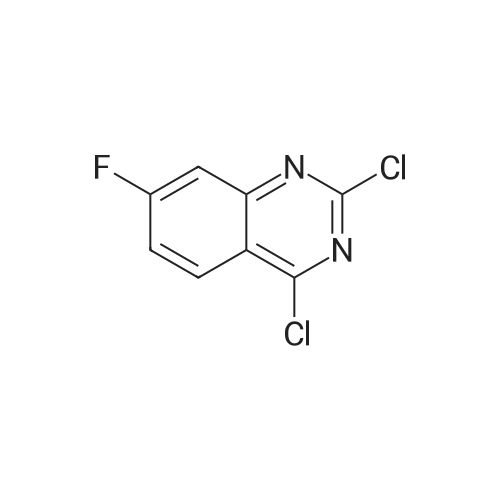
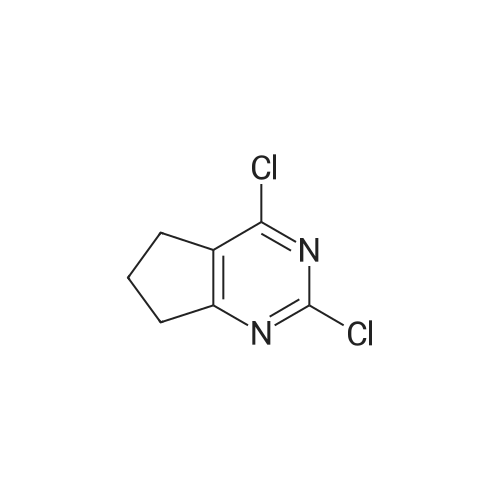
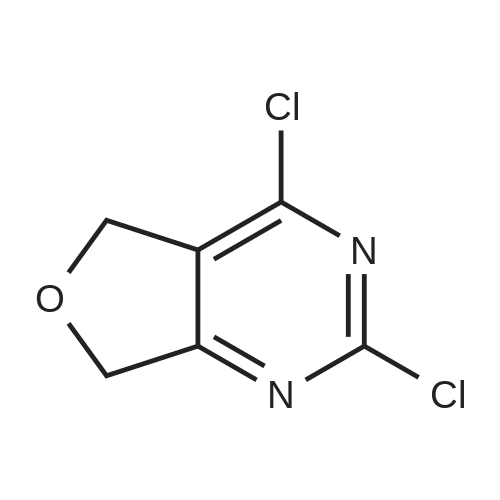
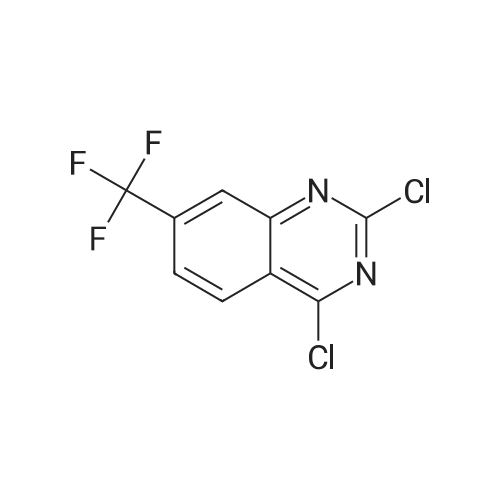
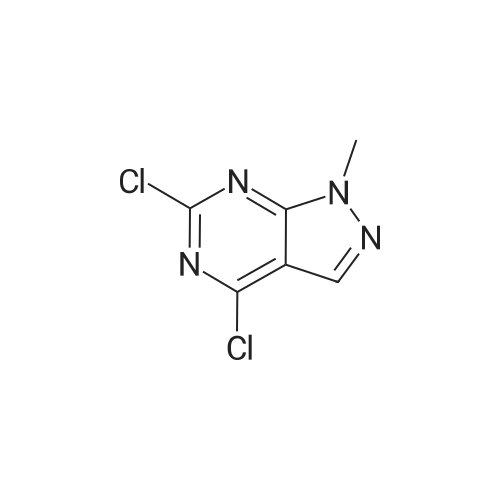
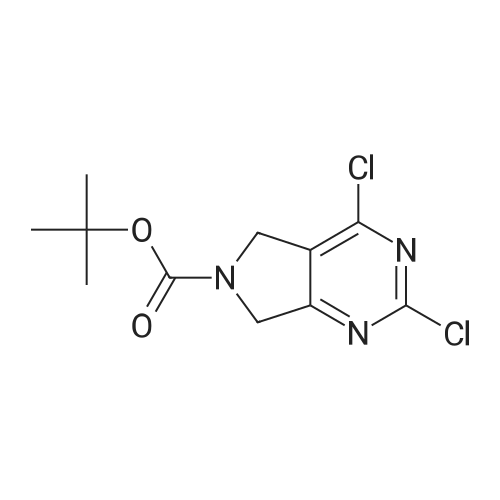
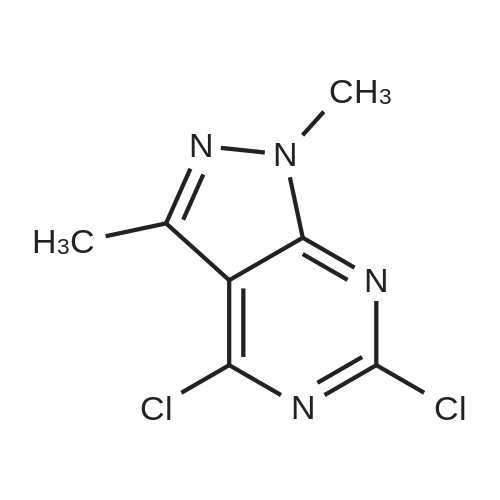
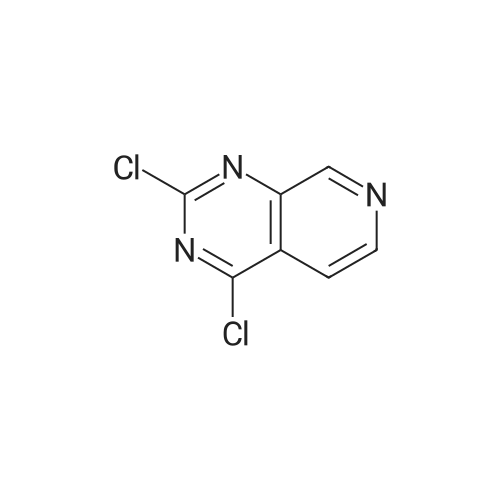
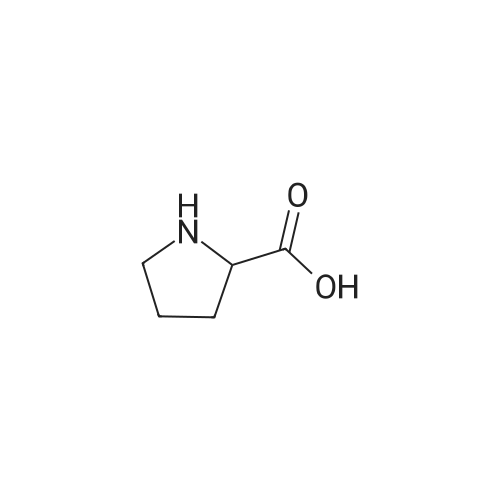
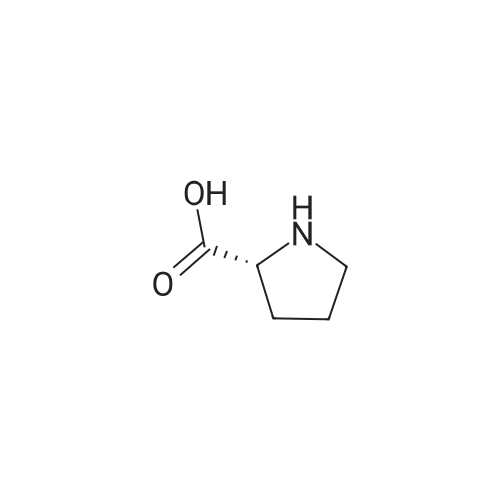
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of Organic Chemistry, 2007, vol. 72, # 8, p. 2737 - 2743
[2] Journal of the Chemical Society, 1958, p. 4458,4462
[3] Journal of Organometallic Chemistry, 1983, vol. 246, # 1, p. 53 - 56
[4] Tetrahedron Letters, 1995, vol. 36, # 43, p. 7885 - 7888
[5] Journal of the Chemical Society - Perkin Transactions 1, 1997, # 19, p. 2891 - 2896
Reference:
[1] Tetrahedron, 1987, vol. 43, # 14, p. 3289 - 3294
[2] Tetrahedron Asymmetry, 1990, vol. 1, # 12, p. 877 - 880
[3] Chemistry - A European Journal, 2017, vol. 23, # 68, p. 17195 - 17198
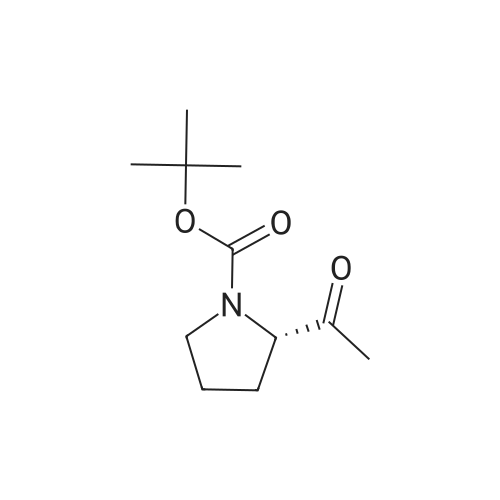
7
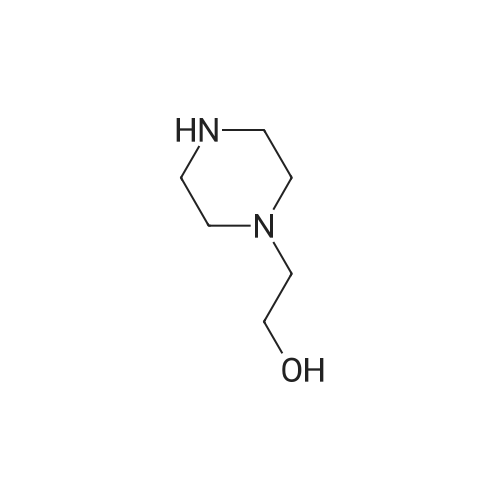
[ 23356-96-9 ]
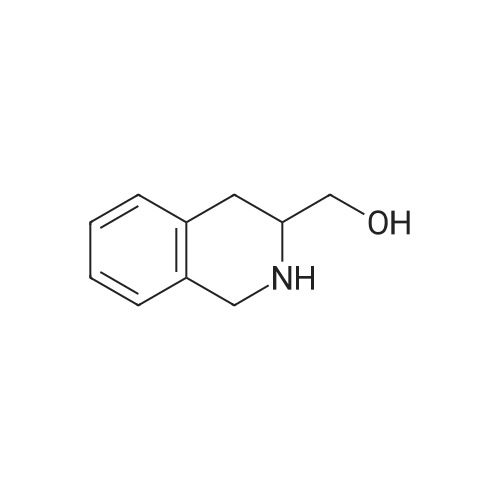
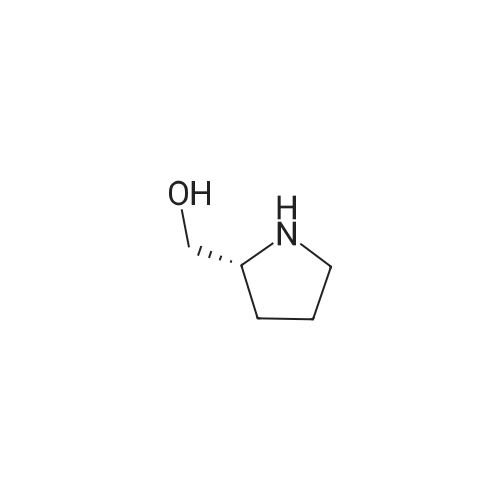
[ 22795-99-9 ]
Reference:
[1] Journal of Medicinal Chemistry, 1991, vol. 34, # 5, p. 1612 - 1624
8
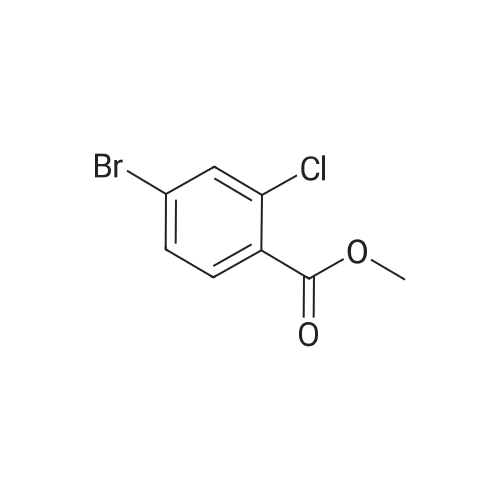
[ 100-44-7 ]
[ 23356-96-9 ]
[ 53912-80-4 ]
Reference:
[1] Journal of Organic Chemistry, 2007, vol. 72, # 8, p. 2737 - 2743
[2] Dyes and Pigments, 2011, vol. 90, # 2, p. 100 - 105
[3] Synlett, 2005, # 8, p. 1235 - 1238
[4] Tetrahedron Letters, 2005, vol. 46, # 20, p. 3579 - 3582
[5] Journal of Organometallic Chemistry, 2007, vol. 692, # 24, p. 5459 - 5473
[6] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1984, # 12, p. 2887 - 2893
[7] RSC Advances, 2015, vol. 5, # 5, p. 3461 - 3464
9
[ 100-52-7 ]
[ 23356-96-9 ]
[ 53912-80-4 ]
Reference:
[1] Synthesis, 2010, # 10, p. 1673 - 1677
10
[ 23356-96-9 ]
[ 53912-80-4 ]
Reference:
[1] Synthesis, 2002, # 3, p. 375 - 380
11
[ 23356-96-9 ]
[ 91550-08-2 ]
Reference:
[1] Bioorganic and Medicinal Chemistry, 2002, vol. 10, # 7, p. 2199 - 2206
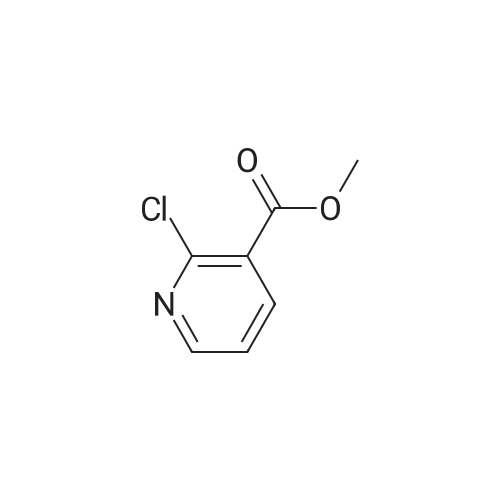
12
[ 23356-96-9 ]
[ 142253-50-7 ]
Reference:
[1] Journal of Medicinal Chemistry, 2000, vol. 43, # 3, p. 342 - 345
[2] Bulletin de la Societe Chimique de France, 1997, vol. 134, # 7, p. 713 - 718
13
[ 18162-48-6 ]
[ 23356-96-9 ]
[ 134756-75-5 ]
Yield
Reaction Conditions
Operation in experiment
76%
With triethylamine In dichloromethane at 20℃;
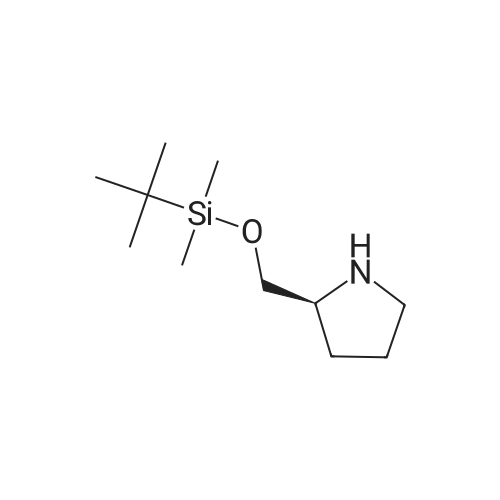
To a solution of (S)-pyrrolidin-2-yl-methanol (0.69 g, 6.82 mmol) in DCM (3 mL) cooled to 0° C. was added TEA (1.38 g, 13.6 mmol) followed by TBDMS-Cl (1.03 g, 6.82 mmol) in DCM (3 mL). The reaction mixture was then stirred at room temperature over night and poured into NH4Cl (20 mL). The aqueous layer was extracted with DCM (2×50 mL). Combined organics were washed with brine, dried over Na2SO4, filtered and evaporated to dryness to give the title compound (1.11 g, 76percent) as a yellow oil. MS: 216.2 (M+H+)
76%
With triethylamine In dichloromethane at 0 - 20℃;
Intermediate A-36-{5-[(S)-2-(tei"i-Butyl-dimethyl-silanyloxymethyl)-pyrrolidin-l-yl]-pyridin-3-yl}-l- methyl-3,4-dihydro-lH-quinolin-2-one [A] (S)-2-(tert-Butyl-dimethyl-silanyloxymethyl)-pyrrolidineTo a solution of (S)-pyrrolidin-2-yl-methanol (0.69 g, 6.82 mmol) in DCM (3 mL) cooled to 0 °C was added TEA (1.38 g, 13.6 mmol) followed by TBDMS-Cl (1.03 g, 6.82 mmol) in DCM (3 mL). The reaction mixture was then stirred at room temperature over night and poured into NH4C1 (20 mL). The aqueous layer was extracted with DCM (2 x 50 mL). Combined organics were washed with brine, dried over Na2S04, filtered and evaporated to dryness to give the title compound (1.11 g, 76percent) as a yellow oil. MS: 216.2 (M+H+).
76%
With triethylamine In dichloromethane at 0 - 20℃; Inert atmosphere
To a solution of (S)-pyrrolidin-2-ylmethanol (0.69 g, 6.82 mmol) in DCM (3 mL) cooled to 0 °C was added TEA (1.38 g, 13.6 mmol) followed by TBDMS-Cl (1.03 g, 6.82 mmol) in DCM (3 mL). The reaction mixture was then stirred at room temperature over night and poured into NH4C1 (20 mL). The aqueous layer was extracted with DCM (2 x 50 mL). Combined organics were washed with brine, dried over Na2S04, filtered and evaporated to dryness to give the title compound (1.1 lg, 76percent) as a yellow oil. MS: 216.2 (M+H+).
76%
With triethylamine In dichloromethane at 0 - 20℃;
[A] (S)-2-(tert-Butyl-dimethyl-silanyloxymethyl)-pyrrolidine To a solution of (S)-pyrrolidin-2-ylmethanol (0.69 g, 6.82 mmol) in DCM (3 mL) cooled to 0° C. was added TEA (1.38 g, 13.6 mmol) followed by TBDMS-Cl (1.03 g, 6.82 mmol) in DCM (3 mL). The reaction mixture was then stirred at room temperature overnight and poured into NH4Cl (20 mL). The aqueous layer was extracted with DCM (2*50 mL). Combined organics were washed with brine, dried over Na2SO4, filtered and evaporated to dryness to give the title compound (1.11 g, 76percent) as a yellow oil. MS: 216.2 (M+H+).
Reference:
[1] Synlett, 2007, # 15, p. 2415 - 2419
[2] Patent: US2013/72679, 2013, A1, . Location in patent: Paragraph 0553; 0554
[3] Patent: WO2013/37779, 2013, A1, . Location in patent: Page/Page column 76; 77
[4] Patent: WO2013/79452, 2013, A1, . Location in patent: Page/Page column 237
[5] Patent: US2013/143863, 2013, A1, . Location in patent: Paragraph 1082; 1083; 1084
[6] Chemical and Pharmaceutical Bulletin, 2007, vol. 55, # 2, p. 328 - 333
14
[ 82911-69-1 ]
[ 23356-96-9 ]
[ 148625-77-8 ]
Yield
Reaction Conditions
Operation in experiment
74%
for 1 h;
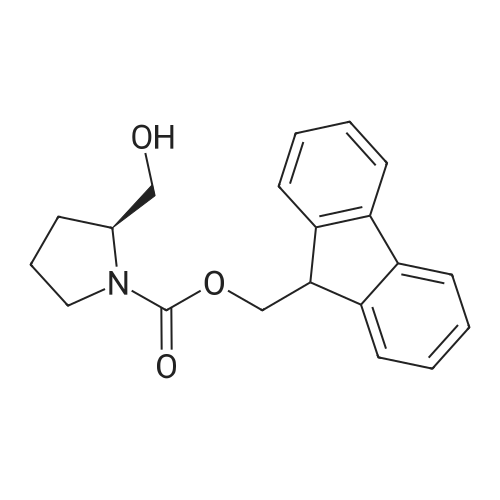
(1) Fmoc-L-Prolinol (Compound 2)L-prolinol (Compound 1) (0.61 g, 6.0 mmol) was dissolved in 70 ml of pure water, thus preparing an L-prolinol aqueous solution. N-(9-Fluorenylmethoxycarbonyloxy)succinimide (Fmoc-OSu) (2.0 g, 6.0 mmol) was dissolved in 10 ml ofTHF. This THF solution was added to the L-prolinol aqueous solution, and this was stirred for 1 hour so as to react the L-prolinol and the Fmoc-OSu. The reaction solution was separated into a liquid fraction and a precipitate fraction. These fractions respectively were subjected to extraction with ethyl acetate, and organic layers respectively were collected therefrom. The thus-obtained organic layers were mixed together, and anhydrous sodium sulfate was added thereto to absorb moisture (hereinafter, this process is referred to as a “drying” process). The organic layers were filtered, and the filtrate obtained was vacuum concentrated. The residual substance obtained was purified by silica gel column chromatography (the eluent: hexane:ethyl acetate=1 :1). Thus, Compound 2 was obtained (1.4 g, yield: 74percent). The result of NMR analysis with respect to this compound is shown below.‘H-NMR (CDC13): ö7.77 (2H, d, J=7.7 Hz, Ar——H), 7.60 (2H, d, J=7.3 Hz,Ar—H), 7.40(2H,t, J=7.5 Hz,Ar—H), 7.31 (2H, t, J=7.6 Hz, Ar—-H), 4.40-4.50 (2H, m, COOCH2), 4.22(1H, t, J=6.5 Hz,Ar-CH), 3.20-3.80 (5H, m, H-5, H-6), 1.75(3H, m, H-3, H-4), 1.40 (1H, m, H-3).
Reference:
[1] Journal of the American Chemical Society, 2006, vol. 128, # 12, p. 4023 - 4034
16
[ 23356-96-9 ]
[ 119020-01-8 ]
Reference:
[1] Nucleosides, Nucleotides and Nucleic Acids, 2001, vol. 20, # 4-7, p. 1377 - 1379
[2] Tetrahedron, 2010, vol. 66, # 51, p. 9703 - 9707
[3] Tetrahedron Letters, 2011, vol. 52, # 5, p. 615 - 618
[4] Journal of the Korean Chemical Society, 2013, vol. 57, # 5, p. 591 - 598
[5] Journal of Labelled Compounds and Radiopharmaceuticals, 2014, vol. 57, # 4, p. 209 - 214
17
[ 23356-96-9 ]
[ 330784-47-9 ]
Yield
Reaction Conditions
Operation in experiment
74%
Stage #1: With N-ethyl-N,N-diisopropylamine In N,N-dimethyl-formamide at 0℃; for 0.25 h; Stage #2: at 20℃; for 4 h;
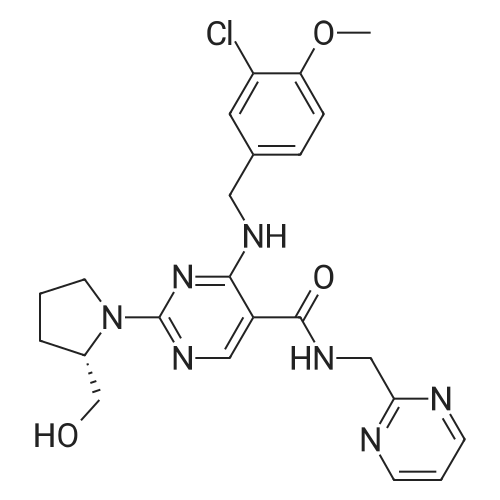
Compound 26 (0.78 g, 1.7 mmol) was dissolved in 1.7 ml anhydrous DMF and anhydrous DIPEA (0.87 g, 6.74 mmol) was added at 0 ° C. After stirring at 0 ° C for 15 min, compound 9 (340 mg, 3.37 mmol). After reaction at 20 ° C for 4 hours, column chromatography purified Compound 1, avanafil (600 mg, 74percent yield).
0.7 g
With triethylamine In ethyl acetate at 20 - 30℃;
0.9g of compound X obtained in step (4) was dissolved in 15ml of ethyl acetate, 196mg of L-prolinol and 0.54ml of triethylamine were directly added into the organic phase, and stirred for 1-16h at 20-30 ° C. 40ml Water and 45ml ethyl acetate. The organic phase was separated, the organic phase was washed three times with 5percent sodium carbonate solution (30ml × 3), then with saturated brine and washed three times with water (30ml × 3), dried over anhydrous sodium sulfate The organic phase was filtered and concentrated under reduced pressure to give 0.7 g of a white solid.
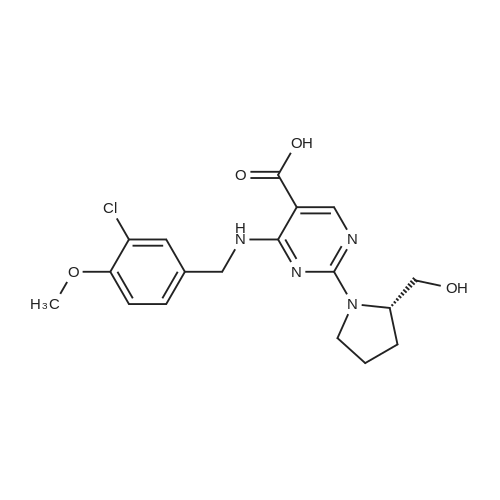
The above prepared 39g ivah that non-intermediate B4-[ (3-chloro-4-benzyl) amino]-2-methylthio-N-(2-pyrimidinyl methyl)-5-pyrimidine carboxamide dissolved in 200 ml acetonitrile is formed in the reaction solution, then adding 13.8gL- dried meat ammonia alcohol and 9.5g pyridine, the reaction stirring under room temperature for 19 hours, evaporate most of the acetonitrile, then adding ethyl acetate, then with water, saturated sodium bicarbonate, the saturated salt water, the organic phase is dried with anhydrous sodium sulfate, filtered, by reduced pressure evaporation to dryness the crude product to 46g, crude product with ethyl acetate/n-heptane is recrystallized, get 37g non- ivah that final product. The purity of 97percent, the yield is 79.5percent
1.1 g
at 20 - 30℃; for 1 h;
215 mg of L-prolinol and 1 ml of triethylamine were directly added to the organic phase of the compound VIII obtained in the step (4), and the mixture was stirred at 20 to 30 ° C for 1 hour. The reaction mixture was added with 50 ml of water and the organic phase was separated. The organic phase was washed three times (30 ml x 3) with 5percent sodium carbonate solution and three times with saturated brine (30 ml x 3). The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give 1.1 g of a white solid by crystallization. Yield in three steps was 77.5percent.
With triethylamine In tetrahydrofuran at 20℃; for 20 h;
The above prepared 41.5g ivah that non-intermediate B2-chloro-4-[ (3-chloro-4-benzyl) amino)-N-(2-pyrimidinyl methyl)-5-pyrimidine carboxamide dissolved in 200 ml of tetrahydrofuran is formed in the reaction solution, then adding 10gL- dried meat ammonia alcohol and 10g triethylamine, the reaction stirred at room temperature 20 hours, evaporate most of the tetrahydrofuran, then added with ethyl acetate, then with water, saturated sodium bicarbonate, the saturated salt water, the organic phase is dried with anhydrous sodium sulfate, filtered, pressure-reducing evaporation did 45.0g crude product, crude product with ethyl acetate/n-heptane is recrystallized, produce a final product 34.8g ivah that non-. The purity is 97.2percent, the yield is 70.3percent.
Stage #1: With (benzotriazo-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate; 1,8-diazabicyclo[5.4.0]undec-7-ene In acetonitrile at 20 - 60℃; for 24 h; Inert atmosphere Stage #2: With sodium hydride In tetrahydrofuran at 65℃; for 5 h;
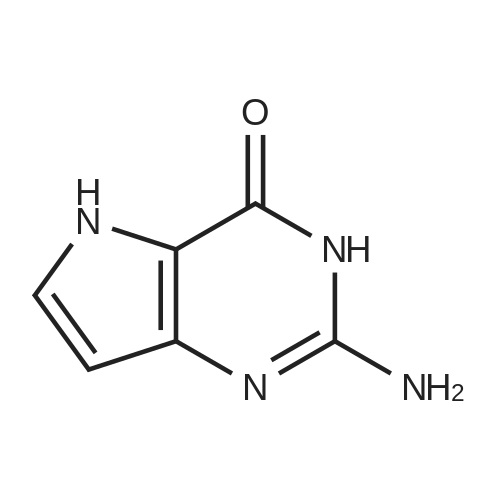
With the protection of nitrogen, 2.0 g of 6-(3-chloro-4-methoxy-benzylamino)-1,2-dihydro-pyrimidin-2-one-5-(N-2-methyl-pyrimidinyl) formamide (VII) (5 mmol), 3.31 g of benzotriazol-1-yloxytris (dimethylamino)-phosphonium hexafluorophosphate (BOP) (7.5 mmol) and 25 mL of acetonitrile in a three-necked flask. 1.15 g of 1,8-diazabicyclo[5.4.0]undec-7-ene(DBU) (7.5 mmol) was added dropwise while stirring, and reacted 12 hours at room temperature; then heated to 60° C. for reaction 12 hours. The solvent was removed by distillation under reduced pressure, then added to 50 mL ethyl acetate to dissolve, and washed with 10 mL of 2M sodium hydroxide. The organic phase was separated, dried and concentrated under reduced pressure, then the residue was dissolved in 50 mL of tetrahydrofuran, added with S-hydroxymethyl pyrrolidine (II) (0.61 g, 6 mmol) and sodium hydride (0.16 g, 6 mmol), heated to 65° C. and stirred for reaction for 5 hours, then the reaction ended with the TLC monitoring. After quenching with saturated brine the reaction, the organic phase was separated, dried and distilled under reduced pressure to recover the solvent. The resulting solid was recrystallized with ethanol to get 1.96 of white solid avanafil (I), with the yield of 81.3percent.
Stage #1: With triethylamine In dichloromethane for 1 h; Cooling with ice Stage #2: With triethylamine In dichloromethane for 2 h; Stage #3: at 20℃;
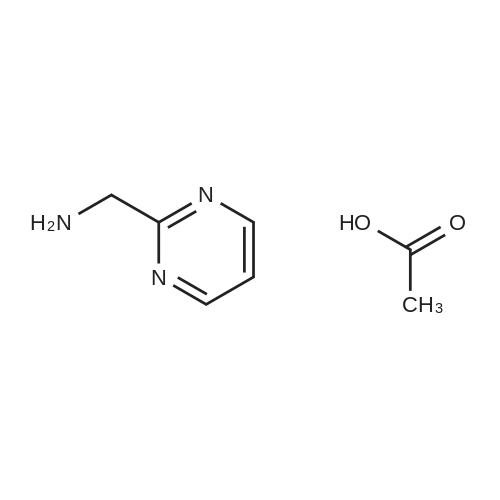
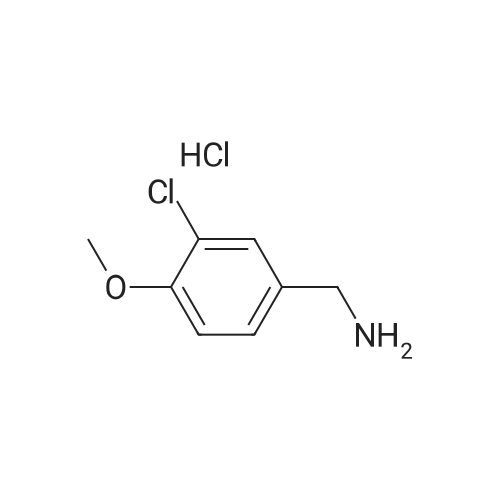
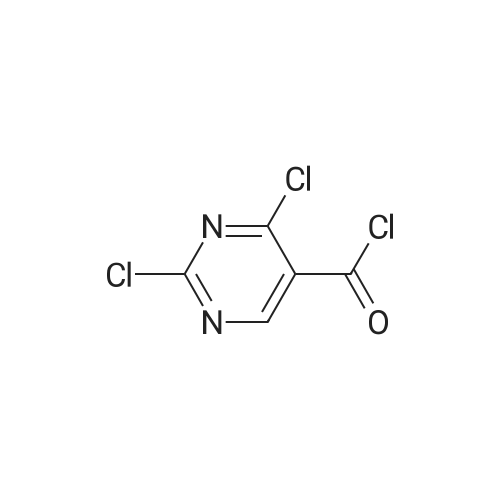
A solution of 2,4-dichloro-5-pyrimidinecarbonyl chloride (0.6 g, 2.8 mmol) in dichloromethane (8 ml) was added to a 50 ml three-necked flask and cooled in an ice bath. The 2-aminomethylpyrimidine acetate (0.48 g, 2.8 mmol) and triethylamine (2.8 mmol) were first dissolved in dichloromethane,And then dropwise dropwise into the reaction solution. Plus finished, the reaction 1 hour,A mixture of 3-chloro-4-methoxybenzylamine hydrochloride (0.59 g, 2.8 mmol) and triethylamine (0.29 g, 2.8 mmol) was added dropwise to the above reaction solution, 2 hours,To the reaction solution was added L-proline alcohol (0.43 g, 4.3 mmol), and the reaction was carried out overnight at room temperature.The reaction solution was poured into ice water, quenched and extracted twice with methylene chloride. The organic phase was combined and washed twice with water,Dried over anhydrous sodium sulfate and concentrated to give the crude product which was purified by column chromatography to give 0.9 g of white solid, and the yield was 64percent.
A solution of 3-chloro-4-methoxybenzylamine hydrochloride (0.59 g, 2.8 mmol)And triethylamine (0.29 g, 2.8 mmol) were added dropwise to the above-mentioned spare solution, and the reaction was carried out for 2 hours. After addition, L-proline alcohol (0.43 g, 4.3 mmol) was added to the reaction solution, The reaction was carried out at room temperature for 4 hours.The reaction solution was poured into ice water, quenched and extracted twice with methylene chloride. The organic phase was combined and washed twice with water, dried over anhydrous sodium sulfate,The resulting crude column was concentrated to give 0.8 g of a white solid, i.e., avanafil, in 32percent yield
To the dichioromethane layer obtained in step i), was added 2.57g of triethylamine followed by slow addition of l25m1 solution of L-prolinol in dichloromethane (2.46g of L-prolinol in l25m1 of dchlromethane). The reaction mixture was maintained overnight. After completion of reaction, the reaction mixture was washed with water followed by evaporation of dichioromethane to obtain an oily mass. The oily mass thus obtained was treated with methanol to yield 8g of Avanafil.
With potassium carbonate; In acetonitrile; at -10 - 0℃; for 2h;Inert atmosphere;
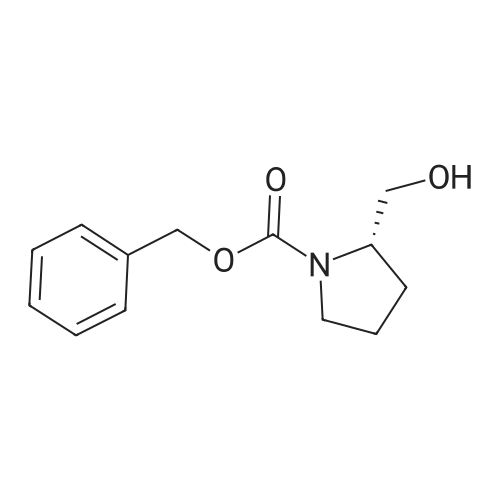
A solution of benzyl chloroformate (3.81 g, 22.44 mmol) in acetonitrile (5 mL) was added slowly to a stirred solution of (S)-prolinol (2.07 g, 20.4 mmol) and finely powdered K2CO3 (6.20 g, 44.88 mmol) in dry acetonitrile (20 mL) at -10 oC under N2 atmosphere. Further, solution was stirred at -10 to 0 oC for 2 h. H2O (50 mL) was added to the reaction mixture and the aqueous layer was extracted with CHCl3 (3 × 20 mL). The combined organic extract was washed successively with H2O (20 mL), 5percent HCl (20 mL), H2O (3 × 20 mL) and then dried over anhyd Na2SO4. The solvent was removed under reduced pressure, the crude product was subjected to column chromatography (SiO2, hexanes/ EtOAc, 6:4) to give (S)-N benzyloxycarbonyl prolinol 14 (3.97 g, 83percent). [alpha]D27 -41.5 (c 0.448, CHCl3), [lit.2[alpha]D27 -40.0 (c 1.05, CH2Cl2)]; 1H NMR (CDCl3, 300 MHz): delta 1.45-2.05 (m, 4H, CH2CH2), 3.36-3.69 (m, 4H, CH2N & CH2OH), 4.01 (m, 1H, CHN), 5.15 (s, 2H, OCH2Ph), 7.26-7.37 (m, 5H, Ar-H); 13C NMR (CDCl3, 75 MHz): delta 24.0 (CH2), 28.4 (CH2), 47.3 (CH2N), 58.7 (CHN), 66.2 (CH2OH), 67.2 (OCH2), 127.9-128.3 (Ar-H), 136.0 (Ar-C), 156.9 (CO).
With sodium hydroxide; In diethyl ether;
EXAMPLE 50 (S)-2-[[[(1,1-Dimethylethyl)dimethylsilyl]oxy]methyl]-1-pyrrolidinecarboxylic acid phenylmethyl ester To a 0° C. solution of 25 g of (S)-2-pyrrolidinemethanol in 150 ml of diethyl ether is added 54.8 g of benzyl chloroformate. At mid point of addition, 49.4 ml of 5N sodium hydroxide is added, simultaneously, and the reaction stirred at 0° C. for 2 hours. Diethyl ether is added, the layers are separated and the aqueous layer reextracted with diethyl ether. The combined organic layers are dried, filtered and concentrated in vacuo. The residue is purified by chromatography (silica gel, 0-10percent methyl alcohol/methylene chloride) to give 59.1 g of (S)-2-hydroxymethyl-1-pyrrolidine carboxylic acid phenylmethyl ester.
With potassium carbonate; In hexane; acetone;
(A) S-N-(Benzyloxycarbonyl)-2-pyrrolidinemethanol Powdered anhydrous potassium carbonate (41 g, 297 mmol) was added with stirring to a solution of S-2-pyrrolidinemethanol (6 g, 59.32 mmol) in acetone (120 ml). The mixture was cooled to 0° C. and benzyl chloroformate (16.94 ml, 118.6 mmol) was added dropwise. After 40 minutes, the reaction mixture was diluted with ethyl acetate and water. The organic layer was separated and the aqueous layer was extracted with ethyl acetate. The organic extracts were combined, dried (magnesium sulfate) and concentrated. The crude oil was chromatographed on a silica gel column and eluted with 25percent ethyl acetate in hexane to obtain the title compound 2 (12.22 g) as a pale yellow oil.
With sodium bicarbonate; In dichloromethane; water;
(1) A solution of carbobenzoxychloride 7.87 g in methylene chloride 50 ml is dropped to a mixture of L-prolinol 4.9 g in methylene chloride 50 ml and sodium hydrogen carbonate 11.6 g in water 50 ml at 0° C. under vigorously agitation. The mixture is stirred for 1 hour at room temperature. The organic layer is separated, washed, dried and concentrated in vacuo to give N-carbobenzoxy-L-prolinol 10.25 g.
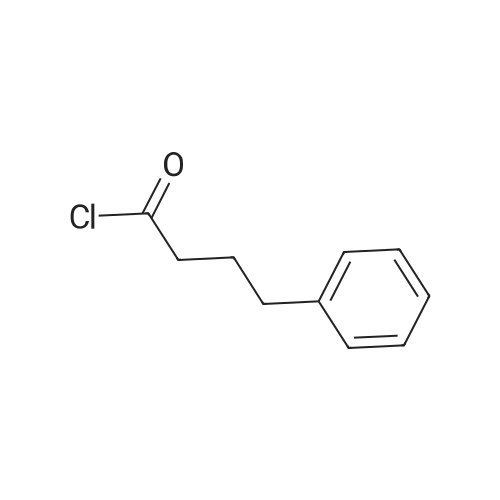
Aqueous sodium hydroxide (1 N, 25 ml) is added to a solution of 2.0 g of L-prolinol (U.S. patent 3,935,280) in 50 ml of CH2 Cl2. After cooling the reaction mixture to 0 C., 4.0 g of 4-<strong>[18496-54-3]phenylbutyric acid chloride</strong> is added and the reaction is stirred vigorously for 4 hours at 0 C., followed by 1 hour at room temperature. The reaction is diluted with an equal volume of CH2 Cl2 and the layers separated. The organic phase is washed with 30 ml of water and dried over Na2 SO4 /K2 CO3. The solvent is evaporated to yield 4.4 g of N-(4-phenylbutanoyl)-L-prolinol having IR peaks at 3280 and 1605 cm-1, [α]D25 =-40.3 (methanol).
With sodium hydroxide; In dichloromethane; water;
Aqueous sodium hydroxide (1N, 25 ml) is added to a solution of 2.0 g of L-prolinol (U.S. Pat. No. 3,935,280) in 50 ml of CH2 Cl2. After cooling the reaction mixture to 0 C., 4.0 g of 4-<strong>[18496-54-3]phenylbutyric acid chloride</strong> is added and the reaction is stirred vigorously for 4 hours at 0 C., followed by 1 hour at room temperature. The reaction is diluted with an equal volume of CH2 Cl2 and the layers separated. The organic phase is washed with 30 ml of water and dried over Na2 SO4 /K2 CO3. The solvent is evaporated to yield 4.4 g of N-(4-phenylbutanoyl)-L-prolinol having IR peaks at 3280 and 1605 cm-1, [α]D25 =-40.3 (methanol).
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide; at 20℃;
4- (benzyloxy) -5-methoxy-2-nitrobenzoic acid (10.20 g, 33.65 mmol) and (S) -pyrrolidin-2-ylmethanol (3.85 g, 38.09 mmol) in dry DMF (150 ml) was added EDC (19.50 g, 101.56 mmol) . The mixture was stirred at RT for overnight, concentrated and purified on SiO 2 column eluted with EtOAc/CH 2Cl 2 (1: 4) to afford the title compound (11.56 g, 89%yield) . MS ESI m/z calcd for C 20H 23N 2O 6 [M+H] + 387.15, found 387.65.
To a solution of (S)-pyrrolidin-2-yl-methanol (0.69 g, 6.82 mmol) in DCM (3 mL) cooled to 0 C. was added TEA (1.38 g, 13.6 mmol) followed by TBDMS-Cl (1.03 g, 6.82 mmol) in DCM (3 mL). The reaction mixture was then stirred at room temperature over night and poured into NH4Cl (20 mL). The aqueous layer was extracted with DCM (2×50 mL). Combined organics were washed with brine, dried over Na2SO4, filtered and evaporated to dryness to give the title compound (1.11 g, 76%) as a yellow oil. MS: 216.2 (M+H+)
76%
With triethylamine; In dichloromethane; at 0 - 20℃;
Intermediate A-36-{5-[(S)-2-(tei"i-Butyl-dimethyl-silanyloxymethyl)-pyrrolidin-l-yl]-pyridin-3-yl}-l- methyl-3,4-dihydro-lH-quinolin-2-one [A] (S)-2-(tert-Butyl-dimethyl-silanyloxymethyl)-pyrrolidineTo a solution of (S)-pyrrolidin-2-yl-methanol (0.69 g, 6.82 mmol) in DCM (3 mL) cooled to 0 C was added TEA (1.38 g, 13.6 mmol) followed by TBDMS-Cl (1.03 g, 6.82 mmol) in DCM (3 mL). The reaction mixture was then stirred at room temperature over night and poured into NH4C1 (20 mL). The aqueous layer was extracted with DCM (2 x 50 mL). Combined organics were washed with brine, dried over Na2S04, filtered and evaporated to dryness to give the title compound (1.11 g, 76%) as a yellow oil. MS: 216.2 (M+H+).
76%
With triethylamine; In dichloromethane; at 0 - 20℃;Inert atmosphere;
To a solution of (S)-pyrrolidin-2-ylmethanol (0.69 g, 6.82 mmol) in DCM (3 mL) cooled to 0 C was added TEA (1.38 g, 13.6 mmol) followed by TBDMS-Cl (1.03 g, 6.82 mmol) in DCM (3 mL). The reaction mixture was then stirred at room temperature over night and poured into NH4C1 (20 mL). The aqueous layer was extracted with DCM (2 x 50 mL). Combined organics were washed with brine, dried over Na2S04, filtered and evaporated to dryness to give the title compound (1.1 lg, 76%) as a yellow oil. MS: 216.2 (M+H+).
76%
With triethylamine; In dichloromethane; at 0 - 20℃;
[A] (S)-2-(tert-Butyl-dimethyl-silanyloxymethyl)-pyrrolidine To a solution of (S)-pyrrolidin-2-ylmethanol (0.69 g, 6.82 mmol) in DCM (3 mL) cooled to 0 C. was added TEA (1.38 g, 13.6 mmol) followed by TBDMS-Cl (1.03 g, 6.82 mmol) in DCM (3 mL). The reaction mixture was then stirred at room temperature overnight and poured into NH4Cl (20 mL). The aqueous layer was extracted with DCM (2*50 mL). Combined organics were washed with brine, dried over Na2SO4, filtered and evaporated to dryness to give the title compound (1.11 g, 76%) as a yellow oil. MS: 216.2 (M+H+).
With 1H-imidazole; In tetrahydrofuran; at 20℃; for 2h;
[0869] To a mixture of L-prolinol (20 g, l97.73mmol), imidazole (4 g, 587.54mmol) in tetrahydrofuran (200 mL) was added Art-butyldimethylchlorosilane (45 g, 298.57mmol), the mixture was stirred for 2 hours at room temperature. Upon completion, the resulting solution was diluted with water, extracted with dichloromethane and the organic layers were combined. The organic layer was dried over anhydrous sodium sulfate and concentrated to afford crude product which directly used in the next step without purification. LC-MS: (ESI, m/z): 216.2 [M+H]+
((2S)-1-[4-(5-methyl-3-phenylisoxazol-4-yl)-phenyl]sulfonyl}pyrrolidin-2-yl)methanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
97%
In tetrahydrofuran; at -78 - 20℃; for 6h;
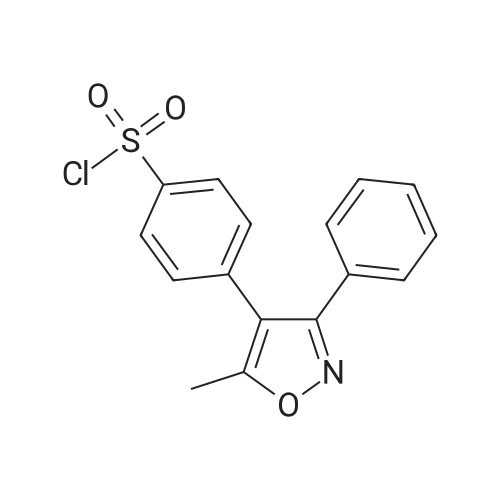
4-(5-Methyl-3-phenylisoxazol-4-yl) benzenesulfonyl chloride (200 mg, 0.60 mmol) was dissolved in THF (3 ml) and the solution was cooled to-78C. After S- (-)-prolinol (182 mg, 1.80 mmol) was added to the solution, the mixture was gradually warmed to room temperature and stirred at room temperature for 6 hours. Ethyl acetate (8 ml) was added thereto, and the mixture was washed with water (3 ml) and subsequently with brine (2 ml), and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane: ethyl acetate = 9: 1-> 1: 1) to give ((2S)-1-[4- (5-methyl-3-phenylisoxazol-4-yl)- phenyl] sulfonyl} pyrrolidin-2-yl) methanol (232 mg, 97%) as a liquid. MS: 399 [M+H] +, APCI (MeOH)
With sodium hydrogencarbonate; In dichloromethane; N,N-dimethyl-formamide;
a (S)-2-(Tetrahydro-pyran-2-yloxymethyl)-pyrrolidine-1-carboxylic acid benzyl ester A solution of L-prolinol (1.00 g, 4.94 mmol) in N,N-dimethylformamide (5 mL) was cooled to 0 C. and treated with sodium hydrogencarbonate (831 mg, 9.87 mmol) and benzyl chloroformate (843 mg, 4.94 mmol). After removal of the ice bath the reaction mixture was stirred 1 h at r.t. then poured onto ice and extracted with ethyl acetate. The organic layer was washed with brine, dried (MgSO4) and evaporated. The residue was taken up in dichloromethane (5 mL), treated with 3,4-dihydro-2H-pyran (1.25 g, 14.8 mmol) and pyridinium toluene-4-sulfonate (1.37 g, 5.43 mmol) and stirred for 48 h at r.t., then evaporated. The residue was partitioned between ethyl acetate and water, the organic layer was washed with brine, dried (MgSO4), and evaporated. Chromatography (SiO2, hexane/ethyl acetate 3:1) yielded the title compound (451 mg, 29%). Colourless liquid, ISP-MS: m/e=320.4 ([M+H]+).
(S)-2-cyclohexyl-1-(2-(hydroxymethyl)pyrrolidin-1-yl)ethanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
With hydrogenchloride; (benzotriazo-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate; In dichloromethane; N-ethyl-N,N-diisopropylamine;
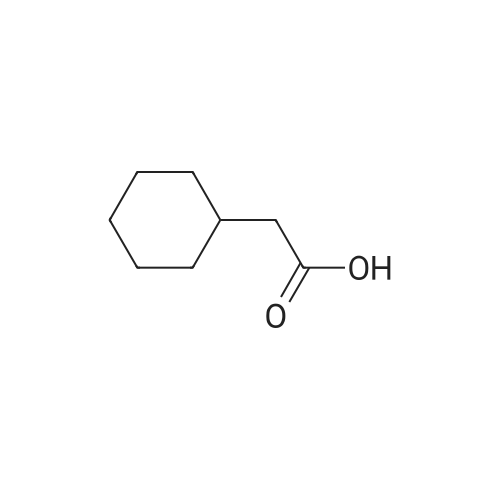
Example 134 (S)-2-cyclohexyl-1-(2-(hydroxymethyl)pyrrolidinyl)ethan-1-one (134) In dichloromethane, 2-cyclohexyl acetic acid (540mg, 3.65mmol, 1.2eq) was dissolved, and BOP (1.61g, 3.65mmol, 1.2eq) was added thereto, followed by stirring the mixture at room temperature for 10 minutes. After adding (S)-pyrrolidin-2-ylmethan-1-ol (0.31 g, 3.04mmol) to the reaction solution, the mixture was stirred while cooling the mixture in ice and diisopropylethylamine was added, followed by stirring the mixture at room temperature for 24 hours. To the reaction solution, 1N hydrochloric acid was added and the resulting mixture was extracted with dichloromethane. Organic layers were combined, washed with saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to obtain 701 mg of (S)-2-cyclohexyl-1-(2-(hydroxymethyl)pyrrolidinyl)ethan-1-one (134) (yield: 85percent). LR-MS(m/z):225(M+) 1H-NMR(300MHz, CDCI3, deltappm): 5.27 (1H, d), 4.23 (1H, m), 3.68-3.42 (4H, m), 2.19 (2H, d), 2.10-1.51 (9H, m), 1.38-0.88 (6H, m)
With sodium cyanoborohydride; acetic acid; In tetrahydrofuran; at 0 - 20℃; for 16h;
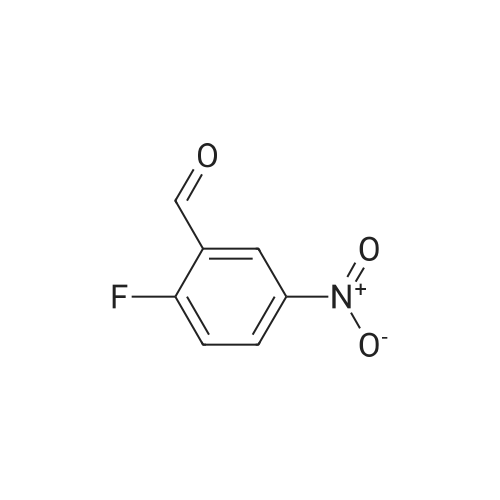
To a solution of <strong>[27996-87-8]2-fluoro-5-nitrobenzaldehyde</strong> (595 mg, 3.52 mmol) in THF (10 ml) was added a solution of (S)-(+)-2-(hydroxymethyl)pyrrolidine in THF (10 ml) followed by 5 acetic acid (251 mul, 4.40 mmol). The mixture was cooled to 0C and sodium cyanoborohydride (276 mg, 4.40 mmol) was added. The cooling bath was removed and the mixture was stirred at RT for 16 h. The mixture was diluted with EtOAc and washed with saturated aqueous sodium hydrogen carbonate. The organic phase was dried (Na2SO4) and the solvent was evaporated. The residue was purified by column chromatography on silica EPO <DP n="45"/>eluting with a gradient of 0-7% methanol in dichloromethane to give an oil (629 mg, 70%). MS ESI m/z M+H+ 255.
With 1-hydroxy-7-aza-benzotriazole; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 20℃; for 3h;
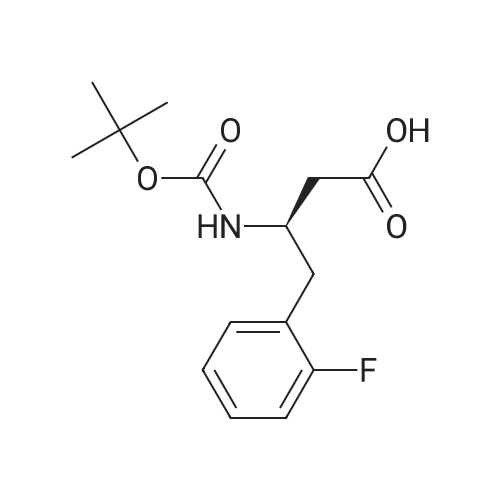
Intermediate A-1: To a mixture of (3R)-3-(Boc-amino)-4-(2-fluorophenyl)butanoic acid (59.1 mg, 0.20 mmol), HOAT (40.8 mg, 0.30 mmol) and EDCI (115 mg, 0.60 mmol) in methylene chloride (1 mL) was added (S)-(+)-2-pyrrolidine methanol. The reaction was stirred at room temperature for 3 hours and concentrated. The residue was partitioned between saturated aqueous sodium bicarbonate and ethyl acetate. The layers were separated and the aqueous layer was extracted with ethyl acetate (2×). The combined organic layers were washed once with saturated aqueous sodium bicarbonate, twice with saturated aqueous ammonium chloride, once with brine, dried with sodium sulfate and concentrated in vacuo to give Intermediate A-1 as a glassy material (74.6 mg, 98% yield): LCMS Method 1: M+H=381; LC Method 1: tR=3.1 mm.
In 1,4-dioxane; at 70 - 80℃; for 12 - 48h;Combinatorial reaction / High throughput screening (HTS);
Example 1. Preparation of a combinatorial library of substituted 2-aminomethyl-5-hydroxy-1H-indole-3-carboxylic acids esters of general formula 1.1. A mixture of 0.357 mmol of ester 2, 0.43 mmol of a secondary amine 3, and 0.43 mmol of formaldehyde in the form of formalin in 3 ml of dioxane is heated at 70-80C at stirring for 12 to 48 hours. Progress of the reaction is monitored by chromato-mass-spectrometry. Upon completion of the reaction, the reaction mass is diluted with water, the residue is filtered and recrystallized from a suitable solvent, or purified by chromatography.
Compound (XVIa), which represents a further preferred embodiment of the first aspect of the present invention, was prepared by reacting compound XIV with compound XVa in the presence of a base as follows:
To a solution of L-proline in methanol:THF (0.10 M; 1:2), at 0° C., is added a solution boran dimethyl sulfide (5 equivalents; 1.0 M) and allowed to stir for overnight. The solvent is then removed and the mixture is resuspended in methylene chloride (0.10 M) and 1.2 equivalents of NaH (30percent) is added at 0° C. and allowed to stir for 1 hour. Next, 1.1 equivlanents of methyliodide is added and allowed to stir at 0° C. for an additional hour. The reaction mixture is then quenched with water, extracted with 100percent ethylacetate, washed over bicarbonate, brine and dried over magnesium sulfate. The crude compound is purifed by flash chromatography to give compound 24.
With N-ethyl-N,N-diisopropylamine In dichloromethane at 100℃; for 0.166667h; Microwave irradiation;
76A
76A. (S)-(1-(2-chloropyridin-4-yl)pyrrolidin-2-yl)methanol A mixture of (S)-pyrrolidin-2-ylmethanol (1.153 g, 11.40 mmol), 2-chloro-4-fluoropyridine (1 g, 7.60 mmol) and N-ethyl-N-isopropylpropan-2-amine (0.983 g, 7.60 mmol) was heated in the microwave oven for 10 min at 100° C. The thick mixture was dissolved in CH2Cl2, which was washed with aqueous 1N HCl, NaHCO3 solution and brine. The combined aqueous solution was adjusted to pH ~8, then it was extracted 3* with CH2Cl2. The combined organic extracts were dried over MgSO4 and concentrated. The residue was purified with silica gel chromatography to yield 76A (yellow oil, 1.58 g, 7.43 mmol, 98% yield). LC-MS calc'd for C10H13ClN2O: 212.68. found [M+H]: 213.1. 1H NMR (400 MHz, chloroform-d) δ ppm 7.90 (1H, d, J=6.24 Hz), 6.46 (1H, d, J=2.24), 6.40 (1H, dd, J=6.24, 2.24 Hz), 3.83-3.96 (1H, m), 3.57-3.75 (2H, m), 3.41-3.52 (1H, m), 3.14-3.25 (1H, m), 1.96-2.24 (4H, m).
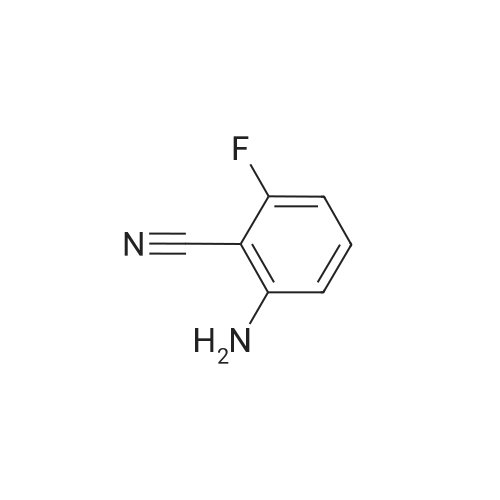
General procedure: To a solution of amino alcohol (0.91 eq.) inanhydrous THF (5 ml) was added NaH as a 60% dispersion in oil (1.0 eq.). Themixture was stirred at 0 C for 30 min, at r.t. for another 30 min beforecooling down to 0 C again. 2-Amino-6-fluorobenzonitrile (1.0 eq.) was addedand the reaction was refluxed for 2 hours before cooling down to r.t. H2O(0.7 ml) was added and the solvents were evaporated in vacuo. The residue was purified by flash column chromatography.
Example 89d (S)-2-amino-6-(pyrrolidin-2-ylmethoxy)benzonitrile Prepared as in Example 24d from (S)-pyrrolidin-2-ylmethanol and <strong>[77326-36-4]2-amino-6-fluorobenzonitrile</strong> as brown solid (51%). MS 218 (MH+).
Example 89d (S)-2-amino-6-(pyrrolidin-2-ylmethoxy)benzonitrile Prepared as in Example 24d from (S)-pyrrolidin-2-ylmethanol and <strong>[77326-36-4]2-amino-6-fluoro-benzonitrile</strong> as brown solid (51%). MS 218 (MH+).
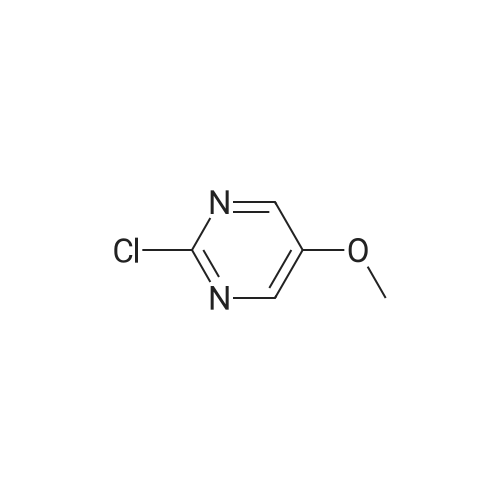
(S)-(1-(5-methoxypyrimidin-2-yl)pyrrolidin-2-yl)methanol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium tert-butylate; In tetrahydrofuran; at 20℃; for 2h;
Step 1 : ( )-(l-(5-Methoxypyrimidin-2-yl)pyrrolidin-2-yl)methanol To a stirring solution of (5)-pyrrolidin-2-ylmethanol (0.202 g, 2 mmol) in THF (2 mL) was added iBuOK (1M in THF) (2 mL, 2 mmol) followed by 2-chloro-5- methoxypyrimidine (0.145 g, 1 mmol). The reaction mixture was stirred at rt for 2 h to give the title compound after purification on silica gel column; MS (ESI) 210 (M + H).
General procedure: To a solution of the corresponding diol (5 mmol) in dry toluene(10 mL), under a nitrogen atmosphere, was added triisopropylborate (1.17 mL, 5.1 mmol) via syringe and the mixture stirredunder mild reflux conditions for approximately 20 min. Whenthe mixture became homogeneous, a solution of amino alcohol indry toluene (10 mL) was added. The mixture was refluxed for20 min and during this time a portion of toluene along with evolvingisopropanol was removed by distillation (about 10 mL). Next,toluene (10 mL) was added to the distillation flask and the contentsevaporated again to make sure all the isopropanol had beenremoved. After stirring for 20 min, the mixture was cooled to roomtemperature and a white precipitate of the spiroborate esterappeared. The product was filtered and recrystallized from tolueneas a white crystalline powder. 4.3.9 (S)-2-[(1,3,2-Dioxaborolan-(4-2',5-3'-(1'R,2'R,3'S,5'R)-pinan)-2-yloxy)-methyl]pyrrolidine 9 White solid; mp 144-146 C; [alpha]D20 = + 22.0 (c 0.5, THF); 1H NMR (CDCl3): delta = 0.82 (s, 3H), 1.24 (s, 3H), 1.28 (s, 3H), 1.49 (d, J = 10.2 Hz, 1H), 1.58-1.70 (m, 1H), 1.71-2.05 (m, 6H), 2.06-2.19 (m, 1H), 2.28 (dt, J = 13.8, 2.1 Hz, 1H), 2.74-2.95 (m, 1H), 3.32-3.54 (m, 2H), 3.82-3.99 (m, 2H), 4.05 (dd, J = 8.7, 2.4 Hz, 1H), 7.08 (br s, 1H); 13C NMR (CDCl3): delta = 24.27, 26.12, 26.79, 27.39, 29.32, 29.76, 37.65, 38.24, 40.19, 46.67, 52.86, 60.56, 65.79, 75.55, 81.58; 11B NMR (CDCl3): delta = 10.98; Anal. calcd for C15H26BNO3: C, 64.53, H, 9.39, N, 5.02; found: C, 64.55, H, 9.27, N, 5.26.
A solution of 2,4-dichloro-5-pyrimidinecarbonyl chloride (0.6 g, 2.8 mmol) in dichloromethane (8 ml) was added to a 50 ml three-necked flask and cooled in an ice bath. The 2-aminomethylpyrimidine acetate (0.48 g, 2.8 mmol) and triethylamine (2.8 mmol) were first dissolved in dichloromethane,And then dropwise dropwise into the reaction solution. Plus finished, the reaction 1 hour,A mixture of <strong>[41965-95-1]3-chloro-4-methoxybenzylamine hydrochloride</strong> (0.59 g, 2.8 mmol) and triethylamine (0.29 g, 2.8 mmol) was added dropwise to the above reaction solution, 2 hours,To the reaction solution was added L-proline alcohol (0.43 g, 4.3 mmol), and the reaction was carried out overnight at room temperature.The reaction solution was poured into ice water, quenched and extracted twice with methylene chloride. The organic phase was combined and washed twice with water,Dried over anhydrous sodium sulfate and concentrated to give the crude product which was purified by column chromatography to give 0.9 g of white solid, and the yield was 64%.
2,4-dichloro-N-(pyrimidin-2-ylmethyl)-5-pyrimidinecarboxamide[ No CAS ]
[ 23356-96-9 ]
[ 41965-95-1 ]
[ 330784-47-9 ]
Yield
Reaction Conditions
Operation in experiment
32%
A solution of <strong>[41965-95-1]3-chloro-4-methoxybenzylamine hydrochloride</strong> (0.59 g, 2.8 mmol)And triethylamine (0.29 g, 2.8 mmol) were added dropwise to the above-mentioned spare solution, and the reaction was carried out for 2 hours. After addition, L-proline alcohol (0.43 g, 4.3 mmol) was added to the reaction solution, The reaction was carried out at room temperature for 4 hours.The reaction solution was poured into ice water, quenched and extracted twice with methylene chloride. The organic phase was combined and washed twice with water, dried over anhydrous sodium sulfate,The resulting crude column was concentrated to give 0.8 g of a white solid, i.e., avanafil, in 32% yield
ethyl (S)-4-((3-chloro-4-methoxybenzyl)amino)-2-(2-(hydroxymethyl)tetrahydropyrrole-1-yl)-5-pyrimidinecarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
70%
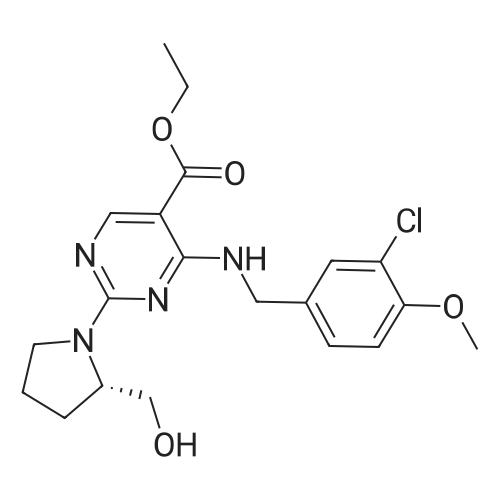
To a 25 ml three-necked flask was added ethyl 2,4-dichloro-5-pyrimidinecarboxylate (150 mg, 0.679 mmol)Triethylamine (270 mg, 2.668 mmol), dichloromethane (2 ml), gradually stirred to dissolve,The ice bath was cooled to 0 C or below, and <strong>[41965-95-1]3-chloro-4-methoxybenzylamine hydrochloride</strong> (145 mg, 0.697 mmol) was added in portions to the reaction solution, and the reaction was carried out for two hours after completion of the addition.L-proline (105 mg, 1.038 mmol) was added dropwise to the reaction solution, and the reaction was continued for 2 hours.The reaction solution was added to water (30 ml) and extracted with dichloromethane.The organic phases were combined, washed twice, dried over anhydrous sodium sulfate, the desiccant was filtered off, and the organic phase was concentrated to the crude product.The crude product was purified by silica gel column chromatography (petroleum ether: ethyl acetate = 6: 1) to give 200 mg of product in 70% yield.
tert-butyl 2,4-dichloro-5-pyrimidinecarboxylic acid[ No CAS ]
[ 23356-96-9 ]
[ 41965-95-1 ]
ethyl (S)-4-((3-chloro-4-methoxybenzyl)amino)-2-(2-(hydroxymethyl)tetrahydropyrrole-1-yl)-5-pyrimidinecarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
75%
To a 25 ml three-necked flask, tert-butyl 2,4-dichloro-5-pyrimidinecarboxylate (169 mg, 0.679 mmol)Triethylamine (270 mg, 2.668 mmol), dichloromethane (2 ml), gradually stirred to dissolve,The ice bath cools below 0 C,3-Chloro-4-methoxybenzylamine hydrochloride (145 mg, 0.697 mmol) was added portionwise to the reaction solution,After feeding the insulation reaction for two hours.To the reaction solution, L-proline (105 mg, 1.038 mmol) was added dropwise,The reaction was continued for 2 hours. The reaction solution was added to water (30 ml) and extracted with dichloromethane.The organic phases were combined, washed twice, dried over anhydrous sodium sulfate, the desiccant was filtered off, and the organic phase was concentrated to the crude product. Crude product by silica gel column chromatography product 228mg, the yield of 75%.
With 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride; In methanol; at 28℃; under 760.051 Torr; for 18h;
At room temperature,Under normal pressure,Weigh 0.5g of 4-ethynylbenzoic acid,According to 4-ethynylbenzoic acid,L-prolinol,The molar ratio of 4-(4,6-dimethoxytriazine)-4-methylmorpholine hydrochloride is 1:1:1.1The ratio of L-prolinol and 4-(4,6-dimethoxytriazine)-4-methylmorpholine hydrochloride is weighed.4-ethynylbenzoic acid,L-prolinol and 4-(4,6-dimethoxytriazine)-4-methylmorpholine hydrochloride were dissolved in 17.5 mL of methanol for amidation reaction.The reaction temperature is 28 C,The reaction time was 18 h.After the reaction is over,Filter the reaction solution,Rotary evaporation,Purified by column chromatography.The eluent used in the column chromatography was ethyl acetate/n-hexane (7/2, V/V).The product was a white flaky solid. The product was named PAA-Val-OH, yield 0.42 g, yield 53.2%.
(S)-(1-(6-benzyl-2-((1-(3,4,5-trimethoxyphenyl)-1H-imidazol-4-yl)amino)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl)pyrrolidin-2-yl)methanol hydrochloride[ No CAS ]
(S)-(1-(2-((1-(3,4,5-trimethoxyphenyl)-1H-imidazol-4-yl)amino)-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)pyrrolidin-2-yl)methanol hydrochloride[ No CAS ]
(S)-(1-(1-methyl-6-((1-(3,4,5-trimethoxyphenyl)-1H-imidazol-4-yl)amino)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)pyrrolidin-2-yl)methanol hydrochloride[ No CAS ]
(S)-tert-butyl 4-(2-(hydroxymethyl)pyrrolidin-1-yl)-2-(1-(3,4,5-trimethoxyphenyl)-1H-imidazol-4-ylamino)-5H-pyrrolo[3,4-d]pyrimidine-6(7H)-carboxylate[ No CAS ]
(S)-(1-(1,3-dimethyl-6-((1-(3,4,5-trimethoxyphenyl)-1H-imidazol-4-yl)amino)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)pyrrolidin-2-yl)methanol hydrochloride[ No CAS ]
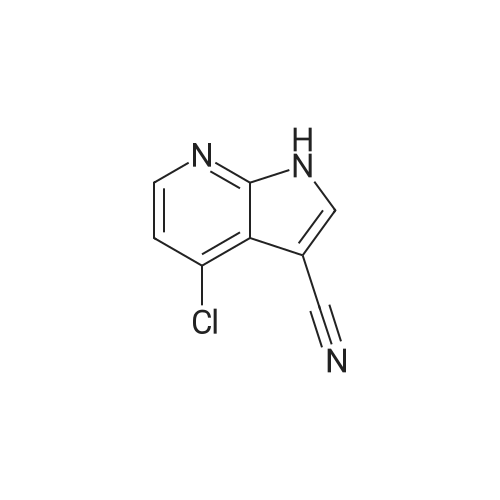
4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
23 mg
In 1-methyl-pyrrolidin-2-one; at 160℃; for 4h;Inert atmosphere;
To a microwave vial was added (S)-(+)-2-Pyrrolidinemethanol (57 mg, 0.56 mmol) and <strong>[920965-87-3]4-chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile</strong> (P5) (50 mg, 0.24 mmol) in NMP (0.5 ml) and heated at 160 00 for 4 hours. The mixture was purified by silica chromatography, eluting with 0-5% MeOH in DCM, to give 4-[(2S)-2- (hydroxymethyl)pyrrolidin-1-yl]-1 H-pyrrolo[2,3-b]pyridine-3-carbonitrile (E38) (23 mg) LCMS ES 243.2 [M+H], Rt = 0.80 mins (Generic Basic Method).
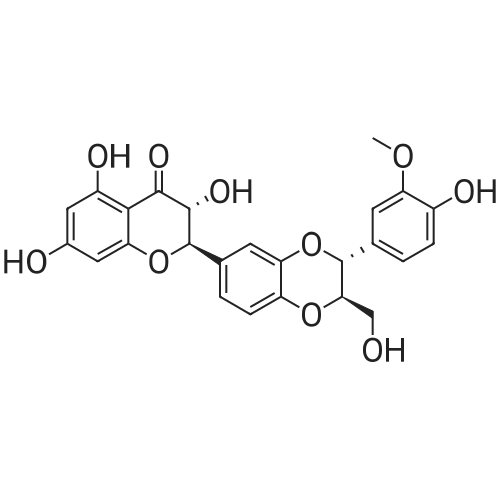
Silybin (5 g, 10.4 mmol), 37% aqueous formaldehyde solution (5.7 g, 62.4 mmol), and L-prolinol (6.30 g, 62.4 mmol) were placed in 50 mL of methanol, stirred at room temperature, and TLC (dichloromethane) : Methanol = 5: 1) The reaction was followed. The reaction was completed in 20 hours, and the reaction was terminated. A solid precipitated during the reaction. After filtration, the solid was slurried with 100 mL of ethanol for 2 h, and filtered under reduced pressure. After drying the solid, 2.2 g of (I-6) was obtained (yield: 30%). The obtained (I-6) was dissolved in a methanol-hydrochloric acid solution (50 mL), stirred and concentrated to dryness to obtain (I-6) hydrochloride.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping