* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
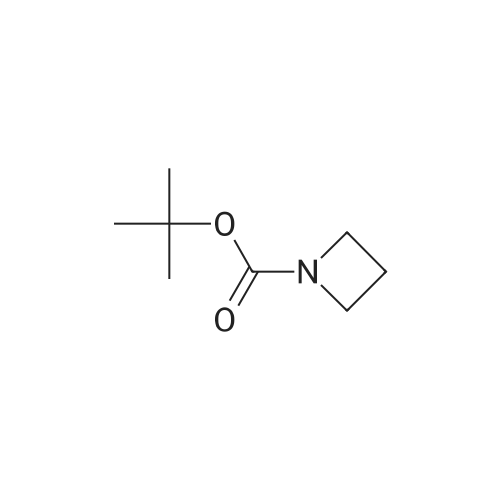
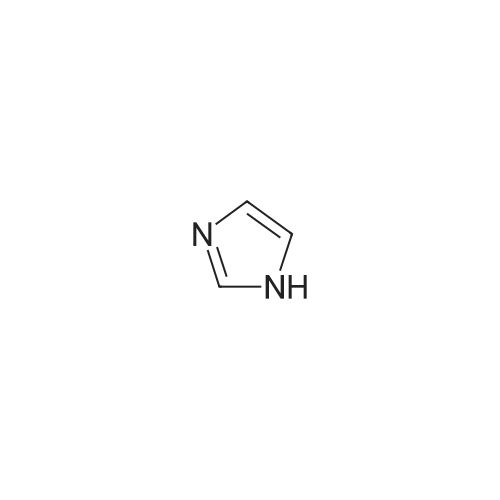
With 1H-imidazole; iodine; triphenylphosphine In toluene at 100℃; for 1 h;
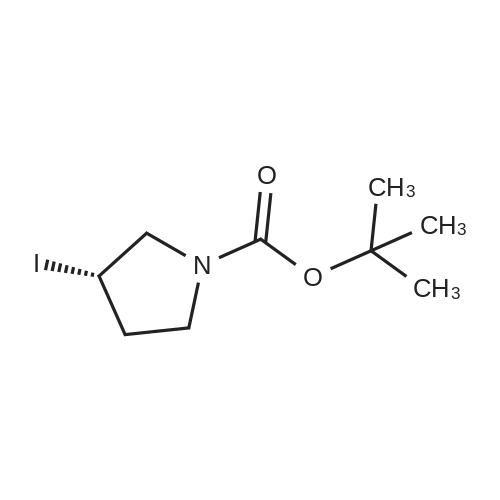
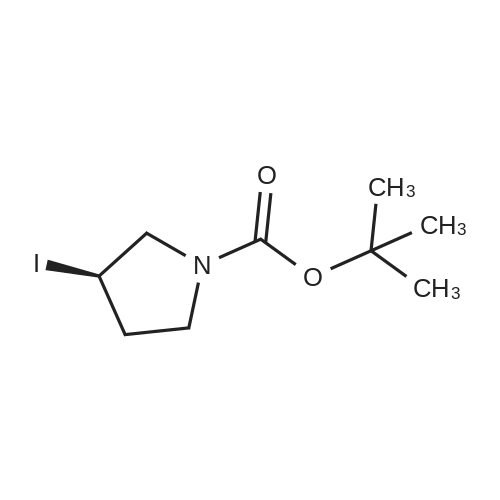
STAP B tert-butyl 3-iodoazetidine- 1 -carboxylate. [Chem.10][0206] To a solution of tert-butyl 3-hydroxyazetidine-l-carboxylate (3.35 g, 19.3 mmol) in toluene (200 mL) was added imidazole (3.95 g, 58.0 mmol), triphenylphosphine (10.1 g, 38.7 mmol) and iodine (7.36 g, 29.0 mmol). The mixture was heated for 1000C for Ih, cooled to room temperature, then poured into sodium bicarbonate aqueous solution (30 mL). Excess triphenylphosphine was destroyed by addition of iodine until iodine coloration persisted in organic layer. The organic layer was separated and washed with saturated sodium thiosulfate aqueous solution, dried over sodium sulfate. The crude product was purified by silica gel column chromatography (hexane - ethyl acetate = 9:1 to 1:1) to gave the title compound (5.42 g, 99percent) as clear oil.[0207] MS (ESI) m/z 284 (M+ 1)+.
95%
With 1H-imidazole; iodine; triphenylphosphine In toluene at 100℃; for 1 h;
A solution of 3-hydroxy-azetidine-i-carboxylic acid tert-butyl ester (3.35 g, 19.34 mmol) in toluene (200 ml) was treated with imidazole (3.95 g, 58.01 mmol), triphenyl- phosphine (10.14 g, 38.65 mmol) and I2 (7.36 g, 28.99 mmol). The mixture was heated at 1000C for 1 h, cooled down to room temperature and next poured into saturated aqueous NaHCO3 (30 ml). Excess triphenylphosphine was destroyed by addition of iodine until I2 coloration persisted in organic layer. The latter was washed with 5percent aqueous Na2S2O3, dried over Na2SO4 and evaporated. Purification of the crude product by flash column chromatography (heptane:ethyl acetate, 2:1) provides the title com.not. pound (5.19 g, 95percent) as a light yellow oil. MS (ESI+) m/z = 227.9 [M-ffiu+H]+1H NMR (400 MHz, CDCI3) : δ (ppm) 1.44 (s, 9H), 4.29 (dd, J = 10.4, 5.4 Hz, 2H), 4.47 (m, 1 H), 4.64 (dd, J = 9.5, 8.0 Hz, 2H).
95%
Stage #1: With 1H-imidazole; iodine; triphenylphosphine In toluene at 100℃; for 1 h; Stage #2: With sodium hydrogencarbonate In water; toluene
A solution of 3-hydroxy-azetidine-1-carboxylic acid tert-butyl ester (3.35 g, 19.34 mmol) in toluene (200 ml) was treated with imidazole (3.95 g, 58.01 mmol), triphenyl- phosphine (10.14 g, 38.65 mmol) and I2 (7.36 g, 28.99 mmol). The mixture was heated at 1000C for 1 h, cooled down to room temperature and subsequently poured into a saturated aqueous solution of NaHCO3 (30 ml). Excess triphenylphosphine was destroyed by addition of iodine until b coloration persisted in organic layer. The latter was washed with an aqueous solution of Na2S2ψ3 (5 percent strength), dried over Na2SU4, filtered and concentrated in vacuo. Purification of the residue by flash column chromatography (heptane: EtOAc, 2:1 ) provides the title compound (5.19 g, 95 percent) as a light yellow oil.MS (ESI+) : m/z = 227.9 [M-tBu+H]+ ; 1H-NMR (400 MHz, CDCI3) : δ = 1.44 (s, 9H), 4.29 (dd, J = 10.4, 5.4 Hz, 2H), 4.47 (m, 1 H), 4.64 ppm (dd, J = 9.5, 8.0 Hz, 2H).
93%
With 1H-imidazole; iodine; triphenylphosphine In toluene at 100℃; for 1 h;
A solution of ieri-butyl 3-hydroxyazetidine-l-carboxylate (25-3) (3.5 g, 0.02 mol) in toluene (200 mL) was treated with imidazole (4.08 g, 0.06 mol), PPh3 (0.6 g, 0.04 mol), and I2 (7.62 g, 0.03 mol). The mixture was heated at 100°C for 1 h and cooled down to room temperature. It was then poured into saturated NaHCC^ solution (30 mL). Excess PPh3 was destroyed by addition of iodine until I2 coloration persisted in organic layer. The mixture was washed with 5percent Na2SC>3 solution, dried over Na2S04, and evaporated in vacuo. The residue was purified on silical gel column to give 199a (5.31 g, 93percent). MS: [M+H]+ 284.
93%
With 1H-imidazole; iodine; triphenylphosphine In toluene at 100℃; for 1 h;
Example 206a tert-Butyl 3-Iodoazetidine-1-carboxylate 206a A solution of tert-butyl 3-hydroxyazetidine-1-carboxylate (3.5 g, 0.020 mol) in toluene (200 mL) was treated with imidazole (4.08 g, 0.060 mol), triphenylphosphine (0.60 g, 0.040 mol), and iodine (7.62 g, 0.030 mol). The mixture was heated at 100°C for 1 h. It was then cooled to room temperature and poured into saturated NaHCO3 solution (30 mL). Excess triphenylphosphine was destroyed by addition of iodine until iodine coloration persisted in organic layer. The mixture was washed with 5percent Na2SO3 solution, dried over Na2SO4, and evaporated in vacuo. The residue was purified by silica-gel column chromatography to afford 206a (5.31 g, 93percent). MS-ESI: [M+H]+284.
91%
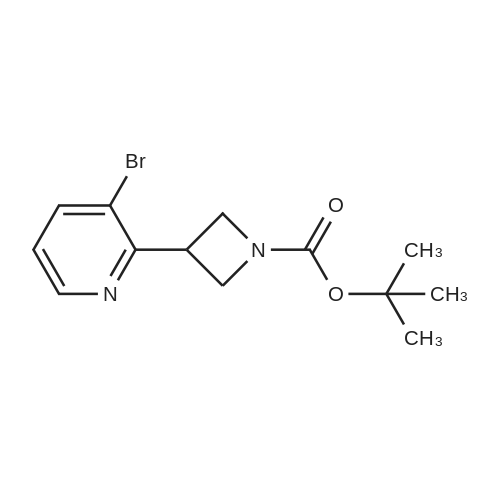
With 1H-imidazole; iodine; triphenylphosphine In toluene at 110℃; for 4 h;
To a solution of tert-butyl 3-hydroxyazetidine-1-carboxylate (400 mg, 2.31 mmol) in toluene (23 ml), imidizole (472 mg, 6.93 mmol), triphenylphosphine (1.21 g, 4.62 mmol) and iodine (879 mg, 3.46 mmol) were added successively and the reaction mixture was heated at 110° C. for 4 h. The cooled reaction mixture was quenched with saturated aqueous NaHCO3 and extracted with Et2O (2*). The Et2O layers were combined and the composite was treated with iodine until a persistent brown color occurred and the mixture was stirred at RT overnight. The Et2O solution was treated with saturated aqueous Na2S2O3 until colorless, the phases were split and the organic phase was dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with 0 to 20percent EtOAc in hexane to give Compound 9a (594 mg, 91percent yield) as a clear colorless oil. MS m/z=284.0, (M+H) 1H NMR (CHLOROFORM-d) δ: 4.57 (t, J=8.4 Hz, 2H), 4.36-4.44 (m, 1H), 4.18-4.26 (m, 2H), 1.37 (s, 9H).
3.23 g
With 1H-imidazole; iodine; triphenylphosphine In toluene at 100℃; for 1.75 h;
(1) To a solution of Compound 1 (2.0 g) in toluene (115 mL) was added imidazole (2.36 g), triphenylphosphine (6.06 g), and iodine (4.4 g), and the mixture was stirred at 100°C for 1 hour and 45 minutes. The reaction mixture was cooled to room temperature, a saturated aqueous solution of sodium hydrogen carbonate was added thereto, stirred, then iodine was added thereto, stirred, and extracted with ethyl acetate. The resultant organic layer was dried, and concentrated under reduced pressure. The residue was purified with silica gel column chromatography (hexane:ethyl acetate=100:0-75:25) to give Compound 2 (3.23 g) as a colorless liquid.
With potassium iodide In N,N-dimethyl-formamide at 110℃; for 16 h;
Potassium iodide (12.9 g, 77.7 mmol) and C6 (6.5 g, 26.0 mmol) were combined in DMF (40 mL). The reaction mixture was stirred at 110° C. for 16 h, then concentrated in vacuo, diluted with water, and extracted with EtOAc. The combined organic layers were washed with water, then washed with saturated aqueous sodium chloride solution and dried over magnesium sulfate. Filtration and removal of solvent in vacuo gave a residue, which was purified by silica gel chromatography (Eluant: 4:1 heptane:EtOAc) to afford C7 as a solid. Yield: 6.2 g, 21.9 mmol, 84percent. LCMS m/z 284.0 (M+1). 1H NMR (400 MHz, CDCl3) δ 1.43 (s, 9H), 4.28 (m, 2H), 4.46 (m, 1H), 4.64 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 2.57, 28.27, 61.49, 80.09, 155.52.
83%
With potassium iodide In N,N-dimethyl-formamide at 110℃; for 16 h;
Tert-butyl 3-((methylsulfonyl)oxy)azetidin-1-carboxylate (2.67 g, 10.62 mmol) was dissolved in 20mL N,N-dimethyl formamide, and potassium iodide (5.3 g, 31.93 mmol) was added. The resultant mixture was heated to 110°C and reacted for 16 h. After the reaction, the solvent was removed by rotary evaporation, and 50mL water was added. After extraction with ethyl acetate (3*30mL), the organic phases were combined, dried with anhydrous sodium sulphate, and filtrated. The solvent was removed by rotary evaporation, and the residue was subjected to silica gel column chromatography (petroleum ether: ethyl acetate=4:1) to obtain the product (2.5g, yield: 83percent).
72%
With potassium iodide In N,N-dimethyl-formamide at 110℃; for 7 h;
Preparation 58 tert-Butyl 3-iodoazetidine-i-carboxylate; tert-Butyl 3-(methylsulfonyloxy)azetidine-1-carboxylate (Preparation 57, 8Og, 0.318 mol) and potassium iodide (159g, 0.96 mol) were mixed in dimethylformamide (500 ml_). The reaction mixture was stirred at 11O0C for 7 hours. The solvent was evaporated and the resulting residue was suspended in water (1 L). The product was extracted with ethyl acetate (80OmL). The combined extracts were washed with water, dried using anhydrous sodium sulfate and concentrated in vacuo. The residue was purified by column chromatography on silica gel eluting with ethyl acetate: hexane (1 :4) to afford the title compound (64.6 g, 72percent). 1H NMR (400 MHz, DMSOd6): δ = 1.38 (s, 9H), 4.05-4.09 (m, 2H), 4.61-4.64 (m, 3H) ppm.
68%
With potassium iodide In dimethyl sulfoxide at 140℃; for 2 h; Inert atmosphere
Production Example 26-3 tert-Butyl 3-iodoazetidine-1-carboxylate Potassium iodide (51.0 g, 307 mind) was added to a solution of tert-butyl 3-((methylsulfonyl)oxy)azetidine-1-carboxylate described in Production Example 26-2 (7.72 g, 30.7 mmol) in dimethylsulfoxide (80 mL) under nitrogen atmosphere at room temperature, and the mixture was stirred at 140° C. for 2 hours. The reaction liquid was diluted with diethyl ether and water. The aqueous layer was extracted with diethyl ether. The combined organic layer was washed serially with an aqueous sodium pyrosulfite solution and a saturated saline solution and then dried over anhydrous sodium sulfate. The drying agent was separated by filtration and then the resultant was concentrated under vacuum. The residue was purified with silica gel column chromatography (n-heptane:ethyl acetate 9:1-1:1) to obtain the title compound (5.91 g, 68percent). 1H-NMR Spectrum (CDCl3) δ (ppm): 1.44 (9H, s), 4.25-4.33 (2H, m), 4.42-4.51 (1H, m), 4.61-4.69 (2H, m).
With potassium iodide In dimethyl sulfoxide; pentane
Preparation 32 1- tert -Butyloxycarbonyl-3-iodoazetidine A mixture of 1-tert-butyloxycarbonyl-3-(methylsulfonyloxy)azetidine (Preparation 31) (28g, 111mmol) and potassium iodide (170g, 1.02mol) in dimethylsulphoxide (250ml) was heated to 140°C and stirred for 2 hours. The reaction mixture was cooled, poured into water (1000ml) and extracted with diethyl ether (x2). The combined organic layers were washed with an aqueous solution of sodium metabisulfite, brine, dried over Na2SO4, filtered and solvent removed under reduced pressure. The residue was purified by column chromatography on silica gel, eluding with a solvent system of ethyl acetate: pentane (1:1, by volume) to give the title compound as a pale yellow oil (19g, 60percent). 1H-NMR (CDCl3): δ = 4.65 (2H, m), 4.50 (1H, m), 4.30 (2H, m), 1.45 (9H, s).
With sodium iodide In tetrahydrofuran; dibutyl ether; acetonitrile at -78 - 24℃;
To a flame-dried 500 mL round bottom flask was added 1-amino-2,3-dibromopropane hydrobromide (17) (7.5 g, 0.025 mol, 1 equiv.) and stirred without solvent to obtain 17 asa fine powder under argon (alternatively, crystals of 17 could be ground to a fine powder by hand before use).Dry THF (75 mL) was added to the flask and cooled to –78 oC. PhLi solution (1.8M in dibutyl ether, 39.8 mL,0.075 mol, 3 equiv.) was slowly added via syringe and the reaction mixture stirred at –78 oC for 2 h. To theresulting mixture was added MeCN (240 mL), NaI (11.4 g, 0.075 mol, 3 equiv.) and Boc2O (11.2 mL, 0.050 mol,2 equiv.) at –78 oC and warmed at rt overnight. The resulting mixture was poured into water (200 mL), washedwith sat. aq. Na2S2O3 (100 mL) and then extracted with ethyl acetate (3 x 150 mL). The combined organiclayers were washed with brine (100 mL), dried over Na2SO4, and concentrated. The crude material waspurified by flash chromatography (silica gel, 5–40percent EtOAc in hexanes) to give the desired product 6 (5.8 g,81percent). Physical State: light yellow oil; Rf = 0.55 (2:8 EtOAc/hexanes, vis. UV); 1H NMR (500 MHz, CDCl3): δ 4.67 –4.60 (m, 2H), 4.50 – 4.43 (m, 1H), 4.33 – 4.24 (m, 2H), 1.44 (s, 9H); 13C NMR (126 MHz, CDCl3): δ 155.7, 80.3,61.7 (br, 2C), 28.4 (3C), 2.7; HRMS (ESI-TOF): calc’d for C8H14INNaO2 [M+Na+] 305.9967; found 305.9950.
With 1,3-Diiodo-5,5-dimethyl-2,4-imidazolidinedione In 1,2-dichloro-ethane for 3 h; Reflux; Irradiation
EXAMPLE 3; Radical iodo-de-caboxylation induced by TV-iodo amides/V-iodo amideR-COOH *- R-lGeneral procedure[00111] Procedure: A mixture of R-COOH (1 mmol), N-iodo amide (1-4 equiv), and solvent (3-6 mL) was refluxed (Δ) for 1-24 h in the dark (NL) or under irradiation with 500 W tungsten lamp (TL) or under fluorescent room lighting (FL).[00112] Treatment: The reaction mixture was cooled to rt, and washed with aq NaHS03 and NaHC03 to destroy excess of iodination agent and dissolve unreacted carboxylic acid. The organic solution was dried (Na2S04), filtered through short silica or alumina pad and concentrated in vacuo to give iodide R-I. 100113J Purification: Optionally, the iodide R-I was further purified by crystallization (if the iodide is crystalline compound), or rectification (if the iodide is liquid compound). Analytical sample of the product was purified by column chromatography./V-iodoamideAlk-COOH *- Alk-I[00114] A mixture of Alk-COOH (1 mmol), N-iodo amide (1-3 equiv), and solvent (4 mL) was refluxed (Δ) in the dark (NL) or under irradiation with 500 W tungsten lamp (TL), or under fluorescent room lighting (FL).
Reference:
[1] Patent: WO2011/154953, 2011, A1, . Location in patent: Page/Page column 28-30
[2] Advanced Synthesis and Catalysis, 2011, vol. 353, # 9, p. 1438 - 1442
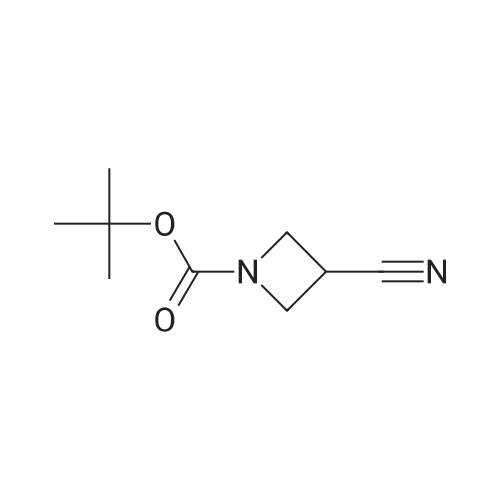
To a 100 mL round bottom flask was added tert-butyl 3-iodoazetidine-1-carboxylate (6) (6.54 g, 0.0231 mol, 1 equiv.) and DMSO (25 mL). NaCN (2.3 g, 0.0462 mol, 2equiv.) was added to the solution in one portion and stirred at 130 oC for 6 h. The resulting mixture was cooledto rt, poured into water (200 mL), and extracted with diethyl ether (5 x 200 mL). The combined organicextracts were washed with brine (50 mL), dried over Na2SO4 and concentrated. The crude compound waspurified by flash chromatography (silica gel, 5–50percent EtOAc in hexanes) to give the desired product 22 (3.27 g,78percent). Physical State: off-white solid (mp 78–80 oC); Rf = 0.40 (2:8 EtOAc/hexanes, vis. KMnO4); 1H NMR (500MHz, CDCl3): δ 4.23 – 4.11 (m, 4H), 3.38 (tt, J 8.9, 6.3 Hz, 1H), 1.43 (s, 9H); 13C NMR (126 MHz, CDCl3): δ 155.6,119.6, 80.8, 52.6 (br, 2C), 28.4 (3C), 17.2.
Stage #1: With chloro-trimethyl-silane; ethylene dibromide In tetrahydrofuran at 20 - 80℃; for 2.5 h; Inert atmosphere Stage #2: With tris-(dibenzylideneacetone)dipalladium(0); trifuran-2-yl-phosphane In tetrahydrofuran at 55℃; for 3 h;
(a) tert-Butyl-3-(4-nitrophenyl) azetidine- 1-carboxylate (16)1,2-Dibromoethane (0.146 mL, 1.69 mmol) was added to a vigorously stirred suspension of zinc dust (0.901 g, 13.8 mmol) in THF (3.5 mL) under a nitrogen atmosphere and the resulting suspension heated at 80 00 for 10 minutes. Trimethylsilyl chloride (0.202 mL, 1.59 mmol) in THF (1.75 mL) was added at roomtemperature and after stirring for 4 minutes a solution of tert-butyl 3-iodoazetidine-1- carboxylate (3.00 g, 10.6 mmol) in THF (3.5 mL) was added dropwise over a period of 15 minutes. The resulting mixture was stirred at room temperature for 2 hours then Pd2(dba)3 (0.155 g, 0.170 mmol) and tri-2-furylphosphine (0.143 g, 0.615 mmol) were added followed by 1-iodo-4-nitrobenzene (2.90 g, 11.7 mmol) in THF (18 mL). Theresulting mixture was heated at 55 00 for 3 hours then quenched at room temperature with a saturated aqueous sodium chloride solution (15 mL). The aqueous phase was extracted with DCM (2 x 15 mL) then the combined organic fractions were dried (magnesium sulfate), filtered and evaporated in vacuo. The residue was purified using silica gel column chromatography (CombiFlash Rf, 40 gSi02 Cartridge, 10-40percent EtOAc in cyclohexane) to give the title compound 16 as anorange oil (2.14 g, 72percent); 1H NMR (300 MHz, CDCI3) O 8.24 (dd, J= 6.8, 1.9 Hz, 2H),7.51 (d, J= 8.6 Hz, 2H), 4.41 (t, J= 8.7 Hz, 2H), 3.98 (dd, J= 8.5, 5.7 Hz, 2H), 3.89-3.81 (5, 1H), 1.49 (5, 9H).
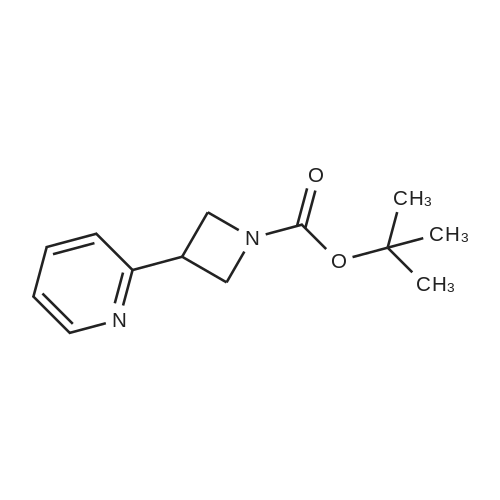
Stage #1: tert-butyl-3-iodoazetidine-1-carboxylate With lithium chloride; zinc In tetrahydrofuran at 110℃; Flow reactor;
Stage #2: 2-bromo-pyridine In tetrahydrofuran at 60℃; Flow reactor;
With bismuth(III) oxide; N-ethyl-N,N-diisopropylamine; para-thiocresol; In dichloromethane;Irradiation;
a), iodo compound 9 (30mg),Bismuth oxide (2.5mg, 5mol%),P-methylthiophenol (39.4 mg, 3 equiv) was weighed into a 5 mL reaction flask.Then add dichloromethane (1ml)And N,N-diisopropylethylamine (55.4 muL, 3 equiv),Reacted under 1W blue LED light,The reaction was terminated after the iodo compound 9 was consumed.Rotating to remove the reaction solvent and volatile substances,Purification by column chromatography gave Compound 9a (14.3 mg, 86%).
TELT-BUTYL 3-IODOAZETIDINE-1-CARBOXYLATE (EP 1176142, pg. 23, ex. 2 (i) ) (2. 0g, 7. 07MMOL) was added to 33% METHYLAMINE in ethanol (45mL) and the reaction mixture heated in a sealed vessel at 100C for 24 hours. The reaction mixture was concentrated in vacuo and the residue partitioned between ethyl acetate and 1 M aqueous sodium hydroxide. The organic layer was separated and washed with brine, dried over magnesium sulphate and concentrated in vacuo. The crude product was purified by column chromatography on silica gel eluting with dichloromethane : methanol : 0.880 ammonia 96: 3.5 : 0.5 to yield the title product. IN NMR (CDC13, 400MHZ) 8 : 1.43 (s, 9H), 1.94 (m, 1 H), 2.41 (s, 3H), 3.49 (m, 1 H), 3.67 (m, 2H), 4.06 (m, 2H). LRMS APCI+ m/z 187 [MH] +
With sodium hydroxide; chloro-trimethyl-silane; zinc;bis(dibenzylideneacetone)-palladium(0); In tetrahydrofuran; water; ethyl acetate;
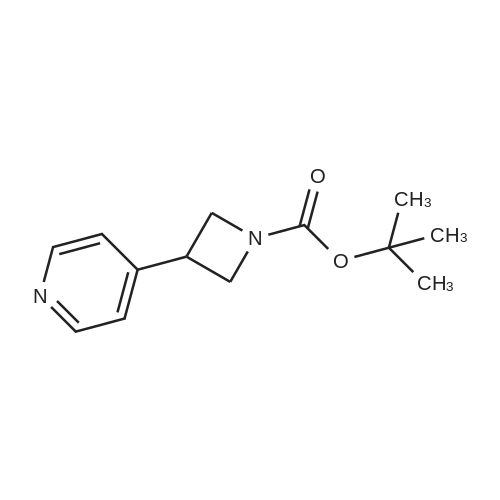
Preparation 33 1- tert -Butyloxycarbonyl-3-(pyridin-4-yl)azetidine 1,2-Dibromoethane (0.07ml, 0.81mmol) was added to a suspension of zinc dust (590mg, 9.07mmol) in tetrahydrofuran (2ml) under nitrogen. The resultant mixture was heated under reflux, to initiate the reaction, for 2 minutes. The reaction mixture was cooled to room temperature then heated under reflux again. After cooling to room temperature, trimethylsilyl chloride (0.10ml, 0.8mmol) was added and the mixture stirred for 30 minutes. A solution of 1-tert-butyloxycarbonyl-3-iodoazetidine (Preparation 32) (2.00g, 7.06mmol) in tetrahydrofuran (5ml) was added portionwise. The temperature of the reaction mixture was maintained between 28-32C during the addition using a cold water bath and then it was stirred at room temperature for 4 hours. A solution of 4-bromopyridine (1.34g, 8.5mmol, obtained from the HCI salt by partitioning the salt between diethyl ether and aqueous sodium hydroxide solution, separation of the organic layer and removal of the solvent therefrom) in tetrahydrofuran (10ml) was added, followed by bis(dibenzylideneacetone)palladium (80mg, 0.09mmol) and tri-(o -furyl)phosphine (65mg, 0.28mmol), and the reaction mixture was stirred at room temperature for 18 hours. After this time, the reaction mixture was partitioned between ethyl acetate and a mixture of <strong>[60-00-4]ethylenediaminetetraacetic acid</strong> (<strong>[60-00-4]EDTA</strong>) (3.8g), sodium hydroxide (1g) and water (100 ml) (pH12-14). The aqueous layer was extracted with ethyl acetate (x3), the combined organic layers were dried over Na2SO4, filtered and solvent removed from the filtrate under reduced pressure. The residue was purified by column chromatography on silica gel eluding with a gradient system of dichloromethane: ethyl acetate (1:1, by volume) gradually changing to ethyl acetate then to ethyl acetate: methanol (90:10, by volume) to afford the title compound (900mg, 55%). 1H-NMR (CDCl3): delta = 8.60 (2H, m), 7.25 (2H, m), 4.35 (2H, m), 3.95 (2H, m), 3.70 (1H, m), 1.45 (9H, s).
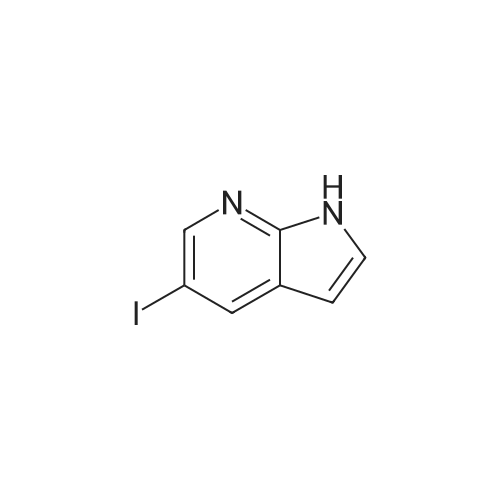
Stage #1: tert-butyl-3-iodoazetidine-1-carboxylate With zinc In ISOPROPYLAMIDE at 20 - 65℃; for 1h;
Stage #2: 5-iodo-1H-pyrrolo[2,3-b]pyridine In ISOPROPYLAMIDE at 85℃; for 5h;
46.a
In an inert atmosphere, zinc dust (223 mg, 3.41 mmol) was vigorously stirred in dimethylacetamide (1.5 ml) and heated to 65°C. Subsequently, trichloromethylsilane (50 μl, 0.38 mmol) and dibromoethane (30 μl, 0.38 mmol) were added, and the reaction mixture was stirred for further 30 minutes at 65°C. 3-lodo-azetidine-1 -carboxylic acid tert-butyl ester (462 mg, 1.89 mmol) in dimethylacetamide (2 ml) was added dropwise to the above prepared solution at 65°C and stirred for 30 minutes. 5-lodo-1 H- pyrrolo[2,3-b]pyridine (697 mg, 2.46 mmol) in dimethylacetamide (4 ml) was added to the reaction mixture. Subsequently, [1.1-bis(diphenylphosphino)ferrocene]dichloro- palladium(ll)-dichlormethane (93 mg, 0.11 mmol) and copper(l)iodide (108 mg, 0.57 mmol) were added. The reaction mixture was heated to 85°C for 5 hours, cooled to room temperature, diluted with ethyl acetate and filtered over Celite. The residue was extracted 3 times with water. The organic layer was dried over magnesium sulphate, filtered, and the solvent was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc:cylcohexane) to afford the title compound (104 mg, 20% yield). ESI-MS [m/z]: 274.1 [M+H]+.
With caesium carbonate In N,N-dimethyl-formamide at 90℃; Inert atmosphere of argon;
22
6-tert-Butyl-8-fluoro-2-(2-hydroxymethyl-3-{1-methyl-5-[5-(1-methyl-azetidin-3-yloxy)-pyridin-2-ylamino]-6-oxo-1,6-dihydro-pyridin-3-yl}-phenyl)-2H-phthalazin-1-one. In a 250 mL round bottom flask, added 6-Bromo-pyridin-3-ol (4.69 g, 26.95 mmol) and 3-Iodo-azetidine-1-carboxylic acid tert-butyl ester (7.63g, 26.95 mmol) and Cs2CO3 (12.26 g, 37.73 mmol) in DMF (10 mL) with stirring under argon. The reaction was heated at 90° C. overnight. Reaction was then poured onto water (200 mL). The crude product was extracted with EtOAc (3*50 mL). The organic extraction was concentrated and purified by flash chromatography with 10% EtOAc in Hexanes to afford product 2.62 g (yield 30%) of 3-(6-Bromo-pyridin-3-yloxy)-azetidine-1-carboxylic acid tert-butyl ester as light brown solid.
With Potassium benzoate; sodium t-butanolate In dimethyl sulfoxide at 50℃; for 4h;
22
Preparation 22: 3-(6-Bromopyridin-3-yloxy)azetidine-l-carboxylic acid tert-butyl ester Sodium tertiary butoxide (276 mg, 2.87 mmol) and potassium benzoate (460 mg, 2.87 mmol) were added to a solution of 6-bromo-pyridin-3-ol (500 mg, 2.87 mmol) and 3-iodo- azetidine-1-carboxylic acid tert-butyl ester in DMSO (10 inL). The resulting reaction mixture was stirred at 5O0C for 2 h. Further 3-iodo-azetidine-l-carboxylic acid tert -butyl ester (407 mg, 1.44 mmol) was added and heating at 5O0C was continued for 2 h. The reaction mixture was cooled to ambient temperature, diluted with EtOAc (100 inL) and washed with saturated aqueous Na2CO3 solution (2 X 30 inL). The combined aqueous washings were extracted with EtOAc (2 X 70 mL) and the combined organic extracts were washed with brine, dried (MgSO4), filtered and concentrated in vacuo. Purification by column chromatography (EtOAc-IH, 1:4) afforded the title compound: RT = 3.52 min; m/z (ES+) = 329.18 [M+H]+.
With Potassium benzoate; sodium t-butanolate In dimethyl sulfoxide at 50℃; for 4h;
7
Preparation 7: 3-(6-Bromopyridin-3-yloxy)azetidine-l-carboxylic acid tert-butyl ester Sodium ferf-butoxide (276 mg, 2.87 mmol) and potassium benzoate (460 mg, 2.87 mmol) were added to a solution of 6-bromopyridin-3-ol (500 mg, 2.87 mmol) and 3-iodo- azetidine-1-carboxylic acid tert-butyl ester (814 mg, 2.87 mmol) in DMSO (10 mL) and the resulting reaction mixture was heated at 5O0C for 4 h. The reaction mixture was cooled to ambient temperature, diluted with EtOAc (100 mL), washed with saturated aqueous Na2CO3 solution (2 X 30 mL) and brine, dried (MgSO4), filtered and concentrated in vacuo. Purification by column chromatography (EtOAc-IH, 1:4) afforded the title compound: RT = 3.52 min; m/z (ES+) = 329.18 [M+H]+ (LCMS protocol 3).
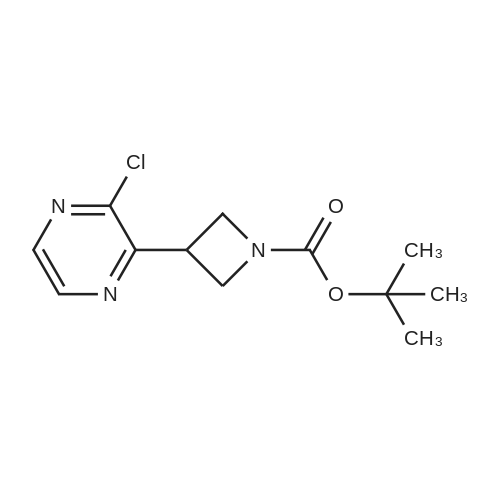
A 12L 3-neck round bottom flask fitted with a magnetic stirrer under nitrogen was charged with zinc dust (745 g, pre-activated, 11.4 mol, 2 eq.) and DMA (2 L, anhydrous). 1, 2-dibromoethane (71 mL, 0.855 mol, 0.15 eq, Aldrich) was then added over 10 minutes, followed by TMSC1 (108 mL, 0.855 mol, 0.15 eq, Acros) over 20 minutes. The reaction mixture was stirred for 25 minutes at room temperature. A solution of N-Boc-3-iodoazetidine (2420 g, 8.55 mol, 1.5 eq, CNH Technologies) in DMA (5 L, anhydrous) was added via a 2L addition funnel over 2 h keeping the internal temperature below 65 C using a water bath. The suspension was stirred for 1 hour at RT at which point it was degassed with nitrogen. Stirring was stopped and the suspension was allowed to stand. A 22L 3-neck round bottom flask fitted with a mechanical stirrer was charged with 2, 3-<strong>[4858-85-9]dichloropyrazine</strong> (850 g, 5.70 mol, 1.0 eq, AK Scientific), PdCl2dppf-CH2Cl2 (140 g, 171 mmol, 0.03 eq, Aldrich), Cul (67.3 g, 353 mmol, 0.062 eq, Aldrich), and DMA (5 L, anhydrous). The solution was degassed with nitrogen. The clear zinc reagent solution above the residual solid zinc was poured into the 22L flask under nitrogen. The brown solution was degassed with nitrogen and heated to 80 C for 16 hours at which point LCMS indicated complete conversion of 2, 3-<strong>[4858-85-9]dichloropyrazine</strong>. The reaction mixture was transferred to brine (8 L) in 50L separatory funnel. Water (8 L) and EtOAc (15 L) were added and the layers were separated. The aqueous layer was extracted with EtOAc (2 x 10 L). The combined organics were washed with water (3 x 10 L) and brine (5 L), dried over sodium sulfate and evaporated. The resulting residue was purified by column chromatography (eluting with hexanes/ethyl acetate = 10: 1) to get 536 g of pure tert-butyl 3-(3-chloropyrazin-2- yl)azetidine- 1 -carboxylate and 121 g of mixed fractions. The impure material was distilled under high vacuum to remove the impurity (N-Boc-azetidine) to give 81 g of pure tert-butyl 3- (3-chloropyrazin-2-yl)azetidine- 1 -carboxylate.Total: 617g, Yield: 40%.
40%
A 12L 3 -neck round bottom flask fitted with a magnetic stirrer under nitrogen was charged with zinc dust (745 g, preactivated according to the above Preparation 1 , 11.4 mol, 2 eq.) and DMA (2 L, anhydrous). 1, 2-dibromoethane (71 mL, 0.855 mol, 0.15 eq, Aldrich) was then added over 10 min, followed by TMSC1 (108 mL, 0.855 mol, 0.15 eq, Acros) over 20 min. The reaction mixture was stirred for 25 min at RT. A solution of N-Boc-3-iodoazetidine (2) (2420 g, 8.55 mol, 1.5 eq, CNH Technologies) in DMA (5 L, anhydrous) was added via a 2L addition funnel over 2 h keeping the internal temperature below 65 C using a water bath. The suspension was stirred for 1 h at RT at which point it was degassed with nitrogen. Stirring was stopped and the suspension was allowed to stand. A 22L 3 -neck round bottom flask fitted with a mechanical stirrer was charged with 2, 3-<strong>[4858-85-9]dichloropyrazine</strong> (6) (850 g, 5.70 mol, 1.0 eq, AK Scientific), PdCl2dppf-CH2Cl2 (140 g, 171 mmol, 0.03 eq, Aldrich), Cul (67.3 g, 353 mmol, 0.062 eq, Aldrich), and DMA (5 L, anhydrous). The solution was degassed with nitrogen. The clear zinc reagent solution above the residual solid zinc was poured into the 22L flask under nitrogen. The brown solution was degassed with nitrogen and heated to 80 C for 16 h at which point LCMS indicated complete conversion of 2, 3-<strong>[4858-85-9]dichloropyrazine</strong> (6). The reaction mixture was transferred to brine (8 L) in 50L separatory funnel. Water (8 L) and EtOAc (15 L) were added and the layers were separated. The aqueous layer was extracted with EtOAc (2 x 10 L). The combined organics were washed with water (3 x 10 L) and brine (5 L), dried over sodium sulfate and evaporated. The resulting residue was purified by column chromatography (eluting with hexanes/ethyl acetate = 10: 1) to obtain 536 g of pure tert-butyl 3-(3-chloropyrazin-2- yl)azetidine-l-carboxylate (7) and 121 g of mixed fractions. The impure material was distilled under high vacuum to remove the impurity (N-Boc-azetidine) to give 81 g of pure tert-butyl 3-(3- chloropyrazin-2-yl)azetidine- 1 -carboxylate (7).Total: 617g, Yield: 40%.
36.5%
To a flame dried 25 mL flask with zinc dust (217 mg, 3.31 mmol) and N,N-dimethylacetamide (2 mL) was added chlorotrimethylsilane (33.5 muL, 0.265 mmol) and 1,2- dibromoethane (22.83 muL, 0.265 mmol). The resulting slurry was stirred 15 min, then tert-butyl 3-iodoazetidine-l-carboxylate (753 mg, 2 66 mmol) was added to the above mixture (mild exotherm). The suspension was stirred at rt 30 min. [00422] The zinc solution was added via syringe to a solution of <strong>[4858-85-9]2,3-<strong>[4858-85-9]dichloropyrazine</strong></strong> (277 mg, 1.862 mmol), (dppf)PdCl2-CH2Cl2 (65.2 mg, 0.080 mmol), and copper(I) iodide (30.4 mg, 0.160 mmol) in N,N-dimethylacetamide (1.0 mL) that was degassed with N2 (3 x). The solution was heated to 80 0C and stirred 1 h. The reaction was quenched with NH4Cl (10 mL) and extracted with EtOAc (3 x 10 mL). The combined organic fractions were dried (MgSO4), concentrated, and purified by ISCO (40 g SiO2, 10-100% EtOAc/Hexane) to give tert-butyl 3-(3- chloropyrazin-2-yl)azetidine-l-carboxylate (262 mg, 0.971 mmol, 36.5 % yield) as a clear, colorless oil.
Zinc powder (150.1 g, 2.30 mol) and molecular sieves (50 g) were combined in a reaction flask and flame-dried under vacuum for 10 mins. Once the flask had returned to room temperature, it was charged with THF (4 L), and 1,2-dibromoethane (24.4 mL, 0.28 mol) was added. The reaction mixture was heated to 50 C. for 10 mins, then allowed to come to ambient temperature, at which time trimethylsilyl chloride (33.5 mL, 0.264 mol) was added {Caution: slightly exothermic}. The mixture was allowed to stir at room temperature for about 18 h. Slow addition of C7 (500 g, 1.77 mol) over 1.5 h was followed by stirring for an additional 18 h. In a separate flask, <strong>[4595-60-2]2-<strong>[4595-60-2]bromopyrimidine</strong></strong> (253 g, 1.59 mol) was combined with molecular sieves (85 g) in THF (1.3 L), and the mixture was degassed. This mixture was treated with tetrakis(triphenylphosphine)palladium(0) (32.7 g, 0.0283 mol), then added to the flask containing the reaction mixture from C7. The reaction was stirred for 25 h, and then filtered through Celite. The filtrate was concentrated under reduced pressure, then partitioned between saturated aqueous sodium carbonate solution (2 L) and EtOAc (2 L). The aqueous layer was extracted with EtOAc (2×2 L), and the combined organic layers were dried over sodium sulfate and concentrated in vacuo. The resulting yellow liquid residue was triturated with methyl tert-butyl ether (500 mL), and the precipitate was removed by filtration. Partial concentration of the filtrate resulted in precipitation of a solid; the mixture at this point was cooled in an ice-water bath. Filtration then provided a solid, which was washed with a minimum quantity of cold methyl tert-butyl ether to afford C33 as a white solid, which was taken directly into the next step. Yield: 131 g, 0.557 mol, 31%. GCMS m/z 180 ([M-tert-butyl]+1); 136 ([M-BOC]+1). 1H NMR (300 MHz, CDCl3) delta 1.45 (s, 9H), 4.0 (m, 1H), 4.3 (m, 4H), 7.2 (t, 1H), 8.75 (d, 2H).
Zn dust (520 mg, 7.95 mmol) was vigorously stirred in THF (2 ml) under nitrogen and 1 ,2 dibromoethane (84 mul, 0.97 mmol) was added. The suspension was then heated at 800C for 8 min and next allowed to cool to room temperature. Trimethylsilyl chloride (115 mul, 0.92 mmol) in THF (1 ml) was then added and the mixture further stirred at room temperature for 45 min. A solution of 3-iodo-azetidine-1 -carboxylic acid tert-butyl ester (1.74 g, 6.14 mmol) in THF (2 ml) was then added dropwise to the solution over a period of 15 min and the reaction mixture stirred at room temperature for 2 h. Pd2(dba)3 (90 mg, 0.10 mmol) and P(2-furyl)3 (85 mg, 0.36 mmol) were then added to the mixture, followed by <strong>[4487-59-6]2-bromo-5-nitropyridine</strong> (1.37 g, 6.74 mmol) in THF (4 ml). The mixture was then heated at 550C for 3 h, cooled to room temperature and quenched with saturated aqueous NaCI. Extraction with CH2CI2, drying (Na2SO4) of the organic phase, filtration and evaporation in vacuo provided the crude material, which was purified by flash col¬ umn chromatography (heptane:ethyl acetate, 3:1) to give the title compound (1.22 g, 71 %) as a light yellow oil. MS (ESI+) m/z = 224.1 [M-fBu+H]+1H NMR (400 MHz, CDCI3) : delta (ppm) 1.47 (s, 9H), 3.99 (m, 1 H), 4.18 (dd, J = 8.2, 6.1 Hz, 2H), 4.35 (t, J = 8.6 Hz, 2H), 7.42 (d, J = 8.5 Hz, 1 H), 8.45 (dd, J = 8.5, 2.6 Hz, 1H), 9.44 (d, J = 2.4 Hz, 1 H).
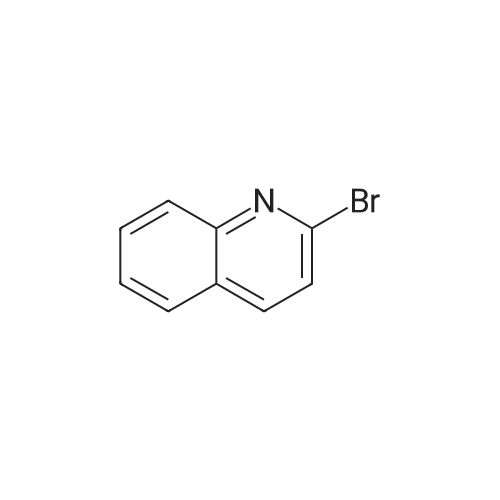
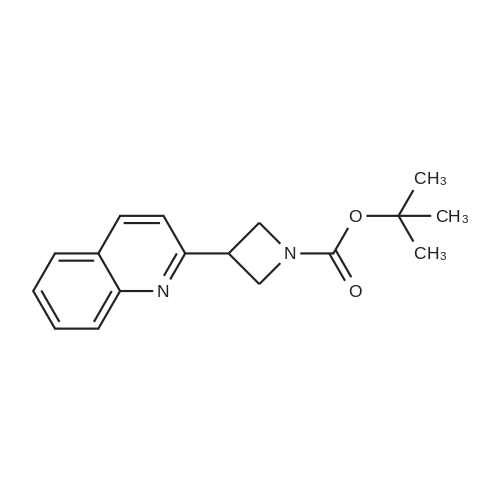
EXAMPLE 57 : 2- [ 1 -(3 -PIPERIDIN AZETIDiN-3 -YLJ-QUINOLINESTEP 1. 3-QUrNOLIN-2-YL-AZETIDrNE- 1 -CARBOXYLIC ACID TERT-BUTYL ESTER; [00220] Zinc dust was slowly added to a stirring solution of aqueous 2N HC1. The material was allowed to stir for 30 min at which point it was filtered, washed with water, MeOH, and diethyl ether. After drying under vacuum overnight at room temperature, the material was ready for use.[00221 ] A 100 mL 3 -neck round bottom flask fitted with a magnetic stirrer and flushed with nitrogen was charged with zinc dust (300 mg, pre-activated, 4.67 mmol, 2.0 eq.) and DMA (10 mL, anhydrous). 1 ,2-Dibromoethane (87 mg, 0.47 mmol, 0.2 eq) was added slowly, followed by TMSCI (51 mg, 0.47 mmol, 0.2 eq). The reaction was stirred for 15 min at room temperature. A solution of tert-butyl 3-iodoazetidine-l-carboxylate (1.0 g, 3.5 mmol, 1.5 eq) in DMA (10 mL, anhydrous) was added dropwise. The suspension was stirred for 1 h at room temperature.[00222 ] A 100 mL 3 -neck round bottom flask fitted with a mechanical stirrer was charged with <strong>[2005-43-8]2-bromo-quinoline</strong> (563 mg, 2.4 mmol, 1.0 eq), PdC12(dppf) (168 mg, 0.24 mmol, 0.1 eq), Cul (46 mg, 0.24 mmol, 0.1 eq), and DMA (20 mL, anhydrous). The dark solution was degassed for 15 min. The clear zinc reagent solution above the residual solid zinc was transferred to the above 100 mL flask by cannulation. The dark solution was degassed and heated to 80 C for 16 h. The reaction was diluted with brine and extracted with EtOAc (3 x 100 mL). The combined organics were washed with water (2 x 100 mL) and brine (100 mL), followed by drying over sodium sulfate. The solution was concentrated and the residue was purified by flash column chromatography (PE: EtOAc = 2: 1) provides the title compound (300 mg, 44% yield) as a light yellow solid.ESI-MS (M+l): 285; calc. for C17H2
[0616] To a stirred suspension of Zn dust (1.70 g, 26.0mmol) in THF (5 mL) under an Argon atmosphere, 1,2-dibromoethane (250 fll) was added at rt. The resulting mixturewas heated at 65 C. for 3 min and allowed to cool to rt.TMSCl (350 fll) was then added and the mixture was stirred atrt for 30 min. tert-Butyl 3-iodoazetidine-1-carboxylate (5.70g, 20.0 mmol) in THF (15 mL) was then added slowly and theresulting mixture was allowed to stir at rt for 45 min. AsolutionofPd2 ( dba )3 (183 mg, 0.200 mmol) andtrifurylphosphine (186 mg, 0.801 mmol) in THF (5 mL) were stirred at rtfor 10 min under an Argon atmosphere and the resultingmixture was added to the organozinc reagent prepared, followed by addition of<strong>[2005-43-8]2-bromoquinoline</strong> (5.00 g, 24.0 mmol).The mixture was then heated at 65 C. for 48 h underArgon.The reaction mixture was allowed to cool to rt and filteredthrough a pad ofdiatomaceous earth. The filtrate was concentrated and the residue obtained was purified by flash columnchromatography on silica gel (0: 1-1:0% EtOA/heptanes) toobtain tert-butyI 3-(quinolin-2-yl)azetidine-1-carboxylate.[0617] To a solution of tert-butyl 3-(quinolin-2-yl)azetidine-1-carboxylate (2.8 g, 9.8 mmol, as prepared above) inDCM (10 mL), TFA (10 mL) was added. The resulting mixture was stirred at rt for 2 h and concentrated to obtain aviscous oil which was dried under reduced pressure. Theresidue obtained was dissolved in DCM (50 mL) and stirredwith saturated NaHC0 3 (50 mL). The DCM layer was separated and the aqueous layer was concentrated. To the residueobtained, 20% iso-PrOH/DCM (50 mL) was added andstirred for 10 min and filtered. This procedure was repeatedthree times. The combined filtrates were dried over Na2 S04 ,filtered, and concentrated to obtain compound 24d as agummy solid. 1 H-NMR(400MHz, CDCI3 ) o(ppm): 8.34 (d,1=8.6 Hz, lH), 7.94-8.01 (m, 2H), 7.76 (s, lH), 7.59 (d, 1=6.7Hz, lH), 7.53 (d, 1=8.6 Hz, lH), 3.89-4.30 (m, 4H), 3.72-3.82(m, lH).
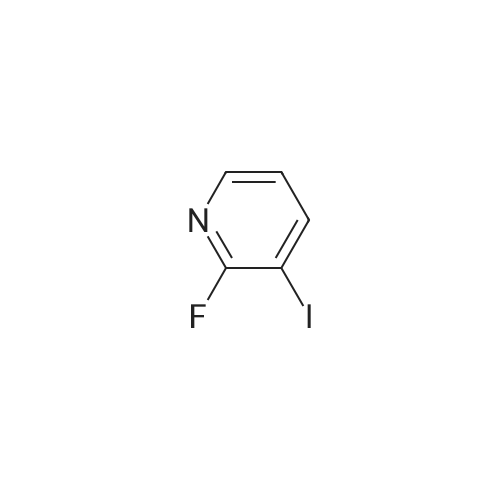
To a 2L 3 necked round bottomed flask fitted with a mechanical stirrer, under nitrogen atmosphere was placed Zinc dust, which was preactivated according to abovePreparation 2, (51.2 g, 0.78 mol, 1.94 eq) and dimethyl acetamide (162 mL). To the above suspension, 1, 2-dibromoethane (12.14 g, 0.0646 mol, 0.16 eq) was added dropwise at RT(exotherm and bubbling were observed), followed by dropwise addition of TMSC1 (6.99 g,0.0646 mol, 0.16 eq). A vigorous reaction (exotherm to 55 C) was observed. To this, a solution of N-Boc-3-iodoazetidine (2) (182.88 g, 0.646 mol, 1.6 eq) in dimethyl acetamide (378 mL) was added dropwise using an addition funnel (exotherm to 50 C was observed). The suspension was stirred for 1.5 h at RT and was then degassed with nitrogen for 15 min. Stirring was stopped and suspension was allowed to stand under nitrogen. To a 5L 3 necked round bottomed flask, fitted with mechanical stirrer, flushed with nitrogen were placed <strong>[113975-22-7]3-iodo-2-fluoropyridine</strong> (24) (90 g,0.404 mol, 1.0 eq), PdCl2dppf.CH2Cl2 (9.88 g, 0.012 mol, 0.03 eq), Cul (4.76 g, 0.025 mol, 0.062 eq ) and dimethyl acetamide (396 mL). The red colored suspension was degassed with nitrogen for 15 min.[00244] The Zinc reagent solution in 2L flask was cannulated into 5L round bottomed flask. The resulting reaction mixture was degassed again with nitrogen for 15 min with stirring and heated to 80C for overnight under nitrogen. LCMS indicates completion of reaction. The reaction was cooled to RT and quenched by addition of brine solution (1 L). To this EtOAc (1 L) and water (1 L) were added and layers were separated. The aqueous layer was extracted with EtOAc (2 x 2 L). The combined EtOAc layers were washed with water (2 L), brine (1 L), dried (Na2S04), filtered, and evaporated. The crude was purified by column chromatography to give 52 g of tert-butyl 3-(2-fluoropyridin-3-yl)azetidine-l-carboxylate (25) (yield: 51%) as an oil, which solidified on standing.1H NMR (300 MHz CDC13) : 8.13 (doublet, J = 4.8 Hz, 1H), 7.83-7.76 (dt, 1H), 7.28-7.20 (dt, 1H), 4.35 (t, J = 8.7 Hz, 2H), 4.05-3.88 (m, 3H), 1.46 (s, 9H). LC-MS: 253 (M+l); calcd for
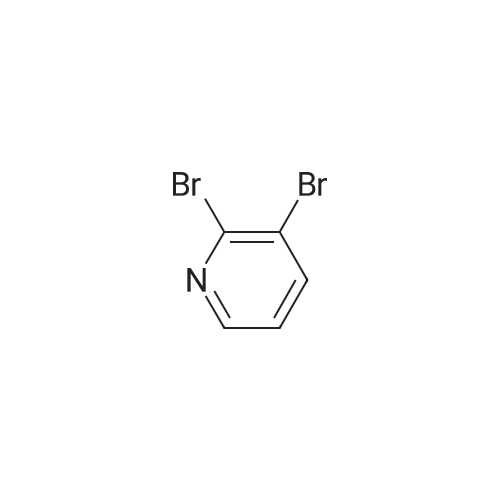
Stage #1: tert-butyl-3-iodoazetidine-1-carboxylate With chloro-trimethyl-silane; ethylene dibromide; zinc In dimethyl amine at 20 - 65℃; for 1.41667h; Inert atmosphere;
Stage #2: 2,3-dibromopyridine With copper(l) iodide In dimethyl amine at 80℃; for 17h; Inert atmosphere;
1.1
A 5L 3 -neck round bottom flask fitted with a magnetic stirrer under nitrogen was charged with zinc dust (138 g, preactivated according to the above Preparation 1, 2.11 mol, 2 eq.) and DMA (370 mL, anhydrous). 1, 2-dibromoethane (13 mL, 0.158 mol, 0.15 eq, Aldrich) was then added over 5 min, followed by TMSCl (20 mL, 0.158 mol, 0.15 eq, Acros) over 5 min. The reaction mixture was stirred for 20 min at RT. A solution of N-Boc-3-iodoazetidine (2) (448 g, 1.583 mol, 1.5 eq, CNH Technologies) in DMA (925 mL, anhydrous) was added over 25 min keeping the internal temperature below 65 °C using a water bath. The suspension was stirred for 1 h at RT at which point it was degassed with nitrogen. Stirring was stopped and the suspension was allowed to stand. A 12L 3 -neck round bottom flask fitted with a mechanical stirrer was charged with 2, 3-dibromopyridine (1) (250 g, 1.055 mol, 1.0 eq, Frontier Scientific),PdCl2dppf-CH2Cl2 (25.8 g, 31.65 mmol, 0.03 eq, Aldrich), Cul (12.5 g, 65.41 mmol, 0.062 eq, Aldrich), and DMA (925 mL, anhydrous). The solution was degassed with nitrogen. The clear zinc reagent solution above the residual solid zinc was poured into the 12L flask under nitrogen. The brown solution was degassed with nitrogen and heated to 80 °C for 17 h at which point LCMS indicated complete conversion of 2, 3-dibromopyridine (1). The reaction mixture was transferred to brine (2 L) in 22L separatory funnel. Water (2 L) and EtOAc (4 L) were added and the layers were separated. The aqueous layer was extracted with EtOAc (2 x 3 L). The combined organics were washed with water (3 x 3 L) and brine (2 L), dried over sodium sulfate and evaporated. The resulting residue was purified by column chromatography (eluting with hexanes/ethyl acetate = 9:1 to 5: 1) to obtain 289 g of impure tert-butyl 3-(3-bromopyridin-2- yl)azetidine-l-carboxylate (3) which was distilled under high vacuum to remove the impurity (N- Boc-azetidine) to give 281 g of pure tert-butyl 3-(3-bromopyridin-2-yl)azetidine-l-carboxylate. Yield: 85%.
A 100 mL 3 -neck round bottom flask fitted with a magnetic stirrer and flushed with nitrogen was charged with zinc dust (813 mg, preactivated according to the abovePreparation 1, 12.7 mmol, 2.0 eq.) and DMA (10 mL, anhydrous). 1, 2-dibromoethane (236 mg, 1.27 mmol, 0.2 eq) was added slowly, followed by TMSC1 (137 mg, 1.27 mmol, 0.2 eq). The reaction was stirred for 15 min at RT. A solution of N-Boc-3-iodoazetidine (2) (2.7 g, 9.5 mmol, 1.5 eq) in DMA (10 mL, anhydrous) was added dropwise. The suspension was stirred for 1 h at RT.[00314] A 100 mL 3 -neck round bottom flask fitted with a mechanical stirrer was charged with <strong>[13721-00-1]2,3-dibromo-quinoline</strong> (97) (1.82 g, 6.4 mmol, 1.0 eq), PdCl2(dppf) (446 mg, 0.64 mmol, 0.1 eq), Cul (121 mg, 0.64 mmol, 0.1 eq), and DMA (20 mL, anhydrous). The dark solution was degassed for 15 min. The clear zinc reagent solution above the residual solid zinc was transferred to the above 100 mL flask by cannulation. The dark solution was degassed and heated to 80 C for 16 h. The reaction was diluted with brine and extracted with EtOAc (3 x 100 mL). The combined organics were washed with water (2 x 100 mL) and brine (100 mL), followed by drying over sodium sulfate. The solution was concentrated and the residue was purified by flash column chromatography (EtOAc :Petro ether = 4: 1) provides the title compound (98) (1.2 g, 3.30 mmol, 52% yield) as a light yellow solid.ESI-MS (M+l): 363 calc. for Ci7Hi9
Stage #1: tert-butyl-3-iodoazetidine-1-carboxylate With chloro-trimethyl-silane; ethylene dibromide; zinc In ISOPROPYLAMIDE Inert atmosphere;
Stage #2: 2-methyl-3-[ethyl(tetrahydro-2H-pyran-4-yl)amino]-5-bromobenzoic acid methyl ester With copper(l) iodide In ISOPROPYLAMIDE at 80℃; for 16h; Inert atmosphere;
44.3
[01487] Step 3: Synthesis of tert-butyl 3-{3-[ethyl(oxan-4-yl)amino]-5-(methoxycarbonyl)-4- methylphenyl} azetidine- l -carboxylate [01488] To a dry flask was added zinc dust (40 mg, 0.61 mmol) followed by anhydrous DMA (2ml) and the vessel was flushed with nitrogen whilst stirring vigorously and heating to 65°C. TMS-C1 (9 μ, 0.07 mmol) and 1 ,2-dibromoethane (6 μ, 0.07 mmol) were added and the reaction was stirred at 65°C for 30mins, followed by the dropwise addition of N-Boc-3- iodoazeticline ( 1 3 mg. 0.47 mmol) as a solution in anhydrous DMA ( 1 ml). The reaction was then cooled to room temperature and methyl 5-bromo-3-[ethyl(oxan-4-yl)amino]-2- methylbenzoate (100 mg, 0.28 mmol) was added as a solution in anhydrous DMA (2ml). The resulting solution was degassed with nitrogen for 5 mins after which Pd(dppf)Cl2-DCM (7 mg, 0.01 mmol) and copper (I) iodide (3 mg, 0.02 mmol) were added as solids. The reaction was heated to 80°C for 16h and then cooled to room temperature followed by the addition of saturated NH4C1 solution (50ml). The aqueous phase was extracted with EtOAc (3x 100ml), and then the combined organic phases were washed with brine (2x50ml), dried with Na2S04, filtered and evaporated. The residue was purified over a 5g Isolute column using 10-30% EtOAc in heptane as the eluent to afford the title compound as a colourless oil (77 mg, 62%>). LC-MS 98%, 2.19min (3.5 minute LC-MS method), m/z=433.2, NMR (500 MHz, CDC13) δ 7.49 (d, J = 1.6 Hz, 1 H), 7.16 (d, J = 1.6 Hz, 1 H), 4.31 (t, J = 8.7 Hz, 2H), 3.94 (dd, J = 14.4, 5.5 Hz, 4H), 3.89 (s, 3H), 3.77 - 3.64 (m, 1 H), 3.31 (td, J = 1 1 A, 2.7 Hz, 2H), 3.06 (q, J = 7.0 Hz, 2H), 2.99 - 2.87 (m, 1 H), 2.46 (s, 3H), 1 .65 (ddd, J = 1 1 .1 , 8.6, 6.0 Hz, 4H), 1 .46 (s, 9H), 0.85 (dd, J = 13.1 , 6.1 Hz, 3H).
tert-butyl 3-(2,6-dichloropyridin-4-yl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
58%
Under a nitrogen atmosphere, zinc dust (6.91 g, 105 mmol) was suspended in N,N-dimethylacetamide (10 mL) and 1,2-dibromoethane (1.08 mL, 12.4 mmol) was added, followed by careful addition of trimethylsilylchloride (1.61 mL, 12.4 mmol) and was added cautiously over 5 min while the flask sat on a bed of ice. The bath was removed, and after stirring for a further 15 min, a solution of N-(tert-butoxycarbonyl)-3-iodoazetidine (25.1 g, 86.9 mmol) in N,N-dimethylacetamide (30 mL) was added over 30 min and stirring was continued for an additional 30 min. In the open atmosphere, this mixture was filtered through Celite as quickly as possible, rinsing with N,N-dimethylacetamide (100 mL). The resulting yellow solution was injected into a separately prepared, nitrogen flushed vessel containing [1,1-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (2.56 g, 3.10 mmol), copper(I) iodide (1.18 g, 6.21 mmol) and <strong>[98027-84-0]2,6-dichloro-4-iodopyridine</strong> (17.0 g, 62.1 mmol) and this mixture was stirred at 80 C. for 19.5 h. After cooling to rt, the mixture was diluted with EtOAc and washed with water (3×). On the third time, filtration through Celite was necessary to break the emulsion, following which, the organics were washed with brine and then dried over MgSO4. After being freed of volatiles, the resultant residue was purified by flash column chromatography (100:0-70:30 heptanes/EtOAc) to afford tert-butyl 3-(2,6-dichloropyridin-4-yl)azetidine-1-carboxylate as a white solid (10.98 g, 58%); 1H NMR (400 MHz, CDCl3) delta 7.22 (s, 2H), 4.35 (dd, J=8.7, 5.6 Hz, 2H), 3.92 (dd, J=8.7, 5.6 Hz, 2H), 3.73-3.61 (m, 1H), 1.47 (s, 9H).
54%
Step 1 : tert-butyl 3-(2,6-dichloropyridin-4-yl)azetidine-l-carboxylate To a suspension of zinc (3.56 g, 3.0 equiv., 53.1 mmol) in DMA (8.3 mL, 88.5 mmol) was added 1,2-dibromoethane (0.37 mL, 0.24 equiv., 4.25 mmol), followed by TMS chloride (0.55 mL, 0.24 equiv., 4.25 mmol). After the reaction ceased down, a solution of l-boc-3-(iodo)azetidine (11.3 g, 2.2 equiv., 39.0 mmol) in DMA (6.6 mL, 70.8 mmol) was added dropwise. After 30 min at 25 C and then 2 h at 50 C, LCMS showed no m/z 228 peak. The mixture was transferred dropwise into a suspension of <strong>[98027-84-0]2,6-dichloro-4-iodopyridine</strong> (5.00 g, 17.7 mmol), [l, -bis(diphenylphosphino)ferrocene]palladium(ii) dichloride dichloromethane adduct (738 mg, 0.050 equiv., 0.885 mmol) and cuprous iodide (337 mg, 0.10 equiv., 1.77 mmol) in DMA (16.5 mL, 177 mmol). DMA ( 16.5 mL, 177 mmol)) was used to rinse the flask. The whole reaction mixture was kept at 80 C in oil-bath for 4 h. DMA was removed at 60 C under vacuum, and the crude was purified via silica gel chromatography (0-50% EtO Ac/Heptanes) to yield 1.73 g (54% based on the conversion) of the title compound. 1H NMR (400 MHz, CDC13) delta (delta) 7.23 (s, 2H), 4.35 (t, J = 8.7 Hz, 2H), 3.92 (dd, J= 8.7, 5.6 Hz, 2H), 3.73 - 3.61 (m, 1H), 1.47 (s, 9H). 13C NMR (101 MHz, CDC13) delta (delta) 157.03, 156.08, 151.09, 121.24, 80.24, 55.31, 32.48, 28.32. LCMS: m z 303 (M+H).
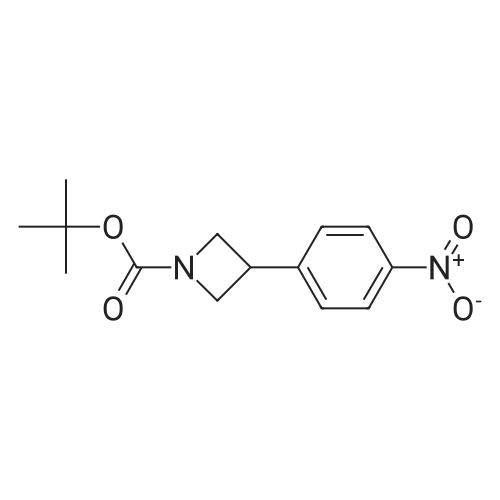
(a) tert-Butyl-3-(4-nitrophenyl) azetidine- 1-carboxylate (16)1,2-Dibromoethane (0.146 mL, 1.69 mmol) was added to a vigorously stirred suspension of zinc dust (0.901 g, 13.8 mmol) in THF (3.5 mL) under a nitrogen atmosphere and the resulting suspension heated at 80 00 for 10 minutes. Trimethylsilyl chloride (0.202 mL, 1.59 mmol) in THF (1.75 mL) was added at roomtemperature and after stirring for 4 minutes a solution of tert-butyl 3-iodoazetidine-1- carboxylate (3.00 g, 10.6 mmol) in THF (3.5 mL) was added dropwise over a period of 15 minutes. The resulting mixture was stirred at room temperature for 2 hours then Pd2(dba)3 (0.155 g, 0.170 mmol) and tri-2-furylphosphine (0.143 g, 0.615 mmol) were added followed by 1-iodo-4-nitrobenzene (2.90 g, 11.7 mmol) in THF (18 mL). Theresulting mixture was heated at 55 00 for 3 hours then quenched at room temperature with a saturated aqueous sodium chloride solution (15 mL). The aqueous phase was extracted with DCM (2 x 15 mL) then the combined organic fractions were dried (magnesium sulfate), filtered and evaporated in vacuo. The residue was purified using silica gel column chromatography (CombiFlash Rf, 40 gSi02 Cartridge, 10-40% EtOAc in cyclohexane) to give the title compound 16 as anorange oil (2.14 g, 72%); 1H NMR (300 MHz, CDCI3) O 8.24 (dd, J= 6.8, 1.9 Hz, 2H),7.51 (d, J= 8.6 Hz, 2H), 4.41 (t, J= 8.7 Hz, 2H), 3.98 (dd, J= 8.5, 5.7 Hz, 2H), 3.89-3.81 (5, 1H), 1.49 (5, 9H).
3-(5-bromopyridin-3-yl)azetidine-1-carboxylic acid tert-butyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
22%
A mixture of 3-bromo-5-(4,4,5,5-tetramethyl-[ 1 ,3,2]dioxaborolan-2-yl)-pyridine (1.18 g,5.8 mmol), Nil2 (200 mg, 0.64 mmol), trans-2-aminocyclohexanol hydrochloride (100 mg,0.66 mmol) and NaHMDS (2.2 g, 12 mmol) in dry iPrOH (10 mL) was stuffed at roomtemperature under N2 for 5 mm. A solution of 3-iodo-azetidine-1-carboxylic acid tert-butylester (1.6 g, 8.8 mmol) in dry iPrOH (1 mL) was then added. The resulting mixture washeated to 80C under microwave irradiation for 50 mm. The reaction solution was concentrated under reduced pressure to give a brown mixture which was purified by flash chromatography to give the title product as colorless oil (400 mg, 22%). MS: 313.1 (M+H).
tert-butyl 3-((4-amino-6-chloropyrimidin-5-yl)oxy)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
56%
With potassium carbonate In N,N-dimethyl-formamide at 100℃; for 16h;
With potassium carbonate In N,N-dimethyl-formamide at 100℃; for 16h;
1.1 (1) tert-Butyl 3-((4-amino-6-chloropyrimidin-5-yl)oxy)azetidine-1-carboxylate, INT 1
(1) tert-Butyl 3-((4-amino-6-chloropyrimidin-5-yl)oxy)azetidine-1-carboxylate, INT 1 To a solution of N-Boc-3-iodoazetidine (6.84 g, 24.16 mmol) in DMF (37 mL) was added 4-amino-6-chloropyrimidin-5-ol (2.00 g, 13.74 mmol) followed by potassium carbonate (5.70 g, 41.24 mmol). The reaction mixture was stirred at 100° C. for 16 hr. The mixture was diluted with EtOAc and washed with saturated aqueous sodium hydrogen carbonate solution. The aqueous layer was back-extracted with EtOAc. The combined organic layers were washed with water (2*) and brine (2*), dried over magnesium sulfate, filtered and concentrated. The crude was dried in vacuum for 30 min. The residue was purified by flash chromatography (DCM/MeOH gradient, 0-5%). The isolated residue was triturated with cyclohexane. The resulting off-white solid was filtered off, rinsed with cyclohexane, and dried in vacuum to afford the title compound INT 1 as an off-white solid. UPLC-MS: MS (ESI): [M+H]+ 301.0, rt=0.83 min. 1H NMR (DMSO-d6): δ (ppm) 7.98 (s, 1H), 7.34 (br s, 2H), 4.93-4.70 (m, 1H), 4.23-3.95 (m, 4H), 1.39 (s, 9H).
With potassium carbonate In N,N-dimethyl-formamide at 100℃; for 16h;
1.1 (1) tert-butyl 3-((4-amino-6-chloropyrimidin-5-yl)oxy)azetidine-1-carboxylate, INT 1
(1) tert-butyl 3-((4-amino-6-chloropyrimidin-5-yl)oxy)azetidine-1-carboxylate, INT 1 To a solution of A/-Boc-3-iodoazetidine (6.84 g, 24.16 mmol) in DMF (37 mL) was added 4-amino-6-chloropyrimidin-5-ol (2.00 g, 13.74 mmol) followed by potassium carbonate (5.70 g, 41 .24 mmol). The reaction mixture was stirred at 100 °C for 16 hr. The mixture was diluted with EtOAc and washed with saturated aqueous sodium hydrogen carbonate solution. The aqueous layer was back-extracted with EtOAc. The combined organic layers were washed with water (2x) and brine (2x), dried over magnesium sulfate, filtered and concentrated. The crude was dried in vacuum for 30 min. The residue was purified by flash chromatography (DCM/MeOH gradient, 0-5%). The isolated residue was triturated with cyclohexane. The resulting off-white solid was filtered off, rinsed with cyclohexane, and dried in vacuum to afford the title compound INT 1 as an off-white solid.UPLC-MS: MS (ESI): [M+H]+301 .0, rt = 0.83 min.1H NMR (DMSO-d6): δ (ppm) 7.98 (s, 1 H), 7.34 (br s, 2H), 4.93-4.70 (m, 1 H), 4.23-3.95 (m, 4H), 1 .39 (s, 9H).
With potassium carbonate In N,N-dimethyl-formamide at 100℃; for 16h;
1.1 (1) tert-Butyl 3-((4-amino-6-chloropyrimidin-5-yl)oxy)azetidine- 1 -carboxylate, INT 1
To a solution of N-Boc-3-iodoazetidine (6.84 g, 24.16 mmol) in DMF (37 mL) was added 4-amino-6- chloropyrimidin-5-ol (2.00 g, 13.74 mmol) followed by potassium carbonate (5.70 g, 41.24 mmol). The reaction mixture was stirred at 100 °C for 16 hr. The mixture was diluted with EtOAc and washed with saturated aqueous sodium hydrogen carbonate solution. The aqueous layer was back-extracted with EtOAc. The combined organic layers were washed with water (2x) and brine (2x), dried over magnesium sulfate, filtered and concentrated. The crude was dried in vacuum for 30 mm. The residue was purified by flash chromatography (DCM/MeOH gradient, 0-5%). The isolated residue was triturated with cyclohexane. The resulting off-white solid was filtered off, rinsed with cyclohexane, anddried in vacuum to afford the title compound INT 1 as an off-white solid.UPLC-MS: MS (ESI): [M+H] 301.0, rt = 0.83 mm. ‘H NMR (DMSO-d6): (ppm) 7.98 (s, 1H), 7.34 (br s, 2H), 4.93-4.70 (m, 1H), 4.23-3.95 (m, 4H), 1.39 (s, 9H).
tert-butyl 3-(5-nitro-6-phenylpyridin-2-yl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With ferrous(II) sulfate heptahydrate; sulfuric acid; dihydrogen peroxide; In water; dimethyl sulfoxide; at 27 - 60℃; for 61h;
Step B: tert-Butyl 3-(5-nitro-6-phenylpyridin-2-yl)azetidine-l-carboxylateAn aqueous solution of 30% H202 (1.31 mL, 15.0 mmol) was added to a stirred solution of <strong>[134896-35-8]3-nitro-2-phenylpyridine</strong> (1.00 g, 5.00 mmol), concentrated aqueous H2S04 solution (0.53 mL, 10 mmol), tert-butyl 3-iodoazetidine-l-carboxylate (2.83 g, 9.99 mmol) and iron(II) sulfate heptahydrate (0.42 g, 1.5 mmol) in DMSO (20 mL). The resulting mixture was stirred at 27 C for 0.5 h before additional iron(II) sulfate heptahydrate (0.42 g, 1.5 mmol) and 30% H202 (1.31 mL, 15.0 mmol) were added. Stirring was continued for 0.5 h before the mixture was charged again with more 30% H202 (1.31 mL, 14.99 mmol) and iron (II) sulfate heptahydrate (0.42 g, 1.50 mmol). The resulting mixture was then heated at 50 C for 60 h. The product mixture was poured into an ice-cold solution of aqueous 15% NaOH solution and the pH was adjusted to > 10. The aqueous mixture was filtered, and the filtrate was extracted with ethyl acetate (150 mL x 3). The combined organic extracts were dried over Na2S04 and concentrated. The residue was purified by flash silica gel chromatography (ISCO; 40 g SepaFlash Silica Flash Column, ethyl acetate in petroleum ether = 0-30%, 40 mL/min, dry loaded) to give the title compound. MS: m/z = 356.1 (M + 1).
With caesium carbonate; In acetonitrile; at 90℃; for 12h;
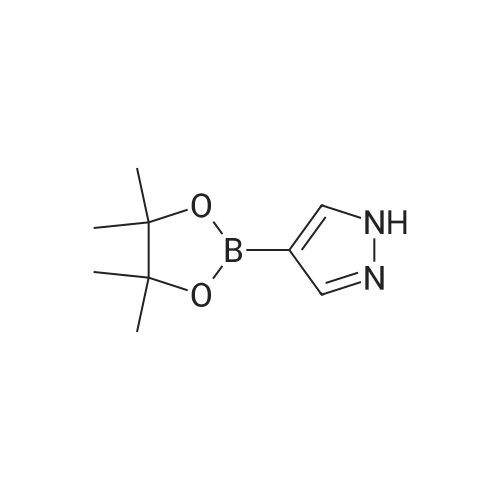
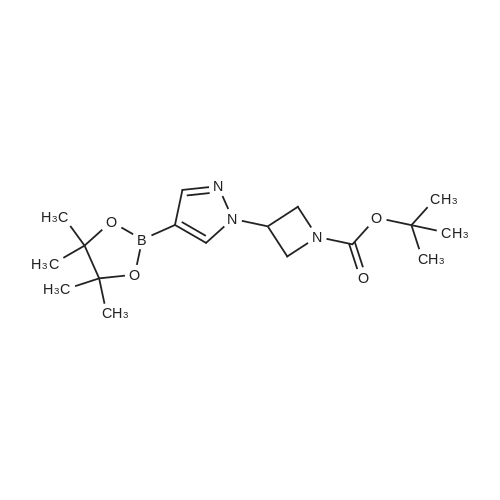
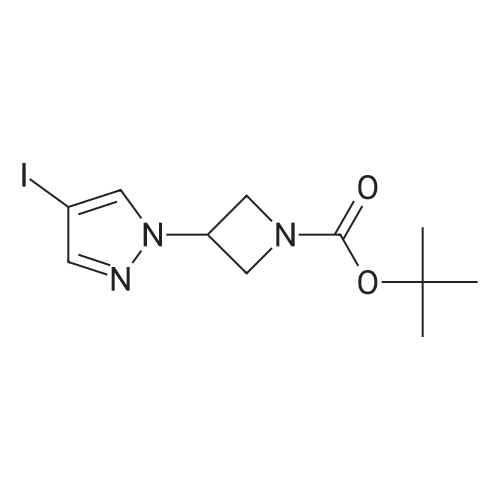
A mixture of 4-(4,4,5,5-tetramethyl- 1,3 ,2-dioxaborolan-2-yl)- 1 H-pyrazole (19.4 mg,0.10 mmol), cesium carbonate (49.0 mg, 0.15 mmol), and tert-butyl 3-iodoazetidine-1- carboxylate (31.0 mg, 0.11 mmol) in acetonitrile (0.5 mL) was stirred and heated at 90 C for 12 hours. The resulting mixture was cooled to room temperature, and the reaction was quenched with saturated aqueous NH4C1, and extracted with DCM (3 x2 mL). The organiclayers were combined, dried over Na2SO4, and concentrated. The crude product was used directly in the next step without further purification.
3-(3,5-difluoro-4-formylphenoxy)azetidine-1-carboxylic acid tert-butyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
93%
With caesium carbonate; In N,N-dimethyl-formamide; at 150℃; for 1h;Inert atmosphere; Microwave irradiation;
To a solution of <strong>[532967-21-8]2,6-difluoro-4-hydroxy-benzaldehyde</strong> 101a (CAS No.: 532967-21-8, 600 mg, 3.79 mmol) in N,N-dimethylformamide (25 mL) under argon were added cesium carbonate (3.09 g, 9.48 mmol) and l-Boc-3-iodoazetidine 101b (CAS No.: 254454-54-1, 2.68 g, 9.48 mmol). The resulting mixture was heated at 150 C under microwave heating for 1 h. The reaction mixture was allowed to cool to ambient temperature, the solid removed by filtration, the filter cake was washed with toluene and the filtrate was concentrated in vacuo. The residue was partitioned between EtOAc and water, the organic phase was separated, washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The crude product was adsorbed onto HMN diatomaceous earth (Isolute, Biotage) and purified by silica gel chromatography (mobile phase: cyclohexane/ethyl acetate, gradient 0% to 30%) to afford 101c as a yellow oil (1.10 g, 93%). 1H NMR (300 MHz, CDC13): delta 10.20 (s, 1H), 6.35 (m, 2H), 4.94 - 4.86 (m, 1H), 4.34 (ddd, 7 = 1.1, 6.4, 9.8 Hz, 2H), 4.05 - 3.98 (m, 2H), 1.45 (s, 9H).
ethyl 5-{1-[(tert-butoxy)carbonyl]azetidin-3-yl}pyridine-3-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
94%
1.3M /PrMgCI.LiCI in THF (6.69 ml, 8.69 mmol) was added dropwise to tertbutyl 3-iodoazetidine-1- carboxylate (1.85 g, 6.52 mmol) in THF (20 ml) at 0C. The reaction mixture was stirred at 0C for 20 min and the resultant solution was added dropwise to a solution of ethyl 5-bromopyridine-3-carboxylate (0.5 g, 2.17 mmol) and FeCb (anhydrous) (0.22 g, 1 .74 mmol) at 0C. The mixture was stirred at 0C for 25 min. The reaction mixture was quenched with saturated NH CI (aq), further diluted with water and DCM and stirred vigourously for 10 min. The mixture was extracted with DCM (3 x 80 ml) aid the combined organic extracts were dried (Na2S04), filtered and evaporated under vacuum. Purification by flash column chromatography (eluting with a gradient of 0-80% EtOAc / heptane) afforded the title compound (0.652 g, 94%) as a yellow oil. 1 H-NMR (CDCI3, 250 MHz): d[ppm]= 9.15 (d, J = 1 .9 Hz, 1 H), 8.72 (d, J = 2.2 Hz, 1 H), 8.35 (t, J = 2.1 Hz, 1 H), 4.52 - 4.37 (m, 4H), 4.02 (dd, J = 8.6, 5.8 Hz, 2H), 3.91 - 3.78 (m, 1 H), 1 .50 (s, 9H), 1.45 (t, J = 7.1 Hz, 3H) HPLCMS (Method M): [m/z]: 307.40 [M+H]+
tert-butyl 3-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
39%
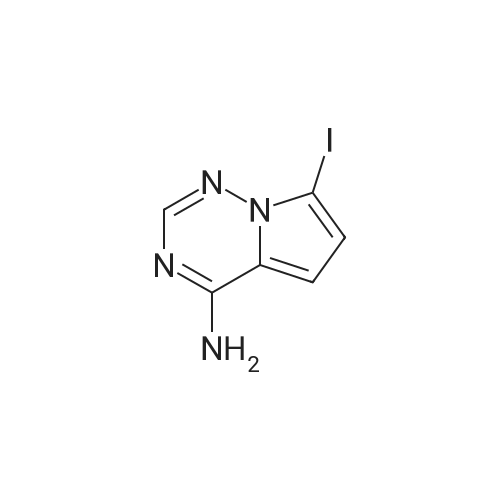
Stage #1: tert-butyl-3-iodoazetidine-1-carboxylate With chloro-trimethyl-silane; zinc In ethylene dibromide; N,N-dimethyl-formamide at 40 - 70℃; for 2h;
Stage #2: 7-iodopyrrolo-[2,1-f][1,2,4]-triazin-4-amine With tris-(dibenzylideneacetone)dipalladium(0); trifuran-2-yl-phosphane In ethylene dibromide; N,N-dimethyl-formamide at 75℃;
94.2 Step 2: tert-Butyl 3-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)azetidine-1-carboxylate
Step 2: tert-Butyl 3-(4-aminopyrrolo[1,2-f][1,2,4]triazin-7-yl)azetidine-1-carboxylate Zinc (0.690 g, 10.5 mmol) was suspended with 1,2-dibromoethane (60 μL, 0.70 mmol) in DMF (20 mL). The resulting mixture was stirred at 70° C. for 10 min and cooled to rt. Chlorotrimethylsilane (89 μL, 0.70 mmol) was added and stirring was continued for 1 h. A solution of tert-butyl 3-iodoazetidine-1-carboxylate (2.5 g, 8.8 mmol) in DMF (10 mL) was then added and the mixture was stirred at 40° C. for 1 h before a mixture of 7-iodopyrrolo[2,1-f][1,2,4]triazin-4-amine (2.4 g, 9.2 mmol), Tris(dibenzylideneacetone)dipalladium(0) (0.80 g, 0.88 mmol) and Tri-(2-furyl)phosphine (0.41 g, 1.8 mmol) in DMF (12 mL) was added. The reaction mixture was then stirred at 75° C. overnight, cooled to rt, and partitioned between EtOAc and saturated NH4Cl solution. The organic layer was separated, washed with water, dried over MgSO4, concentrated and purified via column chromatography (0% to 100% EtOAc in hexanes) to give the product (1.0 g, 39%). LCMS calcd for C14H20N5O2 (M+H)+: m/z=290.2. Found: 290.2.
tert-butyl 3-(naphthalen-2-yl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95.6%
With 1,4-diaza-bicyclo[2.2.2]octane; silver tetrafluoroborate; bis(triphenylphosphane)copper(I) nitrate; triethyloxonium fluoroborate; In N,N-dimethyl-formamide; at 100℃; for 12h;Inert atmosphere;
An appropriate amount of DMF was added to the reaction vessel and purged with nitrogen until the atmosphere was a nitrogen atmosphere. Then 100 mmol of the compound of the above formula (II), 200 mmol of the compound of the above formula (III)7 mmol of bis (triphenylphosphine) cuprous nitrate,100 mmol DABCO and 15 mmol promoter (a mixture of 13 mmol triethyloxonium tetrafluoroborate and 2 mmol AgBF4), warmed to 100 C and reacted at this temperature for 12 hours.After completion of the reaction, saturated brine was added to sufficiently wash the organic phase, the organic phase was separated, washed thoroughly with deionized water and extracted with ether. The upper organic layer was taken, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was passed through a silica gel column Chromatography. The eluent used for column chromatography was a mixture of chloroform and petroleum ether. The volume ratio of the two was 1: 3 to obtain the compound of formula (I). The yield was 95.6%
To a 100 mL round bottom flask was added tert-butyl 3-iodoazetidine-1-carboxylate (6) (6.54 g, 0.0231 mol, 1 equiv.) and DMSO (25 mL). NaCN (2.3 g, 0.0462 mol, 2equiv.) was added to the solution in one portion and stirred at 130 oC for 6 h. The resulting mixture was cooledto rt, poured into water (200 mL), and extracted with diethyl ether (5 x 200 mL). The combined organicextracts were washed with brine (50 mL), dried over Na2SO4 and concentrated. The crude compound waspurified by flash chromatography (silica gel, 5-50% EtOAc in hexanes) to give the desired product 22 (3.27 g,78%). Physical State: off-white solid (mp 78-80 oC); Rf = 0.40 (2:8 EtOAc/hexanes, vis. KMnO4); 1H NMR (500MHz, CDCl3): delta 4.23 - 4.11 (m, 4H), 3.38 (tt, J 8.9, 6.3 Hz, 1H), 1.43 (s, 9H); 13C NMR (126 MHz, CDCl3): delta 155.6,119.6, 80.8, 52.6 (br, 2C), 28.4 (3C), 17.2.
To a flame-dried reaction tube under argon was added tertbutyl3-iodoazetidine-1-carboxylate (6) (83.8 mg, 0.249 mmol, 1 equiv.), potassium acetate (36.6 mg, 0.375mmol, 1.5 equiv.) and dry DMSO (2.5 mL). The mixture was heated at 80 oC and stirred overnight. Completionof the acetoxylation step was monitored by TLC and 1H NMR of the crude reaction mixture. A solution ofpotassium hydroxide (21.0 mg, 0.374 mmol, 1.5 equiv. in 0.8 mL of H2O) was slowly added and the mixturestirred at rt for 30 min. The resulting mixture was diluted with H2O (10 mL) and extracted with ethyl acetate (3x 10 mL). The combined organic layers were washed with brine (5 mL) and dried over Na2SO4 andconcentrated under reduced pressure. The crude material was purified by flash chromatography (silica gel,10-60% EtOAc in hexanes) to give the desired product 24 (31.2 mg, 72%). Physical State: white solid
tert-butyl 3-((2-fluorophenyl)ethynyl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
53%
With bis(η3-allyl-μ-chloropalladium(II)); caesium carbonate; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene In dimethyl sulfoxide at 60℃; for 3h; Inert atmosphere; regioselective reaction;
tert-butyl 2-((2-fluorophenyl)ethynyl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78%
With dichloro(norbornadiene)palladium(II); potassium carbonate; DavePhos In tetrahydrofuran; water; N,N-dimethyl-formamide at 80℃; for 24h; Inert atmosphere; regioselective reaction;
tert-butyl (E)-3-(2,5-dimethoxystyryl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
55%
With 1,1'-bis-(diphenylphosphino)ferrocene; bis(acetylacetonato)palladium(II); N-Methyldicyclohexylamine In N,N-dimethyl acetamide at 90℃; for 8h; Inert atmosphere; stereoselective reaction;
Stage #1: 3-Iodo-azetidine-1-carboxylic acid tert-butyl ester With iodine; zinc In N,N-dimethyl-formamide at 20℃; for 1h; Inert atmosphere;
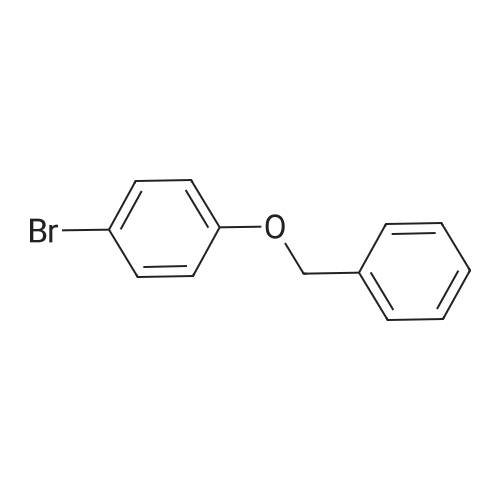
Stage #2: 1-(benzyloxy)-4-bromobenzene With dicyclohexyl({2’,6’-dimethoxy-[1,1‘-biphenyl]-2-yl})phosphane; tris-(dibenzylideneacetone)dipalladium(0) In N,N-dimethyl-formamide at 50℃; for 3h;
1 Step 1: Preparation of tert-butyl 3-(4-(benzyloxy)phenyl)azetidine-1-carboxylate
Into a 1000-mL 3-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed zinc (11 g, 168.25 mmol, 2.95 equiv) in DMF (400 mL), to which was added I2 (2.23 g, 8.79 mmol, 0.15 equiv) at room temperature. The resulting mixture was stirred about 5 min at room temperature, and then was charged with tert-butyl 3- iodoazetidine-1-carboxylate (16.2 g, 57.22 mmol, 1.00 equiv) in portions. The reaction mixture was allowed to stir about 1 h at room temperature. Then to the above mixture was added 1- (benzyloxy)-4-bromobenzene (15 g, 57.01 mmol, 1.00 equiv), SPhos (2.4 g, 5.85 mmol, 0.10 equiv), Pd2(dba)3 (3.6 g, 3.93 mmol, 0.07 equiv). The resulting mixture was stirred for another 3 hours at 50C under nitrogen atmosphere. The reaction was then quenched by the addition of 2000 mL water and the resulting mixture was extracted with ethyl acetate (3 x 700 mL). The organic layers were combined and washed with brine (3 x 700 ml), dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column eluting with ethyl acetate/petroleum ether (0% to 33% gradient). This resulted in 13 g (67.25%) of tert- butyl 3-[4-(benzyloxy)phenyl]azetidine-1-carboxylate as a yellow solid. LC/MS (ESI) m/z: (2711) 340.10 [M+1] +.
5%
Stage #1: 3-Iodo-azetidine-1-carboxylic acid tert-butyl ester With chloro-trimethyl-silane; zinc In tetrahydrofuran; ethylene dibromide at 20 - 65℃; for 1h;
Stage #2: 1-(benzyloxy)-4-bromobenzene With palladium (II) [1,1'-bis(diphenylphosphanyl)ferrocene] dichloride; copper (I) iodide at 70℃; for 4h;
434.1 Step 1: tert-Butyl 3-(4-benzyloxyphenyl)azetidine-1-carboxylate
To a suspension of zinc (251.2 mg, 3.840 mmol) in THF (2 mL) was added 1,2- dibromoethane (approximately 35.69 mg, 16.37 µL, 0.1900 mmol). The mixture was heated to 65 °C and allowed to cool to room temperature over 30 minutes. Chloro(trimethyl)silane (approximately 20.64 mg, 24.11 µL, 0.1900 mmol) was then added, and the mixture was stirred at room temperature for an additional 30 minutes. A solution of tert-butyl 3-iodoazetidine-1- carboxylate (809 mg, 2.858 mmol) in THF (0.8 mL) was added dropwise to the reaction mixture and the mixture was stirred 1 hour at room temperature. The mixture was quickly filtered through a syringe filter into a vessel containing 1-benzyloxy-4-bromo-benzene (500 mg, 1.900 mmol), Pd(dppf)Cl2 (approximately 69.54 mg, 0.09504 mmol) and copper iodide (approximately 181.0 mg, 0.9504 mmol). The reaction mixture was then stirred at 70 °C for 4 hours. The reaction mixture was filtered through Celite and concentrated to 1/3 of the volume under reduced pressure. To the remaining black solution was added EtOAc (25 mL) and a saturated aqueous sodium chloride solution (10 mL). The organic portion was concentrated to dryness under reduced pressure. The residue was purified via silica gel using 0 to 10% EtOAc in hexane to obtain tert-butyl 3-(4-benzyloxyphenyl)azetidine-1-carboxylate (32 mg, 5%).1H NMR (400 MHz, Chloroform-d) δ 77.47 - 7.29 (m, 5H), 7.25 - 7.21 (m, 2H), 7.00 - 6.92 (m, 2H), 5.06 (s, 2H), 4.30 (t, J = 8.7 Hz, 2H), 3.97 - 3.89 (m, 2H), 3.74 - 3.62 (m, 1H), 1.46 (s, 9H). ESI-MS m/z calc.339.18344, found 284.34 (M+1-56)+; Retention time: 3.42 minutes.
Stage #1: 1-(benzyloxy)-4-bromobenzene; 3-Iodo-azetidine-1-carboxylic acid tert-butyl ester With chloro-trimethyl-silane; zinc In ethylene dibromide
Stage #2: With palladium (II) [1,1'-bis(diphenylphosphanyl)ferrocene] dichloride; copper (I) iodide In ethylene dibromide
Stage #1: 3-Iodo-azetidine-1-carboxylic acid tert-butyl ester With chloro-trimethyl-silane; ethylene dibromide; zinc In N,N-dimethyl acetamide at 40℃; for 1h;
Stage #2: 1-(benzyloxy)-4-bromobenzene With copper (I) iodide; dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II) dichloromethane adduct In N,N-dimethyl acetamide at 90℃; for 12h;
90.1 Step 1: tert-butyl 3-(4-(benzyloxy)phenyl)azetidine-1-carboxylate (481-2)
A mixture of Zn (5.54 g, 84.8 mmol) in DMA (42 mL) was heated to 40°C, then 1,2- dibromoethane (2.55 g, 13.6 mmol) in DMA (6 mL) was added, followed by TMSCl (691 mg, 6.4 mmol) in DMA (6 mL), after stirred for 30 min, tert-butyl 3-iodoazetidine-1-carboxylate (12 g, 42.4) in DMA (18 mL) was added to the solution. The mixture was stirred at 40°C for 1 hr. TLC indicated the reaction was completed. The resulting Zinc reagent (0.574 M, 72 mL) solution was added to a mixture of Compound 481-1 (4 g, 15.2 mmol), Pd(dppf)Cl2.CH2Cl2 (621 mg, 760 µmol), CuI (290 mg, 1.5 mmol) in DMA (32 mL) then purged with N2 for three times, and the mixture was stirred at 90°C for 12 hrs. TLC indicated new spot with larger polarity was detected. The reaction mixture was quenched by pouring into sat.NH4Cl (100 mL) and extracted with EtOAc (30 mL x 2). The combined organic layers were washed with brine (20 mL x 3), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue, which was purified by column chromatography to give Compound 481-2 (2.8 g, 8.1 mmol, 53% yield) as yellow oil.
Multi-step reaction with 2 steps
1: caesium carbonate / N,N-dimethyl-formamide / 1 h / 70 °C
2: acetic acid; N-chloro-succinimide; water / 0.33 h / 20 °C
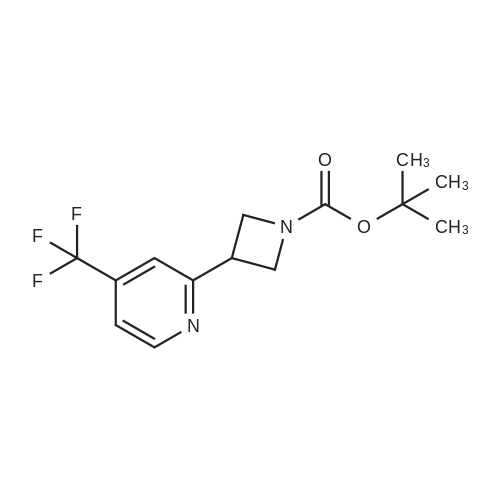
tert-butyl 3-[2-(trifluoromethyl)-4-pyridyl]azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
15%
With N,N,N,N,-tetramethylethylenediamine; chloro(2-dicyclohexylphosphino-2?,4?,6?-triisopropyl-1,1'-biphenyl)[2-(2-aminoethyl)phenyl]palladium(II) methyl tert-butyl ether; sodium caprylate; sodium chloride; zinc; In water; at 45℃; for 36h;Inert atmosphere;
A 20 mL screw-cap vial equipped with a magnetic stir bar and fitted with a teflon blow out septum was charged with Zinc powder (SIGMA ALDRICH, 163 mg, 2.50 mmol), XPhos-Pd- precatalyst {Angew. C em. Int. Ed. 2016, 55, 1849-1853., 43 mg, 5 mmol%), sodium octanoate (FLUOROCHEM, 83 mg, 0.5 mmol), sodium chloride (VWR, 1 16 mg, 2.00 mmol). The reaction tube was evacuated and backfilled with argon (3 times). 4-bromo-2- (trifluoromethyl)pyridine (APOLLO, 123.7mu, 1.00 mmol), tert-butyl 3-iodoazetidine-1- carboxylate (ACTIVATE SCIENTIFIC, 260 mu, 1 .50 mmol), TMEDA (SIGMA-ALDRICH, 377 mu, 2.50 mmol), 1 -octanol (LANCASTER, 237 mu, 1.50 mmol) and degassed water (3.3 mL) were then added. The screw-cap septum was quickly replaced by a new unpunctured septum under a flow of argon and the reaction mixture was stirred at 45 C for 36 h. The tube was cooled to rt and diluted with EtOAc. After shaking the reaction mixture, the contents were filtered through a small pad of Celite. The reaction tube and the Celite bed were washed with an additional 50 mL of EtOAc. The combined filtrates were transferred to a separating funnel and washed with 0.3M aq. solution of HCI (x2) and 0.3N aq. solution of NaOH (x2). The organic layer was separated, dried over (anh) Na2S04, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography using a linear gradient of DCM/MeOH to give title compound (100 mg, 15 %) as yellow oil. 1H NMR (300 MHz, CD2CI2) delta ppm: 8.69 (d, J = 5 Hz, 1 H), 7.69-7.65 (m, 1 H), 7.51 -7.47 (m, 1 H), 4.38 (t, J = 8.7 Hz, 2H), 3.99-3.93 (m, 2H), 3.89-3.76 (m, 1 H), 1 .46 (s, 9H). [ES+ MS] m/z 303 (MH+).
tert-butyl 2-(4-methoxybenzyl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
68%
With dichloro(1,5-cyclooctadiene)palladium(II); tetrabutylammomium bromide; caesium carbonate; ruphos; In 1,4-dioxane; water; at 80℃; for 24h;Inert atmosphere;
General procedure: General Procedure: A glass test tube, which was equipped with a magnetic stir bar and fitted with a stopper, was charged with Pd(COD)Cl2 (14.3 mg, 0.05 mmol) and RuPhos (46.7 mg, 0.10 mmol). Cs2CO3 (488.7 mg, 1.5 mmol, 3.0 equiv.), benzylboronic acid pinacol ester (1.0 mmol, 2.0 equiv.), N-Boc-3-iodo-azetidine (283.1 mg, 0.5 mmol, 1.0 equiv.) and 3 mL 1,4-dioxane/H2O (1:1). The reaction mixture was purged with argon gas three times. Under argon, the reaction mixture was allowed to stir at 80 oC for 24 h. The progress of the reaction was monitored by GC-MS. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate (15 mL) and washed with saturated brine (3×10 mL). The aqueous layer was extracted with ethyl acetate (2×15 mL). The combined organic layers were dried over MgSO4, concentrated in vacuo, and purified by silica gel flash chromatography.
tert-butyl 2-(4-fluorobenzyl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
72%
With dichloro(1,5-cyclooctadiene)palladium(II); tetrabutylammomium bromide; caesium carbonate; ruphos; In 1,4-dioxane; water; at 80℃; for 24h;Inert atmosphere;
General procedure: General Procedure: A glass test tube, which was equipped with a magnetic stir bar and fitted with a stopper, was charged with Pd(COD)Cl2 (14.3 mg, 0.05 mmol) and RuPhos (46.7 mg, 0.10 mmol). Cs2CO3 (488.7 mg, 1.5 mmol, 3.0 equiv.), benzylboronic acid pinacol ester (1.0 mmol, 2.0 equiv.), N-Boc-3-iodo-azetidine (283.1 mg, 0.5 mmol, 1.0 equiv.) and 3 mL 1,4-dioxane/H2O (1:1). The reaction mixture was purged with argon gas three times. Under argon, the reaction mixture was allowed to stir at 80 oC for 24 h. The progress of the reaction was monitored by GC-MS. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate (15 mL) and washed with saturated brine (3×10 mL). The aqueous layer was extracted with ethyl acetate (2×15 mL). The combined organic layers were dried over MgSO4, concentrated in vacuo, and purified by silica gel flash chromatography.
tert-butyl 2-(2-fluorobenzyl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
60%
With dichloro(1,5-cyclooctadiene)palladium(II); tetrabutylammomium bromide; caesium carbonate; ruphos; In 1,4-dioxane; water; at 80℃; for 24h;Inert atmosphere;
General procedure: General Procedure: A glass test tube, which was equipped with a magnetic stir bar and fitted with a stopper, was charged with Pd(COD)Cl2 (14.3 mg, 0.05 mmol) and RuPhos (46.7 mg, 0.10 mmol). Cs2CO3 (488.7 mg, 1.5 mmol, 3.0 equiv.), benzylboronic acid pinacol ester (1.0 mmol, 2.0 equiv.), N-Boc-3-iodo-azetidine (283.1 mg, 0.5 mmol, 1.0 equiv.) and 3 mL 1,4-dioxane/H2O (1:1). The reaction mixture was purged with argon gas three times. Under argon, the reaction mixture was allowed to stir at 80 oC for 24 h. The progress of the reaction was monitored by GC-MS. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate (15 mL) and washed with saturated brine (3×10 mL). The aqueous layer was extracted with ethyl acetate (2×15 mL). The combined organic layers were dried over MgSO4, concentrated in vacuo, and purified by silica gel flash chromatography.
tert-butyl 2-(4-chlorobenzyl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
42%
With dichloro(1,5-cyclooctadiene)palladium(II); tetrabutylammomium bromide; caesium carbonate; ruphos; In 1,4-dioxane; water; at 80℃; for 24h;Inert atmosphere;
General procedure: General Procedure: A glass test tube, which was equipped with a magnetic stir bar and fitted with a stopper, was charged with Pd(COD)Cl2 (14.3 mg, 0.05 mmol) and RuPhos (46.7 mg, 0.10 mmol). Cs2CO3 (488.7 mg, 1.5 mmol, 3.0 equiv.), benzylboronic acid pinacol ester (1.0 mmol, 2.0 equiv.), N-Boc-3-iodo-azetidine (283.1 mg, 0.5 mmol, 1.0 equiv.) and 3 mL 1,4-dioxane/H2O (1:1). The reaction mixture was purged with argon gas three times. Under argon, the reaction mixture was allowed to stir at 80 oC for 24 h. The progress of the reaction was monitored by GC-MS. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate (15 mL) and washed with saturated brine (3×10 mL). The aqueous layer was extracted with ethyl acetate (2×15 mL). The combined organic layers were dried over MgSO4, concentrated in vacuo, and purified by silica gel flash chromatography.
methyl 1-(1-(tert-butoxycarbonyl)azetidin-3-yl)-6-oxo-1,6-dihydropyridine-3-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
270 mg
With caesium carbonate; In N,N-dimethyl-formamide; at 25℃; for 16h;
To a solution of tert-butyl 3-iodoazetidine-l-carboxylate (1.25 g, 4.41 mmol) and <strong>[66171-50-4]methyl 6-hydroxypyridine-3-carboxylate</strong> (0.676 g, 4.42 mmol) in DMF (5 mL), was added Cs2C03(2.16 g, 6.62 mmol). The reaction mixture was stirred at 25 C for 16 hours. TLC showed the reaction was complete. The slurry was diluted with EA, and washed with water and brine. The combined organic layers were dried over anhydrous Na2S04, concentrated under reduced pressure and purified by flash chromatography to give methyl l-{ l-[(tert- butoxy)carbonyl]azetidin-3-yl}-6-oxo-l,6-dihydropyridine-3-carboxylate (270 mg) as a white solid. LC-MS (ESI) found: 309 [M+H]+.
tert-butyl 3-(3-amino-6-chloropyrazin-2-yl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
Zinc dust (activated by the procedure found in WO2011/143365, the disclosure of which is incorporated herein by reference in its entirety) (0.627 g, 9.59 mmol) was charged to a dry flask and suspended in DMA (2.5 mL). 1,2-Dibromoethane (0.031 mL, 0.36 mmol) and TMSCl (0.092 mL, 0.72 mmol) were added and the reaction was stirred for 25 min. tert-Butyl 3-iodoazetidine-1-carboxylate (2.04 g, 7.20 mmol, Oakwood) in DMA (6.0 mL) was added slowly to the mixture which was immersed in a water bath to keep the temperature below 65 C. The mixture was stirred for 1 hour and was degassed by bubbling a stream of nitrogen through the mixture for 5 minutes. (0976) A separate flask was charged with <strong>[76537-18-3]3-bromo-5-chloropyrazin-2-amine</strong> (0.50 g, 2.4 mmol, D-L Chiral Chemicals), dichloro[1,1?-bis(diphenylphosphino)ferrocene]palladium (II) dichloromethane adduct (0.118 g, 0.144 mmol) and copper(I) iodide (0.057 g, 0.30 mmol). DMA (6.0 mL) was added, and the mixture was degassed by bubbling a stream of nitrogen through the mixture for 5 minutes. The solution containing the organozinc in DMA was added, excluding any remaining zinc solids. The reaction mixture was then heated at 80 C. for 30 min. Upon cooling to room temperature, the reaction mixture was partitioned between water and EtOAc. The aqueous layer was extracted with two additional portions of EtOAc. The combined organic extracts were washed with water and brine, dried over sodium sulfate, filtered, and concentrated. The product was purified by flash chromatography, eluting with a gradient from 0-100% EtOAc in hexanes to afford product (0.62 g, 90%). LCMS calculated for C12H18ClN4O2(M+H)+: m/z=285.1, found: 285.1. 1H NMR (400 MHz, CDCl3) delta 7.96-7.90 (s, 1H), 4.78-4.65 (s, 2H), 4.35-4.22 (m, 4H), 3.79-3.69 (p, J=7.4 Hz, 1H), 1.48-1.44 (s, 9H).
90%
[1488] Zinc dust (0.627 g, 9.59 mmol) (activated by the procedure described in WO2011/143365, the disclosure of which is incorporated herein by reference in its entirety) was charged to a dry flask and suspended in DMA (2.5 mL). 1,2-Dibromoethane (0.031 mL, 0.36 mmol) and TMSCl (0.092 mL, 0.72 mmol) were added and the reaction was stirred for 25 min. tert-Butyl 3-iodoazetidine-1-carboxylate (2.04 g, 7.20 mmol) (Oakwood) in DMA (6.0 mL) was added slowly to the mixture which was immersed in a water bath to keep the temperature below 65C. The mixture was stirred for 1 hour and was degassed by bubbling a stream of nitrogen subsurface for 5 minutes.[1489] A separate flask was charged with <strong>[76537-18-3]3-bromo-5-chloropyrazin-2-amine</strong> (0.50 g, 2.4 mmol, D-L Chiral Chemicals), dichloro[1,1'-bis(diphenylphosphino)ferrocene]palladium (II) dichloromethane adduct (0.118 g, 0.144 mmol) and copper(I) iodide (0.057 g, 0.30 mmol). DMA (6.0 mL) was added, and the mixture was degassed by bubbling a stream of nitrogen subsurface for 5 minutes. The solution containing the organozinc in DMA generated above was added, excluding any remaining zinc solids. The reaction was then heated to 80C. for 30 min. Upon cooling to room temperature, the reaction mixture was partitioned between water and EtOAc. The aqueous layer was extracted with two additional portions of EtOAc. The combined organic extracts were washed with water and brine, dried over sodium sulfate, filtered and concentrated. The product was purified by flash chromatography, eluting with a gradient from 0-100% EtOAc in hexanes to afford product (0.62 g, 90%). LCMS calculated for C12H18ClN4O2 (M+H)+: m/z=285.1, found: 285.1. H NMR (400 MHz, CDCl3) delta 7.91 (s, 1H), 4.67 (s, 2H), 4.35-4.22 (m, 4H), 3.71 (p, J=7.4 Hz, 1H), 1.43 (s, 9H).
With 1,1'-bis-(diphenylphosphino)ferrocene; tetrabutyl ammonium fluoride; palladium diacetate In tetrahydrofuran; 1,4-dioxane at 60℃; for 12h; Inert atmosphere; regioselective reaction;
tert-butyl 3-(4-formyl-1H-pyrazol-1-yl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
75.71%
With caesium carbonate; In N,N-dimethyl-formamide;Heating;
To a stirred solution of lH-<strong>[35344-95-7]pyrazole-4-carbaldehyde</strong> 39-2 (1 g, 10.41 mmol) in DMF (10 mL) was added tert-butyl 3-iodoazetidine-l -carboxylate 39-1 (2.95 g, 10.41 mmol) and cesium carbonate (5.09 g, 15.61 mmol) and the reaction mixture was heated at 90C for 16 hours. Volatiles were removed and the residue was diluted with water and ethyl acetate. The layers were separated and the organic part was dried over Na^SCri and evaporated under reduced pressure to afford crude that was purify by column chromatography to afford tert-butyl 3-(4-formyl-l H-pyrazol-l-yl)azetidine- 1-carboxylate 39-3 (2 g, 7.88 mmol, 75.71% yield, 99% purity, 000) as off-white solid. LC MS: ES+ 252 0.
tert-butyl 3-(1H-imidazol-4-yl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With dipotassium peroxodisulfate; N-ethyl-N,N-diisopropylamine In water; dimethyl sulfoxide at 70℃; for 2h; Overall yield = 63 percent; chemoselective reaction;
tert-butyl 3-(2-(4-bromophenyl)-4-(trifluoromethyl)-1H-imidazol-1-yl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
59%
With [2,2]bipyridinyl; copper diacetate In dichloromethane at 110℃; for 18h;
1 Step 1. tert-butyl 3-(2-(4-bromophenyl)-4-(trifluoromethyl)-lH-imidazol-l-yl)azetidine-l- carboxylate
[00199] A mixture of 2-(4-bromophenyl)-4-(trifluoromethyl)-lH-imidazole (1.00 g, 3.44 mmol), tert-butyl 3-iodoazetidine-l-carboxylate (2.92 g, 10.3 mmol), and cesium carbonate (3.36 g, 10.3 mmol) in DMF (20 mL) was stirred for 18 h at 110 °C and then cooled to rt. The mixture was poured into water (20 mL) and then extracted with ethyl acetate (3 x 30 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The resulting crude product was purified by silica gel chromatography (eluting with 0: 100 to 60:40 ethyl acetate/petroleum ether) to afford tert-butyl 3-(2-(4-bromophenyl)-4- (trifluoromethyl)-lH-imidazol-l-yl)azetidine-l-carboxylate as orange oil (900 mg, 59%). LCMS (ESI, m/z) 446, 448 [M+H]+.
methyl 3-(1-(t-butoxycarbonyl)azetidin-3-yl)-1-methyl-1H-indazole-6-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
81.1%
Stage #1: tert-butyl-3-iodoazetidine-1-carboxylate With chloro-trimethyl-silane; ethylene dibromide; zinc In N,N-dimethyl-formamide at 40℃; for 0.5h; Inert atmosphere;
Stage #2: methyl 3-iodo-1-methyl-1H-indazole-6-carboxylate With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; copper(l) iodide In N,N-dimethyl-formamide at 50℃; for 1h; Inert atmosphere;
6.1 1) Preparation of methyl 3-(1-(tert-butoxycarbonyl)azetidin-3-yl)-1-methyl-1H-indazole-6-carboxylate
Add zinc powder (2.1g, 32.1mmol) to dry N,N-dimethylformamide (10mL), under nitrogen protection, place it in an oil bath at 40 and stir, add 1,2-dibromoethyl in sequence Alkane (0.45mL) and trimethylchlorosilane (0.75mL), and stirred at 40 for 0.5 hours, 1-Boc-3-iodoazetidine (4.54g, 16.05mmol) of N,N- The dimethylformamide (5 mL) solution was slowly added to the above reaction solution, reacted at 40°C for 0.5 hours, and the reaction solution was ready for use.The methyl 3-iodo-1-methyl-1H-indazole-6-carboxylate (2.53g, 8mmol, the preparation method can refer to Preparation Example 3), 1,1'-bisdiphenylphosphino two Ferrocene palladium dichloride (506mg) and CuI (506mg) were added to N,N-dimethylformamide (15mL), protected by nitrogen, stirred at 50, and the above-mentioned spare reaction solution was slowly added to it, 50 The reaction was stirred for 1 hour.The reaction solution was added to water (100mL), extracted with ethyl acetate three times (100mL×3), the organic phase was dried over anhydrous sodium sulfate and concentrated, and the residue was subjected to silica gel column chromatography (ethyl acetate: petroleum ether = 1: 4) Purified product (2.24g, yield 81.1%).
tert-butyl 3-[4-[1-(trifluoromethyl)cyclopropyl]phenyl]azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
66%
With nickel(II) chloride ethylene glycol dimethyl ether complex; tris-(trimethylsilyl)silane; [4,4’-bis(1,1-dimethylethyl)-2,2’-bipyridine-N1,N1‘]bis [3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl-N]phenyl-C]iridium(III) hexafluorophosphate; anhydrous sodium carbonate; 4,4'-di-tert-butyl-2,2'-bipyridine; In 1,2-dimethoxyethane; for 1h;Inert atmosphere; Irradiation; Sealed tube;
To a 20 mL vial, equipped with a stir bar, was added <strong>[1227160-18-0]1-bromo-4-(1-(trifluoromethyl)cyclopropyl)benzene</strong> (561 mg, 2.12 mmol, 1.0 equiv; CAS RN 1227160-18-0), tert-butyl 3-iodoazetidine-1-carboxylate (600 mg, 2.12 mmol, 1.0 equiv; CAS RN 254454-54-1), tris(trimethylsilyl)silane (527 mg, 653 μL, 2.12 mmol, 1.0 equiv), photocatalyst bis[3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridyl]phenyl]iridium(1+) 4-tert-butyl-2-(4-tert-butyl-2-pyridyl)pyridine hexafluorophosphate (23.8 mg, 21.2 μmol, 0.01 equiv; Ir[dF(CF3)ppy]2(dtbbpy))PF6, CAS RN 870987-63-6) and anhydrous sodium carbonate (449 mg, 4.24 mmol, 2.0 equiv). The vial was sealed and placed under Ar before dimethoxyethane (9 mL) was added. To a separate vial was added nickel(II) chloride ethylene glycol dimethyl ether complex (4.65 mg, 21.2 μmol, 0.01 equiv; CAS RN 29046-78-4) and 4,4'-di-tert-butyl-2,2'-bipyridine (5.68 mg, 21.2 μmol, 0.01 equiv). The vial was sealed, purged with Ar, and dimethoxyethane (4 mL) was added. The precatalyst solution was sonicated for 5 min, after which 2 mL were syringed into the reaction vessel. The reaction mixture was degassed with Ar and irradiated with a blue LED lamp (420 nm) for 1 h. The reaction was quenched by exposure to air, filtered and the solvent evaporated. The crude reaction mixture was purified by silica gel chromatography using an MPLC system eluting with a gradient of n-heptane:ethyl acetate (100:0 to 70:30) to furnish the title compound as a colorless solid (0.51 g, 66%). MS (ESI): m/z=286.1 [M+2H-tBu]+.
66%
With nickel(II) chloride ethylene glycol dimethyl ether complex; tris-(trimethylsilyl)silane; [4,4’-bis(1,1-dimethylethyl)-2,2’-bipyridine-N1,N1‘]bis [3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl-N]phenyl-C]iridium(III) hexafluorophosphate; anhydrous sodium carbonate; 4,4'-di-tert-butyl-2,2'-bipyridine; In 1,2-dimethoxyethane; for 1h;Sealed tube; Inert atmosphere; Irradiation;
To a 20 mL vial, equipped with a stir bar, was added 1-bromo-4-(1- (trifluoromethyl)cyclopropyl)benzene (561 mg, 2.12 mmol, 1.0 equiv; CAS RN 1227160-18-0), tert-butyl 3-iodoazetidine-1-carboxylate (600 mg, 2.12 mmol, 1.0 equiv; CAS RN 254454-54-1), tris(trimethylsilyl)silane (527 mg, 653 µL, 2.12 mmol, 1.0 equiv), photocatalyst bis[3,5-difluoro- 2-[5-(trifluoromethyl)-2-pyridyl]phenyl]iridium(1+) 4-tert-butyl-2-(4-tert-butyl-2- pyridyl)pyridine hexafluorophosphate (23.8 mg, 21.2 µmol, 0.01 equiv; Ir[dF(CF3)ppy]2(dtbbpy))PF6; CAS RN 870987-63-6) and anhydrous sodium carbonate (449 mg, 4.24 mmol, 2.0 equiv). The vial was sealed and placed under Ar before DME (9 mL) was added. To a separate vial was added nickel(II) chloride ethylene glycol dimethyl ether complex (4.65 mg, 21.2 µmol, 0.01 equiv; CAS RN 29046-78-4) and 4,4'-di-tert-butyl-2,2'-bipyridine (5.68 mg, 21.2 µmol, 0.01 equiv). The vial was sealed, purged with Ar, and DME (4 mL) was added. The precatalyst solution was sonicated for 5 min, after which 2 mL were syringed into the reaction vessel. The reaction mixture was degassed with Ar and irradiated with a blue LED lamp (420 nm) for 1 h. The reaction was quenched by exposure to air, filtered and the solvent evaporated. The crude reaction mixture was purified by silica gel chromatography using a MPLC system eluting with a gradient of n-heptane : ethyl acetate (100 : 0 to 70 : 30) to furnish the title compound as a colorless solid (0.51 g, 66 %). MS (ESI): m/z = 286.1 [M+2H-tBu]+.
tert-butyl 3-(3-chloro-1H-indazol-1-yl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91%
With tetrakis(actonitrile)copper(I) hexafluorophosphate; tributyl-amine; trimethylsilyl(cumyl) peroxide; N,N,N′,N′-tetramethyl-N″-tert-butylguanidine; In acetonitrile; tert-butyl alcohol; at 20℃; for 1h;Sealed tube; Inert atmosphere;
General procedure: An oven-dry tube equipped with a stirring bar was charged with the alkyl iodide (0.20 mmol, 1.0 equiv.), the N-nucleophile (0.30 mmol, 1.5 equiv.) and [Cu(MeCN)4]PF6 (7.5 mg, 0.020 mmol, 10 mol%). The tube was capped with a Supelco aluminium crimp seal with septum (PTFE/butyl) and evacuated and refilled with N2 (three times). Dry and degassed CH3CN (1.0 ml), t-BuOH (1.0 ml), (n-Bu)3N (238 μl, 1.0 mmol, 5.0 equiv.), BTMG (80 μl, 0.40 mmol, 2.0 equiv.) were sequentially added. The resulting solution was treated with cumOOTMS (224 mg, 238 μl, 1.00 mmol, 5.0 equiv.) dropwise under vigorous stirring. After 1 h the tube was opened and the mixture diluted with brine (2 ml) and EtOAc (2 ml). The organic layer was separated and the aqueous layer was extracted with EtOAc (5 ml × 2). The combined organic layers were dried (MgSO4), filtered and evaporated. The crude material was purified by flash column chromatography on silica gel.
3 3.
3. Synthesis of compound t-butyl 3-(4-iodo-1H-pyrazol-1-yl)azetidine-1-carboxylate 4-Iodo-1H-pyrazole (400 mg, 1.40 mmol), N-Boc-3-iodoazetidine (274 mg, 1.41 mmol), and K2CO3 (292 mg, 2.12 mmol) were dissolved in 5 mL DMF. The reaction solution was heated to 85 °C and stirred overnight, and then cooled to room temperature. The reaction solution was added with water and ethyl acetate for extraction. The organic layer was washed with saturated brine, dried, rotatory evaporated to dry, and purified by silica gel column chromatography, to provide compound t-butyl 3-(4-iodo-1H-pyrazol-1-yl)azetidine-1-carboxylate (380 mg, 1.08 mmol), with a yield of 77.0%.
77%
With potassium carbonate In N,N-dimethyl-formamide at 85℃;
3. Synthesis of compound t-butyl 3-(4-iodo-1H-pyrazol-1-yl)azetidine-1-carboxylate
4-Iodo-1H-pyrazole (400 mg, 1.40 mmol), N-Boc-3-iodoazetidine (274 mg, 1.41 mmol), and K2CO3 (292 mg, 2.12 mmol) were dissolved in 5 mL DMF. The reaction solution was heated to 85 °C and stirred overnight, and then cooled to room temperature. The reaction solution was added with water and ethyl acetate for extraction. The organic layer was washed with saturated brine, dried, rotatory evaporated to dry, and purified by silica gel column chromatography, to provide compound t-butyl 3-(4-iodo-1H-pyrazol-1-yl)azetidine-1-carboxylate (380 mg, 1.08 mmol), with a yield of 77.0%.
77%
With potassium carbonate In N,N-dimethyl-formamide at 85℃;
3. Synthesis of compound t-butyl 3-(4-iodo-1H-pyrazol-1-yl)azetidine-1-carboxylate
4-Iodo-1H-pyrazole (400 mg, 1.40 mmol), N-Boc-3-iodoazetidine (274 mg, 1.41 mmol), and K2CO3 (292 mg, 2.12 mmol) were dissolved in 5 mL DMF. The reaction solution was heated to 85 °C and stirred overnight, and then cooled to room temperature. The reaction solution was added with water and ethyl acetate for extraction. The organic layer was washed with saturated brine, dried, rotatory evaporated to dry, and purified by silica gel column chromatography, to provide compound t-butyl 3-(4-iodo-1H-pyrazol-1-yl)azetidine-1-carboxylate (380 mg, 1.08 mmol), with a yield of 77.0%.
tert-butyl 3-(5-bromopyrazin-2-yl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
18%
Stage #1: 3-Iodo-azetidine-1-carboxylic acid tert-butyl ester With chloro-trimethyl-silane; ethylene dibromide; zinc powder In tetrahydrofuran; N,N-dimethyl acetamide at 60℃; for 0.25h; Inert atmosphere;
Stage #2: 2-bromo-5-iodo-pyrazine With copper (I) iodide; dichloro(1,1'-bis(diphenylphosphanyl)ferrocene)palladium(II) dichloromethane adduct In tetrahydrofuran; N,N-dimethyl acetamide at 80℃; for 2h; Inert atmosphere;
1 Step 1: tert-Butyl 3-(5-bromopyrazin-2-yl)azetidine-1-carboxylate
To a suspension of zinc dust (346.4 mg, 5.30 mmol, 1.5 equiv) in THF (8 mL) were added 1,2- dibromoethane (66.4 mg, 30.4 mL, 0.35 mmol, 0.10 equiv) and chlorotrimethylsilane (44.9 mL, 0.35 mmol, 0.10 equiv) and the suspension was stirred at 60 °C for 15 min. A solution of tert- butyl 3-iodoazetidine-1-carboxylate (1.0 g, 613.5 mL, 3.53 mmol, 1.0 equiv, CAS RN 254454- 54-1) in DMA (8 mL) was added and the reaction mixture was stirred at 60 °C for 15 min. The reaction mixture was cooled down to RT and 2-bromo-5-iodo-pyrazine (1.06 g, 3.71 mmol, 1.05 equiv; CAS RN 622392-04-5), 1,1'-bis(diphenylphosphino)ferrocene-palladium(II) dichloride dichloromethane complex (144.2 mg, 0.18 mmol, 0.05 equiv) and copper(I) iodide (34.3 mg, 0.18 mmol, 0.05 equiv) were added and stirring continued at 80 °C for 2 h. The reaction mixture was diluted with ethyl acetate and water and then filtered over Dicalite. The filtrate was extracted with ethyl acetate (3 x 100 mL), the combined organic phase was then washed with a sat. aqueous NaCl solution, dried over MgSO4 and concentrated. The crude product was purified by silica gel chromatography eluting with a gradient of n-heptane : ethyl acetate (100 : 0 to 70 : 30) to give the title compound as a colorless solid (212 mg, 18 %). MS (ESI): m/z = 260.0 [M+2H-tBu]+.
tert-butyl 3-(6-bromo-3-pyridyl)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
38%
With nickel(II) iodide; rac-(1R,2R)-2-aminocyclohexan-1-ol; sodium bis-(trimethyl-silyl)amide In tetrahydrofuran; isopropanol at 20 - 80℃; for 0.666667h; Inert atmosphere; Microwave irradiation;
1 Step 1: tert-Butyl 3-(6-bromo-3-pyridyl)azetidine-1-carboxylate
To a solution of tert-butyl 3-iodoazetidine-1-carboxylate (1.0 g, 3.53 mmol, 1.0 equiv; CAS RN 254454-54-1) in isopropanol (10 mL) were added (6-bromo-3-pyridyl)boronic acid (1.43 g, 7.06 mmol, 2.0 equiv; CAS RN 223463-14-7), rac-(1S,2S)-2-aminocyclohexanol (24.4 mg, 0.21 mmol, 0.06 equiv), nickel(II) iodide (66.2 mg, 0.21 mmol, 0.06 equiv) and sodium bis(trimethylsilyl)amide (3.53 mL, 7.06 mmol, 2.0 equiv; 2 M in THF) and the reaction mixture was stirred under Ar at RT for 10 min before it was heated to 80 °C for 30 min by microwave irradiation. The reaction mixture was poured on water and ethyl acetate and the layers were filtered. The aqueous layer was extracted twice with ethyl acetate. The combined organic layers were washed with water, dried over MgSO4, filtered, treated with silica gel and evaporated. The crude compound was purified by silica gel chromatography using a MPLC system eluting with a gradient of n-heptane : ethyl acetate (100 : 0 to 50 : 50) to get the title compound as a colorless semi-solid (0.44 g, 38 %). MS (ESI): m/z = 315.1 [M+H]+.
tert-butyl 3-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)oxy)azetidine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
36%
With potassium carbonate In N,N-dimethyl-formamide at 70℃;
39.1 Step 1: Preparation of tert-butyl 3-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)oxy)azetidine-1-carboxylate (39- One)
General procedure: 3-(4-hydroxy-1-oxoisoindolin-2-yl)piperidine-2,6-dione (297 mg, 1.14 mmol), tert-butyl 3-iodoazetidine-1-carboxylate (646) mg, 2.28 mmol) and potassium carbonate (315 mg, 2.28 mmol) in N,N-dimethylformamide (3 mL) was stirred at 70°C. After stirring overnight, tert-butyl 3-iodoazetidine-1-carboxylate (200 mg) was further added and stirred for 4 hours. Saturated NH4Cl aqueous solution was added to the reaction mixture, stirred, filtered, washed with water and ethyl acetate, and dried under vacuum to obtain the title compound (150 mg, 36%).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping