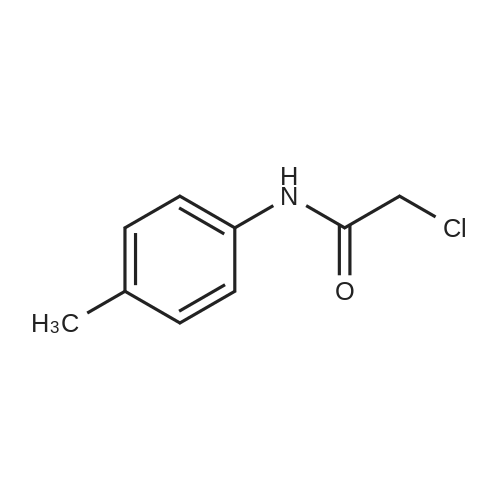
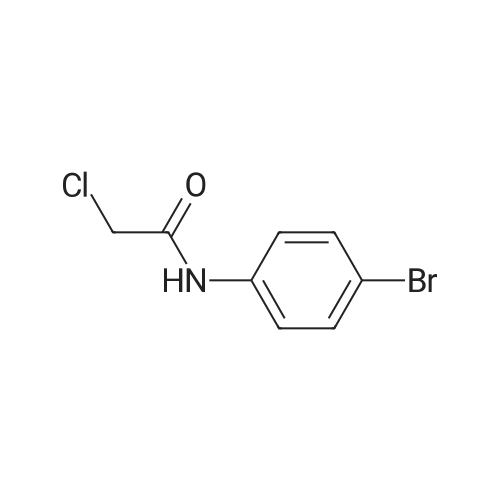
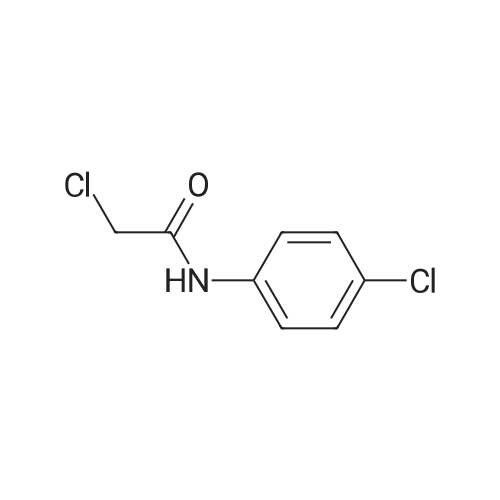
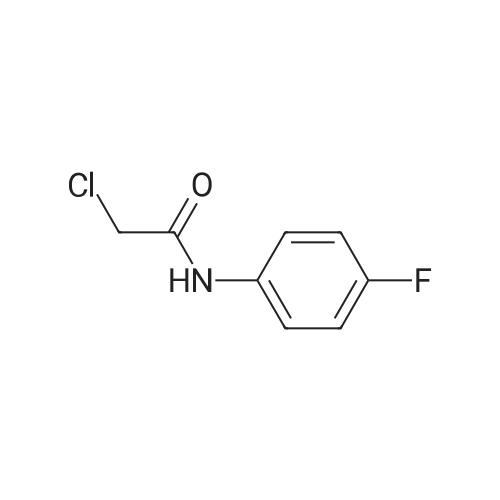
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
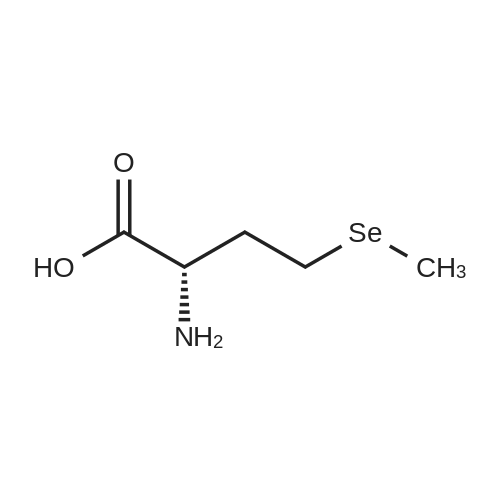
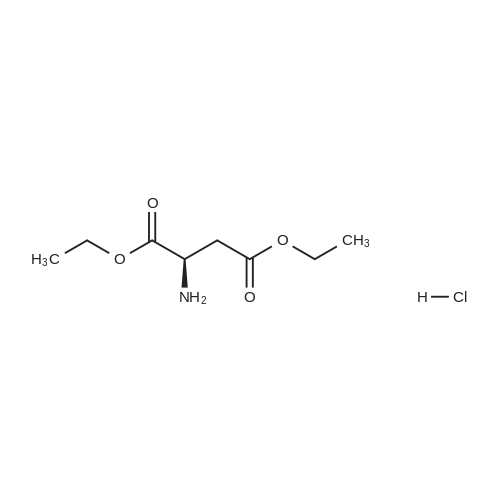
Under a nitrogen atmosphere, 13.75 g of α-amino-γ-butyrolactone hydrochloride was mixed with 40 mL of DMF,Stir evenly. 15.2 g of sodium methyl selenate was dissolved in 20 mL of DMF, added to the above system and refluxed for 2 h. After the reaction was completed, the pH of the system was adjusted to 6.0 with an aqueous acetic acid solution to give a white solid,Solid water and ethanol mixed solution recrystallization, both L-selenomethionine. The total yield was 72.11percentChemical purity> 99percent, ee> 99percent.
Reference:
[1] Patent: CN106928110, 2017, A, . Location in patent: Paragraph 0028; 0030
2
[ 672-15-1 ]
[ 2185-03-7 ]
Yield
Reaction Conditions
Operation in experiment
99%
With hydrogenchloride In water for 0.25 h;
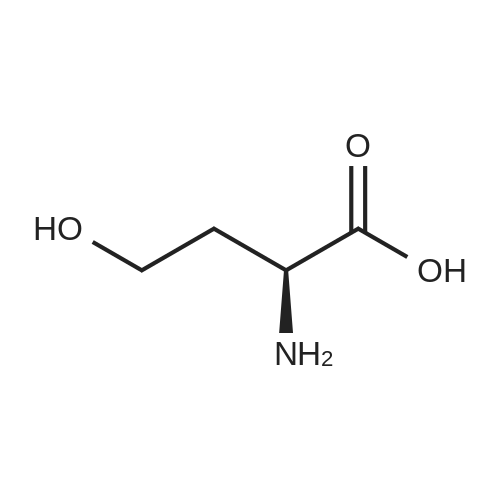
L-Homoserine (0.84 mol) was admixed with 600 ml of concentrated HCl (6.1 mol). The solution was stirred for about 15 minutes until everything had dissolved, and subsequently the water was removed under vacuum over the course of 1.5 hours. The residue was dried. Yield: 99percent of 2-amino-4-butyrolactone hydrochloride salt.
83%
With hydrogenchloride In water at 20℃; Reflux
Synthesis of homoserine lactone (AHL-2), analogs (QS1207; QS0108): (S)-Homoserine lactone hydrochloride: (S)-homoserine (25 g, 0.21 mol) was dissolved in aqueous hydrochloric acid (2.4 M, 322 mL, 0.97 mol, 4.6 equiv). The solution was refluxed for 3 h, and stirred at ambient temperature overnight. Most of the solvent was removed azeotropically with ethanol. Following crystal formation the solution was cooled on ice. The resulting solid was filtered and rinsed three times with cold ethanol. The filtrate was concentrated and cooled, producing additional homoserine lactone. The process was repeated 2 more times. After leaving the white powder on high vacuum line overnight, 24 g (83percent yield) of homoserine lactone hydrochloride was obtained. 1H NMR (D2O) δ 4.59 (t, J=9.2 Hz, 1H), 4.43 (m, 2H), 2.76 (m, 1H), 2.42 (m, 1H), 13C NMR (D2O) δ 178.3, 68.3, 49.4, 27.6.
Reference:
[1] Patent: US7368600, 2008, B2, . Location in patent: Page/Page column 16-17
[2] Tetrahedron Asymmetry, 1995, vol. 6, # 11, p. 2819 - 2828
[3] Patent: US2011/53890, 2011, A1, . Location in patent: Page/Page column 3
[4] Journal of Organic Chemistry, 2016, vol. 81, # 11, p. 4516 - 4529
[5] Tetrahedron Letters, 2010, vol. 51, # 50, p. 6500 - 6502
[6] Tetrahedron, 1993, vol. 49, # 33, p. 7239 - 7250
[7] Organic Letters, 2008, vol. 10, # 24, p. 5521 - 5524
[8] European Journal of Organic Chemistry, 2009, # 25, p. 4242 - 4253
[9] Molecules, 2014, vol. 19, # 10, p. 16349 - 16372
[10] Tetrahedron Asymmetry, 2017, vol. 28, # 9, p. 1163 - 1168
3
[ 63-68-3 ]
[ 2185-03-7 ]
Yield
Reaction Conditions
Operation in experiment
87%
With hydrogenchloride; bromoacetic acid In water at 100℃; for 8 h;
The 1.5L adding water in three mouth bottle, is sequentially added under stirring L - methionine (200g), bromoacetic acid (245.9g) and concentrated hydrochloric acid (134 ml), after adding up to 100 °C, reaction 8 hours. Cooling to room temperature, concentrated under reduced pressure to remove the solvent. To the residue is added ethanol (200 ml), heated to reflux, stirring 1.5 hours. Cooling to room temperature, stirring 10 hours, filtered to get white solid ((S)-3 - amino - γ - butyrolactone hydrochloride) 160.4g, yield: 87percent.
Reference:
[1] Patent: CN106045947, 2016, A, . Location in patent: Paragraph 0027; 0028; 0029; 0030; 0031; 0032; 0033-0040
[2] Tetrahedron Letters, 2009, vol. 50, # 36, p. 5067 - 5070
[3] Chemical and Pharmaceutical Bulletin, 1996, vol. 44, # 12, p. 2322 - 2325
4
[ 63-68-3 ]
[ 96-34-4 ]
[ 2185-03-7 ]
Yield
Reaction Conditions
Operation in experiment
90%
Stage #1: With tetrabutylammomium bromide In water at 90℃; for 5 h; Stage #2: at 0℃; for 1 h;
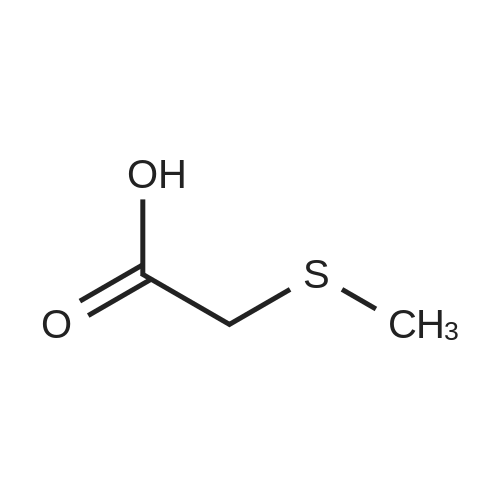
To a 500 ml three-mouth flask is added in L - methionine 59.6g (0.4 µM), chloroethyl acetate 47.7g (0.44 µM), tetrabutyl ammonium bromide 6.44g (0.02 µM), water 200 ml, stir, to be slowly heated to 90 °C, reaction 5h, thin layer chromatographic detection reaction finishes, cooling to room temperature, methylene chloride (100 ml × 3 times) to obtain a water cleaning, water layer by adding 41.7g 37percent hydrochloric acid, stirring 1h, reduced pressure to remove the solvent, then adding 60 ml anhydrous ethanol, stir, cooling to 0 °C stirring 1h, filtering, filters the cake to dry dry to obtain white solid powdery compound III (i.e. L - homoserine lactone hydrochloride) 49.5g, yield 90percent, combined methylene chloride level, vacuum distillation, to obtain by-product methylthio methyl acetate 38g.
Reference:
[1] Patent: CN106083922, 2016, A, . Location in patent: Paragraph 0050; 0051; 0052; 0068-0075; 0078-0081; 0102-0104
5
[ 33693-66-2 ]
[ 2185-03-7 ]
Reference:
[1] Australian Journal of Chemistry, 1985, vol. 38, # 4, p. 621 - 624
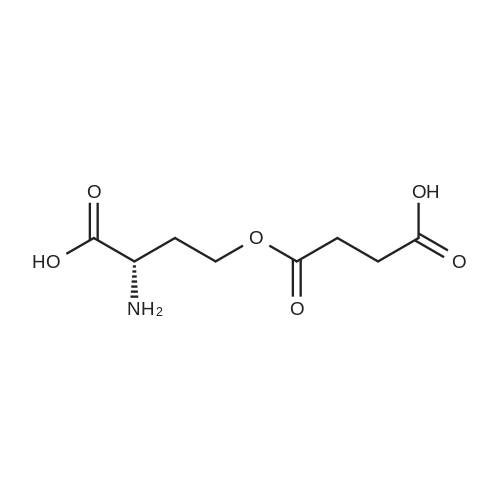
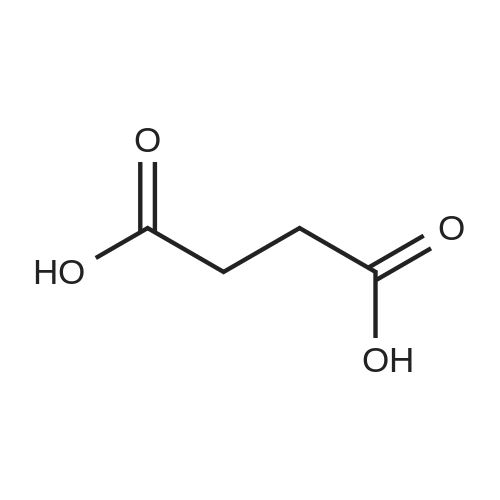
2 g (9.12 mmol) of O-succinyl-L-homoserine was dissolved in 10 ml (120 mmol, 13.2 equivalents) of concentrated hydrochloric acid, and the solution was allowed to react at 50° C. for 2 hours, and then cooled at room temperature for 3 hours. The precipitated solid was filtered, thereby obtaining 0.7 g (5.9 mmol) of succinic acid (SA) crystal (purity: 65percent). The filtrate was concentrated and recrystallized with anhydrous ethanol to yield 1.2 g (8.72 mmol) of homoserine lactone hydrochloride (purity: 95percent). 1H NMR (300 MHz, DMSO) δ 8.83 (2H, brs), 4.46 (1H, t, J=8.8 Hz), 4.36-4.24 (2H, m), 2.61-2.51 (1H, m), 2.30 (1H, t, J=10.3 Hz): Homoserine lactone hydrochloride 1H NMR (300 MHz, D2O) δ 4.36 (1H, t, J=9.0 Hz), 4.29 (2H, q, J=9.0 Hz), 2.69-2.60 (1H, m), 2.36-2.21 (1H, m): Homoserine lactone hydrochloride 1H NMR (300 MHz, D2O) δ 2.47 (4H, s): Succinic acid
Reference:
[1] Patent: CN108774160, 2018, A, . Location in patent: Paragraph 0076; 0078
9
[ 7540-67-2 ]
[ 2185-03-7 ]
[ 64-19-7 ]
Yield
Reaction Conditions
Operation in experiment
8.5 g
for 2 h; Reflux
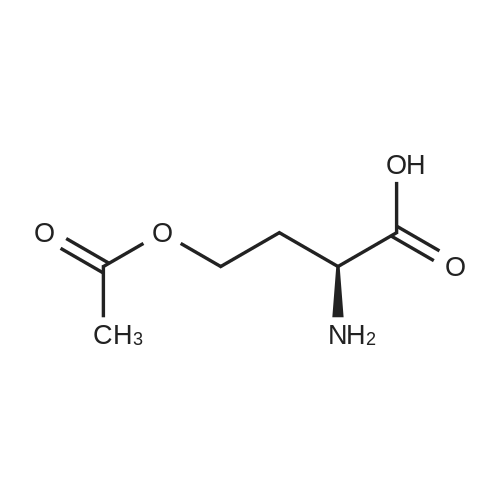
10 g (62.1 mmol) of O-acetyl-L-homoserine was completely dissolved in 50 ml (1.24 M) of water and 5.7 ml (68.3 mmol, 1.1 equivalents) of concentrated hydrochloric acid, and the solution was allowed to react under reflux for 2 hours, followed by removal of the solvent, thereby obtaining 8.5 g (61.8 mmol) of homoserine lactone hydrochloride (purity: 99percent). [0199] 1H NMR (300 MHz, DMSO) δ 8.83 (2H, brs), 4.46 (1H, t, J=8.8 Hz), 4.36-4.24 (2H, m), 2.61-2.51 (1H, m), 2.30 (1H, t, J=10.3 Hz) [0200] 1H NMR (300 MHz, D2O) δ 4.36 (1H, t, J=9.0 Hz), 4.29 (2H, q, J=9.0 Hz), 2.69-2.60 (1H, m), 2.36-2.21 (1H, m)
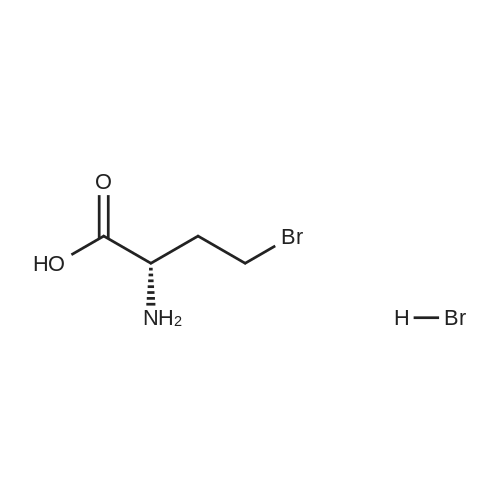
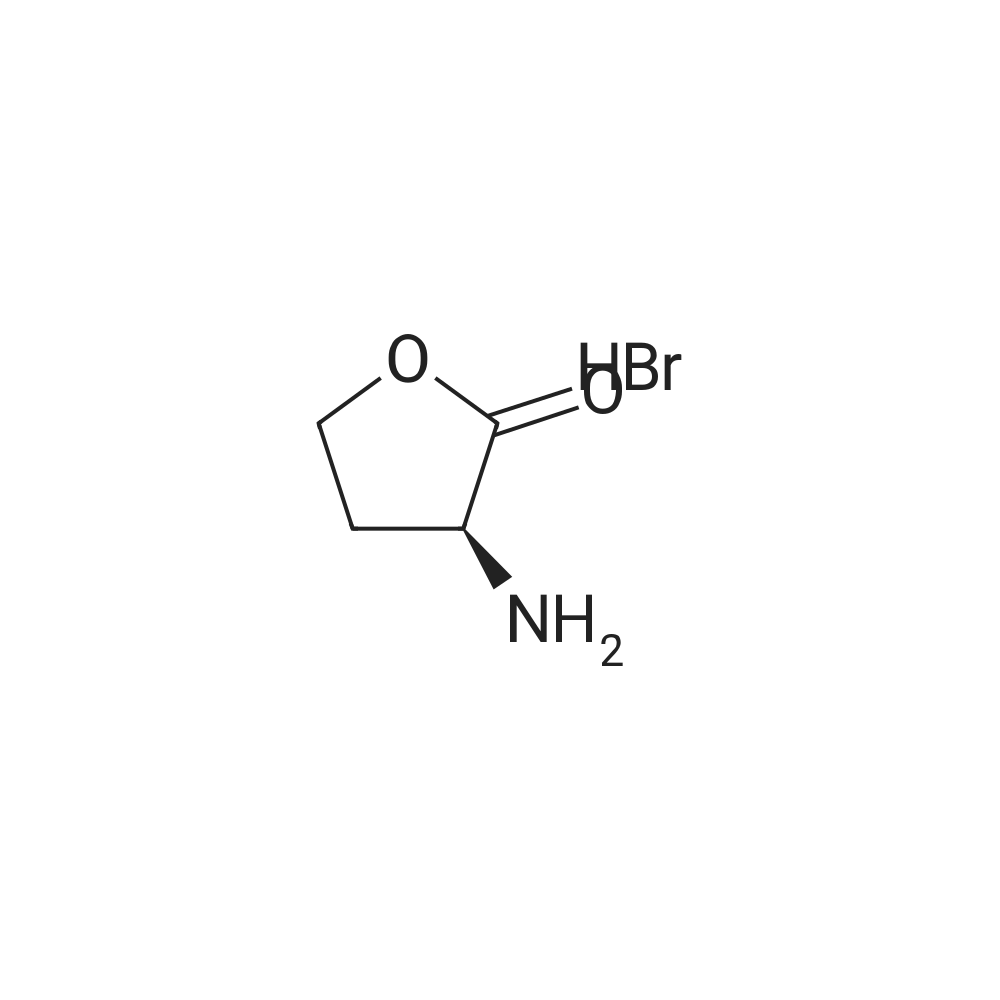
Example 14Compound 322 <n="161"/>[0485] (2R,6S, 13aS, 14aNo., 16aS,Z)-6-(tert-butoxycarbonylamino)- 14a-(2,5- dichlorothiophen-3-ylsuIfonylcarbamoyl)-5, 16-dioxo-2,3,5,6,7}8, 10, 11 , 13a, 14, 14a, 15, 16, 16a- tetradecahydro-1H-cyclopropa[/']pyrroIo[1,2:/][l,6,9]oxadiazacyclopentadecin-2-y] 4- chloroisoindoIine-2-carboxylate, Compound 322, was prepared as shown in the following scheme:.bul. Vi--. HATU, DlEA Step 1: Synthesis of (-S)-2-amino-4-bromobutanoic acid hydrobromide[0486] (5)-3-ammodihydrofuran-2(3H)-one hydrochloride (10.30 g, 74.87 mmol) in 58 mL of 30percent w/w HBr in AcOH was stirred at 65 °C for 30 hours. The solvent was removed under reduced pressure and the resulted solid was suspended in MTBE (200 mL) and stirred for 30 minutes. The solid was collected by filtration and washed with MTBE (200 mL) and dried to give (<S)-2-amino-4-bromobutanoic acid hydrobromide as white solid (19.33 <n="162"/>g, 98percent). 1H NMR (400 MHz, DMSO-/) δ 8.37 (s, 3H), 4.01 (m, 1H), 3.65 (m, 2H), 2.33 (m, 2H).
Reference:
[1] Journal of Medicinal Chemistry, 2014, vol. 57, # 5, p. 1753 - 1769
[2] Patent: WO2008/137779, 2008, A2, . Location in patent: Page/Page column 158-160
[3] Organic Letters, 2008, vol. 10, # 24, p. 5521 - 5524
[4] Tetrahedron, 1985, vol. 41, # 10, p. 1833 - 1845
[5] Journal of Organic Chemistry, 2008, vol. 73, # 1, p. 168 - 176
[6] Molecules, 2014, vol. 19, # 10, p. 16349 - 16372
<strong>[672-15-1]L-Homoserine</strong> (0.84 mol) was admixed with 600 ml of concentrated HCl (6.1 mol). The solution was stirred for about 15 minutes until everything had dissolved, and subsequently the water was removed under vacuum over the course of 1.5 hours. The residue was dried. Yield: 99% of 2-amino-4-butyrolactone hydrochloride salt.
83%
With hydrogenchloride; In water; at 20℃;Reflux;
Synthesis of homoserine lactone (AHL-2), analogs (QS1207; QS0108): (S)-Homoserine lactone hydrochloride: (S)-homoserine (25 g, 0.21 mol) was dissolved in aqueous hydrochloric acid (2.4 M, 322 mL, 0.97 mol, 4.6 equiv). The solution was refluxed for 3 h, and stirred at ambient temperature overnight. Most of the solvent was removed azeotropically with ethanol. Following crystal formation the solution was cooled on ice. The resulting solid was filtered and rinsed three times with cold ethanol. The filtrate was concentrated and cooled, producing additional homoserine lactone. The process was repeated 2 more times. After leaving the white powder on high vacuum line overnight, 24 g (83% yield) of homoserine lactone hydrochloride was obtained. 1H NMR (D2O) delta 4.59 (t, J=9.2 Hz, 1H), 4.43 (m, 2H), 2.76 (m, 1H), 2.42 (m, 1H), 13C NMR (D2O) delta 178.3, 68.3, 49.4, 27.6.
23%
With hydrogenchloride; In water; for 1h;Reflux;
Methyl iodide (1.54 mL, 3.52 g, 24.8 mmol) was added drop wise to a solution of L-(+)-methionine (1.49 g, 10.00 mmol) in H2O/MeOH (28.00 mL/4.00 mL). The resulting two-phase mixture was stirred vigorously for 2 days at room temperature under nitrogen atmosphere. After reaction completion, ~ 60% of the solvent and excess of methyl iodide were removed by rotatory evaporation and the remaining reaction volume was diluted with H2O (15.00 mL) and NaHCO3 (1.50 g, 17.85 mmol) was added slowly to this mixture. Resulted solution was refluxed for 15 h, cooled to room temperature and solvent was removed by rotatory evaporation. Formed residue 37 re-dissolved in 6 N HCl (50.00 mL), treated with 30% H2O2 solution (1.50 mL, 14.70 mmol), and refluxed for 1 h. Upon completion of the reaction, mixture was diluted with H2O (50.00 mL) and washed with diethyl ether (3 × 50.00 mL) to remove iodine as a by-product. Water portion was collected and concentrated in vacuo. To isolate product, formed precipitate was washed with ethanol (2 x 10.00 mL) with sonication and organic portion was collected and concentrated in vacuo. The desired lactone 38 was obtained by recrystallization from a solution of EtOH/H2O (9:1, 10.00 mL) (0.21 g, 23%).1H NMR (400 MHz, D2O) deltaH 2.23 - 2.42 (m, 1H, CH2), 2.60 - 2.75 (m, 1H, CH2), 4.24 - 4.40 (m, 2H, CH2), 4.50 (t, 1H, J = 9.1 Hz, CHNH2).
With hydrogen bromide; acetic acid at 65℃; for 30h;
98%
With hydrogen bromide; acetic acid at 65℃; for 30h;
14.1
Example 14Compound 322 [0485] (2R,6S, 13aS, 14aNo., 16aS,Z)-6-(tert-butoxycarbonylamino)- 14a-(2,5- dichlorothiophen-3-ylsuIfonylcarbamoyl)-5, 16-dioxo-2,3,5,6,7}8, 10, 11 , 13a, 14, 14a, 15, 16, 16a- tetradecahydro-1H-cyclopropa[/']pyrroIo[1,2:/][l,6,9]oxadiazacyclopentadecin-2-y] 4- chloroisoindoIine-2-carboxylate, Compound 322, was prepared as shown in the following scheme:• Vi--. HATU, DlEA Step 1: Synthesis of (-S)-2-amino-4-bromobutanoic acid hydrobromide[0486] (5)-3-ammodihydrofuran-2(3H)-one hydrochloride (10.30 g, 74.87 mmol) in 58 mL of 30% w/w HBr in AcOH was stirred at 65 °C for 30 hours. The solvent was removed under reduced pressure and the resulted solid was suspended in MTBE (200 mL) and stirred for 30 minutes. The solid was collected by filtration and washed with MTBE (200 mL) and dried to give (g, 98%). 1H NMR (400 MHz, DMSO-/) δ 8.37 (s, 3H), 4.01 (m, 1H), 3.65 (m, 2H), 2.33 (m, 2H).
78%
With hydrogen bromide; acetic acid at 100℃; for 5.5h; Sealed bottle;
76%
With hydrogen bromide In acetic acid
With hydrogen bromide at 100℃; for 5.5h;
With hydrogen bromide; acetic acid at 105℃; for 6h;
With hydrogenchloride; bromoacetic acid In water at 100℃; for 8h;
4 Embodiment 4
The 1.5L adding water in three mouth bottle, is sequentially added under stirring L - methionine (200g), bromoacetic acid (245.9g) and concentrated hydrochloric acid (134 ml), after adding up to 100 °C, reaction 8 hours. Cooling to room temperature, concentrated under reduced pressure to remove the solvent. To the residue is added ethanol (200 ml), heated to reflux, stirring 1.5 hours. Cooling to room temperature, stirring 10 hours, filtered to get white solid ((S)-3 - amino - γ - butyrolactone hydrochloride) 160.4g, yield: 87%.
58.2%
Stage #1: L-methionine With chloroacetic acid In water; acetic acid; isopropyl alcohol at 50 - 90℃; for 7h;
Stage #2: With hydrogenchloride In 1,4-dioxane at 20℃; for 3h; Cooling with ice;
With hydrogenchloride; sodium hydrogencarbonate; methyl iodide Multistep reaction;
With triethylamine; In dichloromethane; ethyl acetate;
Step A Preparation of L-N-tert-butoxycarbonyl Homoserine Lactone To a solution of L-homoserine lactone hydrochloride (10 g, 72.7 mmol) and (Boc)2O (19.0 g, 87.2 mmol) in dichloromethane (200 mL) at 0 C. was added Et3N (12.2 mL, 87.2 mmol) dropwise. The solution was warmed to room temperature and stirred for 16 hr. The mixture was poured into 10% citric acid solution, extracted with EtOAc, washed with saturated NaHCO3 solution, then brine, dried (MgSO4) and evaporated in vacuo. Column chromatography (silica gel; hexane:EtOAc 1:1 then pure EtOAc) afforded the title compound as a white solid. Rf (silica): 0.58 (hexane:EtOAc 1:1).
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; triethylamine In DMF (N,N-dimethyl-formamide) at 20℃; for 16h;
5.F
Step F: Preparation of N-(tert-butoxycarbonyl)-2(S)-[2(S)-amino-3(S)-methyl]-pentyloxy-3-phenyl-propionyl-homoserine Lactone To a stirred solution of N-(tert-butoxycarbonyl)-2(S)-[2(S)-amino-3(S)-methyl]-pentyloxy-3-phenylpropionic acid (0.53 g, 1.45 mmol) and 3-hydroxy-1,2,3,-benzotriazin-4(3H)-one (HOOBT) (0.26 g, 1.6 mmol) in DMF (15 ml) at room temperature was added EDC (0.307 g, 1.6 mmol) and L-homoserine lactone hydrochloride (0.219 g, 6.0 mmol). The pH was adjusted to pH=6.5 by addition of NEt3 (the pH was monitored by application of an aliquot of the reaction mixture to a moist strip of pH paper). After stirring at room temperature for 16 hr, the reaction was diluted with EtOAc and washed with saturated NaHCO3 and then brine and dried (NaSO4). Evaporation in vacuo (sufficient to remove DMF) and chromatography over silica gel eluding with 5% acetone in CH2Cl2 afforded the title compound (520 mg, 80%) as a white solid, mp 115-117° C. Rf=0.3 Acetone: CH2Cl2 (5:95). 1H NMR (CDCl3) δ 7.73 (1H, brd, J=5 Hz), 7.40-7.15 (5H, m), 4.68 (1H, dt, J=9 and 7.5 Hz), 4.65-4.35 (2H, m), 4.33-4.18 (1H, m), 4.20 (1H, dd, J=7 and 3 Hz), 3.78 (1H, m), 3.49 (1H, dd, J=7.5 and 4.0 Hz), 3.37 (1H, dd, J=7.5 and 6.5 Hz), 3.15 (1H, dd, J=11.5 and 2 Hz), 2.86 (1H, dd, J=11.5 and 7.5 Hz), 2.68 (1H, m) 2.11 (1H, q, J=9 Hz), 1.55-1.30 (11H, m), 1.07 (1H, m), 0.87 (3H, t, J=6.3 Hz), 0.79 (3H, d, J=6 Hz).
3-hydroxy-1,2,3,-benzotriazin-4(3H)-one (HOOBT)[ No CAS ]
N-(tert-butoxycarbonyl)-2(S)-[2(S)-amino-3(S)-methyl]pentyloxy-3-phenyl-propionic acid[ No CAS ]
[ 2185-03-7 ]
N-(tert-butoxycarbonyl)-2(S)-[2(S)-amino-3(S)-methyl]-pentyloxy-3-phenyl-propionyl-homoserine lactone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80%
With 1,2-dichloro-ethane In dichloromethane; N,N-dimethyl-formamide
1.F Preparation of N-(tert-butoxycarbonyl)-2(S)-[2(S)-amino-3(S)-methyl]-pentyloxy-3-phenyl-propionyl-homoserine lactone
Step F Preparation of N-(tert-butoxycarbonyl)-2(S)-[2(S)-amino-3(S)-methyl]-pentyloxy-3-phenyl-propionyl-homoserine lactone To a stirred solution of N-(tert-butoxycarbonyl)-2(S)-[2(S)-amino-3(S)-methyl]-pentyloxy-3-phenylpropionic acid (0.53 g, 1.45 mmol) and 3-hydroxy-1,2,3,-benzotriazin-4(3H)-one (HOOBT) (0.26 g, 1.6 mmol) in DMF (15 ml) at room temperature was added EDC (0.307 g, 1.6 mmol) and L-homoserine lactone hydrochloride (0.219 g, 6.0 mmol). The pH was adjusted to pH=6.5 by addition of NEt3 (the pH was monitored by application of an aliquot of the reaction mixture to a moist strip of pH paper). After stirring at room temperature for 16 hr, the reaction was diluted with EtOAc and washed with saturated NaHCO3 and then brine and dried (NaSO4). Evaporation in vacuo (sufficient to remove DMF) and chromatography over silica gel eluding with 5 % acetone in CH2 Cl2 afforded the title compound (520 mg, 80%) as a white solid, mp 115°14 117° C. Rf=0.3 Acetone: CH2 Cl2 (5:95). 1 H NMR (CDCl3) δ7.73 (1H, brd, J=5 Hz), 7.40-7.15 (5H, m), 4.68 (1H, dt, J=9 and 7.5 Hz), 4.65-4.35 (2H, m), 4.33-4.18 (1H, m), 4.20 (1H, dd, J=7 and 3 Hz), 3.78 (1H, m), 3.49 (1H, dd, J=7.5 and 4.0 Hz), 3.37 (1H, dd, J=7.5 and 6.5 Hz), 3.15 (1H, dd, J=11.5 and 2 Hz), 2.86 (1H, dd, J=11.5 and 7.5 Hz), 2.68 (1H, m) 2.11 (1H, q, J=9 Hz), 1.55-1.30 1.30 (11H, m), 1.07 (1H, m), 0.87 (3H, t, J=6.3 Hz), 0.79 (3H, d, J=6 Hz).
(1R,2S,5R)-2-isopropyl-5-methyl-cyclohexanecarboxylic acid ((S)-2-oxo-tetrahydro-furan-3-yl)-amide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine In diethyl ether; water at 20℃;
Synthesis of 2-Isopropyl-5-methyl-cyclohexanecarbonyl-L-Hsl; (also known as: (1R,2S,5R)-2-isopropyl-5-methyl-cyclohexanecarboxylic acid (S)-(2-oxo-tetrahydro-furan-3-yl)-amide):(S)-α-amino-γ-butyrolactone hydrochloride was obtained from Aldrich Chemical Co., 500 mg was dissolved in 18 mL diethyether and 1.5 mL double-distilled water. A pinch of the catalyst diaminopyrimidine was added. 0.68 mL of p-menthoyl chloride was then added dropwise, followed by 1.02 mL of triethylamine. The mixture was stirred overnight at room temperature. The precipitate was dissolved with ethyacetate, washed with double-distilled water and dried over sodium sulfate. The organic phase was then evaporated under reduced pressure to yield the final product, which crystallized at room temperature. The expected molecular mass was then confirmed by mass spectroscopy and the absorption spectrum by nuclear magnetic resonance.
With triethylamine In diethyl ether; water at 20℃;
Synthesis of 2-lsopropyl-5-methyl-cyclohexanecarbonyl-L-Hsl(also known as: (1R,2S,5R)-2-isopropyl-5-methyl-cyclohexanecarboxylic acid (S)-(2-oxo- tetrahydro-furan-3-yl)-amide):; (SJ-α-amino-v-butyroIactone hydrochloride was obtained from Aldrich Chemical Co., 500 mg was dissolved in 18 ml_ diethyether and 1.5 ml. double-distilled water. A pinch of the catalyst diaminopyrimidine was added. 0.68 ml_ of p-menthoyl chloride was then added dropwise, followed by 1.02 mL of triethylamine. The mixture was stirred overnight at room temperature. The precipitate was dissolved with ethyacetate, washed with double- distilled water and dried over sodium sulfate. The organic phase was then evaporated under reduced pressure to yield the final product, which crystallized at room temperature. The expected molecular mass was then confirmed by mass spectroscopy and the absorption spectrum by nuclear magnetic resonance.
(S)-N-butyryl homoserine lactone (AHL-2): (S)-homoserine lactone (0.100 g, 0.726 mmol) was dissolved in 5 mL of acetonitrile. N,N-diisopropylethyl amine (0.32 mL, 1.8 mmol) was added, followed by butyrylchloride (0.15 mL, 1.4 mmol). The reaction was stirred overnight. The acetonitrile was evaporated and the crude product was purified by flash chromatography using 80% ethyl acetate-hexanes. 0.1 g of white crystals was obtained (81% yield) (FIG. 1; Scheme 1). 1H NMR (CDCl3) δ 6.72 (d, J=6.2 Hz, 1H), 4.58 (m, 1H), 4.42 (app t, J=8.1 Hz, 1H), 4.24 (m, 1H), 2.71 (m, 1H), 2.20 (m, 3H), 1.63 (sex, J=7.4 Hz, 2H), 0.91 (t, J=7.4 Hz, 3H). 13C NMR (CDCl 3) δ 175.8, 173.6, 65.9, 48.8, 37.8, 29.7, 18.8, 13.5 (16)
63%
Stage #1: L-homoserine lactone hydrochloride With triethylamine In dichloromethane at 0℃; for 0.5h; Inert atmosphere;
Stage #2: butyryl chloride In dichloromethane at 0 - 20℃; for 4h; Inert atmosphere;
Stage #1: L-homoserine lactone hydrochloride With triethylamine In dichloromethane at 5 - 10℃; for 0.166667h;
Stage #2: benzoyl chloride In dichloromethane at 20℃; for 13h;
3.6. (S)-2-Benzoylamino gamma butyrolactone 7
(S)-2-Amino γ-butyrolactone hydrochloride (50 g, 0.37 mol) was taken in MDC (500 ml) and cooled to 5-10 C. Triethylamine (125 ml, 0.90 mol) was then added at 5-10 C over 10 min followed by benzoyl chloride (56.2 g, 0.40 mol) dropwise over a periodof 1 h. The mixture was stirred at rt for 12 h for completion ofthe reaction. The pH of the reaction mixture was adjusted to 2-3 using dilute HCl. The organic layer was separated and washed with ammonium chloride solution twice and concentrated to give the crude product. The crude product was stirred with hexane to precipitatethe product as a white crystalline powder, which was filtered and dried to obtain the product. Yield: 65 g (87% oftheory); Melting point: 165-167°C; [α]27D = -27.8 (c 1, ethanol); 1H NMR (300 MHz, CDCl3) d (ppm) 7.77-7.80 (d, J = 7.2, 2H), 7.35-7.50 (m, 3H), 7.24-7.27 (d, J = 7.8, 1H), 4.77-4.87 (m, 1H), 4.44-4.50 (t, J = 9.0, 1H), 4.26-4.35 (m, 1H), 2.78-2.87 (m, 1H), 2.23-2.40 (m, 1H); 13C NMR (300 MHz, CDCl3) d (ppm) 176.22,167.93, 133.16, 132.25, 128.80, 127.38, 66.47, 49.72, 30.29; Anal.Calcd for C11H11NO3 (FW 205.2099): C, 64.38; H, 5.40; N, 6.83.Found: C, 64.285; H, 5.36; N, 6.83. APCI MS m/z 205.95 (M+H)+.
10.9%
With triethylamine In dichloromethane for 4.5h; Cooling with ice;
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: 2-chloro-N-(2,4-dichlorophenyl)acetamide In acetone at 20℃; for 0.5h;
Preparation of analogues 4a-m.
General procedure: CS2 (0.22ml, 3.64 mmol) was added to the mixture of compound 3 (0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g,2.91 mmol) in acetone (10 ml). The mixture was stirred at room termperature for 0.5 h. Then 2a-m (2.91 mmol) wasadded to the mixture cautiously, the mixture was continued to stir at roomtemperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted withdichloromethane, washed with water, brine, dried with anhydrous Na2SO4and concentrated under reduced pressure. The residue was purified with columnchromatography to obtain analogue 4a-m as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: 2-chloro-N-(2,4-dichlorophenyl)acetamide In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: N-(4-methoxyphenyl)-2-chloroacetamide In acetone at 20℃; for 0.5h;
Preparation of analogues 4a-m.
General procedure: CS2 (0.22ml, 3.64 mmol) was added to the mixture of compound 3 (0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g,2.91 mmol) in acetone (10 ml). The mixture was stirred at room termperature for 0.5 h. Then 2a-m (2.91 mmol) wasadded to the mixture cautiously, the mixture was continued to stir at roomtemperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted withdichloromethane, washed with water, brine, dried with anhydrous Na2SO4and concentrated under reduced pressure. The residue was purified with columnchromatography to obtain analogue 4a-m as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: N-(4-methoxyphenyl)-2-chloroacetamide In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
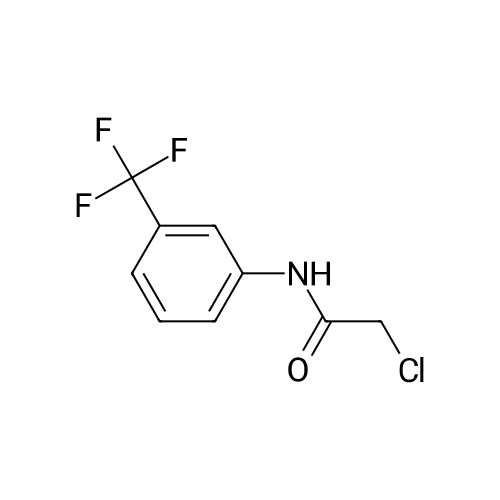
Stage #2: N-chloroacetyl-3-(trifluoromethyl)aniline In acetone at 20℃; for 0.5h;
Preparation of analogues 4a-m.
General procedure: CS2 (0.22ml, 3.64 mmol) was added to the mixture of compound 3 (0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g,2.91 mmol) in acetone (10 ml). The mixture was stirred at room termperature for 0.5 h. Then 2a-m (2.91 mmol) wasadded to the mixture cautiously, the mixture was continued to stir at roomtemperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted withdichloromethane, washed with water, brine, dried with anhydrous Na2SO4and concentrated under reduced pressure. The residue was purified with columnchromatography to obtain analogue 4a-m as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: N-chloroacetyl-3-(trifluoromethyl)aniline In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
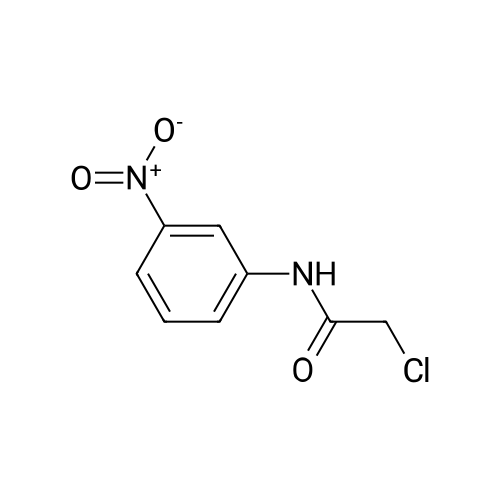
Stage #2: 2-chloro-N-(3-nitrophenyl)acetamide In acetone at 20℃; for 0.5h;
Preparation of analogues 4a-m.
General procedure: CS2 (0.22ml, 3.64 mmol) was added to the mixture of compound 3 (0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g,2.91 mmol) in acetone (10 ml). The mixture was stirred at room termperature for 0.5 h. Then 2a-m (2.91 mmol) wasadded to the mixture cautiously, the mixture was continued to stir at roomtemperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted withdichloromethane, washed with water, brine, dried with anhydrous Na2SO4and concentrated under reduced pressure. The residue was purified with columnchromatography to obtain analogue 4a-m as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: 2-chloro-N-(3-nitrophenyl)acetamide In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
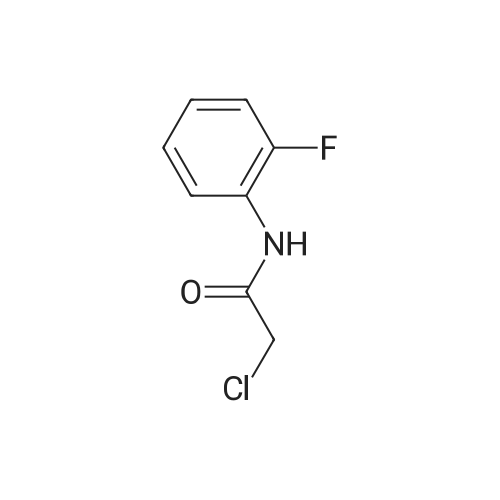
Stage #2: 2-chloro-N-(2-fluorophenyl)acetamide In acetone at 20℃; for 0.5h;
Preparation of analogues 4a-m.
General procedure: CS2 (0.22ml, 3.64 mmol) was added to the mixture of compound 3 (0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g,2.91 mmol) in acetone (10 ml). The mixture was stirred at room termperature for 0.5 h. Then 2a-m (2.91 mmol) wasadded to the mixture cautiously, the mixture was continued to stir at roomtemperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted withdichloromethane, washed with water, brine, dried with anhydrous Na2SO4and concentrated under reduced pressure. The residue was purified with columnchromatography to obtain analogue 4a-m as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: 2-chloro-N-(2-fluorophenyl)acetamide In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: methylene chloride In acetone at 20℃; for 0.5h;
Preparation of analogues 6a-g.
General procedure: CS2 (0.22 ml, 3.64 mmol) wasadded to the mixture of compound 3(0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g, 2.91 mmol) in acetone (10 ml).The mixture was stirred at room termperature for 0.5 h. Then the commercial available halides 5a-g (2.91 mmol)was added to the mixture cautiously, the mixture was continued to stir at room temperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried with anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified withcolumn chromatography to obtain analogue 6a-g as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: chloroethane In acetone at 20℃; for 0.5h;
Preparation of analogues 6a-g.
General procedure: CS2 (0.22 ml, 3.64 mmol) wasadded to the mixture of compound 3(0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g, 2.91 mmol) in acetone (10 ml).The mixture was stirred at room termperature for 0.5 h. Then the commercial available halides 5a-g (2.91 mmol)was added to the mixture cautiously, the mixture was continued to stir at room temperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried with anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified withcolumn chromatography to obtain analogue 6a-g as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: n-Butyl chloride In acetone at 20℃; for 0.5h;
Preparation of analogues 6a-g.
General procedure: CS2 (0.22 ml, 3.64 mmol) wasadded to the mixture of compound 3(0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g, 2.91 mmol) in acetone (10 ml).The mixture was stirred at room termperature for 0.5 h. Then the commercial available halides 5a-g (2.91 mmol)was added to the mixture cautiously, the mixture was continued to stir at room temperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried with anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified withcolumn chromatography to obtain analogue 6a-g as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: 1-Chlorohexane In acetone at 20℃; for 0.5h;
Preparation of analogues 6a-g.
General procedure: CS2 (0.22 ml, 3.64 mmol) wasadded to the mixture of compound 3(0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g, 2.91 mmol) in acetone (10 ml).The mixture was stirred at room termperature for 0.5 h. Then the commercial available halides 5a-g (2.91 mmol)was added to the mixture cautiously, the mixture was continued to stir at room temperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried with anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified withcolumn chromatography to obtain analogue 6a-g as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: n-chlorooctane In acetone at 20℃; for 0.5h;
Preparation of analogues 6a-g.
General procedure: CS2 (0.22 ml, 3.64 mmol) wasadded to the mixture of compound 3(0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g, 2.91 mmol) in acetone (10 ml).The mixture was stirred at room termperature for 0.5 h. Then the commercial available halides 5a-g (2.91 mmol)was added to the mixture cautiously, the mixture was continued to stir at room temperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried with anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified withcolumn chromatography to obtain analogue 6a-g as solid.
General procedure: CS2 (0.22ml, 3.64 mmol) was added to the mixture of compound 3 (0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g,2.91 mmol) in acetone (10 ml). The mixture was stirred at room termperature for 0.5 h. Then 2a-m (2.91 mmol) wasadded to the mixture cautiously, the mixture was continued to stir at roomtemperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted withdichloromethane, washed with water, brine, dried with anhydrous Na2SO4and concentrated under reduced pressure. The residue was purified with columnchromatography to obtain analogue 4a-m as solid.
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: 1-chlorododecane In acetone at 20℃; for 0.5h;
Preparation of analogues 6a-g.
General procedure: CS2 (0.22 ml, 3.64 mmol) wasadded to the mixture of compound 3(0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g, 2.91 mmol) in acetone (10 ml).The mixture was stirred at room termperature for 0.5 h. Then the commercial available halides 5a-g (2.91 mmol)was added to the mixture cautiously, the mixture was continued to stir at room temperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried with anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified withcolumn chromatography to obtain analogue 6a-g as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: myristyl chloride In acetone at 20℃; for 0.5h;
Preparation of analogues 6a-g.
General procedure: CS2 (0.22 ml, 3.64 mmol) wasadded to the mixture of compound 3(0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g, 2.91 mmol) in acetone (10 ml).The mixture was stirred at room termperature for 0.5 h. Then the commercial available halides 5a-g (2.91 mmol)was added to the mixture cautiously, the mixture was continued to stir at room temperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried with anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified withcolumn chromatography to obtain analogue 6a-g as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: 2-chloro-N-(3-methylphenyl)acetamide In acetone at 20℃; for 0.5h;
Preparation of analogues 4a-m.
General procedure: CS2 (0.22ml, 3.64 mmol) was added to the mixture of compound 3 (0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g,2.91 mmol) in acetone (10 ml). The mixture was stirred at room termperature for 0.5 h. Then 2a-m (2.91 mmol) wasadded to the mixture cautiously, the mixture was continued to stir at roomtemperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted withdichloromethane, washed with water, brine, dried with anhydrous Na2SO4and concentrated under reduced pressure. The residue was purified with columnchromatography to obtain analogue 4a-m as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: 2-chloro-N-(3-methylphenyl)acetamide In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: 2-chloro-N-(3-chlorophenyl)acetamide In acetone at 20℃; for 0.5h;
Preparation of analogues 4a-m.
General procedure: CS2 (0.22ml, 3.64 mmol) was added to the mixture of compound 3 (0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g,2.91 mmol) in acetone (10 ml). The mixture was stirred at room termperature for 0.5 h. Then 2a-m (2.91 mmol) wasadded to the mixture cautiously, the mixture was continued to stir at roomtemperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted withdichloromethane, washed with water, brine, dried with anhydrous Na2SO4and concentrated under reduced pressure. The residue was purified with columnchromatography to obtain analogue 4a-m as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: 2-chloro-N-(3-chlorophenyl)acetamide In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: N-chloroacetyl-aniline In acetone at 20℃; for 0.5h;
Preparation of analogues 4a-m.
General procedure: CS2 (0.22ml, 3.64 mmol) was added to the mixture of compound 3 (0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g,2.91 mmol) in acetone (10 ml). The mixture was stirred at room termperature for 0.5 h. Then 2a-m (2.91 mmol) wasadded to the mixture cautiously, the mixture was continued to stir at roomtemperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted withdichloromethane, washed with water, brine, dried with anhydrous Na2SO4and concentrated under reduced pressure. The residue was purified with columnchromatography to obtain analogue 4a-m as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: N-chloroacetyl-aniline In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: N-(2-chlorophenyl)chloroacetamide In acetone at 20℃; for 0.5h;
Preparation of analogues 4a-m.
General procedure: CS2 (0.22ml, 3.64 mmol) was added to the mixture of compound 3 (0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g,2.91 mmol) in acetone (10 ml). The mixture was stirred at room termperature for 0.5 h. Then 2a-m (2.91 mmol) wasadded to the mixture cautiously, the mixture was continued to stir at roomtemperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted withdichloromethane, washed with water, brine, dried with anhydrous Na2SO4and concentrated under reduced pressure. The residue was purified with columnchromatography to obtain analogue 4a-m as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: N-(2-chlorophenyl)chloroacetamide In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: N-(p-tolyl)-2-chloroacetamide In acetone at 20℃; for 0.5h;
Preparation of analogues 4a-m.
General procedure: CS2 (0.22ml, 3.64 mmol) was added to the mixture of compound 3 (0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g,2.91 mmol) in acetone (10 ml). The mixture was stirred at room termperature for 0.5 h. Then 2a-m (2.91 mmol) wasadded to the mixture cautiously, the mixture was continued to stir at roomtemperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted withdichloromethane, washed with water, brine, dried with anhydrous Na2SO4and concentrated under reduced pressure. The residue was purified with columnchromatography to obtain analogue 4a-m as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: N-(p-tolyl)-2-chloroacetamide In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
General procedure: CS2 (0.22ml, 3.64 mmol) was added to the mixture of compound 3 (0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g,2.91 mmol) in acetone (10 ml). The mixture was stirred at room termperature for 0.5 h. Then 2a-m (2.91 mmol) wasadded to the mixture cautiously, the mixture was continued to stir at roomtemperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted withdichloromethane, washed with water, brine, dried with anhydrous Na2SO4and concentrated under reduced pressure. The residue was purified with columnchromatography to obtain analogue 4a-m as solid.
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: 2-chloro-N-(4-chlorophenyl)acetamide In acetone at 20℃; for 0.5h;
Preparation of analogues 4a-m.
General procedure: CS2 (0.22ml, 3.64 mmol) was added to the mixture of compound 3 (0.4 g, 2.91 mmol) and Na3PO4.12H2O (1.104 g,2.91 mmol) in acetone (10 ml). The mixture was stirred at room termperature for 0.5 h. Then 2a-m (2.91 mmol) wasadded to the mixture cautiously, the mixture was continued to stir at roomtemperature for another 0.5 h. Upon completion (monitored by TLC), the solventwas removed under reduced pressure, the residue was extracted withdichloromethane, washed with water, brine, dried with anhydrous Na2SO4and concentrated under reduced pressure. The residue was purified with columnchromatography to obtain analogue 4a-m as solid.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: 2-chloro-N-(4-chlorophenyl)acetamide In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
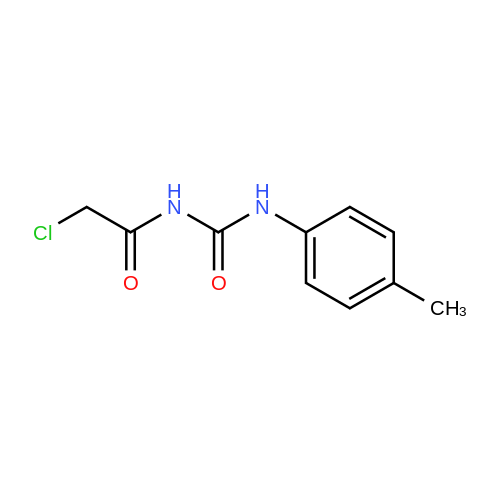
(S)-2-((4-fluorophenyl)amino)-2-oxoethyl (2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
69%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: 2-chloro-N-(4-fluorophenyl)acetamide In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
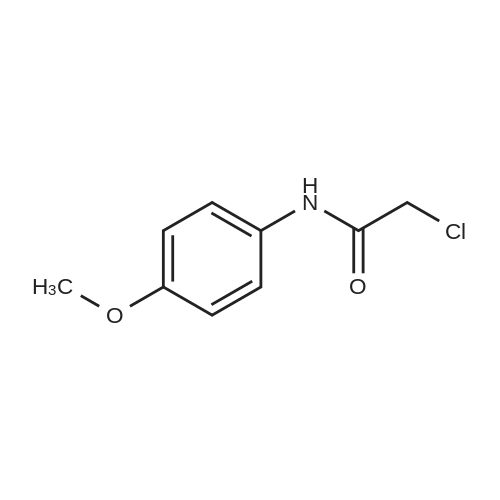
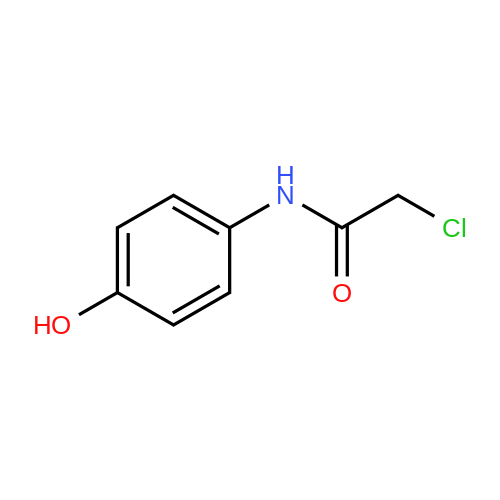
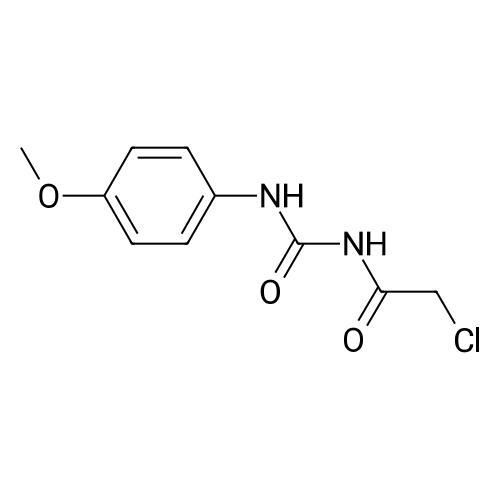
(S)-2-((4-hydroxyphenyl)amino)-2-oxoethyl (2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
64%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: 4-chloroacetamidophenol In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
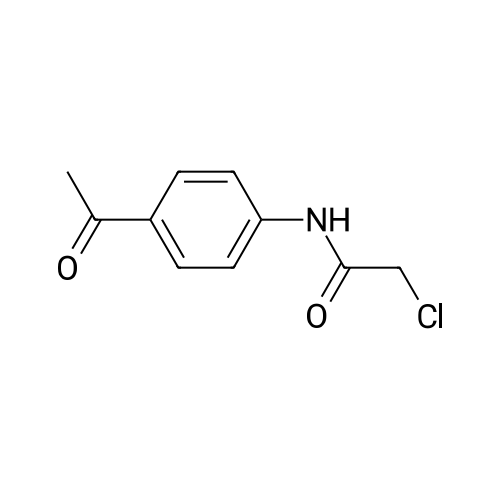
(S)-2-((4-acetylphenyl)amino)-2-oxoethyl (2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
70%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: 4-(2-chloroacetamido)acetophenone In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
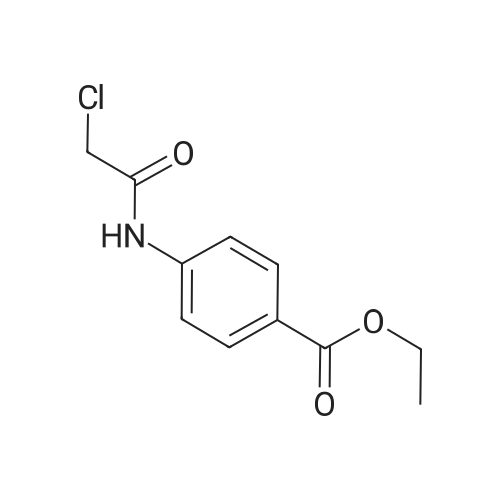
(S)-ethyl 4-(2-(((2-oxotetrahydrofuran-3-yl)carbamothioyl)thio)acetamido)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
60%
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
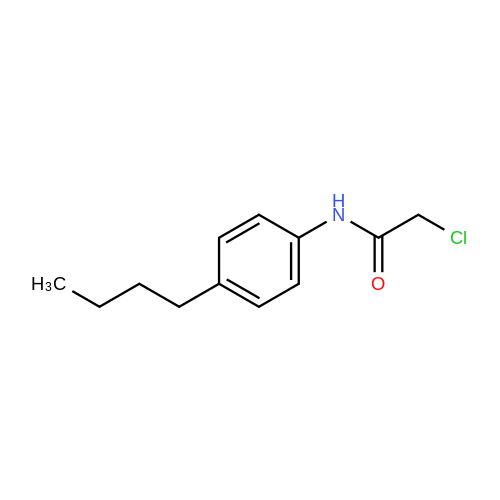
(S)-2-((4-butylphenyl)amino)-2-oxoethyl (2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
71%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: N-(4-butylphenyl)-2-chloroacetamide In acetone at 20℃; for 0.5h;
4.3 Procedure for the synthesis of analogs 4n-r
General procedure: Compounds 4a-m were synthesized previously in our group [23], so only the spectra data of compounds 4n-r were given below. To the mixture of compound 1 (0.4g, 2.91mmol) and Na3PO4·12H2O (1.104g, 2.91mmol) in acetone (10mL) was added CS2 (0.22mL, 3.64mmol). 30min later, acylated aniline (3n-r, 2.91mmol) was added to the mixture slowly, the mixture was stirred at room temperature for another 0.5h. Upon completion (monitored by TLC), the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the residue, which was purified using column chromatography to obtain analogs 4n-r.
(E)-2-chloro-N-(4-(3-(4-chlorophenyl)acryloyl)phenyl)acetamide[ No CAS ]
[ 2185-03-7 ]
(S,E)-2-((4-(3-(4-chlorophenyl)acryloyl)phenyl)amino)-2-oxoethyl (2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
50%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: (E)-2-chloro-N-(4-(3-(4-chlorophenyl)acryloyl)phenyl)acetamide In acetone at 20℃; for 0.5h;
4.5 General procedure for the synthesis of compounds 11a-g
General procedure: To the mixture of compound 1 (0.2g, 1.454mmol) and Na3PO4·12H2O (0.454g, 1.454mmol) in acetone (10mL) was added CS2 (110μL, 1.82mmol). The mixture was stirred at room temperature for 0.5h. Then compounds 10a-g (1.454mmol) were added to the mixture slowly, and stirred at room temperature for another 0.5h. Upon completion, the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified using column chromatography to give analogs 11a-g.
(E)-2-chloro-N-(4-cinnamoylphenyl)acetamide[ No CAS ]
[ 2185-03-7 ]
(S,E)-2-((4-cinnamoylphenyl)amino)-2-oxoethyl (2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
46%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: (E)-2-chloro-N-(4-cinnamoylphenyl)acetamide In acetone at 20℃; for 0.5h;
4.5 General procedure for the synthesis of compounds 11a-g
General procedure: To the mixture of compound 1 (0.2g, 1.454mmol) and Na3PO4·12H2O (0.454g, 1.454mmol) in acetone (10mL) was added CS2 (110μL, 1.82mmol). The mixture was stirred at room temperature for 0.5h. Then compounds 10a-g (1.454mmol) were added to the mixture slowly, and stirred at room temperature for another 0.5h. Upon completion, the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified using column chromatography to give analogs 11a-g.
(E)-N-(4-(3-(4-bromophenyl)acryloyl)phenyl)-2-chloroacetamide[ No CAS ]
[ 2185-03-7 ]
(S,E)-2-((4-(3-(4-bromophenyl)acryloyl)phenyl)amino)-2-oxoethyl (2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
50%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: (E)-N-(4-(3-(4-bromophenyl)acryloyl)phenyl)-2-chloroacetamide In acetone at 20℃; for 0.5h;
4.5 General procedure for the synthesis of compounds 11a-g
General procedure: To the mixture of compound 1 (0.2g, 1.454mmol) and Na3PO4·12H2O (0.454g, 1.454mmol) in acetone (10mL) was added CS2 (110μL, 1.82mmol). The mixture was stirred at room temperature for 0.5h. Then compounds 10a-g (1.454mmol) were added to the mixture slowly, and stirred at room temperature for another 0.5h. Upon completion, the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified using column chromatography to give analogs 11a-g.
(E)-2-chloro-N-(4-(3-(4-fluorophenyl)acryloyl)phenyl)acetamide[ No CAS ]
[ 2185-03-7 ]
(S,E)-2-((4-(3-(4-fluorophenyl)acryloyl)phenyl)amino)-2-oxoethyl (2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
32%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: (E)-2-chloro-N-(4-(3-(4-fluorophenyl)acryloyl)phenyl)acetamide In acetone at 20℃; for 0.5h;
4.5 General procedure for the synthesis of compounds 11a-g
General procedure: To the mixture of compound 1 (0.2g, 1.454mmol) and Na3PO4·12H2O (0.454g, 1.454mmol) in acetone (10mL) was added CS2 (110μL, 1.82mmol). The mixture was stirred at room temperature for 0.5h. Then compounds 10a-g (1.454mmol) were added to the mixture slowly, and stirred at room temperature for another 0.5h. Upon completion, the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified using column chromatography to give analogs 11a-g.
(E)-2-chloro-N-(4-(3-(2-chlorophenyl)acryloyl)phenyl)acetamide[ No CAS ]
[ 2185-03-7 ]
(S,E)-2-((4-(3-(2-chlorophenyl)acryloyl)phenyl)amino)-2-oxoethyl (2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
41%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: (E)-2-chloro-N-(4-(3-(2-chlorophenyl)acryloyl)phenyl)acetamide In acetone at 20℃; for 0.5h;
4.5 General procedure for the synthesis of compounds 11a-g
General procedure: To the mixture of compound 1 (0.2g, 1.454mmol) and Na3PO4·12H2O (0.454g, 1.454mmol) in acetone (10mL) was added CS2 (110μL, 1.82mmol). The mixture was stirred at room temperature for 0.5h. Then compounds 10a-g (1.454mmol) were added to the mixture slowly, and stirred at room temperature for another 0.5h. Upon completion, the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified using column chromatography to give analogs 11a-g.
(E)-2-chloro-N-(4-(3-(3-chlorophenyl)acryloyl)phenyl)acetamide[ No CAS ]
[ 2185-03-7 ]
(S,E)-2-((4-(3-(3-chlorophenyl)acryloyl)phenyl)amino)-2-oxoethyl (2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
42%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: (E)-2-chloro-N-(4-(3-(3-chlorophenyl)acryloyl)phenyl)acetamide In acetone at 20℃; for 0.5h;
4.5 General procedure for the synthesis of compounds 11a-g
General procedure: To the mixture of compound 1 (0.2g, 1.454mmol) and Na3PO4·12H2O (0.454g, 1.454mmol) in acetone (10mL) was added CS2 (110μL, 1.82mmol). The mixture was stirred at room temperature for 0.5h. Then compounds 10a-g (1.454mmol) were added to the mixture slowly, and stirred at room temperature for another 0.5h. Upon completion, the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified using column chromatography to give analogs 11a-g.
(E)-2-chloro-N-(4-(3-(4-nitrophenyl)acryloyl)phenyl)acetamide[ No CAS ]
[ 2185-03-7 ]
(S,E)-2-((4-(3-(4-nitrophenyl)acryloyl)phenyl)amino)-2-oxoethyl (2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
30%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone at 20℃; for 0.5h;
Stage #2: (E)-2-chloro-N-(4-(3-(4-nitrophenyl)acryloyl)phenyl)acetamide In acetone at 20℃; for 0.5h;
4.5 General procedure for the synthesis of compounds 11a-g
General procedure: To the mixture of compound 1 (0.2g, 1.454mmol) and Na3PO4·12H2O (0.454g, 1.454mmol) in acetone (10mL) was added CS2 (110μL, 1.82mmol). The mixture was stirred at room temperature for 0.5h. Then compounds 10a-g (1.454mmol) were added to the mixture slowly, and stirred at room temperature for another 0.5h. Upon completion, the solvent was removed under reduced pressure, the residue was extracted with dichloromethane, washed with water, brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified using column chromatography to give analogs 11a-g.
With triethylamine In acetonitrile at 20℃; Reflux;
4.6 Procedure for the synthesis of OdDHL
To a mixture of malonic acid 15 (4.20g, 40.36mmol) and acetic anhydride (4.96mL, 52.47mmol) was added H2SO4 (0.17mL, 3.23mmol), the mixture was then stirred for about 0.5h in an ice-water bath, followed by addition of acetone 16 (4.16mL, 56.51mmol). After stirring for another 4h, the resulting solution was then allowed to warm to room temperature and kept overnight at the same temperature. Upon completion of the reaction, the precipitate was filtrated, washed with ice water and dried to gain Meldrum's acid 17. To a solution of n-decanoic acid (1.376g, 8mmol) in dry CH2Cl2 were added DMAP (1.024g, 8.4mmol), DCC (1.816g, 8.8mmol) and 17 (1.152g, 8mmol) sequentially, and the reaction mixture was stirred overnight at room temperature. N,N'-dicyclohexylurea formed was removed by filtration, and the filtrate was concentrated under vacuo to give the residue, which was then dissolved in ethyl acetate, washed with 2N hydrochloric acid, dried over MgSO4 and concentrated to give the crude mixture of 19. To a solution of 1 (0.461g, 3.35mmol) and 19 (1.001g) in acetonitrile (50mL) was added Et3N (0.407g, 4.02mmol). The reaction mixture was stirred at room temperature for 4h and then stirred under reflux. Upon completion of the reaction, the reaction mixture was concentrated under vacuo to give the residue, which was then subjected to column chromatography (petroleum ether: acetone=10: 1) to give OdDHL.
(S)-2-chloro-Ν-(2-oxo-tetrahydrofuran-3-yl)acetamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
72%
With sodium hydrogencarbonate In dichloromethane; water for 6h;
1
In 200 ml of water and 30 ml of methanol is added to the mixed solution of 10.12g (67.7mmol) L-methionine, after mixing, slowly under the closed condition to methane instillment iodine about 5.1 ml (1.2eq), the drop finishes, stirring the reaction overnight at room temperature, until the system into a clear transparent shape. After concentrating under reduced pressure, the system for adding dropwisely NaHCO 3 aqueous solution (5.69g dissolved in 80 ml water), the drop finishes, to the system for heating oil bath to reflux the reaction about 6 hours, TLC detection after the reaction is complete, reducing pressure and evaporating the solvent, to obtain bright yellow oily matter, acetone and ethanol (V:V=1:9) recrystallization to obtain a large amount of white solid. The white solid dissolved in 100 ml hydrochloric acid (6mol L -1) solution, heating to reflux about 10h, system is wine red, TLC detection reaction is complete. The system is cooled to room temperature, ethyl ether are used water and methylene chloride to wash nearly colorless, distilling solvent under reduced pressure and with ethanol crystallization is provided, after filtering vacuum drying, to obtain white solid high serine lactone hydrochloride 6.89g, yield 74%. The 1.40g (0.01mol) homoserine lactone hydrochloride was dissolved in 15mL dichloromethane and 15mL water mixed solvent of burning, plus 2.52g NaHCO3 (0.03 mol) clarification system was stirred until dissolved. At room temperature was slowly added dropwise acetyl chloride 920 μl (0.012 mol), the reaction after about 6h, TLC the reaction was complete, dichloromethane layer was fractionated, dried over anhydrous sodium sulfate and the solvent was distilled off under reduced pressure, and dried in vacuo to give a white solid (S) -2- chloro -Ν- (2-oxo-tetrahydrofuran-3-yl) acetamide 1.3g, 72% yield. 100mL round bottom flask was added 0.27g (2mmol) of aminoacetophenone, dissolved in 25mL of ethanol,Successively added 0.52g (2.02mmo 1) under chlorobenzaldehyde and 0.12g of sodium hydroxide, stirred at room temperature overnight. The next day, the reaction mixture was poured into ice-water with stirring to precipitate a yellow solid i.e. a large number of (E)-1-(4-aminophenyl)-3-(4-chlorophenyl)prop-2-en-1-one 0.38g, 74% yield. In 25mL round bottom flask was added 96 mg (0.54 mmol) (3) -2- chloro-4- (2-oxo-tetrahydrofuran-3-yl) acetamide and 270 mg of potassium iodide, stirred and dissolved l0mL DMF added, an oil bath at 60 ° C as The reaction 2h. Then, the reaction system was added 139mg (0.54mmol) (E) -l- (4- aminophenyl) -3- (4-chlorophenyl) prop-2-en-1-one, was heated to 120 ° C for about 6h, the reaction solution was cooled to room temperature, extracted with ethyl acetate, washed with water of DMF was removed, after addition of ethyl acetate was distilled off under reduced pressure, silica gel column chromatography (eluent: acetone / petroleum ether = 2/3), to give a yellow That is a solid title compound 157mg, 73% yield.
72%
Stage #1: L-homoserine lactone hydrochloride With sodium hydrogencarbonate In water for 0.0833333h;
Stage #2: chloroacetyl chloride In dichloromethane; water
4.4 Synthesis of compound 4
To a solution of homoserine lactone hydrochloride 3 (4.0g, 22.52mmol) in water (60mL) was added NaHCO3 (5.67g, 67.58mmol). 5 min later, DCM (60mL) was added to the mixture followed by slow addition of chloroacetyl chloride (2.6mL, 27.02mmol). Upon completion of the reaction indicated by TLC, the DCM layer was separated and evacuated under vacuum to give the white solid, (S)-2-chloro-N-(2-oxotetrahydrofuran-3-yl)acetamide (4), yield: 72%, m.p.121-122°C; 1H NMR (400MHz, DMSO-d6) δ 8.76 (d, J=8.0Hz, 1H, -CO-NH), 4.63 (dd, J=20.0Hz, J=8.0Hz, 1H, -O-CH2), 4.35 (t, J=8.0Hz, 1H, -CH-NH), 4.21 (dd, J=20.0Hz, J=8.0Hz, 1H, -O-CH2), 4.14 (s, 2H, -CH2-Cl), 2.44-2.36 (m, 1H, -O-CH2-CH2-CH), 2.24-2.15 (m, 1H, -O-CH2-CH2-CH); 13C NMR (100MHz, DMSO-d6) δ 175.30, 166.56, 65.80, 48.73, 42.71, 28.41. HRMS (ESI) Calcd for C6H8ClNO3 [M+H]+: 178.0271, found: 178.0273.
69%
With sodium hydrogencarbonate In dichloromethane; water at 20℃; for 6h;
General Procedure for the Synthesis of Compounds 5 and 6
Compounds 5 and 6 were efficiently synthesized from L-homoserine lactone hydrochloride 4 following our previously reported methods.39) The acyl group of 5 and 6 was introduced by condensation with chloroacetyl chloride and 3-chloropropionyl chloride, respectively. (S)-2-Chloro-N-(2-carbonyl Tetrahydrofuran-3-yl)acetamide 5 White solid: (TLC acetone : petroleum ether 2 : 1), (yield 68.2%), mp 121-122 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 8.76 (d, J = 8.0 Hz, 1H, -CO-NH), 4.63 (dd, J = 20.0 Hz, J = 8.0 Hz, 1H, -O-CH2), 4.35 (t, J = 8.0 Hz, 1H, -CH-NH), 4.21 (dd, J = 20.0 Hz, J = 8.0 Hz, 1H, -O-CH2), 4.14 (s, 2H, -CH2-Cl), 2.44-2.36 (m, 1H, -O-CH2-CH2-CH), 2.24-2.15 (m, 1H, -O-CH2-CH2-CH-).
(E)-2-chloro-N-(4-(3-(4-chlorophenyl)acryloyl)phenyl)acetamide[ No CAS ]
[ 2185-03-7 ]
(S,E)-N-(4-(3-(4-chlorophenyl)acryloyl)phenyl)-2-((2-oxotetrahydrofuran-3-yl)amino)acetamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
66%
With potassium carbonate; potassium iodide In acetone for 6h; Reflux;
2 (S,E)-N-(4-(3-(4-chlorophenyl)acryloyl)phenyl)-2-((2-oxotetrahydrofuran-3-yl)amino)acetamide
In 200 ml of water and 30 ml of methanol is added to the mixed solution of 10.12g (67.7mmol) L-methionine, after mixing, slowly under the closed condition to methane instillment iodine about 5.1 ml (1.2eq), the drop finishes, stirring the reaction overnight at room temperature, until the system into a clear transparent shape. After concentrating under reduced pressure, the system for adding dropwisely NaHCO 3 aqueous solution (5.69g dissolved in 80 ml water), the drop finishes, to the system for heating oil bath to reflux the reaction about 6 hours, TLC detection after the reaction is complete, reducing pressure and evaporating the solvent, to obtain bright yellow oily matter, acetone and ethanol (V:V=1:9) recrystallization to obtain a large amount of white solid. The white solid dissolved in 100 ml hydrochloric acid (6mol L -1) solution, heating to reflux about 10h, system is wine red, TLC detection reaction is complete. The system is cooled to room temperature, ethyl ether are used water and methylene chloride to wash nearly colorless, distilling solvent under reduced pressure and with ethanol crystallization is provided, after filtering vacuum drying, to obtain white solid high serine lactone hydrochloride 6.89g, yield 74%. In 100 ml round-bottom flask by adding 0.27g (2mmol) to amino acetophenone, dissolved in 25 ml ethanol, adding 0.52g (2.02mmol) parachlorobenzaldehyde and 0.12g naoh, stir at room temperature overnight. The next day, the reaction solution is poured into the ice water mixture stirring to precipitate in a large amount of yellow solid (E) - 1 - (4-amino-phenyl-3 - (4-chlorophenyl) prop-2-en-1-ones 0.38g, yield 74%. In a 50 ml round-bottom flask by adding 0.51g (E) - 1 - (4-amino-phenyl) - 3 - (4-chlorophenyl) prop-2-en-1-ones, dissolved in 25 ml acetone, adding 332mgK 2 CO 3 (1.2eq) and 180 μ l chloro acetyl chloride (1.2eq); stir r.t 3h the rear, in the reaction solution is poured into the ice water mixture stirring to separate out a large amount of solid, filtered, washing the filter cake, dried under vacuum to get the 0.53g (E) - 2-chloro-N-(4 - (3 - (4-chlorophenyl) acryloyl) phenyl) acetamide solid, yield is 70%. In a 25 ml round-bottom flask by adding 150 mg (0.45mmol) (E) - 2-chloro-N-(4 - (3 - (4-chlorophenyl) acryloyl) phenyl) acetamide and 62 mg (0.45mmol) homoserine lactone hydrochloride, plus 10 ml acetonitrile to stir and dissolve, then adding acid-binding agent K 2 CO 3 187 mg (1.35mmol), catalyst KI15mg (0.09mmol), under the oil bath is heated to about reflux reaction 6h. The tracking of the reaction TLC, oil filtration to K2CO3, after reducing pressure and evaporating acetonitrile, silica gel column chromatography (eluent: acetone/petroleum ether = 1/2), to obtain the yellow solid, that is, a target compound 118 mg, yield 66%.
1
In 200 ml of water and 30 ml of methanol is added to the mixed solution of 10.12g (67.7mmol) L-methionine, after mixing, slowly under the closed condition to methane instillment iodine about 5.1 ml (1.2eq), the drop finishes, stirring the reaction overnight at room temperature, until the system into a clear transparent shape. After concentrating under reduced pressure, the system for adding dropwisely NaHCO 3 aqueous solution (5.69g dissolved in 80 ml water), the drop finishes, to the system for heating oil bath to reflux the reaction about 6 hours, TLC detection after the reaction is complete, reducing pressure and evaporating the solvent, to obtain bright yellow oily matter, acetone and ethanol (V:V=1:9) recrystallization to obtain a large amount of white solid. The white solid dissolved in 100 ml hydrochloric acid (6mol L -1) solution, heating to reflux about 10h, system is wine red, TLC detection reaction is complete. The system is cooled to room temperature, ethyl ether are used water and methylene chloride to wash nearly colorless, distilling solvent under reduced pressure and with ethanol crystallization is provided, after filtering vacuum drying, to obtain white solid high serine lactone hydrochloride 6.89g, yield 74%. The 1.40g (0.01mol) homoserine lactone hydrochloride was dissolved in 15mL dichloromethane and 15mL water mixed solvent of burning, plus 2.52g NaHCO3 (0.03 mol) clarification system was stirred until dissolved. At room temperature was slowly added dropwise acetyl chloride 920 μl (0.012 mol), the reaction after about 6h, TLC the reaction was complete, dichloromethane layer was fractionated, dried over anhydrous sodium sulfate and the solvent was distilled off under reduced pressure, and dried in vacuo to give a white solid (S) -2- chloro -Ν- (2-oxo-tetrahydrofuran-3-yl) acetamide 1.3g, 72% yield. 100mL round bottom flask was added 0.27g (2mmol) of aminoacetophenone, dissolved in 25mL of ethanol,Successively added 0.52g (2.02mmo 1) under chlorobenzaldehyde and 0.12g of sodium hydroxide, stirred at room temperature overnight. The next day, the reaction mixture was poured into ice-water with stirring to precipitate a yellow solid i.e. a large number of (E)-1-(4-aminophenyl)-3-(4-chlorophenyl)prop-2-en-1-one 0.38g, 74% yield. In 25mL round bottom flask was added 96 mg (0.54 mmol) (3) -2- chloro-4- (2-oxo-tetrahydrofuran-3-yl) acetamide and 270 mg of potassium iodide, stirred and dissolved l0mL DMF added, an oil bath at 60 ° C as The reaction 2h. Then, the reaction system was added 139mg (0.54mmol) (E) -l- (4- aminophenyl) -3- (4-chlorophenyl) prop-2-en-1-one, was heated to 120 ° C for about 6h, the reaction solution was cooled to room temperature, extracted with ethyl acetate, washed with water of DMF was removed, after addition of ethyl acetate was distilled off under reduced pressure, silica gel column chromatography (eluent: acetone / petroleum ether = 2/3), to give a yellow That is a solid title compound 157mg, 73% yield.
With hydrogenchloride; dihydrogen peroxide In water Reflux;
With hydrogenchloride; dihydrogen peroxide In water for 12h; Reflux;
Stage #1: L-methionine; methyl chloroacetate With tetrabutylammomium bromide In water at 90℃; for 5h;
Stage #2: With ethanol at 0℃; for 1h;
1.1 A, L - homoserine lactone hydrochloride synthesis
To a 500 ml three-mouth flask is added in L - methionine 59.6g (0.4 µM), chloroethyl acetate 47.7g (0.44 µM), tetrabutyl ammonium bromide 6.44g (0.02 µM), water 200 ml, stir, to be slowly heated to 90 °C, reaction 5h, thin layer chromatographic detection reaction finishes, cooling to room temperature, methylene chloride (100 ml × 3 times) to obtain a water cleaning, water layer by adding 41.7g 37% hydrochloric acid, stirring 1h, reduced pressure to remove the solvent, then adding 60 ml anhydrous ethanol, stir, cooling to 0 °C stirring 1h, filtering, filters the cake to dry dry to obtain white solid powdery compound III (i.e. L - homoserine lactone hydrochloride) 49.5g, yield 90%, combined methylene chloride level, vacuum distillation, to obtain by-product methylthio methyl acetate 38g.
With anhydrous sodium carbonate In water monomer at 5℃; for 1h;
1.2 Second, the synthesis of compound IV
To a 500 ml three-mouth flask compound is added in the III 27.5g (0.2 µM), adding 200 ml water, stirring solution cleaning, cooling to 0 °C, batch add 26.5g (0.25 µM) sodium carbonate, canada finishes, stirring 15min, for dropping funnel slowly dropping 32.6g (0.3 µM) ethyl chloroformate, control temperature 5 °C, after the completion of the dropping, stirring 1h, TLC detection said technological, adjusted to pH=5 for hydrochloric acid system, to evaporate the solvent, adding acetone stirring 30min, filtering to remove the sodium chloride, to remove solvent under reduced pressure, recrystallized with absolute ethanol, to obtain the product 33.9g, yield 98.0%.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate In acetone at 20℃; for 2h;
Stage #2: N-Chloroacetyl-N'-(2-methoxyphenyl) urea In acetone at 20℃; for 6h;
1
Example 1: Preparation of the derivative (2-a) shown in Formula 2, [OO33] A homoserine lactone hydrochloride (150 mg, 1.09 mmol) was dissolved in 8 mL of acetone, and tris Sodium (620 mg, 1.63 mmol) and CS2 (160 yL) were added and stirred at room temperature for 2 h. Then, 2-chloro-N- ((2-methoxyphenyl) aminomethyl is added (291 mg, 1.20 mmol) at room temperature for 6 h, the system evaporated to dryness, extracted with ethyl acetate and water, and the organic layer was washed with water The crude product was separated by silica gel column chromatography (eluent: ethyl acetate / petroleum ether = 1/1) to give a pale yellow solid
51%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate In acetone at 20℃; for 2h;
Stage #2: N-Chloroacetyl-N'-(2-methoxyphenyl) urea In acetone at 20℃; for 6h;
General Procedure for the Synthesis of Compounds 11a-i
General procedure: A solution of compound 4 (150 mg, 1.09 mmol) and Na3PO4·12H2O (620 mg, 1.63 mmol) in acetone was added to CS2 (160 μL) and stirred for 2 h at room temperature. The resulting solution was added to compounds 10a-i (1.20 mmol) to react for 6 h at room temperature. The solvent was removed under vacuum, extracted with ethyl acetate and water. The organic layer was dried over MgSO4 and evaporated under vacuum to give the residue, which was then purified by column chromatography, affording a white solid (11a-i).
In N,N-dimethyl-formamide; for 2h;Inert atmosphere; Reflux;
Under a nitrogen atmosphere, 13.75 g of alpha-amino-gamma-butyrolactone hydrochloride was mixed with 40 mL of DMF,Stir evenly. 15.2 g of sodium methyl selenate was dissolved in 20 mL of DMF, added to the above system and refluxed for 2 h. After the reaction was completed, the pH of the system was adjusted to 6.0 with an aqueous acetic acid solution to give a white solid,Solid water and ethanol mixed solution recrystallization, both L-selenomethionine. The total yield was 72.11%Chemical purity> 99%, ee> 99%.
1.2
At the low temperature, 400 g of a 1,4-dioxane solution having a HCl content of 12% was added to the above systemAfter heating for 10 hours, the mixture was cooled to room temperature, stirred for 2 hours, then decompressed under reduced pressure. The filter cake was washed with 1,4-dioxane to give L-α-amino-γ-butyrolactone hydrochloride Salt, the yield of 79.92%Chemical purity> 99%, optical purity ee> 99%.
2-[(3-amino-6-bromopyrazin-2-yl)formamido]methyl}-6-carboxy-1,3-diethyl-1H-1,3-benzodiazol-3-ium bromide[ No CAS ]
[ 2185-03-7 ]
2-[(3-amino-6-bromopyrazin-2-yl)formamido]methyl}-1,3-diethyl-6-[(3S)-2-oxooxolan-3-yl]carbamoyl}-1H-1,3-benzodiazol-3-ium formate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
21%
Stage #1: 2-[(3-amino-6-bromopyrazin-2-yl)formamido]methyl}-6-carboxy-1,3-diethyl-1H-1,3-benzodiazol-3-ium bromide With 4-methyl-morpholine; O-(1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate In N,N-dimethyl-formamide at 20℃; for 0.0833333h;
Stage #2: L-homoserine lactone hydrochloride In N,N-dimethyl-formamide at 20℃; for 16h;
Stage #3: formic acid In water; N,N-dimethyl-formamide; acetonitrile
18 Example 18 - Synthesis of 2-[(3-amino-6-bromopyrazin-2-yl)formamido]methyl}- 1 ,3-diethyl-6- 3S)-2-oxooxolan-3-yl]carbamoyl}-1 H-1 ,3-benzodiazol-3-ium formate:
A solution of 2-[(3-amino-6-bromopyrazin-2-yl)formamido]methyl}-6-carboxy-1 ,3-diethyl- 1 -/-1 ,3-benzodiazol-3-ium bromide, Example 16 (70 mg, 0.13 mmol), HBTU (55 mg, 0.13 mmol), and 4-methylmorpholine (34 μΙ, 0.27 mmol) in anhydrous DMF (0.5 ml) was stirred at RT for 5 min. (3S)-3-Aminooxolan-2-one hydrochloride (18 mg, 0.13 mmol) was added then the reaction was stirred at RT for 16 h. The reaction mixture was concentrated in vacuo then the crude material was purified by flash column chromatography on C18 (12 g). The column was eluted with MeCN:H20 + 0.1 % formic acid using the following gradient (%MeCN, column volumes): 5%, 2 CV; 5-100%, 20 CV; 100%, 2 CV. The desired fractions were combined and lyophilised to afford the product as an off-white solid (17 mg, 21 %). NMR (500 MHz, DMSO-cfe) δ 9.60 (t, J = 5.2 Hz, 1 H), 9.37 (d, J = 7.8 Hz, 1 H), 8.55 (s, 1 H), 8.42 (s, 1 H), 8.35 (s, 1 H), 8.23 - 8.14 (m, 2H), 7.66 (s, 2H), 5.10 (d, J = 5.3 Hz, 2H), 4.90 - 4.80 (m, 1 H), 4.75 - 4.65 (m, 4H), 4.50 - 4.42 (m, 1 H), 4.36 - 4.27 (m, 1 H), 2.55 - 2.38 (m, 2H + DMSO-cfe), 1.48 - 1.40 (m, 6H). LC/MS (System C): m/z (ESI+) = 530 [M(79Br)+], 532 [M(8 Br)+] R, = 1.61 min, UV purity = 93%.
2-phenoxyethyl (S)-(2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
67%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: phenoxyethyl bromide In acetone
4.7 General procedure for the synthesis of compounds 8a-j
General procedure: Compounds 7a-7j were prepared following the literature reported methods in 54-83% yields [29]. To a solution of compound 3 (102mg, 0.74mmol) and Na3PO4·12H2O (423mg, 1.11mmol) in acetone (10mL) was added CS2 (94μL, 1.55mmol). After 0.5h stirring, the corresponding compounds 7a-7j were added. About 1-2h later, the solvent was removed under vacuum, the resulting residue was dissolved with DCM and then washed with brine. The organic layer was dried over MgSO4 and evacuated under vacuum to give the residue, which was then subject to column chromatography, affording the corresponding 8a-j.
2-(4-nitrophenoxy)ethyl (S)-(2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
53%
General procedure: Compounds 7a-7j were prepared following the literature reported methods in 54-83% yields [29]. To a solution of compound 3 (102mg, 0.74mmol) and Na3PO4·12H2O (423mg, 1.11mmol) in acetone (10mL) was added CS2 (94μL, 1.55mmol). After 0.5h stirring, the corresponding compounds 7a-7j were added. About 1-2h later, the solvent was removed under vacuum, the resulting residue was dissolved with DCM and then washed with brine. The organic layer was dried over MgSO4 and evacuated under vacuum to give the residue, which was then subject to column chromatography, affording the corresponding 8a-j.
2-(naphthalen-1-yloxy)ethyl (S)-(2-oxotetrahydrofuran-3-yl)carbamodithioate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
66%
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate dodecahydrate In acetone for 0.5h;
Stage #2: 1-(2-bromoethoxy)naphthalene In acetone
4.7 General procedure for the synthesis of compounds 8a-j
General procedure: Compounds 7a-7j were prepared following the literature reported methods in 54-83% yields [29]. To a solution of compound 3 (102mg, 0.74mmol) and Na3PO4·12H2O (423mg, 1.11mmol) in acetone (10mL) was added CS2 (94μL, 1.55mmol). After 0.5h stirring, the corresponding compounds 7a-7j were added. About 1-2h later, the solvent was removed under vacuum, the resulting residue was dissolved with DCM and then washed with brine. The organic layer was dried over MgSO4 and evacuated under vacuum to give the residue, which was then subject to column chromatography, affording the corresponding 8a-j.
(S)-4-chloro-2-aminobutanoic acid ethyl ester hydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
80.6%
Stage #1: ethanol; L-homoserine lactone hydrochloride With sulfuric acid for 20h; Reflux;
Stage #2: With thionyl chloride at 10 - 50℃; for 6h;
6
120 g of (S)-2-aminobutyrolactone hydrochloride (0.87 mol) was added to 1200 ml of absolute ethanol. Add 20 ml of sulfuric acid dropwise, After the addition is completed, the mixture is heated to reflux. And after about 20 minutes, the material solids were all dissolved. Continue the reflux reaction for 20 hours. The HPLC was traced to less than 1% of the starting material and the reaction was stopped. The reaction liquid was cooled to 10 ° C in ice water, and the temperature was controlled at 10-20 ° C.200 g (1.65 mol) of thionyl chloride was added dropwise over about 1 hour. After the addition, the mixture was stirred for 2 hours, and slowly heated to 50 ° C. Stirring was continued for 4 hours, and the material was monitored by HPLC to be less than 1.5%, and the reaction was stopped.
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate In acetone at 20℃; for 2h;
Stage #2: 1-chloroacetyl-3-(3-chloro-phenyl)-urea In acetone at 20℃; for 6h;
General Procedure for the Synthesis of Compounds 11a-i
General procedure: A solution of compound 4 (150 mg, 1.09 mmol) and Na3PO4·12H2O (620 mg, 1.63 mmol) in acetone was added to CS2 (160 μL) and stirred for 2 h at room temperature. The resulting solution was added to compounds 10a-i (1.20 mmol) to react for 6 h at room temperature. The solvent was removed under vacuum, extracted with ethyl acetate and water. The organic layer was dried over MgSO4 and evaporated under vacuum to give the residue, which was then purified by column chromatography, affording a white solid (11a-i). 2-(3-(3-Chlorophenyl)ureido)-2-oxoethyl(S)-(2-oxotetrahydrofuran-3-yl)carbamodithi-oate 11a White solid (TLC: petroleum ether : EtOAc 1 : 1), (yield 44%), mp 140-142°C; 1H-NMR (400 MHz, DMSO-d6) δ: 11.04 (s, 1H, -CO-NH-CO-), 10.51 (d, J = 7.9 Hz, 1H, -CS-NH-), 10.40 (s, 1H, Ar-NH-CO-), 7.79 (t, J = 1.9 Hz, 1H, Ar-H), 7.41 (d, J = 8.9 Hz, 1H, Ar-H), 7.34 (t, J = 8.0 Hz, 1H, Ar-H), 7.15 (dd, J = 7.8, 0.8 Hz, 1H, Ar-H), 5.50 (dt, J = 10.9, 8.6 Hz, 1H, -O-CH2-), 4.40 (t, J = 8.4 Hz, 1H, -CH-NH-), 4.33-4.27 (m, 3H, -O-CH2-, -CH2-S-), 2.59-2.55 (m, 1H, -O-CH2-CH2-CH-), 2.22 (m, 1H, -O-CH2-CH2-CH-). 13C-NMR (100 MHz, DMSOd6) δ: 197.2, 173.5, 170.0, 150.4, 136.5, 128.8, 127.4, 121.4, 65.5, 54.7, 30.7, 27.9. HRESI-MS m/z calcd for [M + Na]+ C14H14ClN3O4S2: 410.0006. Found: 409.9986.
(S)-N-((2-methoxyphenyl)carbamoyl)-2-((2-oxotetrahydrofuran-3-yl)amino)acetamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
37%
With sodium hydrogencarbonate In acetonitrile at 75℃; for 8h;
General Procedure for the Synthesis of Compounds 12a-c
General procedure: In a 25 mL round bottom flask, the solution of compound 4 (150 mg, 1.09 mmol) in acetonitrile was added to corresponding compounds 13a-c (1.20 mmol) and NaHCO3 (230 mg, 2.74 mmol). After stirring for 8 h at 75°C, the solvent was removed under vacuum, extracted with ethyl acetate and water. The organic layer was dried over MgSO4 and evaporated under vacuum to give a residue, which was then subject to column chromatography, affording a white solid (12a-c).
(S)-N-((4-methylphenyl)carbamoyl)-2-((2-oxotetrahydrofuran-3-yl)amino)acetamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
56%
With sodium hydrogencarbonate In acetonitrile at 75℃; for 8h;
General Procedure for the Synthesis of Compounds 12a-c
General procedure: In a 25 mL round bottom flask, the solution of compound 4 (150 mg, 1.09 mmol) in acetonitrile was added to corresponding compounds 13a-c (1.20 mmol) and NaHCO3 (230 mg, 2.74 mmol). After stirring for 8 h at 75°C, the solvent was removed under vacuum, extracted with ethyl acetate and water. The organic layer was dried over MgSO4 and evaporated under vacuum to give a residue, which was then subject to column chromatography, affording a white solid (12a-c).
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate In acetone at 20℃; for 2h;
Stage #2: 1-chloroacetyl-3-<i>p</i>-tolyl-urea In acetone at 20℃; for 6h;
General Procedure for the Synthesis of Compounds 11a-i
General procedure: A solution of compound 4 (150 mg, 1.09 mmol) and Na3PO4·12H2O (620 mg, 1.63 mmol) in acetone was added to CS2 (160 μL) and stirred for 2 h at room temperature. The resulting solution was added to compounds 10a-i (1.20 mmol) to react for 6 h at room temperature. The solvent was removed under vacuum, extracted with ethyl acetate and water. The organic layer was dried over MgSO4 and evaporated under vacuum to give the residue, which was then purified by column chromatography, affording a white solid (11a-i).
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate In acetone at 20℃; for 2h;
Stage #2: 2-chloro-N-((4-methoxyphenyl)carbamoyl)acetamide In acetone at 20℃; for 6h;
General Procedure for the Synthesis of Compounds 11a-i
General procedure: A solution of compound 4 (150 mg, 1.09 mmol) and Na3PO4·12H2O (620 mg, 1.63 mmol) in acetone was added to CS2 (160 μL) and stirred for 2 h at room temperature. The resulting solution was added to compounds 10a-i (1.20 mmol) to react for 6 h at room temperature. The solvent was removed under vacuum, extracted with ethyl acetate and water. The organic layer was dried over MgSO4 and evaporated under vacuum to give the residue, which was then purified by column chromatography, affording a white solid (11a-i).
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate In acetone at 20℃; for 2h;
Stage #2: 1-chloroacetyl-3-(2-ethoxy-phenyl)-urea In acetone at 20℃; for 6h;
General Procedure for the Synthesis of Compounds 11a-i
General procedure: A solution of compound 4 (150 mg, 1.09 mmol) and Na3PO4·12H2O (620 mg, 1.63 mmol) in acetone was added to CS2 (160 μL) and stirred for 2 h at room temperature. The resulting solution was added to compounds 10a-i (1.20 mmol) to react for 6 h at room temperature. The solvent was removed under vacuum, extracted with ethyl acetate and water. The organic layer was dried over MgSO4 and evaporated under vacuum to give the residue, which was then purified by column chromatography, affording a white solid (11a-i).
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate In acetone at 20℃; for 2h;
Stage #2: N-Chloracetyl-N'-naphthyl-(1)-harnstoff In acetone at 20℃; for 6h;
General Procedure for the Synthesis of Compounds 11a-i
General procedure: A solution of compound 4 (150 mg, 1.09 mmol) and Na3PO4·12H2O (620 mg, 1.63 mmol) in acetone was added to CS2 (160 μL) and stirred for 2 h at room temperature. The resulting solution was added to compounds 10a-i (1.20 mmol) to react for 6 h at room temperature. The solvent was removed under vacuum, extracted with ethyl acetate and water. The organic layer was dried over MgSO4 and evaporated under vacuum to give the residue, which was then purified by column chromatography, affording a white solid (11a-i).
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate In acetone at 20℃; for 2h;
Stage #2: C12H15ClN2O2 In acetone at 20℃; for 6h;
General Procedure for the Synthesis of Compounds 11a-i
General procedure: A solution of compound 4 (150 mg, 1.09 mmol) and Na3PO4·12H2O (620 mg, 1.63 mmol) in acetone was added to CS2 (160 μL) and stirred for 2 h at room temperature. The resulting solution was added to compounds 10a-i (1.20 mmol) to react for 6 h at room temperature. The solvent was removed under vacuum, extracted with ethyl acetate and water. The organic layer was dried over MgSO4 and evaporated under vacuum to give the residue, which was then purified by column chromatography, affording a white solid (11a-i).
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate In acetone at 20℃; for 2h;
Stage #2: 1-(2-Chloro-acetyl)-3-o-tolyl-urea In acetone at 20℃; for 6h;
General Procedure for the Synthesis of Compounds 11a-i
General procedure: A solution of compound 4 (150 mg, 1.09 mmol) and Na3PO4·12H2O (620 mg, 1.63 mmol) in acetone was added to CS2 (160 μL) and stirred for 2 h at room temperature. The resulting solution was added to compounds 10a-i (1.20 mmol) to react for 6 h at room temperature. The solvent was removed under vacuum, extracted with ethyl acetate and water. The organic layer was dried over MgSO4 and evaporated under vacuum to give the residue, which was then purified by column chromatography, affording a white solid (11a-i).
Stage #1: carbon disulfide; L-homoserine lactone hydrochloride With sodium phosphate In acetone at 20℃; for 2h;
Stage #2: 1-chloroacetyl-3-<i>m</i>-tolyl-urea In acetone at 20℃; for 6h;
General Procedure for the Synthesis of Compounds 11a-i
General procedure: A solution of compound 4 (150 mg, 1.09 mmol) and Na3PO4·12H2O (620 mg, 1.63 mmol) in acetone was added to CS2 (160 μL) and stirred for 2 h at room temperature. The resulting solution was added to compounds 10a-i (1.20 mmol) to react for 6 h at room temperature. The solvent was removed under vacuum, extracted with ethyl acetate and water. The organic layer was dried over MgSO4 and evaporated under vacuum to give the residue, which was then purified by column chromatography, affording a white solid (11a-i).
3-chloro-N-((3S)2-carbonyltetrahydrofuran-3-yl)propanamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
64%
With sodium hydrogencarbonate In dichloromethane; water at 20℃; for 7h;
General Procedure for the Synthesis of Compounds 5 and 6
Compounds 5 and 6 were efficiently synthesized from L-homoserine lactone hydrochloride 4 following our previously reported methods.39) The acyl group of 5 and 6 was introduced by condensation with chloroacetyl chloride and 3-chloropropionyl chloride, respectively.
N-(2-oxotetrahydrofuran-3-yl)-4-(phenylsulfonyl)butanamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
36%
With (benzotriazo-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate; N-ethyl-N,N-diisopropylamine In N,N-dimethyl-formamide at 20℃;
3-nitro-N-(3-oxo-3-((2-oxotetrahydrofuran-3-yl)amino)propyl)benzamide 6b:
General procedure: 3-aminodihydrofuran-2(3H)-one hydrochloride 38 (0.02 g, 0.20 mmol), BOP (0.09 g, 0.20 mmol), and N,N-diisopropylethylamine (0.080 mL) were added to a solution of 40 (0.05 g, 0.20 mmol) in DMF (6.00 mL). The reaction mixture was stirred at room temperature for 16 h. Upon completion of the reaction it was then air dried and diluted with H2O (20.00 mL) and extracted with DCM (2 x 10.00 mL). The white solid crashed out from DCM (0.01 g, 20.6%).
(S)-4-chloro-2-aminobutyric acid ethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
Stage #1: ethanol; L-homoserine lactone hydrochloride With thionyl chloride at 10 - 35℃; for 20.5h;
Stage #2: With sodium carbonate In water
1; 3-17 Example 1
Weigh 10g of the compound of formula (I) hydrochloride (L-homoserine lactone hydrochloride, ee value 99%, 137.56g / mol, 0.073mol) into the reaction vessel, add ethanol (46.07g / mol, 0.886mol) , 0.816g / mL) 50mL, the molar ratio of homoserine lactone hydrochloride to ethanol is 1: 12.1. The temperature of the system was reduced to 10 ° C, and 21.7 g of dichlorosulfoxide (118.97 g / mol, 0.182 mol) was slowly added dropwise. The molar ratio of L-homoserine lactone hydrochloride to dichlorosulfoxide was 1: 2.5. Maintain the temperature of the system at 10 ° C and stir the reaction for 30 min. The temperature was gradually raised to 35 ° C, and the reaction was stirred for 20 hours. During the process, bubbles continued to be generated. The progress of the reaction was monitored by LC-MS to terminate the reaction. Among them, the intermediate product (chlorohomoserine ethyl ester hydrochloride), ester impurities, ether impurities are all LC detection values. The temperature of the system was reduced to room temperature, the remaining dichlorosulfoxide and ethanol were distilled off under reduced pressure, and the solid residue was slurried with a mixed solvent of 30 mL of n-hexane / ethyl acetate (the volume ratio of n-hexane and ethyl acetate was 2: 1), and filtered And drying, to obtain chlorohomoserine ethyl ester hydrochloride (202.08g / mol, 0.0657mol) 13.69g, HPLC purity 97%,The yield based on the reactant L-homoserine lactone hydrochloride was 90%. The chlorohomoserine ethyl ester hydrochloride solid was reacted with saturated sodium carbonate solution, the system pH was adjusted to 7-8, ethyl acetate was added for extraction, a total of 3 extractions, and the amount of ethyl acetate used in the 3 extractions was 30 mL , 10mL and 10mL. The organic phase was collected and concentrated to obtain 10.30 g of ethyl chlorohomoserine compound (165.62 g / mol, 0.0591 mol) as the oily target product of formula (II), HPLC purity 95%, ee value 99%,The yield based on the intermediate product chlorohomoserine ethyl ester hydrochloride was 90%.
(S)-N-(4-cinnamoylphenyl)-2-((2-oxotetrahydrofuran-3-yl)amino)acetamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
61%
With potassium carbonate; potassium iodide In acetonitrile for 6h; Reflux;
Preparation of analogues (VIIIa)-(VIIIm). Generalapproach.
General procedure: To a solution of corresponding chlorinatedcompound (VIIa)-(VIIm) (0.45 mmol) and homoserinelactone hydrochloride (62 mg, 0.45 mmol) dissolvedin acetonitrile (10 mL), K2CO3 (187mg,1.35 mmol) and KI (15 mg, 0.09 mmol) were added.The reaction mixture was refluxed for 6 h. Upon completion(monitored by TLC), the precipitated solidswas filtered off. Removal of the solvent in vacuo gave aresidue that was purified with column chromatographyto afford analogues (VIIIa)-(VIIIm) as solids.
(S)-N-(4-(3-(4-bromophenyl)acryloyl)phenyl)-2-((2-oxotetrahydrofuran-3-yl)amino)acetamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
74%
With potassium carbonate; potassium iodide In acetonitrile for 6h; Reflux;
Preparation of analogues (VIIIa)-(VIIIm). Generalapproach.
General procedure: To a solution of corresponding chlorinatedcompound (VIIa)-(VIIm) (0.45 mmol) and homoserinelactone hydrochloride (62 mg, 0.45 mmol) dissolvedin acetonitrile (10 mL), K2CO3 (187mg,1.35 mmol) and KI (15 mg, 0.09 mmol) were added.The reaction mixture was refluxed for 6 h. Upon completion(monitored by TLC), the precipitated solidswas filtered off. Removal of the solvent in vacuo gave aresidue that was purified with column chromatographyto afford analogues (VIIIa)-(VIIIm) as solids.
(S)-N-(4-(3-([1,1'-biphenyl]-4-yl)acryloyl)phenyl)-2-((2-oxotetrahydrofuran-3-yl)amino)acetamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
55%
With potassium carbonate; potassium iodide In acetonitrile for 6h; Reflux;
Preparation of analogues (VIIIa)-(VIIIm). Generalapproach.
General procedure: To a solution of corresponding chlorinatedcompound (VIIa)-(VIIm) (0.45 mmol) and homoserinelactone hydrochloride (62 mg, 0.45 mmol) dissolvedin acetonitrile (10 mL), K2CO3 (187mg,1.35 mmol) and KI (15 mg, 0.09 mmol) were added.The reaction mixture was refluxed for 6 h. Upon completion(monitored by TLC), the precipitated solidswas filtered off. Removal of the solvent in vacuo gave aresidue that was purified with column chromatographyto afford analogues (VIIIa)-(VIIIm) as solids.
(S)-N-(4-(3-(3,5-dichlorophenyl)acryloyl)phenyl)-2-((2-oxotetrahydrofuran-3-yl)amino)acetamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
64%
With potassium carbonate; potassium iodide In acetonitrile for 6h; Reflux;
Preparation of analogues (VIIIa)-(VIIIm). Generalapproach.
General procedure: To a solution of corresponding chlorinatedcompound (VIIa)-(VIIm) (0.45 mmol) and homoserinelactone hydrochloride (62 mg, 0.45 mmol) dissolvedin acetonitrile (10 mL), K2CO3 (187mg,1.35 mmol) and KI (15 mg, 0.09 mmol) were added.The reaction mixture was refluxed for 6 h. Upon completion(monitored by TLC), the precipitated solidswas filtered off. Removal of the solvent in vacuo gave aresidue that was purified with column chromatographyto afford analogues (VIIIa)-(VIIIm) as solids.
(S)-N-(4-(3-(4-bromo-2-fluorophenyl)acryloyl)phenyl)-2-((2-oxotetrahydrofuran-3-yl)amino)acetamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
57%
With potassium carbonate; potassium iodide In acetonitrile for 6h; Reflux;
Preparation of analogues (VIIIa)-(VIIIm). Generalapproach.
General procedure: To a solution of corresponding chlorinatedcompound (VIIa)-(VIIm) (0.45 mmol) and homoserinelactone hydrochloride (62 mg, 0.45 mmol) dissolvedin acetonitrile (10 mL), K2CO3 (187mg,1.35 mmol) and KI (15 mg, 0.09 mmol) were added.The reaction mixture was refluxed for 6 h. Upon completion(monitored by TLC), the precipitated solidswas filtered off. Removal of the solvent in vacuo gave aresidue that was purified with column chromatographyto afford analogues (VIIIa)-(VIIIm) as solids.
(S)-N-(4-(3-(3,4-dichlorophenyl)acryloyl)phenyl)-2-((2-oxotetrahydrofuran-3-yl)amino)acetamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
62%
With potassium carbonate; potassium iodide In acetonitrile for 6h; Reflux;
Preparation of analogues (VIIIa)-(VIIIm). Generalapproach.
General procedure: To a solution of corresponding chlorinatedcompound (VIIa)-(VIIm) (0.45 mmol) and homoserinelactone hydrochloride (62 mg, 0.45 mmol) dissolvedin acetonitrile (10 mL), K2CO3 (187mg,1.35 mmol) and KI (15 mg, 0.09 mmol) were added.The reaction mixture was refluxed for 6 h. Upon completion(monitored by TLC), the precipitated solidswas filtered off. Removal of the solvent in vacuo gave aresidue that was purified with column chromatographyto afford analogues (VIIIa)-(VIIIm) as solids.
(4S)-1,3-oxazinane-4-carboxylic acid hydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
86%
With hydrogenchloride In water at 20℃; for 17h;
L-Homoserine lactone hydrochloride 21 (2.55 g, 18.5 mmol) wasadded to a mixture of formaldehyde (37% in water, 9.22 mL,90.8 mmol) and hydrochloric acid solution (1.0 mol/L, 1.74 mL) inwater (44.0 mL). The reaction mixture was stirred at room temperaturefor 17 h. The solvent was then distilled under reduced pressure to yielda white solid, which was then suspended in EtOH (100 mL). Afterstirring for 15 min, the insoluble matter was filtered off, and the filtratewas concentrated under reduced pressure to yield approximately 25 mLof crude residue. After the addition of EtOAc (25 mL), the crude residuewas stored at -10 °C overnight. The precipitate was then collected byfiltration to yield (4S)-1,3-oxazinane-4-carboxylic acid hydrochloride asa colorless solid (2.70 g, 86% yield).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






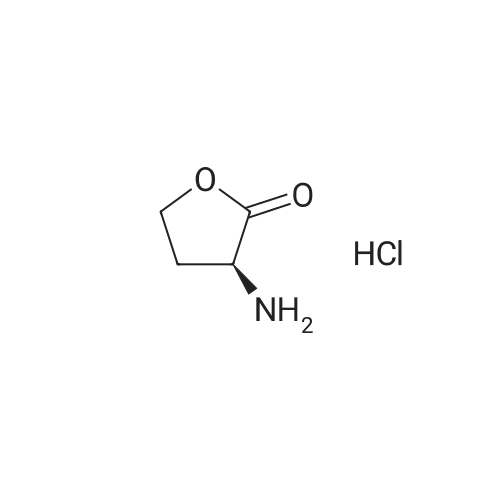

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping