|
With acetyl chloride; at -5℃; for 25h;Reflux; |
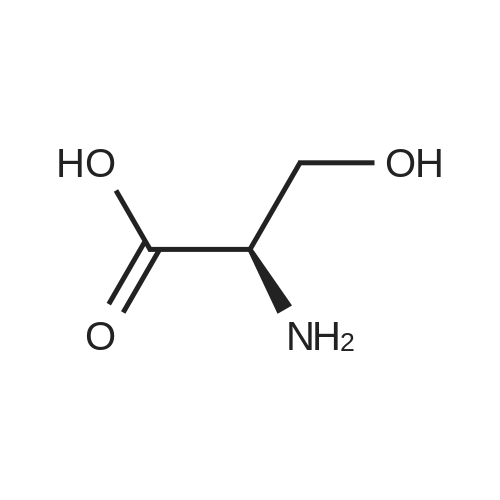
To the suspension of D-serine (105.09 g, 1 .0 moL) in methanol (630 mL) was added acetyl chloride (1 17.75 g, 1.5 moL) slowly and under constant stirring over a 1 hour period at -5C. The reaction mixture was heated to reflux and stirred for 24 hours. The reaction mixture was cooled at 0-5C and charged with sodium bicarbonate (298.20 g, 3.55 moL), sodium iodide (37.47 g, 0.25 moL) and benzyl chloride (259.50 g, 2.05 moL). The reaction mixture was heated to reflux and stirred for 12 hours. The reaction mixture was cooled to room temperature, filtered through a Celite pad and the filter cake was washed with methanol (157 mL). The filtered methanolic solution was added dropwise, to a cooled solution of KOH (224.44 g 4.0 moL) in water (525 mL) at below 5C. The reaction mixture was stirred 0-5C for 20 hours. Water (174 mL) was charged and the pH adjusted to 6.5 to 7.0 by dropwise addition of 30% HCI. The reaction mixture was heated to 20-25C and the pH was adjusted to 3.0 to 3.4 by dropwise addition of 30% HCI. The reaction mixture was stirred at 20-25C for 30 minutes, cooled to 15C and stirred for 30 min. The reaction mixture was filtered and the filter cake was washed three times with water (315 mL).The resulting solid was dried under vacuum at 45-50C for about 12 hours to provide (R)-2-(dibenzylamino)-3- hydroxypropanoic acid as a white solid (228.04 g, yield 80.0%, HPLC purity >99.6%). 1 H NMR (300 MHz, DMSO-d6) delta 12.52 (1 H, brs), 7.47-7.25 (10H, m), 4.76 (bs, 1 H), 3.89-3.82 (2H, m), 3.71-67 (2H, m). 13C NMR (75 MHz, DMSO-d6) delta 172.5, 139.4, 129.3, 129.0, 128.6, 128.3, 127.6, 127.1 , 62.9, 60.3, 54.8. |
|
With thionyl chloride; at 0 - 20℃; for 12h; |
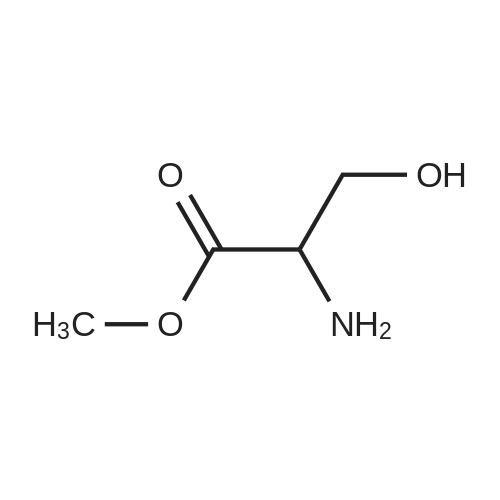
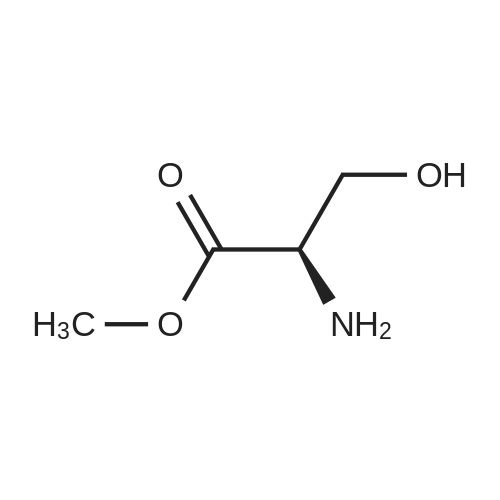
One hundred milliliters of a methanol solution containing D-serine(4.5 g, 43 mmol, 19) was cooled in a salt-ice bath at 0 C, andSOCl2 (18.6 mL, 258 mmol) was added dropwise. The resulting mixturewas stirred for 12 h at ambient temperature and then concentratedin vacuo. After coevaporating the solvent with diethyl ethermultiple times to remove excess SOCl2, the residue was dissolved in100 mL of CH2Cl2, to which Et3N (15.7 mL, 113 mmol) was added at0 C. To this solution was added di-tert-butyl dicarbonate (11.3 g,52 mmol) under stirring, and the resulting mixture was refluxeduntil the starting material was consumed, as determined by TLC(methanol). The reaction mixture was concentrated in vacuo, andthe residue was dissolved in ethyl acetate (100 mL) and thenwashed with saturated NaHCO3 followed by washing with brine(3). The organic layer was dried over Na2SO4, filtered, andconcentrated to afford N-tert-butoxycarbonylserine methyl ester(Boc-NH-Ser-OMe) as the intermediate as an oil, which was used in the subsequent reaction without further purification. The crudeBoc-NH-Ser-OMe was dissolved in a mixture of acetone (100 mL)and 2,2-dimethoxypropane (81.0 mL, 659 mmol). To the resultingmixture was added BF3-Et2O complex (1.1 mL, 9 mmol) at ambienttemperature, and the reaction mixture was stirred for 12 h. Afterdetermining that the reaction was complete by TLC, 1.1 mL ofEt3N was added to the mixture to quench the reaction, and the solventwas removed in vacuo. The brown oil was then partitionedbetween Et2O and saturated NaHCO3 (aq). The aqueous layer wasextracted with Et2O (5), and the organic layers were combined,dried over Na2SO4, and concentrated. The resulting brown oil waspurified by column chromatography (hexane/ethyl acetate = 10:1)to afford 20 as a yellow oil (9.6 g, 85%). |
|
With thionyl chloride; at 0 - 70℃; for 2h; |
To a solution of (R)-2-amino-3-hydroxypropanoic acid (3 g, 28.55 mmol) in MeOH (40 mL) was slowly added thionyl chloride (2.082 mL, 28.55 mmol) at 0C. The mixture was heatedTo a solution of (R)-2-amino-3-hydroxypropanoic acid (3 g, 28.55 mmol) in MeOH (40 mL) was slowly added thionyl chloride (2.082 mL, 28.55 mmol) at 0C. The mixture was heated |
|
|
To a flask containing MeOH (20 mL) was added dropwise AcCl (2.00mL, 28.2 mmol) at 0 C under Ar. After stirring for 15 min at ambient temperature, D-serine (1.03 g, 9.81mmol) was added to the mixture, and the whole was stirred for further 2 h at the same temperature. The volatile solvent was removed in vacuo, and the residue was used for the next step without further purification. To a solution of the obtained D-serine methyl ester in CHCl3 (20 mL) were added Et3N (6.64mL, 47.7 mmol) and TsCl (2.13 g, 11.2 mmol) at 0 C. After stirring for 20 h at ambient temperature, the reaction was quenched by addition of sat. aqueous NaHCO3 solution, and the mixture was extracted with CHCl3 (x2). The extract was washed with brine, dried over Na2SO4, and concentrated in vacuo. Theresidue was purified by column chromatography on silica gel using AcOEt-CHCl3 (6:4, v/v) as eluent togive N-tosyl-D-serine methyl ester (2.19 g, 82%) as a white solid. 1H-NMR (CDCl3; 400 MHz) delta 7.75(2H, d, J = 8.1 Hz), 7.31 (2H, d, J = 8.1 Hz), 5.39 (1H, d, J = 9.2 Hz), 4.05 (1H, ddd, J = 9.2, 3.4, 2.9 Hz),3.97 (1H, dd, J = 9.9, 2.9 Hz), 3.79 (1H, dd, J = 9.9, 3.4 Hz), 3.57 (3H, s), 2.44 (3H, s), 0.84 (9H, s), 0.02(3H, s), 0.01 (3H, s). |
| 22 g |
With thionyl chloride;Inert atmosphere; |
In Step 1, 20 g of S-l is reacted with thionyl chloride in methanol to provide 22 g of S- 2 |
|
With chloro-trimethyl-silane; |
Methyl esters of d-Ile and O-benzyl-Ser derivatives were prepared as described in Li and Sha [38]using 2 mmol of amino acid, 0.5 mL of trimethylsilyl chloride (TMSCl), and 10 mL of methanol. Thereaction mixture was evaporated to dryness and stored in a desiccator. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping