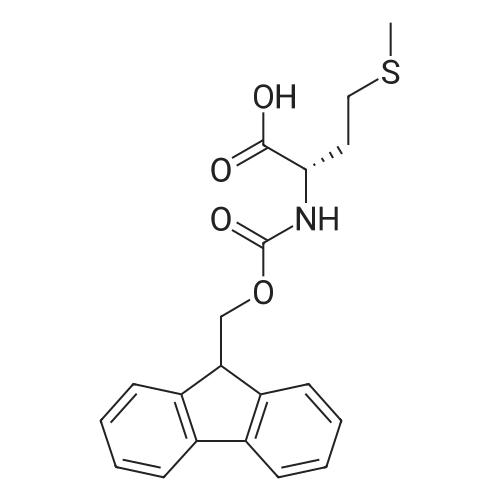
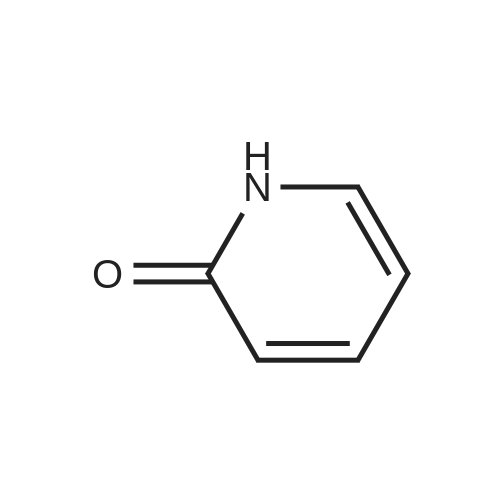
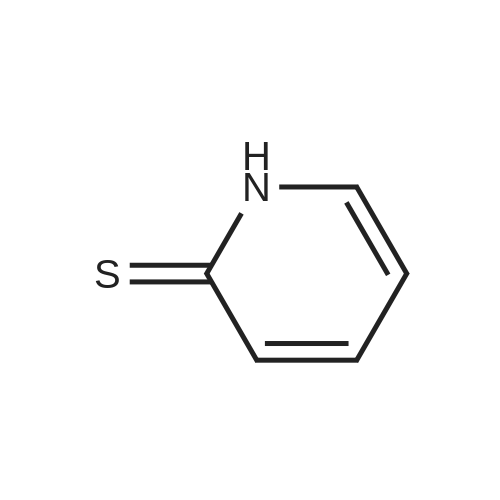
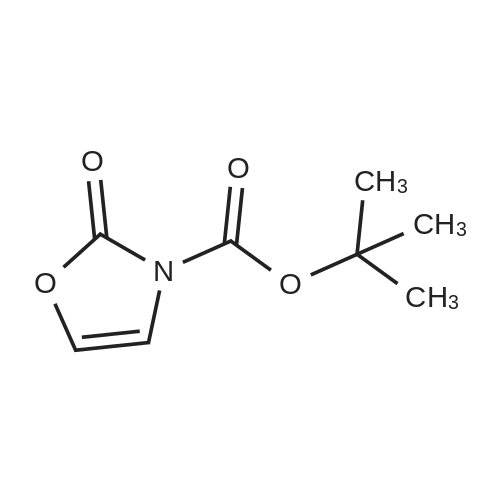
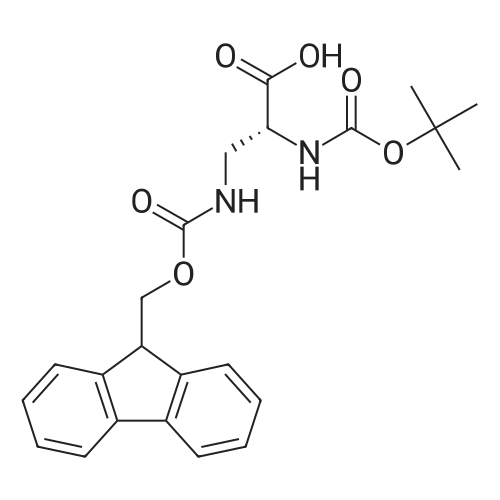
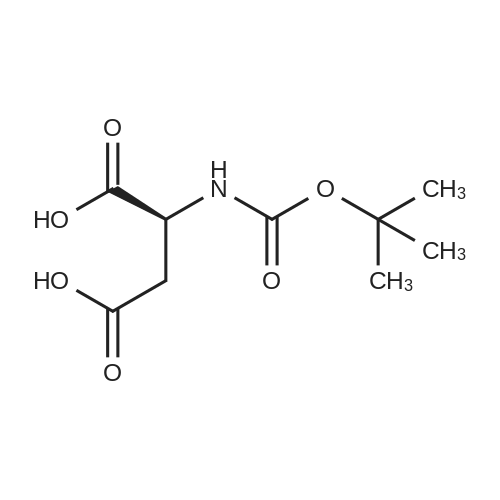
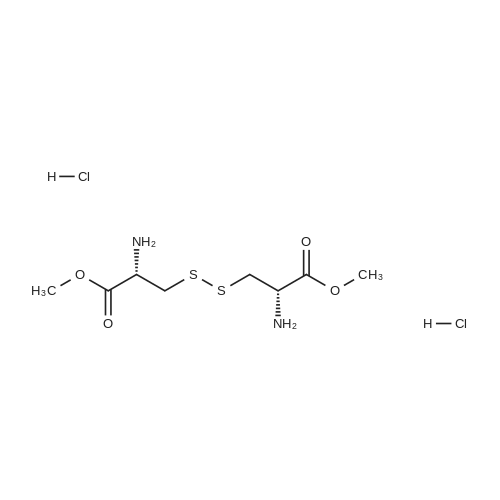
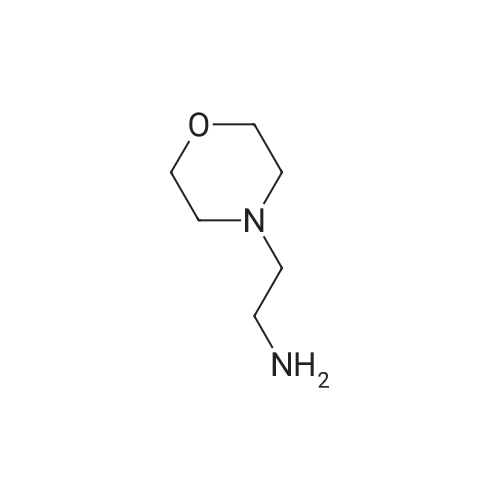
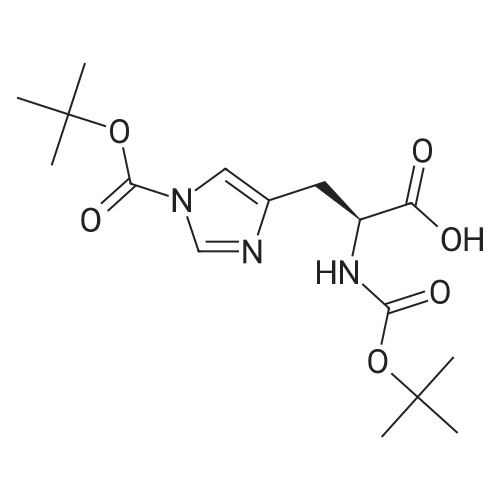
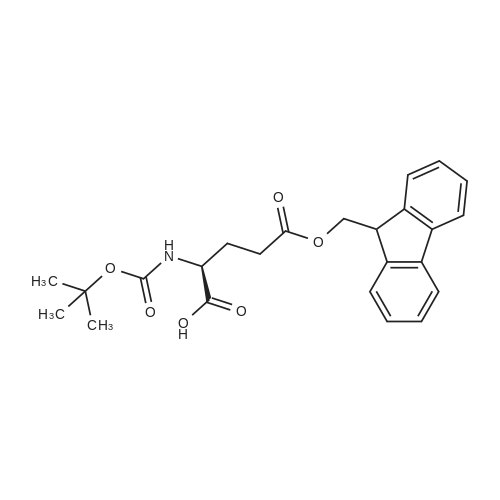
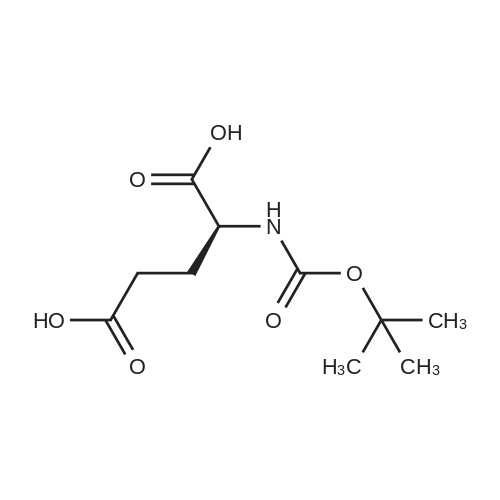
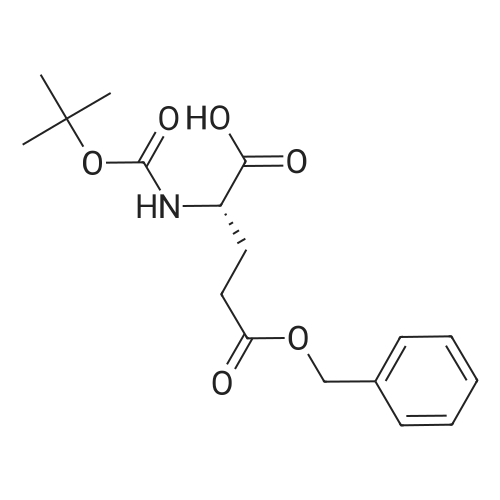
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of the Chemical Society, Dalton Transactions: Inorganic Chemistry (1972-1999), 1984, p. 2305 - 2308
2
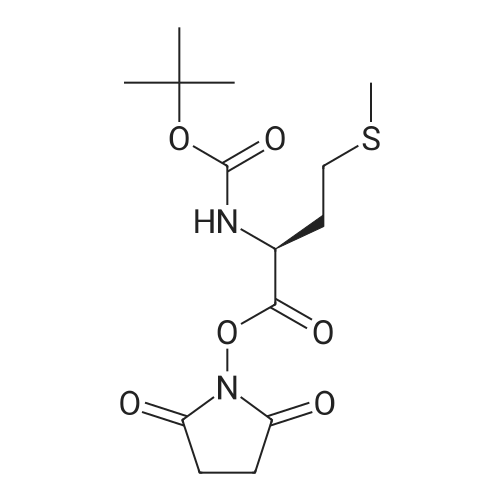
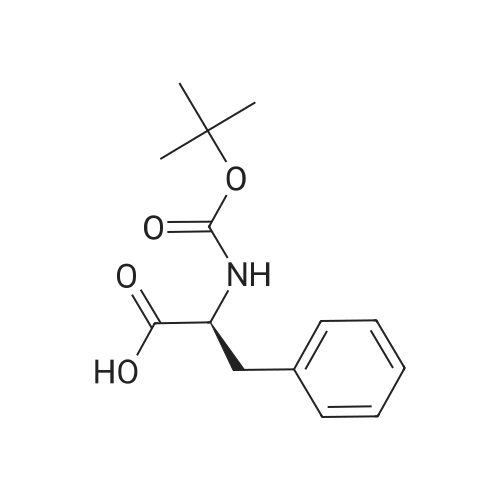
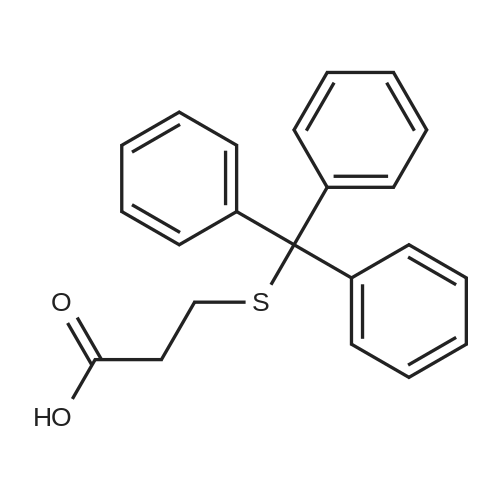
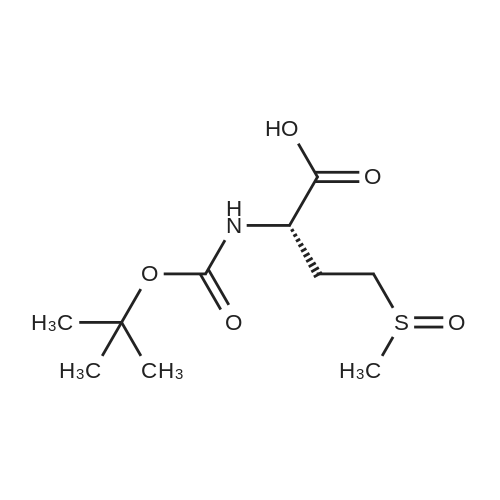
[ 63-68-3 ]
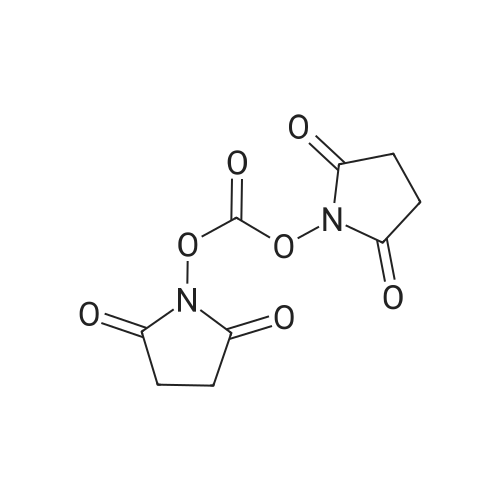
[ 24424-99-5 ]
[ 2488-15-5 ]
Yield
Reaction Conditions
Operation in experiment
95%
With triethylamine; sodium hydroxide In water; acetonitrile at 0 - 25℃; for 12 h;
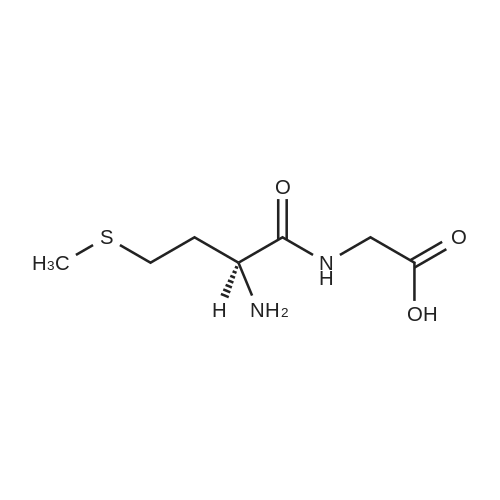
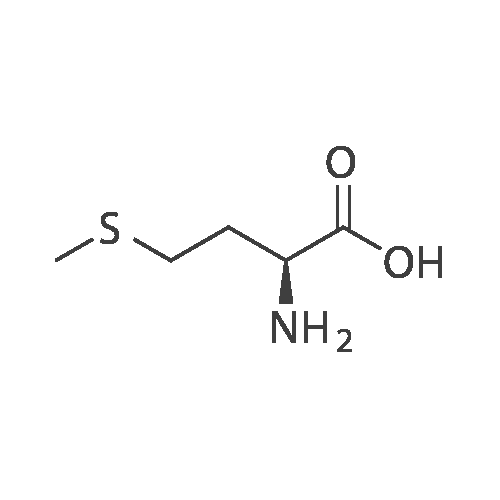
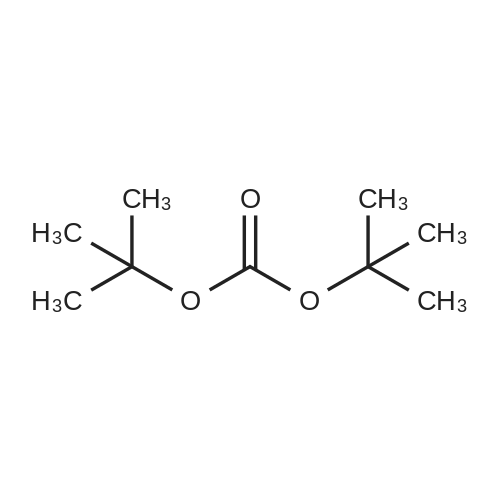
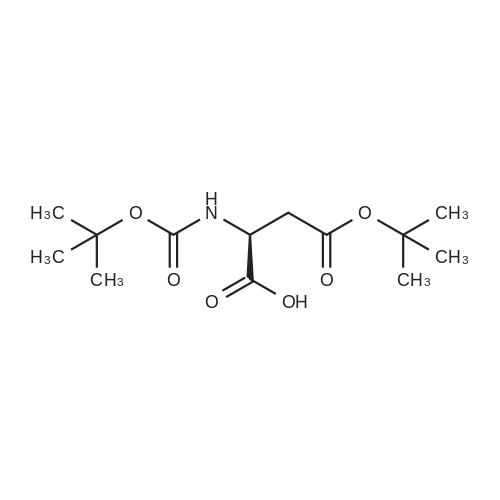
Example 1[0040][0041] 7.2 g Methionine was dissolved in a mixture of 50 mL water and 50 mL acetonitrile. To this solution was added2 g NaOH (0.05 mol). The obtained mixture was cooled down to 0°C and then 10.9 g di-t-butyl dicarbonate (0.05 mol)was added. After addition, the mixture was warmed to room temperature (24-25°C) and reacted for 12 h. Acetonitrilewas removed by distillation. Potassium carbonate was added to the residue and the pH thereof was adjusted to 12. Afterextracted with 50 mL dichloromethane twice, the organic layers were discarded. To the aqueous layer was added 1 Ndilute hydrochloric acid to adjust the pH to 6. After extracted with 50 mL dichloromethane twice, the organic layers werecollected together and washed with 50 mL saturate solution of sodium chloride and then dried over anhydrous sodiumsulfate, followed by concentration to obtain a viscous product (11.4 g). The yield was 95percent.1H NMR (500 MHz, CDCl3) δ 11.62 (br, 1 H), 6.91 (br, 1 H), 4.40 (m. 1H), 2.52 (t, J = 4.8 Hz, 2 H), 2.05 (s, 3H), 1.92∼2.15(m. 2 H), 1.42 (s, 9 H). Ms (M++1): 250.
60%
With sodium hydroxide In water; acetonitrile at 0 - 20℃; for 12 h;
To the solution of L-methionine (43) (2 g, 13.40 mmol) in water (12 mL) and acetonitrile (12 mL) was added NaOH (0.804 g, 20.11 mmol) and boc-anhydride (4.67 mL, 20.11 mmol) at 0 °C. The reaction mixture was stirred at room temperature for 12h. After completion, reaction mixture was concentrated under vacuum to remove acetonitrile and the remaining acqueous layer was diluted with 50 mL water. Aqueous layer was washed with ethylacetate (2 x 50 mL). The aqueous phase was then adjusted to pH ~ 4-5 using aqueous 1N HCl solution and extracted with dichloromethane (4 x 50 mL). Combined organic extracts were washed with brine (100 mL), dried over Sodium sulphate and evaporated under reduced pressure to yield (tert-butoxycarbonyl)-L-methionine (44) as a gummy compound (0.6 g, 8.02 mmol, 60 percent yield) 1H NMR (400 MHz, DMSO-d6): δ 12.47 (br-s, 1H), 7.10 (d, J = 8.0 Hz, 1H), 4.00 (dd, J = 8.3, 4.3 Hz, 1H), 2.60-2.45 (m, 2H), 2.03 (s, 3H), 1.94-1.75 (m, 2H), 1.38 (s, 9H) MS (ESI): 248.2 as [M-1]in -Ve mode.
Reference:
[1] Antimicrobial Agents and Chemotherapy, 2017, vol. 61, # 3,
[2] Tetrahedron Letters, 1994, vol. 35, # 14, p. 2121 - 2124
[3] Patent: EP2647624, 2013, A1, . Location in patent: Paragraph 0040; 0041
[4] Organic Syntheses, 1985, vol. 63, p. 160 - 160
[5] Tetrahedron Letters, 2018, p. 4267 - 4271
[6] Chemistry of Natural Compounds, 1979, vol. 15, p. 471 - 476[7] Khimiya Prirodnykh Soedinenii, 1979, vol. 15, p. 543 - 548
[8] Acta chemica Scandinavica. Series B: Organic chemistry and biochemistry, 1986, vol. 40, # 4, p. 242 - 249
[9] Acta chemica Scandinavica. Series B: Organic chemistry and biochemistry, 1986, vol. 40, # 4, p. 250 - 256
[10] Journal of the Chemical Society - Series Chemical Communications, 1989, # 22, p. 1740 - 1742
[11] Journal of Medicinal Chemistry, 2003, vol. 46, # 16, p. 3502 - 3507
[12] Bioorganic and medicinal chemistry letters, 2004, vol. 14, # 1, p. 275 - 278
[13] Organic letters, 2002, vol. 4, # 23, p. 4005 - 4008
[14] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 23, p. 9984 - 9990
[15] Bioorganic and Medicinal Chemistry, 2010, vol. 18, # 2, p. 887 - 895
[16] Journal of Enzyme Inhibition and Medicinal Chemistry, 2011, vol. 26, # 4, p. 506 - 513
[17] Zeitschrift fur Naturforschung - Section B Journal of Chemical Sciences, 2012, vol. 67, # 7, p. 731 - 746
[18] Molecules, 2014, vol. 19, # 10, p. 16349 - 16372
[19] Chemistry - A European Journal, 2015, vol. 21, # 51, p. 18589 - 18593
[20] Molecules, 2016, vol. 21, # 2,
[21] Organic and Biomolecular Chemistry, 2017, vol. 15, # 40, p. 8493 - 8498
[22] Chemistry - An Asian Journal, 2018, vol. 13, # 4, p. 400 - 403
3
[ 63-68-3 ]
[ 34619-03-9 ]
[ 2488-15-5 ]
Yield
Reaction Conditions
Operation in experiment
95%
Stage #1: With sodium hydroxide In water; acetonitrile at 0 - 45℃; for 12 h; Stage #2: With hydrogenchloride In water
7.2 g Methionine was dissolved in a mixture of 50 mL water and 50 mL acetonitrile. To this solution was added 2 g NaOH (0.05 mol). The obtained mixture was cooled down to 0° C. and then 10.9 g di-t-butyl dicarbonate (0.05 mol) was added. After addition, the mixture was warmed to room temperature (24-25° C.) and reacted for 12 h. Acetonitrile was removed by distillation. Potassium carbonate was added to the residue and the pH thereof was adjusted to 12. After extracted with 50 mL dichloromethane twice, the organic layers were discarded. To the aqueous layer was added 1N dilute hydrochloric acid to adjust the pH to 6. After extracted with 50 mL dichloromethane twice, the organic layers were collected together and washed with 50 mL saturate solution of sodium chloride and then dried over anhydrous sodium sulfate, followed by concentration to obtain a viscous product (11.4 g). The yield was 95percent. 1H NMR (500 MHz, CDCl3) δ 11.62 (br, 1H), 6.91 (br, 1H), 4.40 (m. 1H), 2.52 (t, J=4.8 Hz, 2H), 2.05 (s, 3H), 1.922.15 (m. 2H), 1.42 (s, 9H). Ms (M++1): 250.
Reference:
[1] Chemistry Letters, 1984, p. 237 - 238
[2] Journal of the Chemical Society, Dalton Transactions: Inorganic Chemistry (1972-1999), 1984, p. 2305 - 2308
6
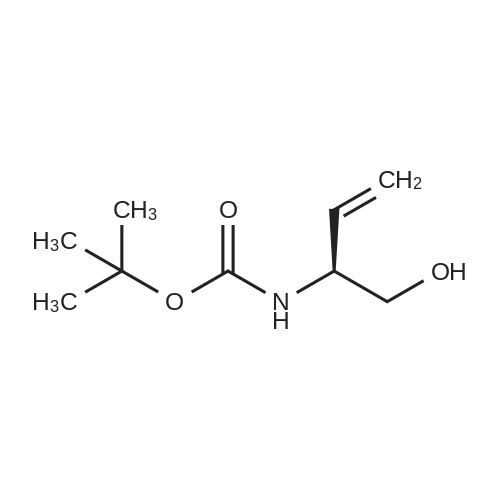
[ 63-68-3 ]
[ 130384-98-4 ]
[ 2488-15-5 ]
Reference:
[1] Synlett, 2011, # 14, p. 2013 - 2016
With sodium tetrahydroborate; iodine In tetrahydrofuran; methanol at 0 - 5℃; for 2 h; Reflux
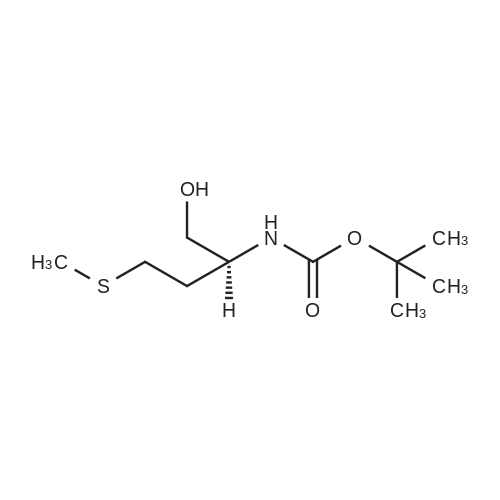
[0060] Into a 500 mL three-necked flask were added the starting compound as shown in the above route (49.8 g, 0.5mol) and 250 mL tetrahydrofuran. The temperature of the reaction mixture was adjusted to about 0-5°C in an ice-saltbath with agitation. To this mixture, sodium borohydride (19 g, 0.5 mol) was added slowly, followed by the addition of50 mL methanol. After addition, 100 mL solution of iodine (127 g, 0.5 mol) in tetrahydrofuran was added. Then, thereaction system was warmed to reflux. The reaction was continued for 2 h under agitation. After the raw material wasconsumed under the detecting of TLC, the temperature of the reaction mixture was adjusted with an ice-water bath.Saturated solution of ammonia chloride was added to quench the reaction. After 100 mL THF was evaporated out by arotary evaporator under reduced pressure, the residue was extracted with ethyl acetate (300 mL32).·The obtainedorganic layer was washed with dilute hydrochloric acid, followed by saturated sodium hydrogen carbonate, and finallyaqueous solution of sodium chloride. After drying and concentrating, a crude product (120 g) was obtained as an oil.The crude product was further treated by column chromatography to obtain a purified product (97.5 g). The yield was 85percent.1H NMR (400 MHz, CDCl3) δ 11.64(br, 1 H), 6.85 (br, 1 H), 4.55∼4.48 (m, 1 H), 2.53 (t, J = 4.9 Hz, 2H), 3.44 (s, 3H),2.05 (s, 3H), 2.02∼1.87 (m, 2H), 1.48 (s, 9H). Ms (M++1): 236.
85%
With methanol; sodium tetrahydroborate; iodine In tetrahydrofuran at 0 - 5℃; for 2 h; Reflux
Into a 500 mL three-necked flask were added the starting compound as shown in the above route (49.8 g, 0.5 mol) and 250 mL tetrahydrofuran. The temperature of the reaction mixture was adjusted to about 0-5° C. in an ice-salt bath with agitation. To this mixture, sodium borohydride (19 g, 0.5 mol) was added slowly, followed by the addition of 50 mL methanol. After addition, 100 mL solution of iodine (127 g, 0.5 mol) in tetrahydrofuran was added. Then, the reaction system was warmed to reflux. The reaction was continued for 2 h under agitation. After the raw material was consumed under the detecting of TLC, the temperature of the reaction mixture was adjusted with an ice-water bath. Saturated solution of ammonia chloride was added to quench the reaction. After 100 mL THF was evaporated out by a rotary evaporator under reduced pressure, the residue was extracted with ethyl acetate (300 mL×2). The obtained organic layer was washed with dilute hydrochloric acid, followed by saturated sodium hydrogen carbonate, and finally aqueous solution of sodium chloride. After drying and concentrating, a crude product (120 g) was obtained as an oil. The crude product was further treated by column chromatography to obtain a purified product (97.5 g). The yield was 85percent. 1H NMR (400 MHz, CDCl3) δ 11.64 (br, 1H), 6.85 (br, 1H), 4.554.48 (m, 1H), 2.53 (t, J=4.9 Hz, 2H), 3.44 (s, 3H), 2.05 (s, 3H), 2.021.87 (m, 2H), 1.48 (s, 9H). Ms (M++1): 236.
Reference:
[1] Antimicrobial Agents and Chemotherapy, 2017, vol. 61, # 3,
[2] Patent: EP2647624, 2013, A1, . Location in patent: Paragraph 0059; 0060
[3] Patent: US2013/281695, 2013, A1, . Location in patent: Paragraph 0108-0110
[4] Organic Letters, 2005, vol. 7, # 5, p. 847 - 849
[5] Chemical and Pharmaceutical Bulletin, 1982, vol. 30, # 5, p. 1921 - 1924
[6] Synthesis, 1990, p. 299 - 301
[7] Journal of Medicinal Chemistry, 2014, vol. 57, # 13, p. 5748 - 5763
[8] Antimicrobial Agents and Chemotherapy, 2018, vol. 62, # 12,
23
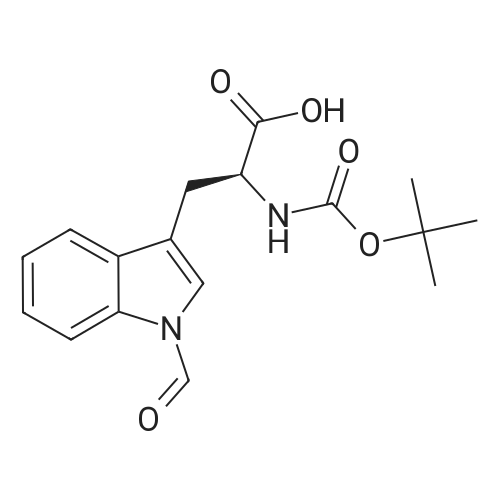
[ 6066-82-6 ]
[ 2488-15-5 ]
[ 3845-64-5 ]
Reference:
[1] Chemische Berichte, 1980, vol. 113, # 11, p. 3511 - 3516
[2] Journal of Organic Chemistry, 1991, vol. 56, # 19, p. 5606 - 5610
[3] Journal of the Chemical Society, Dalton Transactions: Inorganic Chemistry (1972-1999), 1984, p. 2305 - 2308
[4] European Journal of Pharmacology, 2001, vol. 411, # 3, p. 327 - 333
[5] Journal of Medicinal Chemistry, 2003, vol. 46, # 21, p. 4543 - 4551
[6] Journal of the American Chemical Society, 2001, vol. 123, # 6, p. 1023 - 1035
[7] Organic Letters, 2007, vol. 9, # 22, p. 4423 - 4426
[8] Chemistry Letters, 2013, vol. 42, # 6, p. 601 - 603
24
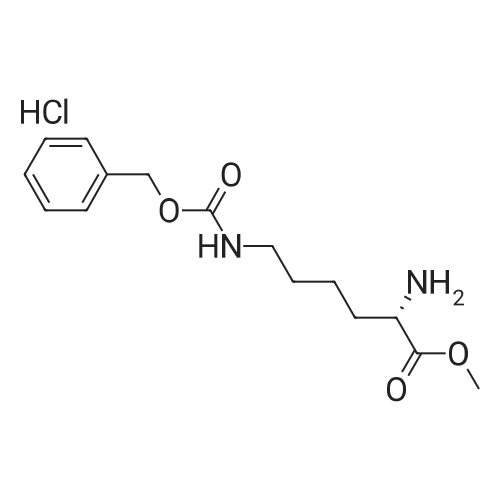
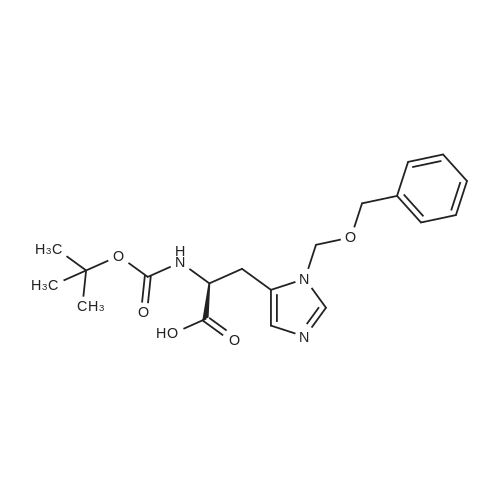
[ 74124-79-1 ]
[ 2488-15-5 ]
[ 3845-64-5 ]
Reference:
[1] Journal of Organic Chemistry, 2014, vol. 79, # 9, p. 4008 - 4017
With benzotriazol-1-ol; N-ethyl-N,N-diisopropylamine; diisopropyl-carbodiimide; In tetrahydrofuran; at 0 - 20℃; for 2h;
Preparation of scaffolds (Figure 7); Figure 7: Boc-(S) Met-(S) Lys-(Cbz)-OMe (compound 15); (S) Lysine-(Cbz)-methyl ester hydrochloride (1 g, 3 mmol), HOBT (0.4 g, 3 mmol), t-butyloxycarbonyl-(S)-Methionine (0.8 g, 3 mmol) and N,N- Diisopropylethylamine (1.05 mL, 6 mmol) were dissolved in dry THF (1.5 mL), the solution was cooled in an ice- water bath and diisopropylcarbodiimide (0.4 g, 3.15 mmol) was added. Stirring was continued for 1 h at 0C and an additional hour at rt. The solvent was evaporated in vacuo. A mixture of EtOAc (15 mL) and sat. NaHCO3 (7.5 mL) was added to the residue and the organic phase was sequentially extracted with 10% citric acid in water, sat NaHCO3 and water (7.5 mL each). The solution was dried over anhydrous Na2SO4, filtered and evaporated to dryness. The residue was triturated with hexane, filtered, washed with hexane and dried. The crude dipeptide derivative compound 15 (Figure 7) (1.80 g,) was purified by chromatography on basic alumina (EtOAc) (1.46 g, 84 % yield) 1H NMR delta 8.89 (bd, 1H), 7.85 (s, 5H), 5.11 (s, 2H), 3.71 (s, 3H), 3.2 (m, 2H ), 2.5 (t, J=6 Hz, 2H), 2.1-2.3 (m. 5H), 2.1-1.1 (m, 8H ), 1.42 (s, 9H).
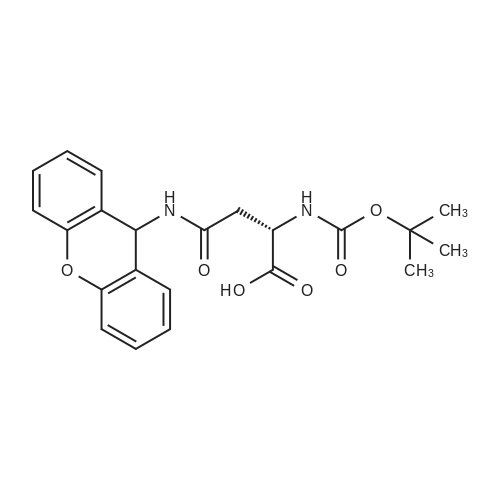
t-Boc-Leu-OCH2-phenylacetamidomethyl resin, t-Boc-Asp-(β-OFm)-OH[ No CAS ]
(1S,4S,7S,10S,13S,16S)-7-Benzyl-10-(1H-indol-3-ylmethyl)-4-isobutyl-16-(2-methylsulfanyl-ethyl)-2,5,8,11,14,17,20-heptaaza-bicyclo[11.5.4]docosane-3,6,9,12,15,18,21-heptaone[ No CAS ]
methyl (6S,9R,14R)-14-((S)-2-((tert-butoxycarbonyl)amino)-4-(methylthio)butanamido)-9-(methoxycarbonyl)-2,2-dimethyl-6-(2-(methylthio)ethyl)-4,7-dioxo-3-oxa-11,12-dithia-5,8-diazapentadecan-15-oate[ No CAS ]
3-<i>tert</i>-butoxycarbonylamino-<i>N</i>-[3-methylsulfanyl-1-(2-morpholin-4-yl-ethylcarbamoyl)-propyl]-succinamic acid <i>tert</i>-butyl ester[ No CAS ]
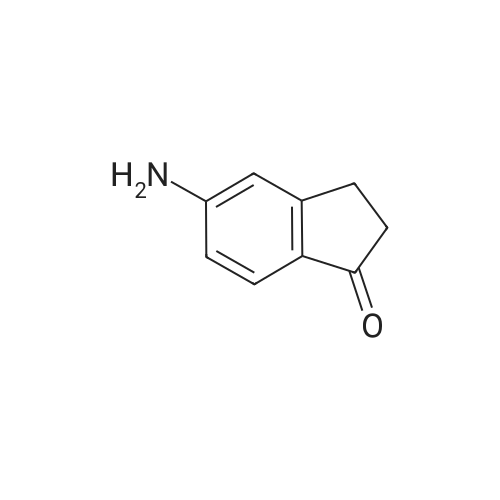
A suspension of N-t-butoxycarbonyl-L-methionine (9.28g) and HATU (15.0g) in anhydrous DCM (200ml) was treated with N,N-diisopropylethylamine (7.14ml) and stirred for 20min. The suspension was treated with <strong>[3470-54-0]5-amino-1-indanone</strong> (5.0g) in 1g portions over 2min. The reaction mixture was stirred at room temperature for 20h and the resulting suspension was partitioned between DCM (2 x 100ml) and saturated aqueous sodium hydrogen carbonate solution (150ml). The organic extracts were combined and washed sequentially with saturated aqueous sodium hydrogen carbonate solution (200ml) and brine. The organic phase was then passed through a hydrophobic frit and evaporated to afford a black oil. After purification on silica gel (Art 9385) eluting with a DCM : MeOH gradient, the column fractions were combined, evaporated to dryness and re-dissolved in DCM (200ml) and washed with saturated aqueous sodium hydrogen carbonate solution (3x 250ml), brine, passed through a hydrophobic frit and evaporated to give the title compound as coloured foam (12.55g, contaminated with (CH3)2NCON(CH3)2). Mass spectrum: Found : MH+ 379 H.p.l.c. Rt 2.97min
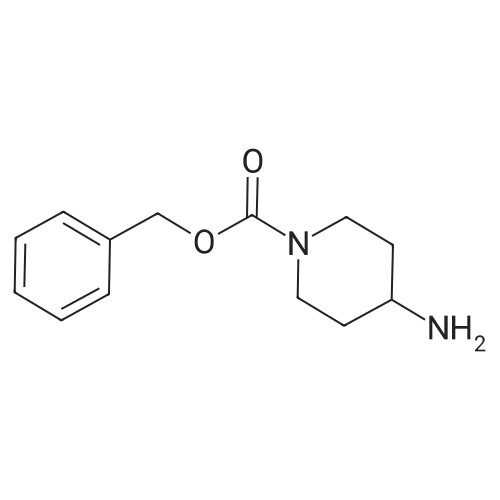
Step A: A solution of HBTU (8.1 g, 21 mmol), (S)-2-(tert- butoxycarbonylamino)-4-(methylthio)butanoic acid (5.3 g, 21 mmol) and DIEA (8.2 mL, 47 mmol) in DMF (50 mL) was stirred at ambient temperature for 30 minutes. Benzyl 4- aminopiperidine-l-carboxylate (5.0 g, 21 mmol) was added and the mixture was stirred at ambient temperature for 18 hours. The mixture was poured into IN NaOH (500 mL) and the combined organic layers were extracted into EtOAc (500 mL). The combined organic layers were washed with IN HC1 (500 mL) and brine (500 mL). The combined organic layers were dried over MgS04, filtered and concentrated in vacuo to yield (S)-benzyl 4-(2-(tert- butoxycarbonylamino)-4-(methylthio)butanamido)piperidine-l-carboxylate (10 g, 21 mmol, 100%).
100%
[00380] Step A: A solution of HBTU (8.1 g, 21 mmol), (S)-2-(tert- butoxycarbonylamino)-4-(methylthio)butanoic acid (5.3 g, 21 mmol) and DIEA (8.2 mL, 47 mmol) in DMF (50 mL) was stirred at ambient temperature for 30 minutes. Benzyl 4- aminopiperidine- 1 -carboxylate (5.0 g, 21 mmol) was added and the mixture stirred at ambient temperature for 18 hours. The mixture was poured into 1 N NaOH (500 mL) and the organics were extracted into EtOAc (500 mL). The organic layer was washed with 1 N HC1 (500 mL) and brine (500 mL), dried over MgS04 and concentrated under vacuum to yield (S)-benzyl 4- (2-(tert-butoxycarbonylamino)-4-(methylthio)butanamido)piperidine-l -carboxylate (10 g, 21 mmol, 100%).
General procedure: 38% Hydrogen peroxide (0.094 mL, 1.2 mmol) was added to a solution of Boc-Met or a methionine-containing peptide in acetic acid (7 mL). The solution was maintained at 20 C for 30 min. After that 5% Pd/C (0.2 g) was charged, and the mixture stirred for another 30 min. The charcoal was filtered off and the filtrate evaporated under vacuum. In the case of Boc-Met, the residue was dissolved in toluene, evaporated under vacuum, and the product dried in a vacuum desiccator over P4O10. In the case of a methionine-containing peptide, the residue was dissolved in water and lyophilized.
Step A: A solution of HBTU (8.1 g, 21 mmol), (S)-2-(tert- butoxycarbonylamino)-4-(methylthio)butanoic acid (5.3 g, 21 mmol) and DIEA (8.2 mL, 47 mmol) in DMF (50 mL) was stirred at ambient temperature for 30 minutes. Benzyl 4- aminopiperidine-1 -carboxylate (5.0 g, 21 mmol) was added and the mixture was stirred at ambient temperature for 18 hours. The mixture was poured into IN NaOH (500 mL) and extracted into EtOAc (500 mL). The combined organic layers were washed with IN HC1 (500 mL) and brine (500 mL), dried over MgS04, filtered and concentrated in vacuo to yield (S)-benzyl 4-(3-(2-fluoro-4-(methylsulfonyl)phenylamino)-2-oxopyrrolidin- 1 -yl)piperidine- 1 -carboxylate (10 g, 21 mmol, 100%).
5,8,11,14,17,20,23,26-octaoxa-2-azanonacosanedioic acid, 1-(9H-fluoren-9-ylmethyl) ester[ No CAS ]
[ 4530-20-5 ]
[ 2488-15-5 ]
[ 7536-58-5 ]
[ 13734-34-4 ]
[ 47355-10-2 ]
[ 57-10-3 ]
C70H111N9O20S[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: The second portion of the CCK4 peptide and the SubP peptide were modified on resin as follows to yield test lipidated peptides (1-SubP-COOH and 1-CCK4-Gly-COOH). Spacers (these are AA's used between the peg linker and the peptide of interest) were introduced on the peptides before pegylation (KGG for SubP and GG for CCK4). The free N-terminus of the peptide on resin was first pegylated with N-Fmoc-PEG8-propionic acid using standard HBTU coupling conditions. The N-Fmoc protecting group was removed by treatment with 10% piperidine in DMF (N,N-Dimethylformamide) for 5 min. Palmitic acid was subsequently coupled with the N-terminal free amine of the pegylated peptide. Peptides were cleaved from the resin using high HF conditions with minor modifications to the usual procedure. For the SubP peptide, longer times were used to ensure removal of Arg(Tos) protecting group (90% anhydrous HF/10% anisole at 0 C. for 2 h). For the CCK-4 peptides, 1,3-propanedithiol was used in the HF cleavage mixture to ensure deprotection of the formyl protecting group and prevent oxidation of methionine to its sulfoxide derivative: 85% anhydrous HF/10% anisole/5% PDT (1,3-propaneithiol) at 0 C. for 2 h) (Matsueda, 1982). Following cleavage from resin, peptides were precipitated with cold Et2O. Unmodified peptides were extracted using 10% AcOH in water and the lipidated peptides were extracted using 10% AcOH in H2O followed by 10% AcOH in 50% EtOH/H2O. Crude peptides were purified by RP-HPLC [Vydac C18, 10 m, 22 mm×250 mm]. The purities of the peptides were assessed by analytical RP-HPLC [Vydac C18, 5 m, 4 mm×250 mm].
Peptides illustrated in FIG. 9 were synthesized using the in-situ neutralization protocol for t-Boc chemistry (Schnolzer et al., In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences, 1992, International journal of peptide and protein research 40:180-193) on PAM resin on a 0.5 mmol scale. Amino acids were used with the following side chain protecting groups: Arg(Tos), Asp(OBzl), Gln(Xan), Lys(Fmoc), Lys(2-Cl-Z) and Trp(For). Peptide coupling reactions were carried out with a 4-fold excess (2.0 mmol) of activated amino acid for at least 15 min. The t-Boc protecting group on the N-terminus was removed using trifluoroacetic acid (TFA). The PAM resin from the CCK4 peptide synthesis was split into two equal portions. One portion of the resin was used for synthesizing non-lipidated peptides. The CCK4 (s-CCK-Gly-COOH) peptide was left unmodified at the N-terminus. This peptide served as positive control for the lipidated counterparts.
5,8,11,14,17,20,23,26-octaoxa-2-azanonacosanedioic acid, 1-(9H-fluoren-9-ylmethyl) ester[ No CAS ]
[ 4530-20-5 ]
[ 15761-39-4 ]
[ 13139-15-6 ]
[ 2488-15-5 ]
[ 13734-34-4 ]
[ 13836-37-8 ]
[ 84624-27-1 ]
[ 54613-99-9 ]
[ 55260-24-7 ]
[ 57-10-3 ]
C111H185N21O28S[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: The second portion of the CCK4 peptide and the SubP peptide were modified on resin as follows to yield test lipidated peptides (1-SubP-COOH and 1-CCK4-Gly-COOH). Spacers (these are AA's used between the peg linker and the peptide of interest) were introduced on the peptides before pegylation (KGG for SubP and GG for CCK4). The free N-terminus of the peptide on resin was first pegylated with N-Fmoc-PEG8-propionic acid using standard HBTU coupling conditions. The N-Fmoc protecting group was removed by treatment with 10% piperidine in DMF (N,N-Dimethylformamide) for 5 min. Palmitic acid was subsequently coupled with the N-terminal free amine of the pegylated peptide. Peptides were cleaved from the resin using high HF conditions with minor modifications to the usual procedure. For the SubP peptide, longer times were used to ensure removal of Arg(Tos) protecting group (90% anhydrous HF/10% anisole at 0 C. for 2 h). For the CCK-4 peptides, 1,3-propanedithiol was used in the HF cleavage mixture to ensure deprotection of the formyl protecting group and prevent oxidation of methionine to its sulfoxide derivative: 85% anhydrous HF/10% anisole/5% PDT (1,3-propaneithiol) at 0 C. for 2 h) (Matsueda, 1982). Following cleavage from resin, peptides were precipitated with cold Et2O. Unmodified peptides were extracted using 10% AcOH in water and the lipidated peptides were extracted using 10% AcOH in H2O followed by 10% AcOH in 50% EtOH/H2O. Crude peptides were purified by RP-HPLC [Vydac C18, 10 m, 22 mm×250 mm]. The purities of the peptides were assessed by analytical RP-HPLC [Vydac C18, 5 m, 4 mm×250 mm].
Peptides illustrated in FIG. 9 were synthesized using the in-situ neutralization protocol for t-Boc chemistry (Schnolzer et al., In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences, 1992, International journal of peptide and protein research 40:180-193) on PAM resin on a 0.5 mmol scale. Amino acids were used with the following side chain protecting groups: Arg(Tos), Asp(OBzl), Gln(Xan), Lys(Fmoc), Lys(2-Cl-Z) and Trp(For). Peptide coupling reactions were carried out with a 4-fold excess (2.0 mmol) of activated amino acid for at least 15 min. The t-Boc protecting group on the N-terminus was removed using trifluoroacetic acid (TFA). The PAM resin from the CCK4 peptide synthesis was split into two equal portions. One portion of the resin was used for synthesizing non-lipidated peptides. The CCK4 (s-CCK-Gly-COOH) peptide was left unmodified at the N-terminus. This peptide served as positive control for the lipidated counterparts.
His-Ser-Gln-Gly-Thr-Phe-Thr-Ser-Asp-Tyr-Ser-Lys-Tyr-Leu-Asp-Ser-Cys-Arg-Ala-Gln-Asp-Phe-Val-Gln-Trp-Leu-Met-Asn-Thr[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
EXAMPLE 1 Synthesis of Glucagon Cys17(1-29) and Similar MonoCys Analogs (0283) 0.2mmole Boc Thr(OBzl) Pam resin (SynChem Inc) in a 60ml reaction vessel and the following sequence was entered and run on a modified Applied Biosystems 430A Peptide Synthesizer using FastBoc HBTU-activated single couplings. (0284) HSQGTFTSDYSKYLDSCRAQDFVQWLMNT (SEQ ID NO: 35) The following side chain protecting groups were used: Arg(Tos), Asp(OcHex), Asn(Xan), Cys(pMeBzl), Glu(OcHex), His(Boc), Lys(2Cl-Z), Ser(Bzl), Thr(Bzl), Trp(CHO), and Tyr(Br-Z). The completed peptidyl resin was treated with 20% piperidine/dimethylformamide to remove the Trp formyl protection then transferred to an HF reaction vessel and dried in vacuo. 1.0ml p-cresol and 0.5 ml dimehyl sulfide were added along with a magnetic stir bar. The vessel was attached to the HF apparatus (Pennisula Labs), cooled in a dry ice/methanol bath, evacuated, and aprox. 10ml liquid hydrogen fluoride was condensed in. The reaction was stirred in an ice bath for 1hr then the HF was removed in vacuo. The residue was suspended in ethyl ether; the solids were filtered, washed with ether, and the peptide extracted into 50 ml aqueous acetic acid. An analytical HPLC was run [0.46 x 5 cm Zorbax C8, 1 ml/min, 45C, 214nm, A buffer of 0.1%TFA, B buffer of 0.1%TFA/90%ACN, gradient=10%B to 80%B over 10min.] with a small sample of the cleavage extract. The remaining extract was loaded onto a 2.2 x 25cm Kromasil C18 preparative reverse phase column and an acetonitrile gradient was run using a Pharmacia FPLC system. 5min fractions were collected while monitoring the UV at 214nm (2.0A). A=0.1%TFA, B=0.1%TFA/50%acetonitrile. Gradient = 30%B to 100%B over 450min. (0285) The fractions containing the purest product (48-52) were combined frozen, and lyophilized to give 30.1mg. An HPLC analysis of the product demonstrated a purity of>90% and MALDI mass spectral analysis demonstrated the desired mass of 3429.7. Glucagon Cys21, Glucagon Cys24, and Glucagon Cys29 were similarly prepared.
EXAMPLE 2 Synthesis of Glucagon-Cex and Other C-Terminal Extended Analogs. (0286) 285mg (0.2mmole) methoxybenzhydrylamine resin (Midwest Biotech) was placed in a 60ml reaction vessel and the following sequence was entered and run on a modified Applied Biosystems 430A peptide synthesizer using FastBoc HBTU-activated single couplings. (0287) HSQGTFTSDYSKYLDSRRAQDFVQWLMNTGPSSGAPPPS (SEQ ID NO: 36) The following side chain protecting groups were used: Arg(Tos), Asp(OcHex), Asn(Xan), Cys(pMeBzl), Glu(OcHex), His(Boc), Lys(2Cl-Z), Ser(Bzl), Thr(Bzl), Trp(CHO), and Tyr(Br-Z). The completed peptidyl resin was treated with 20% piperidine/dimethylformamide to remove the Trp formyl protection then transferred to HF reaction vessel and dried in vacuo. 1.0ml p-cresol and 0.5 ml dimehyl sulfide were added along with a magnetic stir bar. The vessel was attached to the HF apparatus (Pennisula Labs), cooled in a dry ice/methanol bath, evacuated, and aprox. 10ml liquid hydrogen fluoride was condensed in. The reaction was stirred in an ice bath for 1hr then the HF was removed in vacuo. The residue was suspended in ethyl ether; the solids were filtered, washed with ether, and the peptide extracted into 50 ml aqueous acetic acid. An analytical HPLC was run [0.46 x 5 cm Zorbax C8, 1 ml/min, 45C, 214nm, A buffer of 0.1%TFA, B buffer of 0.1%TFA/90%ACN, gradient=10%B to 80%B over 10min.] on an aliquot of the cleavage extract. The extract was loaded onto a 2.2 x 25cm Kromasil C18 preparative reverse phase column and an acetonitrile gradient was run for elution using a Pharmacia FPLC system. 5min fractions were collected while monitoring the UV at 214nm (2.0A). A=0.1%TFA, B=0.1%TFA/50%acetonitrile. Gradient = 30%B to 100%B over 450min. Fractions 58-65 were combined, frozen and lyophilized to give 198.1mg. (0288) HPLC analysis of the product showed a purity of greater than 95%. MALDI mass spectral analysis showed the presence of the desired theoretical mass of 4316.7 with the product as a C-terminal amide. Oxyntomodulin and oxyntomodulin-KRNR were similarly prepared as the C-terminal carboxylic acids starting with the appropriately loaded PAM-resin.
HSQGTFTSDYSKYLDERRAQDFVQWLMNT-NH2, lactam 12-16[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
EXAMPLE 11 Synthesis of Glucagon Lactams (0299) 285 mg (0.2 mmole) methoxybenzhydrylamine resin (Midwest Biotech) was added to a 60 mL reaction vessels and the following sequence was assembled on a modified Applied Biosystems 430A peptide synthesizer using Boc DEPBT-activated single couplings. HSQGTFTSDYSKYLDERRAQDFVQWLMNT-NH2 (12-16 Lactam; SEQ ID NO: 12) (0300) The following side chain protecting groups were used: Arg(Tos), Asp(OcHx), Asn(Xan), Glu(OFm), His(BOM), Lys(Fmoc), Ser(Bzl), Thr(Bzl), Trp(CHO), Tyr(Br-Z). Lys(Cl-Z) was used at position 12 if lactams were constructed from 16-20, 20-24, or 24-28. The completed peptidyl resin was treated with 20% piperidine/dimethylformamide for one hour with rotation to remove the Trp formyl group as well as the Fmoc and OFm protection from Lys12 and Glu16. Upon confirmation of removal by a positive ninhydrin test, the resin was washed with dimethylformamide, followed by dichloromethane and than again with dimethylformamide. The resin was treated with 520 mg (1 mmole) Benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate (PyBOP) in dimethylformamide and diisopropylethylamine (DIEA). The reaction proceeded for 8-10 hours and the cyclization was confirmed by a negative ninhydrin reaction. The resin was washed with dimethylformamide, followed by dichloromethane and subsequently treated with trifluoroacetic acid for 10 minutes. The removal of the Boc group was confirmed by a positive ninhydrin reaction. The resin was washed with dimethylformamide and dichloromethane and dried before being transferred to a hydrofluoric acid (HF) reaction vessel. 500 muL p-cresol was added along with a magnetic stir bar. The vessel was attached to the HF apparatus (Peninsula Labs), cooled in a dry ice/methanol bath, evacuated, and approximately 10 mL of liquid hydrofluoric acid was condensed into the vessel. The reaction was stirred for 1 hour in an ice bath and the HF was subsequently removed in vacuo. The residue was suspended in ethyl ether; the solids were filtered, washed with ether, and the peptide was solubilized with 150 mL 20% acetonitrile/1% acetic acid. (0301) An analytical HPLC analysis of the crude solubilized peptide was conducted under the following conditions [4.6 X 30 mm Xterra C8, 1.50 mL/min, 220 nm, A buffer 0.1% TFA/10% ACN, B buffer 0.1% TFA/100% ACN, gradient 5-95%B over 15 minutes]. The extract was diluted twofold with water and loaded onto a 2.2 X 25 cm Vydac C4 preparative reverse phase column and eluted using an acetonitrile gradient on a Waters HPLC system (A buffer of 0.1% TFA/10% ACN, B buffer of 0.1% TFA/10% CAN and a gradient of 0-100% B over 120 minutes at a flow of 15.00 ml/min. HPLC analysis of the purified peptide demonstrated greater than 95% purity and electrospray ionization mass spectral analysis confirmed a mass of 3506 Da for the 12-16 lactam. Lactams from 16-20, 20-24, and 24-28 were prepared similarly.
4-(4-(3,4-dimethoxybenzyl)-2-carbonyl)tetrahydrofurylmethyl-2-methoxyphenyl-2-(N-t-butoxycarbonyl)amino-4-methylthiobutanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
82%
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In acetonitrile; at 20℃;Cooling with ice;
Weighing 0.20g (0.54mmol) <strong>[26687-82-1]arctigenin</strong> Arctium Fructus Arctii (great burdock achene), 0.27g (1.08mmol) BOC-L-methionine, 0.21g (1.08mmol) 1-ethyl-(3-dimethyl aminopropyl) carbodiimide hydrochloride (EDCI), 0.03g (0.27mmol) 4-dimethylaminopyridine (DMAP) in 100 ml in lessened, by adding 10 ml acetonitrile solution, stirring to dissolve the ice water bath, then room temperature reaction 1-2 hours, to reaction in the detection reaction TLC, solvent evaporating under reduced pressure, to obtain yellowish viscous material. For the filling of the opposite phase YMC column chromatography, using acetonitrile/water (55:45) mixed solvent to elute, collect the required component, evaporating the organic solvent under reduced pressure, freeze-drying, to obtain white powdery compound ARG1.White solid, yield 82%
82%
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In acetonitrile; at 20℃;Cooling with ice;
Weigh 0.20 g (0.54 mmol) Arctiin aglycone, BOC-L-methionine, 0.21g (1.08mmol),1-ethyl-(3-dimethylaminopropyl)carbodiimide hydrochloride, 0.218 (1.08 mmol)(EDCI), 0.03 g (0.27 mmol) of 4-dimethylaminopyridine (DMAP) were placed in a 100 ml vial,Add 10 ml of acetonitrile solution,Ice water bath stirring dissolved,And then room temperature reaction 1-2 hours,TLC detection reaction to the reaction is completed,The solvent was distilled off under reduced pressure,Get a light yellow viscous material. The viscous material was separated by column chromatography using YMC reverse phase packing,Eluting with a mixed solvent of acetonitrile / water (55:45)Collect the required components,The organic solvent was distilled off under reduced pressure,Freeze-dried,A white powdery compound, ARG1, was obtained.
4-(((3S)-4-(3,4-dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenyl-2-(tert-butoxycarbonylamino)-4-(methylthio)butanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
82%
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In acetonitrile; at 20℃;Cooling with ice;
0.22 g (1.08 mmol) of BOC-L-methionine, 0.21 g (1.08 mmol) of 1-ethyl- (3-dimethylaminopropyl) carbodiimide (EDCI), 0.03 g (0.27 mmol) of 4-dimethylaminopyridine (DMAP) were placed in a 100 ml spin flask,Add 10ml of acetonitrile solution, stirring under ice water dissolved,And then room temperature reaction 1-2 hours, TLC reaction to detect the reaction is completed,The solvent was evaporated under reduced pressure to give a pale yellow viscous material.The viscous material was subjected to column chromatography using YMC reverse phase packing,Elution with a mixed solvent of acetonitrile / water (55:45)The desired fractions were collected, the organic solvent was distilled off under reduced pressure,And lyophilized to give compound ARG1 as a white powder.
82%
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In acetonitrile; at 0℃;
General procedure: The following reaction conditions were used to produce <strong>[26687-82-1]arctigenin</strong>: an aminoacid:EDCI:DMAP ratio of 1:2:2:0.5, the mixture was dissolved in acetonitrile for 1-2h at 0 C and the mixture was finally removed under reduced pressure to produce yellow powder. The yellow dope was added to water, washed by stirring, dried, and then concentrated by lyophilization to produce the white crude product. The crude products were chromatographed using silica gel and eluted with acetonitrile/water(55:45) to produce the pure products ARG1?-5?. HCl gas was then used to synthesizethe deprotection derivatives ARG6?-10?. 4-(((3R)-4-(3,4-dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenyl 2-(tert-butoxycarbonylamino)-4-(methylthio)butanoate(ARG1?)White solid (82%). UV(MeOH)lambdamax:221nm. HRESIMS m/z: 626.2394 [M+Na]+(Calcd for C31H41O9NNa 626.2399). ESI-MSm/z: 626.2[M+Na]+,1H-NMR(CDCl3,300MHz)deltappm:6.99(d,J=8.1Hz,1H),6.79(d,J=7.8Hz,1H),6.76(d,J=2.1Hz,1H),6.70(dd,J=1.8Hz,8.1Hz,1H),6.56(dd,J=1.8Hz,7.8Hz,1H),6.51(d,J=1.8Hz,1H),5.24(m,1H),4.73(m,-NH-CH-COO-),3.94-4.21(m,-COOCH2-),3.83(s,3H),2.97-3.03(m,2H),2.70(m,1H),2.38-2.63(m,2H),2.54(m,1H),2.50(m,CH2S-),2.15(s,3H),2.01(m,2H),1.51(s,3H),1.47(s,3H),1.46(s,3H).13C-NMR(CDCl3,300MHz): 138.26,111.90, 150.90, 137.01, 122.45, 120.54,38.11,46.40,178.43,130.22,113.31,149.06,147.91,111.44,121.51,34.58,40.98,71.23,170.52,52.91,32.32,29.77,15.45,157.41,77.22,28.30,28.18,27.24.
General procedure: The relevant Boc-AA1-OH (1.1 equiv), TBTU (1.1 equiv) and DIEA (2.0 equiv) were dissolved in CH2Cl2 anhydrous at room temperature and the mixture stirred for 30 min prior to addition of bromothiazole 5 (1.0 equiv). The mixture was stirred and allowed to proceed at room temperature for further 23h, with periodic monitoring by TLC. The solvent was then reduced at 3mL and the residue was submitted to column chromatography on silica using DCM/Acetone 10:1 (v/v).
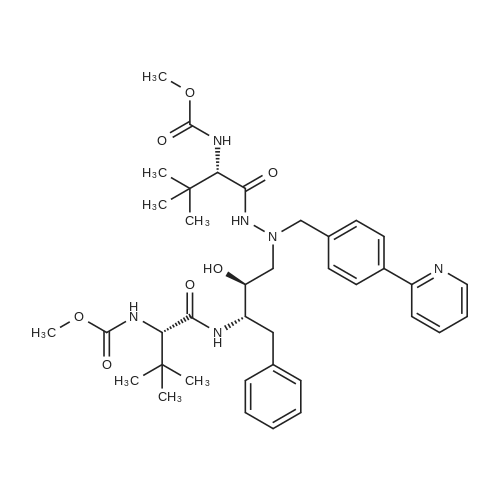
(5S,10S,11S,14S)-11-benzyl-5,14-di-tert-butyl-3,6,13,16-tetraoxo-8-(4-(pyridin-2-yl)benzyl)-2,17-dioxa-4,7,8,12,15-pentaazaoctadecan-10-yl (S)-2-((tert-butoxycarbonyl)amino)-4-(methylthio)butanoate[ No CAS ]
(5S,10S,11S,14S)-11-benzyl-5,14-di-tert-butyl-3,6,13,16-tetraoxo-8-(4-(pyridin-2-yl)benzyl)-2,17-dioxa-4,7,8,12,15-pentaazaoctadecan-10-yl (S)-2-amino-4-(methylthio)butanoate dihydrochloride[ No CAS ]






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping