparations were prepared according to Method H (Preparation 122) using 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole and the appropriate alkyl electrophile.
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃;
A mixture of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (3.0 g, 15.5 mmol), bromomethyl-benzene (3.2 g, 18.7 mmol) and K2CO3 (4.3 g, 31.2 mmol) in DMF (30 mL) was stirred at room temp overnight. After dilution with EtOAc (50 mL) and H2O (50 mL), the organic layer was separated and washed with brine (50 mL), dried over Na2SO4, filtered and concentrated in vacuum. The residue was purified by column chromatography on silica gel eluting with PE/EtOAc (5:1) to give the compound 1-benzyl-4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-pyrazole (3.5 g, 12.3 mmol) as a light yellow solid in 79% yield. 1H NMR (400 MHz, CDCl3): delta 7.81 (s, 1H), 7.66 (s, 1H), 7.37-7.29 (m, 3H), 7.24-7.22 (m, 2H), 5.30 (s, 2H), 1.29 (s, 12H). LCMS (M+H)+ 285.
66%
Compound 244.1. l-Benzyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazole. _:Into a 100-mL three neck round-bottom flask, was placed a solution of 4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (3.60 g, 18.6 mmol) in THF (50 mL). Sodium hydride (742 mg, 18.6 mmol, 60% dispersion in mineral oil) was carefully added in portions at 0 C. The resulting mixture was stirred for 30 min at 0 C, then benzyl bromide (2.21 mL, 18.6 mmol) was added. The resulting mixture was stirred overnight at room temperature, then carefully quenched with water (10 mL). The pH of the mixture was adjusted to 9-10 with aqueous HC1 (2 M) and the aqueous phase was extracted with EtOAc (300 mL). The combined organic layers were washed with brine (2 x 150 mL), dried (Na2S04), filtered and concentrated under vacuum. The residue was purified by silica gel chromatography with ethyl acetate/petroleum ether (1/2) as eluent to yield the title compound as a yellow oil (3.47 g, 66%).
59%
1 -benzyl-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (i34): To a stirred solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (3.0 g, 15.46 mmol) in THF (50 ml_), NaH (0.408 g, 17.01 mmol) was added at 0C and the reaction was stirred for 30 min. Benzyl bromide (2.9 g, 17.01 mmol) was then added at the same temperature and the reaction was stirred at room temperature for 16h. The progress of the reaction was monitored by TLC. After completion, the mixture was diluted with water and the pH adjusted to 7 using 2 M HCI. The aqueous layer was extracted with ethyl acetate. The organic layer was separated, dried over anhydrous sodium sulphate and concentrated under reduced pressure. The crude product was purified by silica gel (100-200 mesh) column chromatography using 8% ethyl acetate in n-hexanes as eluent to afford 1-benzyl-4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 H-pyrazole (i34) (2.6 g, Yield 59%). 1H NMR (400 MHz, DMSO-d6) delta 1 .24 (s, 12H), 5.33 (s, 2H), 7.38-7.20 (m, 5H), 7.60 (s, 1 H), 8.03 (s, 1 H), MS (ESI) m/e (M+1 )+: 285.00
0.28 g
With caesium carbonate; In N,N-dimethyl-formamide; at 20℃;Inert atmosphere;
0.25g 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole was dissolved in 5mL of DMF and 0.63g cesium carbonate was added. To this suspension was added 0.17mL of benzyl bromide and the reaction was stirred overnight at room temperature. The reaction was allowed to settle and the DMF was decanted into a flask. The remaining residue was washed and decanted twice with ethyl acetate and these washes were added to the flask with the DMF. Water was added to the ethyl acetate and DMF and the organic layer removed. The aqueous layer was extracted with ethyl acetate and the combined organic layers were dried over MgS04 and concentrated to give 0.28g of l -benzyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazole which was used without further purification.
With hydrogenchloride; 2,3-dimethyl-2,3-butane diol; In water; at 10 - 25℃; for 4.083h;
1 -( 1 -Ethoxyethyl)-4-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)-l H-pyrazole (60.00 g, 224 mmol) is combined with CPME (120 mL) and 2,3-dimethylbutane-2,3-dtol (26.49 g, 224 mmol). The reaction is cooled to 5-10 C then a solution of anhydrous HQ in CPME (3.1 M, 86.8 mL, 269 mmol) is added over 15 minutes, followed by additional CPME (15 mL). The reaction is stirred at 20-25 C and monitored for completion. After 7 hours, additional HCI solution (3 mL, 9.3 mmol) is added to the reaction and stirring is continued for an additional 15 hours to give 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)-l H-pyrazole hydrochloride salt, which is not isolated.mL) is added over 10 minutes. The reaction mixture temperature increases to 20 C following the addition. Additional CPME (10 mL) is added and the reaction mixture is stirred for 15 minutes then cooled in an ice bath. After 3 hours the reaction mixture is filtered and the solid (triethylamine hydrochloride) is washed with cold CPME (3 x 60 mL). The filtrate and washes are combined to give 426 g of solution containing 102,3 mg of 4- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazo]e/g solution (total 8.15 g, 94% )yield of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyTazole) which is used directly in the next step.
77.3%
With hydrogenchloride; 2,3-dimethyl-2,3-butane diol; In tert-butyl methyl ether; 1,2-dichloro-ethane; at 0 - 20℃;Large scale;
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1) [0166] To a mixture of 2,3-dimethylbutane-2,3-diol (25.0 kg, 211.6 mol) and <strong>[1029716-44-6]1-<strong>[1029716-44-6](1-ethoxyethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole</strong></strong> (24, 55.0 kg, 206.7 mol) in 1,2-dichloroethane (750 kg) was slowly added a solution of HCl in MTBE (25.0 kg, 20-30% of HCl) at 0-5 C. The resulting reaction mixture was then stirred at 10-20 C. for 3-5 hours. After the selective deprotection reaction was complete as monitored by HPLC (1: below 1%), the reaction mixture was degassed and refilled with nitrogen before being cooled to -15 C. The cooled reaction mixture was then added triethylamine (TEA, 30.0 kg, 296.5 mol) to adjust pH to 7-8. The mixture was then gradually warmed to ambient temperature before being treated with water (150 kg). The two phases were separated and the organic layer was washed with brine (60 kg) and dried over sodium sulfate (Na2SO4). The drying reagent, sodium sulfate (Na2SO4), was removed by filtration and the resulting solution was concentrated under reduced pressure at 40-50 C. to a thick oil. The residue was warmed to 60-70 C. and diluted with petroleum ether (100 kg) at the same temperature. The resulting mixture was then gradually cooled to ambient temperature and subsequently to -5 C. and stirred at the same temperature for 3 hours. The solids was collected by centrifugation and dried at 50-60 C. under vacuum to afford the crude desired product (1, 33.75 kg, 40.11 kg theoretical, 84.1%). The crude desired product was then suspended in 1,2-dichloroethane (30 kg) and the resulting mixture was heated to reflux until a clear solution was formed. To the hot solution was then added petroleum ether (150 kg) at the same temperature. The resulting mixture was then gradually cooled to ambient temperature and subsequently to -5 C. and stirred and the same temperature for 3 hours. The solids were collected by centrifugation and dried under vacuum at 50-60 C. to afford 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1, 31.0 kg, 40.11 kg theoretical, 77.3%) as an off-white solid, which is identical in every comparable aspect to the material synthesized by the synthetic method as described above in Example 5.
77.3%
With hydrogenchloride; 2,3-dimethyl-2,3-butane diol; In tert-butyl methyl ether; 1,2-dichloro-ethane; at 0 - 20℃;Large scale;
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1) To a mixture of 2,3-dimethylbutane-2,3-diol (25.0 kg, 211.6 mol) and <strong>[1029716-44-6]1-<strong>[1029716-44-6](1-ethoxyethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole</strong></strong> (24, 55.0 kg, 206.7 mol) in 1,2-dichloroethane (750 kg) was slowly added a solution of HCl in MTBE (25.0 kg, 20-30% of HCl) at 0-5 C. The resulting reaction mixture was then stirred at 10-20 C. for 3-5 hours. After the selective deprotection reaction was complete as monitored by HPLC (1: below 1%), the reaction mixture was degassed and refilled with nitrogen before being cooled to -15 C. The cooled reaction mixture was then added triethylamine (TEA, 30.0 kg, 296.5 mol) to adjust pH to 7-8. The mixture was then gradually warmed to ambient temperature before being treated with water (150 kg). The two phases were separated and the organic layer was washed with brine (60 kg) and dried over sodium sulfate (Na2SO4). The drying reagent, sodium sulfate (Na2SO4), was removed by filtration and the resulting solution was concentrated under reduced pressure at 40-50 C. to a thick oil. The residue was warmed to 60-70 C. and diluted with petroleum ether (100 kg) at the same temperature. The resulting mixture was then gradually cooled to ambient temperature and subsequently to -5 C. and stirred at the same temperature for 3 hours. The solids was collected by centrifugation and dried at 50-60 C. under vacuum to afford the crude desired product (1, 33.75 kg, 40.11 kg theoretical, 84.1%). The crude desired product was then suspended in 1,2-dichloroethane (30 kg) and the resulting mixture was heated to reflux until a clear solution was formed. To the hot solution was then added petroleum ether (150 kg) at the same temperature. The resulting mixture was then gradually cooled to ambient temperature and subsequently to -5 C. and stirred and the same temperature for 3 hours. The solids were collected by centrifugation and dried under vacuum at 50-60 C. to afford 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1, 31.0 kg, 40.11 kg theoretical, 77.3%) as an off-white solid, which is identical in every comparable aspect to the material synthesized by the synthetic method as described above in Example 5.
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃; for 5h;
Synthesis of boronic ester R7.6: 2 g (10.3 mmol) 4-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole and 2.9 mL (20.6 mmol) 4-(iodomethyl)-tetrahydro-2H-pyran are dissolved in 200 mL DMF and 4.274 g (30.9 mmol) K2CO3 are added. The mixture is shaken at 80C for 5 h. After cooling to r.t. the mixture is filtered, the filtrate is concentrated in vacuo to approximately 60 mL. The product is separated using HPLC-MS (Gilson, mass flow 120 mL/min, 10 muiotaeta, 200g Sunfire RP18, ACN/water/TFA). The product fractions are combined and freeze-dried to yield 115 mg product (3.8 %) R7.6.
3.8%
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃; for 5h;
2 g (1 0.3 mmol) 4-( 4,4,5,5-Tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole and 2.9 mL (20.6mmol) 4-(iodomethyl)-tetrahydro-2H-pyran are dissolved in 200 mL DMF and 4.274 g (30.9s mmol) K2C0 3 are added. The mixture is shaken at 80"C for 5 h. After cooling to r.t. the mixture isfiltered, the filtrate is concentrated in vacuo to approximately 60 mL. The product is separatedusing HPLC-MS (Gilson, mass flow 120 mL/min, 10 lJ.m, 200g Sunfire RP18, ACN/waterfTFA).The product fractions arc combined and frecze-d1icd to yield 115 mg product (3.8 %) R6.3.
3.8%
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃; for 5h;
2 g (10.3 mmol) 4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole and 2.9 mL (20.6 mmol) 4-(iodomethyl)-tetrahydro-2H-pyran are dissolved in 200 mL DMF and 4.274 g (30.9 mmol) K2CO3 are added. The mixture is shaken at 80 C. for 5 h. After cooling to r.t. the mixture is filtered, the filtrate is concentrated in vacuo to approximately 60 mL. The product is separated using HPLC-MS (Gilson, mass flow 120 mL/min, 10 mum, 200 g Sunfire RP18, ACN/water/TFA). The product fractions are combined and freeze-dried to yield 115 mg product (3.8%) R6.3
3.8%
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃; for 5h;
2 g (10.3 mmol) 4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole and 2.9 mL (20.6 mmol) 4-(iodomethyl)-tetrahydro-2H-pyran are dissolved in 200 mL DMF and 4.274 g (30.9 mmol) K2CO3 are added. The mixture is shaken at 80 C. for 5 h. After cooling to r.t. the mixture is filtered, the filtrate is concentrated in vacuo to approximately 60 mL. The product is separated using HPLC-MS (Gilson, mass flow 120 mL/min, 10 mum, 200 g Sunfire RP18, ACN/water/TFA). The product fractions are combined and freeze-dried to yield 115 mg product (3.8%) R7.6.
tert-butyl 4-(1H-pyrazol-4-yl)-5,6-dihydropyrido[3,4-d]pyrimidine-7(8H)-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
77%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In 1,4-dioxane; at 150℃; for 1h;Microwave irradiation; Inert atmosphere;
Example 4, Step a: tert-butyl 4-(1H-pyrazol-4-yl)-5,6-dihydropyrido[3,4-d]pyrimidine-7(8H)-carboxylate To a microwave vial was added Intermediate 2 (113 mg, 0.42 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (138 mg, 0.71 mmol) followed by dioxane (4 mL) and 2M Na2CO3 (0.52 mL). To this mixture was added Pd(PPh3)4 (24 mg, 0.02 mmol) and the reaction heated in the microwave for 1 h at 150 C. The reaction was diluted with water and extracted with DCM and EtOAc. The combined organic extracts were dried over Na2SO4, filtered and concentrated in vacuo. Chromatography on SiO2 eluting with EtOAc/Hex afforded the desired product as a slowly crystallizing white solid (97 mg, 77%). MS (ESI) mass calcd. C15H19N5O2, 301.15. m/z found 302.1 [M+H]+. 1H NMR (500 MHz, CDCl3): 8.96 (s, 1H), 8.21 (s, 2H), 4.68 (s, 2H), 3.82-3.70 (m, 2H), 3.07-2.93 (m, 2H), 1.51 (s, 9H).
With caesium carbonate; In N,N-dimethyl-formamide; at 100℃; for 1h;Inert atmosphere; Microwave irradiation; Sealed tube;
Step 1: 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l-(2,2,2-trifluoroethyl)-lH- pyrazole [00249] A 25 mL teflon cap tube was charged with 4-pyrazoleboronic acid pinacol ester (0.94 g, 4.87 mmol), cesium carbonate (2.58 g, 7.31 mmol), 2,2,2 trifluoroethyl methanesulfonate (1.30 g, 0.86 mL, 7.31 mmol) and anhydrous DMF (8 mL). The reagents were capped under N2 and heated at 100 C under microwave irradiation for 1 h. After this time the reaction mixture was cooled to rt and partitioned with EtOAc (60 mL) and water (100 mL). Organic layers were extracted, washed with brine (2 x 65 mL), then dried and filtered (phase separator) to give a pale yellow oil. The crude product was purified by flash silica column chromatography (gradient elution /so-hexane to 33% EtOAc in i'so-hexane) to give the title compound as a colorless oil (0.37 g, 35% - mixture of target material and 2,2,2 trifluoroethyl methanesulfonate 1:2). MS (ES+) consistent with target (M+H)+.
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 20℃; for 88.08h;
4.76 ml (24.0 mmol) of diisopropyl azodicarboxylate are added dropwise to a solution of 3.88 g (20.0 mmol) of pinacolyl pyrazole-4-boronate, 1.78 g (48.0 mmol) of oxetan-3-ol and 6.29 g (24.0 mmol) of triphenylphosphine in 40 ml of THF. The reaction mixture is stirred at room temperature for 16 hours. A further 1.78 g (48.0 mmol) of oxetan-3-ol, 6.29 g (24.0 mmol) of triphenylphosphine and 3.00 ml (15.1 mmol) of diisopropyl azodicarboxylate are then added, and the reaction mixture is stirred at room temperature for 3 days. The reaction mixture is evaporated, and the residue is taken up in cyclohexane. The precipitate formed is filtered off with suction and washed with cyclohexane. The filtrate is evaporated, and the residue is chromatographed on a silica-gel column with cyclohexane/ethyl acetate as eluent: 1-oxetan-3-yl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole as yellow oil; HPLC/MS (A): 2.10 min, [M+H] 251; 1H NMR (400 MHz, DMSO-d6) delta [ppm] 8.07 (s, 1H), 7.72 (s, 1H), 5.60 (p, J=6.9, 1H), 4.89 (m, 4H), 1.25 (s, 12H).
4-(1H-pyrazol-4-yl)-2-(trifluoromethyl)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
84%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,4-dioxane; water; at 90℃;Inert atmosphere;
Step C. 4-(lH-pyrazol-4-yl)-2-(trifluoromethyl)benzonitrile A mixture of 4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-lH-pyrazole (CAS: 269410- 08-4, Sigma- Aldrich) (300 mg, 1.55 mmol), <strong>[101066-87-9]4-iodo-2-(trifluoromethyl)benzonitrile</strong> (300 mg, 1.01 mmol), Pd(PPh3)4 (18.6 mg, 0.016 mmol), K2C03 (891 mg, 6.46 mmol), and water (2 mL) in 1 ,4-dioxane (10 mL) under nitrogen was heated at 90 C overnight. The reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography using petroleum ether :EtO Ac (1 : 1) as eluting solvents to afford 4-(lH-pyrazol-4-yl)-2-(trifluoromethyl)benzonitrile as a white solid (203 mg, 84%). LCMS (ESI) m/z: 238.1 [M+H]+.
tert-butyl 2-methyl-2-(4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-1H-pyrazol-1-yl)propanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
81%
With caesium carbonate; In N,N-dimethyl-formamide; at 20℃; for 16h;
Tert-butyl 2-methyl-2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l- yl)propanoate To a solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (5.0 g, 25.8 mmol) and tert-butyl 2-bromo-2-methylpropanoate (6.32 g, 28.3 mmol) in DMF (50 mL) was added CS2CO3 (12.59 g, 38.65 mmol). The reaction mixture was stirred at room temperature for 16 hours. The mixture was filtered and partitioned between methyl tert-butyl ether (100 mL) and H20 (100 mL). The combined organic layer was separated, washed with brine, dried over anhydrous Na2S04 and concentrated to afford crude product which was purified by flash column chromatography on silica gel eluting with 0-10% EtOAc in hexanes to give the desired product (7.0 g, 81% yield) as a white solid. 1H NMR (400 MHz, CDC13) delta 7.87 (s, 1H), 7.84 (s, 1H), 1.81 (s, 6H), 1.39 (s, 9H), 1.37 (s, 12H).
With potassium carbonate; In acetonitrile; at 65℃; for 16.5h;Inert atmosphere;
0316-1 A mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.867 g), <strong>[592-33-6]3-bromopropyl acetate</strong> (0.970 g), potassium carbonate (1.85 g), and acetonitrile (4 mL) was stirred at 65 C. for 16.5 hours in a nitrogen atmosphere. The reaction mixture was cooled to room temperature, the solid matter was filtered off, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (chloroform-methanol), thereby obtaining 3-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)propyl acetate (1.24 g) as yellow oily substance. 1H-NMR (CDCl3) delta: 7.79 (1H, s), 7.69 (1H, s), 4.23 (2H, t, J=6.6 Hz), 4.05 (2H, t, J=5.9 Hz), 2.26-2.14 (2H, m), 2.05 (3H, s), 1.32 (12H, s).
2-(3-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)oxetan-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
59%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 60℃; for 18h;
To a solution of compound 20-b (500 mg, 5.26 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1.3 g, 6.7 mmol) in acetonitrile (10 mL) was added 1,8-diazabicyclo[5.4.0]undec-7-ene (1.6 g, 10.53 mmol). The mixture was stirred at 60 C. for 18 hours, then concentrated under reduced pressure. 1 N Aqueous hydrochloride solution was added into the residue to adjust pH=3-4, then the mixture was extracted with ethyl acetate (20 mL×3). The organic layers were combined, washed with water (20 mL×3) and saturated brine (20 mL) in sequence, dried over anhydrous sodium sulfate, filtrated, and the filtrate was concentrated under reduced pressure to give yellow oil 20-a (900 mg, yield: 59%), which was used directly for the next step without further purification. LC-MS (ESI): m/z=290 [M+H]+.
2-methyl-1-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)propan-2-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
94%
With caesium carbonate; In N,N-dimethyl-formamide; at 160℃; for 2.5h;Microwave irradiation;
A mixture of 4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -1H-pyrazole (1.01 g, 5.2 mmol) , 1-chloro-2-methylpropan-2-ol (1.1 mL, 11 mmol) and Cs2CO3(4.2 g, 13 mmol) in DMF (15 mL) was stirred under a microwave condition at 160 for 2.5 h. The reaction mixture was concentrated to remove DMF. The residue was diluted with water (20 mL) . The resulting mixture was extracted with DCM (30 mL × 3) . The combined organic layers were dried over anhydrous Na2SO4and concentrated in vacuo. The residue was purified by silica gel column chromatography eluted with PE/EtOAc (v/v) 4/1 to give a light yellow oily product (1.30 g, 94) .[1544]MS (ESI, pos. ion) m/z: 267.1 [M+1]+
With caesium carbonate; In N,N-dimethyl-formamide; at 70℃;
A mixture of 4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -1H-pyrazole (1.5 g, 7.7 mmol) , 2-bromo-N, N-dimethylethanamine hydrobromide (4 g, 20 mmol) and Cs2CO3(9 g, 17.6 mmol) in DMF (15 mL) was stirred at 70 overnight. The reaction mixture was concentrated to remove DMF. The residue was diluted with water (20 mL) . The resulting mixture was extracted with DCM (30 mL × 3) . The combined organic layers were dried over anhydrous Na2SO4and concentrated in vacuo. The residue was purified by silica gel column chromatography eluted with DCM/MeOH (v/v) 25/1 to give a white solid product (0.5 g, 20) .[1634]MS (ESI, pos. ion) m/z: 266.1 [M+1]+.
With caesium carbonate; In N,N-dimethyl-formamide; at 80℃;
[0155] A mixture of 4-( 4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-lH-pyrazole (500 mg, 2.58 mmol), <strong>[135859-47-1]3-hydroxy-3-methylbutyl methanesulfonate</strong> (938 mg, 5.16 mmol)and cesium carbonate (1.68 g, 5.16 mmol) in N,N-dimethylformamide(10 mL) was heated at 80 C. overnight. LCMSand TLC indicated the completion of the reaction. The reactionmixture was cooled to RT and concentrated to give aresidue, which was purified by silica gel chromatography(petroleum ether: ethyl acetate=:) to afford the title compoundas a colorless oil (350 mg, 49% ). MS (ES+)C14H25BN20 3 requires: 280. found: 281 [M+Ht.
2-(1-(ethanesulfonyl)-3-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)azetidine-3-yl)acetonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89.8%
With N,N,N?,N?-tetramethyl-N?-tert-butylguanidine; In tetrahydrofuran; at 65℃; for 3h;
Preparation of 4-(4,4,5 ,5 -tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole solution (107,0 mg/g as a solution in CPME) is conducted using the previously described procedure denoted in Alternate Preparation 5b.The solution of 4-(4,4,5 ,5 -tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole (69.41 g, 79.8 mL, 38.27 mmol) is combined with 2-terf-butyl-l, 1,3,3- tetramethylguanidine (0.54 g, 3.15 mmol) followed by a CPME rinse (6 mL) then heated to about 65 C. 2-(l-(Ethylsulfonylazetidin-3-ylidene)acetonitrile (6.00 g, 31.90 mmol) is dissolved in THF (14 mL) and added over 3 hours followed by a THF rinse (2 mL). The reaction is stirred at about 65 C and monitored for completion (typical completion time is two hours post-addition). The solution is cooled to 25 C and concentrated to a wet residue. 1-Propanol (60 mL) is added and the suspension is again concentrated to a wet solid. The solid is suspended in 1 -propanol (90 mL) and heated to 67 C to form a solution. The solution is cooled to 57 C and seed crystals (0.6 g) of the title compound are added. The resulting slurry is held at a temperature of 57 C for 2 hours then cooled to -3 C over 9 hours and held at that temperature for at least 2 hours. The solids are collected by filtration, washed with cold 1 -propanol (2 chi 12 mL) and dried to give the title compound (10.89 g, 89.8%).
85.78%
With potassium carbonate; In 1,4-dioxane; water; at 20 - 25℃;
1,4-Dioxane (20 mL) was added into a reaction vessel containing a solution of potassium carbonate (4.5 g) in water (30 mL) at about 20C to about 25 C. 2-(l- (Ethylsulfonyl)azetidin-3-ylidene)acetonitrile (2 g; Formula II) and 4-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-2-yl)-lH-pyrazole (2.30 g; Formula VIII) were added into the reaction mixture at about 20C to about 25 C. The reaction mixture was stirred at about 20C to about 25 C for about 16 hours to about 18 hours. On completion of the reaction, 1,4- dioxane was recovered from the reaction mixture under reduced pressure at about 45 C to obtain a residue. Ethyl acetate (20 mL) was added into the residue, and the contents were stirred for about 5 minutes. The organic and aqueous layers were separated. The organic layer was concentrated under reduced pressure at about 45 C to obtain {1-(ethylsulfonyl)-3-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile. Yield: 85.78%. Mass: 381.4 [M + H]+
85%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In acetonitrile; at 20℃; for 2h;
pyrazole-4-boronic acid pinacol ester (14 g, 72.1 mmol) was added to 100 mL of acetonitrile at room temperature, Was added <strong>[1187595-85-2]2-[1-(ethylsulfonyl)-3-azetidinylidene]acetonitrile</strong> (18 g, 96.7 mmol) and DBU (20 mL) After stirring at room temperature for 2 hours, Ethyl acetate (500 mL) and water (500 mL) were added, Extraction, The aqueous layer was extracted once with 50 mL of ethyl acetate, Combined organic layer, And then washed with 50mL saturated sodium chloride once, Dried over anhydrous sodium sulfate, filter, Concentrated under reduced pressure to give 23 g of a yellow solid. Yield 85%
84%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In isopropyl alcohol; acetonitrile; at 60℃; for 4h;
A mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (11) (156 mg, 0.80 mmol, 1.01 equiv.), compound 4 (149 mg, 0.80 mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (0.061 mL, 0.408 mmol) in acetonitrile (20 mL) was heated at 60 C for 4 h. After cooling, the solvent was removed under reduced pressure. The residue was purified by flash chromatography on a silica gel column eluting with methanol in dichloromethane (0-60%) to afford the desired compound 12: Yield 255 mg (84%); m.p. 120-122 C; IR: 3280, 2978, 1639, 1554, 1369 cm-1. Anal. calcd for C16H25BN4O4S: C, 50.54; H,6.63; N, 14.73; found: C, 50.67; H, 6.74; N, 14.62%. MS (m/z): 403 [M+ Na]+; 1H NMR (300 MHz, DMSO-d6): delta 1.22 (t, J = 7.3 Hz, 3H), 1.27(s, 12H), 3.19 (m, J = 7.3 Hz, 2H), 3.59 (s, 2H), 4.14 (d, J = 9.0 Hz, 2H), 4.44 (d, J = 9.0 Hz, 2H), 7.77 (s, 1H), 8.35 (s, 1H); 13C NMR (75 MHz, DMSO-d6): delta 4.6, 24.9, 39.5, 43.2, 55.5, 58.2, 83.2, 116.6, 135.8, 145.6.
84%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In isopropyl alcohol; at 20 - 60℃; for 3.5h;
In isopropanol solvent, was added Compound 10: 1,8-diazabicyclo [5,4,0] undec-7-ene (0.061 mL), Compound 9 (0.156g, 0.80mmol, 1.01equiv) , compound 4 (0.149g, 0.80mmol), at room temperature the formation of a suspension, was refluxed for half an hour, to form a uniform solution.The reaction was refluxed for 3 hours at 60 deg.] C, TLC trace.Cooling to room temperature, vacuum filtration, and purified by column chromatography to give a white solid 0.255 g, 84% yield.
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; caesium carbonate; In water; N,N-dimethyl-formamide; at 150℃; for 1h;Inert atmosphere; Sealed tube; Microwave irradiation;
Step a. To a solution of 6-bromoisoquinolin-3 -amine (0.20 g, 0.89 mmol) and 4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (0.19 g, 0.98 mmol) in DMF : water (4: 1, 5 ml) was prepared in a microwave glass vial. CS2CO3 (0.87 g, 2.68 mmol) was added to the reaction mixture at rt. The reaction mixture was degassed at rt for 15 min before adding Pd(dppf)Cl2 (0.06 g, 0.09 mmol) and the glass vial was sealed. The reaction mixture was subjected to microwave heating at 150C for 1 h. The resulting reaction mixture was poured into water (50 ml) and extracted with EtOAc (3 x 50 ml). The combined organic layer was washed with brine solution (50 ml), dried over Na2SC>4, filtered and concentrated under reduced pressure. The resulting residue was purified by column chromatography (2.2% MeOH in DCM) yielding 6-(lH-pyrazol-4-yl) isoquinolin-3 -amine (0.1 1 g, 0.52 mmol). LCMS: Method A, 1.47 min, MS: ES+ 211.18.
tert-butyl 2-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)propanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
59%
With caesium carbonate; In N,N-dimethyl-formamide; for 120h;
To 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (2.5 g, 12.88 mmol) dissolved in dimethylformamide (30 mL), cesium carbonate (5.25 g, 16.1 mmol) was added, followed by tert-butyl 2-bromopropanoate (2.83 g, 13.52 mmol), and the reaction mixture was stirred for 5 d. The reaction mixture was partitioned between brine and ethyl acetate. The organic layer was washed twice with brine, dried with sodium sulfate, filtered, and concentrated in vacuo. Thecrude residue was purified by silica gel chromatography (eluting with hexanes and ethyl acetate) to provide the title compound (2.43 g, 7.54 mmol, 59%). LCMS M/Z (M+H) 323.
17.6%
With sodium hydride; In N,N-dimethyl-formamide; at 20℃;Inert atmosphere;
To a solution of 4-(4,4,5,5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole (1.0 g, 5.2 mmol) in DMF (25 mL) was added sodium hydride (0.33 g, 8.2 mmol) in small portions under a stream of nitrogen. tert-Butyl 2-bromopropanoate (2.2 g, 10 mmol) was added and the reaction mixture was stirred at room temperature overnight. The reaction mixture was poured into water and the aqueous layer was extracted with EtOAc. The combined organic extracts were washed with water, brine, dried over MgSO4 and concentrated in vacuo. The residue was purified over silica gel (0 to 30% EtOAc in CH2C12) to afford tert-butyl 2-(4-(4,4,5,5- tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazol- 1 -yl)propanoate (0.30 g, 17.6 % yield).
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 96h;
To a stirred solution of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (CAS 269410-08-4; 200 mg, 1.03 mmol, 1 eq.) in DMF (6 mL) are added fert-butyl 2-bromopropanoate (180 pL, 1.08 mmol, 1.05 eq.), K2CO3 (150 mg, 1.08 mmol, 1.05 eq.) and the reaction mixture is stirred at RT for 4 h. Tert-butyl 2-bromopropanoate (17 pL, 0.1 mmol, 0.1 eq.), K2CO3 (14 mg, 0.1 mmol, 0.1 eq.) are added and the reaction mixture is stirred at RT for 4 days. The mixture is concentrated in vacuo, water and DCM are added. The mixture is extracted with DCM, dried by filtration over hydrophobic column and concentrated in vacuo to afford the expected product.
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,4-dioxane; water; for 14h;Heating;
(4-Chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate (10 g; Formula V), water (50 mL), and potassium carbonate (15.5 g) were added into a reaction vessel at ambient temperature. 4-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazole (8.7 g; Formula VIII), 1,4-dioxane (100 mL), and tetrakis(triphenylphosphine)palladium(0) (0.08 g) were added to the reaction mixture. The reaction mixture was heated to a temperature of 80C to 85C, and then stirred at the same temperature for 14 hours. The progress of the reaction was monitored by thin layer chromatography. On completion, ethyl acetate (100 mL) was added to the reaction mixture. The contents were stirred for 1 hour,then filtered through a Hyflo, and then washed with ethyl acetate (40 mL). The organic layer was separated, and then concentrated under reduced pressure to obtain [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3- d]pyrimidin-7-yl]methyl pivalate. Yield: 82.27%
With n-Bu3MgLi; In tetrahydrofuran; at -20 - -10℃;Large scale;
In the reaction kettle, adding tetrahydrofuran 4.5 kg and 1 - BOC - 4 - brompyrazole (2.01 kg, 9.2 muM), stirring 0.5 hours, cooling to -20 C, maintain -10 C -0 C dropwise 3.2molBu3MgLi [preparation method: 1.0eq butyl magnesium chloride in -10 C -0 C lower drop by adding 2.0 equivalent butyl lithium in], TLC detection reaction, maintain the exchange completion -10 C -0 C dropwise dimethylamine fundamental frequency that mellow boron ester (1.61 kg, 9.4 muM) dissolved in 1 kg of the mixed solution in tetrahydrofuran, the completion of the dropping, thermal insulation 2 hours, natural heating stirring overnight. The control temperature of not more than 30 C after adding glacial acetic acid, after detecting the protecting group removal, stop stirring filtration, the mother liquor recovered, solid add triethylamine (1.01 kg, 10 muM) and ethyl acetate 8 kg after, heating to reflux reaction, the proportion of internal standard detecting product is not increased when, after lowering the filtering, the filtrate obtained after the distillation is a kind of white solid, by adding heptane cooling to 0 C beating, filtering, 50 - 60 C vacuum drying to obtain white solid pyrazole -4 - boric acid frequency that alcohol ester 1.36 kg, GC: 99.1%, HNMR consistent with the literature value, yield 76%.
NaH 60percent dispersion in mineral oil (50.0 mg, 1.24 mmol) was suspended in DMF (2 mL) followed by the addition of a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)-1H-pyrazole (200 mg, 1.03 mmol) in DMF (550 muL). The resulting mixture was stirred at r.t. for 1hour. Iodomethane (132 muL, 1.65mmol) was added dropwise and stirring was continued for 2 days. Water was added and the reaction mixture was extracted with EtOAc. The organic layer was washed with water and brine. Dried MgSO4, filtered and concentrated in vacuo. The product was purified by flash chromatography (dry packing) on silica gel using a gradient 0 to 30percent EtOAc in hexanes and afforded the title compound (62.7 mg, 0.28 mmol, 27percent) as a yellow oil.
4-(di-tert-butylphosphino)-N,N-dimethylaniline-dichloropalladium[ No CAS ]
[ 62150-45-2 ]
[ 622-95-7 ]
4-[1-(4-chlorobenzyl)-1H-pyrazol-4-yl]pyridine-2-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium carbonate;
Step 1. 4-[1-(4-Chlorobenzyl)-1H-pyrazol-4-yl]pyridine-2-carbonitrile 4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.60 g, 3.1 mmol, Aldrich) in DMF (16 mL) was treated with 1-(bromomethyl)-4-chlorobenzene (0.70 g, 3.4 mmol, Aldrich) and K2CO3 (1.3 g, 9.3 mmol). After stirring for 2 hours, the mixture was partitioned between water and EtOAc. The organic layer was washed with water, followed by brine, dried over Na2SO4, filtered, and concentrated. The crude product (0.90 g, 2.8 mmol) was combined with <strong>[62150-45-2]4-bromopyridine-2-carbonitrile</strong> (0.45 g, 2.4 mmol, Synthonix), CsF (1 g, 6 mmol), and 4-(di-tert-butylphosphino)-N,N-dimethylaniline-dichloropalladium (2:1) (0.15 g, 0.22 mmol, Aldrich) in 1,4-dioxane (6 mL, 70 mmol) and water (1 mL, 70 mmol). The mixture was degassed and heated to 100° C. for 10 minutes. Upon cooling to room temperature, the mixture was partitioned between water and EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The product was purified by flash chromatography, eluting with a gradient from 0-70percent EtOAc in hexanes. Yield: 0.38 g, 44percent.
With caesium carbonate; In N,N-dimethyl-formamide; at 20℃; for 72h;
2,2-Difluoroethyl trifluoromethanesulfonate (1 .00 g, 4.67 mmol) was added to a mixture of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (0.91 mg, 4.67 mmol) and caesium carbonate (3.04 g, 9.34 mmol) in dry N,N-dimethylformamide (18 mL). The resulting mixture was stirred at room temperature for 72 h. The reaction mixture was diluted with ethyl acetate (150 mL) and washed with brine (3x100 mL). The organic layer was dried with sodium sulfate and concentrated in vacuo. Purification by flash column chromatography (Method L7; 12 g; heptane, 10%-30% ethyl acetate) afforded 0.44 g (1.68 mmol; 36% of theory) of the title compound.GO-MS (Method L9): R1 = 3.58 mm; mlz = 258 M1H NMR (300 MHz, Ohloroform-d, Method M2) 6 7.83 (s, 1H), 7.76 (s, 1H), 6.09 (tt, J = 55.5,4.3 Hz, 1H), 4.47 (td, J = 13.5, 4.3 Hz, 2H), 1.32 (s, 12H).
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃;
4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H-pyrazole (1.00 g) was dissolved in 9.9 m L DMF and potassium carbonate (2.14 g) and 2,2-difluoroethyl trifluoromethanesulfonate (970 p1) were added. This mixture was stirred at 80 D overn ight. The reaction mixture was diluted with ethyl acetate and water. The layers were seperated and the aqueous layer was extracted withethyl acetate. The combined organic layers were dried using a water resistant filter. The filtrate was concentrated under reduced pressure. The crude product was used without further purification.Yield: 1.8 g of 69% pure target compound.LC-MS (Method 2): R = 0.82 mm; MS (ESIpos): m/z = 259 [M+HJ 1H-NMR (400 MHz, DMSO-d6)oe [ppm] = 1.25 (s, 12H), 4.64 (td, 2H), 6.17- 6.53(m, IH), 7.65 (s, IH), 7.99 (s, 1H)
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (300 mg, 1.55 mmol) was dissolved in DMF (5 mL) at 0 C under Ar. NaH (60% in oil, 40.8 mg, 1.70 mmol) was added portionwise, and the reaction mixture was then stirred for 20 minutes at rt. A solution of bromocyclobutane (209 mg, 1.55 mmol) in 1 mL of DMF was added, and stirring was continued 2 h at rt. The reaction mixture was quenched with saturated aq. NH4Cl, and then extracted 3x with EtOAc, washed with brine, dried with Na2SO4, filtered and concentrated. The product was purified by silica gel chromatography to provide pyrazole boronate 20A (40 mg, 5.21 %) . MS (ESI): m/z 249.0 (M+H).
4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (CAS 269410-08-4, 1.20 g, 6.18 mmol) was dissolved in DMF (23 mL) and sodium hydride (0.81 g, 18.6 mmol) was added at room temperature (rt). After 10 min, cyclobutyl bromide (2.51 g, 18.6 mmol) was added and the mixture stirred for 2 g at 50 C and further 16 h at rt. The mixture was partitioned between water and ethyl acetate, extracted with ethyl acetate, and the combined organic layers washed with water, dried (Na2SC>4) and concentrated. The crude product (1.14 g, 87% purity, 64% yield) was used without further purification.
H3Ethyl trifluoromethanesulfonate (1.00 g, 5.61 mmol) was added to a mixture of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.99 g, 5.10 mmol) and caesium carbonate(3.46 g, 10.62 mmol) in dry N,N-dimethylformamide (20 mL) at 0°O. After stirring for 30 mm the ice-water bath was removed. The reaction mixture was stirred at room temperature overnight. lodoethane (0.80 g, 5.10 mmol, 0.41 ml) was added and the reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with ethyl acetate (150 mL) and washed with brine (3x100 mL). The organic layer was dried with sodium sulfate and concentrated in vacuo to afford 1.25 g (4.50 mmol, 68percent of theory) of the title compound. GO-MS (Method L9): R1 = 3.78 mm; mlz = 222 M1H NMR (300 MHz, Ohloroform-d, Method M2) 67.80-7.76 (m, 1H), 7.70 (s, 1H), 4.19 (q, J =7.3 Hz, 2H), 1.47 (t, J = 7.3 Hz, 3H), 1.32 (s, 12H).
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃; for 8h;
3- (Bromomethyl) pyridine (2.67 g, 15.5 mmol)And 4-pyrazole boronic acid pinacol ester(3.01 g, 15.5 mmol) was dissolved in DMF (20 mL)Potassium carbonate (2.2 g, 16 mmol) was added to the system, and the mixture was heated to 80 C. for 8 h.The reaction mixture was poured into water (50 mL) and extracted with ethyl acetate (50 mL × 3)Saturated brine (20 mL), dried over anhydrous sodium sulfate, and the solvent was removed.The residue was subjected to column chromatography (eluent: PE / EtOAc (v / v) = 1 / 1.5)This gave 280 mg of a pale yellow oil, yield: 6.33%.
With potassium carbonate; In N,N-dimethyl-formamide; at 60℃; for 16h;
To a solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.5 g, 2.58 mmol, 1 eq) in DMF (6 mL) was added K2CO3 (712.26 mg, 5.15 mmol, 2 eq) and ethyl 2-chloro-2,2-difluoro-acetate (490.21 mg, 3.09 mmol, 392.16 uL, 1.2 eq). The mixture was stirred at 60 C for 16 h. The reaction mixture was poured into H2O 10 mL, and extracted with EtOAc (10 mLx3). The combined organic layers were washed with brine (10 mLx2), dried over Na2SO4, filtered and concentrated under reduced pressure to afford the title compound (0.4 g, crude) as yellow oil, which was used into the next step without further purification.
1-(2-(4-(4-methoxyphenyl)-1H-pyrazol-1-yl)ethyl)-4-methylpiperazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65%
General procedure: 1) (Cyanomethylene)tributylphosphorane (262 muL, 1.00 mmol) was added to a solution of (tetrahydrofuran-3-yl)methanol (51 mg, 0.50 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (97 mg, 0.5 mmol) in degassed 1,4-dioxane (2 mL) sealed in a microwave tube at rt under nitrogen. The solution was heated to 150°C for 30 min in the microwave reactor and cooled to rt. 2) 1-Bromo-4-methoxybenzene (94 mg, 0.50 mmol), potassium carbonate (207 mg, 1.50 mmol) and [1,1?-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with CH2Cl2 (40.8 mg, 0.05 mmol) were added to the solution. The tube was sealed, evacuated and backfilled with nitrogen. Degassed water (1 mL) was added under nitrogen. The resulting mixture was stirred at 120°C for 20 min. The reaction mixture was diluted with EtOAc (25 mL) and water (15 mL), the layers were separated, and the aqueous layer was extracted with EtOAc (15 mL). The combined organic layers were washed with saturated brine (15 mL). The organic layer was dried with MgSO4, filtered and evaporated to afford the crude product. The crude product was purified by preparative HPLC (Waters XSelect CSH C18 ODB column, 5mu silica, 30mm diameter, 100mm length), using decreasingly polar mixtures of water (containing 1percent by volume NH3OH (28-30percent in H2O)) and MeCN as eluents to afford 4-(4-methoxyphenyl)-1-((tetrahydrofuran-3-yl)methyl)-1H-pyrazole (89mg, 69percent) as a beige solid.
4-(2-(4-(4-methoxyphenyl)-1H-pyrazol-1-yl)ethyl)thiomorpholine 1,1-dioxide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
69%
General procedure: 1) (Cyanomethylene)tributylphosphorane (262 muL, 1.00 mmol) was added to a solution of (tetrahydrofuran-3-yl)methanol (51 mg, 0.50 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (97 mg, 0.5 mmol) in degassed 1,4-dioxane (2 mL) sealed in a microwave tube at rt under nitrogen. The solution was heated to 150°C for 30 min in the microwave reactor and cooled to rt. 2) 1-Bromo-4-methoxybenzene (94 mg, 0.50 mmol), potassium carbonate (207 mg, 1.50 mmol) and [1,1?-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with CH2Cl2 (40.8 mg, 0.05 mmol) were added to the solution. The tube was sealed, evacuated and backfilled with nitrogen. Degassed water (1 mL) was added under nitrogen. The resulting mixture was stirred at 120°C for 20 min. The reaction mixture was diluted with EtOAc (25 mL) and water (15 mL), the layers were separated, and the aqueous layer was extracted with EtOAc (15 mL). The combined organic layers were washed with saturated brine (15 mL). The organic layer was dried with MgSO4, filtered and evaporated to afford the crude product. The crude product was purified by preparative HPLC (Waters XSelect CSH C18 ODB column, 5mu silica, 30mm diameter, 100mm length), using decreasingly polar mixtures of water (containing 1percent by volume NH3OH (28-30percent in H2O)) and MeCN as eluents to afford 4-(4-methoxyphenyl)-1-((tetrahydrofuran-3-yl)methyl)-1H-pyrazole (89mg, 69percent) as a beige solid.
With tetrabutylammomium bromide; potassium carbonate; palladium dichloride; In N,N-dimethyl-formamide; at 130℃; for 16h;Inert atmosphere;
Under a nitrogen atmosphere, 6.28 g of bromobenzene, 7.76 g of pyrazole-4-boronic acid pinacol ester, 0.1 g of PdCl2, 7.10 g of K2CO3, and 2.15 g of tetrabutylammonium bromide were added to a 500 ml four-necked flask. In DMF, heat at 130 C, stir for 16 hours, passThe mixture was controlled by HPLC, cooled to room temperature, filtered, evaporated under reduced pressure, dissolved in dichloromethane, washed with water and concentrated to give white product 5.56 g.The yield is 98.0%,The HPLC purity was 98.0%.
1-[(3,3-difluorocyclobutyl)methyl]-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With di-tert-butyl (E)-azodicarboxylate; triphenylphosphine; In toluene; at 80℃; for 16h;Inert atmosphere;
To a solution of <strong>[681128-39-2](3,3-difluorocyclobutyl)methanol</strong> (770 mg, 6.31 mmol) in toluene (25mL) was added 4-(4,4,5, 5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1H-pyrazole (1.3 g, 6.62 mmol),PPh3 (2.48 g, 9.46 mmol) and DBAD (2.2 g, 9.46 mmol), the mixture was stirred at 80 C for 16 hunder nitrogen atmosphere (balloon). The mixture was concentrated and diluted with EtOAc (60 mL). The mixture was washed with water (20 mL x 3), brine (20 mL), dried over sodium sulfate, filtered and the filtrate was concentrated in vacuum. The residue was purified by combiflash (050% petroleum ether in ethyl acetate) to give the title compound as a white solid. MS: 299(M+ 1)
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; for 48h;
4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (200 mg, 1.01 mmol) and triphenylphosphine (397 mg, 1.52 mmol) were added in a 10 mL round-bottomed flask. THF (2.00 mL) was added followed by the <strong>[98730-77-9]1-(hydroxymethyl)cyclopropanecarbonitrile</strong> (107 muL, 1.26 mmol) and the diisopropyl azodicarboxylate (304 muL, 1.52 mmol). The solution was stirred for 48 hours. Upon completion, the solvent was removed in vacuo to give the title compound (1050 mg, crude) as a pale yellow clear oil, which was used?as is? in subsequent steps.
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; sodium carbonate; In 1,4-dioxane; water; at 85℃; for 16h;Inert atmosphere;
Embodiment 60 2-(2,6-Dimethoxy-4-(2-(2-methylbiphenyl-3-yl)-1H-pyrazol-1-yl)benzylami no)-3-hydroxypropanoic acid 60 Synthetic route Synthesis of compound 60-c 2-Bromo-6-chlorotoluene (2.05g, 10.0mmol) and pyrazole boronic acid ester (2.1g, 11.0mmol) were dissolved in a mixed solvent of 1,4-dioxane (50mL) and water (5mL), followed by addition of [1,1'-bis(diphenylphosphino)ferrocene]palladium dichloride (0.731g, 1.0mmol) and sodium carbonate (3.18g, 30.0mmol). After the reaction system was purged three times with nitrogen, the reaction solution was heated to 85C and stirred for 16 hours. Then the reaction solution was cooled to room temperature, diluted with ethyl acetate (100mL), washed successively with water (50mL x 3) and saturated brine (50mL). The organic phase was dried over anhydrous sodium sulfate and evaporated under reduced pressure. The residue was purified by silica gel column chromatography (petroleum ether: ethyl acetate = 4:1 to 2:1) to give compound 60-c (1.4g, yield 73%). LC-MS (ESI): m/z = 193 [M+H]+.
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 24h;
4-pyrazole boronic acid pinacol ester (3.9 g, 20.0 mmol) was dissolved in 30.0 ml of DMF at room temperature.Potassium carbonate (5.5 g, 40.0 mmol) and <strong>[3395-91-3]methyl 3-bromopropionate</strong> (4.0 g, 24.0 mmol) were added.After stirring at room temperature for 24 hours, TLC was monitored (the ratio of petroleum ether to ethyl acetate was 2:1) still left the reaction starting material. 30 ml of water was added to the reaction mixture, and the mixture was stirred until a clear solution.Then extracted with ethyl acetate (50 ml × 2).The extract was washed with saturated brine (50 ml×2) and dried over anhydrous sodium sulfate.Filtered, and the filtrate was concentrated under reduced pressure.The residue was isolated (5:1 by volume ratio of petroleum ether to ethyl acetate) to obtain intermediate (a) 3.5 g, yield 63%;
tert-butyl 6-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)methyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 0 - 20℃;
A flask was charged with 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.5 g, 2.58 mmol), <strong>[622867-52-1]tert-butyl 6-(hydroxymethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate</strong> (0.339 g, 1.288 mmol), triphenylphosphine (0.743 g, 2.83 mmol), and THF (12 ml). The solution was cooled to 0 C. and DIAD (0.601 ml, 3.09 mmol) was added dropwise. The reaction mixture was stirred overnight at room temperature. The mixture was diluted with ethyl acetate and washed with water, dried and concentrated. The product was purified by column chromatography eluting with Hexane/EtOAc (max. EtOAc 60%) to afford the product. LCMS calculated for C24H35BN3O4(M+H)+: m/z=440.3; found 440.3.
60% sodium hydride (0.873 g, 36.4 mmol) was added to the solution of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (3.482 g, 17.9 mmol) in N,N-dimethylformamide (30 mL) under an ice bath, then the mixture solution was stirred for 10 min. Then oxetan-3-yl methanesulfonate (2.80 g, 18.4 mmol) was added to the above reaction solution, then the reaction solution was stirred at 100 C. for 21 hrs. After the reaction was completed, the mixture solution was cooled to room temperature, diluted with ethyl acetate (15 mL), washed with a saturated ammonium chloride solution (15 mL), and then washed with a saturated brine (15 mL). The organic layer was dried and concentrated, and then separated by column chromatography [eluent: dichloromethane/methanol] to obtain 1-(oxetan-3-yl)-4-(4,4,5,5-tetramethyl)-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.587 g, yield 13%). MS m/z (ESI): 251 [M+H]+.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping